Dermatología
⭳ Abrir artículo (PDF)972.0 KBEste artículo es idéntico en la Edición 3/2000.
Contenido del artículo
X TUMORES CUTANEOS MALIGNOS
X TUMORES CUTANEOS MALIGNOS
DR. ALLAN C. HALPERN
DRA. PATRICIA L. MYSKOWSKI
Los tumores malignos pueden originarse de células de cualquier capa de la piel (queratinocitos, melanocitos, fibroblastos, células endoteliales y adipocitos), así como de células como los linfocitos, que transitan en condiciones normales por la piel. También pueden aparecer metástasis cutáneas de otros sitios primarios. En esta subsección se revisarán los tumores cutáneos malignos más frecuentes en orden de frecuencia.
Tumores malignos de la epidermis
Los cánceres de la epidermis son las neoplasias más frecuentes en los humanos. Se originan en los queratinocitos y los melanocitos de la epidermis y representan una oportunidad única de intervención eficaz por medio tanto de detección temprana como de prevención primaria. Pueden diagnosticarse desde el punto de vista clínico por simple inspección visual y confirmarse histológicamente con una biopsia muy poco invasora.
Tanto el carcinoma basocelular como el epidermoide se originan de los queratinocitos de la epidermis. Debido a que estos dos cánceres comparten muchas características, con frecuencia de agrupan bajo el término de cáncer de piel no melanoma.
El melanoma maligno es una neoplasia que surge de los melanocitos. Aunque puede aparecer en cualquier melanocito del organismo, incluyendo los ojos, la gran mayoría ocurre en la piel. El melanoma maligno cutáneo se ha clasificado en cuatro tipos histogenéticos principales: melanoma léntigo maligno, melanoma de diseminación superficial, melanoma nodular y melanoma lentiginoso acral.
EXPOSICION AL SOL Y CANCER DE PIEL
Varias líneas de evidencia implican a la radiación ultravioleta (UV) en la patogenia de los tres principales cánceres de la epidermis.1 Los datos epidemiológicos relacionan la exposición acumulada al sol con el desarrollo del carcinoma epidermoide y la exposición solar intermitente intensa con el carcinoma basocelular y el melanoma. Los estudios de laboratorio indican que tanto la radiación solar UVA (320 a 400 nm) como la UVB (290 a 320 nm) pueden dañar el ADN en forma directa y oxidativa. Además, la radiación UV puede suprimir el sistema inmunológico cutáneo.2 La asociación de algunos carcinomas epidermoides con carcinógenos químicos y la ocurrencia de melanomas acral lentiginoso y de mucosas en partes no expuestas del organismo destacan la necesidad de realizar estudios para identificar otros agentes etiológicos.
El conocimiento sobre el importante papel que tiene la luz solar en la etiología del cáncer de piel ofrece una oportunidad para realizar prevención primaria por medio del uso de protección solar. Por desgracia, aún se desconoce el espectro de acción, el momento y las dosis de exposición UV exactos involucrados en el desarrollo del cáncer de piel en los humanos. Por ello, los pacientes deben ser instruidos sobre el efecto dañino de exponerse al sol y broncearse. Deben realizarse esfuerzos para disminuir en forma general la exposición al sol por medio de cambios en el comportamiento y el uso de ropa protectora. Las pantallas solares de espectro amplio con un factor de protección solar (SPF) de 15 o más son útiles, pero no deben usarse para aumentar la cantidad de tiempo expuesto en forma directa a la luz solar.3
CANCER DE PIEL NO MELANOMA
El cáncer de piel no melanoma (CPNM) ocurre en forma típica como una lesión rosada en la superficie de piel expuesta al sol. Cualquier lesión rosada que persista o recurra en la misma localización, en especial si se irrita con facilidad por los traumatismos leves, es sospechosa de un CPNM. El clínico debe saber que algunas formas de CPNM se desvanecen con los cambios de estación (i.e., con la menor exposición al sol) o con la aplicación de esteroides tópicos, y deben notificar a los pacientes que cualquier lesión que recurra requiere de atención.
Carcinoma basocelular
El carcinoma basocelular (CBC) es un tumor cutáneo maligno que se origina de los queratinocitos basales de la epidermis.
Epidemiología El CBC es el cáncer de piel más común. Las incidencias reportadas varían desde 3.4 por 100,000 personas por año en afroamericanos a más de 750 por 100,000 por año en caucásicos australianos.4,5 Aunque raro, pueden ocurrir metástasis y muerte por CBC.
Etiología La radiación ultravioleta, en especial la exposición intermitente intensa, parece tener un papel importante en el desarrollo del CBC. Estudios recientes del síndrome de nevos de células basales (síndrome de Gorlin) han permitido nuevos conocimientos sobre la genética del CBC. Se ha identificado un gen denominado patched (parchado), que se descubrió por primera vez en la mosca de la fruta Drosophila, y que parece tener un papel crucial en el desarrollo del CBC. Casi todos los pacientes con síndrome de nevos de células basales parecen heredar una copia mutada de este gen, y los estudios del CBC esporádico sugieren que las mutaciones del gen patched, son un paso necesario, si no es que suficiente, en el desarrollo del CBC.6
Diagnóstico Alrededor del 90 por ciento de los CBC ocurren en la cabeza y el cuello, en formas nodulares y superficiales.
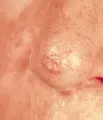
El CBC nodular consiste en una elevación rosada, perlada y translúcida en la superficie de la piel. Se irrita con facilidad, es frágil y se asocia con episodios de ulceración superficial o hemorragia. Cuando la ulceración es prominente puede aparentar una úlcera "de roedor", con un borde perlado y translúcido. Estas lesiones pueden verse más blancas que rosadas y, a la inspección cuidadosa demuestran telangiectasias pequeñas. Tienen una superficie lisa y brillante, y una textura más firme que los nevos dérmicos comunes [ver figura 1].
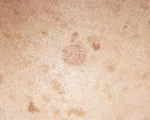
Los CBC superficiales se observan como un parche rosado en la piel. Al
observarlos los más superficiales tienen un borde translúcido,
con áreas que parecen normales o ligeramente fibróticas dentro de
la lesión. Los CBC superficiales suelen localizarse en la parte superior
del tronco, los brazos y las piernas.
Son variantes menos comunes de los CBC las lesiones tipo morfea, pigmentadas y quísticas. Los CBC tipo morfea tienen un patrón infiltrativo que histológica y clínicamene parece una cicatriz. Los CBC pigmentados suelen contener puntos de pigmento azul-negro, aunque pueden mostrar pigmentación importante y total. El pigmento es más característico en la variedad nodular de CBC. Los CBC quísticos son más suaves que los carcinomas nodulares típicos y pueden tener una apariencia azul-gris.
La historia del paciente es muy importante en el diagnóstico del CBC. Cuando se pregunta respecto a lesiones que se irrigan o sangran con facilidad con traumatismos leves los pacientes pueden mostrar al clínico lesiones tempranas que quizá pasarían desapercibidas. Bajo anestesia local debe obtenerse una biopsia de cualquier lesión sospechosa.
Diagnóstico diferencial Los CBC nodulares pueden confundirse con angiofibromas, nevos dérmicos, melanoma amelanótico, metástasis cutáneas y diversos tumores benignos de los anexos (v.gr., tricoepitelioma). Los CBC superficiales simulan a varias dermatosis inflamatorias (v.gr., eccema y tiña) y comparten algunas características clínicas con las queratosis actínicas. Los CBC pigmentados pueden confundirse con facilidad con una neoplasia melanocítica primaria. Los carcinomas quísticos pueden confundirse con tumores de los anexos y lesiones inflamatorias.
Tratamiento El objetivo del tratamiento consiste en erradicar en forma adecuada la lesión y asegurar el mejor resultado tanto cosmético como funcional. Deben tomarse en cuenta muchos factores, como el tamaño, localización y subtipo histológico de las lesiones, así como datos del paciente, incluyendo edad, color y laxitud de la piel, para elegir el tratamiento óptimo.
La gran mayoría de los CBC son susceptibles de tratamiento quirúrgico. Las principales opciones incluyen curetaje y electrodesecación, extirpación, crioterapia, radioterapia y cirugía micrográfica de Mohs.
Un grupo pequeño, pero significativo, de CBC se benefician con la microcirugía de Mohs, que consiste en el examen microscópico de cortes congelados de toda la superficie inferior del tejido extirpado en el momento de la cirugía. Esta técnica puede ser adecuada para las lesiones recurrentes y las lesiones con alta probabilidad de recurrencia. Entre estas se incluyen lesiones mal definidas, lesiones grandes (> 2 cm), lesiones con histología de alto riesgo (i.e., patrón de crecimiento agresivo, patrón de esclerosis o afección perineural) y lesiones que afectan varios planos embrionarios (v.gr., canto ocular o surco nasofacial). La técnica de microcirugía de Mohs ha demostrado asociarse con mayores tasas de curación que los otros tratamientos para las lesiones con riesgo alto.7
La radioterapia puede ser una alternativa eficaz, no dolorosa y bien tolerada, que típicamente se reserva para los pacientes de edad avanzada que son malos candidatos quirúrgicos. Sin embargo, debe evitarse en los pacientes con síndrome de nevos de células basales.
Entre los tratamientos en fase experimental se incluyen la quimioterapia intralesional y la terapia fotodinámica.
Todos los pacientes tratados por CBC tienen riesgo de recurrencia local, así como riesgo significativo de desarrollar otros cánceres de piel. Los enfermos deben ser enseñados para autoexaminar su piel, deben emplear métodos de protección contra el sol y requieren vigilancia médica periódica.
Pronóstico Como ya se mencionó, el riesgo de recurrencia local se relaciona con el tamaño, localización e histología de la lesión. Las metástasis son muy raras: en una serie de 50,000 australianos se reportó una prevalencia de 0.0028%.8 Estas ocurren por vía linfática y hematógena, y los factores de riesgo incluyen síndrome de nevos basales, inmunosupresión y exposición previa a radiación ionizante. Las metástasis no son susceptibles de tratamiento quirúrgico y se asocian con mal pronóstico.
Carcinoma epidermoide
Como el CBC, el carcinoma epidermoide (CE) se origina de los queratinocitos de la epidermis. Desde el punto de vista histológico, las células de los CE bien diferenciados recuerdan a las células de la porción superior de la epidermis.
Epidemiología En 1994 se diagnosticaron en los Estados Unidos entre 150,000 y 250,000 casos nuevos de CE cutáneo.9 La mortalidad calculara por este tipo de cáncer en Estados Unidos en 1988 fue de alrededor de 0.5 por 100,000. Varias líneas de evidencia sugieren un incremento reciente y significativo en la incidencia de CE. Por ejemplo, en Australia, ésta aumento en 51% entre los años de 1985 y 1990.10
Etiología Además de la luz solar, otros agentes etiológicos conocidos que contribuyen al desarrollo de CE cutáneo son la radiación ionizante, los carcinógenos químicos, las quemaduras térmicas y las heridas crónicas que no cicatrizan. Los CE relacionados con exposición solar tienen menor riesgo de metástasis y muerte que los asociados con otras exposiciones. Los factores predisponentes a CE por exposición solar incluyen piel de color claro, tendencia a quemarse e incapacidad para broncearse.
Fisiopatología y patogenia El CE por exposición a luz solar suele asociarse con una lesión precursora denominada queratosis actínica. Estas lesiones ocurren en el cuero cabelludo, cara, superficies extensoras de los antebrazos y dorso de las manos. Son lesiones rosadas de superficie rugosa e irregular. Con frecuencia se sienten más de lo que se ven. La mayoría de los pacientes con queratosis actínicas tienen lesiones múltiples. Se ha calculado que el riesgo de CE en estos pacientes es hasta de 20%.11 El CE también puede aparecer en piel de apariencia normal.
El CE de mucosa oral o genital puede originarse de una lesión precursora denominada leucoplaquia o eritroplaquia. Los CE de mucosas se asocian con riesgo importante de metástasis.
La función inmunológica influye en la progresión del CE. La inmunosupresión que ocurre en pacientes con trasplantes o con linfoma se relaciona con mayor incidencia de CE. En estos pacientes la infección con papilomavirus humano parece tener un papel etiológico asociado a la exposición solar. El CE tiende a ser más agresivo en las personas inmunosuprimidas.
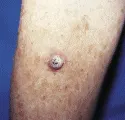
Diagnóstico La mayoría de las lesiones ocurren en áreas del organismo que suelen estar expuestas al sol. Las lesiones son placas firmes y rosadas, con frecuencia con superficie burda y descamativa [ver figura 2]. Se requiere la biopsia para el diagnóstico definitivo.
Diagnóstico diferencial El diagnóstico diferencial del CE
incluye queratoacantoma, enfermedad de Bowen, carcinoma verrucoso, CBC,
queratosis actínica hipertrófica y verrugas comunes.
Los queratoacantomas comparten muchas características con el CBC tanto clínica como histológicamente. Aparecen de novo en piel de apariencia normal y crecen con gran rapidez. Típicamente son elevaciones rosadas, en forma de domo y brillantes en la superficie de la piel, con un tapón queratósico en forma de cráter. Pueden crecer mucho. Aunque los queratoacantomas no se asocian con riesgo de metástasis, destruyen localmente. Se ha demostrado regresión espontánea en el curso de meses.
La enfermedad de Bowen es un CE que se limita a la epidermis. Se presenta como una placa roja, mínimamente elevada y descamativa, con bordes irregulares bien definidos. La asociación reportada antes entre enfermedad de Bowen y neoplasias internas no se ha comprobado.
El CE que no tiene superficie queratósica descamativa puede confundirse con otros tumores de anexos o de la dermis.
Tratamiento Los CE pequeños que evolucionan de una lesión de queratosis actínica pueden tratarse en forma adecuada con curetaje y electrodesecación simple. Las lesiones actínicas de mayor tamaño, así como las lesiones que surgen en áreas de piel no expuesta al sol se tratan mejor con extirpación quirúrgica definitiva con confirmación de bordes negativos. Las lesiones de alto riesgo y mal definidas, en especial las que ocurren en áreas quirúrgicas delicadas de la cara, genitales, manos y pies se tratan mejor por medio de cirugía micrográfica de Mohs.
La radioterapia fraccionada constituye un tratamiento alternativo para el CE primario en los pacientes de edad avanzada que son malos candidatos quirúrgicos. Los beneficios de la radioterapia adyuvante son menos claros, lo mismo que los beneficios de la disección electiva de ganglios linfáticos en pacientes con CE de alto riesgo de cabeza y cuello.
Se han usado quimioterapia citotóxica y modificadores de la respuesta biológica en pacientes con CE avanzado, con tasas de respuesta completa de 25 a 46%, pero con pocos sobrevivientes a largo plazo.
Las queratosis actínicas se tratan con crioterapia, curetaje, quimioterapia tópica con fluorouracilo, desepitelización química y con láser, para prevenir la progresión a CE.
Pronóstico Independientemente del tratamiento empleado, las lesiones con riesgo alto se asocian con una tasa de recurrencia local significativa a los 5 años. Los CE de riesgo alto incluyen los que afectan sitios anatómicos específicos (v.gr., oídos, labios, genitales y otras áreas no expuestas al sol), los mayores de 2 cm de diámetro, los que tienen características histológicas agresivas (profundidad > 4 mm, nivel de Clark IV o mayor, e histología mal diferenciada) y los que ocurren en pacientes inmunosuprimidos.12 La principal ruta de metástasis de los CE es por diseminación linfática a los ganglios regionales. Las tasas reportadas de metástasis varían desde 0.3% en las lesiones pequeñas derivadas de exposición solar, hasta 33% en lesiones más grandes y poco diferenciadas.12 La supervivencia reportada a 5 años para los pacientes con CE y metástasis regionales varía de 25 a 47%.12
MELANOMA MALIGNO
Epidemiología
En los Estados Unidos el riesgo de desarrollar melanoma durante la vida es de alrededor de 1 en 75.13 Entre 1973 y 1994 la incidencia de melanoma aumentó en 121% y la mortalidad asociada en 39%. Actualmente se realiza una detección más temprana, y parece existir una estabilización en las tasas de incidencia en algunos segmentos de la población. Sin embargo, en términos de morbimortalidad, el peso de la enfermedad relacionada con melanoma continúa aumentando. Aunque puede ocurrir melanoma en cualquier persona, es principalmente una enfermedad de la raza blanca. Los melanomas que ocurren en personas de raza negra son típicamente de la variedad acral lentiginosa.
