Reumatología
⭳ Abrir artículo (PDF)609.2 KBEste artículo fue revisado respecto a la Edición 3/2000. Ver esa versión →
Contenido del artículo
IV LUPUS ERITEMATOSO GENERALIZADO
- Manifestaciones clínicas
- FIEBRE E INFECCIONES
- TRASTORNOS MUSCULOESQUELETICOS
- LESIONES EN PIEL
- DISFUNCION RENAL
- ALTERACIONES DEL SISTEMA NERVIOSO
- AFECCION CARDIACA
- AFECCION PULMONAR
- ALTERACIONES GASTROINTESTINALES
- ENFERMEDAD HEPATICA
- SINDROME DE ANTICUERPOS ANTIFOSFOLIPIDO
- EMBARAZO
- OTRAS ALTERACIONES
- Datos de laboratorio
- Patogenia
- Tratamiento
- SALICILATOS
- MEDICAMENTOS ANTIPALUDICOS
- ESTEROIDES Y MEDICAMENTOS CITOTOXICOS
- TRATAMIENTO DE LA ENFERMEDAD RENAL
- TRATAMIENTO DE LA ENFERMEDAD DEL SNC
- Pronóstico
- Lupus eritematoso generalizado inducido por medicamentos
- Lupus discoide
- Enfermedad mixta del tejido conjuntivo
- Paniculitis nodular sistémica
- Síndrome de Sjögren
IV LUPUS ERITEMATOSO GENERALIZADO
DR. DWIGHT R. ROBINSON
El lupus eritematoso generalizado (LEG) tiene grandes variaciones en cuanto a manifestaciones clínicas, modo de presentación y evolución. La evolución de este padecimiento inflamatorio crónico y multisistémico puede variar de benigna a mortal. No existe curación, pero el tratamiento médico disminuye con frecuencia los síntomas del LEG.
La incidencia no se conoce con precisión. En dos grandes estudios se encontró una incidencia anual de alrededor de siete casos por 100,000 personas.1 Un noventa porciento de los casos de LEG se presentan en mujeres; las mujeres de raza negra se ven afectadas con mayor frecuencia que las de raza blanca. Los primeros síntomas suelen aparecer entre los 15 y 25 años de edad.
Manifestaciones clínicas
Un cuadro clínico de fiebre, erupción y artritis en una mujer joven deberá hacer sospechar la posibilidad de LEG. La forma de presentación también puede ser la de una anemia hemolítica aguda, una púrpura trombocitopénica o pericarditis. Es usual la remisión, que puede estar seguida por un intervalo libre de síntomas de varios años de duración. Es factible la recurrencia a largo plazo, manifestada como una serie de formas clínicas. La gravedad varía, desde una enfermedad aguda que afecta de manera simultánea varios sistemas orgánicos hasta una enfermedad leve con una morbilidad insignificante. La evolución es impredecible; es probable que la afección renal o del sistema nervioso central sea un signo de mal pronóstico.1-4
Existen varios factores que pueden precipitar las exacerbaciones, incluyendo el estrés físico o emocional, la luz solar, cirugías, embarazos y ciertos medicamentos. La enfermedad puede recurrir sin causa aparente.
FIEBRE E INFECCIONES
Se presenta fiebre en la mayoría de los pacientes con LEG. Esta suele ser leve, pero puede ser severa durante las llamadas crisis lúpicas. Con frecuencia se observa evidencia de afección multisistémica acompañando a la hipertermia severa en estas crisis, pero también puede ser la fiebre la única manifestación.1-4 La fiebre causada por el LEG debe distinguirse de la ocasionada por una infección añadida. Los pacientes con lupus activo tienen mayor predisposición a sufrir infecciones, sobre todo causadas por organismos oportunistas, y las infecciones son una causa muy importante de morbimortalidad en estos pacientes.5 Aunque las altas dosis de medicamentos inmunosupresores que se emplean en el lupus pueden favorecer las infecciones, un estudio encontró que la frecuencia de infecciones graves, como neumonía y septicemia, se asoció con la actividad del lupus, pero no con el uso de dosis moderadas de esteroides.6 El tratamiento habitual con inmunosupresores, como dosis bajas o moderadas de prednisona (20 mg al día), rara vez contribuye a la mayor propensión a infecciones. Los pacientes tratados con medicamentos inmunosupresores tienen una mayor incidencia de herpes zoster (con una incidencia de 21 porciento en un estudio), pero esta infección pocas veces se disemina.3 Los calosfríos, la leucocitosis neutrofílica y los niveles normales de anticuerpos anti-ADN se asocian más con fiebre causada por infección bacteriana que con la fiebre propia de la actividad del LEG.
TRASTORNOS MUSCULOESQUELETICOS
Ocurre artritis no deformante, una de las manifestaciones más frecuentes del LEG, en alrededor del 90 por ciento de los pacientes. Se manifiesta como artralgias, edema y derrame articular y rigidez matinal. La artritis es simétrica y poliarticular, y afecta con más frecuencia a las manos, muñecas, codos, rodillas y tobillos. Los episodios de artritis suelen ser intermitentes, durando horas a meses y con periodo de remisión variables. 1-4 La artritis semejante a la de la artritis reumatoide, que ocasiona erosiones óseas, es poco frecuente y se asocia con pruebas positivas de factor reumatoide en el suero. Los pacientes con esta forma de artritis tienen menos posibilidad de desarrollar nefritis lúpica que los que no tienen artritis o en los que la artritis es no deformante.7 También ocurre artropatía de Jaccoud, y se asocia con anticuerpos contra ribonucleoproteína U1. Esta enfermedad articular consiste en desviación cubital y deformidades de los dedos en cuello de cisne, que difieren de las mismas deformidades de la artritis reumatoide porque no ocasionan pérdida del cartílago articular ni erosiones óseas.7-8
Puede desarrollarse tendinitis. Los tendones de Aquiles, rotuliano y de la mano rara vez se rompen, y cuando esto ocurre, suele tratarse de pacientes que reciben esteroides.1 Otra forma de afección esquelética es la osteonecrosis o necrosis avascular, la cual puede ser asintomática u ocasionar dolor de gran intensidad. Las grandes articulaciones, por lo general más de una al mismo tiempo, son las afectadas con más frecuencia, sobre todo las de las caderas y rodillas. La osteonecrosis es una complicación conocida del tratamiento con esteroides, pero también puede presentarse como parte del proceso patológico del LEG.9
El análisis del líquido sinovial muestra evidencia de inflamación leve, a pesar del aspecto de las articulaciones en el examen físico. La cuenta de leucocitos suele ser menor a 3,000/mm3, a diferencia de la cuenta celular elevada en la artritis reumatoide. El líquido es viscoso y contiene un buen precipitado de mucina. La concentración de glucosa es parecida a la sérica. El estudio histológico sinovial muestra evidencias de inflamación discreta, a diferencia de la intensa reacción inflamatoria que caracteriza a la artritis reumatoide. Los únicos datos histológicos en el LEG suelen ser la infiltración articular leve por linfocitos y células plasmáticas y cierto edema y necrosis fibrinoide.1
LESIONES EN PIEL
Las lesiones en piel son frecuentes, y con frecuencia un dato diagnóstico importante.1-5,9 Lo más típico es la presencia de eritema en la cara, sobre la región nasal y malar. El eritema "en alas de mariposa" puede ser la primera manifestación de la enfermedad [ver figura 1]. El lupus cutáneo agudo ocurre también en otros sitios, en especial en zonas de exposición al sol. El lupus cutáneo subagudo consiste en una erupción eritematosa y papuloescamosa, que puede ser anular. El lupus cutáneo crónico, que en ocasiones se denomina lupus discoide (LD), puede ocurrir en ausencia de LEG [ver adelante, Lupus discoide]. Consiste en áreas de atrofia con despigmentación central y mayor pigmentación en los bordes junto con telangiectasias, descamación variable y dilatación y taponamiento de los folículos pilosos. Todas las lesiones cutáneas del lupus son precipitadas o exacerbadas por la exposición a la luz solar. Uno a dos tercios de los pacientes con lupus tienen fotosensibilidad, y en estos pacientes los síntomas sistémicos o las erupciones cutáneas pueden exacerbarse después de la exposición al sol. La mayoría de los pacientes con fotosensibilidad tienen anticuerpos anti-Ro en el suero. No se conocen los mecanismos de la fotosensibilidad, pero las evidencias sugieren lesión celular inducida por luz ultravioleta, que causa transferencia de antígenos intracelulares a las superficies celulares, en donde pueden inducir una respuesta autoinmune.5 Son lesiones cutáneas menos específicas del lupus un eritema en parches, con o sin descamación, que aparece en las extremidades, tronco y cara, y parches eritematosos alrededor de las yemas de los dedos, palmas y áreas periungueales, por lo general acompañadas de telangiectasias en los bordes de las uñas de los dedos.
Ocurre alopecia, que se caracteriza por adelgazamiento difuso del cabello en el cuero cabelludo normal, pero también puede presentarse eritema crónico, cicatrización y atrofia del cuero cabelludo. En ausencia de cambios crónicos en la piel cabelluda, la pérdida de cabello, a pesar de que puede ser muy extensa, es reversible.1 La alopecia se asocia con otras manifestaciones cutáneas y con fenómeno de Raynaud, y su presencia se relaciona con el grado de actividad de la enfermedad. La alopecia no se asocia con afección de otros órganos o estructuras específicos, como articulaciones, riñones o sistema nervioso central.10
Pueden observarse ulceraciones de la boca o labios, como áreas pequeñas eritematosas, con edema y dolorosas. La púrpura se asocia con trombocitopenia, pero puede ocurrir aún cuando las plaquetas sean normales como resultado de vasculitis de pequeños vasos.
Aunque las recaídas de la enfermedad sistémica en los pacientes con lupus pueden acompañarse del desarrollo de erupciones cutáneas, los pacientes con enfermedad cutánea no tienen más riesgo de desarrollar afección renal o neurológica que los pacientes sin estas manifestaciones. Las lesiones cutáneas del LEG responden a antimaláricos en la mayoría de los casos. Otros agentes, como dapsona, azatioprina y retinoides, pueden ser benéficos en los pacientes resistentes al tratamiento antimalárico.5
DISFUNCION RENAL
Los pacientes con LEG presentan de manera usual algún grado de afección renal. El examen general de orina puede mostrar proteinuria, piuria, hematuria y cilindros granulares o de eritrocitos. La afección renal más severa puede ocasionar hipoalbuminemia, otras características del síndrome nefrótico o disminución en la filtración glomerular, lo que origina retención de productos nitrogenados, alteraciones electrolíticas y acidosis.1,-4,11
La lesión más benigna de la nefritis lúpica es la de cambios mesangiales mínimos, probablemente presente en la mayoría o en la totalidad de los pacientes lúpicos [ver tabla 1], aunque esta lesión no tiene una morbilidad significativa. La forma proliferativa focal de la lesión renal [ver figura 2] también es benigna y con frecuencia se resuelve. La glomerulonefritis membranosa [ver figura 3] se acompaña de manera característica de uno o todos los elementos que constituyen el síndrome nefrótico. Las remisiones son comunes, de la misma manera que las recaídas, y puede desarrollarse una insuficiencia renal lentamente progresiva. La nefritis proliferativa difusa [ver figura 4] constituye la forma más grave de afección renal, y con frecuencia está asociada con algún grado de insuficiencia renal e hipertensión. Sin embargo, el tipo de patología renal cambia con el paso del tiempo en más de la mitad de los pacientes con enfermedad renal comprobada, un hecho que limita el valor pronóstico que pudiera tener la biopsia renal.
El análisis de las biopsias renales utilizando varios criterios ha originado índices de actividad y cronicidad de la enfermedad renal [ver tabla 2]. Las calificaciones altas del índice de cronicidad (lesiones crónicas, irreversibles) predicen mayor riesgo de insuficiencia renal, sobre todo en personas jóvenes, mientras que las pruebas de función renal anormal y calificaciones altas en el índice de actividad (lesiones activas e individuales) pueden no predecir una mala evolución porque las lesiones pueden ser reversibles.
ALTERACIONES DEL SISTEMA NERVIOSO
En alrededor de la mitad de los pacientes con lupus existe alguna alteración del sistema nervioso central.1-4,11 Incluyendo diversos cambios psicológicos y neurológicos [ver tabla 3]. Las manifestaciones neuropsiquiátricas pueden aparecer en cualquier momento, pero casi siempre lo hacen pronto, durante el primer año de evolución. Por lo general se asocian con evidencia de actividad en otros órganos y sistemas. La depresión reactiva es muy frecuente. Puede ocurrir una psicosis funcional con trastornos del pensamiento y del ánimo característicos de la esquizofrenia, pero es más común el síndrome orgánico cerebral, que ocurre en hasta el 20 por ciento de los pacientes con lupus. Este incluye varios grados de pérdida de memoria, desorientación y deterioro intelectual, y puede ocurrir en ausencia de otros síntomas neurológicos o de actividad sistémica. En algunos pacientes se presenta deterioro lento y progresivo, que causa demencia incapacitante. La psicosis puede ser una complicación del tratamiento con esteroides, aunque también se ha establecido que los problemas psicológicos son una manifestación frecuente del LEG. Además, estas psicosis se asocian con crisis convulsivas y otras evidencias de alteración neurológica. Con frecuencia las crisis convulsivas son de tipo gran mal, aunque se han descrito crisis focales.
Los cambios neurológicos incluyen signos de afección de la vía piramidal. Estas alteraciones, junto con la parálisis espástica, se deben en ocasiones a un accidente cerebrovascular típico. Las alteraciones de pares craneales afectan con frecuencia al séptimo par (manifestada como parálisis facial) y los nervios motores extraoculares (que ocasionan ptosis o diplopia). Las neuropatías periféricas son poco comunes y pueden manifestarse como deficiencias sensitivas o mixtas (sensitivas y motoras). En raras ocasiones puede haber afección múltiple y asimétrica de nervios periféricos, manifestada por un cuadro de mononeuritis múltiple, igual al que ocurre en muchas vasculitis necrosantes. Otras alteraciones neurológicas poco frecuentes incluyen ataxia, corea, escotomas, meningitis aséptica y lesiones de la médula espinal.
Las manifestaciones en el sistema nervioso central son típicamente pasajeras. La psicosis, las convulsiones, la hemiplegia, la diplopia y otros hallazgos, con frecuencia ceden de manera notable y rápida, excepto en el caso poco usual en el que hay una complicación irreversible, como una hemorragia intracerebral grave. Es difícil determinar si el tratamiento con esteroides u otros medicamentos influyen en el pronóstico de la afección a nivel del SNC.
Las pruebas neurológicas revelan pocos cambios que no se hayan ya detectado en el examen físico. Incluso ante alteraciones neurológicas francas, la punción lumbar puede ser normal o demostrar sólo aumento en la concentración de proteínas y pleocitosis leve. El aumento sérico en la concentración de autoanticuerpos contra la proteína ribosomal P se considera específico de LEG y se asocia con depresión grave y psicosis, pero los niveles aumentados de estos anticuerpos no se asocian con enfermedad neurológica no psiquiátrica.12
Las imágenes por resonancia magnética (IRM) pueden demostrar alteraciones asociadas con las manifestaciones neuropsiquiátricas del LEG. Los pacientes con enfermedad difusa del sistema nervioso central, como psicosis, crisis convulsivas generalizadas y disfunción cognoscitiva, tienen áreas de mayor intensidad de señales, que se distribuyen en forma simétrica, en la sustancia blanca subcortical. Estas alteraciones son reversibles. Por el contrario, los enfermos con alteraciones focales tienen áreas de atrofia de distribución asimétrica y aumento en la intensidad de señales en regiones que corresponden a los vasos cerebrales principales. Estos trastornos no son reversibles y se asocian con aumento en los niveles de anticuerpos antifosfolípido [ver adelante, Síndrome de anticuerpos antifosfolípido].11
Los enfermos con LEG que no tienen enfermedad neuropsiquiátrica grave pueden tener disfunción cognoscitiva menos grave que se manifiesta por pérdida de la memoria, cefalea y disminución en la capacidad de trabajo e interacción social. Las pruebas neuropsicológicas de un grupo de pacientes con LEG demostraron un pobre desempeño de tareas complejas de atención y de actividades que requieren capacidad visoespacial. Esta disfunción cognoscitiva fue independiente de la actividad de la enfermedad. del uso de esteroides y de otras patologías neuropsiquiátricas.13
La patología de la afección en el SNC se caracteriza por la escasez de hallazgos histológicos, a pesar de la presencia de manifestaciones clínicas muy notables, excepto en el caso de infarto cerebral secundario a oclusión trombótica o embólica de un vaso importante. El hallazgo más frecuente consiste en un infiltrado celular inflamatorio perivascular leve; es poco usual la presencia de una verdadera vasculitis necrosante en el cerebro. Son comunes los microinfartos, con frecuencia múltiples. Además, puede ocurrir lesión y disfunción neuronal por la presencia de anticuerpos antineurona. Estos se encuentran hasta en el 75 por ciento de los pacientes con síntomas neuropsiquiátricos del lupus y pueden existir también en el 25 por ciento de los enfermos sin trastornos neuropsiquiátricos. Existen anticuerpos contra la proteína Pribosomal en la mayoría de los pacientes con psicosis o depresión.11,13
AFECCION CARDIACA
La pericarditis, que se manifiesta por dolor y frote torácicos, es la manifestación cardiaca más frecuente.1-4,11 Pueden ocurrir derrames pequeños, pero el tamponade es muy poco frecuente. La miocarditis es mucho menos común que la pericarditis. La miocarditis puede simular una cardiomiopatía, ocasionando arritmias ventriculares e insuficiencia cardiaca congestiva. Se observa una gran prevalencia de anticuerpos anti-Ro en los pacientes con miocarditis y alteraciones de la conducción cardiaca.14 La endocarditis de Libman-Sacks, una endocarditis no infecciosa que se caracteriza por vegetaciones verrucosas pequeñas cerca de los anillos de las válvulas cardiacas, se presenta con frecuencia. Estas lesiones suelen ser asintomáticas, pero en algunas ocasiones se complican por tromboembolias, endocarditis infecciosa o disfunción valvular. Se encuentra cardiopatía valvular en el 18 porciento de los pacientes con LEG, la mayoría de los cuales tienen anticuerpos antifosfolípido.15 La coronariopatía ateroesclerótica es una complicación importante en el LEG, y puede causar hasta el tercio de los fallecimientos en este grupo de pacientes. Las causas de enfermedad coronaria incluyen hiperlipidemia, que es agravada por el tratamiento con esteroides, y la hipertensión, que puede ser secundaria a la nefropatía lúpica. La vasculitis coronaria puede causar oclusión de estas arterias, pero es una causa mucho menos frecuente de infarto al miocardio que la ateroesclerosis.5
AFECCION PULMONAR
El dolor torácico pleurítico, otra manifestación común, puede ser de intensidad leve o intensa y estar acompañado de frotes y derrames pleurales.1-4 El análisis del líquido pleural suele revelar la presencia de un exudado que contiene concentraciones de proteínas mayores de 3 g/dl; también pueden observarse trasudados. La cuenta de leucocitos mononucleares puede elevarse hasta varios miles de células por mm3. Los niveles de glucosa en líquido pleural suelen ser similares a los de sangre; en comparación, en la artritis reumatoide los niveles de glucosa en el líquido pleural están reducidos.
La afección del parénquima pulmonar en LEG puede ser aguda o crónica.11,16 Se han descrito tres síndromes agudos. El más común, la neumonitis lúpica aguda, se caracteriza por fiebre de inicio súbito, disnea, hipoxemia e infiltrados alveolares en parches en la radiografía de tórax. El segundo en frecuencia es la hemorragia alveolar, semejante clínicamente a la neumonitis aguda pero con hemorragia en el pulmón, lo que causa disminución en la concentración del hematocrito. Ambos síndromes progresan con rapidez a insuficiencia respiratoria y muerte. El tratamiento con pulsos de metilprednisolona parece ser benéfico, con adición de azatioprina o ciclofosfamida en casos resistentes, aunque no se han realizado estudios controlados. El síndrome menos frecuente de los tres es la hipoxemia aguda reversible, que ocurre en pacientes muy graves con LEG. Se ha postulado que los componentes activados del complemento pueden activar neutrófilos, secuestrándolos en la vasculatura pulmonar. La prednisona es eficaz en pacientes con hipoxemia aguda reversible.
La enfermedad pulmonar crónica ocurre en el lupus eritematoso generalizado como una enfermedad pulmonar intersticial o como hipertensión pulmonar. La enfermedad pulmonar intersticial puede consistir en alveolitis inflamatoria, que responde a tratamiento inmunosupresor, o a fibrosis intersticial, que no lo hace. La evaluación del pulmón afectado por tomografía computada de alta resolucion puede ayudar a distinguir entre la alveolitis inflamatoria y la fibrosis intersiticial.
La hipertensión pulmonar se asocia con alta incidencia de fenómeno de Raynaud, anticuerpos antirribonucleoproteína, factor reumatoide y anticuerpos antifosfolípido. No se conoce un tratamiento eficaz y el pronóstico es malo.
La hipertensión pulmonar en los pacientes con LEG es semejante a la hipertensión pulmonar primaria.11,16 Los pacientes sufren disnea de ejercicio y en ocasiones dolor torácico. Por lo general las radiografías de tórax son normales y no existe evidencia de embolia pulmonar, a pesar de que suelen existir anticuerpos antifosfolípido en estos pacientes.12
ALTERACIONES GASTROINTESTINALES
Son frecuentes la anorexia, náusea, vómito y dolor abdominal, por lo general sin causa aparente. La vasculitis mesentérica puede ocasionar infartos intestinales y perforación. En algunos pacientes se presenta pancreatitis asociada con el tratamiento esteroide. La enteropatía perdedora de proteínas, que responde al tratamiento con esteroides, puede ser una complicación del LEG.18
ENFERMEDAD HEPATICA
De manera ocasional se aprecian en el LEG hepatomegalia y alteraciones mínimas en las pruebas de función hepática, pero esto suele tener poca importancia clínica. Las biopsias de hígado muestran solamente cambios mínimos, inespecíficos.1 Es rara la hepatitis crónica1-3 En algunos pacientes con cirrosis biliar primaria se ha publicado la coexistencia con LEG.
