Contenido del artículo
IV EVALUACION CLINICA DE LAS ENFERMEDADES GLOMERULARES
DR. BURTON D. ROSE
Patología y patogenia
El daño glomerular puede ser ocasionado por una gran variedad de procesos, que se discuten en otra parte de la obra. Sin embargo, es útil revisar algunas consideraciones importantes en relación con la patología, patogenia y características clínicas de las glomerulopatías, así como con su diagnóstico diferencial.
Muchas de las enfermedades glomerulares se definen por los cambios patológicos (más o menos específicos) que se observan bajo el microscopio de luz, de inmunofluorescencia o electrónico. Por ejemplo, bajo microscopio de luz, la enfermedad de cambios minimos se asocia con muy pocas o ninguna alteracion, mientras que la glomerulonefritis membranoproliferativa se caracteriza por engrosamiento de la membrana basal glomerular (MBG) y proliferacion celular dentro del glomérulo.
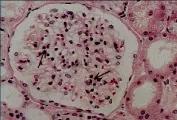
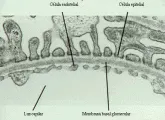
Para comprender lo que significa un cambio membranoso o proliferativo es importante recordar la histologia del glomérulo normal [ver figura 1]. La microscopía de luz muestra asas capilares permeables, paredes capilares delgadas y una o dos células por glomérulo; la porción central del ovillo glomerular contiene tanto células mesangiales como matriz extracelular, que está constituida por material similar a la membrana basal. La anatomía de la pared del capilar glomerular puede definirse mejor por medio de microscopía electrónica, que demuestra tres capas: la célula endotelial fenestrada, la MBG (que permite el paso de agua y solutos pequeños, pero evita la filtración de macromoléculas), y las células epiteliales, con sus podocitos característicos [ver figura 1]
|
|
|
|
|



|
|

Parecen ser varios los factores que determinan el sitio más probable para el deposito inmune1. Se piensa, por ejemplo, que los complejos inmunes circulantes de gran tamaño se atrapan dentro del mesangio y después en el espacio subendotelial, regiones que se encuentran separadas del plasma sólo por la célula endotelial fenestrada [ver porción inferior de figura 1]. La formación de depósitos subepiteliales es más difícil porque las macromoléculas reponsables deben cruzar la MBG. Existen dos factores que influyen en forma importante en la formación de estos depósitos: (1) la formación in situ de complejos antígeno-anticuerpo, en donde el antígeno y el anticuerpo libres (cada uno de los cuales es menor al complejo intacto) se depositan en forma independiente, y (2) la presencia y tipo de la carga antigénica (los antígenos catiónicos pueden unirse a la MBG aniónica y atravesarla con más facilidad que los antígenos con carga negativa o neutra).1
La velocidad con la que se eliminan los complejos inmunes puede también contribuir a la susceptibilidad a la enfermedad glomerular. El C3b, que se forma por la activación de la vía clásica del complemento mediada por complejos inmunes, tiene un papel central para prevenir la enfermedad al limitar el crecimiento de la red de antígeno-anticuerpo y al facilitar la llegada de complejos inmunes al sitio de su depuración en el sistema retículoendotelial. Este último proceso es mediado en parte por la fijación de los complejos a los receptores de C3b en los eritrocitos2. La menor actividad de este sistema se asocia con eliminación deficiente de los complejos inmunológicos y mayor propensión al desarrollo de enfermedad por los mismos. Por ejemplo, se presenta lupus eritematoso generalizado (LEC) en hasta la mitad de los pacientes con deficiencia hereditaria de C2, un componente temprano de la vía clásica del complemento.2 Por lo tanto, el sistema del complemento parece tener una función dual en los padecimientos glomerulares mediados por complejos inmunes; primero, protege al glomérulo al aumentar la velocidad de eliminación de los complejos y, segundo, si los complejos se depositan en el glomérulo, facilita el daño tisular, en parte por la formación local del complejo de ataque a la membrana (C5b-C9).1
El sitio de depósito de los complejos inmunes y las fracciones del sistema del complemento influyen en forma importante en el tipo de lesión glomerular que se produce y en sus manifestaciones clínicas consecuentes.3 La activación del sistema del complemento provoca la formación de C3a y C5a, que pueden atraer a los neutrófilos y células mononucleares circulantes, ocasionando una respuesta infiamatoria local. Esta se manifiesta clínicamente por sedimento urinario activo (ver adelante) y una reducción brusca en la velocidad de filtración glomerular(VFG).