Etiología
Aunque las evidencias epidemiológicas y de ciencias básicas apoyan una asociación entre el melanoma y la exposición a la luz solar, la relación parece ser más compleja. El melanoma léntigo maligno se asocia con exposición crónica y acumulada al sol. El melanoma de diseminación superficial y el nodular parecen asociarse con exposición intensa intermitente, en especial en los jóvenes. El melanoma acral lentiginoso no tiene asociación aparente con la luz del sol. Los estudios básicos y los modelos en animales han implicado a diferentes longitudes de onda de la luz UV en la carcinogénesis del melanoma; la longitud de onda implicada puede variar entre los diversos tipos de melanoma.
Alrededor del 5 por ciento de los melanomas ocurren en pacientes con historia familiar de esta misma neoplasia. Las mutaciones en el gen regulador del ciclo celular p16 (inhibidor 2a de cinasa dependiente de ciclina) se asocian con melanoma en alrededor del 40% de los melanomas de tipo familiar, con asociación del gen al cromosoma 9p.14 Las alteraciones en la reparación del ADN encontradas en pacientes con nevos displásicos sugieren que este es otro mecanismo de predisposición genética al melanoma.
Diagnóstico
Como una lesión pigmentada que ocurre en la superficie de la piel, el melanoma es susceptible de detección temprana por inspección visual simple en una fase fácilmente curable. Si no se trata el melanoma es una de las formas de cáncer más agresivas y con menos respuesta terapéutica.
La detección temprana del melanoma requiere de la inspección cuidadosa de las lesiones pigmentadas en toda la superficie del organismo. A pesar de la intensa asociación del melanoma con la exposición solar, pueden ocurrir lesiones en cualquier sitio de la superficie cutánea. Por lo tanto, el autoexamen del paciente y la revisión del médico deben incluir toda la piel, incluyendo el cuero cabelludo, los genitales y las plantas de los pies. Cualquier lesión pigmentada con cambios recientes o con las características descritas por la nemotecnia ABCD (por asimetría, bordes irregulares, variación del color y diámetro > 6 mm) justifica considerar la posibilidad de un melanoma. Aunque cualquier lunar puede cambiar en forma gradual con el tiempo, cualquiera que cambie de dolor, forma o tamaño en relación con otros lunares merece atención especial [ver figura 3].
Entre las personas de raza blanca se han identificado varios otros factores de
riesgo, como complexión delgada, tendencia a quemarse e incapacidad de
broncearse, presencia de pecas e historia familiar de melanoma. Los marcadores
fenotípicos más fuertes para riesgo de melanoma son los lunares
(nevos), específicamente el aumento en el número de lunares y la
presencia de lunares atípicos (nevos displásicos). El melanoma
puede aparecer en un lunar u originarse de novo en piel de apariencia
normal.
Nevo displásico Varios estudios epidemiológicos han relacionado a los nevos displásicos con riesgo de melanoma. Desde el punto de vista clínico los nevos displásicos son lunares grandes (> 5 mm) con pigmentación abigarrada y bordes mal definidos [ver figura 4]. Histológicamente los nevos displásicos se caracterizan por la presencia de atipia en su arquitectura y atipia citológica dispersa.15 El grado de riesgo de melanoma asociado con el nevo displásico depende del contecto genético. En familias con síndrome de nevos displásicos y melanoma el fenotipo anormal parece heredarse en forma autosómica dominante. Los miembros de estas familias con nevos displásicos tienen un riesgo durante la vida de sufrir melanoma que llega a ser del 100 por ciento.16 Fuera del contexto del melanoma familiar, los nevos displásicos ocurren en el 5 a 10 por ciento de las personas de raza blanca. En esta población los nevos displásicos aún son un marcador de mayor riesgo de melanoma [ver tabla 1].17
Los nevos displásicos representan tanto una oportunidad como un reto en la detección del melanoma. Por un lado su detección permite vigilar en forma eficaz a un grupo de alto riesgo. Por el otro, pueden complicar los intentos de detección de los melanomas al parecer, clínicamente, melanomas tempranos. Aunque algunos nevos displásicos pueden progresar a melanomas, la gran mayoría permanecen benignos. Además, no todos los melanomas que se originan en pacientes con nevos displásicos lo hacen a partir de un lunar previo. La extirpación de todos los nevos displásicos es una medida poco práctica para la prevención de los melanomas. En los pacientes con nevos displásicos la detección de los melanomas se realiza por examen visual por un especialista, aunado con autoexamen y vigilancia para identificar las lesiones que cambian de características.
En la actualidad se desarrollan varios instrumentos que ayudan al diagnóstico del melanoma en pacientes con nevos displásicos. La microscopía epiluminiscente (MEL) consiste en el uso de una especie de otoscopio manual para magnificar una lesión pigmentada mientras se aplica presión y aceite a la superficie. Esta técnica permite la visualización de los patrones de pigmento y las características que no son aparentes por inspección simple. Con entrenamiento y experiencia la MEL puede ser útil para distinguir el melanoma de las lesiones pigmentadas benignas. Sin embargo, si no se tiene experiencia la LEL puede incluso disminuir la certeza diagnóstica.18,19 Otra método para mejorar la detección de melanomas en individuos con riesgo alto es la evaluación ayudada por fotografías. Se usan un grupo basal de fotografías de todo el cuerpo durante el autoexamen y las evaluaciones por el médico para buscar cambios en las lesiones. Este procedimiento elimina la necesidad de extirpar lesiones estables y mejora la sensibilidad del examen para detectar cambios.
Cualquier lesión que origine la sospecha clínica de melanoma requiere de un diagnóstico definitivo. La extirpación total es la técnica preferida para biopsiar las lesiones pigmentadas sospechosas. La biopsia parcial puede causar diagnósticos erróneos por una mala muestra o impedir que el patólogo conozca la arquitectura global y la citología de la lesión. Sin embargo, las biopsias incisionales con buena relación clínico-patológica pueden ser apropiadas para evaluar lesiones grandes y lesiones presentes en áreas delicadas desde el punto de vista quirúrgico. No existen evidencias que sugieran que la biopsia incisional aumenta el riesgo de metástasis.
Diagnóstico diferencial
Los nevos displásicos comparten muchas características con el melanoma temprano de diseminación superficial. Otras lesiones comunes que pueden parecer melanomas son las lentígines, las pecas por el sol, los nevos traumatizados, los angiomas trombosados, los CBC pigmentados, la enfermedad de Bowen pigmentada, los dermatofibromas y las queratosis seborreicas atípicas. Otros dos retos en el diagnóstico diferencial del melanoma merecen mención especial. Los melanomas amelanóticos (melanomas sin pigmento) se presentan como lesiones rosadas que pueden diagnosticarse en forma errónea como CBC o nevos de Spitz. Los nevos de Spitz pueden ser difíciles de distinguir de los melanomas tanto desde el punto de vista clínico como histológico. Ocurren con más frecuencia en niños, pero también se ven en adultos. Como los melanomas nodulares, los nevos de Spitz tienden a aparecer en forma súbita y su color varía de rojo a rojo-café.
Tratamiento
Sitio primario El melanoma cutáneo primario se trata por medio de extirpación quirúrgica definitiva. Las amplias extirpaciones del pasado han cambiado a márgenes de resección más modestos. Los datos de dos estudios aleatorios y prospectivos sobre bordes quirúrgicos en el melanoma cutáneo primario han demostrado la seguridad de dejar bordes de 1 cm para los melanomas de menos de 1 mm de espesor y de 2 cm para aquéllos entre 1 y 4 mm de grosor.20,21 No se han realizado estudios aleatorios sobre bordes de resección para melanomas de más de 4 mm de espesor. La mayoría de los autores recomiendan un borde de 2 a 4 cm para estos melanomas. En lugar de colocar injertos cutáneos, es mejor el cierre primario y los colgajos reconstructivos, desde el punto de vista tanto cosmético como funcional.
Ganglios linfáticos Los pacientes con enfermedad ganglionar regional clínicamente evidente se tratan con disección terapéutica de los ganglios. La realización de disección electiva de ganglios linfáticos (DEGL) en pacientes con melanoma cutáneo primario de espesor intermedio pero sin evidencia clínica de afección regional, ha sido tema de controversia durante mucho tiempo. En general, los datos sobre beneficios en la supervivencia no parecen justificar la morbilidad asociada a este procedimiento.
La biopsia de un ganglio centinela es una alternativa que se usa cada vez más en lugar de la DEGL en pacientes con melanoma cutáneo primario. Esta técnica utiliza la linfografía para identificar los ganglios linfáticos regionales que drenan la piel en el sitio del melanoma primario. En el momento de la extirpación definitiva del melanoma se inyecta un colorante azul con un radioisótopo en la dermis, alrededor del sitio del melanoma. Se realiza una pequeña incisión sobre la mancha identificada en la linfangiografía como área proximal de drenaje linfático regional. Se extirpa el primer ganglio linfático que se identifica que capta el colorante y el radioisótopo (ganglio centinela).
A continuación se evalúa la histología de este ganglio, con frecuencia por medio de técnicas de inmunohistoquímica y en ocasiones empleando la técnica de RCP, que es más sensible. La ausencia de melanoma en el ganglio centinela es muy sensible para descartar la presencia de metástasis en el resto de los ganglios cuando el procedimiento se realiza por un equipo con experiencia. Cuando el ganglio centinela es positivo para melanoma debe realizarse una DEGL completa. Algunos autores consideran que la información proporcionada por la biopsia del ganglio centinela tiene utilidad pronóstica significativa.22 Los pacientes con ganglios centinela positivos son candidatos apropiados para considerar tratamiento adyuvante (ver adelante). En la actualidad se realizan varios estudios multicéntricos para evaluar la utilidad clínica de este procedimiento.23
Metástasis en tránsito Se puede considerar que las metástasis en tránsito se confinan a una sola extremidad por periodos prolongados de tiempo. La amputación no parece mejorar la supervivencia a largo plazo en estos casos y las metástasis en tránsito de crecimiento lento pueden tratarse con cirugía. La enfermedad más extensa debe manejarse con terapia de sensibilización con dinitroclorobenceno (DNCB). Para las metástasis en tránsito extensas limitadas a una extremidad la terapia de perfusión permite una paliación importante, así como salvar la extremidad. Este procedimiento consiste en aislar la vasculatura de la extremidad afectada de la sistémica y perfundir esta extremidad con agentes quimioterapéuticos, agentes biológicos o ambos, a dosis que no podrían tolerarse si se administraran en forma sistémica.24
Metástasis a distancia A pesar del desarrollo de varios enfoques novedosos para el tratamiento de los pacientes con melanoma metastásico, incluyendo quimioterapia múltiple, terapia biológica, inmunoterapia y combinaciones de estos tratamientos, la monoterapia con dacarbacina (DTIC) sigue siendo el esquema original autorizado por la Food and Drug Administration para el tratamiento del melanoma metastásico.25 Se observan respuestas objetivas a la DTIC en alrededor del 5 al 20% de los pacientes, aunque las respuestas completas son raras. La radioterapia puede tener un papel paliativo importante. En ausencia de un tratamiento que haya demostrado ser más eficaz desde el punto de vista clínico, los pacientes con metástasis a distancia deben tener la oportunidad de participar en estudios clínicos de manejos experimentales.
Tratamiento adyuvante Los pacientes con enfermedad cutánea o regional que quirúrgicamente están sin enfermedad pero tienen riesgo de recurrencia o metástasis son candidatos potenciales para el tratamiento adyuvante.26 Se han usado varias terapias adyuvantes para el melanoma, incluyendo inmunoestimuladores como el bacilo Calmette-Guérin, Corynebacterium parvum y levamisol. También se han intentado varios agentes quimioterápicos. Recientemente se ha estudiado la inmunoterapia con citocinas, como los interferones, y la inmunización activa con vacunas. No se ha demostrado en algún estudio clínico aleatorio de terapia adyuvante para el melanoma mejoría significativa en la evolución con una excepción importante: un estudio a gran escala de interferón en dosis altas en pacientes de riesgo alto con melanoma ya resecado demostró un impacto pequeño, pero significativo en la supervivencia global.27 Este estudio constituyó la base para la autorización por la FDA del interferón alfa intravenoso en dosis de 20 millones de unidades/m2 por día durante un mes seguido de la administración subcutánea de 10 millones de unidades/m2 tres veces por semana por 48 semanas más como manejo adyuvante para el melanoma. Los estudios adicionales de interferón en dosis altas, intermedias y bajas no han demostrado efectos semejantes.
En la actualidad se estudian muchas otras estrategias, incluyendo inmunización activa, inmunización pasiva y terapias biológicas, que pueden constituir nuevas oportunidades para los pacientes que son candidatos apropiados para estos estudios.28
Pronóstico
Estadio El factor pronóstico aislado más importante para el melanoma es el estadio de la enfermedad. Se han usado varias clasificaciones de estadificación, todos los sistemas toman en cuenta la clasificación clásica TNM de tamaño del tumor (T), afección de ganglios linfáticos (N) y metástasis a distancia (M). Las diferencias entre los diversos sistemas se relacionan principalmente con la estadificación del sitio primario. Los nuevos esquemas de estadificación intentan considerar los atributos del tumor primario que correlacionan en forma estrecha con la evolución, específicamente el espesor y el nivel de Clark [ver tabla 2].29 Las tasas de supervivencia global a 5 años para los años 1989 a 1994, reportadas por el programa del National Cancer Institute Surveillance, Epidemiology and End Results (NCI SEER) de los EUA, son las siguientes: melanoma cutáneo primario, alrededor del 95 por ciento, con metástasis nodales regionales alrededor del 60 por ciento, y con metástasis a distancia, 12 por ciento.
Atributos del tumor primario Se han identificado varias características del tumor primario que son predictores de la evolución en el melanoma cutáneo primario. El más importante y más consistente es el grosor del tumor según Breslow. Otros parámetros histológicos importantes son el nivel de Clark de invasión, la extensión de la ulceración, la tasa de mitosis, la presencia de linfocitos que infiltran el tumor y la invasión vascular. En los tumores primarios delgados uno de los factores más importantes es la fase de crecimiento.30 El melanoma con fase de crecimiento radial parece no provocar tantas metástasis, mientras que el melanoma con crecimiento vertical (caracterizado por la formación de un nódulo tumoral en la dermis) se asocia con riesgo significativo de metástasis incluso en lesiones menores de 1 mm de espesor.31 Las características del paciente que se asocian con mejor supervivencia incluyen menor edad (< 60 años), sexo femenino y localización del melanoma en una extremidad. Se han desarrollado modelos multivariados para predecir la evolución el melanoma [ver tabla 3].32
Tumores malignos de la dermis
TUMORES METASTASICOS
Ocurren metástasis cutáneas en alrededor del 5 por ciento de los pacientes con tumores sólidos, que suelen asociarse con diseminación de la enfermedad. La frecuencia relativa de metástasis a la piel depende del género y refleja las frecuencias de los cánceres primarios.33 En las mujeres dos tercios de las metástasis provienen de cáncer de mama, pero también son frecuentes las neoplasias de pulmón, colon y recto, melanoma y ovario. En los varones el cáncer de pulmón es el más común, seguido de cáncer del colon, melanoma, CE de la cabeza y el cuello y carcinoma renal.33 La distribución anatómica de las metástasis cutáneas no es aleatoria. Las metástasis cutáneas por cáncer de mama suelen afectar la pared torácica y pueden aparecer como nódulos, linfedema o celulitis. El cuero cabelludo es un sitio frecuente de metástasis, en especial de cáncer de pulmón, riñón (en el varón) y de mama (en la mujer). Los cánceres de cabeza y cuello pueden invadir la piel por extensión local, dando origen a un edema firme, rojo obscuro, de la piel que parece celulitis. Pueden ocurrir metástasis en la pared abdominal de cánceres gastrointestinales o de ovario.33 Desde el punto de vista clínico las metástasis cutáneas casi no dan síntomas, forman pápulas o nódulos dérmicos y son del color de la piel o rosadas. Se diseminan a través de linfáticos o por vía vascular. Las metástasis cutáneas pueden reflejar la histología del tumor primario (v.gr., nódulos negros, café o gris con melanoma metastásico y nódulos vasculares con carcinoma de células renales o tiroideas).
TUMORES PRIMARIOS
Los tumores primarios de la dermis pueden desarrollarse a partir de las múltiples estructuras de la piel, incluyendo glándulas sebáceas (carcinoma sebáceo), tejido conectivo (dermatofibrosarcoma protuberante), músculo liso (leiomiosarcoma) y otros tejidos anexos (carcinoma ecrino).34 La mayoría de estas neoplasias primarias son raras y pueden tener un comportamiento biológico agresivo. Aunque estas neoplasias son muy variadas desde el punto de vista histológico, muchas comparten un cuadro clínico común, presentándose como un nódulo subcutáneo que crece con rapidez, del color de la piel o rosa a rojo y que en ocasiones recuerda un quiste sebáceo.
