Infectología
⭳ Abrir artículo (PDF)1.4 MBEste artículo es idéntico en la Edición 3/2000.
Contenido del artículo
XXXII INFECCIONES POR RETROVIRUS
- Clasificación de los retrovirus
- Estructura y replicación de los retrovirus
- REGULACION DE LA EXPRESION DE HTLV IN VIVO
- ESTRUCTURA Y EXPRESION DEL VIH-1 Y VIH-2
- REGULACION DE LA EXPRESION DEL VIH IN VIVO
- HTLV-I, HTLV-II y transformación maligna por retrovirus
- TRANSFORMACION MALIGNA POR RETROVIRUS ANIMALES
- IDENTIFICACION DE LAS ENFERMEDADES CAUSADAS POR HTLV-I Y VIRUS ASOCIADOS
- EPIDEMIOLOGIA DE LA INFECCION POR HTLV-I
- IDENTIFICACION DEL HTLV-II
- EPIDEMIOLOGIA DE LA INFECCION POR HTLV-II
- TRANSFORMACION MALIGNA POR HTLV-I Y HTLV-II
- HTLV-I Y LLTA
- HTLV-I Y MIELOPATIA CRONICA
- VIH Y SIDA
- DIAGNOSTICO DE LA INFECCION POR VIH
- EPIDEMIOLOGIA DE LA INFECCION POR VIH-1
- PROGRESION DE LA ENFERMEDAD POR VIH
- EPIDEMIOLOGIA DE LA INFECCION POR VIH-2
- IDENTIFICACION DEL VIH-1
- DIVERSIDAD GENETICA DEL VIH-1
- VIGILANCIA DE LA INFECCION POR VIH-1 IN VIVO
- HISTORIA NATURAL DE LA INFECCION POR VIH-1
- MECANISMOS DE DEPLECION DE LAS CELULAS T EN LA INFECCION POR VIH-1
- RESPUESTA INMUNOLOGICA AL VIH-1
- VACUNAS PARA PREVENIR LA INFECCION POR VIH-1
- TRATAMIENTO DE LA INFECCION POR VIH
- Retrovirus y otras enfermedades humanas
- Direcciones futuras en la investigacion de retrovirus
XXXII INFECCIONES POR RETROVIRUS
DR. MARK B. FEINBERG
El potencial patogénico de los retrovirus se reconoció por primera a comienzos del presente siglo gracias a las investigaciones sobre sarcomas y leucemias en las aves. En los años siguientes, se han detectado ejemplos de enfermedad inducida de forma retroviral en una amplia variedad de especies animales.1 Sin embargo, la mayor parte de los intentos para demostrar la relevancia de los retrovirus en la patogenia de la enfermedad humana fueron infructuosos.
Sin embargo, desde 1980 al menos cuatro retrovirus humanos se han descubierto y caracterizado. Estos retrovirus humanos infectan a una célula blanco común, el linfocito T. Además, todos causan infecciones que ponen en peligro la vida, y que persisten a pesar de una respuesta inmune a menudo vigorosa, pero aparentemente ineficaz.
Aunque comparten características comunes, los retrovirus humanos se relacionan con manifestaciones clínicas diferentes. El virus linfotrófico humano de células T tipo 1 (HTLV-I) se identifica como el agente etiológico de una forma agresiva de leucemia conocida como leucemia/linfoide de células T del adulto (LLTA).2, 3 El HTLV-I también se ha relacionado con la mieloneuropatía progresiva denominada paraparesia espástica tropical (PET), o mielopatía relacionada con HTLV-I (MRH).4, 5 El HTLV-II se aisló inicialmente de un individuo con una variante indolente de células T de leucemia de células peludas, pero todavía no se ha relacionado definitivamente con ninguna enfermedad.6 El virus de inmunodeficiencia humana tipo I (VIH-I), identificado por primera vez en 1983, se ha identificado como agente causal de la mayoría de los casos del síndrome de inmunodeficiencia adquirida (SIDA) y sus estados relacionados a nivel mundial.7-9 El retrovirus humano aislado más recientemente, el virus de inmunodeficiencia humana tipo 2 (VIH-2), se identificó en 1986, y se relaciona desde el punto de vista genético y serológico al VIH-1. El VIH-2 es el agente etiológico de un número cada vez mayor de casos de inmunosupresión indistinguible del SIDA relacionado con el VIH-1.
En la actualidad se desconocen los mecanismos precisos por los cuales los retrovirus humanos inducen enfermedad. Sin embargo, es de gran importancia esclarecer dichos mecanismos debido a que esto permitirá el control de la amenaza excepcionalmente seria que representan estos virus, y aumentaría la compresión de los procesos subyacentes a la leucemogénesis y el funcionamiento normal del sistema inmune.
Clasificación de los retrovirus
El término retrovirus se refiere a una familia diversa de virus ARN de cadena simple, con envoltura, cuya replicación y expresión son dependientes de la integración de una forma intermedia de ADN de doble cadena (un provirus) en el genoma de la célula huésped. Los retrovirus se distinguen de los otros virus ARN por la presencia de la enzima transcriptasa inversa, polimerasa de ADN dependiente de ARN codificada en forma viral y que facilita la transferencia de la información genética retroviral desde el ARN al ADN.1, 14 La familia Retroviridae, que incluye a los retrovirus humanos, se divide en tres subfamilias: Oncovirinae, Lentivirinae y Spumavirinae. Oncovirinae, por lo general conocida como oncovirus (término abreviado para virus ARN oncogénicos), puede inducir una amplia variedad de enfermedades malignas en animales, incluyendo muchas de las leucemias y linfomas que ocurren en gatos, ratones silvestres, pájaros, vacas y monos.1,14 HTLV-I y HTLV-II semejan en forma más estrecha a los miembros de esta subfamilia, aunque poseen varios atributos estructurales y funcionales distintivos. Los lentivitus (también llamados retrovirus lentos) causan diversos trastornos no malignos que evolucionan de manera lenta y se presentan en animales, originando varias enfermedades neurológicas, musculoesqueléticas, hematológicas y respiratorias de mamíferos angulados (con cascos) que se caracterizan por un período de incubación largo y una fase sintomática prolongada. Varios estudios han permitido encontrar ciertas similitudes biológicas y morfológicas entre los miembros de esta subfamilia y los retrovirus humanos VIH-1 y VIH-2, y el análisis genético indica en forma clara que el VIH-1 y el VIH-2 son miembros de la subfamilia de los lentivirus. Se ha demostrado que los espumavirus (también conocidos como virus espumosos) inducen infección persistente en varias especies de mamíferos, pero dicha infección no se relaciona con consecuencias patológicas evidentes.
Poco después del descubrimiento del HTLV-I se identificó un virus muy relacionado, denominado virus linfotrófico de células T de los simios tipo 1 (STLV-I), como agente que producía infección natural en muchas especies de monos y grandes simios del Viejo Mundo.15-17 Al igual que el HTLV-I el STLV-I se relaciona con trastornos malignos linfoides espotáneos en huéspedes infectados. Igualmente, la identificación del VIH-1 alentó a los investigadores a realizar estudios serológicos en especies de primates no humanos en búsqueda de una evidencia de infección por otros retrovirus. El análisis de los resultados condujo al descubrimiento de una familia de virus de inmunodeficiencia de los simios (VISs) [ver adelante, Origen del VIH y SIDA].
Estudios epidemiológicos subsecuentes realizados en Africa Occidental demostraron un patrón inesperado de mayor reactividad de ciertos sueros humanos con los antígenos virales de los VIS que con los antígenos virales del VIH-1. Este dato llevó al descubrimiento y aislamiento del VIH-2.10-13 La caracterización molecular del VIH-2, VISmac (obtenido de monos macacos [Macaca mulatta] que sufren una enfermedad parecida al SIDA) y del VISsmm (obtenido de Cercocebus atys sanos) ha indicado que la estructura genética de estos virus se relaciona mucho más entre sí que con la estructura del VIH-1 (ver adelante).19,20 Recientemente se aisló el virus VIScpz, un lentivirus relacionado al VIH-1, de chimpancés salvajes (Pan troglodytes).21,22 La relevancia del VIScpz en el origen de la epidemia por VIH-1 no es clara aún. El estudio continuo de los lentivirus de los simios podrá ofrecer datos interesantes sobre el origen, biología y patogenia de los retrovirus inmunosupresores humanos y proporcionar modelos útiles para evaluar posibles tratamientos y vacunas contra las infecciones por retrovirus.
Estructura y replicación de los retrovirus
Todos los retrovirus con replicación competente contienen al menos tres genes (gag, pol y env) que están localizados en un orden común de izquierda a derecha 5' a 3'.14 El gen gag codifica las proteínas centrales internas de la partícula del virión. El gen pol codifica la polimerasa transcriptasa inversa que copia el ARN del genoma viral a ADN, una ribonucleasa (ARNasa) enzima H que degrada el ARN viral una vez que se ha sintetizado la copia inicial de ADN, y una endonucleasa denominada integrasa que facilita la integración retroviral hacia el cromosoma del huésped. El gen pol codifica también una proteasa necesaria para el procesamiento de los productos maduros de los genes gag y pol a partir de sus precursores poliproteínas durante el ensamble de los viriones. El env codifica la cubierta glucoproteica de superficie que media la unión del retrovirus a la célula blanco huésped. Al final de cada retrovirus se encuentran secuencias repetitivas conocidas como repeticiones terminales prolongadas (RTP). En general, las secuencias que comprenden las RTPs retrovirales no codifican para las proteínas sino que contienen las señales de iniciación y procesamiento de la transcripción al igual que elementos reguladores genéticos centrales para el control de la expresión viral.
El ciclo de replicación retroviral y el proceso de infección comienzan cuando el virus se une a receptores de superficie de células huésped específicas a través de la glucoproteína env [ver figura 1].1, 23 Tras una serie compleja de etapas, el ARN retroviral se convierte en ADN y se incorpora al genoma de la célula huésped de una forma estable llamada provirus. Una vez integrado en el cromosoma de la célula huésped, ocurre producción de ARN viral a través del aparato de transcripción de la célula. Sin embargo, el HTLV-I, HTLV-II, VIH-1, VIH-2 y los virus relacionados de otras especies animales, codifican proteínas que actúan, junto con los procesos celulares, para modular el nivel y carácter de los productos virales producidos en las células infectadas.24-27 La proteínas gag, pol y env son sintetizadas a través de la maquinaria celular del huésped para síntesis proteica y sufren modificación postraslacional (como glucosilación). En un proceso conocido como gemación, un retrovirus intacto que posee una membrana derivada de la membrana plasmática celular se libera de la célula.
Muchos retrovirus patógenos que ocurren en forma natural poseen una
estructura genética relativamente simple que puede tipificarse como la
de los virus de leucosis aviaria (VLA) [ver figura 2]. Los VLA contienen
genes gag, pol y env intactos, por lo que cuentan con toda la
información genética necesaria para su replicación en una
célula huésped.1,14 Los provirus VLA intactos producen
solo dos distintas transcripciones de ARN. La mayor, el ARN mensajero (ARNm) no
dividido, proporciona el ARN genómico incluido en las partículas
virales, pero también funciona como ARNm para la síntesis de gag
y pol. Se produce también un ARNm más pequeño, del que se
han removido las secuencias de gag y pol por medio de un evento
de división de ARN, que sirve como el ARNm para la proteína env.
Los productos genéticos codificados por estos retrovirus relativamente
simples tienen las funciones estructurales o enzimáticas necesarias para
producir los viriones infecciosos, pero la regulación del ARN viral o de
la producción de proteínas depende por completo de funciones
proporcionadas por las células huésped.
Los retrovirus patógenos humanos y los retrovirus relacionados que se encuentran en otras especies animales tienen estructuras genéticas y modos de expresión más complicados.24 Además de producir las proteínas gag, pol y env, elaboran otras proteínas que tienen funciones reguladoras o accesorias importantes. Varias de estas funciones reguladoras son necesarias para la replicación viral e interactúan en forma íntima con factores de la célula huésped que regulan tanto el nivel como las características de los productos genéticos virales producidos en las células infectadas. Además de producir transcritos de ARNm no divididos (genómico y gag-pol) y divididos en una ocasión (env), estos retrovirus más complejos producen una tercera clase de transcritos de ARNm con divisiones múltiples que codifican proteínas con funciones reguladoras importantes para el ciclo vital del virus. Este grado extraordinario de complejidad genómica y su capacidad para la expresión diferencial del gen puede tener un papel importante en la naturaleza persistente y en la génesis de las consecuencias patogénicas específicas de las infecciones por retrovirus humanos.
Estructura del HTLV-I y HTLV-II
HTLV-I y HTLV-II son similares en su organización genómica. Además de contar con los genes gag, pol y env, ambos virus contienen una región codificadora, llamada X, que se localiza cerca de las secuencias terminales del gen env y que se extiende hasta la RPT 3'.27 La homología más grande entre HTLV-I y HTLV-II se encontró en esta región X. El análisis de células infectadas con HTLV-I y HTLV-II ha demostrado que estos virus producen doble empalme de ARN con dos plantillas de lectura abiertas superpuestas. Los genes formados por este fenómeno de empalme poco usual son tax y rex.26, 27
El gen tax codifica una proteína con masa molecular de 40 kd. La proteína se localiza en el núcleo de las células infectadas y desempeña un papel crucial en la expresión y la replicación del HTLV-I y HTLV-II.26 La expresión de esta proteína que actúa junto con factores de transcripción de la célula huésped, origina un notable aumento en la transcripción de ARN viral modulado por polimerasa de ARN a partir de secuencias promotoras específicas presentes en las RPTs del virus, conduciendo a una mayor producción del virus. Este proceso, en el cual un producto difusible actúa a distancia sobre los diversos genes, se conoce como activación transcripcional de acción trans, o transactivación. Además de modular la expresión de los genes virales, las proteínas tax de HTLV-I y HTLV-II activan la función de otros factores de transcripción celular que realizan funciones reguladoras esenciales en la activación y proliferación de las células T. Al hacerlo, la proteína tax puede tener un papel importante en los procesos de inmortalización celular y transformación maligna que ocurren después de la infección viral [ver figura 3].
El gen rex codifica una proteína de 27 kd que, al igual que la proteína tax, se localiza dentro del núcleo de las células infectadas y está implicada en las estrategias de replicación de HTLV-I y HTLV0-II.26, 28 Una segunda proteína, denominada p21, también es codificada por el gen rex, pero su función se desconoce aún. A través de un mecanismo novedoso y no definido del todo, la proteína rex permite la expresión diferencial de las estructuras virales o los genes reguladores. Al actuar por medio de secuencias de ARN específicas derivadas de la RTP 3', la proteína rex parece controlar el transporte de especies específicas de ARN viral desde el núcleo hacia el citoplasma, donde puede traducirse en proteínas virales.26, 28 En ausencia de proteínas rex, sólo los ARN empalmados en forma múltiple (aquellos codificados por las proteínas tac y rex son transportados al citoplasma y disponibles para la traducción; el ARNm empalmado incompletamente que codifica la proteína env, así como el ARN genómico no empalmado que proporciona las transcripciones de ARN de longitud completa y codifica para las proteínas gag y pol, no se expresa de manera funciona. La capacidad de HTLV-I y HTLV-II para expresar preferencialmente proteínas estructurales y reguladoras puede permitir a estos virus la modulación de la cantidad de partículas virales producidas y antígenos virales mostrados. Estos mecanismos puede contribuir al mantenimiento de la infección persistente (ver adelante).
REGULACION DE LA EXPRESION DE HTLV IN VIVO
Tanto HTLV-I como HTLV-II tienen la capacidad de causar infecciones prolongadas que persisten a pesar de la respuesta inmunológica activa del huésped. Es interesante que aunque pueden encontrarse provirus HTLV-I integrados en individuos HTLV-I seropositivos, la gran mayoría de las células infectadas no expresan cantidades detectables de ARNviral, por lo que parecen estar infectadas en forma latente.27,29 No se han identificado aún los reservorios potenciales de la expresión de HTLV-I en personas infectadas, lo que dificulta comprender la manera como este virus se replica in vivo. A diferencia del VIH-1, que muestra una marcada diversidad en su secuencia genética entre los virus aislados de diferentes individuos, los HLV-I aislados en todo el mundo son mucho más homogéneos desde el punto de vista genético.30 La secuencia relativa para el HTLV-I es única entre los retrovirus, que requieren de la enzima transcriptasa inversa para replicar su genoma de ARN, lo que sugiere que el HTLV-I puede emplear una estrategia de replicación diferente. La proliferación de células infectadas inducida por el HTLV-I puede originar la replicación frecuente del genoma proviral por las polimerasas de ADN celulares (mucho más exactas) responsables de la duplicación de los cromosomas de la célula durante la división celular, en lugar de las acciones exclusivas de la transcriptasa inversa viral (que sufre más errores). Sin embargo, la demostración de la expresión in vivo de HTLV-I que puede causar proliferación de células T o inducir respuestas inmunes antivirales, no se ha logrado aún.
ESTRUCTURA Y EXPRESION DEL VIH-1 Y VIH-2
El análisis de secuencias y clonación molecular del VIH-1 han demostrado que es un retrovirus con una complejidad y diversidad genómica sin precedentes. Además de contener los genes característicos gag, pol y env necesarios para la replicación, el genoma del VHI-1 contiene al menos seis genes adicionales: vif, vpr, vpu, tat, rev y nef.24, 25, 31-34
El análisis molecular ha demostrado que las constituciones genéticas del VIH-2 y los VISs son homólogas a las del VIH-1.18, 24, 34 Sin embargo, los genomas del VIH-2 y VISs no contienen el gen vpx. Así mismo, los VISs codifican un gen adicional denominado vpx, el cual no parece tener su contraparte en el VIH-1.
Para facilitar la síntesis de por lo menos nueve productos proteicos distintos a partir de un genoma de menos de 10 kilobases (kb), el VIH-1 y VIH-2 usan un complicado proceso de ruptura diferencial del ARN que origina la producción de más de 30 especies diferentes de ARNm.24,25,28 Los ARNm virales pueden clasificarse por tamaño en tres grupos generales: un grupo de alrededor de 9 kb de transcritos genómicos no divididos, que también dirige la síntesis de las poliproteínas gag y pol, un grupo de ARNm divididos en una sola ocasión de 4.5 kb de longitud que codifican las proteínas env, vif, vpr, vpx y vpu, y un grupo de ARNm con divisiones múltiples de alrededor de 2.0 kb que codifican las proteínas tat, rev y nef.
Los genes tat y rev son indispensables para la replicación viral.24,25 Ambos genes funcionan en una manera trans-acción (i.e., ejercen sus efectos por medio de un producto difusible) para controlar la expresión de otros genes virales. Debido a que se requieren tanto las proteínas tat como rev para la producción de los virus, estas proteínas representan un blanco atractivo para diseñar tratamientos antivirales específicos.
Se sabe relativamente poco acerca de las funciones de los genes vif, vpr, vpu, vpx y nef, pero se conoce que no son indispensables para la replicación in vitro.28,35,36 El conocimiento más detallado de los genes del VIH-1 y VIH-2 estaba limitado antes por la falta de ensayos in vitro para analizar sus funciones. Sin embargo, se han realizado importantes adelantos al respecto en los últimos años, que permiten conocer la función de estos genes en el ciclo vital del virus. El mayor conocimiento de sus funciones proporcionará información útil sobre la compleja biología in vivo de estos virus, y sugerirá nuevos enfoques para interferir en forma específica con su acción.
Los genes tat del VIH-1 y VIH-2 codifican pequeñas proteínas nucleares que amplifican mucho la expresión y producción viral por medio de un mecanismo que aún no se ha definido del todo.37 La proteína tat se une a una estrucutra de asa de ARN denominada el elemento de trans-activación-respuesta (TAR) que se localiza en el extremo 5' de todos los transcritos de ARNm viral. La proteína tat, en combinación con proteínas celulares aún no identificadas, actúa para aumentar el estado estable de los transcritos de ARNm viral, principalmente al estimular la eficacia de la elongación transcripcional a partir de los elementos promotores presentes en los RTP del VIH-1 y VIH-2.37 En ausencia de la proteína tat, la mayoría de los complejos de transcripción ensamblados por los promotores del VIH parecen ser poco productivos y son incapaces de transcribir el templete proviral de ADN más allá de algunos cientos de nucleótidos. Sin embargo, en presencia de la proteína tat los complejos de transcripción se ensamblan en forma mucho más constante, completándose la transcripción del ARN a lo largo de todo el genoma proviral. Aunque la proteína tat puede activar la producción del VIH en modelos de cultivos tisulares de infección por este virus, se desconoce su papel en la regulación de la expresión viral durante la infección natural in vivo.
Los genes rev del VIH-1 y VIH-2 codifican proteínas nucleares que modulan el patrón de transcripción del ARN viral producido en las células infectadas.24,25,28,36 El mecanismo de función de la proteína rev se desconoce, pero parece incluir tanto la supresión de la división del transcrito genómico de ARN como la facilitación del transporte de los ARNm no divididos y divididos del núcleo al citoplasma.28 Por lo tanto, los genes rev del VIH-1 y VIH-2 son análogos a los genes rex del HTLV-I y HTLV-II, aunque los primeros poseen características estructurales y funcionales distintas. Si no se expresa la proteína rev, predominan las especies de ARNm pequeñas y con divisiones múltiples que codifican los genes reguladores del virus (tat, rev y nef). Al mismo tiempo, existen menos transcritos genómicos de longitud total o de una sola división, que juntos proporcionan los genomas virales y la traducción específica de los componentes estructurales (gag, pol y env) y accesorios (vif, vpr, vpx y vpu) del VIH-1 y VIH-2. La función de la proteína rev está mediada por la unión a una secuencia específica de ARN en la región del gen env denominada elemento de respuesta de rev (ERR), que brinda una estructura secundaria estable y compleja para el ARN.
La cantidad relativa de proteína rev expresada después de la infección de una célula blanco por el VIH divide al ciclo viral en las llamadas fases tempranas y tardías.28 Al principio de la infección por VIH de las células blanco existe una cantidad pequeña de proteína rev en las células infectadas, por lo que se expresan preferentemente los ARNm de división múltiple que codifican las proteínas reguladoras del virus. Con el tiempo, la proteína rev puede acumularse y facilitar la transición hacia una expresión predominante de especies ARN de mayor tamaño, que proporcionan los transcritos del ARN genómico y codifican los componentes proteicos estructurales y accesorios. La proteína rev puede coordinar el ensamble y producción del VIH durante un periodo breve, de modo que limita la capacidad del sistema inmune del huésped para identificar y destruir a las células infectadas antes de la liberación de la progenie viral.
El papel de la proteína vif en los ciclos de replicación del VIH-1 y el VIH-2 no se ha definido aún.35,36 Aunque no se requiere de la proteína vif para la replicación viral en ciertas líneas de cultivo de células T, sí es necesaria para la infección viral eficiente de los cultivos de linfocitos y macrófagos de sangre periférica. La proteína vif parece actuar dentro de célula productoras de virus para facilitar el ensamble de partículas virales totalmente infecciosas. Aunque los virus que portan los genes vif defectuosos entran a las células blanco de modo eficaz, son incapaces de completar la síntesis del ADN proviral dentro de la célula recién infectada. Estas observaciones sugieren que la proteína vif influye en la estabilidad, proceso o transporte de los complejos centrales de nucleoproteína del VIH-1 y VIH-2 después de la internalización del virus.
La función de la proteína vpr se desconoce aún.35,36 Está presente en partículas virales, lo que sugiere que tiene algún papel en las fases tempranas de la infección. La mutación del gen vpr disminuye, pero no abole, la replicación del VIH-1 en los macrófagos primarios in vitro. Se ha notificado que la proteína vpr actúa junto con un componente del complejo proteico gag del VIH-1 conocido como proteína matriz para facilitar la entrada del ADN viral al núcelo de las células después de la infección. Esta función puede ser especialmente importante durante la infección de células que no se dividen, como los macrófagos.
De los retrovirus humanos, solo el VIH-1 contiene el gen vpu (de los retrovirus de los simios, solo el VIScpz lo contiene), que se expresa en el mismo transcrito de ARNm del gen env.35,36 La mutación del gen vpu causa menor liberación de partículas virales de la superficie de las células infectadas y mayor gemación en vesículas intracelulares. La proteína vpu también parece inducir la degradación rápida de la molécula CD4 de la célula huésped después de su síntesis en el retículo endoplásmico de las células infectadas. El CD4 sirve como el receptor de superficie celular usado por el complejo glucoproteico env del VIH-1 (gp120-gp41) para entrar a las células blanco. Sin embargo, en las células infectadas, la interacción de moléculasCD4 recién sintetizadas con el precursor del VIH-1 gp160 env en el retículo endoplásmico inhibe el procesamiento y transporte adecuado de la proteína env. Al inducir la degradación de CD4, la proteína vpu puede servir para liberar las moléculas gp160 nacientes, dejando que maduren a complejos funcionales gp120-gp41 en la superficie celular y que se incorporen a las partículas virales en gemación.
El gen vpx es exclusivo del grupo de lentivirus relacionados que consisten en el VIH-2, VISsmm y VISmac.18,35 Se ha propuesto, con base en secuencias semejantes de aminoácidos, que el gen vpx de este grupo puede originarse por duplicación de un gen ancestral vpr. Al igual que la proteína vpr, la proteína vpx existe en las partículas virales, lo que sugiere que funciona en la fase temprana del ciclo de vida del virus. Se sabe que estas similitudes entre las secuencias de aminoácidos de las proteínas vpr y vpx se reflejan también a nivel funcional.
Los genes que codifican la proteína nef se encuentran solo en los lentivirus inmunosupresores de primates.38,39 La proteína nef no es indispensable para la replicación del VIH, y su papel en el ciclo de vida del virus no se ha establecido. Sin embargo, estudios recientes sobre esta proteína han identificado por lo menos dos funciones difeentes que pueden incrementar la eficacia de la replicación viral. La expresión de nef en las células infectadas causa menor expresión en la superficie celular del receptor CD4 al inducir internalización y degradación más rápida de éste.38,39 Aunque el papel de los efectos inducidos por nef sobre la expresión de CD4 en el ciclo viral no se comprenden del todo, el nef puede, al inhibir a CD4, aumentar la infectividad del virus por aumentar el nivel de glucoproteínas virales env incluidas en la superficie de las partículas virales. En forma alterna, se ha propuesto que como el CD4 en la superficie celular disminuye durante la activación normal del linfocito T inducida por antígeno, el nef puede semejar algunos de los aspectos de la activación de la célula T para facilitar la replicación viral. A través de un mecanismo distinto poco conocido, nef actúa dentro de las células infectadas para aumentar la infectividad de las partículas virales producidas.39
Aunque no se requiere del nef para la replicación del VIH-1 en el cultivo de tejidos, estudios recientes de SIDA inducido por VIS en monos rhesus han demostrado la importancia del nef en la biología in vivo de la infección viral. La mutación del gen nef en el VISmac compromete mucho la capacidad del virus para replicarse in vivo. Además, estos virus mutantes persisten a niveles muy bajos en animales infectados, pero no inducen enfermedad. La identificación reciente de un virus mutante nef en un paciente infectado por VIH-1 con una enfermedad excepcionalmente benigna sugiere que nef puede tener un papel semejante para modular in vivo la replicación del VIH-1.40
El mejor conocimiento sobre los productos de los genes reguladores y accesorios de los lentivirus de primates ha producido un cuadro cada vez más coherente de los complejos ciclos de replicación de estos virus. Las proteínas tat, rev y nef son las primeras proteínas virales producidas después de la infección. La proteína tat actúa para amplificar la transcripción del ARN viral, mientras que la proteína rev regula la expresión de las diferentes especies de ARNm y sincroniza la síntesis ordenada de las proteínas involucradas en el ensamble de las partículas virales. La menor expresión de CD4 en células infectadas, que parece necesaria para los ciclos vitales de los virus que usan CD4 como receptor de superficie, se logra por la expresión temprana de la proteína nef, que depura CD4 de la superficie celular. La expresión de la proteína nef continúa durante las fases tardías del ciclo vital, cuando nef actúa para aumentar la infectividad viral. La expresión de la proteína vpu está coordinada con la de la glucoproteína env, y la degradación de las moléculas CD4 recién sintetizadas por la proteína vpu facilita la producción ininterrumpida de la proteína env. La producción de las proteínas vif, vpr y vpx coincide con la de proteínas estructurales del virus, y las proteínas vif, vpr y vpx actúan o se incluyen en el ensamble de las partículas virales. Estas proteínas accesorias pueden ayudar a ensamblar partículas virales infecciosas o actuar en las células recién infectadas para facilitar las fases tempranas de la infección viral.
REGULACION DE LA EXPRESION DEL VIH IN VIVO
El término latencia tiene varias definiciones distintas cuando se aplica a la infección por VIH. Desde el punto de vista clínico, la infección por el VIH se caracteriza por un periodo asintomático (o latente) prolongado antes de que se desarrolle la inmunodeficiencia clínica. Hasta hace poco se suponía que muy pocas células en el individuo infectado portaban o expresaban el VIH y que se llevaba a cabo una replicación viral muy escasa durante ese periodo. Sin embargo, el uso de métodos sensibles para cuantificar la carga viral y su expresión in vivo ha demostrado que la replicación viral es muy activa durante todo el curso clínico de la infección por VIH (ver adelante).41-45 Por ello, el curso de la infección por VIH nunca es completamente quieto o latente.
La disponibilidad de métodos sensibles para medir el número de células infectadas en sangre periférica y tejidos linfoides e identificar células que participen en la síntesis del ARN y las proteínas virales ha permitido, por primera vez, preguntarnos si puede existir el VIH en las células infectadas sin que se exprese. La comparación del número de células infectadas con VIH-1 (i.e., que contienen ADN proviral de VIH-1) con el número de células que producen ARN o proteínas virales sugiere que muchas células infectadas no expresan niveles detectables de productos genéticos del VIH.41,44,46 Se calcula que en la sangre periférica de los pacientes con infección avanzada por VIH el número de células infectadas excede al número de las que producen ARN viral en una relación de por lo menos 10.44,46 Estudios recientes han indicado que la mayor concentración de células que expresan al VIH-1 se encuentra en los tejidos linfáticos, que proporcionan los sitios anatómicos para la respuesta inmunológica.41,42,46 Incluso en estos sitios de densa carga viral, solo una minoría de las células que contienen ADN del VIH-1 muestran evidencia de síntesis de ARN o proteínas virales.46 Una fracción desconocida de las células ADN de VIH positivas pero ARN negativas pueden tener provirus con defectos en la replicación, pero se piensa que el resto contiene provirus competentes para la replicación, pero que no la expresan. Al parecer, las células infectadas que tienen provirus VIH-1 integrados pero que no se expresan (llamadas células infectadas latentes) producen cantidades insuficientes de proteínas virales como para ser detectadas y destruidas por las células T citotóxicas que reconocen los antígenos VIH. De esta manera, existe un reservorio celular de infección por VIH que evita a la respuesta inmunológica, causando persistencia de la infección. Los mecanismos moleculares por los que el VIH-1 logra este estado de expresión atenuada no se conocen. Se sabe muy poco sobre la biología del VIH-2 in vivo, pero se supone que tiene propiedades semejantes a las del VIH-1.
La expresión in vitro del VIH-1 aumenta en forma significativa por agentes que simulen estímulos inmunológicos y por citocinas como el factor de necrosis tumoral-alfa (FNT-alfa), la interleucina-6 (IL-6), la IL-2 y el factor estimulador de colonias de granulocitos y macrófagos (FEC-GM).25,47 El incremento de la expresión viral en estas circunstancias es resultado de la activación de la transcripción del ARN del VIH-1 por miembros de una familia de factores de transcripción inducibles de la célula huésped, conocidos como NF-kB, que se fijan a elementos de secuencia en el RTP del VIH-1.25,48-50 Estos factores de transcripción median la regulación de la expresión de diversos genes que normalmente son activados durante la respuesta del huésped a la infección e inflamación.48 Entre estos genes se encuentran los que codifican a las moléculas de clase I del complejo principal de histocompatibilidad (CPH), la cadena alfa del receptor de IL-2 (IL-2Ra), la IL-2, el FNT-alfa, la IL-6, el interferón beta, el FEC-GM, y ciertos reactantes de fase aguda, como el amiloide sérico A. En la mayoría de las células, proteínas inhibidoras específicas secuestran a los constituyentes NF-kB en el citoplasma en formas inactivas. Diversos estímulos inducen la liberación de los factores NF-kB de sus inhibidores, causando la migración de los complejos ahora activos hacia el núcleo. En el núcleo, los complejos NF-kB tienen un papel importante para controlar la respuesta celular ante los estímulos antigénicos. En forma semejante, un número importante de las funciones de los macrófagos depende de la activación transcripcional mediada por los NF-kB. Al incorporar la regulación mediada por NF-kB en su repertorio regulador, el VIH-1 se asocia en forma íntima al estado de activación de los linfocitos y macrófagos para proporcionar blancos para la infección viral. Entonces, las circunstancias que causan la activación de la respuesta inmunológica pueden también activar la producción del VIH-1.
HTLV-I, HTLV-II y transformación maligna por retrovirus
Los retrovirus son un instrumento atractivo para el estudio de los complejos procesos de transformación a través de los cuales la célula normal se convierte en cancerosa. El genoma retroviral es relativamente simple, y para muchos retrovirus la inducción de afección maligna en animales de laboratorio es reproducible. Los estudios de retrovirus animales han demostrado que aunque las estrategias de transformación utilizadas por los diferentes retrovirus comparten ciertas características, muestran una notable diversidad. Al parecer, los virus linfotróficos de células T humanas tipo I y tipo II usan mecanismos nuevos y todavía mal definidos de transformación maligna que tal vez sean mejor aclarados por el retroceso teórico proporcionado por los retrovirus animales leucemogénicos bien caracterizados.
TRANSFORMACION MALIGNA POR RETROVIRUS ANIMALES
Una clase de retrovirus animal, conocido como retrovirus rápidamente transformante, posee durante el curso de una infección adquirida un gen celular denominado proto-oncogén.51, 52 La incorporación de un proto-oncogén celular en el genoma de uno de los retrovirus rápidamente transformantes suele interrumpir uno o más de los genes necesarios para la replicación, causando que la replicación viral sea defectuosa. Por lo tanto, para causar infección, el retrovirus debe trasmitirse en compañía de un virus cooperador capaz de replicarse, que presta las funciones estructurales y enzimáticas necesarias. La incorporación del proto-oncogén al genoma viral también activa al gen y da al retrovirus la capacidad de transformar en forma aguda a las células tanto in vivo como in vitro. Con una célula blanco adecuada, la expresión del oncogén viral es necesaria y suficiente para la transformación maligna. Los tumores causados por la infección con estos retrovirus son policlonales en derivación y se caracterizan por la integración proviral en sitios al azar en el genoma. Un ejemplo de este tipo de retrovirus rápidamente transformante es el virus de la mielocitomatosis de las aves MC-29, el cual contiene el oncogén myc en su genoma [ver figura 2].
El estudio de los retrovirus rápidamente transformantes ofrece la primera demostración de la existencia de oncogenes celulares y proporciona conocimientos importantes acerca de los mecanismos de transformación maligna de otra clase de retrovirus animales leucemogénicos, los retrovirus lentamente transformantes.52, 53 Los retrovirus de esta clase, los cuales no pueden transformar células in vitro, constituyen la mayor parte de los retrovirus patógenos que ocurren de forma natural. Los retrovirus lentamente transformantes, ejemplificados por el virus de la leucosis aviaria, producen trastornos malignos que poseen un período de latencia de varios meses o incluso más prolongados, a pesar de los altos niveles de expresión viral en todo el huésped (viremia). Aunque la infección está muy diseminada, la conversión maligna de la célula infectada es un fenómeno raro. Los tumores que finalmente surgen son de origen clonal (derivados de un solo progenitor) y se considera que representan la culminación de un proceso genético de múltiples etapas que comenzó con la infección viral. Al revisar los tumores linfoides B producidos de manera característica por el virus de la leucocitos de las aves, se encuentra que su provirus está con frecuencia insertado en el cromosoma de la célula huésped adyacente al oncogén c-myc. En esta situación, la expresión del oncogén c-myc está localizado bajo el control de los elementos de regulación que residen en la RTP retroviral, que origina una expresión elevada o controlada de forma inadecuada. Este fenómeno de activación de un oncogén celular por integración retroviral proximal se conoce como tumorogénesis por inserción promotora.52, 53 Debido a que la mayor parte de los retrovirus lentamente transformantes se integran al azar en los cromosomas de la célula huésped, una explicación para la latencia de la enfermedad maligna característica de esta clase de retrovirus es la rareza con la cual la integración retroviral ocurre en la vecindad de un oncogén.
IDENTIFICACION DE LAS ENFERMEDADES CAUSADAS POR HTLV-I Y VIRUS ASOCIADOS
Los primeros intentos para identificar retrovirus humanos se modelaron sobre el ejemplo de los retrovirus lentamente transformantes, donde puede detectarse una expresión viral abundante in vivo y en líneas celulares tumorales de cultivo. Después de años de resultados negativos, muchos investigadores abandonaron esta línea, mientras que otros comenzaron a desarrollar estrategias para permitir el crecimiento a largo plazo de células hematopoyéticas humanas en cultivo de tejidos. Debido a que muchas células tumorales humanas no crecen in vivo, la atención se centró en la identificación de factores de crecimiento relevantes que pudieran proporcionar un enriquecimiento selectivo para las poblaciones celulares leucémicas y permitir el crecimiento celular en cantidades suficientes para detectar niveles bajos de expresión viral.
Un avance importante fue la identificación en 1976 del factor de crecimiento producido por linfocitos sanguíneos periféricos estimulados por fitohemaglutinina. Esta sustancia, llamada al inicio factor de crecimiento de células T y ahora conocida como interleucina-2 (IL-2), apoya el crecimiento de las células T humanas normales in vitro.54, 55 La IL-2 no se une a las células T normales en reposo, ni induce la proliferación de células T a menos que estas células sean primero activadas por mitógenos o antígenos. Después de la activación, las células T sintetizan receptores de superficie celular para la IL-2; estos receptores fijan IL-2 con gran afinidad y traducen una señal proliferativa.54, 55 Ahora se conoce que el receptor de IL-2 (IL-2R) es un complejo de al menos tres subunidades proteicas llamadas IL-2Rg, IL-2Rb e IL-2Ra, que en combinación proporcionan el receptor funcional de alta afinidad para la IL-2.54, 55 Las células T normales activadas seguirán creciendo en presencia de IL-2, pero después de cuatro a seis semanas sufren inevitablemente un proceso de envejecimiento y mueren.
Se observó que en ciertos pacientes con leucemias de células T maduras que al comienzo se consideró que presentaban variantes agresivas de micosis fungoide o síndrome de Sézary, las células T expresaban receptores de IL-2 y que las células T pudieron proliferar de forma directa en respuesta al IL-2 sin requerir una activación previa. En algunos de estos pacientes los cultivos de células leucémicas siguieron proliferando en ausencia de IL-2 exógena y, evitando el proceso de envejecimiento, se convirtieron en inmortales, es decir, capaces de una reproducción eterna. El examen con microscopía electrónica de estas líneas celulares demostró la presencia de partículas retrovirales, y se detectó actividad de la transcriptasa inversa específica viral en el sobrenadante del cultivo. En las células leucémicas, pero no en los tejidos normales, se identificaron secuencias de ADN que eran homólogas al ARN del virus producidas por las células leucémicas, lo cual indica que el virus se adquirió de manera endógena a través de infección.56, 57 Se identificaron anticuerpos dirigidos contra proteínas retrovirales en el suero de estos pacientes y esto proporcionó un marcador serológico de la infección.56 En 1980 apareció el primer estudio que demuestra el descubrimiento de un auténtico retrovirus humano, denominado HTLV-I.2, 58
Se llevaron a cabo extensos estudios seroepidemiológicos para evaluar la distribución del HTLV-I y para determinar su posible relación con varios trastornos malignos. Estudios iniciales en los Estados Unidos detectaron seropositividad en sólo unos cuantos casos de leucemias y linfomas de células T maduras en adultos. Estos casos con anticuerpos positivos fueron notables porque se manifestaban como leucemias de células T agresivas con fenotipo de célula T cooperadora-inductora CD4+ y pleomorfismo nuclear característico.3, 59 Las manifestaciones clínicas comprendían afectación diseminada de la piel, hipercalcemia y evolución rápidamente mortal.3, 60
Un síndrome clínico similar al observado en los Estados Unidos se ha descrito desde finales de la década de 1970 en Japón, donde se había notado que la proporción de trastornos malignos de células T a células B eran mucho mayor que en los Estados Unidos o Europa. Se observó que el síndrome, denominado leucemia/linfoma de células T del adulto, era prevalente concretamente entre personas nacidas en el suroeste del Japón.3, 61 El examen del suero de los pacientes con LLTA descubrió anticuerpos contra HTLV-I en prácticamente el 100 porciento de las muestras probadas.3 Es más, las áreas geográficas con elevada incidencia de LLTA se correlacionaban estrechamente con regiones donde podía demostrarse una alta prevalencia de HTLV-I desde el punto de vista serológico. Cuando se colocan en tejido celular, las células leucémicas de la mayoría de los pacientes con LLTA originan líneas celulares continuas que producen HTLV-I. Pronto se observó que el HTLV-I podía trasmitirse in vitro a las células T humanas normales y que el virus tenía una especial capacidad para transformar a las células T de modo que crecieran en una forma interminable independiente de IL-2.62,63 Los resultados de estos estudios epidemiológicos y de laboratorio proporcionaron un apoyo importante para la relación etiológica entre el HTLV-I y la LLTA y señaló la primera evidencia de relación causal entre un retrovirus y un trastorno maligno humano.
Los estudios seroepidemiológicos realizados en diversas áreas (v.gr., el Caribe, Sudamérica y Japón), han demostrado que además de su papel eitológico en la LLTA, el HTLV-I se relaciona en forma estrecha con la mieloneuropatía progresiva crónica conocida como paraparesia espástica tropical (PET) o mielopatía relacionada al HTLV-I (MRH).4,5 La PET o MRH son trastornos clínicos progresivos de inicio en el adulto que se caracterizan por paraparesia o paraplegia espástica simétrica que con frecuencia afecta a las extremidades inferiores y por parestesias, hiperreflexia, mínima pérdida sensorial y disfunción vesical frecuente.4 Aunque la PET y la MRH se describieron en un inicio en diferentes regiones geográficas, en la actualidad se consideran idénticas y con frecuencia se refiere a ellas como TEP/MRH. Las muestras de suero y líquido cefalorraquídeo de los pacientes con esta enfermedad contienen anticuerpos con inmunorreactividad característica contra proteínas del HTLV-I. Además, existen linfocitos atípicos que recuerdan a las células de la LLTA en el LCR y la sangre periférica de los pacientes con TEP/MRH, y se ha demostrado la presencia de secuencias provirales de ADN del HTLV-I en los linfocitos de sangre periférica de las personas afectadas.4
Los estudios moleculares han revelado que los HTLV-I aislados de pacientes con TEP/MRH no son obviamente diferentes de los aislados de pacientes con LLTA, y las cepas del virus de pacientes con TEP/MRH y LLTA son genéticamente indistinguibles de los encontrados en portadores asintomáticos. Por lo tanto, es posible que el momento, la vía de adquisición de la infección, o factores del huésped o ambientales, sean los determinantes de la presentación de las enfermedades asociadas con el HTLV-I, y no factores específicos del virus.
Además de la LLTA y la TEP/MRH, se han notificado otros síntomas clínicos en pacientes con infección por HTLV-I.5,64 Se ha descrito una artritis inflamatoria crónica que se asocia con la presencia de linfocitos infectados por HTLV-I en el líquido sinovial en áreas endémicas de infección por este virus en Japón, y que se conoce como artropatía asociada al HTLV-I. Existe una dermatisis infecciosa, caracterizada por eczema asociado con Staphylococcus aureus y estreptococo beta-hemolítico, en niños con infección por HTLV-I en Jamaica. De acuerdo con reportes recientes, una minoría de casos de linfoma cutáneo de células T (LCCT), micosis fungoides y su variante leucémica, el síndrome de Sézary, puede asociarse con infección por HTLV-I.65 Se han publicado también casos de uveítis, polimiositis y panbronquiolitis en individuos infectados por el HTLV-I, lo que sugiere que la investigación futura descubrirá un mayor espectro de enfermedades asociadas con esta infección.
EPIDEMIOLOGIA DE LA INFECCION POR HTLV-I
La infección por HTLV-I se detecta serológicamente.64,66 Debido a que este virus causa una infección persistente, la presencia de anticuerpos contra él indica que una persona tiene una infección activa. Los estudios seroepidemiológicos han descubierto la presencia de HTLV-I en proporciones endémicas en poblaciones geográficamente distantes, incluyendo residentes del suroeste de Japón, el Caribe, Melanesia y partes de Africa.3 Las áreas endémicas de infección por HTLV-I suelen consistir en zonas locales rodeadas por comunidades con baja prevalencia de seropositividad. Los niños en las áreas endémicas suelen tener menos frecuencia de seropositividad que los adultos, y el porcentaje de positividad en los individuos aumenta en forma significativa en relación a la edad. En los grupos de mayor edad las tasas de seropositividad suelen ser mayores en mujeres que en hombres. El incremento en la frecuencia de infección en relación con la edad puede deberse a una mayor oportunidad acumulada para exponerse a la infección o a un efecto de cohorte que refleja tasas históricas más altas de infección por HTLV-I que las observadas actualmente en poblaciones más jóvenes. Estudios recientes de Japón indican que el estado de portador de HTLV-I está disminuyendo en menores de 50 años, y en especial en los grupos jóvenes. Se cree que las mejoras en las condiciones socioeconómicas y los cambios de comportamiento, como la lactancia, han causado una menor trasmisión viral y disminuido la prevalencia de la infección. En las áreas no endémicas, la infección por HTLV-I es más frecuente en inmigrantes de regiones endémicas o en sus parejas sexuales.
El HTLV-I puede trasmitirse de madre a hijo y por contacto sexual, transfusión sanguínea, y uso común de agujas o jeringas contaminadas en los adictos a drogas.3,67-69 Los patrones observados de infección por HTLV-I indican que el virus no es muy contagioso y que la trasmisión viral puede requerir la transferencia de células infectadas de los portadores a los receptores. Estas conclusiones son compatibles con demostraciones de laboratorio de lo difícil que es lograr la trasmisión del virus sin células.
La trasmisión sexual del HTLV-I representa una de las principales vías de diseminación en las áreas endémicas. La trasmisión sexual del virus ocurre más de hombres a mujeres que viceversa, lo cual puede explicar el predominio de la infección en mujeres que ya se mencionó. La trasmisión sexual del virus puede facilitarse por la presencia de otras enfermedades de trasmisión sexual que causen ulceraciones genitales.
La trasmisión del HTLV-I de madre a hijo es frecuente en las áreas endémicas, con tasas de infección de alrededor del 20 porciento en los hijos de madres seropositivas.4,5 Los linfocitos infectados por el HTLV-I se encuentran en la leche materna, y la trasmisión de madre a hijo ocurre sobre todo a través de la lactancia. La sustitución de ésta por biberón en las madres seropositivas puede disminuir en forma significativa el riesgo de trasmisión del virus.
Se ha demostrado la trasmisión viral a través de transfusiones sanguíneas, y esto es motivo de preocupación en las áreas endémicas.61,64 Existe trasmisión viral al transfundir sangre total, paquetes eritrocitarios, o plaquetas (casi siempre contaminados con linfocitos), pero no si se transfunde plasma o sus derivados. En Japón ha ocurrido una importante reducción en las infecciones por HTLV-I secundarias a transfusión y en el número de casos de PET/MRH desde 1987, cuando se instituyó el escrutinio nacional para donadores de sangre en busca de anticuerpos contra HTLV-I.61 En los Estados Unidos se realiza escrutinio en todos los donadores desde 1988. Se ha encontrado evidencia serológica de infección por HTLV-I en alrededor del 0.25 porciento de los donadores de sangre en los Estados Unidos, ocurriendo la mayor prevalencia en las costas del Este y Occidente.70,71 Los estudios que emplean la reacción en cadena de la polimerasa (RCP) para tipificar el virus presente en donadores seropositivos indican que alrededor del 50 porciento de las personas están infectadas por HTLV-I y el otro 50 porciento por HTLV-II.70,72 Entre los donadores de sangre en Estados Unidos, la infección por HTLV-I suele asociarse con haber nacido en un área endémica o haber tenido contacto sexual con personas de un área endémica como Japón o el Caribe.72
La progresión a LLTA es una consecuencia relativamente rara de la infección por HTLV-I. Se calcula que se desarrolla LLTA en dos a cuatro porciento de las personas que viven en regiones en donde el HTLV-I es endémico y en donde es frecuente la infección durante la niñez.5,61,73 La LLTA se ve con más frecuencia entre personas de 40 a 60 años de edad, lo que sugiere que la enfermedad se desarrolla después de un largo periodo latente posinfección, de por lo menos algunas décadas. No existen en la actualidad indicadores pronósticos que identifiquen a los individuos seropositivos que desarrollarán LLTA.