SINDROME DE ANTICUERPOS ANTIFOSFOLIPIDO
El síndrome de anticuerpos antifosfolípido consiste en un estado de hipercoagulabilidad que ocurre en asociación con autoanticuerpos contra fosfolipídos.19,20 Los anticuerpos antifosfolípido no se unen directamente a los fosfolípidos aniónicos, sino que se fijan a epítopes en varias proteínas plasmáticas, incluyendo la ?-glucoproteína I, que forma un complejo con los fosfolípidos. La formación de complejos entre la ?-glucoproteína I y los fosfolípidos causa un cambio conformacional que expone un epítope antes escondido al cual se unen los anticuerpos antifosfolípidos. Los complejos formados entre fosfolípidos y varios factores de la coagulación, incluyendo la protrombina, la proteína C y la proteína S, pueden fijar también anticuerpos antifosfolípido, pero se desconoce aún el mecanismo por el que se presenta la coagulopatía en este síndrome.8,21
Existen anticuerpos antifosfolípido en alrededor de la tercera parte de los pacientes con LEG. Estos anticuerpos pueden desarrollarse también en pacientes con otras enfermedades del tejido conjuntivo, así como con padecimientos infecciosos como sífilis, infección por virus de inmunodeficiencia humana (VIH) y enfermedad de Lyme. También pueden presentarse después del tratamiento con ciertos medicamentos, sobre todo fenotiacinas, procainamida, hidralacina y fenitoína. Sin embargo, los anticuerpos antifosfolípido asociados con infecciones y tratamiento farmacológico existen en bajas concentraciones y no suelen causar enfermedad clínica significativa.
Varias manifestaciones clínicas se asocian con el síndrome de anticuerpos antifosfolípido [ver tabla 4].8,19 El síndrome primario ocurre en pacientes sin LEG y el secundario en pacientes con evidencia clínica de LEG. Los pacientes con la forma primaria pueden tener otros datos, como lívedo reticulares y trombocitopenia crónica [ver tabla 4]. Las complicaciones tromboembólicas se presentan en pacientes con el síndrome primario o con asociación con LEG. Las trombosis, la muerte fetal y la trombocitopenia son las manifestaciones más frecuentes. Las trombosis pueden afectar los lechos vasculares arterial o venoso, y tienden a recurrir después del tratamiento.22 Las trombosis venosas afectan a las extremidades o a los sistemas renal, porta o mesentérico. Con frecuencia las trombosis venosas periféricas ocasionan embolia pulmonar. Las trombosis arteriales, que afectan sobre todo la circulación cerebral pueden causar infartos cerebrales, isquemia cerebral transitoria, isquemia ocular, encefalopatía y demencia por infartos múltiples. Pueden ocurrir trombosis arteriales en múltiples órganos, que suelen ser fatales. Puede ser difícil distinguir la oclusión vascular secundaria a vasculitis en el LEG activo de la vasculopatía no inflamatoria asociada con el síndrome de anticuerpo antifosfolípido. La distinción puede ser importante porque la vasculitis se trata con fármacos inmunosupresores y la coagulopatía del síndrome antifosfolípido con anticoagulantes.5 Las alteraciones neurológicas incluyen cefalea migrañosa y mielitis transversa. El livedo reticularis, un patrón visible, irregular y lineal de venas cutáneas dilatadas, es frecuente. Puede existir necrosis cutánea, así como síndrome de Sneddon (que consiste en la triada de livedo reticularis, enfermedad cerebrovascular e hipertensión lábil). La muerte fetal recurrente, que es hasta dos veces más frecuente en las pacientes con síndrome de anticuerpos antifosfolípido que en la población general, ocurre en el segundo trimestre, y se piensa que se debe a insuficiencia placentaria. Las placentas afectadas muestran vasculopatía y trombosis. En raros casos existe anemia hemolítica con Coombs positivo.
El síndrome de anticuerpos antifosfolípido contribuye a la enfermedad cardiaca en el LEG. La enfermedad valvular en pacientes con LEG difiere de la que se presenta en pacientes con síndrome primario de anticuerpos antifosfolípido. Los pacientes con LEG tienen engrosamiento de las valvas de las válvulas mitral y aórtica y vegetaciones, así como estenosis e insuficiencia valvular, mientras que los pacientes con el síndrome primario tienen engrosamiento valvular sin vegetaciones e insuficiencia valvular en ausencia de estenosis.15 Las trombosis coronarias causadas por el síndrome de anticuerpos antifosfolípido pueden ocasionar infartos del miocardio.
El diagnóstico de síndrome antifosfolípido se hace por la presencia de anticuerpos antifosfolípido asociada a una trombosis arterial, venosa o una pérdida fetal. Los anticuerpos antifosfolípido se determinan por la presencia de títulos altos de anticuerpos de IgG o IgM medidos por ensayo inmunoenzimático (ELISA) o por pruebas positivas para el anticoagulante lúpico, utilizando el tiempo parcial de tromboplastina activado, el tiempo de coagulación de caolín o el tiempo de veneno viperino de Russell. Otras pruebas comerciales no disponibles pueden ser inespecíficas.5
El tratamiento óptimo de las complicaciones del síndrome de anticuerpos antifosfolípido aún no se ha establecido porque las manifestaciones de este padecimiento varían en gravedad desde complicaciones vasculares fatales hasta leves o ausentes. Los pacientes que sufren un evento trombótico importante tienen gran riesgo de recurrencia y por lo general deben ser tratados con warfarina a largo plazo.5 Al parecer se requieren dosis altas de warfarina o heparina, y las dosis bajas de aspirina o anticoagulantes son insuficientes para prevenir las complicaciones. Se han usado esteroides para el tratamiento de la trombosis fulminante, trombocitopenia y muerte fetal recurrente con resultados diversos, pero aún no se ha establecido con claridad cuál es el papel de los esteroides en el tratamiento de este síndrome. En estudios no controlados se ha encontrado que la heparina y la gama-globulina intravenosa son benéficas durante el embarazo.
EMBARAZO
La fertilidad en las pacientes con LEG es normal. Aunque las pacientes con LEG activo pueden tener exacerbaciones durante el embarazo, varios estudios que compararon la frecuencia de estos episodios en pacientes embarazadas con la frecuencia en pacientes controles no embarazadas han concluido que no existe aumento en la frecuencia de exacerbaciones como consecuencia del embarazo en las pacientes con lupus. Las pacientes con nefritis lúpica con frecuencia sufren exacerbación del daño renal durante el embarazo, pero es difícil determinar si ésta es causada por toxemia o por el LEG.5 En forma semejante, ocurren trombocitopenia, artralgias e hipocomplementemia durante el embarazo normal así como durante las exacerbaciones del LEG. Las pacientes con LEG embarazadas pueden sufrir un cuadro de hipertensión, edema y proteinuria de inicio súbito, que es indistinguible de la hipertensión inducida por el embarazo y que es independiente de la presencia de anticuerpos antifosfolípido. La muerte fetal del segundo trimestre se asocia con la presencia de anticuerpos antifosfolípido o con la hipertensión inducida por el embarazo.
El síndrome de lupus neonatal consiste en lesiones lúpicas cutáneas pasajeras y, con menor frecuencia, en trombocitopenia transitoria, anemia y afección hepática. La manifestación más grave del lupus neonatal es el bloqueo cardiaco congénito, que suele ser completo y permanente; el bloqueo cardiaco congénito puede ser responsable de algunas muertes en el periodo neonatal. Las madres y los productos afectados casi siempre presentan autoanticuerpos contra ribonucleoproteínas, por lo general anti-Ro (también llamados SS-A) y con menor frecuencia, anti-La (también conocidos como SS-B) o ambos [ver adelante, Datos de laboratorio, Anticuerpos antinucleares]. El lupus neonatal ocurre en una minoría de pacientes con estos anticuerpos, y el bloqueo congénito cardiaco se presenta en menos del tres porciento de los niños afectados. Sin embargo, en una madre con un niño afectado, el riesgo de lupus neonatal en un segundo producto aumenta a alrededor del 25 porciento, y el de bloqueo cardiaco es de ocho a 16 porciento. Ocurre bloqueo cardiaco congénito en una pequeña proporción de niños cuyas madres tienen anticuerpos contra el componente de 52 kd del antígeno Ro y contra el componente de 48 kd del antígeno La.
El pronóstico a largo plazo en los niños que sobreviven el periodo neonatal es bueno; sin embargo, los niños con bloqueo cardiaco requieren colocación de marcapaso permanente.23 Aunque sólo cerca de la mitad de las madres de los niños afectados presentan síntomas leves de enfermedad autoinmune en el momento del parto, casi todas estas madres manifestarán finalmente síntomas leves.24
OTRAS ALTERACIONES
Puede desarrollarse miopatía, caracterizada por debilidad de los músculos proximales y aumento de la concentración sérica de creatina cinasa. El aumento en la concentración de creatina cinasa y los cambios patológicos del músculo pueden ser idénticos a los observados en la polimiositis, pero en ocasiones predomina la degeneración vacuolar de las fibras musculares.1
Con frecuencia aparece linfadenopatía generalizada y se encuentra esplenomegalia en alrededor del 20 porciento de los pacientes. A menudo se encuentran en el bazo las lesiones características en "aros de cebolla", las cuales consisten en capas concéntricas de tejido fibroso perivascular.1
En unos cuantos pacientes puede desarrollarse síndrome de Sjögren, que afecta a las glándulas salivales y lacrimales y se asocia con queratoconjuntivitis seca. Este síndrome es menos común en el LEG que en la artritis reumatoide y se ha visto en pacientes con LEG y artritis erosiva no deformante.1
Datos de laboratorio
Es frecuente la presencia de anemia, que puede ser el resultado de hemólisis, enfermedad crónica o una combinación de ambas. También es común la leucopenia, pocas veces severa: la cuenta total de leucocitos suele estar por arriba de 2,000/mm3. La cuenta diferencial usualmente es normal, a diferencia de lo que ocurre en el síndrome de Felty. Es frecuente la presencia de trombocitopenia, pero ésta tiene significado clínico sólo cuando la cuenta plaquetaria se reduce por debajo de 50,000/mm3. Los anticuerpos contra proteínas de superficie de las plaquetas, por lo general las glucoproteínas IIb y IIIa, causan fagocitosis de las plaquetas cubiertas por anticuerpos por los macrófagos del sistema retículoendotelial. Las cuentas plaquetarias menores a 20 x 109/L se tratan con esteroides. Puede presentarse trombocitopenia grave acompañada de púrpura y otras manifestaciones hemorragíparas, que requieren tratamiento agresivo. La trombocitopenia leve puede adquirir mayor importancia en presencia del anticoagulante lúpico o inhibidor de LEG. Este último compuesto raramente origina hemorragias clínicas a menos que coexista con otros defectos hemostáticos. Los anticoagulantes circulantes pueden ocasionar prolongación de los tiempos de protrombina y parcial de tromboplastina. Sin embargo, en ocasiones los resultados de estas pruebas estándar de la coagulación son normales y se requieren procedimientos especiales de laboratorio para detectar la presencia de anticoagulantes circulantes en los pacientes con LEG.1
Los pacientes con anticoagulantes circulantes suelen tener una prueba biológica para la sífilis con resultados falsos positivos. Las pruebas serológicas falsas positivas para la sífilis se presentan en alrededor de un 25 porciento de los pacientes con LEG. Tales reacciones son el resultado de la formación de anticuerpos que reaccionan contra el material antigénico que se emplea en las pruebas serológicas para la sífilis (Wasserman, Hinton, Kahn, y pruebas similares). Puede distinguirse a las reacciones falsas positivas de las verdaderamente positivas por la ausencia de una reacción de anticuerpos contra un antígeno específico de treponema (en una prueba de absorción de anticuerpo fluorescente de treponema).1
ANTICUERPOS ANTINUCLEARES
Entre los numerosos autoanticuerpos que aparecen en el LEG algunos de los más característicos son los anticuerpos antinucleares (AAN) [ver figura 5].25
Más de un 90 porciento de los pacientes con LEG tienen pruebas de
anticuerpos antinucleares positivas. Sin embargo, la prueba también es
positiva en muchas otras enfermedades y en algunos pacientes ancianos sin
enfermedad aparente. Aunque la presencia de una prueba repetidamente negativa
hace que el diagnóstico de LEG sea poco probable, de un cinco a un 10
porciento de los pacientes en quienes el diagnóstico está bien
establecido tendrán pruebas negativas. La mayoría de los
pacientes con AAN negativos tienen anticuerpos circulantes contra los
antígenos citoplasmáticos Ro y La y contra el ADN de cadena
simple. Tales pacientes suelen tener una dermatitis fotosensitiva, incidencia
baja de afección renal y del sistema nervioso central, y buen
pronóstico.26
El factor celular del lupus eritematoso (LE), un anticuerpo contra la desoxirribonucleoproteína, reacciona con el núcleo dañado de la célula para formar un cuerpo de hematoxilina, una masa homogénea de material nuclear que tiene apariencia histológica característica. Los leucocitos polimorfonucleares que fagocitan estos cuerpos de hematoxilina se denominan células LE. Los cuerpos de hematoxilina también pueden detectarse en cortes de tejidos. El uso de la prueba de células LE en sangre periférica se ha sustituido en gran parte por otras pruebas de anticuerpos antinucleares.
En los trastornos autoinmunes se han descrito anticuerpos contra muchos determinantes antigénicos situados en el núcleo celular y se han observado patrones típicos de respuesta de anticuerpos en algunos trastornos. Se han caracterizado varios anticuerpos antinucleares, pero estos anticuerpos están dirigidos contra un número limitado de epítopes antigénicos potenciales en el núcleo. Estos epítopes se encuentran en tres estructuras nucleares diferentes: la cromatina, la pequeña ribonucloproteína nuclear (snRNP) y la partícula Ro. La cromatina, un componente principal del núcleo celular, consiste de ADN y de histonas. Se encuentran anticuerpos contra el ADN y las histonas en el LEG pero también se observan en otras enfermedades autoinmunes. Los anticuerpos antinucleares pueden dirigirse contra el ADN desnaturalizado de una sola cadena y contra el ADN nativo de doble cadena, siendo este último más específico para LEG. En los pacientes con LEG se observan anticuerpos contra una o más histonas; las respuestas más potentes se dirigen contra las histonas H1 y H2B. Los individuos con anticuerpos contra el ADN suelen tener también anticuerpos contra histonas.25
Las células contienen ARN en cuatro formas: ARN mensajero, ARN ribosomal, ARN de transferencia y un grupo heterogéneo de moléculas pequeñas de ARN. Cada una de estas moléculas de ARN se relaciona con proteínas específicas para formar ribonucleoproteínas. Debido a que las ribonucleoproteínas son solubles en amortiguadores acuosos, también se les conoce como antígenos nucleares extraíbles (ANE). Se pueden identificar diversas ribonucleoproteínas mediante autoanticuerpos, pero los anticuerpos predominantes en el LEG son anti-U1RNP, anti-Sm y anti-Ro. Todos estos anticuerpos reconocen determinantes situados en las snRNP. Los anticuerpos anti-Sm son específicos de LEG. Los pacientes con anticuerpos anti-Sm casi siempre tienen también anticuerpos anti-U1RNP, pero no sucede a la inversa. Los títulos elevados de anticuerpos anti-R1RNP se relacionan con la enfermedad mixta del tejido conjuntivo.25
La proteína La se une a muchas especies de snRNP y porta un epítope identificable por los anticuerpos anti-La. Los anticuerpos anti-La se acompañan casi siempre de anticuerpos anti-Ro, aunque pueden ocurrir anticuerpos anti-Ro sin la presencia de anticuerpos anti-La. Ni los anticuerpos anti-Ro ni anti-La son específicos de LEG. Estos anticuerpos se observan a menudo en el síndrome de Sjögren y en otros trastornos autoinmunes.25
COMPLEMENTO
La disminución en los niveles séricos del complemento (C) ha sido citada como prueba de un aumento en el consumo de sus componentes durante la formación de complejos inmunes y de daño tisular.1-4 Aunque una parte de la disminución en los niveles séricos de los componentes del complemento puede deberse a disminución en la síntesis más que a aumento en el consumo, el consumo del complemento está incrementado en el LEG. En particular el C3 y las inmunoglobulinas pueden observarse con técnicas de inmunofluorescencia como masas localizadas en el glomérulo renal y otros tejidos dañados.2 Asimismo, se ha detectado properdina en los glomérulos de pacientes lúpicos, lo que indica que hay participación de la vía alterna en la activación del complemento.
La activación de la vía clásica del complemento puede demostrarse clínicamente por la presencia de una reducción en el nivel total de complemento hemolítico (CH50). De manera alterna, la activación de complemento puede determinarse por medio de la medición inmunoquímica del componente C3. El método de hemólisis requiere de niveles funcionantes de todos los componentes del complemento. Sin embargo, debido a que puede haber exceso de varios componentes en los pacientes lúpicos, pueden ocurrir reducciones en algunos componentes (por ejemplo, una reducción del nivel de C3 hasta la mitad), sin que esto cause una alteración en el ensayo por hemólisis. Una amplia variedad de enfermedades que ocasionan inflamación y necrosis tisular causan elevaciones en los niveles séricos de varios componentes del complemento y de otras proteínas plasmáticas que son reactantes de fase aguda. Unas cuantas enfermedades, incluyendo al LEG, ciertas formas de glomerulonefritis y la enfermedad del suero aguda, están asociadas con niveles reducidos de los componentes del complemento en suero.27
Los componentes séricos del complemento no siempre están reducidos en el LEG activo. En un estudio se encontró que los pacientes con LEG y enfermedad extrarrenal activa tenían niveles más elevados de CH50, C4 y C3 y niveles más bajos de complejos inmunes circulantes que los pacientes con enfermedad renal activa. Los anticuerpos anti-ADN no sirvieron para distinguir entre estos dos grupos.27
Se han descrito varias familias con deficiencia hereditaria del componente C2. Los miembros de la familia que son homocigotos tienen niveles no detectables o muy bajos, mientras que los individuos heterocigotos tienen alrededor de la mitad del nivel normal. Se ha encontrado que alrededor de una tercera parte de los individuos homocigotos con deficiencia de C2 tienen LEG o un síndrome similar que podría representar una expresión parcial de la enfermedad. Asimismo, se encontró que aún los heterocigotos para deficiencia de C2 tenían un aumento en la incidencia de LEG. Sin embargo, parece poco probable que se presente una anormalidad significativa en la función del complemento por la disminución en el 50 porciento en el nivel de C2 que se observa en los individuos heterocigotos, sobre todo porque se considera que la cantidad normal de C2 en suero es mayor que la necesaria para que haya actividad hemolítica del complemento. Es probable que el gen para C2 esté en desequilibrio de unión con un gen de la respuesta inmune. Se considera que esta deficiencia de unión está relacionada con las funciones inmunológicas anormales observadas en el LEG.28
PRUEBA DE LA BANDA DEL LUPUS
La prueba de banda de lupus emplea técnicas de inmunofluorescencia para detectar inmunoglobulinas y componentes del complemento en la unión dermoepidérmica. En alrededor de 80 porciento de los pacientes con LEG activo y en 30 a 40 porciento de aquellos con enfermedad inactiva se observa la banda de lupus en la piel que no tiene lesiones. Todas las muestras tomadas de piel con lesiones lúpicas dan resultados positivos, pero el hecho de encontrar una prueba positiva ya sea sobre piel con o sin lesiones no es completamente específico para LEG.1-4
En las lesiones activas de la piel de pacientes con LEG y lupus discorde se ha detectado el complejo terminal del complemento o complejo de ataque a la membrana, compuesto de C5b, C6, C7, C8 y C9. La ausencia de este complejo en la piel no afectada sugiere que se trata de un mediador de las lesiones inflamatorias de la piel.
Patogenia
El LEG parece ser un trastorno de la regulación inmunológica que ocasiona pérdida de tolerancia; esto da como resultado reacciones autoinmunes contra antígenos del huésped que originan inflamación y daño a la vasculatura y otros tejidos.29 La alteración en la inmunidad puede originarse por varios mecanismos; por ejemplo, una deficiencia en las células T, como la disminución en la actividad de las células T supresoras, podría ocasionar una expresión excesiva de las células B. La etiología es aún desconocida, pero el candidato más viable es un agente infeccioso, probablemente viral, que desorganice a la población celular de linfocitos T supresores. Los pacientes con LEG tienen datos de disminución en la inmunidad celular. Pueden alterarse las pruebas de hipersensibilidad tardía en la piel y observarse disminución en la función de las células T in vitro; se ha demostrado la presencia de anticuerpos antilinfocitos en estos pacientes, anormalidad que podría estar relacionada con la linfocitopenia y la hipofunción de las células T.
Sin embargo, este cuadro se ha hecho cada vez más complicado. Las mediciones utilizando anticuerpos monoclonales no han demostrado deficiencia en las células T supresoras en todos los pacientes. En lugar de eso, se ha demostrado que la relación de células T cooperadoras y supresoras presenta amplias variaciones entre los pacientes con LEG cuando se compara con las proporciones que se encuentran en una población control de sujetos normales. Además, en algunos individuos con LEG se ha identificado una deficiencia en un subgrupo específico de células T cooperadoras (también llamadas células T CD4+). Se reconocen dos subgrupos de células T CD4+ con funciones diferentes: los subgrupos 4B4 y 2H4. El subgrupo 4B4 responde a antígenos solubles e induce a los linfocitos B para que secreten inmunoglobulinas. El subgrupo 2H4 (denominado con anterioridad subgrupo de la artritis reumatoide juvenil por la presencia de anticuerpos contra este subgrupo en pacientes con este tipo de artritis) induce a las células T CD8+ a expresar funciones de células T supresoras, incluyendo la supresión de la síntesis de anticuerpos. Los pacientes con LEG y afección renal activa presentan un menor número de células T CD4+ 2H4, observación que se ha relacionado con una función deficiente de las células T supresoras en estos pacientes.30
Los estudios inmunológicos han enfatizado el papel de la apoptosis, un mecanismo fisiológico de muerte celular, en el mantenimiento de la tolerancia. Los defectos en los genes responsables de la regulación de la apoptosis causan enfermedad autoinmune en animales de experimentación . Se ha postulado que la expresión exagerada del gen sintetasa de ácidos grados (fas) causa inhibición de apoptosis de linfocitos autoreactivos en el LEG, pero está hipótesis no se ha comprobado aún.30
Varias observaciones indican que las hormonas sexuales influyen en forma importante en la patogenia del LEG. Apoyan este concepto la frecuencia de la enfermedad en el sexo femenino, los efectos adversos de los estrógenos en hombres y mujeres con la enfermedad y los efectos benéficos de los andrógenos en el lupus murino. Los niveles de varios andrógenos activos están disminuidos en las mujeres con LEG, sobre todo en aquellas con enfermedad activa, tal vez como consecuencia de alteraciones en el metabolismo hormonal.