Esta secuencia de eventos se observa con frecuencia en depósitos mesangiales o subendoteliales, que están separados de la circulación sistémica sólo por la célula endotelial fenestrada. Por el contrario, los depósitos subepiteliales (como los que existen en la nefropatía membranosa) están separados de la circulación por la MBG. Cuando estos existen puede formarse C3a y C5a, pero no tener acceso a las células inflamatorias.3 Por lo tanto, no existirá ni sedimento activo ni insuficiencia renal aguda, siendo la principal manifestación clínica la proteinuria, con frecuencia en rangos nefróticos. El daño local del complejo de ataque de membrana a las células epiteliales del glomérulo y quizá a la MBG parece ser el responsable de este aumento en la excreción de proteínas.
El sitio en que se depositan los complejos inmunes también tiene implicaciones importantes para la duración de la lesión glomerular. Las celulas inflamatorias infiltrantes asociadas con los depósitos mesangiales o subendoteliales tienen función fagocítica, removiendo los depósitos inmunológicos. Como resultado de ello, puede presentarse una resolución relativamente rápida del proceso si el estímulo antigénico desencadenante se controla. Por el contrario, la ausencia de células inflamatorias cuando los depósitos son subepiteliales provoca que la eliminación de los mismos sea muy lenta. Por ejemplo, el oro y la penicilamina pueden inducir nefropatía membranosa, y la suspensión del tratamiento permite la desaparición de la proteinuria en todos los pacientes. Sin embargo, el tiempo promedio de recuperación es casi de un año, y algunos pacientes requieren de más de dos años para recuperarse.4
Datos clínicos y de laboratorio
Existen muchas alteraciones clínicas que pueden observarse en las enfermedades glomerulares (ver tabla 1).5 La patogenia de la hematuria y la proteinuria se analiza en otra subsección y se mencionará sólo en forma breve. Es posible que la hemorragia glomerular sea resultado del paso de eritrocitos a través a dos factores: la selección por tamaño (los poros funcionales impiden la filtración de moléculas más gran des) y por carga eléctrica (los proteoglicanos con carga negativa, como el heparán sulfato, de la MBG retrasan el paso de la albúmina aniónica por repulsión electrostática). Ocurre proteinuria glomerular cuando existe un defecto en alguna de las dos barreras. Por ejemplo, en la enfermedad por cambios mínimos, la proteinuria es causada principalmente por pérdida de la barrera de cargas. En comparación, en la mayoría de las otras enfermedades glomerulares se daña la MBG en forma directa, lo que causa pérdida tanto de la selección por tamaño, como por carga.6-7
|
|
|
Los factores de volumen y de vasoconstriccion contribuyen a la hipertensión que se asocia con frecuencia a las glomerulopatías.5 En el proceso agudo, el aumento en la presión arterial es causado principalmente por expansión de volumen, coexistiendo una supresión adecuada de los agentes vasoconstrictores (v.gr., angiotensina II).8,9 En estos casos, la eliminación de líquidos por medio de diuréticos o diálisis suele normalizar la presión. En los procesos más crónicos, el sistema renina-angiotensina-aldosterona se activa, quizá como resultado de la isquemia regional causada por la cicatrización glomerular.
