Reumatología
⭳ Abrir artículo (PDF)321.9 KBEste artículo fue revisado respecto a la Edición 3/2000. Ver esa versión →
Contenido del artículo
VI MIOPATIAS INFLAMATORIAS IDIOPATICAS
VI MIOPATIAS INFLAMATORIAS IDIOPATICAS
DR. DWIGHT R. ROBINSON
Miositis
La polimiositis es una enfermedad de etiología desconocida que afecta de manera primaria al músculo esquelético. La dermatomiositis es una forma frecuente de polimiositis en la que una erupción cutánea acompaña a la miopatía. La polimiositis y la dermatomiositis (en ocasiones conocidas como miositis) son parte de un grupo de síndromes conocidos como miopatías inflamatorias idiopáticas, que tienen como característica en común debilidad y cambios inflamatorios patológicos en el músculo esquelético. Por lo general, las miopatías inflamatorias idiopáticas son enfermedades graves que tienen respuesta variable al tratamiento.
La polimiositis y la dermatomiositis pueden asociarse con otras enfermedades reumatológicas y con neoplasias malignas. En los Estados Unidos se calcula que la polimiositis y la dermatomiositis tienen una incidencia anual de 2 a 10 casos por millón de personas. La enfermedad es dos veces más frecuente en mujeres que en varones, y las mujeres afroamericanas tienen la incidencia más alta.1-3
CLASIFICACION
Se han propuesto varios sistemas de clasificación para la miositis, que en general se basan en las características comunes de inflamación del músculo esquelético y la debilidad.1-4 En el sistema de clasificación más útil [ver tabla 1], la polimiositis se caracteriza por la ausencia de las características encontradas en las otras formas de miositis. La dermatomiositis se distingue de la polimiositis porque afecta también a la piel. Los pacientes con otras enfermedades reumáticas inflamatorias (v.gr., artritis reumatoide, esclerodermia o lupus eritematoso generalizado [LEG]) pueden desarrollar también debilidad muscular proximal semejante a la observada en la polimiositis. En estos casos la miositis suele ser leve y responde bien al tratamiento.
Miositis y neoplasias
Se ha demostrado en forma clara una relación entre dermatomiositis y neoplasias. Los investigadores concluyeron que la dermatomiositis se asocia con cáncer y es una entidad paraneoplásica con base en un meta análisis que mostró aumento en el riesgo de cáncer tanto antes como después del diagnóstico de la dermatomiositis. También la polimiositis se ha asociado con cáncer, pero el mayor riesgo se asoció solo después del diagnóstico de la miopatía, lo que es compatible o con un sesgo de selección o con una susceptibilidad común de los individuos a la polimiositis y al cáncer.5 La mayoría de los pacientes que tienen una neoplasia subyacente son ancianos, aunque en ocasiones se desarrollan neoplasias en personas jóvenes con dermatomiositis. Los cánceres más comunes son de mama, pulmón, tubo digestivo y útero. El cáncer de ovario puede ser más frecuente en mujeres con polimiositis. El tratamiento exitoso de la neoplasia puede causar remisión o mejoría de la miopatía, la curación es rara. Con más frecuencia ocurre mejoría temporal de la miositis después del tratamiento de la enfermedad maligna y de la administración de esteroides.
Miositis juvenil
El patrón de debilidad muscular en la miositis juvenil es semejante al de los adultos, aunque la forma juvenil tiene algunas características clínicas propias. Los niños se afectan con doble frecuencia que las niñas y alrededor del 90 por ciento de los pacientes tienen afección cutánea.6 Existe incidencia alta de vasculitis, que afecta principalmente la piel y el tubo digestivo, causando ulceración mucosa, perforación y malabsorción, que puede causar absorción no confiable de la prednisona por vía oral. Se presenta calcinosis en alrededor de la tercera parte de todos los pacientes jóvenes, que puede ser uno de los datos más incapacitantes. La calcinosis se asocia con excreción aumentada de proteínas que contienen ácido gama-carboxiglutámico, sustancias que pueden promover la calcificación en los tejidos blandos. Estas proteínas están formadas por la carboxilación catalizada por la vitamina K de residuos de ácido glutámico en las proteínas, pero los anticoagulantes como la warfarina no son útiles contra la calcinosis.1-2 El tratamiento con prednisona o, en casos resistentes a prednisona, methotrexate, parece reducir la morbimortalidad global de los pacientes con miositis juvenil, incluyendo los que tienen calcinosis y vasculitis.
Miositis por cuerpos de inclusión
La miositis por cuerpos de inclusión es una forma de polimiositis que se define por datos patológicos consistentes en vacuolas marginales (que pueden contener depósitos amiloides) y cuerpos de inclusión intranucleares y citoplasmáticos, además de un cuadro clínico característico.7 Existen otros datos patológicos de polimiositis, incluyendo las fibras musculares degenerativas y regenerativas, pero casi no existen infiltrados inflamatorios. La miositis por cuerpos de inclusión suele presentarse en ancianos y tiene un inicio lento y progresivo con niveles de enzimas musculares relativamente bajas, afección asimétrica con debilidad importante en las muñecas, músculos flexores de los dedos y extensores de la rodilla y con mala respuesta al tratamiento inmunosupresor. En ocasiones las biopsias musculares de los pacientes
Con el cuadro clínico típico no tienen inclusiones y solo las biopsias posteriores las revelan. Algunos investigadores han sugerido que los pacientes que tienen polimiositis con mala respuesta al tratamiento en realidad pueden tener miositis por cuerpos de inclusión. Aunque algunos estudios han reportado que la fuerza puede estabilizarse con el mismo tratamiento usado en la polimiositis y la dermatomiositis, la mejoría no es frecuente.1-3,8
ETIOLOGIA Y PATOGENIA
Aunque la etiología de las miopatías inflamatorias es desconocida y la patogenia no está bien caracterizada, es probable que pueda participar una lesión autoinmune. Se ha reportado cierta evidencia de etiología viral, pero el estudio cuidadoso de genomas virales en las biopsias musculares por medio de reacción en cadena de la polimerasa ha dado resultados negativos.9 En la actualidad no existen evidencias definitivas de que los virus tengan un papel etiológico en las miopatías inflamatorias. La patología muscular proporciona evidencia de eventos de tipo inmunológico e indica diferencias en la inmunopatología en los distintos síndromes miopáticos. Se encuentran infiltrados de células T CD8+, principalmente de tipo citotóxico, en la polimiositis y la miositis por cuerpos de inclusión, las células B parecen tener un papel menos importante. Las miofibrillas expresan cantidades aumentadas de antígenos de clase I del complejo principal de histocompatibilidad, lo que al parecer aumenta la citotoxicidad mediada por células T y al microscopio electrónico se aprecian células T fijas a las miofibrillas. Estos datos parecen indicar que las células T citotóxicas reconocen moléculas antigénicas en la superficie de las fibrillas musculares. Por otro lado, en la dermatomiositis el daño microvascular causa degeneración de las fibras musculares en la periferia de los fascículos. Los estudios de inmunofluorescencia muestran depósitos de inmunoglobulinas y complemento en las paredes capilares. La infiltración de linfocitos en el músculo está compuesta principalmente de células B en localización perivascular, y casi todas las células T son CD4+. Estos datos sugieren un proceso de inflamación mediado por complejos inmunes o anticuerpos como mecanismo primario en la dermatomiositis.10 Estas diferencias en los infiltrados linfocíticos apoyan el concepto de que la polimiositis y la dermatomiositis son entidades diferentes.
Reacciones autoinmunes
Se encuentran autoanticuerpos contra antígenos nucleares y citoplasmáticos en hasta el 90 por ciento de los pacientes con una miopatía inflamatoria, que son útiles para distinguir estas enfermedades de otras miopatías. Sin embargo, aunque se han descrito muchos autoanticuerpos diferentes, muchos no son específicos de miositis. Dentro de los inespecíficos, los anticuerpos contra ribonucleoproteína nuclear (nRNP) se observan en el 10 a 15 por ciento de los pacientes con miositis, pero la mayoría de estos tienen datos de LEG o esclerodermia. En forma semejante, varias reacciones serológicas se observan en los casos de síndrome de sobreposición de miositis y esclerodermia, incluyendo anticuerpos contra nRNP, Ro (también llamado SS-A), PM-1 (también llamado PM-Scl), Scl-70, Sm, ADN y La (también denominado SS-B).
Además de estos autoanticuerpos inespecíficos, existen varios autoanticuerpos que se encuentran solo en los pacientes con miositis se asocian con datos característicos de la enfermedad. Estos autoanticuerpos específicos de miositis tienen la propiedad en común de reaccionar con antígenos nucleares y citoplásmicos en moléculas asociadas con aspectos de la síntesis proteica. Se han definido tres grupos de autoanticuerpos, contra la aminoacil-ARN de transferencia (ARNt) sintetasa, contra la partícula de reconocimiento de señales (SRP, por sus siglas en inglés) y contra el antígeno nuclear Mi-2. Cada uno de estos grupos se asocia con un síndrome clínico de miositis específico.