La incidencia de la enfermedad en los familiares en primer grado de los pacientes con lupus es de uno porciento, aproximadamente. El porcentaje de familiares en primer grado que tienen una prueba de anticuerpos antinucleares positiva es más alto; en general, algunos estudios han comunicado una incidencia que varía de un cuatro a un 14 porciento. Sin embargo, el incremento en la frecuencia de las pruebas de anticuerpos antinucleares positivas en familiares no consanguíneos y consanguíneos sugiere que podría haber factores ambientales que tuvieran alguna relación con la enfermedad. La concordancia de LEG en gemelos monocigotos es de alrededor de 30 porciento, mayor que en gemelos dicigotos, en los que la incidencia es semejante a la de otros familiares en primer grado. Estas observaciones indican que hay factores genéticos que contribuyen a la susceptibilidad para padecer LEG, pero que el factor ambiental también contribuye a la enfermedad. Otro hecho más que apoya la presencia de un factor genético en el LEG son las asociaciones de los antígenos de histocompatibilidad HLA-B8 y HLA-DR3 con el LEG espontáneo y de HLA-DR4 con el LEG inducido por hidralacina.31
Una hipótesis plausible acerca de la patogenia del LEG incorpora varios factores.29 Existen datos que apoyan la existencia de una predisposición genética para la enfermedad, misma que produce activación excesiva de las células B; este fenómeno puede ser el resultado de uno o varios mecanismos, incluyendo un defecto intrínseco en las células B, un defecto en las células T cooperadoras que origine una estimulación anormal de las células B o un defecto en las células T supresoras que ocasione una falla para controlar la proliferación de las células B. La activación de las células B puede originarse por factores exógenos o endógenos, incluyendo agentes químicos, medicamentos y antígenos virales o bacterianos. Las manifestaciones individuales de la enfermedad pueden estar determinadas por factores genéticos o de otro tipo que favorezcan la activación de clases específicas de las células productoras de autoanticuerpos.
En el LEG que se presenta en humanos se ha considerado que los complejos inmunes circulantes son responsables de las lesiones tisulares en muchos de los sistemas. El antígeno que se ha implicado con mayor frecuencia es el ADN nativo, que puede encontrarse, junto con componentes del complemento e inmunoglobulinas, en los riñones afectados. La eliminación de los complejos inmunes circulantes se facilita por su unión a los receptores CR1, que se localizan en cerca del 90 porciento en los eritrocitos. Estos receptores funcionan en parte para transportar a los complejos inmunes circulantes al sistema reticuloendotelial, donde son degradados. La cantidad de receptores CR1 en los eritrocitos es un rasgo hereditario, y los individuos de la población normal pueden tener un número elevado, intermedio o bajo de estos receptores. Los estudios de fragmentos de restricción de longitud polimórfica (FRLP) de ADN indican que la herencia de los receptores del complemento CR1 es similar en pacientes con LEG y en controles sanos. Por lo tanto, los niveles reducidos de receptores CR1 en pacientes con LEG no depende de predisposición genética.32
Tratamiento
El tratamiento del LEG es controversial. Debido a que la enfermedad tiene una evolución muy variable y los patrones sintomáticos difieren mucho entre los distintos pacientes, es difícil valorar la respuesta al tratamiento. Debido a la ausencia de estudios cuidadosamente controlados, las recomendaciones para el tratamiento deben, por desgracia, basarse en criterios poco objetivos.1-4
En pacientes con datos de LEG activo está indicado el reposo en cama.1-4 El estrés físico y emocional afectan de manera adversa la evolución de la enfermedad; deberán evitarse situaciones de estrés como cirugías, infecciones y embarazo.
También debe evitarse la exposición a la luz del sol, ya que alrededor de la tercera parte de los pacientes con LEG son fotosensibles.1 La luz solar provoca exacerbaciones tanto de las manifestaciones sistémicas como de las cutáneas. Cuando no es posible evitar la exposición al sol, los pacientes deben usar pantallas solares que contengan ácido aminobenzoico (PABA) con grado elevado de factor protector (FP). Estas preparaciones contienen éster de PABA y benzofenonas, u otras combinaciones de agentes que bloquean tanto el espectro del eritema como el de longitud de onda larga de la luz ultravioleta.
Ni el uso de anticonceptivos orales ni el tratamiento sustitutivo con estrógenos es dañino en las pacientes con LEG. Por otro lado, en un estudio se asoció el tratamiento estrogénico posmenopáusico con un riesgo de alrededor del doble en el desarrollo de LEG.33 Por lo general deben evitarse los estrógenos en pacientes con síndrome antifosfolípido, aunque esto no ha sido evaluado con certeza en estudios clínicos.
SALICILATOS
El tratamiento antinflamatorio con dosis completas de aspirina está indicado en muchos pacientes que tienen LEG activo, especialmente en individuos con fiebre, artritis, pleuritis o pericarditis. La aspirina deberá tomarse en dosis ligeramente por debajo del umbral de toxicidad. El empleo de preparaciones con capa entérica o con amortiguadores puede reducir la frecuente aparición de los efectos colaterales gastrointestinales que tiene la aspirina; ingerir este medicamento con los alimentos también puede ser de ayuda. La dosis diaria total es variable y los adultos jóvenes pueden requerir hasta 6 g por día. La mayoría de los pacientes adultos requerirán entre 3.6 y 4.8 g por día, pero los pacientes mayores de 65 años de edad con frecuencia requieren dosis menores. Los efectos colaterales, aparte de la gastritis, son raros. Algunos pacientes con LEG activo pueden desarrollar datos de hepatitis inducida por aspirina. Sus manifestaciones suelen limitarse a una elevación en los niveles séricos de aminotransferasas y, en algunas ocasiones, de la fosfatasa alcalina, que disminuyen rápidamente una vez que se suspenden los salicilatos. Los efectos antiplaquetarios de la aspirina pocas veces tienen importancia clínica, a menos que existan otras alteraciones de la coagulación. Probablemente debe evitarse el empleo de salicilatos en el caso de trombocitopenia severa y en pacientes con enfermedad renal.
MEDICAMENTOS ANTIPALUDICOS
Se ha demostrado que la cloroquina y la hidroxicloroquina son útiles para el control de algunas de las manifestaciones del LEG, en especial la erupción cutánea y quizá las artralgias y artritis. Un estudio bien controlado demostró que en el LEG leve o inactivo el tratamiento de mantenimiento con hidroxicloroquina es eficaz para reducir la frecuencia de exacerbaciones de actividad.12,34 Estos medicamentos se administran en dosis de 200 a 500 mg, con las que los efectos adversos son poco frecuentes. Pueden ocurrir efectos gastrointestinales colaterales. La agudeza visual puede disminuir por ciclopegia o depósitos corneales, y este efecto es reversible. El efecto adverso más grave es la retinopatía irreversible, que puede causar ceguera. El riesgo de esta complicación es mínimo, sobre todo si el tratamiento no se prolonga más de un año. Puede presentarse miopatía del músculo esquelético, cardiomiopatía y neuropatía periférica con el uso por tiempo prolongado de cloroquina e hidroxicloroquina.
ESTEROIDES Y MEDICAMENTOS CITOTOXICOS
Los esteroides suprimen en forma dramática muchas manifestaciones del LEG. A pesar de su uso diseminado en este padecimiento, sus efectos sobre la supervivencia son desconocidos, y no son claras sus indicaciones de uso. Los pacientes que tienen manifestaciones que no son graves, como artralgias, erupción cutánea, fatiga, fiebre discreta y dolor pleurítico, pueden ser tratados en forma conservadora con reposo, salicilatos y un antipalúdico. Por lo general, es mejor evitar la toxicidad potencial de los esteroides en estos casos. Las manifestaciones más importantes, como anemia hemolítica severa o trombocitopenia, constituyen indicación para tratamiento esteroide. La ciclofosfamida intravenosa, administrada una vez por mes, es eficaz para tratar la trombocitopenia que no puede manejarse en forma satisfactoria por medio de esteroides, danazol o esplenectomía.35
Una crisis lúpica con fiebre elevada, postración y otros síntomas, puede también requerir de esteroides. La fiebre, artritis, erupciones de la piel, pleuritis, pericarditis y otras manifestaciones pueden responder de manera notable. Sin embargo, puede ser difícil reducir la dosis sin precipitar una recaída. Es entonces cuando los pacientes tendrán que enfrentarse a la toxicidad del uso prolongado de estos fármacos. En el LEG y otros padecimientos reumatológicos, los intentos para reducir la toxicidad empleando dosis en días alternos con frecuencia han sido poco satisfactorios, debido a que los síntomas frecuentemente recurren en los días en que no se administra el medicamento. El uso de pulsos de esteroides por vía intravenosa puede asociarse con menor riesgo de osteonecrosis que la administración diaria por vía oral.5
TRATAMIENTO DE LA ENFERMEDAD RENAL
El tratamiento de la enfermedad renal lúpica también es motivo de controversia. No ha habido un estudio con control satisfactorio que compare a los pacientes tratados con esteroides con los pacientes que no reciben tratamiento. No se ha demostrado que los esteroides alteren la evolución final o el desenlace de la glomerulonefritis en el LEG. No obstante, parece razonable administrar un tratamiento limitado con prednisona en los pacientes con glomerulonefritis membranosa o proliferativa difusa.11 El riesgo de que aparezcan complicaciones graves con el tratamiento a largo plazo indica que es poco prudente administrar una dosis mayor de 40 a 60 mg diarios de prednisona en dosis divididas durante más de tres meses. Después de este esquema inicial de tratamiento de tres meses, deberá reducirse la dosis de manera gradual durante periodo de varias semanas hasta llegar a niveles de 10 a 15 mg por día.
La administración de prednisona en dosis leves a moderadas es adecuada para la enfermedad mesangial o proliferativa focal leve. Sin embargo, no es claro si este tratamiento es necesario porque solo el 10 a 15 porciento de los casos progresa a enfermedad más seria, y no se ha demostrado que el tratamiento con prednisona evite la progresión de la enfermedad.9 La glomerulonefritis focal severa o difusa, demostrada por biopsia, se trata en forma más agresiva porque los estudios controlados han demostrado una mejor evolución de la enfermedad renal (aunque no existe un efecto demostrado en la mortalidad) en los pacientes tratados con ciclofosfamida. Con base en la histología renal, la prednisona se administra en dosis de 1 mg/kg/día durante dos meses y después de disminuye hasta una dosis baja de mantenimiento, junto con ciclofosfamida y otro agente inmunosupresor (por lo general azatioprina). La ciclofosfamida es mejor tolerada si se administra en pulsos intravenosos mensuales en lugar de en dosis diaria por vía oral. Se ha recomendado un esquema de seis meses de pulsos mensuales seguido de un pulso cada tres meses durante un año después de la remisión de la enfermedad renal, pero no se sabe si este esquema es el óptimo. Al parecer, el tratamiento en pulsos se asocia con menor riesgo de cáncer de vejiga que la administración oral a largo plazo, pero se ha reportado esterilidad en alrededor de una cuarta parte de las pacientes que han recibido este tratamiento.11 La gravedad de los cambios histológicos crónicos en la biopsia renal correlaciona con el desarrollo de insuficiencia renal. Así mismo, los pacientes con cambios de grado intermedio respondieron mejor al tratamiento inmunosupresor; al parecer los enfermos con cambios crónicos leves no requieren tratamiento y los que presentaron los cambios más graves no respondieron a éste.11
Se ha reportado que la radiación linfoide total mejoró la evolución de la nefritis lúpica en un estudio de 15 pacientes que habían sido refractarios al tratamiento previo con esteroides y citotóxicos.36 No se observaron efectos colaterales graves entre los pacientes del estudio, pero un reporte subsecuente describió complicaciones fatales y ningún beneficio clínico en otros dos pacientes con LEG rebelde a tratamiento que fueron también tratados con radiación linfoide total.37 Por lo tanto, aún debe establecerse cuál es el papel de este método terapéutico en el LEG. Un estudio aleatorio y controlado demostró que la plasmaféresis, aunada al régimen de esteroides y ciclofosfamida, no mejora la evolución en la nefritis lúpica grave.38
En la actualidad la enfermedad renal terminal (ERT) en el LEG se trata en forma eficaz con hemodiálisis por tiempo prolongado y trasplante. Los pacientes con ERT y LEG y los pacientes con ERT causada por otras enfermedades tienen una capacidad semejante para tolerar la diálisis peritoneal o la hemodiálisis, así como igual supervivencia y supervivencia del injerto después de un trasplante. La actividad global de la enfermedad se mitiga durante la diálisis y después del trasplante, aunque no se conoce aún el motivo de esta mejoría.39,40 A pesar de la disminución gradual del tratamiento inmunosupresor, algunos pacientes se recuperaron de la insuficiencia renal y fueron capaces de suspender el tratamiento dialítico. Las muertes fueron poco frecuentes y por lo general se asociaron con tratamiento esteroide a dosis altas. La recurrencia de la nefritis lúpica después del trasplante es rara. La evolución clínica a largo plazo de los pacientes con ERT causada por LEG es semejante a la de los pacientes con nefropatía terminal por otras causas.39,40
TRATAMIENTO DE LA ENFERMEDAD DEL SNC
La justificación para decidir el tratamiento de las complicaciones neurológicas del lupus eritematoso es aún más endeble. Muchas complicaciones aparentemente serias del SNC se resuelven de manera espontánea, haciendo que la evaluación del tratamiento sea difícil. Aunque no existen estudios controlados, la enfermedad neuropsiquiátrica en el LEG puede responder a la prednisona. Cuando no existe mejoría con el tratamiento convencional de prednisona por vía oral, puede emplearse pulsos de metilprednisolona o de ciclofosfamida para los casos graves.11
Pronóstico
El pronóstico para un paciente con LEG es difícil de determinar. La insuficiencia renal, la proteinuria grave y la anemia son signos pronósticos de gravedad. La mortalidad es más elevada en pacientes que pertenecen a un estrato socioeconómico bajo, pero el aumento en la mortalidad no muestra una predilección independiente para personas de una raza u origen étnico en particular.
Las causas más frecuentes de muerte son la enfermedad renal activa (frecuentemente acompañada de afección en otros sistemas) y las infecciones. Sin embargo, el pronóstico del LEG parece estar mejorando, y más de un 80 porciento de los pacientes con glomerulonefritis proliferativa difusa, la forma más grave de afección renal, sobreviven más de cinco años.1 Los factores que se asocian con progresión hacia la insuficiencia renal irreversible son: menor edad, sexo masculino, elevación de la creatinina sérica y cambios histológicos de esclerosis glomerular e intersticial, además de atrofia tubular, en la biopsia renal.13 Las muertes ocasionadas por complicaciones a nivel de SNC y por infarto del miocardio son poco frecuentes. La aparición de enfermedades neoplásicas malignas es rara.
Lupus eritematoso generalizado inducido por medicamentos
Muchos pacientes han desarrollado manifestaciones clínicas y de laboratorio de LEG en relación con ciertos medicamentos. En algunos de ellos el medicamento parece dar lugar a un complejo sintomático idéntico al del LEG, mientras que en otros puede desarrollarse una forma incompleta. Los criterios que se han propuesto para el LEG inducido por medicamentos son: (1) criterios clínicos similares a los propuestos para el diagnóstico de LEG de aparición espontánea, (2) administración del medicamento antes del inicio de los síntomas, usualmente de manera continua, por periodos que varían de tres semanas a dos años, y (3) signos clínicos y síntomas que desaparecen rápidamente después de suspender el medicamento. Los signos clínicos suelen comenzar a a resolverse en término de días, aunque algunos datos de laboratorio pueden tardar meses o aún años en normalizarse.
Una amplia variedad de agentes terapéuticos ha sido relacionada con el síndrome de lupus inducido por medicamentos [ver tabla 5].
El cuadro clínico es similar al de la enfermedad que se presenta de manera espontánea, y la diferencia consiste en que el lupus relacionado con medicamentos es de menor intensidad y las complicaciones renales son menos frecuentes. El LEG inducido por medicamentos es más frecuente en mujeres que en hombres. Los pacientes tienden a ser de edad media y mayores, probablemente debido a que muchos de los medicamentos implicados se emplean con mayor frecuencia en personas en estos grupos de edad.
Se desconoce la incidencia de síndromes de LEG en pacientes expuestos a medicamentos, pero la forma totalmente desarrollada probablemente está presente en el uno porciento o menos, aún entre aquellas personas expuestas a medicamentos de alto riesgo como la hidralacina y la procainamida. Otros pacientes desarrollan síntomas limitados o aislados, como artralgias, que pueden ser manifestaciones del LEG. Se desarrollan pruebas de anticuerpos antinucleares positivas, usualmente no acompañadas por síntomas de LEG, en más del 50 porciento de los pacientes expuestos a medicamentos de alto riesgo. Los anticuerpos contra ADN nativo, presentes en 50 porciento o más de los pacientes con LEG espontáneo, generalmente no se encuentran en pacientes con LEG inducido por medicamentos; las pruebas de estos anticuerpos son de ayuda para hacer esta distinción.
La frecuencia de las manifestaciones del LEG en pacientes que toman medicamentos como la hidralacina o la procainamida hace poco probable que la acción de estos medicamentos sea únicamente la de desenmascarar una diátesis de fondo. Sin embargo, los factores del huésped pueden ser importantes, y los individuos que desarrollan el síndrome completo pueden tener una predisposición innata. La propensión para padecer el síndrome de LEG relacionado con medicamentos como hidralacina o procainamida está relacionada con el fenotipo de acetilador que tenga el paciente. Estos dos medicamentos son metabolizados por medio del sistema hepático de N-acetiltransferasa. Los pacientes que genéticamente son acetiladores lentos metabolizan los medicamentos más lentamente que los pacientes que son acetiladores rápidos. Durante el tratamiento con hidralacina o con procainamida los acetiladores lentos desarrollan positividad para anticuerpos antinucleares de manera más temprana y el síndrome de LEG con mayor frecuencia que los acetiladores rápidos.
Lupus discoide
El lupus discoide crónico es una forma limitada de lupus eritematoso caracterizada por inflamación superficial de la piel. La lesión típica aparece como una lesión elevada, papular y eritematosa, y después se vuelve atrófica, hiperpigmentada o despigmentada. Los estadios posteriores están asociados con pérdida del cabello , escamas y taponamiento folicular, telangiectasias y atrofia. Las lesiones suelen distribuirse como placas irregulares sobre la cara, piel cabelluda, parte superior de los brazos o pecho. La luz solar con frecuencia agrava esta situación.
La mayoría de los pacientes con lupus discoide tienen resultados normales de las pruebas serológicas y no tiene evidencia de afección a nivel sistémico; sin embargo, una minoría no predecible desarrollará la enfermedad generalizada. Las lesiones discoides también se observan con frecuencia en el lupus generalizado.1
Los esteroides, tópicos o en forma de inyección intralesional, o los antipalúdicos orales suelen ser eficaces en el lupus discoide.
Enfermedad mixta del tejido conjuntivo
Se han descrito pacientes que tienen características clínicas que sugieren la presencia de varias enfermedades reumáticas y que no pueden ser colocados con facilidad en alguna de las categorías diagnósticas. Tales pacientes tienen características que sugieren lupus generalizado, esclerodermia y polimiositis, lo que justifica la definición de una enfermedad reumática distinta: la enfermedad mixta del tejido conjuntivo (EMTC). La presencia de títulos elevados de anticuerpos contra antígenos extraíbles del núcleo también distingue a estos pacientes de los que padecen otras enfermedades reumatológicas.41,42
Las características clínicas de la EMTC son diversas.41,42 El rango de edad en el que se presenta es de los 5 a los 80 años y un 80 porciento de los pacientes son mujeres. Casi todos los pacientes tienen artralgias y alrededor de dos terceras partes de ellos tienen una artritis que se parece a la artritis reumatoide, aunque no suele ser deformante. Se desarrolla inflamación de los dedos debida a un incremento en la colágena de la piel y a edema que puede progresar a los cambios crónicos de la esclerodermia con telangiectasias; estos efectos suelen estar limitados a las manos. Pueden existir erupciones similares a las que se observan en el LEG o en la poliomiositis. La disfunción esofágica consiste en una disminución en la amplitud de la presión del esfínter inferior y de la peristalsis en los dos tercios inferiores del esófago, aunque en la mayoría de los pacientes estos hallazgos no están relacionados con síntomas.
También es común encontrar datos de enfermedad pulmonar, como disminución en la capacidad de difusión, por lo general asintomáticos. De manera ocasional, una enfermedad más diseminada puede ocasionar disnea de esfuerzo asociada con la presencia de infiltrados pulmonares en la radiografía de tórax, enfermedad pleural y, en algunas ocasiones, hipertensión pulmonar. Puede observarse pericarditis.
La miositis consiste en una miopatía proximal con hipersensibilidad y debilidad, elevación de los niveles de las enzimas musculares, electromiografías anormales y cambios patológicos observados en la biopsia de músculo. Todos estos cambios son similares a los que se observan en la polimiositis.
La presencia de enfermedad renal es poco común, y cuando está presente, a menudo es leve. Las biopsias de riñón han mostrado cambios mesangiales, nefritis focal y una nefritis membranosa difusa. La tinciones con inmunofluorescencia han identificado depósitos granulares de IgG, C3 y C4 en la membrana basal glomerular.
La afección del sistema nervioso central no es muy frecuente. La neuralgia del trigémino es la complicación neurológica más común. Otros problemas raros incluyen confusión transitoria, infarto cerebral, meningitis aséptica y convulsiones. La meningitis aséptica en la EMTC responde a los esteroides. Tambien pueden ocurrir neuropatía periférica y psicosis.
Las alteraciones de laboratorio más frecuentes consisten en velocidad de sedimentación elevada, leucopenia e hipergamaglobulinemia difusa. En raras ocasiones existe anemia hemolítica importante con prueba de Coombs positiva y trombocitopenia. Las pruebas de factor reumatoide son positivas en alrededor de la mitad de los pacientes.
En los pacientes con EMTC las pruebas de anticuerpos antinucleares representan los estudios diagnósticos más útiles. Suelen encontrarse títulos elevados de anticuerpos antinucleares fluorescentes con patrón moteado [ver figura 5], que tienden a permanecer constantes sin importar la actividad de la enfermedad. El dato más característico es un resultado fuertemente positivo para anticuerpos contra antígenos RNP (ver antes, Datos de laboratorio, Anticuerpos antinucleares). El suero que contiene anticuerpos contra el antígeno RNPU1 sólo es muy característico en la EMTC. Sin embargo, los anticuerpos anti-RNPU1 no son totalmente específicos de la EMTC.
