Medicina cardiovascular
⭳ Abrir artículo (PDF)553.1 KBEste artículo es idéntico en la Edición 3/2000.
Contenido del artículo
XV CARDIOPATIAS CONGENITAS
- Etiología y consideraciones diagnósticas generales
- SOPLOS INOCENTES CONTRA SOPLOS SIGNIFICATIVOS
- ESTUDIOS DIAGNOSTICOS NO INVASIVOS
- DIAGNOSTICO DE ANOMALIAS CARDIACAS CONGENITAS ESPECIFICAS
- Anomalías cardiacas congénitas en adultos
- PERSISTENCIA DEL CONDUCTO ARTERIOSO
- DEFECTOS DEL TABIQUE INTERAURICULAR
- PERSISTENCIA DEL FORAMEN OVAL
- DEFECTOS DEL TABIQUE INTERVENTRICULAR
- ESTENOSIS VALVULAR PULMONAR
- CARDIOPATIA CONGENITA CIANOGENA
- COARTACION AORTICA
- ALTERACIONES DE LAS ARTERIAS CORONARIAS Y COMUNICACIONES ANORMALES
- Problemas generales en el tratamiento
- ENDOCARDITIS INFECCIOSA
- ABSCESO CEREBRAL
- ARRITMIAS CARDIACAS
- ALTERACIONES VALVULARES
- ALTERACIONES MIOCARDICAS
- HIPERTENSION ARTERIAL
- COMPLICACIONES RELACIONADAS CON PROTESIS
- COMPLICACIONES RELACIONADAS CON EL EMBARAZO
- POLICITEMIA Y FLEBOTOMIA
- COMPLICACIONES RELACIONADAS CON EL EJERCICIO Y LAS ACTIVIDADES ATLETICAS
- PROBLEMAS PSICOSOCIALES
- CONSEJO GENETICO
XV CARDIOPATIAS CONGENITAS
DR. E. WILLIAM HANCOCK
Etiología y consideraciones diagnósticas generales
Las cardiopatías congénitas ocurren en cerca del uno por ciento de los recién nacidos. En el pasado se pensaba que la mayoría de las cardiopatías congénitas eran de causa multifactorial, tanto ambiental como genética,1 y que menos del 10 por ciento eran predominántemente genéticas. Sin embargo, los estudios actuales sugieren que los defectos genéticos constituyen una proporción mucho mayor, y que los defectos de un gen son la causa en muchos casos.2 Un defecto de un solo gen puede causar formas aparentemente muy diversas de cardiopatía, y un solo tipo de malformación cardiaca puede deberse a mutaciones genéticas en diferentes sitios.
Otras formas de cardiopatía tienen también bases genéticas pero no se clasifican como congénitas por manifestarse en etapas más tardías de la vida. Entre estas se incluyen algunos tipos de cardiomiopatía, cardiopatía valvular, enfermedades de la aorta y arritmias cardiacas primarias.
Cerca del dos por ciento de los casos de cardiopatía congénita son sobre todo el resultado de factores externos o ambientales, que incluyen la infección por rubéola y la ingestión de sustancias como el litio y el alcohol.3
El diagnóstico resulta aparente durante la primera semana de vida en la mitad de los pacientes y antes de los cinco años en la mayoría de los casos restantes. Alrededor de 25,000 niños con cardiopatía congénita nacen en los Estados Unidos cada año, y cerca del 85 por ciento de estos sobrevive hasta la edad adulta, en ocasiones gracias al tratamiento quirúrgico realizado en la niñez.4 Como resultado, existe una población cada vez más grande de adultos jóvenes con cardiopatías congénitas. El manejo de la cardiopatía congénita en la edad adulta se ha convertido en una nueva subespecialidad, provocando el desarrollo de unidades integrales de tratamiento ambulatorio dedicadas al tratamiento a largo plazo de estos pacientes en instituciones médicas generales.5,6
SOPLOS INOCENTES CONTRA SOPLOS SIGNIFICATIVOS
Los adolescentes y adultos jóvenes a menudo presentan soplos sistólicos fisiológicos que reflejan una alta velocidad de expulsión normal a través de las válvulas aórtica o pulmonar. Los soplos de expulsión fisiológicos generalmente son de intensidad grado 1 o 2, de duración relativamente corta, y no se trasmiten ampliamente ni se acompañan de frémito
Los soplos sistólicos patológicos debidos a cardiopatías congénitas casi siempre se acompañan de fenómenos ausculatorios, como desdoblamiento anormal del segundo ruido o un chasquido sistólico de expulsión temprano. El electrocardiograma es un instrumento importante en la evaluación de las cardiopatías congénitas porque la mayoría de las lesiones causan sobrecargas hemodinámicas que se reflejan por hipertrofia auricular o ventricular. La radiografía de tórax también es importante porque revela aumento o disminución anormal en el flujo sanguíneo pulmonar.
La presencia de un soplo sistólico relativamente leve en un paciente asintomático que no tiene alteraciones en el ECG o en la radiografía de tórax rara vez indica una cardiopatía congénita importante.
ESTUDIOS DIAGNOSTICOS NO INVASIVOS
La ecocardiografía tiene gran valor en la evaluación de las anomalías cardiacas congénitas y debe ser el primer estudio diagnóstico especializado si la historia clínica, el examen físico, la radiografía de tórax y el electrocardiograma sugieren la presencia de cardiopatía congénita. El ecocardiograma proporciona información sobre la anatomía cardiaca, el tamaño de las cavidades, las conexiones de los grandes vasos, anomalías de las válvulas y obstrucciones subvalvulares. Las imágenes ecocardiográficas se reforzan con inyecciones intravenosas de solución salina a través de una vena periférica del brazo; dichas inyecciones producen burbujas microscópicas que proveen contraste y permiten la visualización de cortocircuitos cardiacos, de izquierda a derecha y de derecha a izquierda.
El estudio con ultrasonido Doppler es útil para detectar defectos septales y para evaluar la cantidad de sangre que se desvía a través de un defecto. El tamaño del cortocircuito a través de un defecto septal también puede evaluarse con este método al comparar la velocidad de flujo a través de la aorta y a través de la pulmonar. La ecocardiografía transesofágica es de especial valor para estudiar defectos septales, aunque también permite observar otras lesiones en forma exacta.8 Los estudios de ultrasonido Doppler son útiles también para evaluar estenosis e insuficiencia valvulares.
La tomografía computada y la imagen por resonancia magnética proporcionan una buena representación de las anomalías anatómicas de la cardiopatía congénita y ofrece ventajas sobre la ecocardiografía para demostrar anomalías que afecten los grandes vasos. La IRM es de especial utilidad en adultos con cardiopatía congénita, en los que la ecocardiografia puede no ser satisfactoria por interferencia de la pared torácica y de los pulmones. Comparado con los adultos, los ecocardiogramas dan mejor resultado en los niños porque su pared torácica y pulmones son de menor tamaño, además de que los niños no toleran con facilidad el confinamiento necesario para la IRM. Este estudio ofrece información sobre gradientes valvulares, velocidad de flujo y detalles anatómicos.9 La ecocardiografía y la IRM son investigaciones complementarias, cada una con ventajas particulares en situaciones específicas.10
DIAGNOSTICO DE ANOMALIAS CARDIACAS CONGENITAS ESPECIFICAS
El análisis cuidadoso del examen físico, la radiografía de tórax y el electrocardiograma generalmente permitirán un diagnóstico preliminar preciso que indicará el uso selectivo de estudios concluyentes no invasivos e invasivos.
El cateterismo cardiaco y la angiografía selectiva son las técnicas diagnósticas más concluyentes disponibles en la actualidad en pacientes con cardiopatía congénita. No obstante, los estudios no invasivos a menudo ofrecen información equivalente a la obtenida a partir del cateterismo cardiaco y suficiente para el plan quirúrgico.11
La cirugía paliativa y curativa es el tipo de tratamiento más empleado para las cardiopatías congénitas. Se calcula que se realizan alrededor de 20,000 cirugías de este tipo en los Estados Unidos cada año. Sin embargo, muchas lesiones pueden repararse por técnicas de cateterismo intervencionista. En algunos casos es claro que el cateterismo es el procedimiento de elección, mientras que en otros existe controversia al comparar con la cirugía. Existe un grupo de situaciones en las que el cateterismo intervencionista no ha demostrado del todo su utilidad, a pesar de ser técnicamente factible [ver tabla 1].11
Anomalías cardiacas congénitas en adultos
PERSISTENCIA DEL CONDUCTO ARTERIOSO
El conducto arterioso persistente suele diagnosticarse en la infancia o niñez temprana por el soplo continuo característico que produce. Por lo general está indicado su cierre por el riesgo de endocarditis infecciosa. Se ha logrado con bastante éxito el cierre no quirúrgico por medio de cateterismo cardiaco.
DEFECTOS DEL TABIQUE INTERAURICULAR
Los defectos del tabique interauricular son las lesiones congénitas de importancia más frecuentes en adultos. A menudo se diagnostican hasta la edad adulta, aún en la actualidad, ya que rara vez producen síntomas en la niñez y los signos físicos fácilmente se confunden con los datos encontrados en niños normales.
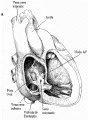
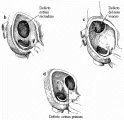
Desde el punto de vista anatómico, los defectos del tabique auricular se clasifican en tres tipos: ostium secundum, seno venoso, y ostium primum [ver figura 1]. Estos defectos del tabique auricular se acompañan de cortocircuitos de izquierda a derecha a nivel auricular, con sobrecarga de volumen en el ventrículo derecho. La sangre sobrecircula en forma crónica a través de los pulmones a niveles normales de presión intracardiaca. El aumento del flujo a través de la válvula pulmonar produce un soplo pulmonar sistólico de expulsión característico. La válvula pulmonar se cierra tardíamente por la reducción de la impedancia en el sistema arterial pulmonar, produciendo un desdoblamiento amplio del segundo ruido cardiaco, el otro dato característico de los defectos del tabique interauricular. El desdoblamiento se mantiene relativamente "fijo" con respecto a la respiración: los componentes aórtico y pulmonar permanecen audiblemente separados durante la espiración. La radiografía de tórax generalmente revela cardiomegalia y signos de aumento de la circulación pulmonar, como aumento del tronco pulmonar y de los trayectos vasculares pulmonares. La gravedad relativa de estas alteraciones es un reflejo del tamaño del corto circuito de izquierda a derecha. El electrocardiograma muestra un patrón característico para este defecto [ver figura 2].
