Nefrología
⭳ Abrir artículo (PDF)1.3 MBEste artículo es idéntico en la Edición 3/2000.
Contenido del artículo
V GLOMERULONEFRITIS Y SINDROME NEFROTICO
V GLOMERULONEFRITIS Y SINDROME NEFROTICO
DR. CECIL H. COGGINS
DR. HELMUT G. RENNKE
DR. BURTON D. ROSE
En este capítulo se discuten las principales características clínicas de las enfermedades glomerulares individuales, que se catalogan de acuerdo con su forma de aparición en: glomerulonefritis difusa, glomerulonefritis focal y síndrome nefrótico. Las manifestaciones renales de las enfermedades sistémicas, como la diabetes mellitus, el lupus eritematoso generalizado (LEG), las vasculitis necrosantes y la amiloidosis no se incluyen en esta subsección.
Glomerulonefritis difusa
Los trastornos relacionados con la glomerulonefritis difusa suelen manifestarse por datos de síndrome nefrítico: sedimento urinario activo que contiene eritrocitos, leucocitos y cilindros granulares y celulares, grados variables de proteinuria, que pueden alcanzar el rango nefrótico (> 3 g/día), concentración elevada de creatinina plasmática y, con frecuencia, hipertensión arterial y edema.
GLOMERULONEFRITIS POSINFECCIOSA
Glomerulonefritis posestreptocócica
La glomerulonefritis posestreptocócica es la enfermedad típica relacionada con el síndrome nefrítico agudo (ver adelante).1 Las manifestaciones clínicas se caracterizan por aparecer después de una infección por estreptococos ß-hemolíticos del grupo A, de los cuales algunas cepas son especialmente nefritogénicas en particular, como el tipo 12 (relacionado con faringitis) y el tipo 49 (relacionado con impétigo).2,3
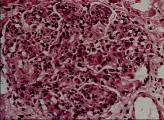
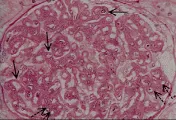
En la microscopía de luz la glomerulonefritis posinfecciosa se caracteriza por aumento focal o difuso en la celularidad glomerular, que produce cierre de la luz capilar. En este trastorno se observa infiltración de neutrófilos circulantes y de células mononucleares y proliferación de células endoteliales y mensagiales glomerulares, que puede complicarse por la formación de medias lunas en los casos graves [ver figura 1a]. La microscopía por inmunofluorescencia muestra de manera característica un patrón granular con IgG y complemento, que indica depósito de complejos inmunes. Sin embargo, el cambio más distintivo lo constituye el depósito de medias lunas subepiteliales, o en forma de giba, observado por microscopía electrónica [ver figura 1b]. Estos depósitos inician una reacción mediada por complemente que origina daño glomerular. Se supone que los complejos inmunes incluyen un antígeno relacionado a los estreptococos, aunque no se ha definido el antígeno específico.
La incidencia de la glomerulonefritis posestreptocócica ha disminuido en
los Estados Unidos y Europa, pero sigue siendo frecuente en muchas otras partes
del mundo. Los estudios epidemiológicos de la glomerulonefritis
postestreptocócica indican que la incidencia de enfermedad renal es de
cerca del cinco al 10 porciento en pacientes con faringitis y del 25 porciento
en enfermos con infección cutánea.2,3 El periodo de
latencia entre la aparición de la infección y los datos
clínicos de enfermedad renal varían de acuerdo con el sitio de la
infección, siendo en promedio de 10 días en enfermos con
faringitis y de 21 días en el caso del impétigo.4 La
administración de penicilina no parece evitar la enfermedad glomerular,
aunque sí controla los síntomas locales e impide la
diseminación de la infección a los contactos cercanos.
Las manifestaciones clínicas de la glomerulonefritis posestreptocócica varían desde la hematuria asintomática y proteinuria leve hasta el síndrome nefrítico típico con hematuria franca, proteinuria, oliguria, edema, hipertensión arterial e insuficiencia renal. Los niveles de complemento sérico por lo general están bajos; los títulos de antiestreptolisina O y sobre todo de anti-DNasa B se encuentran elevados. El diagnóstico diferencial para los pacientes que presentan enfermedad posestreptocócica incluye otras causas de síndrome nefrítico.