La PET/MRH se desarrolla en menos del uno porciento de las personas infectadas por HTLV-I.4,5,61,74 La enfermedad ocurre con más frecuencia en mujeres que en hombres y comienza alrededor de la cuarta década de la vida. El periodo de latencia para el desarrollo de este padecimiento parece más corto que el de la LLTA, en especial en los pacientes que adquieren la infección por transfusión sanguínea. Se ha reportado un intervalo promedio de 3.3 años entre los receptores de sangre contaminada con HTLV-I y el desarrollo de PET/MRH.4,5
IDENTIFICACION DEL HTLV-II
Un segundo retrovirus humano exógeno distinto, denominado HTLV-II, se aisló de un paciente con una variante de leucemia de células peludas de células T en 1982.3 El HTLV-I y el HTLV-II comparten una organización genómica común y conservan la secuencia estructural de genes, pero son filogenéticamente diferentes. El análisis de la secuencia de nucleótidos del HTLV-II ha demostrado una homología particularmente significativa con el HTLV-I en la región X. Además, el HTLV-II comparte con el HTLV-I la capacidad de transformar en forma aguda las células T para que tengan crecimiento autónomo e inmortal in vitro.27
Desde su descubrimiento en 1982, el HTLV-II se ha aislado de otro paciente con una variante atípica de leucemia de células peludas.75 Sin embargo, los estudios subsecuentes no han podido demostrar una asociación etiológica del HTLV-II con este trastorno u otras enfermedades linfoproliferativas.76 Se han reportado algunos casos de una enfermedad neurológica parecida a la PET/MRH en personas infectadas por HTLV-II, pero el papel del virus en éstos no es claro.77 Debido a la similitud en la estructura genética y al comportamiento in vitro del HTLV-I y el HTLV-II, la aparente falta de patogenicidad del HTLV-II parece sorprendente. Una posible explicación para esta diferencia es el descubrimiento reciente de que mientras que el HTLV-I tiene un tropismo preferencial por células T CD4+ in vivo, el HTLV-II infecta preferentemente células T CD8+.78
EPIDEMIOLOGIA DE LA INFECCION POR HTLV-II
Las proteínas del HTLV-II pueden ser serológicamente distintas a las del HTLV-I, pero el suero de la mayoría de las personas infectadas ha demostrado tener anticuerpos que tienen intensa reacción cruzada en el ensayo de inmunoabsorción ligada a enzimas (ELISA) que se emplea para escrutinio de la infección por HTLV-I.64,66 Hasta hace poco, principalmente por la falta de pruebas serológicas para distinguir con certeza la infección por HTLV-II de la HTLV-I, existía poca información sobre la epidemiología de la infección por HTLV-II. Sin embargo, los inmunoensayos que emplean proteínas recombinantes específicas del virus o péptidos sintéticos y los enfoques de amplificación de genes que usan la RCP permiten distinguir entre las dos infecciones. Debido a la muy diferente implicación clínica de estas dos infecciones, es importante identificar el tipo de virus en los individuos que tienen prueba positiva para anticuerpos anti HTLV.64
La aplicación de nuevos métodos para discriminar entre la infección por HTLV-I y HTLV-II ha descubierto que los adictos a drogas intravenosas representa el mayor reservorio de infección por este virus en los Estados Unidos.68,79,80 Los estudios serológicos de personas de diferentes áreas urbanas han demostrado una seroprevalencia de HTLV de ocho a 24 porciento, la gran mayoría de estos casos son de infección por HTLV-II. Las tasas más altas de seroprevalencia se encuentran a lo largo de las costas Este y Oeste, y en el área de Nueva Orleans. A diferencia de la infección por VIH-1, las infecciones por HTLV-II son prevalentes entre los adictos a drogas intravenosas desde principios de los años 70, e incluso entonces pudieron no ser recientes en este grupo de población.81
Recientemente se ha demostrado también que el HTLV-II es prevalente en varias tribus indias americanas, incluyendo los indios Guaymi de Panamá y los nativos americanos de Florida y Nuevo México.80 En estas poblaciones la prevalencia de HTLV-II es de alrededor de dos porciento o menos. Debido al aislamiento prolongado de muchas de estas poblaciones, es posible que la infección por HTLV-II sea muy antigua, proveniente de los antecesores de estas personas, que migraron a America desde Asia.
Aunque se han estudiado mucho menos, los mecanismos de trasmisión del HTLV-II parecen ser idénticos a los del HTLV-I.80 La alta prevalencia de HTLV-II entre adictos a drogas intravenosas parece ser causada por compartir los equipos de inyección. El HTLV-II también puede trasmitirse por vía sexual, como es el caso de la infección con HTLV-I, VIH-1 o VIH-2, y se asocia un mayor riesgo de trasmisión viral si coexiste otra enfermedad de trasmisión sexual en el receptor. El contacto sexual con un adicto a drogas ha sido identificado como el factor de riesgo más frecuente para la infección por HTLV en mujeres seropositivas identificadas por el escrutinio que se realiza a los donadores de sangre en los Estados Unidos. Al igual que la trasmisión por HTLV-I, puede ocurrir trasmisión de HTLV-II por transfusión de sangre total, paquete de eritrocitos o plaquetas. Las pruebas de escrutinio autorizadas en la actualidad para anticuerpos contra HTLV-I no detectan los anticuerpos contra HTLV-II en algunas muestras, y han ocurrido casos de trasmisión de HTLV-II por transfusión de sangre de donadores que tuvieron escrutinio negativo para HTLV-I.82 Se ha reportado trasmisión de madre a hijo, pero es rara o inexistente si no existe lactancia.83
TRANSFORMACION MALIGNA POR HTLV-I Y HTLV-II
El fenómeno de transformación in vitro por HTLV-I y HTLV-II de las células T derivadas de donantes normales ha recibido mucha atención. Las células T transformadas in vitro comparten varias características sobresalientes con las células de LLTA, y la delineación del mecanismo de transformación in vitro brindaría conocimientos importantes sobre la naturaleza de la leucemogénesis por los retrovirus humanos. Al igual que las células de LLTA, la mayor parte de las líneas celulares inmortales derivadas de la infección in vitro de las células T de donadores sanos consisten en células T CD4+, que poseen núcleos lobulados y tienden a formar células gigantes multinucleadas.3 Con frecuencia las células T transformadas proliferan en ausencia de IL-2 exógena; sin embargo, al igual que las células de LLTA, expresan altos niveles de IL-2R. Las células T transformadas también expresan en forma consistente receptores de transferrina y antígenos HLA de clase II (HLA-DR), lo cual indica que están en estado activado.
El HTLV-I y HTLV-II utilizan mecanismos únicos de transformación que los colocan aparte de los retrovirus animales rápida y lentamente transformantes. Ambos retrovirus humanos pueden transformar de forma aguda células in vitro, pero ni el HTLV-I ni el HTLV-II contienen los oncogenes derivados de las células que portan los retrovirus rápidamente transformantes. Es más, aunque se considera que la LLTA surge después de un período de latencia posinfección prolongado, el mecanismo patogénico del HTLV-I difiere del de los retrovirus lentamente transformantes. La mayor parte de las células tumorales LLTA contienen un provirus sencillo integrado de forma monoclonal, y expresan un gen rearreglado Vbeta del receptor de la célula T, lo que indica que las células malignas son de origen clonal y surgen de una sola célula infectada. Sin embargo, no se encuentra el HTLV-I integrado en los sitios preferenciales de los genomas de las células T malignas, lo que sugiere que el virus no induce leucemia por la activación vía promotor-inserción de un oncogén celular.
La naturaleza de la transformación de las celulas T in vitro por HTLV-I y HTLV-II ha conducido a la suposición de que estos virus portan, además de los genes necesarios para la replicación, un gen que codifica productos proteicos cuya expresión dota a las células T con un potencial de crecimiento ilimitado. Se ha postulado que la proteína tax que estimula el incremento de transcripción de ARN viral a través del proceso de activación transcripcional de acción trans (ver antes), puede actuar también sobre genes celulares involucrados en el control de la proliferación de células T, proporcionando de esta manera un estímulo inapropiado para el crecimiento26, 27 [ver figura 3].
De acuerdo a este modelo de transactivación de la leucemogénesis, la infección viral y la consecuente expresión de la proteína tax origina la transcripción constitutiva de los genes celulares que en condiciones normales son estrechamente regulados y cuya expresión persistente conduce a la proliferación descontrolada26, 27 [ver figura 3]. Un importante componente de este modelo se ha validado mediante experimentos que muestran que la inducción del gen tax en las células T origina la activación y expresión sostenida del gen que codifica el receptor de alta afinidad de IL-2a y, bajo ciertas condiciones, el propio gen de IL-2.26, 27 Asimismo, la expresión de tax también puede originar la elaboración de citocinas específicas de granulocitos y macrófagos (FEC-GM) y la linfotoxina (factor de necrosis tumoral beta), que puede contribuir a las manifestaciones clínicas típicas del HTLV-I relacionadas con enfermedades malignas y neurológicas.26, 27 Por último, la proteína tax activa la expresión de la proteína relacionada a la hormona paratiroidea (PrPTH), que puede ser la causa de la hipercalcemia que se observa en muchos pacientes con LLTA.84
La sugestión de que la inmortalización de la célula T in vitro se facilita por un fenómeno de transactivación mediada por el producto del gen tax está apoyada por las demostraciones recientes tanto de la capacidad transformadora del gen tax en modelos de cultivo tisular usando fibroblastos de roedores como en la apariencia de los tumores mesenquimatosos y la leucemia linfocítica en ratones transgénicos tax.26,27,85 Además, el uso de los métodos de transferencia de genes para introducir las regiones tax y rex del HTLV-I a células T humanas primarias ha provocado la inmortalización de estás células T in vitro.86 Sin embargo, no se sabe si la expresión de la proteína tax es suficiente para la inmortalización de la célula T, y la relación entre los proceso de inmortalización in vitro y leucemogénesis in vivo no se comprende del todo aún. Además, el modelo no explica por completo el potencial leucemogénico in vivo del HTLV-I, porque a pesar de sus similitudes existen diferencias sustanciales entre las células T transformadas in vitro y las células leucémicas LLTA. Mientras que la expresión viral en las infectadas puede documentarse con rapidez in vitro, la gran mayoría de las células tumorales LLTA no expresan el virus in vivo. Por consiguiente, aunque la expresión viral puede ser necesaria para comenzar el proceso leucémico, no parece requerirse para el mantenimiento de la enfermedad. El cuadro se complica más por la observación de que las células leucémicas de LLTA poseen con frecuencia evidentes rearreglos cromosómicos (aunque no consistentes), mientras que las líneas de células T in vitro por lo general son euploides y de potencial maligno poco claro.27, 87
Estudios epidemiológicos han demostrado que la mayoría de los individuos infectados con HTLV-I no adquieren LLTA y que si se desarrollan manifestaciones clínicas existe un período de latencia prolongado entre infección y presentación leucémica (ver antes). Estos complejos datos de laboratorio y epidemiológicos indican que la expresión viral puede ocurrir en algún punto en el curso de una infección y predisponer a las células T infectadas a alteraciones genéticas adicionales, implicando a cofactores todavía no identificados necesarios para la evolución maligna. Aun falta mucho por conocer acerca del papel que las influencias genéticas, inmunológicas y ambientales desempeñan en la modulación del curso clínico entre infección por HTLV-I y la presentación maligna de la LLTA.
HTLV-I Y LLTA
La LLTA se caracteriza por la proliferación agresiva de células malignas inductoras-cooperadoras que tienen un inmunofenotipo CD4+, CD25+ (IL-2Ra).27,29 Las manifestaciones clínicas de la LLTA son muy características y frecuentemente incluyen linfadenopatía, hepatomegalia, esplenomegalia, lesiones óseas líticas, hipercalcemia, afección cutánea y diseminación leucémica.5,59,60 La supervivencia promedio desde el diagnóstico inicial es de 11 meses.
Las diversas presentaciones clínicas de la enfermedad, incluyendo el síndrome conocido como LLTA latente y LLTA crónica, son notables porque pueden tener alguna explicación en el mecanismo de la leucemogénesis en la infección por HTLV-I.88 La LLTA se presenta con frecuencia con manifestaciones cutáneas premonitorias, pero en pacientes con esta enfermedad la cuenta de leucocitos y el calcio sérico son normales. Estos pacientes pueden sufrir progresión aguda a LLTA franca. La LLTA crónica se caracteriza por linfocitosis de las células T con evidencia de células leucémicas circulantes, linfadenopatía, hepatoesplenomegalia, y lesiones cutáneas, pero sin hipercalcemia. La apariencia de LLTA crónica se asocia con frecuencia con progresión de la enfermedad y mal pronóstico. Se considera que el desarrollo de la LLTA es un proceso progresivo que comienza con la infección asintomática por HTLV-I y en ciertas personas es seguida de la aparición de condiciones preleucémicas como la LLTA crónica o latente, cualquiera de las cuales puede evolucionar a LLTA franca. Sin embargo, en muchos pacientes la LLTA franca se presenta sin un síndrome preleucémico que le anteceda.
Los patrones de integración proviral del HTLV-I cambian con la transformación preleucémica. Se observa integración policlonal en las células T infectadas durante el estado de portador asintomático, mientras que las células leucémicas transformadas portan provirus integrados en forma monoclonal. También se observan alteraciones cromosómicas con mayor frecuencia en las presentaciones clínicas más agresivas. El mayor conocimiento de estas entidades patológicas diversas puede contribuir a una mayor apreciación de los eventos tempranos en la patogenia de las enfermedades malignas que se asocian con el HTLV-I.
El tratamiento con los esquemas terapéuticos convencionales es bastante ineficaz en la LLTA, y con frecuencia se observan respuestas limitadas y recaídas tempranas.5,64 Está en estudio el tratamiento experimental de la LLTA, en el que las células tumorales son el blanco por la expresión constitucional del IL-2R. En estudios clínicos de pacientes con LLTA se está evaluando la eficacia terapéutica de anticuerpos monoclonales conjugados a una toxina que reconocen el IL-2R y proteínas de fusión Il-2-toxina, que se ha demostrado destruyen a las células infectadas por HTLV-I en cultivos de tejidos.89
HTLV-I Y MIELOPATIA CRONICA
La presentación clínica de la PET/MRH se caracteriza por el desarrollo de paraparesia o paraplegia espástica progresiva de las extremidades inferiores.4 Los síntomas incluyen debilidad de las extremidades inferiores, rigidez y parestesias, así como dolor lumbar persistente, urgencia e incontinencia urinarias y alteraciones intestinales. La debilidad y espasticidad de los músculos proximales de las piernas pueden causar una marcha lenta en tijera. El inicio de la enfermedad suele ser insidioso, y la progresión de la enfermedad se da en varios años. Al examen físico se observa evidencia de lesiones bilaterales de las vías piramidales, incluyendo hiperreflexia, respuestas plantares extensoras y clonus de los tobillos. Se encuentran cambios mínimos en los nervios sensoriales. Aunque la PET/MRH comparte muchas características con la esclerosis múltiple, puede distinguirse de ésta por la ausencia de afección a los nervios craneales, la conservación de la función cognoscitiva y la rareza con la que ocurre debilidad de las extremidades superiores. Aún más, los signos y síntomas de la PET/MRH, a diferencia de los de la esclerosis múltiple, no oscilan.
Los mecanismos patogénicos de la PET/MRH no se han definido, pero se cree que incluyen proceso inflamatorios y quizá autoinmunes.4,5 En los pacientes con PET/MRH, el dato patológico más prominente es la atrofia moderada a severa de la médula espinal. Es evidente un proceso inflamatorio crónico, principalmente meningomielitis de la médula espinal torácica inferior. Se encuentran células B, T y macrófagos infiltrando alrededor de los vasos sanguíneos parenquimatosos y en el parénquima tanto de la sustancia gris como blanca. El daño al tejido parenquimatoso se manifiesta como destrucción tanto de mielina como de axones, hialinosis de los vasos sanguíneos y presencia de fibrosis meníngea y cicatrización glial. Existen células T CD8+ positivas en las lesiones inflamatorias, que se acompañan de un alto grado de expresión de antígenos HLA de clase I, pero no es claro si éstas reaccionan contra los antígenos HTLV-I, autoantígenos u otros blancos. Aunque algunos investigadores han sugerido que la PET/MRH puede ser causada por una respuesta inmunológica citotóxica a las células infectadas por el HTLV-I en el sistema nervioso central, no puede detectarse la expresión del ARN del HTLV-I en las áreas afectadas.4,5 Una hipótesis alternativa es que la infección por HTLV-I inicia de alguna manera un proceso autoinmune que causa la destrucción de la mielina.4,5 Sin embargo, no existe evidencia clara que apoye esta hipótesis.
Un porcentaje significativo de células T CD4+ de la sangre periférica de pacientes con PET/MRH muestra un fenotipo activado (positivo para antígeno de clase I del HLA e IL-2Ra [CD25]) in vivo, y manifiesta un grado importante de proliferación espontánea in vitro.4,5 En la mayoría de los casos los pacientes con PET/MRH demuestran títulos de anticuerpos contra HTLV-I en el suero y LCR que son significativamente mayores que los demostrados por pacientes con LLTA o portadores seropositivos. También se encuentran niveles altos de células T citotóxicas específicas para HTLV-I en la sangre y LCR de los pacientes con PET/MRH. Las intensas respuestas humoral y celular antiviral en la PET/MRH pueden reflejar activación inmunológica crónica inducida por la replicación viral activa. Algo que apoya este concepto es el hecho de que a pesar de las respuestas antivirales intensas, los pacientes con PET/MRH tienen una carga mucho mayor de células infectadas por el HTLV-I que los portadores asintomáticos.4,5 En los pacientes con PET/MRH se encuentra el HTLV-I integrado en una forma policlonal en un número significativo (alrededor del 10 al 20 porciento) de células T de sangre periférica. A pesar de esta carga viral apreciable, la expresión del ARN del HTLV-I no puede detectarse en la gran mayoría de las células infectadas.
El tratamiento de la PET/MRH intenta basarse en el concepto de que la respuesta inmunológica del huésped tiene un papel importante en la patogenia de la enfermedad.4,5 El tratamiento con esteroides ayuda a amilorar los síntomas de la mielopatía en el 50 porciento o más de los pacientes con PET/MRH. Sin embargo, la mejoría puede ser temporal y en el seguimiento a largo plazo no se observan diferencias entre los pacientes tratados y los no tratados. Es interesante que los beneficios del tratamiento esteroideo pueden ser más evidentes en pacientes que adquieren la infección por HTLV-I a través de transfusión sanguínea y en los que se encuentran en la fase temprana de la enfermedad. El danazol, un andrógeno sintético, puede mejorar también en forma temporal los síntomas. Tratamientos más agresivos, como la plasmaféresis, la ciclofosfamida o el interferón alfa, pueden causar mejoría a corto plazo sin beneficios evidentes a largo plazo. Se está evaluando, en pacientes con PET/MRH, el uso de anticuerpos monoclonales que reconocen el IL-2Ra que se expresa en muchas células infectadas por el HTLV-I.
VIH Y SIDA
El SIDA se describió por primera vez en mayo de 1981 y se caracteriza por infecciones oportunistas poco usuales que ponen en peligro la vida y diversas enfermedades malignas en pacientes que no presentan otra causa conocida de inmunodeficiencia.90-92 El síndrome se manifiesta por diversas alteraciones inmunológicas características y una amplia constelación de datos clínicos, incluyendo neumonía por Pneumocystis carinii, muchas otras infecciones oportunistas causadas por virus, hongos, micobacterias y protozoarios, alteraciones neurológicas importantes, linfomas de células B, pérdida de peso intratable y una variante agresiva del sarcoma de Kaposi.93 La definición de casos usada por los Centros para el Control y Prevención de las Enfermedades (CDC) de los EUA para la vigilancia epidemiológica, se modificó cuando se identificó que el VIH-1 era el agente infeccioso causal del SIDA y varias veces después para incluir el mayor conocimiento sobre el espectro de las enfermedades asociadas al VIH.94,95 Con el conocimiento de que la depleción de las células T CD4+ es la característica básica de la inmunodeficiencia inducida por el VIH, la definición de SIDA se ha modificado para incluir a cualquier individuo infectado por el VIH-1 que tenga una cuenta de células T CD4+ menor de 200 células /µl o un porcentaje de células T CD4+ menor de 14.96,97 Aunque la definición de casos de los CDC es útil para estudios epidemiológicos, para fines clínicos es mejor considerar a la enfermedad por VIH-1 como un espectro continuo que va de la infección primaria a la infección asintomática y después a la inmunodeficiencia avanzada. Las características clínicas del SIDA se analizan en otra subsección.
DIAGNOSTICO DE LA INFECCION POR VIH
La evaluación de la respuesta serológica al VIH-1 es una medida indispensable para el diagnóstico de la infección por VIH, para realizar análisis epidemiológicos de la prevalencia de la infección y para el escrutinio de sangre y donadores de órganos con objeto de minimizar el riesgo de trasmisión.66,98 El escrutinio inicial del suero en busca de anticuerpos reactivos contra el VIH-1 emplea una prueba sensible de ELISA.66,98 La especificidad de las muestras reactivas se confirma por medio de una técnica de hibridización en filtro conocida como Western blot. Las pruebas de escrutinio autorizadas en la actualidad tienen tasas muy bajas de falsas positivas y falsas negativas. La mayoría de las personas recién infectadas producen niveles de anticuerpos detectables uno a dos meses después de la infección, y más del 95 porciento tienen una prueba positiva de anticuerpos antiVIH a los seis meses de la infección inicial. Puede no identificarse a las personas infectadas por el VIH-1 durante el periodo llamado de ventana, que ocurre pronto después de la infección, cuando no se han formado aún niveles detectables de anticuerpos. En la práctica se piensa que la magnitud de este error es pequeño, pero las tasas de fracaso pueden depender de la prevalencia de factores de riesgo en la población estudiada. La infección por VIH-1 puede diagnosticarse antes de la producción de anticuerpos antivirales por medio de un ensayo de captura del antígeno que detecta el antígeno viral central p24 en el suero o por RCP para identificar secuencias ADN o ARN del virus en muestras de células de sangre periférica o plasma, respectivamente. Debido a que la presencia del virus puede confirmarse por cultivo viral o por técnicas de RCP con gran frecuencia en las personas seropositivas y ya que la infección persiste por el resto de la vida, la seropositividad para VIH-1 debe considerarse como marcador de infección viral activa.99,100
Ciertos estudios iniciales que usaron técnicas de aislamiento del virus y amplificación genética por RCP sugirieron que un porcentaje de los pacientes con riesgo alto de infección por VIH podían infectarse y permanecer seronegativos durante periodos prolongados, quizá más de dos años.101 Sin embargo, estos datos han sido refutados por estudios subsecuentes más extensos.102 En forma semejante, tampoco se han confirmado algunos reportes de personas que revirtieron de un estado seropositivo a seronegativo para el VIH-1 a pesar de resultados de RCP persistentemente positivos.103,104
EPIDEMIOLOGIA DE LA INFECCION POR VIH-1
Desde su descripción inicial como una entidad clínica poco usual, el SIDA se ha convertido con rapidez en una pandemia, con casos reportados en más de 170 países de todo el mundo.105,106 Debido al largo periodo asintomático entre la infección inicial por el VIH-1 y el desarrollo de enfermedad clínica, el virus se ha diseminado en forma no aparente y extensa en varias poblaciones. Se sabe que la infección por VIH-1 es mucho más prevalente que el SIDA clínico u otras manifestaciones clínicas de la infección. Se calcula que más de 14 millones de personas se han infectado por el virus desde el inicio de la pandemia, alrededor de ocho millones de hombres, cinco millones de mujeres y un millón de niños.105,106 La Organización Mundial de la Salud (OMS) calcula que más de 2.5 millones de casos de SIDA y un millón de muertes han ocurrido hasta la fecha. Las proyecciones recientes sugieren que hasta 40 millones de personas podrán ifnectarse por el VIH-1 para el año 2000.105
Más del 60 porciento de los casos de SIDA en todo el mundo ocurren en la región sub-Sahara de Africa. Las infecciones continúan aumentando en Latinoamérica, el Caribe, Norteamérica, el Medio Oriente, Europa Oriental y Asia.106 Aunque la infección por VIH-1 fue introducida a los países de Asia hasta hace poco, se ha diseminado con gran rapidez, en especial en los países del suroeste, como Tailandia y la India.
Durante la década de los 80 la infección por VIH-1 destacó como una de las principales causas de muerte en los Estados Unidos, se reportaron más de 339,000 casos de SIDA y casi 200,000 fallecimientos para octubre de 1993.107 En 1992 la infección se convirtió en la principal causa de muere entre varones de 25 a 44 años de edad, y en la actualidad es la cuarta causa de muerte en mujeres del mismo grupo de edad.108 Se calcula que alrededor de un millón de personas están infectadas en este momento en los Estados Unidos, la mayoría de los cuales están asintomáticas y muchas no saben de su infección.
El VIH-1 se trasmite a través de varias vías: sexual, de personas infectadas a sus parejas homo o heterosexuales, parenteral, a receptores de transfusiones con sangre infectadas o sus productos o por la costumbre entre los adictos a drogas intravenosas de compartir las jeringas con personas infectadas, y perinatal, de madres infectadas a sus hijos.102 También ha ocurrido trasmisión del VIH-1 por trasplante de órganos e inseminación artificial con semen de una persona infectada.109,110 Muchos estudios epidemiológicos han fracaso en demostrar la trasmisión del VIH-1 por contacto casual con una persona infectada.102
Los análisis epidemiológicos indican que el semen, las secreciones cervicales y vaginales, y la sangre o sus derivados son los principales vehículos, aunque no los únicos, para la trasmisión del virus. En la actualidad no se conocen del todo los factores que determinan la capacidad de una persona infectada para trasmitir el virus. En forma semejante, tampoco se han definido las variables que pueden influir en la propensión de una persona a la infección por el VIH-1 después de la exposición. Los estudios realizados en diferentes sitios de todo el mundo indican que la ruptura de superficies mucosas por otras enfermedades de trasmisión sexual, en especial la sífilis y el chancroide que causan ulceraciones genitales, puede facilitar en mucho la infección por VIH-1.102,111 Como resultado, la prevalencia de enfermedades de trasmisión sexual en una población de personas con riesgo de infección por VIH-1 puede alterar en forma significativa la eficacia de la diseminación viral.
Los patrones predominantes de trasmisión del VIH-1 varían en las diferentes partes del mundo.102,106 En los Estados Unidos y otros países industrializados, el patrón principal ha sido la trasmisión sexual entre varones homosexuales y bisexuales. Sin embargo, la diseminación por agujas entre adictos a drogas continúa ocurriendo, y la tasa de trasmisión heterosexual está aumentando. Aunque los varones homosexuales y bisexuales constituyen aún la mayor proporción de los infectados por el VIH-1, los adictos a drogas forman el segundo grupo en tamaño. La trasmisión por adictos a drogas, ya sea en forma directa al compartir las jeringas o indirecta por contacto heterosecual o perinatal causa en la actualidad la mayor parte de las infecciones por VIH-1 entre mujeres y niños.
En la región sub-Sahara de Africa, Sudamérica, Asia y el Caribe, el VIH-1 se disemina predominantemente por trasmisión heterosexual y perinatal.105,106 En estas regiones el número de varones y mujeres infectados es aproximadamente igual. Por lo tanto, en todo el mundo es el contacto sexual, principalmente heterosexual, el modo predominante de trasmisión del VIH-1, causando más del 80 porciento de todas las infecciones.105,106 Los motivos subyacentes para la variación mundial observada en los patrones de trasmisión del VIH-1 no se conocen del todo, pero se piensa que incluyen gran cantidad de factores complejos, como comportamiento personal, prácticas sociales, prevalencia de enfermedades coexistentes de trasmisión sexual y la fecha de introducción del VIH-1 a la población.
Debido a las tasas altas de infección por VIH-1 en mujeres en edad reproductiva en los países en desarrollo, la infección adquirida por vía perinatal es responsable de alrededor del 20 porciento de todos los casos de SIDA en estas áreas.105,106 Como resultado, la epidemia creciente de SIDA pediátrico anulará mucho del progreso logrado en las últimas décadas para mejorar la supervivencia de los niños en los países en vías de desarrollo. La tasa de trasmisión observada de madre a hijo varía desde 13 porciento hasta un 45 porciento en estudios de cohorte realizados en Europa y Africa, respectivamente, con un promedio de alrededor del 25 porciento.112,113 Los motivos de esta variación se desconocen, pero es probable que se relacionen con diferencias en la fase de la enfermedad materna y en lo adecuado de los cuidados médicos prenatales y perinatales.
La trasmisión del VIH-1 de una madre a su hijo puede ocurrir en útero por el paso trasplacentario del virus, durante el parto o posnatal a través de la lactancia.114 Para los niños que nacen de madres seropositivas, la transferencia placentaria de anticuerpos maternos complica el diagnóstico de la infección por métodos serológicos, e impide calcular el número exacto de niños infectados in útero contra los infectados durante o después del nacimiento. Otros métodos diagnósticos, incluyendo el cultivo del VIH-1, los ensayos de inmunocomplejos disociados (ICD) del antígeno nuclear p24 y la RCP permiten el diagnóstico temprano de la infección por VIH-1.115,117 La presencia de un antígeno central VIH-1 p24 y el cultivo de virus al nacer en un grupo de neonatos nacidos de madres infectadas indica la trasmisión in útero. Sin embargo, se cree que la mayoría de las infecciones se adquieren durante el nacimiento por el contacto con sangre o secreciones contaminadas. Entre los gemelos de madres infectadas por VIH, se reporta una tasa más alta de infección en el niño que nace primero incluso en casos de cesárea, lo que sugiere que factores relacionados con el alumbramiento modifican el riesgo de infección.118 Diversos factores asociados con la enfermedad avanzada por VIH-1 en la madre, incluyendo antigenemia p24, niveles altos de ARN del VIH-1 en el plasma, y cuentas bajas de linfocitos T CD4+, correlacionan con un mayor riesgo de infección para el neonato.119-122 Se han demostrado casos de trasmisión posnatal, que se supone son resultado de la lactancia. Sin embargo, estos casos son poco comunes y se refieren sobre todo a madres que estaban recién infectadas y cuya leche podía haber tenido una cantidad sustancial de VIH-1.
Un estudio importante ha demostrado que cuando se administra zidovudina (AZT) a las mujeres embarazadas infectadas por VIH-1 durante el segundo y tercer trimestres del embarazo, se continúa intraparto y después se administra a los infantes durante las primeras seis semanas de vida, el riesgo de trasmisión materno-infante se reduce en forma significativa (ver adelante).123 La disponibilidad de una intervención eficaz para disminuir la trasmisión perinatal del VIH-1 ha originado un mayor interés por realizar escrutinio en todas las mujeres embarazadas en busca de presencia de infección por VIH-1.124,125
Se calcula que, antes de 1985, cuando se inició la prueba de anticuerpos en donadores de sangre en los Estados Unidos, 10,000 o más personas se infectaron por el VIH-1 a través de sangre o sus derivados contaminados por el virus.102,126 La trasmisión del VIH-1 por transfusión de sangre es muy eficaz, y por lo menos el 90 porciento de los receptores de componentes sanguíneos VIH-1 positivos se infectan. Varios miles de hemofílicos también se infectaron por el VIH-1 durante este periodo después de recibir concentrados de factores de coagulación contaminados. Los casos de trasmisión del VIH-1 por transfusión se han relacionado a la recepción de sangre total, paquete de eritrocitos, leucocitos, plaquetas y plasma. Las preparaciones de gamaglobulina, inmunoglobulina Rho, globulina inmune contra la hepatitis B y de vacuna contra la hepatitis B derivada del plasma no se han asociado con trasmisión del VIH-1. Los procedimientos usados para preparar estos productos parecen eliminar la infectividad del virus.
Con la introducción del escrutinio a donadores de sangre en busca de infección por VIH-1, la trasmisión asociada a transfusiones se redujo en forma significativa.102,127,128 Se han demostrado muy pocos casos de trasmisión por sangre que tuvo resultados negativos para anticuerpos antivirales en la prueba de escrutinio; además, se ha calculado que existe una (o menos) unidad de sangre infectada y seronegativa por cada 40,000 a 225,000 unidades de sangre que se transfunden.126,128,129 Debido a la rareza con la que donadores de sangre infectados pero seronegativos escapan a los estudios de escrutinio actuales, trabajos recientes indican que el uso de los ensayos de antígeno central p24 del VIH-1 o RCP para identificar a estos individuos que se encuentran en el periodo denominado de ventana antes de la seroconversión, no mejoran en forma importante la seguridad de las transfusiones de sangre en los Estados Unidos.130-132 El tratamiento con calor de los concentrados de factores de la coagulación para inactivar el VIH-1 proporciona una medida adicional de seguridad para las personas con hemofilia. Sin embargo, se requiere el uso intensivo de los métodos actuales para proteger a los receptores de sangre de la infección por VIH-1, como autoexclusión de los donadores, exclusión confidencial y pruebas de escrutinio rigurosas en busca de anticuerpos anti-VIH-1, para maximizar la seguridad de los productos sanguíneos y reducir el mínimo el ya muy pequeño riesgo de infección asociada a las transfusiones. A diferencia de lo que sucede en los países industrializados, la trasmisión del VIH-1 por sangre o sus derivados contaminados sigue siendo un gran problema en los países en vías de desarrollo por la falta de recursos adecuados para estudiar a todos los donadores y de bancos de sangre apropiados.
Aunque varios trabajadores del área de la salud se han infectado por la exposición a sangre de personas infectadas por el VIH-1, los estudios a gran escala sugieren que el riesgo de trasmisión de este virus por la exposición laboral es pequeño (0.34 porciento por exposición)., aunque varía con la severidad de la exposición.133,134 El uso de zidovudina para limitar la trasmisión después de la exposición de alto riesgo tiene un beneficio incierto, ya que han ocurrido infecciones a pesar de su uso.135-137 Aún no se establece si otros agentes antivirales o sus combinaciones serán más eficaces para evitar la trasmisión del VIH-1 después de la exposición laboral. Aunque los estudios de trasmisión laboral del virus han demostrado un riesgo bajo de trasmisión después de la exposición a un inóculo infectado pequeño, también han identificado una gran frecuencia de exposiciónes percutáneas y cutáneas, principalmente entre estudiantes de medicina y personal de base.138 Por lo tanto, a menos que disminuya la frecuencia de exposición ocupacional, continuarán viéndose casos que pudieron prevenirse de infección en trabajadores de la salud. La aplicación rigurosa de las medidas de control de infecciones (llamadas precauciones universales) para limitar la exposición a los líquidos corporales del paciente, independientemente de la probabilidad de infección por VIH-1, aumentan al máximo la seguridad para los trabajadores de la salud y reducen la posible discriminación contra pacientes que pueden considerarse probables portadores del virus.97,133,139
En 1990 surgieron controversias sobre la trasmisión del VIH-1 en el área de trabajo por personal de salud cuando los CDC notificaron que un dentista con SIDA había trasmitido la infección a cinco pacientes sometidos a procedimientos dentales invasivos.140,141 No se conoce el modo preciso de trasmisión del VIH en estos pacientes. Los estudios subsecuentes de pacientes de varios dentistas, médicos y cirujanos infectados fueron incapaces de demostrar algún caso de trasmisión del VIH. Por lo tanto, el riesgo de trasmisión de trabajadores de la salud infectados a sus pacientes es tan bajo que no puede medirse.102,141a,141b Sin embargo, ha surgido un debate público sobre el escrutinio para los trabajadores de la salud y la restricción de la práctica clínica en los seropositivos. Se ha afirmado que el entrenamiento cuidadoso de todos los trabajadores de la salud en las prácticas adecuadas de control de infecciones y la adherencia rigurosa al uso de las precauciones universales cuando se atiende a todos los pacientes constituye una medida más eficaz y menos discriminatoria.139,142
PROGRESION DE LA ENFERMEDAD POR VIH
Se calcula que por lo menos el 25 a 50 porciento de las personas infectadas por VIH-1 tienen progresión al SIDA clínico en un periodo de cinco a 10 años después de la infección.102,143,144 Un mayor porcentaje de casos tiene una duración de la infección subclínica mayor, y los estudios epidemiológicos han indicado que la posibilidad de desarrollar SIDA es directamente proporcional a la duración de la infección por el VIH-1. La progresión a SIDA también se asocia con la tasa de reducción de las cuentas de linfocitos T CD4+.143,145,146 Aunque las cuentas de células T CD4+ disminuyen con el tiempo en virtualmente todos los pacientes infectados por el VIH-1, lo hacen con tasas sustancialmente diferentes. El SIDA se desarrolla más rápido en individuos con una pendiente de disminución de linfocitos T CD4+ más pronunciada que en los que tienen tasas de reducción más lentas. En forma semejante, existe considerable variación en el tiempo que tarda la enfermedad clínca en manifestarse en los diferentes pacientes. Una minoría de las personas tienen una evolución con SIDA franco en uno a dos años después de la infección, mientras que en el otro extremo del espectro, se han observado pequeñas cohortes de pacientes infectados con infecciones asintomáticas prolongadas (> 10 años).147-149 Las contribuciones relativas del huésped y de los factores virales que son responsables de estas variaciones clínicas son un tema muy importante de estudio en el futuro.149,150
La progresión de la enfermedad por VIH-1 tiene dos patrones en los niños infectados por sus madres.151,152 En el primer año de vida el 20 porciento de los lactantes desarrollan inmunodeficiencia grave, que con frecuencia se asocia con complicaciones del SNC. El resto de los niños manifiesta una enfermedad con evolución más lentamente progresiva, semejante a la observada en los adultos. El riesgo de desarrollar enfermedad temprana y severa es mayor en los neonatos que son seropositivos para el VIH-1 al nacer por cultivo viral, RCP o ensayo del antígeno central P24 en suero, lo que indica infección in útero. El riesgo de progresión rápida es significativamente mayor en los hijos de madres con enfermedad avanzada en el momento del nacimiento del niño. Aún no se sabe si este mayor riesgo es resultado de una mayor carga viral materna o de la presencia de virus más virulentos en una persona con enfermedad en fase tardía.
La posibilidad de que factores genéticos o ambientales influyan en la evolución de la inmunosupresión inducida por el VIH-1 y en la progresión al SIDA es un tema de gran interés. Se ha reportado que personas que expresan ciertas combinaciones deHLA están predispuestas a progresión más rápida de la enfermedad, lo que sugiere que la eficacia de la respuesta inmunológica del huésped contra el VIH-1 puede influir en la tasa de progresión del SIDA.183 Sin embargo, se requiere más estudios sobre los factores del huésped que modifican la progresión de la enfermedad y sobre la aplicación de nuevos métodos para la tipificación de HLA de alta resolución. La identificación de cofactores ambientales podría permitir el desarrollo de intervenciones para limitar la progresión en personas infectadas. Debido a que la replicación del VIH-1 en cultivo de tejidos se facilita por el estímulo inmunológico de las células blanco, es posible que la exposición a antígenos ambientales o a infecciones comunes pueda activar también la expresión del VIH-1 in vivo, contribuyendo a la pérdida acelerada de células T CD4+. Sin embargo, esta afirmación no se ha demostrado del todo, y los estudios epidemiológicos no han identificado factores ambientales que correlacionen en forma clara con el desarrollo de la enfermedad. Sin embargo, es interesante que los estudios sugieren que la administración de isoniacida a pacientes infectados por VIH-1 con pruebas cutáneas positivas a tuberculina155 y la administración de aciclovir a pacientes infectados por VIH con afección extensa a herpesvirus155 puede retrasar la progresión del SIDA o prolongar la supervivencia. Es posible que estos beneficios resulten de limitar las acciones de estímulo inmunológico crónico por los agentes infecciosos, lo que podría aumentar la replicación del VIH-1 y así contribuir a la progresión de la enfermedad por este virus.
EPIDEMIOLOGIA DE LA INFECCION POR VIH-2
La infección por VIH-2 es más prevalente en ciertas regiones de Africa Occidental. También se ha reportado cada vez más en otras partes del mundo, incluyendo Portugal, Francia, Alemania, Brasil e India.156,157 Aunque en la mayoría de los países la presencia del VIH-2 puede asociarse con lazos a Africa Occidental, en la actualidad se ha reportado diseminación hacia la población indígena en Francia, Portugal y la India.
Se considera que la prevalencia de infección por VIH-2 en los Estados Unidos es muy baja en la actualidad, aunque han ocurrido casos de infección y de SIDA asociado al VIH-2.156 En todos los casos en los que se ha contado con la historia del paciente, la persona infectada había vivido en Africa Occidental o había tenido una pareja sexual de esa región. No se han identificado aún casos de infección por VIH-2 en adictos a drogas intravenosas, varones homosexuales o receptores de transfusiones. Debido a que el VIH-2 se relaciona solo en modo distante al VIH-1, con conservación de alrededor del 50 porciento de los aminoácidos en las proteínas gag y pol y menos del 30 porciento de conservación en los productos del gen env, su presencia no se detecta en forma eficaz por los estudios serológicos que se usan en la actualidad para detectar la infección por el VIH-1.66,156 Por lo tanto, debido a la presencia de VIH-2 en los Estados Unidos y a que solo alrededor del 80 porciento de los sueros de individuos infectados por VIH-2 reaccionan en el ELISA del VIH-1, la Food and Drug Administration recomendó en junio de 1992 que todas las unidades de sangre donadas sean analizadas en forma específica en busca de evidencia serológica de infección por VIH-2.156
Al igual que el VIH-1, el VIH-2 se trasmite por exposición a sangre infectada.156 Aunque la trasmisión heterosexual representa el principal modo de trasmisión en Africa Occidental, evidencias recientes indican que la diseminación heterosexual del VIH-2 sucede con una tasa menor que la del VIH-1.158 En forma semejante, la trasmisión perinatal del VIH-2 es menos frecuente, y menos del 10 porciento de los lactantes nacidos de madres infectadas por VIH-2 adquieren la infección. Existen dos posibles razones para esta aparente menor tasa de trasmisión del VIH-2: el virus puede ser en forma inherente menos infeccioso que el VIH-1, o los individuos infectados pueden tener una carga viral menor. Sin embargo, aún se tiene poca información al respecto.
La infección por VIH-2 se asocia con el desarrollo de inmunodeficiencia progresiva, susceptibilidad a infecciones oportunistas, enfermedad del SNC y emaciación.156 Aunque la infección por VIH-2 puede causar SIDA, los estudios sugieren que los individuos infectados por este virus tienen un periodo asintomático más largo y en consecuencia una menor incidencia de progresión a SIDA que los infectados por el VIH-1.156,159,160 No se sabe si el VIH-2 es un poco menos virulento o si la respuesta inmunológica del huésped es más capaz de amilorar las consecuencias patógenas de esta infección viral. Se requieren estudios adicionales para esclarecer mejor estos aspectos. En la actualidad existe poca información sobre la utilidad del tratamiento antiviral en las personas infectadas por el VIH-2.
Origen del VIH y del SIDA
La epidemia del SIDA se manifestó como una enfermedad aparentemente nueva en los Estados Unidos en 1981.90-92 Desde la identificación y la definición del SIDA como entidad clínica, estudios retrospectivos han demostrado un notable incremento de casos de afecciones relacionadas con el SIDA en Africa central a finales de la década de 1970, con algunos casos aislados ocurridos en los 60.161 Después del descubrimiento del VIH-1 como el agente etiológico del SIDA, los estudios serológicos que utilizaron muestras de sangre almacenada revelaron que la diseminación epidémica del VIH-1 había ocurrido en áreas de Africa central a mediados o finales de los años 70. La reactividad serológica más temprana con VIH-1 se detectó en una muestra recolectada en 1959. Sin embargo, este reporte se ha puesto en duda de modo reciente.162 Los registros seroepidemiológicos del VIH-2 están menos bien documentados, pero parecen haberse presentado casos de infección en Africa Occidental desde por lo menos 1966.162
La aparición de una nueva y devastadora enfermedad y los descubrimientos de los VIH-1, VIH-2 y un número creciente de virus relacionados en simios, han resultado tema de discusión con respecto al origen de los VIH y el SIDA.161-162 Se han propuesto numerosas hipótesis para explicar la historia filogenética del VIH-1 y VIH-2, incluyendo la infección relativamente reciente de poblaciones humanas con un virus simiano, la mutación de un virus humano no patógeno preexistente a una forma más virulenta, o la diseminación epidémica de un virus inmunosupresor humano no apreciado y secuestrado con anterioridad desde el punto de vista geográfico. Los retrovirus no dejan el equivalente a un registro fósil, y la magnitud de la epidemia continúa expandiéndose, borrando los márgenes epidemiológicos previos. Puede ser que al final sea imposible reconstruir en forma definitiva la historia evolutiva del VIH y el principio de su diseminación.
Aunque la información disponible no permite delimitar en forma clara el origen de la pandemia de SIDA, han surgido datos orientadores a partir de estudios genéticos comparativos. Los análisis recientes de las secuencias de nucleótidos de un gran número de VIH-1, VIH-2 y VIS han apoyado la hipótesis de que los VIH pueden haberse originado de un reservorio diverso de lentivirus de primates no humanos.20,162 Como en el caso de muchos otros agentes infecciosos, los VIS no parecen causar enfermedad en sus huéspedes naturales. El hecho de que el VIH-1 y el VIH-2 ocasionen enfermedad en los humanos puede reflejar la introducción reciente de estos virus a la población humana. La evidencia de un origen simiano del VIH-2 es bastante obvia; sin embargo, no se ha identificado un progenitor directo del VIH-1.
Los estudios seroepidemiológicos revelan que varias especies de monos africanos, incluyendo los monos mangabeys/negros, los monos verdes africanos (Cercopithecus aethiops) y los mandriles (Papio [Mandrillus] sphinx), tienen altas tasas de infección natural por VIS (VISsmm, VISagm, y VISmnd, respectivamente).18 Pueden encontrarse evidencias de infección por VIS en el 25 a 50 porciento de los animales salvajes, y la infección no se asocia con ninguna enfermedad. Los VIS aislados de los animales infectados pueden clasificarse en tres grupos diferentes por análisis de la secuencia genética. Las secuencias de nucleótidos de los VISsmm, VISagm y VISmnd tienen cierta similitud entre sí, aunque cada virus está equidistante a los otros dentro de la escala evolutiva.18,20 El VISmac, que se ha aislado de macacos que sufren una enfermedad parecida al SIDA, está relacionado en forma estrecha al VISsmm, y ambos pertenecen al grupo genético que incluye al VIH-2. El virus VISmac patógeno no se encuentra en los macacos salvajes, sino que parece originarse del VISsmm, el virus de los sooty mangabey aparentemente inofensivo, por trasmisión zoonótica en cautiverio. Es interesante que las evidencias genéticas y la coincidencia geográfica indican que los mangabeys/negros han sido fuente también de la infección zoonótica de los humanos en Africa Occidental, con el origen subsecuente del VIH-2.19 El habitat natural de los mangabeys/negros en Africa Occidental coincide con la misma región geográfica en donde el VIH-2 es endémico, y se ha demostrado contacto estrecho entre los mangabeys y los humanos. Aunque no se han demostrado casos de trasmisión del VISsmm de un mangabey infectado a un humano, sí se han reportado recientemente infecciones en personas que trabajan en el laboratorio con este virus.163,164
El VIH-1 difiere del VIH-2 en que no se ha encontrado un progenitor obvio en primates no humanos, y el VIH no se replica en monos inoculados en forma experimental. Recientemente se aisló de los chimpancés el virus VIScpz, que parece estar más relacionado al VIH-1 desde el punto de vista de secuencia de nucleótidos que cualquier otro VIS o que el VIH-2.20,21,162,165 El VIScpz comparte una organización genómica común con el VIH-1, pero su secuencia de nucleótidos es muy divergente. Es interesante que se han identificado grupos de VIH-1 poco usuales (denominados subtipo O) cuya secuencia de nucleótidos indica que están equidistantes del VIScpz y de tipos más comunes de VIH-1.166,167 Aunque este dato puede indicar que el VIH-1 y el VIScpz tienen un origen común, la infección de los chimpancés por virus VIS parece bastante rara. Se requieren más estudios sobre la diversidad de los virus aislados en primates humanos y no humanos para comprender mejor la historia evolutiva del VIH-1.
