Neurología
⭳ Abrir artículo (PDF)567.1 KBEste artículo es idéntico en la Edición 3/2000.
Contenido del artículo
III ENFERMEDAD DEL MUSCULO Y DE LA UNION NEUROMUSCULAR
- Estudio de los pacientes con enfermedad neuromuscular
- Distrofias musculares
- PRINCIPIOS SOBRE LAS PROTEINAS DEL CITOESQUELETO MUSCULAR
- DISTROFINOPATIAS
- Distrofia muscular de Duchenne
- Distrofia muscular de Becker
- Mujeres portadoras y distrofinopatía en mujeres
- Cardiomiopatía dilatada ligada al X
- Síntomas miopáticos leves, inespecíficos
- SARCOGLICANOPATIAS EN LAS DISTROFIAS MUSCULARES DE LAS CINTURAS DE LAS EXTREMIDADES
- MEROSINOPATIAS Y DISTROFIAS MUSCULARES CONGENITAS
- OTRAS DISTROFIAS
- Hipertermia maligna
- Miopatías metabólicas
- PRINCIPIOS GENERALES DE LOS REQUERIMIENTOS DE ENERGIA DEL MUSCULO
- GLUCOGENOSIS
- DEFICIENCIA DE MALTASA ACIDA
- MIOPATIAS POR ALMACENAMIENTO DE LIPIDOS Y TRASTORNOS DEL METABOLISMO DE LOS LIPIDOS QUE AFECTAN AL MUSCULO
- Miopatías mitocondriales y encefalopatías
- DELECIONES ESPORADICAS DEL ADNmt: SINDROME DE KEARNS-SAYRE Y OFTALMOPLEGIA EXTERNA PROGRESIVA CRONICA
- MUTACIONES PUNTIFORMES DEL ADNMT HEREDADAS POR VIA MATERNA
- Trastornos de los canales de iones, parálisis periódica familiar y miotonías no distróficas
- Miopatías tóxicas inducidas por drogas
- PRINCIPIOS GENERALES
- MIOPATIA POR ZIDOVUDINA Y TOXICIDAD DE OTROS ANALOGOS DE NUCLEOSIDOS
- MIOPATIA POR AGENTES REDUCTORES DEL COLESTEROL
- MIOPATIA POR ESTEROIDES Y AGENTES BLOQUEADORES (MIOPATIA DE LA ENFERMEDAD GRAVE)
- INTOXICACION POR VITAMINA E
- MIOPATIA POR CLOROQUINA
- MIOPATIA POR COLCHICINA
- ESTEROIDES
- MIOPATIA ALCOHOLICA
- MIOPATIA INFLAMATORIA INDUCIDA POR DROGAS
- Trastornos de la trasmisión neuromuscular
III ENFERMEDAD DEL MUSCULO Y DE LA UNION
NEUROMUSCULAR
DR. MARINOS C. DALAKAS
Estudio de los pacientes con enfermedad neuromuscular
Las enfermedades del músculo y de la unión neuromuscular constituyen un grupo heterogéneo de padecimientos adquiridos y hereditarios. Los síntomas de presentación son variados e incluyen fatiga, debilidad del músculo esquelético, atrofia, calambres musculares y alteración de los músculos respiratorios, faríngeos, faciales y oculares. En general, los músculos proximales se afectan en forma más selectiva y severa que los distales. Por lo tanto, los pacientes presentan dificultad para subir las escaleras, levantarse de una silla baja, correr, peinarse e incorporarse o volterarse en la cama. La mayoría de las miopatías afectan solo a los músculos esqueléticos, pero pueden alterarse también el músculo liso y el cardiaco. Los pacientes con miopatías no tienen trastornos sensoriales o disfunción autonómica porque la función de los nervios periféricos y el sistema nervioso autónomo está conservada.
El examen clínico debe enfocarse a evaluar los datos musculares y excluir enfermedades que pueden presentarse con síntomas y signos miopáticos como los síndromes de neurona motora, las neuropatías motoras y las enfermedades psicógenas. Se requiere una historia familiar completa y el examen de los familiares para excluir un padecimiento hereditario. Los estudios de laboratorio útiles incluyen (1) estudios para excluir una enfermedad sistémica, factores exógenos, toxinas o virus que puedan inducir miopatía, (2) electromiografía (EMG) para localizar la lesión al músculo o la unión neuromuscular y excluir alteraciones de la neurona motora o de los nervios periféricos, (3) determinación de niveles séricos de enzimas musculares, (4) medición de los autoanticuerpos específicos dirigidos contra antígenos musculares conocidos o posibles, (5) biopsia muscular para determinar la histoquímica enzimática, inmunopatología, microscopía electrónica y medición bioquímica de enzimas o proteínas específicas del músculo, (6) prueba de ejercicio con isquemia para medir la producción de lactato y amoniaco en las miopatías metabólicas, (7) estudios genéticos y (8) estudios de imagen muscular, según el problema específico que se esté investigando.
La mayoría de las miopatías son incapacitantes o catastróficas si no se tratan. Por lo tanto, el diagnóstico debe establecerse con rapidez para iniciar un tratamiento temprano. Para los pacientes con trastornos intratables es indispensable administrar tratamiento de apoyo, rehabilitación y consejo genético.
En esta subsección se describen las miopatías y trastornos de la unión neuromuscular más comunes, con énfasis en el cuadro clínico, patogenia, diagnóstico y tratamiento.
Distrofias musculares
Las distrofias musculares constituyen un grupo heterogéneo de enfermedades congénitas del músculo que se caracterizan por debilidad muscular severa, atrofia, elevación de los niveles de enzimas musculares en suero y cambios destructivos en la citoarquitectura de las fibras musculares. La clasificación tradicional de las distrofias musculares en de Duchene, de Becker, de cintura escapular y pélvica y congénita ha cambiado porque se han identificado varios defectos genéticos en las proteínas musculares que parecen ser responsables de algunas de estas enfermedades y porque se ha demostrado ausencia o deficiencia de proteínas musculares específicas en ciertas distrofias.
Los estudios moleculares, bioquímicos e inmunocitoquímicos1,2 han identificado que el complejo de proteína asociada a la distrofina (PAD) constituye la proteína transmembrana clave que une a la proteína distrofina del citoesqueleto con la proteína de la matriz extracelular laminina-2 [ver figura 1]. Las deficiencias en ciertos componentes de este sistema se asocian con síndromes clínicos específicos. En la actualidad las distrofias musculares se clasifican de acuerdo con los genes involucrados y la proteína defectuosa, que puede ser un componente del citoesqueleto, la membrana o la matriz extracelular.
PRINCIPIOS SOBRE LAS PROTEINAS DEL CITOESQUELETO MUSCULAR
Las proteínas musculares más importantes implicadas en las distrofias musculares son la distrofina, el complejo distroglucano, que se asocia con la proteína de la lámina basal, la merosina y el complejo sarcoglucano [ver figura 1]. La distrofina, una proteína del citoesqueleto de 427 kd con forma de bastón, constituye el cinco porciento de todas las proteínas del citoesqueleto del sarcolema y sirve para anclar a la actina F (la forma filamentosa de la actina) a la membrana plasmática (sarcolema) del músculo.1 La distrofina parece reforzar y estabilizar la membrana plasmática durante la tensión de la contracción muscular al mantener un enlace mecánico entre el citoesqueleto y la matriz extracelular. La deficiencia o ausencia de distrofina se asocia con diversas distrofinopatías, de las que el prototipo es la distrofia muscular de Duchenne.3-7
El complejo distroglucano consiste en alfa-distroglucanos, proteínas de la matriz celular, siendo la más relevante desde el punto de vista clínico la alfa2-laminina (o merosina, la proteína involucrada en la distrofia muscular congénita), y el beta-distroglucano, que fija alfa distroglucano y distrofina. No se ha identificado a ningún paciente con defectos primarios en los distroglucanos. El complejo sarcoglucano está formado por cuatro proteínas transmembrana, incluyendo la 50 kd adhalina (alfa-sarcoglucano), la glucoproteína asociada a distrofina 43 kd (beta-sarcoglucano), una proteína de 35 kg (gama-sarcoglucano) y una proteína de 95 kg (delta-sarcoglucano). Los componentes transmembrana del complejo sarcoglucano son específicos del músculo esquelético y cardiaco y se afectan en forma selectiva en pacientes con distrofias musculares recesivas de las cinturas de las extremidades, que se conocen como sarcoglucanopatías. Las distrofias musculares se dividen en distrofinopatías, sarcoglucanopatías y merosinopatías2, de acuerdo con la proteína faltante o deficiente.
Debido a que se han identificado los genes para varias formas de distrofias, en la actualidad se emplea una clasificación genética: (1) distrofias musculares recesivas ligadas al cromosoma X (Duchenne y Becker), (2) distrofias musculares congénitas con deficiencia de merosina, (3) una distrofia muscular de las cinturas de las extremidades (DMCE1) , autosómico dominante y rara, con mutación localizada en el cromosoma 5, y (4) todas las distrofias musculares de las cinturas de las extremidades recesivas (DMCE2). Las DMCE2 se subdividen en la DMCE2A, causada por deficiencia de calpaína, la DMCEB, que no se ha caracterizado, la DMCE2C (deficiencia de gama-sarcoglucano), la DMCE2D (deficiencia de alfa-sarcoglucano), la DMCE2E (deficiencia de beta-sarcoglucano) y la DMCE2F (deficiencia de delta-sarcoglucano).8
DISTROFINOPATIAS
Distrofia muscular de Duchenne
La distrofia muscular de Duchenne (DMD), un padecimiento recesivo ligado al cromosoma X, es causado por mutaciones en el gen de la distrofina en el brazo corto del cromosoma X en la posición Xp21. El gen de la distrofina ocupa más de 2,000 kb de ADN. En el 65 a 70 porciento de los casos la DMD es resultado de deleciones largas (varios kilobases) en el gen de la distrofina, con una falta consecuente de distrofina muscular. Las deleciones, que se detectan en el ADN extraído de linfocitos de sangre periférica, interrumpen el marco de lectura de las tripletas de codones de ARN mensajero (ARNm), lo que produce las formas severas de DMD. Las duplicaciones parciales del gen, detectadas por análisis de Southern blot, causan el seis porciento de las mutaciones de la distrofina, y las mutaciones pequeñas (v.gr., mutaciones puntiformes y errores de división) que producen una proteína distrofina trucada, provocan hasta el 30 porciento de los casos de DMD.5 También son frecuentes las mutaciones espontáneas.3-5 La ausencia de distrofina debilita e interrumpe la membrana de sarcolema, permitiendo la entrada de calcio, lo que ocasiona necrosis de la fibra muscular.
La DMD ocurre en uno de cada 3,000 varones nacidos. Los niños afectados inician sus síntomas después de que comienzan a caminar, por lo general entre los dos y tres años de edad. La marcha vacilante, la postura lordótica, la hipertrofia de las pantorrillas, las contracturas articulares y el caminar sobre los dedos de los pies son las primeras manifestaciones. Estas son seguidas de debilidad y atrofia muscular progresivas, lo que dificulta el levantarse del piso o una silla baja, subir escaleras y elevar los brazos. El retraso en el vaciamiento gástrico puede causar episodios súbitos de vómito y dolor abdominal.5 La enfermedad es lentamente progresiva. Para los 12 años de edad los niños afectados están confinados a una silla de ruedas, y alrededor de los 25 fallecen por complicaciones de insuficiencia respiratoria. Aunque la DMD es un padecimiento del músculo esquelético, el músculo cardiaco se afecta con frecuencia y en las etapas tardías ocurren insuficiencia cardiaca congestiva y arritmias. También se presenta afección no progresiva del sistema nervioso central, que se manifiesta por irritabilidad, hiperactividad o disfunción cognoscitiva.
Los estudios de laboratorio de rutina de los pacientes con DMD muestran al inicio elevación de la creatina cinasa (CK) sérica hasta de 20,000 unidades, con una disminución constante al depletarse la masa muscular. La biopsia de músculo revela una miopatía destructiva severa, con gran cantidad de células T CD8+ y macrófagos (que invaden las fibras musculares), aumento en la cantidad de tejido conectivo y fibras musculares hipercontraídas.7 Para fines diagnósticos, la ausencia de distrofina (o menos del tres porciento de la concentración normal) se demuestra por inmunicitoquímica usando anticuerpos antidistrofina en los cortes de la biopsia muscular y por inmunoblots preparados de las muestras de biopsia.
Tratamiento La presencia de inflamación endomisial ha provocado que se usen esteroides para tratar a la DMD. En un estudio controldo, el uso de esteroides causó mejoría temporal y leve.9 Sin embargo, su uso a largo plazo está restringido por los severos efectos colaterales, en especial obesidad, fracturas, osteoporosis, diabetes e hipertensión. El deflazacort, un esteroide con menos efectos mineralocorticoides, parece un poco más seguro. En una forma modificada de terapia génica, se inyectaron mioblastos humanos que contenían distrofina normal en músculos de pacientes con DMD, pero el procedimiento no cambió la función muscular de los receptores.10 Es posible que las futuras terapias génicas sean eficaces si se encuentran vectores apropiados para insertar en forma eficaz el gen en el músculo. Hasta entonces, el tratamiento es totalmente sintomático, con énfasis en proporcionar terapia respiratoria y física sistemática para el paciente y apoyo psicológico tanto para el paciente como para la familia. El consejo genético es muy importante.
Distrofia muscular de Becker
La distrofia muscular de Becker (DMB) y la DMD son padecimientos alélicos, pero la primera inicia en forma más tardía y progresa con más lentitud. La edad de inicio es muy variable. Los casos pueden detectarse tan pronto como a los tres años o tan tarde como a los 70 años de edad, con promedio de edad de 12 años.5 El espectro de la expresión fenotípica de la DMB es muy amplio. Las formas leves se manifiestan solo por calambres musculares, intolerancia al ejercicio, mioglobinuria, elevación asintomática de los niveles de CK en suero, debilidad muscular leve o miopatía del cuadríceps.5,11 El dolor en la pantorrilla al ejercicio es un síntoma inicial frecuente, lo mismo que las hipertrofias de las pantorrillas. La mayoría de los pacientes pierden la capacidad de deambular a los 40 años (con rango de 10 a 70 años de edad). La edad de muerte tembién varía, de 23 a 89 años (en promedio 42 años). Los pacientes presentan debilidad muscular proximal y niveles de CK hasta de 20 veces lo normal. Los datos de la biopsia muscular son semejantes a los observados en los pacientes con DMD, pero no son tan severos. En los pacientes menores de ocho años de edad la presentación de la DMB suele ser indistinguible de la DMD. Las manifestaciones cardiacas son comunes, su severidad no se relaciona con la miopatía y la cardiomiopatía puede ser severa.
Alrededor del 65 porciento de los pacientes con DMB tienen deleciones dentro del marco de lectura del gen de la distrofina y la proteína producida suele estar truncada y ser solo semifuncional.5,11 Los casos de duplicación del gen de la distrofina han ocasionado una distrofina más larga y en forma de bastón. La inmunocitoquímica del músculo usando anticuerpos antidistrofina demuestra tinción atenuada del sarcolema (no ausencia, como en la DMD) y revela fragmentación de la membrana en el área inmunoteñida. Los inmunoblots detectan una cantidad disminuida de una distrofina menor o mayor de lo normal. No existe un tratamiento eficaz para la DMB.
Mujeres portadoras y distrofinopatía en mujeres
La historia cuidadosa y el examen clínico de las mujeres portadoras asintomáticas puede revelar debilidad muscular leve, calambres, hipertrofia aislada de las pantorrillas y fatiga, con elevación de los niveles de CK. La biopsia de músculo revela fibras negativas para distrofina. Cuando se amplifican los exones específicos de deleción dentro del gen de la distrofina en los heterocigotos por medio de la reacción en cadena de la polimerasa, las detecciones se detectan como una reducción del 50 porciento en la intensidad de la banda de ADN amplificado, comparado con la banda para exones nativos.5 Este método detecta alrededor del 98 porciento de las deleciones. Sin embargo, el mosaicismo gonadal materno es responsable de otras mutaciones de DMD apenas detectadas en hasta el 20 porciento de los casos de diagnóstico reciente de DMD; esto significa que las mutaciones se encuentran solo en los oocitos.11 Las mujeres con estas mutaciones pueden producir varones afectados o mujeres portadoras, por lo que las mujeres e hijas de las madres de pacientes con DMD y DMB deben estudiarse en forma independiente para detectar a las portadoras.11 La manifestación de la DMD en las mujeres heterocigotas ocurre cuando el cromosoma X paterno normal que porta el gen de la distrofina se inactiva en una gran proporción de células embriónicas (hipótesis de Lyon). La enfermedad en estas mujeres puede ser tan severa como en los hombres.
Cardiomiopatía dilatada ligada al X
La cardiomiopatía dilatada ligada al X (CDLX) se debe a la deficiencia de distrofina en el músculo cardiaco, pero no en el esquelético. Se presenta como un trastorno cardiaco progresivo, ocurriendo insuficiencia cardiaca congestiva en la segunda o tercera década de la vida. Los portadores femeninos con manifestaciones tienen una cardiomiopatía de inicio lento que ocurre alrededor de la quinta década de la vida. Se ha propuesto que la causa de la enfermedad es una deleción cerca del exón 1 del gen de la distrofina, que afecta la expresión o función de la misma en el músculo cardiaco.5,11
Síntomas miopáticos leves, inespecíficos
Las distrofinopatías pueden presentarse como debilidad miopática inespecífica leve, aumento en la concentración de CK, intolerancia al ejercicio, calambres musculares, hipertrofia aislada de las pantorrillas o debilidad aislada del cuadríceps.
SARCOGLICANOPATIAS EN LAS DISTROFIAS MUSCULARES DE LAS CINTURAS DE LAS EXTREMIDADES
Se demostró por primera vez deficiencia de alfa-sarcoglucano en una distrofia muscular autosómica recesiva severa de la infancia (DMARSI) en familias del norte de Africa.1,5,11 La presentación de la DMARSI es semejante a la forma severa de DMD pero afecta a varones y mujeres. En la DMARSI los tres componentes del complejo sarcoglucano están ausentes, pero el complejo distroglucano es normal, lo que sugiere una verdadera sarcoglucanopatía.1,12 Se han mapeado mutaciones en los genes para el alfa, beta, gama y delta-sarcoglucano en los cromosomas 17q12, 4, 13q12 y 5q33, respectivamente.8,12 Se han identificado mutaciones en los genes de sarcoglucanos en varias familias europeas y americanas con distrofias musculares de las cinturas de las extremidades (DMCE) autosómico recesivas que incluyen deleciones sin sentido, en contra sentido y de marco de lectura y que ocasionan fenotipos de DMCE de severidad clínica variable. Estas mutaciones causan un ensamble inapropiado de los sarcoglucanos con los otros complejos proteicos de distroglucanos, interrumpiendo el enlace entre el sarcolema y la matriz extracelular.
Los hombres y las mujeres prsentan debilidad muscular proximal leve a severa (en especial en las piernas), elevación de la CK a alrededor de 3,000 unidades, cambios distróficos en el músculo y con frecuencia hipertrofia de pantorrillas. La edad de inicio es variable. La inmunocitoquímica y el análisis de inmunoblot de las muestras de biopsia muscular para la distrofina son normales, pero el sarcoglucano está ausente o muy deficiente. Ocurre deficiencia en las proteínas sarcoglucanos (fácilmente detectable usando anticuerpos contra alfa-sarcoglucano) en el 20 porciento de los pacientes con DMCE que tienen distrofina normal. Se han reportado mutaciones en los genes respectivos en hasta el 60 porciento de los pacientes.
MEROSINOPATIAS Y DISTROFIAS MUSCULARES CONGENITAS
Las distrofias musculares congénitas (DMC) son un grupo heterogéneo de trastornos determinados genéticamente. Se caracterizan por cambios distróficos en el músculo, elevación leve a importante de la CK, grados variables de debilidad muscular, contracturas articulares que afectan los codos, caderas, rodillas y tobillos (conocidas como artrogrifosis cuando existen desde el nacimiento) y progresión que comienza al nacer o en la infancia temprana. Tres formas de DMC se asocian también con alteraciones estructurales del cerebro, malformaciones oculares y deterioro intelectual. Estas incluyen el tipo Fukuyama (observado en Japón), el síndrome de Walker-Warburg (distrofia muscular displasia cerebro-ocular) y la enfermedad músculo-ojo-cerebro del tipo finlandés.13 Sin embargo, en la forma clásica de DMC no existe malformación cerebral o retraso mental. La enfermedad puede ocurrir también en adultos jóvenes.
Más del 40 porciento de los niños con la forma clásica de DMC tienen deficiencia de merosina en la lámina basal del músculo esquelético.13 La enfermedad se asocia con leucoencefalopatía definida por IRM (sin signos evidentes de deterioro intelectual), hipotonía neonatal, niveles elevados de CK en suero, retraso en el desarrollo motor, neuropatía axonal y debilidad de los músculos respiratorios. La DMC atribuida a deficiencia de merosina se asocia con el cromosoma 6q22. En los pacientes con merosina negativa, el complejo DAG está alterado y en consecuencia se destruye la membrana muscular.