Carcinoma de células de Merkel Esta neoplasia dérmica tiene origen neuroendócrino. Suele aparecer como una pápula o nódulo dérmico rojo a violáceo en la cabeza y cuello de los pacientes ancianos, aunque se afectan todos los grupos de edad.34 El tratamiento de elección es la extirpación local amplia con o sin DEGL. Son frecuentes las recurrencias locales y ocurren metástasis a distancia en más de la tercera parte de los pacientes.34 La quimioterapia de las metástasis no suele dar buenos resultados.34
Enfermedad de Paget Esta neoplasia rara de la piel asociada con un adenocarcinoma subyacente,33,34 suele presentarse como una dermatisis eritematosa, con frecuencia eccematosa, unilateral en la mama, afectando el pezón y la areola. El diagnóstico diferencial incluye eccema, psoriasis, dermatitis de contacto e impétigo. Por este motivo es importante realizar biopsia de las dermatitis inflamatorias del pezón y la areola que no se resuelven con facilidad. En la enfermedad de Paget la biopsia revela células de Paget típicas, que tiñen en forma leve, dentro de la epidermis. La resección quirúrgica apropiada de la neoplasia cutánea y subyacente es el tratamiento de elección; con frecuencia ocurren metástasis a ganglios linfáticos.
Enfermedad de Paget extramamaria La enfermedad extramamaria de Paget es aún menos frecuente que la enfermedad de Paget y típicamente se presenta como placas rojas, con frecuencia ulceradas, en las regiones perineales de personas ancianas.33,34 Las lesiones pueden ser pruriginosas o asintomáticas, con frecuencia de larga evolución, y pueden diagnosticarse de modo equívoco como psoriasis, dermatitis de contacto o infecciones micóticas crónicas. Los tumores asociados subyacentes incluyen carcinomas rectales y genitourinarios. Incluso sin una neoplasia interna asociada la enfermedad extramamaria de Paget es difícil de tratar, con una alta tasa de recurrencia local.34
Angiosarcoma El angiosarcoma es una neoplasia vascular rara y con frecuencia muy agresiva.34 Puede aparecer como parches o nódulos multicéntricos rojos o púrpura en una extremidad linfedematosa, como un brazo linfedematoso después de una mastectomía (síndrome de Stewart-Treves). Otra forma de presentación consiste en parches o placas violáceas en la cabeza o cuello (en especial el cuero cabelludo) de personas ancianas. El angiosarcoma tiene mal pronóstico, causando metástasis pulmonares a pesar de la cirugía o radiación.34
Dermatofibrosarcoma protuberante El dermatofibrosarcoma protuberante es una neoplasia de crecimiento lento, agresiva en forma local, que rara vez metastatiza, pero sí recurre. Las lesiones típicamente son nódulos firmes, rojo-café o morado, por lo general en el tronco o extremidades, en zonas no expuestas al sol. El diagnóstico diferencial incluye queloides y dermatofibroma benigno. Suele afectar principalmente adultos jóvenes, aunque el tumor puede ocurrir a cualquier edad. La extirpación local con o sin cirugía micrográfica de Mohs ofrece la mejor posibilidad de curación.34
SARCOMA DE KAPOSI
El sarcoma de Kaposi (SK) es una neoplasia cutánea multicéntrica que tiene diversas variantes clínicas.35 A pesar de su nombre, el SK no es un verdadero sarcoma. Aunque no se ha definido con claridad la célula de origen, parece que deriva del endotelio linfático.35
Epidemiología
En su forma clásica el SK es una enfermedad indolente de varones ancianos con ascendencia mediterránea o de Europa oriental, que se manifiesta por nódulos y placas violáceas en las extremidades inferiores.35,36 Antes de la aparición del SIDA el trastorno era raro en los Estados Unidos, con una incidencia anual ajustada por edad de 0.29 por 100,000 habitantes en los varones y 0.07 por 100,000 habitantes en las mujeres.36 A finales de los años 60 se reconoció una segunda forma del SK, endémica, en Africa Central. El SK de Africa Central, que típicamente afecta adultos jóvenes y niños, tiene una evolución más agresiva que el SK clásico, con afección frecuente a hueso, ganglios linfáticos y vísceras.35
En 1981 el SK fue el primer tumor reconocido como parte del SIDA. Afecta principalmente a varones homosexuales y fue el padecimiento que definió al SIDA en el 30 a 40 por ciento de los pacientes durante los primeros años de la epidemia del VIH.36 Los varones homosexuales infectados por VIH tienen un incremento de 73,000 veces en la incidencia de SK, y las mujeres y varones no homosexuales infectados de 10,000 veces, si se compara con la población general de los Estados Unidos.36 Se ha descrito una cuarta variante de SK en pacientes inmunosuprimidos en forma iatrogénica, en especial receptores de trasplantes. Los varones se afectan un poco más que las mujeres.35
Etiología
La epidemiología del SK ha sugerido a un agente infeccioso como causal o cofactor.35,36 Las evidencias actuales implican a un herpesvirus humano novedoso: el herpesvirus del sarcoma de Kaposi (HVSK), también conocido como herpesvirus humano tipo 8 (HVH-8).37,39 El HVH-8 se ha detectado en todas las variantes de SK,37 y el desarrollo de anticuerpos contra antígenos nucleares del HVH-8 en pacientes infectados por VIH es predictivo del desarrollo de SK.38 El aislamiento y propagación del herpesvirus humano a partir de SK asociado a VIH que tiene secuencias de ADN idénticas al HVH-8 apoya aún más el posible papel del herpesvirus humano.39 Sin embargo, aún quedan muchas dudas respecto al HVH-8. También se la ha asociado con linfomas en cavidades corporales,36 enfermedad de Castleman,35,36, linfadenopatía angioblástica,35 y diversas lesiones de la piel en receptores de trasplantes de órganos.35,36 El mecanismo por el que el HVH-8 podría causar la tumorigénesis en el SK no es claro, por lo tanto, se ha sugerido que el HVH-8 es un virus ubicuo en ciertas poblaciones.35-39
Los factores del huésped, en especial la inmunosupresión, son cruciales en algunas poblaciones con SK.35,36 El VIH puede tener un papel indirecto en el desarrollo de SK a través de la depleción de células CD4 y la mayor producción de citocinas como la interleucina-1 (IL-1) y la IL-6.35,36 Los fármacos inmunosupresores, en especial la ciclosporina, la azatioprina y la prednisona, aumentan el riesgo de desarrollar SK, sobre todo en receptores de trasplante de riñón e hígado.36 Se ha observado regresión espontánea del SK después de suspender la ciclosporina y los esteroides.36
A pesar de la prevalencia de SK en algunos grupos étnicos, el papel de cualquier factor genético posible no es claro. Se ha puesto en duda la mayor incidencia del HLA-DR5 en pacientes con SK clásico.36 El SK familiar es muy raro,35 lo que sugiere que los factores genéticos solos no son los responsables.
Por último, el género parece constituir un factor de riesgo significativo, en especial en el SK clásico, en el que la relación varón a mujer puede variar de 3:1 a 10:1.35,36 Las explicaciones para este predominio en varones han incluido los efectos inhibitorios de las hormonas femeninas (v.gr., gonadrotropina coriónica humana-beta), pero no se conocen los mecanismos exactos.35
Diagnóstico
Las manifestaciones clínicas del SK difieren entre las variantes de este trastorno.35,36 En el SK clásico aparecen máculas o parches rojo-púrpura claro o nódulos púrpura primero en los pies, en especial en las plantas. En raras ocasiones existe linfadenopatía (en especial inguinal). Las lesiones pueden desarrollarse también en brazos y áreas genitales. Al progresar la enfermedad las lesiones coalescen para formar placas violáceas.
El SK asociado al VIH suele presentarse con lesiones cutáneas, pero las primeras pueden aparecer en la mucosa oral o los ganglios linfáticos. En contraste a las lesiones de SK clásico, las asociadas con VIH suelen comenzar en la parte superior del cuerpo (cara, tronco o brazos). Lo más típico es que las lesiones de SK asociado a VIH sean pápulas púrpura o rojas, con frecuencia ovales, que siguen los pliegues de la piel en una distribución semejante a la pitiriasis rosada.35,36 Las lesiones varían de manchas rosas a placas púrpura obscuro [ver figura 5] o pueden parecer equímosis, en especial en pacientes con cuentas bajas de células T CD4+. Las lesiones orales suelen consistir en placas o nódulos rojo-púrpura en el paladar, las encías o la mucosa oral. Los pacientes con piel más obscura pueden tener lesiones púrpura obscuro o negras, así como placas hiperpigmentadas.35
Al progresar el SK asociado al VIH puede desarrollarse linfedema en los pies, escroto, genitales y regiones periorbitarias, así como linfadenopatía (en especial inguinal). Las lesiones gastrointestinales pueden ser submucosas y asintomáticas, aunque pueden provocar hemorragia gastrointestinal. El SK pulmonar se asocia con mal pronóstico.40
La evaluación de laboratorio de los pacientes con SK debe incluir detección de anticuerpos contra VIH, citología hemática completa, sangre oculta en heces y radiografía de tórax. Está indicado solicitar cuenta de células T CD4+ en los pacientes VIH-positivos. Se realizará una historia clínica y examen físico completos, con especial atención a la presencia de infecciones oportunistas en pacientes infectados por VIH o inmunosuprimidos por otros motivos.
En todo paciente con sospecha de SK debe realizarse biopsia de piel. La histopatología de todas las variantes es parecida: células en forma de huso dentro de la dermis con eritrocitos extravasados presentes en las hendiduras entre los espacios vasculares irregulares.41
Diagnóstico diferencial
El diagnóstico diferencial clínico del SK incluye dermatofibroma, púrpura, granuloma piógeno, angiomatosis bacilar, melanoma metastásico y CBC. Otras entidades histopatológicas que pueden recordar al SK son el angiosarcoma y la dermatitis por estasis.35,36
Tratamiento
Sarcoma de Kaposi clásico El tratamiento del SK es paliativo. En el SK clásico, en el que la enfermedad es indolente y los pacientes son ancianos, rara vez se justifica el tratamiento sistémico agresivo.35 En lugar de ello, la radioterapia es el tratamiento de elección.35,42 El SK es muy radiosensible: se han usado dosis aisladas de 800 cGy para la paliación rápida en pacientes con mal pronóstico.43 Las dosis totales de 800 a 3,500 cGy causan remisiones completas en el 56 por ciento de los casos y parciales en el 46 por ciento, y más de la mitad de los pacientes no requieren tratamiento posterior hasta por 13 años.42 Se ha sugerido un esquema de tratamiento equivalente a 3,000 cGy en 10 fracciones durante 2 semanas.42
Para los pacientes con SK clásico que tienen solo una a dos pápulas la biopsia excisional puede ser suficiente tanto para diagnóstico como para tratamiento. La crioterapia con nitrógeno líquido puede ser útil en la pápulas aisladas. El tratamiento sistémico puede estar indicado en casos de afección cutánea extensa o compromiso visceral. Con frecuencia se emplea quimioterapia con un solo agente, con alcaloides de la vinca (vincristina o vinblastina). También puede ser eficaz el interferón alfa recombinante en dosis bajas; sin embargo, los efectos adversos (fiebre, calosfríos, mialgias y fatiga) pueden no ser bien tolerados por los pacientes ancianos.35
Sarcoma de Kaposi asociado al VIH Aunque el SK es más agresivo en los pacientes infectados por VIH, la magnitud de la inmunosupresión y la presencia de infecciones oportunistas y otras enfermedades sistémicas tienen gran importancia en la estadificación, pronóstico y elección del tratamiento apropiado.35,36 Las características clínicas que tradicionalmente se asocian con una evolución más favorable incluyen cuentas de células T CD4+ mayores de 200 células/mm3, ausencia de enfermedad sistémica, SK limitado a la piel o ganglios linfáticos y SK oral mínimo (i.e., no nodular). Los factores de mal pronóstico son cuenta de células T CD4+ menor de 200 células/mm3, linfedema asociado al SK, SK visceral, ulcerado u oral nodular, e infección oportunista.44 Con la aparición del tratamiento antirretroviral muy activo (TARMA), los médicos que tratan a pacientes con SK asociado al VIH tienen en la actualidad la oportunidad de influir e incluso revertir la inmunosupresión al modificar tanto la carga viral de VIH como la cuenta de células T CD4+. Se ha observado regresión del SK después del inicio del TARMA,45 con frecuencia durante los primeros meses de tratamiento.
El tratamiento local es una medida adecuada en pacientes con SK y enfermedad limitada, los que tienen complicaciones infecciosas y los que no pueden tolerar el tratamiento sistémico.43 Se ha demostrado que el tratamiento antitumoral específico mejora la supervivencia global.43 La radioterapia es eficaz en el SK asociado a VIH en dosis semejantes a las usadas para el SK clásico (ver antes). Sin embargo, las respuestas del SK asociado a VIH son breves.35 También han sido útiles las inyecciones intralesionales de vinblastina o interferón para lesiones seleccionadas.35,43 La crioterapia con nitrógeno líquido es eficaz en lesiones pequeñas;43 sin embargo, está contraindicada en pacientes con piel obscura en los que la hipopigmentación postratamiento puede ser cosméticamente mucho peor que la lesión original de SK.
El tratamiento sistémico incluye quimioterapia convencional y modificadores de la respuesta biológica. Los esquemas quimioterápicos con un solo agente permiten tasas de respuesta de 30 a 70 porciento.36 La quimioterapia combinada (v.gr., doxorrubicina, bleomicina y vincristina) se complica con supresión intensa de la médula ósea e infecciones oportunistas frecuentes. La disponibilidad de factores estimuladores de colonias hematopoyéticas (v.gr., factor estimulador de colonias de granulocitos) y la profilaxis contra Pneumocystis carinii han disminuido estas complicaciones.
El desarrollo de antraciclinas encapsuladas en liposomas (v.gr., doxorrubicina y daunorrubicina) representa una nueva opción importante en el tratamiento del SK.36,46 En un estudio aleatorio de 258 pacientes con SK asociado a SIDA avanzado, los que recibieron doxorrubicina en liposomas tuvieron tasas de respuesta significativamente mayores que los que recibieron la combinación estándar de doxorrubicina, bleomicina y vincristina.46 El paclitaxel también se asoció con buena respuesta en pacientes con SK avanzado.40
Los enfoques de investigación promisorios para el SK asociado al VIH incluyen tratamiento combinado con agentes retrovirales y terapia antitumoral específica,36 además del tratamiento potencial contra el HVH-8.
Complicaciones
Las infecciones bacterianas y la sepsis complican con frecuencia los tumores ulcerados de piernas y pies. Pueden participar infecciones por oportunistas, en especial en pacientes con cuentas de células T CD4+ muy bajas.
Pronóstico
Como ya se mencionó, el tratamiento del SK es paliativo. La cuenta de células T CD4+ es el factor más importante para predecir la supervivencia en el SK asociado al VIH.44 Los tumores grandes,44 el linfedema y el SK pulmonar también indican mal pronóstico.35,46
Linfoma cutáneo
Los linfomas pueden ser de origen celular B o T y afectar la piel en forma primaria o secundaria. Los linfomas de células B, en especial los linfomas no Hodgkin, pueden afectar la piel en forma secundaria en la enfermedad avanzada. Aparecen típicamente como placas o nódulos subcutáneos rojo-púrpura. Los linfomas primarios de células B de la piel son muy raros, aparecen como nódulos rojos que con frecuencia permanecen localizados a la piel, pero que pueden progresar hacia una afección sistémica. La gran mayoría de los linfomas cutáneos primarios son linfomas cutáneos de células T (LCCT).