Los estudios genéticos del VIH-1 de diferentes localizaciones geográficas pueden proporcionar claves sobre la evolución de la pandemia del SIDA.162 La determinación de las secuencias de gag y env de muchos VIH-1 ha permitido identificar ocho diferentes subtipos (designados de la A a la H y conocidos en conjunto como grupo principal M) en los principales centros de la pandemia del SIDA.34,162 El subtipo O del VIH-1, muy diferente, se identificó por primera vez en Camerún y Gabón, y representa un noveno subtipo, considerándose un grupo distinto.166,167 Los estudios actuales podrán identificar subtipos adicionales por sus secuencias virales. Diversos países en Africa, Asia y Sudamérica tienen subtipos de VIH-1 cocirculantes. En los Estados Unidos solo se ha reportado el subtipo B, probablemente como resultado de un intenso efecto fundador. En la actualidad no existen evidencias de que los subtipos individuales del VIH-1 difieran entre sí en sus propiedades biológicas o potencial patógeno. Además de ayudar en los estudios de epidemiología y evolución del VIH-1, el estudio de estos subtipos proporcionará la información práctica necesaria para asegurar que las pruebas serológicas disponibles identifican con sensibilidad la infección por todos los subtipos virales,168 y guiarán el desarrollo de las vacunas en diferentes partes del mundo.169
Aunque el origen de las infecciones por VIH sigue siendo un enigma, diversos factores parecen responsables de su diseminación mundial. Los focos de infección del VIH antes de que el virus se diseminara no están bien definidos. En algunas áreas rurales de Africa, en donde las condiciones sociales no favorecen la rápida diseminación del virus, la seroprevalencia a VIH-1 permaneció baja y relativamente estable en el mismo periodo en el que en otros sitios se incrementó.159 Sin embargo, durante los últimos 30 años, las presiones económicas, los cambios sociales y la marcada inestabilidad política han causado una migración masiva de habitantes rurales a áreas urbanas en Africa.159 Los cambios demográficos secundarios han creado un ambiente que facilitó la diseminación epidémica de enfermedades de trasmisión sexual, lo que a su vez ayudó a desencadenar la epidemia porVIH-1. El comercio internacional y el turismo proporcionaron oportunidades para la diseminación de la infección en todo el mundo. El sureste de Asia fué la última región en la que se introdujo el SIDA, pero la alta densidad de población y los comportamientos de riesgo prevalentes han causado que la infección se disemine con rapidez. Se cree que cambios sociales semejantes son los responsables del incremento en la infección endémica de VIH-2 en Africa Occidental, y su diseminación eventual a Europa, América y la India. El motivo de que el VIH-1 se disemine con más rapidez y en forma más amplia que el VIH-2 no se conoce; este dato puede reflejar diferencias biológicas entre los virus u otros factores. Sin embargo, independientemente de la historia evolutiva aparentemente distinta y de las diferencias biológicas potenciales entre los dos virus, la emergencia coincidente de la epidemia de VIH-1y de VIH-2 sugiere una compleja interrelación entre factores sociales y la diseminación de patógenos infecciosos.159
IDENTIFICACION DEL VIH-1
Los métodos utilizados para identificar el VIH-1 se basaron en las técnicas utilizadas para el aislamiento del HTLV-I y HTLV-II. Sin embargo, en contraste con los cultivos de células infectadas por HTLV-I y HTLV-II, la proliferación persistente de células T no continuó cuando los linfocitos de personas con SIDA o estados relacionados con el SIDA se colocaron in vitro. En lugar de ello, después de un periodo de crecimiento a corto plazo, se observó muerte celular extensa en los cultivos, junto con la presencia de actividad de transcriptasa y de partículas parecidas a retrovirus.7, 8, 170
Un adelanto importante fue el descubrimiento de que el VIH-1 podía propagarse en ciertas líneas celulares leucémicas T establecidas sin inducir efecto citopático.7, 170 Entonces el retrovirus podía crecer en grandes cantidades, permitiendo así su caracterización detallada y la preparación de reactivos para su uso en análisis epidemiológicos. El escrutinio del suero de pacientes con SIDA mostró casi el 100 porciento de reactividad con el VIH-1, mientras que el suero control de personas normales fue negativo en forma uniforme.171, 172 El análisis de personas con estados relacionados al SIDA o en alto riesgo de SIDA demostró muy elevada prevalencia de anticuerpos contra el VIH-1. Aún más, el VIH-1 podía aislarse de los linfocitos de la mayoría de las personas con SIDA o estados relacionados al SIDA al igual que de individuos asintomáticos seropositivos para VIH-1. En los estudios iniciales, la imposibilidad para aislar el virus de todas las personas seropositivas se debió a las limitaciones técnicas de los métodos empleados. Sin embargo, los métodos actuales permiten aislar el VIH-1 de la gran mayoría de las personas seropositivas.99,173
El VIH-1 puede trasmitirse con facilidad in vitro a cultivos de linfocitos de donantes normales. Poco después de la infección se observa un efecto citopático considerable, que se manifiesta por fusión celular, formación de células gigantes multinucleadas conocidas como sincicio y muerte celular. El cuidadoso examen de cultivos infectados revela que el blanco primario de la infección es la célula T inductora-colaboradora CD4+ y que las consecuencias citopáticas de la infección viral son más pronunciadas en esta población celular.74 La molécula CD4 (también llamada T4) es una glucoproteína que se encuentra predominantemente sobre la superficie de las células T. Es necesaria la interacción entre la molécula de superficie de la célula T CD4+ y las moléculas clase II del complejo principal de histocompatibilidad (CPH) expresada sobre la superficie de las células presentadoras de antígenos, para la generación de una respuesta inmune celular eficaz.174 Las células T CD4+ son la principal fuente de linfocinas, que brindan señales inductoras para la activación y orquestación de múltiples ramas de la respuesta inmune.
La naturaleza de este citotropismo peculiar no se comprendió al principio pero estaba de acuerdo con la pérdida de células T CD4+ in vivo que caracteriza a la inmunodeficiencia del SIDA. Desde entonces se ha demostrado que la misma molécula de superficie CD4, que desde el punto de vista fenotípico define a la población inductora-cooperadora de células T, es de hecho el receptor de superficie utilizado por el VIH-1 para entrar en las células blanco.174 Por ello, el rango de tipos celulares susceptibles a la infección por VIH-1, in vitro e in vivo, es paralelo con aquellas células que expresan el receptor de superficie CD4. Además, en la actualidad se sabe que el proceso de fusión celular que causa la formación característica del sincicio después de la infección por VIH-1 en las células T susceptibles, es una consecuencia directa de la interacción, específica y de alta afinidad, entre la molécula T CD4 y la glucoproteína env del VIH-1174 [ver figura 4].
El VIH-2 y los VISs comparten un citotropismo similar y sus
glucoproteínas de la envoltura viral también se considera que se
unen a la molécula CD4 en el comienzo de la infección. Lass
células que expresan moléculas CD4 sobre su superficie incluyen
ciertos miembros de los linajes monocitos-macrófago y linfoide T. Las
infecciones por VIH-1 de estos tipos celulares se ha observado in vivo en la
circulación periférica y tejidos linfáticos así
como en el cerebro y otros tejidos extralinfáticos, como los
pulmones.
DIVERSIDAD GENETICA DEL VIH-1
Se han obtenido gran cantidad de virus VIH-1 de regiones en todo el mundo. En muchos de ellos se ha determinado la secuencia de nucleótidos, y la comparación de estas secuencias revela un sorprendente grado de diversidad genómica, que es más marcada en la región del gen env.34,175 También existe un grado semejante de diversidad genómica entre los VIH-2 aislados.34 La diversidad del VIH-1 y el VIH-2 contrasta en forma marcada con la homogeneidad genética en todo el mundo de los HTLV-I, y se piensa que refleja el acúmulo de mutaciones que suceden en el proceso de transcripción inversa durante los numerosos ciclos de replicación en un huésped infectado por el VIH.175 A diferencia de las polimerasas celulares, que tienen una función endógena llamada de lectura probada para maximizar la fidelidad de la replicación, la transcriptasa inversa retroviral es incapaz de reparar los errores introducidos durante el proceso de síntesis del ADN. Se calcula que, por ello, de una a 10 mutaciones puntiformes se incorporan al genoma del VIH-1 durante cada ronda de replicación.36,176 Se introducen variaciones adicionales en la secuencia genómica viral por recombinación durante la síntesis viral, que es causada por cambio de templetes entre las dos moléculas de ARN viral que existen juntas en la partícula del virus VIH-1. Se sabe que la replicación del VIH-1 es un proceso muy activo en las persnas infectadas, y que proporciona numerosas oportunidades para la generación de la diversidad de la secuencia viral.
En un individuo infectado, cada genoma de VIH-1 puede tener una secuencia diferente de la de los otros, y los virus deben considerarse más como poblaciones, llamadas cuasiespecies, de variantes relacionadas en forma estrecha.175 Las variantes del VIH-1 presentes en estas cuasiespecies pueden mostrar diferencias marcadas con respecto a sus características biológicas, incluyendo el tropismo celular, el potencial citopático, la tasa de replicación, la susceptibilidad a medicamentos antivirales y las propiedades de antígeno de superficie que pueden permitirles escapar de ser depurados por el sistema inmunológico del huésped.
Mientras que muchos de los errores que ocurren en la transcripción inversa causan virus incompetentes para replicarse, ciertas áreas del genoma del VIH, como la secuencia que codifica el gen env, pueden adaptarse con más facilidad a las alteraciones de los nucleótidos.174,177 Las regiones del gen env que muestran una divergencia importante en la secuencia de nucleótidos se intercalan con dominios que se conservan bien en los diferentes VIH. Las comparaciones de las secuencias predichas de aminoácidos para las glucoproteínas gp120 env del VIH-1 de los diversos virus han revelado cinco regiones discontinuas, designadas como V1 a V5, que contienen residuos de aminoácidos muy variables.178 La variación genética en el gen env de los virus aislados determina su tropismo relativo a las células T o macrófagos, y su capacidad relativa para inducir la formación de sincicio en las células infectadas.178-180 Las secuencias variables del gen env influyen también en la susceptibilidad de un virus determinado para ser neutralizado por anticuerpos y preparaciones recombinantes de CD4 soluble.178
In vivo se ha observado la generación de la diversidad genética del VIH-1 y la selección de variantes virales específicas con el tiempo.175,177 Pueden identificarse en un mismo paciente y al mismo tiempo variantes virales relacionadas, pero distinguibles entre sí, y la representación relativa de las secuencias variantes específicas puede cambiar en forma significativa durante el curso de la infección.175,181,182 La imposición de una presión selectiva, como la administración de un medicamento antiviral, sobre el reservorio heterogéneo de diferentes VIH-1 in vivo, puede ocasionar crecimiento rápido de las variantes cuya replicación se inhiba en forma menos eficaz.45 En varios casos de trasmisión viral, se ha seguido tanto en el donador como en el receptor la evolución de la diversidad de secuencias del VIH-1.183-185 Después de la infección, el desarrollo de las variantes observadas parece depender de factores específicos del huésped en particular. El sistema inmunológico puede ser el principal factor que influya en la naturaleza y evolución de la diversidad genética del VIH-1. Sin embargo, aún no se conoce el papel preciso de la selección inmune en este proceso, y es posible que también participen otros factores. Por lo menos en algunos casos, la diversidad en la secuencia de nucleótidos entre los VIH-1 aislados puede traducirse también en diversidad de los antígenos de reconocimiento (ver adelante).
VIGILANCIA DE LA INFECCION POR VIH-1 IN VIVO
La infección por VIH-1 inicia un proceso citopático persistente y progresivo que culmina en la destrucción casi completa de las células T subtipo CD4+.41,44 Los primeros esfuerzos por sintetizar un modelo coherente sobre las profundas consecuencias patogénicas de la infección por VIH-1 se confundieron por la idea de que muy pocas células de los individuos infectados portan o expresan el genoma viral. Sin embargo, la aplicación reciente de métodos sensibles para detectar el VIH-1 ha demostrado que la carga viral es mucho mayor de lo que se pensaba antes, y que aumenta de modo significativo al progresar la enfermedad [ver figura 5]. Esta nueva apreciación sobre la dinámica de población del VIH-1 proporciona importantes datos sobre la patogenia de la enfermedad.41,44,45,186-188 La magnitud de la replicación durante la evolución clínica de la infección por VIH-1 sugiere que las consecuencias citopáticas directas de la expresión viral pueden ser las responsables de la depleción de linfocitos T. Además, la fluctuación en el grado de producción viral que ocurre en los diferentes estadios de la enfermedad indica que el sistema inmunológico puede ser capaz de restringir la replicación del VIH-1, por lo menos en forma temporal.
Hasta hace poco los métodos disponibles para vigilar el nivel de replicación viral en los pacientes infectados por el VIH-1 estaban limitados por problemas de insuficiente sensibilidad y reproducibilidad. Sin embargo, los adelantos en los métodos usados para cuantificar el nivel de VIH-1 en el plasma o células de sangre periférica de los pacientes infectados han proporcionado nuevos enfoques para dilucidar los aspectos virológicos de la historia natural de la enfermedad por este virus. Dos nuevos métodos son importantes para medir la concentración de ARNm del VIH-1 en las partículas virales en el plasma: una se basa en la amplificación del blanco (métodos cuantitativos de RCP) y la otra en la amplificación de la señal (el llamado ensayo de ADN ramificado [ADNr]).43,189,190 Al parecer, el grado de viremia (medido por la cantidad de ARN del VIH-1 en el plasma) refleja con exactitud la extensión de la replicación viral activa.43,45,187,188 Además, estudios recientes de cinética indican que la vida media del virus en el plasma (alrededor de dos días o menos) varía poco entre los pacientes con diferentes cargas virales o en diferentes estadios de la enfermedad. La tasa y magnitud de la reducción del virus en plasma observado en estos estudios implica que la vida media promedio de una célula infectada es de menos de uno a dos días [ver figura 6]. Estos datos indican que la concentración estable en plasma de VIH-1 está en función de la magnitud de la replicación y producción viral activa.187,188 En muchas maneras, la aplicación de estos nuevos métodos para vigilar la replicación del VIH-1 in vivo ha modificado las nociones sobre los mecanismos patogénicos de la enfermedad por VIH-1 [ver figura 5].
HISTORIA NATURAL DE LA INFECCION POR VIH-1
Los mecanismos de trasmisión del VIH-1 no se conocen del todo. El virus puede trasmitirse por transferencia de células infectadas, del virus libre o ambos, aunque la contribución precisa de estos componentes a un determinado episodio de infección probablemente depende de la naturaleza y vía de exposición. Si un donador trasmisor del virus tiene enfermedad avanzada, el receptor tiene más probabilidad de infectarse.120 Aún más, la infección por un donador en fase avanzada se asocia con mayor incidencia de infección aguda sintomática, con evolución más rápida a SIDA. Este fenómeno puede ser causado por mayor carga viral en el trasmisor, mayor virulencia de la cepa viral trasmitida o ambos. Las cargas virales en plasma y asociadas con células aumentan con la progresión de la enfermedad, y los estudios demuestran que se detecta la presencia del ARN de VIH-1 en forma más fácil en el semen de los varones con cuentas bajas de células T CD4+.41,44,191,192
Desde el sitio de entrada del virus, ya sea a través de las mucosas o directamente al torrente sanguíneo, el virus VIH-1 libre o las células infectadas se dirigen al tejido linfoide. La presencia de células blanco susceptibles, incluyendo células T, células dendríticas y macrófagos, y el ambiente inmunológicamente activo en los tejidos linfáticos permite nuevos ciclos de replicación del VIH-1 e infección de las células del huésped. El análisis de los VIH-1 aislados del trasmisor y del receptor indica que la población viral muy heterogénea del donador sufre un proceso de selección que permite que la infección se establezca por un subgrupo pequeño de VIH-1.193,194 En el nuevo huésped, recién infectado, predomina una población genéticamente homogénea de virus que son principalmente citotrópicos para los macrófagos y que no inducen sincicios (NIS) (ver adelante). No se sabe si la ventaja selectiva de esta subpoblación de VIH-1 es causada por la trasmisión de un número limitado de variantes virales o por la replicación preferente de ciertos subtipos en el nuevo huésped. Tampoco se ha demostrado que la infección con variantes específicas cause manifestaciones clínicas especiales, pero el tropismo y potencial patogénico de ciertas cepas sugiere que la patogenicidad selectiva puede contribuir a las diferentes manifestaciones clínicas de la infección por VIH-1 y a los diversos grados de progresión de la enfermedad en los distintos pacientes.
El rango y gravedad de los síntomas de la infección por VIH-1 varía en forma muy considerable. Alrededor del 40 a 60 porciento de los pacientes sufren un síndrome viral agudo que consiste en fiebre, cefalea, linfadenopatía, mialgias y erupción durante la infección primaria, cuando ocurren altos grados de replicación del VIH-1 antes del desarrollo de una respuesta inmunológica antiviral [ver figura 5].195-198 Coincidente con la resolución de los síntomas de la infección aguda, que dura alrededor de un mes, caen en forma brusca los niveles de viremia en plasma y el número de células T CD4+ infectadas en sangre periférica. La disminución en la replicación del VIH-1 correlaciona con la aparición de una respuesta de células T citotóxicas contra el VIH-1.199-201
En contraste con la historia natural de la infección primaria sintomática, los aspectos virológicos e inmunológicos de la infección primaria asintomática no se conocen. Los individuos que tienen síntomas clínicos severos durante la infección primaria parecen tener más probabilidad de sufrir infección primaria sintomática.202 Este fenómeno puede ser especialmente cierto en pacientes que sufren un alto nivel de linfocitosis T CD8+, los que tienen una respuesta inmunológica celular antiviral deteriorada o con restricción clonal, o los que montan una respuesta serológica limitada al antígeno central p24 del VIH-1.201,202
Una vez que ceden los síntomas de la infección primaria, el paciente entra en una fase clínicamente asintomática o de síntomas mínimos. Aunque la respuesta inmunológica antiviral puede limitar la replicación del VIH-1 durante este periodo de latencia clínica, parece no ser totalmente eficaz, persistiendo la producción viral, lo que se hace evidente por la existencia continua de VIH-1 en el plasma [ver figura 5] y los tejidos linfoides [ver figura 7].41,43,44,187,191,192 La replicación activa y continua del VIH-1 en los individuos asintomáticos ocasiona la reducción lentamente progresiva de las células T CD4+. La evidencia de una mayor carga viral en el plasma y de concentraciones elevadas de ADN y ARN del VIH-1 en las células de sangre periférica correlaciona con una depleción más acelerada en las células T CD4+, lo mismo que en la progresión de la enfermedad, y es probable que refleje la falla progresiva de la respuesta inmunológica para contener la replicación del VIH-1.41,44,203
Además de los cambios en los grados de replicación viral
observados en fases suscesivas de la enfermedad, el fenotipo replicativo de los
virus VIH-1 puede también relacionarse con la progresión de la
enfermedad. La manifestación citopática más obvia de la
infección por VIH-1 en cultivo de tejidos es la fusión y muerte
subsecuente de las células T susceptibles. Los VIH-1 aislados de
individuos infectados pueden tener capacidades muy distintas para fusionar a
las células T CD4+ en sincicios [ver figura 4]: las
cepas que inducen sincicios (IS) suelen replicarse más y ser más
citopáticas in vitro que sus contrapartes NIS. Diversos estudios han
indicado que la aparición de cepas IS durante el curso de la
infección por VIH puede presagiar una tasa acelerada de pérdida
de las células T CD4+ y deterioro
clínico.204 Sin embargo, también se ha demostrado que
pueden aislarse virus IS de solo el 50 a 60 porciento de los pacientes con
SIDA, por lo que la presencia de virus IS no es necesaria para el desarrollo de
la inmunodeficiencia clínica. No se sabe si los cambios observados en la
capacidad de replicación viral, la citopatogenicidad y el tropismo
celular que se observan durante el curso de la infección por VIH-1
representan las causas o las consecuencias de la inmunosupresión en
evolución.
La mayoría de las evaluaciones inmunológicas y virológicas en pacientes infectados por VIH-1 se han enfocado a los linfocitos de sangre periférica. Sin embargo, se calcula que estas células representan alrededor del dos porciento del reservorio de linfocitos en todo el organismo. Se ha señalado la importancia de los órganos linfoides como reservorios de la infección por VIH-1 por la demostración de que el porcentajde de linfocitos T CD4+ infectados por VIH-1 puede ser hasta 10 veces mayor en los ganglios linfáticos que en sangre periférica42,46 [ver figura 7]. Además, los linfocitos infectados por el VIH-1 que se encuentran en los órganos linfoides expresan más ARN viral que sus contrapartes en sangre periférica. Los hallazgos de que las células activadas proporcionan blancos preferenciales para la infección y producción del VIH-1 y que mucha de la carga viral se localiza en los tejidos linfáticos que participan en la generación de la respuesta inmune sugiere que las células inmunológicamente activadas pueden también sufir las consecuencias patogénicas de la infección por VIH-1.
En las fases tempranas de la enfermedad se encuentran cantidades sorprendentes de partículas virales en los ganglios linfáticos asociados con las células dendríticas foliculares (CDF) que tienen un papel indispensable en el procesamiento y presentación de los antígenos.41,44 No parece ser que las CDF se infecten, pero sí pueden facilitar la infección de las células T CD4+ que migran a través de los folículos linfoides durante el curso de una respuesta inmunológica o de la recirculación normal. Con el tiempo ocurre la destrucción gradual de la red de CDF, lo que reduce la capacidad de atrapamiento del virus y de antígenos. Al progresar la enfermedad, la arquitectura normal de los ganglios linfáticos se distorsiona, lo que puede comprometer la función inmunológica normal. No se sabe si la disrrupción de la red de CDF es causada por el efecto tóxico directo de la infección por VIH-1 o por la entrada de células T citotóxicas CD8+ que han sido observadas cerca de los sitios de destrucción de las CDF.
MECANISMOS DE DEPLECION DE LAS CELULAS T EN LA INFECCION POR VIH-1
Aún no se define el mecanismo exacto por el que la infección por VIH-1 causa depleción progresiva y deterioro funcional de las células T CD4+, pero es probable que incluya diversos mecanismos directos e indirectos.41,44,205 El reconocimiento reciente de que durante el curso de la enfermedad persiste una expresión y replicación considerable del VIH-1 apoya la hipótesis de que los efectos citopáticos directos de la replicación viral son los causantes de mucho del compromiso inmunológico observado. Los estudios intervencionistas que han usado medicamentos antivirales potentes pero de efecto temporal, proporcionan evidencia directa de que un mayor nivel de replicación del VIH-1 correlaciona con tasas aceleradas de recambio en las poblaciones de linfocitos T CD4+ [ver figura 6].45,187,188
La infección por VIH-1 causa efectos citopáticos directos en las células T CD4+ in vitro que pueden observarse tanto en la célula aislada como por la formación de sincicios [ver figura 4]. In vivo la formación directa de sincicios se observa muy rara vez (excepto en los cerebros de algunos pacientes infectados), quizá por la rápida depuración de cualquier célula gigante multinucleada aberrante de la circulación. Las consecuencias patológicas de la infección por VIH-1 no se distribuyen en forma igual entre todas las células T CD4+, sino que se enfocan en forma preferente a las células proliferantes o activadas inmunológicamente. Los pacientes con infección asintomática tienen defectos específicos en la función de las células T incluso antes de que se observe disminución en su número.206 Estos defectos son más importantes en la población de células T de memoria que participan en el reconocimiento y respuesta en la segunda exposición a un antígeno específico. Al parecer esta subpoblación de células T es el blanco preferencial de la infección por VIH-1 in vivo.207 No se conoce la explicación precisa para el compromiso numérico o funcional de esta importante subpoblación de células inmunes efectoras. Sin embargo, suponiendo que las células T de memoria reconocen y proliferan en respuesta a antígenos que son comunes en el ambiente, la activación frecuente de estás células en un huésped infectado por el VIH-1 puede causar depleción paradójica de la población de células respondedoras. Por lo tanto, el repertorio de reactividad de las células T a un antígeno específico puede restringirse en forma progresiva en la infección por VIH-1 avanzada, lo que predispone al paciente a sufrir infecciones oportunistas.
Se ha sugerido que además de causar defectos preferentes en las células T de memoria, la infección por VIH-1 puede alterar el balance apropiado entre las subpoblaciones especializadas de células T CD4+ conocidas como TH1 y TH2, que se definen por su patrones particulares de producción de citocinas y su papel para inducir respuestas inmunes celulares y humorales, respectivamente.208,209 Sin embargo, varios estudios han puesto en duda este aspecto,208,210,211 y deben realizarse más estudios para definir las alteraciones potenciales en las subpoblaciones de células T CD4+ o los patrones alterados de producción de citocinas que pueden acompañar a la infección por VIH-1.
Se ha demostrado que la infección por VIH-1 inicia respuestas tanto celulares como humorales hacia los antígenos codificados por el virus (ver adelante). Aunque la destrucción inmunológica de los linfocitos T y los macrófagos infectados es indispensable para que el huésped controle la infección, puede, en el caso de una infección crónica y persistente, acelerar la deficiencia del sistema inmunológico que sigue a la infección por el VIH-1.174,212 Las células infectadas por el virus expresan antígenos virales que las convierten en blanco para la depuración inmunológica. Además, se ha sugerido que la unión de gp120 (liberado de las células infectadas y de las partículas virales) a la superficie de células no infectadas que portan CD4, las hace susceptibles a la destrucción inadecuada por las respuestas inmunológicas antivirales del huésped por un mecanismo denominado de espectador inocente. No se conoce aún la importancia in vivo de este mecanismo para la depleción de células T.
También se ha sugerido que la infección por VIH-1 causa destrucción indirecta de las células T CD4+ al sensibilizarlas para sufrir apoptosis, o muerte celular programada.205 La apoptosis es un mecanismo fisiológico normal por el que se eliminan ciertas clases de células durante el desarrollo de los linfocitos (v.gr., la deleción de las células T reactivas a los antígenos del huésped) o al terminar una respuesta inmunológica. Varios estudios han descrito mayores grados de apoptosis cuando las células T de individuos infectados por el VIH-1 se activan en cultivos de tejidos.41,205,213,214 El número de linfocitos CD4+ que sufre apoptosis excede en mucho al de los infectados por el virus. Los linfocitos CD8+, que no son blanco de la infección por VIH-1, también son sensibilizados para sufrir apoptosis. No se conoce del todo el mecanismo por el que se activa la apoptosis en las células T de los individuos infectados, ni se ha determinado el significado in vivo de este fenómeno descrito en cultivo de tejidos.
La depleción númérica de las células T CD4+ es la alteración inmunológica más obvia que sigue a la infección, pero se han descrito también varios defectos en la función de las células T, las células B y los monocitos.47 No se sabe si estos defectos tienen un papel primario en la inmunodeficiencia inducida por el VIH-1 o si reflejan la alteración en la regulación causada por la depleción de las células T CD4+. In vitro, las células T de los individuos infectados demuestran también menor reactividad a antígenos y una menor capacidad para traducir las señales de proliferación después de la estimulación. Se ha sugerido que las señales inapropiadas de las células T que son resultado de la unión de complejos de gp120-anti-gp120 al CD4 pueden causar la inducción de un estado de no respuesta inmunológica o anergia de las células T CD4+ en las personas infectadas por VIH-1.
La depleción de las células T CD4+ observada después de la infección por VIH-1 debe considerarse dentro del contexto de la dinámica de población de los linfocitos T, esto es, el balance entre la producción y la destrucción de las células T. Por desgracia, la capacidad de la población de las células T para autorrenovarse no se comprende del todo. No se sabe hasta qué grado la expansión periférica de células T maduras o la maduración de nuevas células T a partir de sus progenitores contribuye a mantener el reservorio total de linfocitos T en el adulto. Sin embargo, evidencias recientes sugieren que la contribución del timo para mantener la concentración de las células T es muy limitada después de los 20 años de edad.215 En cualquier caso, es probable que la infección por VIH-1 altere ambas fuentes de producción de células T, de modo que el remplazo celular no compence la pérdida. Se conoce que el timo es un órgano blanco importante en la infección y citopatología de la infección por VIH-1, y es probable que esto limite el desarrollo eficaz de células T.
Puede ser que, por mecanismos directos o indirectos, la depleción de las poblaciones de células T CD4+ maduras y la disrupción de la arquitectura de los órganos linfoides después de la infección por VIH-1, implique alteración en la expansión del reservorio periférico de células T. Al parecer el sistema inmunológico humano tiene mecanismos homeostáticos por los que se mantiene un número normal de células T CD4+. Cuando ocurre depleción de éstos por la infección por el VIH-1, los mecanismos compensatorios intentan normalizar las cifras celulares, lo que provoca mayor proliferación de linfocitos T CD4+ maduros del reservorio periférico. Por desgracia, estas células activas desde el punto de vista de proliferación constituyen blancos fértiles para la infección por VIH-1 y en sí pueden infectarse y destruirse [ver figura 6]. En ausencia de un remplazo adecuado de células T, puede presentarse una reducción definitiva en el número de estas células.
RESPUESTA INMUNOLOGICA AL VIH-1
La influencia del sistema inmunológico humano para modular la evolución de la infección por VIH-1 no se conoce del todo. Sin embargo, la persistencia y naturaleza casi siempre progresiva de la infección sugiere que el sistema inmunológico es relativamente ineficaz. Esta impotencia inmunológica puede ser resultado de una estregia viral para evadir el reconocimiento antigénico, del fracaso para montar una respuesta neutralizante humoral o celular, o por infección y destrucción viral de la población T CD4+, que tiene un papel crítico en la eliminación de las infecciones incipientes.
Aunque las investigaciones seroepidemiológicas han demostrado que existen anticuerpos antiVIH-1 específicos en el suero de las personas con SIDA o riesgo de SIDA, no se sabe si estos anticuerpos tienen un efecto protector in vivo.178 Los anticuerpos obtenidos del suero de las personas infectadas son capaces de neutralizar la infectividad del VIH-1 en cultivo de tejidos, pero los títulos de estos anticuerpos en las personas infectadas son muy bajos. Aún más, los anticuerpos neutralizantes no parecen inhibir en forma eficaz la trasmisión viral a través del contacto célula-célula. La reducción inicial en la viremia del VIH-1 que ocurre con la resolución de la infección primaria se observa antes de que se desarrollen anticuerpos, y la presencia de anticuerpos neutralizantes no parece proteger contra la progresión de la enfermedad. Por lo tanto, no es claro el significado biológico de la respuesta neutralizante de anticuerpos en la modulación del curso de la infección por VIH-1.
Los anticuerpos que bloquean la infección por VIH-1 en cultivo de tejidos son específicos para dos dominios distintos de la glucoproteína env del VIH-1.178 Un dominio consiste en las regiones conservadas de la glucoproteína env que se requieren para unir a la molécula CD4. Debido a la naturaleza conservada de este dominio, los anticuerpos dirigidos contra él neutralizan diversas cepas de VIH-1 y se conocen como grupo específicos. Un segundo tipo de anticuerpos neutralizantes reconoce una estructura de asa en el tercer dominio hipervariable (o V3) de la glucoproteína env que parece tener un papel crítico en los pasos de la infección viral que ocurren después de la unión a CD4. Los anticuerpos dirigidos contra el dominio V3 bloquean la fusión del virus a la membrana celular, pero no inhiben la fijación viral al CD4. Los anticuerpos neutralizantes que reconocen el dominio V3 se denominan tipo específicos, porque bloquean la infección solo por cepas son secuencias de V3 semejantes a las de la variante VIH-1 que los originó.178 Las respuestas iniciales de anticuerpos neutralizantes producidas en etapas tempranas de la infección por VIH-1 forman anticuerpos que reconocen sobre todo los determinantes de la regiónV3, y son tipo específicos. En una fase más tardía se desarrollan anticuerpos neutralizantes con reactividad más amplia dirigidos contra la región de unión CD4.178 La mutación de las secuencias del gen env del VIH-1 ocasiona la aparición de cepas resistentes a los anticuerpos neutralizantes en cultivo de tejidos e in vivo. Sin embargo, no se ha determinado la importancia de estos hallazgos en la persistencia de la infección por VIH-1.
Se ha sugerido que ciertos anticuerpos antivirales pueden facilitar, en lugar de prevenir, la infección por VIH-1 de las células de linaje monocito-macrófago a través deun mecanismo que involucra la captación mediada por el receptor Fc de los complejos inmunes que contienen el virus. Los anticuerpos que facilitan la infección viral se conocen como anticuerpos promotores, y pueden identificarse por medio de ensayos en cultivo de tejidos.217 En la actualidad no existe evidencia clara de que los anticuerpos promotores contribuyan a la diseminación de la infección in vivo por VIH-1.
Mucho del análisis inicial de la reacción del huésped a la infección por VIH-1 se ha enfocado a la respuesta inmune humoral. Sin embargo, la inmunidad celular tiene un papel crítico en la eliminación de muchos otros tipos de infección viral, por lo que la magnitud en la que sus componentes puede reconocer y limitar la infección del VIH-1 es de gran interés. Aunque existe relativamente poca información sobre el papel de la inmunidad celular para modular la evolución de la infección por VIH-1, evidencias recientes indican que la rápida declinación en la replicación del VIH-1 observada después de la infección primaria correlaciona de modo temporal con la aparición de células T citotóxicas específicas.199,200,218 Además, se ha observado conservación de una respuesta citotóxica de células T contra el VIH-1 en algunos individuos que permanecen asintomáticos a pesar de tener infección de larga evolución.147,148 Se han obtenido células T citotóxicas reactivas contra los antígenos del VIH-1 en sangre periférica, LCR y lavado broncoalveolar de pacientes infectados.219 Estas células T reconocen los antígenos VIH-1 en el contexto de las moléculas de clase I del HLA del huéped y destruyen las células infectadas autólogas, pero no las heterólogas. Lo más frecuente es que reaccionen contra los determinantes antigénicos de las proteínas gag, env, transcriptasa inversa y nev del VIH-1. Los estudios indican que la diversidad de secuencia entre las variantes del VIH-1 que aparece durante el curso de una infección in vivo puede causar variabilidad en el reconocimiento antigénico y en la destrucción por las células T citotóxicas, pero no se sabe si esta variabilidad ayuda a las células infectadas a escapar de la depuración inmunológica.220,221
Además de las células T citotóxicas, la respuesta inmune celular al VIH-1 incluye una población de células efectoras que tienen receptores Fc. Estas células destruyen a las células infectadas por el virus en presencia de anticuerpos antivirales, por un proceso conocido como citotoxicidad celular mediada por anticuerpos (CCMA).222 Los anticuerpos que dirigen el proceso de CCMA se dirigen principalmente contra la glucoproteína gp120 env del VIH-1, y las células efectoras responsables muestran los marcadores fenotípicos de las células asesinas naturales (células NK).
La complejidad de la respuesta inmunológica a la infección por el VIH-1 y las diferencias inmunológicas potenciales que pueden existir en el espectro de este padecimiento representan temas muy importantes de estudio. El vigor de la respuesta inmune inicial contra el VIH-1 puede influir en la evolución subsecuente de la enfermedad (ver adelante). En fases posteriores, la pérdida de reactividad de anticuerpos contra la proteína nuclear p24 del VIH-1 y la aparición subsecuente de este antígeno en el suero se ha asociado con mal pronóstico.223 Sin embargo, no se sabe si la pérdida de la reactividad inmunológica en estos casos es la causa o la consecuencia de la inmunosupresión progresiva. Aún debe establecerse el significado de estos datos y las variaciones adicionales posibles en la reactividad inmunológica con otros productos virales. Es crucial aumentar nuestros conocimientos sobre la respuesta inmunológica contra el VIH-1 si deseamos prevenir y tratar la enfermedad.
VACUNAS PARA PREVENIR LA INFECCION POR VIH-1
La velocidad de la diseminación mundial del VIH-1 ha hecho que se ponga especial atención al desarrollo de una vacuna para prevenir la infección. Por desgracia, los prospectos para una vacuna segura y eficaz han sido limitados en el concepto y en la práctica por varias complejidades biológicas.169,225-227 Además, el establecimiento de la eficacia en cualquier candidato a ser vacuna se complica por aspectos éticos, legales y de responsabilidad.169,225,228,229
La falla en la inmunidad natural durante el curso de la infección por VIH-1 distingue a esta infección de muchas de otras infecciones virales en los humanos. Los mecanismos por los que el VIH-1 evade la depuración inmunológica no se conocen del todo, pero es probable que incluyan tanto a su compleja capacidad genética reguladora, que facilita el establecimiento de una fase latente, y la tremenda complejidad y diversidad de secuencias, que permite que el virus escape de las respuestas inmunológicas del huésped. Existen precedentes de vacunas útiles solo en las enfermedades, como la polio o el sarampión, en las que se desarrolla inmunidad protectora en el curso de la infección natural. Para ser eficaz, cualquier vacuna viral debe prevenir la adquisición del agente infeccioso o originar una respuesta inmune suficiente para facilitar la eliminación de las células infectadas antes del desarrollo de la enfermedad. Un hecho importante pero con frecuencia poco apreciado es que la mayoría de las vacunas previenen la enfermedad, no la infección. Sin embargo, el sistema inmunológico parece incapaz de eliminar la infección por VIH-1 una vez que esta se establece. Como resultado, las vacunas candidatos deben satisfacer gran cantidad de requerimientos, incluyendo la capacidad de inducir respuestas inmunológicas que neutralicen en forma eficaz la infectividad del VIH-1 libre y eliminen a las células infectadas antes de que se establezca la infección sistémica. El desarrollo de la vacuna también puede estar limitado por ramificaciones inmunológicas por la gran diversidad genética del VIH-1, y una vacuna eficaz debe proteger contra diversas variantes virales.169,225,227
Debido a que el contacto sexual por el que se depositan virus o células infectadas en mucosas es la causa de la infección en más del 80 porciento de todas las infecciones por VIH-1, la vacuna eficaz debe inducir inmunidad protectora tanto sistémica como en mucosas.230 Por desgracia, la mayoría de las vacunas que se desarrollan en la actualidad generan inmunidad sistémica, más que mucosa, y no limitan en forma eficaz el establecimiento de la infección a través de la principal vía de trasmisión del VIH-1.230
La definición de las correlaciones potenciales de la respuesta inmune, una herramienta esencial en el desarrollo de las vacunas, no ha sido posible en el caso del VIH-1 por la falta de un modelo animal de SIDA inducido por VIH-1 y de una cohorte de individuos que sean naturalmente resistentes a la infección por VIH-1.169,225-227 Con la esperanza de descubrir los factores inmunológicos del huésped que son responsables de la protección ante la infección o enfermedad, se ha puesto especial atención a los pocos individuos con infección por VIH-1 de muy larga duración y no progresiva, y a los que parecen ser resistentes a la infección a pesar de numerosas exposiciones. También se han reportado casos raros de individuos en quienes aparentemente se ha eliminado la infección por el VIH-1,230a pero es necesario investigar más sobre estos reportes para establecer la frecuencia y validez de los datos. La identificación de factores que pudieran proteger de la infección después de la exposición o del desarrollo de la enfermedad podrían ayudar a guiar el desarrollo de la vacuna, tarea que parece aún muy difícil.147-149,231
La glucoproteína env del VIH-1 ha sido identificada como el componente viral crucial reconocido por los anticuerpos neutralizantes, por lo que puede ser el inmunógeno principal en las vacunas contra el virus. Los anticuerpos neutralizantes podrían ser suficientes para limitar la infección por VIH-1 libre, pero es poco probable que detengan la infección después de la exposición a células infectadas.169,225,227 Por lo tanto, debido a que al parecer la trasmisión del VIH ocurre sobre todo por la transferencia de células infectadas, es lógico suponer que la vcuna eficaz contra el VIH-1 debe estimular a las células T citotóxicas específicas contra el virus. La evaluación de la importancia relativa de las respuestas humoral y celular en las posibles vacunas no puede evaluarse por estudios en humanos, y la mayoría de los esfuerzos experimentales se han realizado en modelos de primates no humanos con SIDA.
Las vacunas que se estudian en la actualidad en modelos animales para proteger contra las infecciones por VIH y VIS incluyen al virus inactivado completo, a antígenos proteicos recombinantes, a péptidos sintéticos o a virus de la vaccinina u otros poxvirus.169,225,227 En la mayoría de los estudios experimentales realizados hasta la fecha los animales han sido inmunizados por vía experimental y se les ha administrado dosis bajas de virus libre homólogo o muy relacionado en dosis bajas en el momento de la máxima respuesta inmunológica inducida por la vacuna. Los paradigmas experimentales empleados en estos estudios han sido criticados por no corresponder a las circunstancias de trasmisión del VIH-1 en los humanos. Además, los estudios de vacunas empleadas en modelos animales han proporcionado poca información sobre la inmunidad protectora necesaria para el desarrollo de una vacuna contra VIH-1.
La inmunización repetida con virus íntegros inactivados ha demostrado proteger a los monos rhesus contra el reto intravenoso con VIS patógenos vivos.227 Sin embargo, en los casos en los que se observó protección contra el virus libre, esta protección no duró muchó más allá del pico de la inmunidad máxima, y no se logró protección contra la exposición por mucosas. El éxito de estos estudios parece ser resultado de respuestas inmunológicas no esperadas contra antígenos celulares humanos contenidos en la preparación de la vacuna contra VIS y en el reservorio de virus, ya que ambos se propagan en líneas celulares T humanas. La inmunización con subunidades recombinantes de antígenos de VIS, análogos a las vacunas probadas en la actualidad en humanos, han sido incapaces de proteger a los monos susceptibles de un inóculo experimental con VIS virulentos.227 Aunque este resultado puede proporcionar la base para la definición de la correlación experimental de la inmunidad protectora contra los lentivirus, las dudas sobre la seguridad de este enfoque en la preparación de una vcuna contra el VIH-1 puede limitar su uso en humanos.223
Los chimpancés son los únicos primates no humanos que pueden infectarse por el VIH-1, pero su utilidd como un modelo animal exacto para SIDA es limitado porque no desarrollan enfermedad clínica después de la infección. Se ha demostrado que los chimpancés inmunizados contra una versión recombinante de glucoproteína 120gp env o un virus vaccinia recombinante que contiene todo el gen env del VIH, desarrollan niveles mesurables de anticuerpos específicos contra el VIH-1 y respuestas inmunológicas mediadas por células T.227,234 Las respuestas de anticuerpos neutralizantes inducidas por la vacuna son tipo específico y temporales. La inmunización de los chimpancés con los antígenos recombinantes de glucoproteína env que se han probado también en humanos se asocian con un éxito variable y limitado para proteger a los animales inmunizados contra un inóculo intravenoso del VIH-1 homólogo.33,210,227,235,236
Las vacunas probadas en la actualidad en voluntarios humanos no infectados incluyen la glucoproteína gp 120 env del VIH-1, producida en forma purificada por métodos de ADN recombinante, péptidos sintéticos derivados de gp120 y el precursor glucoproteína gp160 env insertado en el genoma de un virus vaccinia u otro vector poxvirus atenuado.169,227 Los estudios de fase I de estas vacunas indican que son seguras, e inmunogénicas en grado variable.169,227 En la actualidad se realiza un estudio de fase II que incluye a adultos con riesgo alto de infección y un estudio de fase I en lactantes nacidos de madres seropositivas.
Las vacunas de glucoproteína env recombinante pueden originar anticuerpos que neutralizan la infectividad del VIH-1 libre en cultivo de tejidos. La inducción de títulos apreciables de anticuerpos neutralizantes por las vacunas VIH-1 requiere con frecuencia de múltiples inmunizaciones, y los niveles de anticuerpos desaparecen algunos meses después. 169,227 Debido a que los inmunógenos de la proteína env del VIH-1 no se expresan en las células del individuo inmunizado, no despiertan una respuesta de células T citotóxicas. A diferencia de los anticuerpos neutralizantes que se observan en los individuos infectados por el VIH-1 y que neutralizan un amplio rango de cepas de VIH-1, los anticuerpos inducidos por la vacuna se dirigen principalmente contra la secuencia de la región específica V3 de la proteína env de la cepa de VIH-1 usada como inmunógeno en la vacuna.169,178,227 En consecuencia, estos anticuerpos son tipo específicos, y neutralizan solo cepas de VIH-1 muy relacionadas. El reconocimiento de que los anticuerpos inducidos por la vacuna no neutralizan las principales cepas de VIH-1, a diferencia de las cepas de laboratorio que se han propagado en líneas celulares T inmortalizadas, constituye una gran desventaja. Si los antígenos recombinantes de la proteína env van a convertirse alguna vez en el origen de la vacuna, debemos comprender los mecanismos moleculares que explican las limitaciones de las vacunas de primera generación y diseñar nuevos enfoques para inducir anticuerpos reactivos de espectro más amplio que neutralicen en forma eficaz a los principales grupos de VIH-1.
Los virus recombinantes de vaccinia que expresan gp160 sí despiertan respuestas de células T citotóxicas, pero muy pocos o ningún anticuerpo neutralizante anti-VIH-1.169,226,227 La inmunogenicidad de las vacunas basadas en virus vaccinia es limitada en personas que ya han sido antes inmunizadas contra la viruela. Además, debido a las dudas sobre la seguridad de las vacunas de virus vivos en personas inmunosuprimidas, estas vacunas pueden no ser adecuadas en las regiones del mundo en las que no es práctico realizar escrutinio anti-VIH-1.
A pesar de las evidencias obtenidas en las respuestas inmunológicas inducidas por vaccinia, algunos participantes de los estudios iniciales de vacuna anti-VIH-1 se infectaron posteriormente por el virus debido a la exposición por comportamientos de riesgo alto. Aunque estos casos de infección indican que las vacunas no son totalmente eficaces para prevenir la infección, no se ha evaluado en estudios preliminares su eficacia. En vista de la limitación de vacunas eficaces disponibles en la actualidad, en los Estados Unidos se han pospuesto recientemente los planes para realizar estudios de fase III de eficacia en gran escala.
Como resultado de los múltiples obstáculos que complican el desarrollo de las vacunas, pocos especialistas son optimistas y esperan que se disponga de una vacuna eficaz a corto plazo. Por lo tanto, deben enfatizarse las medidas de salud pública dirigidas a limitar la diseminación de la infección por el VIH-1.
TRATAMIENTO DE LA INFECCION POR VIH
Se considera que el compromiso inmunológico y neurológico lentamente evolutivo observado en la infección por VIH-1 es la manifestación del daño acumulado debido al bajo nivel pero persiste de replicación viral. Si la replicación viral demuestra ser la causa del compromiso clínico y si los sistemas afectados del huésped poseen suficiente potencial regenerativo, entonces los medicamentos que inhiben la replicación viral pueden prevenir o invertir las manifestaciones clínicas de las infecciones por VIH-1. Los esfuerzos actuales de investigación se dirigen a definir estrategias terapéuticas que limiten en forma más eficaz el grado de replicación del VIH-1 en los pacientes tratados y que mantengan la supresión de la replicación viral durante periodos prolongados.
El comportamiento biológico del VIH-1 impone varios prerrequisitos en la selección y utilidad de fármacos antivirales potenciales. Debido a la propensión del VIH-1 a infectar el sistema nervioso central y la capacidad del virus para establecer una infección persistente, los medicamentos propuestos deben cruzar la barrera hematoencefálica y poseer un índice terapéutico compatible con la administración a largo plazo. Aún más, debido a la propensión del VIH-1 para infectar células en el SNC, los medicamentos candidatos deben cruzar la barrera hematoencefálica. Por último, debido a que los linfocitos CD4+ y los macrófagos que son los blancos del VIH-1 in vivo pueden mostrar diferentes patrones de metabolismo de fármacos, debe contarse con agentes antivirales eficaces para lograr concentraciones adecuadas de sus metabolistos activos en cada uno de estos sitios cruciales de replicación viral.
Para disminuir la toxicidad y aumentar al máximo la eficacia, los fármacos deben limitar de forma importante la replicación viral pero producir interferencia mínima con el metabolismo de la célula huésped. Aunque el VIH-1 utiliza componentes de la maquinaria de la célula huésped para la síntesis y el procesamiento de su RNA y proteínas constituyentes, también depende de funciones biológicas únicas de varios productos específicos de virus para la replicación, incluyendo la enzima transcriptasa inversa y las proteínas tat y rev, que proveen blancos potenciales para terapéuticas farmacológicas eficaces. A pesar que cualquier etapa del ciclo de replicación del VIH-1, desde la unión inicial al receptor hasta las etapas culminantes del ensamble y maduración del virión, ofrece un punto potencial para la intervención terapéutica [ver figura 8], es probable que el diseño de fármacos antivirales eficaces se base en el mejor conocimiento de la función genética y la biología celular del VIH-1.237, 238
Incluso cuando se identifican los agentes antivirales, ocurren in vivo numerosos ciclos de replicación viral y la subsecuente variación extensa de las secuencias del VIH-1 proporciona un gran sustrato para generar y seleccionar variantes virales que son resistentes a un determinado agente antiviral. De hecho, es probable que exista un número sustancial de variantes virales resistentes a medicamentos que ya están presentes en la persona infectada por el VIH-1 antes de que se inicie el tratamiento.45 Es seguro que puede esperarse desarrollo de resistencia a fármacos cuando se administran fármacos que bloquean en forma incompleta la replicación viral durante periodos prolongados. Estudios recientes indican que la aparición de virus mutantes resistentes a medicamentos es la principal causa del fracaso de todos los tratamientos farmacológicos existentes hasta la fecha. Los virus resistentes a medicamentos que surgen durante el tratamiento se replican menos bien, o están menos adaptados que sus contrapartes naturales, por lo que puede lograrse una carga viral más baja que la que existía antes del tratamiento [ver figura 6]. Se espera que en el futuro se identifiquen combinaciones de medicamentos que seleccionen mutaciones en la proteína blanco del VIH-1 y que comprometan la replicación viral en tal grado que los virus mutantes sean menos patógenos o, en forma ideal, incapaces de replicarse.
Además de estos retos en el desarollo de medicamentos, ha sido muy difícil probar en forma amplia los medicamentos antivirales contra el VIH-1. Los esfuerzos iniciales se limitaban a medir en forma directa la inhibición de la replicación del VIH-1 inducida por el medicamento en los pacientes infectados y usaban como criterios medidas inadecuadas, por ejemplo la concentración de linfocitos T CD4+, para vigilar la eficacia clínica.165,173,239,240 El uso de puntos clínicos finales, como el diagnóstico o muerte por SIDA, como criterios eficaces, implican necesariamente un proceso lento, y estos puntos finales ocurren solo después de un avance considerable de la infección por VIH-1. Como resultado, la evaluación de los medicamentos antivirales ha progresado con lentitud, y en la actualidad persiste gran incertidumbre sobre cuando iniciar y cuando modificar el tratamiento. La aplicación de métodos de desarrollo reciente, en especial los que miden los niveles de ARN del VIH-1 en plasma, para medir la replicaciones en pacientes infectados debe ayudar a aclarar muchas de las dudas actuales sobre la actividad y el uso óptimo de los medicamentos antivirales actualmente disponibles y acelerar las pruebas con nuevos agentes.173,189,241
La mayoría de los medicamentos que se han estudiado como inhibidores de la replicación del VIH-1 están dirigidos contra la enzima transcriptasa inversa. Los inhibidores de esta enzima pueden prevenir la diseminación del virus a las nuevas células blanco, pero no modificar la producción viral de células que portan ya los genomas provirales integrados. La zidovudina (3'-azido-3'-desoxitimidina), la didenosina (2'2'-dideoxinosina, también conocida como ddI), la zalcitabina (2',3'-dideoxicitidina, también conocida como ddC), la stavudina (2',3'-didehidro-3'-desoxitimidina, también conocida como d4T) y la lamiduvina (2',3'-didesoxi-3'-tiacitidina, también conocida como 3TC) son análogos de nucleósidos que inhiben la capacidad de la transcriptasa inversa para sintetizar copias complementarias del ADN del VIH-1 en etapas tempranas del ciclo de replicación viral. La estructura molecular de estos análogos de nucleósidos es tal, que una vez que se incorporan a la copia naciente de ARN del VIH-1, la polimerización del ADN se termina en forma prematura y no puede ya extenderse la cadena de ADN. Estos análogos de nucleósido inhiben también la actividad de la transcriptasa inversa del VIH-2. Con la zidovudina u otra monoterapia análoga en pacientes sin tratamiento previo, con frecuencia se observan reducciones en las concentraciones plasmáticas de ARN del VIH-1 de alrededor de 0.6 a 0.7 log10. Aunque estos cambios son mesurables, su magnitud indica que estos agentes bloquean la replicación in vivo del VIH-1 en forma incompleta, lo que puede explicar el modesto beneficio clínico observado con la monoterapia con analogos de nucleósidos (ver adelante). Las combinaciones de los análogos de nucleósidos, como la zidovudina y la lamivudina inhiben in vivo la replicación del VIH-1 en forma más potente, con reducciones del ARN de VIH-1 en plasma de alrededor de 1.5 log10.