SINDROME NEFROTICO
El síndrome nefrótico se caracteriza por un aumento marcado en la permeabilidad del glomérulo a macromoléculas, causando proteinuria importante y diversas alteraciones secundarias (ver tabla l). Por ejemplo, la hipoalbuminemia parece ser causada por pérdida de albúmina en la orina y, quizá en parte, por la reabsorción y catabolismo subsecuente de parte de la albúmina filtrada en las células tubulares proximales. Además, el aumento compensatorio esperado de la síntesis hepática de albúmina suele estar ausente o disminuido, tal vez debido a desnutrición secundaria a las pérdidas proteicas a través de la orina.10
Edema Se ha propuesto que el edema nefrótico está ocasionado principalmente por la disminución en la presión oncótica del plasma. Esto disminuye el gradiente de presión oncótica transcapilar, facilitando el paso de líquido del espacio vascular hacia el intersticio. La depleción consecuente de volumen ocasiona mayor retención renal de sodio y, por tanto, más edema. Sin embargo, algunos estudios realizados en animales y en humanos sugieren que la hipoalbuminemia no causa edema en forma directa a menos de que la concentración plasmática de albúmina sea menor de 2 g/dl. En la mayor parte de los casos, la retención de líquidos está causada por un aumento primario de la reabsorción distal de sodio (inducido por un mecanismo desconocido)11 y el volumen plasmático es normal o está aumentado, y no reducido.12 Como resultado de ello, los pacientes con síndrome nefrótico no tienen en forma típica concentraciones altas de renina o disminuidas de péptido auricular natriurético, como se esperaría si estuvieran hipovolémicos.9
La poca influencia aparente de la hipoalbuminemia leve o moderada en la distribución de líquidos es resultado de un cambio relativamente pequeño en el gradiente transcapilar de presión oncótica. Aunque la presion oncótica del plasma disminuye, existe una reducción paralela en la presión oncótica intersticial, que es ocasionada por reducción en la cantidad de la proteína que cruza la pared capilar y por aumento del flujo linfático [ver figura 2a].13
En vista de que el volumen plasmático tiende a conservarse, el tratamiento diurético para el edema suele ser bien tolerado.12 Sin embargo, puede ocurrir depleción del volumen intravascular y empeoramiento de la azoemia en los pacientes sometidos a diuresis excesiva o que tienen hipoalbuminemia importante.
Hiperlipidemia El síndrome nefrótico se caracteriza por elevación moderada a severa en las concentraciones plasmáticas de colesterol y trigliceridos.14,15 Por ejemplo, no es raro encontrar en estos casos cifras de colesterol plasmático mayores a 400 a 500 mg/dl.
Existen dos factores que contribuyen a la hiperlipidemia nefrótica, el aumento en la producción hepática de lipoproteínas que contienen apoproteína B y colesterol, y la disminución en el metabolismo periférico de las lipoproteínas. No se conoce por qué aumenta la síntesis hepática de lipoproteínas, pero es claro que la reducción en la presión oncótica del plasma influye en forma importante. La gravedad de la hiperlipidemia es inversa, y está muy relacionada al grado de hipoalbuminemia.16 Es más, la infusión de albúmina o dextrán para elevar en forma brusca la presión oncótica del plasma provoca una disminución rápida en la concentración de lípidos. 14
Por el contrario, el deterioro en el metabolismo, más que el aumento en la síntesis, es el factor responsable de la hipertrigliceridemia.15-17 El defecto en la depuración de lípidos está muy relacionado con la eliminación renal de albúmina, y no así con la presión oncótica del plasma. Este hallazgo sugiere que la pérdida urinaria de un regulador del metabolismo de lípidos, aún no identificado y que en condiciones normales no se filtra, influye en forma primordial en la hipertrigliceridemia del síndrome nefrótico.15
Los pacientes con sindrome nefrótico persistente e hipercolesterolemia marcada tienen quizá mayor propensión a la enfermedad vascular ateroesclerótica, sobre todo si también son hipertensos. 14,18 Existen observaciones experimentales que sugieren que la hiperlipidemia puede también acelerar la progresión del daño glomerular, quizá al promover dentro del glomérulo un equivalente a la ateroesclerosis.19
En el pasado, el tratamiento médico de este problema estaba limitado por la falta aparente de eficacia de la dieta, y por los efectos colaterales asociados a muchos de los fármacos usados para reducir las concentraciones séricas de lípidos.14,20 Sin embargo, los inhibidores de la reductasa de 3-hidroxi-3 metil-glutaril coenzima A (HMG-CoA), como el lovastatin o el simvastatin, pueden disminuir en forma significativa las concentraciones séricas de lípidos y suelen ser bien tolerados.14,10,21
Una preocupación con el uso de los inhibidores de la reductasa de HMG-CoA ha sido el desarrollo de rabdiomiolisis, que puede asociarse con insuficiencia renal aguda por mioglobinuria. La rabdomiolisis es poco frecuente con la monoterapia, pero ocurre hasta en el cinco porciento de los enfermos que toman también gemfibrozil y en el 30 porciento de los que reciben ciclosporina.22 En consecuencia, puede ser conveniente evitar la combinación de los inhibidores de la reductasa de HMG-CoA con estos fármacos. Las complicaciones asociadas al uso de inhibidores de la reductasa de HMG-CoA y ciclosporina son especialmente importantes en pacientes hiperlipidémicos con trasplante renal.