Los defectos del tabique interauricular producen dos complicaciones
importantes; la hipertensión arterial pulmonar y la insuficiencia del
ventrículo derecho. La hipertensión arterial pulmonar se produce
por la elevada resistencia vascular pulmonar, y se desarrolla después de
la adolescencia en cerca del 15 por ciento de los pacientes. En la
mayoría de los casos más severos, ocurre arteriopatía
plexiforme irreversible, similar a la observada en el síndrome de
Eisenmenger o en la hipertensión pulmonar primaria. Como resultado de la
hipertensión pulmonar, el cortocircuito de izquierda a derecha primero
disminuye, luego se vuelve bidireccional y por último se invierte; se
desarrolla sobrecarga de presión en el ventrículo derecho, se
reduce el flujo sanguíneo pulmonar y el paciente presenta
cianosis.
La insuficiencia ventricular derecha se desarrolla como resultado de la sobrecarga de volumen prolongada y suele afectar a pacientes mayores de 40 años. Suele acompañarse de aleteo o fibrilación auricular, y a menudo se relaciona con insuficiencia tricuspídea. Finalmente se desarrolla un síndrome de insuficiencia cardiaca congestiva derecha e izquierda, y en este estadio puede ser difícil diferenciar clínicamente los defectos del tabique interauricular con otras enfermedades como cardiomiopatías y valvulopatía mitral.
Tratamiento
La corrección quirúrgica de los defectos del tabique interauricular es un procedimiento muy seguro y eficaz. Por lo tanto, la cirugía profiláctica está indicada en todos los enfermos en los que el flujo sanguíneo pulmonar sea dos veces mayor o más, que la circulación sistémica. Casi todos los pacientes en quienes se puede diagnosticar clínicamente esta alteración muestran por lo menos, este grado de cortocircuito de izquierda a derecha. La cirugía está contraindicada cuando la hipertensión pulmonar se acerca al nivel de la presión sistémica, porque en tales pacientes la mortalidad operatoria es alta y la resistencia vascular pulmonar elevada no disminuye después de la cirugía. Sin embargo, si la hipertensión pulmonar no es excesiva, la cirugía que se realiza cuando existe insuficiencia cardiaca congestiva da resultado satisfactorios, aún en pacientes ancianos.12 Es probable que el resultado de la cirugía sea bueno si la resistencia pulmonar total es menor de 15 U/m2. El cierre no quirúrgico usando cateterismo y un equipo semejante a una sombrilla ha sido exitoso en pacientes con defectos menores de 2 cm de diámetro, casi todos los pacientes han sido lactantes o niños pequeños. Este método está aun en etapa de investigación.13
Las variantes anatómicas asociadas con los defectos del tabique interauricular, como las venas pulmonares anómalas, son difíciles de diagnósticar clínicamente, pero generalmente se pueden evaluar y corregir sin dificultad en la cirugía. Sin embargo, la corrección quirúrgica de los defectos del tabique interauricular tipo ostium primum, también llamados defectos del colchón endocárdico, es difícil. Las hendiduras valvulares mitral y tricúspide relacionadas con este defecto a menudo requieren reparación, y la posición baja del defecto septal, que se extiende hasta el tabique interventricular, representa problemas adicionales. La reparación quirúrgica de este defecto se asocia con riesgo de bloqueo cardiaco en vista de que las suturas se pueden colocar en forma inadvertida en los tejidos especializados de conducción del tabique interventricular.
PERSISTENCIA DEL FORAMEN OVAL
La persistencia del foramen oval, que consiste en un pequeño defecto del tabique auricular, existe en el 20 a 30 porciento de la población adulta en general. Suele asociarse con una fosa oval más grande y mayor redundancia del colgajo de tejido que cubre el foramen del que se observa en la mayoría de las personas normales. En algunos casos la redundancia es tan grande como para crear la apariencia de un aneurisma del tabique auricular. La persistencia del foramen oval, el defecto septal auricular pequeño y el aneurisma del tabique auricular no causan alteraciones hemodinámicas detectables, pero permiten la ocurrencia de embolias paradójicas de derecha a izquierda en algunas circunstancias. Estas lesiones se detectan por ecocardio-grafía, con frecuencia en pacientes con eventos cerebro-vasculares sin otra causa evidente. En ocasiones está indicado el cierre del defecto, aunque no se han establecido lineamientos claros de tratamiento para esta condición.14,15
DEFECTOS DEL TABIQUE INTERVENTRICULAR
El defecto del tabique interventricular es la anomalía cardiaca congénita más frecuente en niños. Se ve raramente en adultos, porque los defectos sustanciales del tabique interventricular que no se corrigen quirúrgicamente se asocian con una mortalidad elevada. Además, la incidencia de cierre espontáneo de los defectos del tabique interventricular es relativamente alta; el cierre ocurre con particular frecuencia en la infancia, pero también a mayor edad.
Los defectos del tabique ventricular que aparecen en adultos como anomalías aisladas generalmente miden menos de 1 cm de diámetro. Como la apertura es bastante pequeña, se puede mantener la presión sistólica normal en el ventrículo derecho y en la arteria pulmonar. El cortocircuito de izquierda a derecha es demasiado pequeño para sobrecargar de manera importante a cualquiera de los ventrículos. Cuando se encuentran en adolescentes y adultos jóvenes, algunos de estos defectos probablemente tengan un cierre espontáneo. En la mayoría de los casos, el riesgo de hipertensión pulmonar y de insuficiencia cardiaca congestiva tardía es muy remoto.
Generalmente se efectúa el cierre quirúrgico para evitar la endocarditis infecciosa [ver adelante, Problemas generales en el tratamiento]. No se ha establecido la incidencia de esta complicación; de cualquier manera, la cirugía como medida profiláctica parece ser muy eficaz.
ESTENOSIS VALVULAR PULMONAR
La estenosis valvular pulmonar se puede identificar con facilidad con base en los signos físicos característicos. Este defecto se acompaña de un soplo holosistólico expulsivo pulmonar rudo, iniciado por un chasquido pulmonar de expulsión y seguido por un segundo ruido ampliamente desdoblado, con un componente pulmonar débil o inaudible. Generalmente se demuestra en la radiografía simple de tórax la dilatación posestenótica de la arteria pulmonar. El electrocardiograma a menudo ayudará a evaluar el grado de estenosis valvular, la evidencia de hipertrofia ventricular derecha y el signo de la P pulmonale indican crecimiento auricular derecho y reflejan obstrucción severa del flujo de salida del ventrículo derecho [ver figura 3]. Si también existe un defecto del tabique interauricular o un foramen oval asociado, el desarrollo de un cortocircuito invertido a nivel auricular puede causar cianosis; sin embargo, esta complicación es menos frecuente en adultos que en niños.
Es poco probable encontrar formas graves de estenosis valvular pulmonar en adultos, en vista de que la cirugía correctiva ha estado ampliamente disponible durante más de 25 años. La forma moderada, caracterizada por una presión sistólica del ventrículo derecho de entre 50 y 80 mm Hg, suele ser bien tolerada, y la indicación quirúrgica es dicutible en enfermos asintomáticos.16 Las formas leves, en las que la presión sistólica del ventrículo derecho es menor de 50 mm Hg, tienen poca importancia clínica.
El tratamiento de la estenosis valvular pulmonar mediante la dilatación con balón por cateterismo ha demostrado ser muy eficaz y ha remplazado en gran parte a la valvulotomía quirúrgica.17
CARDIOPATIA CONGENITA CIANOGENA
Tetralogía de Fallot
Los componentes anatómicos esenciales de la tetralogía de Fallot son el defecto del tabique interventricular y la estenosis pulmonar [ver figura 4]. Generalmente el grado de estenosis es suficiente para causar un cortocircuito de derecha a izquierda, cianosis y disminución del flujo sanguíneo pulmonar. La estenosis pulmonar puede ser valvular o infudibular; la forma infundibular generalmente predomina en adultos. Las otras dos características anatómicas comprendidas en la tetralogía de Fallot son aorta cabalgada e hipertrofia ventricular derecha. Existen variaciones en el grado de dextroposición de la aorta, pero este defecto anatómico raramente tiene importancia funcional.
La técnica quirúrgica totalmente correctiva suele realizarse en la infancia o niñez. Los pacientes que sobreviven hasta la edad adulta sin operación o con una operación paliativa de cortocircuito en que una arteria sistémica se anastomosa a la arteria pulmonar, pueden desarrollar una tolerancia satisfactoria al esfuerzo, cianosis mínima o nula y tener un pronóstico aparentemente bueno. Sin embargo, tales pacientes pueden desarrollar incapacidad progresiva como resultado del cierre gradual del cortocircuito y de la evolución de la estenosis pulmonar infundibular. Es más, si la anomalía no se repara pueden surgir complicaciones como endocarditis infecciosa, tromboembolia cerebral, y absceso cerebral [ver adelante, Problemas generales en el tratamiento]. La corrección total de la tetralogía de Fallot implica un riesgo de mortalidad operatoria menor del 5 por ciento, y los resultados tardíos generalmente son buenos, a pesar de que ocurra insuficiencia valvular pulmonar y menor contracción del ventrículo derecho en muchos pacientes. Los resultados a largo plazo son mejores si la cirugía se realiza cuando el paciente tiene menos de cinco años.18 La muerte súbita causada por arritmias es la causa más común de mortalidad tardía.
