Neumología
⭳ Abrir artículo (PDF)591.2 KBEste artículo es idéntico en la Edición 3/2000.
Contenido del artículo
X EDEMA PULMONAR
X EDEMA PULMONAR
DR. GERALD W. STATON, JR.
DR. ROLAND H. INGRAM, JR.
El edema pulmonar agudo puede dividirse en forma amplia en dos categorías: edema pulmonar causado por aumento en la presión capilar (edema hidrostático o cardiogénico) y edema pulmonar causado por aumento en la permeabilidad capilar (síndrome de insuficiencia respiratoria progresiva aguda). En algunos casos ocurren tanto aumento en la presión como en la permeabilidad capilar.
Patogenia
El edema pulmonar, que consiste en el acúmulo anormal y difuso de líquido extravascular en el tejido pulmonar y en los espacios aéreos, es el tipo más frecuente de enfermedad parenquimatosa difusa aguda y no infecciosa. La patogenia del edema pulmonar puede comprenderse mejor examinando la ecuación de Starling, que define los determinantes del flujo de líquido a través de la membrana capilar pulmonar:
en donde Q es el líquido que fluye, K es el coeficiente de filtración (directamente proporcional al área de superficie endotelial e inversamente proporcional al grosor de la pared capilar); Pcap, la presión hidrostática intravascular (capilar); Pint, la presión hidrostática intersticial; sigma, el coeficiente de reflejo de las proteínas (i.e., el grado de permeabilidad a macromoléculas); tcap, la presión coloidosmótica intravascular; y tint, la presión coloidosmótica intersticial. La primera parte de la ecuación, K (Pcap - Pint), representa la fuerza hidrostática, y la segunda porción, sigma(tcap - tint), representa la fuerza coloidosmótica. La alteración en cualquiera de los factores de la ecuación de Starling puede causar aumento en la salida transvascular de líquido. Sin embargo, en la práctica clínica solo dos de estos factores causan en forma frecuente edema pulmonar: el aumento en la presión capilar que ocasiona edema pulmonar cardiogénico o hidrostático, o la disminución del coeficiente de reflejo, que se asocia con edema pulmonar no cardiogénico o por permeabilidad.
La principal defensa contra el edema pulmonar depende del sistema linfático intrapulmonar. En condiciones normales el líquido que se filtra a través de la membrana capilar es eliminado por los vasos linfáticos. La reserva linfática es tal que incluso un aumento de 4 a 6 veces en la salida transvascular de líquido puede ser tolerada sin que aumente el agua dentro del pulmón. Una vez que la reserva linfática es superada, aumenta el agua pulmonar, primero en el intersticio alrededor de las vías respiratorias, después en el intersticio alrededor del alveolo y después dentro del alveolo. La evidencia clínica o radiográfica de edema pulmonar implica, por lo tanto, un gran aumento en la salidad transcapilar de líquido.
Características clínicas
El edema pulmonar, sea por aumento en la presión capilar o por aumento en la permealibidad, suele manifestarse en la radiografía de tórax estándar anteroposterior por opacidades alveolares simétricas y bilaterales que afectan los cuatro cuadrantes. Es frecuente la distribución perihiliar predominante, y en ocasiones existe una delimitación muy clara entre el área central de edema pulmonar y la periferia, lo que ocasiona un patrón denominado en alas de murciélago o de mariposa. Este patrón es más típico del edema pulmonar cardiogénico que del causado por aumento en la permeabilidad [ver figura 1]. Esta línea de delimitación no corresponde a límites anatómicos, pero puede ser causada por gradientes fisiológicos de ventilación, perfusión y flujo linfático de las porciones centrales del pulmón hacia la periferia. Con menos frecuencia el edema es muy asimétrico o totalmente unilateral. El edema asimétrico puede ser explicado por el hecho de que el paciente estaba recostado sobre el lado afectado cuando se desarrolló el padecimiento, o por la presencia de obstrucción vascular (tromboembolia) o enfisema en las áreas no afectadas del pulmón. Sin embargo, con frecuencia no se encuentra explicación para el edema pulmonar asimétrico o unilateral.
Algunas características que pueden observarse en la radiografía portátil del tórax ayudan a distinguir entre el edema pulmonar cardiogénico y el causado por mayor permeabilidad. En un estudio, dos de los datos más útiles fueron el grosor del pedículo vascular central y la presencia de broncogramas aéreos discretos. Un pedículo vascular ancho sugiere aumento en la presión capilar pulmonar, y los broncogramas aéreos visibles son más frecuentes en el edema pulmonar por aumento en la permeabilidad. Otras características radiográficas, como el tamaño de la silueta cardiaca y la presencia o ausencia de derrames pleurales, tienen menor valor para hacer el diagnóstico diferencial. Por desgracia, en un caso individual no puede distinguirse con confianza entre el edema pulmonar hidrostático o por mayor permeabilidad sin utilizar pruebas cruentas.
Edema pulmonar debido a aumento en la presión capilar
PATOGENIA
El edema pulmonar que ocurre por aumento en la presión capilar suele asociarse con baja concentración de proteínas en el líquido del edema, y puede ser secundario a disfunción sistólica del ventrículo izquierdo, alteración de la válvula mitral, hipervolemia asociada con función cardiaca izquierda normal (v.gr., en un paciente con insuficiencia renal) u obstrucción pulmonar venosa. La causa más frecuente de edema pulmonar hidrostático es la disfunción del ventrículo izquierdo. En la cardiomiopatía congestiva, la función sistólica del ventrículo izquierdo se deteriora, el ventrículo se dilata y la presión final del ventrículo izquierdo (PDFVI) aumenta. Este aumento en la PDFVI causa aumento en la presión capilar. Otros tipos de enfermedad cardiaca pueden también aumentar la PDFVI, a pesar de que exista una función sistólica normal y euvolemia, al disminuir la complianza del ventrículo izquierdo. La reducción de la complianza puede ser permanente (v.gr., en la hipertrofia ventricular izquierda o en la cardiopatía restrictiva por enfermedad cardiaca infiltrativa) o transitoria (v.gr., por isquemia miocárdica). El aumento en la presión capilar con una PDFVI normal no es frecuente, pero ocurre en la estenosis mitral o como resultado de la obstrucción del flujo en las venas pulmonares (enfermedad venoclusiva pulmonar).
CARACTERISTICAS CLINICAS
Las características clínicas principales del edema pulmonar cardiogénico son disnea extrema, taquipnea y signos de importante actividad simpática, como taquicardia, hipertensión y diaforesis. La hipotensión es menos común, pero puede observarse si el edema pulmonar es secundario a un infarto del miocardio agudo extenso. La disnea rara vez se alivia al corregir la hipoxemia, lo que sugiere que la causa puede ser la activación de receptores intrapulmonares de estiramiento, más que la hipoxemia en sí. En el edema pulmonar cardiogénico puede observarse cianosis central si la hipoxemia arterial es importante, pero lo más común es que la cianosis sea de tipo periférico debida a vasoconstricción cutánea intensa. Es común el uso de los músculos accesorios de la respiración por el gran aumento del trabajo respiratorio. El esfuerzo requerido para respirar suele ser tan grande que con frecuencia se necesita intubación endotraqueal y ventilación mecánica para corregir o prevenir el desarrollo de insuficiencia respiratoria francamente hipercápnica. Al inicio del padecimiento la auscultación torácica puede revelar sibilancias causadas por edema de las vías respiratorias, después se escuchan estertores difusos.
Ciertos datos radiográficos sugieren aumento en la presión capilar (v.gr., cefalización de la circulación, ensanchamiento del pedículo vascular o líneas B de Kerley), pero ésta puede establecerse con mayor exactitud midiendo la presión de oclusión de la arteria pulmonar (la llamada presión en cuña) con un catéter que tiene un balón y se coloca en la arteria pulmonar. La presión en cuña es una medida de la presión en las venas pulmonares, y la presión venosa pulmonar se aproxima a la de los capilares pulmonares. Por lo tanto, el aumento en la presión en cuña sugiere que la elevación de la presión capilar es la causa del edema pulmonar, mientras que una presión en cuña por debajo del límite para la formación del edema pulmonar implica alteración en la permeabilidad capilar. En condiciones normales, el límite de la presión en cuña para edema pulmonar es de alrededor de 22 a 34 mm Hg. Puede existir un límite mayor (v.gr., > 25 a 30 mm Hg) cuando la presión capilar está aumentada en forma crónica, quizá porque se produce hipertrofia linfática.
Aunque la medición de la presión en cuña es útil para ayudar a aclarar la etiología del edema pulmonar, deben mencionarse ciertos detalles. Una presión en cuña por debajo del límite para edema pulmonar hidrostático (v.gr., < 18 mm Hg) no excluye el aumento en la presión capilar como causa del edema pulmonar. Esta situación se presenta sobre todo cuando el aumento en la presión es causado por una disminución temporal en la complianza del ventrículo izquierdo o por isquemia del musculo papilar con insuficiencia mitral aguda en un paciente con un infarto agudo al miocardio. Para el momento en que se coloca el catéter pulmonar la presión en cuña suele haberse normalizado. También ocurren aumentos importantes y transitorios en la presión capilar en el edema pulmonar neurogénico y con los aumentos súbitos en la presión arterial.
En los pacientes con edema pulmonar causado por la poco frecuente enfermedad venoclusiva pulmonar, la presión capilar es mucho más alta que la presión en las venas pulmonares; por lo tanto, la presión en cuña subestima la presión que determina la filtración de líquido a través de la membrana capilar. Por último, a pesar de la aparente lógica para el uso de cáteteres pulmonares, debe mencionarse que no se ha atribuido ningún efecto benéfico en la evolución por su uso. De hecho, un estudio reciente de gran número de pacientes en unidades de cuidados intensivos ha sugerido que los pacientes que tuvieron catéteres arteriales pulmonares sufrieron mayor mortalidad y mayor gasto económico que los pacientes que no.1 Este reporte ha sido criticado, pero es el único análisis publicado sobre el efecto del uso de estos catéteres en la evolución de los pacientes de la UCI.