OTRAS DISTROFIAS
Distrofia muscular de Emery-Dreifuss
La distrofia muscular de Emery-Dreifuss (DMED) es un padecimiento recesivo ligado al cromosoma X que se ha mapeado en Xq28.14 Se han identificado mutaciones en esta región que han demostrado codificar una proteína llamada emerina, una proteína de la membrana nuclear.15 La DMED se presenta con afección muscular humeroperoneal (con consumo y debilidad prominente del bíceps, tríceps, tibial anterior y peroneos), y progresa en forma lenta con un patrón escapulopelviperoneo (con afección de los músculos pectoral y de la pelvis). La DMED se caracteriza por el desarrollo temprano de contracciones en los codos, flexores, tendón de Aquiles, cuello y columna, que puede ocurrir antes de que exista debilidad significativa y afección cardiaca, que se manifiesta por defectos en la conducción con bradicardia y prolongación del intervalo PR. La DMED comienza en las primeras dos décadas de la vida y su detección pronta con colocación de marcapaso puede prevenir la muerte súbita o los ataques de síncope. Las biopsias musculares revelan datos miopáticos inespecíficos. La CK puede estar ligeramente elevada. No existe un tratamiento específico, pero la terapia física puede retrasar el desarrollo de las contracturas. En la actualidad existen pruebas genéticas para identificar a los portadores, que pueden tener trastornos de la conducción cardiaca. Una prueba diagnóstica útil emplea emerina, y es posible demostrar la ausencia de esta proteína por medio de inmunocitoquímica y Western blot en muestras de biopsias musculares de los pacientes y sus familiares.
Distrofia muscular facioescapulohumeral y escapuloperonea
La enfermedad suele comenzar durante la segunda década de la vida y se presenta como debilidad de los músculos de la cara (en especial del orbicular de los ojos). Se conserva la función de los músculos extraoculares, maseteros y faríngeos.16 La debilidad temprana de los músculos escapulares produce una apariencia alada de las escápulas con apariencia de que los hombros se van hacia adelante. Existe siempre debilidad de los músculos tibiales anteriores, con pie caido. La debilidad progresa en forma lenta y existen largos periodos de estabilidad. La progresión ocurre en forma descendente: la afección de la cintura escapular es seguida de afección del bíceps, tríceps y músculos de la cintura pélvica.
La distrofia muscular fascioescapulohumeral (DFEH) se asocia al cromosoma 4q. Es una enfermedad autosómico dominante con penetrancia completa. Sin embargo, en algunos miembros de la familia solo ocurre afección facial mínima. Los niveles de CK en suero están ligeramente elevados y el músculo cardiaco no se afecta. Los datos de la biopsia son variables. Una gran mayoría de los pacientes tiene alteraciones en los capilares de la retina, desprendimiento de retina y deterioro de la audición.16
Distrofia muscular oculofaringea
La distrofia muscular oculofaríngea (DMOF) es un padecimiento autosómico dominante poco frecuente que se manifiesta entre la cuarta y la sexta décadas de la vida. Se caracteriza por ptosis y disfagia, pudiendo ser ambas severas. Puede ocurrir debilidad leve de los músculos distales. La mutación responsable de la DMOF se asocia en forma estrecha al locus del gen de la cadena pesada de la miosina en el cromosoma 14q11.2.17
Distrofia miotónica
La distrofia miotónica (DM) es la distrofia muscular del adulto más común. Tiene una incidencia de uno por 8,000 personas y una prevalencia de cinco por 100,000 habitantes.18 La miopatía tiene una distribución característica: (1) ptosis de los párpados con afección de los músculos extraoculares, (2) atrofia de los maseteros y músculos temporales, lo que ocasiona una configuración facial angosta típica, (3) atrofia del músculo esternocleidomastoideo conservación relativa de los músculos poseriores del cuello y de la cintura escapular (signo clínico que diferencía a la DM de la DFEH), (4) atrofia de grupos musculares distales, con ligera afección proximal en las fases tempranas de la enfermedad, y (5) afección de los músculos del paladar y faríngeos, que puede producir disartria y disfagia.
La miotonía, definida como una relajación lenta después de una contracción muscular normal, es un signo clínico importante. Para despertar la miotonía durante el examen clínico, el paciente da un apretón de manos firme y después se le pide que suelte con rapidez. Será evidente su incapacidad para hacerlo. La percusión de la eminencia tenar o del extensor de los dedos también muestra la relajación lenta característica de la miotonía. Las características sistémicas incluyen defectos de la conducción cardiaca, disfunción mental leve (con frecuencia con comportamiento y expresiones tontas o inapropiadas), atrofia testicular, calviece frontal, cataratas, afección del tubo digestivo (con retraso en el movimiento y vaciamiento), hipersomnia y menor respuesta a la hipoxia, que causa menor concentración y apatía. Los niños de las madres afectadas pueden tener menos movimientos fetales y síntomas tempranos de hipotonía severa, dificultad para alimentarse, debilidad facial bilateral e insuficiencia respiratoria (distrofia miotónica congénita). La DM debe distinguirse de la miotonía congénita, que tiene patrones de herencia dominantes y recesivos, se manifiesta por miotonía e hipertrofia muscular, pero no se asocia con debilidad muscular, atrofia o síntomas sistémicos.
El diagnóstico clínico de la DM se confirma por la EMG, que muestra descargas miotónicas. En los casos difíciles el examen con lámpara de hendidura puede mostrar la formación temprana de cataratas. La expresión clínica de la DM es variable y las personas afectadas pueden no ser diagnosticadas sino hasta que tienen hijos. La edad de inicio es cada vez más temprana en las generaciones sucesivas (anticipación). No es poco frecuente ver familias en las que la abuela solo tuvo cataratas tempranas, y la hija, que no sabía que estaba afectada, tuvo un hijo con distrofia miotónica congénita severa. La DM se debe a mutaciones en la región 3' no traducida del gen de la proteina cinasa de miotonina en el cromosoma 19.19,20 En las personas con afección leve, una región de repetición polimórfica CTG (citosina, timina, guanina) en la proteína cinasa se expande de 50 a 80 veces. En las personas con afección severa pueden existir más de 2,000 repeticiones. El tamaño de la expansión de la CTG aumenta por generaciones, lo que explica la anticipación.19,20 La medición de la longitud de la expansión de la CTG es una prueba genética que puede usarse para confirmar la presencia de DM en familiares, para diagnóstico prenatal o para consejo genético eficaz de personas asintomáticas con riesgo de DM.
El tratamiento es sintomático. Son indispensables el apoyo emocional y la educación sobre las precauciones para evitar caídas y lesiones. Los medicamentos como la quinidina, la procainamida, la fenitoína y los beta bloqueadores pueden ayudar a la miotonía, pero no a la debilidad.
Hipertermia maligna
La hipertermia maligna (HM) ocurre durante la anestesia general. Se caracteriza por un incremento rápido del metabolismo aeróbico y anaeróbico, durante el que la temperatura corporal puede ser superior a 43 ¡C (109.4 ¡F). Además, las concentraciones de bióxido de carbono y lactato aumentan (la concentración de bióxido de carbono arterial [PaCO2] puede ser superior a 100 mm Hg), y el pH de la sangre puede ser menor a 7. La HM se caracteriza por taquicardia, rigidez (causada por contractura muscular que puede progresar a rigidez o muerte), liberación excesiva de mioglobina del músculo y mioglobinuria, mayor permeabilidad muscular (con aumento en la concentración sérica de K+, Ca2+ y Na+), y edema muscular.21 El trismus o espasmo muscular de los maseteros que ocurre en un paciente durante la inducción de la anestesia puede indicar HM.21 La HM debe distinguirse de (1) la rabdomiolisis sola asociada con la anestesia, (2) las reacciones tóxicas a medicamentos, (3) la porfiria, (4) la tormenta tiroidea precipitada por cirugía y anestesia, y (5) el síndrome neuroléptico maligno (SNM) precipitado por fármacos psicoactivos (v.gr., haloperidol y fenotiacinas) que bloquean las vías dopaminérgicas centrales.
Entre los desencadenantes de la HM se incluyen los anestésicos volátiles potentes, como halotano, enfluorano, desfluorano, ciclopropano, éter y succinilcolina.21 Aunque ocurre HM en algunos pacientes sin enfermedad muscular conocida, los pacientes en riesgo incluyen los que tienen alteraciones musculoesqueléticas congénitas múltiples, dislocación congénita de cadera aislada, o enfermedad nculear, y algunos de los que tienen distrofia musular de Duchenne o de Becker.
La HM se hereda en forma autosómica dominante; en muchos pedigrees el locus se encuentra en el cromosoma 19q13 en el sitio del receptor de rianodine (RyR).21,22 La HM parece estar precipitada por la incapacidad para controlar las concentraciones de calcio dentro de las fibras musculares por un retículo sarcoplásmico (RS) disfuncionante y mutaciones en el gen RyR. El RyR es el canal de liberación de calcio del RS y une la brecha que existe entre el RS y el túbulo transverso. Las mutaciones en el receptor afectan la comunicación entre el RS y el túbulo transverso de modo que ocurre liberación acelerada del calcio desde el RS cuando tiene lugar la despolarización del túbulo transverso.
La prueba in vitro de contracción con cafeína-halotano, que se realiza en biopsia de músculo, se emplea para investigar la posibilidad de HM en los pacientes.23 La sensibilidad y específidad de esta prueba puede ser tan alta como 97 y 80 porciento, respectivamente.21,23
El dantroleno es el tratamiento eficaz para la HM. Este fármaco disminuye la liberación de calcio del RS sin alterar la recaptura de calcio. El uso de dantroleno ha disminuido la mortalidad de esta condición a siete porciento.21 El episodio agudo se la HM se trata en forma sintomática. Debe administrarse dantroleno intravenoso (2 a 10 mg/kg cada cinco minutos) al inicio del episodio, cuando aún existe una perfusión muscular adecuada. Para los pacientes que se sabe son susceptibles a la HM puede administrarse dantroleno en dosis de 2 mg/kg 10 a 15 minutos antes de la anestesia. La mejor manera de evitar la HM en personas susceptibles es empleando agentes anestésicos seguros (v.gr., oxido nitroso y tiopental) y relajantes musculares no despolarizantes.
Miopatías metabólicas
PRINCIPIOS GENERALES DE LOS REQUERIMIENTOS DE ENERGIA DEL MUSCULO
En el reposo la energía muscular deriva principalmente de la oxidación de los ácidos grasos libres (AGL). Durante el ejercicio aeróbico de gran intensidad el glucógeno es la principal fuente de combustible para la fosforilación oxidativa. Sin embargo, las reservas de glucógeno muscular se depletan después de 90 minutos de ejercicio,24 y si éste se prolonga, aumenta la utilización de ácidos grasos libres y glucosa sanguínea. Debido a que la disponibilidad de AAG del tejido adiposo es casi ilimitada, una persona sana normal puede realizar ejercicio moderado por muchas horas. En los pacientes con miopatías metabólicas los síntomas se vuelven evidentes durante las actividades que requieren mayor demanda metabólica, como el ejercicio. Las miopatías metabólicas son causadas por defectos en la ruptura del glucógeno o la utilización de AGL. Las glucogenosis se deben a defectos en el metabolismo y utilización del glucógeno o de la glucosa por el músculo. Se clasifican según la secuencia del defecto enzimático a lo largo de la vía glucogenolítica o glucolítica [ver figura 2]. En esta sección solo se analizarán los trastornos más comunes.
GLUCOGENOSIS
Deficiencia de fosforilasa muscular
La deficiencia de fosforilasa muscular (también conocida como enfermedad de McArdle) es el prototipo de las glucogenosis: la ruptura del glucógeno está inhibida, lo que causa reducción de piruvato y menor cantidad de energía. Este padecimiento autosómico recesivo se presenta como intolerancia al ejercicio y mioglobinuria en pacientes mayores de 15 años. Si los pacientes descansan un poco después de presentar las mialgias y rigidez inducidas por el ejercicio, pueden reanudar su actividad con mejor resistencia (fenómeno de segundo aire), debido a mayor movilización y utilización de los AGL y la glucosa. En etapas más tardías puede desarrollarse debilidad muscular permanente. La concentración de CK en reposo suele estar aumentada. La prueba de ejercicio con isquemia en el antebrazo no muestra aumento del lactato venoso. En la biopsia muscular se encuentra ausencia de fosforilasa, presencia de vacuolas de subsrcolema y mayor acúmulo de glucógeno. El defecto es causado por mutaciones en la isoforma muscular de la fosforilasa en el cromosoma 11q13. No existe tratamiento disponible pero el entrenamiento con ejercicio aeróbico y una dieta alta en proteínas pueden ser útiles.
Deficiencia de fosfofructocinasa
La fosfofructocinasa (FFC) es una enzima con tres subunidades estructurales genéticamente diferentes: M, expresada en el músculo, eritrocitos, corazón y cerebro, L, expresada en el hígado y los eritrocitos, y P, que se expresa en las plaquetas. La deficiencia de FFC es un padecimiento autosómico recesivo. Las mutaciones en la subunidad M, localizada en el cromosoma 1, causan síntomas miopáticos y hemólisis crónica, además de aumento en la concentración de bilirrubina sérica y en la cuenta de leucocitos. Debido a que la deficiencia de FFC es un defecto glucolítico, las consecuencias funcionales de la deficiencia de FFC son semejantes a las observadas en la enfermedad de McArdle.24 Debe sospecharse deficiencia de FFC en los pacientes con intolerancia al ejercicio, náusea y mioglobinuria. Se desarrolla debilidad muscular permanente en etapas tardías de la vida. La historia de hemólisis leve, compensada y crónica, una cuenta alta de reticulocitos, el aumento en la bilirrubina y la hiperuricemia, también indican deficiencia de FFC, en especial en ciertos grupos étnicos, como los japoneses y los judíos Ashkenazi.
DEFICIENCIA DE MALTASA ACIDA
La deficiencia de maltasa ácida (DMA) es una enfermedad por almacenamiento de glucógeno autosómica recesiva causada por deficiencia de la alfa-glucosidasa, una enzima condificada en un gen localizado en el cromosoma 17q23.25 Las mutaciones o deleciones pequeñas que causan una ruptura anormal afectan la expresión de la alfa-glucosidasa. Existen tres formas clínicas de DMA: infantil, de la infancia y del adulto. La forma infantil (enfermedad de Pompe) se presenta dentro de los primeros meses de vida como hipotonía, debilidad y crecimiento del corazón, lengua e hígado, ocurriendo cambios respiratorios y cardiovasculares que causan la muerte antes de los dos años. La forma de la infancia se manifiesta por miopatía, con retraso de las habilidades motoras, debilidad muscular proximal, afección de los músculos respiratorios y crecimiento de las pantorrillas.25 La enfermedad causa la muerte para la segunda década de la vida.
La forma adulta de la DMA se presenta en personas mayores de 20 años como debilidad muscular proximal que recuerda a la polimiositis o a la distrofia de las cinturas de las extremidades. La debilidad de los músculos respiratorios puede ser el síntoma de presentación en una tercera parte de los adultos con DMA. El acúmulo de glucógeno ocurre principalmente en el músculo. Sin embargo, la enzima es deficiente en el músculo, hígado, corazón y fibroblastos cultivados. El paciente tiene elevación de la CK sérica, la EMG muestra descargas miotónicas prominentes (sin miotonía clínica), en especial en los músculos paraespinales, y la biopsia muscular muestra múltiples vacuolas con alta concentración de glucógeno que reaccionan en forma intensa con la fosfatasa ácida, lo que indica mayor actividad lisosomal. Debido a que la utilización de glucógeno y glucosa no se alteran, la DMA causa debilidad estable, pero no intolerancia al ejercicio o mioglobinuria. La fibra muscular sufre un proceso autofágico por alteraciones en los lisosomas.25 El diagnóstico se confirma por ensayo bioquímico del nivel enzimático en el músculo, cultivo de fibroblastos, linfocitos u orina. En algunas familias es posible realizar análisis de genética molecular. No existe un tratamiento específico.
MIOPATIAS POR ALMACENAMIENTO DE LIPIDOS Y TRASTORNOS DEL METABOLISMO DE LOS LIPIDOS QUE AFECTAN AL MUSCULO
Los lípidos como fuente de energía para el músculo
Durante el ejercicio rápido y extenuante la energía para el músculo es aportada por vía anaeróbica por la glucosa derivada de la degradación del glucógeno. Poco después de que comience el ejercicio aumenta la circulación, el aporte de glucosa, AGL y oxígeno hacia el músculo. Durante el ejercicio sostenido los ácidos grados de cadena larga (AGCL) son la principal fuente de energía, derivan de los alimentos o, durante el ayuno, del tejido adiposo. Los AGCL requieren ser transportados primero hacia la mitocondria. La transferencia de AGCL a través de la membrana mitocondrial interna requiere de L-carnitina y dos enzimas, las carnitina palmitoiltransferasas (CPT I y CPT II), que se localizan en las membranas mitocondriales externa e interna, respectivamente. Dentro de la mitocondria la §-oxidación se facilita primero por las deshidrogenasas de acil coenzima A (acil-CoA) y después por la transferencia de electrones a través de flavoproteínas hacia las proteínas de la cadena respiratoria.26,27 Las miopatías por almacenamiento de lípidos son causadas por menor oxidación de los ácidos grasos por las mitocondrias, que se debe a defectos en (1) la carnitina y la CPT, que alteran el transporte de ácidos grasos a través de la membrana mitocondrial, (2) las enzimas asociadas con la §-oxidación y (3) las proteínas de la cadena respiratoria y las flavoproteínas que transfieren electrones.26,27
Deficiencia de carnitina primaria y secundaria
La carnitina deriva principalmente de la dieta, pero el 25 porciento se sintetiza en el hígado a partir de lisina y metionina.26 La carnitina es crucial para la oxidación de los AGCL. La consecuencia de la deficiencia de carnitina es la disfunción del hígado, corazón y tejidos musculares, que son muy dependientes de la oxidación de AGCL. La deficiencia primaria de carnitina (DPC) es causada por ausencia de receptores de carnitina de alta afinidad funcionales, lo que trae como consecuencia un transporte deficiente de la carnitina a través de las membranas celulares. Se considera que los defectos en el transporte de lípidos a la mitocondria asociados con DPC causan un desplazamiento hacia la vía glucolítica. Esto causa acúmulo de acil-CoA en la mitocondria, que esterifica a la canitina libre para formar acilcarnitinas. Estas acil-CoA, son fácilmente liberadas de la mitocondria y excretadas en la orina.26,28
Los pacientes con DPC sufren cardiomiopatía progresiva, episodios de hipoglucemia hipocetósica (por disfunción hepática) y debilidad miopática proximal. Los lípidos se acumulan y forman gotas pequeñas.
Sin embargo, las causas más comunes de deficiencia de carnitina son secundariasy se deben a (1) §-oxidación defectuosa, que se asocia con acidurias orgánicas, (2) disfunción mitocondrial y defectos en las proteínas de la cadena respiratoria, (3) enfermedad renal, como el síndrome de Fanconi, la cistinosis nefropática o la hemodiálisis crónica, y (4) tratamiento con fármacos, en especial zidovudina (AZT) y valproato.26,28
Los suplementos con carnitina producen resultados variables.
Deficiencia de carnitina palmitoiltransferasa
En los lactantes la deficiencia de CPT I se manifiesta como síndrome de Reye, con encefalopatía hepática, hipoglucemia hipocetótica e hiperamonemia. En los adultos, el síndrome de deficiencia de carnitina suele causar deficiencia de CPT II. El gen para la CPT II se localiza en el cromosoma 11p11-p13, y se han descrito mutaciones sin sentido.27 La deficiencia de CPT II representan la causa más común de mioglobinuria en adultos jóvenes. Los pacientes presentan ataques de rigidez muscular, calambres, mialgias y mioglobinuria horas después de realizar ejercicio prolongado o constante, en especial después del ayuno o cuando la energía del músculo depende de la utilización de los AGCL y no de la utilización de glucógeno o glucosa. Los pacientes con deficiencia de CPT-II no tienen menor tolerancia al ejercicio, fenómeno de segundo aire o signos de alarma como mialgias que eviten que continúen haciendo ejercicio. Entre los ataques la fuerza muscular y la CK sérica son normales. El diagnóstico se establece midiendo la actividad de la CPT II en el músculo.
No es claro el motivo por el que la deficiencia de CPT causa ataques intermitentes de mioglobinuria y por qué no se acumulan lípidos en el músculo. No existe tratamiento para evitar los ataques mioglobinúricos. Sin embargo, se recomiendan una dieta alta en carbohidratos, baja en lípidos, comidas frecuentes e ingesta extra de carbohidratos antes y durante el ejercicio sostenido.
Miopatías mitocondriales y encefalopatías
Estas enfermedades constituyen un grupo diverso de trastornos que afectan no solo al músculo y al sistema nervioso, sino también otros órganos. La principal función de la motocondria es generar energía para la célula al producir trifosfato de adenosina (ATP) por medio de la fosforilación xoidativa (FO). La FO se basa en cinco complejos enzimáticos, que incluyen coenzimas y transición de componentes metálicos (hierro, cobre), localizados en la membrana mitocondrial interna. Estos complejos (designados como I, II, III, IV [citocromo oxidasa] y V) colectan y transfieren en forma secuencial electrones derivados del catabolismo de grasas, proteínas y carbohidratos, al O2. El acoplamiento de la oxidación y fosforilación por un gradiente de protones a través de la membrana mitocondrial interna permite la fosforilación del difosfato de adenosina (ADP) para producir ATP.