Anticuerpos contra la aminoacil-ARNt sintetasa Cada aminoacil ARNt-sintetasa cataliza el acomplamiento de un aminoácido a su ARNt específico. Una vez formado, el aminoacil-ARNt puede donar, bajo dirección del ARN mensajero (ARNm) su aminoácido a las cadenas polipéptidicas que van formándose en los ribosomas. En los pacientes con polimiositis o dermatomiositis ocurren autoanticuerpos contra cinco de las 20 amioacil-ARNt sintetasas, la mayoría de los pacientes tienen autoanticuerpos contra la histidil-ARNt sintetasa, también llamados anticuerpos anti-Jo-1. Aunque suele disponerse de pruebas para detectarlos anticuerpos anti-histidil sintetasa, las pruebas específicas para detectar otros autoanticuerpos específicos de miositis solo están disponibles en centros especializados.
Los pacientes con cualquiera de los autoanticuerpos contra las aminoacil-ARNt sintetasas tienen características clínicas que se denominan en forma colectiva como el síndrome antisintetasa. Estos pacientes con frecuencia tienen miositis (90 por ciento), artritis (90 por ciento), enfermedad pulmonar intersticial (ILD, 80 por ciento), fiebre (80 por ciento) y una erupción acompañada por hiperqueratosis de las manos (las llamadas manos del mecánico, 70 por ciento) y fenómeno de Raynaud (60 por ciento).11 La enfermedad suele ser severa, con inicio en la primavera y responde solo en forma parcial al tratamiento. El síndrome antisintetasa causa alrededor del 20 a 25 por ciento de los casos de pacientes con miositis.
Anticuerpos anti-SRP La SRP es un complejo de varias proteínas citoplásmicas
que facilitan la traslocación de los nuevos polipéptidos recién sintetizados a través del retículo endoplásmico. Los pacientes con autoanticuerpos contra la SRP tienen miositis severa con mialgias y afección cardiaca. La enfermedad suele iniciar en el otoño. Menos del 5 por ciento de los pacientes tienen miositis de este tipo, que ocurre con más frecuencia en mujeres afroamericanas, responde mal al tratamiento y tiene mal pronóstico.
Anticuerpos anti-Mi-2 El antígeno reconocido por estos autoanticuerpos es una proteína nuclear cuya función se desconoce. Los pacientes con anticuerpos anti-Mi-2 tienen dermatomiositis con una erupción cutánea prominente que se distribuye en hombros, porción superior del tórax y parte alta de la espalda (el llamado signo del mantón), pápulas de Gottron y engrosamiento periungueal. Cinco a 10 por ciento de estos pacientes con miositis tienen esta forma de enfermedad, que tiene buena respuesta al tratamiento y buen pronóstico.
DIAGNOSTICO
Manifestaciones clínicas
Debilidad muscular La debilidad afecta a los músculos estriados voluntarios y con una mayor frecuencia a los grupos musculares proximales que a los distales. Los síntomas se relacionan con debilidad de las cinturas escapular y del hombro (v.gr., dificultad para levantarse de la silla, para subir escaleras y para alcanzar cosas que están por encima de la cabeza). Con frecuencia están afectados los flexores del cuello y, en casos graves, los músculos distales. La parálisis de los músculos faríngeos produce disfagia, voz nasal y aspiración de los materiales ingeridos. La participación de otros músculos inervados por los pares craneales es menos obvia; la paresia de los músculos extraoculares es rara. Los músculos intercostales, diafragmáticos y del tronco pueden afectarse, produciendo insuficiencia ventilatoria, aunque la debilidad del diafragma se asocia con deficiencia de maltasa ácida, más que con miositis.12
La gravedad y rapidez de la debilidad muscular varía, pero la enfermedad suele progresar de manera gradual en un periodo de semanas a meses. La evolución fulminante de la enfermedad es poco frecuente: en términos de unos días a semanas aparece miolisis aguda con mioglobinuria, debilidad generalizada grave e insuficiencia respiratoria. En otros pacientes la debilidad muscular puede progresar gradualmente en el plazo de varios años. Se ha descrito la mejoría espontánea de la miositis, pero es probable que esto ocurra con poca frecuencia.1-3
Cambios en la piel La erupción característica de la dermatomiositis es eritematosa, con o sin la presencia de escamas y atrofia. Se presenta en cara, cuello, parte superior del tórax y sobre las superficies extensoras de las articulaciones, como las de las manos y codos. Puede aparecer edema periorbitario y también puede encontrarse eritema en heliotropo, que consiste en una coloración violácea o lila, principalmente de los párpados. Ocasionalmente, la erupción puede ser más diseminada o adoptar formas diferentes [ver figura 1]. También ocurren eritema y telangiectasias en las áreas periungueales. La vasculitis cutánea puede producir nódulos dolorosos, infartos y ulceraciones en los dedos y otras áreas. Esta vasculitis cutánea se ha asociado con neoplasias subyacentes.13
Los cambios cutáneos típicos de la dermatomiositis pueden ocurrir incluso en ausencia de evidencia clínica de afección muscular, y en este caso se denomina a la enfermedad dermatomiositis amiopática.14 Sin embargo, un estudio demostró que las técnicas de imagen por resonancia magnética (IRM) pueden revelar cambios que indican una alteración metabólica después del ejercicio en pacientes con este trastorno.15
Afección pulmonar Ocurre afección pulmonar en casi el 50 por ciento de los pacientes que tienen miositis. La neumonía es la forma más común de enfermedad pulmonar en estos pacientes. Puede desarrollarse neumonía por aspiración por una combinación de disfunción de los músculos faríngeos y tos ineficaz causada por debilidad de los músculos respiratorios. El uso de medicamentos inmunosupresores puede ocasionar neumonía por organismos oportunistas y reacciones pulmonares reversibles asociadas al tratamiento con metotrexate y ciclofosfamida.
Se presenta enfermedad pulmonar intersiticial (EPI) en hasta el 30 por ciento de los pacientes. Esta puede ser leve a severa y de evolución lenta o rápidamente progresiva. El padecimiento puede ser asintomático o causar síntomas consistentes en disnea progresiva al ejercicio y tos no productiva asociados con estertores crepitantes basales. Las pruebas de función respiratoria revelan hipoxemia, reducción del volumen pulmonar y menor capacidad de difusión. Suele encontrarse una de tres formas histológicas de EPI: neumonía intersticial, daño alveolar difuso o bronquiolitis obliterante con o sin neumonía organizada. La EPI ocurre con o sin afección cutánea. No existe correlación entre el desarrollo de la EPI y la gravedad de la afección muscular, y la EPI puede preceder o ser subsecuente al inicio de la debilidad muscular.1-3 La EPI se asocia con alta mortalidad y poca respuesta al tratamiento de la miositis, pero se ha reportado que el tratamiento con esteroides en bolo o ciclofosfamida en bolos o diario puede ser útil.1-3
Alteraciones cardiacas La afección cardiaca es más frecuente de lo que se detectaba en el pasado. Se manifiesta por (1) alteraciones de la conducción con grados variables de bloqueo cardiaco, incluyendo bloqueos de rama y cambios en el segmento ST-T, (2) taquiarritmias auriculares y ventriculares y síndrome del seno enfermo, y (3) miocarditis o fibrosis miocárdica que causa insuficiencia cardiaca. Las pruebas diagnósticas sensibles, incluyendo el ecocardiograma, el monitor Holter, la gamagrafía perfusoria y los estudios de ventanas y reservorios sanguíneos, indican afección cardiaca en la mayoría de los pacientes con miositis.
Otras manifestaciones En ocasiones los pacientes tienen fenómeno de Raynaud y artralgias. Puede ocurrir artritis leve, inflamatoria y simétrica que afecta las manos, muñecas y rodillas. Además, en ocasiones se desarrollan deformidades articulares por subluxación, sin las erosiones yuxtarticulares óseas típicas de la artritis reumatoide. La disfagia puede ser causada por afección de los músculos esqueléticos de la faringe o del músculo liso esofágico, alterando la nutrición. Los pacientes adultos con miositis rara vez tienen afección del tubo digestivo bajo.1-3
Pruebas de laboratorio
Enzimas musculares La necrosis del músculo ocasiona la liberación hacia el suero de varias enzimas intracelulares. Los niveles de estas enzimas son de utilidad en el diagnóstico y como indicadores de la eficacia del tratamiento. Las enzimas cuantificadas con mayor frecuencia son la creatinina cinasa (CK), las transaminasas, la aldolasa y la dehidrogenasa láctica. La CK constituye el indicador más específico de lesión muscular, debido a que su presencia se encuentra limitada al tejido muscular y nervioso. La determinación de las isoenzimas de CK puede ayudar a valorar la importancia de las elevaciones de esta enzima. La CK ocurre en tres formas que difieren en los distintos tejidos. Dos formas de CK son homodímeros de subunidades M o B, y una es un heterodímero M y B. La MM predomina en el músculo esquelético y su elevación indica daño a este tejido. La MB se encuentra principalmente en el músculo cardiaco, en donde constituye el 20 a 25 por ciento de la CK cardiaca total, aunque también existe MB en el músculo esquelético en regeneración. La BB se localiza principalmente en el sistema nervioso central y el músculo liso. La elevación en los niveles séricos de aldolasa, una aminotransferasa, o deshidrogenasa láctica no es específica de las lesiones musculares porque estas enzimas existen en muchos tejidos.