TRATAMIENTO
Después de que la hipoxemia es aliviada con oxígeno suplementario, el tratamiento del edema pulmonar causado por aumento en la presión capilar se dirige a disminuir las presiones de llenado del ventrículo izquierdo y a mejorar la función cardiaca. La morfina, los diuréticos y la nitroglicerina sublingual o intravenosa aumentan la capacitancia venosa y disminuyen la precarga. La reducción de la precarga con medicamentos vasodilatadores, como nitroprusiato intravenoso (que también reduce la precarga), o inhibidores de la enzima convertidora de angiotensina mejoran el gasto cardiaco. En los pacientes muy graves o que responden con lentitud a estas medidas la ventilación no invasiva con aplicación nasal de presión positiva al final de la espiración o la intubación y aplicación de ventilación con presión positiva pueden mejorar la oxigenación y disminuir el retorno venoso.
Edema pulmonar causado por aumento en la permeabilidad capilar: Síndrome de insuficiencia respiratoria progresiva aguda
El síndrome de insuficiencia respiratoria progresiva aguda (SIRPA) se caracteriza por lesión difusa al endotelio pulmonar, que causa edema pulmonar por aumento importante en la permeabilidad capilar al agua, solutos y macromoléculas. A diferencia del edema pulmonar que es causado por alteración en las fuerzas de Starling, el edema pulmonar en el SIRPA se caracteriza por aumento en la concentración de proteínas en el líquido de edema, que llega a ser hasta de 80 a 90 porciento de las proteínas séricas. Aún más, la presencia de muchos neutrófilos y sus productos secretorios (v.gr., elastasa y colagenasa) en el líquido del lavado broncoalveolar demuestra la respuesta inflamatoria subyacente que no es característica del edema pulmonar cardiogénico.
Muchas lesiones hematógenas y aéreas pueden producir SIRPA [ver tabla 1]. El riesgo de desarrollar SIRPA como resultado de una de ellas puede aumentar mucho si existe historia de abuso de alcohol.3
PATOGENIA
El daño alveolar difuso (DAD) es el término descriptivo para la secuencia de cambios inespecíficos, pero predecibles, que ocurren en forma característica en los pacientes con SIRPA2. Por lo general, el agente o proceso causal no puede ser determinado por los datos histopatológicos. Además, las alteraciones pueden resolverse en cualquier momento de la evolución de la enfermedad. La apariencia histológica del DAD varía durante el periodo entre el evento precipitante y la biopsia o autopsia de acuerdo con tres fases: una fase exudativa aguda (días 0 a 7), una fase proliferativa subaguda o no organizada (días 7 a 14) y una fase crónica (después del día 14) [ver figura 2].
La primera parte de la fase aguda exudativa se caracteriza por edema intersticial e intralveolar, infiltración neutrofílica, hemorragia y depósito de fibrina. Se deposita una mezcla de fibrina y detritos celulares en el espacio alveolar para formar las llamadas membranas hialinas que son prominentes 3 a 7 días después de la lesión. El desprendimiento de las células de la superficie alveolar deja una membrana basal denudada que es muy importante para la reparación o fibrosis subsecuente. El infiltrado intersticial de células inflamatorias se vuelve más intenso alrededor del día 7 y persiste durante toda la fase proliferativa.
La denudación de la membrana basal causa que los neumocitos tipo II proliferen (días 3 a 7), produciendo un patrón de hiperplasia de las células superficiales alveolares. En los pacientes en los que se resuelve el síndrome, estas células proliferantes se diferencían a neumocitos tipo I, restableciendo el lado epitelial de la pared alveolocapilar y normalizando el intercambio de gases.
La fase proliferativa del SIRPA se caracteriza por inflamación intersticial y proliferación de fibroblastos, al inicio en el intersticio. Los fibroblastos invaden los espacios alveolares a través de los defectos de la membrana basal, un proceso que produce áreas de fibrosis intralveolar. Durante esta fase las membranas hialinas desaparecen como resultado de fagocitosis o de organización con incorporación del exudado en acúmulos intralveolares de fibroblastos proliferantes.
La fase crónica del SIRPA se manifiesta por regiones de fibrosis intensa, regiones focales de expansión excesiva y obliteración vascular pulmonar. Desde el punto de vista histológico, esta fase de la enfermedad puede ser semejante a la fibrosis pulmonar idiopática (FPI). Sin embargo, a diferencia de este padecimiento, la fase crónica del SIRPA puede mejorar con el tiempo si el paciente sobrevive.
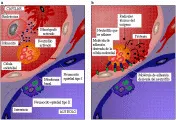
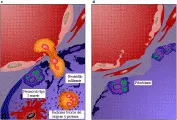
Aunque los mecanismos que causan el SIRPA han sido estudiados en forma muy intensa, aún no se comprenden del todo.4 Los dos sitios de lesión temprana clave en el proceso son el endotelio del lecho capilar pulmonar y el epitelio alveolar [ver figura 3].
El endotelio de los capilares pulmonares parece ser el primer sitio de
lesión en el SIRPA que es causado por sepsis u otros trastornos
sistémicos. La endotoxina estimula la liberación de varios
mediadores, como fragmentos activados del complemento, factores de la
coagulación y factor de necrosis tumoral (FNT), factor activador de
plaquetas, sustancias antinflamatorias como la interleucina-10 (IL-10),
citocinas (v.gr., IL-1, IL-6 e IL-8 y otras), antagonistas de receptores de
citocinas, surfactante, autoanticuerpos anticitocinas, antioxidantes y
antiproteasas. Si existe un desequilibrio a favor de los mediadores
inflamatorios,5 éstos actúan como
quimiotácticos y secretagogos para los neutrófilos y plaquetas,
causando agregación y embolización de estas células en la
vasculatura pulmonar. Los agregados intravasculares de neutrófilos y
plaquetas obstruyen el flujo sanguíneo local y reducen el volumen eficaz
y el área de superficie del lecho capilar. Las plaquetas liberan
prostaglandinas como el tromboxano A2 y pueden, en asociación
con la obstrucción local, contribuir a la hipertensión pulmonar
que ocurre con frecuencia en los pacientes con SIRPA.
A través de la expresión de moléculas de adhesión en la superficie celular (selectinas e integrinas), los neutrófilos se adhieren al endotelio y liberan oxidantes dañinos, enzimas proteolíticas y metabolitos del ácido araquidónico, lo que causa disfunción de las células endoteliales y destrucción y denudación del lado endotelial de la membrana basal. Además, la endotoxina puede lesionar las células endoteliales en forma directa. Los macrófagos, que tienen capacidades secretoras semejantes a las de los neutrófilos pueden participar en esta lesión, y esto podría explicar el desarrollo de SIRPA en los pacientes con neutropenia. El aumento en la permeabilidad de la membrana alveolocapilar permite al plasma y a las células entrar a los espacios intersticiales del pulmón y, al final, al alveolo. En el resto del organismo ocurre un tipo de daño microvascular semejante, que puede participar en la disfunción orgánica múltiple asociada al SIRPA causado por sepsis u otros padecimientos.
El segundo sitio de lesión en el SIRPA temprano, el epitelio alveolar, es muy importante para la fisiología pulmonar normal porque las uniones estrechas entre las células del epitelio alveolar evitan que el líquido intersticial penetre a los espacios alveolares. Las células tienen un sistema activo de transporte de sodio sensible a la acción agonista beta que puede ayudar a eliminar líquido de los espacios alveolares, y los neumocitos tipo II producen surfactante. A las pocas horas de la lesión pulmonar aguda los neumocitos tipo I mueren, denudando el lado alveolar de la membrana basal. Se desconoce la causa exacta de esta fase de la lesión pulmonar; sin embargo, existen neutrófilos y macrófagos alveolares activados en los espacios alveolares en etapas tempranas del padecimiento que quizá participen en el daño. Se ha encontrado elastasa y otras enzimas proteolíticas de los neutrófilos y macrófagos en el líquido de lavado bronquioalveolar de pacientes con SIRPA. Aunque el plasma contiene varias proteínas antiproteolíticas, estas pueden inactivarse por los oxidantes originados por las células inflamatorias, lo que permite a las enzimas proteolíticas activarse en forma local. Los oxidantes, como el peróxido de hidrógeno y los radicales hidroxilo pueden lesionar también las células en forma directa. Además, los macrófagos producen sustancias procoagulantes, como factor tisular y factor VII, que activan factores de la coagulación que alcanzan los espacios intersticial y alveolar y producen fibrina.6 La fibrina inhibe el surfactante y, con otros detritos celulares, produce las membranas hialinas que se observan en las fases tempranas del SIRPA. La producción de surfactante por las células tipo II restantes puede también disminuir por acción del FNT.7 La combinación de menor producción e inhibición causa una profunda deficiencia de surfactante.8
Una vez que ha ocurrido la lesión pulmonar aguda, el proceso de reparación puede causar resolución completa o fibrosis progresiva. El factor crítico para ello es la gravedad de la lesión en el lado epitelial de la membrana alveolocapilar. Los neumocitos epiteliales II comienzan a proliferar desde la fase más temprana del proceso de reparación en un intento por recuperar la membrana basal denudada. Si el daño a la membrana basal no es grave, es más probable que las células tipo II tengan éxito. Una vez reparada la membrana basal, los neumocitos epiteliales participan en la remoción de líquido y detritos de los espacios alveolares, así como en la síntesis de nuevo surfactante. Con el tiempo, las células nuevas se diferencían en células tipo I y restablecen la delgada estructura de la membrana que es crítica para el proceso de intercambio de gases.
Si las células epiteliales y la membrana basal han sido lesionadas en forma grave, ocurrirá fibrosis intersticial e intralveolar aguda y progresiva. La fibrosis se presenta cuando los fibroblastos, que son controlados por factores de crecimiento (muchos derivados de los macrófagos) y que usan probablemente una matriz de fibrina y fibronectina como base, invaden el exudado, y en él proliferan y sintetizan colágena (tipo I), elastina y otros componentes de la matriz. La arquitectura pulmonar normal es remplazada en forma progresiva por tejido fibroso que deteriora en forma severa el intercambio de gases.