Otros defectos que producen cianosis
Además de la tetralogía de Fallot, las cardiopatías congénitas cianógenas en adultos incluyen el síndrome de Eisenmenger y varias malformaciones complejas como transposición de los grandes vasos, ventrículo único, ventrículo derecho de doble salida, tronco arterioso persistente, atresia pulmonar y atresia tricuspídea. En todas estas cardiopatías, los defectos asociados del tabique interauricular o interventricular permiten la supervivencia hasta la edad adulta. Muchas de estas anomalías se pueden corregir quirúrgicamente con la construcción de conductos, para crear nuevas vías de salida desde el ventrículo derecho hasta la arteria pulmonar (operación de Rastelli), la creación de derivaciones de la aurícula derecha a la arteria pulmonar (procedimiento de Fontan) o el intercambio directo de los grandes vasos. La enfermedad vascular pulmonar es la principal limitante para el éxito en estos casos.
El término síndrome de Eisenmenger se aplica en sentido amplio a aquellos defectos en los que la enfermedad de los vasos pulmonares ha progresado al punto en que la sangre se desvía en forma predominante de derecha a izquierda.19 Este síndrome comprende la categoría principal de cardiopatías congénitas cianógenas que no se pueden corregir quirúrgicamente. El tratamiento de la enfermedad vascular pulmonar es el principal problema no resuelto en las cardiopatías congénitas. La corrección quirúrgica antes de que se desarrolle hipertensión pulmonar grave es el procedimiento más eficaz. La operación debe realizarse antes de que el paciente cumpla dos años de edad.
Desde 1981 se ha usado con éxito el transplante de corazón y pulmón para el tratamiento de cardiopatías congénitas que de otra manera son inoperables. Aunque el trasplante de corazón aislado puede desempeñar algún papel en los enfermos con defectos anatómicos intracardiacos para quienes no está indicada la cirugía correctiva, tal situación es relativamente rara. La cardiopatía congénita inoperable más frecuente se debe a la presencia de enfermedad severa de los vasos pulmonares, para la cual no hay tratamiento eficaz, además del transplante. La aplicación clínica del transplante de corazón y pulmones se ha visto limitada por la falta de donadores adecuados y por la aparición de bronquiolitis obliterativa en los receptores. Se considera que la bronquiolitis representa una manifestación de reacción de rechazo crónico. El trasplante de un solo pulmón, combinado con el cierre del defecto septal, es otra opción que debe considerarse en el tratamiento de las cardiopatías congénitas que se complican por hipertensión pulmonar severa.21
COARTACION AORTICA
La coartación aórtica suele producir obstrucción moderada al flujo sanguíneo, aunque ocasionalmente hay obstrucción completa; en algunos casos, llamados seudocoartación, la obstrucción es insignificante. La presión sistólica se eleva porque se está expulsando un volumen por latido normal hacia una aorta proximal con capacitancia disminuida. Existe un aumento brusco de la presión sistólica durante el ejercicio; esta elevación es mucho más notable en pacientes con coartación que en enfermos con hipertensión esencial con presiones arteriales en reposo parecidas. La disminución, el retardo o la ausencia de pulsos femorales orientan hacia el diagnóstico de coartación. La radiografía de tórax casi siempre revela un botón aórtico y una aorta torácica descendente anormales [ver figura 5]; ocasionalmente se pueden observar muescas en las costillas. La coartación puede observarse por medio de TC o IRM y el gradiente de presión puede calcularse con base en los estudios de ultrasonido Doppler. El gradiente de presión puede también determinarse en forma confiable comparando las presiones sistólicas obtenidas con un esfigmomanómetro y una sonda Doppler, que detecta la velocidad de flujo sanguíneo por el ultrasonido reflejado en la arteria braquial y la arteria tibial posterior. La diferencia de presión del brazo es de más de 35 mm Hg en reposo en comparación con la del tobillo en todos pacientes en los que la luz de la coartación es menor del 50 por ciento de lo normal.22
La reparación quirúrgica de la coartación aórtica
suele tener resultados muy satisfactorios y debe realizarse siempre que exista
hipertensión asociada. Ocasionalmente, cuando la presión
sistólica en reposo está en el límite, una
elevación extrema durante el ejercicio puede ser una indicación
de cirugía.
La dilatación con balón a través de cateterismo en ocasiones resulta eficaz en el tratamiento de la coartación de la aorta, aunque la aplicación clínica de este procedimiento se ha limitado por el desarrollo ocasional de aneurismas en el sitio de la dilatación. La dilatación con balón es más eficaz para tratar la recurrencia de la coartación en pacientes sometidos antes a tratamiento quirúrgico.23 El seguimiento a largo plazo después del tratamiento quirúrgico de la coartación muestra una incidencia significativa de hipertensión, en especial en los pacientes que tenían más de cinco a 10 años en el momento de la cirugía.24
ALTERACIONES DE LAS ARTERIAS CORONARIAS Y COMUNICACIONES ANORMALES
Otras anomalías que afectan a la aorta y sus ramas además de la cortación, raramente son importantes después de la niñez; sin embargo, algunas anomalías de las arterias coronarias son notables. El origen anómalo de la arteria coronaria izquierda a partir de la arteria pulmonar generalmente produce un síndrome de infarto del miocardio en niños, con insuficiencia cardiaca congestiva que conduce a la muerte en el primer año de vida. Sin embargo, algunos individuos con este defecto presentan datos mínimos o no presentan datos anormales durante la niñez y sobreviven hasta la edad adulta. En dichos pacientes se han desarrollado vasos colaterales prominentes entre las arterias coronarias derecha e izquierda y un cortocircuito de izquierda a derecha lleva sangre de la arteria coronaria derecha a través de la coronaria izquierda a la arteria pulmonar; este defecto se relaciona con lesión isquémica crónica del ventrículo izquierdo.
Otra malformación importante es el origen anómalo de la arteria coronaria izquierda a partir del seno de Valsalva derecho; en ocasiones esta alteración se asocia con angina de pecho, infarto del miocardio, y muerte súbita en adultos jóvenes.25
Problemas generales en el tratamiento
ENDOCARDITIS INFECCIOSA
La endocarditis infecciosa es una complicación más frecuente de las cardiopatías congénitas en adultos que en niños.26 La susceptibilidad a esta enfermedad varía mucho según la malformación cardiaca [ver tabla 2]. Los pacientes con válvula aórtica bicúspide y regurgitación aórtica asociada son especialmente susceptibles a la endocarditis infecciosa. Por otro lado, los pacientes con comunicación interauricular no lo son, probablemente porque esta anomalía no se relaciona con un flujo sanguíneo en chorro de alta velocidad. Alrededor de la mitad de los casos de endocarditis infecciosa que ocurre como complicación de lesiones congénitas se desarrollan en pacientes que han sido sometidos antes a cirugía cardiaca, incluyendo los que tienen válvulas y conductos protésicos. Al igual que los pacientes con cardiopatía reumática, los pacientes con lesiones congénitas que predispongan a endocarditis infecciosa deben recibir antibióticos profilácticos al someterse a procedimientos quirúrgicos y dentales.
El síndrome de endocarditis infecciosa derecha, que disemina la infección en los pulmones, se observa principalmente en pacientes con endocarditis de la válvula tricúspide que son adictos a drogas intravenosas, pero también ocurre en ocasiones en pacientes con cardiopatías congénitas. Los pacientes con comunicación inter-ventri-cular tienen mayor riesgo de desarrollar este trastorno.
La prevención de la endocarditis infecciosa es una razón suficiente para recomendar el cierre quirúrgico del conducto arterioso persistente, al menos en niños y adultos jóvenes, aún cuando el conducto sea muy pequeño. Sigue siendo controvertido si se debe llevar a cabo o no el cierre quirúrgico en pacientes con comunicación interventricular; sin embargo, en estos enfermos debe evitarse hasta donde sea posible la endocarditis infecciosa porque es probable que produzca complicaciones graves como insuficiencia aórtica.28
ABSCESO CEREBRAL
El desarrollo de abscesos cerebrales es un riesgo importante en pacientes con cardiopatías congénitas cianógenas. La mayor parte de estos casos de absceso cerebral ocurren en niños, aunque también se desarrollan en adultos. El inicio de un absceso cerebral a menudo se parece al de la enfermedad vascular cerebral, tanto clínicamente como en su aspecto en la TC. Se debe considerar el uso de antibióticos en el plan de tratamiento inicial cuando un paciente con cardiopatía congénita cianógena desarrolla lo que parece ser una enfermedad vascular cerebral. Los cambios seriados subsecuentes en los hallazgos clínicos y en la TC, mostrarán si existen un infarto isquémico o un absceso cerebral.
ARRITMIAS CARDIACAS
Las arritmias cardiacas no suelen ser una característica notable de las cardiopatías congénitas en niños, pero son más frecuentes en adultos. Las arritmias se relacionan con la cicatrización del miocardio y el daño específico a marcapasos y tejidos de conducción como resultado de las incisiones que se practican en el corazón, así como por los efectos a largo plazo de la sobrecarga hemodinámica. Ocasionalmente ocurren muertes súbitas cardiacas relacionadas con arritmias en pacientes con antecedente de cirugía previa por alguna cardiopatía congénita.28
Los procedimientos quirúrgicos practicados en la región del nodo auriculoventricular (AV) generalmente incluyen el cierre de un defecto del tabique interventricular como parte de la corrección de la tetralogía de Fallot. Dicha cirugía puede ocasionar un bloqueo AV que requiera colocación de marcapaso; en otros casos, la cirugía puede ocasionar bloqueo de rama, puede ocurrir la combinación de bloqueo de rama derecha y desviación del eje a la izquierda (bloqueo del fascículo izquierdo anterior) y ser precursora de un bloqueo AV completo. Estos pacientes requieren control cuidadoso con atención a los hallazgos clínicos o electrocardiográficos que puedan indicar progresión del bloqueo AV. En estas circunstancias también pueden ocurrir arritmias ventriculares como complicación tardía. En algunos casos los estudios de electrofisiología intracardiaca pueden revelar la presencia de taquicardia ventricular inducible que no se detectó por observación clínica, electrocadiografía en ejercicio o vigilancia electrocardiográfica ambulatoria.