El LCCT es un padecimiento raro de la piel que suele conocerse como micosis fungoides (MF).47 Las MF son en realidad el subtipo más grande de los LCCT, aunque los términos suelen usarse en forma indistinta. La variante leucémica del LCCT es el síndrome de Sézary.47 Otra variante del LCCT se asocia con el HTLV-1 y es parte del espectro de los linfoma/leucemia de células T del adulto y de los linfomas de células T periféricos.47
EPIDEMIOLOGIA
Alrededor de 1,000 nuevos casos de LCCT se diagnostican en los Estados Unidos cada año.47,48 La incidencia ha aumentado, de 0.19 por 100,000 habitantes en 1973 a 0.42 por 100,000 habitantes en 1984.47 El LCCT afecta principalmente a adultos de edad media, siendo la edad promedio de presentación los 50 años.47,48 La relación entre varón y mujer es de alrededor de 2:1, y los pacientes de raza negra se afectan el doble que los de raza blanca.47,48
ETIOLOGIA
La susceptibilidad del huésped y un antígeno ambiental, quizá viral, parecen tener una función importante en la patogenia del LCCT.49 Los factores genéticos pueden relacionarse con los antígenos del complejo de histocompatibilidad, y se ha observado mayor frecuencia de HLA-DR5 en pacientes con LCCT.49 La estimulación antigénica crónica (v.gr., por infección) puede participar también.48
Los virus son los candidatos antigénicos principales, considerando la aparente asociación del HTLV-1 con el linfoma de células T periféricas.48-50 El linfoma de células T periféricas asociado al HTLV-1 tiene características clínicas únicas, incluyendo hipercalcemia y lesiones óseas.48
DIAGNOSTICO
Manifestaciones clínicas
Las manifestaciones clínicas de las MF aparecen durante muchos meses a años. En un estudio la duración promedio de los síntomas antes del diagnóstico fue de 7.5 años.51 Aparecen manchas eritematosos planas, con frecuencia descamativas y en ocasiones atróficas, principalmente en el tronco y los muslos [ver figura 6]. Las lesiones son asintomáticas o tienen prurito leve y pueden remitir en forma espontánea o responder al tratamiento esteroideo tópico. Los pacientes pueden también reportar mejoría después de la exposición al sol. Las lesiones, denominadas de parapsoriasis, progresan a LCCT en el 10 a 30 por ciento de los casos, según seguimientos a largo plazo (< 10 años).48
Al progresar la MF las manchas tienden a crecer y engrosarse hasta formar
placas. El dolor pueden volverse rojo obscuro, en las personas de piel obscura
las lesiones pueden al inicio ser hiper o hipopigmentadas y adquirir un halo
eritematoso o violáceo. En las MF avanzadas pueden dearrollarse
tumores48 o transformarse en un linfoma de células
grandes.52 En alrededor del 10 por ciento de los casos los tumores
son la presentación inicial del LCCT.47,48 La eritrodermia
generalizada con células T atípicas circulantes (en el
síndrome de Sézary) es el tipo de presentación en el 5 por
ciento de los pacientes con LCCT.48
El examen físico de los pacientes con probable LCCT incluye examen completo de la piel, incluyendo clasificación de las lesiones (mancha, placa o tumor) y la extensión de la superficie corporal afectada. Deben examinarse los ganglios linfáticos, el hígado y el bazo.
Biopsia de piel
Se requiere biopsia de la piel para hacer el diagnóstico definitivo de MF. La presencia de células linfoides atípicas con nucleos cerebriformes con múltiples circunvoluciones en racimos dentro de la epidermis (microabascesos de Pautrier)47,48 y los infiltrados linfocíticos en banda en la dermis superior son diagnósticos de LCCT.47 La célula maligna es una célula T, y la mayoría de las células expresan los marcadores de las células T en general, CD2, CD3 y CD5.48 El uso de estudios sobre rearreglos genéticos del receptor de la célula T para confirmar la clonalidad en la fase temprana de la enfermedad puede ayudar al diagnóstico.48 Ni los estudios inmunofenotípicos ni la microscopía electrónica pueden considerarse como definitivos para el diagnóstico de las MF, se requiere correlación clinicopatológica.
Estudios de laboratorio
La evaluación de laboratorio del LCCT incluye citología hemática completa, cuenta de eosinófilos, cuenta de células de Sézary, deshidrogenasa láctica (DHL) pruebas de función hepática. La biopsia de médula ósea no es necesaria en ausencia de células leucémicas circulantes. Debe considerarse investigar HTLV-1 en pacientes con factores de riesgo o presentaciones atípicas. La biopsia de ganglio linfático es adecuada si existen ganglios palpables, en especial mayores de 2 cm. Puede ser importante realizar tomografía abdominal computada o radiografía de tórax en los pacientes con tumores o sospecha de afección visceral.
DIAGNOSTICO DIFERENCIAL
En sus fases tempranas las MF pueden parecer diversos trastornos inflamatorios benignos (v.gr., reacción a fármacos, eccema, psoriasis o dermatitis de contacto). Es necesario descartar estos padecimientos antes de iniciar el tratamiento.
TRATAMIENTO
Tratamiento tópico
El tratamiento tópico es la base del tratamiento para las manchas o placas tempranas (en fase IA, IB y IIA).53 El tratamiento agresivo inicial con radio y quimioterapia no ha demostrado ser superior a las medidas locales para controlar la enfermedad o mejorar la supervivencia en pacientes con enfermedad limitada.54 Un enfoque lógico para la enfermedad temprana (o histológicamente dudosa) consiste en administrar esteroides tópicos.58 La mostaza nitrogenada (mecloretamina) tópica, en forma acuosa o en ungüento, es la quimioterapia tópica empleada con más frecuencia y permite remisiones completas iniciales en la enfermedad en mancha o placa en el 60 a 80 por ciento de los pacientes.47,53 El tratamiento debe continuarse por periodos prolongados (hasta por 3 años después de desaparecidas las lesiones). Se desarrolla dermatitis de contacto en alrededor de la tercera parte de los pacientes.
La solución de carmustina (BCNU), aplicada a las lesiones en forma diaria, es otro esquema útil. El tratamiento suele durar 8 a 16 semanas, pero se ha continuado hasta por 6 meses.53 Debido a que la absorción sistémica puede provocar supresión de la médula ósea, deben realizarse biometrías hemáticas periódicas.47,53
Radiación ultravioleta
La radiación ultravioleta se administra en diferentes formas para el LCCT, desde luz ultravioleta hasta radiación ionizante. La UVB es útil en la enfermedad en estadio I, con una tasa de remisión clínica completa de 71 por ciento en un estudio. El tiempo promedio de remisión fue de 5 meses y la duración de la misma fue de 22 meses.56
Otro método eficaz en las MF es la combinación de psoralenos y luz ultravioleta A (PUVA). En un estudio el 95 por ciento de los pacientes con LCCT en estadio I tuvieron remisión clínica completa, con una duración promedio de la respuesta de 43 meses.57
Radioterapia
La radiación total de la piel con haz de electrones (RTPHE) aplica radioterapia a la superficie de la piel sin una dosis interna significativa. Es especialmente útil en la enfermedad en placas. Las dosis típicas son de 2,400 a 3,600 cGy, fraccionadas en varias semanas con un haz de radiación de electrones de 4 a 9 meV.58 La remisión completa se relaciona con el estadio, de la siguiente manera: IA, 84 a 96 por ciento, IB 56 a 81 por ciento, IIA 63 a 74 por ciento, IIB 24 a 53 por ciento, III 26 a 50 por ciento y IVA 8 a 33 por ciento.10 Se logró una supervivencia libre de recaídas del 50 por ciento a 5 años con la enfermedad IA, pero la mayoría de los pacientes con enfermedad más avanzada sufren recaídas para los 5 años.58
Tratamiento sistémico
El tratamiento sistémico para el LCCT se considera como tratamiento primario en la enfermedad avanzada (estadios III a IVB)48,54,59,60 y como terapia secuencial para promover respuestas más duraderas en la enfermedad temprana.48,59,60
La fotoféresis extracorpórea parece ser más útil en el LCCT eritrodérmico y en el síndrome de Sézary.47,60 En este tratamiento el paciente es sometido a fotoféresis extracorpórea con radiación UVA a los leucocitos después de la ingestión oral de 8-metoxipsoraleno.
El tumor avanzado y el LCCT visceral han sido tratados con quimioterapia de uno o varios agentes, incluyendo metotrexate, análogos de adenosina, interferón alfa y retinoides.48,59 El tratamiento experimental incluye anticuerpos monoclonales, retinoides y citocinas.48
Tratamiento combinado
El tratamiento agresivo temprano del LCCT (RTPHE seguido de quimioterapia combinada con ciclofosfamida, doxorrubicina, etopósido y vincristina) no proporciona ventajas en la supervivencia comparado con el tratamiento tópico secuencial.54 En forma semejante, la adición de quimioterapia sistémica (doxorrubicina y ciclofosfamida) o fotoféresis extracorpórea después de una respuesta completa a la RTPHE no tuvo impacto en la supervivencia en micosis fungoides tempranas ni impacto sobre la supervivencia libre de recaídas en todos los estadios.60 Otros esquemas incluyen interferón alfa y retinoides (isotretinoín) con RTPHE, seguido de mostaza nitrogenada tópica.59 La heterogeneidad de los esquemas de tratamiento combinado en el LCCT hace prácticamente imposible comparar los resultados.
COMPLICACIONES
Las complicaciones más serias del LCCT son las infeciones. La sepsis por tumores cutáneos ulcerados es una causa común de muerte. Puede ocurrir LCCT visceral, lo mismo que transformación a un linfoma de células grandes en algunos pacientes con LCCT (probabilidad del 39 por ciento después de 12 años).52 En los pacientes con enfermedad temprana y que sobreviven por tiempo prolongado, el tratamiento local (v.gr., RTPHE o PUVA) puede contribuir al desarrollo de otros cánceres de piel (v.gr., CBC o CE) y cataratas.
PRONOSTICO
La estadificación del LCCT se basa en la evaluación del tipo y extensión de las lesiones cutáneas y de la afección a ganglios linfáticos, sangre periférica y vísceras.48,51 Se han realizado muchos intentos por clasificar el LCCT en grupos pronósticos útiles. Un estudio inicial y aún válido que usó el sistema TNM identificó tres grupos principales: pacientes con riesgo adecuado (estadios IA, IB y IIA, con enfermedad cutánea solo en placas y sin afección a ganglios linfáticos, sangre o vísceras [supervivencia promedio > 12 años]), pacientes con riesgo intermedio (estadios IIB, III y IVA, con tumores cutáneos, eritrodermia o enfermedad en placas y afección ganglionar o sanguínea pero sin daño visceral o destrucción ganglionar [supervivencia promedio de 5 años], y pacientes con riesgo elevado (estadio IVB con afección visceral y destrucción ganglionar [supervivencia promedio 2.5 años]).47,51
La eosinofilia también se asocia con supervivencia corta.51 Estudios recientes han demostrado que, en general, los pacientes más jóvenes (< 35 años) con micosis fungoides tienden a presentar enfermedad en estadio más temprano.61 Otros estudios a largo plazo han revelado que los pacientes en estadio IA no tienen reducción en su expectancia de vida y que menos del 10 por ciento de estos pacientes progresan a estadios más avanzados.62 La supervivencia de los pacientes con MF en mancha o placa generalizada (estadios IB o IIA) es significativamente peor que la de la población control de raza, edad y sexo equiparable a los 11.7 años. El género y la raza no parecen tener efecto sobre la supervivencia, pero los pacientes de mayor edad (> 58 años) tienen supervivencias más cortas.63
DR. ALLAN C. HALPERN
DRA. PATRICIA L. MYSKOWSKI
Los tumores malignos pueden originarse de células de cualquier capa de la piel (queratinocitos, melanocitos, fibroblastos, células endoteliales y adipocitos), así como de células como los linfocitos, que transitan en condiciones normales por la piel. También pueden aparecer metástasis cutáneas de otros sitios primarios. En esta subsección se revisarán los tumores cutáneos malignos más frecuentes en orden de frecuencia.
Tumores malignos de la epidermis
Los cánceres de la epidermis son las neoplasias más frecuentes en los humanos. Se originan en los queratinocitos y los melanocitos de la epidermis y representan una oportunidad única de intervención eficaz por medio tanto de detección temprana como de prevención primaria. Pueden diagnosticarse desde el punto de vista clínico por simple inspección visual y confirmarse histológicamente con una biopsia muy poco invasora.
Tanto el carcinoma basocelular como el epidermoide se originan de los queratinocitos de la epidermis. Debido a que estos dos cánceres comparten muchas características, con frecuencia de agrupan bajo el término de cáncer de piel no melanoma.
El melanoma maligno es una neoplasia que surge de los melanocitos. Aunque puede aparecer en cualquier melanocito del organismo, incluyendo los ojos, la gran mayoría ocurre en la piel. El melanoma maligno cutáneo se ha clasificado en cuatro tipos histogenéticos principales: melanoma léntigo maligno, melanoma de diseminación superficial, melanoma nodular y melanoma lentiginoso acral.
EXPOSICION AL SOL Y CANCER DE PIEL
Varias líneas de evidencia implican a la radiación ultravioleta (UV) en la patogenia de los tres principales cánceres de la epidermis.1 Los datos epidemiológicos relacionan la exposición acumulada al sol con el desarrollo del carcinoma epidermoide y la exposición solar intermitente intensa con el carcinoma basocelular y el melanoma. Los estudios de laboratorio indican que tanto la radiación solar UVA (320 a 400 nm) como la UVB (290 a 320 nm) pueden dañar el ADN en forma directa y oxidativa. Además, la radiación UV puede suprimir el sistema inmunológico cutáneo.2 La asociación de algunos carcinomas epidermoides con carcinógenos químicos y la ocurrencia de melanomas acral lentiginoso y de mucosas en partes no expuestas del organismo destacan la necesidad de realizar estudios para identificar otros agentes etiológicos.
El conocimiento sobre el importante papel que tiene la luz solar en la etiología del cáncer de piel ofrece una oportunidad para realizar prevención primaria por medio del uso de protección solar. Por desgracia, aún se desconoce el espectro de acción, el momento y las dosis de exposición UV exactos involucrados en el desarrollo del cáncer de piel en los humanos. Por ello, los pacientes deben ser instruidos sobre el efecto dañino de exponerse al sol y broncearse. Deben realizarse esfuerzos para disminuir en forma general la exposición al sol por medio de cambios en el comportamiento y el uso de ropa protectora. Las pantallas solares de espectro amplio con un factor de protección solar (SPF) de 15 o más son útiles, pero no deben usarse para aumentar la cantidad de tiempo expuesto en forma directa a la luz solar.3
CANCER DE PIEL NO MELANOMA
El cáncer de piel no melanoma (CPNM) ocurre en forma típica como una lesión rosada en la superficie de piel expuesta al sol. Cualquier lesión rosada que persista o recurra en la misma localización, en especial si se irrita con facilidad por los traumatismos leves, es sospechosa de un CPNM. El clínico debe saber que algunas formas de CPNM se desvanecen con los cambios de estación (i.e., con la menor exposición al sol) o con la aplicación de esteroides tópicos, y deben notificar a los pacientes que cualquier lesión que recurra requiere de atención.
Carcinoma basocelular
El carcinoma basocelular (CBC) es un tumor cutáneo maligno que se origina de los queratinocitos basales de la epidermis.
Epidemiología El CBC es el cáncer de piel más común. Las incidencias reportadas varían desde 3.4 por 100,000 personas por año en afroamericanos a más de 750 por 100,000 por año en caucásicos australianos.4,5 Aunque raro, pueden ocurrir metástasis y muerte por CBC.
Etiología La radiación ultravioleta, en especial la exposición intermitente intensa, parece tener un papel importante en el desarrollo del CBC. Estudios recientes del síndrome de nevos de células basales (síndrome de Gorlin) han permitido nuevos conocimientos sobre la genética del CBC. Se ha identificado un gen denominado patched (parchado), que se descubrió por primera vez en la mosca de la fruta Drosophila, y que parece tener un papel crucial en el desarrollo del CBC. Casi todos los pacientes con síndrome de nevos de células basales parecen heredar una copia mutada de este gen, y los estudios del CBC esporádico sugieren que las mutaciones del gen patched, son un paso necesario, si no es que suficiente, en el desarrollo del CBC.6
Diagnóstico Alrededor del 90 por ciento de los CBC ocurren en la cabeza y el cuello, en formas nodulares y superficiales.
El CBC nodular consiste en una elevación rosada, perlada y translúcida en la superficie de la piel. Se irrita con facilidad, es frágil y se asocia con episodios de ulceración superficial o hemorragia. Cuando la ulceración es prominente puede aparentar una úlcera "de roedor", con un borde perlado y translúcido. Estas lesiones pueden verse más blancas que rosadas y, a la inspección cuidadosa demuestran telangiectasias pequeñas. Tienen una superficie lisa y brillante, y una textura más firme que los nevos dérmicos comunes [ver figura 1].
|
|
Son variantes menos comunes de los CBC las lesiones tipo morfea, pigmentadas y quísticas. Los CBC tipo morfea tienen un patrón infiltrativo que histológica y clínicamene parece una cicatriz. Los CBC pigmentados suelen contener puntos de pigmento azul-negro, aunque pueden mostrar pigmentación importante y total. El pigmento es más característico en la variedad nodular de CBC. Los CBC quísticos son más suaves que los carcinomas nodulares típicos y pueden tener una apariencia azul-gris.
La historia del paciente es muy importante en el diagnóstico del CBC. Cuando se pregunta respecto a lesiones que se irrigan o sangran con facilidad con traumatismos leves los pacientes pueden mostrar al clínico lesiones tempranas que quizá pasarían desapercibidas. Bajo anestesia local debe obtenerse una biopsia de cualquier lesión sospechosa.