CARACTERISTICAS CLINICAS
Los signos clínicos del edema pulmonar causado por aumento en la permeabilidad son un poco diferentes de los del edema pulmonar cardiogénico. Suele existir menor actividad simpática y, cuando existe cianosis, es causada por hipoxemia arterial. Cuando la sepsis es la causa del edema pulmonar la piel puede estar caliente en lugar de fría, húmeda y pálida, y el pulso puede ser hiperdinámico.
Independientemente de la etiología y patogenia subyacentes, las características clínicas y fisiológicas del SIRPA son semejantes. Los datos clínicos incluyen disnea y taquipnea, con un patrón respiratorio caracterizado por disminución del volumen corriente y aumento de la frecuencia respiratoria. Suele requerirse de la ventilación mecánica por la incapacidad del paciente para mantener una ventilación por minuto adecuada, por el aumento importante en el trabajo respiratorio y por la hipoxemia grave que suele ser refractaria al aumento en la cantidad de oxígeno inspirado.
Es típico que la radiografía de tórax demuestre un proceso de ocupación alveolar difuso y simétrico. Cuando se realiza tomografía computada, la distribución de los espacios ocupados suele observarse menos simétrica. En las radiografías que se realizan en posición supina suele apreciarse un mayor grado de consolidación en las zonas posteriores del pulmón; sin embargo, esta distribución puede revertirse cambiando al paciente a la posición prona durante algunas horas. En esta última posición aumenta la consolidación en las áreas anteriores (dependientes de la gravedad) y las posteriores muestran mejor ventilación. Este dato demuestra la contribución de las fuezas de Starling (i.e., presión capilar) a la gravedad del edema en el caso de aumento en la permeabilidad.
El intercambio gaseoso en el SIRPA se caracteriza al principio por hipoxemia que es relativamente refractaria al aumento en las concentraciones de oxígeno inspirado, lo que implica la presencia de cortocircuitos intrapulmonares. Estas son consecuencia principalmente de colapso alveolar difuso con microatelectasias. Se cree que el colapso alveolar difuso es causado por alteraciones en el surfactante, una sustancia que normalmente ayuda a mantener la distensión alveolar al reducir la tensión superficial en la interfase aire-líquido del pulmón.
Al principio la presión arterial de bióxido de carbono (PaCO2) es baja o se mantiene dentro del rango normal, con sólo un pequeño aumento en la ventilación minuto. Sin embargo, la relación espacio muerto a volumen corriente (VD/VT) tiende a aumentar durante el tiempo, por lo que se requiere una ventilación por minuto cada vez mayor para mantener una PaCO2 normal. En algunos casos el aumento en la VD/VT es tan importante que no puede evitarse la hipercapnia a pesar de que la ventilación por minuto se aumente al máximo. El aumento en el espacio muerto fisiológico se debe a daño en el lecho capilar pulmonar, con lo que se crean regiones de alta ventilación en relación con la perfusión. Otra causa del aumento en el requerimiento de la ventilación por minuto en el SIRPA es la presencia de una mayor producción de bióxido de carbono por el hipermetabolismo. La alta relación VD/VT del SIRPA se resuelve en forma muy lenta y, junto con la reducción en la complianza pulmonar que impone una carga sobre la respiración espontánea, es responsable de que con frecuencia se requiera ventilación mecánica durante periodos muy prolongados. Es frecuente que el apoyo ventiltorio deba continuarse a pesar de que la hipoxemia haya mejorado lo suficiente para que sólo se requiera un modesto aumento en la concentración inspirada de oxígeno.
La mecánica pulmonar en el SIRPA se caracteriza principalmente por reducción en la complianza pulmonar (mayor elastancia), por lo que se requieren mayores presiones transpulmonares para alcanzar una ventilación corriente normal. En las etapas tempranas del SIRPA, cuando predomina el edema, mucho de la presión de distensión que se necesita para inflar el pulmón es necesaria para abrir los aveolos colapsados. De hecho, la complianza pulmonar (la pendiente de la curva presión-volumen) puede estar dentro del rango normal si se mide después de que estos alveolos colapsados han sido distendidos. Sin embargo, existe una reducción significativa en la complianza al evolucionar el padecimiento, cuando predomina la fibrosis alveolar. Esta reducción aparente puede ser causada por un engrosamiento difuso de la membrana alveolocapilar como resultado de fibrosis o por pérdida de un gran porcentaje de unidades alveolocapilares disponibles para la ventilación. Se ha sugerido que la mecánica pulmonar alterada en el SIRPA se conceptualiza mejor si se considera como causada por un pulmón pequeño, más que por un pulmón rígido.9 También se observa en el SIRPA aumento en la resistencia de las vías aéreas que responde a broncodilatadores inhalados.10
Además de su efecto sobre la relación VD/VT, los cambios en la vasculatura pulmonar en el SIRPA son también resultado de aumento en las resistencias de los vasos pulmonares y de hipertensión pulmonar. De hecho, la presión arterial pulmonar está elevada en casi todos los pacientes con SIRPA moderado a severo. La etiología de la hipertensión pulmonar en este síndrome parece ser multifactorial; sin embargo, una causa importante puede ser la presencia de pequeños trombos en la arteria pulmonar. Estos trombos pueden demostrarse por medio de una angiografía pulmonar realizada en la cama del paciente. La patogenia de los trombos en el SIRPA no es clara, pero es probable que se formen in situ.
Muchos pacientes con SIRPA tienen ciertos indicios de coagulación intravascular acelerada.11 Sin embargo, a pesar de la importancia aparente de los trombos para producir hipertensión pulmonar, no se ha determinado si la anticoagulación es benéfica en los pacientes con SIRPA.
El manejo de los pacientes con SIRPA se discute con detalle en la Subsección VIII.
EVOLUCION
Aunque el grado de lesión pulmonar en el SIRPA se relaciona con el pronóstico, otros parámetros predicen en forma más exacta la evolución. Esta observación no es sorprendente porque el SIRPA es casi siempre parte de un síndrome de respuesta inflamatoria sistémica. Con frecuencia no es clara la causa del síndrome, pero las posibilidades incluyen bronconeumonía, traslocación de productos bacterianos a través del intestino y liberación persistente de mediadores endógenos en ausencia de infección activa. La sepsis, que con frecuencia se asocia con vasodilatación que no responde a vasoconstrictores, es la causa más frecuente de muerte durante la enfermedad. Como resultado de la disponibilidad de técnicas excelentes para el apoyo ventilatorio, la insuficiencia respiratoria es causa de muerte en solo el 10 porciento de los casos en fase temprana y el 18 porciento de los más avanzados. Este hecho destaca la importancia de la disfunción en otros sistemas, como la falla hemodinámica con choque refractario o la insuficiencia renal progresiva, como causa de morbimortalidad. Los datos sugieren que la evolución de los pacientes con SIRPA puede estar mejorando, quizá como consecuencia de un mejor tratamiento.12
La mortalidad está determinada sobre todo por el número de sistemas que fallan. De acuerdo con Knaus y Wagner, la mortalidad aumenta en forma proporcional con el número de órganos disfuncionantes y el número de días de la disfunción13 [ver figura 4]. Existe una mayor mortalidad en los pacientes mayores de 65 años. Por ejemplo, cuando ha existido un solo órgano insuficiente durante 5 días en un paciente menor de 65 años la mortalidad es de 27 porciento, y en un paciente mayor de 65 años, de 48 porciento. De igual modo, la falla de dos órganos durante 2 días causa una mortalidad del 47 porciento en enfermos menores de 65 años y de 73 porciento en pacientes mayores de esa edad. La disfunción de tres o más órganos durante 5 días en pacientes de cualquier edad se asocia con una mortalidad de 97 porciento. Estos datos proporcionan una guía útil para tomar decisiones en pacientes con enfermedad catastrófica.
Si el paciente sobrevive a la enfermedad aguda que causó el SIRPA, el pronóstico para la recuperación de la función pulmonar es bueno. Los factores que se asocian con mala evolución funcional pulmonar son la gravedad del SIRPA, la menor complianza y la duración de la ventilación con presión positiva.14,15 La función pulmonar mejorará con rapidez durante las primeras semanas, y después en forma más lenta durante un periodo de hasta 2 años. Los signos y síntomas comunes incluyen disnea con el ejercicio, tos, sibilancias y estertores persistentes. Las pruebas de función pulmonar pueden demostrar la presencia de enfermedad restrictiva, obstructiva, con frecuencia con mayor reactividad de las vías respiratorias, o menor capacidad de difusión. Un año o más después del inicio del SIRPA, más del 75 porciento de los pacientes tienen función respiratoria normal o sufren solo daño leve.
Causas diversas de edema pulmonar
LESIONES NEUROLOGICAS
El edema pulmonar puede ser secundario a diversas lesiones al sistema nervioso central, incluyendo las convulsiones tipo gran mal, el traumatismo craneoencefálico, la hemorragia subaracnoidea, la hemorragia intracerebral y el hematoma subdural. El común denominador en estos padecimientos del SNC es que son graves y ocurren en un momento específico en el tiempo. Lo más frecuente es que el edema pulmonar sea agudo, ocurriendo minutos a horas después del evento en el SNC. En ocasiones puede presentarse una evolución más tardía y gradual, en un periodo de varios días. La patogenia del edema pulmonar neurogénico no está bien comprendida. La forma aguda, que es la más frecuente, puede ser causada en parte por una actividad simpática intensa asociada con hipertensión sistémica, acúmulo central del volumen sanguíneo y constricción de las venas pulmonares. Esta combinación de factores puede ocasionar un aumento extraordinario, casi siempre transitorio, en la presión capilar pulmonar, y edema pulmonar con patrón cardiogénico predominante. Se cree que el aumento en la permeabilidad causado por lesión mecánica inducida por presión a los capilares pulmonares o quizá el control del SNC sobre la permeabilidad de los capilares pulmonares puede participar en el edema pulmonar neurogénico.16
Clínicamente, el edema pulmonar neurogénico suele diagnosticarse por su asociación con una lesión dramática del SNC. El principal diagnóstico diferencial es la lesión por aspiración pulmonar. A diferencia del edema neurogénico, la neumonitis química secundaria a aspiración suele persistir por más de algunos días y se complica con una infección bacteriana secundaria. Si el proceso pulmonar se aclara con rapidez (i.e, en unos días) el diagnóstico más probable será edema pulmonar neurogénico. El tratamiento del edema pulmonar neurogénico es sobre todo de sostén. No deben usarse diuréticos si no existe hipervolemia, por el riesgo de desarrollar una hipotensión hipovolémica que podría agravar la lesión del SNC en el caso de aumento de la presión intracraneal.