ALTERACIONES VALVULARES
Las técnicas quirúrgicas son eficaces para cerrar defectos septales severos y aliviar estenosis valvulares o subvalvulares graves. Sin embargo, las alteraciones valvulares menos severas a menudo quedan sin corregir después de la cirugía, causando estenosis recurrente de la válvula aórtica o insuficiencia en cualquiera de las válvulas cardiacas como complicación tardía del tratamiento de pacientes con alteraciones valvulares.
ALTERACIONES MIOCARDICAS
En niños con cardiopatías congénitas, el miocardio suele ser normal y los defectos hemodinámicos se pueden analizar esencialmente en términos de sobrecarga de presión y de volumen, o ambas. Sin embargo, en adultos, sobre todo los que se han sometido a cirugía correctiva, a menudo se encuentra deterioro miocárdico. Las incisiones en los ventrículos y otras secuelas quirúrgicas participan en el desarrollo de las alteraciones miocárdicas. En estos pacientes, la dilatación, la hipertrofia y el deterioro funcional como resultado de la sobrecarga hemodinámica prolongada también producen deterioro miocárdico. Puede existir además disfunción miocárdica por perfusión de las arterias coronarias con sangre con menor saturación de oxígeno.
HIPERTENSION ARTERIAL
Los pacientes que han sido sometidos a corrección quirúrgica por coartación aórtica desarrollan con mucha frecuencia hipertensión arterial subsecuente. La frecuencia es particularmente elevada cuando la cirugía se lleva a cabo durante la adolescencia o edad adulta. La reparación quirúrgica de la coartación solo puede ser parcialmente satisfactoria, y aún después de cirugía correctiva, puede recurrir la coartación. Estos pacientes necesitan evaluación cuidadosa en busca de posible coartación residual y de hipertensión esencial.24,29
Puede ocurrir disección de la aorta como complicación tardía de la hipertensión en pacientes con coartación aórtica. También ocurre disección aórtica en pacientes que han sido intervenidos por coartación o valvulopatía aórtica, sin hipertensión arterial.
COMPLICACIONES RELACIONADAS CON PROTESIS
Con frecuencia se usan válvulas artificiales y parches protésicos en la reparación quirúrgica de defectos cardiacos congénitos más complejos. Estos procedimientos a menudo requieren la colocación de válvulas artificiales en cavidades derechas del corazón. Las válvulas porcinas son preferibles a las prótesis mecánicas para este propósito, incluso en los niños.30
Los problemas que ocurren en pacientes con válvulas protésicas son similares a los que se presentan en adultos con valvulopatías. Las complicaciones relacionadas con las válvulas protésicas son mayores en los pacientes jóvenes por las dificultades en la anticoagulación, la posibilidad de embarazo y la tendencia de los sustitutos valvulares porcinos de sufrir degeneración cálcica acelerada en estos pacientes. Aunque los parches y conductos protésicos se asocian complicaciones que las válvulas protésicas, dichas prótesis también pueden causar complicaciones tardías.
COMPLICACIONES RELACIONADAS CON EL EMBARAZO
El embarazo es bien tolerado en la mayoría de las pacientes con cardiopatías congénitas que presentan síntomas mínimos antes de embarazarse. Una excepción lo constituyen las pacientes con síndrome de Marfán, una enfermedad que predispone a la disección de la aorta durante el embarazo. El riesgo de complicaciones por el embarazo es más elevado en mujeres con síndrome de Eisenmenger; en estos casos la mortalidad materna puede ser mayor del 50 porciento [ver tabla 3].31,32
POLICITEMIA Y FLEBOTOMIA
Los pacientes con cardiopatías congénitas cianógenas, sobre todo aquellos con síndrome de Eisenmenger, típicamente desarrollan policitemia secundaria. Esta alteración es la respuesta fisiológica a la oxigenación subnormal de la sangre, en ocasiones capaz de producir sintomatología. Puede aparecer cefalea y otros síntomas congestivos cuando el volumen sanguíneo es excesivo, y las complicaciones tromboembólicas representan un riesgo cuando el hematocrito alcanza el 65 a 70 por ciento, nivel al cual la viscosidad de la sangre se eleva considerablemente. La flebotomía puede estar indicada en pacientes sintomáticos con hematocrito en el rango de 50 a 65 por ciento o como medida profiláctica a niveles superiores. Sin embargo, se debe tomar en cuenta la posibilidad de que la flebotomía empeore la disnea en ejercicio. Los pacientes con policitemia secundaria pueden tener deficiencia de hierro relativa; como consecuencia, la elevación en el hematocrito es mayor que la elevación en la concentración de hemoglobina.33 En estos casos se puede indicar el tratamiento con hierro y flebotomía simultáneos para disminuir el hematocrito sin afectar el nivel de hemoglobina. De esta forma se disminuye el volumen y la viscosidad de la sangre sin alterar la capacidad de transporte de oxígeno.
Los pacientes con policitemia secundaria pueden tener defectos de coagulación causados por deficiencia de los factores dependientes de la vitamina K sintetizados en el hígado (factores II, VII y X). El tratamiento con vitamina K es ineficaz, y su corrección requiere administración de los factores faltantes. Los defectos en la agregación plaquetaria pueden producir una tendencia hemorrágica, que se corrige disminuyendo el volumen de eritrocitos. Estos defectos de la coagulación son importantes sobre todo como posible fuente de complicaciones hemorrágicas después de la cirugía cardiaca.
Las concentraciones sanguíneas de ácido úrico con frecuencia están elevadas en pacientes con cardiopatías congénitas cianógenas, particularmente en hombres adultos. Aunque ni la gota ni las nefropatías ocurren con frecuencia, puede ser prudente usar alopurinol en forma profiláctica en algunos pacientes.
COMPLICACIONES RELACIONADAS CON EL EJERCICIO Y LAS ACTIVIDADES ATLETICAS
Los pacientes que presentan cardiopatías congénitas, sin tomar en consideración si se han sometido a cirugía correctiva temprana, pueden estar en riesgo de sufrir complicaciones inducidas por el ejercicio. Las normas o guías para la orientación de adolescentes y adultos jóvenes deben tomar en cuenta el tipo de cardiopatía que presentan, así como la clase de ejercicio o actividad atlética que van a desarrollar. Deben considerarse los aspectos estáticos (i.e., isométricos) y dinámicos (i.e., isotónicos) del ejercicio para calcular la tensión a la que se somete el corazón. El riesgo de que aparezcan arritmias inducidas por el ejercicio, como la taquicardia ventricular, a menudo debe evaluarse por medio de prueba de esfuerzo realizada en banda sin fin o mediante vigilancia electrocardiográfica ambulatoria.34,35
Una conferencia del American College of Cardiology realizada en Bethesda describió los lineamientos específicos para la participación en competencias atléticas para 24 diagnósticos y categorías funcionales de cardiopatía congénita.34
PROBLEMAS PSICOSOCIALES
Los adultos jóvenes con cardiopatías congénitas pueden tener problemas psicosociales significativos.35 Su desarrollo educativo y social puede haberse limitado por la enfermedad y por protección paterna excesiva. Aunque el 70 por ciento de los pacientes tienen una capacidad funcional satisfactoria y solo el 10 por ciento sufre incapacidad importante, los pacientes con cardiopatía congénita con frecuencia tienen dificultades para encontrar empleo. Uno de los obstáculos para esto es la reserva de los patrones para proporcionar un seguro médico que cubra cardiopatías previas. Los servicios de orientación y apoyo pueden ser útiles para ayudar a resolver estos problemas.
CONSEJO GENETICO
Debido al mayor número de pacientes con cardiopatías congénitas que sobreviven hasta la edad adulta, ha surgido la necesidad de orientar sobre el riesgo de cardiopatías en la descendencia de estos pacientes. El riesgo varía según las diferentes malformaciones, pero en la mayoría de ellas está aumentado [ver tabla 4].4 La ecocardiografía fetal es útil para identificar las malformaciones intracardiacas importantes después de las 18 semanas de gestación.
Bibliografía
DR. E. WILLIAM HANCOCK
Etiología y consideraciones diagnósticas generales
Las cardiopatías congénitas ocurren en cerca del uno por ciento de los recién nacidos. En el pasado se pensaba que la mayoría de las cardiopatías congénitas eran de causa multifactorial, tanto ambiental como genética,1 y que menos del 10 por ciento eran predominántemente genéticas. Sin embargo, los estudios actuales sugieren que los defectos genéticos constituyen una proporción mucho mayor, y que los defectos de un gen son la causa en muchos casos.2 Un defecto de un solo gen puede causar formas aparentemente muy diversas de cardiopatía, y un solo tipo de malformación cardiaca puede deberse a mutaciones genéticas en diferentes sitios.
Otras formas de cardiopatía tienen también bases genéticas pero no se clasifican como congénitas por manifestarse en etapas más tardías de la vida. Entre estas se incluyen algunos tipos de cardiomiopatía, cardiopatía valvular, enfermedades de la aorta y arritmias cardiacas primarias.