El grado de elevación de los niveles séricos de las enzimas musculares revela el grado de afectación del músculo y la rapidez de su destrucción. Por lo tanto, el comienzo agudo de la miopatía puede estar relacionado con valores séricos elevados de enzimas musculares, mientras que en la enfermedad de progresión lenta los niveles séricos pueden ser normales o ligeramente elevados, aun cuando la debilidad muscular sea severa. Sin embargo, todos los pacientes con polimiositis muestran niveles séricos elevados de una o más enzimas musculares en algún momento de la evolución de la enfermedad; de ahí que ante la ausencia de esta elevación, el diagnóstico deberá hacerse con precaución. Tales enzimas también son de ayuda para valorar la evolución de la enfermedad y su respuesta al tratamiento.1-3
La Academia Americana de Dermatología ha publicado guías sobre las manifestaciones cutáneas de la dermatomiositis.16
Electromiografía e IRM En la mayoría de los pacientes la electromiografía muestra potenciales de unidad motora polifásicos y de baja amplitud, lo que indica falta de sincronía en la contracción entre las fibras musculares dentro de la unidad motora. Este hallazgo se correlaciona con la distribución normalmente irregular de la generación muscular que se observa en el examen histopatológico. La presencia de fibrilaciones y de irritabilidad durante la inserción son evidencia de irritabilidad de la membrana. Esos hallazgos son característicos de la polimiositis, pero también pueden presentarse en otros procesos miopáticos.
La IRM proporciona una manera no invasiva de evaluar grandes volúmenes de masa muscular, evitando los errores que pueden ocurrir al tomar la muestra en una biopsia muscular.15 Detecta la atrofia con sustitución grasa del músculo afectado y las áreas de inflamación. La IRM es una técnica con buena relación costo eficacia para aumentar la exactitud de la biopsia muscular en pacientes con sospecha de polimiositis.17,18
Histopatología Aunque las características histológicas de la polimiositis varían, los principales cambios incluyen datos de degeneración de las fibras y pérdida de estriaciones cruzadas, cambios hialinos y granulares, núcleos picnóticos y fragmentación celular [ver figura 2]. Con frecuencia estos cambios son irregulares, y las fibras necróticas pueden estar diseminadas entre fibras normales. La regeneración de las fibras musculares es característica, como se demuestra por los cambios basófilos en el citoplasma y los numerosos núcleos grandes. Los cambios inflamatorios en el músculo consisten en infiltración de linfocitos, células plasmáticas e histiocitos con edema intersticial, pero estos cambios no siempre se observan, especialmente en casos crónicos. La miositis juvenil se acompaña de lesiones vasculares características en los músculos y la piel. Esta vasculopatía incluye infiltración por células inflamatorias, cambios proliferativos y degenerativos de las células endoteliales e inclusiones celulares.1-3
Criterios diagnósticos
La presencia de cuatro criterios diagnósticos mayores establece el diagnóstico de polimiositis [ver tabla 2]; el diagnóstico es de probabilidad si se cumplen tres criterios. El diagnóstico de miositis por cuerpos de inclusión se establece por los resultados de microscopía de luz y electrónica de las biopsias musculares. Debe realizarse biopsia muscular en todos los pacientes adultos antes de iniciar el tratamiento. Pueden requerirse varias biopsias si la primera no es diagnóstica y se emplearán tinciones especiales para buscar miositis por cuerpos de inclusión y miopatías mitocondriales y metabólicas.2,12 La IRM puede ser una guía útil para elegir el sitio de biopsia muscular.
Diagnóstico diferencial
Deben excluirse ciertas enfermedades similares a la polimiositis [ver tabla 3]. Las miopatías metabólicas se analizan en detalle en otra sección. La sarcoidosis puede producir una miopatía granulomatosa. Una miopatía de tipo inflamatorio similar a la polimiositis se presenta en pacientes con síndrome de inmunodeficiencia adquirida (SIDA). Además, la zidovudina (AZT), usada con frecuencia para tratar pacientes con SIDA, puede causar miopatía por disfunción mitocondrial.19 Varios fármacos, incluyendo colchicina y cloroquina, producen cambios miopáticos, generalmente reversibles cuando se suspende el medicamento. La intoxicación con cocaína puede causar mioglobinuria, insuficiencia renal y hepática, coagulación intravascular diseminada y muerte.1-3
TRATAMIENTO
Los lineamientos para el tratamiento de las miopatías inflamatorias idiopáticas no se han establecido del todo por varios motivos. Los padecimientos son poco frecuentes, lo que dificulta acumular un número suficiente de pacientes para realizar estudios aleatorios y controlados. Además, ciertas formas de la enfermedad tienen curso lento y prolongado, por lo que requieren periodos largos de observación. Por último, no existe una clasificación aceptada de modo uniforme de esta enfermedad, por lo que las comparaciones de tratamientos administrados a grupos diferentes de pacientes en diferentes momentos y lugares puede no ser válida.4 Como ejemplos, en el pasado la polimiositis y la dermatomiositis se incluían en la misma categoría y la miositis por cuerpos de inclusión no se había definido. En la actualidad es claro que las diferentes formas de estas enfermedades varían en su pronóstico y respuesta al tratamiento.
Esteroides
Los corticoides son los fármacos de elección para el tratamiento inicial de la miositis, aunque nunca se ha realizado un estudio rigurosamente controlado para comprobar su eficacia. Aunque en ocasiones los pacientes mejoran con dosis bajas de esteroides, o incluso sin ellos, la mayoría de los pacientes requiere dosis altas de esteroides por periodos de tiempo relativamente prolongados. El tratamiento debe iniciarse con prednisona en dosis de 1 mg/kg/día divida en varias dosis. Un esfoque estándar consiste en mantener esta dosis durante 3 meses o hasta que los niveles séricos de enzimas musculares se normalicen o exista mejoría clínica. Después del periodo inicial, la prednisona puede administrarse en una sola dosis diaria matutina para minimizar la toxicidad y reducirse gradualmente 20 a 25 por ciento cada mes. Con este esquema se alcanza una dosis de mantenimiento de 5 a 10 mg diarios en alrededor de 6 a 8 meses. Se considera que no existe respuesta el tratamiento esteroideo si no ocurre mejoría después de los tres meses iniciales.
Un esquema alternativo de tratamiento consiste en 60 a 80 mg de prednisona al día en una dosis durante 3 a 4 semanas, con reducción posterior en 12 semanas hasta alcanzar 80 a 100 mg cada tercer día. Al disminuir la dosis 5 o 10 mg cada 3 o 4 semanas se alcanza una dosis de mantenimiento de 20 a 25 mg cada tercer día. Se considera que el tratamiento fracasó si no existe mejoría después de 4 meses de tratamiento.20
Por último, varios reportes han sugerido que los pulsos intravenosos de metilprednisolona tienen ventajas sobre la prednisona oral diaria. Las dosis varían de 0.5 a 1.0 g/día por 1 a 3 días, repetidos en intervalos de 1 a 4 semanas. Aunque los resultados no están demostrados, los pacientes tratados con este esquema tuvieron mayor frecuencia y rapidez para lograr la remisión, con menos toxicidad por esteroides, que los pacientes tratados con esteroide oral diario.9
Agentes inmunosupresores
La adición de otro agente inmunosupresor generalmente se reserva para los pacientes que han recibido el tratamiento con prednisona descrito antes y no tienen una respuesta satisfactoria. Varios factores han hecho que algunos investigadores modifiquen este esquema. El tratamiento intensivo con esteroides se asocia en forma invariable con efectos adversos, algunos de los cuales son severos. Estos incluyen apariencia cushinoide, fracturas por compresión, necrosis avascular, cataratas e infecciones. Hasta el 40 por ciento de los pacientes tienen enfermedad resistente a los esteroides, en especial los que tienen debilidad de más de 9 meses antes de iniciar el tratamiento. Aunque no son concluyentes, las evidencias actuales apoyan que la evolución puede mejorar agregando un segundo agente, por lo general metotrexate o azatioprina, junto con el inicio del esteroide. Esto puede estar indicado específicamente en pacientes con deterioro rápido, dificultad en la deglución o EPI.