EXPOSICION A GRANDES ALTURAS
Se ha demostrado que se presenta edema pulmonar por altura entre los 2,500 y 5,000 m (8,202 a 16,404 pies) sobre el nivel del mar, con una incidencia de 0.5 a 15 porciento, dependiendo de factores como la edad, el sexo y la velocidad de ascenso.17 También puede ocurrir en personas que residen a gran altura y que regresan después de algunos días de estancia en un sitio a una altitud más baja.17 El inicio de los síntomas suele ser gradual, pero ocurre entre 48 y 96 horas de estar a gran altura. El edema pulmonar fulminante puede ser precedido de síntomas menos severos de enfermedad aguda de las montañas. La distribución del edema pulmonar puede ser difusa o asimétrica. La principal alteración fisiopatológica del edema pulmonar por altura parece ser el aumento en la permeabilidad capilar. El mecanismo de este aumento de permeabilidad es incierto, pero se ha sugerido que la importante vasoconstricción pulmonar inducida por hipoxia causa aumento en la perfusión de las porciones menos obstruidas del lecho vascular, lo que ocasiona lesión endotelial.17 La evidencia de este mecanismo proviene de la observación de que individuos que han experimentado edema pulmonar de altura tienen una respuesta de vasoconstricción pulmonar hipóxica más exagerada que individuos que no han tenido este padecimiento. Es posible que en el mecanismo participen mediadores edematogénicos liberados a partir de las células inflamatorias o endoteliales.17
El riesgo de edema pulmonar puede disminuirse ascendiendo en forma lenta y estable. Además, se ha demostrado que la nifedipina previene el edema pulmonar por altura en personas susceptibles.17 Cuando se presentan síntomas de enfermedad aguda de las montañas, el descenso también disminuye el riesgo de edema pulmonar. Una vez que el edema pulmonar se ha desarrollado, es obligatoria la administración de oxígeno y el descenso inmediato, que pueden salvar la vida. Incluso el descender unos cientos de metros puede ser benéfico. Se ha demostrado que la inhalación de óxido nítrico mejora la oxigenación arterial y puede ser útil en pacientes que no pueden ser evacuados a una altitud menor.18 Los individuos que han sufrido edema pulmonar por altura tienen mayor riesgo de recurrencia y deben evitar las grandes alturas.
REEXPANSION DE UN PULMON COLAPSADO
La reexpansión rápida de un pulmón colapsado puede causar edema ipsilateral o, en ocasiones, bilateral.19 El riesgo de edema pulmonar por reexpansión después de evacuar un neumotórax o un derrame pleural se relaciona con la cantidad de aire o líquido en el espacio pleural, la duración del colapso y la rapidez de la reexpansión. El desarrollo de una presión pleural negativa muy alta durante la eliminación del aire o líquido, que causa reducción marcada en la presión hidrostática del intersticio, puede ser importante en la patogenia del edema pulmonar por reexpansión. La alta concentración de proteínas en el líquido del edema sugiere que existe aumento en la permeabilidad de la membrana. Esta mayor permeabilidad puede ser causada en parte por estiramiento mecánico y deformación de los poros endoteliales o por formación de radicales tóxicos del oxígeno durante la reperfusión del pulmón que se expande con rapidez.19 La depleción del surfactante en el pulmón colapsado puede participar en la reducción de la presión hidrostática intersticial durante la reexpansión. El riesgo de edema por reexpansión pulmonar es muy bajo durante la evacuación de un neumotórax que ha estado presente durante un día o menos. En el caso de un neumotórax que tiene más de un día de duración, la evacuación por medio de un sello de agua, más que por la aplicación de presión negativa, puede disminuir el riesgo de edema. La eliminación del líquido pleural no suele causar edema pulmonar a menos que se extraiga 1.0 a 1.5 L de líquido en forma rápida. Se ha sugerido que puede extraerse cualquier cantidad de líquido con seguridad si la presión pleural se mantiene a un nivel por arriba de -20 cm H2O.20 Sin embargo, no es seguro que esta técnica prevenga siempre el edema pulmonar por reexpansión; por lo tanto, es aconsejable eliminar los derrames en forma gradual y durante varias horas. El tratamiento del edema pulmonar por reexpansión es de sostén. No existen evidencias de que los diuréticos sean benéficos.
OBSTRUCCION DE LAS VIAS RESPIRATORIAS SUPERIORES
Se ha reportado que ocurre edema pulmonar después de episodios de obstrucción de las vías respiratorias superiores causados por laringoespasmo posextubación, tumores, estrangulación o apnea obstructiva del sueño.20 Se piensa que la patogenia está relacionada con el desarrollo de presiones negativas intrapleurales muy altas (-50 a -100 cm H2O) por el esfuerzo inspiratorio vigoroso contra la vía respiratoria obstruída (maniobra de Müller). La presión negativa intrapleural tan alta disminuye la presión hidrostática intersticial, aumenta el retorno venoso e impone un aumento en la poscarga del ventrículo izquierdo. Además, esta presión puede causar activación simpática intensa, hipertensión sistémica y acúmulo central del volumen sanguíneo. Estos factores en conjunto pueden causar edema pulmonar agudo al aumentar el gradiente de presión transcapilar (i.e., la diferencia entre la presión capilar y la presión hidrostática intersticial). El padecimiento se resuelve con rapidez.
MEDICAMENTOS
Puede ocurrir edema pulmonar no cardiogénico después de la administración de diversas sustancias [ver tabla 2]. Puede presentarse edema pulmonar agudo después de la inyección intravenosa de heroína y otros narcóticos.21 Debido a que el líquido de edema tiene una alta concentración de proteínas, se ha sugerido que un defecto en la permeabilidad puede ser un factor patogénico, pero este hallazgo podría ser causado por un aumento extremo y transitorio en la presión capilar provocado por un mecanismo neurogénico. El inicio suele ser pocas horas después del uso de narcóticos, pero en ocasiones puede retrasarse hasta por 24 horas. Además de las características clínicas y radiográficas del edema pulmonar, existen los signos típicos de la intoxicación por narcóticos, como constricción pupilar, disminución en la respiración y alteración de las funciones mentales. La fiebre y la leucocitosis no necesariamente indican la presencia de una infección. Como en el caso del edema pulmonar, el principal diagnóstico diferencial es la broncoaspiración por la alteración en el nivel de conciencia.
El tratamiento es de sostén y debe incluir la intubación con ventilación mecánica, tanto para garantizar un adecuado apoyo ventilatorio como para proteger las vías respiratorias contra la aspiración. El papel de la naloxona es dudoso. Es un hecho que un paciente que sufre una sobredosis de narcóticos y presenta hipotensión o bradicardia que ponen en peligro la vida debe recibir naloxona. Si se administra naloxona a un paciente que no responde, está hipopneico y que no requiere intubación por edema pulmonar, puede evitarse la intubación. Por el contrario, un enfermo que se intuba en forma urgente por edema de pulmón y que después se estabiliza desde el punto de vista hemodinámico, puede ser manejado mejor dejando que la intoxicación por narcóticos se revierta en forma gradual, y no en forma precipitada. No existen evidencias de que la naloxona ayude a agilizar la resolución del edema pulmonar inducido por narcóticos. De hecho, se ha reportado que la naloxona causa edema pulmonar.21 Aún más, la reversión aguda de la intoxicación por narcóticos en un adicto crónico puede causar agitación, actividad simpática importante y una evolución clínica menos estable.
La cocaína causa también edema pulmonar agudo, por lo general cuando se usa la cocaína base libre.22 La fisiopatología del proceso no es clara. Al igual que la heroína, la cocaína provoca edema pulmonar con alta concentración de proteínas, lo que sugiere lesión de las células endoteliales y aumento en la permeabilidad capilar. Sin embargo, como se ha sugerido en el caso de la heroína, es posible que la cocaína produzca activación simpática extrema con aumento importante en la presión capilar que pudiera provocar un incremento transitorio en la fuga de proteínas a través de la membrana capilar. La cocaína causa también vasoconstricción coronaria, con isquemia o infarto del miocardio, lo que puede ocasionar edema pulmonar.
Reconocimientos
Figura 2 Talar Agasyan.
Figura 3 Dana Burns-Pizer.
Figura 4 Talar Agasyan.
Bibliografía
DR. GERALD W. STATON, JR.
DR. ROLAND H. INGRAM, JR.
El edema pulmonar agudo puede dividirse en forma amplia en dos categorías: edema pulmonar causado por aumento en la presión capilar (edema hidrostático o cardiogénico) y edema pulmonar causado por aumento en la permeabilidad capilar (síndrome de insuficiencia respiratoria progresiva aguda). En algunos casos ocurren tanto aumento en la presión como en la permeabilidad capilar.
Patogenia
El edema pulmonar, que consiste en el acúmulo anormal y difuso de líquido extravascular en el tejido pulmonar y en los espacios aéreos, es el tipo más frecuente de enfermedad parenquimatosa difusa aguda y no infecciosa. La patogenia del edema pulmonar puede comprenderse mejor examinando la ecuación de Starling, que define los determinantes del flujo de líquido a través de la membrana capilar pulmonar:
| Q | = | K (Pcap - Pint) | - | sigma(tcap - tint) |
en donde Q es el líquido que fluye, K es el coeficiente de filtración (directamente proporcional al área de superficie endotelial e inversamente proporcional al grosor de la pared capilar); Pcap, la presión hidrostática intravascular (capilar); Pint, la presión hidrostática intersticial; sigma, el coeficiente de reflejo de las proteínas (i.e., el grado de permeabilidad a macromoléculas); tcap, la presión coloidosmótica intravascular; y tint, la presión coloidosmótica intersticial. La primera parte de la ecuación, K (Pcap - Pint), representa la fuerza hidrostática, y la segunda porción, sigma(tcap - tint), representa la fuerza coloidosmótica. La alteración en cualquiera de los factores de la ecuación de Starling puede causar aumento en la salida transvascular de líquido. Sin embargo, en la práctica clínica solo dos de estos factores causan en forma frecuente edema pulmonar: el aumento en la presión capilar que ocasiona edema pulmonar cardiogénico o hidrostático, o la disminución del coeficiente de reflejo, que se asocia con edema pulmonar no cardiogénico o por permeabilidad.