La evolución de la EMTC es variable, y en algunos pacientes crónica. Las características compatibles con LEG, esclerodermia y polimiositis pueden aparecer de manera simultánea o ir apareciendo en término de muchos meses o años. El tratamiento con esteroides ha tenido al parecer un éxito razonable, aunque hasta la fecha no han habido estudios controlados. Los síntomas leves pueden responder a los medicamentos antinflamatorios no esteroides. Las alteraciones en la función pulmonar y esofágica mejoran con el tratamiento con esteroides.
Aunque por lo general la enfermedad tiene un curso benigno con buen pronóstico, pueden ocurrir complicaciones graves con consecuencias incluso fatales. La afección pulmonar es común y la hipertensión pulmonar puede ser refractaria al tratamiento. Puede ocurrir afección renal, por glomerulonefritis por complejos inmunes o por una vasculopatía semejante a la de la esclerodermia, en alrededor del 25 porciento de los pacientes, en ocasiones con insuficiencia renal secundaria.
Paniculitis nodular sistémica
La paniculitis nodular sistémica, también llamada enfermedad de Weber-Christian, se caracteriza por la presencia de ataques recurrentes de grupos de nódulos subcutáneos, rojos, dolorosos, hipersensibles, que se localizan con mayor frecuencia en las piernas y nalgas.43 Los brotes están acompañados por fiebre, leucocitosis y eosinofilia. Los ataques son autolimitados y tienen una duración de dos a tres semanas. Pueden quedar como secuelas áreas pigmentadas, ligeramente deprimidas. En la enfermedad grave puede estar afectada cualquier parte del cuerpo excepto la cara; pueden desarrollarse sitios de afección extracutánea, como el mesenterio. En una variante de esta enfermedad las lesiones pueden sufrir licuefacción y drenar un material blanco, viscoso, compuesto por tejido adiposo necrótico. La localización de los nódulos inflamatorios en áreas periarticulares origina una aparente artritis aguda en una o más articulaciones, pero no existen estudios del líquido o tejido sinovial.
Los estudios de las lesiones subcutáneas muestran un infiltrado de neutrófilos y eosinófilos acompañado de cambios leucocitoclásticos y grados variables de necrosis de células grasas; las células grasas necróticas están rodeadas por histiocitos, células espumosas y células gigantes.
DIAGNOSTICO DIFERENCIAL
La paniculitis relacionada con LEG también se manifiesta por nódulos inflamatorios subcutáneos que pueden evolucionar hasta formar lesiones crónicas deprimidas con drenaje de grasa necrótica. Las características histológicas de la paniculitis lúpica incluyen un infiltrado linfocítico subcutáneo, vasculitis y depósitos de complejos inmunes en las paredes de los vasos. La paniculitis nodular generalizada y el eritema nodoso difieren en sus características clínicas e histológicas. La paniculitis nodular generalizada puede distinguirse de la vasculitis nodular en que las lesiones de esta última consisten en nódulos subcutáneos, firmes, crónicos y ligeramente hipersensibles, que aparecen con más frecuencia en la parte posterior de las piernas. Estos nódulos tienden a evolucionar hasta formar ulceraciones y cicatrices.
La paniculitis nodular generalizada puede acompañarse de pancreatitis o adenocarcinoma de páncreas. En este caso las lesiones subcutáneas pueden asociarse con poliserositis y artritis.
Ocasionalmente se emplean los medicamentos antinflamatorios no esteroides y los esteroides como tratamiento para la panicuitis nodular generalizada, pero no se ha comprobado la eficacia de estos medicamentos.
Síndrome de Sjögren
El síndrome de Sjögren es una enfermedad autoinmune inflamatoria crónica de las glándulas exócrinas que se relaciona con frecuencia con otros trastornos autoinmunes.44,45 Más del 90 porciento de los pacientes son mujeres mayores de 50 años, aunque el síndrome puede ocurrir en niños y adultos jóvenes. Se calcula que en los Estados Unidos entre dos y tres millones de individuos presentan la enfermedad. Sin embargo, la prevalencia exacta se desconoce porque los síntomas del síndrome pueden ser sutiles y por lo tanto pasan desapercibidos. Aproximadamente el 10 al 15 porciento de los pacientes con artritis reumatoide y un menor número de individuos con otras enfermedades reumáticas (v.gr., lupus eritematoso generalizado y esclerodermia) presentan datos clínicos de síndrome de Sjögren; otros pacientes con trastornos reumáticos se mantienen asintomáticos y la evidencia de síndrome de Sjögren sólo es aparente mediante examen histológico.
La etiología del síndrome de Sjögren es desconocida. Existen algunas evidencias de que puede existir una infección por un retrovirus de tipo A. Al parecer esta infección es la responsable del defecto en las señales intracelulares que se observan en las células T CD4+ que predominan en los tejidos afectados de los pacientes con esta enfermedad.46
MANIFESTACIONES CLINICAS
Las manifestaciones clínicas más frecuentes están relacionadas con el síndrome sicca, que consiste en queratoconjuntivitis seca (resequedad en los ojos) causada por afección de las glándulas lacrimales y por xerostomía (resequedad en la boca) producida por afección de las glándulas salivales.44,45 Los síntomas del síndrome pueden desarrollarse en forma lenta e insidiosa o con rapidez. Cuando el comienzo es rápido, la enfermedad se acompaña de parotiditis episódica. La queratoconjutivitis produce sensación de cuerpo extraño en los ojos, acúmulo variable de secreciones espesas cerca del canto interno de los párpados, menor lagrimeo, ojos rojos y fotofobia. Una película de secreciones espesas sobre la córnea puede interferir con la visión, y la sequedad excesiva puede producir ulceración corneal y pérdida grave de la vista.
La insuficiencia en la salivación produce dificultad en la masticación. La deglución de los alimentos requiere la ingestión frecuente de agua u otros líquidos al comer. Pueden desarrollarse caries dentales severas y ruptura de reparaciones dentales. Con menor frecuencia la sequedad de las vías respiratorias superiores y del árbol traqueobronquial puede causar epistaxis, disfonía, bronquitis o neumonía. En cerca de la mitad de los pacientes ocurre crecimiento de las glándulas parótidas.
La presencia de queratoconjuntivitis seca se establece con certeza por medio de examen con lámpara de hendidura después de teñir la córnea con colorante rosa de bengala, demostrándose queratitis filamentosa y áreas de epitelio corneal denudado. También es útil la prueba de Schirmer, que mide la capacidad de las lágrimas para humedecer una tira de papel filtro cuando se coloca un extremo de este papel dentro del párpado inferior.
Los pacientes con síndrome de Sjögren pueden presentar muchas otras manifestaciones. Se ha descrito la resequedad de la piel y de las mucosas de los genitales. La afección de órganos no exócrinos puede ocasionar tiroiditis autoinmune, neuropatía craneana y periférica y miositis. Otras alteraciones atribuibles al síndrome de Sjögren incluyen el desarrollo de pancreatitis aguda y crónica, de cirrosis biliar con anticuerpos antimitocondriales y diversos trastornos tubulares renales que incluyen la acidosis tubular renal, la diabetes insípida nefrogénica o el síndrome de Fanconi completo. La glomerulonefritis es poco frecuente. En un estudio se encontraron alteraciones neuropsiquiátricas en la mayoría de los pacientes con síndrome de Sjögren, sin que hubiera una enfermedad reumática asociada.47 Las manifestaciones psiquiátricas incluyen depresión y trastornos de la personalidad sin deterioro funcional serio. Las principales anomalías neurológicas son defectos cognoscitivos leves, descargas electroencefalográficas focales y neuropatías por atrapamiento. La elevada frecuencia de afección neurológica sutil indica que las manifestaciones psiquiátricas son de origen orgánico. Estas observaciones difieren de las alteraciones neuropsiquiátricas del lupus eritematoso generalizado; en este último las manifestaciones más comunes son la psicosis y las crisis convulsivas. En un estudio, las alteraciones neuropsiquiátricas en los pacientes con síndrome de Sjögren se parecían con frecuencia a las que se observan en pacientes con esclerosis múltiple.48 En los individuos con síndrome de Sjögren también ocurren con mayor frecuencia reacciones de hipersensibilidad a diversos medicamentos.
Ocurre artritis reumatoide asociada, por lo general del tipo clásico, en cerca de la mitad de los pacientes con síndrome de Sjögren. En otros casos se desarrolla una artritis inflamatoria no erosiva, leve, de predominio en las rodillas y los codos. La poliomiositis, el LEG y la esclerodermia pueden asociarse con el síndrome de Sjögren. El fenómeno de Raynaud ocurre solo o acompañado de otras enfermedades reumáticas. Los síndrome vasculíticos asociados pueden afectar vasos de mediano calibre, como en la poliarteritis nodosa, o vasos de pequeño calibre, en ocasiones con hiperglobulinemia o crioglobulinemia.
DATOS PATOLOGICOS
Las glándulas salivales mayores y menores, las glándulas lacrimales y otras glándulas exócrinas del aparato respiratorio, del aparato digestivo y de la vagina son infiltradas con células B, T y células plasmáticas, con predominio de linfocitos T CD4+. Existe atrofia del tejido acinar secretorio glandular y de algunas de las células que revisten los conductos salivales. La proliferación de las células epimioepiteliales en los conductos produce agrupamientos característicos denominados islotes epimioepiteliales. La arquitectura lobular por lo general no se encuentra muy distorsionada, a menos de que ocurran cambios de seudolinfoma o de linfoma maligno.
ALTERACIONES DE LABORATORIO
Puede medirse la velocidad de flujo salival para establecer si el paciente presenta síndrome de Sjögren. Se coloca jugo de limón en la lengua para estimular el flujo de saliva; a continuación se recolecta la saliva desde el orificio del conducto parotídeo en recipientes especiales. En el síndrome de Sjögren se encuentran velocidades de flujo menores de 0.5 ml por minuto. Las alteraciones de las glándulas salivales principales también pueden establecerse mediante sialografía secretora. En esta técnica se obtienen radiografías de los conductos parotídeos después de inyectar cantidades pequeñas de medio de contraste radiopaco en el orificio de los conductos parotídeos. Los datos patológicos incluyen distorsión del patrón de arborización normal de los conductos y vaciamiento reducido del medio de contraste a partir de los conductos. De manera alternativa, la función salival puede calcularse mediante gamagrafía, que mide la captación y liberación de un radioisótopo como el pertecnectato de tecnecio-99m después de la estimulación de las glándulas salivales.
Los síntomas de queratoconjuntivitis seca y xerostomía suelen sugerir el diagnóstico de síndrome de Sjögren, que puede establecerse en la mayoría de los casos por medio de tinción de la córnea con rosa de bengala y gamagrafía salival. El diagnóstico definitivo del síndrome se realiza por medio de una biopsia del labio inferior, con estudio histológico de las glándulas salivales menores. Este procedimiento cuantifica el infiltrado linfocitario y la distorsión de la arquitectura de las glándulas salivales menores. Sin embargo, si tanto la tinción con rosa de bengala como la gamagrafía salival dan resultados positivos, puede realizarse diagnóstico de síndrome de Sjögren sin necesidad de biopsia de labio.49
La velocidad de sedimentación globular suele encontrarse elevada y la hipergamaglobulinemia es común. Existe factor reumatoide positivo, que es un anticuerpo contra IgG, en más del 90 porciento de los pacientes con síndrome de Sjögren. Un alto porcentaje de los factores reumatoides de estos pacientes comparten una característica estructural común que a menudo no está presente en los factores reumatoides observados en pacientes con otras enfermedades. Los anticuerpos monoclonales identifican un idiotipo de reacción cruzada común sobre el segmento variable de las cadenas ligeras de inmunoglobulinas tipo k en los factores reumatoides en el síndrome de Sjögren. Además, las células B que originan las células productoras de esta clase de factores reumatoides, se acumulan de manera selectiva en las lesiones de las glándulas salivales, pero no se encuentran en los tejidos sinoviales ni en la sangre. Es posible que la presencia de este idiotipo identifique clonas de células B que poseen una frecuencia aumentada de transformación neoplásica, y esta observación tal vez explique la asociación de linfoma no-Hodgkin con el síndrome de Sjögren.
El 70 porciento de los pacientes presentan anticuerpos antinucleares, que suelen mostrar un patrón homogéneo o moteado en la inmunofluorescencia. En la mayoría de los casos se encuentran anticuerpos contra el antígeno nucleoproteico SS-B, también conocido como La, y aproximadamente en la mitad de los pacientes se detectan anticuerpos contra el antígeno SS-A, también llamado Ro. Cuando el síndrome de Sjögren se asocia con artritis reumatoide, los anticuerpos anti-SS-A y SS-B se observan en menos del 10 porciento de los pacientes. Se han identificado otros autoanticuerpos, pero cada uno se encuentra presente sólo en una minoría de los pacientes, lo que ilustra la naturaleza general (inespecífica de un órgano en particular) de la autoinmunidad en el síndrome de Sjögren. Estos incluyen anticuerpos contra las células parietales gástricas, tiroides, músculo liso, células pancreáticas y mitocondrias. Pueden detectarse anticuerpos contra antígenos del epitelio de los conductos salivales en pacientes con síndrome de Sjögren, pero por lo general sólo en asociación con artritis reumatoide.
Las evidencias de una base genética para la predisposición a la enfermedad consisten en una mayor prevalencia de los antígenos de histocompatibilidad HLA-DR3 y el supertipo MT2 del sistema HLA (DRw52). El síndrome de Sjögren primario (i.e., en el que no existe ninguna otra enfermedad asociada) se relaciona en forma muy estrecha con el antígeno de histocompatibilidad HLA-DR3, según estudios realizados en poblaciones caucásicas. Las evidencias indican que el alelo HLA-DR3 no sólo se encuentra en desequilibrio de unión, sino que constituye en sí un factor importante de susceptibilidad para la enfermedad.50 El síndrome de Sjögren en el sexo masculino también se relaciona con un aumento en la prevalencia MT2, pero con una prevalencia normal de DR3. Las manifestaciones clínicas del síndrome de Sjögren son similares en hombres y mujeres, pero en el sexo masculino existe menor incidencia de pruebas positivas para factor reumatoide y de anticuerpos contra SS-A.
TRATAMIENTO
Por lo general el síndrome de Sjögren debe tratarse de manera conservadora.44,45 Las preparaciones de lágrimas artificiales suelen ser eficaces para la queratoconjuntivitis seca. Otras complicaciones, como la ulceración corneal, requieren atención oftalmológica intensiva. Los agentes mucolíticos pueden ser necesarios para la eliminación de las secreciones espesas y pegajosas, y los antibióticos están indicados en caso de infección local. La xerostomía por lo general se trata aumentando la cantidad y la frecuencia de líquidos orales, aunque en ocasiones son de utilidad las preparaciones de saliva artificial. La candidiasis oral puede requerir la aplicación tópica de nistatina. Es importante la higiene dental cuidadosa, incluyendo el enjuage bucal escrupuloso, el cepillado dental, los programas de control de placas, las aplicaciones de fluoruro y la reducción de la ingestión de sacarosa. Las soluciones nasales salinas y el aumento en la humedad pueden ser de utilidad. Las otras enfermedades reumáticas asociadas deberán tratarse de la misma forma que cuando no coexisten con el síndrome de Sjögren. Los esteroides y los medicamentos citotóxicos no suelen estar indicados en el síndrome de Sjögren no complicado.
SINDROME DE LINFOCITOSIS INFILTRATIVA DIFUSA
En los pacientes con infección por VIH ocurre un síndrome de linfocitosis infiltrativa difusa muy semejante al síndrome de Sjögren. Los pacientes con este trastorno sufren crecimiento parotídeo bilateral, xerostomía y, con menos frecuencia, queratoconjuntivitis seca, además de una gran prevalencia de infiltración con linfocitos CD8 en tejidos extraglandulares.51 Esta infiltración suele causar insuficiencia respiratoria por neumonitis intersticial linfocítica; con menos frecuencia los síntomas se relacionan con la presencia de infiltrados en el sistema nervioso central y en el tubo digestivo. El síndrome de linfocitosis infiltrativa difusa asociado con infección por VIH difiere también del síndrome de Sjögren en que en pocas ocasiones se encuentran anticuerpos antinucleares o factores reumatoides positivos. Además, se relaciona en forma intensa con la presencia de HLA-DR5 en los enfermos de raza negra.
El síndrome de linfocitosis infiltrativa difusa puede distinguirse del síndrome de Sjögren por medio del estudio de una muestra de biopsia de glándula salival del labio. Los estudios inmunohistoquímicos de este tejido pueden detectar diferencias entre los antígenos de superficie en los linfocitos y células epiteliales de estos dos padecimientos.
El tratamiento de la neumonitis infiltrativa linfocítica con medicamentos inmunosupresores ha sido eficaz y bien tolerado, mientras que el tratamiento de otras enfermedades autoinmunes, como el síndrome de Reiter, en pacientes con infección por VIH se ha asociado con infecciones oportunistas graves.
LINFOMA ASOCIADO CON SINDROME DE SJÖGREN
En el síndrome de Sjögren la infiltración linfoide de los tejidos salivales es constante. El desarrollo de linfoma debe sospecharse cuando la proliferación linfoide se vuelve extrema, provocando crecimiento firme y prominente de las glándulas salivales. Asimismo, puede desarrollarse hiperplasia linfoide en sitios extrasalivales, incluyendo los ganglios linfáticos regionales próximos a la cabeza y cuello, al igual que los que se encuentran en regiones distantes. La infiltración de linfocitos en otros órganos, como riñones o pulmones, puede alterar la función de dichos órganos. La proliferación linfoide excesiva, también conocida como seudolinfoma, tiene un aspecto histológico pleomórfico, con distorsión de la arquitectura de los ganglios linfáticos. Puede desarrollarse un linfoma maligno franco, independientemente de que exista seudolinfoma. En un estudio se calculó que el riesgo de linfoma maligno en pacientes con síndrome de Sjögren fue 44 veces mayor de lo normal. Las neoplasias por lo general son linfomas no-Hodgkin de la estirpe de las células B y a menudo producen IgMk.53
Los trastornos linfoproliferativos que se asocian con el síndrome de Sjögren pueden requerir tratamiento agresivo. El seudolinfoma suele responder en forma satisfactoria a dosis bajas de esteroides y ciclofosfamida, pero el linfoma maligno evidente requiere el uso de esquemas terapéuticos más agresivos.
Bibliografía
DR. DWIGHT R. ROBINSON
El lupus eritematoso generalizado (LEG) tiene grandes variaciones en cuanto a manifestaciones clínicas, modo de presentación y evolución. La evolución de este padecimiento inflamatorio crónico y multisistémico puede variar de benigna a mortal. No existe curación, pero el tratamiento médico disminuye con frecuencia los síntomas del LEG.
La incidencia no se conoce con precisión. En dos grandes estudios se encontró una incidencia anual de alrededor de siete casos por 100,000 personas.1 Un noventa porciento de los casos de LEG se presentan en mujeres; las mujeres de raza negra se ven afectadas con mayor frecuencia que las de raza blanca. Los primeros síntomas suelen aparecer entre los 15 y 25 años de edad.
Manifestaciones clínicas
Un cuadro clínico de fiebre, erupción y artritis en una mujer joven deberá hacer sospechar la posibilidad de LEG. La forma de presentación también puede ser la de una anemia hemolítica aguda, una púrpura trombocitopénica o pericarditis. Es usual la remisión, que puede estar seguida por un intervalo libre de síntomas de varios años de duración. Es factible la recurrencia a largo plazo, manifestada como una serie de formas clínicas. La gravedad varía, desde una enfermedad aguda que afecta de manera simultánea varios sistemas orgánicos hasta una enfermedad leve con una morbilidad insignificante. La evolución es impredecible; es probable que la afección renal o del sistema nervioso central sea un signo de mal pronóstico.1-4
Existen varios factores que pueden precipitar las exacerbaciones, incluyendo el estrés físico o emocional, la luz solar, cirugías, embarazos y ciertos medicamentos. La enfermedad puede recurrir sin causa aparente.
FIEBRE E INFECCIONES
Se presenta fiebre en la mayoría de los pacientes con LEG. Esta suele ser leve, pero puede ser severa durante las llamadas crisis lúpicas. Con frecuencia se observa evidencia de afección multisistémica acompañando a la hipertermia severa en estas crisis, pero también puede ser la fiebre la única manifestación.1-4 La fiebre causada por el LEG debe distinguirse de la ocasionada por una infección añadida. Los pacientes con lupus activo tienen mayor predisposición a sufrir infecciones, sobre todo causadas por organismos oportunistas, y las infecciones son una causa muy importante de morbimortalidad en estos pacientes.5 Aunque las altas dosis de medicamentos inmunosupresores que se emplean en el lupus pueden favorecer las infecciones, un estudio encontró que la frecuencia de infecciones graves, como neumonía y septicemia, se asoció con la actividad del lupus, pero no con el uso de dosis moderadas de esteroides.6 El tratamiento habitual con inmunosupresores, como dosis bajas o moderadas de prednisona (20 mg al día), rara vez contribuye a la mayor propensión a infecciones. Los pacientes tratados con medicamentos inmunosupresores tienen una mayor incidencia de herpes zoster (con una incidencia de 21 porciento en un estudio), pero esta infección pocas veces se disemina.3 Los calosfríos, la leucocitosis neutrofílica y los niveles normales de anticuerpos anti-ADN se asocian más con fiebre causada por infección bacteriana que con la fiebre propia de la actividad del LEG.