Los agentes usados con más frecuencia en los pacientes que son resistentes al tratamiento esteroideo son el metotrexate y la azatioprina. El metotrexate puede administrarse por vía oral en dosis inicial es de 7.5 a 10 mg a la semana, que se aumenta en forma gradual hasta 25 mg por semana. En forma alternativa, el metotrexate puede administrarse por vía intravenosa en dosis de 15 mg por semana y aumentarse a 60 mg por semana. Al aumentar la dosis de metotrexate se disminuye la de prednisona. La reducción en las enzimas musculares indica que ocurrirá mejoría en la fuerza muscular. Por lo general ambos esquemas de tratamiento con metotrexate son bien tolerados por los pacientes con miositis. Los reportes han sugerido que el metotrexate puede disminuir los requerimientos de esteroides y beneficiar a los pacientes con más rapidez que la azatioprina. La toxicidad es semejante a la de los pacientes con artritis reumatoide que reciben este medicamento. Puede ocurrir enfermedad hepática en pacientes con miositis tratados con metotrexate, y estos pacientes deben ser vigilados con pruebas diferentes a los niveles de aminotransferasa, incluyendo gama-glutamiltranspeptidasa, porque los niveles de aminotransferasas pueden reflejar el daño muscular. El metotrexate puede mejorar la EPI, pero también causa toxicidad pulmonar y está relativamente contraindicado en pacientes con miositis y EPI. En algunos pacientes con miositis tratados con metotrexate, linfomas asociados al virus Epstein-Barr sufrieron remisión al suspender el metotrexate.2,3
En un estudio prospectivo, controlado y doble ciego, la azatioprina demostró ser eficaz en los pacientes con miositis. Además, los pacientes que fueron tratados con una combinación de prednisona y azatioprina tuvieron mejor evolución funcional y requirieron menos prednisona que los enfermos que fueron tratados solo con prednisona. Se ha reportado que la azatioprina es eficaz en pacientes con miositis, pero se requiere tratamiento por lo menos durante 6 meses para que ocurra la mejoría. Este fármaco debe iniciarse con dosis de 25 a 50 mg/día aumentando después a un máximo de 200 mg diarios para minimizar la toxicidad gastrointestinal. Sus efectos tóxicos consistieron en alteraciones gastrointestinales, supresión de la médula ósea, desarrollo de infecciones y quizá de neoplasias. En los pacientes en los que el tratamiento con esteroides y metotrexate o azatioprina ha fracasado, la combinación de metotrexate y azatioprina parece promisoria y se recomienda en lugar de los agentes alquilantes más tóxicos.2,3
Se han usado varios otros agentes y tratamientos para la miositis resistente a esteroides. Debido a su toxicidad, los agentes alquilantes se usan con poca frecuencia. Se ha administrado ciclofosfamida tanto en pulsos intravenosos como por vía oral diaria, pero las evidencias respecto a la eficacia de ambos modos de administración son controversiales. Se ha reportado cierto éxito en pacientes con EPI asociada y en niños con miositis y vasculitis. También se ha empleado el clorambucil en algunos enfermos que no respondieron a los esquemas habituales de tratamiento.2,3
La ciclosporina parece tener algún papel en el tratamiento de las miopatías inflamatorias; sin embargo, es importante balancear los beneficios potenciales de este fármaco contra los riesgos de toxicidad.21 Los efectos tóxicos incluyen hipertensión y daño renal, así como una miopatía relacionada a hipomagnesemia. Estudios no controlados con ciclosporina han reportado mejoría en pacientes con polimiositis y dermatomiositis del adulto y juvenil, mientras que otros estudios no han encontrado buenos resultados. Un estudio preliminar de tacrolimus en pacientes con polimiositis refractaria ha sido alentador.22
La globulina inmune intravenosa parece ser útil en pacientes tanto con dermatomiositis como con polimiositis. Un estudio controlado de globulina inmune IV en pacientes con dermatomiositis demostró eficacia cuando se administró a una dosis de 1 g/kg/día por 2 días, repetida cada 24 días por 3 meses.23 Los reportes han descrito beneficios en los pacientes con polimiositis o dermatomiositis juvenil;24 sin embargo, en un estudio controlado de globulina inmune IV en pacientes con miositis por cuerpos de inclusión no se estableció la eficacia de este tratamiento, quizá por el tamaño pequeño de la muestra.25 Se requieren estudios adecuados a largo plazo, incluyendo la relación costo-beneficio, antes de recomendar la globulina inmune IM como tratamiento de elección.26
En un estudio controlado la plasmaféresis no fue eficaz en pacientes con miositis crónica, aunque reportes anecdóticos han descrito éxito en los pacientes con afección aguda.2,3
La fisioterapia tiene un papel muy importante en la rehabilitación de los pacientes con miositis. Durante la fase de enfermedad inflamatoria activa se requieren ejercicios pasivos de rango de movimiento para evitar contracturas. Una vez que se ha controlado el componente inflamatorio de la enfermedad, los ejercicios activos de resistencia son útiles para recobrar la fuerza muscular.1-3,27
PRONOSTICO
La supervivencia de los pacientes con polimiositis ha mejorado en los últimos años y puede ser de casi 75 por ciento 8 años después del diagnóstico. La supervivencia más corta se asoció con edad mayor de 45 años en el momento del diagnóstico, afección cardiaca, pulmonar, disfagia y, en algunas series, enfermedades malignas. La polimiositis juvenil suele tener un buen pronóstico. No obstante, la morbimortalidad sigue siendo importante en todos los grupos de edad, ya sea secundaria a la enfermedad por sí misma o a las complicaciones derivadas del tratamiento. En la actualidad, la mortalidad general es cercana al 20 por ciento durante un periodo de 5 años. En varias series, sólo una minoría de las causas de muerte se atribuyeron a enfermedad maligna. Las remisiones completas y permanentes son poco frecuentes. En un estudio se observó que los pacientes que provenían de un medio ambiente rural parecían tener una enfermedad de menor gravedad y respuestas al tratamiento más completas que los pacientes de un medio ambiente urbano. Una posible explicación para este hallazgo es que los investigadores en centros médicos urbanos grandes seleccionan pacientes con enfermedad más severa que la observada en pacientes de otras áreas.1-3
Enfermedades asociadas con mioglobinuria
Muchas enfermedades que producen lesiones del músculo esquelético, incluyendo enfermedades graves consideradas en el diagnóstico diferencial de la polimiositis, se caracterizan por mioglobinuria [ver tabla 4]. La mioglobinuria es la excreción urinaria de mioglobina, una proteína que transporta el oxígeno dentro de células del músculo esquelético. Las lesiones graves del músculo esquelético pueden producir rabdomiolisis, desintegración del tejido muscular que se manifiesta por mioglobinemia y mioglobinuria. Los episodios graves de mioglobinuria pueden inducir insuficiencia renal aguda.
La formación idiopática de mioglobinuria, también denominada mioglobinuria paroxística, se caracteriza por episodios agudos de dolores musculares, hipersensibilidad y debilidad que duran 2 a 3 días o más. Los paroxismos suelen ser precipitados por el ejercicio; la enfermedad puede aparecer durante la niñez o en etapas posteriores de la vida. Raramente se desarrolla debilidad muscular crónica y la insuficiencia renal aguda que aparece durante un episodio agudo puede ser mortal. El síndrome idiopático probablemente representa un grupo de enfermedades que no se ha caracterizado completamente; algunas de ellas son claramente familiares. En algunos pacientes que se habían considerado como portadores de mioglobinuria idiopática se han identificado deficiencias enzimáticas. Se considera que algunos defectos metabólicos, como la deficiencia de carnitina palmitoiltransferasa y las deficiencias enzimáticas que producen las glucogenosis son la causa de algunos casos de mioglobinuria.
El diagnóstico de mioglobinuria se basa en la siguiente tríada: prueba de la ortotoluidina u otras pruebas para detección de sangre en orina positivas en ausencia de eritrocitos; cilindros pigmentados en el sedimento urinario y elevación de la CK y otras enzimas musculares en suero. La rabdomiolisis puede elevar el nivel sérico de CK más de 100 veces. También deben emplearse pruebas específicas para identificar de manera definitiva al pigmento urinario como mioglobina. El tratamiento está dirigido hacia la causa primaria de la mioglobinuria, cuando ésta existe. Además, se recomienda reposo absoluto, líquidos y alcalinización.1-3
Reconocimientos
Figura 2 Fotografías cortesía del Dr. E. P. Richardson, Departamento de Neuropatología, Massachusetts General Hospital, Boston.
DR. DWIGHT R. ROBINSON
Miositis
La polimiositis es una enfermedad de etiología desconocida que afecta de manera primaria al músculo esquelético. La dermatomiositis es una forma frecuente de polimiositis en la que una erupción cutánea acompaña a la miopatía. La polimiositis y la dermatomiositis (en ocasiones conocidas como miositis) son parte de un grupo de síndromes conocidos como miopatías inflamatorias idiopáticas, que tienen como característica en común debilidad y cambios inflamatorios patológicos en el músculo esquelético. Por lo general, las miopatías inflamatorias idiopáticas son enfermedades graves que tienen respuesta variable al tratamiento.
La polimiositis y la dermatomiositis pueden asociarse con otras enfermedades reumatológicas y con neoplasias malignas. En los Estados Unidos se calcula que la polimiositis y la dermatomiositis tienen una incidencia anual de 2 a 10 casos por millón de personas. La enfermedad es dos veces más frecuente en mujeres que en varones, y las mujeres afroamericanas tienen la incidencia más alta.1-3
CLASIFICACION
Se han propuesto varios sistemas de clasificación para la miositis, que en general se basan en las características comunes de inflamación del músculo esquelético y la debilidad.1-4 En el sistema de clasificación más útil [ver tabla 1], la polimiositis se caracteriza por la ausencia de las características encontradas en las otras formas de miositis. La dermatomiositis se distingue de la polimiositis porque afecta también a la piel. Los pacientes con otras enfermedades reumáticas inflamatorias (v.gr., artritis reumatoide, esclerodermia o lupus eritematoso generalizado [LEG]) pueden desarrollar también debilidad muscular proximal semejante a la observada en la polimiositis. En estos casos la miositis suele ser leve y responde bien al tratamiento.
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
Miositis y neoplasias
Se ha demostrado en forma clara una relación entre dermatomiositis y neoplasias. Los investigadores concluyeron que la dermatomiositis se asocia con cáncer y es una entidad paraneoplásica con base en un meta análisis que mostró aumento en el riesgo de cáncer tanto antes como después del diagnóstico de la dermatomiositis. También la polimiositis se ha asociado con cáncer, pero el mayor riesgo se asoció solo después del diagnóstico de la miopatía, lo que es compatible o con un sesgo de selección o con una susceptibilidad común de los individuos a la polimiositis y al cáncer.5 La mayoría de los pacientes que tienen una neoplasia subyacente son ancianos, aunque en ocasiones se desarrollan neoplasias en personas jóvenes con dermatomiositis. Los cánceres más comunes son de mama, pulmón, tubo digestivo y útero. El cáncer de ovario puede ser más frecuente en mujeres con polimiositis. El tratamiento exitoso de la neoplasia puede causar remisión o mejoría de la miopatía, la curación es rara. Con más frecuencia ocurre mejoría temporal de la miositis después del tratamiento de la enfermedad maligna y de la administración de esteroides.