La principal defensa contra el edema pulmonar depende del sistema linfático intrapulmonar. En condiciones normales el líquido que se filtra a través de la membrana capilar es eliminado por los vasos linfáticos. La reserva linfática es tal que incluso un aumento de 4 a 6 veces en la salida transvascular de líquido puede ser tolerada sin que aumente el agua dentro del pulmón. Una vez que la reserva linfática es superada, aumenta el agua pulmonar, primero en el intersticio alrededor de las vías respiratorias, después en el intersticio alrededor del alveolo y después dentro del alveolo. La evidencia clínica o radiográfica de edema pulmonar implica, por lo tanto, un gran aumento en la salidad transcapilar de líquido.
Características clínicas
El edema pulmonar, sea por aumento en la presión capilar o por aumento en la permealibidad, suele manifestarse en la radiografía de tórax estándar anteroposterior por opacidades alveolares simétricas y bilaterales que afectan los cuatro cuadrantes. Es frecuente la distribución perihiliar predominante, y en ocasiones existe una delimitación muy clara entre el área central de edema pulmonar y la periferia, lo que ocasiona un patrón denominado en alas de murciélago o de mariposa. Este patrón es más típico del edema pulmonar cardiogénico que del causado por aumento en la permeabilidad [ver figura 1]. Esta línea de delimitación no corresponde a límites anatómicos, pero puede ser causada por gradientes fisiológicos de ventilación, perfusión y flujo linfático de las porciones centrales del pulmón hacia la periferia. Con menos frecuencia el edema es muy asimétrico o totalmente unilateral. El edema asimétrico puede ser explicado por el hecho de que el paciente estaba recostado sobre el lado afectado cuando se desarrolló el padecimiento, o por la presencia de obstrucción vascular (tromboembolia) o enfisema en las áreas no afectadas del pulmón. Sin embargo, con frecuencia no se encuentra explicación para el edema pulmonar asimétrico o unilateral.
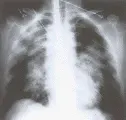
|
| Figura 1 |
| Patrón radiográfico en alas de murciélago |
Algunas características que pueden observarse en la radiografía portátil del tórax ayudan a distinguir entre el edema pulmonar cardiogénico y el causado por mayor permeabilidad. En un estudio, dos de los datos más útiles fueron el grosor del pedículo vascular central y la presencia de broncogramas aéreos discretos. Un pedículo vascular ancho sugiere aumento en la presión capilar pulmonar, y los broncogramas aéreos visibles son más frecuentes en el edema pulmonar por aumento en la permeabilidad. Otras características radiográficas, como el tamaño de la silueta cardiaca y la presencia o ausencia de derrames pleurales, tienen menor valor para hacer el diagnóstico diferencial. Por desgracia, en un caso individual no puede distinguirse con confianza entre el edema pulmonar hidrostático o por mayor permeabilidad sin utilizar pruebas cruentas.
Edema pulmonar debido a aumento en la presión capilar
PATOGENIA
El edema pulmonar que ocurre por aumento en la presión capilar suele asociarse con baja concentración de proteínas en el líquido del edema, y puede ser secundario a disfunción sistólica del ventrículo izquierdo, alteración de la válvula mitral, hipervolemia asociada con función cardiaca izquierda normal (v.gr., en un paciente con insuficiencia renal) u obstrucción pulmonar venosa. La causa más frecuente de edema pulmonar hidrostático es la disfunción del ventrículo izquierdo. En la cardiomiopatía congestiva, la función sistólica del ventrículo izquierdo se deteriora, el ventrículo se dilata y la presión final del ventrículo izquierdo (PDFVI) aumenta. Este aumento en la PDFVI causa aumento en la presión capilar. Otros tipos de enfermedad cardiaca pueden también aumentar la PDFVI, a pesar de que exista una función sistólica normal y euvolemia, al disminuir la complianza del ventrículo izquierdo. La reducción de la complianza puede ser permanente (v.gr., en la hipertrofia ventricular izquierda o en la cardiopatía restrictiva por enfermedad cardiaca infiltrativa) o transitoria (v.gr., por isquemia miocárdica). El aumento en la presión capilar con una PDFVI normal no es frecuente, pero ocurre en la estenosis mitral o como resultado de la obstrucción del flujo en las venas pulmonares (enfermedad venoclusiva pulmonar).
CARACTERISTICAS CLINICAS
Las características clínicas principales del edema pulmonar cardiogénico son disnea extrema, taquipnea y signos de importante actividad simpática, como taquicardia, hipertensión y diaforesis. La hipotensión es menos común, pero puede observarse si el edema pulmonar es secundario a un infarto del miocardio agudo extenso. La disnea rara vez se alivia al corregir la hipoxemia, lo que sugiere que la causa puede ser la activación de receptores intrapulmonares de estiramiento, más que la hipoxemia en sí. En el edema pulmonar cardiogénico puede observarse cianosis central si la hipoxemia arterial es importante, pero lo más común es que la cianosis sea de tipo periférico debida a vasoconstricción cutánea intensa. Es común el uso de los músculos accesorios de la respiración por el gran aumento del trabajo respiratorio. El esfuerzo requerido para respirar suele ser tan grande que con frecuencia se necesita intubación endotraqueal y ventilación mecánica para corregir o prevenir el desarrollo de insuficiencia respiratoria francamente hipercápnica. Al inicio del padecimiento la auscultación torácica puede revelar sibilancias causadas por edema de las vías respiratorias, después se escuchan estertores difusos.
Ciertos datos radiográficos sugieren aumento en la presión capilar (v.gr., cefalización de la circulación, ensanchamiento del pedículo vascular o líneas B de Kerley), pero ésta puede establecerse con mayor exactitud midiendo la presión de oclusión de la arteria pulmonar (la llamada presión en cuña) con un catéter que tiene un balón y se coloca en la arteria pulmonar. La presión en cuña es una medida de la presión en las venas pulmonares, y la presión venosa pulmonar se aproxima a la de los capilares pulmonares. Por lo tanto, el aumento en la presión en cuña sugiere que la elevación de la presión capilar es la causa del edema pulmonar, mientras que una presión en cuña por debajo del límite para la formación del edema pulmonar implica alteración en la permeabilidad capilar. En condiciones normales, el límite de la presión en cuña para edema pulmonar es de alrededor de 22 a 34 mm Hg. Puede existir un límite mayor (v.gr., > 25 a 30 mm Hg) cuando la presión capilar está aumentada en forma crónica, quizá porque se produce hipertrofia linfática.
Aunque la medición de la presión en cuña es útil para ayudar a aclarar la etiología del edema pulmonar, deben mencionarse ciertos detalles. Una presión en cuña por debajo del límite para edema pulmonar hidrostático (v.gr., < 18 mm Hg) no excluye el aumento en la presión capilar como causa del edema pulmonar. Esta situación se presenta sobre todo cuando el aumento en la presión es causado por una disminución temporal en la complianza del ventrículo izquierdo o por isquemia del musculo papilar con insuficiencia mitral aguda en un paciente con un infarto agudo al miocardio. Para el momento en que se coloca el catéter pulmonar la presión en cuña suele haberse normalizado. También ocurren aumentos importantes y transitorios en la presión capilar en el edema pulmonar neurogénico y con los aumentos súbitos en la presión arterial.
En los pacientes con edema pulmonar causado por la poco frecuente enfermedad venoclusiva pulmonar, la presión capilar es mucho más alta que la presión en las venas pulmonares; por lo tanto, la presión en cuña subestima la presión que determina la filtración de líquido a través de la membrana capilar. Por último, a pesar de la aparente lógica para el uso de cáteteres pulmonares, debe mencionarse que no se ha atribuido ningún efecto benéfico en la evolución por su uso. De hecho, un estudio reciente de gran número de pacientes en unidades de cuidados intensivos ha sugerido que los pacientes que tuvieron catéteres arteriales pulmonares sufrieron mayor mortalidad y mayor gasto económico que los pacientes que no.1 Este reporte ha sido criticado, pero es el único análisis publicado sobre el efecto del uso de estos catéteres en la evolución de los pacientes de la UCI.
TRATAMIENTO
Después de que la hipoxemia es aliviada con oxígeno suplementario, el tratamiento del edema pulmonar causado por aumento en la presión capilar se dirige a disminuir las presiones de llenado del ventrículo izquierdo y a mejorar la función cardiaca. La morfina, los diuréticos y la nitroglicerina sublingual o intravenosa aumentan la capacitancia venosa y disminuyen la precarga. La reducción de la precarga con medicamentos vasodilatadores, como nitroprusiato intravenoso (que también reduce la precarga), o inhibidores de la enzima convertidora de angiotensina mejoran el gasto cardiaco. En los pacientes muy graves o que responden con lentitud a estas medidas la ventilación no invasiva con aplicación nasal de presión positiva al final de la espiración o la intubación y aplicación de ventilación con presión positiva pueden mejorar la oxigenación y disminuir el retorno venoso.
Edema pulmonar causado por aumento en la permeabilidad capilar: Síndrome de insuficiencia respiratoria progresiva aguda
El síndrome de insuficiencia respiratoria progresiva aguda (SIRPA) se caracteriza por lesión difusa al endotelio pulmonar, que causa edema pulmonar por aumento importante en la permeabilidad capilar al agua, solutos y macromoléculas. A diferencia del edema pulmonar que es causado por alteración en las fuerzas de Starling, el edema pulmonar en el SIRPA se caracteriza por aumento en la concentración de proteínas en el líquido de edema, que llega a ser hasta de 80 a 90 porciento de las proteínas séricas. Aún más, la presencia de muchos neutrófilos y sus productos secretorios (v.gr., elastasa y colagenasa) en el líquido del lavado broncoalveolar demuestra la respuesta inflamatoria subyacente que no es característica del edema pulmonar cardiogénico.