TRASTORNOS MUSCULOESQUELETICOS
Ocurre artritis no deformante, una de las manifestaciones más frecuentes del LEG, en alrededor del 90 por ciento de los pacientes. Se manifiesta como artralgias, edema y derrame articular y rigidez matinal. La artritis es simétrica y poliarticular, y afecta con más frecuencia a las manos, muñecas, codos, rodillas y tobillos. Los episodios de artritis suelen ser intermitentes, durando horas a meses y con periodo de remisión variables. 1-4 La artritis semejante a la de la artritis reumatoide, que ocasiona erosiones óseas, es poco frecuente y se asocia con pruebas positivas de factor reumatoide en el suero. Los pacientes con esta forma de artritis tienen menos posibilidad de desarrollar nefritis lúpica que los que no tienen artritis o en los que la artritis es no deformante.7 También ocurre artropatía de Jaccoud, y se asocia con anticuerpos contra ribonucleoproteína U1. Esta enfermedad articular consiste en desviación cubital y deformidades de los dedos en cuello de cisne, que difieren de las mismas deformidades de la artritis reumatoide porque no ocasionan pérdida del cartílago articular ni erosiones óseas.7-8
Puede desarrollarse tendinitis. Los tendones de Aquiles, rotuliano y de la mano rara vez se rompen, y cuando esto ocurre, suele tratarse de pacientes que reciben esteroides.1 Otra forma de afección esquelética es la osteonecrosis o necrosis avascular, la cual puede ser asintomática u ocasionar dolor de gran intensidad. Las grandes articulaciones, por lo general más de una al mismo tiempo, son las afectadas con más frecuencia, sobre todo las de las caderas y rodillas. La osteonecrosis es una complicación conocida del tratamiento con esteroides, pero también puede presentarse como parte del proceso patológico del LEG.9
El análisis del líquido sinovial muestra evidencia de inflamación leve, a pesar del aspecto de las articulaciones en el examen físico. La cuenta de leucocitos suele ser menor a 3,000/mm3, a diferencia de la cuenta celular elevada en la artritis reumatoide. El líquido es viscoso y contiene un buen precipitado de mucina. La concentración de glucosa es parecida a la sérica. El estudio histológico sinovial muestra evidencias de inflamación discreta, a diferencia de la intensa reacción inflamatoria que caracteriza a la artritis reumatoide. Los únicos datos histológicos en el LEG suelen ser la infiltración articular leve por linfocitos y células plasmáticas y cierto edema y necrosis fibrinoide.1
LESIONES EN PIEL
Las lesiones en piel son frecuentes, y con frecuencia un dato diagnóstico importante.1-5,9 Lo más típico es la presencia de eritema en la cara, sobre la región nasal y malar. El eritema "en alas de mariposa" puede ser la primera manifestación de la enfermedad [ver figura 1]. El lupus cutáneo agudo ocurre también en otros sitios, en especial en zonas de exposición al sol. El lupus cutáneo subagudo consiste en una erupción eritematosa y papuloescamosa, que puede ser anular. El lupus cutáneo crónico, que en ocasiones se denomina lupus discoide (LD), puede ocurrir en ausencia de LEG [ver adelante, Lupus discoide]. Consiste en áreas de atrofia con despigmentación central y mayor pigmentación en los bordes junto con telangiectasias, descamación variable y dilatación y taponamiento de los folículos pilosos. Todas las lesiones cutáneas del lupus son precipitadas o exacerbadas por la exposición a la luz solar. Uno a dos tercios de los pacientes con lupus tienen fotosensibilidad, y en estos pacientes los síntomas sistémicos o las erupciones cutáneas pueden exacerbarse después de la exposición al sol. La mayoría de los pacientes con fotosensibilidad tienen anticuerpos anti-Ro en el suero. No se conocen los mecanismos de la fotosensibilidad, pero las evidencias sugieren lesión celular inducida por luz ultravioleta, que causa transferencia de antígenos intracelulares a las superficies celulares, en donde pueden inducir una respuesta autoinmune.5 Son lesiones cutáneas menos específicas del lupus un eritema en parches, con o sin descamación, que aparece en las extremidades, tronco y cara, y parches eritematosos alrededor de las yemas de los dedos, palmas y áreas periungueales, por lo general acompañadas de telangiectasias en los bordes de las uñas de los dedos.
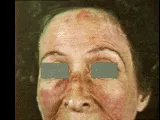
|
| Figura 1 |
| Eritema en alas de mariposa |
Ocurre alopecia, que se caracteriza por adelgazamiento difuso del cabello en el cuero cabelludo normal, pero también puede presentarse eritema crónico, cicatrización y atrofia del cuero cabelludo. En ausencia de cambios crónicos en la piel cabelluda, la pérdida de cabello, a pesar de que puede ser muy extensa, es reversible.1 La alopecia se asocia con otras manifestaciones cutáneas y con fenómeno de Raynaud, y su presencia se relaciona con el grado de actividad de la enfermedad. La alopecia no se asocia con afección de otros órganos o estructuras específicos, como articulaciones, riñones o sistema nervioso central.10
Pueden observarse ulceraciones de la boca o labios, como áreas pequeñas eritematosas, con edema y dolorosas. La púrpura se asocia con trombocitopenia, pero puede ocurrir aún cuando las plaquetas sean normales como resultado de vasculitis de pequeños vasos.
Aunque las recaídas de la enfermedad sistémica en los pacientes con lupus pueden acompañarse del desarrollo de erupciones cutáneas, los pacientes con enfermedad cutánea no tienen más riesgo de desarrollar afección renal o neurológica que los pacientes sin estas manifestaciones. Las lesiones cutáneas del LEG responden a antimaláricos en la mayoría de los casos. Otros agentes, como dapsona, azatioprina y retinoides, pueden ser benéficos en los pacientes resistentes al tratamiento antimalárico.5
DISFUNCION RENAL
Los pacientes con LEG presentan de manera usual algún grado de afección renal. El examen general de orina puede mostrar proteinuria, piuria, hematuria y cilindros granulares o de eritrocitos. La afección renal más severa puede ocasionar hipoalbuminemia, otras características del síndrome nefrótico o disminución en la filtración glomerular, lo que origina retención de productos nitrogenados, alteraciones electrolíticas y acidosis.1,-4,11
La lesión más benigna de la nefritis lúpica es la de cambios mesangiales mínimos, probablemente presente en la mayoría o en la totalidad de los pacientes lúpicos [ver tabla 1], aunque esta lesión no tiene una morbilidad significativa. La forma proliferativa focal de la lesión renal [ver figura 2] también es benigna y con frecuencia se resuelve. La glomerulonefritis membranosa [ver figura 3] se acompaña de manera característica de uno o todos los elementos que constituyen el síndrome nefrótico. Las remisiones son comunes, de la misma manera que las recaídas, y puede desarrollarse una insuficiencia renal lentamente progresiva. La nefritis proliferativa difusa [ver figura 4] constituye la forma más grave de afección renal, y con frecuencia está asociada con algún grado de insuficiencia renal e hipertensión. Sin embargo, el tipo de patología renal cambia con el paso del tiempo en más de la mitad de los pacientes con enfermedad renal comprobada, un hecho que limita el valor pronóstico que pudiera tener la biopsia renal.
|
|
|
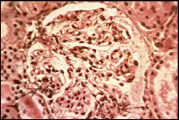
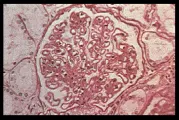
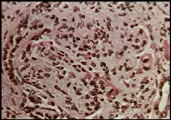
|
|
|||||||||||||||||||||||||
|
El análisis de las biopsias renales utilizando varios criterios ha originado índices de actividad y cronicidad de la enfermedad renal [ver tabla 2]. Las calificaciones altas del índice de cronicidad (lesiones crónicas, irreversibles) predicen mayor riesgo de insuficiencia renal, sobre todo en personas jóvenes, mientras que las pruebas de función renal anormal y calificaciones altas en el índice de actividad (lesiones activas e individuales) pueden no predecir una mala evolución porque las lesiones pueden ser reversibles.
|
||||||
Nota: cada hallazgo se califica en 3 puntos, la necrosis fibrinoide y las medias lunas celulares se multiplican por un factor 2; el índice máximo de actividad es de 24 y el índice máximo de cronicidad es de 12 |
ALTERACIONES DEL SISTEMA NERVIOSO
En alrededor de la mitad de los pacientes con lupus existe alguna alteración del sistema nervioso central.1-4,11 Incluyendo diversos cambios psicológicos y neurológicos [ver tabla 3]. Las manifestaciones neuropsiquiátricas pueden aparecer en cualquier momento, pero casi siempre lo hacen pronto, durante el primer año de evolución. Por lo general se asocian con evidencia de actividad en otros órganos y sistemas. La depresión reactiva es muy frecuente. Puede ocurrir una psicosis funcional con trastornos del pensamiento y del ánimo característicos de la esquizofrenia, pero es más común el síndrome orgánico cerebral, que ocurre en hasta el 20 por ciento de los pacientes con lupus. Este incluye varios grados de pérdida de memoria, desorientación y deterioro intelectual, y puede ocurrir en ausencia de otros síntomas neurológicos o de actividad sistémica. En algunos pacientes se presenta deterioro lento y progresivo, que causa demencia incapacitante. La psicosis puede ser una complicación del tratamiento con esteroides, aunque también se ha establecido que los problemas psicológicos son una manifestación frecuente del LEG. Además, estas psicosis se asocian con crisis convulsivas y otras evidencias de alteración neurológica. Con frecuencia las crisis convulsivas son de tipo gran mal, aunque se han descrito crisis focales.
|
||||||||||||||||||||||||
|
Los cambios neurológicos incluyen signos de afección de la vía piramidal. Estas alteraciones, junto con la parálisis espástica, se deben en ocasiones a un accidente cerebrovascular típico. Las alteraciones de pares craneales afectan con frecuencia al séptimo par (manifestada como parálisis facial) y los nervios motores extraoculares (que ocasionan ptosis o diplopia). Las neuropatías periféricas son poco comunes y pueden manifestarse como deficiencias sensitivas o mixtas (sensitivas y motoras). En raras ocasiones puede haber afección múltiple y asimétrica de nervios periféricos, manifestada por un cuadro de mononeuritis múltiple, igual al que ocurre en muchas vasculitis necrosantes. Otras alteraciones neurológicas poco frecuentes incluyen ataxia, corea, escotomas, meningitis aséptica y lesiones de la médula espinal.
Las manifestaciones en el sistema nervioso central son típicamente pasajeras. La psicosis, las convulsiones, la hemiplegia, la diplopia y otros hallazgos, con frecuencia ceden de manera notable y rápida, excepto en el caso poco usual en el que hay una complicación irreversible, como una hemorragia intracerebral grave. Es difícil determinar si el tratamiento con esteroides u otros medicamentos influyen en el pronóstico de la afección a nivel del SNC.
Las pruebas neurológicas revelan pocos cambios que no se hayan ya detectado en el examen físico. Incluso ante alteraciones neurológicas francas, la punción lumbar puede ser normal o demostrar sólo aumento en la concentración de proteínas y pleocitosis leve. El aumento sérico en la concentración de autoanticuerpos contra la proteína ribosomal P se considera específico de LEG y se asocia con depresión grave y psicosis, pero los niveles aumentados de estos anticuerpos no se asocian con enfermedad neurológica no psiquiátrica.12
Las imágenes por resonancia magnética (IRM) pueden demostrar alteraciones asociadas con las manifestaciones neuropsiquiátricas del LEG. Los pacientes con enfermedad difusa del sistema nervioso central, como psicosis, crisis convulsivas generalizadas y disfunción cognoscitiva, tienen áreas de mayor intensidad de señales, que se distribuyen en forma simétrica, en la sustancia blanca subcortical. Estas alteraciones son reversibles. Por el contrario, los enfermos con alteraciones focales tienen áreas de atrofia de distribución asimétrica y aumento en la intensidad de señales en regiones que corresponden a los vasos cerebrales principales. Estos trastornos no son reversibles y se asocian con aumento en los niveles de anticuerpos antifosfolípido [ver adelante, Síndrome de anticuerpos antifosfolípido].11
Los enfermos con LEG que no tienen enfermedad neuropsiquiátrica grave pueden tener disfunción cognoscitiva menos grave que se manifiesta por pérdida de la memoria, cefalea y disminución en la capacidad de trabajo e interacción social. Las pruebas neuropsicológicas de un grupo de pacientes con LEG demostraron un pobre desempeño de tareas complejas de atención y de actividades que requieren capacidad visoespacial. Esta disfunción cognoscitiva fue independiente de la actividad de la enfermedad. del uso de esteroides y de otras patologías neuropsiquiátricas.13
La patología de la afección en el SNC se caracteriza por la escasez de hallazgos histológicos, a pesar de la presencia de manifestaciones clínicas muy notables, excepto en el caso de infarto cerebral secundario a oclusión trombótica o embólica de un vaso importante. El hallazgo más frecuente consiste en un infiltrado celular inflamatorio perivascular leve; es poco usual la presencia de una verdadera vasculitis necrosante en el cerebro. Son comunes los microinfartos, con frecuencia múltiples. Además, puede ocurrir lesión y disfunción neuronal por la presencia de anticuerpos antineurona. Estos se encuentran hasta en el 75 por ciento de los pacientes con síntomas neuropsiquiátricos del lupus y pueden existir también en el 25 por ciento de los enfermos sin trastornos neuropsiquiátricos. Existen anticuerpos contra la proteína Pribosomal en la mayoría de los pacientes con psicosis o depresión.11,13
AFECCION CARDIACA
La pericarditis, que se manifiesta por dolor y frote torácicos, es la manifestación cardiaca más frecuente.1-4,11 Pueden ocurrir derrames pequeños, pero el tamponade es muy poco frecuente. La miocarditis es mucho menos común que la pericarditis. La miocarditis puede simular una cardiomiopatía, ocasionando arritmias ventriculares e insuficiencia cardiaca congestiva. Se observa una gran prevalencia de anticuerpos anti-Ro en los pacientes con miocarditis y alteraciones de la conducción cardiaca.14 La endocarditis de Libman-Sacks, una endocarditis no infecciosa que se caracteriza por vegetaciones verrucosas pequeñas cerca de los anillos de las válvulas cardiacas, se presenta con frecuencia. Estas lesiones suelen ser asintomáticas, pero en algunas ocasiones se complican por tromboembolias, endocarditis infecciosa o disfunción valvular. Se encuentra cardiopatía valvular en el 18 porciento de los pacientes con LEG, la mayoría de los cuales tienen anticuerpos antifosfolípido.15 La coronariopatía ateroesclerótica es una complicación importante en el LEG, y puede causar hasta el tercio de los fallecimientos en este grupo de pacientes. Las causas de enfermedad coronaria incluyen hiperlipidemia, que es agravada por el tratamiento con esteroides, y la hipertensión, que puede ser secundaria a la nefropatía lúpica. La vasculitis coronaria puede causar oclusión de estas arterias, pero es una causa mucho menos frecuente de infarto al miocardio que la ateroesclerosis.5
AFECCION PULMONAR
El dolor torácico pleurítico, otra manifestación común, puede ser de intensidad leve o intensa y estar acompañado de frotes y derrames pleurales.1-4 El análisis del líquido pleural suele revelar la presencia de un exudado que contiene concentraciones de proteínas mayores de 3 g/dl; también pueden observarse trasudados. La cuenta de leucocitos mononucleares puede elevarse hasta varios miles de células por mm3. Los niveles de glucosa en líquido pleural suelen ser similares a los de sangre; en comparación, en la artritis reumatoide los niveles de glucosa en el líquido pleural están reducidos.
La afección del parénquima pulmonar en LEG puede ser aguda o crónica.11,16 Se han descrito tres síndromes agudos. El más común, la neumonitis lúpica aguda, se caracteriza por fiebre de inicio súbito, disnea, hipoxemia e infiltrados alveolares en parches en la radiografía de tórax. El segundo en frecuencia es la hemorragia alveolar, semejante clínicamente a la neumonitis aguda pero con hemorragia en el pulmón, lo que causa disminución en la concentración del hematocrito. Ambos síndromes progresan con rapidez a insuficiencia respiratoria y muerte. El tratamiento con pulsos de metilprednisolona parece ser benéfico, con adición de azatioprina o ciclofosfamida en casos resistentes, aunque no se han realizado estudios controlados. El síndrome menos frecuente de los tres es la hipoxemia aguda reversible, que ocurre en pacientes muy graves con LEG. Se ha postulado que los componentes activados del complemento pueden activar neutrófilos, secuestrándolos en la vasculatura pulmonar. La prednisona es eficaz en pacientes con hipoxemia aguda reversible.
La enfermedad pulmonar crónica ocurre en el lupus eritematoso generalizado como una enfermedad pulmonar intersticial o como hipertensión pulmonar. La enfermedad pulmonar intersticial puede consistir en alveolitis inflamatoria, que responde a tratamiento inmunosupresor, o a fibrosis intersticial, que no lo hace. La evaluación del pulmón afectado por tomografía computada de alta resolucion puede ayudar a distinguir entre la alveolitis inflamatoria y la fibrosis intersiticial.
La hipertensión pulmonar se asocia con alta incidencia de fenómeno de Raynaud, anticuerpos antirribonucleoproteína, factor reumatoide y anticuerpos antifosfolípido. No se conoce un tratamiento eficaz y el pronóstico es malo.
La hipertensión pulmonar en los pacientes con LEG es semejante a la hipertensión pulmonar primaria.11,16 Los pacientes sufren disnea de ejercicio y en ocasiones dolor torácico. Por lo general las radiografías de tórax son normales y no existe evidencia de embolia pulmonar, a pesar de que suelen existir anticuerpos antifosfolípido en estos pacientes.12
ALTERACIONES GASTROINTESTINALES
Son frecuentes la anorexia, náusea, vómito y dolor abdominal, por lo general sin causa aparente. La vasculitis mesentérica puede ocasionar infartos intestinales y perforación. En algunos pacientes se presenta pancreatitis asociada con el tratamiento esteroide. La enteropatía perdedora de proteínas, que responde al tratamiento con esteroides, puede ser una complicación del LEG.18
ENFERMEDAD HEPATICA
De manera ocasional se aprecian en el LEG hepatomegalia y alteraciones mínimas en las pruebas de función hepática, pero esto suele tener poca importancia clínica. Las biopsias de hígado muestran solamente cambios mínimos, inespecíficos.1 Es rara la hepatitis crónica1-3 En algunos pacientes con cirrosis biliar primaria se ha publicado la coexistencia con LEG.
SINDROME DE ANTICUERPOS ANTIFOSFOLIPIDO
El síndrome de anticuerpos antifosfolípido consiste en un estado de hipercoagulabilidad que ocurre en asociación con autoanticuerpos contra fosfolipídos.19,20 Los anticuerpos antifosfolípido no se unen directamente a los fosfolípidos aniónicos, sino que se fijan a epítopes en varias proteínas plasmáticas, incluyendo la ?-glucoproteína I, que forma un complejo con los fosfolípidos. La formación de complejos entre la ?-glucoproteína I y los fosfolípidos causa un cambio conformacional que expone un epítope antes escondido al cual se unen los anticuerpos antifosfolípidos. Los complejos formados entre fosfolípidos y varios factores de la coagulación, incluyendo la protrombina, la proteína C y la proteína S, pueden fijar también anticuerpos antifosfolípido, pero se desconoce aún el mecanismo por el que se presenta la coagulopatía en este síndrome.8,21
Existen anticuerpos antifosfolípido en alrededor de la tercera parte de los pacientes con LEG. Estos anticuerpos pueden desarrollarse también en pacientes con otras enfermedades del tejido conjuntivo, así como con padecimientos infecciosos como sífilis, infección por virus de inmunodeficiencia humana (VIH) y enfermedad de Lyme. También pueden presentarse después del tratamiento con ciertos medicamentos, sobre todo fenotiacinas, procainamida, hidralacina y fenitoína. Sin embargo, los anticuerpos antifosfolípido asociados con infecciones y tratamiento farmacológico existen en bajas concentraciones y no suelen causar enfermedad clínica significativa.
Varias manifestaciones clínicas se asocian con el síndrome de anticuerpos antifosfolípido [ver tabla 4].8,19 El síndrome primario ocurre en pacientes sin LEG y el secundario en pacientes con evidencia clínica de LEG. Los pacientes con la forma primaria pueden tener otros datos, como lívedo reticulares y trombocitopenia crónica [ver tabla 4]. Las complicaciones tromboembólicas se presentan en pacientes con el síndrome primario o con asociación con LEG. Las trombosis, la muerte fetal y la trombocitopenia son las manifestaciones más frecuentes. Las trombosis pueden afectar los lechos vasculares arterial o venoso, y tienden a recurrir después del tratamiento.22 Las trombosis venosas afectan a las extremidades o a los sistemas renal, porta o mesentérico. Con frecuencia las trombosis venosas periféricas ocasionan embolia pulmonar. Las trombosis arteriales, que afectan sobre todo la circulación cerebral pueden causar infartos cerebrales, isquemia cerebral transitoria, isquemia ocular, encefalopatía y demencia por infartos múltiples. Pueden ocurrir trombosis arteriales en múltiples órganos, que suelen ser fatales. Puede ser difícil distinguir la oclusión vascular secundaria a vasculitis en el LEG activo de la vasculopatía no inflamatoria asociada con el síndrome de anticuerpo antifosfolípido. La distinción puede ser importante porque la vasculitis se trata con fármacos inmunosupresores y la coagulopatía del síndrome antifosfolípido con anticoagulantes.5 Las alteraciones neurológicas incluyen cefalea migrañosa y mielitis transversa. El livedo reticularis, un patrón visible, irregular y lineal de venas cutáneas dilatadas, es frecuente. Puede existir necrosis cutánea, así como síndrome de Sneddon (que consiste en la triada de livedo reticularis, enfermedad cerebrovascular e hipertensión lábil). La muerte fetal recurrente, que es hasta dos veces más frecuente en las pacientes con síndrome de anticuerpos antifosfolípido que en la población general, ocurre en el segundo trimestre, y se piensa que se debe a insuficiencia placentaria. Las placentas afectadas muestran vasculopatía y trombosis. En raros casos existe anemia hemolítica con Coombs positivo.
|
|
|
El síndrome de anticuerpos antifosfolípido contribuye a la enfermedad cardiaca en el LEG. La enfermedad valvular en pacientes con LEG difiere de la que se presenta en pacientes con síndrome primario de anticuerpos antifosfolípido. Los pacientes con LEG tienen engrosamiento de las valvas de las válvulas mitral y aórtica y vegetaciones, así como estenosis e insuficiencia valvular, mientras que los pacientes con el síndrome primario tienen engrosamiento valvular sin vegetaciones e insuficiencia valvular en ausencia de estenosis.15 Las trombosis coronarias causadas por el síndrome de anticuerpos antifosfolípido pueden ocasionar infartos del miocardio.
El diagnóstico de síndrome antifosfolípido se hace por la presencia de anticuerpos antifosfolípido asociada a una trombosis arterial, venosa o una pérdida fetal. Los anticuerpos antifosfolípido se determinan por la presencia de títulos altos de anticuerpos de IgG o IgM medidos por ensayo inmunoenzimático (ELISA) o por pruebas positivas para el anticoagulante lúpico, utilizando el tiempo parcial de tromboplastina activado, el tiempo de coagulación de caolín o el tiempo de veneno viperino de Russell. Otras pruebas comerciales no disponibles pueden ser inespecíficas.5
El tratamiento óptimo de las complicaciones del síndrome de anticuerpos antifosfolípido aún no se ha establecido porque las manifestaciones de este padecimiento varían en gravedad desde complicaciones vasculares fatales hasta leves o ausentes. Los pacientes que sufren un evento trombótico importante tienen gran riesgo de recurrencia y por lo general deben ser tratados con warfarina a largo plazo.5 Al parecer se requieren dosis altas de warfarina o heparina, y las dosis bajas de aspirina o anticoagulantes son insuficientes para prevenir las complicaciones. Se han usado esteroides para el tratamiento de la trombosis fulminante, trombocitopenia y muerte fetal recurrente con resultados diversos, pero aún no se ha establecido con claridad cuál es el papel de los esteroides en el tratamiento de este síndrome. En estudios no controlados se ha encontrado que la heparina y la gama-globulina intravenosa son benéficas durante el embarazo.