No existe tratamiento específico para la glomerulonefritis posestreptocócica, que se caracteriza por ser una enfermedad autolimitada. La proteinuria leve y la hematuria microscópica pueden persistir durante meses o incluso años. La proteinuria persistente en rango nefrótico es muy poco común, pero puede ocurrir, en especial en pacientes con gran cantidades de gibas subepiteliales.5 Por lo tanto, debe sospecharse uno de los trastornos nefróticos, como nefropatía membranosa, en los casos en que el paciente es revisado por primera vez en esta etapa tardía.
En la mayoría de los casos ocurre la recuperación completa, en especial en niños y cuando la enfermedad se adquiere durante una epidemia de infección estreptocócica.4,6 En algunos casos, sobretodo esporádicos y en adultos, ocurre solo recuperación parcial, aunque esto no es frecuente. Cuando la enfermedad aguda es severa, con oliguria prolongada, la insuficiencia renal puede ser permanente. Existen controversias acerca de la persistencia de las alteraciones renales a largo plazo. Se han presentado algunos casos de hipertensión, disminución de la velocidad de filtración glomerular (VFG) sin insuficiencia renal clínica y alteraciones histológicas, incluyendo esclerosis mesangial, que ocurren varios años después de la enfermedad aguda (sin embargo, es difícil establecer en estos casos si el diagnóstico original en realidad fue glomerulonefritis posestreptocócica).
Otras formas de glomerulonefritis posinfecciosa
El síndrome nefrítico agudo también puede ocurrir después de infecciones bacterianas (estafilocócicas, neumocócicas o estreptocócicas del grupo C), virales o parasitarias.9-12 Un ejemplo es la nefritis relacionada con endocarditis bacteriana o con derivaciones auriculoventriculares infectadas.13-15 Las manifestaciones renales de estos dos trastornos pueden ser parecidas, histológica y clínicamente, a las que se observan en la glomerulonefritis posestreptocócica (con gibas subepiteliales) o en la glomerulonefritis membranoproliferativa tipo I (con depósitos mensangiales y subendoteliales). La gravedad de las manifestaciones clínicas se relaciona sobre todo con la duración de la infección antes de iniciar el tratamiento antimicrobiano adecuado. En general, el control de la infección, incluyendo la extracción del cortocircuito infectado, conduce a la resolución rápida de la enfermedad, con recuperación de la función renal a los límites basales. Sin embargo, puede ocurrir insuficiencia renal irreversible, sobre todo cuando el tratamiento se ha retrasado.14
En pacientes con endocarditis la afección glomerular debe distinguirse de las embolias renales y de la nefritis intersticial aguda inducida por antimicrobianos. Las embolias renales se sospechan por dolor unilateral en los flancos y por otros síntomas generales; el diagnóstico puede confirmarse por la presencia de defectos de perfusión local o por gamagrafía con radioisótopos. La nefritis intersticial debe sospecharse por la aparición tardía de la enfermedad (por lo general más de 10 días después de la infección). Las manifestaciones de la nefritis intersticial incluyen eosinofilia, exantema y eosinofiluria. En comparación, la glomerulonefritis suele encontrarse en su grado máximo de gravedad antes de iniciar el tratamiento.
GLOMERULONEFRITIS MEMBRANOPROLIFERATIVA
La glomerulonefritis membranoproliferativa (GNMP), también llamada glomerulonefritis hipocomplementémica, glomerulonefritis lobular y glomerulonefritis mesangiocapilar, puede manifestarse como nefritis aguda o con una evolución más indolente relacionada con el síndrome nefrótico.16-18 La mayoría de los pacientes son niños o adultos jóvenes, y la GNMP puede causar el cinco a 10 porciento de los casos de síndrome nefrótico en este grupo. Durante su evolución, la mayoría de los pacientes puede tener cifras bajas de la fracción C3 del complemento.
La microscopía de luz revela paredes capilares glomerulares engrosadas y proliferación celular mesangial [ver figura 2a]. Por microscopía electrónica se han identificado dos subgrupos de GNMP. La enfermedad tipo I se define por dos características: (1) división de la membrana basal por la interposición de matriz mesangial, que produce una apariencia en vías de ferrocarril cuando la muestra de biopsia se tiñe con el reactivo ácido periódico de Schiff (PAS) o se tiñe con plata, y (2) depósitos de complejos inmunes subendoteliales y mesangiales notables. En la enfermedad tipo II la interposición de la matriz mesangial es menos obvia, y se observan depósitos prominentes, densos, en forma de listón (en lugar de complejos aislados) en la membrana basal; de aquí el nombre alternativo de enfermedad de depósitos densos [ver figura 2b]. Se desconoce el origen de estos depósitos.