Estado de hipercoagulabilidad Los pacientes con síndrome nefrótico tienen también mayor incidencia de tromboembolias arteriales y venosas, en especial de trombosis de las venas profundas y de las venas renales.23,24 Entre el 10 y el 40 porciento de los enfermos pueden sufrir esta complicación, y el mecanismo de la trombosis no es bien conocido. Las menores concentraciones de antitrombina III (causadas por pérdidas urinarias), el aumento en el fibrinógeno y la mayor activación plaquetaria han sido descritos como factores desencadentantes, pero su importancia relativa no se conoce, aunque las alteraciones plaquetarias parecen ser el factor patogénico primario.23,25
La trombosis de la vena renal puede ser uni o bilateral. Puede ocurrir infarto renal, que causa dolor en el flanco, hematuria y concentraciones elevadas de deshidrogenasa láctica en plasma. Sin embargo, la trombosis de la vena renal es con frecuencia un proceso insidioso, que no provoca síntomas ni cambios en la función renal.24 En estos casos, el diagnóstico, que se establece por venografía renal o ultrasonido Doppler,26 sólo se hace si el paciente sufre una embolia pulmonar o si es evaluado en forma prospectiva. Por motivos desconocidos, la trombosis de la vena renal parece ser más frecuente en la nefropatía membranosa. 23 Algunos estudios han encontrado que del 25 al 50 porciento de los pacientes con este tipo de nefropatía sufren trombosis de la vena renal, aunque otras investigaciones han notificado una incidencia mucho menor.
El tratamiento óptimo de la trombosis de la vena renal es aún tema de controversia. Algunos investigadores han propuesto realizar venografías renales prospectivas a pacientes con nefropatía membranosa y proteinuria importante, incluso en ausencia de signos o síntomas sugestivos de trombosis, como una embolia pulmonar. Sin embargo, esta actitud no es recomendable porque ningún estudio ha comparado el riesgo de la trombosis de la vena renal no diagnosticada ni tratada con el de la anticoagulación a largo plazo, y porque pueden desarrollarse lesiones trombóticas subsecuentes en pacientes con un estudio radiográfico inicial normal.
Por otro lado, el tratamiento de la trombosis de la vena renal o de otros episodios tromboembólicos consiste en la anticoagulación con heparina y después con warfarina. Es posible que la warfarina deba continuarse mientras el paciente presente proteinuria en rangos nefróticos, porque el estado hipercoagulable persistirá.