La mitocondria contiene su propio ADN extracromosómico (ADN mt), que es diferente al ADN nuclear. Es una molécula circular de doble cadena que codifica 24 ARN estructurales, 2 ARN ribosomales (ARNr), 22 ARN de transferencia (ARNt) y 13 ARNm. Los ARNm codifican varios polipéptidos de la cadena respiratoria. El resto de las subunidades de la FO y otras proteínas mitocondriales son codificadas por genes nucleares. La organización del ADNmt es muy compacta, no tiene intrones. Por lo tanto, las mutaciones aleatorias en el ADNmt suelen afectar una secuencia que codifica, con un padecimiento resultante. Además, el ADNmt es susceptible de sufrir daño por radicales del oxígeno por su proximidad a la producción de estos radicales por la FO y porque sus mecanismos de reparación son mínimos.29,30 Todo el ADNmt de cada persona es heredado de la madre (herencia no mendeliana) porque el espermatozoide contribuye solo con su ADN nuclear que pasa al cigoto durante la fertilización. En ocasiones pueden ocurrir trastornos de la FO por mutaciones en algunos genes de la FO codificados en el nucleo, y estas enfermedades siguen un patrón de herencia mendeliano.
Las biopsias musculares de los pacientes con defectos de la FO son anormales y revelan fibras rojas rasgadas en la tinción tricrómica o fibras azules rastadas en la tinción con succinato deshidrogenasa. Esto es resultado del acúmulo de mitocondrias en la periferia de las fibras musculares. Al microscopio electrónico la mitocondria tiene inclusiones paracristalinas o crestas anormales.30 Al estudiar el ADNmt se detectan mutaciones, deleciones o depleciones específicas.
DELECIONES ESPORADICAS DEL ADNmt: SINDROME DE KEARNS-SAYRE Y OFTALMOPLEGIA EXTERNA PROGRESIVA CRONICA
El síndrome de Kearns-Sayre (SKS) se presenta en pacientes menores de 20 años y se caracteriza por oftalmoplegia, ptosis, retinitis pigmentosa y debilidad miopática. Son comunes la estatura corta, los defectos en la conducción cardiaca, los niveles elevados de proteínas en el líquido cefalorraquídeo, los síndromes cerebelosos, la hipoacusia sensorineural y los niveles elevados de lactato en suero. La biopsia muscular revela fibras rojas rasgadas. Cuando la enfermedad comienza en personas mayores de 20 años y la oftalmoplegia domina el fenotipo, se clasifica como oftalmoplegia externa progresiva crónica (OEPC).31 El SKS y la OEPC más limitada se caracterizan por una sola deleción larga del ADNmt de entre nueve y 50 pares de bases. Con más frecuencia la deleción ocurre en forma esporádica y rara vez se hereda por vía materna. La FO es defectuosa y las actividades de los complejos I y IV se reducen. Las variantes de OEPC, caracterizadas por deleciones múltiples del ADNmt en los genes de la FO codificados por el ADN nuclear pueden trasmitirse en forma autosómica dominante o recesiva.
MUTACIONES PUNTIFORMES DEL ADNMT HEREDADAS POR VIA MATERNA
Síndrome MELAS
La presencia de encefalomiopatía mitocondrial (crisis convulsivas focales o generalizadas y migrañas recurrentes), acidosis láctica y episodios semejantes a eventos cerebrovasculares se combinan para formar el síndrome MELAS (por las siglas en inglés, n. del t.). La biopsia muscular de los pacientes afectados revela fibras rojas rasgadas. Los pacientes pueden tener también hipoacusia, estatura corta, cardiomiopatía, diabetes o degeneración pigmentaria de la retina que recuerda al SKS o a la OEPC. Hasta el 80 porciento de los enfermos tienen mutaciones puntiformes del ADNmt en el gen ARNt leucina.32 Estas mutaciones son heteroplásmicas, lo mismo que las otras mutaciones del ADNmt, y pueden coexistir el ADNmt normal y mutado en la misma célula. Normalmente una célula contiene entre dos y 10 moléculas de ADNmt, lo que permite que una mutación que de otra forma sería letal (i.e., deterioro letal de la FO) persista en organismos viables.
Síndrome MERRF El síndrome MERRF (por las siglas en inglés, n. del t.) consiste en epilepsia mioclónica y miopatía con fibras rojas rasgadas. Además, son comunes la ataxia, demencia, sordera, debilidad, atrofia muscular y alteraciones cardiacas, aunque su expresión es variable y depende del grado de heteroplasmía. Alrededor del 80 porciento de los pacientes con MERRF tienen una mutación en el gen ARNt lisina.33
Neuropatía óptica hereditaria de Leber Una enfermedad de adultos jóvenes cada vez más reconocida (con mayor prevalencia en hombres que en mujeres), la neuropatía óptica hereditaria de Leber (NOHL), se manifiesta por pérdida de la visión indolor, subaguda y bilateral. Se han observado varias mutaciones en el ADNmt. Algunos pacientes con NOHL y diferentes mutaciones pueden tener otros trastornos asociados, como encefalopatía, sordera, ataxia, mielopatía o distonía.34
Trastornos de los canales de iones, parálisis periódica familiar y miotonías no distróficas
PRINCIPIOS GENERALES DE EXCITABILIDAD DE LA MEMBRANA
Después de la excitación de la unión neuromuscular, los potenciales de acción se propagan de acuerdo a flujos de iones a través de la membrana sarcolema, que dependen de la apertura (activación) y cierre (inactivación) de los canales iónicos apropiados. En las células musculares y nerviosas la apertura del canal de voltaje de Na+ causa un rápido incremento en la permeabilidad del Na+ y despolarización de la membrana. Para que la membrana inicie el siguiente potencial de acción los canales de Na+ deben cerrarse. Los canales de voltaje de K+ se abren y salen iones K+ fuera de la célula, creando un voltaje hiperpolarizado a través de la membrana celular. Los canales de Cl- contribuyen a la repolarización al estabilizar el potencial de membrana.
Las alteraciones en la excitabilidad de la membrana pueden ocasionar miotonía y parálisis periódica.35 La miotonía, como un síntoma de las miotonías no distróficas, ocurre en la mitonía congénita, una enfermedad autosómico dominante o recesiva que se manifiesta por hipertrofia muscular sin ataques paralíticos. La miotonía se presenta como rigidez no dolorosa después de un periodo de inactividad, que mejora después del movimiento continuo (fenómeno de calentamiento). Es causada por mutaciones en las proteínas del canal de cloro.36 Cuando se desarrolla miotonía después de la exposición al frío y empeora con el ejercicio (miotonía paradójica) o se asocia con ataques de debilidad, el padecimiento se conoce como paramiotonía congénita (PC). La PC es causada por mutaciones en el canal de Na+.
Los otros trastornos de la excitabilidad de la membrana son la hipocalemia y la parálisis periódica hipercalémica. Los ataques paralíticos se asocian con hipercalemia (que puede coexistir con PC), debida a mutaciones en los canales del Na+, o con hipocalemia, causada por mutaciones en el gen para el receptor de dihidropiridina, que funciona como un canal de voltaje de calcio.35,36
Trastornos de los canales de sodio
Los trastornos de los canales de sodio incluyen la parálisis periódica hipercalémica (PPhiperK), la paramiotonía congénita y la miotonía de los canales de sodio. Se han asociado con el gen SCN4A, que codifica la isoforma adulta del canal de Na+, en el cromosoma 17q13.1-13.37,38 La PPhiperK y la PC son trastornos alélicos del mismo gen. La miotonía de los canales de sodio, llamada antes miotoía congénita con respuesta a acetazolamida o miotonía fluctuans, también se asocia al mismo locus. Se presenta como miotonía con fenómeno de calentamiento y sin debilidad después del enfriamento. Después de la ingestión de K+ la miotonía aumenta.39 Se han identificado varias mutaciones sin sentido en pacientes con este trastorno.
La PPhiperK y la PC son enfermedades autosómico dominantes. Se caracterizan por ataques de debilidad que comienzan en la infancia o niñez temprana. Los episodios son precipitados por el resposo después del ejercicio, estrés, administración de K+ y ciertos alimentos. El frío puede inducir miotonía paradójica en algunos pacientes. Los que tienen PC pueden tener también ataques episódicos que no se asocian con PPhiperK.35 Puede ocurrir debilidad entre los ataques, que se presenta después de varios años como una miopatía. Algunos pacientes con PPhiperK pueden tener arritmias cardiacas con prolongación del QT o ectopia ventricular y características dismórficas. La frecuencia de los ataques e incluso la debilidad entre los mismos responde a los inhibidores de la anhidrasa carbónica como la acetazolamida y la diclorfenamida.35
Trastornos de los canales del calcio
Los trastornos de los canales del calcio se manifiestan como parálisis periódica hipocalémica (PPhipoK), una enfermedad autosómico dominante localizada en el cromosoma 1q31-q32 cerca del gen que codifica al receptor de dihidropiridina muscular.36,40 La PPhipoK se manifiesta por parálisis episódica que comienza en la primera o segunda década de la vida, por lo general durante el sueño. Esta es precipitada por el consumo de carbohidratos, el reposo después del ejercicio y la excitación. Durante los ataques la concentración de K+ en suero disminuye y ocurre retención urinaria de Na+ y agua. Entre los ataques muchos pacientes desarrollan una miopatía permanente y lentamente progresiva. La enfermedad debe distinguirse de todas las causas de hipocalemia secundaria que causan debilidad, incluyendo uso de diuréticos, enfermedad renal, hiperaldosteronismo, intoxicación por orozuz, abuso de laxantes y enfermedades gastrointestinales perdedoras de potasio.35 En la hipercalemia secundaria no ocurren ataques paralíticos a menos que la concentración de K+ sea menor de 3 mEq (casi siempre menos de 2.5 mEq). Por el contrario, los ataques en la PPhipoK ocurren incluso con concentraciones de K+ cercanas a lo normal. La PPhipoK puede asociarse con tirotoxicosis, en especial en descendientes de asiáticos. En este caso la PPhipoK responde al uso de propranolol o a la normalización de la función tiroidea. Aunque la PPhipoK se hereda con patrón dominante, una tercera parte de los pacientes puede tener una enfermedad esporádica. Puede requerirse para el diagnóstico la prueba de provocación con infusión de insulina y glucosa con monitoreo cardiaco y respiratorio. La frecuencia de los ataques y la debilidad entre los mismos responde al uso de acetazolamida y diclorfenamida.41
Trastornos de los canales del cloro
Los trastornos de los canales del cloro incluyen la miotonía congénita autosómico dominante (enfermedad de Thomsen), que ocurre en la quinta década de la vida y se asocia con hipertrofia muscular y miotonía congénita autosómico recesiva (enfermedad de Becker), que se presenta en niños menores de tres años. La miotonía mejora con el ejercicio. Los pacientes con estos trastornos no tienen parálisis periódica o debilidad, aunque en la forma de Becker puede existir debilidad temporal, que mejora con el ejercicio. Son datos característicos la miotonía por percusión y la miotonía generalizada, referida por el paciente como rigidez. Ambos padecimientos se han relacionado con mutaciones diferente en el gen del canal del cloro en el cromosoma 7q35.36
Neuromiotonía autoinmune adquirida
La mioquimia, o músculo que se retuerce durante el reposo, la menor relajación muscular con rigidez en reposo, los calambres dolorosos y el aumento en la sudoración caracterizan a esta enfermedad adquirida. Es causada por hiperactividad de las terminales periféricas de los nervios motores y correctamente debe denominarse neuromiotonía porque la actividad continua de la fibra muscular es abolida por curare y no por bloqueo del nervio proximal. La neuromiotonía ocurre en forma esporádica, pero han ocurrido casos familiares que con frecuencia se asocian con neuropatía. También se han observado asociaciones con miastenia gravis y timoma. Un anticuerpo dirigido contra los canales de K+ sensibles a o-dendrotoxina puede ser el responsable de la enfermedad.42 El tratamiento es sintomático (fenitoína, carbamacepina y mexiletine) pero en casos resistentes puede requerirse inmunoglobulina intravenosa o plasmaféresis.
Miopatías tóxicas inducidas por drogas
PRINCIPIOS GENERALES
Aunque las miopatías inducidas por drogas no son raras, son difíciles de identificar en la práctica clínica. El médico debe sospechar una miopatía tóxica en un paciente que no tenía una enfermedad muscular previa, cuya miopatía se desarrolla en forma aguda o subaguda (en ocasiones lentamente) con debilidad y dolor muscular, que presenta mioglobinuria después de la administración del posible agente miotóxico, o que mejora al suspender el medicamento tóxico [ver tabla 1].43 Los agentes miotóxicos pueden causar miopatía al afectar en forma directa a los organelos musculares, como la mitocondria, induciendo una reacción inmunológica, o al inducir efectos sistémicos secundarios como alteraciones de los electrolitos, deprivación nutricional (v.gr., el agente compite con vitaminas) y malabsorción. Varios medicamentos pueden ser miotóxicos tanto solos como al combinarlos con otros [ver tabla 1], y el clínico debe estar alerta respecto al potencial miotóxico de los medicamentos más recientes.
MIOPATIA POR ZIDOVUDINA Y TOXICIDAD DE OTROS ANALOGOS DE NUCLEOSIDOS
Las características clínicas de la miopatía por zidovudina incluyen debilidad muscular proximal, mialgias (sobre todo en los muslos y pantorrillas), fatiga, cambios miopáticos en la EMG y elevación de los niveles de CK en suero, que aumenta más con el ejercicio.44 La pérdida de peso y la elevación en la concentración de lactato en suero pueden anteceder al inicio de miopatía por zidovudina. La zidovudina puede causar síntomas miopáticos después de un año de su administración. En un seguimiento de 20 pacientes con miopatía por zidovudina, cuatro a seis semanas después de suspender el medicamento las mialgias se resolvieron, mejoró o se normalizó la fuerza y la actividad espontánea en la EMG disminuyó o desapareció.45
Las características histológicas típicas de la miopatía por zidovudina incluyen fibras rojas rasgadas, sugestivas de alteraciones mitocondriales, y muchas fibras negativas para detección de oxidasa de citocromo c. La zidovudina, un terminador de la cadena de ADN, inhibe a la gama-polimerasa presente en la matriz mitocondrial, con terminación de la síntesis de ADNmt, por lo que se presenta depleción hasta del 78 porciento del ADNmt muscular.46 La zidovudina es el único análogo de nucleósidos miotóxico usado en el tratamiento del síndrome de inmunodeficiencia adquirida (SIDA). La dideoxicitosina , la dideoxinosina y la lamivudina causan neuropatía axonal dolorosa, pero no miopatía. Aunque los inhibidores de la proteasa de uso reciente no se han asociado con miotoxicidad, se requiere una observación cuidadosa para determinar si ésta ocurre después de su uso prolongado.
MIOPATIA POR AGENTES REDUCTORES DEL COLESTEROL
Varios agentes reductores del colesterol [ver tabla 1] causan una miopatía casi siempre reversible que se caracteriza por debilidad proximal, mialgias, elevación de la concentración de CK en suero y cambios miopáticos en la EMG. Debido a que el colesterol es el principal esterol constituyente de las membranas musculares, la reducción del reservorio normal de colesterol disponible para la síntesis de membrana puede aumentar la fluidez de la membrana, causando un sarcolema inestable, descargas miotónicas y, en casos avanzados, rabdomiolisis. El EMG presenta potenciales de fibrilación y complejos miotónicos o descargas repetitivas.43,44 Puede ocurrir recuperación gradual después de suspender el agente agresor.
Aunque son comunes el aumento temporal en la concentración sérica de CK y las mialgias después del tratamiento con lovastatin, el medicamento se ha asociado con síntomas miopáticos clínicos en menos del cinco porciento de los pacientes tratados. El uso de medicamentos combinados aumenta el riesgo de miopatía. La coadministración de ciclosporina y lovastatin en pacientes con trasplantes de corazón o riñón e hiperlipidemia parece aumentar la incidencia de miopatía con rabdomiolisis.44
MIOPATIA POR ESTEROIDES Y AGENTES BLOQUEADORES (MIOPATIA DE LA ENFERMEDAD GRAVE)
Los pacientes con parálisis prolongada inducida por agentes bloqueadores no despolarizantes como el pancuronio pueden manifestar una miopatía aguda después de suspender la ventilación mecánica. La mayoría de estos pacientes recibieron dosis altas de esteroides por un estado asmático u otro padecimiento grave por el que se indujo parálisis artificial para asegurar una higiene pulmonar adecuada.47 Aunque la combinación de esteroides y agentes bloqueadores se ha implicado en forma consistente en el desarrollo de miopatía de la enfermedad grave, en ocasiones la exposición a uno de estos agentes puede ser mínima.44 El cuadro clínico se caracteriza por debilidad severa y generalizada de las extremidades, incapacidad para retirar el ventilador mecánico, atrofia muscular, elevación normal o moderada de la concentración de CK en suero y cambios miopáticos en la EMG. Por lo general la debilidad mejora en forma lenta.48 La miopatía por agentes bloqueadores-esteroides suele coexistir con una neuropatía axonal, constituyen la neuromiopatía de la enfermedad grave.48 El diagnóstico diferencial debe incluir los defectos de trasmisión neuromucular, el síndrome de Guillain-Barré, la miopatía inflamatoria o necrosante aguda y la parálisis periódica.44 La biopsia muscular muestra alteraciones morfológicas francas, con pérdida de los filamentos gruesos, lo que se caracteriza por áreas centrales vacías sin signos de necrosis, inflamación o fagocitosis. Con la tincion de la trifosfatasa de adenosina (ATPasa) se observan áreas de palidez central en muchas fibras. La microscopía electrónica revela pérdida selectiva y extensa de los miofilamentos gruesos, con conservación de los miofilamentos delgados y las líneas Z. Es necesario tener precaución al usar dosis altas de esteroides en pacientes que reciben agentes paralizantes por periodos prolongados.
INTOXICACION POR VITAMINA E
Se ha reportado una miopatía necrosante con debilidad muscular proximal y elevación del nivel de CK en suero en pacientes automedicados con dosis altas de vitamina E.49
MIOPATIA POR CLOROQUINA
El antimalárico cloroquina es empleado con frecuencia por los reumatólogos para el tratamiento de varias enfermedades difusas del tejido conectivo. Puede ocasionar degeneración macular y corneal, neuropatía periférica y miopatía. La miopatía se observa con la administración prolongada de dosis altas (500 mg diarios). Esta tiene características morfológicas interesantes que recuerdan a las que ocurren en la deficiencia de maltasa ácida: múltiples vacuolas con material positivo a fosfatasa ácida, cuerpos mieloides dentro de las vacuolas y lisosomas grandes con aumento en la actividad enzimática.
MIOPATIA POR COLCHICINA
La colchicina interfiere con el crecimiento de los microtúbulos, afectando la mitosis. Después de su uso prolongado, causa una miopatía vacuolar y una neuropatía axonal, con frecuencia en pacientes entre los 50 y 70 años de edad y que tienen gota e insuficiencia renal leve crónica.50 Los síntomas incluyen debilidad proximal, elevación de la concentración de CK, afección neurosensorial distal y arreflexia. Los síntomas se resuelven cuatro a seis semanas después de suspender el medicamento.
ESTEROIDES
Los pacientes con hiperadrenocorticismo (i.e., síndrome de Cushing) pueden desarrollar debilidad. Pueden ocurrir cambios semejantes durante la administración crónica de prednisona (casi siempre en dosis mayores de 20 mg al día) o dexametasona. La debilidad miopática inducida por esteroides es leve, respeta los músculos flexores del cuello,44 y puede agravar la debilidad causada por el trastorno inmune o la neoplasia para la que se administró el esteroide. La disminución de la dosis del medicamento revierte la miopatía. La concentración sérica de CK es normal y la EMG no proporciona información específica.
MIOPATIA ALCOHOLICA
Los alcohólicos desarrollan principalmente una neuropatía axonal periférica y en ocasiones miopatía aguda o crónica. Precedida por edema muscular y dolor, la miopatía aguda se presenta como rabdomiolisis y mioglobinuria y es dolorosa.43 Puede recurrir si el paciente continúa bebiendo. La miopatía aguda en los alcohólicos puede relacionarse también con hipocalemia (< 2.5 mEq). Esta miopatía no es dolorosa, se asocia con edema muscular y es rápidamente reversible. La debilidad muscular proximal en alcohólicos crónicos suele ser multifactorial y no necesariamente un proceso miopático; por ejemplo, puede deberse a desnutrición, inactividad o enfermedad neurogénica.43 Desde el punto de vista histológico, es común la atrofia de las fibras tipo II. Algunos bebedores crónicos pueden sufrir elevación asintomática de la concentración de CK (hasta 20 veces el normal) que se agrava por la actividad física.
MIOPATIA INFLAMATORIA INDUCIDA POR DROGAS
Los pacientes con enfermedad de Wilson, artritis reumatoide o esclerodermia pueden desarrollar polimiositis o miastenia gravis durante el tratamiento con D-penicilamina. La enfermedad responde a la suspensión del medicamento o la administración de esteroides.
En reportes aislados y no confirmados se ha asociado el uso de procainamida, propiltiouracilo y cimetidina con un padecimiento semejante a polimiositis. Los autores no han observado ningún caso.
El L-triptofano contaminadofue responsable de un brote de un síndrome caracterizado por fascitis eosinofílica, miositis, engrosamiento de la piel, neuropatía axonal y otras manifestaciones sistémicas. Un proceso autoinmune contra los fibroblastos en la matriz extracelular desencadenado por el contaminante fue implicado como causa del síndrome, al que se denominó síndrome de mialgia-eosinofilia.51 La enfermedad ha dejado varios pacientes con engrosamiento residual de la piel, mialgia y fatiga.
El interferón alfa (IFN-alfa), usado cada vez más para algunas neoplasias y hepatitis, causa fatiga severa, artralgias y en ocasiones mialgia. Se desconoce el mecanismo exacto de la fatiga muscular y las mialgias. Es bien conocida la toxicidad al SNC que causan las dosis altas de IFN-alfa.
La citocina interleucina-2, usada para el carcinoma de células renales, puede exacerbar la polimiositis o dermatomiositis previas.
