Oncología
⭳ Abrir artículo (PDF)173.1 KBEste artículo es idéntico en la Edición 3/2000.
Contenido del artículo
XIII TUMORES OSEOS
XIII TUMORES OSEOS
DR. STEPHEN M. KRANE
DR. LEE SIMON
Fisiopatología
Las neoplasias óseas se originan de los linajes mesenquimatoso, hematopoyético y vascular. Las células mesenquimatosas incluyen condrocitos, osteoblastos y fibroblastos del estroma; las células hematopoyéticas son los precursores de los osteoclastos, y las células vasculares (endoteliales y de músculo liso) son componentes de los vasos sanguíneos. La índole de las neoplasias óseas con frecuencia refleja el fenotipo normal de las células que los componen, como su morfología y organización multicelular y el tipo de matriz extracelular que producen.
La localización de los tumores óseos y la reacción que inducen en el tejido que los rodea influyen en su evolución. Por ejemplo, el cartílago articular o la placa de crecimiento parecen proporcionar una barrera a la diseminación del tejido neoplásico. Aunque las lesiones benignas en ocasiones se relacionan con la formación de hueso nuevo denso en el tejido circundante, las neoplasias malignas, así como las benignas, suelen inducir la resorción del hueso que los rodea. Parece ser que el tumor o las células normales que infiltran el tumor, como los monocitos-macrófagos, producen sustancias que directa o indirectamente estimulan a los osteoclastos para la resorción ósea; estas sustancias pueden también inducir el reclutamiento de los osteoclastos y causar su diferenciación a partir de células precursoras.1 Las sustancias que podrían actuar de esta manera son los factores transformadores del crecimiento, la interleucina-1, el factor de necrosis tumoral (FNT) y un péptido derivado del tumor relacionado con la hormona paratiroidea que comparte algunas secuencias con la porción amino terminal de esta hormona .2,3
Clasificación de los tumores óseos
MIELOMA MULTIPLE
El tumor maligno de hueso más frecuente en los adultos es el mieloma múltipe, que se origina de células del linaje de los linfocitos. Se diagnostican aproximadamente 10,000 casos nuevos de mieloma múltiple cada año en los Estados Unidos. Las células del mieloma producen una linfotoxina con estructura similar al FNT y que se une al mismo receptor que el FNT. Esta linfotoxina puede ser responsable del mayor número de osteoclastos que se observan en el hueso no afectado en la periferia de las lesiones del mieloma, que casi siempre son osteolíticas.
Los linfomas malignos pueden ser primarios de hueso, en donde de manera típica inducen una respuesta osteolítica; sin embargo, la enfermedad de Hodgkin puede producir lesiones esclerosas en el hueso.
TUMOR DE EWING
El tumor o sarcoma de Ewing es un tumor maligno poco frecuente que casi siempre ocurre en individuos menores de 30 años.4 Los sitios afectados con más frecuencia son la pelvis, el fémur y la tibia. En términos generales, el tumor de Ewing es considerado el más letal de todos los tumores óseos; las metástasis, que con frecuencia afectan el pulmón, la pleura o los ganglios linfáticos, están presentes en el momento del diagnóstico en el 15 a 35 porciento de los pacientes, dependiendo de la serie revisada.4,5 El examen histológico demuestra que estas neoplasias están compuestas por pequeñas células redondas que varían poco en tamaño y forma y que poseen mínimo estroma. Aunque la célula que da origen al tumor de Ewing se desconoce, la evidencia obtenida por medio de tinción específica para enolasa neuronal y la utilización de anticuerpos Leu-7 monoclonales sugiere que esta célula podría ser de origen neuroectodérmico.6 Además, los pacientes con sarcoma de Ewing, así como los que tienen tumores neuroectodérmicos periféricos malignos, tienen una traslocación característica entre los cromosomas 11 y 22,7,8 que parece causar la producción de un oncogén de fusión.9
Antes de 1970, sólo un cinco porciento de los pacientes tratados con cirugía o radiación solas alcanzaban una supervivencia prolongada. La terapéutica multimodal con cirugía, radiación local y quimioterapia adyuvante (con frecuencia una combinación de ciclofosfamida y doxorrubicina) ha permitido una supervivencia libre de enfermedad a largo plazo en alrededor del 50 porciento de los pacientes, aún en aquellos individuos con metástasis evidentes en el momento del diagnóstico.4,5,10 El pronóstico para los pacientes con linfoma no Hodgkin del hueso tratado, lesión que en ocasiones puede ser confundida con el tumor de Ewing, es mucho mejor que el de este último, y por lo tanto estas dos lesiones deben diferenciarse con claridad.12
CONDROSARCOMAS
Los condrosarcomas son tumores óseos primarios compuestos por células neoplásicas en proliferación relacionadas con los condrocitos; estos tumores afectan principalmente adultos jóvenes.13 El examen histológico con frecuencia demuestra conjuntos de células con núcleos pletóricos y a menudo múltiples, parecidos a las células del cartílago. La interpretación de estos cambios histológicos es difícil debido a que otras lesiones de comportamiento clínico benigno, como los condromas de la mano, también pueden ser hipercelulares y semejan lesiones malignas. Los tumores de una variedad especialmente maligna se asocian con frecuencia con alteraciones en los cromosomas 17 o 13, o con alteraciones del gen supresor de tumores p53.14-16
Los condrosarcomas se encuentran con frecuencia en la cintura pélvica y en las costillas, en la cintura escapular y en la epífisis superior del fémur y del húmero. Las lesiones crecen en forma lenta y a menudo destruyen el hueso adyacente. El examen radiológico puede mostrar zonas moteadas con densidad aumentada que indican mineralización de la matriz cartilaginosa o la presencia de hueso mineralizado.