Diagnostico diferencial
La mayor parte de las enfermedades glomerulares se diagnostican por biopsia renal porque la historia clínica del paciente no es de utilidad a menos de que exista un padecimiento sistémico, como LEG, o una infección estreptocócica reciente. Sin embargo, el examen general de orina, los hallazgos clínicos (v.gr., hipertensión o edema) y la edad del paciente suelen ser útiles para limitar el diagnóstico diferencial a algunos padecimientos.6
EXAMEN GENERAL DE ORINA
Las enfermedades glomerurales pueden producir tres patrones diferentes de alteraciones urinarias: nefróticas, nefríticas y crónicas (ver tabla 2). Debido a que estos patrones, en especial los dos primeros, son causados típicamente por padecimientos distintos, identificarlos tiene gran importancia clínica (ver tabla 3).
|
||||||
|
|
|||||||||||||||
* Aunque la glomerulonefritis posinfecciosa se asocia en forma típica con un sedimento activo durante la fase aguda, a inflamación desaparece antes que el daño a la pared capilar, causando en ocaciones un sedimento benigno con proteinuria que suele normalizarse en forma espontánea en seis a 12 meses.
|
|||||||||||||||
Sedimento nefrótico
El sedimento nefrótico se caracteriza principalmente por proteinuria severa (por lo general mayor de 2.5 g/día) y lipiduria (gotas libres de grasa, cuerpos grasos ovales y cilindros grasos). El grado de proteinuria suele medirse en la orina de 24 horas. Sin embargo, la excreción de proteínas (medida en gramos por 1.73 m2 de superficie corporal) puede medirse en forma más simple a partir de la relación entre la proteína total y la creatinina (mg/mg) en una muestra aleatoria de orina .27 Por lo tanto, si la concentración total de proteínas es de 350 mg/dlmg/dl, y la concentración de creatinina es de 100 mg/dl, la relación será de 3.5, lo que representa alrededor de 3.5 g de proteína excretada al día por 1.73 m2 de superficie corporal. También puede observarse hematuria microscópica leve, pero no existen hematuria importante ni cilindros granulares.
Este grupo de manifestaciones son causadas por enfermedades que aumentan la permeabilidad glomerular al dañar la MBG, como la enfermedad de cambios mínimos y la nefropatía diabética, o por nefropatías membranosas, que se caracterizan por depósitos inmunes subepiteliales. Sin embargo, no se encuentra proliferación celular, infiltración leucocitaria ni necrosis en estos padecimientos porque se requiere la presencia de depósitos mesangiales o subendoteliales para que se atraigan células inflamatorias (ver antes).3 Es esta falta de inflamación la responsable del sedimento urinario benigno.
Sedimento nefrítico
El sedimento nefrítico, a diferencia del nefrótico, se caracteriza por hematuria, piuria y cilindros celulares (incluyendo eritrocitarios) y granulares. El grado de proteinuria es variable, puede estar dentro de límites nefróticos, pero es el sedimento activo lo que distingue a las glomerulopatías nefríticas de los padecimientos con sedimento nefrótico. Suele existir una buena relación clinicopatológica entre los hallazgos del examen de orina y los histológicos porque los padecimientos nefríticos se asocian con cambios inflamatorios marcados en la biopsia renal, como proliferación celular y areas de necrosis.
La glomerulonefritis posestreptocócica representa una de las enfermedades en que puede existir componente nefrítico y nefrótico. El sedimento urinario activo y la insuficiencia renal aguda son causadas por depósitos inmunes subendoteliales, mientras que la proteinuria es secundaria a las gibas subepiteliales características de este padecimiento [ver figura 2d]. La proteinuria tiende a persistir durante un periodo mayor que el sedimento activo por una depuración más lenta de los depósitos subepiteliales.3,28 Los pacientes que se encuentren en esta etapa tardía parecerán tener un padecimiento puramente nefrótico.28
Los padecimientos nefríticos pueden ser divididos en focales y difusos. Bajo microscopía de luz, las glomerulonefritis focales se caracterizan por afección de menos del 50 porciento de los glomérulos, mientras que las difusas presentan lesión en más del 50 porciento de los mismos. La enfermedad foca] suele asociarse con manifestaciones clínicas relativamente leves. Estos pacientes sufren, en forma típica, hematuria y proteinuria tomáticas, o episodios recurrentes de hematuria. Las manifestaciones más graves de la enfermedad glomerular (v.gr., insuficiencia renal, hipertensión, edema y síndrome nefrótico) ocurren principalmente glomerulonefritis difusas o en las vasculitis (ver tabla 3).