Trastornos de la trasmisión neuromuscular
UNION Y TRASMISION NEUROMUSCULAR NORMAL
Las principales áreas de la unión neuromuscular son (1) la región presináptica, que activa zonas que contienen dos filas paralelas de canales de calcio dependientes de voltaje (CCDV) y vesículas sinápticas que contienen acetilcolina (AC), (2) el espacio sináptico y (3) la región posináptica, que consiste en pliegues de unión que contienen receptores de acetilcolina (RAC) [ver figura 3]. Desde el punto de vista funcional, la trasmisión neuromuscular depende de la liberación de AC de las terminales de los nervios motores, la interacción de la AC con los RAC y la despolarización resultante de la fibra muscular. En un estado de reposo, existe liberación aleatoria de AC desde una vesícula en particular. La AC se une al RAC y abre un canal catiónico selectivo, que permite que entre Na+ a la región de la placa terminal. Esto crea un potencial de despolarización denominado potencial miniatura de placa terminal (PMPT).52 A continuación la AC se disocia del RAC y es rápidamente hidrolizada por la acetilcolinesterasa (ACE). Cuando la ACE es inhibida, como ocurre durante el tratamiento con medicamentos anticolinesterasa como la piridostigmina, más moléculas de AC se unen a RAC y abren múltiples canales iónicos.
Cuando el potencial de acción alcanza la porción terminal del nervio se abren los CCDV y fluyen iones de Ca2+ hacia la terminal nerviosa. El ingreso de Ca2+ ocasiona la fusión y exocitosis de las vesículas sinápticas y la liberación de moléculas de AC (cada vesícula contiene de 6,000 a 10,000 moléculas de AC) que difunden y se diseminan en los pliegues de unión. La liberación simultánea de AC por la fusión de 50 a 300 vesículas causa un potencial de placa terminal (PPT). Si el PPT excede cierto umbral, un potencial de acción muscular (PAM) origina la contracción del músculo. En condiciones normales las interacciones entre la AC liberada y el RAC son más que suficientes para producir PPT capaces de desencadenar un PAM. La diferencia entre la amplitud del PPT actual y la amplitud requerida para desencadenar un PAM se denomina margen de seguridad.
Existen tres padecimientos principales que afectan la trasmisión neuromuscular: la miastenia gravis (MG), el síndrome miasténico de Eaton-Lambert (SMEL) y varios síndromes miasténicos congénitos.
MIASTENIA GRAVIS
La miastenia gravis es el prototipo de las enfermedades autoinmunes. Satisface todos los criterios inmunológicos:5254 se conoce el antígeno, el RAC, pueden detectarse anticuerpos específicos contra éste y medirse en el suero de los pacientes afectados, la enfermedad puede trasmitirse a animales de experimentación a través de la IgG patogénica de los pacientes, la inmunoglobulina se une al RAC, causando destrucción dependiente del complemento, y la eliminación de los anticuerpos ocasiona mejoría clínica.
En la MG los anticuerpos dirigidos contra el RAC disminuyen el número de receptores disponibles. En consecuencia, la AC liberada no puede fijarse en forma suficiente a los RAC, por lo que el PPT es pequeño e insuficiente para desencadenar un PAM y la trasmisión neuromuscular falla. Cuando ocurre falla de la trasmisión neuromuscular en muchas uniones neuromusculares, el músculo no puede activarse del todo, y esto causa fatiga y debilidad. Durante la estimulación nerviosa repetida en personas sanas la cantidad de AC liberada disminuye después de los primeros impulsos, pero la trasmisión neuromucular se conserva porque el margen de seguridad es suficiente. En los pacientes con MG el PPT es de sí pequeño. Durante la estimulación nerviosa repetida el PPT disminuye más porque la menor cantidad de AC liberada en los estímulos posteriores no es suficiente para activar un número adecuado de RAC, con lo que se decrementa más el PAM. Los estudios de estimulación nerviosa repetida se usan como diagnóstico al demostrar la menor trasmisión neuromuscular en los pacientes con MG.
Epidemiología
La prevalencia de la MG es de alrededor de cuatro a seis casos por 100,000 habitantes. La incidencia anual varía, pero se reconocen dos picos, uno se asocia con mujeres en la segunda o tercera décadas de la vida y el otro con varones de más de 60 años. La enfermedad ocurre 50 porciento más en mujeres que en hombres. La edad promedio de inicio es de 28 años en las mujeres y 42 en los varones. Debido a que la MG afecta al RAC nicotínico en la unión neuromuscular, solo se altera el sistema motor.
Puede desarrollarse también MG clásica en niños, que responde bien a timectomía o anticolinesterasas. La MG puede relacionarse con timoma en hasta el 15 porciento de los adultos, o puede presentarse después de extirpar el timoma.52-54 Además, hasta el cinco porciento de los pacientes con MG tienen otros padecimientos autoinmunes sistémicos, el 15 porciento presenta otros autoanticuerpos, el 20 porciento tiene enfermedad tiroidea y hasta el cinco porciento puede tener historia familiar de otra enfermedad autoinmune. El embarazo tiene un efecto variable, pero en la mayoría de los pacientes la debilidad aumenta en el periodo posparto. Alrededor del 12 porciento de los niños que nacen de madres con MG desarrollan MG neonatal temporal como resultado de la transferencia placentaria de anticuerpos anti-RAC circulantes. Estos niños desarrollan debilidad, trastornos para alimentarse, problemas respiratorios, llanto débil y debilidad facial en las primeras horas después de nacidos. La condición dura hasta tres semanas, coincidiendo con la vida media de la IgG.
Inmunopatogenia de la MG
Autoanticuerpos La MG es mediada por autoanticuerpos patógenos que se unen al RAC y causan pérdida funcional de los receptores y destrucción focal de los pliegues de unión posináptica, interfiriendo con la despolarización de la membrana posináptica [ver figura 4].
Entre el 10 y 20 porciento de los pacientes con MG, en especial los que tienen formas leves, de la infancia o localizadas (v.gr., oculares), no tienen anticuerpos detectables. La mayoría de estos pacientes tienen una MG autoinmune clásica, pero los anticuerpos son de baja afinidad o están dirigidos contra epítopes que no están presentes en los extractos solubles del RAC que se usan en los ensayos estándar. Si el ensayo de unión a RAC es negativo, debe realizarse un inmunoensayo que usa anticuerpos para detectar al RAC presente en las células musculares en el cultivo de tejido.
Otros anticuerpos asociados con la MG incluyen los anticuerpos contra células estriacionales. Estos se encuentran en hasta el 80 porciento de los pacientes con timoma y MG, 25 porciento de los enfermos con timoma sin MG, 30 porciento de los adultos con MG (55 porciento cuando la MG comienza después de los 60 años), 6 porciento de los pacientes con SMEL, 3 porciento de los enfermos con cáncer pulmonar y con frecuencia en pacientes con enfermedad hepática autoinmune.55 Son frecuentes los anticuerpos contra microsomas tiroideos y tiroglobulina en los pacientes con MG ocular y con SMEL sin cáncer.
Inmunorregulación El RAC es un antígeno dependiente de células T. En la MG las células T CD4+ están sensibilizadas y responden a la estimulación con RAC con péptidos inmunodominantes sintéticos del RAC.56,57 Debido a que los linfocitos de las personas sanas también responden a estos péptidos, aunque en menor grado, las células T específicas contra RAC son parte del repertorio inmune normal. Esto sugiere que la MG es una enfermedad causada por alteración en la inmunorregulación. Se ha postulado que antígenos microbianos (v.gr., Escherichia coli y Klebsiella) o virus herpes simple, inducen la MG porque estas bacterias y virus comparten una secuencia homóloga con péptidos de la subunidad alfa del RAC.
Alrededor del 75 porciento de los pacientes con MG tienen alteraciones tímicas, sea hiperplasia con formación de centros germinales o timoma (en el 15 porciento de todos los pacientes con MG). La extirpación del timo causa mejoría clínica o remisión. Las evidencias adicionales que implican un papel para el timo en la MG se basan en diversos aspectos: (1) el timo de las personas afectadas contiene una mayor población de células B que pueden secretar anticuerpos contra el RAC que el timo de una persona normal sana, (2) el timo de una persona afectada posee células mieloides que contienen RAC cercanos a las células dendríticas interdigitantes, y (3) el número de células sensibilizadas contra el RAC es mayor en el timo de una persona afectada que en el de una persona normal sana.52,53
En la MG la ruptura de la autotolerancia puede comenzar en el timo. Los RAC en las células mieloides pueden ser detectadas por las células dendríticas del timo que presentan el antígeno a las células T CD4+, las que a su vez estimulan a las células B para que produzcan anticuerpos contra el RAC. Estos anticuerpos reconocen y reaccionan con los RAC en el músculo esquelético.
La MG se asocia con frecuencia con otros padecimientos autoinmunes, incluyendo enfermedades difusas del tejido conectivo, polimiositis, pénfigo, enfermedades tiroideas autoinmunes y enfermedad injerto contra huésped. El uso de la D-penicilamina ha inducido MG clásica, que mejora al suspender el fármaco.
Manifestaciones clínicas
La MG se manifiesta por debilidad muscular y fatiga. Es característica la debilidad para elevar los párpados y mover los músculos extraoculares, lo que causa ptosis asimétrica y diplopia. Los extensores del cuello suelen ser débiles y la cabeza cae. La debilidad de los músculos facial y bulbar puede ocasionar una mueca cuando el paciente intenta sonreir, lenguaje nasal, disártrico, disfónico y de bajo volumen, y disfagia, que puede ocasionar reflujo y ahogamiento. La debilidad de los músculos proximales y la fatiga producen dificultad para caminar, subir escaleras, peinarse y cargar objetos. Puede haber debilidad significativa de los músculos respiratorios. Los síntomas fluctúan y son menores por la mañana. La fatiga empeora hacia el final del día o con la actividad repetida. Si se le pide a un paciente que abduzca los brazos, se observa que los baja en foma gradual. Si se le pide que hable en forma continua, la voz se volverá ronca, nasal, lenta y al final inaudible. La sensación y la función cognoscitiva son normales. Los reflejos tendinosos, en especial los de los músculos no atróficos, suelen estar exaltados o ser normales. Los dedos de los pies ven hacia abajo. Los síntomas empeoran durante o antes del periodo menstrual y durante las infecciones virales o bacterianas.
Diagnóstico
Además de demostrar la fatiga y debilidad a la exploración, el diagnóstico clínico puede confirmarse por medio de la prueba de edrofonio o neostigmina. Estos medicamentos inhiben a la ACE, lo que permite a la AC interactuar en forma repetida con los RAC restantes, con mejoría consecuente de la fuerza. Los estudios de estimulación nerviosa repetida demuestran una reducción rápida (mayor del 12 porciento) en la amplitud del PAM. La EMG de una fibra demuestra mayor irritabilidad o bloqueo en más del 90 porciento de los pacientes. Debe realizarse una IRM del tórax en busca de hiperplasia tímica o timoma. La MG debe distinguirse de otras enfermedades o trastornos inducidos por farmacos que presentan cuadros clínicos parecidos, como el botulismo, la intoxicación por organofosfatados, la reacción tóxica por D-penicilamina, la miopatía mitocondrial, la lesión compresiva de los nervios cranealeas, el SMEA y los síndromes miasténicos congénitos.
El diagnóstico serológico de MG se realiza detectando anticuerpos contra el RAC con un ensayo de radioinmunoprecipitación. Estos anticuerpos están presentes en hasta el 90 porciento de los pacientes con MG generalizada y hasta 70 porciento de los que tienen MG ocular. Se encuentran también en el 30 porciento de los pacientes con enfermedad hepática autoinmune, 10 porciento de enfermos con anemia perniciosa y hasta el 13 porciento de los que tienen SMEL.55 En general, los títulos de anticuerpos contra RAC no correlacionan con la severidad clínica.
Tratamiento
El término gravis en la actualidad no es correcto porque la MG responde bastante bien a los tratamientos disponibles. Aunque la secuencia de aplicación de las diversas modalidades terapéuticas puede diferir entre los médicos, la elección inicial en los casos leves suele ser un medicamento anticolinesterasa cada cuatro horas mientras el paciente está despierto. En la mayoría de las instituciones se realiza timectomía por vía transesternal después de la pubertad y hasta los 60 años de edad.
La prednisona es el tratamiento inmunoterapéutico de elección. Suelen administrarse 60 a 80 mg al día en una dosis única por la mañana durante un periodo inicial de tres a cuatro semanas. Después se reduce la dosis en forma gradual y en días alternos durante un periodo de ocho semanas a razón de 10 mg por semana (más rápido si se observan efectos adversos severos) hasta que se alcance la dosis más baja posible que controle la enfermedad. Algunos médicos prefieren iniciar con la dosis más baja de prednisona (i.e., 40 mg/día) y aumentarla en forma gradual para evitar un empeoramiento temporal que se ha observado en algunas ocasiones. La plasmaféresis o la Ig intravenosa suele reservarse para las crisis y los casos severos, y antes de la timectomía. La ciclosporina o azatioprina pueden usarse como tratamiento de segunda elección. Por ejemplo, la azatioprina es un inmunosupresor eficaz que se administra por vía oral en dosis de hasta 3 mg/kg.
SINDROME MIASTENICO DE EATON-LAMBERT
En el SMEL los anticuerpos específicos contra los CCDV causan modulación antigénica y depleción de los CCDV. Esto restringe el ingreso de Ca2+ en la terminal del nervio motor y reduce la liberación de AC durante el impulso nervioso. La cantidad de AC generada puede originar solo un PPT pequeño, que no es capaz de desencadenar un PAM; esto ocasiona falla en la trasmisión neuromuscular y debilidad muscular. Una observación importante en el SMEL es la asociación frecuente con cáncer pulmonar de células pequeñas (CPCP), que también expresa CCDV.54
El SMEL ocurre con más frecuencia en hombres que en mujeres. Alrededor del 70 porciento de los hombres con SMEL tienen CPCP, comparado con el 20 porciento de las mujeres. El SMEL puede preceder a la detección de neoplasias hasta por tres años. Las neoplasias son raras en las personas menores de 40 años.
La enfermedad se presenta como debilidad muscular proximal, aumento en la fatiga y síntomas oculares temporales de ptosis o diplopia. Es frecuente la sintomatología autonómica, con boca y ojos secos, impotencia sexual, constipación y reflejos pupilares anormales. Es común la hiporreflexia. Un signo típico es el aumento en la fuerza muscular y los reflejos pocos segundos después de un esfuerzo máximo sostenido. El diagnóstico se confirma por los estudios de estimulación nerviosa repetitiva que demuestran un aumento dramático en la amplitud del PAM hasta la estimulación tetánica. La EMG de una sola fibra muestra mayor irritabilidad y bloqueo, que mejora al aumentar la frecuencia de estimulación. Usando péptidos radiomarcados de toxinas de víbora de cascabel, pueden demostrarse anticuerpos contra los canales de calcio tipo P o tipo Q en hasta el 95 porciento de los pacientes con SMEL.54 Se observan anticuerpos contra los canales tipo N en hasta el 75 porciento de los pacientes con SMEL, en especial los que tienen cáncer pulmonar.58 La búsqueda de CPCP, en especial en los varones mayores de 40 años, debe ser parte indispensable del proceso diagnóstico.
La región presináptica de las placas terminales motoras en los pacientes con SMEL muestra depleción marcada de las partículas de la zona activa que representan los CCDV. Los CCDV en el SMEL no se localizan en filas paralelas, sino que se agregan como resultado de su interacción con los anticuerpos IgG específicos. La IgG del paciente [la fracción F(abÕ)2], al inyectarse a ratones, trasmite los cambios electrofisiológicos y morfológicos asociados con la enfermedad. El complemento no participa en el proceso. El CPCP expresa CCDV del tipo N, L o P. Se cree que las células tumorales desencadenan autoanticuerpos contra determinantes de la superficie, que reaccionan en forma cruzada con epítopes similares en las regiones presinápticas de las uniones neuromosculares.
En el SMEL asociado con CPCP, el foco primario consiste en tratar el tumor porque los síntomas del SMEL mejoran al remitir el cáncer. El medicamento de elección para el SMEL es la 3,4-diaminopiridina. Este medicamento prolonga la duración del potencial de acción presináptico al bloquear las corrientes externas de K+. Esto permite una mayor entrada de Ca2+ hacia la terminal nerviosa, con mayor liberación de AC. El fármaco alivia la fatiga y la debilidad en los pacientes con SMEL y es bien tolerado en dosis de 40 a 60 mg al día. En ocasiones se agrega piridostigmina, y la prednisona ayuda mucho. La azatioprina, la plasmaféresis y la Ig intravenosa constituyen otros recursos terapéuticos que se administran como agentes ahorradores de esteroides o para mejorar la fuerza de los pacientes que no responden bien a la 3,4-diaminopiridina y la prednisona.
SINDROMES MIASTENICOS CONGENITOS
Los síndromes miasténicos congénitos son un grupo heterogéneo de síndromes que causan falla de la trasmisión neuromuscular que no es de tipo autoinmune sino resultado de defectos genéticos en varias moléculas, enzimas o canales en la unión neuromuscular.
Se sospecha un síndrome miasténico congénito cuando un niño, infante o, en ocasiones, adulto joven, presenta ptosis fluctuante, fatiga, mayor debilidad durante el ejercicio constante (todos síntomas comunes de los defectos de trasmisión neuromuscular), retraso en el desarrollo motor, ausencia de MG autoinmune y una historia familiar positiva.
Reconocimientos
Figuras 1, 3 y 4 Tom Moore
Figura 2 Dana-Burns-Pizer
DR. MARINOS C. DALAKAS
Estudio de los pacientes con enfermedad neuromuscular
Las enfermedades del músculo y de la unión neuromuscular constituyen un grupo heterogéneo de padecimientos adquiridos y hereditarios. Los síntomas de presentación son variados e incluyen fatiga, debilidad del músculo esquelético, atrofia, calambres musculares y alteración de los músculos respiratorios, faríngeos, faciales y oculares. En general, los músculos proximales se afectan en forma más selectiva y severa que los distales. Por lo tanto, los pacientes presentan dificultad para subir las escaleras, levantarse de una silla baja, correr, peinarse e incorporarse o volterarse en la cama. La mayoría de las miopatías afectan solo a los músculos esqueléticos, pero pueden alterarse también el músculo liso y el cardiaco. Los pacientes con miopatías no tienen trastornos sensoriales o disfunción autonómica porque la función de los nervios periféricos y el sistema nervioso autónomo está conservada.
El examen clínico debe enfocarse a evaluar los datos musculares y excluir enfermedades que pueden presentarse con síntomas y signos miopáticos como los síndromes de neurona motora, las neuropatías motoras y las enfermedades psicógenas. Se requiere una historia familiar completa y el examen de los familiares para excluir un padecimiento hereditario. Los estudios de laboratorio útiles incluyen (1) estudios para excluir una enfermedad sistémica, factores exógenos, toxinas o virus que puedan inducir miopatía, (2) electromiografía (EMG) para localizar la lesión al músculo o la unión neuromuscular y excluir alteraciones de la neurona motora o de los nervios periféricos, (3) determinación de niveles séricos de enzimas musculares, (4) medición de los autoanticuerpos específicos dirigidos contra antígenos musculares conocidos o posibles, (5) biopsia muscular para determinar la histoquímica enzimática, inmunopatología, microscopía electrónica y medición bioquímica de enzimas o proteínas específicas del músculo, (6) prueba de ejercicio con isquemia para medir la producción de lactato y amoniaco en las miopatías metabólicas, (7) estudios genéticos y (8) estudios de imagen muscular, según el problema específico que se esté investigando.
La mayoría de las miopatías son incapacitantes o catastróficas si no se tratan. Por lo tanto, el diagnóstico debe establecerse con rapidez para iniciar un tratamiento temprano. Para los pacientes con trastornos intratables es indispensable administrar tratamiento de apoyo, rehabilitación y consejo genético.
En esta subsección se describen las miopatías y trastornos de la unión neuromuscular más comunes, con énfasis en el cuadro clínico, patogenia, diagnóstico y tratamiento.
Distrofias musculares
Las distrofias musculares constituyen un grupo heterogéneo de enfermedades congénitas del músculo que se caracterizan por debilidad muscular severa, atrofia, elevación de los niveles de enzimas musculares en suero y cambios destructivos en la citoarquitectura de las fibras musculares. La clasificación tradicional de las distrofias musculares en de Duchene, de Becker, de cintura escapular y pélvica y congénita ha cambiado porque se han identificado varios defectos genéticos en las proteínas musculares que parecen ser responsables de algunas de estas enfermedades y porque se ha demostrado ausencia o deficiencia de proteínas musculares específicas en ciertas distrofias.
Los estudios moleculares, bioquímicos e inmunocitoquímicos1,2 han identificado que el complejo de proteína asociada a la distrofina (PAD) constituye la proteína transmembrana clave que une a la proteína distrofina del citoesqueleto con la proteína de la matriz extracelular laminina-2 [ver figura 1]. Las deficiencias en ciertos componentes de este sistema se asocian con síndromes clínicos específicos. En la actualidad las distrofias musculares se clasifican de acuerdo con los genes involucrados y la proteína defectuosa, que puede ser un componente del citoesqueleto, la membrana o la matriz extracelular.