Los denominados inhibidores no nucleósidos de la transcriptasa inversa (INNTI), como el nevirapine y las piridonas actúan por un mecanismo diferente, uniéndose en forma directa al complejo enzimático e inhibiendo que la enzima tome una conformación activa necesaria para la polimerización del ADN. Los INNTI son inhibidores muy específicos y no competitivos del VIH-1 y no inhiben la replicación de los otros retrovirus, incluyendo el VIH-2. Los INNTI pueden inhibir la replicación del VIH-1 en forma potente, pero solo temporal, en pacientes tratados, disminuyendo los niveles de ARN del VIH-1 de 1.6 a 2.0 log10, dos a cuatro semanas después de iniciado el tratamiento.188
Por desgracia, la monoterpia con análogos de nucleósidos o INNTI en pacientes infectados por el VIH-1 ha causado, sin excepción, la aparición de virus resistentes a medicamentos.237,242-244 La resistencia farmacológica aparece en algunas semanas después de iniciado el tratamiento con los INNTI y en meses a años después del uso de análogos de nucleósidos. La caracterización genética de los virus resistentes ha demostrado que existen mutaciones específicas en la región que codifica la transcriptasa inversa y que estas mutaciones se asocian con la resistencia observada a los fármacos individuales.237,242,245
El desarrollo de resistencia farmacológica por el VIH-1 durante el tratamiento con análogos de nucleósidos se ha estudiado en forma más amplia con la zidovudina.237,242,245 La sustitución de los aminácidos en cinco posiciones en la transcriptasa inversa del VIH-1 ha sido implicada en la resistencia a la zidovudina. Estas sustituciones se acumulan durante el tratamiento, y los niveles más altos de resistencia (reducción de hasta 100 veces en la sensibilidad in vitro) se observan en virus que tienen sustituciones en cuatro o cinco posiciones. La resistencia a la zidovudina se desarrolla con más rapidez en personas con infección avanzada, quizá porque tienen una carga viral mayor, replicación más extensa y una diversidad genética viral más grande aunada a la inmunodeficiencia progresiva. Los virus resistentes a la zidovudina también pueden trasmitirse entre los individuos.246 Estos virus resistentes suelen ser sensibles a didanocina, zalcitabina y los INNTI.237,242,245
La rápida aparición de altos niveles de resistencia con los INNTI refleja que solo se requiere la alteración de un aminoácido en la transcriptasa inversa del VIH-1 para disminuir la sensibilidad a estos agentes.242,244,247 Por lo tanto, aunque los INNTI son bien tolerados y muestran actividad antiviral detectable in vivo, el rápido desarrollo de resistencia farmacológica limita mucho su utilidad como monoterapia.
Se ha sugerido que pueden usarse combinaciones de inhibidores de la transcriptasa inversa para retrasar la aparición de virus resistentes a los agentes antivirales individuales, o que este tipo de tratamiento podría provocar la aparición de mutaciones múltiples que disminuyeran o abolieran la replicación del VIH-1 in vivo.237,242,245 Aunque se han encontrado cepas de VIH-1 que son simultáneamente resistentes a varios inhibidores de la transcriptasa inversa en pacientes tratados, estudios recientes de tratamiento combinado con zidovudina y lamivudina han identificado un sinergismo antiviral no esperado que causa reducción sostenida en los niveles de ARN del VIH-1 y elevación en el número de linfocitos T CD4+. Este efecto parece ser causado por la aparición de mutaciones de la transcriptasa inversa que confieren resistencia a la lamivudina. Estas mutaciones sirven para evitar la aparición de mutaciones que causen resistencia a la zidovudina en pacientes que no la han recibido o para revertir la resistencia en enfermos previamente tratados y que portaban mutaciones que conferían resistencia.
La zidovudina es un inhibidor eficaz de la replicación del VIH-1 in vitro.237 In vivo, la zidovudina se convierte en su forma activa trifosfato por las enzimas celulares. Los estudios clínicos iniciales sugieren que la zidovudina mejora la supervivencia y la función neurológica en personas con enfermedad avanzada (células T CD4+ <200/µl), y fue el primer medicamento antiviral autorizado por la FDA para el manejo de la infección por VIH.248 Se ha reportado que el tratamiento con zidovudina se asocia con reducción en los niveles de antígeno central p24 circulante en suero y con incrementos temporales en el número de linfocitos CD4+ en sangre periférica. En estudios más recientes la zidovudina parece detener la progresión a SIDA en personas asintomáticas con disminución del número absoluto de linfocitos T CD4+ (<500 células T CD4+/µl).249,250 El menor riesgo de progresión de la enfermedad reportado en estos estudios es transitorio, durando menos de dos años en la mayoría de los pacientes, y parece existir mayor beneficio en pacientes que tenían cuentas de células T CD4+ más altas al ingresar al estudio.251 Sin embargo, a diferencia de estos resultados, un estudio reciente controlado con placebo de zidovudina en pacientes asintomáticos infectados por VIH no encontró diferencia entre los grupos de tratamiento en relación con supervivencia o progresión a enfermedad avanzada después de un tiempo promedio de estudio de tres años.252 Como resultado, ha resultado difícil emitir recomendaciones definitivas sobre cuando iniciar el tratamiento con zidovudina.253 Los clínicos y los pacientes deben analizar en cada caso los beneficios potenciales del tratamiento con este agente y sus efectos adversos.254
La utilidad clínica de la zidovudina en el tratamiento de los individuos con infección avanzada está limitada por la inducción de anemia y neutropenia en un número significativo de pacientes.255 Sin embargo, la toxicidad de la zidovudina se ha reducido al disminuir la dosis usada en forma habitual, y en la actualidad se observan menos efectos adversos en personas con infección temprana o asintomática. El tratamiento prolongado con zidovudina puede causar una miopatía que se asocia con niveles elevados de creatina cinasa en suero y que se piensa es resultado de la inhibición inducida por medicamentos de la replicación mitocondrial del ADN.
Es importante que se ha reportado que el uso de la zidovudina en pacientes embarazadas disminuye en forma significativa la tasa de trasmisión del VIH-1 a sus hijos123: cuando se administra el medicamento a una mujer embarazada preparto y transparto y a los recién nacidos durante las primeras seis semanas de vida, la tasa de trasmisión materno-infantil se reduce en alrededor de dos terceras partes. Este estudio incluyó mujeres embarazadas con enfermedad ligeramente sintomática y ausencia o mínimo tratamiento previo con zidovudina. No se sabe si pueden lograrse resultados semejantes en mujeres en otros estadios de la enfermedad y hasta qué grado la resistencia a la zidovudina limita la eficacia de esta terapéutica.
Las evidencias preliminares señalan que la capacidad de la zidovudina para disminuir el riesgo de trasmisión del VIH-1 de la madre al infante se relaciona con el grado en el que la carga viral materna disminuye después del inicio del tratamiento. Por lo general la zidovudina es bien tolerada durante el embarazo, y no se ha asociado con estrés fetal, parto prematuro o malformaciones congénitas cuando se usa de acuerdo con las recomendaciones actuales. Los estudios futuros ayudarán a definir el momento óptimo para la administración del tratamiento antiviral con objeto de limitar la trasmisión materno-fetal y si otros medicamentos antivirales o estrategias diferentes de manejo pueden ser incluso más eficaces.
La didanosina fue el segundo medicamento autorizado por la FDA para el tratamiento de la infección por VIH.237 Debido a que la didanosina no parece ser superior a la zidovudina como tratamiento inicial, su uso fue autorizado en personas que no pueden tolerar la zidovudina o que han recibido tratamientos prolongados o ineficaces con ésta. La didanosina es un inhibidor activo de la transcripción inversa y de la replicación por VIH-1 in vitro. En las células humanas, la didanosina se metaboliza a su forma activa, 2',3'-dideoxiadenosina-5'-trifosfato (ddATP). Debido a que la ddATP tiene una vida media intracelular muy larga (>12 horas), la didanosina requiere una dosificación menos frecuente que la zidovudina para mantener niveles terapéuticos.
En los estudios de fase I de didanosina, los pacientes han presentado mejoría de la función inmunológica, aumento de peso moderado, disminución en los niveles de antígeno central p24 e incrementos temporales en la cuentas de células T CD4+.237 Los estudios subsecuentes indican que el cambio de zidovudina a didanosina puede ser benéfico en pacientes con enfermedad avanzada y tratados previamente cn zidovudina.256,257 Sin embargo, se desconoce el momento adecuado para modificar el tratamiento. Durante el tratamiento, también aparecen cepas de VIH-1 resistentes a didanosina, y algunas de éstas son resistentes también a zalcitabina.
Los principales efectos adversos de la didanosina incluyen el desarrollo de pancreatitis (que puede variar desde levemente sintomática y con incremento moderado de la amilasa hasta una enfermedad fatal) en cinco a 10 porciento de los pacientes tratados y el desarrollo de neuropatía periférica en alrededor del 15 al 30 porciento. También se reportan con frecuencia cefaleas y diarrea, y a muchos pacientes les desagrada el sabor del medicamento.
La zalcitabina, el tercer análogo de nucleósido autorizado en los Estados Unidos
para el tratamiento de la infección por VIH-1, se recomendó al inicio para usarse solo en combinación con zidovudina en pacientes adultos que tuvieran menos de 300 células T CD4+/µl y que sufrieran un deterioro clínico o inmunológico importante. Posteriormente se ha autorizado su uso como monoterapia en pacientes en los que han fracasado otros medicamentos antivirales o que no pueden tolerarlos. Desde el punto de vista molar, la zalcitabina es el más potente de los análogos de nucleósidos autorizados para inhibir la replicación del VIH-1 en cultivo de tejidos. Como en el caso de otros análogos de nucleósidos, la actividad inhibitoria de la zalcitabina depende de la conversión intracelular de una forma trifosfato activa.
Los estudios de fase I han indicado que la zalcitabina, al igual que la zidovudina y la didanosina, pueden disminuir la antigenemia p24 e incrementar en forma temporal la cuenta absoluta de linfocitos T CD4+.258 Un estudio de comparación directa de zalcitabina y zidovudina en pacientes con enfermedad avanzada por VIH-1 se terminó en forma prematura cuando ocurrieron más muertes en el grupo tratado con zalcitabina.259 Sin embargo, un trabajo en el que se combinaron los dos medicamentos reportó que los pacientes tratados con la combinación tuvieron una elevación de células T CD4+ mayor y más prolongada que los que fueron manejados solo con zidovudina.260 Aunque la combinación de zidovudina y zalcitabina no parece ser más eficaz que cada medicamento aislado en pacientes con enfermedad avanzada (cuentas de células T CD4+<150/µl), puede ser de mayor beneficio en enfermos en fase temprana (cuentas de células T CD4+ > 150/µl), sin que se haya determinado su papel preciso. Al parecer la zalcitabina es tan eficaz como la didanosina para el tratamiento de pacientes que no responden o no pueden tolerar la zidovudina.261 La principal toxicidad de la zalcitabina es el desarrollo de neuropatía periférica dolorosa.
La stavudina es el más reciente de los análogos de nucleósidos autorizado para el tratamiento de la infección por VIH-1.262,263 En la actualidad está autorizado para emplearse en adultos que no han mejorado con el tratamiento con otros agentes antivirales o que no los toleran. Los estudios preliminares han indicado que su perfil de actividad es semejante al de la zidovudina, pero puede causar incrementos temporales más importantes en la cuenta de linfocitos T CD4+. Su principal efecto tóxico es la neuropatía periférica.262,263
La lamivudina, el análogo de nucleósidos de desarrollo más reciente, parecer tener utilidad limitada cuando se usa solo por el surgimiento rápido de virus resistentes a medicamentos. Sin embargo, estudios recientes de lamivudina en combinación con zidovudina han demostrado que pueden lograrse periodos prolongados de supresión de la replicación del VIH-1 por periodos que duran seis meses o más. Aunque el beneficio clínico final de esta combinación no se ha establecido, estos resultados han despertado de nuevo el interés por los análogos de nucleósidos y por la identificación de tratamientos combinados más eficaces.
Aunque el desarrollo de agentes adicionales para inhibir la transcriptasa inversa del VIH-1 continúa en forma activa, se ha enfocado la atención al desarrollo de medios farmacológicos para interferir con otros pasos esenciales en el ciclo de replicación del VIH-1.237,238 Se espera que al dirigirse a muchos procesos del ciclo de vida del virus se logren estrategias terapéuticas más eficaces para el tratamiento del VIH-1.
La proteasa del VIH-1 proporciona un blanco atractivo para el desarrollo de medicamentos antivirales.182,264 La proteasa viral fragmenta las proteínas precursoras gag y pol del VIH-1 para formar las proteínas funcionales maduras durante o poco después de la gemación de partículas virales de las células infectadas. Por lo tanto, la ruptura por la proteasa es un paso esencial en la maduración de las partículas de VIH-1, y la mutación experimental de la proteasa causa la producción de partículas virales no infecciosas que contienen un centro intacto (gag) y precursores no separados gag-pol (transcriptasa inversa, ARNasa H e integrasa). La proteasa del VIH-1 es una aspartil proteasa que consiste en dos subunidades idénticas cuya estructura tridimensional se ha definido por análisis de cristalografía por rayos X.182,265 Nuestro conocimiento sobre la estructura de la proteasa del VIH-1, combinado con la experiencia extensa de la industria farmacéutica para desarrollar inhibidores de proteasas (como el uso muy difundido de los inhibidores de la enzima convertidora de angiotensina [ECA]), ha facilitado el desarrollo rápida de diversos agentes que inhiben en forma eficaz la función de la proteasa viral.
Los primeros inhibidores de la proteasa del VIH-1 que se desarrollaron y probaron en humanos son los análogos de sustrato con base peptídica, que son resistentes a la digestión por la proteasa del VIH-1 y que actúan estabilizando la enzima en un estado de transición inactivo. Aunque muchos de estos inhibidores son activos contra la proteasa del VIH-1, tienen escasa biodisponibilidad oral, quizá en parte por su caracter peptídico. Recientemente se han desarrollado otros inhibidores de la proteasa, incluyendo inhibidores no peptídicos, que actúan por diferentes mecanismos para inhibir la actividad catalítica. Ciertos de los inhibidores no peptídicos de la proteasa tienen actividad antiviral adecuada y buena biodisponibilidad por vía oral. Los estudios realizados han demostrado que los inhibidores de la proteasa pueden inhibir en forma potencial la replicación del VIH-1 y frustrar en forma eficaz la producción de virus infecciosos en células infectadas en forma crónica en cultivo de tejidos.237,238 También se ha demostrado que varios inhibidores de la proteasa tienen un efecto antiviral potente in vivo. Estos agentes pueden actuar en una forma aditiva o sinérgica cuando se usan con los inhibidores de la transcriptasa inversa, lo que sugiere que la combinación de medicamentos que actúan en diferentes niveles del ciclo del virus puede ser útil.237,238 Sin embargo, el desarrollo de inhibidores de la proteasa se ha complicado por su compleja y cara síntesis química, que los convierte en medicamentos demasiado costosos como para administrarlos en forma crónica.
El saquinavir se encuentra en este momento en estudios de fase III. Los estudios de fase II indican que una triple combinación del saquinavir, la zidovudina y la zalcitabina tiene mayor actividad antiviral que los tratamientos dobles o la monoterapia. Otros dos inhibidores de la proteasa, el L-735,524 (también conocido como MK-639) y el ABT-538, son potentes inhibidores de la replicación del VIH-1187,188 que han demostrado disminuir en forma significativa la carga viral y aumentar las cuentas de linfocitos T CD4+ durante periodos de seis meses o más. Se ha demostrado que el MK-639 y el ABT-538 disminuyen las concentraciones de ARN del VIH-1 en alrededor de 2.0 log10 o más en pacientes tratados, demostrando una potente inhibición in vivo de la replicación viral. Los pacientes con cuentas de linfocitos T CD4+ menores (<50 células T CD4+/µl) y una carga viral más alta sufren en forma típica una respuesta menos dramática, que se mantiene por periodos cortos.
Como en el caso de los inhibidores de la transcriptasa inversa, se desarrolla resistencia viral a los inhibidores de la proteasa en los pacientes tratados, y ésta correlaciona con la reducción del efecto antiviral.266 Por la presión selectiva impuesta por el tratamiento farmacológico, se acumulan con el tiempo mutaciones en la proteasa del VIH-1 que causan reducción progresiva en la sensibilidad al medicamento y una resistencia cruzada a otros inhibidores de la proteasa. En la actualidad se ha determinado la estructura cristalizada de varias proteasas obtenidas de virus resistentes a medicamentos, y parece ser que pronto se definirá el mecanismo de resistencia viral a los inhibidores de la proteasa. Se espera que los nuevos conocimientos permitan establecer estrategias para superar la resistencia a estos agentes, de modo que puedan usarse con eficacia en el tratamiento de la infección por VIH-1.266
Las proteínas tat y rev del VIH-1, que son indispensables para la replicación viral, proporcionan un blanco importante para el desarrollo de medicamentos antivirales. Sin embargo, la elaboración de inhibidores de estas proteínas requerirá una mayor comprensión de sus mecanismos de acción. Las áreas activas de investigación se enfocan a determinar la base estructural de la interacción de estas proteínas virales reguladoras con sus ARN blancos y a la identificación de proteínas celulares que interactúan con las proteínas tat y rev y ayudan a mediar sus actividades.
Por lo menos se ha identificado un inhibidor de la proteína tat por medio de métodos de escrutinio de fármacos.267 El agente, conocido como Ro24-7429, es un miembro de la familia de las benzodiacepinas que actúa a través de un mecanismo no bien definido para inhibir la función de la proteína tat y la replicación del VIH-1 en el cultivo tisular. Por desgracia, los estudios de fase I de este compuesto en los pacientes infectados por SIDA han demostrado una toxicidad inaceptable y ninguna evidencia de efecto antiviral. Aunque los experimentos clínicos preliminares con este inhibidor han sido desalentadoras, la inhibición de la función de la proteína tat sigue siendo un blanco terapéutico interesante.
La interacción entre la glucoproteína gp 120 env del VIH-1 y el receptor celular CD4 es un paso indispensable para iniciar la infección viral. La región precisa de la molécula CD4 que se fija a la gp120 se ha localizado en un dominio de la porción extracelular del CD4 que porta una secuencia homóloga con las regiones variables de las inmunoglobulinas. Se han preparado versiones de ingeniería genética del gen CD4 en las que se deleciona la región responsable de la inserción de membrana, originando la secreción de una proteína conocida como CD4 soluble. En los estudios iniciales en cultivo de tejidos que han usado cepas de VIH-1 de laboratorio, se encontró que el CD4 soluble se une con alta afinidad al gp120 en la superficie de la partículas del virus VIH-1, evitando la unión viral a los receptores de la superficie celular, e induce cambios conformacionales irreversibles en la proteína env, que convierten a la partícula viral en no infecciosa. Por desgracia, los resultados de varios estudios clínicos con CD4 soluble y sus derivados no han mostrado evidencia de beneficio clínico, ni efecto sobre los marcadores de replicación viral en los pacientes infectados.237,238 En un principio no se conocían los motivos de la falta de eficacia de esta estrategia promisoria, pero se ha encontrado que las principales cepas de VIH-1 aisladas en la clínica son mucho menos susceptibles a la inhibición por CD4 soluble que las cepas de laboratorio.198,268,269 Aunque este intento inicial para inhibir la interacción virus-receptor fue desalentador, la interacción gp120-CD4 sigue siendo un blanco atractivo para el diseño de fármacos. La definición reciente de la estructura tridimensional de la región del CD4 que fija a la gp 120 ayudará en estos esfuerzos.270,271 Por desgracia, la estructura terciaria del gp120 ha demostrado ser fugaz.
Además del desarrollo de agentes farmacológicos para interferir con la replicación del VIH-1, se ha intentado aumentar la respuesta inmunológica antiviral de los individuos infectados. Un enfoque para esto, conocido como vacunación terapéutica, incluye la inmunización de los pacientes infectados con antígenos del VIH-1 recombinantes (v.gr., gp120) o preparaciones de virus inactivos. Los estudios de inmunización preliminar usaron una versión recombinante de la proteína env del VIH-1, y sugirieron que puede despertarse una respuesta inmune antiviral de significado aún incierto.272 Sin embargo, estudios más recientes no han podido demostrar evidencia de disminución en la carga viral o retraso en la progresión clínica. Un reporte reciente sobre el uso de una vacuna inactivada del VIH-1 con depleción de la proteína env afirma que esta vacuna causa un ligero retraso en la pérdida de células T CD4+.273 Sin embargo, se requieren estudios adicionales que confirmen y amplíen esta observación para establecer el beneficio de esta terapia.
También se están explorando agentes naturales conocidos como modificadores de la respuesta biológica como medios potenciales para limitar la replicación del VIH-1 o reconstituir la función inmunológica deteriorada. El interferón alfa inhibe en forma parcial la replicación in vitro del VIH-1 y se ha reportado que disminuye los niveles de antígeno central p24 y aumenta en forma temporal las cuentas absolutas de linfocitos T CD4+ en las peronas infectadas por VIH-1.274.275 Los resultados preliminares indican que el tratamiento intermitente con IL-2 puede aumentar las cuentas de células T CD4+ en algunos pacientes en los que estos niveles están solo ligeramente disminuidos.276 Sin embargo, los incrementos temporales en la viremia plasmática después de la infusión de IL-2 tienen un significado incierto, y no se sabe si el aumento secundario en las células T CD4+ causa mejoría de la función inmunológica. La función de la compleja red de citocinas in vivo se comprende poco aún, y el éxito de los modificadores de la respuesta biológica para el tratamiento de la infección por VIH-1 requerirá mayor conocimiento sobre la complejidad de esta red de citocinas in vivo.
Ninguno de los medicamentos disponibles en la actualidad contra el SIDA ha demostrado detener en forma eficaz la replicación viral in vivo o evitar el desarrollo subsecuente de la enfermedad en personas infectadas por el VIH-1. La identificación de agentes de este tipo parece una tarea muy difícil. Sin embargo, los intentos iniciales por tratar las infecciones por retrovirus humanos han proporcionado mucha información para los esfuerzos futuros. Nuestro mayor conocimiento de la historia natural de la infección por el VIH-1 facilitará el desarrollo y aplicación de intervenciones terapéuticas contra esta enfermedad.
Retrovirus y otras enfermedades humanas
Una área de investigación activa, aunque controvertida, se orienta a la posibilidad de que existan nuevos retrovirus humanos no descubiertos que sean los agentes etiológicos de enfermedades humanas consideradas idiopáticas. Algunos estudios indican que parece existir una relación entre el HTLV-1 o retrovirus relacionados y la esclerosis múltiple.277-279 Se ha observado la presencia de anticuerpos reactivos contra la proteína gag del HTLV-I en varios pacientes con esclerosis múltiple. Estudios que utilizan la reacción en cadena de la polimerasa han identificado secuencias genómicas relacionadas con HTLV-I en un porcentaje significativo de individuos con esclerosis múltiple, y tales secuencias son muy raras en las poblaciones controles. La importancia de estos estudios para la identificación del agente etiológico de la esclerosis múltiple es muy incierto, y se espera un mayor análisis de dichas observaciones.152 Se ha propuesto la etiología retroviral para la enfermedad de Kawasaki,280 pero evaluaciones posteriores no han encontrado apoyo para esta suposición.281, 282 De igual manera, una investigación de RCP en pacientes con síndrome de fatiga crónica reportó que en muchos casos podían demostrarse secuencias del HTLV-II.283 Sin embargo, otros estudios no han podido corroborar estos resultados.228
Recientemente se ha dado gran publicidad y se originó gran preocupación por el reporte de un retrovirus no detectado por las pruebas serológicas para VIH-1, VIH-2, HTLV-I y HTLV-II que podría asociarse con el síndrome recién definido conocido como linfopenia T CD4+ idiopática (LCI), que se reportó por vez primera en 1992 a 1993.284,285 La LCI es un síndrome muy raro caracterizado por una cuenta absoluta de células T CD4+ menor de 300 células/µl o de menos del 20 porciento del total de las células T en más de una ocasión, con reacciones serológicas negativas para los retrovirus de las familias VIH y HTLV y sin una causa que explique la inmunodeficiencia.284 A pesar de la atención brindada a la descripción inicial de la LCI, las investigaciones subsecuentes han indicado con claridad que el síndrome no se asocia con una etiología retroviral y, de hecho, no es un padecimiento infeccioso.284,285 Además, la LCI no parece ser una entidad clínica de reciente aparición, sino que se reconoce más en la actualidad por la disponibilidad para medir el número de células T CD4+ en pacientes con evidencia clínica de inmunodeficiencia.
Cualquiera que sea la dirección que tomen las investigaciones futuras en las infecciones retrovirales, la experiencia derivada del estudio del HTLV-I, HTLV-II, VIH-1 y VIH-2 ha mejorado en mucho el conocimiento sobre el potencial patógeno de los retrovirus humanos, y proporcionado herramientas para buscar patógenos retrovirales adicionales.
Direcciones futuras en la investigacion de retrovirus
Desde 1980, cuando los retrovirus humanos eran considerados como entidades hipotéticas y estudiados por sólo unos cuantos laboratorios científicos, estos virus se han identificado como los agentes etiológicos de importantes enfermedades humanas, y la investigación sobre los mismos es abrumadora. Aunque las modalidades actuales de tratamiento alivian muy poco la gravedad de la enfermedad humana inducida por retrovirus, nuevos estudios basados en la biología característica de esta clase de virus y guiados por el mayor conocimiento de los blancos moleculares para la intervención farmacológica se encuentran en el horizonte. Además, el mayor conocimiento sobre la historia natural y la patogenia de la infección con retrovirus humanos puede facilitar el desarrollo y aplicación de las intervenciones para prevenir o tratar las enfermedades asociadas. El pesimismo respecto al pronóstico actual de los pacientes con LLTA y el SIDA puede ser compensado por una apreciación esperanzada de los grandes adelantos logrados en el conocimiento de los retrovirus patógenos en un periodo tan corto.
Bibliografía
DR. MARK B. FEINBERG
El potencial patogénico de los retrovirus se reconoció por primera a comienzos del presente siglo gracias a las investigaciones sobre sarcomas y leucemias en las aves. En los años siguientes, se han detectado ejemplos de enfermedad inducida de forma retroviral en una amplia variedad de especies animales.1 Sin embargo, la mayor parte de los intentos para demostrar la relevancia de los retrovirus en la patogenia de la enfermedad humana fueron infructuosos.
Sin embargo, desde 1980 al menos cuatro retrovirus humanos se han descubierto y caracterizado. Estos retrovirus humanos infectan a una célula blanco común, el linfocito T. Además, todos causan infecciones que ponen en peligro la vida, y que persisten a pesar de una respuesta inmune a menudo vigorosa, pero aparentemente ineficaz.
Aunque comparten características comunes, los retrovirus humanos se relacionan con manifestaciones clínicas diferentes. El virus linfotrófico humano de células T tipo 1 (HTLV-I) se identifica como el agente etiológico de una forma agresiva de leucemia conocida como leucemia/linfoide de células T del adulto (LLTA).2, 3 El HTLV-I también se ha relacionado con la mieloneuropatía progresiva denominada paraparesia espástica tropical (PET), o mielopatía relacionada con HTLV-I (MRH).4, 5 El HTLV-II se aisló inicialmente de un individuo con una variante indolente de células T de leucemia de células peludas, pero todavía no se ha relacionado definitivamente con ninguna enfermedad.6 El virus de inmunodeficiencia humana tipo I (VIH-I), identificado por primera vez en 1983, se ha identificado como agente causal de la mayoría de los casos del síndrome de inmunodeficiencia adquirida (SIDA) y sus estados relacionados a nivel mundial.7-9 El retrovirus humano aislado más recientemente, el virus de inmunodeficiencia humana tipo 2 (VIH-2), se identificó en 1986, y se relaciona desde el punto de vista genético y serológico al VIH-1. El VIH-2 es el agente etiológico de un número cada vez mayor de casos de inmunosupresión indistinguible del SIDA relacionado con el VIH-1.
En la actualidad se desconocen los mecanismos precisos por los cuales los retrovirus humanos inducen enfermedad. Sin embargo, es de gran importancia esclarecer dichos mecanismos debido a que esto permitirá el control de la amenaza excepcionalmente seria que representan estos virus, y aumentaría la compresión de los procesos subyacentes a la leucemogénesis y el funcionamiento normal del sistema inmune.
Clasificación de los retrovirus
El término retrovirus se refiere a una familia diversa de virus ARN de cadena simple, con envoltura, cuya replicación y expresión son dependientes de la integración de una forma intermedia de ADN de doble cadena (un provirus) en el genoma de la célula huésped. Los retrovirus se distinguen de los otros virus ARN por la presencia de la enzima transcriptasa inversa, polimerasa de ADN dependiente de ARN codificada en forma viral y que facilita la transferencia de la información genética retroviral desde el ARN al ADN.1, 14 La familia Retroviridae, que incluye a los retrovirus humanos, se divide en tres subfamilias: Oncovirinae, Lentivirinae y Spumavirinae. Oncovirinae, por lo general conocida como oncovirus (término abreviado para virus ARN oncogénicos), puede inducir una amplia variedad de enfermedades malignas en animales, incluyendo muchas de las leucemias y linfomas que ocurren en gatos, ratones silvestres, pájaros, vacas y monos.1,14 HTLV-I y HTLV-II semejan en forma más estrecha a los miembros de esta subfamilia, aunque poseen varios atributos estructurales y funcionales distintivos. Los lentivitus (también llamados retrovirus lentos) causan diversos trastornos no malignos que evolucionan de manera lenta y se presentan en animales, originando varias enfermedades neurológicas, musculoesqueléticas, hematológicas y respiratorias de mamíferos angulados (con cascos) que se caracterizan por un período de incubación largo y una fase sintomática prolongada. Varios estudios han permitido encontrar ciertas similitudes biológicas y morfológicas entre los miembros de esta subfamilia y los retrovirus humanos VIH-1 y VIH-2, y el análisis genético indica en forma clara que el VIH-1 y el VIH-2 son miembros de la subfamilia de los lentivirus. Se ha demostrado que los espumavirus (también conocidos como virus espumosos) inducen infección persistente en varias especies de mamíferos, pero dicha infección no se relaciona con consecuencias patológicas evidentes.
Poco después del descubrimiento del HTLV-I se identificó un virus muy relacionado, denominado virus linfotrófico de células T de los simios tipo 1 (STLV-I), como agente que producía infección natural en muchas especies de monos y grandes simios del Viejo Mundo.15-17 Al igual que el HTLV-I el STLV-I se relaciona con trastornos malignos linfoides espotáneos en huéspedes infectados. Igualmente, la identificación del VIH-1 alentó a los investigadores a realizar estudios serológicos en especies de primates no humanos en búsqueda de una evidencia de infección por otros retrovirus. El análisis de los resultados condujo al descubrimiento de una familia de virus de inmunodeficiencia de los simios (VISs) [ver adelante, Origen del VIH y SIDA].
Estudios epidemiológicos subsecuentes realizados en Africa Occidental demostraron un patrón inesperado de mayor reactividad de ciertos sueros humanos con los antígenos virales de los VIS que con los antígenos virales del VIH-1. Este dato llevó al descubrimiento y aislamiento del VIH-2.10-13 La caracterización molecular del VIH-2, VISmac (obtenido de monos macacos [Macaca mulatta] que sufren una enfermedad parecida al SIDA) y del VISsmm (obtenido de Cercocebus atys sanos) ha indicado que la estructura genética de estos virus se relaciona mucho más entre sí que con la estructura del VIH-1 (ver adelante).19,20 Recientemente se aisló el virus VIScpz, un lentivirus relacionado al VIH-1, de chimpancés salvajes (Pan troglodytes).21,22 La relevancia del VIScpz en el origen de la epidemia por VIH-1 no es clara aún. El estudio continuo de los lentivirus de los simios podrá ofrecer datos interesantes sobre el origen, biología y patogenia de los retrovirus inmunosupresores humanos y proporcionar modelos útiles para evaluar posibles tratamientos y vacunas contra las infecciones por retrovirus.
Estructura y replicación de los retrovirus
Todos los retrovirus con replicación competente contienen al menos tres genes (gag, pol y env) que están localizados en un orden común de izquierda a derecha 5' a 3'.14 El gen gag codifica las proteínas centrales internas de la partícula del virión. El gen pol codifica la polimerasa transcriptasa inversa que copia el ARN del genoma viral a ADN, una ribonucleasa (ARNasa) enzima H que degrada el ARN viral una vez que se ha sintetizado la copia inicial de ADN, y una endonucleasa denominada integrasa que facilita la integración retroviral hacia el cromosoma del huésped. El gen pol codifica también una proteasa necesaria para el procesamiento de los productos maduros de los genes gag y pol a partir de sus precursores poliproteínas durante el ensamble de los viriones. El env codifica la cubierta glucoproteica de superficie que media la unión del retrovirus a la célula blanco huésped. Al final de cada retrovirus se encuentran secuencias repetitivas conocidas como repeticiones terminales prolongadas (RTP). En general, las secuencias que comprenden las RTPs retrovirales no codifican para las proteínas sino que contienen las señales de iniciación y procesamiento de la transcripción al igual que elementos reguladores genéticos centrales para el control de la expresión viral.
El ciclo de replicación retroviral y el proceso de infección comienzan cuando el virus se une a receptores de superficie de células huésped específicas a través de la glucoproteína env [ver figura 1].1, 23 Tras una serie compleja de etapas, el ARN retroviral se convierte en ADN y se incorpora al genoma de la célula huésped de una forma estable llamada provirus. Una vez integrado en el cromosoma de la célula huésped, ocurre producción de ARN viral a través del aparato de transcripción de la célula. Sin embargo, el HTLV-I, HTLV-II, VIH-1, VIH-2 y los virus relacionados de otras especies animales, codifican proteínas que actúan, junto con los procesos celulares, para modular el nivel y carácter de los productos virales producidos en las células infectadas.24-27 La proteínas gag, pol y env son sintetizadas a través de la maquinaria celular del huésped para síntesis proteica y sufren modificación postraslacional (como glucosilación). En un proceso conocido como gemación, un retrovirus intacto que posee una membrana derivada de la membrana plasmática celular se libera de la célula.
|
|
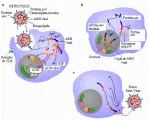

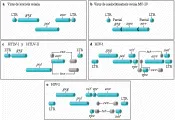
|
| Figura 2 |
| Genes retrovirales |
Los retrovirus patógenos humanos y los retrovirus relacionados que se encuentran en otras especies animales tienen estructuras genéticas y modos de expresión más complicados.24 Además de producir las proteínas gag, pol y env, elaboran otras proteínas que tienen funciones reguladoras o accesorias importantes. Varias de estas funciones reguladoras son necesarias para la replicación viral e interactúan en forma íntima con factores de la célula huésped que regulan tanto el nivel como las características de los productos genéticos virales producidos en las células infectadas. Además de producir transcritos de ARNm no divididos (genómico y gag-pol) y divididos en una ocasión (env), estos retrovirus más complejos producen una tercera clase de transcritos de ARNm con divisiones múltiples que codifican proteínas con funciones reguladoras importantes para el ciclo vital del virus. Este grado extraordinario de complejidad genómica y su capacidad para la expresión diferencial del gen puede tener un papel importante en la naturaleza persistente y en la génesis de las consecuencias patogénicas específicas de las infecciones por retrovirus humanos.
Estructura del HTLV-I y HTLV-II
HTLV-I y HTLV-II son similares en su organización genómica. Además de contar con los genes gag, pol y env, ambos virus contienen una región codificadora, llamada X, que se localiza cerca de las secuencias terminales del gen env y que se extiende hasta la RPT 3'.27 La homología más grande entre HTLV-I y HTLV-II se encontró en esta región X. El análisis de células infectadas con HTLV-I y HTLV-II ha demostrado que estos virus producen doble empalme de ARN con dos plantillas de lectura abiertas superpuestas. Los genes formados por este fenómeno de empalme poco usual son tax y rex.26, 27
El gen tax codifica una proteína con masa molecular de 40 kd. La proteína se localiza en el núcleo de las células infectadas y desempeña un papel crucial en la expresión y la replicación del HTLV-I y HTLV-II.26 La expresión de esta proteína que actúa junto con factores de transcripción de la célula huésped, origina un notable aumento en la transcripción de ARN viral modulado por polimerasa de ARN a partir de secuencias promotoras específicas presentes en las RPTs del virus, conduciendo a una mayor producción del virus. Este proceso, en el cual un producto difusible actúa a distancia sobre los diversos genes, se conoce como activación transcripcional de acción trans, o transactivación. Además de modular la expresión de los genes virales, las proteínas tax de HTLV-I y HTLV-II activan la función de otros factores de transcripción celular que realizan funciones reguladoras esenciales en la activación y proliferación de las células T. Al hacerlo, la proteína tax puede tener un papel importante en los procesos de inmortalización celular y transformación maligna que ocurren después de la infección viral [ver figura 3].
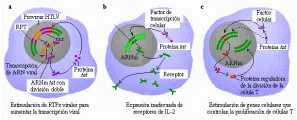
|
| Figura 3 |
| Mecanismo de transactivación |
El gen rex codifica una proteína de 27 kd que, al igual que la proteína tax, se localiza dentro del núcleo de las células infectadas y está implicada en las estrategias de replicación de HTLV-I y HTLV0-II.26, 28 Una segunda proteína, denominada p21, también es codificada por el gen rex, pero su función se desconoce aún. A través de un mecanismo novedoso y no definido del todo, la proteína rex permite la expresión diferencial de las estructuras virales o los genes reguladores. Al actuar por medio de secuencias de ARN específicas derivadas de la RTP 3', la proteína rex parece controlar el transporte de especies específicas de ARN viral desde el núcleo hacia el citoplasma, donde puede traducirse en proteínas virales.26, 28 En ausencia de proteínas rex, sólo los ARN empalmados en forma múltiple (aquellos codificados por las proteínas tac y rex son transportados al citoplasma y disponibles para la traducción; el ARNm empalmado incompletamente que codifica la proteína env, así como el ARN genómico no empalmado que proporciona las transcripciones de ARN de longitud completa y codifica para las proteínas gag y pol, no se expresa de manera funciona. La capacidad de HTLV-I y HTLV-II para expresar preferencialmente proteínas estructurales y reguladoras puede permitir a estos virus la modulación de la cantidad de partículas virales producidas y antígenos virales mostrados. Estos mecanismos puede contribuir al mantenimiento de la infección persistente (ver adelante).
REGULACION DE LA EXPRESION DE HTLV IN VIVO
Tanto HTLV-I como HTLV-II tienen la capacidad de causar infecciones prolongadas que persisten a pesar de la respuesta inmunológica activa del huésped. Es interesante que aunque pueden encontrarse provirus HTLV-I integrados en individuos HTLV-I seropositivos, la gran mayoría de las células infectadas no expresan cantidades detectables de ARNviral, por lo que parecen estar infectadas en forma latente.27,29 No se han identificado aún los reservorios potenciales de la expresión de HTLV-I en personas infectadas, lo que dificulta comprender la manera como este virus se replica in vivo. A diferencia del VIH-1, que muestra una marcada diversidad en su secuencia genética entre los virus aislados de diferentes individuos, los HLV-I aislados en todo el mundo son mucho más homogéneos desde el punto de vista genético.30 La secuencia relativa para el HTLV-I es única entre los retrovirus, que requieren de la enzima transcriptasa inversa para replicar su genoma de ARN, lo que sugiere que el HTLV-I puede emplear una estrategia de replicación diferente. La proliferación de células infectadas inducida por el HTLV-I puede originar la replicación frecuente del genoma proviral por las polimerasas de ADN celulares (mucho más exactas) responsables de la duplicación de los cromosomas de la célula durante la división celular, en lugar de las acciones exclusivas de la transcriptasa inversa viral (que sufre más errores). Sin embargo, la demostración de la expresión in vivo de HTLV-I que puede causar proliferación de células T o inducir respuestas inmunes antivirales, no se ha logrado aún.
ESTRUCTURA Y EXPRESION DEL VIH-1 Y VIH-2
El análisis de secuencias y clonación molecular del VIH-1 han demostrado que es un retrovirus con una complejidad y diversidad genómica sin precedentes. Además de contener los genes característicos gag, pol y env necesarios para la replicación, el genoma del VHI-1 contiene al menos seis genes adicionales: vif, vpr, vpu, tat, rev y nef.24, 25, 31-34
El análisis molecular ha demostrado que las constituciones genéticas del VIH-2 y los VISs son homólogas a las del VIH-1.18, 24, 34 Sin embargo, los genomas del VIH-2 y VISs no contienen el gen vpx. Así mismo, los VISs codifican un gen adicional denominado vpx, el cual no parece tener su contraparte en el VIH-1.
Para facilitar la síntesis de por lo menos nueve productos proteicos distintos a partir de un genoma de menos de 10 kilobases (kb), el VIH-1 y VIH-2 usan un complicado proceso de ruptura diferencial del ARN que origina la producción de más de 30 especies diferentes de ARNm.24,25,28 Los ARNm virales pueden clasificarse por tamaño en tres grupos generales: un grupo de alrededor de 9 kb de transcritos genómicos no divididos, que también dirige la síntesis de las poliproteínas gag y pol, un grupo de ARNm divididos en una sola ocasión de 4.5 kb de longitud que codifican las proteínas env, vif, vpr, vpx y vpu, y un grupo de ARNm con divisiones múltiples de alrededor de 2.0 kb que codifican las proteínas tat, rev y nef.
Los genes tat y rev son indispensables para la replicación viral.24,25 Ambos genes funcionan en una manera trans-acción (i.e., ejercen sus efectos por medio de un producto difusible) para controlar la expresión de otros genes virales. Debido a que se requieren tanto las proteínas tat como rev para la producción de los virus, estas proteínas representan un blanco atractivo para diseñar tratamientos antivirales específicos.
Se sabe relativamente poco acerca de las funciones de los genes vif, vpr, vpu, vpx y nef, pero se conoce que no son indispensables para la replicación in vitro.28,35,36 El conocimiento más detallado de los genes del VIH-1 y VIH-2 estaba limitado antes por la falta de ensayos in vitro para analizar sus funciones. Sin embargo, se han realizado importantes adelantos al respecto en los últimos años, que permiten conocer la función de estos genes en el ciclo vital del virus. El mayor conocimiento de sus funciones proporcionará información útil sobre la compleja biología in vivo de estos virus, y sugerirá nuevos enfoques para interferir en forma específica con su acción.
Los genes tat del VIH-1 y VIH-2 codifican pequeñas proteínas nucleares que amplifican mucho la expresión y producción viral por medio de un mecanismo que aún no se ha definido del todo.37 La proteína tat se une a una estrucutra de asa de ARN denominada el elemento de trans-activación-respuesta (TAR) que se localiza en el extremo 5' de todos los transcritos de ARNm viral. La proteína tat, en combinación con proteínas celulares aún no identificadas, actúa para aumentar el estado estable de los transcritos de ARNm viral, principalmente al estimular la eficacia de la elongación transcripcional a partir de los elementos promotores presentes en los RTP del VIH-1 y VIH-2.37 En ausencia de la proteína tat, la mayoría de los complejos de transcripción ensamblados por los promotores del VIH parecen ser poco productivos y son incapaces de transcribir el templete proviral de ADN más allá de algunos cientos de nucleótidos. Sin embargo, en presencia de la proteína tat los complejos de transcripción se ensamblan en forma mucho más constante, completándose la transcripción del ARN a lo largo de todo el genoma proviral. Aunque la proteína tat puede activar la producción del VIH en modelos de cultivos tisulares de infección por este virus, se desconoce su papel en la regulación de la expresión viral durante la infección natural in vivo.
Los genes rev del VIH-1 y VIH-2 codifican proteínas nucleares que modulan el patrón de transcripción del ARN viral producido en las células infectadas.24,25,28,36 El mecanismo de función de la proteína rev se desconoce, pero parece incluir tanto la supresión de la división del transcrito genómico de ARN como la facilitación del transporte de los ARNm no divididos y divididos del núcleo al citoplasma.28 Por lo tanto, los genes rev del VIH-1 y VIH-2 son análogos a los genes rex del HTLV-I y HTLV-II, aunque los primeros poseen características estructurales y funcionales distintas. Si no se expresa la proteína rev, predominan las especies de ARNm pequeñas y con divisiones múltiples que codifican los genes reguladores del virus (tat, rev y nef). Al mismo tiempo, existen menos transcritos genómicos de longitud total o de una sola división, que juntos proporcionan los genomas virales y la traducción específica de los componentes estructurales (gag, pol y env) y accesorios (vif, vpr, vpx y vpu) del VIH-1 y VIH-2. La función de la proteína rev está mediada por la unión a una secuencia específica de ARN en la región del gen env denominada elemento de respuesta de rev (ERR), que brinda una estructura secundaria estable y compleja para el ARN.
La cantidad relativa de proteína rev expresada después de la infección de una célula blanco por el VIH divide al ciclo viral en las llamadas fases tempranas y tardías.28 Al principio de la infección por VIH de las células blanco existe una cantidad pequeña de proteína rev en las células infectadas, por lo que se expresan preferentemente los ARNm de división múltiple que codifican las proteínas reguladoras del virus. Con el tiempo, la proteína rev puede acumularse y facilitar la transición hacia una expresión predominante de especies ARN de mayor tamaño, que proporcionan los transcritos del ARN genómico y codifican los componentes proteicos estructurales y accesorios. La proteína rev puede coordinar el ensamble y producción del VIH durante un periodo breve, de modo que limita la capacidad del sistema inmune del huésped para identificar y destruir a las células infectadas antes de la liberación de la progenie viral.
El papel de la proteína vif en los ciclos de replicación del VIH-1 y el VIH-2 no se ha definido aún.35,36 Aunque no se requiere de la proteína vif para la replicación viral en ciertas líneas de cultivo de células T, sí es necesaria para la infección viral eficiente de los cultivos de linfocitos y macrófagos de sangre periférica. La proteína vif parece actuar dentro de célula productoras de virus para facilitar el ensamble de partículas virales totalmente infecciosas. Aunque los virus que portan los genes vif defectuosos entran a las células blanco de modo eficaz, son incapaces de completar la síntesis del ADN proviral dentro de la célula recién infectada. Estas observaciones sugieren que la proteína vif influye en la estabilidad, proceso o transporte de los complejos centrales de nucleoproteína del VIH-1 y VIH-2 después de la internalización del virus.
La función de la proteína vpr se desconoce aún.35,36 Está presente en partículas virales, lo que sugiere que tiene algún papel en las fases tempranas de la infección. La mutación del gen vpr disminuye, pero no abole, la replicación del VIH-1 en los macrófagos primarios in vitro. Se ha notificado que la proteína vpr actúa junto con un componente del complejo proteico gag del VIH-1 conocido como proteína matriz para facilitar la entrada del ADN viral al núcelo de las células después de la infección. Esta función puede ser especialmente importante durante la infección de células que no se dividen, como los macrófagos.
De los retrovirus humanos, solo el VIH-1 contiene el gen vpu (de los retrovirus de los simios, solo el VIScpz lo contiene), que se expresa en el mismo transcrito de ARNm del gen env.35,36 La mutación del gen vpu causa menor liberación de partículas virales de la superficie de las células infectadas y mayor gemación en vesículas intracelulares. La proteína vpu también parece inducir la degradación rápida de la molécula CD4 de la célula huésped después de su síntesis en el retículo endoplásmico de las células infectadas. El CD4 sirve como el receptor de superficie celular usado por el complejo glucoproteico env del VIH-1 (gp120-gp41) para entrar a las células blanco. Sin embargo, en las células infectadas, la interacción de moléculasCD4 recién sintetizadas con el precursor del VIH-1 gp160 env en el retículo endoplásmico inhibe el procesamiento y transporte adecuado de la proteína env. Al inducir la degradación de CD4, la proteína vpu puede servir para liberar las moléculas gp160 nacientes, dejando que maduren a complejos funcionales gp120-gp41 en la superficie celular y que se incorporen a las partículas virales en gemación.
El gen vpx es exclusivo del grupo de lentivirus relacionados que consisten en el VIH-2, VISsmm y VISmac.18,35 Se ha propuesto, con base en secuencias semejantes de aminoácidos, que el gen vpx de este grupo puede originarse por duplicación de un gen ancestral vpr. Al igual que la proteína vpr, la proteína vpx existe en las partículas virales, lo que sugiere que funciona en la fase temprana del ciclo de vida del virus. Se sabe que estas similitudes entre las secuencias de aminoácidos de las proteínas vpr y vpx se reflejan también a nivel funcional.