Sedimento crónico
La enfermedad glomerular crónica se caracteriza por remplazo de las areas de inflamación aguda por fibrosis. Como resultado de ello, las alteraciones urinarias disminuyen, aunque puede existir insuficiencia renal progresiva. Por lo tanto, la proteinuria tiende a disminuir por una reducción en la VFG, y la hematuria, piuria y cilindros celulares son sustituidos por cilindros a céreos. En algunos pacientes los hallazgos urinarios pueden ser tan inespecíficos que será dificil asegurar que existe un padecimiento glomerular.
EDAD DEL PACIENTE
Aunque las diferentes enfermedades glomerulares pueden ocurrir a cualquier edad, muchos de estos procesos son más frecuentes en algunos grupos específicos de edad. Por ejemplo, el LEG y la glomerulonefritis membranoproliferativa idiopática afectan principalmente a pacientes entre los 15 y 40 años de edad, mientras que la amiloidosis primaria se observa, de manera típica, en enfermos mayores de 50 años.
A consecuencia de ello, el diagnóstico diferencial se facilita si se considera la edad del paciente en combinación con el patrón de cambios urinarios (ver tabla 3). Por ejemplo, en pacientes mayores de 40 años, la glomerulonefritis focal es causada, con más frecuencia, por nefropatía por IgA o por enferme membrana basal delgada, mientras que la glomerulonefritis difusa suele ser secundaria a una glomerulonefritis rápidamente progresiva (con medias lunas), va o glomerulonefritis fibrilar. Sin embargo, el diagnóstico diferencial clínico debe ser confirmado por 1 renal en la mayoría de los pacientes.
La identificación del diagnóstico más probable permitirá también elegir las pruebas de laboratorio más adecuadas. Por ejemplo, deberán realizarse determinaciones de anticuerpos antinucleares y anti-ADN si se so LEG; cultivo faríngeo y títulos de anti-ADNasa E so de probable glomerulonefritis posestreptocócica; hemocultivos para la endocarditis bacteriana; glucemia en diabetes mellitus e inmunoelectroforesis sérica urinaria para detectar paraproteínas si se sospecha amiloidosis primaria. La medición de las concentraciones plasmáticas del complemento también son útiles, aunque menos específicas, porque ocurre hipocomplementaria en muchos de los padecimientos nefríticos difusos, como la nefritis lúpica, la glomerulonefritis posinfecciosa, la glomerulonefritis membranoproliferativa y la vasculitis asociada con crioglobulinemia mixta.
Tabla 2 Adaptada de "Identification and Clinical Significance of
Urinary Casts," por G. E. Schreiner, en Archives of Internal Medicine
99:356, 1957. @ 1957 American Medical Association. Usado con autorización.
Tabla 3 De Pathophysiology of Renal Disease, 2o ed., por B. D. Rose. Mc Graw-Hill Book Co., New York, 1987, p. 139.
Bibliografía
- Couser WG: Mechanisms of glomerular injury in immmune-complex disease. Kidney Int 28:569, 1985
- Schifferli JA, Ng YC, Peters DK: The role of complement and its receptors in the elimination of immune complexes. N Engl J Med 315:488, 1986 [PMID 2942776]
- Fries JW, Mendrick DL, Rennke HG: Determinants of immune complex-mediated glomerulonephritis. Kidney Int 34:333, 1988 [PMID 2971836]
- Hall CL, Fothergill NJ, Blackwell NM, et al: The natural course of gold nephropathy: long term study of 21 patients. Br Med J 295:745, 1987
- Rose BD: Pathophysiology of Renal Disease, 2nd ed. McGraw-Hill Book Co, New York, 1987, p 139
- Myers BD, Okarma TB, Friedman S, et al: Mechanisms of proteinuria in human glomerulonephritis. J Clin Invest 70:732, 1982 [PMID 6181095]
- Guasch A, Hashimoto H, Sibley RK, et al: Glomerular dysfunction in nephrotic humans with minimal changes or focal glomerulosclerosis. Am J Physiol 260:F728, 1991 [PMID 1709791]
- Rodriguez-Iturbe B, Baggio B, Colina-Chourio J, et al: Studies on the renin-aldosterone system in the acute nephritic syndrome. Kidney Int 19:445, 1981 [PMID 7017245]
- Rodriguez-Iturbe B, Colic D, Parra G, et al: Atrial natriuretic factor in the acute nephritic and nephrotic syndromes. Kidney Int 38:512, 1990 [PMID 2146429]
- Kaysen GA: Albumin metabolism in the nephrotic syndrome: the effect of dietary protein intake. Am J Kidney Dis 12:461, 1988
-
Ichikawa I, Rennke HG, Hoyer JR, et al: Role for intrarenal
mechanisms in the impaired salt excretion of experimental nephrotic syndrome.