EMBARAZO
La fertilidad en las pacientes con LEG es normal. Aunque las pacientes con LEG activo pueden tener exacerbaciones durante el embarazo, varios estudios que compararon la frecuencia de estos episodios en pacientes embarazadas con la frecuencia en pacientes controles no embarazadas han concluido que no existe aumento en la frecuencia de exacerbaciones como consecuencia del embarazo en las pacientes con lupus. Las pacientes con nefritis lúpica con frecuencia sufren exacerbación del daño renal durante el embarazo, pero es difícil determinar si ésta es causada por toxemia o por el LEG.5 En forma semejante, ocurren trombocitopenia, artralgias e hipocomplementemia durante el embarazo normal así como durante las exacerbaciones del LEG. Las pacientes con LEG embarazadas pueden sufrir un cuadro de hipertensión, edema y proteinuria de inicio súbito, que es indistinguible de la hipertensión inducida por el embarazo y que es independiente de la presencia de anticuerpos antifosfolípido. La muerte fetal del segundo trimestre se asocia con la presencia de anticuerpos antifosfolípido o con la hipertensión inducida por el embarazo.
El síndrome de lupus neonatal consiste en lesiones lúpicas cutáneas pasajeras y, con menor frecuencia, en trombocitopenia transitoria, anemia y afección hepática. La manifestación más grave del lupus neonatal es el bloqueo cardiaco congénito, que suele ser completo y permanente; el bloqueo cardiaco congénito puede ser responsable de algunas muertes en el periodo neonatal. Las madres y los productos afectados casi siempre presentan autoanticuerpos contra ribonucleoproteínas, por lo general anti-Ro (también llamados SS-A) y con menor frecuencia, anti-La (también conocidos como SS-B) o ambos [ver adelante, Datos de laboratorio, Anticuerpos antinucleares]. El lupus neonatal ocurre en una minoría de pacientes con estos anticuerpos, y el bloqueo congénito cardiaco se presenta en menos del tres porciento de los niños afectados. Sin embargo, en una madre con un niño afectado, el riesgo de lupus neonatal en un segundo producto aumenta a alrededor del 25 porciento, y el de bloqueo cardiaco es de ocho a 16 porciento. Ocurre bloqueo cardiaco congénito en una pequeña proporción de niños cuyas madres tienen anticuerpos contra el componente de 52 kd del antígeno Ro y contra el componente de 48 kd del antígeno La.
El pronóstico a largo plazo en los niños que sobreviven el periodo neonatal es bueno; sin embargo, los niños con bloqueo cardiaco requieren colocación de marcapaso permanente.23 Aunque sólo cerca de la mitad de las madres de los niños afectados presentan síntomas leves de enfermedad autoinmune en el momento del parto, casi todas estas madres manifestarán finalmente síntomas leves.24
OTRAS ALTERACIONES
Puede desarrollarse miopatía, caracterizada por debilidad de los músculos proximales y aumento de la concentración sérica de creatina cinasa. El aumento en la concentración de creatina cinasa y los cambios patológicos del músculo pueden ser idénticos a los observados en la polimiositis, pero en ocasiones predomina la degeneración vacuolar de las fibras musculares.1
Con frecuencia aparece linfadenopatía generalizada y se encuentra esplenomegalia en alrededor del 20 porciento de los pacientes. A menudo se encuentran en el bazo las lesiones características en "aros de cebolla", las cuales consisten en capas concéntricas de tejido fibroso perivascular.1
En unos cuantos pacientes puede desarrollarse síndrome de Sjögren, que afecta a las glándulas salivales y lacrimales y se asocia con queratoconjuntivitis seca. Este síndrome es menos común en el LEG que en la artritis reumatoide y se ha visto en pacientes con LEG y artritis erosiva no deformante.1
Datos de laboratorio
Es frecuente la presencia de anemia, que puede ser el resultado de hemólisis, enfermedad crónica o una combinación de ambas. También es común la leucopenia, pocas veces severa: la cuenta total de leucocitos suele estar por arriba de 2,000/mm3. La cuenta diferencial usualmente es normal, a diferencia de lo que ocurre en el síndrome de Felty. Es frecuente la presencia de trombocitopenia, pero ésta tiene significado clínico sólo cuando la cuenta plaquetaria se reduce por debajo de 50,000/mm3. Los anticuerpos contra proteínas de superficie de las plaquetas, por lo general las glucoproteínas IIb y IIIa, causan fagocitosis de las plaquetas cubiertas por anticuerpos por los macrófagos del sistema retículoendotelial. Las cuentas plaquetarias menores a 20 x 109/L se tratan con esteroides. Puede presentarse trombocitopenia grave acompañada de púrpura y otras manifestaciones hemorragíparas, que requieren tratamiento agresivo. La trombocitopenia leve puede adquirir mayor importancia en presencia del anticoagulante lúpico o inhibidor de LEG. Este último compuesto raramente origina hemorragias clínicas a menos que coexista con otros defectos hemostáticos. Los anticoagulantes circulantes pueden ocasionar prolongación de los tiempos de protrombina y parcial de tromboplastina. Sin embargo, en ocasiones los resultados de estas pruebas estándar de la coagulación son normales y se requieren procedimientos especiales de laboratorio para detectar la presencia de anticoagulantes circulantes en los pacientes con LEG.1
Los pacientes con anticoagulantes circulantes suelen tener una prueba biológica para la sífilis con resultados falsos positivos. Las pruebas serológicas falsas positivas para la sífilis se presentan en alrededor de un 25 porciento de los pacientes con LEG. Tales reacciones son el resultado de la formación de anticuerpos que reaccionan contra el material antigénico que se emplea en las pruebas serológicas para la sífilis (Wasserman, Hinton, Kahn, y pruebas similares). Puede distinguirse a las reacciones falsas positivas de las verdaderamente positivas por la ausencia de una reacción de anticuerpos contra un antígeno específico de treponema (en una prueba de absorción de anticuerpo fluorescente de treponema).1
ANTICUERPOS ANTINUCLEARES
Entre los numerosos autoanticuerpos que aparecen en el LEG algunos de los más característicos son los anticuerpos antinucleares (AAN) [ver figura 5].25
|
|
|
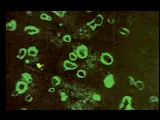
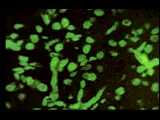
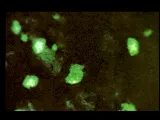
El factor celular del lupus eritematoso (LE), un anticuerpo contra la desoxirribonucleoproteína, reacciona con el núcleo dañado de la célula para formar un cuerpo de hematoxilina, una masa homogénea de material nuclear que tiene apariencia histológica característica. Los leucocitos polimorfonucleares que fagocitan estos cuerpos de hematoxilina se denominan células LE. Los cuerpos de hematoxilina también pueden detectarse en cortes de tejidos. El uso de la prueba de células LE en sangre periférica se ha sustituido en gran parte por otras pruebas de anticuerpos antinucleares.
En los trastornos autoinmunes se han descrito anticuerpos contra muchos determinantes antigénicos situados en el núcleo celular y se han observado patrones típicos de respuesta de anticuerpos en algunos trastornos. Se han caracterizado varios anticuerpos antinucleares, pero estos anticuerpos están dirigidos contra un número limitado de epítopes antigénicos potenciales en el núcleo. Estos epítopes se encuentran en tres estructuras nucleares diferentes: la cromatina, la pequeña ribonucloproteína nuclear (snRNP) y la partícula Ro. La cromatina, un componente principal del núcleo celular, consiste de ADN y de histonas. Se encuentran anticuerpos contra el ADN y las histonas en el LEG pero también se observan en otras enfermedades autoinmunes. Los anticuerpos antinucleares pueden dirigirse contra el ADN desnaturalizado de una sola cadena y contra el ADN nativo de doble cadena, siendo este último más específico para LEG. En los pacientes con LEG se observan anticuerpos contra una o más histonas; las respuestas más potentes se dirigen contra las histonas H1 y H2B. Los individuos con anticuerpos contra el ADN suelen tener también anticuerpos contra histonas.25
Las células contienen ARN en cuatro formas: ARN mensajero, ARN ribosomal, ARN de transferencia y un grupo heterogéneo de moléculas pequeñas de ARN. Cada una de estas moléculas de ARN se relaciona con proteínas específicas para formar ribonucleoproteínas. Debido a que las ribonucleoproteínas son solubles en amortiguadores acuosos, también se les conoce como antígenos nucleares extraíbles (ANE). Se pueden identificar diversas ribonucleoproteínas mediante autoanticuerpos, pero los anticuerpos predominantes en el LEG son anti-U1RNP, anti-Sm y anti-Ro. Todos estos anticuerpos reconocen determinantes situados en las snRNP. Los anticuerpos anti-Sm son específicos de LEG. Los pacientes con anticuerpos anti-Sm casi siempre tienen también anticuerpos anti-U1RNP, pero no sucede a la inversa. Los títulos elevados de anticuerpos anti-R1RNP se relacionan con la enfermedad mixta del tejido conjuntivo.25
La proteína La se une a muchas especies de snRNP y porta un epítope identificable por los anticuerpos anti-La. Los anticuerpos anti-La se acompañan casi siempre de anticuerpos anti-Ro, aunque pueden ocurrir anticuerpos anti-Ro sin la presencia de anticuerpos anti-La. Ni los anticuerpos anti-Ro ni anti-La son específicos de LEG. Estos anticuerpos se observan a menudo en el síndrome de Sjögren y en otros trastornos autoinmunes.25
COMPLEMENTO
La disminución en los niveles séricos del complemento (C) ha sido citada como prueba de un aumento en el consumo de sus componentes durante la formación de complejos inmunes y de daño tisular.1-4 Aunque una parte de la disminución en los niveles séricos de los componentes del complemento puede deberse a disminución en la síntesis más que a aumento en el consumo, el consumo del complemento está incrementado en el LEG. En particular el C3 y las inmunoglobulinas pueden observarse con técnicas de inmunofluorescencia como masas localizadas en el glomérulo renal y otros tejidos dañados.2 Asimismo, se ha detectado properdina en los glomérulos de pacientes lúpicos, lo que indica que hay participación de la vía alterna en la activación del complemento.
La activación de la vía clásica del complemento puede demostrarse clínicamente por la presencia de una reducción en el nivel total de complemento hemolítico (CH50). De manera alterna, la activación de complemento puede determinarse por medio de la medición inmunoquímica del componente C3. El método de hemólisis requiere de niveles funcionantes de todos los componentes del complemento. Sin embargo, debido a que puede haber exceso de varios componentes en los pacientes lúpicos, pueden ocurrir reducciones en algunos componentes (por ejemplo, una reducción del nivel de C3 hasta la mitad), sin que esto cause una alteración en el ensayo por hemólisis. Una amplia variedad de enfermedades que ocasionan inflamación y necrosis tisular causan elevaciones en los niveles séricos de varios componentes del complemento y de otras proteínas plasmáticas que son reactantes de fase aguda. Unas cuantas enfermedades, incluyendo al LEG, ciertas formas de glomerulonefritis y la enfermedad del suero aguda, están asociadas con niveles reducidos de los componentes del complemento en suero.27
Los componentes séricos del complemento no siempre están reducidos en el LEG activo. En un estudio se encontró que los pacientes con LEG y enfermedad extrarrenal activa tenían niveles más elevados de CH50, C4 y C3 y niveles más bajos de complejos inmunes circulantes que los pacientes con enfermedad renal activa. Los anticuerpos anti-ADN no sirvieron para distinguir entre estos dos grupos.27
Se han descrito varias familias con deficiencia hereditaria del componente C2. Los miembros de la familia que son homocigotos tienen niveles no detectables o muy bajos, mientras que los individuos heterocigotos tienen alrededor de la mitad del nivel normal. Se ha encontrado que alrededor de una tercera parte de los individuos homocigotos con deficiencia de C2 tienen LEG o un síndrome similar que podría representar una expresión parcial de la enfermedad. Asimismo, se encontró que aún los heterocigotos para deficiencia de C2 tenían un aumento en la incidencia de LEG. Sin embargo, parece poco probable que se presente una anormalidad significativa en la función del complemento por la disminución en el 50 porciento en el nivel de C2 que se observa en los individuos heterocigotos, sobre todo porque se considera que la cantidad normal de C2 en suero es mayor que la necesaria para que haya actividad hemolítica del complemento. Es probable que el gen para C2 esté en desequilibrio de unión con un gen de la respuesta inmune. Se considera que esta deficiencia de unión está relacionada con las funciones inmunológicas anormales observadas en el LEG.28
PRUEBA DE LA BANDA DEL LUPUS
La prueba de banda de lupus emplea técnicas de inmunofluorescencia para detectar inmunoglobulinas y componentes del complemento en la unión dermoepidérmica. En alrededor de 80 porciento de los pacientes con LEG activo y en 30 a 40 porciento de aquellos con enfermedad inactiva se observa la banda de lupus en la piel que no tiene lesiones. Todas las muestras tomadas de piel con lesiones lúpicas dan resultados positivos, pero el hecho de encontrar una prueba positiva ya sea sobre piel con o sin lesiones no es completamente específico para LEG.1-4
En las lesiones activas de la piel de pacientes con LEG y lupus discorde se ha detectado el complejo terminal del complemento o complejo de ataque a la membrana, compuesto de C5b, C6, C7, C8 y C9. La ausencia de este complejo en la piel no afectada sugiere que se trata de un mediador de las lesiones inflamatorias de la piel.
Patogenia
El LEG parece ser un trastorno de la regulación inmunológica que ocasiona pérdida de tolerancia; esto da como resultado reacciones autoinmunes contra antígenos del huésped que originan inflamación y daño a la vasculatura y otros tejidos.29 La alteración en la inmunidad puede originarse por varios mecanismos; por ejemplo, una deficiencia en las células T, como la disminución en la actividad de las células T supresoras, podría ocasionar una expresión excesiva de las células B. La etiología es aún desconocida, pero el candidato más viable es un agente infeccioso, probablemente viral, que desorganice a la población celular de linfocitos T supresores. Los pacientes con LEG tienen datos de disminución en la inmunidad celular. Pueden alterarse las pruebas de hipersensibilidad tardía en la piel y observarse disminución en la función de las células T in vitro; se ha demostrado la presencia de anticuerpos antilinfocitos en estos pacientes, anormalidad que podría estar relacionada con la linfocitopenia y la hipofunción de las células T.
Sin embargo, este cuadro se ha hecho cada vez más complicado. Las mediciones utilizando anticuerpos monoclonales no han demostrado deficiencia en las células T supresoras en todos los pacientes. En lugar de eso, se ha demostrado que la relación de células T cooperadoras y supresoras presenta amplias variaciones entre los pacientes con LEG cuando se compara con las proporciones que se encuentran en una población control de sujetos normales. Además, en algunos individuos con LEG se ha identificado una deficiencia en un subgrupo específico de células T cooperadoras (también llamadas células T CD4+). Se reconocen dos subgrupos de células T CD4+ con funciones diferentes: los subgrupos 4B4 y 2H4. El subgrupo 4B4 responde a antígenos solubles e induce a los linfocitos B para que secreten inmunoglobulinas. El subgrupo 2H4 (denominado con anterioridad subgrupo de la artritis reumatoide juvenil por la presencia de anticuerpos contra este subgrupo en pacientes con este tipo de artritis) induce a las células T CD8+ a expresar funciones de células T supresoras, incluyendo la supresión de la síntesis de anticuerpos. Los pacientes con LEG y afección renal activa presentan un menor número de células T CD4+ 2H4, observación que se ha relacionado con una función deficiente de las células T supresoras en estos pacientes.30
Los estudios inmunológicos han enfatizado el papel de la apoptosis, un mecanismo fisiológico de muerte celular, en el mantenimiento de la tolerancia. Los defectos en los genes responsables de la regulación de la apoptosis causan enfermedad autoinmune en animales de experimentación . Se ha postulado que la expresión exagerada del gen sintetasa de ácidos grados (fas) causa inhibición de apoptosis de linfocitos autoreactivos en el LEG, pero está hipótesis no se ha comprobado aún.30
Varias observaciones indican que las hormonas sexuales influyen en forma importante en la patogenia del LEG. Apoyan este concepto la frecuencia de la enfermedad en el sexo femenino, los efectos adversos de los estrógenos en hombres y mujeres con la enfermedad y los efectos benéficos de los andrógenos en el lupus murino. Los niveles de varios andrógenos activos están disminuidos en las mujeres con LEG, sobre todo en aquellas con enfermedad activa, tal vez como consecuencia de alteraciones en el metabolismo hormonal.
La incidencia de la enfermedad en los familiares en primer grado de los pacientes con lupus es de uno porciento, aproximadamente. El porcentaje de familiares en primer grado que tienen una prueba de anticuerpos antinucleares positiva es más alto; en general, algunos estudios han comunicado una incidencia que varía de un cuatro a un 14 porciento. Sin embargo, el incremento en la frecuencia de las pruebas de anticuerpos antinucleares positivas en familiares no consanguíneos y consanguíneos sugiere que podría haber factores ambientales que tuvieran alguna relación con la enfermedad. La concordancia de LEG en gemelos monocigotos es de alrededor de 30 porciento, mayor que en gemelos dicigotos, en los que la incidencia es semejante a la de otros familiares en primer grado. Estas observaciones indican que hay factores genéticos que contribuyen a la susceptibilidad para padecer LEG, pero que el factor ambiental también contribuye a la enfermedad. Otro hecho más que apoya la presencia de un factor genético en el LEG son las asociaciones de los antígenos de histocompatibilidad HLA-B8 y HLA-DR3 con el LEG espontáneo y de HLA-DR4 con el LEG inducido por hidralacina.31
Una hipótesis plausible acerca de la patogenia del LEG incorpora varios factores.29 Existen datos que apoyan la existencia de una predisposición genética para la enfermedad, misma que produce activación excesiva de las células B; este fenómeno puede ser el resultado de uno o varios mecanismos, incluyendo un defecto intrínseco en las células B, un defecto en las células T cooperadoras que origine una estimulación anormal de las células B o un defecto en las células T supresoras que ocasione una falla para controlar la proliferación de las células B. La activación de las células B puede originarse por factores exógenos o endógenos, incluyendo agentes químicos, medicamentos y antígenos virales o bacterianos. Las manifestaciones individuales de la enfermedad pueden estar determinadas por factores genéticos o de otro tipo que favorezcan la activación de clases específicas de las células productoras de autoanticuerpos.
En el LEG que se presenta en humanos se ha considerado que los complejos inmunes circulantes son responsables de las lesiones tisulares en muchos de los sistemas. El antígeno que se ha implicado con mayor frecuencia es el ADN nativo, que puede encontrarse, junto con componentes del complemento e inmunoglobulinas, en los riñones afectados. La eliminación de los complejos inmunes circulantes se facilita por su unión a los receptores CR1, que se localizan en cerca del 90 porciento en los eritrocitos. Estos receptores funcionan en parte para transportar a los complejos inmunes circulantes al sistema reticuloendotelial, donde son degradados. La cantidad de receptores CR1 en los eritrocitos es un rasgo hereditario, y los individuos de la población normal pueden tener un número elevado, intermedio o bajo de estos receptores. Los estudios de fragmentos de restricción de longitud polimórfica (FRLP) de ADN indican que la herencia de los receptores del complemento CR1 es similar en pacientes con LEG y en controles sanos. Por lo tanto, los niveles reducidos de receptores CR1 en pacientes con LEG no depende de predisposición genética.32
Tratamiento
El tratamiento del LEG es controversial. Debido a que la enfermedad tiene una evolución muy variable y los patrones sintomáticos difieren mucho entre los distintos pacientes, es difícil valorar la respuesta al tratamiento. Debido a la ausencia de estudios cuidadosamente controlados, las recomendaciones para el tratamiento deben, por desgracia, basarse en criterios poco objetivos.1-4
En pacientes con datos de LEG activo está indicado el reposo en cama.1-4 El estrés físico y emocional afectan de manera adversa la evolución de la enfermedad; deberán evitarse situaciones de estrés como cirugías, infecciones y embarazo.
También debe evitarse la exposición a la luz del sol, ya que alrededor de la tercera parte de los pacientes con LEG son fotosensibles.1 La luz solar provoca exacerbaciones tanto de las manifestaciones sistémicas como de las cutáneas. Cuando no es posible evitar la exposición al sol, los pacientes deben usar pantallas solares que contengan ácido aminobenzoico (PABA) con grado elevado de factor protector (FP). Estas preparaciones contienen éster de PABA y benzofenonas, u otras combinaciones de agentes que bloquean tanto el espectro del eritema como el de longitud de onda larga de la luz ultravioleta.
Ni el uso de anticonceptivos orales ni el tratamiento sustitutivo con estrógenos es dañino en las pacientes con LEG. Por otro lado, en un estudio se asoció el tratamiento estrogénico posmenopáusico con un riesgo de alrededor del doble en el desarrollo de LEG.33 Por lo general deben evitarse los estrógenos en pacientes con síndrome antifosfolípido, aunque esto no ha sido evaluado con certeza en estudios clínicos.
SALICILATOS
El tratamiento antinflamatorio con dosis completas de aspirina está indicado en muchos pacientes que tienen LEG activo, especialmente en individuos con fiebre, artritis, pleuritis o pericarditis. La aspirina deberá tomarse en dosis ligeramente por debajo del umbral de toxicidad. El empleo de preparaciones con capa entérica o con amortiguadores puede reducir la frecuente aparición de los efectos colaterales gastrointestinales que tiene la aspirina; ingerir este medicamento con los alimentos también puede ser de ayuda. La dosis diaria total es variable y los adultos jóvenes pueden requerir hasta 6 g por día. La mayoría de los pacientes adultos requerirán entre 3.6 y 4.8 g por día, pero los pacientes mayores de 65 años de edad con frecuencia requieren dosis menores. Los efectos colaterales, aparte de la gastritis, son raros. Algunos pacientes con LEG activo pueden desarrollar datos de hepatitis inducida por aspirina. Sus manifestaciones suelen limitarse a una elevación en los niveles séricos de aminotransferasas y, en algunas ocasiones, de la fosfatasa alcalina, que disminuyen rápidamente una vez que se suspenden los salicilatos. Los efectos antiplaquetarios de la aspirina pocas veces tienen importancia clínica, a menos que existan otras alteraciones de la coagulación. Probablemente debe evitarse el empleo de salicilatos en el caso de trombocitopenia severa y en pacientes con enfermedad renal.