Cerca del dos por ciento de los casos de cardiopatía congénita son sobre todo el resultado de factores externos o ambientales, que incluyen la infección por rubéola y la ingestión de sustancias como el litio y el alcohol.3
El diagnóstico resulta aparente durante la primera semana de vida en la mitad de los pacientes y antes de los cinco años en la mayoría de los casos restantes. Alrededor de 25,000 niños con cardiopatía congénita nacen en los Estados Unidos cada año, y cerca del 85 por ciento de estos sobrevive hasta la edad adulta, en ocasiones gracias al tratamiento quirúrgico realizado en la niñez.4 Como resultado, existe una población cada vez más grande de adultos jóvenes con cardiopatías congénitas. El manejo de la cardiopatía congénita en la edad adulta se ha convertido en una nueva subespecialidad, provocando el desarrollo de unidades integrales de tratamiento ambulatorio dedicadas al tratamiento a largo plazo de estos pacientes en instituciones médicas generales.5,6
SOPLOS INOCENTES CONTRA SOPLOS SIGNIFICATIVOS
Los adolescentes y adultos jóvenes a menudo presentan soplos sistólicos fisiológicos que reflejan una alta velocidad de expulsión normal a través de las válvulas aórtica o pulmonar. Los soplos de expulsión fisiológicos generalmente son de intensidad grado 1 o 2, de duración relativamente corta, y no se trasmiten ampliamente ni se acompañan de frémito
Los soplos sistólicos patológicos debidos a cardiopatías congénitas casi siempre se acompañan de fenómenos ausculatorios, como desdoblamiento anormal del segundo ruido o un chasquido sistólico de expulsión temprano. El electrocardiograma es un instrumento importante en la evaluación de las cardiopatías congénitas porque la mayoría de las lesiones causan sobrecargas hemodinámicas que se reflejan por hipertrofia auricular o ventricular. La radiografía de tórax también es importante porque revela aumento o disminución anormal en el flujo sanguíneo pulmonar.
La presencia de un soplo sistólico relativamente leve en un paciente asintomático que no tiene alteraciones en el ECG o en la radiografía de tórax rara vez indica una cardiopatía congénita importante.
ESTUDIOS DIAGNOSTICOS NO INVASIVOS
La ecocardiografía tiene gran valor en la evaluación de las anomalías cardiacas congénitas y debe ser el primer estudio diagnóstico especializado si la historia clínica, el examen físico, la radiografía de tórax y el electrocardiograma sugieren la presencia de cardiopatía congénita. El ecocardiograma proporciona información sobre la anatomía cardiaca, el tamaño de las cavidades, las conexiones de los grandes vasos, anomalías de las válvulas y obstrucciones subvalvulares. Las imágenes ecocardiográficas se reforzan con inyecciones intravenosas de solución salina a través de una vena periférica del brazo; dichas inyecciones producen burbujas microscópicas que proveen contraste y permiten la visualización de cortocircuitos cardiacos, de izquierda a derecha y de derecha a izquierda.
El estudio con ultrasonido Doppler es útil para detectar defectos septales y para evaluar la cantidad de sangre que se desvía a través de un defecto. El tamaño del cortocircuito a través de un defecto septal también puede evaluarse con este método al comparar la velocidad de flujo a través de la aorta y a través de la pulmonar. La ecocardiografía transesofágica es de especial valor para estudiar defectos septales, aunque también permite observar otras lesiones en forma exacta.8 Los estudios de ultrasonido Doppler son útiles también para evaluar estenosis e insuficiencia valvulares.
La tomografía computada y la imagen por resonancia magnética proporcionan una buena representación de las anomalías anatómicas de la cardiopatía congénita y ofrece ventajas sobre la ecocardiografía para demostrar anomalías que afecten los grandes vasos. La IRM es de especial utilidad en adultos con cardiopatía congénita, en los que la ecocardiografia puede no ser satisfactoria por interferencia de la pared torácica y de los pulmones. Comparado con los adultos, los ecocardiogramas dan mejor resultado en los niños porque su pared torácica y pulmones son de menor tamaño, además de que los niños no toleran con facilidad el confinamiento necesario para la IRM. Este estudio ofrece información sobre gradientes valvulares, velocidad de flujo y detalles anatómicos.9 La ecocardiografía y la IRM son investigaciones complementarias, cada una con ventajas particulares en situaciones específicas.10
DIAGNOSTICO DE ANOMALIAS CARDIACAS CONGENITAS ESPECIFICAS
El análisis cuidadoso del examen físico, la radiografía de tórax y el electrocardiograma generalmente permitirán un diagnóstico preliminar preciso que indicará el uso selectivo de estudios concluyentes no invasivos e invasivos.
El cateterismo cardiaco y la angiografía selectiva son las técnicas diagnósticas más concluyentes disponibles en la actualidad en pacientes con cardiopatía congénita. No obstante, los estudios no invasivos a menudo ofrecen información equivalente a la obtenida a partir del cateterismo cardiaco y suficiente para el plan quirúrgico.11
La cirugía paliativa y curativa es el tipo de tratamiento más empleado para las cardiopatías congénitas. Se calcula que se realizan alrededor de 20,000 cirugías de este tipo en los Estados Unidos cada año. Sin embargo, muchas lesiones pueden repararse por técnicas de cateterismo intervencionista. En algunos casos es claro que el cateterismo es el procedimiento de elección, mientras que en otros existe controversia al comparar con la cirugía. Existe un grupo de situaciones en las que el cateterismo intervencionista no ha demostrado del todo su utilidad, a pesar de ser técnicamente factible [ver tabla 1].11
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
Anomalías cardiacas congénitas en adultos
PERSISTENCIA DEL CONDUCTO ARTERIOSO
El conducto arterioso persistente suele diagnosticarse en la infancia o niñez temprana por el soplo continuo característico que produce. Por lo general está indicado su cierre por el riesgo de endocarditis infecciosa. Se ha logrado con bastante éxito el cierre no quirúrgico por medio de cateterismo cardiaco.
DEFECTOS DEL TABIQUE INTERAURICULAR
Los defectos del tabique interauricular son las lesiones congénitas de importancia más frecuentes en adultos. A menudo se diagnostican hasta la edad adulta, aún en la actualidad, ya que rara vez producen síntomas en la niñez y los signos físicos fácilmente se confunden con los datos encontrados en niños normales.
Desde el punto de vista anatómico, los defectos del tabique auricular se clasifican en tres tipos: ostium secundum, seno venoso, y ostium primum [ver figura 1]. Estos defectos del tabique auricular se acompañan de cortocircuitos de izquierda a derecha a nivel auricular, con sobrecarga de volumen en el ventrículo derecho. La sangre sobrecircula en forma crónica a través de los pulmones a niveles normales de presión intracardiaca. El aumento del flujo a través de la válvula pulmonar produce un soplo pulmonar sistólico de expulsión característico. La válvula pulmonar se cierra tardíamente por la reducción de la impedancia en el sistema arterial pulmonar, produciendo un desdoblamiento amplio del segundo ruido cardiaco, el otro dato característico de los defectos del tabique interauricular. El desdoblamiento se mantiene relativamente "fijo" con respecto a la respiración: los componentes aórtico y pulmonar permanecen audiblemente separados durante la espiración. La radiografía de tórax generalmente revela cardiomegalia y signos de aumento de la circulación pulmonar, como aumento del tronco pulmonar y de los trayectos vasculares pulmonares. La gravedad relativa de estas alteraciones es un reflejo del tamaño del corto circuito de izquierda a derecha. El electrocardiograma muestra un patrón característico para este defecto [ver figura 2].
|
|
|
|


La insuficiencia ventricular derecha se desarrolla como resultado de la sobrecarga de volumen prolongada y suele afectar a pacientes mayores de 40 años. Suele acompañarse de aleteo o fibrilación auricular, y a menudo se relaciona con insuficiencia tricuspídea. Finalmente se desarrolla un síndrome de insuficiencia cardiaca congestiva derecha e izquierda, y en este estadio puede ser difícil diferenciar clínicamente los defectos del tabique interauricular con otras enfermedades como cardiomiopatías y valvulopatía mitral.
Tratamiento
La corrección quirúrgica de los defectos del tabique interauricular es un procedimiento muy seguro y eficaz. Por lo tanto, la cirugía profiláctica está indicada en todos los enfermos en los que el flujo sanguíneo pulmonar sea dos veces mayor o más, que la circulación sistémica. Casi todos los pacientes en quienes se puede diagnosticar clínicamente esta alteración muestran por lo menos, este grado de cortocircuito de izquierda a derecha. La cirugía está contraindicada cuando la hipertensión pulmonar se acerca al nivel de la presión sistémica, porque en tales pacientes la mortalidad operatoria es alta y la resistencia vascular pulmonar elevada no disminuye después de la cirugía. Sin embargo, si la hipertensión pulmonar no es excesiva, la cirugía que se realiza cuando existe insuficiencia cardiaca congestiva da resultado satisfactorios, aún en pacientes ancianos.12 Es probable que el resultado de la cirugía sea bueno si la resistencia pulmonar total es menor de 15 U/m2. El cierre no quirúrgico usando cateterismo y un equipo semejante a una sombrilla ha sido exitoso en pacientes con defectos menores de 2 cm de diámetro, casi todos los pacientes han sido lactantes o niños pequeños. Este método está aun en etapa de investigación.13
Las variantes anatómicas asociadas con los defectos del tabique interauricular, como las venas pulmonares anómalas, son difíciles de diagnósticar clínicamente, pero generalmente se pueden evaluar y corregir sin dificultad en la cirugía. Sin embargo, la corrección quirúrgica de los defectos del tabique interauricular tipo ostium primum, también llamados defectos del colchón endocárdico, es difícil. Las hendiduras valvulares mitral y tricúspide relacionadas con este defecto a menudo requieren reparación, y la posición baja del defecto septal, que se extiende hasta el tabique interventricular, representa problemas adicionales. La reparación quirúrgica de este defecto se asocia con riesgo de bloqueo cardiaco en vista de que las suturas se pueden colocar en forma inadvertida en los tejidos especializados de conducción del tabique interventricular.