Diagnóstico diferencial Los CBC nodulares pueden confundirse con angiofibromas, nevos dérmicos, melanoma amelanótico, metástasis cutáneas y diversos tumores benignos de los anexos (v.gr., tricoepitelioma). Los CBC superficiales simulan a varias dermatosis inflamatorias (v.gr., eccema y tiña) y comparten algunas características clínicas con las queratosis actínicas. Los CBC pigmentados pueden confundirse con facilidad con una neoplasia melanocítica primaria. Los carcinomas quísticos pueden confundirse con tumores de los anexos y lesiones inflamatorias.
Tratamiento El objetivo del tratamiento consiste en erradicar en forma adecuada la lesión y asegurar el mejor resultado tanto cosmético como funcional. Deben tomarse en cuenta muchos factores, como el tamaño, localización y subtipo histológico de las lesiones, así como datos del paciente, incluyendo edad, color y laxitud de la piel, para elegir el tratamiento óptimo.
La gran mayoría de los CBC son susceptibles de tratamiento quirúrgico. Las principales opciones incluyen curetaje y electrodesecación, extirpación, crioterapia, radioterapia y cirugía micrográfica de Mohs.
Un grupo pequeño, pero significativo, de CBC se benefician con la microcirugía de Mohs, que consiste en el examen microscópico de cortes congelados de toda la superficie inferior del tejido extirpado en el momento de la cirugía. Esta técnica puede ser adecuada para las lesiones recurrentes y las lesiones con alta probabilidad de recurrencia. Entre estas se incluyen lesiones mal definidas, lesiones grandes (> 2 cm), lesiones con histología de alto riesgo (i.e., patrón de crecimiento agresivo, patrón de esclerosis o afección perineural) y lesiones que afectan varios planos embrionarios (v.gr., canto ocular o surco nasofacial). La técnica de microcirugía de Mohs ha demostrado asociarse con mayores tasas de curación que los otros tratamientos para las lesiones con riesgo alto.7
La radioterapia puede ser una alternativa eficaz, no dolorosa y bien tolerada, que típicamente se reserva para los pacientes de edad avanzada que son malos candidatos quirúrgicos. Sin embargo, debe evitarse en los pacientes con síndrome de nevos de células basales.
Entre los tratamientos en fase experimental se incluyen la quimioterapia intralesional y la terapia fotodinámica.
Todos los pacientes tratados por CBC tienen riesgo de recurrencia local, así como riesgo significativo de desarrollar otros cánceres de piel. Los enfermos deben ser enseñados para autoexaminar su piel, deben emplear métodos de protección contra el sol y requieren vigilancia médica periódica.
Pronóstico Como ya se mencionó, el riesgo de recurrencia local se relaciona con el tamaño, localización e histología de la lesión. Las metástasis son muy raras: en una serie de 50,000 australianos se reportó una prevalencia de 0.0028%.8 Estas ocurren por vía linfática y hematógena, y los factores de riesgo incluyen síndrome de nevos basales, inmunosupresión y exposición previa a radiación ionizante. Las metástasis no son susceptibles de tratamiento quirúrgico y se asocian con mal pronóstico.
Carcinoma epidermoide
Como el CBC, el carcinoma epidermoide (CE) se origina de los queratinocitos de la epidermis. Desde el punto de vista histológico, las células de los CE bien diferenciados recuerdan a las células de la porción superior de la epidermis.
Epidemiología En 1994 se diagnosticaron en los Estados Unidos entre 150,000 y 250,000 casos nuevos de CE cutáneo.9 La mortalidad calculara por este tipo de cáncer en Estados Unidos en 1988 fue de alrededor de 0.5 por 100,000. Varias líneas de evidencia sugieren un incremento reciente y significativo en la incidencia de CE. Por ejemplo, en Australia, ésta aumento en 51% entre los años de 1985 y 1990.10
Etiología Además de la luz solar, otros agentes etiológicos conocidos que contribuyen al desarrollo de CE cutáneo son la radiación ionizante, los carcinógenos químicos, las quemaduras térmicas y las heridas crónicas que no cicatrizan. Los CE relacionados con exposición solar tienen menor riesgo de metástasis y muerte que los asociados con otras exposiciones. Los factores predisponentes a CE por exposición solar incluyen piel de color claro, tendencia a quemarse e incapacidad para broncearse.
Fisiopatología y patogenia El CE por exposición a luz solar suele asociarse con una lesión precursora denominada queratosis actínica. Estas lesiones ocurren en el cuero cabelludo, cara, superficies extensoras de los antebrazos y dorso de las manos. Son lesiones rosadas de superficie rugosa e irregular. Con frecuencia se sienten más de lo que se ven. La mayoría de los pacientes con queratosis actínicas tienen lesiones múltiples. Se ha calculado que el riesgo de CE en estos pacientes es hasta de 20%.11 El CE también puede aparecer en piel de apariencia normal.
El CE de mucosa oral o genital puede originarse de una lesión precursora denominada leucoplaquia o eritroplaquia. Los CE de mucosas se asocian con riesgo importante de metástasis.
La función inmunológica influye en la progresión del CE. La inmunosupresión que ocurre en pacientes con trasplantes o con linfoma se relaciona con mayor incidencia de CE. En estos pacientes la infección con papilomavirus humano parece tener un papel etiológico asociado a la exposición solar. El CE tiende a ser más agresivo en las personas inmunosuprimidas.
Diagnóstico La mayoría de las lesiones ocurren en áreas del organismo que suelen estar expuestas al sol. Las lesiones son placas firmes y rosadas, con frecuencia con superficie burda y descamativa [ver figura 2]. Se requiere la biopsia para el diagnóstico definitivo.
|
|
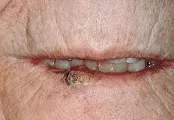
Los queratoacantomas comparten muchas características con el CBC tanto clínica como histológicamente. Aparecen de novo en piel de apariencia normal y crecen con gran rapidez. Típicamente son elevaciones rosadas, en forma de domo y brillantes en la superficie de la piel, con un tapón queratósico en forma de cráter. Pueden crecer mucho. Aunque los queratoacantomas no se asocian con riesgo de metástasis, destruyen localmente. Se ha demostrado regresión espontánea en el curso de meses.
La enfermedad de Bowen es un CE que se limita a la epidermis. Se presenta como una placa roja, mínimamente elevada y descamativa, con bordes irregulares bien definidos. La asociación reportada antes entre enfermedad de Bowen y neoplasias internas no se ha comprobado.
El CE que no tiene superficie queratósica descamativa puede confundirse con otros tumores de anexos o de la dermis.
Tratamiento Los CE pequeños que evolucionan de una lesión de queratosis actínica pueden tratarse en forma adecuada con curetaje y electrodesecación simple. Las lesiones actínicas de mayor tamaño, así como las lesiones que surgen en áreas de piel no expuesta al sol se tratan mejor con extirpación quirúrgica definitiva con confirmación de bordes negativos. Las lesiones de alto riesgo y mal definidas, en especial las que ocurren en áreas quirúrgicas delicadas de la cara, genitales, manos y pies se tratan mejor por medio de cirugía micrográfica de Mohs.
La radioterapia fraccionada constituye un tratamiento alternativo para el CE primario en los pacientes de edad avanzada que son malos candidatos quirúrgicos. Los beneficios de la radioterapia adyuvante son menos claros, lo mismo que los beneficios de la disección electiva de ganglios linfáticos en pacientes con CE de alto riesgo de cabeza y cuello.
Se han usado quimioterapia citotóxica y modificadores de la respuesta biológica en pacientes con CE avanzado, con tasas de respuesta completa de 25 a 46%, pero con pocos sobrevivientes a largo plazo.
Las queratosis actínicas se tratan con crioterapia, curetaje, quimioterapia tópica con fluorouracilo, desepitelización química y con láser, para prevenir la progresión a CE.
Pronóstico Independientemente del tratamiento empleado, las lesiones con riesgo alto se asocian con una tasa de recurrencia local significativa a los 5 años. Los CE de riesgo alto incluyen los que afectan sitios anatómicos específicos (v.gr., oídos, labios, genitales y otras áreas no expuestas al sol), los mayores de 2 cm de diámetro, los que tienen características histológicas agresivas (profundidad > 4 mm, nivel de Clark IV o mayor, e histología mal diferenciada) y los que ocurren en pacientes inmunosuprimidos.12 La principal ruta de metástasis de los CE es por diseminación linfática a los ganglios regionales. Las tasas reportadas de metástasis varían desde 0.3% en las lesiones pequeñas derivadas de exposición solar, hasta 33% en lesiones más grandes y poco diferenciadas.12 La supervivencia reportada a 5 años para los pacientes con CE y metástasis regionales varía de 25 a 47%.12
MELANOMA MALIGNO
Epidemiología
En los Estados Unidos el riesgo de desarrollar melanoma durante la vida es de alrededor de 1 en 75.13 Entre 1973 y 1994 la incidencia de melanoma aumentó en 121% y la mortalidad asociada en 39%. Actualmente se realiza una detección más temprana, y parece existir una estabilización en las tasas de incidencia en algunos segmentos de la población. Sin embargo, en términos de morbimortalidad, el peso de la enfermedad relacionada con melanoma continúa aumentando. Aunque puede ocurrir melanoma en cualquier persona, es principalmente una enfermedad de la raza blanca. Los melanomas que ocurren en personas de raza negra son típicamente de la variedad acral lentiginosa.
Etiología
Aunque las evidencias epidemiológicas y de ciencias básicas apoyan una asociación entre el melanoma y la exposición a la luz solar, la relación parece ser más compleja. El melanoma léntigo maligno se asocia con exposición crónica y acumulada al sol. El melanoma de diseminación superficial y el nodular parecen asociarse con exposición intensa intermitente, en especial en los jóvenes. El melanoma acral lentiginoso no tiene asociación aparente con la luz del sol. Los estudios básicos y los modelos en animales han implicado a diferentes longitudes de onda de la luz UV en la carcinogénesis del melanoma; la longitud de onda implicada puede variar entre los diversos tipos de melanoma.
Alrededor del 5 por ciento de los melanomas ocurren en pacientes con historia familiar de esta misma neoplasia. Las mutaciones en el gen regulador del ciclo celular p16 (inhibidor 2a de cinasa dependiente de ciclina) se asocian con melanoma en alrededor del 40% de los melanomas de tipo familiar, con asociación del gen al cromosoma 9p.14 Las alteraciones en la reparación del ADN encontradas en pacientes con nevos displásicos sugieren que este es otro mecanismo de predisposición genética al melanoma.
Diagnóstico
Como una lesión pigmentada que ocurre en la superficie de la piel, el melanoma es susceptible de detección temprana por inspección visual simple en una fase fácilmente curable. Si no se trata el melanoma es una de las formas de cáncer más agresivas y con menos respuesta terapéutica.
La detección temprana del melanoma requiere de la inspección cuidadosa de las lesiones pigmentadas en toda la superficie del organismo. A pesar de la intensa asociación del melanoma con la exposición solar, pueden ocurrir lesiones en cualquier sitio de la superficie cutánea. Por lo tanto, el autoexamen del paciente y la revisión del médico deben incluir toda la piel, incluyendo el cuero cabelludo, los genitales y las plantas de los pies. Cualquier lesión pigmentada con cambios recientes o con las características descritas por la nemotecnia ABCD (por asimetría, bordes irregulares, variación del color y diámetro > 6 mm) justifica considerar la posibilidad de un melanoma. Aunque cualquier lunar puede cambiar en forma gradual con el tiempo, cualquiera que cambie de dolor, forma o tamaño en relación con otros lunares merece atención especial [ver figura 3].
|
|
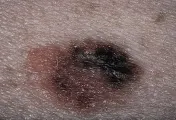
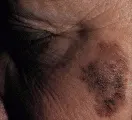
|
|
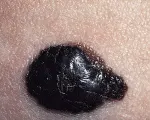
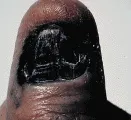
Nevo displásico Varios estudios epidemiológicos han relacionado a los nevos displásicos con riesgo de melanoma. Desde el punto de vista clínico los nevos displásicos son lunares grandes (> 5 mm) con pigmentación abigarrada y bordes mal definidos [ver figura 4]. Histológicamente los nevos displásicos se caracterizan por la presencia de atipia en su arquitectura y atipia citológica dispersa.15 El grado de riesgo de melanoma asociado con el nevo displásico depende del contecto genético. En familias con síndrome de nevos displásicos y melanoma el fenotipo anormal parece heredarse en forma autosómica dominante. Los miembros de estas familias con nevos displásicos tienen un riesgo durante la vida de sufrir melanoma que llega a ser del 100 por ciento.16 Fuera del contexto del melanoma familiar, los nevos displásicos ocurren en el 5 a 10 por ciento de las personas de raza blanca. En esta población los nevos displásicos aún son un marcador de mayor riesgo de melanoma [ver tabla 1].17
|
|

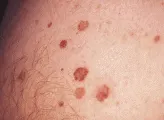
|
||||||||||||||||||||||||||||||||||||
* Ajustado en forma mutual y para edad, sexo, institución, patrón de referencia, nevos displásicos < 5 mm. quemaduras, pecas, baño solar, cicatrices, nevos extirpados e historia familiar de carcinoma (intervalo de confianza = 95%). |
Los nevos displásicos representan tanto una oportunidad como un reto en la detección del melanoma. Por un lado su detección permite vigilar en forma eficaz a un grupo de alto riesgo. Por el otro, pueden complicar los intentos de detección de los melanomas al parecer, clínicamente, melanomas tempranos. Aunque algunos nevos displásicos pueden progresar a melanomas, la gran mayoría permanecen benignos. Además, no todos los melanomas que se originan en pacientes con nevos displásicos lo hacen a partir de un lunar previo. La extirpación de todos los nevos displásicos es una medida poco práctica para la prevención de los melanomas. En los pacientes con nevos displásicos la detección de los melanomas se realiza por examen visual por un especialista, aunado con autoexamen y vigilancia para identificar las lesiones que cambian de características.
En la actualidad se desarrollan varios instrumentos que ayudan al diagnóstico del melanoma en pacientes con nevos displásicos. La microscopía epiluminiscente (MEL) consiste en el uso de una especie de otoscopio manual para magnificar una lesión pigmentada mientras se aplica presión y aceite a la superficie. Esta técnica permite la visualización de los patrones de pigmento y las características que no son aparentes por inspección simple. Con entrenamiento y experiencia la MEL puede ser útil para distinguir el melanoma de las lesiones pigmentadas benignas. Sin embargo, si no se tiene experiencia la LEL puede incluso disminuir la certeza diagnóstica.18,19 Otra método para mejorar la detección de melanomas en individuos con riesgo alto es la evaluación ayudada por fotografías. Se usan un grupo basal de fotografías de todo el cuerpo durante el autoexamen y las evaluaciones por el médico para buscar cambios en las lesiones. Este procedimiento elimina la necesidad de extirpar lesiones estables y mejora la sensibilidad del examen para detectar cambios.
Cualquier lesión que origine la sospecha clínica de melanoma requiere de un diagnóstico definitivo. La extirpación total es la técnica preferida para biopsiar las lesiones pigmentadas sospechosas. La biopsia parcial puede causar diagnósticos erróneos por una mala muestra o impedir que el patólogo conozca la arquitectura global y la citología de la lesión. Sin embargo, las biopsias incisionales con buena relación clínico-patológica pueden ser apropiadas para evaluar lesiones grandes y lesiones presentes en áreas delicadas desde el punto de vista quirúrgico. No existen evidencias que sugieran que la biopsia incisional aumenta el riesgo de metástasis.
Diagnóstico diferencial
Los nevos displásicos comparten muchas características con el melanoma temprano de diseminación superficial. Otras lesiones comunes que pueden parecer melanomas son las lentígines, las pecas por el sol, los nevos traumatizados, los angiomas trombosados, los CBC pigmentados, la enfermedad de Bowen pigmentada, los dermatofibromas y las queratosis seborreicas atípicas. Otros dos retos en el diagnóstico diferencial del melanoma merecen mención especial. Los melanomas amelanóticos (melanomas sin pigmento) se presentan como lesiones rosadas que pueden diagnosticarse en forma errónea como CBC o nevos de Spitz. Los nevos de Spitz pueden ser difíciles de distinguir de los melanomas tanto desde el punto de vista clínico como histológico. Ocurren con más frecuencia en niños, pero también se ven en adultos. Como los melanomas nodulares, los nevos de Spitz tienden a aparecer en forma súbita y su color varía de rojo a rojo-café.