Los condrosarcomas a menudo se tratan con resección radical; pueden ocurrir recurrencias tardías, y con frecuencia los tumores han dado metástasis antes de la cirugía. En los pacientes con condrosarcomas que se originan en la pared torácica son factores pronósticos adversos la ocurrencia de metástasis en el momento del diagnóstico o durante la evolución, la edad mayor de 50 años, la resección incompleta y la recurrencia local.17
OSTEOSARCOMAS
Los osteosarcomas son tumores primarios de hueso que se derivan de células del linaje osteoblástico.18-20 Estos tumores son raros, sobre todo en adultos; sólo se diagnostican cerca de 1,500 casos nuevos cada año. Con mayor frecuencia los osteosarcomas ocurren en adolescentes, en la etapa de crecimiento acelerado. La lesión primaria suele encontrarse en aquellas porciones de los huesos largos en las que la velocidad de crecimiento es mayor, como el hueso metafisario de la porción distal del fémur, la porción proximal de la tibia y la porción proximal del húmero. Menos del 10 porciento de los osteosarcomas primarios ocurren en el cráneo y mandíbula, vértebras o pelvis.
Un porcentaje importante de osteosarcomas en adultos se asocian con la enfermedad de Paget del hueso o con sitios de radiación previa.. La prevalencia de osteosarcoma en la enfermedad de Paget es menor del uno porciento.21-23 Se ha propuesto que estos sarcomas se desarrollan en los sitios de las fracturas óseas de la enfermedad de Paget, pero esta relación no se ha confirmado. Los sarcomas en la enfermedad de Paget se presentan con mayor frecuencia en la pelvis, fémur, húmero, cráneo y huesos faciales, y sólo en raras ocasiones en las vértebras. Los cambios malignos siempre parecen originarse dentro de los focos de la enfermedad de Paget.
El aspecto histológico de los osteosarcomas, incluyendo los que ocurren en la enfermedad de Paget, es muy variado, desde el predominio fibroso hasta el óseo perfecto. Casi siempre se encuentra hueso bien formado, al menos focos pequeños, y esta característica distingue los osteosarcomas de los fibrosarcomas, que pueden originarse en los tejidos blandos adyacentes al hueso.
Patogenia
Aunque se desconoce la causa del osteosarcoma, la exposición a la radiación ionizante y la presencia de enfermedad de Paget son factores predisponentes en adultos, y los factores genéticos también parecen tener un papel importante. Por ejemplo, se ha notado un aumento importante en la incidencia de osteosarcoma en los hermanos de los pacientes afectados.24 Además, existe un gran aumento en la incidencia de osteosarcoma entre los individuos que han padecido retinoblastoma, una neoplasia que se considera posee una base genética.25,26 La pérdida de material genético del brazo largo del cromosarcoma 13, que se observa a menudo en individuos con retinoblastoma, también se ha descrito en pacientes con osteosarcomas.27 Se ha propuesto que puede encontrarse un alelo que predispone al tumor en el locus de retinoblastoma, y que la inactivación del alelo normal en un retinoblasto sensible provoca el retinoblastoma. Del mismo modo, una mutación somática que produjera homocicidad para dicho alelo en una célula ósea adecuada podría conducir al osteosarcoma. Es probable que las mutaciones en genes supresores de tumores, como el gen de retinoblastoma (Rb) y el p53 sean importantes en la patogenia del osteosarcoma.28 El gen p53 se localiza en el brazo corto del cromosoma 17, la expresión de la proteína p53 se asocia con transformación maligna en varios modelos tumorales experimentales, y clínicamente se asocia con otras neoplasias.16,29
Diagnóstico
La presencia de un tumor óseo suele manifestarse por la aparición de una tumoración de tejidos blandos o deformidad en una extremidad, dolor local, o la llamada fractura patológica (esto es, una fractura que ocurre en un paciente sin un traumatismo que lo justifique). En ocasiones las lesiones óseas o aún las metástasis pulmonares pueden ser detectadas como datos incidentales en una radiografía, y en casos raros el aumento en la actividad de la fosfatasa alcalina sérica proporciona el primer signo de un osteosarcoma oculto. El diagnóstico y las decisiones acerca del tratamiento deben tomarse en conjunto por cirujanos ortopedistas , patólogos y radiólogos, debido a que la mayoría de los médicos han tenido poca experiencia en la interpretación de las alteraciones observadas en las imágenes radiológicas, la tomografía computada (TC) y la imágen por resonancia magnética (IRM), o de los cambios histológicos que se observan en la biopsia.18 Pueden surgir problemas en el diagnóstico debido a que ciertas enfermedades no malignas producen cambios compatibles con tumores óseos. Por ejemplo, los signos radiológicos en la miositis osificante traumática pueden semejar a los que se observan en el osteosarcoma, y los componentes celulares de esta lesión en ocasiones pueden sugerir cáncer. De hecho, algunos individuos han sufrido amputaciones innecesarias.30
La fosfatasa alcalina, una enzima que en condiciones normales se encuentra sólo en los osteoblastos del hueso, suele estar aumentada en el tejido osteosarcomatoso; el nivel sérico de esta enzima puede también estar aumentado, sobre todo en los pacientes con tumores de predominio osteogénico. La extirpación del tumor óseo provoca disminución en el nivel sérico de la fosfatasa alcalina, pero con frecuencia el nivel se eleva de nuevo cuando las lesiones recurren en forma local o dan metástasis.