PERSISTENCIA DEL FORAMEN OVAL
La persistencia del foramen oval, que consiste en un pequeño defecto del tabique auricular, existe en el 20 a 30 porciento de la población adulta en general. Suele asociarse con una fosa oval más grande y mayor redundancia del colgajo de tejido que cubre el foramen del que se observa en la mayoría de las personas normales. En algunos casos la redundancia es tan grande como para crear la apariencia de un aneurisma del tabique auricular. La persistencia del foramen oval, el defecto septal auricular pequeño y el aneurisma del tabique auricular no causan alteraciones hemodinámicas detectables, pero permiten la ocurrencia de embolias paradójicas de derecha a izquierda en algunas circunstancias. Estas lesiones se detectan por ecocardio-grafía, con frecuencia en pacientes con eventos cerebro-vasculares sin otra causa evidente. En ocasiones está indicado el cierre del defecto, aunque no se han establecido lineamientos claros de tratamiento para esta condición.14,15
DEFECTOS DEL TABIQUE INTERVENTRICULAR
El defecto del tabique interventricular es la anomalía cardiaca congénita más frecuente en niños. Se ve raramente en adultos, porque los defectos sustanciales del tabique interventricular que no se corrigen quirúrgicamente se asocian con una mortalidad elevada. Además, la incidencia de cierre espontáneo de los defectos del tabique interventricular es relativamente alta; el cierre ocurre con particular frecuencia en la infancia, pero también a mayor edad.
Los defectos del tabique ventricular que aparecen en adultos como anomalías aisladas generalmente miden menos de 1 cm de diámetro. Como la apertura es bastante pequeña, se puede mantener la presión sistólica normal en el ventrículo derecho y en la arteria pulmonar. El cortocircuito de izquierda a derecha es demasiado pequeño para sobrecargar de manera importante a cualquiera de los ventrículos. Cuando se encuentran en adolescentes y adultos jóvenes, algunos de estos defectos probablemente tengan un cierre espontáneo. En la mayoría de los casos, el riesgo de hipertensión pulmonar y de insuficiencia cardiaca congestiva tardía es muy remoto.
Generalmente se efectúa el cierre quirúrgico para evitar la endocarditis infecciosa [ver adelante, Problemas generales en el tratamiento]. No se ha establecido la incidencia de esta complicación; de cualquier manera, la cirugía como medida profiláctica parece ser muy eficaz.
ESTENOSIS VALVULAR PULMONAR
La estenosis valvular pulmonar se puede identificar con facilidad con base en los signos físicos característicos. Este defecto se acompaña de un soplo holosistólico expulsivo pulmonar rudo, iniciado por un chasquido pulmonar de expulsión y seguido por un segundo ruido ampliamente desdoblado, con un componente pulmonar débil o inaudible. Generalmente se demuestra en la radiografía simple de tórax la dilatación posestenótica de la arteria pulmonar. El electrocardiograma a menudo ayudará a evaluar el grado de estenosis valvular, la evidencia de hipertrofia ventricular derecha y el signo de la P pulmonale indican crecimiento auricular derecho y reflejan obstrucción severa del flujo de salida del ventrículo derecho [ver figura 3]. Si también existe un defecto del tabique interauricular o un foramen oval asociado, el desarrollo de un cortocircuito invertido a nivel auricular puede causar cianosis; sin embargo, esta complicación es menos frecuente en adultos que en niños.

|
| Figura 3 |
| Estenosis de la válvula pulmonar |
Es poco probable encontrar formas graves de estenosis valvular pulmonar en adultos, en vista de que la cirugía correctiva ha estado ampliamente disponible durante más de 25 años. La forma moderada, caracterizada por una presión sistólica del ventrículo derecho de entre 50 y 80 mm Hg, suele ser bien tolerada, y la indicación quirúrgica es dicutible en enfermos asintomáticos.16 Las formas leves, en las que la presión sistólica del ventrículo derecho es menor de 50 mm Hg, tienen poca importancia clínica.
El tratamiento de la estenosis valvular pulmonar mediante la dilatación con balón por cateterismo ha demostrado ser muy eficaz y ha remplazado en gran parte a la valvulotomía quirúrgica.17
CARDIOPATIA CONGENITA CIANOGENA
Tetralogía de Fallot
Los componentes anatómicos esenciales de la tetralogía de Fallot son el defecto del tabique interventricular y la estenosis pulmonar [ver figura 4]. Generalmente el grado de estenosis es suficiente para causar un cortocircuito de derecha a izquierda, cianosis y disminución del flujo sanguíneo pulmonar. La estenosis pulmonar puede ser valvular o infudibular; la forma infundibular generalmente predomina en adultos. Las otras dos características anatómicas comprendidas en la tetralogía de Fallot son aorta cabalgada e hipertrofia ventricular derecha. Existen variaciones en el grado de dextroposición de la aorta, pero este defecto anatómico raramente tiene importancia funcional.
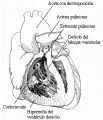
|
| Figura 4 |
| Tetralogía de Fallot |
La técnica quirúrgica totalmente correctiva suele realizarse en la infancia o niñez. Los pacientes que sobreviven hasta la edad adulta sin operación o con una operación paliativa de cortocircuito en que una arteria sistémica se anastomosa a la arteria pulmonar, pueden desarrollar una tolerancia satisfactoria al esfuerzo, cianosis mínima o nula y tener un pronóstico aparentemente bueno. Sin embargo, tales pacientes pueden desarrollar incapacidad progresiva como resultado del cierre gradual del cortocircuito y de la evolución de la estenosis pulmonar infundibular. Es más, si la anomalía no se repara pueden surgir complicaciones como endocarditis infecciosa, tromboembolia cerebral, y absceso cerebral [ver adelante, Problemas generales en el tratamiento]. La corrección total de la tetralogía de Fallot implica un riesgo de mortalidad operatoria menor del 5 por ciento, y los resultados tardíos generalmente son buenos, a pesar de que ocurra insuficiencia valvular pulmonar y menor contracción del ventrículo derecho en muchos pacientes. Los resultados a largo plazo son mejores si la cirugía se realiza cuando el paciente tiene menos de cinco años.18 La muerte súbita causada por arritmias es la causa más común de mortalidad tardía.
Otros defectos que producen cianosis
Además de la tetralogía de Fallot, las cardiopatías congénitas cianógenas en adultos incluyen el síndrome de Eisenmenger y varias malformaciones complejas como transposición de los grandes vasos, ventrículo único, ventrículo derecho de doble salida, tronco arterioso persistente, atresia pulmonar y atresia tricuspídea. En todas estas cardiopatías, los defectos asociados del tabique interauricular o interventricular permiten la supervivencia hasta la edad adulta. Muchas de estas anomalías se pueden corregir quirúrgicamente con la construcción de conductos, para crear nuevas vías de salida desde el ventrículo derecho hasta la arteria pulmonar (operación de Rastelli), la creación de derivaciones de la aurícula derecha a la arteria pulmonar (procedimiento de Fontan) o el intercambio directo de los grandes vasos. La enfermedad vascular pulmonar es la principal limitante para el éxito en estos casos.
El término síndrome de Eisenmenger se aplica en sentido amplio a aquellos defectos en los que la enfermedad de los vasos pulmonares ha progresado al punto en que la sangre se desvía en forma predominante de derecha a izquierda.19 Este síndrome comprende la categoría principal de cardiopatías congénitas cianógenas que no se pueden corregir quirúrgicamente. El tratamiento de la enfermedad vascular pulmonar es el principal problema no resuelto en las cardiopatías congénitas. La corrección quirúrgica antes de que se desarrolle hipertensión pulmonar grave es el procedimiento más eficaz. La operación debe realizarse antes de que el paciente cumpla dos años de edad.
Desde 1981 se ha usado con éxito el transplante de corazón y pulmón para el tratamiento de cardiopatías congénitas que de otra manera son inoperables. Aunque el trasplante de corazón aislado puede desempeñar algún papel en los enfermos con defectos anatómicos intracardiacos para quienes no está indicada la cirugía correctiva, tal situación es relativamente rara. La cardiopatía congénita inoperable más frecuente se debe a la presencia de enfermedad severa de los vasos pulmonares, para la cual no hay tratamiento eficaz, además del transplante. La aplicación clínica del transplante de corazón y pulmones se ha visto limitada por la falta de donadores adecuados y por la aparición de bronquiolitis obliterativa en los receptores. Se considera que la bronquiolitis representa una manifestación de reacción de rechazo crónico. El trasplante de un solo pulmón, combinado con el cierre del defecto septal, es otra opción que debe considerarse en el tratamiento de las cardiopatías congénitas que se complican por hipertensión pulmonar severa.21
COARTACION AORTICA
La coartación aórtica suele producir obstrucción moderada al flujo sanguíneo, aunque ocasionalmente hay obstrucción completa; en algunos casos, llamados seudocoartación, la obstrucción es insignificante. La presión sistólica se eleva porque se está expulsando un volumen por latido normal hacia una aorta proximal con capacitancia disminuida. Existe un aumento brusco de la presión sistólica durante el ejercicio; esta elevación es mucho más notable en pacientes con coartación que en enfermos con hipertensión esencial con presiones arteriales en reposo parecidas. La disminución, el retardo o la ausencia de pulsos femorales orientan hacia el diagnóstico de coartación. La radiografía de tórax casi siempre revela un botón aórtico y una aorta torácica descendente anormales [ver figura 5]; ocasionalmente se pueden observar muescas en las costillas. La coartación puede observarse por medio de TC o IRM y el gradiente de presión puede calcularse con base en los estudios de ultrasonido Doppler. El gradiente de presión puede también determinarse en forma confiable comparando las presiones sistólicas obtenidas con un esfigmomanómetro y una sonda Doppler, que detecta la velocidad de flujo sanguíneo por el ultrasonido reflejado en la arteria braquial y la arteria tibial posterior. La diferencia de presión del brazo es de más de 35 mm Hg en reposo en comparación con la del tobillo en todos pacientes en los que la luz de la coartación es menor del 50 por ciento de lo normal.22
|
|

La dilatación con balón a través de cateterismo en ocasiones resulta eficaz en el tratamiento de la coartación de la aorta, aunque la aplicación clínica de este procedimiento se ha limitado por el desarrollo ocasional de aneurismas en el sitio de la dilatación. La dilatación con balón es más eficaz para tratar la recurrencia de la coartación en pacientes sometidos antes a tratamiento quirúrgico.23 El seguimiento a largo plazo después del tratamiento quirúrgico de la coartación muestra una incidencia significativa de hipertensión, en especial en los pacientes que tenían más de cinco a 10 años en el momento de la cirugía.24
ALTERACIONES DE LAS ARTERIAS CORONARIAS Y COMUNICACIONES ANORMALES
Otras anomalías que afectan a la aorta y sus ramas además de la cortación, raramente son importantes después de la niñez; sin embargo, algunas anomalías de las arterias coronarias son notables. El origen anómalo de la arteria coronaria izquierda a partir de la arteria pulmonar generalmente produce un síndrome de infarto del miocardio en niños, con insuficiencia cardiaca congestiva que conduce a la muerte en el primer año de vida. Sin embargo, algunos individuos con este defecto presentan datos mínimos o no presentan datos anormales durante la niñez y sobreviven hasta la edad adulta. En dichos pacientes se han desarrollado vasos colaterales prominentes entre las arterias coronarias derecha e izquierda y un cortocircuito de izquierda a derecha lleva sangre de la arteria coronaria derecha a través de la coronaria izquierda a la arteria pulmonar; este defecto se relaciona con lesión isquémica crónica del ventrículo izquierdo.