Tratamiento
Sitio primario El melanoma cutáneo primario se trata por medio de extirpación quirúrgica definitiva. Las amplias extirpaciones del pasado han cambiado a márgenes de resección más modestos. Los datos de dos estudios aleatorios y prospectivos sobre bordes quirúrgicos en el melanoma cutáneo primario han demostrado la seguridad de dejar bordes de 1 cm para los melanomas de menos de 1 mm de espesor y de 2 cm para aquéllos entre 1 y 4 mm de grosor.20,21 No se han realizado estudios aleatorios sobre bordes de resección para melanomas de más de 4 mm de espesor. La mayoría de los autores recomiendan un borde de 2 a 4 cm para estos melanomas. En lugar de colocar injertos cutáneos, es mejor el cierre primario y los colgajos reconstructivos, desde el punto de vista tanto cosmético como funcional.
Ganglios linfáticos Los pacientes con enfermedad ganglionar regional clínicamente evidente se tratan con disección terapéutica de los ganglios. La realización de disección electiva de ganglios linfáticos (DEGL) en pacientes con melanoma cutáneo primario de espesor intermedio pero sin evidencia clínica de afección regional, ha sido tema de controversia durante mucho tiempo. En general, los datos sobre beneficios en la supervivencia no parecen justificar la morbilidad asociada a este procedimiento.
La biopsia de un ganglio centinela es una alternativa que se usa cada vez más en lugar de la DEGL en pacientes con melanoma cutáneo primario. Esta técnica utiliza la linfografía para identificar los ganglios linfáticos regionales que drenan la piel en el sitio del melanoma primario. En el momento de la extirpación definitiva del melanoma se inyecta un colorante azul con un radioisótopo en la dermis, alrededor del sitio del melanoma. Se realiza una pequeña incisión sobre la mancha identificada en la linfangiografía como área proximal de drenaje linfático regional. Se extirpa el primer ganglio linfático que se identifica que capta el colorante y el radioisótopo (ganglio centinela).
A continuación se evalúa la histología de este ganglio, con frecuencia por medio de técnicas de inmunohistoquímica y en ocasiones empleando la técnica de RCP, que es más sensible. La ausencia de melanoma en el ganglio centinela es muy sensible para descartar la presencia de metástasis en el resto de los ganglios cuando el procedimiento se realiza por un equipo con experiencia. Cuando el ganglio centinela es positivo para melanoma debe realizarse una DEGL completa. Algunos autores consideran que la información proporcionada por la biopsia del ganglio centinela tiene utilidad pronóstica significativa.22 Los pacientes con ganglios centinela positivos son candidatos apropiados para considerar tratamiento adyuvante (ver adelante). En la actualidad se realizan varios estudios multicéntricos para evaluar la utilidad clínica de este procedimiento.23
Metástasis en tránsito Se puede considerar que las metástasis en tránsito se confinan a una sola extremidad por periodos prolongados de tiempo. La amputación no parece mejorar la supervivencia a largo plazo en estos casos y las metástasis en tránsito de crecimiento lento pueden tratarse con cirugía. La enfermedad más extensa debe manejarse con terapia de sensibilización con dinitroclorobenceno (DNCB). Para las metástasis en tránsito extensas limitadas a una extremidad la terapia de perfusión permite una paliación importante, así como salvar la extremidad. Este procedimiento consiste en aislar la vasculatura de la extremidad afectada de la sistémica y perfundir esta extremidad con agentes quimioterapéuticos, agentes biológicos o ambos, a dosis que no podrían tolerarse si se administraran en forma sistémica.24
Metástasis a distancia A pesar del desarrollo de varios enfoques novedosos para el tratamiento de los pacientes con melanoma metastásico, incluyendo quimioterapia múltiple, terapia biológica, inmunoterapia y combinaciones de estos tratamientos, la monoterapia con dacarbacina (DTIC) sigue siendo el esquema original autorizado por la Food and Drug Administration para el tratamiento del melanoma metastásico.25 Se observan respuestas objetivas a la DTIC en alrededor del 5 al 20% de los pacientes, aunque las respuestas completas son raras. La radioterapia puede tener un papel paliativo importante. En ausencia de un tratamiento que haya demostrado ser más eficaz desde el punto de vista clínico, los pacientes con metástasis a distancia deben tener la oportunidad de participar en estudios clínicos de manejos experimentales.
Tratamiento adyuvante Los pacientes con enfermedad cutánea o regional que quirúrgicamente están sin enfermedad pero tienen riesgo de recurrencia o metástasis son candidatos potenciales para el tratamiento adyuvante.26 Se han usado varias terapias adyuvantes para el melanoma, incluyendo inmunoestimuladores como el bacilo Calmette-Guérin, Corynebacterium parvum y levamisol. También se han intentado varios agentes quimioterápicos. Recientemente se ha estudiado la inmunoterapia con citocinas, como los interferones, y la inmunización activa con vacunas. No se ha demostrado en algún estudio clínico aleatorio de terapia adyuvante para el melanoma mejoría significativa en la evolución con una excepción importante: un estudio a gran escala de interferón en dosis altas en pacientes de riesgo alto con melanoma ya resecado demostró un impacto pequeño, pero significativo en la supervivencia global.27 Este estudio constituyó la base para la autorización por la FDA del interferón alfa intravenoso en dosis de 20 millones de unidades/m2 por día durante un mes seguido de la administración subcutánea de 10 millones de unidades/m2 tres veces por semana por 48 semanas más como manejo adyuvante para el melanoma. Los estudios adicionales de interferón en dosis altas, intermedias y bajas no han demostrado efectos semejantes.
En la actualidad se estudian muchas otras estrategias, incluyendo inmunización activa, inmunización pasiva y terapias biológicas, que pueden constituir nuevas oportunidades para los pacientes que son candidatos apropiados para estos estudios.28
Pronóstico
Estadio El factor pronóstico aislado más importante para el melanoma es el estadio de la enfermedad. Se han usado varias clasificaciones de estadificación, todos los sistemas toman en cuenta la clasificación clásica TNM de tamaño del tumor (T), afección de ganglios linfáticos (N) y metástasis a distancia (M). Las diferencias entre los diversos sistemas se relacionan principalmente con la estadificación del sitio primario. Los nuevos esquemas de estadificación intentan considerar los atributos del tumor primario que correlacionan en forma estrecha con la evolución, específicamente el espesor y el nivel de Clark [ver tabla 2].29 Las tasas de supervivencia global a 5 años para los años 1989 a 1994, reportadas por el programa del National Cancer Institute Surveillance, Epidemiology and End Results (NCI SEER) de los EUA, son las siguientes: melanoma cutáneo primario, alrededor del 95 por ciento, con metástasis nodales regionales alrededor del 60 por ciento, y con metástasis a distancia, 12 por ciento.
|
|||||||||||||||||
is- in situ, M0-no metástasis a distancia, M1-metástasis a distancia, N-ganglios linfáticos, pT-tumor primario |
Atributos del tumor primario Se han identificado varias características del tumor primario que son predictores de la evolución en el melanoma cutáneo primario. El más importante y más consistente es el grosor del tumor según Breslow. Otros parámetros histológicos importantes son el nivel de Clark de invasión, la extensión de la ulceración, la tasa de mitosis, la presencia de linfocitos que infiltran el tumor y la invasión vascular. En los tumores primarios delgados uno de los factores más importantes es la fase de crecimiento.30 El melanoma con fase de crecimiento radial parece no provocar tantas metástasis, mientras que el melanoma con crecimiento vertical (caracterizado por la formación de un nódulo tumoral en la dermis) se asocia con riesgo significativo de metástasis incluso en lesiones menores de 1 mm de espesor.31 Las características del paciente que se asocian con mejor supervivencia incluyen menor edad (< 60 años), sexo femenino y localización del melanoma en una extremidad. Se han desarrollado modelos multivariados para predecir la evolución el melanoma [ver tabla 3].32
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
* Intervalo de confianza =95%.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tumores malignos de la dermis
TUMORES METASTASICOS
Ocurren metástasis cutáneas en alrededor del 5 por ciento de los pacientes con tumores sólidos, que suelen asociarse con diseminación de la enfermedad. La frecuencia relativa de metástasis a la piel depende del género y refleja las frecuencias de los cánceres primarios.33 En las mujeres dos tercios de las metástasis provienen de cáncer de mama, pero también son frecuentes las neoplasias de pulmón, colon y recto, melanoma y ovario. En los varones el cáncer de pulmón es el más común, seguido de cáncer del colon, melanoma, CE de la cabeza y el cuello y carcinoma renal.33 La distribución anatómica de las metástasis cutáneas no es aleatoria. Las metástasis cutáneas por cáncer de mama suelen afectar la pared torácica y pueden aparecer como nódulos, linfedema o celulitis. El cuero cabelludo es un sitio frecuente de metástasis, en especial de cáncer de pulmón, riñón (en el varón) y de mama (en la mujer). Los cánceres de cabeza y cuello pueden invadir la piel por extensión local, dando origen a un edema firme, rojo obscuro, de la piel que parece celulitis. Pueden ocurrir metástasis en la pared abdominal de cánceres gastrointestinales o de ovario.33 Desde el punto de vista clínico las metástasis cutáneas casi no dan síntomas, forman pápulas o nódulos dérmicos y son del color de la piel o rosadas. Se diseminan a través de linfáticos o por vía vascular. Las metástasis cutáneas pueden reflejar la histología del tumor primario (v.gr., nódulos negros, café o gris con melanoma metastásico y nódulos vasculares con carcinoma de células renales o tiroideas).
TUMORES PRIMARIOS
Los tumores primarios de la dermis pueden desarrollarse a partir de las múltiples estructuras de la piel, incluyendo glándulas sebáceas (carcinoma sebáceo), tejido conectivo (dermatofibrosarcoma protuberante), músculo liso (leiomiosarcoma) y otros tejidos anexos (carcinoma ecrino).34 La mayoría de estas neoplasias primarias son raras y pueden tener un comportamiento biológico agresivo. Aunque estas neoplasias son muy variadas desde el punto de vista histológico, muchas comparten un cuadro clínico común, presentándose como un nódulo subcutáneo que crece con rapidez, del color de la piel o rosa a rojo y que en ocasiones recuerda un quiste sebáceo.
Carcinoma de células de Merkel Esta neoplasia dérmica tiene origen neuroendócrino. Suele aparecer como una pápula o nódulo dérmico rojo a violáceo en la cabeza y cuello de los pacientes ancianos, aunque se afectan todos los grupos de edad.34 El tratamiento de elección es la extirpación local amplia con o sin DEGL. Son frecuentes las recurrencias locales y ocurren metástasis a distancia en más de la tercera parte de los pacientes.34 La quimioterapia de las metástasis no suele dar buenos resultados.34
Enfermedad de Paget Esta neoplasia rara de la piel asociada con un adenocarcinoma subyacente,33,34 suele presentarse como una dermatisis eritematosa, con frecuencia eccematosa, unilateral en la mama, afectando el pezón y la areola. El diagnóstico diferencial incluye eccema, psoriasis, dermatitis de contacto e impétigo. Por este motivo es importante realizar biopsia de las dermatitis inflamatorias del pezón y la areola que no se resuelven con facilidad. En la enfermedad de Paget la biopsia revela células de Paget típicas, que tiñen en forma leve, dentro de la epidermis. La resección quirúrgica apropiada de la neoplasia cutánea y subyacente es el tratamiento de elección; con frecuencia ocurren metástasis a ganglios linfáticos.
Enfermedad de Paget extramamaria La enfermedad extramamaria de Paget es aún menos frecuente que la enfermedad de Paget y típicamente se presenta como placas rojas, con frecuencia ulceradas, en las regiones perineales de personas ancianas.33,34 Las lesiones pueden ser pruriginosas o asintomáticas, con frecuencia de larga evolución, y pueden diagnosticarse de modo equívoco como psoriasis, dermatitis de contacto o infecciones micóticas crónicas. Los tumores asociados subyacentes incluyen carcinomas rectales y genitourinarios. Incluso sin una neoplasia interna asociada la enfermedad extramamaria de Paget es difícil de tratar, con una alta tasa de recurrencia local.34
Angiosarcoma El angiosarcoma es una neoplasia vascular rara y con frecuencia muy agresiva.34 Puede aparecer como parches o nódulos multicéntricos rojos o púrpura en una extremidad linfedematosa, como un brazo linfedematoso después de una mastectomía (síndrome de Stewart-Treves). Otra forma de presentación consiste en parches o placas violáceas en la cabeza o cuello (en especial el cuero cabelludo) de personas ancianas. El angiosarcoma tiene mal pronóstico, causando metástasis pulmonares a pesar de la cirugía o radiación.34
Dermatofibrosarcoma protuberante El dermatofibrosarcoma protuberante es una neoplasia de crecimiento lento, agresiva en forma local, que rara vez metastatiza, pero sí recurre. Las lesiones típicamente son nódulos firmes, rojo-café o morado, por lo general en el tronco o extremidades, en zonas no expuestas al sol. El diagnóstico diferencial incluye queloides y dermatofibroma benigno. Suele afectar principalmente adultos jóvenes, aunque el tumor puede ocurrir a cualquier edad. La extirpación local con o sin cirugía micrográfica de Mohs ofrece la mejor posibilidad de curación.34
SARCOMA DE KAPOSI
El sarcoma de Kaposi (SK) es una neoplasia cutánea multicéntrica que tiene diversas variantes clínicas.35 A pesar de su nombre, el SK no es un verdadero sarcoma. Aunque no se ha definido con claridad la célula de origen, parece que deriva del endotelio linfático.35
Epidemiología
En su forma clásica el SK es una enfermedad indolente de varones ancianos con ascendencia mediterránea o de Europa oriental, que se manifiesta por nódulos y placas violáceas en las extremidades inferiores.35,36 Antes de la aparición del SIDA el trastorno era raro en los Estados Unidos, con una incidencia anual ajustada por edad de 0.29 por 100,000 habitantes en los varones y 0.07 por 100,000 habitantes en las mujeres.36 A finales de los años 60 se reconoció una segunda forma del SK, endémica, en Africa Central. El SK de Africa Central, que típicamente afecta adultos jóvenes y niños, tiene una evolución más agresiva que el SK clásico, con afección frecuente a hueso, ganglios linfáticos y vísceras.35
En 1981 el SK fue el primer tumor reconocido como parte del SIDA. Afecta principalmente a varones homosexuales y fue el padecimiento que definió al SIDA en el 30 a 40 por ciento de los pacientes durante los primeros años de la epidemia del VIH.36 Los varones homosexuales infectados por VIH tienen un incremento de 73,000 veces en la incidencia de SK, y las mujeres y varones no homosexuales infectados de 10,000 veces, si se compara con la población general de los Estados Unidos.36 Se ha descrito una cuarta variante de SK en pacientes inmunosuprimidos en forma iatrogénica, en especial receptores de trasplantes. Los varones se afectan un poco más que las mujeres.35
Etiología
La epidemiología del SK ha sugerido a un agente infeccioso como causal o cofactor.35,36 Las evidencias actuales implican a un herpesvirus humano novedoso: el herpesvirus del sarcoma de Kaposi (HVSK), también conocido como herpesvirus humano tipo 8 (HVH-8).37,39 El HVH-8 se ha detectado en todas las variantes de SK,37 y el desarrollo de anticuerpos contra antígenos nucleares del HVH-8 en pacientes infectados por VIH es predictivo del desarrollo de SK.38 El aislamiento y propagación del herpesvirus humano a partir de SK asociado a VIH que tiene secuencias de ADN idénticas al HVH-8 apoya aún más el posible papel del herpesvirus humano.39 Sin embargo, aún quedan muchas dudas respecto al HVH-8. También se la ha asociado con linfomas en cavidades corporales,36 enfermedad de Castleman,35,36, linfadenopatía angioblástica,35 y diversas lesiones de la piel en receptores de trasplantes de órganos.35,36 El mecanismo por el que el HVH-8 podría causar la tumorigénesis en el SK no es claro, por lo tanto, se ha sugerido que el HVH-8 es un virus ubicuo en ciertas poblaciones.35-39
Los factores del huésped, en especial la inmunosupresión, son cruciales en algunas poblaciones con SK.35,36 El VIH puede tener un papel indirecto en el desarrollo de SK a través de la depleción de células CD4 y la mayor producción de citocinas como la interleucina-1 (IL-1) y la IL-6.35,36 Los fármacos inmunosupresores, en especial la ciclosporina, la azatioprina y la prednisona, aumentan el riesgo de desarrollar SK, sobre todo en receptores de trasplante de riñón e hígado.36 Se ha observado regresión espontánea del SK después de suspender la ciclosporina y los esteroides.36
A pesar de la prevalencia de SK en algunos grupos étnicos, el papel de cualquier factor genético posible no es claro. Se ha puesto en duda la mayor incidencia del HLA-DR5 en pacientes con SK clásico.36 El SK familiar es muy raro,35 lo que sugiere que los factores genéticos solos no son los responsables.