Los genes que codifican la proteína nef se encuentran solo en los lentivirus inmunosupresores de primates.38,39 La proteína nef no es indispensable para la replicación del VIH, y su papel en el ciclo de vida del virus no se ha establecido. Sin embargo, estudios recientes sobre esta proteína han identificado por lo menos dos funciones difeentes que pueden incrementar la eficacia de la replicación viral. La expresión de nef en las células infectadas causa menor expresión en la superficie celular del receptor CD4 al inducir internalización y degradación más rápida de éste.38,39 Aunque el papel de los efectos inducidos por nef sobre la expresión de CD4 en el ciclo viral no se comprenden del todo, el nef puede, al inhibir a CD4, aumentar la infectividad del virus por aumentar el nivel de glucoproteínas virales env incluidas en la superficie de las partículas virales. En forma alterna, se ha propuesto que como el CD4 en la superficie celular disminuye durante la activación normal del linfocito T inducida por antígeno, el nef puede semejar algunos de los aspectos de la activación de la célula T para facilitar la replicación viral. A través de un mecanismo distinto poco conocido, nef actúa dentro de las células infectadas para aumentar la infectividad de las partículas virales producidas.39
Aunque no se requiere del nef para la replicación del VIH-1 en el cultivo de tejidos, estudios recientes de SIDA inducido por VIS en monos rhesus han demostrado la importancia del nef en la biología in vivo de la infección viral. La mutación del gen nef en el VISmac compromete mucho la capacidad del virus para replicarse in vivo. Además, estos virus mutantes persisten a niveles muy bajos en animales infectados, pero no inducen enfermedad. La identificación reciente de un virus mutante nef en un paciente infectado por VIH-1 con una enfermedad excepcionalmente benigna sugiere que nef puede tener un papel semejante para modular in vivo la replicación del VIH-1.40
El mejor conocimiento sobre los productos de los genes reguladores y accesorios de los lentivirus de primates ha producido un cuadro cada vez más coherente de los complejos ciclos de replicación de estos virus. Las proteínas tat, rev y nef son las primeras proteínas virales producidas después de la infección. La proteína tat actúa para amplificar la transcripción del ARN viral, mientras que la proteína rev regula la expresión de las diferentes especies de ARNm y sincroniza la síntesis ordenada de las proteínas involucradas en el ensamble de las partículas virales. La menor expresión de CD4 en células infectadas, que parece necesaria para los ciclos vitales de los virus que usan CD4 como receptor de superficie, se logra por la expresión temprana de la proteína nef, que depura CD4 de la superficie celular. La expresión de la proteína nef continúa durante las fases tardías del ciclo vital, cuando nef actúa para aumentar la infectividad viral. La expresión de la proteína vpu está coordinada con la de la glucoproteína env, y la degradación de las moléculas CD4 recién sintetizadas por la proteína vpu facilita la producción ininterrumpida de la proteína env. La producción de las proteínas vif, vpr y vpx coincide con la de proteínas estructurales del virus, y las proteínas vif, vpr y vpx actúan o se incluyen en el ensamble de las partículas virales. Estas proteínas accesorias pueden ayudar a ensamblar partículas virales infecciosas o actuar en las células recién infectadas para facilitar las fases tempranas de la infección viral.
REGULACION DE LA EXPRESION DEL VIH IN VIVO
El término latencia tiene varias definiciones distintas cuando se aplica a la infección por VIH. Desde el punto de vista clínico, la infección por el VIH se caracteriza por un periodo asintomático (o latente) prolongado antes de que se desarrolle la inmunodeficiencia clínica. Hasta hace poco se suponía que muy pocas células en el individuo infectado portaban o expresaban el VIH y que se llevaba a cabo una replicación viral muy escasa durante ese periodo. Sin embargo, el uso de métodos sensibles para cuantificar la carga viral y su expresión in vivo ha demostrado que la replicación viral es muy activa durante todo el curso clínico de la infección por VIH (ver adelante).41-45 Por ello, el curso de la infección por VIH nunca es completamente quieto o latente.
La disponibilidad de métodos sensibles para medir el número de células infectadas en sangre periférica y tejidos linfoides e identificar células que participen en la síntesis del ARN y las proteínas virales ha permitido, por primera vez, preguntarnos si puede existir el VIH en las células infectadas sin que se exprese. La comparación del número de células infectadas con VIH-1 (i.e., que contienen ADN proviral de VIH-1) con el número de células que producen ARN o proteínas virales sugiere que muchas células infectadas no expresan niveles detectables de productos genéticos del VIH.41,44,46 Se calcula que en la sangre periférica de los pacientes con infección avanzada por VIH el número de células infectadas excede al número de las que producen ARN viral en una relación de por lo menos 10.44,46 Estudios recientes han indicado que la mayor concentración de células que expresan al VIH-1 se encuentra en los tejidos linfáticos, que proporcionan los sitios anatómicos para la respuesta inmunológica.41,42,46 Incluso en estos sitios de densa carga viral, solo una minoría de las células que contienen ADN del VIH-1 muestran evidencia de síntesis de ARN o proteínas virales.46 Una fracción desconocida de las células ADN de VIH positivas pero ARN negativas pueden tener provirus con defectos en la replicación, pero se piensa que el resto contiene provirus competentes para la replicación, pero que no la expresan. Al parecer, las células infectadas que tienen provirus VIH-1 integrados pero que no se expresan (llamadas células infectadas latentes) producen cantidades insuficientes de proteínas virales como para ser detectadas y destruidas por las células T citotóxicas que reconocen los antígenos VIH. De esta manera, existe un reservorio celular de infección por VIH que evita a la respuesta inmunológica, causando persistencia de la infección. Los mecanismos moleculares por los que el VIH-1 logra este estado de expresión atenuada no se conocen. Se sabe muy poco sobre la biología del VIH-2 in vivo, pero se supone que tiene propiedades semejantes a las del VIH-1.
La expresión in vitro del VIH-1 aumenta en forma significativa por agentes que simulen estímulos inmunológicos y por citocinas como el factor de necrosis tumoral-alfa (FNT-alfa), la interleucina-6 (IL-6), la IL-2 y el factor estimulador de colonias de granulocitos y macrófagos (FEC-GM).25,47 El incremento de la expresión viral en estas circunstancias es resultado de la activación de la transcripción del ARN del VIH-1 por miembros de una familia de factores de transcripción inducibles de la célula huésped, conocidos como NF-kB, que se fijan a elementos de secuencia en el RTP del VIH-1.25,48-50 Estos factores de transcripción median la regulación de la expresión de diversos genes que normalmente son activados durante la respuesta del huésped a la infección e inflamación.48 Entre estos genes se encuentran los que codifican a las moléculas de clase I del complejo principal de histocompatibilidad (CPH), la cadena alfa del receptor de IL-2 (IL-2Ra), la IL-2, el FNT-alfa, la IL-6, el interferón beta, el FEC-GM, y ciertos reactantes de fase aguda, como el amiloide sérico A. En la mayoría de las células, proteínas inhibidoras específicas secuestran a los constituyentes NF-kB en el citoplasma en formas inactivas. Diversos estímulos inducen la liberación de los factores NF-kB de sus inhibidores, causando la migración de los complejos ahora activos hacia el núcleo. En el núcleo, los complejos NF-kB tienen un papel importante para controlar la respuesta celular ante los estímulos antigénicos. En forma semejante, un número importante de las funciones de los macrófagos depende de la activación transcripcional mediada por los NF-kB. Al incorporar la regulación mediada por NF-kB en su repertorio regulador, el VIH-1 se asocia en forma íntima al estado de activación de los linfocitos y macrófagos para proporcionar blancos para la infección viral. Entonces, las circunstancias que causan la activación de la respuesta inmunológica pueden también activar la producción del VIH-1.
HTLV-I, HTLV-II y transformación maligna por retrovirus
Los retrovirus son un instrumento atractivo para el estudio de los complejos procesos de transformación a través de los cuales la célula normal se convierte en cancerosa. El genoma retroviral es relativamente simple, y para muchos retrovirus la inducción de afección maligna en animales de laboratorio es reproducible. Los estudios de retrovirus animales han demostrado que aunque las estrategias de transformación utilizadas por los diferentes retrovirus comparten ciertas características, muestran una notable diversidad. Al parecer, los virus linfotróficos de células T humanas tipo I y tipo II usan mecanismos nuevos y todavía mal definidos de transformación maligna que tal vez sean mejor aclarados por el retroceso teórico proporcionado por los retrovirus animales leucemogénicos bien caracterizados.
TRANSFORMACION MALIGNA POR RETROVIRUS ANIMALES
Una clase de retrovirus animal, conocido como retrovirus rápidamente transformante, posee durante el curso de una infección adquirida un gen celular denominado proto-oncogén.51, 52 La incorporación de un proto-oncogén celular en el genoma de uno de los retrovirus rápidamente transformantes suele interrumpir uno o más de los genes necesarios para la replicación, causando que la replicación viral sea defectuosa. Por lo tanto, para causar infección, el retrovirus debe trasmitirse en compañía de un virus cooperador capaz de replicarse, que presta las funciones estructurales y enzimáticas necesarias. La incorporación del proto-oncogén al genoma viral también activa al gen y da al retrovirus la capacidad de transformar en forma aguda a las células tanto in vivo como in vitro. Con una célula blanco adecuada, la expresión del oncogén viral es necesaria y suficiente para la transformación maligna. Los tumores causados por la infección con estos retrovirus son policlonales en derivación y se caracterizan por la integración proviral en sitios al azar en el genoma. Un ejemplo de este tipo de retrovirus rápidamente transformante es el virus de la mielocitomatosis de las aves MC-29, el cual contiene el oncogén myc en su genoma [ver figura 2].
El estudio de los retrovirus rápidamente transformantes ofrece la primera demostración de la existencia de oncogenes celulares y proporciona conocimientos importantes acerca de los mecanismos de transformación maligna de otra clase de retrovirus animales leucemogénicos, los retrovirus lentamente transformantes.52, 53 Los retrovirus de esta clase, los cuales no pueden transformar células in vitro, constituyen la mayor parte de los retrovirus patógenos que ocurren de forma natural. Los retrovirus lentamente transformantes, ejemplificados por el virus de la leucosis aviaria, producen trastornos malignos que poseen un período de latencia de varios meses o incluso más prolongados, a pesar de los altos niveles de expresión viral en todo el huésped (viremia). Aunque la infección está muy diseminada, la conversión maligna de la célula infectada es un fenómeno raro. Los tumores que finalmente surgen son de origen clonal (derivados de un solo progenitor) y se considera que representan la culminación de un proceso genético de múltiples etapas que comenzó con la infección viral. Al revisar los tumores linfoides B producidos de manera característica por el virus de la leucocitos de las aves, se encuentra que su provirus está con frecuencia insertado en el cromosoma de la célula huésped adyacente al oncogén c-myc. En esta situación, la expresión del oncogén c-myc está localizado bajo el control de los elementos de regulación que residen en la RTP retroviral, que origina una expresión elevada o controlada de forma inadecuada. Este fenómeno de activación de un oncogén celular por integración retroviral proximal se conoce como tumorogénesis por inserción promotora.52, 53 Debido a que la mayor parte de los retrovirus lentamente transformantes se integran al azar en los cromosomas de la célula huésped, una explicación para la latencia de la enfermedad maligna característica de esta clase de retrovirus es la rareza con la cual la integración retroviral ocurre en la vecindad de un oncogén.
IDENTIFICACION DE LAS ENFERMEDADES CAUSADAS POR HTLV-I Y VIRUS ASOCIADOS
Los primeros intentos para identificar retrovirus humanos se modelaron sobre el ejemplo de los retrovirus lentamente transformantes, donde puede detectarse una expresión viral abundante in vivo y en líneas celulares tumorales de cultivo. Después de años de resultados negativos, muchos investigadores abandonaron esta línea, mientras que otros comenzaron a desarrollar estrategias para permitir el crecimiento a largo plazo de células hematopoyéticas humanas en cultivo de tejidos. Debido a que muchas células tumorales humanas no crecen in vivo, la atención se centró en la identificación de factores de crecimiento relevantes que pudieran proporcionar un enriquecimiento selectivo para las poblaciones celulares leucémicas y permitir el crecimiento celular en cantidades suficientes para detectar niveles bajos de expresión viral.
Un avance importante fue la identificación en 1976 del factor de crecimiento producido por linfocitos sanguíneos periféricos estimulados por fitohemaglutinina. Esta sustancia, llamada al inicio factor de crecimiento de células T y ahora conocida como interleucina-2 (IL-2), apoya el crecimiento de las células T humanas normales in vitro.54, 55 La IL-2 no se une a las células T normales en reposo, ni induce la proliferación de células T a menos que estas células sean primero activadas por mitógenos o antígenos. Después de la activación, las células T sintetizan receptores de superficie celular para la IL-2; estos receptores fijan IL-2 con gran afinidad y traducen una señal proliferativa.54, 55 Ahora se conoce que el receptor de IL-2 (IL-2R) es un complejo de al menos tres subunidades proteicas llamadas IL-2Rg, IL-2Rb e IL-2Ra, que en combinación proporcionan el receptor funcional de alta afinidad para la IL-2.54, 55 Las células T normales activadas seguirán creciendo en presencia de IL-2, pero después de cuatro a seis semanas sufren inevitablemente un proceso de envejecimiento y mueren.
Se observó que en ciertos pacientes con leucemias de células T maduras que al comienzo se consideró que presentaban variantes agresivas de micosis fungoide o síndrome de Sézary, las células T expresaban receptores de IL-2 y que las células T pudieron proliferar de forma directa en respuesta al IL-2 sin requerir una activación previa. En algunos de estos pacientes los cultivos de células leucémicas siguieron proliferando en ausencia de IL-2 exógena y, evitando el proceso de envejecimiento, se convirtieron en inmortales, es decir, capaces de una reproducción eterna. El examen con microscopía electrónica de estas líneas celulares demostró la presencia de partículas retrovirales, y se detectó actividad de la transcriptasa inversa específica viral en el sobrenadante del cultivo. En las células leucémicas, pero no en los tejidos normales, se identificaron secuencias de ADN que eran homólogas al ARN del virus producidas por las células leucémicas, lo cual indica que el virus se adquirió de manera endógena a través de infección.56, 57 Se identificaron anticuerpos dirigidos contra proteínas retrovirales en el suero de estos pacientes y esto proporcionó un marcador serológico de la infección.56 En 1980 apareció el primer estudio que demuestra el descubrimiento de un auténtico retrovirus humano, denominado HTLV-I.2, 58
Se llevaron a cabo extensos estudios seroepidemiológicos para evaluar la distribución del HTLV-I y para determinar su posible relación con varios trastornos malignos. Estudios iniciales en los Estados Unidos detectaron seropositividad en sólo unos cuantos casos de leucemias y linfomas de células T maduras en adultos. Estos casos con anticuerpos positivos fueron notables porque se manifestaban como leucemias de células T agresivas con fenotipo de célula T cooperadora-inductora CD4+ y pleomorfismo nuclear característico.3, 59 Las manifestaciones clínicas comprendían afectación diseminada de la piel, hipercalcemia y evolución rápidamente mortal.3, 60
Un síndrome clínico similar al observado en los Estados Unidos se ha descrito desde finales de la década de 1970 en Japón, donde se había notado que la proporción de trastornos malignos de células T a células B eran mucho mayor que en los Estados Unidos o Europa. Se observó que el síndrome, denominado leucemia/linfoma de células T del adulto, era prevalente concretamente entre personas nacidas en el suroeste del Japón.3, 61 El examen del suero de los pacientes con LLTA descubrió anticuerpos contra HTLV-I en prácticamente el 100 porciento de las muestras probadas.3 Es más, las áreas geográficas con elevada incidencia de LLTA se correlacionaban estrechamente con regiones donde podía demostrarse una alta prevalencia de HTLV-I desde el punto de vista serológico. Cuando se colocan en tejido celular, las células leucémicas de la mayoría de los pacientes con LLTA originan líneas celulares continuas que producen HTLV-I. Pronto se observó que el HTLV-I podía trasmitirse in vitro a las células T humanas normales y que el virus tenía una especial capacidad para transformar a las células T de modo que crecieran en una forma interminable independiente de IL-2.62,63 Los resultados de estos estudios epidemiológicos y de laboratorio proporcionaron un apoyo importante para la relación etiológica entre el HTLV-I y la LLTA y señaló la primera evidencia de relación causal entre un retrovirus y un trastorno maligno humano.
Los estudios seroepidemiológicos realizados en diversas áreas (v.gr., el Caribe, Sudamérica y Japón), han demostrado que además de su papel eitológico en la LLTA, el HTLV-I se relaciona en forma estrecha con la mieloneuropatía progresiva crónica conocida como paraparesia espástica tropical (PET) o mielopatía relacionada al HTLV-I (MRH).4,5 La PET o MRH son trastornos clínicos progresivos de inicio en el adulto que se caracterizan por paraparesia o paraplegia espástica simétrica que con frecuencia afecta a las extremidades inferiores y por parestesias, hiperreflexia, mínima pérdida sensorial y disfunción vesical frecuente.4 Aunque la PET y la MRH se describieron en un inicio en diferentes regiones geográficas, en la actualidad se consideran idénticas y con frecuencia se refiere a ellas como TEP/MRH. Las muestras de suero y líquido cefalorraquídeo de los pacientes con esta enfermedad contienen anticuerpos con inmunorreactividad característica contra proteínas del HTLV-I. Además, existen linfocitos atípicos que recuerdan a las células de la LLTA en el LCR y la sangre periférica de los pacientes con TEP/MRH, y se ha demostrado la presencia de secuencias provirales de ADN del HTLV-I en los linfocitos de sangre periférica de las personas afectadas.4
Los estudios moleculares han revelado que los HTLV-I aislados de pacientes con TEP/MRH no son obviamente diferentes de los aislados de pacientes con LLTA, y las cepas del virus de pacientes con TEP/MRH y LLTA son genéticamente indistinguibles de los encontrados en portadores asintomáticos. Por lo tanto, es posible que el momento, la vía de adquisición de la infección, o factores del huésped o ambientales, sean los determinantes de la presentación de las enfermedades asociadas con el HTLV-I, y no factores específicos del virus.
Además de la LLTA y la TEP/MRH, se han notificado otros síntomas clínicos en pacientes con infección por HTLV-I.5,64 Se ha descrito una artritis inflamatoria crónica que se asocia con la presencia de linfocitos infectados por HTLV-I en el líquido sinovial en áreas endémicas de infección por este virus en Japón, y que se conoce como artropatía asociada al HTLV-I. Existe una dermatisis infecciosa, caracterizada por eczema asociado con Staphylococcus aureus y estreptococo beta-hemolítico, en niños con infección por HTLV-I en Jamaica. De acuerdo con reportes recientes, una minoría de casos de linfoma cutáneo de células T (LCCT), micosis fungoides y su variante leucémica, el síndrome de Sézary, puede asociarse con infección por HTLV-I.65 Se han publicado también casos de uveítis, polimiositis y panbronquiolitis en individuos infectados por el HTLV-I, lo que sugiere que la investigación futura descubrirá un mayor espectro de enfermedades asociadas con esta infección.
EPIDEMIOLOGIA DE LA INFECCION POR HTLV-I
La infección por HTLV-I se detecta serológicamente.64,66 Debido a que este virus causa una infección persistente, la presencia de anticuerpos contra él indica que una persona tiene una infección activa. Los estudios seroepidemiológicos han descubierto la presencia de HTLV-I en proporciones endémicas en poblaciones geográficamente distantes, incluyendo residentes del suroeste de Japón, el Caribe, Melanesia y partes de Africa.3 Las áreas endémicas de infección por HTLV-I suelen consistir en zonas locales rodeadas por comunidades con baja prevalencia de seropositividad. Los niños en las áreas endémicas suelen tener menos frecuencia de seropositividad que los adultos, y el porcentaje de positividad en los individuos aumenta en forma significativa en relación a la edad. En los grupos de mayor edad las tasas de seropositividad suelen ser mayores en mujeres que en hombres. El incremento en la frecuencia de infección en relación con la edad puede deberse a una mayor oportunidad acumulada para exponerse a la infección o a un efecto de cohorte que refleja tasas históricas más altas de infección por HTLV-I que las observadas actualmente en poblaciones más jóvenes. Estudios recientes de Japón indican que el estado de portador de HTLV-I está disminuyendo en menores de 50 años, y en especial en los grupos jóvenes. Se cree que las mejoras en las condiciones socioeconómicas y los cambios de comportamiento, como la lactancia, han causado una menor trasmisión viral y disminuido la prevalencia de la infección. En las áreas no endémicas, la infección por HTLV-I es más frecuente en inmigrantes de regiones endémicas o en sus parejas sexuales.
El HTLV-I puede trasmitirse de madre a hijo y por contacto sexual, transfusión sanguínea, y uso común de agujas o jeringas contaminadas en los adictos a drogas.3,67-69 Los patrones observados de infección por HTLV-I indican que el virus no es muy contagioso y que la trasmisión viral puede requerir la transferencia de células infectadas de los portadores a los receptores. Estas conclusiones son compatibles con demostraciones de laboratorio de lo difícil que es lograr la trasmisión del virus sin células.
La trasmisión sexual del HTLV-I representa una de las principales vías de diseminación en las áreas endémicas. La trasmisión sexual del virus ocurre más de hombres a mujeres que viceversa, lo cual puede explicar el predominio de la infección en mujeres que ya se mencionó. La trasmisión sexual del virus puede facilitarse por la presencia de otras enfermedades de trasmisión sexual que causen ulceraciones genitales.
La trasmisión del HTLV-I de madre a hijo es frecuente en las áreas endémicas, con tasas de infección de alrededor del 20 porciento en los hijos de madres seropositivas.4,5 Los linfocitos infectados por el HTLV-I se encuentran en la leche materna, y la trasmisión de madre a hijo ocurre sobre todo a través de la lactancia. La sustitución de ésta por biberón en las madres seropositivas puede disminuir en forma significativa el riesgo de trasmisión del virus.
Se ha demostrado la trasmisión viral a través de transfusiones sanguíneas, y esto es motivo de preocupación en las áreas endémicas.61,64 Existe trasmisión viral al transfundir sangre total, paquetes eritrocitarios, o plaquetas (casi siempre contaminados con linfocitos), pero no si se transfunde plasma o sus derivados. En Japón ha ocurrido una importante reducción en las infecciones por HTLV-I secundarias a transfusión y en el número de casos de PET/MRH desde 1987, cuando se instituyó el escrutinio nacional para donadores de sangre en busca de anticuerpos contra HTLV-I.61 En los Estados Unidos se realiza escrutinio en todos los donadores desde 1988. Se ha encontrado evidencia serológica de infección por HTLV-I en alrededor del 0.25 porciento de los donadores de sangre en los Estados Unidos, ocurriendo la mayor prevalencia en las costas del Este y Occidente.70,71 Los estudios que emplean la reacción en cadena de la polimerasa (RCP) para tipificar el virus presente en donadores seropositivos indican que alrededor del 50 porciento de las personas están infectadas por HTLV-I y el otro 50 porciento por HTLV-II.70,72 Entre los donadores de sangre en Estados Unidos, la infección por HTLV-I suele asociarse con haber nacido en un área endémica o haber tenido contacto sexual con personas de un área endémica como Japón o el Caribe.72
La progresión a LLTA es una consecuencia relativamente rara de la infección por HTLV-I. Se calcula que se desarrolla LLTA en dos a cuatro porciento de las personas que viven en regiones en donde el HTLV-I es endémico y en donde es frecuente la infección durante la niñez.5,61,73 La LLTA se ve con más frecuencia entre personas de 40 a 60 años de edad, lo que sugiere que la enfermedad se desarrolla después de un largo periodo latente posinfección, de por lo menos algunas décadas. No existen en la actualidad indicadores pronósticos que identifiquen a los individuos seropositivos que desarrollarán LLTA.
La PET/MRH se desarrolla en menos del uno porciento de las personas infectadas por HTLV-I.4,5,61,74 La enfermedad ocurre con más frecuencia en mujeres que en hombres y comienza alrededor de la cuarta década de la vida. El periodo de latencia para el desarrollo de este padecimiento parece más corto que el de la LLTA, en especial en los pacientes que adquieren la infección por transfusión sanguínea. Se ha reportado un intervalo promedio de 3.3 años entre los receptores de sangre contaminada con HTLV-I y el desarrollo de PET/MRH.4,5
IDENTIFICACION DEL HTLV-II
Un segundo retrovirus humano exógeno distinto, denominado HTLV-II, se aisló de un paciente con una variante de leucemia de células peludas de células T en 1982.3 El HTLV-I y el HTLV-II comparten una organización genómica común y conservan la secuencia estructural de genes, pero son filogenéticamente diferentes. El análisis de la secuencia de nucleótidos del HTLV-II ha demostrado una homología particularmente significativa con el HTLV-I en la región X. Además, el HTLV-II comparte con el HTLV-I la capacidad de transformar en forma aguda las células T para que tengan crecimiento autónomo e inmortal in vitro.27
Desde su descubrimiento en 1982, el HTLV-II se ha aislado de otro paciente con una variante atípica de leucemia de células peludas.75 Sin embargo, los estudios subsecuentes no han podido demostrar una asociación etiológica del HTLV-II con este trastorno u otras enfermedades linfoproliferativas.76 Se han reportado algunos casos de una enfermedad neurológica parecida a la PET/MRH en personas infectadas por HTLV-II, pero el papel del virus en éstos no es claro.77 Debido a la similitud en la estructura genética y al comportamiento in vitro del HTLV-I y el HTLV-II, la aparente falta de patogenicidad del HTLV-II parece sorprendente. Una posible explicación para esta diferencia es el descubrimiento reciente de que mientras que el HTLV-I tiene un tropismo preferencial por células T CD4+ in vivo, el HTLV-II infecta preferentemente células T CD8+.78
EPIDEMIOLOGIA DE LA INFECCION POR HTLV-II
Las proteínas del HTLV-II pueden ser serológicamente distintas a las del HTLV-I, pero el suero de la mayoría de las personas infectadas ha demostrado tener anticuerpos que tienen intensa reacción cruzada en el ensayo de inmunoabsorción ligada a enzimas (ELISA) que se emplea para escrutinio de la infección por HTLV-I.64,66 Hasta hace poco, principalmente por la falta de pruebas serológicas para distinguir con certeza la infección por HTLV-II de la HTLV-I, existía poca información sobre la epidemiología de la infección por HTLV-II. Sin embargo, los inmunoensayos que emplean proteínas recombinantes específicas del virus o péptidos sintéticos y los enfoques de amplificación de genes que usan la RCP permiten distinguir entre las dos infecciones. Debido a la muy diferente implicación clínica de estas dos infecciones, es importante identificar el tipo de virus en los individuos que tienen prueba positiva para anticuerpos anti HTLV.64
La aplicación de nuevos métodos para discriminar entre la infección por HTLV-I y HTLV-II ha descubierto que los adictos a drogas intravenosas representa el mayor reservorio de infección por este virus en los Estados Unidos.68,79,80 Los estudios serológicos de personas de diferentes áreas urbanas han demostrado una seroprevalencia de HTLV de ocho a 24 porciento, la gran mayoría de estos casos son de infección por HTLV-II. Las tasas más altas de seroprevalencia se encuentran a lo largo de las costas Este y Oeste, y en el área de Nueva Orleans. A diferencia de la infección por VIH-1, las infecciones por HTLV-II son prevalentes entre los adictos a drogas intravenosas desde principios de los años 70, e incluso entonces pudieron no ser recientes en este grupo de población.81
Recientemente se ha demostrado también que el HTLV-II es prevalente en varias tribus indias americanas, incluyendo los indios Guaymi de Panamá y los nativos americanos de Florida y Nuevo México.80 En estas poblaciones la prevalencia de HTLV-II es de alrededor de dos porciento o menos. Debido al aislamiento prolongado de muchas de estas poblaciones, es posible que la infección por HTLV-II sea muy antigua, proveniente de los antecesores de estas personas, que migraron a America desde Asia.
Aunque se han estudiado mucho menos, los mecanismos de trasmisión del HTLV-II parecen ser idénticos a los del HTLV-I.80 La alta prevalencia de HTLV-II entre adictos a drogas intravenosas parece ser causada por compartir los equipos de inyección. El HTLV-II también puede trasmitirse por vía sexual, como es el caso de la infección con HTLV-I, VIH-1 o VIH-2, y se asocia un mayor riesgo de trasmisión viral si coexiste otra enfermedad de trasmisión sexual en el receptor. El contacto sexual con un adicto a drogas ha sido identificado como el factor de riesgo más frecuente para la infección por HTLV en mujeres seropositivas identificadas por el escrutinio que se realiza a los donadores de sangre en los Estados Unidos. Al igual que la trasmisión por HTLV-I, puede ocurrir trasmisión de HTLV-II por transfusión de sangre total, paquete de eritrocitos o plaquetas. Las pruebas de escrutinio autorizadas en la actualidad para anticuerpos contra HTLV-I no detectan los anticuerpos contra HTLV-II en algunas muestras, y han ocurrido casos de trasmisión de HTLV-II por transfusión de sangre de donadores que tuvieron escrutinio negativo para HTLV-I.82 Se ha reportado trasmisión de madre a hijo, pero es rara o inexistente si no existe lactancia.83
TRANSFORMACION MALIGNA POR HTLV-I Y HTLV-II
El fenómeno de transformación in vitro por HTLV-I y HTLV-II de las células T derivadas de donantes normales ha recibido mucha atención. Las células T transformadas in vitro comparten varias características sobresalientes con las células de LLTA, y la delineación del mecanismo de transformación in vitro brindaría conocimientos importantes sobre la naturaleza de la leucemogénesis por los retrovirus humanos. Al igual que las células de LLTA, la mayor parte de las líneas celulares inmortales derivadas de la infección in vitro de las células T de donadores sanos consisten en células T CD4+, que poseen núcleos lobulados y tienden a formar células gigantes multinucleadas.3 Con frecuencia las células T transformadas proliferan en ausencia de IL-2 exógena; sin embargo, al igual que las células de LLTA, expresan altos niveles de IL-2R. Las células T transformadas también expresan en forma consistente receptores de transferrina y antígenos HLA de clase II (HLA-DR), lo cual indica que están en estado activado.
El HTLV-I y HTLV-II utilizan mecanismos únicos de transformación que los colocan aparte de los retrovirus animales rápida y lentamente transformantes. Ambos retrovirus humanos pueden transformar de forma aguda células in vitro, pero ni el HTLV-I ni el HTLV-II contienen los oncogenes derivados de las células que portan los retrovirus rápidamente transformantes. Es más, aunque se considera que la LLTA surge después de un período de latencia posinfección prolongado, el mecanismo patogénico del HTLV-I difiere del de los retrovirus lentamente transformantes. La mayor parte de las células tumorales LLTA contienen un provirus sencillo integrado de forma monoclonal, y expresan un gen rearreglado Vbeta del receptor de la célula T, lo que indica que las células malignas son de origen clonal y surgen de una sola célula infectada. Sin embargo, no se encuentra el HTLV-I integrado en los sitios preferenciales de los genomas de las células T malignas, lo que sugiere que el virus no induce leucemia por la activación vía promotor-inserción de un oncogén celular.
La naturaleza de la transformación de las celulas T in vitro por HTLV-I y HTLV-II ha conducido a la suposición de que estos virus portan, además de los genes necesarios para la replicación, un gen que codifica productos proteicos cuya expresión dota a las células T con un potencial de crecimiento ilimitado. Se ha postulado que la proteína tax que estimula el incremento de transcripción de ARN viral a través del proceso de activación transcripcional de acción trans (ver antes), puede actuar también sobre genes celulares involucrados en el control de la proliferación de células T, proporcionando de esta manera un estímulo inapropiado para el crecimiento26, 27 [ver figura 3].
De acuerdo a este modelo de transactivación de la leucemogénesis, la infección viral y la consecuente expresión de la proteína tax origina la transcripción constitutiva de los genes celulares que en condiciones normales son estrechamente regulados y cuya expresión persistente conduce a la proliferación descontrolada26, 27 [ver figura 3]. Un importante componente de este modelo se ha validado mediante experimentos que muestran que la inducción del gen tax en las células T origina la activación y expresión sostenida del gen que codifica el receptor de alta afinidad de IL-2a y, bajo ciertas condiciones, el propio gen de IL-2.26, 27 Asimismo, la expresión de tax también puede originar la elaboración de citocinas específicas de granulocitos y macrófagos (FEC-GM) y la linfotoxina (factor de necrosis tumoral beta), que puede contribuir a las manifestaciones clínicas típicas del HTLV-I relacionadas con enfermedades malignas y neurológicas.26, 27 Por último, la proteína tax activa la expresión de la proteína relacionada a la hormona paratiroidea (PrPTH), que puede ser la causa de la hipercalcemia que se observa en muchos pacientes con LLTA.84
La sugestión de que la inmortalización de la célula T in vitro se facilita por un fenómeno de transactivación mediada por el producto del gen tax está apoyada por las demostraciones recientes tanto de la capacidad transformadora del gen tax en modelos de cultivo tisular usando fibroblastos de roedores como en la apariencia de los tumores mesenquimatosos y la leucemia linfocítica en ratones transgénicos tax.26,27,85 Además, el uso de los métodos de transferencia de genes para introducir las regiones tax y rex del HTLV-I a células T humanas primarias ha provocado la inmortalización de estás células T in vitro.86 Sin embargo, no se sabe si la expresión de la proteína tax es suficiente para la inmortalización de la célula T, y la relación entre los proceso de inmortalización in vitro y leucemogénesis in vivo no se comprende del todo aún. Además, el modelo no explica por completo el potencial leucemogénico in vivo del HTLV-I, porque a pesar de sus similitudes existen diferencias sustanciales entre las células T transformadas in vitro y las células leucémicas LLTA. Mientras que la expresión viral en las infectadas puede documentarse con rapidez in vitro, la gran mayoría de las células tumorales LLTA no expresan el virus in vivo. Por consiguiente, aunque la expresión viral puede ser necesaria para comenzar el proceso leucémico, no parece requerirse para el mantenimiento de la enfermedad. El cuadro se complica más por la observación de que las células leucémicas de LLTA poseen con frecuencia evidentes rearreglos cromosómicos (aunque no consistentes), mientras que las líneas de células T in vitro por lo general son euploides y de potencial maligno poco claro.27, 87
Estudios epidemiológicos han demostrado que la mayoría de los individuos infectados con HTLV-I no adquieren LLTA y que si se desarrollan manifestaciones clínicas existe un período de latencia prolongado entre infección y presentación leucémica (ver antes). Estos complejos datos de laboratorio y epidemiológicos indican que la expresión viral puede ocurrir en algún punto en el curso de una infección y predisponer a las células T infectadas a alteraciones genéticas adicionales, implicando a cofactores todavía no identificados necesarios para la evolución maligna. Aun falta mucho por conocer acerca del papel que las influencias genéticas, inmunológicas y ambientales desempeñan en la modulación del curso clínico entre infección por HTLV-I y la presentación maligna de la LLTA.
HTLV-I Y LLTA
La LLTA se caracteriza por la proliferación agresiva de células malignas inductoras-cooperadoras que tienen un inmunofenotipo CD4+, CD25+ (IL-2Ra).27,29 Las manifestaciones clínicas de la LLTA son muy características y frecuentemente incluyen linfadenopatía, hepatomegalia, esplenomegalia, lesiones óseas líticas, hipercalcemia, afección cutánea y diseminación leucémica.5,59,60 La supervivencia promedio desde el diagnóstico inicial es de 11 meses.
Las diversas presentaciones clínicas de la enfermedad, incluyendo el síndrome conocido como LLTA latente y LLTA crónica, son notables porque pueden tener alguna explicación en el mecanismo de la leucemogénesis en la infección por HTLV-I.88 La LLTA se presenta con frecuencia con manifestaciones cutáneas premonitorias, pero en pacientes con esta enfermedad la cuenta de leucocitos y el calcio sérico son normales. Estos pacientes pueden sufrir progresión aguda a LLTA franca. La LLTA crónica se caracteriza por linfocitosis de las células T con evidencia de células leucémicas circulantes, linfadenopatía, hepatoesplenomegalia, y lesiones cutáneas, pero sin hipercalcemia. La apariencia de LLTA crónica se asocia con frecuencia con progresión de la enfermedad y mal pronóstico. Se considera que el desarrollo de la LLTA es un proceso progresivo que comienza con la infección asintomática por HTLV-I y en ciertas personas es seguida de la aparición de condiciones preleucémicas como la LLTA crónica o latente, cualquiera de las cuales puede evolucionar a LLTA franca. Sin embargo, en muchos pacientes la LLTA franca se presenta sin un síndrome preleucémico que le anteceda.
Los patrones de integración proviral del HTLV-I cambian con la transformación preleucémica. Se observa integración policlonal en las células T infectadas durante el estado de portador asintomático, mientras que las células leucémicas transformadas portan provirus integrados en forma monoclonal. También se observan alteraciones cromosómicas con mayor frecuencia en las presentaciones clínicas más agresivas. El mayor conocimiento de estas entidades patológicas diversas puede contribuir a una mayor apreciación de los eventos tempranos en la patogenia de las enfermedades malignas que se asocian con el HTLV-I.
El tratamiento con los esquemas terapéuticos convencionales es bastante ineficaz en la LLTA, y con frecuencia se observan respuestas limitadas y recaídas tempranas.5,64 Está en estudio el tratamiento experimental de la LLTA, en el que las células tumorales son el blanco por la expresión constitucional del IL-2R. En estudios clínicos de pacientes con LLTA se está evaluando la eficacia terapéutica de anticuerpos monoclonales conjugados a una toxina que reconocen el IL-2R y proteínas de fusión Il-2-toxina, que se ha demostrado destruyen a las células infectadas por HTLV-I en cultivos de tejidos.89
HTLV-I Y MIELOPATIA CRONICA
La presentación clínica de la PET/MRH se caracteriza por el desarrollo de paraparesia o paraplegia espástica progresiva de las extremidades inferiores.4 Los síntomas incluyen debilidad de las extremidades inferiores, rigidez y parestesias, así como dolor lumbar persistente, urgencia e incontinencia urinarias y alteraciones intestinales. La debilidad y espasticidad de los músculos proximales de las piernas pueden causar una marcha lenta en tijera. El inicio de la enfermedad suele ser insidioso, y la progresión de la enfermedad se da en varios años. Al examen físico se observa evidencia de lesiones bilaterales de las vías piramidales, incluyendo hiperreflexia, respuestas plantares extensoras y clonus de los tobillos. Se encuentran cambios mínimos en los nervios sensoriales. Aunque la PET/MRH comparte muchas características con la esclerosis múltiple, puede distinguirse de ésta por la ausencia de afección a los nervios craneales, la conservación de la función cognoscitiva y la rareza con la que ocurre debilidad de las extremidades superiores. Aún más, los signos y síntomas de la PET/MRH, a diferencia de los de la esclerosis múltiple, no oscilan.
Los mecanismos patogénicos de la PET/MRH no se han definido, pero se cree que incluyen proceso inflamatorios y quizá autoinmunes.4,5 En los pacientes con PET/MRH, el dato patológico más prominente es la atrofia moderada a severa de la médula espinal. Es evidente un proceso inflamatorio crónico, principalmente meningomielitis de la médula espinal torácica inferior. Se encuentran células B, T y macrófagos infiltrando alrededor de los vasos sanguíneos parenquimatosos y en el parénquima tanto de la sustancia gris como blanca. El daño al tejido parenquimatoso se manifiesta como destrucción tanto de mielina como de axones, hialinosis de los vasos sanguíneos y presencia de fibrosis meníngea y cicatrización glial. Existen células T CD8+ positivas en las lesiones inflamatorias, que se acompañan de un alto grado de expresión de antígenos HLA de clase I, pero no es claro si éstas reaccionan contra los antígenos HTLV-I, autoantígenos u otros blancos. Aunque algunos investigadores han sugerido que la PET/MRH puede ser causada por una respuesta inmunológica citotóxica a las células infectadas por el HTLV-I en el sistema nervioso central, no puede detectarse la expresión del ARN del HTLV-I en las áreas afectadas.4,5 Una hipótesis alternativa es que la infección por HTLV-I inicia de alguna manera un proceso autoinmune que causa la destrucción de la mielina.4,5 Sin embargo, no existe evidencia clara que apoye esta hipótesis.
Un porcentaje significativo de células T CD4+ de la sangre periférica de pacientes con PET/MRH muestra un fenotipo activado (positivo para antígeno de clase I del HLA e IL-2Ra [CD25]) in vivo, y manifiesta un grado importante de proliferación espontánea in vitro.4,5 En la mayoría de los casos los pacientes con PET/MRH demuestran títulos de anticuerpos contra HTLV-I en el suero y LCR que son significativamente mayores que los demostrados por pacientes con LLTA o portadores seropositivos. También se encuentran niveles altos de células T citotóxicas específicas para HTLV-I en la sangre y LCR de los pacientes con PET/MRH. Las intensas respuestas humoral y celular antiviral en la PET/MRH pueden reflejar activación inmunológica crónica inducida por la replicación viral activa. Algo que apoya este concepto es el hecho de que a pesar de las respuestas antivirales intensas, los pacientes con PET/MRH tienen una carga mucho mayor de células infectadas por el HTLV-I que los portadores asintomáticos.4,5 En los pacientes con PET/MRH se encuentra el HTLV-I integrado en una forma policlonal en un número significativo (alrededor del 10 al 20 porciento) de células T de sangre periférica. A pesar de esta carga viral apreciable, la expresión del ARN del HTLV-I no puede detectarse en la gran mayoría de las células infectadas.
El tratamiento de la PET/MRH intenta basarse en el concepto de que la respuesta inmunológica del huésped tiene un papel importante en la patogenia de la enfermedad.4,5 El tratamiento con esteroides ayuda a amilorar los síntomas de la mielopatía en el 50 porciento o más de los pacientes con PET/MRH. Sin embargo, la mejoría puede ser temporal y en el seguimiento a largo plazo no se observan diferencias entre los pacientes tratados y los no tratados. Es interesante que los beneficios del tratamiento esteroideo pueden ser más evidentes en pacientes que adquieren la infección por HTLV-I a través de transfusión sanguínea y en los que se encuentran en la fase temprana de la enfermedad. El danazol, un andrógeno sintético, puede mejorar también en forma temporal los síntomas. Tratamientos más agresivos, como la plasmaféresis, la ciclofosfamida o el interferón alfa, pueden causar mejoría a corto plazo sin beneficios evidentes a largo plazo. Se está evaluando, en pacientes con PET/MRH, el uso de anticuerpos monoclonales que reconocen el IL-2Ra que se expresa en muchas células infectadas por el HTLV-I.
VIH Y SIDA
El SIDA se describió por primera vez en mayo de 1981 y se caracteriza por infecciones oportunistas poco usuales que ponen en peligro la vida y diversas enfermedades malignas en pacientes que no presentan otra causa conocida de inmunodeficiencia.90-92 El síndrome se manifiesta por diversas alteraciones inmunológicas características y una amplia constelación de datos clínicos, incluyendo neumonía por Pneumocystis carinii, muchas otras infecciones oportunistas causadas por virus, hongos, micobacterias y protozoarios, alteraciones neurológicas importantes, linfomas de células B, pérdida de peso intratable y una variante agresiva del sarcoma de Kaposi.93 La definición de casos usada por los Centros para el Control y Prevención de las Enfermedades (CDC) de los EUA para la vigilancia epidemiológica, se modificó cuando se identificó que el VIH-1 era el agente infeccioso causal del SIDA y varias veces después para incluir el mayor conocimiento sobre el espectro de las enfermedades asociadas al VIH.94,95 Con el conocimiento de que la depleción de las células T CD4+ es la característica básica de la inmunodeficiencia inducida por el VIH, la definición de SIDA se ha modificado para incluir a cualquier individuo infectado por el VIH-1 que tenga una cuenta de células T CD4+ menor de 200 células /µl o un porcentaje de células T CD4+ menor de 14.96,97 Aunque la definición de casos de los CDC es útil para estudios epidemiológicos, para fines clínicos es mejor considerar a la enfermedad por VIH-1 como un espectro continuo que va de la infección primaria a la infección asintomática y después a la inmunodeficiencia avanzada. Las características clínicas del SIDA se analizan en otra subsección.
DIAGNOSTICO DE LA INFECCION POR VIH
La evaluación de la respuesta serológica al VIH-1 es una medida indispensable para el diagnóstico de la infección por VIH, para realizar análisis epidemiológicos de la prevalencia de la infección y para el escrutinio de sangre y donadores de órganos con objeto de minimizar el riesgo de trasmisión.66,98 El escrutinio inicial del suero en busca de anticuerpos reactivos contra el VIH-1 emplea una prueba sensible de ELISA.66,98 La especificidad de las muestras reactivas se confirma por medio de una técnica de hibridización en filtro conocida como Western blot. Las pruebas de escrutinio autorizadas en la actualidad tienen tasas muy bajas de falsas positivas y falsas negativas. La mayoría de las personas recién infectadas producen niveles de anticuerpos detectables uno a dos meses después de la infección, y más del 95 porciento tienen una prueba positiva de anticuerpos antiVIH a los seis meses de la infección inicial. Puede no identificarse a las personas infectadas por el VIH-1 durante el periodo llamado de ventana, que ocurre pronto después de la infección, cuando no se han formado aún niveles detectables de anticuerpos. En la práctica se piensa que la magnitud de este error es pequeño, pero las tasas de fracaso pueden depender de la prevalencia de factores de riesgo en la población estudiada. La infección por VIH-1 puede diagnosticarse antes de la producción de anticuerpos antivirales por medio de un ensayo de captura del antígeno que detecta el antígeno viral central p24 en el suero o por RCP para identificar secuencias ADN o ARN del virus en muestras de células de sangre periférica o plasma, respectivamente. Debido a que la presencia del virus puede confirmarse por cultivo viral o por técnicas de RCP con gran frecuencia en las personas seropositivas y ya que la infección persiste por el resto de la vida, la seropositividad para VIH-1 debe considerarse como marcador de infección viral activa.99,100
Ciertos estudios iniciales que usaron técnicas de aislamiento del virus y amplificación genética por RCP sugirieron que un porcentaje de los pacientes con riesgo alto de infección por VIH podían infectarse y permanecer seronegativos durante periodos prolongados, quizá más de dos años.101 Sin embargo, estos datos han sido refutados por estudios subsecuentes más extensos.102 En forma semejante, tampoco se han confirmado algunos reportes de personas que revirtieron de un estado seropositivo a seronegativo para el VIH-1 a pesar de resultados de RCP persistentemente positivos.103,104
EPIDEMIOLOGIA DE LA INFECCION POR VIH-1
Desde su descripción inicial como una entidad clínica poco usual, el SIDA se ha convertido con rapidez en una pandemia, con casos reportados en más de 170 países de todo el mundo.105,106 Debido al largo periodo asintomático entre la infección inicial por el VIH-1 y el desarrollo de enfermedad clínica, el virus se ha diseminado en forma no aparente y extensa en varias poblaciones. Se sabe que la infección por VIH-1 es mucho más prevalente que el SIDA clínico u otras manifestaciones clínicas de la infección. Se calcula que más de 14 millones de personas se han infectado por el virus desde el inicio de la pandemia, alrededor de ocho millones de hombres, cinco millones de mujeres y un millón de niños.105,106 La Organización Mundial de la Salud (OMS) calcula que más de 2.5 millones de casos de SIDA y un millón de muertes han ocurrido hasta la fecha. Las proyecciones recientes sugieren que hasta 40 millones de personas podrán ifnectarse por el VIH-1 para el año 2000.105
Más del 60 porciento de los casos de SIDA en todo el mundo ocurren en la región sub-Sahara de Africa. Las infecciones continúan aumentando en Latinoamérica, el Caribe, Norteamérica, el Medio Oriente, Europa Oriental y Asia.106 Aunque la infección por VIH-1 fue introducida a los países de Asia hasta hace poco, se ha diseminado con gran rapidez, en especial en los países del suroeste, como Tailandia y la India.
Durante la década de los 80 la infección por VIH-1 destacó como una de las principales causas de muerte en los Estados Unidos, se reportaron más de 339,000 casos de SIDA y casi 200,000 fallecimientos para octubre de 1993.107 En 1992 la infección se convirtió en la principal causa de muere entre varones de 25 a 44 años de edad, y en la actualidad es la cuarta causa de muerte en mujeres del mismo grupo de edad.108 Se calcula que alrededor de un millón de personas están infectadas en este momento en los Estados Unidos, la mayoría de los cuales están asintomáticas y muchas no saben de su infección.
El VIH-1 se trasmite a través de varias vías: sexual, de personas infectadas a sus parejas homo o heterosexuales, parenteral, a receptores de transfusiones con sangre infectadas o sus productos o por la costumbre entre los adictos a drogas intravenosas de compartir las jeringas con personas infectadas, y perinatal, de madres infectadas a sus hijos.102 También ha ocurrido trasmisión del VIH-1 por trasplante de órganos e inseminación artificial con semen de una persona infectada.109,110 Muchos estudios epidemiológicos han fracaso en demostrar la trasmisión del VIH-1 por contacto casual con una persona infectada.102
Los análisis epidemiológicos indican que el semen, las secreciones cervicales y vaginales, y la sangre o sus derivados son los principales vehículos, aunque no los únicos, para la trasmisión del virus. En la actualidad no se conocen del todo los factores que determinan la capacidad de una persona infectada para trasmitir el virus. En forma semejante, tampoco se han definido las variables que pueden influir en la propensión de una persona a la infección por el VIH-1 después de la exposición. Los estudios realizados en diferentes sitios de todo el mundo indican que la ruptura de superficies mucosas por otras enfermedades de trasmisión sexual, en especial la sífilis y el chancroide que causan ulceraciones genitales, puede facilitar en mucho la infección por VIH-1.102,111 Como resultado, la prevalencia de enfermedades de trasmisión sexual en una población de personas con riesgo de infección por VIH-1 puede alterar en forma significativa la eficacia de la diseminación viral.