Otra malformación importante es el origen anómalo de la arteria coronaria izquierda a partir del seno de Valsalva derecho; en ocasiones esta alteración se asocia con angina de pecho, infarto del miocardio, y muerte súbita en adultos jóvenes.25
Problemas generales en el tratamiento
ENDOCARDITIS INFECCIOSA
La endocarditis infecciosa es una complicación más frecuente de las cardiopatías congénitas en adultos que en niños.26 La susceptibilidad a esta enfermedad varía mucho según la malformación cardiaca [ver tabla 2]. Los pacientes con válvula aórtica bicúspide y regurgitación aórtica asociada son especialmente susceptibles a la endocarditis infecciosa. Por otro lado, los pacientes con comunicación interauricular no lo son, probablemente porque esta anomalía no se relaciona con un flujo sanguíneo en chorro de alta velocidad. Alrededor de la mitad de los casos de endocarditis infecciosa que ocurre como complicación de lesiones congénitas se desarrollan en pacientes que han sido sometidos antes a cirugía cardiaca, incluyendo los que tienen válvulas y conductos protésicos. Al igual que los pacientes con cardiopatía reumática, los pacientes con lesiones congénitas que predispongan a endocarditis infecciosa deben recibir antibióticos profilácticos al someterse a procedimientos quirúrgicos y dentales.
|
||||
|
El síndrome de endocarditis infecciosa derecha, que disemina la infección en los pulmones, se observa principalmente en pacientes con endocarditis de la válvula tricúspide que son adictos a drogas intravenosas, pero también ocurre en ocasiones en pacientes con cardiopatías congénitas. Los pacientes con comunicación inter-ventri-cular tienen mayor riesgo de desarrollar este trastorno.
La prevención de la endocarditis infecciosa es una razón suficiente para recomendar el cierre quirúrgico del conducto arterioso persistente, al menos en niños y adultos jóvenes, aún cuando el conducto sea muy pequeño. Sigue siendo controvertido si se debe llevar a cabo o no el cierre quirúrgico en pacientes con comunicación interventricular; sin embargo, en estos enfermos debe evitarse hasta donde sea posible la endocarditis infecciosa porque es probable que produzca complicaciones graves como insuficiencia aórtica.28
ABSCESO CEREBRAL
El desarrollo de abscesos cerebrales es un riesgo importante en pacientes con cardiopatías congénitas cianógenas. La mayor parte de estos casos de absceso cerebral ocurren en niños, aunque también se desarrollan en adultos. El inicio de un absceso cerebral a menudo se parece al de la enfermedad vascular cerebral, tanto clínicamente como en su aspecto en la TC. Se debe considerar el uso de antibióticos en el plan de tratamiento inicial cuando un paciente con cardiopatía congénita cianógena desarrolla lo que parece ser una enfermedad vascular cerebral. Los cambios seriados subsecuentes en los hallazgos clínicos y en la TC, mostrarán si existen un infarto isquémico o un absceso cerebral.
ARRITMIAS CARDIACAS
Las arritmias cardiacas no suelen ser una característica notable de las cardiopatías congénitas en niños, pero son más frecuentes en adultos. Las arritmias se relacionan con la cicatrización del miocardio y el daño específico a marcapasos y tejidos de conducción como resultado de las incisiones que se practican en el corazón, así como por los efectos a largo plazo de la sobrecarga hemodinámica. Ocasionalmente ocurren muertes súbitas cardiacas relacionadas con arritmias en pacientes con antecedente de cirugía previa por alguna cardiopatía congénita.28
Los procedimientos quirúrgicos practicados en la región del nodo auriculoventricular (AV) generalmente incluyen el cierre de un defecto del tabique interventricular como parte de la corrección de la tetralogía de Fallot. Dicha cirugía puede ocasionar un bloqueo AV que requiera colocación de marcapaso; en otros casos, la cirugía puede ocasionar bloqueo de rama, puede ocurrir la combinación de bloqueo de rama derecha y desviación del eje a la izquierda (bloqueo del fascículo izquierdo anterior) y ser precursora de un bloqueo AV completo. Estos pacientes requieren control cuidadoso con atención a los hallazgos clínicos o electrocardiográficos que puedan indicar progresión del bloqueo AV. En estas circunstancias también pueden ocurrir arritmias ventriculares como complicación tardía. En algunos casos los estudios de electrofisiología intracardiaca pueden revelar la presencia de taquicardia ventricular inducible que no se detectó por observación clínica, electrocadiografía en ejercicio o vigilancia electrocardiográfica ambulatoria.
ALTERACIONES VALVULARES
Las técnicas quirúrgicas son eficaces para cerrar defectos septales severos y aliviar estenosis valvulares o subvalvulares graves. Sin embargo, las alteraciones valvulares menos severas a menudo quedan sin corregir después de la cirugía, causando estenosis recurrente de la válvula aórtica o insuficiencia en cualquiera de las válvulas cardiacas como complicación tardía del tratamiento de pacientes con alteraciones valvulares.
ALTERACIONES MIOCARDICAS
En niños con cardiopatías congénitas, el miocardio suele ser normal y los defectos hemodinámicos se pueden analizar esencialmente en términos de sobrecarga de presión y de volumen, o ambas. Sin embargo, en adultos, sobre todo los que se han sometido a cirugía correctiva, a menudo se encuentra deterioro miocárdico. Las incisiones en los ventrículos y otras secuelas quirúrgicas participan en el desarrollo de las alteraciones miocárdicas. En estos pacientes, la dilatación, la hipertrofia y el deterioro funcional como resultado de la sobrecarga hemodinámica prolongada también producen deterioro miocárdico. Puede existir además disfunción miocárdica por perfusión de las arterias coronarias con sangre con menor saturación de oxígeno.
HIPERTENSION ARTERIAL
Los pacientes que han sido sometidos a corrección quirúrgica por coartación aórtica desarrollan con mucha frecuencia hipertensión arterial subsecuente. La frecuencia es particularmente elevada cuando la cirugía se lleva a cabo durante la adolescencia o edad adulta. La reparación quirúrgica de la coartación solo puede ser parcialmente satisfactoria, y aún después de cirugía correctiva, puede recurrir la coartación. Estos pacientes necesitan evaluación cuidadosa en busca de posible coartación residual y de hipertensión esencial.24,29
Puede ocurrir disección de la aorta como complicación tardía de la hipertensión en pacientes con coartación aórtica. También ocurre disección aórtica en pacientes que han sido intervenidos por coartación o valvulopatía aórtica, sin hipertensión arterial.
COMPLICACIONES RELACIONADAS CON PROTESIS
Con frecuencia se usan válvulas artificiales y parches protésicos en la reparación quirúrgica de defectos cardiacos congénitos más complejos. Estos procedimientos a menudo requieren la colocación de válvulas artificiales en cavidades derechas del corazón. Las válvulas porcinas son preferibles a las prótesis mecánicas para este propósito, incluso en los niños.30
Los problemas que ocurren en pacientes con válvulas protésicas son similares a los que se presentan en adultos con valvulopatías. Las complicaciones relacionadas con las válvulas protésicas son mayores en los pacientes jóvenes por las dificultades en la anticoagulación, la posibilidad de embarazo y la tendencia de los sustitutos valvulares porcinos de sufrir degeneración cálcica acelerada en estos pacientes. Aunque los parches y conductos protésicos se asocian complicaciones que las válvulas protésicas, dichas prótesis también pueden causar complicaciones tardías.
COMPLICACIONES RELACIONADAS CON EL EMBARAZO
El embarazo es bien tolerado en la mayoría de las pacientes con cardiopatías congénitas que presentan síntomas mínimos antes de embarazarse. Una excepción lo constituyen las pacientes con síndrome de Marfán, una enfermedad que predispone a la disección de la aorta durante el embarazo. El riesgo de complicaciones por el embarazo es más elevado en mujeres con síndrome de Eisenmenger; en estos casos la mortalidad materna puede ser mayor del 50 porciento [ver tabla 3].31,32
|
|
* Cardiopatía congénita leve o corregida por cirugía.
|
POLICITEMIA Y FLEBOTOMIA
Los pacientes con cardiopatías congénitas cianógenas, sobre todo aquellos con síndrome de Eisenmenger, típicamente desarrollan policitemia secundaria. Esta alteración es la respuesta fisiológica a la oxigenación subnormal de la sangre, en ocasiones capaz de producir sintomatología. Puede aparecer cefalea y otros síntomas congestivos cuando el volumen sanguíneo es excesivo, y las complicaciones tromboembólicas representan un riesgo cuando el hematocrito alcanza el 65 a 70 por ciento, nivel al cual la viscosidad de la sangre se eleva considerablemente. La flebotomía puede estar indicada en pacientes sintomáticos con hematocrito en el rango de 50 a 65 por ciento o como medida profiláctica a niveles superiores. Sin embargo, se debe tomar en cuenta la posibilidad de que la flebotomía empeore la disnea en ejercicio. Los pacientes con policitemia secundaria pueden tener deficiencia de hierro relativa; como consecuencia, la elevación en el hematocrito es mayor que la elevación en la concentración de hemoglobina.33 En estos casos se puede indicar el tratamiento con hierro y flebotomía simultáneos para disminuir el hematocrito sin afectar el nivel de hemoglobina. De esta forma se disminuye el volumen y la viscosidad de la sangre sin alterar la capacidad de transporte de oxígeno.