Miositis juvenil
El patrón de debilidad muscular en la miositis juvenil es semejante al de los adultos, aunque la forma juvenil tiene algunas características clínicas propias. Los niños se afectan con doble frecuencia que las niñas y alrededor del 90 por ciento de los pacientes tienen afección cutánea.6 Existe incidencia alta de vasculitis, que afecta principalmente la piel y el tubo digestivo, causando ulceración mucosa, perforación y malabsorción, que puede causar absorción no confiable de la prednisona por vía oral. Se presenta calcinosis en alrededor de la tercera parte de todos los pacientes jóvenes, que puede ser uno de los datos más incapacitantes. La calcinosis se asocia con excreción aumentada de proteínas que contienen ácido gama-carboxiglutámico, sustancias que pueden promover la calcificación en los tejidos blandos. Estas proteínas están formadas por la carboxilación catalizada por la vitamina K de residuos de ácido glutámico en las proteínas, pero los anticoagulantes como la warfarina no son útiles contra la calcinosis.1-2 El tratamiento con prednisona o, en casos resistentes a prednisona, methotrexate, parece reducir la morbimortalidad global de los pacientes con miositis juvenil, incluyendo los que tienen calcinosis y vasculitis.
Miositis por cuerpos de inclusión
La miositis por cuerpos de inclusión es una forma de polimiositis que se define por datos patológicos consistentes en vacuolas marginales (que pueden contener depósitos amiloides) y cuerpos de inclusión intranucleares y citoplasmáticos, además de un cuadro clínico característico.7 Existen otros datos patológicos de polimiositis, incluyendo las fibras musculares degenerativas y regenerativas, pero casi no existen infiltrados inflamatorios. La miositis por cuerpos de inclusión suele presentarse en ancianos y tiene un inicio lento y progresivo con niveles de enzimas musculares relativamente bajas, afección asimétrica con debilidad importante en las muñecas, músculos flexores de los dedos y extensores de la rodilla y con mala respuesta al tratamiento inmunosupresor. En ocasiones las biopsias musculares de los pacientes
Con el cuadro clínico típico no tienen inclusiones y solo las biopsias posteriores las revelan. Algunos investigadores han sugerido que los pacientes que tienen polimiositis con mala respuesta al tratamiento en realidad pueden tener miositis por cuerpos de inclusión. Aunque algunos estudios han reportado que la fuerza puede estabilizarse con el mismo tratamiento usado en la polimiositis y la dermatomiositis, la mejoría no es frecuente.1-3,8
ETIOLOGIA Y PATOGENIA
Aunque la etiología de las miopatías inflamatorias es desconocida y la patogenia no está bien caracterizada, es probable que pueda participar una lesión autoinmune. Se ha reportado cierta evidencia de etiología viral, pero el estudio cuidadoso de genomas virales en las biopsias musculares por medio de reacción en cadena de la polimerasa ha dado resultados negativos.9 En la actualidad no existen evidencias definitivas de que los virus tengan un papel etiológico en las miopatías inflamatorias. La patología muscular proporciona evidencia de eventos de tipo inmunológico e indica diferencias en la inmunopatología en los distintos síndromes miopáticos. Se encuentran infiltrados de células T CD8+, principalmente de tipo citotóxico, en la polimiositis y la miositis por cuerpos de inclusión, las células B parecen tener un papel menos importante. Las miofibrillas expresan cantidades aumentadas de antígenos de clase I del complejo principal de histocompatibilidad, lo que al parecer aumenta la citotoxicidad mediada por células T y al microscopio electrónico se aprecian células T fijas a las miofibrillas. Estos datos parecen indicar que las células T citotóxicas reconocen moléculas antigénicas en la superficie de las fibrillas musculares. Por otro lado, en la dermatomiositis el daño microvascular causa degeneración de las fibras musculares en la periferia de los fascículos. Los estudios de inmunofluorescencia muestran depósitos de inmunoglobulinas y complemento en las paredes capilares. La infiltración de linfocitos en el músculo está compuesta principalmente de células B en localización perivascular, y casi todas las células T son CD4+. Estos datos sugieren un proceso de inflamación mediado por complejos inmunes o anticuerpos como mecanismo primario en la dermatomiositis.10 Estas diferencias en los infiltrados linfocíticos apoyan el concepto de que la polimiositis y la dermatomiositis son entidades diferentes.
Reacciones autoinmunes
Se encuentran autoanticuerpos contra antígenos nucleares y citoplasmáticos en hasta el 90 por ciento de los pacientes con una miopatía inflamatoria, que son útiles para distinguir estas enfermedades de otras miopatías. Sin embargo, aunque se han descrito muchos autoanticuerpos diferentes, muchos no son específicos de miositis. Dentro de los inespecíficos, los anticuerpos contra ribonucleoproteína nuclear (nRNP) se observan en el 10 a 15 por ciento de los pacientes con miositis, pero la mayoría de estos tienen datos de LEG o esclerodermia. En forma semejante, varias reacciones serológicas se observan en los casos de síndrome de sobreposición de miositis y esclerodermia, incluyendo anticuerpos contra nRNP, Ro (también llamado SS-A), PM-1 (también llamado PM-Scl), Scl-70, Sm, ADN y La (también denominado SS-B).
Además de estos autoanticuerpos inespecíficos, existen varios autoanticuerpos que se encuentran solo en los pacientes con miositis se asocian con datos característicos de la enfermedad. Estos autoanticuerpos específicos de miositis tienen la propiedad en común de reaccionar con antígenos nucleares y citoplásmicos en moléculas asociadas con aspectos de la síntesis proteica. Se han definido tres grupos de autoanticuerpos, contra la aminoacil-ARN de transferencia (ARNt) sintetasa, contra la partícula de reconocimiento de señales (SRP, por sus siglas en inglés) y contra el antígeno nuclear Mi-2. Cada uno de estos grupos se asocia con un síndrome clínico de miositis específico.
Anticuerpos contra la aminoacil-ARNt sintetasa Cada aminoacil ARNt-sintetasa cataliza el acomplamiento de un aminoácido a su ARNt específico. Una vez formado, el aminoacil-ARNt puede donar, bajo dirección del ARN mensajero (ARNm) su aminoácido a las cadenas polipéptidicas que van formándose en los ribosomas. En los pacientes con polimiositis o dermatomiositis ocurren autoanticuerpos contra cinco de las 20 amioacil-ARNt sintetasas, la mayoría de los pacientes tienen autoanticuerpos contra la histidil-ARNt sintetasa, también llamados anticuerpos anti-Jo-1. Aunque suele disponerse de pruebas para detectarlos anticuerpos anti-histidil sintetasa, las pruebas específicas para detectar otros autoanticuerpos específicos de miositis solo están disponibles en centros especializados.
Los pacientes con cualquiera de los autoanticuerpos contra las aminoacil-ARNt sintetasas tienen características clínicas que se denominan en forma colectiva como el síndrome antisintetasa. Estos pacientes con frecuencia tienen miositis (90 por ciento), artritis (90 por ciento), enfermedad pulmonar intersticial (ILD, 80 por ciento), fiebre (80 por ciento) y una erupción acompañada por hiperqueratosis de las manos (las llamadas manos del mecánico, 70 por ciento) y fenómeno de Raynaud (60 por ciento).11 La enfermedad suele ser severa, con inicio en la primavera y responde solo en forma parcial al tratamiento. El síndrome antisintetasa causa alrededor del 20 a 25 por ciento de los casos de pacientes con miositis.
Anticuerpos anti-SRP La SRP es un complejo de varias proteínas citoplásmicas
que facilitan la traslocación de los nuevos polipéptidos recién sintetizados a través del retículo endoplásmico. Los pacientes con autoanticuerpos contra la SRP tienen miositis severa con mialgias y afección cardiaca. La enfermedad suele iniciar en el otoño. Menos del 5 por ciento de los pacientes tienen miositis de este tipo, que ocurre con más frecuencia en mujeres afroamericanas, responde mal al tratamiento y tiene mal pronóstico.
Anticuerpos anti-Mi-2 El antígeno reconocido por estos autoanticuerpos es una proteína nuclear cuya función se desconoce. Los pacientes con anticuerpos anti-Mi-2 tienen dermatomiositis con una erupción cutánea prominente que se distribuye en hombros, porción superior del tórax y parte alta de la espalda (el llamado signo del mantón), pápulas de Gottron y engrosamiento periungueal. Cinco a 10 por ciento de estos pacientes con miositis tienen esta forma de enfermedad, que tiene buena respuesta al tratamiento y buen pronóstico.