MEDICAMENTOS ANTIPALUDICOS
Se ha demostrado que la cloroquina y la hidroxicloroquina son útiles para el control de algunas de las manifestaciones del LEG, en especial la erupción cutánea y quizá las artralgias y artritis. Un estudio bien controlado demostró que en el LEG leve o inactivo el tratamiento de mantenimiento con hidroxicloroquina es eficaz para reducir la frecuencia de exacerbaciones de actividad.12,34 Estos medicamentos se administran en dosis de 200 a 500 mg, con las que los efectos adversos son poco frecuentes. Pueden ocurrir efectos gastrointestinales colaterales. La agudeza visual puede disminuir por ciclopegia o depósitos corneales, y este efecto es reversible. El efecto adverso más grave es la retinopatía irreversible, que puede causar ceguera. El riesgo de esta complicación es mínimo, sobre todo si el tratamiento no se prolonga más de un año. Puede presentarse miopatía del músculo esquelético, cardiomiopatía y neuropatía periférica con el uso por tiempo prolongado de cloroquina e hidroxicloroquina.
ESTEROIDES Y MEDICAMENTOS CITOTOXICOS
Los esteroides suprimen en forma dramática muchas manifestaciones del LEG. A pesar de su uso diseminado en este padecimiento, sus efectos sobre la supervivencia son desconocidos, y no son claras sus indicaciones de uso. Los pacientes que tienen manifestaciones que no son graves, como artralgias, erupción cutánea, fatiga, fiebre discreta y dolor pleurítico, pueden ser tratados en forma conservadora con reposo, salicilatos y un antipalúdico. Por lo general, es mejor evitar la toxicidad potencial de los esteroides en estos casos. Las manifestaciones más importantes, como anemia hemolítica severa o trombocitopenia, constituyen indicación para tratamiento esteroide. La ciclofosfamida intravenosa, administrada una vez por mes, es eficaz para tratar la trombocitopenia que no puede manejarse en forma satisfactoria por medio de esteroides, danazol o esplenectomía.35
Una crisis lúpica con fiebre elevada, postración y otros síntomas, puede también requerir de esteroides. La fiebre, artritis, erupciones de la piel, pleuritis, pericarditis y otras manifestaciones pueden responder de manera notable. Sin embargo, puede ser difícil reducir la dosis sin precipitar una recaída. Es entonces cuando los pacientes tendrán que enfrentarse a la toxicidad del uso prolongado de estos fármacos. En el LEG y otros padecimientos reumatológicos, los intentos para reducir la toxicidad empleando dosis en días alternos con frecuencia han sido poco satisfactorios, debido a que los síntomas frecuentemente recurren en los días en que no se administra el medicamento. El uso de pulsos de esteroides por vía intravenosa puede asociarse con menor riesgo de osteonecrosis que la administración diaria por vía oral.5
TRATAMIENTO DE LA ENFERMEDAD RENAL
El tratamiento de la enfermedad renal lúpica también es motivo de controversia. No ha habido un estudio con control satisfactorio que compare a los pacientes tratados con esteroides con los pacientes que no reciben tratamiento. No se ha demostrado que los esteroides alteren la evolución final o el desenlace de la glomerulonefritis en el LEG. No obstante, parece razonable administrar un tratamiento limitado con prednisona en los pacientes con glomerulonefritis membranosa o proliferativa difusa.11 El riesgo de que aparezcan complicaciones graves con el tratamiento a largo plazo indica que es poco prudente administrar una dosis mayor de 40 a 60 mg diarios de prednisona en dosis divididas durante más de tres meses. Después de este esquema inicial de tratamiento de tres meses, deberá reducirse la dosis de manera gradual durante periodo de varias semanas hasta llegar a niveles de 10 a 15 mg por día.
La administración de prednisona en dosis leves a moderadas es adecuada para la enfermedad mesangial o proliferativa focal leve. Sin embargo, no es claro si este tratamiento es necesario porque solo el 10 a 15 porciento de los casos progresa a enfermedad más seria, y no se ha demostrado que el tratamiento con prednisona evite la progresión de la enfermedad.9 La glomerulonefritis focal severa o difusa, demostrada por biopsia, se trata en forma más agresiva porque los estudios controlados han demostrado una mejor evolución de la enfermedad renal (aunque no existe un efecto demostrado en la mortalidad) en los pacientes tratados con ciclofosfamida. Con base en la histología renal, la prednisona se administra en dosis de 1 mg/kg/día durante dos meses y después de disminuye hasta una dosis baja de mantenimiento, junto con ciclofosfamida y otro agente inmunosupresor (por lo general azatioprina). La ciclofosfamida es mejor tolerada si se administra en pulsos intravenosos mensuales en lugar de en dosis diaria por vía oral. Se ha recomendado un esquema de seis meses de pulsos mensuales seguido de un pulso cada tres meses durante un año después de la remisión de la enfermedad renal, pero no se sabe si este esquema es el óptimo. Al parecer, el tratamiento en pulsos se asocia con menor riesgo de cáncer de vejiga que la administración oral a largo plazo, pero se ha reportado esterilidad en alrededor de una cuarta parte de las pacientes que han recibido este tratamiento.11 La gravedad de los cambios histológicos crónicos en la biopsia renal correlaciona con el desarrollo de insuficiencia renal. Así mismo, los pacientes con cambios de grado intermedio respondieron mejor al tratamiento inmunosupresor; al parecer los enfermos con cambios crónicos leves no requieren tratamiento y los que presentaron los cambios más graves no respondieron a éste.11
Se ha reportado que la radiación linfoide total mejoró la evolución de la nefritis lúpica en un estudio de 15 pacientes que habían sido refractarios al tratamiento previo con esteroides y citotóxicos.36 No se observaron efectos colaterales graves entre los pacientes del estudio, pero un reporte subsecuente describió complicaciones fatales y ningún beneficio clínico en otros dos pacientes con LEG rebelde a tratamiento que fueron también tratados con radiación linfoide total.37 Por lo tanto, aún debe establecerse cuál es el papel de este método terapéutico en el LEG. Un estudio aleatorio y controlado demostró que la plasmaféresis, aunada al régimen de esteroides y ciclofosfamida, no mejora la evolución en la nefritis lúpica grave.38
En la actualidad la enfermedad renal terminal (ERT) en el LEG se trata en forma eficaz con hemodiálisis por tiempo prolongado y trasplante. Los pacientes con ERT y LEG y los pacientes con ERT causada por otras enfermedades tienen una capacidad semejante para tolerar la diálisis peritoneal o la hemodiálisis, así como igual supervivencia y supervivencia del injerto después de un trasplante. La actividad global de la enfermedad se mitiga durante la diálisis y después del trasplante, aunque no se conoce aún el motivo de esta mejoría.39,40 A pesar de la disminución gradual del tratamiento inmunosupresor, algunos pacientes se recuperaron de la insuficiencia renal y fueron capaces de suspender el tratamiento dialítico. Las muertes fueron poco frecuentes y por lo general se asociaron con tratamiento esteroide a dosis altas. La recurrencia de la nefritis lúpica después del trasplante es rara. La evolución clínica a largo plazo de los pacientes con ERT causada por LEG es semejante a la de los pacientes con nefropatía terminal por otras causas.39,40
TRATAMIENTO DE LA ENFERMEDAD DEL SNC
La justificación para decidir el tratamiento de las complicaciones neurológicas del lupus eritematoso es aún más endeble. Muchas complicaciones aparentemente serias del SNC se resuelven de manera espontánea, haciendo que la evaluación del tratamiento sea difícil. Aunque no existen estudios controlados, la enfermedad neuropsiquiátrica en el LEG puede responder a la prednisona. Cuando no existe mejoría con el tratamiento convencional de prednisona por vía oral, puede emplearse pulsos de metilprednisolona o de ciclofosfamida para los casos graves.11
Pronóstico
El pronóstico para un paciente con LEG es difícil de determinar. La insuficiencia renal, la proteinuria grave y la anemia son signos pronósticos de gravedad. La mortalidad es más elevada en pacientes que pertenecen a un estrato socioeconómico bajo, pero el aumento en la mortalidad no muestra una predilección independiente para personas de una raza u origen étnico en particular.
Las causas más frecuentes de muerte son la enfermedad renal activa (frecuentemente acompañada de afección en otros sistemas) y las infecciones. Sin embargo, el pronóstico del LEG parece estar mejorando, y más de un 80 porciento de los pacientes con glomerulonefritis proliferativa difusa, la forma más grave de afección renal, sobreviven más de cinco años.1 Los factores que se asocian con progresión hacia la insuficiencia renal irreversible son: menor edad, sexo masculino, elevación de la creatinina sérica y cambios histológicos de esclerosis glomerular e intersticial, además de atrofia tubular, en la biopsia renal.13 Las muertes ocasionadas por complicaciones a nivel de SNC y por infarto del miocardio son poco frecuentes. La aparición de enfermedades neoplásicas malignas es rara.
Lupus eritematoso generalizado inducido por medicamentos
Muchos pacientes han desarrollado manifestaciones clínicas y de laboratorio de LEG en relación con ciertos medicamentos. En algunos de ellos el medicamento parece dar lugar a un complejo sintomático idéntico al del LEG, mientras que en otros puede desarrollarse una forma incompleta. Los criterios que se han propuesto para el LEG inducido por medicamentos son: (1) criterios clínicos similares a los propuestos para el diagnóstico de LEG de aparición espontánea, (2) administración del medicamento antes del inicio de los síntomas, usualmente de manera continua, por periodos que varían de tres semanas a dos años, y (3) signos clínicos y síntomas que desaparecen rápidamente después de suspender el medicamento. Los signos clínicos suelen comenzar a a resolverse en término de días, aunque algunos datos de laboratorio pueden tardar meses o aún años en normalizarse.
Una amplia variedad de agentes terapéuticos ha sido relacionada con el síndrome de lupus inducido por medicamentos [ver tabla 5].
|
|||||||||||||
|
El cuadro clínico es similar al de la enfermedad que se presenta de manera espontánea, y la diferencia consiste en que el lupus relacionado con medicamentos es de menor intensidad y las complicaciones renales son menos frecuentes. El LEG inducido por medicamentos es más frecuente en mujeres que en hombres. Los pacientes tienden a ser de edad media y mayores, probablemente debido a que muchos de los medicamentos implicados se emplean con mayor frecuencia en personas en estos grupos de edad.
Se desconoce la incidencia de síndromes de LEG en pacientes expuestos a medicamentos, pero la forma totalmente desarrollada probablemente está presente en el uno porciento o menos, aún entre aquellas personas expuestas a medicamentos de alto riesgo como la hidralacina y la procainamida. Otros pacientes desarrollan síntomas limitados o aislados, como artralgias, que pueden ser manifestaciones del LEG. Se desarrollan pruebas de anticuerpos antinucleares positivas, usualmente no acompañadas por síntomas de LEG, en más del 50 porciento de los pacientes expuestos a medicamentos de alto riesgo. Los anticuerpos contra ADN nativo, presentes en 50 porciento o más de los pacientes con LEG espontáneo, generalmente no se encuentran en pacientes con LEG inducido por medicamentos; las pruebas de estos anticuerpos son de ayuda para hacer esta distinción.
La frecuencia de las manifestaciones del LEG en pacientes que toman medicamentos como la hidralacina o la procainamida hace poco probable que la acción de estos medicamentos sea únicamente la de desenmascarar una diátesis de fondo. Sin embargo, los factores del huésped pueden ser importantes, y los individuos que desarrollan el síndrome completo pueden tener una predisposición innata. La propensión para padecer el síndrome de LEG relacionado con medicamentos como hidralacina o procainamida está relacionada con el fenotipo de acetilador que tenga el paciente. Estos dos medicamentos son metabolizados por medio del sistema hepático de N-acetiltransferasa. Los pacientes que genéticamente son acetiladores lentos metabolizan los medicamentos más lentamente que los pacientes que son acetiladores rápidos. Durante el tratamiento con hidralacina o con procainamida los acetiladores lentos desarrollan positividad para anticuerpos antinucleares de manera más temprana y el síndrome de LEG con mayor frecuencia que los acetiladores rápidos.
Lupus discoide
El lupus discoide crónico es una forma limitada de lupus eritematoso caracterizada por inflamación superficial de la piel. La lesión típica aparece como una lesión elevada, papular y eritematosa, y después se vuelve atrófica, hiperpigmentada o despigmentada. Los estadios posteriores están asociados con pérdida del cabello , escamas y taponamiento folicular, telangiectasias y atrofia. Las lesiones suelen distribuirse como placas irregulares sobre la cara, piel cabelluda, parte superior de los brazos o pecho. La luz solar con frecuencia agrava esta situación.
La mayoría de los pacientes con lupus discoide tienen resultados normales de las pruebas serológicas y no tiene evidencia de afección a nivel sistémico; sin embargo, una minoría no predecible desarrollará la enfermedad generalizada. Las lesiones discoides también se observan con frecuencia en el lupus generalizado.1
Los esteroides, tópicos o en forma de inyección intralesional, o los antipalúdicos orales suelen ser eficaces en el lupus discoide.
Enfermedad mixta del tejido conjuntivo
Se han descrito pacientes que tienen características clínicas que sugieren la presencia de varias enfermedades reumáticas y que no pueden ser colocados con facilidad en alguna de las categorías diagnósticas. Tales pacientes tienen características que sugieren lupus generalizado, esclerodermia y polimiositis, lo que justifica la definición de una enfermedad reumática distinta: la enfermedad mixta del tejido conjuntivo (EMTC). La presencia de títulos elevados de anticuerpos contra antígenos extraíbles del núcleo también distingue a estos pacientes de los que padecen otras enfermedades reumatológicas.41,42
Las características clínicas de la EMTC son diversas.41,42 El rango de edad en el que se presenta es de los 5 a los 80 años y un 80 porciento de los pacientes son mujeres. Casi todos los pacientes tienen artralgias y alrededor de dos terceras partes de ellos tienen una artritis que se parece a la artritis reumatoide, aunque no suele ser deformante. Se desarrolla inflamación de los dedos debida a un incremento en la colágena de la piel y a edema que puede progresar a los cambios crónicos de la esclerodermia con telangiectasias; estos efectos suelen estar limitados a las manos. Pueden existir erupciones similares a las que se observan en el LEG o en la poliomiositis. La disfunción esofágica consiste en una disminución en la amplitud de la presión del esfínter inferior y de la peristalsis en los dos tercios inferiores del esófago, aunque en la mayoría de los pacientes estos hallazgos no están relacionados con síntomas.
También es común encontrar datos de enfermedad pulmonar, como disminución en la capacidad de difusión, por lo general asintomáticos. De manera ocasional, una enfermedad más diseminada puede ocasionar disnea de esfuerzo asociada con la presencia de infiltrados pulmonares en la radiografía de tórax, enfermedad pleural y, en algunas ocasiones, hipertensión pulmonar. Puede observarse pericarditis.
La miositis consiste en una miopatía proximal con hipersensibilidad y debilidad, elevación de los niveles de las enzimas musculares, electromiografías anormales y cambios patológicos observados en la biopsia de músculo. Todos estos cambios son similares a los que se observan en la polimiositis.
La presencia de enfermedad renal es poco común, y cuando está presente, a menudo es leve. Las biopsias de riñón han mostrado cambios mesangiales, nefritis focal y una nefritis membranosa difusa. La tinciones con inmunofluorescencia han identificado depósitos granulares de IgG, C3 y C4 en la membrana basal glomerular.
La afección del sistema nervioso central no es muy frecuente. La neuralgia del trigémino es la complicación neurológica más común. Otros problemas raros incluyen confusión transitoria, infarto cerebral, meningitis aséptica y convulsiones. La meningitis aséptica en la EMTC responde a los esteroides. Tambien pueden ocurrir neuropatía periférica y psicosis.
Las alteraciones de laboratorio más frecuentes consisten en velocidad de sedimentación elevada, leucopenia e hipergamaglobulinemia difusa. En raras ocasiones existe anemia hemolítica importante con prueba de Coombs positiva y trombocitopenia. Las pruebas de factor reumatoide son positivas en alrededor de la mitad de los pacientes.
En los pacientes con EMTC las pruebas de anticuerpos antinucleares representan los estudios diagnósticos más útiles. Suelen encontrarse títulos elevados de anticuerpos antinucleares fluorescentes con patrón moteado [ver figura 5], que tienden a permanecer constantes sin importar la actividad de la enfermedad. El dato más característico es un resultado fuertemente positivo para anticuerpos contra antígenos RNP (ver antes, Datos de laboratorio, Anticuerpos antinucleares). El suero que contiene anticuerpos contra el antígeno RNPU1 sólo es muy característico en la EMTC. Sin embargo, los anticuerpos anti-RNPU1 no son totalmente específicos de la EMTC.
La evolución de la EMTC es variable, y en algunos pacientes crónica. Las características compatibles con LEG, esclerodermia y polimiositis pueden aparecer de manera simultánea o ir apareciendo en término de muchos meses o años. El tratamiento con esteroides ha tenido al parecer un éxito razonable, aunque hasta la fecha no han habido estudios controlados. Los síntomas leves pueden responder a los medicamentos antinflamatorios no esteroides. Las alteraciones en la función pulmonar y esofágica mejoran con el tratamiento con esteroides.
Aunque por lo general la enfermedad tiene un curso benigno con buen pronóstico, pueden ocurrir complicaciones graves con consecuencias incluso fatales. La afección pulmonar es común y la hipertensión pulmonar puede ser refractaria al tratamiento. Puede ocurrir afección renal, por glomerulonefritis por complejos inmunes o por una vasculopatía semejante a la de la esclerodermia, en alrededor del 25 porciento de los pacientes, en ocasiones con insuficiencia renal secundaria.
Paniculitis nodular sistémica
La paniculitis nodular sistémica, también llamada enfermedad de Weber-Christian, se caracteriza por la presencia de ataques recurrentes de grupos de nódulos subcutáneos, rojos, dolorosos, hipersensibles, que se localizan con mayor frecuencia en las piernas y nalgas.43 Los brotes están acompañados por fiebre, leucocitosis y eosinofilia. Los ataques son autolimitados y tienen una duración de dos a tres semanas. Pueden quedar como secuelas áreas pigmentadas, ligeramente deprimidas. En la enfermedad grave puede estar afectada cualquier parte del cuerpo excepto la cara; pueden desarrollarse sitios de afección extracutánea, como el mesenterio. En una variante de esta enfermedad las lesiones pueden sufrir licuefacción y drenar un material blanco, viscoso, compuesto por tejido adiposo necrótico. La localización de los nódulos inflamatorios en áreas periarticulares origina una aparente artritis aguda en una o más articulaciones, pero no existen estudios del líquido o tejido sinovial.
Los estudios de las lesiones subcutáneas muestran un infiltrado de neutrófilos y eosinófilos acompañado de cambios leucocitoclásticos y grados variables de necrosis de células grasas; las células grasas necróticas están rodeadas por histiocitos, células espumosas y células gigantes.
DIAGNOSTICO DIFERENCIAL
La paniculitis relacionada con LEG también se manifiesta por nódulos inflamatorios subcutáneos que pueden evolucionar hasta formar lesiones crónicas deprimidas con drenaje de grasa necrótica. Las características histológicas de la paniculitis lúpica incluyen un infiltrado linfocítico subcutáneo, vasculitis y depósitos de complejos inmunes en las paredes de los vasos. La paniculitis nodular generalizada y el eritema nodoso difieren en sus características clínicas e histológicas. La paniculitis nodular generalizada puede distinguirse de la vasculitis nodular en que las lesiones de esta última consisten en nódulos subcutáneos, firmes, crónicos y ligeramente hipersensibles, que aparecen con más frecuencia en la parte posterior de las piernas. Estos nódulos tienden a evolucionar hasta formar ulceraciones y cicatrices.
La paniculitis nodular generalizada puede acompañarse de pancreatitis o adenocarcinoma de páncreas. En este caso las lesiones subcutáneas pueden asociarse con poliserositis y artritis.
Ocasionalmente se emplean los medicamentos antinflamatorios no esteroides y los esteroides como tratamiento para la panicuitis nodular generalizada, pero no se ha comprobado la eficacia de estos medicamentos.
Síndrome de Sjögren
El síndrome de Sjögren es una enfermedad autoinmune inflamatoria crónica de las glándulas exócrinas que se relaciona con frecuencia con otros trastornos autoinmunes.44,45 Más del 90 porciento de los pacientes son mujeres mayores de 50 años, aunque el síndrome puede ocurrir en niños y adultos jóvenes. Se calcula que en los Estados Unidos entre dos y tres millones de individuos presentan la enfermedad. Sin embargo, la prevalencia exacta se desconoce porque los síntomas del síndrome pueden ser sutiles y por lo tanto pasan desapercibidos. Aproximadamente el 10 al 15 porciento de los pacientes con artritis reumatoide y un menor número de individuos con otras enfermedades reumáticas (v.gr., lupus eritematoso generalizado y esclerodermia) presentan datos clínicos de síndrome de Sjögren; otros pacientes con trastornos reumáticos se mantienen asintomáticos y la evidencia de síndrome de Sjögren sólo es aparente mediante examen histológico.
La etiología del síndrome de Sjögren es desconocida. Existen algunas evidencias de que puede existir una infección por un retrovirus de tipo A. Al parecer esta infección es la responsable del defecto en las señales intracelulares que se observan en las células T CD4+ que predominan en los tejidos afectados de los pacientes con esta enfermedad.46
MANIFESTACIONES CLINICAS
Las manifestaciones clínicas más frecuentes están relacionadas con el síndrome sicca, que consiste en queratoconjuntivitis seca (resequedad en los ojos) causada por afección de las glándulas lacrimales y por xerostomía (resequedad en la boca) producida por afección de las glándulas salivales.44,45 Los síntomas del síndrome pueden desarrollarse en forma lenta e insidiosa o con rapidez. Cuando el comienzo es rápido, la enfermedad se acompaña de parotiditis episódica. La queratoconjutivitis produce sensación de cuerpo extraño en los ojos, acúmulo variable de secreciones espesas cerca del canto interno de los párpados, menor lagrimeo, ojos rojos y fotofobia. Una película de secreciones espesas sobre la córnea puede interferir con la visión, y la sequedad excesiva puede producir ulceración corneal y pérdida grave de la vista.
La insuficiencia en la salivación produce dificultad en la masticación. La deglución de los alimentos requiere la ingestión frecuente de agua u otros líquidos al comer. Pueden desarrollarse caries dentales severas y ruptura de reparaciones dentales. Con menor frecuencia la sequedad de las vías respiratorias superiores y del árbol traqueobronquial puede causar epistaxis, disfonía, bronquitis o neumonía. En cerca de la mitad de los pacientes ocurre crecimiento de las glándulas parótidas.