Muchas lesiones hematógenas y aéreas pueden producir SIRPA [ver tabla 1]. El riesgo de desarrollar SIRPA como resultado de una de ellas puede aumentar mucho si existe historia de abuso de alcohol.3
|
||||||||||||||||||
|
PATOGENIA
El daño alveolar difuso (DAD) es el término descriptivo para la secuencia de cambios inespecíficos, pero predecibles, que ocurren en forma característica en los pacientes con SIRPA2. Por lo general, el agente o proceso causal no puede ser determinado por los datos histopatológicos. Además, las alteraciones pueden resolverse en cualquier momento de la evolución de la enfermedad. La apariencia histológica del DAD varía durante el periodo entre el evento precipitante y la biopsia o autopsia de acuerdo con tres fases: una fase exudativa aguda (días 0 a 7), una fase proliferativa subaguda o no organizada (días 7 a 14) y una fase crónica (después del día 14) [ver figura 2].
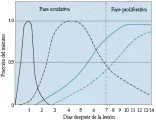
|
| Figura 2 |
| Cambios patológicos en el SIRPA |
La primera parte de la fase aguda exudativa se caracteriza por edema intersticial e intralveolar, infiltración neutrofílica, hemorragia y depósito de fibrina. Se deposita una mezcla de fibrina y detritos celulares en el espacio alveolar para formar las llamadas membranas hialinas que son prominentes 3 a 7 días después de la lesión. El desprendimiento de las células de la superficie alveolar deja una membrana basal denudada que es muy importante para la reparación o fibrosis subsecuente. El infiltrado intersticial de células inflamatorias se vuelve más intenso alrededor del día 7 y persiste durante toda la fase proliferativa.
La denudación de la membrana basal causa que los neumocitos tipo II proliferen (días 3 a 7), produciendo un patrón de hiperplasia de las células superficiales alveolares. En los pacientes en los que se resuelve el síndrome, estas células proliferantes se diferencían a neumocitos tipo I, restableciendo el lado epitelial de la pared alveolocapilar y normalizando el intercambio de gases.
La fase proliferativa del SIRPA se caracteriza por inflamación intersticial y proliferación de fibroblastos, al inicio en el intersticio. Los fibroblastos invaden los espacios alveolares a través de los defectos de la membrana basal, un proceso que produce áreas de fibrosis intralveolar. Durante esta fase las membranas hialinas desaparecen como resultado de fagocitosis o de organización con incorporación del exudado en acúmulos intralveolares de fibroblastos proliferantes.
La fase crónica del SIRPA se manifiesta por regiones de fibrosis intensa, regiones focales de expansión excesiva y obliteración vascular pulmonar. Desde el punto de vista histológico, esta fase de la enfermedad puede ser semejante a la fibrosis pulmonar idiopática (FPI). Sin embargo, a diferencia de este padecimiento, la fase crónica del SIRPA puede mejorar con el tiempo si el paciente sobrevive.
Aunque los mecanismos que causan el SIRPA han sido estudiados en forma muy intensa, aún no se comprenden del todo.4 Los dos sitios de lesión temprana clave en el proceso son el endotelio del lecho capilar pulmonar y el epitelio alveolar [ver figura 3].
|
|
A través de la expresión de moléculas de adhesión en la superficie celular (selectinas e integrinas), los neutrófilos se adhieren al endotelio y liberan oxidantes dañinos, enzimas proteolíticas y metabolitos del ácido araquidónico, lo que causa disfunción de las células endoteliales y destrucción y denudación del lado endotelial de la membrana basal. Además, la endotoxina puede lesionar las células endoteliales en forma directa. Los macrófagos, que tienen capacidades secretoras semejantes a las de los neutrófilos pueden participar en esta lesión, y esto podría explicar el desarrollo de SIRPA en los pacientes con neutropenia. El aumento en la permeabilidad de la membrana alveolocapilar permite al plasma y a las células entrar a los espacios intersticiales del pulmón y, al final, al alveolo. En el resto del organismo ocurre un tipo de daño microvascular semejante, que puede participar en la disfunción orgánica múltiple asociada al SIRPA causado por sepsis u otros padecimientos.
El segundo sitio de lesión en el SIRPA temprano, el epitelio alveolar, es muy importante para la fisiología pulmonar normal porque las uniones estrechas entre las células del epitelio alveolar evitan que el líquido intersticial penetre a los espacios alveolares. Las células tienen un sistema activo de transporte de sodio sensible a la acción agonista beta que puede ayudar a eliminar líquido de los espacios alveolares, y los neumocitos tipo II producen surfactante. A las pocas horas de la lesión pulmonar aguda los neumocitos tipo I mueren, denudando el lado alveolar de la membrana basal. Se desconoce la causa exacta de esta fase de la lesión pulmonar; sin embargo, existen neutrófilos y macrófagos alveolares activados en los espacios alveolares en etapas tempranas del padecimiento que quizá participen en el daño. Se ha encontrado elastasa y otras enzimas proteolíticas de los neutrófilos y macrófagos en el líquido de lavado bronquioalveolar de pacientes con SIRPA. Aunque el plasma contiene varias proteínas antiproteolíticas, estas pueden inactivarse por los oxidantes originados por las células inflamatorias, lo que permite a las enzimas proteolíticas activarse en forma local. Los oxidantes, como el peróxido de hidrógeno y los radicales hidroxilo pueden lesionar también las células en forma directa. Además, los macrófagos producen sustancias procoagulantes, como factor tisular y factor VII, que activan factores de la coagulación que alcanzan los espacios intersticial y alveolar y producen fibrina.6 La fibrina inhibe el surfactante y, con otros detritos celulares, produce las membranas hialinas que se observan en las fases tempranas del SIRPA. La producción de surfactante por las células tipo II restantes puede también disminuir por acción del FNT.7 La combinación de menor producción e inhibición causa una profunda deficiencia de surfactante.8
Una vez que ha ocurrido la lesión pulmonar aguda, el proceso de reparación puede causar resolución completa o fibrosis progresiva. El factor crítico para ello es la gravedad de la lesión en el lado epitelial de la membrana alveolocapilar. Los neumocitos epiteliales II comienzan a proliferar desde la fase más temprana del proceso de reparación en un intento por recuperar la membrana basal denudada. Si el daño a la membrana basal no es grave, es más probable que las células tipo II tengan éxito. Una vez reparada la membrana basal, los neumocitos epiteliales participan en la remoción de líquido y detritos de los espacios alveolares, así como en la síntesis de nuevo surfactante. Con el tiempo, las células nuevas se diferencían en células tipo I y restablecen la delgada estructura de la membrana que es crítica para el proceso de intercambio de gases.
Si las células epiteliales y la membrana basal han sido lesionadas en forma grave, ocurrirá fibrosis intersticial e intralveolar aguda y progresiva. La fibrosis se presenta cuando los fibroblastos, que son controlados por factores de crecimiento (muchos derivados de los macrófagos) y que usan probablemente una matriz de fibrina y fibronectina como base, invaden el exudado, y en él proliferan y sintetizan colágena (tipo I), elastina y otros componentes de la matriz. La arquitectura pulmonar normal es remplazada en forma progresiva por tejido fibroso que deteriora en forma severa el intercambio de gases.
CARACTERISTICAS CLINICAS
Los signos clínicos del edema pulmonar causado por aumento en la permeabilidad son un poco diferentes de los del edema pulmonar cardiogénico. Suele existir menor actividad simpática y, cuando existe cianosis, es causada por hipoxemia arterial. Cuando la sepsis es la causa del edema pulmonar la piel puede estar caliente en lugar de fría, húmeda y pálida, y el pulso puede ser hiperdinámico.
Independientemente de la etiología y patogenia subyacentes, las características clínicas y fisiológicas del SIRPA son semejantes. Los datos clínicos incluyen disnea y taquipnea, con un patrón respiratorio caracterizado por disminución del volumen corriente y aumento de la frecuencia respiratoria. Suele requerirse de la ventilación mecánica por la incapacidad del paciente para mantener una ventilación por minuto adecuada, por el aumento importante en el trabajo respiratorio y por la hipoxemia grave que suele ser refractaria al aumento en la cantidad de oxígeno inspirado.
Es típico que la radiografía de tórax demuestre un proceso de ocupación alveolar difuso y simétrico. Cuando se realiza tomografía computada, la distribución de los espacios ocupados suele observarse menos simétrica. En las radiografías que se realizan en posición supina suele apreciarse un mayor grado de consolidación en las zonas posteriores del pulmón; sin embargo, esta distribución puede revertirse cambiando al paciente a la posición prona durante algunas horas. En esta última posición aumenta la consolidación en las áreas anteriores (dependientes de la gravedad) y las posteriores muestran mejor ventilación. Este dato demuestra la contribución de las fuezas de Starling (i.e., presión capilar) a la gravedad del edema en el caso de aumento en la permeabilidad.
El intercambio gaseoso en el SIRPA se caracteriza al principio por hipoxemia que es relativamente refractaria al aumento en las concentraciones de oxígeno inspirado, lo que implica la presencia de cortocircuitos intrapulmonares. Estas son consecuencia principalmente de colapso alveolar difuso con microatelectasias. Se cree que el colapso alveolar difuso es causado por alteraciones en el surfactante, una sustancia que normalmente ayuda a mantener la distensión alveolar al reducir la tensión superficial en la interfase aire-líquido del pulmón.
Al principio la presión arterial de bióxido de carbono (PaCO2) es baja o se mantiene dentro del rango normal, con sólo un pequeño aumento en la ventilación minuto. Sin embargo, la relación espacio muerto a volumen corriente (VD/VT) tiende a aumentar durante el tiempo, por lo que se requiere una ventilación por minuto cada vez mayor para mantener una PaCO2 normal. En algunos casos el aumento en la VD/VT es tan importante que no puede evitarse la hipercapnia a pesar de que la ventilación por minuto se aumente al máximo. El aumento en el espacio muerto fisiológico se debe a daño en el lecho capilar pulmonar, con lo que se crean regiones de alta ventilación en relación con la perfusión. Otra causa del aumento en el requerimiento de la ventilación por minuto en el SIRPA es la presencia de una mayor producción de bióxido de carbono por el hipermetabolismo. La alta relación VD/VT del SIRPA se resuelve en forma muy lenta y, junto con la reducción en la complianza pulmonar que impone una carga sobre la respiración espontánea, es responsable de que con frecuencia se requiera ventilación mecánica durante periodos muy prolongados. Es frecuente que el apoyo ventiltorio deba continuarse a pesar de que la hipoxemia haya mejorado lo suficiente para que sólo se requiera un modesto aumento en la concentración inspirada de oxígeno.