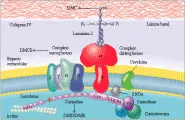
|
| Figura 1 |
| Complejo distroglucano en el sarcolema. |
PRINCIPIOS SOBRE LAS PROTEINAS DEL CITOESQUELETO MUSCULAR
Las proteínas musculares más importantes implicadas en las distrofias musculares son la distrofina, el complejo distroglucano, que se asocia con la proteína de la lámina basal, la merosina y el complejo sarcoglucano [ver figura 1]. La distrofina, una proteína del citoesqueleto de 427 kd con forma de bastón, constituye el cinco porciento de todas las proteínas del citoesqueleto del sarcolema y sirve para anclar a la actina F (la forma filamentosa de la actina) a la membrana plasmática (sarcolema) del músculo.1 La distrofina parece reforzar y estabilizar la membrana plasmática durante la tensión de la contracción muscular al mantener un enlace mecánico entre el citoesqueleto y la matriz extracelular. La deficiencia o ausencia de distrofina se asocia con diversas distrofinopatías, de las que el prototipo es la distrofia muscular de Duchenne.3-7
El complejo distroglucano consiste en alfa-distroglucanos, proteínas de la matriz celular, siendo la más relevante desde el punto de vista clínico la alfa2-laminina (o merosina, la proteína involucrada en la distrofia muscular congénita), y el beta-distroglucano, que fija alfa distroglucano y distrofina. No se ha identificado a ningún paciente con defectos primarios en los distroglucanos. El complejo sarcoglucano está formado por cuatro proteínas transmembrana, incluyendo la 50 kd adhalina (alfa-sarcoglucano), la glucoproteína asociada a distrofina 43 kd (beta-sarcoglucano), una proteína de 35 kg (gama-sarcoglucano) y una proteína de 95 kg (delta-sarcoglucano). Los componentes transmembrana del complejo sarcoglucano son específicos del músculo esquelético y cardiaco y se afectan en forma selectiva en pacientes con distrofias musculares recesivas de las cinturas de las extremidades, que se conocen como sarcoglucanopatías. Las distrofias musculares se dividen en distrofinopatías, sarcoglucanopatías y merosinopatías2, de acuerdo con la proteína faltante o deficiente.
Debido a que se han identificado los genes para varias formas de distrofias, en la actualidad se emplea una clasificación genética: (1) distrofias musculares recesivas ligadas al cromosoma X (Duchenne y Becker), (2) distrofias musculares congénitas con deficiencia de merosina, (3) una distrofia muscular de las cinturas de las extremidades (DMCE1) , autosómico dominante y rara, con mutación localizada en el cromosoma 5, y (4) todas las distrofias musculares de las cinturas de las extremidades recesivas (DMCE2). Las DMCE2 se subdividen en la DMCE2A, causada por deficiencia de calpaína, la DMCEB, que no se ha caracterizado, la DMCE2C (deficiencia de gama-sarcoglucano), la DMCE2D (deficiencia de alfa-sarcoglucano), la DMCE2E (deficiencia de beta-sarcoglucano) y la DMCE2F (deficiencia de delta-sarcoglucano).8
DISTROFINOPATIAS
Distrofia muscular de Duchenne
La distrofia muscular de Duchenne (DMD), un padecimiento recesivo ligado al cromosoma X, es causado por mutaciones en el gen de la distrofina en el brazo corto del cromosoma X en la posición Xp21. El gen de la distrofina ocupa más de 2,000 kb de ADN. En el 65 a 70 porciento de los casos la DMD es resultado de deleciones largas (varios kilobases) en el gen de la distrofina, con una falta consecuente de distrofina muscular. Las deleciones, que se detectan en el ADN extraído de linfocitos de sangre periférica, interrumpen el marco de lectura de las tripletas de codones de ARN mensajero (ARNm), lo que produce las formas severas de DMD. Las duplicaciones parciales del gen, detectadas por análisis de Southern blot, causan el seis porciento de las mutaciones de la distrofina, y las mutaciones pequeñas (v.gr., mutaciones puntiformes y errores de división) que producen una proteína distrofina trucada, provocan hasta el 30 porciento de los casos de DMD.5 También son frecuentes las mutaciones espontáneas.3-5 La ausencia de distrofina debilita e interrumpe la membrana de sarcolema, permitiendo la entrada de calcio, lo que ocasiona necrosis de la fibra muscular.
La DMD ocurre en uno de cada 3,000 varones nacidos. Los niños afectados inician sus síntomas después de que comienzan a caminar, por lo general entre los dos y tres años de edad. La marcha vacilante, la postura lordótica, la hipertrofia de las pantorrillas, las contracturas articulares y el caminar sobre los dedos de los pies son las primeras manifestaciones. Estas son seguidas de debilidad y atrofia muscular progresivas, lo que dificulta el levantarse del piso o una silla baja, subir escaleras y elevar los brazos. El retraso en el vaciamiento gástrico puede causar episodios súbitos de vómito y dolor abdominal.5 La enfermedad es lentamente progresiva. Para los 12 años de edad los niños afectados están confinados a una silla de ruedas, y alrededor de los 25 fallecen por complicaciones de insuficiencia respiratoria. Aunque la DMD es un padecimiento del músculo esquelético, el músculo cardiaco se afecta con frecuencia y en las etapas tardías ocurren insuficiencia cardiaca congestiva y arritmias. También se presenta afección no progresiva del sistema nervioso central, que se manifiesta por irritabilidad, hiperactividad o disfunción cognoscitiva.
Los estudios de laboratorio de rutina de los pacientes con DMD muestran al inicio elevación de la creatina cinasa (CK) sérica hasta de 20,000 unidades, con una disminución constante al depletarse la masa muscular. La biopsia de músculo revela una miopatía destructiva severa, con gran cantidad de células T CD8+ y macrófagos (que invaden las fibras musculares), aumento en la cantidad de tejido conectivo y fibras musculares hipercontraídas.7 Para fines diagnósticos, la ausencia de distrofina (o menos del tres porciento de la concentración normal) se demuestra por inmunicitoquímica usando anticuerpos antidistrofina en los cortes de la biopsia muscular y por inmunoblots preparados de las muestras de biopsia.
Tratamiento La presencia de inflamación endomisial ha provocado que se usen esteroides para tratar a la DMD. En un estudio controldo, el uso de esteroides causó mejoría temporal y leve.9 Sin embargo, su uso a largo plazo está restringido por los severos efectos colaterales, en especial obesidad, fracturas, osteoporosis, diabetes e hipertensión. El deflazacort, un esteroide con menos efectos mineralocorticoides, parece un poco más seguro. En una forma modificada de terapia génica, se inyectaron mioblastos humanos que contenían distrofina normal en músculos de pacientes con DMD, pero el procedimiento no cambió la función muscular de los receptores.10 Es posible que las futuras terapias génicas sean eficaces si se encuentran vectores apropiados para insertar en forma eficaz el gen en el músculo. Hasta entonces, el tratamiento es totalmente sintomático, con énfasis en proporcionar terapia respiratoria y física sistemática para el paciente y apoyo psicológico tanto para el paciente como para la familia. El consejo genético es muy importante.
Distrofia muscular de Becker
La distrofia muscular de Becker (DMB) y la DMD son padecimientos alélicos, pero la primera inicia en forma más tardía y progresa con más lentitud. La edad de inicio es muy variable. Los casos pueden detectarse tan pronto como a los tres años o tan tarde como a los 70 años de edad, con promedio de edad de 12 años.5 El espectro de la expresión fenotípica de la DMB es muy amplio. Las formas leves se manifiestan solo por calambres musculares, intolerancia al ejercicio, mioglobinuria, elevación asintomática de los niveles de CK en suero, debilidad muscular leve o miopatía del cuadríceps.5,11 El dolor en la pantorrilla al ejercicio es un síntoma inicial frecuente, lo mismo que las hipertrofias de las pantorrillas. La mayoría de los pacientes pierden la capacidad de deambular a los 40 años (con rango de 10 a 70 años de edad). La edad de muerte tembién varía, de 23 a 89 años (en promedio 42 años). Los pacientes presentan debilidad muscular proximal y niveles de CK hasta de 20 veces lo normal. Los datos de la biopsia muscular son semejantes a los observados en los pacientes con DMD, pero no son tan severos. En los pacientes menores de ocho años de edad la presentación de la DMB suele ser indistinguible de la DMD. Las manifestaciones cardiacas son comunes, su severidad no se relaciona con la miopatía y la cardiomiopatía puede ser severa.
Alrededor del 65 porciento de los pacientes con DMB tienen deleciones dentro del marco de lectura del gen de la distrofina y la proteína producida suele estar truncada y ser solo semifuncional.5,11 Los casos de duplicación del gen de la distrofina han ocasionado una distrofina más larga y en forma de bastón. La inmunocitoquímica del músculo usando anticuerpos antidistrofina demuestra tinción atenuada del sarcolema (no ausencia, como en la DMD) y revela fragmentación de la membrana en el área inmunoteñida. Los inmunoblots detectan una cantidad disminuida de una distrofina menor o mayor de lo normal. No existe un tratamiento eficaz para la DMB.
Mujeres portadoras y distrofinopatía en mujeres
La historia cuidadosa y el examen clínico de las mujeres portadoras asintomáticas puede revelar debilidad muscular leve, calambres, hipertrofia aislada de las pantorrillas y fatiga, con elevación de los niveles de CK. La biopsia de músculo revela fibras negativas para distrofina. Cuando se amplifican los exones específicos de deleción dentro del gen de la distrofina en los heterocigotos por medio de la reacción en cadena de la polimerasa, las detecciones se detectan como una reducción del 50 porciento en la intensidad de la banda de ADN amplificado, comparado con la banda para exones nativos.5 Este método detecta alrededor del 98 porciento de las deleciones. Sin embargo, el mosaicismo gonadal materno es responsable de otras mutaciones de DMD apenas detectadas en hasta el 20 porciento de los casos de diagnóstico reciente de DMD; esto significa que las mutaciones se encuentran solo en los oocitos.11 Las mujeres con estas mutaciones pueden producir varones afectados o mujeres portadoras, por lo que las mujeres e hijas de las madres de pacientes con DMD y DMB deben estudiarse en forma independiente para detectar a las portadoras.11 La manifestación de la DMD en las mujeres heterocigotas ocurre cuando el cromosoma X paterno normal que porta el gen de la distrofina se inactiva en una gran proporción de células embriónicas (hipótesis de Lyon). La enfermedad en estas mujeres puede ser tan severa como en los hombres.
Cardiomiopatía dilatada ligada al X
La cardiomiopatía dilatada ligada al X (CDLX) se debe a la deficiencia de distrofina en el músculo cardiaco, pero no en el esquelético. Se presenta como un trastorno cardiaco progresivo, ocurriendo insuficiencia cardiaca congestiva en la segunda o tercera década de la vida. Los portadores femeninos con manifestaciones tienen una cardiomiopatía de inicio lento que ocurre alrededor de la quinta década de la vida. Se ha propuesto que la causa de la enfermedad es una deleción cerca del exón 1 del gen de la distrofina, que afecta la expresión o función de la misma en el músculo cardiaco.5,11
Síntomas miopáticos leves, inespecíficos
Las distrofinopatías pueden presentarse como debilidad miopática inespecífica leve, aumento en la concentración de CK, intolerancia al ejercicio, calambres musculares, hipertrofia aislada de las pantorrillas o debilidad aislada del cuadríceps.
SARCOGLICANOPATIAS EN LAS DISTROFIAS MUSCULARES DE LAS CINTURAS DE LAS EXTREMIDADES
Se demostró por primera vez deficiencia de alfa-sarcoglucano en una distrofia muscular autosómica recesiva severa de la infancia (DMARSI) en familias del norte de Africa.1,5,11 La presentación de la DMARSI es semejante a la forma severa de DMD pero afecta a varones y mujeres. En la DMARSI los tres componentes del complejo sarcoglucano están ausentes, pero el complejo distroglucano es normal, lo que sugiere una verdadera sarcoglucanopatía.1,12 Se han mapeado mutaciones en los genes para el alfa, beta, gama y delta-sarcoglucano en los cromosomas 17q12, 4, 13q12 y 5q33, respectivamente.8,12 Se han identificado mutaciones en los genes de sarcoglucanos en varias familias europeas y americanas con distrofias musculares de las cinturas de las extremidades (DMCE) autosómico recesivas que incluyen deleciones sin sentido, en contra sentido y de marco de lectura y que ocasionan fenotipos de DMCE de severidad clínica variable. Estas mutaciones causan un ensamble inapropiado de los sarcoglucanos con los otros complejos proteicos de distroglucanos, interrumpiendo el enlace entre el sarcolema y la matriz extracelular.
Los hombres y las mujeres prsentan debilidad muscular proximal leve a severa (en especial en las piernas), elevación de la CK a alrededor de 3,000 unidades, cambios distróficos en el músculo y con frecuencia hipertrofia de pantorrillas. La edad de inicio es variable. La inmunocitoquímica y el análisis de inmunoblot de las muestras de biopsia muscular para la distrofina son normales, pero el sarcoglucano está ausente o muy deficiente. Ocurre deficiencia en las proteínas sarcoglucanos (fácilmente detectable usando anticuerpos contra alfa-sarcoglucano) en el 20 porciento de los pacientes con DMCE que tienen distrofina normal. Se han reportado mutaciones en los genes respectivos en hasta el 60 porciento de los pacientes.
MEROSINOPATIAS Y DISTROFIAS MUSCULARES CONGENITAS
Las distrofias musculares congénitas (DMC) son un grupo heterogéneo de trastornos determinados genéticamente. Se caracterizan por cambios distróficos en el músculo, elevación leve a importante de la CK, grados variables de debilidad muscular, contracturas articulares que afectan los codos, caderas, rodillas y tobillos (conocidas como artrogrifosis cuando existen desde el nacimiento) y progresión que comienza al nacer o en la infancia temprana. Tres formas de DMC se asocian también con alteraciones estructurales del cerebro, malformaciones oculares y deterioro intelectual. Estas incluyen el tipo Fukuyama (observado en Japón), el síndrome de Walker-Warburg (distrofia muscular displasia cerebro-ocular) y la enfermedad músculo-ojo-cerebro del tipo finlandés.13 Sin embargo, en la forma clásica de DMC no existe malformación cerebral o retraso mental. La enfermedad puede ocurrir también en adultos jóvenes.
Más del 40 porciento de los niños con la forma clásica de DMC tienen deficiencia de merosina en la lámina basal del músculo esquelético.13 La enfermedad se asocia con leucoencefalopatía definida por IRM (sin signos evidentes de deterioro intelectual), hipotonía neonatal, niveles elevados de CK en suero, retraso en el desarrollo motor, neuropatía axonal y debilidad de los músculos respiratorios. La DMC atribuida a deficiencia de merosina se asocia con el cromosoma 6q22. En los pacientes con merosina negativa, el complejo DAG está alterado y en consecuencia se destruye la membrana muscular.
OTRAS DISTROFIAS
Distrofia muscular de Emery-Dreifuss
La distrofia muscular de Emery-Dreifuss (DMED) es un padecimiento recesivo ligado al cromosoma X que se ha mapeado en Xq28.14 Se han identificado mutaciones en esta región que han demostrado codificar una proteína llamada emerina, una proteína de la membrana nuclear.15 La DMED se presenta con afección muscular humeroperoneal (con consumo y debilidad prominente del bíceps, tríceps, tibial anterior y peroneos), y progresa en forma lenta con un patrón escapulopelviperoneo (con afección de los músculos pectoral y de la pelvis). La DMED se caracteriza por el desarrollo temprano de contracciones en los codos, flexores, tendón de Aquiles, cuello y columna, que puede ocurrir antes de que exista debilidad significativa y afección cardiaca, que se manifiesta por defectos en la conducción con bradicardia y prolongación del intervalo PR. La DMED comienza en las primeras dos décadas de la vida y su detección pronta con colocación de marcapaso puede prevenir la muerte súbita o los ataques de síncope. Las biopsias musculares revelan datos miopáticos inespecíficos. La CK puede estar ligeramente elevada. No existe un tratamiento específico, pero la terapia física puede retrasar el desarrollo de las contracturas. En la actualidad existen pruebas genéticas para identificar a los portadores, que pueden tener trastornos de la conducción cardiaca. Una prueba diagnóstica útil emplea emerina, y es posible demostrar la ausencia de esta proteína por medio de inmunocitoquímica y Western blot en muestras de biopsias musculares de los pacientes y sus familiares.
Distrofia muscular facioescapulohumeral y escapuloperonea
La enfermedad suele comenzar durante la segunda década de la vida y se presenta como debilidad de los músculos de la cara (en especial del orbicular de los ojos). Se conserva la función de los músculos extraoculares, maseteros y faríngeos.16 La debilidad temprana de los músculos escapulares produce una apariencia alada de las escápulas con apariencia de que los hombros se van hacia adelante. Existe siempre debilidad de los músculos tibiales anteriores, con pie caido. La debilidad progresa en forma lenta y existen largos periodos de estabilidad. La progresión ocurre en forma descendente: la afección de la cintura escapular es seguida de afección del bíceps, tríceps y músculos de la cintura pélvica.
La distrofia muscular fascioescapulohumeral (DFEH) se asocia al cromosoma 4q. Es una enfermedad autosómico dominante con penetrancia completa. Sin embargo, en algunos miembros de la familia solo ocurre afección facial mínima. Los niveles de CK en suero están ligeramente elevados y el músculo cardiaco no se afecta. Los datos de la biopsia son variables. Una gran mayoría de los pacientes tiene alteraciones en los capilares de la retina, desprendimiento de retina y deterioro de la audición.16
Distrofia muscular oculofaringea
La distrofia muscular oculofaríngea (DMOF) es un padecimiento autosómico dominante poco frecuente que se manifiesta entre la cuarta y la sexta décadas de la vida. Se caracteriza por ptosis y disfagia, pudiendo ser ambas severas. Puede ocurrir debilidad leve de los músculos distales. La mutación responsable de la DMOF se asocia en forma estrecha al locus del gen de la cadena pesada de la miosina en el cromosoma 14q11.2.17
Distrofia miotónica
La distrofia miotónica (DM) es la distrofia muscular del adulto más común. Tiene una incidencia de uno por 8,000 personas y una prevalencia de cinco por 100,000 habitantes.18 La miopatía tiene una distribución característica: (1) ptosis de los párpados con afección de los músculos extraoculares, (2) atrofia de los maseteros y músculos temporales, lo que ocasiona una configuración facial angosta típica, (3) atrofia del músculo esternocleidomastoideo conservación relativa de los músculos poseriores del cuello y de la cintura escapular (signo clínico que diferencía a la DM de la DFEH), (4) atrofia de grupos musculares distales, con ligera afección proximal en las fases tempranas de la enfermedad, y (5) afección de los músculos del paladar y faríngeos, que puede producir disartria y disfagia.
La miotonía, definida como una relajación lenta después de una contracción muscular normal, es un signo clínico importante. Para despertar la miotonía durante el examen clínico, el paciente da un apretón de manos firme y después se le pide que suelte con rapidez. Será evidente su incapacidad para hacerlo. La percusión de la eminencia tenar o del extensor de los dedos también muestra la relajación lenta característica de la miotonía. Las características sistémicas incluyen defectos de la conducción cardiaca, disfunción mental leve (con frecuencia con comportamiento y expresiones tontas o inapropiadas), atrofia testicular, calviece frontal, cataratas, afección del tubo digestivo (con retraso en el movimiento y vaciamiento), hipersomnia y menor respuesta a la hipoxia, que causa menor concentración y apatía. Los niños de las madres afectadas pueden tener menos movimientos fetales y síntomas tempranos de hipotonía severa, dificultad para alimentarse, debilidad facial bilateral e insuficiencia respiratoria (distrofia miotónica congénita). La DM debe distinguirse de la miotonía congénita, que tiene patrones de herencia dominantes y recesivos, se manifiesta por miotonía e hipertrofia muscular, pero no se asocia con debilidad muscular, atrofia o síntomas sistémicos.
El diagnóstico clínico de la DM se confirma por la EMG, que muestra descargas miotónicas. En los casos difíciles el examen con lámpara de hendidura puede mostrar la formación temprana de cataratas. La expresión clínica de la DM es variable y las personas afectadas pueden no ser diagnosticadas sino hasta que tienen hijos. La edad de inicio es cada vez más temprana en las generaciones sucesivas (anticipación). No es poco frecuente ver familias en las que la abuela solo tuvo cataratas tempranas, y la hija, que no sabía que estaba afectada, tuvo un hijo con distrofia miotónica congénita severa. La DM se debe a mutaciones en la región 3' no traducida del gen de la proteina cinasa de miotonina en el cromosoma 19.19,20 En las personas con afección leve, una región de repetición polimórfica CTG (citosina, timina, guanina) en la proteína cinasa se expande de 50 a 80 veces. En las personas con afección severa pueden existir más de 2,000 repeticiones. El tamaño de la expansión de la CTG aumenta por generaciones, lo que explica la anticipación.19,20 La medición de la longitud de la expansión de la CTG es una prueba genética que puede usarse para confirmar la presencia de DM en familiares, para diagnóstico prenatal o para consejo genético eficaz de personas asintomáticas con riesgo de DM.
El tratamiento es sintomático. Son indispensables el apoyo emocional y la educación sobre las precauciones para evitar caídas y lesiones. Los medicamentos como la quinidina, la procainamida, la fenitoína y los beta bloqueadores pueden ayudar a la miotonía, pero no a la debilidad.