Los pacientes con policitemia secundaria pueden tener defectos de coagulación causados por deficiencia de los factores dependientes de la vitamina K sintetizados en el hígado (factores II, VII y X). El tratamiento con vitamina K es ineficaz, y su corrección requiere administración de los factores faltantes. Los defectos en la agregación plaquetaria pueden producir una tendencia hemorrágica, que se corrige disminuyendo el volumen de eritrocitos. Estos defectos de la coagulación son importantes sobre todo como posible fuente de complicaciones hemorrágicas después de la cirugía cardiaca.
Las concentraciones sanguíneas de ácido úrico con frecuencia están elevadas en pacientes con cardiopatías congénitas cianógenas, particularmente en hombres adultos. Aunque ni la gota ni las nefropatías ocurren con frecuencia, puede ser prudente usar alopurinol en forma profiláctica en algunos pacientes.
COMPLICACIONES RELACIONADAS CON EL EJERCICIO Y LAS ACTIVIDADES ATLETICAS
Los pacientes que presentan cardiopatías congénitas, sin tomar en consideración si se han sometido a cirugía correctiva temprana, pueden estar en riesgo de sufrir complicaciones inducidas por el ejercicio. Las normas o guías para la orientación de adolescentes y adultos jóvenes deben tomar en cuenta el tipo de cardiopatía que presentan, así como la clase de ejercicio o actividad atlética que van a desarrollar. Deben considerarse los aspectos estáticos (i.e., isométricos) y dinámicos (i.e., isotónicos) del ejercicio para calcular la tensión a la que se somete el corazón. El riesgo de que aparezcan arritmias inducidas por el ejercicio, como la taquicardia ventricular, a menudo debe evaluarse por medio de prueba de esfuerzo realizada en banda sin fin o mediante vigilancia electrocardiográfica ambulatoria.34,35
Una conferencia del American College of Cardiology realizada en Bethesda describió los lineamientos específicos para la participación en competencias atléticas para 24 diagnósticos y categorías funcionales de cardiopatía congénita.34
PROBLEMAS PSICOSOCIALES
Los adultos jóvenes con cardiopatías congénitas pueden tener problemas psicosociales significativos.35 Su desarrollo educativo y social puede haberse limitado por la enfermedad y por protección paterna excesiva. Aunque el 70 por ciento de los pacientes tienen una capacidad funcional satisfactoria y solo el 10 por ciento sufre incapacidad importante, los pacientes con cardiopatía congénita con frecuencia tienen dificultades para encontrar empleo. Uno de los obstáculos para esto es la reserva de los patrones para proporcionar un seguro médico que cubra cardiopatías previas. Los servicios de orientación y apoyo pueden ser útiles para ayudar a resolver estos problemas.
CONSEJO GENETICO
Debido al mayor número de pacientes con cardiopatías congénitas que sobreviven hasta la edad adulta, ha surgido la necesidad de orientar sobre el riesgo de cardiopatías en la descendencia de estos pacientes. El riesgo varía según las diferentes malformaciones, pero en la mayoría de ellas está aumentado [ver tabla 4].4 La ecocardiografía fetal es útil para identificar las malformaciones intracardiacas importantes después de las 18 semanas de gestación.
- Nora JJ: Causes of congenital heart disease: old and new modes, mechanisms, and models. Am Heart J 125:1409, 1993
- Payne RM, Johnson MC, Grant JW, et al: Toward a molecular understanding of congenital heart disease. Circulation 91:494, 1995 [PMID 7805255]
- Zierler S: Maternal drugs and congenital heart disease. Obstet Gynecol 65:155, 1985
- Perloff JK: Congenital heart disease in adults: a new cardiovascular subspecialty. Circulation 84:1881, 1991
- Wilson NJ, Neutze JM: Adult congenital heart disease: principles and management guidelines (pt I). Aust N Z J Med 23:498, 1993 [PMID 8297281]
- Wilson NJ, Neutze JM: Adult congenital heart disease: principles and management guidelines (pt II). Aust N Z J Med 23:697, 1993 [PMID 7511373]
- Gross GW, Steiner RM: Radiographic manifestations of congenital heart disease in the adult patient. Radiol Clin North Am 29:293, 1991 [PMID 1998053]
- Daniel WG, Mugge A: Transesophageal echocardiography. N Engl J Med 332:1268, 1995 [PMID 7708072]
- Wexler L, Higgins CB: The use of magnetic resonance imaging in adult congenital heart disease. American Journal of Cardiac Imaging 9:15, 1995 [PMID 7894229]
- Simpson IA, Sahn DJ: Adult congenital heart disease: use of transthoracic echocardiography versus magnetic resonance imaging scanning. American Journal of Cardiac Imaging 9:29, 1995
- Landzberg MJ, Lock JE: Interventional catheter procedures used in congenital heart disease. Cardiol Clin 11:569, 1993 [PMID 8252560]
- Murphy JG, Gersh BJ, McGoon MD, et al: Long-term outcome after surgical repair of isolated atrial septal defect: follow-up at 27 to 32 years. N Engl J Med 323:1645, 1990 [PMID 2233961]
- Lloyd TR, Rao PS, Beekman RH III, et al: Atrial septal defect occlusion with the buttoned device (a multi-institutional U.S. trial). Am J Cardiol 73:286, 1994
- Louie EK, Konstadt SN, Rao TLK, et al: Transesophageal echocardiographic diagnosis of right to left shunting across the foramen ovale in adults without prior stroke. J Am Coll Cardiol 21:1231, 1993 [PMID 8459082]
- Zabalgoitia M, Norris LP, Garcia M: Atrial septal aneurysm as a potential source of neurological ischemic events. American Journal of Cardiac Imaging 8:39, 1994 [PMID 8130614]
- Johnson LW, Grossman W, Dalen JE, et al: Pulmonic stenosis in the adult: long-term follow-up results. N Engl J Med 287:1159, 1972 [PMID 5082217]
- Stanger P, Cassidy SC, Girod DA, et al: Balloon pulmonary valvuloplasty: results of the valvuloplasty and angioplasty of congenital anomalies registry. Am J Cardiol 65:775, 1990
- Murphy JG, Gersh BJ, Mair DD, et al: Long-term outcome in patients undergoing surgical repair of tetralogy of Fallot. N Engl J Med 329:593, 1993 [PMID 7688102]
- Collins-Nakai RL, Rabinovitch M: Pulmonary vascular obstructive disease. Cardiol Clin 11:675, 1993 [PMID 8252567]
- Otulana BA, Higenbottam TW, Wallwork J: Causes of exercise limitation after heart-lung transplantation. J Heart Lung Transplant 11:S244, 1992 [PMID 1515447]
- Starnes VA, Stinson EB, Oyer PE, et al: Single lung transplantation: a new therapeutic option for patients with pulmonary hypertension. Transplant Proc 23:129, 1991 [PMID 1989188]
- Engvall J, Sonnhag C, Nylander E, et al: Arm-ankle systolic blood pressure difference at rest and after exercise in the assessment of aortic coarctation. Br Heart J 73:270, 1995
- Hellenbrand WE, Allen HD, Golinko RJ, et al: Balloon angioplasty for aortic recoarctation: results of valvuloplasty and angioplasty of congenital anomalies registry. Am J Cardiol 65:793, 1990 [PMID 2180265]
- Cohen M, Fuster V, Steele PM, et al: Coarctation of the aorta: long-term follow-up and prediction of outcome after surgical correction. Circulation 80:840, 1989 [PMID 2791247]
- Kragel AH, Roberts WC: Anomalous origin of either the right or left main coronary artery from the aorta with subsequent coursing between aorta and pulmonary trunk: analysis of 32 necropsy cases. Am J Cardiol 62:771, 1988 [PMID 3421178]
- Freed MD: Infective endocarditis in the adult with congenital heart disease. Cardiol Clin 11:589, 1993
- Awadallah SM, Kavey R-E, Byrum CJ, et al: The changing pattern of infective endocarditis in childhood. Am J Cardiol 68:90, 1991 [PMID 2058565]
- Chandar JS, Wolff GS, Garson A Jr, et al: Ventricular arrhythmias in postoperative tetralogy of Fallot. Am J Cardiol 65:655, 1990 [PMID 1689935]
- Kavey R-E, Cotton JL, Blackman MS: Atenolol therapy for exercise-induced hypertension after aortic coarctation repair. Am J Cardiol 66:1233, 1990
- Ilbawi MN, Idriss FS, DeLeon SY, et al: Valve replacement in children: guidelines for selection of prosthesis and timing of surgical intervention. Ann Thorac Surg 44:398, 1987
- Mendelson MA: Pregnancy in the woman with congenital heart disease. American Journal of Cardiac Imaging 9:44, 1995
- Sciscione AC, Callan NA: Pregnancy and contraception. Cardiol Clin 11:701, 1993 [PMID 8252569]
- Perloff JK: Systemic complications of cyanosis in adults with congenital heart disease: hematologic derangements, renal function, and urate metabolism. Cardiol Clin 11:689, 1993
- Graham TP Jr, Bricker JT, James FW, et al: Task Force 1: Congenital heart disease. J Am Coll Cardiol 24:867, 1994 [PMID 7930218]
- Hart EM, Garson A Jr: Psychosocial concerns of adults with congenital heart disease. Cardiol Clin 11:711, 1993 [PMID 8252570]