La mecánica pulmonar en el SIRPA se caracteriza principalmente por reducción en la complianza pulmonar (mayor elastancia), por lo que se requieren mayores presiones transpulmonares para alcanzar una ventilación corriente normal. En las etapas tempranas del SIRPA, cuando predomina el edema, mucho de la presión de distensión que se necesita para inflar el pulmón es necesaria para abrir los aveolos colapsados. De hecho, la complianza pulmonar (la pendiente de la curva presión-volumen) puede estar dentro del rango normal si se mide después de que estos alveolos colapsados han sido distendidos. Sin embargo, existe una reducción significativa en la complianza al evolucionar el padecimiento, cuando predomina la fibrosis alveolar. Esta reducción aparente puede ser causada por un engrosamiento difuso de la membrana alveolocapilar como resultado de fibrosis o por pérdida de un gran porcentaje de unidades alveolocapilares disponibles para la ventilación. Se ha sugerido que la mecánica pulmonar alterada en el SIRPA se conceptualiza mejor si se considera como causada por un pulmón pequeño, más que por un pulmón rígido.9 También se observa en el SIRPA aumento en la resistencia de las vías aéreas que responde a broncodilatadores inhalados.10
Además de su efecto sobre la relación VD/VT, los cambios en la vasculatura pulmonar en el SIRPA son también resultado de aumento en las resistencias de los vasos pulmonares y de hipertensión pulmonar. De hecho, la presión arterial pulmonar está elevada en casi todos los pacientes con SIRPA moderado a severo. La etiología de la hipertensión pulmonar en este síndrome parece ser multifactorial; sin embargo, una causa importante puede ser la presencia de pequeños trombos en la arteria pulmonar. Estos trombos pueden demostrarse por medio de una angiografía pulmonar realizada en la cama del paciente. La patogenia de los trombos en el SIRPA no es clara, pero es probable que se formen in situ.
Muchos pacientes con SIRPA tienen ciertos indicios de coagulación intravascular acelerada.11 Sin embargo, a pesar de la importancia aparente de los trombos para producir hipertensión pulmonar, no se ha determinado si la anticoagulación es benéfica en los pacientes con SIRPA.
El manejo de los pacientes con SIRPA se discute con detalle en la Subsección VIII.
EVOLUCION
Aunque el grado de lesión pulmonar en el SIRPA se relaciona con el pronóstico, otros parámetros predicen en forma más exacta la evolución. Esta observación no es sorprendente porque el SIRPA es casi siempre parte de un síndrome de respuesta inflamatoria sistémica. Con frecuencia no es clara la causa del síndrome, pero las posibilidades incluyen bronconeumonía, traslocación de productos bacterianos a través del intestino y liberación persistente de mediadores endógenos en ausencia de infección activa. La sepsis, que con frecuencia se asocia con vasodilatación que no responde a vasoconstrictores, es la causa más frecuente de muerte durante la enfermedad. Como resultado de la disponibilidad de técnicas excelentes para el apoyo ventilatorio, la insuficiencia respiratoria es causa de muerte en solo el 10 porciento de los casos en fase temprana y el 18 porciento de los más avanzados. Este hecho destaca la importancia de la disfunción en otros sistemas, como la falla hemodinámica con choque refractario o la insuficiencia renal progresiva, como causa de morbimortalidad. Los datos sugieren que la evolución de los pacientes con SIRPA puede estar mejorando, quizá como consecuencia de un mejor tratamiento.12
La mortalidad está determinada sobre todo por el número de sistemas que fallan. De acuerdo con Knaus y Wagner, la mortalidad aumenta en forma proporcional con el número de órganos disfuncionantes y el número de días de la disfunción13 [ver figura 4]. Existe una mayor mortalidad en los pacientes mayores de 65 años. Por ejemplo, cuando ha existido un solo órgano insuficiente durante 5 días en un paciente menor de 65 años la mortalidad es de 27 porciento, y en un paciente mayor de 65 años, de 48 porciento. De igual modo, la falla de dos órganos durante 2 días causa una mortalidad del 47 porciento en enfermos menores de 65 años y de 73 porciento en pacientes mayores de esa edad. La disfunción de tres o más órganos durante 5 días en pacientes de cualquier edad se asocia con una mortalidad de 97 porciento. Estos datos proporcionan una guía útil para tomar decisiones en pacientes con enfermedad catastrófica.
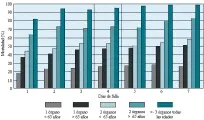
|
| Figura 4 |
| Tasas de mortalidad en SIRPA |
Si el paciente sobrevive a la enfermedad aguda que causó el SIRPA, el pronóstico para la recuperación de la función pulmonar es bueno. Los factores que se asocian con mala evolución funcional pulmonar son la gravedad del SIRPA, la menor complianza y la duración de la ventilación con presión positiva.14,15 La función pulmonar mejorará con rapidez durante las primeras semanas, y después en forma más lenta durante un periodo de hasta 2 años. Los signos y síntomas comunes incluyen disnea con el ejercicio, tos, sibilancias y estertores persistentes. Las pruebas de función pulmonar pueden demostrar la presencia de enfermedad restrictiva, obstructiva, con frecuencia con mayor reactividad de las vías respiratorias, o menor capacidad de difusión. Un año o más después del inicio del SIRPA, más del 75 porciento de los pacientes tienen función respiratoria normal o sufren solo daño leve.
Causas diversas de edema pulmonar
LESIONES NEUROLOGICAS
El edema pulmonar puede ser secundario a diversas lesiones al sistema nervioso central, incluyendo las convulsiones tipo gran mal, el traumatismo craneoencefálico, la hemorragia subaracnoidea, la hemorragia intracerebral y el hematoma subdural. El común denominador en estos padecimientos del SNC es que son graves y ocurren en un momento específico en el tiempo. Lo más frecuente es que el edema pulmonar sea agudo, ocurriendo minutos a horas después del evento en el SNC. En ocasiones puede presentarse una evolución más tardía y gradual, en un periodo de varios días. La patogenia del edema pulmonar neurogénico no está bien comprendida. La forma aguda, que es la más frecuente, puede ser causada en parte por una actividad simpática intensa asociada con hipertensión sistémica, acúmulo central del volumen sanguíneo y constricción de las venas pulmonares. Esta combinación de factores puede ocasionar un aumento extraordinario, casi siempre transitorio, en la presión capilar pulmonar, y edema pulmonar con patrón cardiogénico predominante. Se cree que el aumento en la permeabilidad causado por lesión mecánica inducida por presión a los capilares pulmonares o quizá el control del SNC sobre la permeabilidad de los capilares pulmonares puede participar en el edema pulmonar neurogénico.16
Clínicamente, el edema pulmonar neurogénico suele diagnosticarse por su asociación con una lesión dramática del SNC. El principal diagnóstico diferencial es la lesión por aspiración pulmonar. A diferencia del edema neurogénico, la neumonitis química secundaria a aspiración suele persistir por más de algunos días y se complica con una infección bacteriana secundaria. Si el proceso pulmonar se aclara con rapidez (i.e, en unos días) el diagnóstico más probable será edema pulmonar neurogénico. El tratamiento del edema pulmonar neurogénico es sobre todo de sostén. No deben usarse diuréticos si no existe hipervolemia, por el riesgo de desarrollar una hipotensión hipovolémica que podría agravar la lesión del SNC en el caso de aumento de la presión intracraneal.
EXPOSICION A GRANDES ALTURAS
Se ha demostrado que se presenta edema pulmonar por altura entre los 2,500 y 5,000 m (8,202 a 16,404 pies) sobre el nivel del mar, con una incidencia de 0.5 a 15 porciento, dependiendo de factores como la edad, el sexo y la velocidad de ascenso.17 También puede ocurrir en personas que residen a gran altura y que regresan después de algunos días de estancia en un sitio a una altitud más baja.17 El inicio de los síntomas suele ser gradual, pero ocurre entre 48 y 96 horas de estar a gran altura. El edema pulmonar fulminante puede ser precedido de síntomas menos severos de enfermedad aguda de las montañas. La distribución del edema pulmonar puede ser difusa o asimétrica. La principal alteración fisiopatológica del edema pulmonar por altura parece ser el aumento en la permeabilidad capilar. El mecanismo de este aumento de permeabilidad es incierto, pero se ha sugerido que la importante vasoconstricción pulmonar inducida por hipoxia causa aumento en la perfusión de las porciones menos obstruidas del lecho vascular, lo que ocasiona lesión endotelial.17 La evidencia de este mecanismo proviene de la observación de que individuos que han experimentado edema pulmonar de altura tienen una respuesta de vasoconstricción pulmonar hipóxica más exagerada que individuos que no han tenido este padecimiento. Es posible que en el mecanismo participen mediadores edematogénicos liberados a partir de las células inflamatorias o endoteliales.17
El riesgo de edema pulmonar puede disminuirse ascendiendo en forma lenta y estable. Además, se ha demostrado que la nifedipina previene el edema pulmonar por altura en personas susceptibles.17 Cuando se presentan síntomas de enfermedad aguda de las montañas, el descenso también disminuye el riesgo de edema pulmonar. Una vez que el edema pulmonar se ha desarrollado, es obligatoria la administración de oxígeno y el descenso inmediato, que pueden salvar la vida. Incluso el descender unos cientos de metros puede ser benéfico. Se ha demostrado que la inhalación de óxido nítrico mejora la oxigenación arterial y puede ser útil en pacientes que no pueden ser evacuados a una altitud menor.18 Los individuos que han sufrido edema pulmonar por altura tienen mayor riesgo de recurrencia y deben evitar las grandes alturas.