Los signos radiológicos de los osteosarcomas varían en relación a varios factores: fenotipo celular, velocidad de proliferación de las células neoplásicas, tipo de la matriz extracelular producida y grado de mineralización, localización del tumor en el hueso y respuesta del hueso y tejidos blandos que lo rodean. Algunos tumores tienen aspecto radiopaco debido a la presencia de espículas de hueso organizado, mientras otros son radiolúcidos y contienen sólo pequeñas áreas de mineralización amorfa. Pueden existir interrupciones corticales del hueso, y en ocasiones se observa formación de hueso nuevo en el periostio en múltiples capas. El tumor puede invadir los tejidos blandos, penetrando en forma completa la corteza del hueso.
Puede utilizarse la TC para determinar de manera precisa la extensión de la lesión primaria, la presencia de invasión cortical y la magnitud de la afección de los tejidos blandos; en algunos casos este estudio puede proporcionar información acerca de la naturaleza de la lesión misma, esto es, el tipo específico de tumor óseo presente. Además, la TC es más útil que la radiografía convencional para determinar la existencia y extensión de las metástasis. La IRM puede proporcionar una mayor precisión diagnóstica que la TC sola, y puede ser muy valiosa en la clasificación por etapas del cáncer de hueso, en particular del osteosarcoma, y en la evaluación de la extensión de las metástasis y de la respuesta al tratamiento.31
Estadificación y pronóstico
En la actualidad se ha adoptado un sistema uniforme para la estadificación de los osteosarcomas, que toma en cuenta factores como el grado histológico del tumor, la extensión más allá del compartimiento óseo y la presencia de metástasis a distancia. Este sistema de clasificación permite una mejor planeación de los procedimientos quirúrgicos y el uso adecuado de la quimioterapia.18
El grado de aneuploidia y poliploidia de las células en los tumores óseos malignos, determinado por citometría de flujo, se relaciona de manera importante con el grado de malignidad establecido por los patólogos expertos en hueso.18,32 El aumento en la actividad de la fosfatasa alcalina en el suero o en el tejido del tumor se asocia a mal pronóstico.13 Por el contrario, una menor aneuploidia, niveles de fosfatasa alcalina más bajos y ciertas características histológicas, como la variante intramedular, se relacionan con un pronóstico mejor.33
En los adolescentes los osteosarcomas crecen con rapidez y con frecuencia alcanzan el doble de su volumen en un mes. Las recaídas que se presentan después de que se ha logrado el control de la lesión primaria a menudo aparecen dentro del primer año, y rara vez después del segundo año. Antes de 1972 el único tratamiento para los niños o adolescentes con osteosarcomas era la amputación. El pronóstico era malo, y la supervivencia a cinco años se había mantenido alrededor de 20 porciento durante varias décadas.20 Sin embargo, al inicio de la década de 1970, la introducción de los programas de quimioterapia adyuvante combinada con cirugía mejoró en forma notable el pronóstico para los adolescentes con osteosarcoma: en este grupo de pacientes, la supervivencia libre de enfermedad a los cuatro o cinco años después del tratamiento ha aumentado a 80 porciento o más. En las naciones en vías de desarrollo, donde existe un acceso limitado al tratamiento agresivo, la tasa de supervivencia a cinco años todavía es del 30 porciento, semejante a la que prevalecía en naciones industrializadas antes del uso de la quimioterapia adyuvante.34
Los adultos con osteosarcomas relacionados con la enfermedad de Paget o con exposición previa a radiación ionizante tienen un pronóstico peor. Estos tumores no suelen ser extirpables, y no se ha demostrado que la quimioterapia adyuvante sea eficaz para prolongar la supervivencia en los pacientes con osteosarcomas relacionados con la enfermedad de Paget.22
Tratamiento
La cirugía radical aislada no logra la curación en cerca del 80 porciento de los enfermos con osteosarcoma. Debido a que la amputación erradica la masa del tumor en individuos sin metástasis macroscópicas, es probable que el alto índice de fracasos sea causado por la presencia de metástasis microscópicas no detectadas. El pulmón es el sitio más frecuente de estas metástasis. Se han introducido varios programas de quimioterapia adyuvante agresiva en un esfuerzo para erradicar estos focos microscópicos. Estos programas han sido modificados para tratar los problemas especiales que presentan los tumores con datos que indican un peor pronóstico. Los métodos incluyen dosis altas de methotrexate y rescate con leucovorín, solo o combinado con doxorrubicina, bleomicina, ciclofosfamida, dactinomicina o cisplatino. Un tratamiento experimental a base de un tripéptido muramil modificado para activar a los macrófagos a destruir a las células tumorales, ha dado resultados promisorios en estudios en animales, y puede llegar a emplearse como tratamiento adyuvante para incrementar la eficacia del la quimioterapia tradicional en los pacientes con osteosarcomas.35
En general, se acepta que es necesario practicar resecciones extensas, radicales o amputaciones para lograr el control local.10,18,20,36-47 En la actualidad se recomienda la administración de quimioterapia antes de la cirugía (quimioterapia neoadyuvante). La demora en la cirugía para administrar quimioterapia no aumenta el riesgo de metástasis. Los programas de quimioterapia neoadyuvante tienen varias ventajas, como la erradicación temprana de las micrometástasis y la identificación de posibles candidatos para procedimientos de conservación de una extremidad. Además, el tipo de respuesta del tumor primario al tratamiento inicial con quimioterapia puede ayudar en la elección de la quimioterapia posterior.