Hipertermia maligna
La hipertermia maligna (HM) ocurre durante la anestesia general. Se caracteriza por un incremento rápido del metabolismo aeróbico y anaeróbico, durante el que la temperatura corporal puede ser superior a 43 ¡C (109.4 ¡F). Además, las concentraciones de bióxido de carbono y lactato aumentan (la concentración de bióxido de carbono arterial [PaCO2] puede ser superior a 100 mm Hg), y el pH de la sangre puede ser menor a 7. La HM se caracteriza por taquicardia, rigidez (causada por contractura muscular que puede progresar a rigidez o muerte), liberación excesiva de mioglobina del músculo y mioglobinuria, mayor permeabilidad muscular (con aumento en la concentración sérica de K+, Ca2+ y Na+), y edema muscular.21 El trismus o espasmo muscular de los maseteros que ocurre en un paciente durante la inducción de la anestesia puede indicar HM.21 La HM debe distinguirse de (1) la rabdomiolisis sola asociada con la anestesia, (2) las reacciones tóxicas a medicamentos, (3) la porfiria, (4) la tormenta tiroidea precipitada por cirugía y anestesia, y (5) el síndrome neuroléptico maligno (SNM) precipitado por fármacos psicoactivos (v.gr., haloperidol y fenotiacinas) que bloquean las vías dopaminérgicas centrales.
Entre los desencadenantes de la HM se incluyen los anestésicos volátiles potentes, como halotano, enfluorano, desfluorano, ciclopropano, éter y succinilcolina.21 Aunque ocurre HM en algunos pacientes sin enfermedad muscular conocida, los pacientes en riesgo incluyen los que tienen alteraciones musculoesqueléticas congénitas múltiples, dislocación congénita de cadera aislada, o enfermedad nculear, y algunos de los que tienen distrofia musular de Duchenne o de Becker.
La HM se hereda en forma autosómica dominante; en muchos pedigrees el locus se encuentra en el cromosoma 19q13 en el sitio del receptor de rianodine (RyR).21,22 La HM parece estar precipitada por la incapacidad para controlar las concentraciones de calcio dentro de las fibras musculares por un retículo sarcoplásmico (RS) disfuncionante y mutaciones en el gen RyR. El RyR es el canal de liberación de calcio del RS y une la brecha que existe entre el RS y el túbulo transverso. Las mutaciones en el receptor afectan la comunicación entre el RS y el túbulo transverso de modo que ocurre liberación acelerada del calcio desde el RS cuando tiene lugar la despolarización del túbulo transverso.
La prueba in vitro de contracción con cafeína-halotano, que se realiza en biopsia de músculo, se emplea para investigar la posibilidad de HM en los pacientes.23 La sensibilidad y específidad de esta prueba puede ser tan alta como 97 y 80 porciento, respectivamente.21,23
El dantroleno es el tratamiento eficaz para la HM. Este fármaco disminuye la liberación de calcio del RS sin alterar la recaptura de calcio. El uso de dantroleno ha disminuido la mortalidad de esta condición a siete porciento.21 El episodio agudo se la HM se trata en forma sintomática. Debe administrarse dantroleno intravenoso (2 a 10 mg/kg cada cinco minutos) al inicio del episodio, cuando aún existe una perfusión muscular adecuada. Para los pacientes que se sabe son susceptibles a la HM puede administrarse dantroleno en dosis de 2 mg/kg 10 a 15 minutos antes de la anestesia. La mejor manera de evitar la HM en personas susceptibles es empleando agentes anestésicos seguros (v.gr., oxido nitroso y tiopental) y relajantes musculares no despolarizantes.
Miopatías metabólicas
PRINCIPIOS GENERALES DE LOS REQUERIMIENTOS DE ENERGIA DEL MUSCULO
En el reposo la energía muscular deriva principalmente de la oxidación de los ácidos grasos libres (AGL). Durante el ejercicio aeróbico de gran intensidad el glucógeno es la principal fuente de combustible para la fosforilación oxidativa. Sin embargo, las reservas de glucógeno muscular se depletan después de 90 minutos de ejercicio,24 y si éste se prolonga, aumenta la utilización de ácidos grasos libres y glucosa sanguínea. Debido a que la disponibilidad de AAG del tejido adiposo es casi ilimitada, una persona sana normal puede realizar ejercicio moderado por muchas horas. En los pacientes con miopatías metabólicas los síntomas se vuelven evidentes durante las actividades que requieren mayor demanda metabólica, como el ejercicio. Las miopatías metabólicas son causadas por defectos en la ruptura del glucógeno o la utilización de AGL. Las glucogenosis se deben a defectos en el metabolismo y utilización del glucógeno o de la glucosa por el músculo. Se clasifican según la secuencia del defecto enzimático a lo largo de la vía glucogenolítica o glucolítica [ver figura 2]. En esta sección solo se analizarán los trastornos más comunes.
|
| Figura 2 |
| Esquema del metabolismo del glucógeno, glucólisis y ácidos grasos. |
GLUCOGENOSIS
Deficiencia de fosforilasa muscular
La deficiencia de fosforilasa muscular (también conocida como enfermedad de McArdle) es el prototipo de las glucogenosis: la ruptura del glucógeno está inhibida, lo que causa reducción de piruvato y menor cantidad de energía. Este padecimiento autosómico recesivo se presenta como intolerancia al ejercicio y mioglobinuria en pacientes mayores de 15 años. Si los pacientes descansan un poco después de presentar las mialgias y rigidez inducidas por el ejercicio, pueden reanudar su actividad con mejor resistencia (fenómeno de segundo aire), debido a mayor movilización y utilización de los AGL y la glucosa. En etapas más tardías puede desarrollarse debilidad muscular permanente. La concentración de CK en reposo suele estar aumentada. La prueba de ejercicio con isquemia en el antebrazo no muestra aumento del lactato venoso. En la biopsia muscular se encuentra ausencia de fosforilasa, presencia de vacuolas de subsrcolema y mayor acúmulo de glucógeno. El defecto es causado por mutaciones en la isoforma muscular de la fosforilasa en el cromosoma 11q13. No existe tratamiento disponible pero el entrenamiento con ejercicio aeróbico y una dieta alta en proteínas pueden ser útiles.
Deficiencia de fosfofructocinasa
La fosfofructocinasa (FFC) es una enzima con tres subunidades estructurales genéticamente diferentes: M, expresada en el músculo, eritrocitos, corazón y cerebro, L, expresada en el hígado y los eritrocitos, y P, que se expresa en las plaquetas. La deficiencia de FFC es un padecimiento autosómico recesivo. Las mutaciones en la subunidad M, localizada en el cromosoma 1, causan síntomas miopáticos y hemólisis crónica, además de aumento en la concentración de bilirrubina sérica y en la cuenta de leucocitos. Debido a que la deficiencia de FFC es un defecto glucolítico, las consecuencias funcionales de la deficiencia de FFC son semejantes a las observadas en la enfermedad de McArdle.24 Debe sospecharse deficiencia de FFC en los pacientes con intolerancia al ejercicio, náusea y mioglobinuria. Se desarrolla debilidad muscular permanente en etapas tardías de la vida. La historia de hemólisis leve, compensada y crónica, una cuenta alta de reticulocitos, el aumento en la bilirrubina y la hiperuricemia, también indican deficiencia de FFC, en especial en ciertos grupos étnicos, como los japoneses y los judíos Ashkenazi.
DEFICIENCIA DE MALTASA ACIDA
La deficiencia de maltasa ácida (DMA) es una enfermedad por almacenamiento de glucógeno autosómica recesiva causada por deficiencia de la alfa-glucosidasa, una enzima condificada en un gen localizado en el cromosoma 17q23.25 Las mutaciones o deleciones pequeñas que causan una ruptura anormal afectan la expresión de la alfa-glucosidasa. Existen tres formas clínicas de DMA: infantil, de la infancia y del adulto. La forma infantil (enfermedad de Pompe) se presenta dentro de los primeros meses de vida como hipotonía, debilidad y crecimiento del corazón, lengua e hígado, ocurriendo cambios respiratorios y cardiovasculares que causan la muerte antes de los dos años. La forma de la infancia se manifiesta por miopatía, con retraso de las habilidades motoras, debilidad muscular proximal, afección de los músculos respiratorios y crecimiento de las pantorrillas.25 La enfermedad causa la muerte para la segunda década de la vida.
La forma adulta de la DMA se presenta en personas mayores de 20 años como debilidad muscular proximal que recuerda a la polimiositis o a la distrofia de las cinturas de las extremidades. La debilidad de los músculos respiratorios puede ser el síntoma de presentación en una tercera parte de los adultos con DMA. El acúmulo de glucógeno ocurre principalmente en el músculo. Sin embargo, la enzima es deficiente en el músculo, hígado, corazón y fibroblastos cultivados. El paciente tiene elevación de la CK sérica, la EMG muestra descargas miotónicas prominentes (sin miotonía clínica), en especial en los músculos paraespinales, y la biopsia muscular muestra múltiples vacuolas con alta concentración de glucógeno que reaccionan en forma intensa con la fosfatasa ácida, lo que indica mayor actividad lisosomal. Debido a que la utilización de glucógeno y glucosa no se alteran, la DMA causa debilidad estable, pero no intolerancia al ejercicio o mioglobinuria. La fibra muscular sufre un proceso autofágico por alteraciones en los lisosomas.25 El diagnóstico se confirma por ensayo bioquímico del nivel enzimático en el músculo, cultivo de fibroblastos, linfocitos u orina. En algunas familias es posible realizar análisis de genética molecular. No existe un tratamiento específico.
MIOPATIAS POR ALMACENAMIENTO DE LIPIDOS Y TRASTORNOS DEL METABOLISMO DE LOS LIPIDOS QUE AFECTAN AL MUSCULO
Los lípidos como fuente de energía para el músculo
Durante el ejercicio rápido y extenuante la energía para el músculo es aportada por vía anaeróbica por la glucosa derivada de la degradación del glucógeno. Poco después de que comience el ejercicio aumenta la circulación, el aporte de glucosa, AGL y oxígeno hacia el músculo. Durante el ejercicio sostenido los ácidos grados de cadena larga (AGCL) son la principal fuente de energía, derivan de los alimentos o, durante el ayuno, del tejido adiposo. Los AGCL requieren ser transportados primero hacia la mitocondria. La transferencia de AGCL a través de la membrana mitocondrial interna requiere de L-carnitina y dos enzimas, las carnitina palmitoiltransferasas (CPT I y CPT II), que se localizan en las membranas mitocondriales externa e interna, respectivamente. Dentro de la mitocondria la §-oxidación se facilita primero por las deshidrogenasas de acil coenzima A (acil-CoA) y después por la transferencia de electrones a través de flavoproteínas hacia las proteínas de la cadena respiratoria.26,27 Las miopatías por almacenamiento de lípidos son causadas por menor oxidación de los ácidos grasos por las mitocondrias, que se debe a defectos en (1) la carnitina y la CPT, que alteran el transporte de ácidos grasos a través de la membrana mitocondrial, (2) las enzimas asociadas con la §-oxidación y (3) las proteínas de la cadena respiratoria y las flavoproteínas que transfieren electrones.26,27
Deficiencia de carnitina primaria y secundaria
La carnitina deriva principalmente de la dieta, pero el 25 porciento se sintetiza en el hígado a partir de lisina y metionina.26 La carnitina es crucial para la oxidación de los AGCL. La consecuencia de la deficiencia de carnitina es la disfunción del hígado, corazón y tejidos musculares, que son muy dependientes de la oxidación de AGCL. La deficiencia primaria de carnitina (DPC) es causada por ausencia de receptores de carnitina de alta afinidad funcionales, lo que trae como consecuencia un transporte deficiente de la carnitina a través de las membranas celulares. Se considera que los defectos en el transporte de lípidos a la mitocondria asociados con DPC causan un desplazamiento hacia la vía glucolítica. Esto causa acúmulo de acil-CoA en la mitocondria, que esterifica a la canitina libre para formar acilcarnitinas. Estas acil-CoA, son fácilmente liberadas de la mitocondria y excretadas en la orina.26,28
Los pacientes con DPC sufren cardiomiopatía progresiva, episodios de hipoglucemia hipocetósica (por disfunción hepática) y debilidad miopática proximal. Los lípidos se acumulan y forman gotas pequeñas.
Sin embargo, las causas más comunes de deficiencia de carnitina son secundariasy se deben a (1) §-oxidación defectuosa, que se asocia con acidurias orgánicas, (2) disfunción mitocondrial y defectos en las proteínas de la cadena respiratoria, (3) enfermedad renal, como el síndrome de Fanconi, la cistinosis nefropática o la hemodiálisis crónica, y (4) tratamiento con fármacos, en especial zidovudina (AZT) y valproato.26,28
Los suplementos con carnitina producen resultados variables.
Deficiencia de carnitina palmitoiltransferasa
En los lactantes la deficiencia de CPT I se manifiesta como síndrome de Reye, con encefalopatía hepática, hipoglucemia hipocetótica e hiperamonemia. En los adultos, el síndrome de deficiencia de carnitina suele causar deficiencia de CPT II. El gen para la CPT II se localiza en el cromosoma 11p11-p13, y se han descrito mutaciones sin sentido.27 La deficiencia de CPT II representan la causa más común de mioglobinuria en adultos jóvenes. Los pacientes presentan ataques de rigidez muscular, calambres, mialgias y mioglobinuria horas después de realizar ejercicio prolongado o constante, en especial después del ayuno o cuando la energía del músculo depende de la utilización de los AGCL y no de la utilización de glucógeno o glucosa. Los pacientes con deficiencia de CPT-II no tienen menor tolerancia al ejercicio, fenómeno de segundo aire o signos de alarma como mialgias que eviten que continúen haciendo ejercicio. Entre los ataques la fuerza muscular y la CK sérica son normales. El diagnóstico se establece midiendo la actividad de la CPT II en el músculo.
No es claro el motivo por el que la deficiencia de CPT causa ataques intermitentes de mioglobinuria y por qué no se acumulan lípidos en el músculo. No existe tratamiento para evitar los ataques mioglobinúricos. Sin embargo, se recomiendan una dieta alta en carbohidratos, baja en lípidos, comidas frecuentes e ingesta extra de carbohidratos antes y durante el ejercicio sostenido.
Miopatías mitocondriales y encefalopatías
Estas enfermedades constituyen un grupo diverso de trastornos que afectan no solo al músculo y al sistema nervioso, sino también otros órganos. La principal función de la motocondria es generar energía para la célula al producir trifosfato de adenosina (ATP) por medio de la fosforilación xoidativa (FO). La FO se basa en cinco complejos enzimáticos, que incluyen coenzimas y transición de componentes metálicos (hierro, cobre), localizados en la membrana mitocondrial interna. Estos complejos (designados como I, II, III, IV [citocromo oxidasa] y V) colectan y transfieren en forma secuencial electrones derivados del catabolismo de grasas, proteínas y carbohidratos, al O2. El acoplamiento de la oxidación y fosforilación por un gradiente de protones a través de la membrana mitocondrial interna permite la fosforilación del difosfato de adenosina (ADP) para producir ATP.
La mitocondria contiene su propio ADN extracromosómico (ADN mt), que es diferente al ADN nuclear. Es una molécula circular de doble cadena que codifica 24 ARN estructurales, 2 ARN ribosomales (ARNr), 22 ARN de transferencia (ARNt) y 13 ARNm. Los ARNm codifican varios polipéptidos de la cadena respiratoria. El resto de las subunidades de la FO y otras proteínas mitocondriales son codificadas por genes nucleares. La organización del ADNmt es muy compacta, no tiene intrones. Por lo tanto, las mutaciones aleatorias en el ADNmt suelen afectar una secuencia que codifica, con un padecimiento resultante. Además, el ADNmt es susceptible de sufrir daño por radicales del oxígeno por su proximidad a la producción de estos radicales por la FO y porque sus mecanismos de reparación son mínimos.29,30 Todo el ADNmt de cada persona es heredado de la madre (herencia no mendeliana) porque el espermatozoide contribuye solo con su ADN nuclear que pasa al cigoto durante la fertilización. En ocasiones pueden ocurrir trastornos de la FO por mutaciones en algunos genes de la FO codificados en el nucleo, y estas enfermedades siguen un patrón de herencia mendeliano.
Las biopsias musculares de los pacientes con defectos de la FO son anormales y revelan fibras rojas rasgadas en la tinción tricrómica o fibras azules rastadas en la tinción con succinato deshidrogenasa. Esto es resultado del acúmulo de mitocondrias en la periferia de las fibras musculares. Al microscopio electrónico la mitocondria tiene inclusiones paracristalinas o crestas anormales.30 Al estudiar el ADNmt se detectan mutaciones, deleciones o depleciones específicas.
DELECIONES ESPORADICAS DEL ADNmt: SINDROME DE KEARNS-SAYRE Y OFTALMOPLEGIA EXTERNA PROGRESIVA CRONICA
El síndrome de Kearns-Sayre (SKS) se presenta en pacientes menores de 20 años y se caracteriza por oftalmoplegia, ptosis, retinitis pigmentosa y debilidad miopática. Son comunes la estatura corta, los defectos en la conducción cardiaca, los niveles elevados de proteínas en el líquido cefalorraquídeo, los síndromes cerebelosos, la hipoacusia sensorineural y los niveles elevados de lactato en suero. La biopsia muscular revela fibras rojas rasgadas. Cuando la enfermedad comienza en personas mayores de 20 años y la oftalmoplegia domina el fenotipo, se clasifica como oftalmoplegia externa progresiva crónica (OEPC).31 El SKS y la OEPC más limitada se caracterizan por una sola deleción larga del ADNmt de entre nueve y 50 pares de bases. Con más frecuencia la deleción ocurre en forma esporádica y rara vez se hereda por vía materna. La FO es defectuosa y las actividades de los complejos I y IV se reducen. Las variantes de OEPC, caracterizadas por deleciones múltiples del ADNmt en los genes de la FO codificados por el ADN nuclear pueden trasmitirse en forma autosómica dominante o recesiva.
MUTACIONES PUNTIFORMES DEL ADNMT HEREDADAS POR VIA MATERNA
Síndrome MELAS
La presencia de encefalomiopatía mitocondrial (crisis convulsivas focales o generalizadas y migrañas recurrentes), acidosis láctica y episodios semejantes a eventos cerebrovasculares se combinan para formar el síndrome MELAS (por las siglas en inglés, n. del t.). La biopsia muscular de los pacientes afectados revela fibras rojas rasgadas. Los pacientes pueden tener también hipoacusia, estatura corta, cardiomiopatía, diabetes o degeneración pigmentaria de la retina que recuerda al SKS o a la OEPC. Hasta el 80 porciento de los enfermos tienen mutaciones puntiformes del ADNmt en el gen ARNt leucina.32 Estas mutaciones son heteroplásmicas, lo mismo que las otras mutaciones del ADNmt, y pueden coexistir el ADNmt normal y mutado en la misma célula. Normalmente una célula contiene entre dos y 10 moléculas de ADNmt, lo que permite que una mutación que de otra forma sería letal (i.e., deterioro letal de la FO) persista en organismos viables.
Síndrome MERRF El síndrome MERRF (por las siglas en inglés, n. del t.) consiste en epilepsia mioclónica y miopatía con fibras rojas rasgadas. Además, son comunes la ataxia, demencia, sordera, debilidad, atrofia muscular y alteraciones cardiacas, aunque su expresión es variable y depende del grado de heteroplasmía. Alrededor del 80 porciento de los pacientes con MERRF tienen una mutación en el gen ARNt lisina.33
Neuropatía óptica hereditaria de Leber Una enfermedad de adultos jóvenes cada vez más reconocida (con mayor prevalencia en hombres que en mujeres), la neuropatía óptica hereditaria de Leber (NOHL), se manifiesta por pérdida de la visión indolor, subaguda y bilateral. Se han observado varias mutaciones en el ADNmt. Algunos pacientes con NOHL y diferentes mutaciones pueden tener otros trastornos asociados, como encefalopatía, sordera, ataxia, mielopatía o distonía.34
Trastornos de los canales de iones, parálisis periódica familiar y miotonías no distróficas
PRINCIPIOS GENERALES DE EXCITABILIDAD DE LA MEMBRANA
Después de la excitación de la unión neuromuscular, los potenciales de acción se propagan de acuerdo a flujos de iones a través de la membrana sarcolema, que dependen de la apertura (activación) y cierre (inactivación) de los canales iónicos apropiados. En las células musculares y nerviosas la apertura del canal de voltaje de Na+ causa un rápido incremento en la permeabilidad del Na+ y despolarización de la membrana. Para que la membrana inicie el siguiente potencial de acción los canales de Na+ deben cerrarse. Los canales de voltaje de K+ se abren y salen iones K+ fuera de la célula, creando un voltaje hiperpolarizado a través de la membrana celular. Los canales de Cl- contribuyen a la repolarización al estabilizar el potencial de membrana.
Las alteraciones en la excitabilidad de la membrana pueden ocasionar miotonía y parálisis periódica.35 La miotonía, como un síntoma de las miotonías no distróficas, ocurre en la mitonía congénita, una enfermedad autosómico dominante o recesiva que se manifiesta por hipertrofia muscular sin ataques paralíticos. La miotonía se presenta como rigidez no dolorosa después de un periodo de inactividad, que mejora después del movimiento continuo (fenómeno de calentamiento). Es causada por mutaciones en las proteínas del canal de cloro.36 Cuando se desarrolla miotonía después de la exposición al frío y empeora con el ejercicio (miotonía paradójica) o se asocia con ataques de debilidad, el padecimiento se conoce como paramiotonía congénita (PC). La PC es causada por mutaciones en el canal de Na+.