Por último, el género parece constituir un factor de riesgo significativo, en especial en el SK clásico, en el que la relación varón a mujer puede variar de 3:1 a 10:1.35,36 Las explicaciones para este predominio en varones han incluido los efectos inhibitorios de las hormonas femeninas (v.gr., gonadrotropina coriónica humana-beta), pero no se conocen los mecanismos exactos.35
Diagnóstico
Las manifestaciones clínicas del SK difieren entre las variantes de este trastorno.35,36 En el SK clásico aparecen máculas o parches rojo-púrpura claro o nódulos púrpura primero en los pies, en especial en las plantas. En raras ocasiones existe linfadenopatía (en especial inguinal). Las lesiones pueden desarrollarse también en brazos y áreas genitales. Al progresar la enfermedad las lesiones coalescen para formar placas violáceas.
El SK asociado al VIH suele presentarse con lesiones cutáneas, pero las primeras pueden aparecer en la mucosa oral o los ganglios linfáticos. En contraste a las lesiones de SK clásico, las asociadas con VIH suelen comenzar en la parte superior del cuerpo (cara, tronco o brazos). Lo más típico es que las lesiones de SK asociado a VIH sean pápulas púrpura o rojas, con frecuencia ovales, que siguen los pliegues de la piel en una distribución semejante a la pitiriasis rosada.35,36 Las lesiones varían de manchas rosas a placas púrpura obscuro [ver figura 5] o pueden parecer equímosis, en especial en pacientes con cuentas bajas de células T CD4+. Las lesiones orales suelen consistir en placas o nódulos rojo-púrpura en el paladar, las encías o la mucosa oral. Los pacientes con piel más obscura pueden tener lesiones púrpura obscuro o negras, así como placas hiperpigmentadas.35
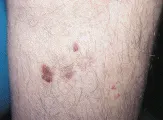
|
| Figura 5 |
| Sarcoma de Kaposi |
Al progresar el SK asociado al VIH puede desarrollarse linfedema en los pies, escroto, genitales y regiones periorbitarias, así como linfadenopatía (en especial inguinal). Las lesiones gastrointestinales pueden ser submucosas y asintomáticas, aunque pueden provocar hemorragia gastrointestinal. El SK pulmonar se asocia con mal pronóstico.40
La evaluación de laboratorio de los pacientes con SK debe incluir detección de anticuerpos contra VIH, citología hemática completa, sangre oculta en heces y radiografía de tórax. Está indicado solicitar cuenta de células T CD4+ en los pacientes VIH-positivos. Se realizará una historia clínica y examen físico completos, con especial atención a la presencia de infecciones oportunistas en pacientes infectados por VIH o inmunosuprimidos por otros motivos.
En todo paciente con sospecha de SK debe realizarse biopsia de piel. La histopatología de todas las variantes es parecida: células en forma de huso dentro de la dermis con eritrocitos extravasados presentes en las hendiduras entre los espacios vasculares irregulares.41
Diagnóstico diferencial
El diagnóstico diferencial clínico del SK incluye dermatofibroma, púrpura, granuloma piógeno, angiomatosis bacilar, melanoma metastásico y CBC. Otras entidades histopatológicas que pueden recordar al SK son el angiosarcoma y la dermatitis por estasis.35,36
Tratamiento
Sarcoma de Kaposi clásico El tratamiento del SK es paliativo. En el SK clásico, en el que la enfermedad es indolente y los pacientes son ancianos, rara vez se justifica el tratamiento sistémico agresivo.35 En lugar de ello, la radioterapia es el tratamiento de elección.35,42 El SK es muy radiosensible: se han usado dosis aisladas de 800 cGy para la paliación rápida en pacientes con mal pronóstico.43 Las dosis totales de 800 a 3,500 cGy causan remisiones completas en el 56 por ciento de los casos y parciales en el 46 por ciento, y más de la mitad de los pacientes no requieren tratamiento posterior hasta por 13 años.42 Se ha sugerido un esquema de tratamiento equivalente a 3,000 cGy en 10 fracciones durante 2 semanas.42
Para los pacientes con SK clásico que tienen solo una a dos pápulas la biopsia excisional puede ser suficiente tanto para diagnóstico como para tratamiento. La crioterapia con nitrógeno líquido puede ser útil en la pápulas aisladas. El tratamiento sistémico puede estar indicado en casos de afección cutánea extensa o compromiso visceral. Con frecuencia se emplea quimioterapia con un solo agente, con alcaloides de la vinca (vincristina o vinblastina). También puede ser eficaz el interferón alfa recombinante en dosis bajas; sin embargo, los efectos adversos (fiebre, calosfríos, mialgias y fatiga) pueden no ser bien tolerados por los pacientes ancianos.35
Sarcoma de Kaposi asociado al VIH Aunque el SK es más agresivo en los pacientes infectados por VIH, la magnitud de la inmunosupresión y la presencia de infecciones oportunistas y otras enfermedades sistémicas tienen gran importancia en la estadificación, pronóstico y elección del tratamiento apropiado.35,36 Las características clínicas que tradicionalmente se asocian con una evolución más favorable incluyen cuentas de células T CD4+ mayores de 200 células/mm3, ausencia de enfermedad sistémica, SK limitado a la piel o ganglios linfáticos y SK oral mínimo (i.e., no nodular). Los factores de mal pronóstico son cuenta de células T CD4+ menor de 200 células/mm3, linfedema asociado al SK, SK visceral, ulcerado u oral nodular, e infección oportunista.44 Con la aparición del tratamiento antirretroviral muy activo (TARMA), los médicos que tratan a pacientes con SK asociado al VIH tienen en la actualidad la oportunidad de influir e incluso revertir la inmunosupresión al modificar tanto la carga viral de VIH como la cuenta de células T CD4+. Se ha observado regresión del SK después del inicio del TARMA,45 con frecuencia durante los primeros meses de tratamiento.
El tratamiento local es una medida adecuada en pacientes con SK y enfermedad limitada, los que tienen complicaciones infecciosas y los que no pueden tolerar el tratamiento sistémico.43 Se ha demostrado que el tratamiento antitumoral específico mejora la supervivencia global.43 La radioterapia es eficaz en el SK asociado a VIH en dosis semejantes a las usadas para el SK clásico (ver antes). Sin embargo, las respuestas del SK asociado a VIH son breves.35 También han sido útiles las inyecciones intralesionales de vinblastina o interferón para lesiones seleccionadas.35,43 La crioterapia con nitrógeno líquido es eficaz en lesiones pequeñas;43 sin embargo, está contraindicada en pacientes con piel obscura en los que la hipopigmentación postratamiento puede ser cosméticamente mucho peor que la lesión original de SK.
El tratamiento sistémico incluye quimioterapia convencional y modificadores de la respuesta biológica. Los esquemas quimioterápicos con un solo agente permiten tasas de respuesta de 30 a 70 porciento.36 La quimioterapia combinada (v.gr., doxorrubicina, bleomicina y vincristina) se complica con supresión intensa de la médula ósea e infecciones oportunistas frecuentes. La disponibilidad de factores estimuladores de colonias hematopoyéticas (v.gr., factor estimulador de colonias de granulocitos) y la profilaxis contra Pneumocystis carinii han disminuido estas complicaciones.
El desarrollo de antraciclinas encapsuladas en liposomas (v.gr., doxorrubicina y daunorrubicina) representa una nueva opción importante en el tratamiento del SK.36,46 En un estudio aleatorio de 258 pacientes con SK asociado a SIDA avanzado, los que recibieron doxorrubicina en liposomas tuvieron tasas de respuesta significativamente mayores que los que recibieron la combinación estándar de doxorrubicina, bleomicina y vincristina.46 El paclitaxel también se asoció con buena respuesta en pacientes con SK avanzado.40
Los enfoques de investigación promisorios para el SK asociado al VIH incluyen tratamiento combinado con agentes retrovirales y terapia antitumoral específica,36 además del tratamiento potencial contra el HVH-8.
Complicaciones
Las infecciones bacterianas y la sepsis complican con frecuencia los tumores ulcerados de piernas y pies. Pueden participar infecciones por oportunistas, en especial en pacientes con cuentas de células T CD4+ muy bajas.
Pronóstico
Como ya se mencionó, el tratamiento del SK es paliativo. La cuenta de células T CD4+ es el factor más importante para predecir la supervivencia en el SK asociado al VIH.44 Los tumores grandes,44 el linfedema y el SK pulmonar también indican mal pronóstico.35,46
Linfoma cutáneo
Los linfomas pueden ser de origen celular B o T y afectar la piel en forma primaria o secundaria. Los linfomas de células B, en especial los linfomas no Hodgkin, pueden afectar la piel en forma secundaria en la enfermedad avanzada. Aparecen típicamente como placas o nódulos subcutáneos rojo-púrpura. Los linfomas primarios de células B de la piel son muy raros, aparecen como nódulos rojos que con frecuencia permanecen localizados a la piel, pero que pueden progresar hacia una afección sistémica. La gran mayoría de los linfomas cutáneos primarios son linfomas cutáneos de células T (LCCT).
El LCCT es un padecimiento raro de la piel que suele conocerse como micosis fungoides (MF).47 Las MF son en realidad el subtipo más grande de los LCCT, aunque los términos suelen usarse en forma indistinta. La variante leucémica del LCCT es el síndrome de Sézary.47 Otra variante del LCCT se asocia con el HTLV-1 y es parte del espectro de los linfoma/leucemia de células T del adulto y de los linfomas de células T periféricos.47
EPIDEMIOLOGIA
Alrededor de 1,000 nuevos casos de LCCT se diagnostican en los Estados Unidos cada año.47,48 La incidencia ha aumentado, de 0.19 por 100,000 habitantes en 1973 a 0.42 por 100,000 habitantes en 1984.47 El LCCT afecta principalmente a adultos de edad media, siendo la edad promedio de presentación los 50 años.47,48 La relación entre varón y mujer es de alrededor de 2:1, y los pacientes de raza negra se afectan el doble que los de raza blanca.47,48
ETIOLOGIA
La susceptibilidad del huésped y un antígeno ambiental, quizá viral, parecen tener una función importante en la patogenia del LCCT.49 Los factores genéticos pueden relacionarse con los antígenos del complejo de histocompatibilidad, y se ha observado mayor frecuencia de HLA-DR5 en pacientes con LCCT.49 La estimulación antigénica crónica (v.gr., por infección) puede participar también.48
Los virus son los candidatos antigénicos principales, considerando la aparente asociación del HTLV-1 con el linfoma de células T periféricas.48-50 El linfoma de células T periféricas asociado al HTLV-1 tiene características clínicas únicas, incluyendo hipercalcemia y lesiones óseas.48
DIAGNOSTICO
Manifestaciones clínicas
Las manifestaciones clínicas de las MF aparecen durante muchos meses a años. En un estudio la duración promedio de los síntomas antes del diagnóstico fue de 7.5 años.51 Aparecen manchas eritematosos planas, con frecuencia descamativas y en ocasiones atróficas, principalmente en el tronco y los muslos [ver figura 6]. Las lesiones son asintomáticas o tienen prurito leve y pueden remitir en forma espontánea o responder al tratamiento esteroideo tópico. Los pacientes pueden también reportar mejoría después de la exposición al sol. Las lesiones, denominadas de parapsoriasis, progresan a LCCT en el 10 a 30 por ciento de los casos, según seguimientos a largo plazo (< 10 años).48
|
|
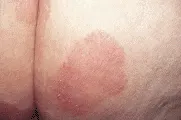
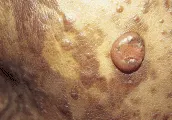
El examen físico de los pacientes con probable LCCT incluye examen completo de la piel, incluyendo clasificación de las lesiones (mancha, placa o tumor) y la extensión de la superficie corporal afectada. Deben examinarse los ganglios linfáticos, el hígado y el bazo.
Biopsia de piel
Se requiere biopsia de la piel para hacer el diagnóstico definitivo de MF. La presencia de células linfoides atípicas con nucleos cerebriformes con múltiples circunvoluciones en racimos dentro de la epidermis (microabascesos de Pautrier)47,48 y los infiltrados linfocíticos en banda en la dermis superior son diagnósticos de LCCT.47 La célula maligna es una célula T, y la mayoría de las células expresan los marcadores de las células T en general, CD2, CD3 y CD5.48 El uso de estudios sobre rearreglos genéticos del receptor de la célula T para confirmar la clonalidad en la fase temprana de la enfermedad puede ayudar al diagnóstico.48 Ni los estudios inmunofenotípicos ni la microscopía electrónica pueden considerarse como definitivos para el diagnóstico de las MF, se requiere correlación clinicopatológica.
Estudios de laboratorio
La evaluación de laboratorio del LCCT incluye citología hemática completa, cuenta de eosinófilos, cuenta de células de Sézary, deshidrogenasa láctica (DHL) pruebas de función hepática. La biopsia de médula ósea no es necesaria en ausencia de células leucémicas circulantes. Debe considerarse investigar HTLV-1 en pacientes con factores de riesgo o presentaciones atípicas. La biopsia de ganglio linfático es adecuada si existen ganglios palpables, en especial mayores de 2 cm. Puede ser importante realizar tomografía abdominal computada o radiografía de tórax en los pacientes con tumores o sospecha de afección visceral.
DIAGNOSTICO DIFERENCIAL
En sus fases tempranas las MF pueden parecer diversos trastornos inflamatorios benignos (v.gr., reacción a fármacos, eccema, psoriasis o dermatitis de contacto). Es necesario descartar estos padecimientos antes de iniciar el tratamiento.
TRATAMIENTO
Tratamiento tópico
El tratamiento tópico es la base del tratamiento para las manchas o placas tempranas (en fase IA, IB y IIA).53 El tratamiento agresivo inicial con radio y quimioterapia no ha demostrado ser superior a las medidas locales para controlar la enfermedad o mejorar la supervivencia en pacientes con enfermedad limitada.54 Un enfoque lógico para la enfermedad temprana (o histológicamente dudosa) consiste en administrar esteroides tópicos.58 La mostaza nitrogenada (mecloretamina) tópica, en forma acuosa o en ungüento, es la quimioterapia tópica empleada con más frecuencia y permite remisiones completas iniciales en la enfermedad en mancha o placa en el 60 a 80 por ciento de los pacientes.47,53 El tratamiento debe continuarse por periodos prolongados (hasta por 3 años después de desaparecidas las lesiones). Se desarrolla dermatitis de contacto en alrededor de la tercera parte de los pacientes.
La solución de carmustina (BCNU), aplicada a las lesiones en forma diaria, es otro esquema útil. El tratamiento suele durar 8 a 16 semanas, pero se ha continuado hasta por 6 meses.53 Debido a que la absorción sistémica puede provocar supresión de la médula ósea, deben realizarse biometrías hemáticas periódicas.47,53
Radiación ultravioleta
La radiación ultravioleta se administra en diferentes formas para el LCCT, desde luz ultravioleta hasta radiación ionizante. La UVB es útil en la enfermedad en estadio I, con una tasa de remisión clínica completa de 71 por ciento en un estudio. El tiempo promedio de remisión fue de 5 meses y la duración de la misma fue de 22 meses.56
Otro método eficaz en las MF es la combinación de psoralenos y luz ultravioleta A (PUVA). En un estudio el 95 por ciento de los pacientes con LCCT en estadio I tuvieron remisión clínica completa, con una duración promedio de la respuesta de 43 meses.57
Radioterapia
La radiación total de la piel con haz de electrones (RTPHE) aplica radioterapia a la superficie de la piel sin una dosis interna significativa. Es especialmente útil en la enfermedad en placas. Las dosis típicas son de 2,400 a 3,600 cGy, fraccionadas en varias semanas con un haz de radiación de electrones de 4 a 9 meV.58 La remisión completa se relaciona con el estadio, de la siguiente manera: IA, 84 a 96 por ciento, IB 56 a 81 por ciento, IIA 63 a 74 por ciento, IIB 24 a 53 por ciento, III 26 a 50 por ciento y IVA 8 a 33 por ciento.10 Se logró una supervivencia libre de recaídas del 50 por ciento a 5 años con la enfermedad IA, pero la mayoría de los pacientes con enfermedad más avanzada sufren recaídas para los 5 años.58
Tratamiento sistémico
El tratamiento sistémico para el LCCT se considera como tratamiento primario en la enfermedad avanzada (estadios III a IVB)48,54,59,60 y como terapia secuencial para promover respuestas más duraderas en la enfermedad temprana.48,59,60
La fotoféresis extracorpórea parece ser más útil en el LCCT eritrodérmico y en el síndrome de Sézary.47,60 En este tratamiento el paciente es sometido a fotoféresis extracorpórea con radiación UVA a los leucocitos después de la ingestión oral de 8-metoxipsoraleno.