En pacientes con metástasis pulmonares aisladas se considera que la resección de las metástasis junto con quimioterapia es un enfoque razonable, en especial si las metástasis se limitan a un solo lóbulo. El éxito de la quimioterapia combinada con procedimientos de conservación de la extremidad, que utilizan aloinjertos óseos o prótesis como la segmentaria de metal para la parte distal del fémur, han permitido evoluciones favorables en el 60 a 80 porciento de los pacientes con osteosarcomas.46-49 Son factores de mal pronóstico la afección importante del paquete neurovascular, la presencia de fracturas patológicas, la existencia de infección, el esqueleto con edad inmadura y la diseminación local extensa hacia el músculo.
DR. STEPHEN M. KRANE
DR. LEE SIMON
Fisiopatología
Las neoplasias óseas se originan de los linajes mesenquimatoso, hematopoyético y vascular. Las células mesenquimatosas incluyen condrocitos, osteoblastos y fibroblastos del estroma; las células hematopoyéticas son los precursores de los osteoclastos, y las células vasculares (endoteliales y de músculo liso) son componentes de los vasos sanguíneos. La índole de las neoplasias óseas con frecuencia refleja el fenotipo normal de las células que los componen, como su morfología y organización multicelular y el tipo de matriz extracelular que producen.
La localización de los tumores óseos y la reacción que inducen en el tejido que los rodea influyen en su evolución. Por ejemplo, el cartílago articular o la placa de crecimiento parecen proporcionar una barrera a la diseminación del tejido neoplásico. Aunque las lesiones benignas en ocasiones se relacionan con la formación de hueso nuevo denso en el tejido circundante, las neoplasias malignas, así como las benignas, suelen inducir la resorción del hueso que los rodea. Parece ser que el tumor o las células normales que infiltran el tumor, como los monocitos-macrófagos, producen sustancias que directa o indirectamente estimulan a los osteoclastos para la resorción ósea; estas sustancias pueden también inducir el reclutamiento de los osteoclastos y causar su diferenciación a partir de células precursoras.1 Las sustancias que podrían actuar de esta manera son los factores transformadores del crecimiento, la interleucina-1, el factor de necrosis tumoral (FNT) y un péptido derivado del tumor relacionado con la hormona paratiroidea que comparte algunas secuencias con la porción amino terminal de esta hormona .2,3
Clasificación de los tumores óseos
MIELOMA MULTIPLE
El tumor maligno de hueso más frecuente en los adultos es el mieloma múltipe, que se origina de células del linaje de los linfocitos. Se diagnostican aproximadamente 10,000 casos nuevos de mieloma múltiple cada año en los Estados Unidos. Las células del mieloma producen una linfotoxina con estructura similar al FNT y que se une al mismo receptor que el FNT. Esta linfotoxina puede ser responsable del mayor número de osteoclastos que se observan en el hueso no afectado en la periferia de las lesiones del mieloma, que casi siempre son osteolíticas.
Los linfomas malignos pueden ser primarios de hueso, en donde de manera típica inducen una respuesta osteolítica; sin embargo, la enfermedad de Hodgkin puede producir lesiones esclerosas en el hueso.
TUMOR DE EWING
El tumor o sarcoma de Ewing es un tumor maligno poco frecuente que casi siempre ocurre en individuos menores de 30 años.4 Los sitios afectados con más frecuencia son la pelvis, el fémur y la tibia. En términos generales, el tumor de Ewing es considerado el más letal de todos los tumores óseos; las metástasis, que con frecuencia afectan el pulmón, la pleura o los ganglios linfáticos, están presentes en el momento del diagnóstico en el 15 a 35 porciento de los pacientes, dependiendo de la serie revisada.4,5 El examen histológico demuestra que estas neoplasias están compuestas por pequeñas células redondas que varían poco en tamaño y forma y que poseen mínimo estroma. Aunque la célula que da origen al tumor de Ewing se desconoce, la evidencia obtenida por medio de tinción específica para enolasa neuronal y la utilización de anticuerpos Leu-7 monoclonales sugiere que esta célula podría ser de origen neuroectodérmico.6 Además, los pacientes con sarcoma de Ewing, así como los que tienen tumores neuroectodérmicos periféricos malignos, tienen una traslocación característica entre los cromosomas 11 y 22,7,8 que parece causar la producción de un oncogén de fusión.9
Antes de 1970, sólo un cinco porciento de los pacientes tratados con cirugía o radiación solas alcanzaban una supervivencia prolongada. La terapéutica multimodal con cirugía, radiación local y quimioterapia adyuvante (con frecuencia una combinación de ciclofosfamida y doxorrubicina) ha permitido una supervivencia libre de enfermedad a largo plazo en alrededor del 50 porciento de los pacientes, aún en aquellos individuos con metástasis evidentes en el momento del diagnóstico.4,5,10 El pronóstico para los pacientes con linfoma no Hodgkin del hueso tratado, lesión que en ocasiones puede ser confundida con el tumor de Ewing, es mucho mejor que el de este último, y por lo tanto estas dos lesiones deben diferenciarse con claridad.12
CONDROSARCOMAS
Los condrosarcomas son tumores óseos primarios compuestos por células neoplásicas en proliferación relacionadas con los condrocitos; estos tumores afectan principalmente adultos jóvenes.13 El examen histológico con frecuencia demuestra conjuntos de células con núcleos pletóricos y a menudo múltiples, parecidos a las células del cartílago. La interpretación de estos cambios histológicos es difícil debido a que otras lesiones de comportamiento clínico benigno, como los condromas de la mano, también pueden ser hipercelulares y semejan lesiones malignas. Los tumores de una variedad especialmente maligna se asocian con frecuencia con alteraciones en los cromosomas 17 o 13, o con alteraciones del gen supresor de tumores p53.14-16
Los condrosarcomas se encuentran con frecuencia en la cintura pélvica y en las costillas, en la cintura escapular y en la epífisis superior del fémur y del húmero. Las lesiones crecen en forma lenta y a menudo destruyen el hueso adyacente. El examen radiológico puede mostrar zonas moteadas con densidad aumentada que indican mineralización de la matriz cartilaginosa o la presencia de hueso mineralizado.