Los otros trastornos de la excitabilidad de la membrana son la hipocalemia y la parálisis periódica hipercalémica. Los ataques paralíticos se asocian con hipercalemia (que puede coexistir con PC), debida a mutaciones en los canales del Na+, o con hipocalemia, causada por mutaciones en el gen para el receptor de dihidropiridina, que funciona como un canal de voltaje de calcio.35,36
Trastornos de los canales de sodio
Los trastornos de los canales de sodio incluyen la parálisis periódica hipercalémica (PPhiperK), la paramiotonía congénita y la miotonía de los canales de sodio. Se han asociado con el gen SCN4A, que codifica la isoforma adulta del canal de Na+, en el cromosoma 17q13.1-13.37,38 La PPhiperK y la PC son trastornos alélicos del mismo gen. La miotonía de los canales de sodio, llamada antes miotoía congénita con respuesta a acetazolamida o miotonía fluctuans, también se asocia al mismo locus. Se presenta como miotonía con fenómeno de calentamiento y sin debilidad después del enfriamento. Después de la ingestión de K+ la miotonía aumenta.39 Se han identificado varias mutaciones sin sentido en pacientes con este trastorno.
La PPhiperK y la PC son enfermedades autosómico dominantes. Se caracterizan por ataques de debilidad que comienzan en la infancia o niñez temprana. Los episodios son precipitados por el resposo después del ejercicio, estrés, administración de K+ y ciertos alimentos. El frío puede inducir miotonía paradójica en algunos pacientes. Los que tienen PC pueden tener también ataques episódicos que no se asocian con PPhiperK.35 Puede ocurrir debilidad entre los ataques, que se presenta después de varios años como una miopatía. Algunos pacientes con PPhiperK pueden tener arritmias cardiacas con prolongación del QT o ectopia ventricular y características dismórficas. La frecuencia de los ataques e incluso la debilidad entre los mismos responde a los inhibidores de la anhidrasa carbónica como la acetazolamida y la diclorfenamida.35
Trastornos de los canales del calcio
Los trastornos de los canales del calcio se manifiestan como parálisis periódica hipocalémica (PPhipoK), una enfermedad autosómico dominante localizada en el cromosoma 1q31-q32 cerca del gen que codifica al receptor de dihidropiridina muscular.36,40 La PPhipoK se manifiesta por parálisis episódica que comienza en la primera o segunda década de la vida, por lo general durante el sueño. Esta es precipitada por el consumo de carbohidratos, el reposo después del ejercicio y la excitación. Durante los ataques la concentración de K+ en suero disminuye y ocurre retención urinaria de Na+ y agua. Entre los ataques muchos pacientes desarrollan una miopatía permanente y lentamente progresiva. La enfermedad debe distinguirse de todas las causas de hipocalemia secundaria que causan debilidad, incluyendo uso de diuréticos, enfermedad renal, hiperaldosteronismo, intoxicación por orozuz, abuso de laxantes y enfermedades gastrointestinales perdedoras de potasio.35 En la hipercalemia secundaria no ocurren ataques paralíticos a menos que la concentración de K+ sea menor de 3 mEq (casi siempre menos de 2.5 mEq). Por el contrario, los ataques en la PPhipoK ocurren incluso con concentraciones de K+ cercanas a lo normal. La PPhipoK puede asociarse con tirotoxicosis, en especial en descendientes de asiáticos. En este caso la PPhipoK responde al uso de propranolol o a la normalización de la función tiroidea. Aunque la PPhipoK se hereda con patrón dominante, una tercera parte de los pacientes puede tener una enfermedad esporádica. Puede requerirse para el diagnóstico la prueba de provocación con infusión de insulina y glucosa con monitoreo cardiaco y respiratorio. La frecuencia de los ataques y la debilidad entre los mismos responde al uso de acetazolamida y diclorfenamida.41
Trastornos de los canales del cloro
Los trastornos de los canales del cloro incluyen la miotonía congénita autosómico dominante (enfermedad de Thomsen), que ocurre en la quinta década de la vida y se asocia con hipertrofia muscular y miotonía congénita autosómico recesiva (enfermedad de Becker), que se presenta en niños menores de tres años. La miotonía mejora con el ejercicio. Los pacientes con estos trastornos no tienen parálisis periódica o debilidad, aunque en la forma de Becker puede existir debilidad temporal, que mejora con el ejercicio. Son datos característicos la miotonía por percusión y la miotonía generalizada, referida por el paciente como rigidez. Ambos padecimientos se han relacionado con mutaciones diferente en el gen del canal del cloro en el cromosoma 7q35.36
Neuromiotonía autoinmune adquirida
La mioquimia, o músculo que se retuerce durante el reposo, la menor relajación muscular con rigidez en reposo, los calambres dolorosos y el aumento en la sudoración caracterizan a esta enfermedad adquirida. Es causada por hiperactividad de las terminales periféricas de los nervios motores y correctamente debe denominarse neuromiotonía porque la actividad continua de la fibra muscular es abolida por curare y no por bloqueo del nervio proximal. La neuromiotonía ocurre en forma esporádica, pero han ocurrido casos familiares que con frecuencia se asocian con neuropatía. También se han observado asociaciones con miastenia gravis y timoma. Un anticuerpo dirigido contra los canales de K+ sensibles a o-dendrotoxina puede ser el responsable de la enfermedad.42 El tratamiento es sintomático (fenitoína, carbamacepina y mexiletine) pero en casos resistentes puede requerirse inmunoglobulina intravenosa o plasmaféresis.
Miopatías tóxicas inducidas por drogas
PRINCIPIOS GENERALES
Aunque las miopatías inducidas por drogas no son raras, son difíciles de identificar en la práctica clínica. El médico debe sospechar una miopatía tóxica en un paciente que no tenía una enfermedad muscular previa, cuya miopatía se desarrolla en forma aguda o subaguda (en ocasiones lentamente) con debilidad y dolor muscular, que presenta mioglobinuria después de la administración del posible agente miotóxico, o que mejora al suspender el medicamento tóxico [ver tabla 1].43 Los agentes miotóxicos pueden causar miopatía al afectar en forma directa a los organelos musculares, como la mitocondria, induciendo una reacción inmunológica, o al inducir efectos sistémicos secundarios como alteraciones de los electrolitos, deprivación nutricional (v.gr., el agente compite con vitaminas) y malabsorción. Varios medicamentos pueden ser miotóxicos tanto solos como al combinarlos con otros [ver tabla 1], y el clínico debe estar alerta respecto al potencial miotóxico de los medicamentos más recientes.
|
||
|
MIOPATIA POR ZIDOVUDINA Y TOXICIDAD DE OTROS ANALOGOS DE NUCLEOSIDOS
Las características clínicas de la miopatía por zidovudina incluyen debilidad muscular proximal, mialgias (sobre todo en los muslos y pantorrillas), fatiga, cambios miopáticos en la EMG y elevación de los niveles de CK en suero, que aumenta más con el ejercicio.44 La pérdida de peso y la elevación en la concentración de lactato en suero pueden anteceder al inicio de miopatía por zidovudina. La zidovudina puede causar síntomas miopáticos después de un año de su administración. En un seguimiento de 20 pacientes con miopatía por zidovudina, cuatro a seis semanas después de suspender el medicamento las mialgias se resolvieron, mejoró o se normalizó la fuerza y la actividad espontánea en la EMG disminuyó o desapareció.45
Las características histológicas típicas de la miopatía por zidovudina incluyen fibras rojas rasgadas, sugestivas de alteraciones mitocondriales, y muchas fibras negativas para detección de oxidasa de citocromo c. La zidovudina, un terminador de la cadena de ADN, inhibe a la gama-polimerasa presente en la matriz mitocondrial, con terminación de la síntesis de ADNmt, por lo que se presenta depleción hasta del 78 porciento del ADNmt muscular.46 La zidovudina es el único análogo de nucleósidos miotóxico usado en el tratamiento del síndrome de inmunodeficiencia adquirida (SIDA). La dideoxicitosina , la dideoxinosina y la lamivudina causan neuropatía axonal dolorosa, pero no miopatía. Aunque los inhibidores de la proteasa de uso reciente no se han asociado con miotoxicidad, se requiere una observación cuidadosa para determinar si ésta ocurre después de su uso prolongado.
MIOPATIA POR AGENTES REDUCTORES DEL COLESTEROL
Varios agentes reductores del colesterol [ver tabla 1] causan una miopatía casi siempre reversible que se caracteriza por debilidad proximal, mialgias, elevación de la concentración de CK en suero y cambios miopáticos en la EMG. Debido a que el colesterol es el principal esterol constituyente de las membranas musculares, la reducción del reservorio normal de colesterol disponible para la síntesis de membrana puede aumentar la fluidez de la membrana, causando un sarcolema inestable, descargas miotónicas y, en casos avanzados, rabdomiolisis. El EMG presenta potenciales de fibrilación y complejos miotónicos o descargas repetitivas.43,44 Puede ocurrir recuperación gradual después de suspender el agente agresor.
Aunque son comunes el aumento temporal en la concentración sérica de CK y las mialgias después del tratamiento con lovastatin, el medicamento se ha asociado con síntomas miopáticos clínicos en menos del cinco porciento de los pacientes tratados. El uso de medicamentos combinados aumenta el riesgo de miopatía. La coadministración de ciclosporina y lovastatin en pacientes con trasplantes de corazón o riñón e hiperlipidemia parece aumentar la incidencia de miopatía con rabdomiolisis.44
MIOPATIA POR ESTEROIDES Y AGENTES BLOQUEADORES (MIOPATIA DE LA ENFERMEDAD GRAVE)
Los pacientes con parálisis prolongada inducida por agentes bloqueadores no despolarizantes como el pancuronio pueden manifestar una miopatía aguda después de suspender la ventilación mecánica. La mayoría de estos pacientes recibieron dosis altas de esteroides por un estado asmático u otro padecimiento grave por el que se indujo parálisis artificial para asegurar una higiene pulmonar adecuada.47 Aunque la combinación de esteroides y agentes bloqueadores se ha implicado en forma consistente en el desarrollo de miopatía de la enfermedad grave, en ocasiones la exposición a uno de estos agentes puede ser mínima.44 El cuadro clínico se caracteriza por debilidad severa y generalizada de las extremidades, incapacidad para retirar el ventilador mecánico, atrofia muscular, elevación normal o moderada de la concentración de CK en suero y cambios miopáticos en la EMG. Por lo general la debilidad mejora en forma lenta.48 La miopatía por agentes bloqueadores-esteroides suele coexistir con una neuropatía axonal, constituyen la neuromiopatía de la enfermedad grave.48 El diagnóstico diferencial debe incluir los defectos de trasmisión neuromucular, el síndrome de Guillain-Barré, la miopatía inflamatoria o necrosante aguda y la parálisis periódica.44 La biopsia muscular muestra alteraciones morfológicas francas, con pérdida de los filamentos gruesos, lo que se caracteriza por áreas centrales vacías sin signos de necrosis, inflamación o fagocitosis. Con la tincion de la trifosfatasa de adenosina (ATPasa) se observan áreas de palidez central en muchas fibras. La microscopía electrónica revela pérdida selectiva y extensa de los miofilamentos gruesos, con conservación de los miofilamentos delgados y las líneas Z. Es necesario tener precaución al usar dosis altas de esteroides en pacientes que reciben agentes paralizantes por periodos prolongados.
INTOXICACION POR VITAMINA E
Se ha reportado una miopatía necrosante con debilidad muscular proximal y elevación del nivel de CK en suero en pacientes automedicados con dosis altas de vitamina E.49
MIOPATIA POR CLOROQUINA
El antimalárico cloroquina es empleado con frecuencia por los reumatólogos para el tratamiento de varias enfermedades difusas del tejido conectivo. Puede ocasionar degeneración macular y corneal, neuropatía periférica y miopatía. La miopatía se observa con la administración prolongada de dosis altas (500 mg diarios). Esta tiene características morfológicas interesantes que recuerdan a las que ocurren en la deficiencia de maltasa ácida: múltiples vacuolas con material positivo a fosfatasa ácida, cuerpos mieloides dentro de las vacuolas y lisosomas grandes con aumento en la actividad enzimática.
MIOPATIA POR COLCHICINA
La colchicina interfiere con el crecimiento de los microtúbulos, afectando la mitosis. Después de su uso prolongado, causa una miopatía vacuolar y una neuropatía axonal, con frecuencia en pacientes entre los 50 y 70 años de edad y que tienen gota e insuficiencia renal leve crónica.50 Los síntomas incluyen debilidad proximal, elevación de la concentración de CK, afección neurosensorial distal y arreflexia. Los síntomas se resuelven cuatro a seis semanas después de suspender el medicamento.
ESTEROIDES
Los pacientes con hiperadrenocorticismo (i.e., síndrome de Cushing) pueden desarrollar debilidad. Pueden ocurrir cambios semejantes durante la administración crónica de prednisona (casi siempre en dosis mayores de 20 mg al día) o dexametasona. La debilidad miopática inducida por esteroides es leve, respeta los músculos flexores del cuello,44 y puede agravar la debilidad causada por el trastorno inmune o la neoplasia para la que se administró el esteroide. La disminución de la dosis del medicamento revierte la miopatía. La concentración sérica de CK es normal y la EMG no proporciona información específica.
MIOPATIA ALCOHOLICA
Los alcohólicos desarrollan principalmente una neuropatía axonal periférica y en ocasiones miopatía aguda o crónica. Precedida por edema muscular y dolor, la miopatía aguda se presenta como rabdomiolisis y mioglobinuria y es dolorosa.43 Puede recurrir si el paciente continúa bebiendo. La miopatía aguda en los alcohólicos puede relacionarse también con hipocalemia (< 2.5 mEq). Esta miopatía no es dolorosa, se asocia con edema muscular y es rápidamente reversible. La debilidad muscular proximal en alcohólicos crónicos suele ser multifactorial y no necesariamente un proceso miopático; por ejemplo, puede deberse a desnutrición, inactividad o enfermedad neurogénica.43 Desde el punto de vista histológico, es común la atrofia de las fibras tipo II. Algunos bebedores crónicos pueden sufrir elevación asintomática de la concentración de CK (hasta 20 veces el normal) que se agrava por la actividad física.
MIOPATIA INFLAMATORIA INDUCIDA POR DROGAS
Los pacientes con enfermedad de Wilson, artritis reumatoide o esclerodermia pueden desarrollar polimiositis o miastenia gravis durante el tratamiento con D-penicilamina. La enfermedad responde a la suspensión del medicamento o la administración de esteroides.
En reportes aislados y no confirmados se ha asociado el uso de procainamida, propiltiouracilo y cimetidina con un padecimiento semejante a polimiositis. Los autores no han observado ningún caso.
El L-triptofano contaminadofue responsable de un brote de un síndrome caracterizado por fascitis eosinofílica, miositis, engrosamiento de la piel, neuropatía axonal y otras manifestaciones sistémicas. Un proceso autoinmune contra los fibroblastos en la matriz extracelular desencadenado por el contaminante fue implicado como causa del síndrome, al que se denominó síndrome de mialgia-eosinofilia.51 La enfermedad ha dejado varios pacientes con engrosamiento residual de la piel, mialgia y fatiga.
El interferón alfa (IFN-alfa), usado cada vez más para algunas neoplasias y hepatitis, causa fatiga severa, artralgias y en ocasiones mialgia. Se desconoce el mecanismo exacto de la fatiga muscular y las mialgias. Es bien conocida la toxicidad al SNC que causan las dosis altas de IFN-alfa.
La citocina interleucina-2, usada para el carcinoma de células renales, puede exacerbar la polimiositis o dermatomiositis previas.
Trastornos de la trasmisión neuromuscular
UNION Y TRASMISION NEUROMUSCULAR NORMAL
Las principales áreas de la unión neuromuscular son (1) la región presináptica, que activa zonas que contienen dos filas paralelas de canales de calcio dependientes de voltaje (CCDV) y vesículas sinápticas que contienen acetilcolina (AC), (2) el espacio sináptico y (3) la región posináptica, que consiste en pliegues de unión que contienen receptores de acetilcolina (RAC) [ver figura 3]. Desde el punto de vista funcional, la trasmisión neuromuscular depende de la liberación de AC de las terminales de los nervios motores, la interacción de la AC con los RAC y la despolarización resultante de la fibra muscular. En un estado de reposo, existe liberación aleatoria de AC desde una vesícula en particular. La AC se une al RAC y abre un canal catiónico selectivo, que permite que entre Na+ a la región de la placa terminal. Esto crea un potencial de despolarización denominado potencial miniatura de placa terminal (PMPT).52 A continuación la AC se disocia del RAC y es rápidamente hidrolizada por la acetilcolinesterasa (ACE). Cuando la ACE es inhibida, como ocurre durante el tratamiento con medicamentos anticolinesterasa como la piridostigmina, más moléculas de AC se unen a RAC y abren múltiples canales iónicos.
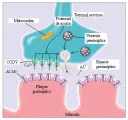
|
| Figura 3 |
| Trasmisión neuromuscular |
Cuando el potencial de acción alcanza la porción terminal del nervio se abren los CCDV y fluyen iones de Ca2+ hacia la terminal nerviosa. El ingreso de Ca2+ ocasiona la fusión y exocitosis de las vesículas sinápticas y la liberación de moléculas de AC (cada vesícula contiene de 6,000 a 10,000 moléculas de AC) que difunden y se diseminan en los pliegues de unión. La liberación simultánea de AC por la fusión de 50 a 300 vesículas causa un potencial de placa terminal (PPT). Si el PPT excede cierto umbral, un potencial de acción muscular (PAM) origina la contracción del músculo. En condiciones normales las interacciones entre la AC liberada y el RAC son más que suficientes para producir PPT capaces de desencadenar un PAM. La diferencia entre la amplitud del PPT actual y la amplitud requerida para desencadenar un PAM se denomina margen de seguridad.
Existen tres padecimientos principales que afectan la trasmisión neuromuscular: la miastenia gravis (MG), el síndrome miasténico de Eaton-Lambert (SMEL) y varios síndromes miasténicos congénitos.
MIASTENIA GRAVIS
La miastenia gravis es el prototipo de las enfermedades autoinmunes. Satisface todos los criterios inmunológicos:5254 se conoce el antígeno, el RAC, pueden detectarse anticuerpos específicos contra éste y medirse en el suero de los pacientes afectados, la enfermedad puede trasmitirse a animales de experimentación a través de la IgG patogénica de los pacientes, la inmunoglobulina se une al RAC, causando destrucción dependiente del complemento, y la eliminación de los anticuerpos ocasiona mejoría clínica.
En la MG los anticuerpos dirigidos contra el RAC disminuyen el número de receptores disponibles. En consecuencia, la AC liberada no puede fijarse en forma suficiente a los RAC, por lo que el PPT es pequeño e insuficiente para desencadenar un PAM y la trasmisión neuromuscular falla. Cuando ocurre falla de la trasmisión neuromuscular en muchas uniones neuromusculares, el músculo no puede activarse del todo, y esto causa fatiga y debilidad. Durante la estimulación nerviosa repetida en personas sanas la cantidad de AC liberada disminuye después de los primeros impulsos, pero la trasmisión neuromucular se conserva porque el margen de seguridad es suficiente. En los pacientes con MG el PPT es de sí pequeño. Durante la estimulación nerviosa repetida el PPT disminuye más porque la menor cantidad de AC liberada en los estímulos posteriores no es suficiente para activar un número adecuado de RAC, con lo que se decrementa más el PAM. Los estudios de estimulación nerviosa repetida se usan como diagnóstico al demostrar la menor trasmisión neuromuscular en los pacientes con MG.
Epidemiología
La prevalencia de la MG es de alrededor de cuatro a seis casos por 100,000 habitantes. La incidencia anual varía, pero se reconocen dos picos, uno se asocia con mujeres en la segunda o tercera décadas de la vida y el otro con varones de más de 60 años. La enfermedad ocurre 50 porciento más en mujeres que en hombres. La edad promedio de inicio es de 28 años en las mujeres y 42 en los varones. Debido a que la MG afecta al RAC nicotínico en la unión neuromuscular, solo se altera el sistema motor.
Puede desarrollarse también MG clásica en niños, que responde bien a timectomía o anticolinesterasas. La MG puede relacionarse con timoma en hasta el 15 porciento de los adultos, o puede presentarse después de extirpar el timoma.52-54 Además, hasta el cinco porciento de los pacientes con MG tienen otros padecimientos autoinmunes sistémicos, el 15 porciento presenta otros autoanticuerpos, el 20 porciento tiene enfermedad tiroidea y hasta el cinco porciento puede tener historia familiar de otra enfermedad autoinmune. El embarazo tiene un efecto variable, pero en la mayoría de los pacientes la debilidad aumenta en el periodo posparto. Alrededor del 12 porciento de los niños que nacen de madres con MG desarrollan MG neonatal temporal como resultado de la transferencia placentaria de anticuerpos anti-RAC circulantes. Estos niños desarrollan debilidad, trastornos para alimentarse, problemas respiratorios, llanto débil y debilidad facial en las primeras horas después de nacidos. La condición dura hasta tres semanas, coincidiendo con la vida media de la IgG.
Inmunopatogenia de la MG
Autoanticuerpos La MG es mediada por autoanticuerpos patógenos que se unen al RAC y causan pérdida funcional de los receptores y destrucción focal de los pliegues de unión posináptica, interfiriendo con la despolarización de la membrana posináptica [ver figura 4].

|
| Figura 4 |
| Destrucción del RAC en la miastenia gravis por anticuerpos |
Entre el 10 y 20 porciento de los pacientes con MG, en especial los que tienen formas leves, de la infancia o localizadas (v.gr., oculares), no tienen anticuerpos detectables. La mayoría de estos pacientes tienen una MG autoinmune clásica, pero los anticuerpos son de baja afinidad o están dirigidos contra epítopes que no están presentes en los extractos solubles del RAC que se usan en los ensayos estándar. Si el ensayo de unión a RAC es negativo, debe realizarse un inmunoensayo que usa anticuerpos para detectar al RAC presente en las células musculares en el cultivo de tejido.