Los patrones predominantes de trasmisión del VIH-1 varían en las diferentes partes del mundo.102,106 En los Estados Unidos y otros países industrializados, el patrón principal ha sido la trasmisión sexual entre varones homosexuales y bisexuales. Sin embargo, la diseminación por agujas entre adictos a drogas continúa ocurriendo, y la tasa de trasmisión heterosexual está aumentando. Aunque los varones homosexuales y bisexuales constituyen aún la mayor proporción de los infectados por el VIH-1, los adictos a drogas forman el segundo grupo en tamaño. La trasmisión por adictos a drogas, ya sea en forma directa al compartir las jeringas o indirecta por contacto heterosecual o perinatal causa en la actualidad la mayor parte de las infecciones por VIH-1 entre mujeres y niños.
En la región sub-Sahara de Africa, Sudamérica, Asia y el Caribe, el VIH-1 se disemina predominantemente por trasmisión heterosexual y perinatal.105,106 En estas regiones el número de varones y mujeres infectados es aproximadamente igual. Por lo tanto, en todo el mundo es el contacto sexual, principalmente heterosexual, el modo predominante de trasmisión del VIH-1, causando más del 80 porciento de todas las infecciones.105,106 Los motivos subyacentes para la variación mundial observada en los patrones de trasmisión del VIH-1 no se conocen del todo, pero se piensa que incluyen gran cantidad de factores complejos, como comportamiento personal, prácticas sociales, prevalencia de enfermedades coexistentes de trasmisión sexual y la fecha de introducción del VIH-1 a la población.
Debido a las tasas altas de infección por VIH-1 en mujeres en edad reproductiva en los países en desarrollo, la infección adquirida por vía perinatal es responsable de alrededor del 20 porciento de todos los casos de SIDA en estas áreas.105,106 Como resultado, la epidemia creciente de SIDA pediátrico anulará mucho del progreso logrado en las últimas décadas para mejorar la supervivencia de los niños en los países en vías de desarrollo. La tasa de trasmisión observada de madre a hijo varía desde 13 porciento hasta un 45 porciento en estudios de cohorte realizados en Europa y Africa, respectivamente, con un promedio de alrededor del 25 porciento.112,113 Los motivos de esta variación se desconocen, pero es probable que se relacionen con diferencias en la fase de la enfermedad materna y en lo adecuado de los cuidados médicos prenatales y perinatales.
La trasmisión del VIH-1 de una madre a su hijo puede ocurrir en útero por el paso trasplacentario del virus, durante el parto o posnatal a través de la lactancia.114 Para los niños que nacen de madres seropositivas, la transferencia placentaria de anticuerpos maternos complica el diagnóstico de la infección por métodos serológicos, e impide calcular el número exacto de niños infectados in útero contra los infectados durante o después del nacimiento. Otros métodos diagnósticos, incluyendo el cultivo del VIH-1, los ensayos de inmunocomplejos disociados (ICD) del antígeno nuclear p24 y la RCP permiten el diagnóstico temprano de la infección por VIH-1.115,117 La presencia de un antígeno central VIH-1 p24 y el cultivo de virus al nacer en un grupo de neonatos nacidos de madres infectadas indica la trasmisión in útero. Sin embargo, se cree que la mayoría de las infecciones se adquieren durante el nacimiento por el contacto con sangre o secreciones contaminadas. Entre los gemelos de madres infectadas por VIH, se reporta una tasa más alta de infección en el niño que nace primero incluso en casos de cesárea, lo que sugiere que factores relacionados con el alumbramiento modifican el riesgo de infección.118 Diversos factores asociados con la enfermedad avanzada por VIH-1 en la madre, incluyendo antigenemia p24, niveles altos de ARN del VIH-1 en el plasma, y cuentas bajas de linfocitos T CD4+, correlacionan con un mayor riesgo de infección para el neonato.119-122 Se han demostrado casos de trasmisión posnatal, que se supone son resultado de la lactancia. Sin embargo, estos casos son poco comunes y se refieren sobre todo a madres que estaban recién infectadas y cuya leche podía haber tenido una cantidad sustancial de VIH-1.
Un estudio importante ha demostrado que cuando se administra zidovudina (AZT) a las mujeres embarazadas infectadas por VIH-1 durante el segundo y tercer trimestres del embarazo, se continúa intraparto y después se administra a los infantes durante las primeras seis semanas de vida, el riesgo de trasmisión materno-infante se reduce en forma significativa (ver adelante).123 La disponibilidad de una intervención eficaz para disminuir la trasmisión perinatal del VIH-1 ha originado un mayor interés por realizar escrutinio en todas las mujeres embarazadas en busca de presencia de infección por VIH-1.124,125
Se calcula que, antes de 1985, cuando se inició la prueba de anticuerpos en donadores de sangre en los Estados Unidos, 10,000 o más personas se infectaron por el VIH-1 a través de sangre o sus derivados contaminados por el virus.102,126 La trasmisión del VIH-1 por transfusión de sangre es muy eficaz, y por lo menos el 90 porciento de los receptores de componentes sanguíneos VIH-1 positivos se infectan. Varios miles de hemofílicos también se infectaron por el VIH-1 durante este periodo después de recibir concentrados de factores de coagulación contaminados. Los casos de trasmisión del VIH-1 por transfusión se han relacionado a la recepción de sangre total, paquete de eritrocitos, leucocitos, plaquetas y plasma. Las preparaciones de gamaglobulina, inmunoglobulina Rho, globulina inmune contra la hepatitis B y de vacuna contra la hepatitis B derivada del plasma no se han asociado con trasmisión del VIH-1. Los procedimientos usados para preparar estos productos parecen eliminar la infectividad del virus.
Con la introducción del escrutinio a donadores de sangre en busca de infección por VIH-1, la trasmisión asociada a transfusiones se redujo en forma significativa.102,127,128 Se han demostrado muy pocos casos de trasmisión por sangre que tuvo resultados negativos para anticuerpos antivirales en la prueba de escrutinio; además, se ha calculado que existe una (o menos) unidad de sangre infectada y seronegativa por cada 40,000 a 225,000 unidades de sangre que se transfunden.126,128,129 Debido a la rareza con la que donadores de sangre infectados pero seronegativos escapan a los estudios de escrutinio actuales, trabajos recientes indican que el uso de los ensayos de antígeno central p24 del VIH-1 o RCP para identificar a estos individuos que se encuentran en el periodo denominado de ventana antes de la seroconversión, no mejoran en forma importante la seguridad de las transfusiones de sangre en los Estados Unidos.130-132 El tratamiento con calor de los concentrados de factores de la coagulación para inactivar el VIH-1 proporciona una medida adicional de seguridad para las personas con hemofilia. Sin embargo, se requiere el uso intensivo de los métodos actuales para proteger a los receptores de sangre de la infección por VIH-1, como autoexclusión de los donadores, exclusión confidencial y pruebas de escrutinio rigurosas en busca de anticuerpos anti-VIH-1, para maximizar la seguridad de los productos sanguíneos y reducir el mínimo el ya muy pequeño riesgo de infección asociada a las transfusiones. A diferencia de lo que sucede en los países industrializados, la trasmisión del VIH-1 por sangre o sus derivados contaminados sigue siendo un gran problema en los países en vías de desarrollo por la falta de recursos adecuados para estudiar a todos los donadores y de bancos de sangre apropiados.
Aunque varios trabajadores del área de la salud se han infectado por la exposición a sangre de personas infectadas por el VIH-1, los estudios a gran escala sugieren que el riesgo de trasmisión de este virus por la exposición laboral es pequeño (0.34 porciento por exposición)., aunque varía con la severidad de la exposición.133,134 El uso de zidovudina para limitar la trasmisión después de la exposición de alto riesgo tiene un beneficio incierto, ya que han ocurrido infecciones a pesar de su uso.135-137 Aún no se establece si otros agentes antivirales o sus combinaciones serán más eficaces para evitar la trasmisión del VIH-1 después de la exposición laboral. Aunque los estudios de trasmisión laboral del virus han demostrado un riesgo bajo de trasmisión después de la exposición a un inóculo infectado pequeño, también han identificado una gran frecuencia de exposiciónes percutáneas y cutáneas, principalmente entre estudiantes de medicina y personal de base.138 Por lo tanto, a menos que disminuya la frecuencia de exposición ocupacional, continuarán viéndose casos que pudieron prevenirse de infección en trabajadores de la salud. La aplicación rigurosa de las medidas de control de infecciones (llamadas precauciones universales) para limitar la exposición a los líquidos corporales del paciente, independientemente de la probabilidad de infección por VIH-1, aumentan al máximo la seguridad para los trabajadores de la salud y reducen la posible discriminación contra pacientes que pueden considerarse probables portadores del virus.97,133,139
En 1990 surgieron controversias sobre la trasmisión del VIH-1 en el área de trabajo por personal de salud cuando los CDC notificaron que un dentista con SIDA había trasmitido la infección a cinco pacientes sometidos a procedimientos dentales invasivos.140,141 No se conoce el modo preciso de trasmisión del VIH en estos pacientes. Los estudios subsecuentes de pacientes de varios dentistas, médicos y cirujanos infectados fueron incapaces de demostrar algún caso de trasmisión del VIH. Por lo tanto, el riesgo de trasmisión de trabajadores de la salud infectados a sus pacientes es tan bajo que no puede medirse.102,141a,141b Sin embargo, ha surgido un debate público sobre el escrutinio para los trabajadores de la salud y la restricción de la práctica clínica en los seropositivos. Se ha afirmado que el entrenamiento cuidadoso de todos los trabajadores de la salud en las prácticas adecuadas de control de infecciones y la adherencia rigurosa al uso de las precauciones universales cuando se atiende a todos los pacientes constituye una medida más eficaz y menos discriminatoria.139,142
PROGRESION DE LA ENFERMEDAD POR VIH
Se calcula que por lo menos el 25 a 50 porciento de las personas infectadas por VIH-1 tienen progresión al SIDA clínico en un periodo de cinco a 10 años después de la infección.102,143,144 Un mayor porcentaje de casos tiene una duración de la infección subclínica mayor, y los estudios epidemiológicos han indicado que la posibilidad de desarrollar SIDA es directamente proporcional a la duración de la infección por el VIH-1. La progresión a SIDA también se asocia con la tasa de reducción de las cuentas de linfocitos T CD4+.143,145,146 Aunque las cuentas de células T CD4+ disminuyen con el tiempo en virtualmente todos los pacientes infectados por el VIH-1, lo hacen con tasas sustancialmente diferentes. El SIDA se desarrolla más rápido en individuos con una pendiente de disminución de linfocitos T CD4+ más pronunciada que en los que tienen tasas de reducción más lentas. En forma semejante, existe considerable variación en el tiempo que tarda la enfermedad clínca en manifestarse en los diferentes pacientes. Una minoría de las personas tienen una evolución con SIDA franco en uno a dos años después de la infección, mientras que en el otro extremo del espectro, se han observado pequeñas cohortes de pacientes infectados con infecciones asintomáticas prolongadas (> 10 años).147-149 Las contribuciones relativas del huésped y de los factores virales que son responsables de estas variaciones clínicas son un tema muy importante de estudio en el futuro.149,150
La progresión de la enfermedad por VIH-1 tiene dos patrones en los niños infectados por sus madres.151,152 En el primer año de vida el 20 porciento de los lactantes desarrollan inmunodeficiencia grave, que con frecuencia se asocia con complicaciones del SNC. El resto de los niños manifiesta una enfermedad con evolución más lentamente progresiva, semejante a la observada en los adultos. El riesgo de desarrollar enfermedad temprana y severa es mayor en los neonatos que son seropositivos para el VIH-1 al nacer por cultivo viral, RCP o ensayo del antígeno central P24 en suero, lo que indica infección in útero. El riesgo de progresión rápida es significativamente mayor en los hijos de madres con enfermedad avanzada en el momento del nacimiento del niño. Aún no se sabe si este mayor riesgo es resultado de una mayor carga viral materna o de la presencia de virus más virulentos en una persona con enfermedad en fase tardía.
La posibilidad de que factores genéticos o ambientales influyan en la evolución de la inmunosupresión inducida por el VIH-1 y en la progresión al SIDA es un tema de gran interés. Se ha reportado que personas que expresan ciertas combinaciones deHLA están predispuestas a progresión más rápida de la enfermedad, lo que sugiere que la eficacia de la respuesta inmunológica del huésped contra el VIH-1 puede influir en la tasa de progresión del SIDA.183 Sin embargo, se requiere más estudios sobre los factores del huésped que modifican la progresión de la enfermedad y sobre la aplicación de nuevos métodos para la tipificación de HLA de alta resolución. La identificación de cofactores ambientales podría permitir el desarrollo de intervenciones para limitar la progresión en personas infectadas. Debido a que la replicación del VIH-1 en cultivo de tejidos se facilita por el estímulo inmunológico de las células blanco, es posible que la exposición a antígenos ambientales o a infecciones comunes pueda activar también la expresión del VIH-1 in vivo, contribuyendo a la pérdida acelerada de células T CD4+. Sin embargo, esta afirmación no se ha demostrado del todo, y los estudios epidemiológicos no han identificado factores ambientales que correlacionen en forma clara con el desarrollo de la enfermedad. Sin embargo, es interesante que los estudios sugieren que la administración de isoniacida a pacientes infectados por VIH-1 con pruebas cutáneas positivas a tuberculina155 y la administración de aciclovir a pacientes infectados por VIH con afección extensa a herpesvirus155 puede retrasar la progresión del SIDA o prolongar la supervivencia. Es posible que estos beneficios resulten de limitar las acciones de estímulo inmunológico crónico por los agentes infecciosos, lo que podría aumentar la replicación del VIH-1 y así contribuir a la progresión de la enfermedad por este virus.
EPIDEMIOLOGIA DE LA INFECCION POR VIH-2
La infección por VIH-2 es más prevalente en ciertas regiones de Africa Occidental. También se ha reportado cada vez más en otras partes del mundo, incluyendo Portugal, Francia, Alemania, Brasil e India.156,157 Aunque en la mayoría de los países la presencia del VIH-2 puede asociarse con lazos a Africa Occidental, en la actualidad se ha reportado diseminación hacia la población indígena en Francia, Portugal y la India.
Se considera que la prevalencia de infección por VIH-2 en los Estados Unidos es muy baja en la actualidad, aunque han ocurrido casos de infección y de SIDA asociado al VIH-2.156 En todos los casos en los que se ha contado con la historia del paciente, la persona infectada había vivido en Africa Occidental o había tenido una pareja sexual de esa región. No se han identificado aún casos de infección por VIH-2 en adictos a drogas intravenosas, varones homosexuales o receptores de transfusiones. Debido a que el VIH-2 se relaciona solo en modo distante al VIH-1, con conservación de alrededor del 50 porciento de los aminoácidos en las proteínas gag y pol y menos del 30 porciento de conservación en los productos del gen env, su presencia no se detecta en forma eficaz por los estudios serológicos que se usan en la actualidad para detectar la infección por el VIH-1.66,156 Por lo tanto, debido a la presencia de VIH-2 en los Estados Unidos y a que solo alrededor del 80 porciento de los sueros de individuos infectados por VIH-2 reaccionan en el ELISA del VIH-1, la Food and Drug Administration recomendó en junio de 1992 que todas las unidades de sangre donadas sean analizadas en forma específica en busca de evidencia serológica de infección por VIH-2.156
Al igual que el VIH-1, el VIH-2 se trasmite por exposición a sangre infectada.156 Aunque la trasmisión heterosexual representa el principal modo de trasmisión en Africa Occidental, evidencias recientes indican que la diseminación heterosexual del VIH-2 sucede con una tasa menor que la del VIH-1.158 En forma semejante, la trasmisión perinatal del VIH-2 es menos frecuente, y menos del 10 porciento de los lactantes nacidos de madres infectadas por VIH-2 adquieren la infección. Existen dos posibles razones para esta aparente menor tasa de trasmisión del VIH-2: el virus puede ser en forma inherente menos infeccioso que el VIH-1, o los individuos infectados pueden tener una carga viral menor. Sin embargo, aún se tiene poca información al respecto.
La infección por VIH-2 se asocia con el desarrollo de inmunodeficiencia progresiva, susceptibilidad a infecciones oportunistas, enfermedad del SNC y emaciación.156 Aunque la infección por VIH-2 puede causar SIDA, los estudios sugieren que los individuos infectados por este virus tienen un periodo asintomático más largo y en consecuencia una menor incidencia de progresión a SIDA que los infectados por el VIH-1.156,159,160 No se sabe si el VIH-2 es un poco menos virulento o si la respuesta inmunológica del huésped es más capaz de amilorar las consecuencias patógenas de esta infección viral. Se requieren estudios adicionales para esclarecer mejor estos aspectos. En la actualidad existe poca información sobre la utilidad del tratamiento antiviral en las personas infectadas por el VIH-2.
Origen del VIH y del SIDA
La epidemia del SIDA se manifestó como una enfermedad aparentemente nueva en los Estados Unidos en 1981.90-92 Desde la identificación y la definición del SIDA como entidad clínica, estudios retrospectivos han demostrado un notable incremento de casos de afecciones relacionadas con el SIDA en Africa central a finales de la década de 1970, con algunos casos aislados ocurridos en los 60.161 Después del descubrimiento del VIH-1 como el agente etiológico del SIDA, los estudios serológicos que utilizaron muestras de sangre almacenada revelaron que la diseminación epidémica del VIH-1 había ocurrido en áreas de Africa central a mediados o finales de los años 70. La reactividad serológica más temprana con VIH-1 se detectó en una muestra recolectada en 1959. Sin embargo, este reporte se ha puesto en duda de modo reciente.162 Los registros seroepidemiológicos del VIH-2 están menos bien documentados, pero parecen haberse presentado casos de infección en Africa Occidental desde por lo menos 1966.162
La aparición de una nueva y devastadora enfermedad y los descubrimientos de los VIH-1, VIH-2 y un número creciente de virus relacionados en simios, han resultado tema de discusión con respecto al origen de los VIH y el SIDA.161-162 Se han propuesto numerosas hipótesis para explicar la historia filogenética del VIH-1 y VIH-2, incluyendo la infección relativamente reciente de poblaciones humanas con un virus simiano, la mutación de un virus humano no patógeno preexistente a una forma más virulenta, o la diseminación epidémica de un virus inmunosupresor humano no apreciado y secuestrado con anterioridad desde el punto de vista geográfico. Los retrovirus no dejan el equivalente a un registro fósil, y la magnitud de la epidemia continúa expandiéndose, borrando los márgenes epidemiológicos previos. Puede ser que al final sea imposible reconstruir en forma definitiva la historia evolutiva del VIH y el principio de su diseminación.
Aunque la información disponible no permite delimitar en forma clara el origen de la pandemia de SIDA, han surgido datos orientadores a partir de estudios genéticos comparativos. Los análisis recientes de las secuencias de nucleótidos de un gran número de VIH-1, VIH-2 y VIS han apoyado la hipótesis de que los VIH pueden haberse originado de un reservorio diverso de lentivirus de primates no humanos.20,162 Como en el caso de muchos otros agentes infecciosos, los VIS no parecen causar enfermedad en sus huéspedes naturales. El hecho de que el VIH-1 y el VIH-2 ocasionen enfermedad en los humanos puede reflejar la introducción reciente de estos virus a la población humana. La evidencia de un origen simiano del VIH-2 es bastante obvia; sin embargo, no se ha identificado un progenitor directo del VIH-1.
Los estudios seroepidemiológicos revelan que varias especies de monos africanos, incluyendo los monos mangabeys/negros, los monos verdes africanos (Cercopithecus aethiops) y los mandriles (Papio [Mandrillus] sphinx), tienen altas tasas de infección natural por VIS (VISsmm, VISagm, y VISmnd, respectivamente).18 Pueden encontrarse evidencias de infección por VIS en el 25 a 50 porciento de los animales salvajes, y la infección no se asocia con ninguna enfermedad. Los VIS aislados de los animales infectados pueden clasificarse en tres grupos diferentes por análisis de la secuencia genética. Las secuencias de nucleótidos de los VISsmm, VISagm y VISmnd tienen cierta similitud entre sí, aunque cada virus está equidistante a los otros dentro de la escala evolutiva.18,20 El VISmac, que se ha aislado de macacos que sufren una enfermedad parecida al SIDA, está relacionado en forma estrecha al VISsmm, y ambos pertenecen al grupo genético que incluye al VIH-2. El virus VISmac patógeno no se encuentra en los macacos salvajes, sino que parece originarse del VISsmm, el virus de los sooty mangabey aparentemente inofensivo, por trasmisión zoonótica en cautiverio. Es interesante que las evidencias genéticas y la coincidencia geográfica indican que los mangabeys/negros han sido fuente también de la infección zoonótica de los humanos en Africa Occidental, con el origen subsecuente del VIH-2.19 El habitat natural de los mangabeys/negros en Africa Occidental coincide con la misma región geográfica en donde el VIH-2 es endémico, y se ha demostrado contacto estrecho entre los mangabeys y los humanos. Aunque no se han demostrado casos de trasmisión del VISsmm de un mangabey infectado a un humano, sí se han reportado recientemente infecciones en personas que trabajan en el laboratorio con este virus.163,164
El VIH-1 difiere del VIH-2 en que no se ha encontrado un progenitor obvio en primates no humanos, y el VIH no se replica en monos inoculados en forma experimental. Recientemente se aisló de los chimpancés el virus VIScpz, que parece estar más relacionado al VIH-1 desde el punto de vista de secuencia de nucleótidos que cualquier otro VIS o que el VIH-2.20,21,162,165 El VIScpz comparte una organización genómica común con el VIH-1, pero su secuencia de nucleótidos es muy divergente. Es interesante que se han identificado grupos de VIH-1 poco usuales (denominados subtipo O) cuya secuencia de nucleótidos indica que están equidistantes del VIScpz y de tipos más comunes de VIH-1.166,167 Aunque este dato puede indicar que el VIH-1 y el VIScpz tienen un origen común, la infección de los chimpancés por virus VIS parece bastante rara. Se requieren más estudios sobre la diversidad de los virus aislados en primates humanos y no humanos para comprender mejor la historia evolutiva del VIH-1.
Los estudios genéticos del VIH-1 de diferentes localizaciones geográficas pueden proporcionar claves sobre la evolución de la pandemia del SIDA.162 La determinación de las secuencias de gag y env de muchos VIH-1 ha permitido identificar ocho diferentes subtipos (designados de la A a la H y conocidos en conjunto como grupo principal M) en los principales centros de la pandemia del SIDA.34,162 El subtipo O del VIH-1, muy diferente, se identificó por primera vez en Camerún y Gabón, y representa un noveno subtipo, considerándose un grupo distinto.166,167 Los estudios actuales podrán identificar subtipos adicionales por sus secuencias virales. Diversos países en Africa, Asia y Sudamérica tienen subtipos de VIH-1 cocirculantes. En los Estados Unidos solo se ha reportado el subtipo B, probablemente como resultado de un intenso efecto fundador. En la actualidad no existen evidencias de que los subtipos individuales del VIH-1 difieran entre sí en sus propiedades biológicas o potencial patógeno. Además de ayudar en los estudios de epidemiología y evolución del VIH-1, el estudio de estos subtipos proporcionará la información práctica necesaria para asegurar que las pruebas serológicas disponibles identifican con sensibilidad la infección por todos los subtipos virales,168 y guiarán el desarrollo de las vacunas en diferentes partes del mundo.169
Aunque el origen de las infecciones por VIH sigue siendo un enigma, diversos factores parecen responsables de su diseminación mundial. Los focos de infección del VIH antes de que el virus se diseminara no están bien definidos. En algunas áreas rurales de Africa, en donde las condiciones sociales no favorecen la rápida diseminación del virus, la seroprevalencia a VIH-1 permaneció baja y relativamente estable en el mismo periodo en el que en otros sitios se incrementó.159 Sin embargo, durante los últimos 30 años, las presiones económicas, los cambios sociales y la marcada inestabilidad política han causado una migración masiva de habitantes rurales a áreas urbanas en Africa.159 Los cambios demográficos secundarios han creado un ambiente que facilitó la diseminación epidémica de enfermedades de trasmisión sexual, lo que a su vez ayudó a desencadenar la epidemia porVIH-1. El comercio internacional y el turismo proporcionaron oportunidades para la diseminación de la infección en todo el mundo. El sureste de Asia fué la última región en la que se introdujo el SIDA, pero la alta densidad de población y los comportamientos de riesgo prevalentes han causado que la infección se disemine con rapidez. Se cree que cambios sociales semejantes son los responsables del incremento en la infección endémica de VIH-2 en Africa Occidental, y su diseminación eventual a Europa, América y la India. El motivo de que el VIH-1 se disemine con más rapidez y en forma más amplia que el VIH-2 no se conoce; este dato puede reflejar diferencias biológicas entre los virus u otros factores. Sin embargo, independientemente de la historia evolutiva aparentemente distinta y de las diferencias biológicas potenciales entre los dos virus, la emergencia coincidente de la epidemia de VIH-1y de VIH-2 sugiere una compleja interrelación entre factores sociales y la diseminación de patógenos infecciosos.159
IDENTIFICACION DEL VIH-1
Los métodos utilizados para identificar el VIH-1 se basaron en las técnicas utilizadas para el aislamiento del HTLV-I y HTLV-II. Sin embargo, en contraste con los cultivos de células infectadas por HTLV-I y HTLV-II, la proliferación persistente de células T no continuó cuando los linfocitos de personas con SIDA o estados relacionados con el SIDA se colocaron in vitro. En lugar de ello, después de un periodo de crecimiento a corto plazo, se observó muerte celular extensa en los cultivos, junto con la presencia de actividad de transcriptasa y de partículas parecidas a retrovirus.7, 8, 170
Un adelanto importante fue el descubrimiento de que el VIH-1 podía propagarse en ciertas líneas celulares leucémicas T establecidas sin inducir efecto citopático.7, 170 Entonces el retrovirus podía crecer en grandes cantidades, permitiendo así su caracterización detallada y la preparación de reactivos para su uso en análisis epidemiológicos. El escrutinio del suero de pacientes con SIDA mostró casi el 100 porciento de reactividad con el VIH-1, mientras que el suero control de personas normales fue negativo en forma uniforme.171, 172 El análisis de personas con estados relacionados al SIDA o en alto riesgo de SIDA demostró muy elevada prevalencia de anticuerpos contra el VIH-1. Aún más, el VIH-1 podía aislarse de los linfocitos de la mayoría de las personas con SIDA o estados relacionados al SIDA al igual que de individuos asintomáticos seropositivos para VIH-1. En los estudios iniciales, la imposibilidad para aislar el virus de todas las personas seropositivas se debió a las limitaciones técnicas de los métodos empleados. Sin embargo, los métodos actuales permiten aislar el VIH-1 de la gran mayoría de las personas seropositivas.99,173
El VIH-1 puede trasmitirse con facilidad in vitro a cultivos de linfocitos de donantes normales. Poco después de la infección se observa un efecto citopático considerable, que se manifiesta por fusión celular, formación de células gigantes multinucleadas conocidas como sincicio y muerte celular. El cuidadoso examen de cultivos infectados revela que el blanco primario de la infección es la célula T inductora-colaboradora CD4+ y que las consecuencias citopáticas de la infección viral son más pronunciadas en esta población celular.74 La molécula CD4 (también llamada T4) es una glucoproteína que se encuentra predominantemente sobre la superficie de las células T. Es necesaria la interacción entre la molécula de superficie de la célula T CD4+ y las moléculas clase II del complejo principal de histocompatibilidad (CPH) expresada sobre la superficie de las células presentadoras de antígenos, para la generación de una respuesta inmune celular eficaz.174 Las células T CD4+ son la principal fuente de linfocinas, que brindan señales inductoras para la activación y orquestación de múltiples ramas de la respuesta inmune.
La naturaleza de este citotropismo peculiar no se comprendió al principio pero estaba de acuerdo con la pérdida de células T CD4+ in vivo que caracteriza a la inmunodeficiencia del SIDA. Desde entonces se ha demostrado que la misma molécula de superficie CD4, que desde el punto de vista fenotípico define a la población inductora-cooperadora de células T, es de hecho el receptor de superficie utilizado por el VIH-1 para entrar en las células blanco.174 Por ello, el rango de tipos celulares susceptibles a la infección por VIH-1, in vitro e in vivo, es paralelo con aquellas células que expresan el receptor de superficie CD4. Además, en la actualidad se sabe que el proceso de fusión celular que causa la formación característica del sincicio después de la infección por VIH-1 en las células T susceptibles, es una consecuencia directa de la interacción, específica y de alta afinidad, entre la molécula T CD4 y la glucoproteína env del VIH-1174 [ver figura 4].
|
|
|


DIVERSIDAD GENETICA DEL VIH-1
Se han obtenido gran cantidad de virus VIH-1 de regiones en todo el mundo. En muchos de ellos se ha determinado la secuencia de nucleótidos, y la comparación de estas secuencias revela un sorprendente grado de diversidad genómica, que es más marcada en la región del gen env.34,175 También existe un grado semejante de diversidad genómica entre los VIH-2 aislados.34 La diversidad del VIH-1 y el VIH-2 contrasta en forma marcada con la homogeneidad genética en todo el mundo de los HTLV-I, y se piensa que refleja el acúmulo de mutaciones que suceden en el proceso de transcripción inversa durante los numerosos ciclos de replicación en un huésped infectado por el VIH.175 A diferencia de las polimerasas celulares, que tienen una función endógena llamada de lectura probada para maximizar la fidelidad de la replicación, la transcriptasa inversa retroviral es incapaz de reparar los errores introducidos durante el proceso de síntesis del ADN. Se calcula que, por ello, de una a 10 mutaciones puntiformes se incorporan al genoma del VIH-1 durante cada ronda de replicación.36,176 Se introducen variaciones adicionales en la secuencia genómica viral por recombinación durante la síntesis viral, que es causada por cambio de templetes entre las dos moléculas de ARN viral que existen juntas en la partícula del virus VIH-1. Se sabe que la replicación del VIH-1 es un proceso muy activo en las persnas infectadas, y que proporciona numerosas oportunidades para la generación de la diversidad de la secuencia viral.
En un individuo infectado, cada genoma de VIH-1 puede tener una secuencia diferente de la de los otros, y los virus deben considerarse más como poblaciones, llamadas cuasiespecies, de variantes relacionadas en forma estrecha.175 Las variantes del VIH-1 presentes en estas cuasiespecies pueden mostrar diferencias marcadas con respecto a sus características biológicas, incluyendo el tropismo celular, el potencial citopático, la tasa de replicación, la susceptibilidad a medicamentos antivirales y las propiedades de antígeno de superficie que pueden permitirles escapar de ser depurados por el sistema inmunológico del huésped.
Mientras que muchos de los errores que ocurren en la transcripción inversa causan virus incompetentes para replicarse, ciertas áreas del genoma del VIH, como la secuencia que codifica el gen env, pueden adaptarse con más facilidad a las alteraciones de los nucleótidos.174,177 Las regiones del gen env que muestran una divergencia importante en la secuencia de nucleótidos se intercalan con dominios que se conservan bien en los diferentes VIH. Las comparaciones de las secuencias predichas de aminoácidos para las glucoproteínas gp120 env del VIH-1 de los diversos virus han revelado cinco regiones discontinuas, designadas como V1 a V5, que contienen residuos de aminoácidos muy variables.178 La variación genética en el gen env de los virus aislados determina su tropismo relativo a las células T o macrófagos, y su capacidad relativa para inducir la formación de sincicio en las células infectadas.178-180 Las secuencias variables del gen env influyen también en la susceptibilidad de un virus determinado para ser neutralizado por anticuerpos y preparaciones recombinantes de CD4 soluble.178
In vivo se ha observado la generación de la diversidad genética del VIH-1 y la selección de variantes virales específicas con el tiempo.175,177 Pueden identificarse en un mismo paciente y al mismo tiempo variantes virales relacionadas, pero distinguibles entre sí, y la representación relativa de las secuencias variantes específicas puede cambiar en forma significativa durante el curso de la infección.175,181,182 La imposición de una presión selectiva, como la administración de un medicamento antiviral, sobre el reservorio heterogéneo de diferentes VIH-1 in vivo, puede ocasionar crecimiento rápido de las variantes cuya replicación se inhiba en forma menos eficaz.45 En varios casos de trasmisión viral, se ha seguido tanto en el donador como en el receptor la evolución de la diversidad de secuencias del VIH-1.183-185 Después de la infección, el desarrollo de las variantes observadas parece depender de factores específicos del huésped en particular. El sistema inmunológico puede ser el principal factor que influya en la naturaleza y evolución de la diversidad genética del VIH-1. Sin embargo, aún no se conoce el papel preciso de la selección inmune en este proceso, y es posible que también participen otros factores. Por lo menos en algunos casos, la diversidad en la secuencia de nucleótidos entre los VIH-1 aislados puede traducirse también en diversidad de los antígenos de reconocimiento (ver adelante).
VIGILANCIA DE LA INFECCION POR VIH-1 IN VIVO
La infección por VIH-1 inicia un proceso citopático persistente y progresivo que culmina en la destrucción casi completa de las células T subtipo CD4+.41,44 Los primeros esfuerzos por sintetizar un modelo coherente sobre las profundas consecuencias patogénicas de la infección por VIH-1 se confundieron por la idea de que muy pocas células de los individuos infectados portan o expresan el genoma viral. Sin embargo, la aplicación reciente de métodos sensibles para detectar el VIH-1 ha demostrado que la carga viral es mucho mayor de lo que se pensaba antes, y que aumenta de modo significativo al progresar la enfermedad [ver figura 5]. Esta nueva apreciación sobre la dinámica de población del VIH-1 proporciona importantes datos sobre la patogenia de la enfermedad.41,44,45,186-188 La magnitud de la replicación durante la evolución clínica de la infección por VIH-1 sugiere que las consecuencias citopáticas directas de la expresión viral pueden ser las responsables de la depleción de linfocitos T. Además, la fluctuación en el grado de producción viral que ocurre en los diferentes estadios de la enfermedad indica que el sistema inmunológico puede ser capaz de restringir la replicación del VIH-1, por lo menos en forma temporal.

|
| Figura 5 |
| Historia natural de la infección por VIH |
Hasta hace poco los métodos disponibles para vigilar el nivel de replicación viral en los pacientes infectados por el VIH-1 estaban limitados por problemas de insuficiente sensibilidad y reproducibilidad. Sin embargo, los adelantos en los métodos usados para cuantificar el nivel de VIH-1 en el plasma o células de sangre periférica de los pacientes infectados han proporcionado nuevos enfoques para dilucidar los aspectos virológicos de la historia natural de la enfermedad por este virus. Dos nuevos métodos son importantes para medir la concentración de ARNm del VIH-1 en las partículas virales en el plasma: una se basa en la amplificación del blanco (métodos cuantitativos de RCP) y la otra en la amplificación de la señal (el llamado ensayo de ADN ramificado [ADNr]).43,189,190 Al parecer, el grado de viremia (medido por la cantidad de ARN del VIH-1 en el plasma) refleja con exactitud la extensión de la replicación viral activa.43,45,187,188 Además, estudios recientes de cinética indican que la vida media del virus en el plasma (alrededor de dos días o menos) varía poco entre los pacientes con diferentes cargas virales o en diferentes estadios de la enfermedad. La tasa y magnitud de la reducción del virus en plasma observado en estos estudios implica que la vida media promedio de una célula infectada es de menos de uno a dos días [ver figura 6]. Estos datos indican que la concentración estable en plasma de VIH-1 está en función de la magnitud de la replicación y producción viral activa.187,188 En muchas maneras, la aplicación de estos nuevos métodos para vigilar la replicación del VIH-1 in vivo ha modificado las nociones sobre los mecanismos patogénicos de la enfermedad por VIH-1 [ver figura 5].

|
| Figura 6 |
| Efecto del tratamiento antiviral sobre la carga viral |
HISTORIA NATURAL DE LA INFECCION POR VIH-1
Los mecanismos de trasmisión del VIH-1 no se conocen del todo. El virus puede trasmitirse por transferencia de células infectadas, del virus libre o ambos, aunque la contribución precisa de estos componentes a un determinado episodio de infección probablemente depende de la naturaleza y vía de exposición. Si un donador trasmisor del virus tiene enfermedad avanzada, el receptor tiene más probabilidad de infectarse.120 Aún más, la infección por un donador en fase avanzada se asocia con mayor incidencia de infección aguda sintomática, con evolución más rápida a SIDA. Este fenómeno puede ser causado por mayor carga viral en el trasmisor, mayor virulencia de la cepa viral trasmitida o ambos. Las cargas virales en plasma y asociadas con células aumentan con la progresión de la enfermedad, y los estudios demuestran que se detecta la presencia del ARN de VIH-1 en forma más fácil en el semen de los varones con cuentas bajas de células T CD4+.41,44,191,192
Desde el sitio de entrada del virus, ya sea a través de las mucosas o directamente al torrente sanguíneo, el virus VIH-1 libre o las células infectadas se dirigen al tejido linfoide. La presencia de células blanco susceptibles, incluyendo células T, células dendríticas y macrófagos, y el ambiente inmunológicamente activo en los tejidos linfáticos permite nuevos ciclos de replicación del VIH-1 e infección de las células del huésped. El análisis de los VIH-1 aislados del trasmisor y del receptor indica que la población viral muy heterogénea del donador sufre un proceso de selección que permite que la infección se establezca por un subgrupo pequeño de VIH-1.193,194 En el nuevo huésped, recién infectado, predomina una población genéticamente homogénea de virus que son principalmente citotrópicos para los macrófagos y que no inducen sincicios (NIS) (ver adelante). No se sabe si la ventaja selectiva de esta subpoblación de VIH-1 es causada por la trasmisión de un número limitado de variantes virales o por la replicación preferente de ciertos subtipos en el nuevo huésped. Tampoco se ha demostrado que la infección con variantes específicas cause manifestaciones clínicas especiales, pero el tropismo y potencial patogénico de ciertas cepas sugiere que la patogenicidad selectiva puede contribuir a las diferentes manifestaciones clínicas de la infección por VIH-1 y a los diversos grados de progresión de la enfermedad en los distintos pacientes.
El rango y gravedad de los síntomas de la infección por VIH-1 varía en forma muy considerable. Alrededor del 40 a 60 porciento de los pacientes sufren un síndrome viral agudo que consiste en fiebre, cefalea, linfadenopatía, mialgias y erupción durante la infección primaria, cuando ocurren altos grados de replicación del VIH-1 antes del desarrollo de una respuesta inmunológica antiviral [ver figura 5].195-198 Coincidente con la resolución de los síntomas de la infección aguda, que dura alrededor de un mes, caen en forma brusca los niveles de viremia en plasma y el número de células T CD4+ infectadas en sangre periférica. La disminución en la replicación del VIH-1 correlaciona con la aparición de una respuesta de células T citotóxicas contra el VIH-1.199-201
En contraste con la historia natural de la infección primaria sintomática, los aspectos virológicos e inmunológicos de la infección primaria asintomática no se conocen. Los individuos que tienen síntomas clínicos severos durante la infección primaria parecen tener más probabilidad de sufrir infección primaria sintomática.202 Este fenómeno puede ser especialmente cierto en pacientes que sufren un alto nivel de linfocitosis T CD8+, los que tienen una respuesta inmunológica celular antiviral deteriorada o con restricción clonal, o los que montan una respuesta serológica limitada al antígeno central p24 del VIH-1.201,202
Una vez que ceden los síntomas de la infección primaria, el paciente entra en una fase clínicamente asintomática o de síntomas mínimos. Aunque la respuesta inmunológica antiviral puede limitar la replicación del VIH-1 durante este periodo de latencia clínica, parece no ser totalmente eficaz, persistiendo la producción viral, lo que se hace evidente por la existencia continua de VIH-1 en el plasma [ver figura 5] y los tejidos linfoides [ver figura 7].41,43,44,187,191,192 La replicación activa y continua del VIH-1 en los individuos asintomáticos ocasiona la reducción lentamente progresiva de las células T CD4+. La evidencia de una mayor carga viral en el plasma y de concentraciones elevadas de ADN y ARN del VIH-1 en las células de sangre periférica correlaciona con una depleción más acelerada en las células T CD4+, lo mismo que en la progresión de la enfermedad, y es probable que refleje la falla progresiva de la respuesta inmunológica para contener la replicación del VIH-1.41,44,203
|
|

|
|
La mayoría de las evaluaciones inmunológicas y virológicas en pacientes infectados por VIH-1 se han enfocado a los linfocitos de sangre periférica. Sin embargo, se calcula que estas células representan alrededor del dos porciento del reservorio de linfocitos en todo el organismo. Se ha señalado la importancia de los órganos linfoides como reservorios de la infección por VIH-1 por la demostración de que el porcentajde de linfocitos T CD4+ infectados por VIH-1 puede ser hasta 10 veces mayor en los ganglios linfáticos que en sangre periférica42,46 [ver figura 7]. Además, los linfocitos infectados por el VIH-1 que se encuentran en los órganos linfoides expresan más ARN viral que sus contrapartes en sangre periférica. Los hallazgos de que las células activadas proporcionan blancos preferenciales para la infección y producción del VIH-1 y que mucha de la carga viral se localiza en los tejidos linfáticos que participan en la generación de la respuesta inmune sugiere que las células inmunológicamente activadas pueden también sufir las consecuencias patogénicas de la infección por VIH-1.
En las fases tempranas de la enfermedad se encuentran cantidades sorprendentes de partículas virales en los ganglios linfáticos asociados con las células dendríticas foliculares (CDF) que tienen un papel indispensable en el procesamiento y presentación de los antígenos.41,44 No parece ser que las CDF se infecten, pero sí pueden facilitar la infección de las células T CD4+ que migran a través de los folículos linfoides durante el curso de una respuesta inmunológica o de la recirculación normal. Con el tiempo ocurre la destrucción gradual de la red de CDF, lo que reduce la capacidad de atrapamiento del virus y de antígenos. Al progresar la enfermedad, la arquitectura normal de los ganglios linfáticos se distorsiona, lo que puede comprometer la función inmunológica normal. No se sabe si la disrrupción de la red de CDF es causada por el efecto tóxico directo de la infección por VIH-1 o por la entrada de células T citotóxicas CD8+ que han sido observadas cerca de los sitios de destrucción de las CDF.
MECANISMOS DE DEPLECION DE LAS CELULAS T EN LA INFECCION POR VIH-1
Aún no se define el mecanismo exacto por el que la infección por VIH-1 causa depleción progresiva y deterioro funcional de las células T CD4+, pero es probable que incluya diversos mecanismos directos e indirectos.41,44,205 El reconocimiento reciente de que durante el curso de la enfermedad persiste una expresión y replicación considerable del VIH-1 apoya la hipótesis de que los efectos citopáticos directos de la replicación viral son los causantes de mucho del compromiso inmunológico observado. Los estudios intervencionistas que han usado medicamentos antivirales potentes pero de efecto temporal, proporcionan evidencia directa de que un mayor nivel de replicación del VIH-1 correlaciona con tasas aceleradas de recambio en las poblaciones de linfocitos T CD4+ [ver figura 6].45,187,188
La infección por VIH-1 causa efectos citopáticos directos en las células T CD4+ in vitro que pueden observarse tanto en la célula aislada como por la formación de sincicios [ver figura 4]. In vivo la formación directa de sincicios se observa muy rara vez (excepto en los cerebros de algunos pacientes infectados), quizá por la rápida depuración de cualquier célula gigante multinucleada aberrante de la circulación. Las consecuencias patológicas de la infección por VIH-1 no se distribuyen en forma igual entre todas las células T CD4+, sino que se enfocan en forma preferente a las células proliferantes o activadas inmunológicamente. Los pacientes con infección asintomática tienen defectos específicos en la función de las células T incluso antes de que se observe disminución en su número.206 Estos defectos son más importantes en la población de células T de memoria que participan en el reconocimiento y respuesta en la segunda exposición a un antígeno específico. Al parecer esta subpoblación de células T es el blanco preferencial de la infección por VIH-1 in vivo.207 No se conoce la explicación precisa para el compromiso numérico o funcional de esta importante subpoblación de células inmunes efectoras. Sin embargo, suponiendo que las células T de memoria reconocen y proliferan en respuesta a antígenos que son comunes en el ambiente, la activación frecuente de estás células en un huésped infectado por el VIH-1 puede causar depleción paradójica de la población de células respondedoras. Por lo tanto, el repertorio de reactividad de las células T a un antígeno específico puede restringirse en forma progresiva en la infección por VIH-1 avanzada, lo que predispone al paciente a sufrir infecciones oportunistas.
Se ha sugerido que además de causar defectos preferentes en las células T de memoria, la infección por VIH-1 puede alterar el balance apropiado entre las subpoblaciones especializadas de células T CD4+ conocidas como TH1 y TH2, que se definen por su patrones particulares de producción de citocinas y su papel para inducir respuestas inmunes celulares y humorales, respectivamente.208,209 Sin embargo, varios estudios han puesto en duda este aspecto,208,210,211 y deben realizarse más estudios para definir las alteraciones potenciales en las subpoblaciones de células T CD4+ o los patrones alterados de producción de citocinas que pueden acompañar a la infección por VIH-1.
Se ha demostrado que la infección por VIH-1 inicia respuestas tanto celulares como humorales hacia los antígenos codificados por el virus (ver adelante). Aunque la destrucción inmunológica de los linfocitos T y los macrófagos infectados es indispensable para que el huésped controle la infección, puede, en el caso de una infección crónica y persistente, acelerar la deficiencia del sistema inmunológico que sigue a la infección por el VIH-1.174,212 Las células infectadas por el virus expresan antígenos virales que las convierten en blanco para la depuración inmunológica. Además, se ha sugerido que la unión de gp120 (liberado de las células infectadas y de las partículas virales) a la superficie de células no infectadas que portan CD4, las hace susceptibles a la destrucción inadecuada por las respuestas inmunológicas antivirales del huésped por un mecanismo denominado de espectador inocente. No se conoce aún la importancia in vivo de este mecanismo para la depleción de células T.
También se ha sugerido que la infección por VIH-1 causa destrucción indirecta de las células T CD4+ al sensibilizarlas para sufrir apoptosis, o muerte celular programada.205 La apoptosis es un mecanismo fisiológico normal por el que se eliminan ciertas clases de células durante el desarrollo de los linfocitos (v.gr., la deleción de las células T reactivas a los antígenos del huésped) o al terminar una respuesta inmunológica. Varios estudios han descrito mayores grados de apoptosis cuando las células T de individuos infectados por el VIH-1 se activan en cultivos de tejidos.41,205,213,214 El número de linfocitos CD4+ que sufre apoptosis excede en mucho al de los infectados por el virus. Los linfocitos CD8+, que no son blanco de la infección por VIH-1, también son sensibilizados para sufrir apoptosis. No se conoce del todo el mecanismo por el que se activa la apoptosis en las células T de los individuos infectados, ni se ha determinado el significado in vivo de este fenómeno descrito en cultivo de tejidos.
La depleción númérica de las células T CD4+ es la alteración inmunológica más obvia que sigue a la infección, pero se han descrito también varios defectos en la función de las células T, las células B y los monocitos.47 No se sabe si estos defectos tienen un papel primario en la inmunodeficiencia inducida por el VIH-1 o si reflejan la alteración en la regulación causada por la depleción de las células T CD4+. In vitro, las células T de los individuos infectados demuestran también menor reactividad a antígenos y una menor capacidad para traducir las señales de proliferación después de la estimulación. Se ha sugerido que las señales inapropiadas de las células T que son resultado de la unión de complejos de gp120-anti-gp120 al CD4 pueden causar la inducción de un estado de no respuesta inmunológica o anergia de las células T CD4+ en las personas infectadas por VIH-1.
La depleción de las células T CD4+ observada después de la infección por VIH-1 debe considerarse dentro del contexto de la dinámica de población de los linfocitos T, esto es, el balance entre la producción y la destrucción de las células T. Por desgracia, la capacidad de la población de las células T para autorrenovarse no se comprende del todo. No se sabe hasta qué grado la expansión periférica de células T maduras o la maduración de nuevas células T a partir de sus progenitores contribuye a mantener el reservorio total de linfocitos T en el adulto. Sin embargo, evidencias recientes sugieren que la contribución del timo para mantener la concentración de las células T es muy limitada después de los 20 años de edad.215 En cualquier caso, es probable que la infección por VIH-1 altere ambas fuentes de producción de células T, de modo que el remplazo celular no compence la pérdida. Se conoce que el timo es un órgano blanco importante en la infección y citopatología de la infección por VIH-1, y es probable que esto limite el desarrollo eficaz de células T.
Puede ser que, por mecanismos directos o indirectos, la depleción de las poblaciones de células T CD4+ maduras y la disrupción de la arquitectura de los órganos linfoides después de la infección por VIH-1, implique alteración en la expansión del reservorio periférico de células T. Al parecer el sistema inmunológico humano tiene mecanismos homeostáticos por los que se mantiene un número normal de células T CD4+. Cuando ocurre depleción de éstos por la infección por el VIH-1, los mecanismos compensatorios intentan normalizar las cifras celulares, lo que provoca mayor proliferación de linfocitos T CD4+ maduros del reservorio periférico. Por desgracia, estas células activas desde el punto de vista de proliferación constituyen blancos fértiles para la infección por VIH-1 y en sí pueden infectarse y destruirse [ver figura 6]. En ausencia de un remplazo adecuado de células T, puede presentarse una reducción definitiva en el número de estas células.
RESPUESTA INMUNOLOGICA AL VIH-1
La influencia del sistema inmunológico humano para modular la evolución de la infección por VIH-1 no se conoce del todo. Sin embargo, la persistencia y naturaleza casi siempre progresiva de la infección sugiere que el sistema inmunológico es relativamente ineficaz. Esta impotencia inmunológica puede ser resultado de una estregia viral para evadir el reconocimiento antigénico, del fracaso para montar una respuesta neutralizante humoral o celular, o por infección y destrucción viral de la población T CD4+, que tiene un papel crítico en la eliminación de las infecciones incipientes.