Los condrosarcomas a menudo se tratan con resección radical; pueden ocurrir recurrencias tardías, y con frecuencia los tumores han dado metástasis antes de la cirugía. En los pacientes con condrosarcomas que se originan en la pared torácica son factores pronósticos adversos la ocurrencia de metástasis en el momento del diagnóstico o durante la evolución, la edad mayor de 50 años, la resección incompleta y la recurrencia local.17
OSTEOSARCOMAS
Los osteosarcomas son tumores primarios de hueso que se derivan de células del linaje osteoblástico.18-20 Estos tumores son raros, sobre todo en adultos; sólo se diagnostican cerca de 1,500 casos nuevos cada año. Con mayor frecuencia los osteosarcomas ocurren en adolescentes, en la etapa de crecimiento acelerado. La lesión primaria suele encontrarse en aquellas porciones de los huesos largos en las que la velocidad de crecimiento es mayor, como el hueso metafisario de la porción distal del fémur, la porción proximal de la tibia y la porción proximal del húmero. Menos del 10 porciento de los osteosarcomas primarios ocurren en el cráneo y mandíbula, vértebras o pelvis.
Un porcentaje importante de osteosarcomas en adultos se asocian con la enfermedad de Paget del hueso o con sitios de radiación previa.. La prevalencia de osteosarcoma en la enfermedad de Paget es menor del uno porciento.21-23 Se ha propuesto que estos sarcomas se desarrollan en los sitios de las fracturas óseas de la enfermedad de Paget, pero esta relación no se ha confirmado. Los sarcomas en la enfermedad de Paget se presentan con mayor frecuencia en la pelvis, fémur, húmero, cráneo y huesos faciales, y sólo en raras ocasiones en las vértebras. Los cambios malignos siempre parecen originarse dentro de los focos de la enfermedad de Paget.
El aspecto histológico de los osteosarcomas, incluyendo los que ocurren en la enfermedad de Paget, es muy variado, desde el predominio fibroso hasta el óseo perfecto. Casi siempre se encuentra hueso bien formado, al menos focos pequeños, y esta característica distingue los osteosarcomas de los fibrosarcomas, que pueden originarse en los tejidos blandos adyacentes al hueso.
Patogenia
Aunque se desconoce la causa del osteosarcoma, la exposición a la radiación ionizante y la presencia de enfermedad de Paget son factores predisponentes en adultos, y los factores genéticos también parecen tener un papel importante. Por ejemplo, se ha notado un aumento importante en la incidencia de osteosarcoma en los hermanos de los pacientes afectados.24 Además, existe un gran aumento en la incidencia de osteosarcoma entre los individuos que han padecido retinoblastoma, una neoplasia que se considera posee una base genética.25,26 La pérdida de material genético del brazo largo del cromosarcoma 13, que se observa a menudo en individuos con retinoblastoma, también se ha descrito en pacientes con osteosarcomas.27 Se ha propuesto que puede encontrarse un alelo que predispone al tumor en el locus de retinoblastoma, y que la inactivación del alelo normal en un retinoblasto sensible provoca el retinoblastoma. Del mismo modo, una mutación somática que produjera homocicidad para dicho alelo en una célula ósea adecuada podría conducir al osteosarcoma. Es probable que las mutaciones en genes supresores de tumores, como el gen de retinoblastoma (Rb) y el p53 sean importantes en la patogenia del osteosarcoma.28 El gen p53 se localiza en el brazo corto del cromosoma 17, la expresión de la proteína p53 se asocia con transformación maligna en varios modelos tumorales experimentales, y clínicamente se asocia con otras neoplasias.16,29
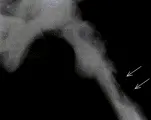
|
| Figura 1 |
| Osteosarcoma en la enfermedad de Paget |
Diagnóstico
La presencia de un tumor óseo suele manifestarse por la aparición de una tumoración de tejidos blandos o deformidad en una extremidad, dolor local, o la llamada fractura patológica (esto es, una fractura que ocurre en un paciente sin un traumatismo que lo justifique). En ocasiones las lesiones óseas o aún las metástasis pulmonares pueden ser detectadas como datos incidentales en una radiografía, y en casos raros el aumento en la actividad de la fosfatasa alcalina sérica proporciona el primer signo de un osteosarcoma oculto. El diagnóstico y las decisiones acerca del tratamiento deben tomarse en conjunto por cirujanos ortopedistas , patólogos y radiólogos, debido a que la mayoría de los médicos han tenido poca experiencia en la interpretación de las alteraciones observadas en las imágenes radiológicas, la tomografía computada (TC) y la imágen por resonancia magnética (IRM), o de los cambios histológicos que se observan en la biopsia.18 Pueden surgir problemas en el diagnóstico debido a que ciertas enfermedades no malignas producen cambios compatibles con tumores óseos. Por ejemplo, los signos radiológicos en la miositis osificante traumática pueden semejar a los que se observan en el osteosarcoma, y los componentes celulares de esta lesión en ocasiones pueden sugerir cáncer. De hecho, algunos individuos han sufrido amputaciones innecesarias.30
La fosfatasa alcalina, una enzima que en condiciones normales se encuentra sólo en los osteoblastos del hueso, suele estar aumentada en el tejido osteosarcomatoso; el nivel sérico de esta enzima puede también estar aumentado, sobre todo en los pacientes con tumores de predominio osteogénico. La extirpación del tumor óseo provoca disminución en el nivel sérico de la fosfatasa alcalina, pero con frecuencia el nivel se eleva de nuevo cuando las lesiones recurren en forma local o dan metástasis.