Otros anticuerpos asociados con la MG incluyen los anticuerpos contra células estriacionales. Estos se encuentran en hasta el 80 porciento de los pacientes con timoma y MG, 25 porciento de los enfermos con timoma sin MG, 30 porciento de los adultos con MG (55 porciento cuando la MG comienza después de los 60 años), 6 porciento de los pacientes con SMEL, 3 porciento de los enfermos con cáncer pulmonar y con frecuencia en pacientes con enfermedad hepática autoinmune.55 Son frecuentes los anticuerpos contra microsomas tiroideos y tiroglobulina en los pacientes con MG ocular y con SMEL sin cáncer.
Inmunorregulación El RAC es un antígeno dependiente de células T. En la MG las células T CD4+ están sensibilizadas y responden a la estimulación con RAC con péptidos inmunodominantes sintéticos del RAC.56,57 Debido a que los linfocitos de las personas sanas también responden a estos péptidos, aunque en menor grado, las células T específicas contra RAC son parte del repertorio inmune normal. Esto sugiere que la MG es una enfermedad causada por alteración en la inmunorregulación. Se ha postulado que antígenos microbianos (v.gr., Escherichia coli y Klebsiella) o virus herpes simple, inducen la MG porque estas bacterias y virus comparten una secuencia homóloga con péptidos de la subunidad alfa del RAC.
Alrededor del 75 porciento de los pacientes con MG tienen alteraciones tímicas, sea hiperplasia con formación de centros germinales o timoma (en el 15 porciento de todos los pacientes con MG). La extirpación del timo causa mejoría clínica o remisión. Las evidencias adicionales que implican un papel para el timo en la MG se basan en diversos aspectos: (1) el timo de las personas afectadas contiene una mayor población de células B que pueden secretar anticuerpos contra el RAC que el timo de una persona normal sana, (2) el timo de una persona afectada posee células mieloides que contienen RAC cercanos a las células dendríticas interdigitantes, y (3) el número de células sensibilizadas contra el RAC es mayor en el timo de una persona afectada que en el de una persona normal sana.52,53
En la MG la ruptura de la autotolerancia puede comenzar en el timo. Los RAC en las células mieloides pueden ser detectadas por las células dendríticas del timo que presentan el antígeno a las células T CD4+, las que a su vez estimulan a las células B para que produzcan anticuerpos contra el RAC. Estos anticuerpos reconocen y reaccionan con los RAC en el músculo esquelético.
La MG se asocia con frecuencia con otros padecimientos autoinmunes, incluyendo enfermedades difusas del tejido conectivo, polimiositis, pénfigo, enfermedades tiroideas autoinmunes y enfermedad injerto contra huésped. El uso de la D-penicilamina ha inducido MG clásica, que mejora al suspender el fármaco.
Manifestaciones clínicas
La MG se manifiesta por debilidad muscular y fatiga. Es característica la debilidad para elevar los párpados y mover los músculos extraoculares, lo que causa ptosis asimétrica y diplopia. Los extensores del cuello suelen ser débiles y la cabeza cae. La debilidad de los músculos facial y bulbar puede ocasionar una mueca cuando el paciente intenta sonreir, lenguaje nasal, disártrico, disfónico y de bajo volumen, y disfagia, que puede ocasionar reflujo y ahogamiento. La debilidad de los músculos proximales y la fatiga producen dificultad para caminar, subir escaleras, peinarse y cargar objetos. Puede haber debilidad significativa de los músculos respiratorios. Los síntomas fluctúan y son menores por la mañana. La fatiga empeora hacia el final del día o con la actividad repetida. Si se le pide a un paciente que abduzca los brazos, se observa que los baja en foma gradual. Si se le pide que hable en forma continua, la voz se volverá ronca, nasal, lenta y al final inaudible. La sensación y la función cognoscitiva son normales. Los reflejos tendinosos, en especial los de los músculos no atróficos, suelen estar exaltados o ser normales. Los dedos de los pies ven hacia abajo. Los síntomas empeoran durante o antes del periodo menstrual y durante las infecciones virales o bacterianas.
Diagnóstico
Además de demostrar la fatiga y debilidad a la exploración, el diagnóstico clínico puede confirmarse por medio de la prueba de edrofonio o neostigmina. Estos medicamentos inhiben a la ACE, lo que permite a la AC interactuar en forma repetida con los RAC restantes, con mejoría consecuente de la fuerza. Los estudios de estimulación nerviosa repetida demuestran una reducción rápida (mayor del 12 porciento) en la amplitud del PAM. La EMG de una fibra demuestra mayor irritabilidad o bloqueo en más del 90 porciento de los pacientes. Debe realizarse una IRM del tórax en busca de hiperplasia tímica o timoma. La MG debe distinguirse de otras enfermedades o trastornos inducidos por farmacos que presentan cuadros clínicos parecidos, como el botulismo, la intoxicación por organofosfatados, la reacción tóxica por D-penicilamina, la miopatía mitocondrial, la lesión compresiva de los nervios cranealeas, el SMEA y los síndromes miasténicos congénitos.
El diagnóstico serológico de MG se realiza detectando anticuerpos contra el RAC con un ensayo de radioinmunoprecipitación. Estos anticuerpos están presentes en hasta el 90 porciento de los pacientes con MG generalizada y hasta 70 porciento de los que tienen MG ocular. Se encuentran también en el 30 porciento de los pacientes con enfermedad hepática autoinmune, 10 porciento de enfermos con anemia perniciosa y hasta el 13 porciento de los que tienen SMEL.55 En general, los títulos de anticuerpos contra RAC no correlacionan con la severidad clínica.
Tratamiento
El término gravis en la actualidad no es correcto porque la MG responde bastante bien a los tratamientos disponibles. Aunque la secuencia de aplicación de las diversas modalidades terapéuticas puede diferir entre los médicos, la elección inicial en los casos leves suele ser un medicamento anticolinesterasa cada cuatro horas mientras el paciente está despierto. En la mayoría de las instituciones se realiza timectomía por vía transesternal después de la pubertad y hasta los 60 años de edad.
La prednisona es el tratamiento inmunoterapéutico de elección. Suelen administrarse 60 a 80 mg al día en una dosis única por la mañana durante un periodo inicial de tres a cuatro semanas. Después se reduce la dosis en forma gradual y en días alternos durante un periodo de ocho semanas a razón de 10 mg por semana (más rápido si se observan efectos adversos severos) hasta que se alcance la dosis más baja posible que controle la enfermedad. Algunos médicos prefieren iniciar con la dosis más baja de prednisona (i.e., 40 mg/día) y aumentarla en forma gradual para evitar un empeoramiento temporal que se ha observado en algunas ocasiones. La plasmaféresis o la Ig intravenosa suele reservarse para las crisis y los casos severos, y antes de la timectomía. La ciclosporina o azatioprina pueden usarse como tratamiento de segunda elección. Por ejemplo, la azatioprina es un inmunosupresor eficaz que se administra por vía oral en dosis de hasta 3 mg/kg.
SINDROME MIASTENICO DE EATON-LAMBERT
En el SMEL los anticuerpos específicos contra los CCDV causan modulación antigénica y depleción de los CCDV. Esto restringe el ingreso de Ca2+ en la terminal del nervio motor y reduce la liberación de AC durante el impulso nervioso. La cantidad de AC generada puede originar solo un PPT pequeño, que no es capaz de desencadenar un PAM; esto ocasiona falla en la trasmisión neuromuscular y debilidad muscular. Una observación importante en el SMEL es la asociación frecuente con cáncer pulmonar de células pequeñas (CPCP), que también expresa CCDV.54
El SMEL ocurre con más frecuencia en hombres que en mujeres. Alrededor del 70 porciento de los hombres con SMEL tienen CPCP, comparado con el 20 porciento de las mujeres. El SMEL puede preceder a la detección de neoplasias hasta por tres años. Las neoplasias son raras en las personas menores de 40 años.
La enfermedad se presenta como debilidad muscular proximal, aumento en la fatiga y síntomas oculares temporales de ptosis o diplopia. Es frecuente la sintomatología autonómica, con boca y ojos secos, impotencia sexual, constipación y reflejos pupilares anormales. Es común la hiporreflexia. Un signo típico es el aumento en la fuerza muscular y los reflejos pocos segundos después de un esfuerzo máximo sostenido. El diagnóstico se confirma por los estudios de estimulación nerviosa repetitiva que demuestran un aumento dramático en la amplitud del PAM hasta la estimulación tetánica. La EMG de una sola fibra muestra mayor irritabilidad y bloqueo, que mejora al aumentar la frecuencia de estimulación. Usando péptidos radiomarcados de toxinas de víbora de cascabel, pueden demostrarse anticuerpos contra los canales de calcio tipo P o tipo Q en hasta el 95 porciento de los pacientes con SMEL.54 Se observan anticuerpos contra los canales tipo N en hasta el 75 porciento de los pacientes con SMEL, en especial los que tienen cáncer pulmonar.58 La búsqueda de CPCP, en especial en los varones mayores de 40 años, debe ser parte indispensable del proceso diagnóstico.
La región presináptica de las placas terminales motoras en los pacientes con SMEL muestra depleción marcada de las partículas de la zona activa que representan los CCDV. Los CCDV en el SMEL no se localizan en filas paralelas, sino que se agregan como resultado de su interacción con los anticuerpos IgG específicos. La IgG del paciente [la fracción F(abÕ)2], al inyectarse a ratones, trasmite los cambios electrofisiológicos y morfológicos asociados con la enfermedad. El complemento no participa en el proceso. El CPCP expresa CCDV del tipo N, L o P. Se cree que las células tumorales desencadenan autoanticuerpos contra determinantes de la superficie, que reaccionan en forma cruzada con epítopes similares en las regiones presinápticas de las uniones neuromosculares.
En el SMEL asociado con CPCP, el foco primario consiste en tratar el tumor porque los síntomas del SMEL mejoran al remitir el cáncer. El medicamento de elección para el SMEL es la 3,4-diaminopiridina. Este medicamento prolonga la duración del potencial de acción presináptico al bloquear las corrientes externas de K+. Esto permite una mayor entrada de Ca2+ hacia la terminal nerviosa, con mayor liberación de AC. El fármaco alivia la fatiga y la debilidad en los pacientes con SMEL y es bien tolerado en dosis de 40 a 60 mg al día. En ocasiones se agrega piridostigmina, y la prednisona ayuda mucho. La azatioprina, la plasmaféresis y la Ig intravenosa constituyen otros recursos terapéuticos que se administran como agentes ahorradores de esteroides o para mejorar la fuerza de los pacientes que no responden bien a la 3,4-diaminopiridina y la prednisona.
SINDROMES MIASTENICOS CONGENITOS
Los síndromes miasténicos congénitos son un grupo heterogéneo de síndromes que causan falla de la trasmisión neuromuscular que no es de tipo autoinmune sino resultado de defectos genéticos en varias moléculas, enzimas o canales en la unión neuromuscular.
Se sospecha un síndrome miasténico congénito cuando un niño, infante o, en ocasiones, adulto joven, presenta ptosis fluctuante, fatiga, mayor debilidad durante el ejercicio constante (todos síntomas comunes de los defectos de trasmisión neuromuscular), retraso en el desarrollo motor, ausencia de MG autoinmune y una historia familiar positiva.
Figuras 1, 3 y 4 Tom Moore
Figura 2 Dana-Burns-Pizer
Bibliografía
- Campbell KP: Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell 80:675, 1995
- Matsumura K, Tome FMS, Collin H, et al: Deficiencies of the 50K dystrophin-associated glycoprotein in severe childhood autosomal recessive muscular dystrophy. Nature 359:320, 1992 [PMID 1406935]
- Hoffman EP, Brown RH, Kunkel LM: Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51:919, 1987 [PMID 3319190]
- Koenig M, Hoffman EP, Bertelson CJ: Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in mouse and affected individuals. Cell 50:509, 1987 [PMID 3607877]
- Engel AG, Yamamoto M, Fischbeck KH: Dystrophinopathies. Myology. Engel AG, Franzini-Armstrong C, Eds. McGraw-Hill, New York, 1994, p 1133
- Matsumura K, Nonaka I, Tome FMS, et al: Mild deficiency of dystrophin-associated proteins in Becker muscular dystrophy patients having in-frame deletions in the rod domain of dystrophin. Am J Hum Genet 53:409, 1993 [PMID 8328458]
- Sunada Y, Campbell KP: Dystrophin glycoprotein complex: molecular organization and critical role in skeletal muscle. Curr Opin Neurol 8:379, 1995 [PMID 8542044]
- Duggan DJ, Gorospe R, Fanin M, et al: Mutations in the sarcoglycan genes in patients with myopathy. N Engl J Med 336:618, 1997 [PMID 9032047]
- Mendell JR, Moxley RT, Griggs RC, et al: Randomized, double-blind six-month trial of prednisone in Duchenne's muscular dystrophy. N Engl J Med 320:1592, 1989 [PMID 2657428]
- Karpati G, Ajdukovic D, Arnold D, et al: Myoblast transfer in Duchenne dystrophy. Ann Neurol 34:8, 1993 [PMID 8517684]
- Mendell JR, Sahenk Z, Prior TW: The childhood muscular dystrophies: diseases sharing a common pathogenesis of membrane instability. J Child Neurol 10:150, 1995 [PMID 7782608]
- Campbell KP: Adhalin gene mutations and autosomal recessive limb-girdle muscular dystrophy. Ann Neurol 38:353, 1995
- Arahata K, lshii H, Hayashi YK: Congenital muscular dystrophies. Curr Opin Neurol 8:385, 1995
- Grimm T, Janka M: Emery-Dreifuss muscular dystrophy. Myology. Engel AG, Franzini-Armstrong C, Eds. McGraw-Hill, New York, 1994, p 1188
- Manilal S, Nguyen TM, Sewry CA, et al: The Emery-Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum Mol Genet 6:810, 1996
- Munsat TL: Facioscapulohumeral disease and the scapuloperoneal syndrome. Myology. Engel AG, Franzini-Armstrong C, Eds. McGraw-Hill, New York, 1994, p 1220
- Tome FMS, Fardeau M: Oculopharyngeal muscular dystrophy. Myology. Engel AG, Franzini-Armstrong C, Eds. McGraw-Hill, New York, 1994, p 1233
- Harper PS, Rudel R: Myotonic dystrophy. Myology. Engel AG, Franzini-Armstrong C, Eds. McGraw-Hill, New York, 1994, p 1192
- Ashizawa T, Dubel JR, Dunne PW, et al: Anticipation in myotonic dystrophy: complex relationships between clinical findings and structure of the GCT repeat. Neurology 42:1877, 1992 [PMID 1407566]
- Tsilfidis C, MacKenzie AK, Mettler G, et al: Correlation between CTG trinucleotide repeat length and frequency of severe congenital myotonic dystrophy. Nat Genet 1:192, 1992 [PMID 1303233]
- Gronert GA: Malignant hyperthermia. Myology. Engel AG, Franzini-Armstrong C, Eds. McGraw-Hill, New York, 1994, p 1661
- MacLennan DH, Phillips MS: Malignant hyperthermia. Science 256:789, 1993
- MacKenzie AK, Allen G, Lahey D, et al: A comparison of the caffeine halothane muscle contracture test with the molecular genetic diagnosis of malignant hyperthermia. Anesthesiology 75:4, 1991
- Martin A, Haller RG, Barohn R: Metabolic myopathies. Curr Opin Rheumatol 6:552, 1994 [PMID 7865373]
- Engel AG, Hirschhorn R: Acid maltase deficiency. Myology. Engel AG, Franzini-Armstrong C, Eds. McGraw-Hill, New York, 1994, p 1553
- Di Donato S: Disorders of lipid metabolism affecting skeletal muscle: carnitine deficiency syndromes, defects in the catabolic pathway, and Chanarin disease. Myology. Engel AG, Franzini-Armstrong C, Eds. McGraw-Hill, New York, 1994, p 1587
- Zierz S: Carnitine palmitoyltransferase deficiency. Myology. Engel AG, Franzini-Armstrong C, Eds. McGraw-Hill, New York, 1994, p 1577
- Dalakas MC, Leon-Monzon ME, Bernardini I, et al: The zidovudine-induced mitochondrial myopathy is associated with muscle carnitine deficiency and lipid storage. Ann Neurol 35:482, 1994 [PMID 8154877]
- Johns DR: Mitochondrial DNA and disease. N Engl J Med 333:638, 1995
- Wallace DC: Diseases of the mitochondrial DNA. Annu Rev Biochem 61:1175, 1992
- Holt IJ, Harding AK, Morgan-Hughes JA: Deletions of mitochondrial DNA in patients with mitochondrial myopathies. Nature 331:717,1988 [PMID 2830540]
- Moraes CT, Ricci E, Bonilla E, et al: The mitochondrial tRNA(Leu [UUR])muta-tion in mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS): genetic, biochemical, and morphological correlations in skeletal muscle. Am J Hum Genet 50:934, 1992 [PMID 1315123]
- Wallace DC, Zheng X, Lott MT, et al: Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological and biochemical characterization of a mitochondrial DNA disease. Cell 55:601, 1988 [PMID 3180221]
- Holt IJ, Miller DH, Harding AK, et al: Genetic heterogeneity and mitochondrial DNA heteroplasmy in Leber's hereditary optic neuropathy. J Med Genet 26:739, 1989 [PMID 2575667]
- Lehmann-Horn F, Engel AG, Ricker K, et al: The periodic paralyses and para myotonia congenita. Myology. Engel AG, Franzini-Armstrong C, Eds. McGraw-Hill, New York, 1994, p 1303
- George AL: Molecular genetics of ion channel disease. Kidney Int 48:118, 1995
- Ebers GC, George AL, Barchi RL, et al: Paramyotonia congenita and hyperkalemic periodic paralysis are linked to the adult muscle sodium channel gene. Ann Neurol 30:810, 1991 [PMID 1686388]
- Ptacek U, Gouw L, Kwiecinski H, et al: Sodium channel mutations in paramyotonia congenita and hyperkalemic periodic paralysis. Ann Neurol 33:300, 1993
- Ricker K, Moxley RT, Heine R, et al: Myotonia fluctuans. Arch Neurol 51:1095, 1994 [PMID 7980103]
- Ptacek LJ, Tawil R, Griggs RC, et al: Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell 77:863, 1994 [PMID 8004673]
- Dalakas MC, Engel WK: Treatment of "permanent" muscle weakness in familial hypokalemic periodic paralysis. Muscle Nerve 6:182, 1983 [PMID 6855804]
- Sinha S, Newsom-Davis J, Mills K, et al: Autoimmune aetiology for acquired neuromyotonia (Isaac's syndrome). Lancet 2:75, 1991
- Victor M, Sieb JP: Myopathies due to drugs, toxins, and nutritional deficiency. Myology. Engel AG, Franzini-Armstrong C, Eds. McGraw-Hill, New York, 1994, p 1697
- Dalakas MC: Inflammatory and toxic myopathies: Curr Opin Neurol Neurosurg 5:645, 1992
- Mhiri C, Baudrimont M, Bonne G, et al: Zidovudine myopathy: a distinctive order associated with mitochondrial dysfunction. Ann Neurol 29:606, 1991 [PMID 1892364]
- Lewis W, Dalakas MC: Mitochondrial toxicity of antiviral drugs. Nat Med 1:417, 1995 [PMID 7585087]
- Danon MJ, Carpenter S: Myopathy with thick filament loss following prolonged paralysis with vecuronium during steroid treatment. Muscle Nerve 14:1131, 1991 [PMID 1684024]
- Hirano M, Ott B, Raps E, et al: Acute quadriplegic myopathy: a complication of steroids, nondepolarizing blocking agents or both. Neurology 42:2082, 1992 [PMID 1436516]
- Neville HE, Ringel SP, Guggenheim MA, et al: Ultrastructural and histochemical abnormalities of skeletal muscle in patients with chronic vitamin E deficiency. Neurology 33:483, 1983 [PMID 6188077]
- Kuncl RW: Colchicine myopathy and neuropathy. N Engl J Med 316:1562, 1987
-
Illa I, Dinsmore S, Dalakas MC: Immune-mediated mechanisms
and immune activation of fibroblasts in the pathogenesis of eosinophiliamyalgia
syndrome induced by l-tryptophan. Hum Pathol 24:702, 1993 [PMID
8100551]
-
Drachman DB. Myasthenia gravis (review). N Engl J Med 330:1797,
1994
-
Engel AG: Acquired autoimmune myasthenia gravis. Myology.
Engel AG, Franzini-Armstrong C, Eds. McGraw-Hill, New York, 1994, p 1769
-
Vincent V, Newsom-Davis J: Immunology of the motor
nerve terminal. Immunology of Neuromuscular Disease. Hohlfeld R , Ed. Kluwer
Academic Publishers, Lancaster, England, 1994, p 147
-
Lennon VA: Serological diagnosis of myasthenia gravis
and the Lambert-Eaton myasthenic syndrome. Handbook of Myasthenia Gravis.
Lisak R, Ed. Marcel Dekker, New York, 1994, p 149
-
Engel AG: Acquired autoimmune myasthenia gravis. Myology.
Engel AG, Franzini-Armstrong C, Eds. McGraw-Hill, New York, 1994, p 1769
-
Drachman DB: Myasthenia gravis. N Engl J Med 330:1797,
1994
- Lennon VA, Kryzer TJ, Griesmann GE, et al: Calcium-channel antibodies in Lambert-Eaton myasthenic syndrome and other paraneoplastic syndromes. N Engl J Med 332:1467, 1995