DIAGNOSTICO
Manifestaciones clínicas
Debilidad muscular La debilidad afecta a los músculos estriados voluntarios y con una mayor frecuencia a los grupos musculares proximales que a los distales. Los síntomas se relacionan con debilidad de las cinturas escapular y del hombro (v.gr., dificultad para levantarse de la silla, para subir escaleras y para alcanzar cosas que están por encima de la cabeza). Con frecuencia están afectados los flexores del cuello y, en casos graves, los músculos distales. La parálisis de los músculos faríngeos produce disfagia, voz nasal y aspiración de los materiales ingeridos. La participación de otros músculos inervados por los pares craneales es menos obvia; la paresia de los músculos extraoculares es rara. Los músculos intercostales, diafragmáticos y del tronco pueden afectarse, produciendo insuficiencia ventilatoria, aunque la debilidad del diafragma se asocia con deficiencia de maltasa ácida, más que con miositis.12
La gravedad y rapidez de la debilidad muscular varía, pero la enfermedad suele progresar de manera gradual en un periodo de semanas a meses. La evolución fulminante de la enfermedad es poco frecuente: en términos de unos días a semanas aparece miolisis aguda con mioglobinuria, debilidad generalizada grave e insuficiencia respiratoria. En otros pacientes la debilidad muscular puede progresar gradualmente en el plazo de varios años. Se ha descrito la mejoría espontánea de la miositis, pero es probable que esto ocurra con poca frecuencia.1-3
Cambios en la piel La erupción característica de la dermatomiositis es eritematosa, con o sin la presencia de escamas y atrofia. Se presenta en cara, cuello, parte superior del tórax y sobre las superficies extensoras de las articulaciones, como las de las manos y codos. Puede aparecer edema periorbitario y también puede encontrarse eritema en heliotropo, que consiste en una coloración violácea o lila, principalmente de los párpados. Ocasionalmente, la erupción puede ser más diseminada o adoptar formas diferentes [ver figura 1]. También ocurren eritema y telangiectasias en las áreas periungueales. La vasculitis cutánea puede producir nódulos dolorosos, infartos y ulceraciones en los dedos y otras áreas. Esta vasculitis cutánea se ha asociado con neoplasias subyacentes.13
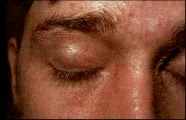
|
| Figura 1 |
| Dermatomiositis |
Los cambios cutáneos típicos de la dermatomiositis pueden ocurrir incluso en ausencia de evidencia clínica de afección muscular, y en este caso se denomina a la enfermedad dermatomiositis amiopática.14 Sin embargo, un estudio demostró que las técnicas de imagen por resonancia magnética (IRM) pueden revelar cambios que indican una alteración metabólica después del ejercicio en pacientes con este trastorno.15
Afección pulmonar Ocurre afección pulmonar en casi el 50 por ciento de los pacientes que tienen miositis. La neumonía es la forma más común de enfermedad pulmonar en estos pacientes. Puede desarrollarse neumonía por aspiración por una combinación de disfunción de los músculos faríngeos y tos ineficaz causada por debilidad de los músculos respiratorios. El uso de medicamentos inmunosupresores puede ocasionar neumonía por organismos oportunistas y reacciones pulmonares reversibles asociadas al tratamiento con metotrexate y ciclofosfamida.
Se presenta enfermedad pulmonar intersiticial (EPI) en hasta el 30 por ciento de los pacientes. Esta puede ser leve a severa y de evolución lenta o rápidamente progresiva. El padecimiento puede ser asintomático o causar síntomas consistentes en disnea progresiva al ejercicio y tos no productiva asociados con estertores crepitantes basales. Las pruebas de función respiratoria revelan hipoxemia, reducción del volumen pulmonar y menor capacidad de difusión. Suele encontrarse una de tres formas histológicas de EPI: neumonía intersticial, daño alveolar difuso o bronquiolitis obliterante con o sin neumonía organizada. La EPI ocurre con o sin afección cutánea. No existe correlación entre el desarrollo de la EPI y la gravedad de la afección muscular, y la EPI puede preceder o ser subsecuente al inicio de la debilidad muscular.1-3 La EPI se asocia con alta mortalidad y poca respuesta al tratamiento de la miositis, pero se ha reportado que el tratamiento con esteroides en bolo o ciclofosfamida en bolos o diario puede ser útil.1-3
Alteraciones cardiacas La afección cardiaca es más frecuente de lo que se detectaba en el pasado. Se manifiesta por (1) alteraciones de la conducción con grados variables de bloqueo cardiaco, incluyendo bloqueos de rama y cambios en el segmento ST-T, (2) taquiarritmias auriculares y ventriculares y síndrome del seno enfermo, y (3) miocarditis o fibrosis miocárdica que causa insuficiencia cardiaca. Las pruebas diagnósticas sensibles, incluyendo el ecocardiograma, el monitor Holter, la gamagrafía perfusoria y los estudios de ventanas y reservorios sanguíneos, indican afección cardiaca en la mayoría de los pacientes con miositis.
Otras manifestaciones En ocasiones los pacientes tienen fenómeno de Raynaud y artralgias. Puede ocurrir artritis leve, inflamatoria y simétrica que afecta las manos, muñecas y rodillas. Además, en ocasiones se desarrollan deformidades articulares por subluxación, sin las erosiones yuxtarticulares óseas típicas de la artritis reumatoide. La disfagia puede ser causada por afección de los músculos esqueléticos de la faringe o del músculo liso esofágico, alterando la nutrición. Los pacientes adultos con miositis rara vez tienen afección del tubo digestivo bajo.1-3
Pruebas de laboratorio
Enzimas musculares La necrosis del músculo ocasiona la liberación hacia el suero de varias enzimas intracelulares. Los niveles de estas enzimas son de utilidad en el diagnóstico y como indicadores de la eficacia del tratamiento. Las enzimas cuantificadas con mayor frecuencia son la creatinina cinasa (CK), las transaminasas, la aldolasa y la dehidrogenasa láctica. La CK constituye el indicador más específico de lesión muscular, debido a que su presencia se encuentra limitada al tejido muscular y nervioso. La determinación de las isoenzimas de CK puede ayudar a valorar la importancia de las elevaciones de esta enzima. La CK ocurre en tres formas que difieren en los distintos tejidos. Dos formas de CK son homodímeros de subunidades M o B, y una es un heterodímero M y B. La MM predomina en el músculo esquelético y su elevación indica daño a este tejido. La MB se encuentra principalmente en el músculo cardiaco, en donde constituye el 20 a 25 por ciento de la CK cardiaca total, aunque también existe MB en el músculo esquelético en regeneración. La BB se localiza principalmente en el sistema nervioso central y el músculo liso. La elevación en los niveles séricos de aldolasa, una aminotransferasa, o deshidrogenasa láctica no es específica de las lesiones musculares porque estas enzimas existen en muchos tejidos.
El grado de elevación de los niveles séricos de las enzimas musculares revela el grado de afectación del músculo y la rapidez de su destrucción. Por lo tanto, el comienzo agudo de la miopatía puede estar relacionado con valores séricos elevados de enzimas musculares, mientras que en la enfermedad de progresión lenta los niveles séricos pueden ser normales o ligeramente elevados, aun cuando la debilidad muscular sea severa. Sin embargo, todos los pacientes con polimiositis muestran niveles séricos elevados de una o más enzimas musculares en algún momento de la evolución de la enfermedad; de ahí que ante la ausencia de esta elevación, el diagnóstico deberá hacerse con precaución. Tales enzimas también son de ayuda para valorar la evolución de la enfermedad y su respuesta al tratamiento.1-3
La Academia Americana de Dermatología ha publicado guías sobre las manifestaciones cutáneas de la dermatomiositis.16
Electromiografía e IRM En la mayoría de los pacientes la electromiografía muestra potenciales de unidad motora polifásicos y de baja amplitud, lo que indica falta de sincronía en la contracción entre las fibras musculares dentro de la unidad motora. Este hallazgo se correlaciona con la distribución normalmente irregular de la generación muscular que se observa en el examen histopatológico. La presencia de fibrilaciones y de irritabilidad durante la inserción son evidencia de irritabilidad de la membrana. Esos hallazgos son característicos de la polimiositis, pero también pueden presentarse en otros procesos miopáticos.
La IRM proporciona una manera no invasiva de evaluar grandes volúmenes de masa muscular, evitando los errores que pueden ocurrir al tomar la muestra en una biopsia muscular.15 Detecta la atrofia con sustitución grasa del músculo afectado y las áreas de inflamación. La IRM es una técnica con buena relación costo eficacia para aumentar la exactitud de la biopsia muscular en pacientes con sospecha de polimiositis.17,18
Histopatología Aunque las características histológicas de la polimiositis varían, los principales cambios incluyen datos de degeneración de las fibras y pérdida de estriaciones cruzadas, cambios hialinos y granulares, núcleos picnóticos y fragmentación celular [ver figura 2]. Con frecuencia estos cambios son irregulares, y las fibras necróticas pueden estar diseminadas entre fibras normales. La regeneración de las fibras musculares es característica, como se demuestra por los cambios basófilos en el citoplasma y los numerosos núcleos grandes. Los cambios inflamatorios en el músculo consisten en infiltración de linfocitos, células plasmáticas e histiocitos con edema intersticial, pero estos cambios no siempre se observan, especialmente en casos crónicos. La miositis juvenil se acompaña de lesiones vasculares características en los músculos y la piel. Esta vasculopatía incluye infiltración por células inflamatorias, cambios proliferativos y degenerativos de las células endoteliales e inclusiones celulares.1-3
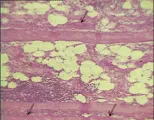
|
| Figura 2 |
| Histología en la polimiositis |
Criterios diagnósticos
La presencia de cuatro criterios diagnósticos mayores establece el diagnóstico de polimiositis [ver tabla 2]; el diagnóstico es de probabilidad si se cumplen tres criterios. El diagnóstico de miositis por cuerpos de inclusión se establece por los resultados de microscopía de luz y electrónica de las biopsias musculares. Debe realizarse biopsia muscular en todos los pacientes adultos antes de iniciar el tratamiento. Pueden requerirse varias biopsias si la primera no es diagnóstica y se emplearán tinciones especiales para buscar miositis por cuerpos de inclusión y miopatías mitocondriales y metabólicas.2,12 La IRM puede ser una guía útil para elegir el sitio de biopsia muscular.