Los signos radiológicos de los osteosarcomas varían en relación a varios factores: fenotipo celular, velocidad de proliferación de las células neoplásicas, tipo de la matriz extracelular producida y grado de mineralización, localización del tumor en el hueso y respuesta del hueso y tejidos blandos que lo rodean. Algunos tumores tienen aspecto radiopaco debido a la presencia de espículas de hueso organizado, mientras otros son radiolúcidos y contienen sólo pequeñas áreas de mineralización amorfa. Pueden existir interrupciones corticales del hueso, y en ocasiones se observa formación de hueso nuevo en el periostio en múltiples capas. El tumor puede invadir los tejidos blandos, penetrando en forma completa la corteza del hueso.
Puede utilizarse la TC para determinar de manera precisa la extensión de la lesión primaria, la presencia de invasión cortical y la magnitud de la afección de los tejidos blandos; en algunos casos este estudio puede proporcionar información acerca de la naturaleza de la lesión misma, esto es, el tipo específico de tumor óseo presente. Además, la TC es más útil que la radiografía convencional para determinar la existencia y extensión de las metástasis. La IRM puede proporcionar una mayor precisión diagnóstica que la TC sola, y puede ser muy valiosa en la clasificación por etapas del cáncer de hueso, en particular del osteosarcoma, y en la evaluación de la extensión de las metástasis y de la respuesta al tratamiento.31
Estadificación y pronóstico
En la actualidad se ha adoptado un sistema uniforme para la estadificación de los osteosarcomas, que toma en cuenta factores como el grado histológico del tumor, la extensión más allá del compartimiento óseo y la presencia de metástasis a distancia. Este sistema de clasificación permite una mejor planeación de los procedimientos quirúrgicos y el uso adecuado de la quimioterapia.18
El grado de aneuploidia y poliploidia de las células en los tumores óseos malignos, determinado por citometría de flujo, se relaciona de manera importante con el grado de malignidad establecido por los patólogos expertos en hueso.18,32 El aumento en la actividad de la fosfatasa alcalina en el suero o en el tejido del tumor se asocia a mal pronóstico.13 Por el contrario, una menor aneuploidia, niveles de fosfatasa alcalina más bajos y ciertas características histológicas, como la variante intramedular, se relacionan con un pronóstico mejor.33
En los adolescentes los osteosarcomas crecen con rapidez y con frecuencia alcanzan el doble de su volumen en un mes. Las recaídas que se presentan después de que se ha logrado el control de la lesión primaria a menudo aparecen dentro del primer año, y rara vez después del segundo año. Antes de 1972 el único tratamiento para los niños o adolescentes con osteosarcomas era la amputación. El pronóstico era malo, y la supervivencia a cinco años se había mantenido alrededor de 20 porciento durante varias décadas.20 Sin embargo, al inicio de la década de 1970, la introducción de los programas de quimioterapia adyuvante combinada con cirugía mejoró en forma notable el pronóstico para los adolescentes con osteosarcoma: en este grupo de pacientes, la supervivencia libre de enfermedad a los cuatro o cinco años después del tratamiento ha aumentado a 80 porciento o más. En las naciones en vías de desarrollo, donde existe un acceso limitado al tratamiento agresivo, la tasa de supervivencia a cinco años todavía es del 30 porciento, semejante a la que prevalecía en naciones industrializadas antes del uso de la quimioterapia adyuvante.34
Los adultos con osteosarcomas relacionados con la enfermedad de Paget o con exposición previa a radiación ionizante tienen un pronóstico peor. Estos tumores no suelen ser extirpables, y no se ha demostrado que la quimioterapia adyuvante sea eficaz para prolongar la supervivencia en los pacientes con osteosarcomas relacionados con la enfermedad de Paget.22
Tratamiento
La cirugía radical aislada no logra la curación en cerca del 80 porciento de los enfermos con osteosarcoma. Debido a que la amputación erradica la masa del tumor en individuos sin metástasis macroscópicas, es probable que el alto índice de fracasos sea causado por la presencia de metástasis microscópicas no detectadas. El pulmón es el sitio más frecuente de estas metástasis. Se han introducido varios programas de quimioterapia adyuvante agresiva en un esfuerzo para erradicar estos focos microscópicos. Estos programas han sido modificados para tratar los problemas especiales que presentan los tumores con datos que indican un peor pronóstico. Los métodos incluyen dosis altas de methotrexate y rescate con leucovorín, solo o combinado con doxorrubicina, bleomicina, ciclofosfamida, dactinomicina o cisplatino. Un tratamiento experimental a base de un tripéptido muramil modificado para activar a los macrófagos a destruir a las células tumorales, ha dado resultados promisorios en estudios en animales, y puede llegar a emplearse como tratamiento adyuvante para incrementar la eficacia del la quimioterapia tradicional en los pacientes con osteosarcomas.35
En general, se acepta que es necesario practicar resecciones extensas, radicales o amputaciones para lograr el control local.10,18,20,36-47 En la actualidad se recomienda la administración de quimioterapia antes de la cirugía (quimioterapia neoadyuvante). La demora en la cirugía para administrar quimioterapia no aumenta el riesgo de metástasis. Los programas de quimioterapia neoadyuvante tienen varias ventajas, como la erradicación temprana de las micrometástasis y la identificación de posibles candidatos para procedimientos de conservación de una extremidad. Además, el tipo de respuesta del tumor primario al tratamiento inicial con quimioterapia puede ayudar en la elección de la quimioterapia posterior.