Los dos tipos de GNMP también tienen una patogenia y manifestaciones
clínicas distintas, incluyendo la patogenia de la hipocomplementemia
[ver tabla 1]. En la enfermedad tipo I el complemento se activa por la
vía clásica, que al parecer se inicia por el depósito de
complejos inmunes. Por el contrario, la hipocomplementemia de la enfermedad
tipo II se produce sobre todo por activación de la vía alterna y
se relaciona de manera característica con la presencia del factor
activador del complemento circulante denominado factor C3 nefrítico
(FC3NeF).18 Las evidencias indican que el FC3NeF es un anticuerpo de
IgG que se une a la convertasa de C3 de la vía alterna del complemento
y prolonga su vida media.19 El efecto neto es el constante
rompimiento de C3 y la hipocomplementemia persistente. El papel
patogénico de este factor tal vez sea limitado, ya que la
evolución de la enfermedad no puede predecirse con base en los niveles
de C3 o de FC3NeF. Por lo general, la enfermedad tipo II es menos frecuente,
ocurre en pacientes más jóvenes y progresa en forma más
constante a insuficiencia renal que la enfermedad tipo I.
Aunque la GNMP suele ser idiopática, puede relacionarse con algunos trastornos generalizados. Es común que los pacientes con lipodistrofia parcial presenten el tipo II de la GNMP y FC3NeF.20 El patrón membranoproliferativo también se ha observado en otros trastornos relacionados con la formación de complejos inmunes, incluyendo LEG, crioglobulinemia mixta, infecciones en derivaciones, endocarditis bacteriana, linfoma linfocítico crónico y esquistosomiasis.22
El diagnóstico de GNMP sólo puede confirmarse por biopsia renal. El dato clínico principal que indica la presencia de este trastorno es hipocomplementemia sin datos clínicos o serológicos de LEG o de infección estreptocócica reciente.
Al igual que con otras glomerulopatías, la gravedad de la enfermedad en el momento de aparición es un dato pronóstico importante. Los siguientes datos se relacionan con un riesgo mayor de evolución a insuficiencia renal terminal: síndrome nefrótico persistente, hipertensión arterial, insuficiencia renal, presencia de medias lunas (ver adelante) o de esclerosis en la biopsia, y la presencia de FC3NeF.18 En los pacientes con hematuria o proteinuria asintomática que presentan lesiones focales y segmentarias en la biopsia renal el pronóstico suele ser mejor.23
El tratamiento óptimo de la GNMP no se ha establecido. En un estudio retrospectivo, no controlado, de niños con enfermedad tipo I, el posible beneficio de la prednisona a dosis bajas para mantener la VFG fue cotrarrestado por un empeoramiento de la hipertensión.24,25 No existen en la actualidad estudios controlados de esteroides en adultos o en pacientes con enfermedad de tipo II. El tratamiento antiplaquetario, con aspirina o dipiridamol, puede ser benéfico a corto plazo (un año) para mantener la VFG, pero está en duda el beneficio a largo plazo.26 Los estudios controlados de ciclofosfamida, dipiridamol y warfarina no han demostrado beneficio a largo plazo.27,28
En resumen, ninguno de los tratamientos probados tienen un efecto benéfico demostrado a largo plazo. En los casos de síndrome nefrótico, algunos nefrólogos recomiendan, además de las medidas generales, administrar tratamiento a largo plazo en dosis moderadas y días alternos de esteroides en los pacientes en los que no es difícil controlar la hipertensión.
Las lesiones histológicas de la GNMP tipo I, y sobre todo del tipo II, con frecuencia recurren en los riñones trasplantados; sin embargo, esta afección a menudo es silenciosa, con mínimas manifestaciones clínicas o asintomática.29.30 Por lo tanto, el riesgo de recurrencia no impide que el trasplante tenga éxito en estos pacientes.31 La pérdida del injerto por GNMP recurrente ocurre en menos del 10 porciento de los casos, la mayoría de los cuales presentaron evolución relativamente rápida de su enfermedad inicial.