|
|
Nota: ver referencias 1-3 |
Diagnóstico diferencial
Deben excluirse ciertas enfermedades similares a la polimiositis [ver tabla 3]. Las miopatías metabólicas se analizan en detalle en otra sección. La sarcoidosis puede producir una miopatía granulomatosa. Una miopatía de tipo inflamatorio similar a la polimiositis se presenta en pacientes con síndrome de inmunodeficiencia adquirida (SIDA). Además, la zidovudina (AZT), usada con frecuencia para tratar pacientes con SIDA, puede causar miopatía por disfunción mitocondrial.19 Varios fármacos, incluyendo colchicina y cloroquina, producen cambios miopáticos, generalmente reversibles cuando se suspende el medicamento. La intoxicación con cocaína puede causar mioglobinuria, insuficiencia renal y hepática, coagulación intravascular diseminada y muerte.1-3
|
||||||||||||||||||
Nota: ver referencias 1-3. |
TRATAMIENTO
Los lineamientos para el tratamiento de las miopatías inflamatorias idiopáticas no se han establecido del todo por varios motivos. Los padecimientos son poco frecuentes, lo que dificulta acumular un número suficiente de pacientes para realizar estudios aleatorios y controlados. Además, ciertas formas de la enfermedad tienen curso lento y prolongado, por lo que requieren periodos largos de observación. Por último, no existe una clasificación aceptada de modo uniforme de esta enfermedad, por lo que las comparaciones de tratamientos administrados a grupos diferentes de pacientes en diferentes momentos y lugares puede no ser válida.4 Como ejemplos, en el pasado la polimiositis y la dermatomiositis se incluían en la misma categoría y la miositis por cuerpos de inclusión no se había definido. En la actualidad es claro que las diferentes formas de estas enfermedades varían en su pronóstico y respuesta al tratamiento.
Esteroides
Los corticoides son los fármacos de elección para el tratamiento inicial de la miositis, aunque nunca se ha realizado un estudio rigurosamente controlado para comprobar su eficacia. Aunque en ocasiones los pacientes mejoran con dosis bajas de esteroides, o incluso sin ellos, la mayoría de los pacientes requiere dosis altas de esteroides por periodos de tiempo relativamente prolongados. El tratamiento debe iniciarse con prednisona en dosis de 1 mg/kg/día divida en varias dosis. Un esfoque estándar consiste en mantener esta dosis durante 3 meses o hasta que los niveles séricos de enzimas musculares se normalicen o exista mejoría clínica. Después del periodo inicial, la prednisona puede administrarse en una sola dosis diaria matutina para minimizar la toxicidad y reducirse gradualmente 20 a 25 por ciento cada mes. Con este esquema se alcanza una dosis de mantenimiento de 5 a 10 mg diarios en alrededor de 6 a 8 meses. Se considera que no existe respuesta el tratamiento esteroideo si no ocurre mejoría después de los tres meses iniciales.
Un esquema alternativo de tratamiento consiste en 60 a 80 mg de prednisona al día en una dosis durante 3 a 4 semanas, con reducción posterior en 12 semanas hasta alcanzar 80 a 100 mg cada tercer día. Al disminuir la dosis 5 o 10 mg cada 3 o 4 semanas se alcanza una dosis de mantenimiento de 20 a 25 mg cada tercer día. Se considera que el tratamiento fracasó si no existe mejoría después de 4 meses de tratamiento.20
Por último, varios reportes han sugerido que los pulsos intravenosos de metilprednisolona tienen ventajas sobre la prednisona oral diaria. Las dosis varían de 0.5 a 1.0 g/día por 1 a 3 días, repetidos en intervalos de 1 a 4 semanas. Aunque los resultados no están demostrados, los pacientes tratados con este esquema tuvieron mayor frecuencia y rapidez para lograr la remisión, con menos toxicidad por esteroides, que los pacientes tratados con esteroide oral diario.9
Agentes inmunosupresores
La adición de otro agente inmunosupresor generalmente se reserva para los pacientes que han recibido el tratamiento con prednisona descrito antes y no tienen una respuesta satisfactoria. Varios factores han hecho que algunos investigadores modifiquen este esquema. El tratamiento intensivo con esteroides se asocia en forma invariable con efectos adversos, algunos de los cuales son severos. Estos incluyen apariencia cushinoide, fracturas por compresión, necrosis avascular, cataratas e infecciones. Hasta el 40 por ciento de los pacientes tienen enfermedad resistente a los esteroides, en especial los que tienen debilidad de más de 9 meses antes de iniciar el tratamiento. Aunque no son concluyentes, las evidencias actuales apoyan que la evolución puede mejorar agregando un segundo agente, por lo general metotrexate o azatioprina, junto con el inicio del esteroide. Esto puede estar indicado específicamente en pacientes con deterioro rápido, dificultad en la deglución o EPI.
Los agentes usados con más frecuencia en los pacientes que son resistentes al tratamiento esteroideo son el metotrexate y la azatioprina. El metotrexate puede administrarse por vía oral en dosis inicial es de 7.5 a 10 mg a la semana, que se aumenta en forma gradual hasta 25 mg por semana. En forma alternativa, el metotrexate puede administrarse por vía intravenosa en dosis de 15 mg por semana y aumentarse a 60 mg por semana. Al aumentar la dosis de metotrexate se disminuye la de prednisona. La reducción en las enzimas musculares indica que ocurrirá mejoría en la fuerza muscular. Por lo general ambos esquemas de tratamiento con metotrexate son bien tolerados por los pacientes con miositis. Los reportes han sugerido que el metotrexate puede disminuir los requerimientos de esteroides y beneficiar a los pacientes con más rapidez que la azatioprina. La toxicidad es semejante a la de los pacientes con artritis reumatoide que reciben este medicamento. Puede ocurrir enfermedad hepática en pacientes con miositis tratados con metotrexate, y estos pacientes deben ser vigilados con pruebas diferentes a los niveles de aminotransferasa, incluyendo gama-glutamiltranspeptidasa, porque los niveles de aminotransferasas pueden reflejar el daño muscular. El metotrexate puede mejorar la EPI, pero también causa toxicidad pulmonar y está relativamente contraindicado en pacientes con miositis y EPI. En algunos pacientes con miositis tratados con metotrexate, linfomas asociados al virus Epstein-Barr sufrieron remisión al suspender el metotrexate.2,3
En un estudio prospectivo, controlado y doble ciego, la azatioprina demostró ser eficaz en los pacientes con miositis. Además, los pacientes que fueron tratados con una combinación de prednisona y azatioprina tuvieron mejor evolución funcional y requirieron menos prednisona que los enfermos que fueron tratados solo con prednisona. Se ha reportado que la azatioprina es eficaz en pacientes con miositis, pero se requiere tratamiento por lo menos durante 6 meses para que ocurra la mejoría. Este fármaco debe iniciarse con dosis de 25 a 50 mg/día aumentando después a un máximo de 200 mg diarios para minimizar la toxicidad gastrointestinal. Sus efectos tóxicos consistieron en alteraciones gastrointestinales, supresión de la médula ósea, desarrollo de infecciones y quizá de neoplasias. En los pacientes en los que el tratamiento con esteroides y metotrexate o azatioprina ha fracasado, la combinación de metotrexate y azatioprina parece promisoria y se recomienda en lugar de los agentes alquilantes más tóxicos.2,3
Se han usado varios otros agentes y tratamientos para la miositis resistente a esteroides. Debido a su toxicidad, los agentes alquilantes se usan con poca frecuencia. Se ha administrado ciclofosfamida tanto en pulsos intravenosos como por vía oral diaria, pero las evidencias respecto a la eficacia de ambos modos de administración son controversiales. Se ha reportado cierto éxito en pacientes con EPI asociada y en niños con miositis y vasculitis. También se ha empleado el clorambucil en algunos enfermos que no respondieron a los esquemas habituales de tratamiento.2,3
La ciclosporina parece tener algún papel en el tratamiento de las miopatías inflamatorias; sin embargo, es importante balancear los beneficios potenciales de este fármaco contra los riesgos de toxicidad.21 Los efectos tóxicos incluyen hipertensión y daño renal, así como una miopatía relacionada a hipomagnesemia. Estudios no controlados con ciclosporina han reportado mejoría en pacientes con polimiositis y dermatomiositis del adulto y juvenil, mientras que otros estudios no han encontrado buenos resultados. Un estudio preliminar de tacrolimus en pacientes con polimiositis refractaria ha sido alentador.22
La globulina inmune intravenosa parece ser útil en pacientes tanto con dermatomiositis como con polimiositis. Un estudio controlado de globulina inmune IV en pacientes con dermatomiositis demostró eficacia cuando se administró a una dosis de 1 g/kg/día por 2 días, repetida cada 24 días por 3 meses.23 Los reportes han descrito beneficios en los pacientes con polimiositis o dermatomiositis juvenil;24 sin embargo, en un estudio controlado de globulina inmune IV en pacientes con miositis por cuerpos de inclusión no se estableció la eficacia de este tratamiento, quizá por el tamaño pequeño de la muestra.25 Se requieren estudios adecuados a largo plazo, incluyendo la relación costo-beneficio, antes de recomendar la globulina inmune IM como tratamiento de elección.26
En un estudio controlado la plasmaféresis no fue eficaz en pacientes con miositis crónica, aunque reportes anecdóticos han descrito éxito en los pacientes con afección aguda.2,3
La fisioterapia tiene un papel muy importante en la rehabilitación de los pacientes con miositis. Durante la fase de enfermedad inflamatoria activa se requieren ejercicios pasivos de rango de movimiento para evitar contracturas. Una vez que se ha controlado el componente inflamatorio de la enfermedad, los ejercicios activos de resistencia son útiles para recobrar la fuerza muscular.1-3,27
PRONOSTICO
La supervivencia de los pacientes con polimiositis ha mejorado en los últimos años y puede ser de casi 75 por ciento 8 años después del diagnóstico. La supervivencia más corta se asoció con edad mayor de 45 años en el momento del diagnóstico, afección cardiaca, pulmonar, disfagia y, en algunas series, enfermedades malignas. La polimiositis juvenil suele tener un buen pronóstico. No obstante, la morbimortalidad sigue siendo importante en todos los grupos de edad, ya sea secundaria a la enfermedad por sí misma o a las complicaciones derivadas del tratamiento. En la actualidad, la mortalidad general es cercana al 20 por ciento durante un periodo de 5 años. En varias series, sólo una minoría de las causas de muerte se atribuyeron a enfermedad maligna. Las remisiones completas y permanentes son poco frecuentes. En un estudio se observó que los pacientes que provenían de un medio ambiente rural parecían tener una enfermedad de menor gravedad y respuestas al tratamiento más completas que los pacientes de un medio ambiente urbano. Una posible explicación para este hallazgo es que los investigadores en centros médicos urbanos grandes seleccionan pacientes con enfermedad más severa que la observada en pacientes de otras áreas.1-3
Enfermedades asociadas con mioglobinuria
Muchas enfermedades que producen lesiones del músculo esquelético, incluyendo enfermedades graves consideradas en el diagnóstico diferencial de la polimiositis, se caracterizan por mioglobinuria [ver tabla 4]. La mioglobinuria es la excreción urinaria de mioglobina, una proteína que transporta el oxígeno dentro de células del músculo esquelético. Las lesiones graves del músculo esquelético pueden producir rabdomiolisis, desintegración del tejido muscular que se manifiesta por mioglobinemia y mioglobinuria. Los episodios graves de mioglobinuria pueden inducir insuficiencia renal aguda.