El tumor avanzado y el LCCT visceral han sido tratados con quimioterapia de uno o varios agentes, incluyendo metotrexate, análogos de adenosina, interferón alfa y retinoides.48,59 El tratamiento experimental incluye anticuerpos monoclonales, retinoides y citocinas.48
Tratamiento combinado
El tratamiento agresivo temprano del LCCT (RTPHE seguido de quimioterapia combinada con ciclofosfamida, doxorrubicina, etopósido y vincristina) no proporciona ventajas en la supervivencia comparado con el tratamiento tópico secuencial.54 En forma semejante, la adición de quimioterapia sistémica (doxorrubicina y ciclofosfamida) o fotoféresis extracorpórea después de una respuesta completa a la RTPHE no tuvo impacto en la supervivencia en micosis fungoides tempranas ni impacto sobre la supervivencia libre de recaídas en todos los estadios.60 Otros esquemas incluyen interferón alfa y retinoides (isotretinoín) con RTPHE, seguido de mostaza nitrogenada tópica.59 La heterogeneidad de los esquemas de tratamiento combinado en el LCCT hace prácticamente imposible comparar los resultados.
COMPLICACIONES
Las complicaciones más serias del LCCT son las infeciones. La sepsis por tumores cutáneos ulcerados es una causa común de muerte. Puede ocurrir LCCT visceral, lo mismo que transformación a un linfoma de células grandes en algunos pacientes con LCCT (probabilidad del 39 por ciento después de 12 años).52 En los pacientes con enfermedad temprana y que sobreviven por tiempo prolongado, el tratamiento local (v.gr., RTPHE o PUVA) puede contribuir al desarrollo de otros cánceres de piel (v.gr., CBC o CE) y cataratas.
PRONOSTICO
La estadificación del LCCT se basa en la evaluación del tipo y extensión de las lesiones cutáneas y de la afección a ganglios linfáticos, sangre periférica y vísceras.48,51 Se han realizado muchos intentos por clasificar el LCCT en grupos pronósticos útiles. Un estudio inicial y aún válido que usó el sistema TNM identificó tres grupos principales: pacientes con riesgo adecuado (estadios IA, IB y IIA, con enfermedad cutánea solo en placas y sin afección a ganglios linfáticos, sangre o vísceras [supervivencia promedio > 12 años]), pacientes con riesgo intermedio (estadios IIB, III y IVA, con tumores cutáneos, eritrodermia o enfermedad en placas y afección ganglionar o sanguínea pero sin daño visceral o destrucción ganglionar [supervivencia promedio de 5 años], y pacientes con riesgo elevado (estadio IVB con afección visceral y destrucción ganglionar [supervivencia promedio 2.5 años]).47,51
La eosinofilia también se asocia con supervivencia corta.51 Estudios recientes han demostrado que, en general, los pacientes más jóvenes (< 35 años) con micosis fungoides tienden a presentar enfermedad en estadio más temprano.61 Otros estudios a largo plazo han revelado que los pacientes en estadio IA no tienen reducción en su expectancia de vida y que menos del 10 por ciento de estos pacientes progresan a estadios más avanzados.62 La supervivencia de los pacientes con MF en mancha o placa generalizada (estadios IB o IIA) es significativamente peor que la de la población control de raza, edad y sexo equiparable a los 11.7 años. El género y la raza no parecen tener efecto sobre la supervivencia, pero los pacientes de mayor edad (> 58 años) tienen supervivencias más cortas.63
Bibliografía
-
Marks R: An overview of skin cancers: incidence and causation.
Cancer 75:607, 1995
-
Grossman D, Leffell DJ: The molecular basis of nonmelanoma
skin cancer: new understanding. Arch Dermatol 133:1263, 1997 [PMID
9382565]
-
Naylor MF, Farmer KC: The case for sunscreens: a review of
their use in preventing actinic damage and neoplasia. Arch Dermatol 133:1146,
1997 [PMID
9301593]
-
Stenbeck KD, Baland KP, Williams MJ, et al: Patterns of treated
non-melanoma skin cancer in Queensland-the region with the highest incidence
rates in the world. Med J Aust 153: 511, 1990 [PMID
2233471]
-
Scotto J, Fears TR, Fraumeni, JF: Incidence of non-melanoma
skin cancer in the United States (NIH Publication No. 83-2433). U.S. Public
Health Service, Bethesda, Maryland, 1983.
-
Gailani MR, Bale AE: Developmental genes and cancer: role
of patched in basal cell carcinoma of the skin. J Natl Cancer Inst 89:1103,
1997 [PMID
9262247]
-
Shriner DL, McCoy DK, Goldberg DJ, et al: Mohs micrographic
surgery. J Am Acad Dermatol 39:79, 1998 [PMID
9674401]
-
Paver K, Royser K, Burry N, et al: The incidence of basal
cell carcinoma and their metastases in Australia and New Zealand. Australas
J Dermatol 14:53, 1973 [PMID
4753676]
-
Miller DL, Weinstock MA: Nonmelanoma skin cancer in the United
States: incidence. J Am Acad Dermatol 30:774, 1994 [PMID
8176018]
-
Staples M, Marks R, Giles G: Trends in the incidence of non-melanocytic
skin cancer (NMSC) treated in Australia 1985-1995: are primary prevention
programs starting to have an effect? Int J Cancer 78:144, 1998
-
Elder D, Elenitsas R, Jaworsky C, et al: Tumors and cysts
of the epidermis. Lever's Histopathology of the Skin, 8th ed. Lippincott-Raven,
Philadelphia, 1997, p 685
-
Rowe DE, Carroll RJ, Day CL Jr: Prognostic factors for local
recurrence, metastasis, and survival rates in squamous cell carcinoma of
the skin, ear, and lip: implications for treatment modality selection.
J Am Acad Dermatol 26:976, 1992 [PMID
1607418]
-
Hall HI, Miller DR, Rogers JD, et al: Update on the incidence
and mortality from melanoma in the United States. J Am Acad Dermatol 40:35,
1999 [PMID
9922010]
-
Haluska FG, Hodi FS: Molecular genetics of familial cutaneous
melanoma. J Clin Oncol 16:670, 1998 [PMID
9469357]
-
Elder DE, Clark WH Jr, Elenitsas R, et al: The early and
intermediate precursor lesions of tumor progression in the melanocytic
system: common acquired nevi and atypical (dysplastic) nevi. Semin Diagn
Pathol 10:18, 1993 [PMID
8506414]
-
Carey WP Jr, Thompson CJ, Synnestvedt M, et al: Dysplastic
nevi as a melanoma risk factor in patients with familial melanoma. Cancer
74:3118, 1994 [PMID
7982177]
-
Tucker MA, Halpern A, Holly EA, et al: Clinically recognized
dysplastic nevi: a central risk factor for cutaneous melanoma. JAMA 277:1439,
1997 [PMID
9145715]
-
Binder M, Puespoeck-Schwarz M, Steiner A, et al: Epiluminescence
microscopy of small pigmented skin lesions: short-term formal training
improves the diagnostic performance of dermatologists. J Am Acad Dermatol
36:197, 1997 [PMID
9039168]
-
Binder M, Schwarz M, Winkler A, et al: Epiluminescence microscopy:
a useful tool for the diagnosis of pigmented skin lesions for formally
trained dermatologists. Arch Dermatol 131:286, 1995 [PMID
7887657]
-
Balch CM, Urist MM, Karakousis CP, et al: Efficacy of 2-cm
surgical margins for intermediate-thickness melanomas (1 to 4 mm): results
of a multi-institutional randomized surgical trial. Ann Surg 218:262, 1993
[PMID
8373269]
-
Veronesi U, Cascinelli N, Adamus J, et al: Thin stage I primary
cutaneous malignant melanoma: comparison of excision with margins of 1
or 3 cm. N Engl J Med 318:1159, 1988 [PMID
3079582]
-
Piepkorn M, Weinstock MA, Barnhill RL: Theoretical and empirical
arguments in relation to elective lymph node dissection for melanoma. Arch
Dermatol 133:995, 1997 [PMID
9267247]
-
Kelley MC, Ollila DW, Morton DL: Lymphatic mapping and sentinel
lymphadenectomy for melanoma. Semin Surg Oncol 14:283, 1998 [PMID
9588721]
-
Thompson JF, Hunt JA, Shannon KF, et al: Frequency and duration
of remission after isolated limb perfusion for melanoma. Arch Surg 132:903,
1997 [PMID
9267277]
-
Houghton A, Coit D, Bloomer W, et al: NCCN melanoma practice
guidelines. Oncology 12:153, 1998
-
Agarwala SS, Kirkwood JM: Adjuvant interferon treatment for
melanoma. Hematol Oncol Clin North Am 12:823, 1998 [PMID
9759581]
-
Kirkwood JM, Strawderman MH, Ernstoff MS, et al: Interferon
alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the
Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol 14:7, 1996
[PMID
8558223]
-
Wolchok JD, Livingston PO, Houghton AN: Vaccines and other
adjuvant therapies for melanoma. Hematol Oncol Clin North Am 12:835, 1998
[PMID
9759582]
-
Buzaid A, Ross MI, Balch CM, et al: Critical analysis of
the current American Joint Committee on Cancer staging system for cutaneous
melanoma and proposal of a new staging system. J Clin Oncol 15:1039, 1997
[PMID
9060544]
-
Guerry D 4th, Synnestvedt M, Elder DE, et al: Lessons from
tumor progression: the invasive radial growth phase of melanoma is common,
incapable of metastasis, and indolent. J Invest Dermatol 100:342S, 1993
[PMID
8440920]
-
Elder DE, Van Belle P, Elenitsas R, et al: Neoplastic progression
and prognosis in melanoma. Semin Cutan Med Surg 15:336, 1996 [PMID
9069601]
-
Schuchter L, Schultz DJ, Synnestvedt M, et al: A prognostic
model for predicting 10-year survival in patients with primary melanoma.
Ann Intern Med 125:369, 1996 [PMID
8702087]
-
Schwartz RA: Cutaneous metastatic disease. J Am Acad Dermatol
33:161, 1995
-
Demetrius RW, Randle HW: High-risk nonmelanoma skin cancers.
Dermatol Surg 24:1272, 1998 [PMID
9865191]
-
Myskowski PL, Ahkami R: Advances in Kaposi's sarcoma. Dermatol
Clin 15:177, 1997 [PMID
9001870]
-
Krown SE: Acquired immunodeficiency syndrome-associated Kaposi's
sarcoma: biology and management. Med Clin North Am 81:471, 1997
-
Moore PS, Change Y: Detection of herpesvirus-like DNA sequences
in Kaposi's sarcoma in patients with and without HIV infection. N Engl
J Med 332:1181, 1995 [PMID
7700310]
-
Gao SJ, Kingsley L, Hoover DR, et al: Seroconversion to antibodies
against Kaposi's sarcoma-associated herpesvirus-related latent nuclear
antigen before the development of Kaposi's sarcoma. N Engl J Med 335:233,
1996 [PMID
8657239]
-
Foreman KE, Friborg J Jr, Kong WP, et al: Propagation of
a human herpesvirus from AIDS-associated Kaposi's sarcoma. N Engl J Med
336:163, 1997 [PMID
8988896]
-
Welles L, Saville MW, Lietzau J, et al: Phase II trial with
dose titration of paclitaxel for the therapy of human immunodeficiency
virus-associated Kaposi's sarcoma. J Clin Oncol 16:1112, 1998 [PMID
9508198]
-
Niedt GW, Myskowski PL, Urmacher C, et al: Histologic predictors
of survival in acquired immunodeficiency syndrome-associated Kaposi's sarcoma.
Hum Pathol 23:1419, 1992 [PMID
1468779]
-
Cooper JS, Sacco J, Newall J: The duration of local control
of classic (non-AIDS-associated) Kaposi's sarcoma by radiotherapy. J Am
Acad Dermatol 19:59, 1988 [PMID
3403746]
-
Webster GF: Local therapy for mucocutaneous Kaposi's sarcoma
in patients with acquired immunodeficiency syndrome. Dermatol Surg 21:205,
1995
-
Krown SE, Testa MA, Huang J: AIDS-related Kaposi's sarcoma:
prospective validation of the AIDS Clinical Trials Group staging classification.
J Clin Oncol 15:3085, 1997 [PMID
9294471]
-
Murphy M, Armstrong D, Sepkowitz KA, et al: Regression of
AIDS-related Kaposi's sarcoma following treatment with an HIV-1 protease
inhibitor. AIDS 11:261, 1997 [PMID
9030382]
-
Northfelt DW, Dezube BJ, Thommes JA, et al: Peglated-liposomal
doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment
of AIDS-related Kaposi's sarcoma: results of a randomized phase III clinical
trial. J Clin Oncol 16:2445, 1998 [PMID
9667262]
-
Lorincz AL: Cutaneous T-cell lymphoma (mycosis fungoides).
Lancet 347:871, 1996
-
Diamandidou E, Cohen PR, Kurzrock R: Mycosis fungoides and
Sezary syndrome. Blood 88:2385, 1996 [PMID
8839829]
-
Jackow CM, McHam JB, Friss A, et al: HLA-DR5 and DQB1*03
class II alleles are associated with cutaneous T-cell lymphoma. J Invest
Dermatol 107:373, 1996 [PMID
8751973]
-
Kikuchi A, Nishikawa T, Yamaguchi K: Absence of human T-cell
lymphotropic virus type I in cutaneous T-cell lymphoma. N Engl J Med 336:296,
1997 [PMID
9005322]
-
Sausville EA, Eddy JL, Makuch RW, et al: Histopathologic
staging at initial diagnosis of mycosis fungoides and the Sezary syndrome:
definition of three distinctive prognostic groups. Ann Intern Med 109:372,
1988 [PMID
3408055]
-
Diamandidou E, Colome-Grimmer M, Fayad L, et al: Transformation
of mycosis fungoides/Sezary syndrome: clinical characteristics and prognosis.
Blood 92:1150, 1998 [PMID
9694702]
-
Ramsay DL, Meller JA, Zackheim HS: Topical treatment of early
cutaneous T-cell lymphoma. Hematol Oncol Clin North Am 9:1031, 1995 [PMID
8522483]
-
Kaye FJ, Bunn PA, Steinberg SM, et al: A randomized trial
comparing combination electron-beam radiation and chemotherapy with topical
therapy in the initial treatment of mycosis fungoides. N Engl J Med 321:1784,
1989 [PMID
2594037]
-
Zackheim HS, Kashani-Sabet M, Amin S: Topical corticosteroids
for mycosis fungoides: experience in 79 patients. Arch Dermatol 134:949,
1998 [PMID
9722724]
-
Ramsey DL, Lish KM, Yalowitz CB, et al: Ultraviolet-B phototherapy
for early-stage cutaneous T-cell lymphoma. Arch Dermatol 128:931, 1992
-
Herrmann JJ, Roenigk HH Jr, Hurria A, et al: Treatment of
mycosis fungoides with photochemotherapy (PUVA): long-term follow-up. J
Am Acad Dermatol 33:234, 1995 [PMID
7622650]
-
Jones GW, Hoppe RT, Glatstein E: Electron beam treatment
for cutaneous T-cell lymphoma. Hematol Oncol Clin North Am 9:1057, 1995
[PMID
8522484]
-
Duvic M, Lemak NA, Redman JR, et al: Combined modality therapy
for cutaneous T-cell lymphoma. J Am Acad Dermatol 34:1022, 1996 [PMID
8647968]
-
Wilson LD, Licata AL, Braverman IM, et al: Systemic chemotherapy
and extracorporeal photochemotherapy for T3 and T4 cutaneous T-cell lymphoma
patients who have achieved a complete response to total skin electron beam
therapy. Int J Radiat Oncol Biol Phys 32:987, 1995 [PMID
7607973]
-
Crowley JJ, Nikko A, Varghese A, et al: Mycosis fungoides
in young patients: clinical characteristics and outcome. J Am Acad Dermatol
38:696, 1998 [PMID
9591813]
-
Kim YH, Jensen RA, Watanabe GL, et al: Clinical stage IA
(limited patch and plaque) mycosis fungoides: a long-term outcome analysis.
Arch Dermatol 132:1309, 1996 [PMID
8915308]
-
Kim YH, Chow S, Varghese A, et al: Clinical characteristics
and long-term outcome of patients with generalized patch and/or plaque
(T2) mycosis fungoides. Arch Dermatol 135:26, 1999