Aunque las investigaciones seroepidemiológicas han demostrado que existen anticuerpos antiVIH-1 específicos en el suero de las personas con SIDA o riesgo de SIDA, no se sabe si estos anticuerpos tienen un efecto protector in vivo.178 Los anticuerpos obtenidos del suero de las personas infectadas son capaces de neutralizar la infectividad del VIH-1 en cultivo de tejidos, pero los títulos de estos anticuerpos en las personas infectadas son muy bajos. Aún más, los anticuerpos neutralizantes no parecen inhibir en forma eficaz la trasmisión viral a través del contacto célula-célula. La reducción inicial en la viremia del VIH-1 que ocurre con la resolución de la infección primaria se observa antes de que se desarrollen anticuerpos, y la presencia de anticuerpos neutralizantes no parece proteger contra la progresión de la enfermedad. Por lo tanto, no es claro el significado biológico de la respuesta neutralizante de anticuerpos en la modulación del curso de la infección por VIH-1.
Los anticuerpos que bloquean la infección por VIH-1 en cultivo de tejidos son específicos para dos dominios distintos de la glucoproteína env del VIH-1.178 Un dominio consiste en las regiones conservadas de la glucoproteína env que se requieren para unir a la molécula CD4. Debido a la naturaleza conservada de este dominio, los anticuerpos dirigidos contra él neutralizan diversas cepas de VIH-1 y se conocen como grupo específicos. Un segundo tipo de anticuerpos neutralizantes reconoce una estructura de asa en el tercer dominio hipervariable (o V3) de la glucoproteína env que parece tener un papel crítico en los pasos de la infección viral que ocurren después de la unión a CD4. Los anticuerpos dirigidos contra el dominio V3 bloquean la fusión del virus a la membrana celular, pero no inhiben la fijación viral al CD4. Los anticuerpos neutralizantes que reconocen el dominio V3 se denominan tipo específicos, porque bloquean la infección solo por cepas son secuencias de V3 semejantes a las de la variante VIH-1 que los originó.178 Las respuestas iniciales de anticuerpos neutralizantes producidas en etapas tempranas de la infección por VIH-1 forman anticuerpos que reconocen sobre todo los determinantes de la regiónV3, y son tipo específicos. En una fase más tardía se desarrollan anticuerpos neutralizantes con reactividad más amplia dirigidos contra la región de unión CD4.178 La mutación de las secuencias del gen env del VIH-1 ocasiona la aparición de cepas resistentes a los anticuerpos neutralizantes en cultivo de tejidos e in vivo. Sin embargo, no se ha determinado la importancia de estos hallazgos en la persistencia de la infección por VIH-1.
Se ha sugerido que ciertos anticuerpos antivirales pueden facilitar, en lugar de prevenir, la infección por VIH-1 de las células de linaje monocito-macrófago a través deun mecanismo que involucra la captación mediada por el receptor Fc de los complejos inmunes que contienen el virus. Los anticuerpos que facilitan la infección viral se conocen como anticuerpos promotores, y pueden identificarse por medio de ensayos en cultivo de tejidos.217 En la actualidad no existe evidencia clara de que los anticuerpos promotores contribuyan a la diseminación de la infección in vivo por VIH-1.
Mucho del análisis inicial de la reacción del huésped a la infección por VIH-1 se ha enfocado a la respuesta inmune humoral. Sin embargo, la inmunidad celular tiene un papel crítico en la eliminación de muchos otros tipos de infección viral, por lo que la magnitud en la que sus componentes puede reconocer y limitar la infección del VIH-1 es de gran interés. Aunque existe relativamente poca información sobre el papel de la inmunidad celular para modular la evolución de la infección por VIH-1, evidencias recientes indican que la rápida declinación en la replicación del VIH-1 observada después de la infección primaria correlaciona de modo temporal con la aparición de células T citotóxicas específicas.199,200,218 Además, se ha observado conservación de una respuesta citotóxica de células T contra el VIH-1 en algunos individuos que permanecen asintomáticos a pesar de tener infección de larga evolución.147,148 Se han obtenido células T citotóxicas reactivas contra los antígenos del VIH-1 en sangre periférica, LCR y lavado broncoalveolar de pacientes infectados.219 Estas células T reconocen los antígenos VIH-1 en el contexto de las moléculas de clase I del HLA del huéped y destruyen las células infectadas autólogas, pero no las heterólogas. Lo más frecuente es que reaccionen contra los determinantes antigénicos de las proteínas gag, env, transcriptasa inversa y nev del VIH-1. Los estudios indican que la diversidad de secuencia entre las variantes del VIH-1 que aparece durante el curso de una infección in vivo puede causar variabilidad en el reconocimiento antigénico y en la destrucción por las células T citotóxicas, pero no se sabe si esta variabilidad ayuda a las células infectadas a escapar de la depuración inmunológica.220,221
Además de las células T citotóxicas, la respuesta inmune celular al VIH-1 incluye una población de células efectoras que tienen receptores Fc. Estas células destruyen a las células infectadas por el virus en presencia de anticuerpos antivirales, por un proceso conocido como citotoxicidad celular mediada por anticuerpos (CCMA).222 Los anticuerpos que dirigen el proceso de CCMA se dirigen principalmente contra la glucoproteína gp120 env del VIH-1, y las células efectoras responsables muestran los marcadores fenotípicos de las células asesinas naturales (células NK).
La complejidad de la respuesta inmunológica a la infección por el VIH-1 y las diferencias inmunológicas potenciales que pueden existir en el espectro de este padecimiento representan temas muy importantes de estudio. El vigor de la respuesta inmune inicial contra el VIH-1 puede influir en la evolución subsecuente de la enfermedad (ver adelante). En fases posteriores, la pérdida de reactividad de anticuerpos contra la proteína nuclear p24 del VIH-1 y la aparición subsecuente de este antígeno en el suero se ha asociado con mal pronóstico.223 Sin embargo, no se sabe si la pérdida de la reactividad inmunológica en estos casos es la causa o la consecuencia de la inmunosupresión progresiva. Aún debe establecerse el significado de estos datos y las variaciones adicionales posibles en la reactividad inmunológica con otros productos virales. Es crucial aumentar nuestros conocimientos sobre la respuesta inmunológica contra el VIH-1 si deseamos prevenir y tratar la enfermedad.
VACUNAS PARA PREVENIR LA INFECCION POR VIH-1
La velocidad de la diseminación mundial del VIH-1 ha hecho que se ponga especial atención al desarrollo de una vacuna para prevenir la infección. Por desgracia, los prospectos para una vacuna segura y eficaz han sido limitados en el concepto y en la práctica por varias complejidades biológicas.169,225-227 Además, el establecimiento de la eficacia en cualquier candidato a ser vacuna se complica por aspectos éticos, legales y de responsabilidad.169,225,228,229
La falla en la inmunidad natural durante el curso de la infección por VIH-1 distingue a esta infección de muchas de otras infecciones virales en los humanos. Los mecanismos por los que el VIH-1 evade la depuración inmunológica no se conocen del todo, pero es probable que incluyan tanto a su compleja capacidad genética reguladora, que facilita el establecimiento de una fase latente, y la tremenda complejidad y diversidad de secuencias, que permite que el virus escape de las respuestas inmunológicas del huésped. Existen precedentes de vacunas útiles solo en las enfermedades, como la polio o el sarampión, en las que se desarrolla inmunidad protectora en el curso de la infección natural. Para ser eficaz, cualquier vacuna viral debe prevenir la adquisición del agente infeccioso o originar una respuesta inmune suficiente para facilitar la eliminación de las células infectadas antes del desarrollo de la enfermedad. Un hecho importante pero con frecuencia poco apreciado es que la mayoría de las vacunas previenen la enfermedad, no la infección. Sin embargo, el sistema inmunológico parece incapaz de eliminar la infección por VIH-1 una vez que esta se establece. Como resultado, las vacunas candidatos deben satisfacer gran cantidad de requerimientos, incluyendo la capacidad de inducir respuestas inmunológicas que neutralicen en forma eficaz la infectividad del VIH-1 libre y eliminen a las células infectadas antes de que se establezca la infección sistémica. El desarrollo de la vacuna también puede estar limitado por ramificaciones inmunológicas por la gran diversidad genética del VIH-1, y una vacuna eficaz debe proteger contra diversas variantes virales.169,225,227
Debido a que el contacto sexual por el que se depositan virus o células infectadas en mucosas es la causa de la infección en más del 80 porciento de todas las infecciones por VIH-1, la vacuna eficaz debe inducir inmunidad protectora tanto sistémica como en mucosas.230 Por desgracia, la mayoría de las vacunas que se desarrollan en la actualidad generan inmunidad sistémica, más que mucosa, y no limitan en forma eficaz el establecimiento de la infección a través de la principal vía de trasmisión del VIH-1.230
La definición de las correlaciones potenciales de la respuesta inmune, una herramienta esencial en el desarrollo de las vacunas, no ha sido posible en el caso del VIH-1 por la falta de un modelo animal de SIDA inducido por VIH-1 y de una cohorte de individuos que sean naturalmente resistentes a la infección por VIH-1.169,225-227 Con la esperanza de descubrir los factores inmunológicos del huésped que son responsables de la protección ante la infección o enfermedad, se ha puesto especial atención a los pocos individuos con infección por VIH-1 de muy larga duración y no progresiva, y a los que parecen ser resistentes a la infección a pesar de numerosas exposiciones. También se han reportado casos raros de individuos en quienes aparentemente se ha eliminado la infección por el VIH-1,230a pero es necesario investigar más sobre estos reportes para establecer la frecuencia y validez de los datos. La identificación de factores que pudieran proteger de la infección después de la exposición o del desarrollo de la enfermedad podrían ayudar a guiar el desarrollo de la vacuna, tarea que parece aún muy difícil.147-149,231
La glucoproteína env del VIH-1 ha sido identificada como el componente viral crucial reconocido por los anticuerpos neutralizantes, por lo que puede ser el inmunógeno principal en las vacunas contra el virus. Los anticuerpos neutralizantes podrían ser suficientes para limitar la infección por VIH-1 libre, pero es poco probable que detengan la infección después de la exposición a células infectadas.169,225,227 Por lo tanto, debido a que al parecer la trasmisión del VIH ocurre sobre todo por la transferencia de células infectadas, es lógico suponer que la vcuna eficaz contra el VIH-1 debe estimular a las células T citotóxicas específicas contra el virus. La evaluación de la importancia relativa de las respuestas humoral y celular en las posibles vacunas no puede evaluarse por estudios en humanos, y la mayoría de los esfuerzos experimentales se han realizado en modelos de primates no humanos con SIDA.
Las vacunas que se estudian en la actualidad en modelos animales para proteger contra las infecciones por VIH y VIS incluyen al virus inactivado completo, a antígenos proteicos recombinantes, a péptidos sintéticos o a virus de la vaccinina u otros poxvirus.169,225,227 En la mayoría de los estudios experimentales realizados hasta la fecha los animales han sido inmunizados por vía experimental y se les ha administrado dosis bajas de virus libre homólogo o muy relacionado en dosis bajas en el momento de la máxima respuesta inmunológica inducida por la vacuna. Los paradigmas experimentales empleados en estos estudios han sido criticados por no corresponder a las circunstancias de trasmisión del VIH-1 en los humanos. Además, los estudios de vacunas empleadas en modelos animales han proporcionado poca información sobre la inmunidad protectora necesaria para el desarrollo de una vacuna contra VIH-1.
La inmunización repetida con virus íntegros inactivados ha demostrado proteger a los monos rhesus contra el reto intravenoso con VIS patógenos vivos.227 Sin embargo, en los casos en los que se observó protección contra el virus libre, esta protección no duró muchó más allá del pico de la inmunidad máxima, y no se logró protección contra la exposición por mucosas. El éxito de estos estudios parece ser resultado de respuestas inmunológicas no esperadas contra antígenos celulares humanos contenidos en la preparación de la vacuna contra VIS y en el reservorio de virus, ya que ambos se propagan en líneas celulares T humanas. La inmunización con subunidades recombinantes de antígenos de VIS, análogos a las vacunas probadas en la actualidad en humanos, han sido incapaces de proteger a los monos susceptibles de un inóculo experimental con VIS virulentos.227 Aunque este resultado puede proporcionar la base para la definición de la correlación experimental de la inmunidad protectora contra los lentivirus, las dudas sobre la seguridad de este enfoque en la preparación de una vcuna contra el VIH-1 puede limitar su uso en humanos.223
Los chimpancés son los únicos primates no humanos que pueden infectarse por el VIH-1, pero su utilidd como un modelo animal exacto para SIDA es limitado porque no desarrollan enfermedad clínica después de la infección. Se ha demostrado que los chimpancés inmunizados contra una versión recombinante de glucoproteína 120gp env o un virus vaccinia recombinante que contiene todo el gen env del VIH, desarrollan niveles mesurables de anticuerpos específicos contra el VIH-1 y respuestas inmunológicas mediadas por células T.227,234 Las respuestas de anticuerpos neutralizantes inducidas por la vacuna son tipo específico y temporales. La inmunización de los chimpancés con los antígenos recombinantes de glucoproteína env que se han probado también en humanos se asocian con un éxito variable y limitado para proteger a los animales inmunizados contra un inóculo intravenoso del VIH-1 homólogo.33,210,227,235,236
Las vacunas probadas en la actualidad en voluntarios humanos no infectados incluyen la glucoproteína gp 120 env del VIH-1, producida en forma purificada por métodos de ADN recombinante, péptidos sintéticos derivados de gp120 y el precursor glucoproteína gp160 env insertado en el genoma de un virus vaccinia u otro vector poxvirus atenuado.169,227 Los estudios de fase I de estas vacunas indican que son seguras, e inmunogénicas en grado variable.169,227 En la actualidad se realiza un estudio de fase II que incluye a adultos con riesgo alto de infección y un estudio de fase I en lactantes nacidos de madres seropositivas.
Las vacunas de glucoproteína env recombinante pueden originar anticuerpos que neutralizan la infectividad del VIH-1 libre en cultivo de tejidos. La inducción de títulos apreciables de anticuerpos neutralizantes por las vacunas VIH-1 requiere con frecuencia de múltiples inmunizaciones, y los niveles de anticuerpos desaparecen algunos meses después. 169,227 Debido a que los inmunógenos de la proteína env del VIH-1 no se expresan en las células del individuo inmunizado, no despiertan una respuesta de células T citotóxicas. A diferencia de los anticuerpos neutralizantes que se observan en los individuos infectados por el VIH-1 y que neutralizan un amplio rango de cepas de VIH-1, los anticuerpos inducidos por la vacuna se dirigen principalmente contra la secuencia de la región específica V3 de la proteína env de la cepa de VIH-1 usada como inmunógeno en la vacuna.169,178,227 En consecuencia, estos anticuerpos son tipo específicos, y neutralizan solo cepas de VIH-1 muy relacionadas. El reconocimiento de que los anticuerpos inducidos por la vacuna no neutralizan las principales cepas de VIH-1, a diferencia de las cepas de laboratorio que se han propagado en líneas celulares T inmortalizadas, constituye una gran desventaja. Si los antígenos recombinantes de la proteína env van a convertirse alguna vez en el origen de la vacuna, debemos comprender los mecanismos moleculares que explican las limitaciones de las vacunas de primera generación y diseñar nuevos enfoques para inducir anticuerpos reactivos de espectro más amplio que neutralicen en forma eficaz a los principales grupos de VIH-1.
Los virus recombinantes de vaccinia que expresan gp160 sí despiertan respuestas de células T citotóxicas, pero muy pocos o ningún anticuerpo neutralizante anti-VIH-1.169,226,227 La inmunogenicidad de las vacunas basadas en virus vaccinia es limitada en personas que ya han sido antes inmunizadas contra la viruela. Además, debido a las dudas sobre la seguridad de las vacunas de virus vivos en personas inmunosuprimidas, estas vacunas pueden no ser adecuadas en las regiones del mundo en las que no es práctico realizar escrutinio anti-VIH-1.
A pesar de las evidencias obtenidas en las respuestas inmunológicas inducidas por vaccinia, algunos participantes de los estudios iniciales de vacuna anti-VIH-1 se infectaron posteriormente por el virus debido a la exposición por comportamientos de riesgo alto. Aunque estos casos de infección indican que las vacunas no son totalmente eficaces para prevenir la infección, no se ha evaluado en estudios preliminares su eficacia. En vista de la limitación de vacunas eficaces disponibles en la actualidad, en los Estados Unidos se han pospuesto recientemente los planes para realizar estudios de fase III de eficacia en gran escala.
Como resultado de los múltiples obstáculos que complican el desarrollo de las vacunas, pocos especialistas son optimistas y esperan que se disponga de una vacuna eficaz a corto plazo. Por lo tanto, deben enfatizarse las medidas de salud pública dirigidas a limitar la diseminación de la infección por el VIH-1.
TRATAMIENTO DE LA INFECCION POR VIH
Se considera que el compromiso inmunológico y neurológico lentamente evolutivo observado en la infección por VIH-1 es la manifestación del daño acumulado debido al bajo nivel pero persiste de replicación viral. Si la replicación viral demuestra ser la causa del compromiso clínico y si los sistemas afectados del huésped poseen suficiente potencial regenerativo, entonces los medicamentos que inhiben la replicación viral pueden prevenir o invertir las manifestaciones clínicas de las infecciones por VIH-1. Los esfuerzos actuales de investigación se dirigen a definir estrategias terapéuticas que limiten en forma más eficaz el grado de replicación del VIH-1 en los pacientes tratados y que mantengan la supresión de la replicación viral durante periodos prolongados.
El comportamiento biológico del VIH-1 impone varios prerrequisitos en la selección y utilidad de fármacos antivirales potenciales. Debido a la propensión del VIH-1 a infectar el sistema nervioso central y la capacidad del virus para establecer una infección persistente, los medicamentos propuestos deben cruzar la barrera hematoencefálica y poseer un índice terapéutico compatible con la administración a largo plazo. Aún más, debido a la propensión del VIH-1 para infectar células en el SNC, los medicamentos candidatos deben cruzar la barrera hematoencefálica. Por último, debido a que los linfocitos CD4+ y los macrófagos que son los blancos del VIH-1 in vivo pueden mostrar diferentes patrones de metabolismo de fármacos, debe contarse con agentes antivirales eficaces para lograr concentraciones adecuadas de sus metabolistos activos en cada uno de estos sitios cruciales de replicación viral.
Para disminuir la toxicidad y aumentar al máximo la eficacia, los fármacos deben limitar de forma importante la replicación viral pero producir interferencia mínima con el metabolismo de la célula huésped. Aunque el VIH-1 utiliza componentes de la maquinaria de la célula huésped para la síntesis y el procesamiento de su RNA y proteínas constituyentes, también depende de funciones biológicas únicas de varios productos específicos de virus para la replicación, incluyendo la enzima transcriptasa inversa y las proteínas tat y rev, que proveen blancos potenciales para terapéuticas farmacológicas eficaces. A pesar que cualquier etapa del ciclo de replicación del VIH-1, desde la unión inicial al receptor hasta las etapas culminantes del ensamble y maduración del virión, ofrece un punto potencial para la intervención terapéutica [ver figura 8], es probable que el diseño de fármacos antivirales eficaces se base en el mejor conocimiento de la función genética y la biología celular del VIH-1.237, 238
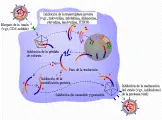
|
| Figura 8 |
| Intervención farmacológica en el ciclo viral del VIH |
Incluso cuando se identifican los agentes antivirales, ocurren in vivo numerosos ciclos de replicación viral y la subsecuente variación extensa de las secuencias del VIH-1 proporciona un gran sustrato para generar y seleccionar variantes virales que son resistentes a un determinado agente antiviral. De hecho, es probable que exista un número sustancial de variantes virales resistentes a medicamentos que ya están presentes en la persona infectada por el VIH-1 antes de que se inicie el tratamiento.45 Es seguro que puede esperarse desarrollo de resistencia a fármacos cuando se administran fármacos que bloquean en forma incompleta la replicación viral durante periodos prolongados. Estudios recientes indican que la aparición de virus mutantes resistentes a medicamentos es la principal causa del fracaso de todos los tratamientos farmacológicos existentes hasta la fecha. Los virus resistentes a medicamentos que surgen durante el tratamiento se replican menos bien, o están menos adaptados que sus contrapartes naturales, por lo que puede lograrse una carga viral más baja que la que existía antes del tratamiento [ver figura 6]. Se espera que en el futuro se identifiquen combinaciones de medicamentos que seleccionen mutaciones en la proteína blanco del VIH-1 y que comprometan la replicación viral en tal grado que los virus mutantes sean menos patógenos o, en forma ideal, incapaces de replicarse.
Además de estos retos en el desarollo de medicamentos, ha sido muy difícil probar en forma amplia los medicamentos antivirales contra el VIH-1. Los esfuerzos iniciales se limitaban a medir en forma directa la inhibición de la replicación del VIH-1 inducida por el medicamento en los pacientes infectados y usaban como criterios medidas inadecuadas, por ejemplo la concentración de linfocitos T CD4+, para vigilar la eficacia clínica.165,173,239,240 El uso de puntos clínicos finales, como el diagnóstico o muerte por SIDA, como criterios eficaces, implican necesariamente un proceso lento, y estos puntos finales ocurren solo después de un avance considerable de la infección por VIH-1. Como resultado, la evaluación de los medicamentos antivirales ha progresado con lentitud, y en la actualidad persiste gran incertidumbre sobre cuando iniciar y cuando modificar el tratamiento. La aplicación de métodos de desarrollo reciente, en especial los que miden los niveles de ARN del VIH-1 en plasma, para medir la replicaciones en pacientes infectados debe ayudar a aclarar muchas de las dudas actuales sobre la actividad y el uso óptimo de los medicamentos antivirales actualmente disponibles y acelerar las pruebas con nuevos agentes.173,189,241
La mayoría de los medicamentos que se han estudiado como inhibidores de la replicación del VIH-1 están dirigidos contra la enzima transcriptasa inversa. Los inhibidores de esta enzima pueden prevenir la diseminación del virus a las nuevas células blanco, pero no modificar la producción viral de células que portan ya los genomas provirales integrados. La zidovudina (3'-azido-3'-desoxitimidina), la didenosina (2'2'-dideoxinosina, también conocida como ddI), la zalcitabina (2',3'-dideoxicitidina, también conocida como ddC), la stavudina (2',3'-didehidro-3'-desoxitimidina, también conocida como d4T) y la lamiduvina (2',3'-didesoxi-3'-tiacitidina, también conocida como 3TC) son análogos de nucleósidos que inhiben la capacidad de la transcriptasa inversa para sintetizar copias complementarias del ADN del VIH-1 en etapas tempranas del ciclo de replicación viral. La estructura molecular de estos análogos de nucleósidos es tal, que una vez que se incorporan a la copia naciente de ARN del VIH-1, la polimerización del ADN se termina en forma prematura y no puede ya extenderse la cadena de ADN. Estos análogos de nucleósido inhiben también la actividad de la transcriptasa inversa del VIH-2. Con la zidovudina u otra monoterapia análoga en pacientes sin tratamiento previo, con frecuencia se observan reducciones en las concentraciones plasmáticas de ARN del VIH-1 de alrededor de 0.6 a 0.7 log10. Aunque estos cambios son mesurables, su magnitud indica que estos agentes bloquean la replicación in vivo del VIH-1 en forma incompleta, lo que puede explicar el modesto beneficio clínico observado con la monoterapia con analogos de nucleósidos (ver adelante). Las combinaciones de los análogos de nucleósidos, como la zidovudina y la lamivudina inhiben in vivo la replicación del VIH-1 en forma más potente, con reducciones del ARN de VIH-1 en plasma de alrededor de 1.5 log10.
Los denominados inhibidores no nucleósidos de la transcriptasa inversa (INNTI), como el nevirapine y las piridonas actúan por un mecanismo diferente, uniéndose en forma directa al complejo enzimático e inhibiendo que la enzima tome una conformación activa necesaria para la polimerización del ADN. Los INNTI son inhibidores muy específicos y no competitivos del VIH-1 y no inhiben la replicación de los otros retrovirus, incluyendo el VIH-2. Los INNTI pueden inhibir la replicación del VIH-1 en forma potente, pero solo temporal, en pacientes tratados, disminuyendo los niveles de ARN del VIH-1 de 1.6 a 2.0 log10, dos a cuatro semanas después de iniciado el tratamiento.188
Por desgracia, la monoterpia con análogos de nucleósidos o INNTI en pacientes infectados por el VIH-1 ha causado, sin excepción, la aparición de virus resistentes a medicamentos.237,242-244 La resistencia farmacológica aparece en algunas semanas después de iniciado el tratamiento con los INNTI y en meses a años después del uso de análogos de nucleósidos. La caracterización genética de los virus resistentes ha demostrado que existen mutaciones específicas en la región que codifica la transcriptasa inversa y que estas mutaciones se asocian con la resistencia observada a los fármacos individuales.237,242,245
El desarrollo de resistencia farmacológica por el VIH-1 durante el tratamiento con análogos de nucleósidos se ha estudiado en forma más amplia con la zidovudina.237,242,245 La sustitución de los aminácidos en cinco posiciones en la transcriptasa inversa del VIH-1 ha sido implicada en la resistencia a la zidovudina. Estas sustituciones se acumulan durante el tratamiento, y los niveles más altos de resistencia (reducción de hasta 100 veces en la sensibilidad in vitro) se observan en virus que tienen sustituciones en cuatro o cinco posiciones. La resistencia a la zidovudina se desarrolla con más rapidez en personas con infección avanzada, quizá porque tienen una carga viral mayor, replicación más extensa y una diversidad genética viral más grande aunada a la inmunodeficiencia progresiva. Los virus resistentes a la zidovudina también pueden trasmitirse entre los individuos.246 Estos virus resistentes suelen ser sensibles a didanocina, zalcitabina y los INNTI.237,242,245
La rápida aparición de altos niveles de resistencia con los INNTI refleja que solo se requiere la alteración de un aminoácido en la transcriptasa inversa del VIH-1 para disminuir la sensibilidad a estos agentes.242,244,247 Por lo tanto, aunque los INNTI son bien tolerados y muestran actividad antiviral detectable in vivo, el rápido desarrollo de resistencia farmacológica limita mucho su utilidad como monoterapia.
Se ha sugerido que pueden usarse combinaciones de inhibidores de la transcriptasa inversa para retrasar la aparición de virus resistentes a los agentes antivirales individuales, o que este tipo de tratamiento podría provocar la aparición de mutaciones múltiples que disminuyeran o abolieran la replicación del VIH-1 in vivo.237,242,245 Aunque se han encontrado cepas de VIH-1 que son simultáneamente resistentes a varios inhibidores de la transcriptasa inversa en pacientes tratados, estudios recientes de tratamiento combinado con zidovudina y lamivudina han identificado un sinergismo antiviral no esperado que causa reducción sostenida en los niveles de ARN del VIH-1 y elevación en el número de linfocitos T CD4+. Este efecto parece ser causado por la aparición de mutaciones de la transcriptasa inversa que confieren resistencia a la lamivudina. Estas mutaciones sirven para evitar la aparición de mutaciones que causen resistencia a la zidovudina en pacientes que no la han recibido o para revertir la resistencia en enfermos previamente tratados y que portaban mutaciones que conferían resistencia.
La zidovudina es un inhibidor eficaz de la replicación del VIH-1 in vitro.237 In vivo, la zidovudina se convierte en su forma activa trifosfato por las enzimas celulares. Los estudios clínicos iniciales sugieren que la zidovudina mejora la supervivencia y la función neurológica en personas con enfermedad avanzada (células T CD4+ <200/µl), y fue el primer medicamento antiviral autorizado por la FDA para el manejo de la infección por VIH.248 Se ha reportado que el tratamiento con zidovudina se asocia con reducción en los niveles de antígeno central p24 circulante en suero y con incrementos temporales en el número de linfocitos CD4+ en sangre periférica. En estudios más recientes la zidovudina parece detener la progresión a SIDA en personas asintomáticas con disminución del número absoluto de linfocitos T CD4+ (<500 células T CD4+/µl).249,250 El menor riesgo de progresión de la enfermedad reportado en estos estudios es transitorio, durando menos de dos años en la mayoría de los pacientes, y parece existir mayor beneficio en pacientes que tenían cuentas de células T CD4+ más altas al ingresar al estudio.251 Sin embargo, a diferencia de estos resultados, un estudio reciente controlado con placebo de zidovudina en pacientes asintomáticos infectados por VIH no encontró diferencia entre los grupos de tratamiento en relación con supervivencia o progresión a enfermedad avanzada después de un tiempo promedio de estudio de tres años.252 Como resultado, ha resultado difícil emitir recomendaciones definitivas sobre cuando iniciar el tratamiento con zidovudina.253 Los clínicos y los pacientes deben analizar en cada caso los beneficios potenciales del tratamiento con este agente y sus efectos adversos.254
La utilidad clínica de la zidovudina en el tratamiento de los individuos con infección avanzada está limitada por la inducción de anemia y neutropenia en un número significativo de pacientes.255 Sin embargo, la toxicidad de la zidovudina se ha reducido al disminuir la dosis usada en forma habitual, y en la actualidad se observan menos efectos adversos en personas con infección temprana o asintomática. El tratamiento prolongado con zidovudina puede causar una miopatía que se asocia con niveles elevados de creatina cinasa en suero y que se piensa es resultado de la inhibición inducida por medicamentos de la replicación mitocondrial del ADN.
Es importante que se ha reportado que el uso de la zidovudina en pacientes embarazadas disminuye en forma significativa la tasa de trasmisión del VIH-1 a sus hijos123: cuando se administra el medicamento a una mujer embarazada preparto y transparto y a los recién nacidos durante las primeras seis semanas de vida, la tasa de trasmisión materno-infantil se reduce en alrededor de dos terceras partes. Este estudio incluyó mujeres embarazadas con enfermedad ligeramente sintomática y ausencia o mínimo tratamiento previo con zidovudina. No se sabe si pueden lograrse resultados semejantes en mujeres en otros estadios de la enfermedad y hasta qué grado la resistencia a la zidovudina limita la eficacia de esta terapéutica.
Las evidencias preliminares señalan que la capacidad de la zidovudina para disminuir el riesgo de trasmisión del VIH-1 de la madre al infante se relaciona con el grado en el que la carga viral materna disminuye después del inicio del tratamiento. Por lo general la zidovudina es bien tolerada durante el embarazo, y no se ha asociado con estrés fetal, parto prematuro o malformaciones congénitas cuando se usa de acuerdo con las recomendaciones actuales. Los estudios futuros ayudarán a definir el momento óptimo para la administración del tratamiento antiviral con objeto de limitar la trasmisión materno-fetal y si otros medicamentos antivirales o estrategias diferentes de manejo pueden ser incluso más eficaces.
La didanosina fue el segundo medicamento autorizado por la FDA para el tratamiento de la infección por VIH.237 Debido a que la didanosina no parece ser superior a la zidovudina como tratamiento inicial, su uso fue autorizado en personas que no pueden tolerar la zidovudina o que han recibido tratamientos prolongados o ineficaces con ésta. La didanosina es un inhibidor activo de la transcripción inversa y de la replicación por VIH-1 in vitro. En las células humanas, la didanosina se metaboliza a su forma activa, 2',3'-dideoxiadenosina-5'-trifosfato (ddATP). Debido a que la ddATP tiene una vida media intracelular muy larga (>12 horas), la didanosina requiere una dosificación menos frecuente que la zidovudina para mantener niveles terapéuticos.
En los estudios de fase I de didanosina, los pacientes han presentado mejoría de la función inmunológica, aumento de peso moderado, disminución en los niveles de antígeno central p24 e incrementos temporales en la cuentas de células T CD4+.237 Los estudios subsecuentes indican que el cambio de zidovudina a didanosina puede ser benéfico en pacientes con enfermedad avanzada y tratados previamente cn zidovudina.256,257 Sin embargo, se desconoce el momento adecuado para modificar el tratamiento. Durante el tratamiento, también aparecen cepas de VIH-1 resistentes a didanosina, y algunas de éstas son resistentes también a zalcitabina.
Los principales efectos adversos de la didanosina incluyen el desarrollo de pancreatitis (que puede variar desde levemente sintomática y con incremento moderado de la amilasa hasta una enfermedad fatal) en cinco a 10 porciento de los pacientes tratados y el desarrollo de neuropatía periférica en alrededor del 15 al 30 porciento. También se reportan con frecuencia cefaleas y diarrea, y a muchos pacientes les desagrada el sabor del medicamento.
La zalcitabina, el tercer análogo de nucleósido autorizado en los Estados Unidos
para el tratamiento de la infección por VIH-1, se recomendó al inicio para usarse solo en combinación con zidovudina en pacientes adultos que tuvieran menos de 300 células T CD4+/µl y que sufrieran un deterioro clínico o inmunológico importante. Posteriormente se ha autorizado su uso como monoterapia en pacientes en los que han fracasado otros medicamentos antivirales o que no pueden tolerarlos. Desde el punto de vista molar, la zalcitabina es el más potente de los análogos de nucleósidos autorizados para inhibir la replicación del VIH-1 en cultivo de tejidos. Como en el caso de otros análogos de nucleósidos, la actividad inhibitoria de la zalcitabina depende de la conversión intracelular de una forma trifosfato activa.
Los estudios de fase I han indicado que la zalcitabina, al igual que la zidovudina y la didanosina, pueden disminuir la antigenemia p24 e incrementar en forma temporal la cuenta absoluta de linfocitos T CD4+.258 Un estudio de comparación directa de zalcitabina y zidovudina en pacientes con enfermedad avanzada por VIH-1 se terminó en forma prematura cuando ocurrieron más muertes en el grupo tratado con zalcitabina.259 Sin embargo, un trabajo en el que se combinaron los dos medicamentos reportó que los pacientes tratados con la combinación tuvieron una elevación de células T CD4+ mayor y más prolongada que los que fueron manejados solo con zidovudina.260 Aunque la combinación de zidovudina y zalcitabina no parece ser más eficaz que cada medicamento aislado en pacientes con enfermedad avanzada (cuentas de células T CD4+<150/µl), puede ser de mayor beneficio en enfermos en fase temprana (cuentas de células T CD4+ > 150/µl), sin que se haya determinado su papel preciso. Al parecer la zalcitabina es tan eficaz como la didanosina para el tratamiento de pacientes que no responden o no pueden tolerar la zidovudina.261 La principal toxicidad de la zalcitabina es el desarrollo de neuropatía periférica dolorosa.
La stavudina es el más reciente de los análogos de nucleósidos autorizado para el tratamiento de la infección por VIH-1.262,263 En la actualidad está autorizado para emplearse en adultos que no han mejorado con el tratamiento con otros agentes antivirales o que no los toleran. Los estudios preliminares han indicado que su perfil de actividad es semejante al de la zidovudina, pero puede causar incrementos temporales más importantes en la cuenta de linfocitos T CD4+. Su principal efecto tóxico es la neuropatía periférica.262,263
La lamivudina, el análogo de nucleósidos de desarrollo más reciente, parecer tener utilidad limitada cuando se usa solo por el surgimiento rápido de virus resistentes a medicamentos. Sin embargo, estudios recientes de lamivudina en combinación con zidovudina han demostrado que pueden lograrse periodos prolongados de supresión de la replicación del VIH-1 por periodos que duran seis meses o más. Aunque el beneficio clínico final de esta combinación no se ha establecido, estos resultados han despertado de nuevo el interés por los análogos de nucleósidos y por la identificación de tratamientos combinados más eficaces.
Aunque el desarrollo de agentes adicionales para inhibir la transcriptasa inversa del VIH-1 continúa en forma activa, se ha enfocado la atención al desarrollo de medios farmacológicos para interferir con otros pasos esenciales en el ciclo de replicación del VIH-1.237,238 Se espera que al dirigirse a muchos procesos del ciclo de vida del virus se logren estrategias terapéuticas más eficaces para el tratamiento del VIH-1.
La proteasa del VIH-1 proporciona un blanco atractivo para el desarrollo de medicamentos antivirales.182,264 La proteasa viral fragmenta las proteínas precursoras gag y pol del VIH-1 para formar las proteínas funcionales maduras durante o poco después de la gemación de partículas virales de las células infectadas. Por lo tanto, la ruptura por la proteasa es un paso esencial en la maduración de las partículas de VIH-1, y la mutación experimental de la proteasa causa la producción de partículas virales no infecciosas que contienen un centro intacto (gag) y precursores no separados gag-pol (transcriptasa inversa, ARNasa H e integrasa). La proteasa del VIH-1 es una aspartil proteasa que consiste en dos subunidades idénticas cuya estructura tridimensional se ha definido por análisis de cristalografía por rayos X.182,265 Nuestro conocimiento sobre la estructura de la proteasa del VIH-1, combinado con la experiencia extensa de la industria farmacéutica para desarrollar inhibidores de proteasas (como el uso muy difundido de los inhibidores de la enzima convertidora de angiotensina [ECA]), ha facilitado el desarrollo rápida de diversos agentes que inhiben en forma eficaz la función de la proteasa viral.
Los primeros inhibidores de la proteasa del VIH-1 que se desarrollaron y probaron en humanos son los análogos de sustrato con base peptídica, que son resistentes a la digestión por la proteasa del VIH-1 y que actúan estabilizando la enzima en un estado de transición inactivo. Aunque muchos de estos inhibidores son activos contra la proteasa del VIH-1, tienen escasa biodisponibilidad oral, quizá en parte por su caracter peptídico. Recientemente se han desarrollado otros inhibidores de la proteasa, incluyendo inhibidores no peptídicos, que actúan por diferentes mecanismos para inhibir la actividad catalítica. Ciertos de los inhibidores no peptídicos de la proteasa tienen actividad antiviral adecuada y buena biodisponibilidad por vía oral. Los estudios realizados han demostrado que los inhibidores de la proteasa pueden inhibir en forma potencial la replicación del VIH-1 y frustrar en forma eficaz la producción de virus infecciosos en células infectadas en forma crónica en cultivo de tejidos.237,238 También se ha demostrado que varios inhibidores de la proteasa tienen un efecto antiviral potente in vivo. Estos agentes pueden actuar en una forma aditiva o sinérgica cuando se usan con los inhibidores de la transcriptasa inversa, lo que sugiere que la combinación de medicamentos que actúan en diferentes niveles del ciclo del virus puede ser útil.237,238 Sin embargo, el desarrollo de inhibidores de la proteasa se ha complicado por su compleja y cara síntesis química, que los convierte en medicamentos demasiado costosos como para administrarlos en forma crónica.
El saquinavir se encuentra en este momento en estudios de fase III. Los estudios de fase II indican que una triple combinación del saquinavir, la zidovudina y la zalcitabina tiene mayor actividad antiviral que los tratamientos dobles o la monoterapia. Otros dos inhibidores de la proteasa, el L-735,524 (también conocido como MK-639) y el ABT-538, son potentes inhibidores de la replicación del VIH-1187,188 que han demostrado disminuir en forma significativa la carga viral y aumentar las cuentas de linfocitos T CD4+ durante periodos de seis meses o más. Se ha demostrado que el MK-639 y el ABT-538 disminuyen las concentraciones de ARN del VIH-1 en alrededor de 2.0 log10 o más en pacientes tratados, demostrando una potente inhibición in vivo de la replicación viral. Los pacientes con cuentas de linfocitos T CD4+ menores (<50 células T CD4+/µl) y una carga viral más alta sufren en forma típica una respuesta menos dramática, que se mantiene por periodos cortos.
Como en el caso de los inhibidores de la transcriptasa inversa, se desarrolla resistencia viral a los inhibidores de la proteasa en los pacientes tratados, y ésta correlaciona con la reducción del efecto antiviral.266 Por la presión selectiva impuesta por el tratamiento farmacológico, se acumulan con el tiempo mutaciones en la proteasa del VIH-1 que causan reducción progresiva en la sensibilidad al medicamento y una resistencia cruzada a otros inhibidores de la proteasa. En la actualidad se ha determinado la estructura cristalizada de varias proteasas obtenidas de virus resistentes a medicamentos, y parece ser que pronto se definirá el mecanismo de resistencia viral a los inhibidores de la proteasa. Se espera que los nuevos conocimientos permitan establecer estrategias para superar la resistencia a estos agentes, de modo que puedan usarse con eficacia en el tratamiento de la infección por VIH-1.266
Las proteínas tat y rev del VIH-1, que son indispensables para la replicación viral, proporcionan un blanco importante para el desarrollo de medicamentos antivirales. Sin embargo, la elaboración de inhibidores de estas proteínas requerirá una mayor comprensión de sus mecanismos de acción. Las áreas activas de investigación se enfocan a determinar la base estructural de la interacción de estas proteínas virales reguladoras con sus ARN blancos y a la identificación de proteínas celulares que interactúan con las proteínas tat y rev y ayudan a mediar sus actividades.
Por lo menos se ha identificado un inhibidor de la proteína tat por medio de métodos de escrutinio de fármacos.267 El agente, conocido como Ro24-7429, es un miembro de la familia de las benzodiacepinas que actúa a través de un mecanismo no bien definido para inhibir la función de la proteína tat y la replicación del VIH-1 en el cultivo tisular. Por desgracia, los estudios de fase I de este compuesto en los pacientes infectados por SIDA han demostrado una toxicidad inaceptable y ninguna evidencia de efecto antiviral. Aunque los experimentos clínicos preliminares con este inhibidor han sido desalentadoras, la inhibición de la función de la proteína tat sigue siendo un blanco terapéutico interesante.
La interacción entre la glucoproteína gp 120 env del VIH-1 y el receptor celular CD4 es un paso indispensable para iniciar la infección viral. La región precisa de la molécula CD4 que se fija a la gp120 se ha localizado en un dominio de la porción extracelular del CD4 que porta una secuencia homóloga con las regiones variables de las inmunoglobulinas. Se han preparado versiones de ingeniería genética del gen CD4 en las que se deleciona la región responsable de la inserción de membrana, originando la secreción de una proteína conocida como CD4 soluble. En los estudios iniciales en cultivo de tejidos que han usado cepas de VIH-1 de laboratorio, se encontró que el CD4 soluble se une con alta afinidad al gp120 en la superficie de la partículas del virus VIH-1, evitando la unión viral a los receptores de la superficie celular, e induce cambios conformacionales irreversibles en la proteína env, que convierten a la partícula viral en no infecciosa. Por desgracia, los resultados de varios estudios clínicos con CD4 soluble y sus derivados no han mostrado evidencia de beneficio clínico, ni efecto sobre los marcadores de replicación viral en los pacientes infectados.237,238 En un principio no se conocían los motivos de la falta de eficacia de esta estrategia promisoria, pero se ha encontrado que las principales cepas de VIH-1 aisladas en la clínica son mucho menos susceptibles a la inhibición por CD4 soluble que las cepas de laboratorio.198,268,269 Aunque este intento inicial para inhibir la interacción virus-receptor fue desalentador, la interacción gp120-CD4 sigue siendo un blanco atractivo para el diseño de fármacos. La definición reciente de la estructura tridimensional de la región del CD4 que fija a la gp 120 ayudará en estos esfuerzos.270,271 Por desgracia, la estructura terciaria del gp120 ha demostrado ser fugaz.
Además del desarrollo de agentes farmacológicos para interferir con la replicación del VIH-1, se ha intentado aumentar la respuesta inmunológica antiviral de los individuos infectados. Un enfoque para esto, conocido como vacunación terapéutica, incluye la inmunización de los pacientes infectados con antígenos del VIH-1 recombinantes (v.gr., gp120) o preparaciones de virus inactivos. Los estudios de inmunización preliminar usaron una versión recombinante de la proteína env del VIH-1, y sugirieron que puede despertarse una respuesta inmune antiviral de significado aún incierto.272 Sin embargo, estudios más recientes no han podido demostrar evidencia de disminución en la carga viral o retraso en la progresión clínica. Un reporte reciente sobre el uso de una vacuna inactivada del VIH-1 con depleción de la proteína env afirma que esta vacuna causa un ligero retraso en la pérdida de células T CD4+.273 Sin embargo, se requieren estudios adicionales que confirmen y amplíen esta observación para establecer el beneficio de esta terapia.
También se están explorando agentes naturales conocidos como modificadores de la respuesta biológica como medios potenciales para limitar la replicación del VIH-1 o reconstituir la función inmunológica deteriorada. El interferón alfa inhibe en forma parcial la replicación in vitro del VIH-1 y se ha reportado que disminuye los niveles de antígeno central p24 y aumenta en forma temporal las cuentas absolutas de linfocitos T CD4+ en las peronas infectadas por VIH-1.274.275 Los resultados preliminares indican que el tratamiento intermitente con IL-2 puede aumentar las cuentas de células T CD4+ en algunos pacientes en los que estos niveles están solo ligeramente disminuidos.276 Sin embargo, los incrementos temporales en la viremia plasmática después de la infusión de IL-2 tienen un significado incierto, y no se sabe si el aumento secundario en las células T CD4+ causa mejoría de la función inmunológica. La función de la compleja red de citocinas in vivo se comprende poco aún, y el éxito de los modificadores de la respuesta biológica para el tratamiento de la infección por VIH-1 requerirá mayor conocimiento sobre la complejidad de esta red de citocinas in vivo.
Ninguno de los medicamentos disponibles en la actualidad contra el SIDA ha demostrado detener en forma eficaz la replicación viral in vivo o evitar el desarrollo subsecuente de la enfermedad en personas infectadas por el VIH-1. La identificación de agentes de este tipo parece una tarea muy difícil. Sin embargo, los intentos iniciales por tratar las infecciones por retrovirus humanos han proporcionado mucha información para los esfuerzos futuros. Nuestro mayor conocimiento de la historia natural de la infección por el VIH-1 facilitará el desarrollo y aplicación de intervenciones terapéuticas contra esta enfermedad.
Retrovirus y otras enfermedades humanas
Una área de investigación activa, aunque controvertida, se orienta a la posibilidad de que existan nuevos retrovirus humanos no descubiertos que sean los agentes etiológicos de enfermedades humanas consideradas idiopáticas. Algunos estudios indican que parece existir una relación entre el HTLV-1 o retrovirus relacionados y la esclerosis múltiple.277-279 Se ha observado la presencia de anticuerpos reactivos contra la proteína gag del HTLV-I en varios pacientes con esclerosis múltiple. Estudios que utilizan la reacción en cadena de la polimerasa han identificado secuencias genómicas relacionadas con HTLV-I en un porcentaje significativo de individuos con esclerosis múltiple, y tales secuencias son muy raras en las poblaciones controles. La importancia de estos estudios para la identificación del agente etiológico de la esclerosis múltiple es muy incierto, y se espera un mayor análisis de dichas observaciones.152 Se ha propuesto la etiología retroviral para la enfermedad de Kawasaki,280 pero evaluaciones posteriores no han encontrado apoyo para esta suposición.281, 282 De igual manera, una investigación de RCP en pacientes con síndrome de fatiga crónica reportó que en muchos casos podían demostrarse secuencias del HTLV-II.283 Sin embargo, otros estudios no han podido corroborar estos resultados.228
Recientemente se ha dado gran publicidad y se originó gran preocupación por el reporte de un retrovirus no detectado por las pruebas serológicas para VIH-1, VIH-2, HTLV-I y HTLV-II que podría asociarse con el síndrome recién definido conocido como linfopenia T CD4+ idiopática (LCI), que se reportó por vez primera en 1992 a 1993.284,285 La LCI es un síndrome muy raro caracterizado por una cuenta absoluta de células T CD4+ menor de 300 células/µl o de menos del 20 porciento del total de las células T en más de una ocasión, con reacciones serológicas negativas para los retrovirus de las familias VIH y HTLV y sin una causa que explique la inmunodeficiencia.284 A pesar de la atención brindada a la descripción inicial de la LCI, las investigaciones subsecuentes han indicado con claridad que el síndrome no se asocia con una etiología retroviral y, de hecho, no es un padecimiento infeccioso.284,285 Además, la LCI no parece ser una entidad clínica de reciente aparición, sino que se reconoce más en la actualidad por la disponibilidad para medir el número de células T CD4+ en pacientes con evidencia clínica de inmunodeficiencia.
Cualquiera que sea la dirección que tomen las investigaciones futuras en las infecciones retrovirales, la experiencia derivada del estudio del HTLV-I, HTLV-II, VIH-1 y VIH-2 ha mejorado en mucho el conocimiento sobre el potencial patógeno de los retrovirus humanos, y proporcionado herramientas para buscar patógenos retrovirales adicionales.
Direcciones futuras en la investigacion de retrovirus
Desde 1980, cuando los retrovirus humanos eran considerados como entidades hipotéticas y estudiados por sólo unos cuantos laboratorios científicos, estos virus se han identificado como los agentes etiológicos de importantes enfermedades humanas, y la investigación sobre los mismos es abrumadora. Aunque las modalidades actuales de tratamiento alivian muy poco la gravedad de la enfermedad humana inducida por retrovirus, nuevos estudios basados en la biología característica de esta clase de virus y guiados por el mayor conocimiento de los blancos moleculares para la intervención farmacológica se encuentran en el horizonte. Además, el mayor conocimiento sobre la historia natural y la patogenia de la infección con retrovirus humanos puede facilitar el desarrollo y aplicación de las intervenciones para prevenir o tratar las enfermedades asociadas. El pesimismo respecto al pronóstico actual de los pacientes con LLTA y el SIDA puede ser compensado por una apreciación esperanzada de los grandes adelantos logrados en el conocimiento de los retrovirus patógenos en un periodo tan corto.