La presencia de queratoconjuntivitis seca se establece con certeza por medio de examen con lámpara de hendidura después de teñir la córnea con colorante rosa de bengala, demostrándose queratitis filamentosa y áreas de epitelio corneal denudado. También es útil la prueba de Schirmer, que mide la capacidad de las lágrimas para humedecer una tira de papel filtro cuando se coloca un extremo de este papel dentro del párpado inferior.
Los pacientes con síndrome de Sjögren pueden presentar muchas otras manifestaciones. Se ha descrito la resequedad de la piel y de las mucosas de los genitales. La afección de órganos no exócrinos puede ocasionar tiroiditis autoinmune, neuropatía craneana y periférica y miositis. Otras alteraciones atribuibles al síndrome de Sjögren incluyen el desarrollo de pancreatitis aguda y crónica, de cirrosis biliar con anticuerpos antimitocondriales y diversos trastornos tubulares renales que incluyen la acidosis tubular renal, la diabetes insípida nefrogénica o el síndrome de Fanconi completo. La glomerulonefritis es poco frecuente. En un estudio se encontraron alteraciones neuropsiquiátricas en la mayoría de los pacientes con síndrome de Sjögren, sin que hubiera una enfermedad reumática asociada.47 Las manifestaciones psiquiátricas incluyen depresión y trastornos de la personalidad sin deterioro funcional serio. Las principales anomalías neurológicas son defectos cognoscitivos leves, descargas electroencefalográficas focales y neuropatías por atrapamiento. La elevada frecuencia de afección neurológica sutil indica que las manifestaciones psiquiátricas son de origen orgánico. Estas observaciones difieren de las alteraciones neuropsiquiátricas del lupus eritematoso generalizado; en este último las manifestaciones más comunes son la psicosis y las crisis convulsivas. En un estudio, las alteraciones neuropsiquiátricas en los pacientes con síndrome de Sjögren se parecían con frecuencia a las que se observan en pacientes con esclerosis múltiple.48 En los individuos con síndrome de Sjögren también ocurren con mayor frecuencia reacciones de hipersensibilidad a diversos medicamentos.
Ocurre artritis reumatoide asociada, por lo general del tipo clásico, en cerca de la mitad de los pacientes con síndrome de Sjögren. En otros casos se desarrolla una artritis inflamatoria no erosiva, leve, de predominio en las rodillas y los codos. La poliomiositis, el LEG y la esclerodermia pueden asociarse con el síndrome de Sjögren. El fenómeno de Raynaud ocurre solo o acompañado de otras enfermedades reumáticas. Los síndrome vasculíticos asociados pueden afectar vasos de mediano calibre, como en la poliarteritis nodosa, o vasos de pequeño calibre, en ocasiones con hiperglobulinemia o crioglobulinemia.
DATOS PATOLOGICOS
Las glándulas salivales mayores y menores, las glándulas lacrimales y otras glándulas exócrinas del aparato respiratorio, del aparato digestivo y de la vagina son infiltradas con células B, T y células plasmáticas, con predominio de linfocitos T CD4+. Existe atrofia del tejido acinar secretorio glandular y de algunas de las células que revisten los conductos salivales. La proliferación de las células epimioepiteliales en los conductos produce agrupamientos característicos denominados islotes epimioepiteliales. La arquitectura lobular por lo general no se encuentra muy distorsionada, a menos de que ocurran cambios de seudolinfoma o de linfoma maligno.
ALTERACIONES DE LABORATORIO
Puede medirse la velocidad de flujo salival para establecer si el paciente presenta síndrome de Sjögren. Se coloca jugo de limón en la lengua para estimular el flujo de saliva; a continuación se recolecta la saliva desde el orificio del conducto parotídeo en recipientes especiales. En el síndrome de Sjögren se encuentran velocidades de flujo menores de 0.5 ml por minuto. Las alteraciones de las glándulas salivales principales también pueden establecerse mediante sialografía secretora. En esta técnica se obtienen radiografías de los conductos parotídeos después de inyectar cantidades pequeñas de medio de contraste radiopaco en el orificio de los conductos parotídeos. Los datos patológicos incluyen distorsión del patrón de arborización normal de los conductos y vaciamiento reducido del medio de contraste a partir de los conductos. De manera alternativa, la función salival puede calcularse mediante gamagrafía, que mide la captación y liberación de un radioisótopo como el pertecnectato de tecnecio-99m después de la estimulación de las glándulas salivales.
Los síntomas de queratoconjuntivitis seca y xerostomía suelen sugerir el diagnóstico de síndrome de Sjögren, que puede establecerse en la mayoría de los casos por medio de tinción de la córnea con rosa de bengala y gamagrafía salival. El diagnóstico definitivo del síndrome se realiza por medio de una biopsia del labio inferior, con estudio histológico de las glándulas salivales menores. Este procedimiento cuantifica el infiltrado linfocitario y la distorsión de la arquitectura de las glándulas salivales menores. Sin embargo, si tanto la tinción con rosa de bengala como la gamagrafía salival dan resultados positivos, puede realizarse diagnóstico de síndrome de Sjögren sin necesidad de biopsia de labio.49
La velocidad de sedimentación globular suele encontrarse elevada y la hipergamaglobulinemia es común. Existe factor reumatoide positivo, que es un anticuerpo contra IgG, en más del 90 porciento de los pacientes con síndrome de Sjögren. Un alto porcentaje de los factores reumatoides de estos pacientes comparten una característica estructural común que a menudo no está presente en los factores reumatoides observados en pacientes con otras enfermedades. Los anticuerpos monoclonales identifican un idiotipo de reacción cruzada común sobre el segmento variable de las cadenas ligeras de inmunoglobulinas tipo k en los factores reumatoides en el síndrome de Sjögren. Además, las células B que originan las células productoras de esta clase de factores reumatoides, se acumulan de manera selectiva en las lesiones de las glándulas salivales, pero no se encuentran en los tejidos sinoviales ni en la sangre. Es posible que la presencia de este idiotipo identifique clonas de células B que poseen una frecuencia aumentada de transformación neoplásica, y esta observación tal vez explique la asociación de linfoma no-Hodgkin con el síndrome de Sjögren.
El 70 porciento de los pacientes presentan anticuerpos antinucleares, que suelen mostrar un patrón homogéneo o moteado en la inmunofluorescencia. En la mayoría de los casos se encuentran anticuerpos contra el antígeno nucleoproteico SS-B, también conocido como La, y aproximadamente en la mitad de los pacientes se detectan anticuerpos contra el antígeno SS-A, también llamado Ro. Cuando el síndrome de Sjögren se asocia con artritis reumatoide, los anticuerpos anti-SS-A y SS-B se observan en menos del 10 porciento de los pacientes. Se han identificado otros autoanticuerpos, pero cada uno se encuentra presente sólo en una minoría de los pacientes, lo que ilustra la naturaleza general (inespecífica de un órgano en particular) de la autoinmunidad en el síndrome de Sjögren. Estos incluyen anticuerpos contra las células parietales gástricas, tiroides, músculo liso, células pancreáticas y mitocondrias. Pueden detectarse anticuerpos contra antígenos del epitelio de los conductos salivales en pacientes con síndrome de Sjögren, pero por lo general sólo en asociación con artritis reumatoide.
Las evidencias de una base genética para la predisposición a la enfermedad consisten en una mayor prevalencia de los antígenos de histocompatibilidad HLA-DR3 y el supertipo MT2 del sistema HLA (DRw52). El síndrome de Sjögren primario (i.e., en el que no existe ninguna otra enfermedad asociada) se relaciona en forma muy estrecha con el antígeno de histocompatibilidad HLA-DR3, según estudios realizados en poblaciones caucásicas. Las evidencias indican que el alelo HLA-DR3 no sólo se encuentra en desequilibrio de unión, sino que constituye en sí un factor importante de susceptibilidad para la enfermedad.50 El síndrome de Sjögren en el sexo masculino también se relaciona con un aumento en la prevalencia MT2, pero con una prevalencia normal de DR3. Las manifestaciones clínicas del síndrome de Sjögren son similares en hombres y mujeres, pero en el sexo masculino existe menor incidencia de pruebas positivas para factor reumatoide y de anticuerpos contra SS-A.
TRATAMIENTO
Por lo general el síndrome de Sjögren debe tratarse de manera conservadora.44,45 Las preparaciones de lágrimas artificiales suelen ser eficaces para la queratoconjuntivitis seca. Otras complicaciones, como la ulceración corneal, requieren atención oftalmológica intensiva. Los agentes mucolíticos pueden ser necesarios para la eliminación de las secreciones espesas y pegajosas, y los antibióticos están indicados en caso de infección local. La xerostomía por lo general se trata aumentando la cantidad y la frecuencia de líquidos orales, aunque en ocasiones son de utilidad las preparaciones de saliva artificial. La candidiasis oral puede requerir la aplicación tópica de nistatina. Es importante la higiene dental cuidadosa, incluyendo el enjuage bucal escrupuloso, el cepillado dental, los programas de control de placas, las aplicaciones de fluoruro y la reducción de la ingestión de sacarosa. Las soluciones nasales salinas y el aumento en la humedad pueden ser de utilidad. Las otras enfermedades reumáticas asociadas deberán tratarse de la misma forma que cuando no coexisten con el síndrome de Sjögren. Los esteroides y los medicamentos citotóxicos no suelen estar indicados en el síndrome de Sjögren no complicado.
SINDROME DE LINFOCITOSIS INFILTRATIVA DIFUSA
En los pacientes con infección por VIH ocurre un síndrome de linfocitosis infiltrativa difusa muy semejante al síndrome de Sjögren. Los pacientes con este trastorno sufren crecimiento parotídeo bilateral, xerostomía y, con menos frecuencia, queratoconjuntivitis seca, además de una gran prevalencia de infiltración con linfocitos CD8 en tejidos extraglandulares.51 Esta infiltración suele causar insuficiencia respiratoria por neumonitis intersticial linfocítica; con menos frecuencia los síntomas se relacionan con la presencia de infiltrados en el sistema nervioso central y en el tubo digestivo. El síndrome de linfocitosis infiltrativa difusa asociado con infección por VIH difiere también del síndrome de Sjögren en que en pocas ocasiones se encuentran anticuerpos antinucleares o factores reumatoides positivos. Además, se relaciona en forma intensa con la presencia de HLA-DR5 en los enfermos de raza negra.
El síndrome de linfocitosis infiltrativa difusa puede distinguirse del síndrome de Sjögren por medio del estudio de una muestra de biopsia de glándula salival del labio. Los estudios inmunohistoquímicos de este tejido pueden detectar diferencias entre los antígenos de superficie en los linfocitos y células epiteliales de estos dos padecimientos.
El tratamiento de la neumonitis infiltrativa linfocítica con medicamentos inmunosupresores ha sido eficaz y bien tolerado, mientras que el tratamiento de otras enfermedades autoinmunes, como el síndrome de Reiter, en pacientes con infección por VIH se ha asociado con infecciones oportunistas graves.
LINFOMA ASOCIADO CON SINDROME DE SJÖGREN
En el síndrome de Sjögren la infiltración linfoide de los tejidos salivales es constante. El desarrollo de linfoma debe sospecharse cuando la proliferación linfoide se vuelve extrema, provocando crecimiento firme y prominente de las glándulas salivales. Asimismo, puede desarrollarse hiperplasia linfoide en sitios extrasalivales, incluyendo los ganglios linfáticos regionales próximos a la cabeza y cuello, al igual que los que se encuentran en regiones distantes. La infiltración de linfocitos en otros órganos, como riñones o pulmones, puede alterar la función de dichos órganos. La proliferación linfoide excesiva, también conocida como seudolinfoma, tiene un aspecto histológico pleomórfico, con distorsión de la arquitectura de los ganglios linfáticos. Puede desarrollarse un linfoma maligno franco, independientemente de que exista seudolinfoma. En un estudio se calculó que el riesgo de linfoma maligno en pacientes con síndrome de Sjögren fue 44 veces mayor de lo normal. Las neoplasias por lo general son linfomas no-Hodgkin de la estirpe de las células B y a menudo producen IgMk.53
Los trastornos linfoproliferativos que se asocian con el síndrome de Sjögren pueden requerir tratamiento agresivo. El seudolinfoma suele responder en forma satisfactoria a dosis bajas de esteroides y ciclofosfamida, pero el linfoma maligno evidente requiere el uso de esquemas terapéuticos más agresivos.
- Wallace DJ, Hahn BH, Quismorio FP Jr, et al: Dubois' Lupus Erythematosus, 4th ed. Lea & Febiger, Philadelphia, 1993
- Rothfield NF: Systemic lupus erythematosus: clinical aspects and treatment. Arthritis and Allied Conditions: A Textbook of Rheumatology, 12th ed. McCarty DJ, Koopman WJ, Eds. Lea & Febiger, Philadelphia, 1993, p 911
- Gladman DD, Urowitz MB: Systemic lupus erythematosus. Rheumatology. Klippel JH, Dieppe PA, Eds. Mosby-Year Book Europe, London,1994
- Schur PH: Clinical features of SLE. Textbook of Rheumatology, 4th ed. Kelley WN, Harris ED, Ruddy S, et al, Eds. WB Saunders Co, Philadelphia, 1993, p 1017
- Boumpas DT, Fessler BJ, Austin HA, et al: Systemic lupus erythematosus: emerging concepts. II: Dermatologic and joint disease, the antiphospholipid antibody syndrome, pregnancy and hormonal therapy, morbidity and mortality, and pathogenesis. Ann Intern Med 123:42, 1995
- Duffy KN, Duffy CM, Gladman DD: Infection and disease activity in systemic lupus erythematosus: a review of hospitalized patients. J RheumatoI 18:1180, 1991
- Kaplan D, Ginzler EM, Feldman J: Arthritis and nephritis in patients with systemic lupus erythematosus. J RheumatoI 18:223, 1991
- Reilly PA, Evison G, McHugh NJ, et al: Arthropathy of hands and feet in systemic lupus erythematosus. J RheumatoI 17:777, 1990
- MiIls JA: Systemic lupus erythematosus. N Engl J Med 330:1871, 1994
- Wysenbeek AJ, Leibovici L, Amit M, et al: Alopecia in systemic lupus erythematosus: relation to disease manifestations. J RheumatoI 18:1185, 1991
- Boumpas DT, Austin HA, Fessler BJ, et al: Systemic lupus erythematosus: emerging concepts. I: Renal, neuropsychiatric, cardiovascular, pulmonary, and hematologic disease. Ann Intern Med 122:940, 1995
- Schneebaum AB, Singleton JD, West SG, et al: Association of psychiatric manifestations with antibodies to ribosomaI P proteins in systemic lupus erythematosus. Am J Med 90:54, 1991
- Ginsburg KS, Wright EA, Larson MG, et al: A controlled study of the prevalence of cognitive dysfunction in randomly selected patients with systemic lupus erythematosus. Arthritis Rheum 35:776, 1992
- Logar D, Kveder T, Rozman B, et al: Possible association between antiRo antibodies and myocarditis or cardiac conduction defects in adults with systemic lupus erythematosus. Ann Rheum Dis 49:627, 1990
- Galve E, Ordi J, Barquinero J, et al: Valvular heart disease in the primary antiphospholipid syndrome. Ann Intern Med 116:293, 1992
- Weinrib L, Sharma OP, Quismorio FP Jr: A long-term study of interstitial lung disease in systemic lupus erythematosus. Semin Arthritis Rheum 20:48, 1990
- Asherson RA, Higenbottam TW, Dinh Xuan AT, et al: Pulrnonary hypertension in a lupus clinic: experience with twenty-four patients. J RheumatoI 17:1292, 1990
- Tsutsumi A, Sugiyama T, Matsumura R, et al: Protein losing enteropathy associated with collagen diseases. Ann Rheum Dis 50:178, 1991
- Harris EN, Khamashta MA, Hughes GRV: Antiphospholipid antibody syndrome. Arthritis and Allied Conditions: A Textbook of Rheumatology, 12th ed. McCarty DJ, Koopman WJ, Eds. Lea & Febiger, Philadelphia, 1993
- Matsuura E, Igarashi Y, Yashuda T, et al: Anticardiolipin antibodies recognize ??-glycoprotein I structure altered by interacting with an oxygen modified solid phase surface. J Exp Med 179:457, 1994
- Harris EN: The antiphospholipid syndrome: diagnosis, management, and pathogenesis. Clin Rev AlIergy ImmunoI 13:39, 1995
- Rosove MH, Brewer PMC: Antiphospholipid thrombosis: clinical course after the first thrombotic event in 70 patients. Ann Intern Med 117:303, 1992
- Provost TT, Watson R, Gammon WR, et al: The neonatal lupus syndrome associated with U1RNP (nRNP) antibodies. N Engl J Med 316:1135, 1987
- McCune AB, Weston WL, Lee LA: Maternal and fetal outcome in neonatal lupus erythematosus. Ann Intern Med 106:518, 1987
- Tan EM: Autoantibodies in systemic lupus erythematosus. Arthritis and Allied Conditions: A Textbook of Rheumatology, 12th ed. McCarty DJ, Koopman WJ, Eds. Lea & Febiger, Philadelphia, 1993
- Maddison PJ, Provost TT, Reichlin M: Serological findings in patients with "ANA-negative" systemic lupus erythematosus. Medicine (Baltimore) 60:87, 1981
- Lloyd W, Schur PH: Immune complexes, complement, and anti-DNA in exacerbations of systemic lupus erythematosus (SLE). Medicine (Baltimore) 60:208, 1981
- Glass D, Raum D, Gibson D, et al: Inherited deficiency of the second component of complement: rheumatic disease associations. J Clin Invest 58:853, 1976
- Woods VL: Systemic lupus erythematosus and related syndromes. Textbook of Rheumatology, 4th ed. Kelley WN, Harris ED, Ruddy S, et al, Eds. WB Saunders Co, Philadelphia, 1993
- Morimoto C, Scholossman SF: Antilymphocyte antibodies and systemic lupus erythematosus. Arthritis Rheum 30:225, 1987
- Russell AS: Genetic factors in systemic lupus erythematosus. Semin Arthritis Rheum 10:255, 1981
- Moldenhauer F, David J, Fielder AHL, et al: Inherited deficiency of erythrocyte complement receptor type 1 does not cause susceptibility to systemic lupus erythematosus. Arthritis Rheum 30:961, 1987
- Sanchez-Guerrero J, Liang MH, Karlson EW, et al: Postmenopausal estrogen therapy and the risk for developing systemic lupus erythematosus. Ann Intern Med 122:430, 1995
- The Canadian Hydroxychloroquine Study Group: A randomized study of the effect of withdrawing hydroxychloroquine sulfate in systemic lupus erythematosus. N Engl J Med 324:150, 1991
- Boumpas DT, Barez S, Klippel JH, et al: Intermittent cyclophosphamide for the treatment of autoimmune thrombocytopenia in systemic lupus erythematosus. Ann Intern Med 112:674, 1990
- Strober S, Farinas MC, Field EH, et al: Lupus nephritis after total lymphoid irradiation: persistent improvement and reduction of steroid therapy. Ann Intern Med 107:689, 1987
- Ben-Chetrit E, Gross DJ, Braverman A, et al: Total lymphoid irradiation in refractory systemic lupus erythematosus. Ann Intern Med 105:58, 1986
- Lewis EJ, Hunsicker LG, Lan S-P, et al: A controlled trial of plasmapheresis therapy in severe lupus nephritis. N Engl J Med 326:1373, 1992
- Nossent HC, Swaak TJG, Berden JHM, et al: Systemic lupus erythematosus after renal transplantation: patient and graft survival and disease activity. Ann Intern Med 114:183, 1991
- Cheigh JS, Hong K, Stenzel KH, et al: Systemic lupus erythematosus in patients with end-stage renal disease: long-term follow-up on the prognosis of patients and the evolution of lupus activity. Am J Kidney Dis 16:189, 1990
- Bennett RM: Mixed connective tissue disease and other overlap syndromes. Textbook of Rheumatology, Kelley WN, Harris ED Jr, Ruddy S, et al, Eds. WB Saunders Co, Philadelphia, 1985, p 1115
- Reichlin M: Introduction to systemic rheumatic diseases: nosology and overlap syndromes. Arthritis and AlIied Conditions: A Textbook of Rheumatology, 1Oth ed. McCarty DJ, Ed. Lea & Febiger, Philadelphia, 1985, p 907
- De Moragas JM: Panniculitis. Dermatology in General Medicine, 2nd ed. Fitzpatrick TB, Eisen AZ, Wolff K, et al, Eds. McGraw-Hill Book Co, New York, 1979, p 784
- Whaley K, Alspaugh MA: SjÖgren's syndrome. Textbook of Rheumatology. Kelley WN, Harris ED Jr, Ruddy S, et al, Eds. WB Saunders Co, Philadelphia, 1985, p 956
- Talal N: SjÖgren's syndrome and connective tissue disease with other immunologic disorders. Arthritis and Allied Conditions: A Textbook of Rheumatology, 10th ed. McCarty DJ, Ed. Lea & Febiger, Philadelphia, 1985, p 1037
- Flescher E, Dauphinée MJ, Fossum D, et al: Signal transduction in SjÖgren's syndrome T cells. Arthritis Rheum 35:1068, 1992
- Malinow KL, Molina R, Gordon B, et al: Neuropsychiatric dysfunction in primary SjÖgren's syndrome. Ann Intern Med 103:344, 1985
- AIexander EL, Malinow K, Lejewski JE, et al: Primary SjÖgren's syndrome with central nervous system disease mimicking multiple sclerosis. Ann Intern Med 104:323, 1986
- CoIl J, Porta M, Rubiés-Prat J, et al: SjÖgren's syndrome: a stepwise approach to the use of diagnostic tests. Ann Rheum Dis 51:607, 1992
- Foster H, Stephenson A, Walker D, et al: Linkage studies of HLA and primary SjÖgren's syndrome in multicase famiIies. Arthritis Rheum 36:473, 1993
- Itescu S, Brancato LJ, Buxbaum J, et al: A diffuse infiltrative CD8 lymphocytosis syndrome in human immunodeficiency virus (HIV) infection: a host immune response associated with HLA-DR5. Ann Intern Med 112:3, 1990
- CoIl J, Tomás S, Corominas JM, et al: Sicca syndrome associated with human immunodeficiency virus infection: an immunohistochemical study. Arthritis Rheum 36:875, 1993
- Zulman J, Jaffe R, Talal N: Evidence that the malignant Iymphoma of SjÖgren's syndrome is a monoclonal B-ceIl neoplasm. N Engl J Med 229:1215, 1978