En pacientes con metástasis pulmonares aisladas se considera que la resección de las metástasis junto con quimioterapia es un enfoque razonable, en especial si las metástasis se limitan a un solo lóbulo. El éxito de la quimioterapia combinada con procedimientos de conservación de la extremidad, que utilizan aloinjertos óseos o prótesis como la segmentaria de metal para la parte distal del fémur, han permitido evoluciones favorables en el 60 a 80 porciento de los pacientes con osteosarcomas.46-49 Son factores de mal pronóstico la afección importante del paquete neurovascular, la presencia de fracturas patológicas, la existencia de infección, el esqueleto con edad inmadura y la diseminación local extensa hacia el músculo.
Bibliografía
- Mundy GR: Role of cytokines in bone resorption. J Cell Biochem 53:296, 1993
- Strewler GJ, Stern PH, Jacobs JW, et al: Parathyroid hormonelike protein from human renal carcinoma cells: structural and functional homology with parathyroid hormone. J Clin Invest 80:1803, 1987 [PMID 3680530]
- Strewler GJ, Nissenson RA: Peptide mediators of hypercalcemia in malignancy. Annu Rev Med 41:35, 1990 [PMID 2184736]
- Bacci G, Picci P, Gherlinzoni F, et al: Localized Ewing's sarcoma of bone: ten years' experience at the Istituto Ortopedico Rizzoli in 124 cases treated with multimodal therapy. Eur J Cancer Clin Oncol 21:163, 1985 [PMID 3987754]
- Hayes FA, Thompson EI, Parvey L, et al: Metastatic Ewing's sarcoma: remission induction and survival. J Clin Oncol 5:1199, 1987 [PMID 3625245]
- Kawaguchi K, Koike M: Neuron-specific enolase and Leu-7 immunoreactive small round-cell neoplasm: the relationship to Ewing's sarcoma in bone and soft tissue. Am J Clin Pathol 86:79, 1986
- Roessner A, Jurgens H: Round cell tumours of bone. Pathol Res Pract 189:111, 1993 [PMID 8183732]
- Stephenson CF, Bridge JA, Sandberg AA: Cytogenetic and pathologic aspects of Ewing's sarcoma and neuroectodermal tumors. Hum Pathol 23:1270, 1992 [PMID 1330877]
- Dunn T, Praissman L, Hagag N, et al: ERG gene is translocated in an Ewing's sarcoma cell line. Cancer Genet Cytogenet 76:19, 1994 [PMID 8076344]
- Mazanet R, Antman KH: Adjuvant therapy for sarcomas. Semin Oncol 18:603, 1991 [PMID 1775977]
- Dirix LY, Van Oosterom AT: The role of ifosfamide in the treatment of adult soft tissue sarcomas, Ewing's sarcoma, and osteosarcoma: a review. Semin Oncol 17(suppl 4):50, 1990 [PMID 2185547]
- Bacci G, Jaffe N, Emiliani E, et al: Therapy for primary non-Hodgkin's lymphoma of bone and a comparison of results with Ewing's sarcoma: ten years' experience at the Istituto Ortopedico Rizzoli. Cancer 57:1468, 1986 [PMID 3948127]
- Bjornsson JW, Unni KK, Dahlin DC, et al: Clear cell chondrosarcoma of bone: observations in 47 cases. Am J Surg Pathol 8:223, 1984
- Bridge JA, DeBoer J, Travis J, et al: Simultaneous interphase cytogenetic analysis and fluorescence immunophenotyping of dedifferentiated chondrosarcoma: implications for histopathogenesis. Am J Pathol 144:215, 1994 [PMID 8311109]
- Orndal C, Mandahl N, Rydholm A, et al: Chromosome aberrations and cytogenetic intratumor heterogeneity in chondrosarcomas. J Cancer Res Clin Oncol 120:51, 1993 [PMID 8270609]
- Wadayama B, Toguchida J, Yamaguchi T, et al: p53 expression and its relationship to DNA alterations in bone and soft tissue sarcomas. Br J Cancer 68:1134, 1993 [PMID 8260365]
- Burt M, Fulton M, Wessner-Dunlap S, et al: Primary bony and cartilaginous sarcomas of chest wall: results of therapy. Ann Thorac Surg 54:226, 1992 [PMID 1637209]
- Mankin HJ, Gebhardt MC: Advances in the management of bone tumors. Clin Orthop 200:73, 1985
- Yunis EJ, Barnes L: The histologic diversity of osteosarcoma. Pathol Annu 21(pt 1):121, 1986 [PMID 3510412]
- Goorin AM, Abelson HT, Frei E III: Osteosarcoma: fifteen years later. N Engl J Med 313:1637, 1985
- Wick MR, Siegal GP, Unni KK, et al: Sarcomas of bone complicating osteitis deformans (Paget's disease): fifty years' experience. Am J Surg Pathol 5:47, 1981 [PMID 6941703]
- Healey JH, Buss D: Radiation and pagetic osteogenic sarcomas. Clin Orthop 270: 128, 1991 [PMID 1884531]