- Temin HM: Origin and general nature of retroviruses. The Retroviridae, Vol1. Levy JA, Ed. Plenum Press, New York, 1992, p 1
- Poiesz BJ, Ruscetti FW, Gazdar AF,et al: Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci USA 77:7415, 1980
- Sugamura K, Hinuma Y: Human retroviruses: HTLV-1 and HTLV-II. The Retroviridae, Vol 2. Levy JA, Ed. Plenum Press, New York, 1993, p 399
- Gessain A, Gout O: Chronic myelopathy associated with human T-Iymphotropic virus type I (HTLV-I). Ann Intem Med 117:933, 1992
- Höllsberg P, Hafler DA: Pathogenesis of diseases induced by human lymphotropic virus type I infection. N Engl J Med 328:1173, 1993
- Kalyanaraman VS, Sarngadharan MG, Robert-Guroff M, et al: A new subtype of human T-cell leukemia virus (HTLV-II) associated with a T-cell variant of hairy cell leukemia. Science 218:571, 1982
- Gallo RC, Salahuddin SZ, Popovic M, et al: Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AlDS and at risk for AIDS. Science 224:500, 1984
- Barre-Sinoussi F, Chermann JC, Rey F, et al: Isolation of a T lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220:868, 1983
- Levy JA, Hoffman AD, Kramer SM, et al: Isolation of lymphocytopathic retroviruses from San Francisco patients with AIDS. Science 225:840, 1984
- Kanki PJ, Barin F, M'Boup S, et.al: New human T-Iymphotropic retrovirus related to simian T-Iymphotropic virus type III (STLV-IIIAGM). Science 232:238, 1986
- Kanki PJ, M'Boup S, Ricard D, et al: Human T-Iymphotropic virus type.4 and the human immunodeficiency virus in West Africa. Science 236:827, 1987
- Clavel F,Guetard D, Brun-Vézinet F, et al: Isolation of a new human retrovirus from West African patients with AIDS. Science 233:343, 1986
- Clavel F, Mansinho K, Chamaret S, et al: Human immunodefidency virus type 2 infection associated with AIDS in West Africa. N Engl J Med 316:1180, 1987
- Coffin JM: Structure and classification of retroviruses. The Retroviridae, Vol 1. Levy JA, Ed. Plenum Press, New York, 1992, p 19
- Miyoshi I, Fujishita M, Taguchi H, et al: Natural infection in nonhuman primates with adult T-cell leukemia virus or a closely related agent. Int J Cancer 32:333, 1983
- Hunsmann G, Schneider J, Schmitt J, et al: Detection of serum antibodies to adult T-cell leukemia virus in non-human primates and in people from Africa. Int J Cancer 32:329, 1983
- Homma T, Kanki PJ, King NW Jr, et al: Lymphoma in macaques: association with virus of human T lymphotrophic family. Science 225:716, 1984
- Desrosiers RC: The simian immunodeficiency viruses. Annu Rev ImmunoI 8:557, 1990
- Gao F, Yue L, White AT, et al: Human infection by genetically diverse SIVSM-related HIV-2 in West Africa. Nature 358:495, 1992
- Johnson PR, Myers G, Hirsch VM: Genetic diversity and phylogeny of nonhuman primate lentiviruses. Annual Review of AIDS Research. Koff W, Ed. Marcel Dekker, NewYork, 1991, p 47
- Huet T, Cheynier R, Meyerhans A, et al: Genetic organization of a chimpanzee lentivirus related to HIV-1. Nature 345:356, 1990
- Peeters M, Fransen K, Delaporte E, et al: Isolation and characterization of a new chimpanzee lentivirus (simian immunodeficiency virus isolate cpz-ant) from a wild-captured chimpanzee. AIDS 6:447, 1992
- Weiss RA: Cellular receptors and viral glycoproteins involved in retrovirus entry. The Retroviridae, Vol 2. Levy JA, Ed. Plenum Press, New York, 1993, p 1
- Cullen BR: An introduction to human retroviruses. Human Retroviruses. Cullen BR, Ed. Oxford University Press, New York, 1993, p 1
- Greene WC: The molecular biology of human immunodeficiency virus type 1 infection. N Engl J Med 324:308, 1991
- Smith MR, Greene WC: Molecular biology of the type I human T-cell leukemia virus (HTLV-I) and adult T-cell leukemia. J Clin Invest 87:761, 1991
- Green PL, Chen ISY: Molecular features of the human T-cell leukemia virus: mechanisms of transformation and leukemogenicity. The Retroviridae, Vo13. Levy JA, Ed. Plenum Press, New York, 1994, p 277
- Parslow TG: Post-transcriptional regulation of human retroviral gene expression. Human Retroviruses. Cullen BR, Ed. Oxford University Press, New York, 1993, p 101
- Kinoshita T, Shimoyama M, Tobinai K, et al: Detection of mRNA for the tax 1/rex 1 gene of human T-cell leukemia virus type I in fresh peripheral blood mononuclear cells of adult T-cell leukemia patients and viral carriers by using the polymerase chain reaction. Proc Natl Acad Sci USA 86:5620,1989
- Paine E, Garcia J, Philpott TC, et al: Limited sequence variation in human T-lymphotropic virus type 1 isolates from North American and African patients. Virology 182:111, 1991
- Gallo R, Wong-Staal F, Montagnier L, et al: HIV / HTLV gene nomenclature (letter). Nature 333:504, 1988
- Cullen BR: Does HIV-l Tat induce a change in viral initiation rights? Cell 73:417, 1993
- Girard M, Kieny M-P, Pinter A, et al: Immunization of chimpanzees confers protection against challenge with human immunodeficiency virus. Proc Natl Acad Sci USA 88:542, 1991
- Human Retroviruses and AIDS 1993: A Compilation and Analysis of Nucleic Acid and Amino Acid Sequences, Vo1s 1 and 2. Myers G, Korber B, Wain-Hobson S, et al, Eds. Los Alamos National Laboratory, Los Alamos, New Mexico, 1993
- Gibbs JS, Desrosiers RC: Auxiliary proteins of the primate immunodeficiency viruses. Human Retroviruses. Cullen BR, Ed. Oxford University Press, New York, 1993, p 137
- Young JAT: The replication cycle of HIV-1. The AIDS Knowledge Base: A Textbook on HIV Disease from the University of California, San Francisco, and the San Francisco General Hospital, 2nd ed. Cohen PT, Sande MA, Volberding PA, Eds. Little, Brown & Co, Boston, 1994, p 3.1-1
- Frankel AD: Activation of HIV transcription by Tat. Curr Opin Genet Dev 2:293, 1992
- Cullen BR: The role of Nef in the replication cycle of the human and simian immunodeficiency viruses. Virology 205:1, 1994
- Miller MD, Feinberg MB, Greene WC: The HIV-1 nef gene acts as a positive viral infectivity factor. Trends in Microbiology 2:294, 1994
- Kirchhoff F, Greenough TC, Brettler DB, et al: Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV -1 infection. N Engl J Med 332:228, 1995
- Pantaleo G, Graziosi C, Fauci AS: New concepts in the immunopathogenesis of human immunodeficiency virus infection. N Engl J Med 328:327, 1993
- Pantaleo G, Graziosi C, Demarest JF, et al: HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature 362:355, 1993
- Piatak M Jr, Saag MS, Yang LC, et al: High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science 259:1749, 1993
- Weiss RA: How does HIV cause AIDS? Science 260:1273,1993
- Coffin JM: HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267:483, 1995
- Embretson J, Zupancic M, Ribas JL, et al: Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature 362:359, 1993
- Fauci AS: Multifactorial nature of human immunodeficiency virus disease: implications for therapy. Science 262:1011, 1993
- Baeuerle P A, Henkel T: Function and activation of NF-kappa B in the immune system. Annu Rev ImmunoI 12:141, 1994
- Gaynor R: Cellular transcription factors involved in the regulation of HIV-1 gene expression. AIDS 6:347, 1992
- Nabel GJ: The role of cellular transcription factors in the regulation of human immunodeficiency virus gene expression. Human Retroviruses. Cullen BR, Ed. Oxford University Press, New York, 1993, p 49
- Bishop JM: The molecular genetics of cancer. Science 235:305, 1987
- Fan H: Retroviruses and their role in cancer. The Retroviridae, Vol 3. Levy JA, Ed. Plenum Press, New York, 1994, p 313
- Teich N, Wyke J, Mak T, et al: Pathogenesis of retrovirus-induced disease. RNA Tumor Viruses: Molecular Biology of Tumor Viruses, 2nd ed. Weiss R, Teich N, Varmus H,et al, Eds.Cold Spring Harbor Laboratory, Cold spring Harbor, New York, 1984, p 785
- Minami Y, Kono T, Miyazaki T, et al: The IL-2 receptor complex: its structure, function, and target genes. Annu Rev Immunol 11:245, 1993
- Taniguchi T, Minami Y: The IL-2/IL-2 receptor system: a current overview. CeIl 73:5, 1993
- Robert-Guroff M, Nakao Y, Notake K, et al: Natural antibodies to human retrovirus HTLV in a cluster of Japanese patients with adult T cell leukemia. Science 215:975, 1982
- Yoshida M, Seiki M, Yamaguchi K, et al: Monoclonal integration of human T-cell leukemia provirus in all primary tumors of adult T-cell leukemia suggests causative role of human T-cell leukemia virus in the disease. Proc Natl Acad Sci USA 81:2534, 1984
- Popovic M, Sarin Ps, Robert-Guroff M, et al: Isolation and transmission of human retrovirus (human t-cell leukemia virus). Science 219:856, 1983
- Blayney DW, Jaffe ES, Fisher RI, et al: The human T-cell leukemia / lymphoma virus, lymphoma, lytic bone lesions, and hypercalcemia. Ann Intem Med 98:144, 1983
- Bunn PA Jr, Schechter GP, Jaffe E, et al: Clinical course of retrovirus-associated adult T-cell lymphoma in the United States. N Engl J Med 309:257, 1983
- Yamaguchi K: Human T-lymphotropic virus type 1 in Japan. Lancet 343:213, 1994
- Popovic M, Lange-Wantzin G, Sarin PS, et al: Transformation of human umbilical cord blood T cells by human T-cell leukemia / lymphoma virus. Proc Natl Acad Sci USA 80:5402, 1983
- Miyoshi I, Kubonishi I, Yoshimoto S, et al: Type C virus particles in a cord T-cell line derived by co-cultivating normal human cord leukocytes and human leukaemic T cells. Nature 294:770, 1981
- Centers for Disease Control and Prevention and the USPHS Working Group: Guidelines for counseling persons infected with human T-Iymphotropic virus type I (HTLV-I) and type II (HTLV-II). Ann Intern Med 118:448, 1993
- Hall WW: Human T cell lymphotropic virus type I and cutaneous T cell leukernia / lymphoma. J Exp Med 180:1581, 1994
- Constantine NT: Serologic tests for the retroviruses: approaching a decade of evolution. AIDS 7:1, 1993
- Khabbaz RF, Onorato IM, Cannon RO, et al: Seroprevalence of HTLV-I and HTLV-II among intravenous drug users and persons in clinics for sexually transmitted diseases. N Engl J Med 326:375, 1992
- Feigal E, Murphy E, Vranizan K, et al: Human T cell lymphotropic virus types I and II in intravenous drug users in San Francisco: risk factors associated with seropositivity. J Infect Dis 164:36, 1991
- Murphy EL, Figueroa JP, Gibbs WN, et al: Sexual transmission of human T-Iymphotropic virus type I (HTLV-I). Ann Intern Med 111:555, 1989
- Lee HH, Swanson P, Rosenblatt JD, et al: Relative prevalence and risk factors of HTLV-I and HTLV-II infection in US blood donors. Lancet 337:1435, 1991
- Williams AE, Fang CT, Slamon DJ, et al: Seroprevalence and epidemiological correlates of HTLV-I infection in US blood donors. Science 240:643, 1988
- Gebretsadik T, Murphy EL: Counseling and medical evaluation of HTLV-I- and HTLV-II-infected patients. AIDS Clinical Review, 1993/1994. Volberding P, Jacobson MA, Eds. Marcel Dekker, New York, 1993, p 19
- Murphy EL, Hanchard B, Figueroa JP, et al: Modelling the risk of adult T-cell leukemia / lyrnphoma in persons infected with human T-Iymphotropic virus type I. Int J Cancer 43:250, 1989
- Kaplan JE, Osame M, Kubota H, et al: The risk of development of HTLV-I-associated myelopathy / tropical spastic paraparesis among persons infected with HTLV-I. J Acquir Immune Defic Syndr 3:1096, 1990
- Rosenblatt JD, Golde DW, Wachsman W, et al: A second isolate of HTLV-II associated with atypical hairy-cell leukemia. N Engl J Med 315:372, 1986
- Hjelle B, Mills R, Swenson S, et al: Incidence of hairy cell leukemia, mycosis fungoides, and chronic lymphocytic leukemia in first known HTLV-II-endemic population. J Infect Dis 163:435, 1991
- Hjelle B, Appenzeller O, Mills R, et al: Chronic neurodegenerative disease associated with HTLV-II infection. Lancet 339:645, 1992
- Ijichi S, Ramundo MB, Takahashi H, et al: In vivo cellular tropism of human T cell leukemia virus type II (HTLV-II). J Exp Med 176:293, 1992
- Lee H, Swanson P, Shorty VS, et al: High rate of HTLV-II infection in seropositive i.v. drug abusers in New Orleans. Science 244:471, 1989
- Weiss SH: The evolving epidemiology of human T lymphotropic virus type II. J Infect Dis 169:1080, 1994
- Biggar RJ, Buskell-Bales Z, Yakshe PN, et al: Antibody to human retroviruses among drug users in three East Coast American cities, 1972-1976. J Infect Dis 163:57, 1991
- Rios M, Khabbaz RF, Kaplan JE, et al: Transmission of human T cell lymphotropic virus (HTLV) type II by transfusion of HTLV-I-screened blood products. J Infect Dis 170:206, 1994
- Lal RB, Owen SM, Segurado AAC, et al: Mother-to-child transmission of human T-lymphotropic virus type II (HTL V-II). Ann Intern Med 120:300, 1994
- Watanabe T, Yamaguchi K, Takatsuki K, et al: Constitutive expression of parathyroid hormone-related protein gene in human T cell leukemia virus type 1 (HTLV-1) carriers and adult T cell leukemia patients that can be trans-activated by HTLV-1 tax gene. J Exp Med 172:759, 1990
- Grossman WJ, Kimata JT, Wong F-H, et al: Developmentof leukemia in mice transgenic for the tax gene of human T-cell leukemia virus type I. Proc Natl Acad Sci USA 92:1057, 1995
- Grassmann R, Berchtold S, Radant I, et al: Role of human T-cell leukemia virus type 1 X region proteins in immortalization of primary human lymphocytes in culture. J Virol 66:4570, 1992
- Hall WW, Liu CR, Schneewind O, et al: Deleted HTLV-I provirus in blood and cutaneous lesions of patients with mycosis fungoides. Science 253:317, 1991
- Shimoyama M, Members of the Lymphoma Study Group: Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-Iymphoma: a report from the Lymphoma Study Group (1984-87). Br J HaematoI 79:428, 1991
- Waldmann TA: Human T-cell lymphotropic virus type I-associated adult T-cell leukemia. JAMA 273:735, 1995
- Pneumocystis pneumonia-Los Angeles. MMWR 30:250, 1981
- Gabuzda DH, Lawrence K, Langhoff E, et al: Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J ViroI 66:6489, 1992
- Gottlieb MS, Schroff R, Schanker HM, et al: Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men: evidence of a new acquired cellular immunodeficiency. N Engl J Med 305:1425, 1981
- Hoover DR, Saah AJ, Bacellar H, et al: Clinical manifestations of AIDS in the era of Pneumocystis prophylaxis: Multicenter AIDS Cohort Study. N Engl J Med 329:1922, 1993
- Revision of the CDC surveillance case definition for acquired immunodeficiency syndrome: Council of State and Territorial Epidemiologists; AIDS Program, Center for Infectious Diseases. MMWR 36(suppl1):lS, 1987
- 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR 41(RR-17):1, 1992
- Buehler JW, Ward JW: A new definition for AIDS surveillance (editorial). Ann Intem Med 118:390, 1993
- American College of Physicians and Infectious Diseases Society of America: Human immunodeficiency virus (HIV) infection. Ann Intern Med 120:310, 1994
- Wilber JC: HIV-antibody testing: methodology. The AIDS Knowledge Base: A Textbook on HIV Disease from the University of California, San Francisco, and the San Francisco General Hospital, 2nd ed. Cohen PT, Sande MA, Volberding PA, Eds. Little, Brown & Co, Boston, 1994, p 2.2-1
- Jackson JB, Kwok SY, Sninsky JJ, et al: Human immunodeficiency virus type 1 detected in all seropositive symptomatic and asymptomatic individuals. J Clin Microbiol 28:16, 1990
- Ou CY, Kwok S, Mitchell SW, et al: DNA amplification for direct detection of HIV-1 in DNA of peripheral blood mononuclear cells. Science 239:295, 1988
- Imagawa DT, Lee MH, Wolinsky SM, et al: Human immunodeficiency virus type 1 infection in homosexual men who remain seronegative for prolonged periods. N Engl J Med 320:1458, 1989
- Buehler JW, Petersen LR, Jaffe HW: Current trends in the epidemiology of HIV / AIDS. The Medical Management of AIDS, 4th ed. Sande AM, Volberding PA, Eds. WB Saunders Co, Philadelphia, 1995, p 3
- Farzadegan H, Polis MA, Wolinsky SM, et al: Loss of human immunodeficiency virus type 1 (HIV-1) antibodies with evidence of viral infection in asymptomatic homosexual men: a report from the Multicenter AIDS Cohort Study. Ann Intem Med 108:785, 1988
- Roy MJ, Damato JJ, Burke DS: Absence of true seroreversion of HIV-1 antibody in seroreactive individuals. JAMA 269:2876, 1993
- Merson MH: Slowing the spread of HIV: agenda for the 1990s. Science 260:1266, 1993
- The AIDS Knowledge Base: A Textbook on HIV Disease from the University of California, San Francisco, and the San Francisco General Hospital, 2nd ed. Cohen PT, Sande MA, Volberding PA, Eds. Little, Brown & Co, Boston, 1994
- Projections of the number of persons diagnosed with AIDS and the number of immunosuppressed HIV-infected persons-United States, 1992-1994. MMWR42:17, 1993
- Update: mortality attributable to HIV infection among persons aged 25-44 years-United States, 1991 and 1992. MMWR 42:869, 1993
- Centers for Disease Control and Prevention: Guidelines for preventing transmission of human immunodeficiency virus through transplantation of human tissue and organs. MMWR 43(RR-8):1, 1994
- Kumar P, Pearson JE, Martin DH, et al: Transmission of human immunodeficiency virus by transplantation of a renal allograft, with development of the acquired immunodeficiency syndrome. Ann Intern Med 106:244, 1987
- de Vincenzi I: A longitudinal study of human immunodeficiency virus transmission by heterosexual partners: European study Group on Heterosexual Transmission of HIV- N Engl J Med 331:341, 1994
- European Collaborative study: Children born to women with HIV-1 infection: natural history and risk of transmission. Lancet 337:253, 1991
- Blanche S, Rouzioux C, Guihard Moscato M-L, et al: A prospective study of infants born to women seropositive for human immunodeficiency virus type 1- N Engl J Med 320:1643, 1989
- Pizzo PA, Butler KM: In the vertical transmission of HIV, timing may be everything (editorial). N Engl J Med 325:652, 1991
- Burgard M, Mayaux MJ, Blanche S, et al: The use of viral culture and p24 antigen testing to diagnose human immunodeficiency virus infection in neonates: The HIV Infection in Newborns French Collaborative Study Group. N Engl J Med 327:1192, 1992
- Miles SA, Balden E, Magpantay L, et al: Rapid serologic testing with immune-complex-dissociated HIV p24 antigen for early detection of HIV infection in neonates: Southern California Pediatric AIDS Consortium. N Engl J Med 328:297, 1993
- Rogers MF, Ou C-Y, Rayfield M, et al: Use of the polymerase chain reaction for early detection of the proviral sequences of human immunodeficiency virus in infants born to seropositive mothers. N Engl J Med 320:1649, 1989
- Goedert JJ, Duliege AM,Amos CI,et al: High risk of HIV-1 infection for first-born twins: The International Registry of HIV-Exposed Twins. Lancet 338:1471, 1991
- Blanche S, Mayaux MJ, Rouzioux C, et al: Relation of the course of HIV infection in children to the severity of the disease in their mothers at delivery. N Engl J Med 330:308, 1994
- Staprans SI, Feinberg MB: Natural history and immunopathogenesis of HIV-1 disease. The AIDS Knowledge Base: A Textbook on HIV Disease from the University of California, San Francisco, and the San Francisco General Hospital, 2nd de. Cohen PT, sande MA, Volberding P A, Eds. Little, Brown & Co, Boston, 1994, p 3.4-1
- St Louis ME, Kamenga M, Brown C, et al: Risk for perinatal HIV-1 transmission according to maternal immunologic, virologic, and placental factors. JAMA 269:2853, 1993
- Weiser B, Nachman S, Tropper P, et al: Quantitation of human immunodeficiency virus type 1 during pregnancy: relationship of viral titer to mother-to-child transmission and stability of viral load. Proc Natl Acad Sci USA 91:8037, 1994
- Connor EM, Sperling RS, Gelber R, et al: Reduction of maternal-infant transmission of human immunodeficiency virus type 1 with zidovudine treatment. N Engl J Med 331:1173, 1994
- Rogers MF, Jaffe HW: Reducing the risk of maternal-infant transmission of HIV: a door is opened. N Engl J Med 331:1222, 1994
- Bayer R: Ethical challenges posed by zidovudine treatment to reduce vertical transmission of HIV. N Engl J Med 331:1223, 1994
- Cohen ND, Muñoz A, Reitz BA, et al: Transmission of retroviruses by transfusion of screened blood in patients undergoing cardiac surgery. N Engl J Med 320:1172, 1989
- Menitove JE: The decreasing risk of transfusion-associated AIDS. N Engl J Med 321:966, 1989
- Heymann SJ, Brewer TF, Fineberg HV , et al: How safe is safe enough? New infections and the US blood supply (editorial). Ann Intern Med 117:612, 1992
- Nelson KE, Donahue JG, Muñoz A, et al: Transmission of retroviruses from seronegative donors by transfusion during cardiac surgery: a multicenter study of HIV-1 and HTLV-I / II infections. Ann Intern Med 117:554, 1992
- Alter HJ, Epstein JS, Swenson SG, et al: Prevalence of human immunodeficiency virus type 1 p24 antigen in US blood donors-an assessment of the efficacy of testing in donor screening- N Engl J Med 323:1312, 1990
- Busch MP, Taylor PE, Lenes BA, et al: Screening of selected male blood donors for p24 antigen of human immunodeficiency virus type 1. N Engl J Med 323:1308, 1990
- Busch MP, Eble BE, Khayam-Bashi H, et al: Evaluation of screened blood donations for human immunodeficiency virus type 1 infection by culture and DNA amplification of pooled cells. N Engl J Med 325:1, 1991
- Becker CE, Cone JE, Gerberding J: Occupational infection with human immunodeficiency virus (HIV): risks and risk reduction Ann Intern Med 110:653, 1989
- Henderson DK, Fahey BJ, Willy M, et al: Risk for occupational transmission of human immunodeficiency virus type 1 (HIV-1) associated with clinical exposures: a prospective evaluation. Ann Intem Med 113:740, 1990
- Gerberding J: Is antiretroviral treatment after percutaneous HlV exposure justified? (editorial). Ann Intern Med 118:979, 1993
- Tindall B, Gaines H, Imrie A, et al: Zidovudine in the management of primary HlV-1 infection. AIDS 5:477, 1991
- Tokars JI, Marcus R, Culver DH,etal: Surveillance of HIV infection and zidovudine use among health care workers after occupational exposure to HIV-infected blood. Ann Intern Med 118:913, 1993
- Fahey BJ, Koziol DE, Banks SM, et al: Frequency of nonparenteral occupational exposures to blood and body fluids before and after universal precautions training. Am J Med 90:145, 1991
- Brennan TA: Transmission of the human immunodeficiency virus in the health care setting-time for action. N Engl J Med 324:1504, 1991
- Ou C-Y, Ciesielski CA, Myers G, et al: Molecular epidemiology of HIV transmission in a dental practice. Science 256:1165, 1992
- Ciesielski C, Marianos D, Ou C-Y, et al: Transmission of human immunodeficiency virus in a dental practice. Ann Intern Med 116:798, 1992
| 141a | Owens DK, Harris RA, Scott PM, et al: Screening surgeons
for HlV infection: a cost-effectiveness analysis. Ann Intern Med 122:641,
1995
|
| 141b | Robert LM, Chamberland ME, Cleveland JL, et al: Investigations
of patients of health care workers infected with HlV: the Centers for Disease
Control and Prevention database. Ann Intern Med 122:653, 1995
|
- Klein RS: Universal precautions for preventing occupational exposures to human immunodeficiency virus type 1 (editorial). Am J Med 90:141, 1991
- Phair JP: Estimating prognosis in HIV-1 infection (editorial). Ann Intern Med 118:742, 1993
- Polis MA, Masur H: Predicting the progression to AIDS (editorial). Am J Med 89:701, 1990
- De Gruttola V, Wulfsohn M, Fischl MA, et al: Modeling the relationship between survival and CD4 lymphocytes in patients with AIDS and AIDS-related complex. J Acquir Immune Defic Syndr 6:359, 1993
- Stein DS, Korvick JA, Vermund SH: CD4+ lymphocyte cell enumeration for prediction of clinical course of human immunodeficiency virus disease: a review. J Infect Dis 165:352, 1992
- Cao Y, Qin L, Zhang L, et al: Virologic and immunologic characterization of long-term survivors of human immunodeficiency virus type 1 infection. N Engl J Med 332:201, 1995
- Pantaleo G, Menzo S, Vaccarezza M, et al: Studies in subjects with long-term nonprogressive human immunodeficiency virus infection. N Engl J Med 332:209, 1995
- Baltimore D: Lessons from people with nonprogressive HIV infection (editorial). N Engl J Med 332:259, 1995
- Schrager LK, Young JM, Fowler MG, et al: Long-term survivors of HIV-1 infection: definitions and research challenges. AIDS 8(suppl 1):S95, 1994
- Scott GB, Hutto C, Makuch RW, et al: Survival in children with perinatally acquired human immunodeficiency virus type 1 infection. N Engl J Med 321:1791, 1989
- Duliege AM, Messiah A, Blanche S, et al: Natural history of human immunodeficiency virus type 1 infection in children: prognostic value of laboratory tests on the bimodal progression of the disease. Pediatr Infect Dis J 11:630, 1992
- Kaslow RA, Mann DL: The role of the major histocompatibility complex in human immunodeficiency virus infection-ever more complex? (editorial). J Infect Dis 169:1332, 1994
- Pape JW, Jean SS, Ho JL, et al: Effect of isoniazid prophylaxis on incidence of active tuberculosis and progression of HIV infection. Lancet 342:268, 1993
- Stein DS, Graham NM, Park LP, et al: The effect of the interaction of acyclovir with zidovudine on progression to AIDS and survival: analysis of data in the Multicenter AIDS Cohort Study. Ann Intern Med 121:100, 1994
- Markovitz DM: Infection with the human immunodeficiency virus type 2. Ann Intern Med 118:211, 1993
- AIDS in the World. Mann JM, Tarantola DJM, Netter TW, Eds. Harvard University Press, Cambridge, Massachusetts, 1992
- Kanki PJ, Travers KU, MBoup S, et al: Slower heterosexual spread of HIV-2 than HIV-1. Lancet 343:943, 1994
- Quinn TC: Population migration and the spread of types 1 and 2 human immunodeficiency viruses. Proc Natl Acad Sci USA 91:2407, 1994
- Marlink R, Kanki P, Thior I, et al: Reduced rate of disease development after HIV.2 infection as compared to HIV-1. Science 265:1587, 1994
- Grmek MD: History of AIDS: Emergence and Origin of a Modern Pandemic. Princeton University Press, Princeton, New Jersey, 1990
- Myers G, Maclnnes K, Myers L: Phylogenetic moments in the AIDS epidemic. Emerging Viruses. Morse SS, Ed. Oxford University Press, Oxford, England, 1993, chapter 12
- Khabbaz RF , Heneine W, George JR, et al: Brief report: infection of a laboratory worker with simian immunodeficiency virus. N Engl J Med 330:172, 1994
- Essex M: Simian immunodeficiency virus in people (editorial). N Engl J Med 330:209, 1994
- Machado SG, Gail MH, Ellenberg SS: On the use of laboratory markers as surrogates for clinical endpoints in the evaluation of treatment for HIV infection. J Acquir Immune Defic Syndr 3:1065, 1990
- De Leys R, Vanderborght B, Vanden Haesevelde M, et al: Isolation and partial characterization of an unusual human immunodeficiency retrovirus from two persons of west-central African origin. J Virol 64:1207, 1990
- Gürtler LG, Hauser PH, Eberle J, et al: A new subtype of human immunodeficiency virus type 1 (MVP-5180) from Cameroon. J Virol 68:1581, 1994
- Loussert-Ajaka I, Ly TD Chaix ML, et al: HIV-1 / HIV-2 seronegativity in HIV-1 subtype O infected patients. Lancet 343:1393, 1994
- Haynes BF: Scientific and social issues of human immunodeficiency virus vaccine development. Science 260:l279, 1993
- Popovic M, Sarngadharan MG, Read E, et al: Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science 224:497, 1984
- Sarngadharan MG, Popovic M, Bruch L, et al: Antibodies reactive with human T-lymphotropic retroviruses (HTLV-III) in the serum of patients with AIDS. Science 224:506, 1984
- Brun-Vezinet F, Rouzioux C, Barre-Sinoussi F, et al: Detection of IgG antibodies to lymphadenopathy-associated virus in patients with AIDS or lymphadenopathy syndrome. Lancet 1:1253, 1984
- Elbeik T, Feinberg MB: HIV isolation and quantitation methods. The AIDS Knowledge Base: A Textbook on HIV Disease from the University of California, San Francisco, and the San Francisco General Hospital, 2nd ed. Cohen PT, Sande MA, Volberding PA, Eds. Little, Brown & Co, Boston, 1994, p 2.4-1
- Capon DJ, Ward RH: The CD4-gp 120 interaction and AIDS pathogenesis. Annu Rev lmmunoI 9:649, 1991
- Wain-Hobson S: Human immunodeficiency virus type 1 quasispecies in vivoand ex vivo. Curr Top Microbiol lmmunoI 176:181, 1992
- Coffin JM: Genetic diversity and evolution of retroviruses. Curr Top Microbiol lmmunoI 176:143, 1992
- Wain-Hobson S: Is antigenic variation of HIV important for AIDS and what might be expected in the future? The Evolutionary Biology of Viruses. Morse SS, Ed. Raven Press, New York, 1994, p 185
- Burns DPW, Desrosiers RC: Envelope sequence variation, neutralizing antibodies, and primate lentivirus persistence. Curr Top Microbiol lmmunoI 188:185, 1994
- Planelles V, Li Q-X, Chen ISY: The biological and molecular basis for cell tropism in HIV. Human Retroviruses. Cullen BR, Ed. Oxford University Press, New York, 1993, p 17
- Hwang SS, Boyle TJ, Lyerly HK,et al: ldentification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science 253:71, 1991
- Pang S, Shlesinger Y, Daar ES, et al: Rapid generation of sequence variation during primary HIV-1 infection. AIDS 6:453, 1992
- Wlodawer A, Erickson JW: Structure-based inhibitors of HIV-1 protease. Annu Rev Biochem 62:543, 1993
- Burger H, Weiser B, Flaherty K, et al: Evolution of human immunodeficiency virus type 1 nucleotide sequence diversity among close contacts. Proc Natl Acad Sci USA 88:11236, 1991
- Balfe P, Simmonds P, Ludlam CA, et al: Concurrent evolution of human immunodefidency virus type 1 in patients infected from the same source: rate of sequence change and low frequency of inactivating mutations. J Virol 64:6221, 1990
- Wolfs TF, Zwart G, Bakker M, et al: HIV-1 genomic RNA diversification following sexual and parenteral virus transmission. Virology 189:103, 1992
- Baltimore D, Feinberg MB: HIV revealed: toward a natural history of the infection (editorial). N Engl J Med 321:1673, 1989
- Ho DD,Neumann AU,Perelson AS, etal:Rapid tumover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123, 1995
- Wei X, Ghosh SK, Taylor ME, et al: Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373:117, 1995
- Sninsky JJ, Kwok S: The application of quantitative polymerase chain reaction to therapeutic monitoring. AIDS 7(suppI 2):S29 , 1993
- Urdea MS, Wilber JC, Yeghiazarian T, et al: Direct and quantitative detection of HIV-1 RNA in human plasma with a branched DNA signal amplification assay. AIDS 7(suppI 2):S11, 1993
- Coombs RW, Collier AC, Allain JP, et al: Plasma viremia in human immunodeficiency virus infection. N Engl J Med 321:1626, 1989
- Ho DD, Moudgil T, Alam M: Quantitation of human immunodeficiency virus type 1 in the blood of infected persons. N Engl J Med 321:1621, 1989
- Roos MT, Lange JM, de Goede RE, et al: Viral phenotype and immune response in primary human immunodeficiency virus type 1 infection. J Infect Dis 165:427, 1992
- Zhang LQ, MacKenzie P, Cleland A, et al: Selection for specific sequences in the extemal envelope protein of human immunodeficiency virus type 1 upon primary infection. J ViroI 67:3345, 1993
- Clark SJ, Saag MS, Decker WD, et al: High titers of cytopathic virus in plasma of patients with symptomatic primary HIV-1 infection. N Engl J Med 324:954, 1991
- Feinberg J: The acute HIV seroconversion syndrome. Current Opinion in Infectious Diseases 5:221, 1992
- Tindall B, Cooper DA: Primary HIV infection: host responses and intervention strategies. AIDS 5:1,1991
- Daar ES, Moudgil T, Meyer RD, et al: Transient high levels of virernia in patients with primary human immunodeficiency virus type 1 infection. N Engl J Med 324:961, 1991
- Koup RA, Safrit JT, Cao Y, et al: Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J ViroI 68:4650, 1994
- Safrit JT, Andrews CA, Zhu T, et al: Characterization of human immunodeficiency virus type 1-specific cytotoxic T lymphocyte clones isolated during acute seroconversion: recognition of autologous virus sequences within a conserved immunodominant epitope. J Exp Med 179:463, 1994
- Pantaleo G, Demarest JF, Soudeyns H, et al: Major expansion of CD8+ T cells with a predominant V beta usage during the primary immune response to HIV. Nature 370:463, 1994
- Keet IP, Krijnen P; Koot M, et al: Predictors of rapid progression to AIDS in HIV-1 seroconverters. AIDS 7:51, 1993
- Mellors JW, Kingsley LA, Rinaldo CR Jr, et al: Quantitation of HIV-1 RNA in plasma predicts outcome after seroconversion. Ann Intern Med 122:573, 1995
- Koot M, Keet IPM, Vos AHV, et al: Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann Intern Med 118:681, 1993
- Finkel TH, Banda NK: Indirect mechanisms of HIV pathogenesis: how does HIV kill T cells? Curr Opin ImmunoI 6:605, 1994
- Shearer GM, Clerici M: Early T-helper cell defects in HIV infection (editorial). AIDS 5:245, 1991
- Schnittman SM, Lane HC, Greenhouse J, et al: Preferential infection ofCD4+ memory T cells by human immunodeficiency virus type 1: evidence for a role in the selective T-cell functional defects observed in infected individuals. Proc Natl Acad Sci USA 87:6058, 1990
- Mosmann TR: Cytokine patterns during the progression to AIDS. Sience 265:193, 1994
- Clerici M, Shearer GM: A TH1->TH2 switch is a critical step in the etiology of HIV infection. Immunol Today 14:107, 1993
- Graziosi C, Pantaleo G, Gantt KR, et al: Lack of evidence for the dichotomy of TH1 and TH2 predominance in HIV-infected individuals. Science 265:248, 1994
- Maggi E, Mazzetti M, Ravina A, et al: Ability of HIV to promote a TH1 to TH0 shift and to replicate preferentially in TH2 and TH0 cells. Science 265:244, 1994
- Lyerly HK, Matthews TJ, Langlois AJ, et al: Human T-cell lymphotropic virus IIIB glycoprotein (gp 120) bound to CD4 determinants on normal lymphocytes and expressed by infected cells serves as target for immune attack. Proc Natl Acad Sci USA 84:4601, 1987
- Meyaard L, Otto SA, Jonker RR, et al: Programmed death of T cells in HIV-1 infection. Science 257:217, 1992
- Groux H, Torpier G, Monte D, et al: Activation-induced death by apoptosis in CD4+ T cells from human immunodeficiency virus-infected asymptomatic individuals. J Exp Med 175:331, 1992
- Mackall CL, Fleisher TA, Brown MR, et al: Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. N Engl J Med 332:143, 1995
- McCune JM: HIV-1: the infective process in vivo. Cell 64:351, 1991
- Homsy J, Meyer M, Tateno M, et al: The Fc and not CD4 receptor mediates antibody enhancement of HIV infection in human cells. Science 244:1357, 1989
- Koenig S, Woods RM, Brewah YA, et al: Characterization of MHC class I restricted cytotoxic T cell responses to tax in HTL V-1 infected patients with neurologic disease. J ImmunoI 151:3874, 1993
- Buseyne F, Riviere Y: HIV -specific CD8+ T-cell immune responses and viral replication. AIDS 7(suppI 2):S81, 1993
- Koup RA: Virus escape from CTL recognition. J Exp Med 180:779, 1994
- Phillips RE, Rowland-Jones S, Nixon DF, et al: Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354:453, 1991
- Takeda A, Tuazon CU, Ennis FA: Antibody-enhanced infection by HIV-1 via Fc receptor-mediated entry. Science 242:580, 1988
- Weber JN,Clapham PR, Weiss RA,et al: Human immunodeficiency virus infection in two cohorts of homosexual men: neutralising sera and association of anti-gag antibody with prognosis. Lancet 1:119, 1987
- Carmichael A, Jin X, Sissons P, et al: Quantitative analysis of the human immunodeficiency virus type 1 (HIV-1)-specific cytotoxic Tlymphocyte (CTL) response at differentstages of HIV-1 infection: differential CTL responses to HIV-1 and Epstein-Barr virus in late disease. J Exp Med 177:249, 1993
- Hoth DF, Bolognesi DP, Corey L, et al: HIV vaccine development: a progress report. Ann Intern Med 8:603, 1994
- Karzon DT, Bolognesi DP, Koff WC: Development of a vaccine for the prevention of AIDS, a critical appraisal. Vaccine 10:1039, 1992
- Letvin NL: Vaccines against human immunodefidency virus-progress and prospects. N Engl J Med 329:1400, 1993
- Levine PH, Jacobson S, Pocinki AG, et al: Clinical, epidemiologic, and virologic studies in four clusters of the chronic fatigue syndrome. Arch Intern Med 152:1611, 1992
- Lurie P, Bishaw M, Chesney M, et al: Ethical, behavioral, and social aspects of HIV vaccine trials in developing countries. JAMA 271:295, 1994
- Forrest BD: Mucosal immunity: challenges for HIV vaccines. AIDS Research Reviews, Vo1 3. Koff W, Wong-Staal F, Kennedy R, Eds. Marcel Dekker, New York, 1993, p 327
| 230a | Bryson YJ, Pang S, Wei LS, et al: Clearance of HIV
infection in a perinatally infected infant. N Engl J Med 332:833, 1995
|
- Rowland-Jones S, Sutton J, Ariyoshi K, etal: HIV-specific cytotoxic T-cells in HIV-exposed but uninfected Gambian women. Nature Medicine 1:59, 1995
- Daniel MD, Kirchhoff F, Czajak SC, et al: Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science 258:1938, 1992
- Baba TW , Jeong YS, Penninck D, et al: Pathogenicity of live, attenuated SIV after mucosal infection of neonatal macaques. Science 267:1820, 1995
- Berman PW, Gregory TJ, Riddle L, et al: Protection of chimpanzees from infection by HIV-1 after vaccination with recombinant glycoprotein gp120 but not gp160. Nature 345:622, 1990
- Fultz PN, Nara P, Barre-Sinoussi F, et al: Vaccine protection of chimpanzees against challenge with HIV-1-infected peripheral blood mononuclear cells. Science 256:1687, 1992
- Hu SL, Fultz PN, McClure HM, et al: Effect of immunization with a vaccinia-HIV env recombinant on HIV infection of chimpanzees. Nature 328:721, 1987
- Hirsch MS, D'Aquila RT: Therapy for human immunodefidency virus infection. N Engl J Med 328:1686, 1993
- Johnston MI, Hoth DF: Present status and future prospects for HIV therapies. Science 260:1286, 1993
- Hammer S, Crumpacker C, D'Aquila R, et al: Use of virologic assays for detection of human immunodeficiency virus in clinical trials: recommendations of the AIDS Clinical Trials Group Virology Committee. J Clin MicrobioI 31:2557, 1993
- Choi S, Lagakos SW, Schooley RT, et al: CD4+ lymphocytes are an incomplete surrogate marker for clinical progression in persons with asymptomatic HIV infection taking zidovudine. Ann Intern Med 118:674, 1993
- Mulder J, McKinney N, Christopherson C, et al: Rapid and simple PCR assay for quantitation of human immunodeficiency virus type 1 RNA in plasma: application to acute retroviral infection. J Clin MicrobioI 32:292, 1994
- Erice A, Balfour HH Jr: Resistance of human immunodeficiency virus type 1 to antiretroviral agents: a review. Clin Infect Dis 18:149, 1994
- Richman DD: HIV drug resistance. AIDS Res Hum Retroviruses 8:1065, 1992
- Saag MS, Emini EA, Laskin OL, et al: A short-term clinical evaluation of L-697,661, a non-nucleoside inhibitor of H1V-1 reverse transcriptase. N Engl J Med 329:1065, 1993
- Richman DD: Playing chess with reverse transcriptase. Nature 361:588, 1993
- Erice A, Mayers DL, Strike DG, et al: Brief report: primary infection with zidovudine-resistant human immunodeficiency virus type 1. N Engl J Med 328:1163, 1993
- Richman DD, Havlir D, Corbeil J, et al: Nevirapine resistance mutations of human immunodeficiency virus type 1 selected during therapy. J ViroI 68:1660, 1994
- Fischl MA, Richman DD, Grieco MH, et al: The efficacy of azidothymidine (AZT) in the treatrnent of patients with AIDS and AIDS-related complex: a double-blind, placebo-controlled trial. N Engl J Med 317:185, 1987
- Volberding PA, Lagakos SW, Koch MA, et al: Zidovudine in asymptomatic human immunodeficiency virus infection: a controlled trial in persons with fewer than 500 CD4-positive cells per cubic millimeter. N Engl J Med 322:941, 1990
- Cooper DA, Gatell JM, Kroon S, et al: Zidovudine in persons with asymptomatic HIV irifection and CD4+ cell counts greater than 400 per cubic millimeter. N Engl J Med 329:297, 1993
- Volberding PA, Lagakos SW, Grimes JM, et al: The duration of zidovudine benefit in persons with asymptomatic HIV infection: prolonged evaluation of protocol 019 of the AIDS Clinical Trials Group. JAMA 272:437, 1994
- Concorde Coordinating Committee: MRC / ANRs randomised double-blind controlled trial of immediate and deferred zidovudine in symptom-free HIV infection. Lancet 343:871, 1994
- Sande MA, Carpenter CCJ, Cobbs CG, et al: Antiretroviral therapy for adult HIV-infected patients: recommendations from a state-of-the-art conference. JAMA 270:2583, 1993
- Lenderking WR, Gelber RD, Cotton DJ, et al: Evaluation of the quality of life associated with zidovudine treatment in asymptomatic human immunodeficiency virus infection. N Engl J Med 330:738, 1994
- Richman DD, Fischl MA, Grieco MH, et al: The toxicity of azidothymidine (AZT) in the treatrnent of patients with AIDS and AIDS-related complex: a double-blind, placebo-controlled trial. N Engl J Med 317:192, 1987
- Kahn JO, Lagakos SW, Richman DD, et al: A controlled trial comparing continued zidovudine with didanosine in human immunodeficiency virus irifection. N Engl J Med 327:581, 1992
- Spruance SL, Pavia AT, Peterson D, et al: Didanosine compared with continuation of zidovudine in HIV-infected patients with signs of clinical deterioration while receiving zidovudine: a randomized, double-blind clinical trial. Ann Intern Med 120:360, 1994
- Merigan TC, Skowron G, Bozzette SA, et al: Circulating p24 antigen levels and responses to dideoxycytidine in human immunodeficiency virus (HIV) irifections: a phase I and II study. Ann Intern Med l10:189, 1989
- Fischl MA, Olson RM, Follansbee SE, et al: Zalcitabine compared with zidovudine in patients with advanced HIV-1 infection who received previous zidovudine therapy. Ann Intern Med 118:762, 1993
- Meng T-C, Fischi MA, Boota AM, et al: Combination therapy with zidovudine and dideoxycytidine in patients with advanced human immunodeficiency virus irifection: a phase I / II study. Ann Intern Med 116:13, 1992
- Abrams DI, Goldman AI Launer C, et al: A comparative trial of didanosine or zalcitabine after treatment with zidovudine in patients with human immunodeficiency virus infection. N Engl J Med 330:657, 1994
- Skowron G: Biologic effects and safety of stavudine: overview of phase I and II clinical trials. J Infect Dis 171(suppI 2):S113, 1995
- Petersen EA, Ramírez-Ronda CH, Hardy WD, et al: Dose-related activity of stavudine in patients infected with human immunodeficiency virus. J Infect Dis 17l(suppI 2):S131, 1995
- Robins T, Plattner J: HIV protease inhibitors: their anti-HIV activity and potential role in treatment. J Acquir Immune Defic Syndr 6:162, 1993
- Miller M, Schneider J, Sathyanarayana BK, et al: Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2.3 A resolution. Science 246:1149, 1989
- Condra JH, Schleif WA, Blahy OM, et al: In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature 374:569, 1995
- Hsu MC, Schutt AD, Holly M, et al: Inhibition of HIV replication in acute and chronic infections in vitro by a Tat antagonist. Science 254:1799, 1991
- Ashkenazi A, Smith DH, Marsters SA, et al: Resistance of primary isolates of human immunodeficiency virus type 1 to soluble CD4 is independent of CD4-rgp 120 binding affinity. Proc Natl Acad Sci USA 88:7056, 1991
- Moore JP, McKeatingJA, Huang Y, et al: Virions of primary human immunodeficiency virus type 1 isoIates resistant to soluble CD4 (sCD4) neutralization differ in sCD4 binding and glycoprotein gp120 retention from sCD4-sensitive isolates. J ViroI 66:235, 1992
- RyuS-E,Kwong PD, Truneh A, etal: Crystal structure of an HIV-binding recombinant fragment of human CD4. Nature 348:419, 1990
- Wang J, Yan Y, Garrett TPJ, et al: Atomic structure of a fragment of human CD4 containing two immunoglobulin-like domains. Nature 348:411, 1990
- Redfield RR, Birx DL, Ketter N, et al: A phase I evaluation of the safety and immunogenicity of vaccination with recombinant gp 160 in patients with early human immunodeficiency virus infection. N Engl J Med 324.1677, 1991
- Trauger RJ, Ferre F, Daigle AE, et al: Effect of immunization with inactivated gp120-depleted human immunodeficiency virus type 1 (HIV-1) immunogen on HIV-1 immunity, viral DNA, and percentage of CD4 cells. J Infect Dis 169:1256, 1994
- Lane HC, Kovacs JA, Feinberg J, et al: Anti-retroviral effects of interferon-alpha inAIDS-associated Kaposi'ssarcoma. Lancet 2:1218, 1988
- Schnittman SM, Vogel S, Baseler M, et al. A phase I study of interferon-alpha 2b in combination with interleukin-2 in patients with human immunodeficiency virus infection. J Infect Dis 169:98l, 1994
- Kovacs JA, Baseler M, Dewar RJ, et al: Increases in CD4 T lymphocytes with intermittent courses of interleukin-2 in patients with human immunodeficiency virus infection: a preliminary study. N Engl J Med 332:567, 1995
- Koprowski H, DeFreitas EC, Harper ME, et al: Multiple sclerosis and human T-cell lymphotropic retroviruses. Nature 318:154, 1985
- Greenberg SJ, Ehrlich GD, Abbott MA, et al: Detection of sequences honiologous to human retroviral DNA in multiple sclerosis by gene amplification. Proc Natl Acad Sci USA 86:2878, 1989
- Reddy EP, Sandberg-Woilheim M, Mettus RV, et al: Amplification and molecular cloning of HTLV-I sequences from DNA of multiple sclerosis patients. Science 243:529, 1989
- Burns JC, Geha RS, Schneeberger EE, et al: Polymerase activity in lymphocyte culture supernatants from patients with Kawasaki disease. Nature 323:814, 1986
- Melish ME, Marchette NJ, Kaplan JC, et al: Absence of significant RNA-dependent DNA polymerase activity in Iymphocytes from patients with Kawasaki syndrome. Nature 337:288, 1989
- Rowley A, Castro B, Levy J, et al: Failure to confirm the presence of a retrovirus in cultured lymphocytes from patients with Kawasaki syndrome. Pediatr Res 29:417, 1991
- DeFreitas E, Hilliard B, Cheney PR, et al: Retroviral sequences reIated to human T-lymphotropic virus type II in patients with chronic fatigue immune dysfunction syndrome. Proc Natl Acad Sci USA 88:2922, 1991
- Smith DK, Neal JJ, Holmberg SD: Unexplained opportunistic infections and CD4+ T-lymphocytopenia without HIV infection: an investigation of cases in the United States. N Engl J Med 328:373, 1993
- Fauci AS: CD4+ T-lymphocytopenia without HIV infection-no lights, no camera, just facts (editorial). N Engl J Med 328:429, 1993