J Clin Invest 71:91, 1983 [PMID
6848563]
-
Geers AB, Koomans HA, Roos JC, et al: Preservation of blood
volume during edema removal in nephrotic subjects. Kidney Int 28:652, 1985
[PMID
4087686]
-
Koomans HA, Kortlandt W, Geers AB, et al: Lowered protein
content of tissue fluid in patients with the nephrotic syndrome: observations
during disease and recovery. Nephron 40:391, 1985 [PMID
4022206]
-
Appel G: Lipid abnormalities in renal disease. Kidney
Int 39:169, 1991
-
Kaysen GA: Hyperlipidemia in the nephrotic syndrome.
Kidney Int 39(suppl 31):S8, 1991
-
Joven J, Villabona C, Vilella E, et al: Abnormalities of
lipoprotein metabolism in patients with the nephrotic syndrome. N Engl
J Med 323:579, 1990 [PMID
2381443]
-
Warwick GL, Packard CJ, Demant T: Metabolism of apolipoprotein
B-containing lipoproteins in subjects with nephrotic-range proteinuria.
Kidney Int 40:129, 1991 [PMID
1921148]
-
Mallick NP, Short CD: The nephrotic syndrome and ischemic
heart disease. Nephron 27:54, 1981 [PMID
7266702]
-
Keane WF, Mulcahy WS, Kasiske BL, et al: Hyperlipidemia
and progressive renal disease. Kidney Int 39(suppl 31):S41, 1991
-
D'Amico G, Gentile MG: Pharmacological and dietary
treatment of lipid abnormalities in nephrotic patients. Kidney Int 39(suppl
31):S65, 1991
-
Rabelink AJ, Hene RJ, Erkelens DW, et al: Effects of
simvastatin and cholestyramine on lipoprotein profile in hyperlipidaemia
of nephrotic syndrome. Lancet 2:1335, 1988
-
Pierce LR, Wysowski DK, Gross TP: Myopathy and rhabdomyolysis
with lovastatin-gemfibrozil combination therapy. JAMA 264:71, 1990 [PMID
2355431]
-
Bernard DB: Extrarenal complications of the nephrotic
syndrome. Kidney Int 33:1884, 1988
-
Llach F: Hypercoagulability, renal vein thrombosis,
and other thrombotic complications of the nephrotic syndrome. Kidney Int
28:429, 1985
-
Robert A, Olmer M, Sampol J, et al: Clinical correlation
between hypercoagulability and thrombo-embolic phenomena. Kidney Int 31:830,
1987 [PMID
2437353]
-
Avasthi PS, Greene ER, Scholler C, et al: Noninvasive
diagnosis of renal vein thrombosis by ultrasonic echo-Doppler flowmetry.
Kidney Int 23:882, 1983 [PMID
6224961]
-
Ginsberg JM, Chang BS, Matarese RA, et al: Use of single
voided urine samples to estimate quantitative proteinuria. N Engl J Med
309:1543, 1983 [PMID
6656849]
- Sorger K, Gessler U, Hubner FK, et al: Follow-up studies of three subtypes of acute postinfectious glomerulonephritis ascertained by renal biopsy. Clin Nephrol 27:111, 1987 [PMID 3552342]