REEXPANSION DE UN PULMON COLAPSADO
La reexpansión rápida de un pulmón colapsado puede causar edema ipsilateral o, en ocasiones, bilateral.19 El riesgo de edema pulmonar por reexpansión después de evacuar un neumotórax o un derrame pleural se relaciona con la cantidad de aire o líquido en el espacio pleural, la duración del colapso y la rapidez de la reexpansión. El desarrollo de una presión pleural negativa muy alta durante la eliminación del aire o líquido, que causa reducción marcada en la presión hidrostática del intersticio, puede ser importante en la patogenia del edema pulmonar por reexpansión. La alta concentración de proteínas en el líquido del edema sugiere que existe aumento en la permeabilidad de la membrana. Esta mayor permeabilidad puede ser causada en parte por estiramiento mecánico y deformación de los poros endoteliales o por formación de radicales tóxicos del oxígeno durante la reperfusión del pulmón que se expande con rapidez.19 La depleción del surfactante en el pulmón colapsado puede participar en la reducción de la presión hidrostática intersticial durante la reexpansión. El riesgo de edema por reexpansión pulmonar es muy bajo durante la evacuación de un neumotórax que ha estado presente durante un día o menos. En el caso de un neumotórax que tiene más de un día de duración, la evacuación por medio de un sello de agua, más que por la aplicación de presión negativa, puede disminuir el riesgo de edema. La eliminación del líquido pleural no suele causar edema pulmonar a menos que se extraiga 1.0 a 1.5 L de líquido en forma rápida. Se ha sugerido que puede extraerse cualquier cantidad de líquido con seguridad si la presión pleural se mantiene a un nivel por arriba de -20 cm H2O.20 Sin embargo, no es seguro que esta técnica prevenga siempre el edema pulmonar por reexpansión; por lo tanto, es aconsejable eliminar los derrames en forma gradual y durante varias horas. El tratamiento del edema pulmonar por reexpansión es de sostén. No existen evidencias de que los diuréticos sean benéficos.
OBSTRUCCION DE LAS VIAS RESPIRATORIAS SUPERIORES
Se ha reportado que ocurre edema pulmonar después de episodios de obstrucción de las vías respiratorias superiores causados por laringoespasmo posextubación, tumores, estrangulación o apnea obstructiva del sueño.20 Se piensa que la patogenia está relacionada con el desarrollo de presiones negativas intrapleurales muy altas (-50 a -100 cm H2O) por el esfuerzo inspiratorio vigoroso contra la vía respiratoria obstruída (maniobra de Müller). La presión negativa intrapleural tan alta disminuye la presión hidrostática intersticial, aumenta el retorno venoso e impone un aumento en la poscarga del ventrículo izquierdo. Además, esta presión puede causar activación simpática intensa, hipertensión sistémica y acúmulo central del volumen sanguíneo. Estos factores en conjunto pueden causar edema pulmonar agudo al aumentar el gradiente de presión transcapilar (i.e., la diferencia entre la presión capilar y la presión hidrostática intersticial). El padecimiento se resuelve con rapidez.
MEDICAMENTOS
Puede ocurrir edema pulmonar no cardiogénico después de la administración de diversas sustancias [ver tabla 2]. Puede presentarse edema pulmonar agudo después de la inyección intravenosa de heroína y otros narcóticos.21 Debido a que el líquido de edema tiene una alta concentración de proteínas, se ha sugerido que un defecto en la permeabilidad puede ser un factor patogénico, pero este hallazgo podría ser causado por un aumento extremo y transitorio en la presión capilar provocado por un mecanismo neurogénico. El inicio suele ser pocas horas después del uso de narcóticos, pero en ocasiones puede retrasarse hasta por 24 horas. Además de las características clínicas y radiográficas del edema pulmonar, existen los signos típicos de la intoxicación por narcóticos, como constricción pupilar, disminución en la respiración y alteración de las funciones mentales. La fiebre y la leucocitosis no necesariamente indican la presencia de una infección. Como en el caso del edema pulmonar, el principal diagnóstico diferencial es la broncoaspiración por la alteración en el nivel de conciencia.
|
|
||
|
El tratamiento es de sostén y debe incluir la intubación con ventilación mecánica, tanto para garantizar un adecuado apoyo ventilatorio como para proteger las vías respiratorias contra la aspiración. El papel de la naloxona es dudoso. Es un hecho que un paciente que sufre una sobredosis de narcóticos y presenta hipotensión o bradicardia que ponen en peligro la vida debe recibir naloxona. Si se administra naloxona a un paciente que no responde, está hipopneico y que no requiere intubación por edema pulmonar, puede evitarse la intubación. Por el contrario, un enfermo que se intuba en forma urgente por edema de pulmón y que después se estabiliza desde el punto de vista hemodinámico, puede ser manejado mejor dejando que la intoxicación por narcóticos se revierta en forma gradual, y no en forma precipitada. No existen evidencias de que la naloxona ayude a agilizar la resolución del edema pulmonar inducido por narcóticos. De hecho, se ha reportado que la naloxona causa edema pulmonar.21 Aún más, la reversión aguda de la intoxicación por narcóticos en un adicto crónico puede causar agitación, actividad simpática importante y una evolución clínica menos estable.
La cocaína causa también edema pulmonar agudo, por lo general cuando se usa la cocaína base libre.22 La fisiopatología del proceso no es clara. Al igual que la heroína, la cocaína provoca edema pulmonar con alta concentración de proteínas, lo que sugiere lesión de las células endoteliales y aumento en la permeabilidad capilar. Sin embargo, como se ha sugerido en el caso de la heroína, es posible que la cocaína produzca activación simpática extrema con aumento importante en la presión capilar que pudiera provocar un incremento transitorio en la fuga de proteínas a través de la membrana capilar. La cocaína causa también vasoconstricción coronaria, con isquemia o infarto del miocardio, lo que puede ocasionar edema pulmonar.
Figura 2 Talar Agasyan.
Figura 3 Dana Burns-Pizer.
Figura 4 Talar Agasyan.
Bibliografía
-
Speroff T, Dawson NV, et al: The effectiveness of right heart
catheterization in the care of critically ill patients. SUPPORT Investigators.
JAMA 276:889, 1996
-
Kollef MH, Schuster DP: The acute respiratory distress syndrome.
N Engl J Med 332:27, 1995 [PMID
7646623]
-
Moss M, Bucher B, Moore FA, et al: The role of chronic alcohol
abuse in the development of acute respiratory distress syndrome in adults.
JAMA 275:50, 1996 [PMID
8531287]
-
Pittet JF, Mackersie RC, Martin TR, et al: Biological markers
of acute lung injury: prognostic and pathogenetic significance. Am J Respir
Crit Care Med 155:1187, 1997 [PMID
9105054]
-
Meduri GU: The role of the host defence response in the progression
and outcome of ARDS: pathophysiological correlations and response to glucocorticoid
treatment. Eur Respir J 9:2650, 1996
-
Fuchs-Buder T, de Moerloose P, Ricou B, et al: Time course
of procoagulant activity and D dimer in bronchoalveolar fluid of patients
at risk for or with acute respiratory distress syndrome. Am J Respir Crit
Care Med 153:163, 1996 [PMID
8542111]
-
Arias-Diaz J, Vara E, Garc'a C, et al: Tumor necrosis factor-a-induced
inhibition of phosphatidylcholine synthesis by human type II pneumocytes
is partially mediated by prostaglandins. J Clin Invest 94:244, 1994 [PMID
8040266]
-
Gunther A, Siebert C, Schmidt R, et al: Surfactant alterations
in severe pneumonia, acute respiratory distress syndrome, and cardiogenic
lung edema. Am J Respir Crit Care Med 153:176, 1996 [PMID
8542113]
-
Pelosi P, Crotti S, Brazzi L, et al: Computed tomography
in adult respiratory distress syndrome: what has it taught us? Eur Respir
J 9:1055, 1996
-
Wright PE, Carmichael LC, Bernard GR: Effect of bronchodilators
on lung mechanics in the acute respiratory distress syndrome (ARDS). Chest
106:1517, 1994 [PMID
7956413]
-
Hasegawa N, Husari AW, Hart WT, et al: Role of the coagulation
system in ARDS. Chest 105:268, 1994 [PMID
8275746]
-
Milberg JA, Davis DR, Steinberg KP, et al: Improved survival
of patients with acute respiratory distress syndrome (ARDS): 1983-1993.
Jama 273:306, 1995 [PMID
7815658]
-
Knaus W, Wagner DP: Multiple systems organ failure: epidemiology
and prognosis. Crit Care Clin 5:221, 1989 [PMID
2650815]
-
Suchyta MR, Elliott CG, Jensen RL, et al: Predicting the
presence of pulmonary function impairment in adult respiratory distress
syndrome survivors. Respiration 60:103, 1993 [PMID
8341851]
-
McHugh LG, Milberg JA, Whitcomb ME, et al: Recovery of function
in survivors of the acute respiratory distress syndrome. Am J Respir Crit
Care Med 150:90, 1994 [PMID
8025779]
-
Simon RP: Neurogenic pulmonary edema. Neurol Clin 11:309,
1993
-
Hultgren HN: High-altitude pulmonary edema: current concepts.
Annu Rev Med 47:267, 1996
-
Scherrer U, Vollenweider L, Delabays A, et al: Inhaled nitric
oxide for high-altitude pulmonary edema. N Engl J Med 334:624, 1996 [PMID
8592525]
-
Tarver RD, Broderick LS, Conces DJ Jr: Reexpansion pulmonary
edema. J Thoracic Imaging 11:198, 1996
-
Kollef MH, Pluss J: Noncardiogenic pulmonary edema following
upper airway obstruction: 7 cases and a review of the literature. Medicine
(Baltimore) 70:91, 1991 [PMID
2005779]
-
Parsons PE: Respiratory failure as a result of drugs,
overdoses, and poisonings. Clin Chest Med 15:93, 1994
-
Albertson TE, Walby WF, Derlet RW: Stimulant-induced pulmonary
toxicity. Chest 108:1140, 1995 [PMID
7555128]
- Katzenstein A-LA, Askin FB: Acute lung injury patterns:diffuse alveolar damage, acute interstitial pneumonia, bronchitis obliterans-organizing pneumonia. Surgical Pathology of Non-neoplastic Lung Disease. Katzenstein A-LA, Ed. WB Saunders Co, Philadelphia, 1990