|
||
|
La formación idiopática de mioglobinuria, también denominada mioglobinuria paroxística, se caracteriza por episodios agudos de dolores musculares, hipersensibilidad y debilidad que duran 2 a 3 días o más. Los paroxismos suelen ser precipitados por el ejercicio; la enfermedad puede aparecer durante la niñez o en etapas posteriores de la vida. Raramente se desarrolla debilidad muscular crónica y la insuficiencia renal aguda que aparece durante un episodio agudo puede ser mortal. El síndrome idiopático probablemente representa un grupo de enfermedades que no se ha caracterizado completamente; algunas de ellas son claramente familiares. En algunos pacientes que se habían considerado como portadores de mioglobinuria idiopática se han identificado deficiencias enzimáticas. Se considera que algunos defectos metabólicos, como la deficiencia de carnitina palmitoiltransferasa y las deficiencias enzimáticas que producen las glucogenosis son la causa de algunos casos de mioglobinuria.
El diagnóstico de mioglobinuria se basa en la siguiente tríada: prueba de la ortotoluidina u otras pruebas para detección de sangre en orina positivas en ausencia de eritrocitos; cilindros pigmentados en el sedimento urinario y elevación de la CK y otras enzimas musculares en suero. La rabdomiolisis puede elevar el nivel sérico de CK más de 100 veces. También deben emplearse pruebas específicas para identificar de manera definitiva al pigmento urinario como mioglobina. El tratamiento está dirigido hacia la causa primaria de la mioglobinuria, cuando ésta existe. Además, se recomienda reposo absoluto, líquidos y alcalinización.1-3
Figura 2 Fotografías cortesía del Dr. E. P. Richardson, Departamento de Neuropatología, Massachusetts General Hospital, Boston.
Bibliografía
- Medsger TA, Oddis CV: Inflammatory muscle disease: clinical features. Rheumatology. Klippel JH, Dieppe PA, Brooks P, et al, Eds. Mosby, St. Louis, 1994, p 6.12.1
- Miller FW: Inflammatory myopathies: polymyositis, dermatomyositis, and related conditions. Arthritis and Allied Conditions: A Textbook of Rheumatology, 13th ed. Koopman WJ, Ed. Williams & Wilkins, Baltimore, 1997, p 1407
- Wortmann RL: Inflammatory diseases of muscle. Textbook of Rheumatology, 5th ed. Kelley WN, Ruddy S, Harris ED Jr, et al, Eds. WB Saunders Co, Philadelphia, 1997, p 177
- Targoff IN, Miller FW, Medsger TA Jr, et al: Classification criteria for the idiopathic inflammatory myopathies. Curr Opin Rheumatol 9:527, 1997 [PMID 9375282]
- Zantos A, Zhang Y, Felson D: The overall and temporal association of cancer with polymyositis and dermatomyositis. J Rheumatol 21:1855, 1994
- Pachman LM: Inflammatory myopathy in children. Rheum Dis Clin North Am 20:919, 1994
- Calabrese LH, CHou SM: Inclusion body myositis. Rheum Dis Clin North Am 20:955, 1994 [PMID 7855331]
- Amato AA, Gronseth GS, Jackson CE, et al: Inclusion body myositis: clinical and pathological boundaries. Ann Neurol 40:581, 1996 [PMID 8871577]
- Leff RL, Love LA, Miller FW, et al: Viruses in idiopathic inflammatory myopathies: absence of candidate viral genomes in muscle. Lancet 339:1192, 1992 [PMID 1349938]
- Hohlfeld R, Engel AG, Goebels N, et al: Cellular immune mechanisms in inflammatory myopathies. Curr Opin Rheumatol 9:520, 1997 [PMID 9375281]
- Love LA, Leff RL, Fraser DD, et al: A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 70:360, 1991 [PMID 1659647]
- Adams EM, Plotz PH: The treatment of myositis: how to approach resistant disease. Rheum Dis Clin North Am 21:179, 1995 [PMID 7732167]
- Callen JP: Relationship of cancer to inflammatory muscle diseases: dermatomyositis, polymyositis, and inclusion body myositis. Rheum Dis Clin North Am 20:943, 1994
- Stonecipher MR, Jorizzo JL, White WL, et al: Cutaneous changes of dermatomyositis in patients with normal muscle enzymes: dermatomyositis sine myositis? J Am Acad Dermatol 28:951, 1993
- Park JH, Olsen NJ, King L Jr, et al: Use of magnetic resonance imaging and P-31 magnetic resonance spectroscopy to detect and quantify muscle dysfunction in the amyopathic and myopathic variants of dermatomyositis. Arthritis Rheum 38:68, 1995 [PMID 7818575]
- Drake LA, Dinehart SM, Farmer ER, et al: Guidelines of care for dermatomyositis. American Academy of Dermatology. J Am Acad Dermatol 34:824, 1996
- Schweitzer ME, Fort J: Cost-effectiveness of MR imaging in evaluating polymyositis. AJR Am J Roentgenol 165:1469, 1995 [PMID 7484589]
- Olsen NJ, Park JH: Inflammatory myopathies: issues in diagnosis and management. Arthritis Care Res 10:200, 1997 [PMID 9335632]
- Peters BS, Winer J, Landon DN, et al: Mitochondrial myopathy associated with chronic zidovudine therapy in AIDS. Q J Med 86:5, 1993 [PMID 8438050]
- Joffe MM, Love LA, Leff RL, et al: Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 94:379, 1993 [PMID 8386437]
- Langford CA, Klippel JH, Balow JE, et al: Use of cytotoxic agents and cyclosporine in the treatment of autoimmune disease. Part 1: rheumatologic and renal diseases. Ann Intern Med 128:1021, 1998
- Villalba L, Adams M: Update on therapy for refractory dermatomyositis and polymysitis. Curr Opin Rheumatol 8:544, 1996 [PMID 9018458]
- Dalakas MC, Illa I, Dambrosia JM, et al: A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med 329:1993, 1993 [PMID 8247075]
- Dalakas MC: Intravenous immune globulin therapy for neurologic diseases. Ann Intern Med 126:721, 1997
- Dalakas MC, Sonies B, Dambrosia J, et al: Treatment of inclusion-body myositis with IVIg: a double-blind, placebo-controlled study. Neurology 48:712, 1997 [PMID 9065553]
- van der Meche FG, van Doorn PA: The current place of high-dose immunoglobulins in the treatment of neuromuscular disorders. Muscle Nerve 20:136, 1997 [PMID 9040650]
- Escalante A, Miller L, Beardmore TD: Resistive exercise in the rehabilitation of polymyositis/dermatomyositis. J Rheumatol 20:1340, 1993 [PMID 8230016]