- Huvos AG: Osteogenic sarcoma of bones and soft tissues in older persons: a clinicopathologic analysis of 117 patients older than 60 years. Cancer 57:1442, 1986
- Colyer RA: Osteogenic sarcoma in siblings. Johns Hopkins Medical Journal 145: 131, 1979
- Abramson DH, Ellsworth RM, Kitchin FD, et al: Second nonocular tumors in retinoblastoma survivors: are they radiation-induced? Ophthalmology 91:1351, 1984
- Helton KJ, Fletcher BD, Kun LE, et al: Bone tumors other than osteosarcoma after retinoblastoma. Cancer 71:2847, 1993 [PMID 8467462]
- Dryja TP, Rapaport JM, Epstein J, et al: Chromosome 13 homozygosity in osteosarcoma without retinoblastoma. Am J Hum Genet 38:59, 1986 [PMID 3004203]
- Fuchs N, Winkler K: Osteosarcoma. Curr Opin Oncol 5:667, 1993 [PMID 8364083]
- Masuda H, Miller C, Koeffler HP, et al: Rearrangement of the p53 gene in human osteogenic sarcomas. Proc Natl Acad Sci USA 84:7716, 1987 [PMID 2823272]
- Unni KK, McLeod RA, Dahlin DC: Conditions that simulate primary neoplasms of bone. Pathol Annu 15(pt 1):91, 1980
- Fletcher BD: Response of osteosarcoma and Ewing sarcoma to chemotherapy: imaging evaluation. Am J Roentgenol 157:825, 1991
- Alho A, Skjeldal S, Melvik JE, et al: The clinical importance of DNA synthesis and aneuploidy in bone and soft tissue tumours. Anticancer Res 13:2383, 1993 [PMID 8135471]
- Davis AM, Bell RS, Goodwin PJ: Prognostic factors in osteosarcoma: a critical review. J Clin Oncol 12:423, 1994 [PMID 8113851]
-
Susnerwala SS, Pande SC, Dinshaw KA, et al: Osteosarcoma:
experience of the Tata Memorial Hospital, Bombay, India. Cancer Treat Res
62:365, 1993 [PMID
8096751]
-
Kleinerman ES, Maeda M, Jaffe N: Liposome-encapsulated
muramyl tripeptide: a new biologic response modifier for the treatment
of osteosarcoma. Cancer Treat Res 62: 101, 1993 [PMID
8096724]
-
Picci P, Ferrari S, Bacci G, et al: Treatment recommendations
for osteosarcoma and adult soft tissue sarcomas. Drugs 47:82, 1994 [PMID
7510623]
-
Sutow WW, Herson J, Perez C: Survival after metastasis
in osteosarcoma. Natl Cancer Inst Monogr 56:227, 1981
-
Han M-T, Telander RL, Pairolero PC, et al: Aggressive
thoracotomy for pulmonary metastatic osteogenic sarcoma in children and
young adolescents. J Pediatr Surg 16:928, 1981
-
Rosen G, Caparros B, Huvos AG, et al: Preoperative
chemotherapy for osteogenic sarcoma: selection of postoperative adjuvant
chemotherapy based on the response of the primary tumor to preoperative
chemotherapy. Cancer 49:1221, 1982 [PMID
6174200]
-
Rosen G, Marcove RC, Caparros B, et al: Primary osteogenic
sarcoma: the rationale for preoperative chemotherapy and delayed surgery.
Cancer 43:2163, 1979 [PMID
88251]
-
Simon MA, Nachman J: The clinical utility of preoperative
therapy for sarcomas. J Bone Joint Surg [Am] 68:1458, 1986 [PMID
3536937]
-
Bacci G, Springfield D, Capanna R, et al: Neoadjuvant
chemotherapy for osteosarcoma of the extremity. Clin Orthop 224:268, 1987
[PMID
3478163]
-
Aboulafia AJ, Malawer MM: Surgical management of pelvic
and extremity osteosarcoma. Cancer 71:3358, 1993 [PMID
8490883]
-
Winkler K, Bielack SS, Delling G, et al: Treatment
of osteosarcoma: experience of the Cooperative Osteosarcoma Study Group
(COSS). Cancer Treat Res 62:269, 1993 [PMID
7682088]
-
Antman KH: Chemotherapy of advanced sarcomas of bone
and soft tissue. Semin Oncol 19(suppl 12):13, 1992
-
Veth RPH: IIB Æsteosarcoma:
current management, local control, and survival statistics-the Netherlands.
Clin Orthop 270:67, 1991
-
Meyer WH, Malawer MM: Osteosarcoma: clinical features
and evolving surgical and chemotherapeutic strategies. Pediatr Clin North
Am 38:317, 1991 [PMID
2006080]
-
Simon MA, Aschliman MA, Thomas N, et al: Limb-salvage
treatment versus amputation for osteosarcoma of the distal end of the femur.
J Bone Joint Surg [Am] 68: 1331, 1986 [PMID
3465732]
-
Mankin HJ, Gebhardt MC, Tomford WW: The use of frozen
cadaveric allografts in the management of patients with bone tumors of
the extremities. Orthop Clin North Am 18: 275, 1987 [PMID
3550576]