GLOMERULONEFRITIS EN MEDIAS LUNAS, O RAPIDAMENTE PROGRESIVA
A diferencia del inicio súbito del síndrome nefrítico que se observa en la glomerulonefritis aguda, con resolución en varias semanas, algunos pacientes sufren deterioro progresivo en semanas a meses que llega hasta insuficiencia renal. El examen general de orina revela proteinuria, en ocasiones en rango nefrótico, y sedimento activo con eritrocitos dismórficos y cilindros eritrocitarios. En los pacientes con glomerulonefritis rápidamente progresiva (GNRP), la biopsia suele mostrar medias luns celulares que cubren la capa parietal de la cápsula de Bowman en más del 50 porciento del glomérulo [ver figura 3]. Las medias lunas constituyen una respuesta inespecífica al daño glomerular y pueden ocurrir en cualquier enfermedad glomerular grave. El evento inicial en la formación de medias lunas es el daño a la pared capilar glomerular que permite al fibrinógeno o a la fibrina entrar al espacio de Bowman.32 La entrada de macrófagos y la proliferación secundaria de células epiteliales acompañan estas lesiones, y estos son los componentes celulares de la media luna. La insuficiencia renal se produce sobre todo por compresión del penacho capilar por la media luna expandida.
El examen de muestras de biopsia renal por microscopía de luz es insuficiente para confirmar el diagnóstico. Por ejemplo, la identificación de depósitos mensagiales prominentes de IgA en la microscopía de inmunofluorescencia es diagnóstica de nefropatía por IgA de fondo, y la identificación de gibas subepiteliales por microscopía electrónica es diagnóstica de glomerulonefritis posinfecciosa [ver figura 1b].
Los pacientes con evolución clínica de GNRP y biopsias que muestran medias lunas celulares abundantes por microscopía de luz pueden subdividirse en tres grupos patogénicos con base en la inmunofluorescencia.33-36 Alrededor del 50 porciento tendrá evidencia mínima o nula de depósito de inmunoglobulinas en el glomérulo (GNRP pauci-inmune), el 40 porciento mostrará depósitos de complejos inmunes, y alrededor del 10 porciento presentará un patrón de anticuerpos dirigidos contra un componente de la membrana basal del capilar glomerular (enfermedad antimembrana basal glomerular o anti-MBG). La presencia de cilindros hemáticos y de leucocitos en la orina ayuda a distinguir la GNRP de otras causas de insuficiencia renal aguda, como la nefritis intersticial aguda, la obstrucción de las vías urinarias o la enfermedad ateroembólica.
GNRP sin anticuerpos o pauci-inmune
Los pacientes con GNRP sin anticuerpos o pauci-inmune suelen tener anticuerpos anticitoplasma de neutrófilo (ANCA, por sus siglas en inglés), lo que indica que la GNRP es parte de un síndrome de vasculitis. Los que tienen ANCA dirigidos contra la proteinasa 3 leucocitaria suelen tener granulomatosis de Wegener, mientras que los que tienen anticuerpos antimieloperoxidasa con más frecuencia tienen poliarteritis microscópica. El tratamiento inmediato e intensivo con esteroides y alquilantes, en ocasiones aunado a plasmaféresis, suele conservar la función e interrumpir lo que de otro modo sería una evolución progresiva hacia el deterioro.
GNRP por complejos inmunes
La mayoría de los pacientes con GNRP mediada por complejos inmunes tiene evidencia de una enfermedad sistémica, incluyendo LES, púrpura de Schönlein-Henoch, endocarditis bacteriana subaguda o infección de derivaciones, y crioglobulinemia mixta. Algunos parecen tener medias lunas sobrepuestas a otras enfermedades renales primarias, incluyendo glomerulonefritis membranoproliferativa, nefropatía por IgA y nefropatía membranosa. Algunos tienen un padecimiento que parece idiopático, aunque se han implicado a la penicilamina, la sífilis y las neoplasias en casos aislados.22 También es posible que exista cierta predisposición genética, el trastorno se ha asociado con el fenotipo BfF de la properdina y con el haplotipo HLA-DR2 o MT3.37 El pronóstico y tratamiento del padecimiento es semejante al de la GNRP sin anticuerpos.
Enfermedad por anticuerpos anti-MBG
El Dr. Ernest W. Goodpasture describió por primera vez la combinación de glomerulonefritis y hemorragia pulmonar. En la actualidad se ha demostrado que este grupo de síntomas puede producirse por diversos trastornos;38 por lo tanto, el término síndrome de Goodpasture debe limitarse a la entidad patológica específica, observada con más frecuencia en hombres jóvenes, en la cual la presencia de anticuerpos circulantes contra la MBG explica la glomerulonefritis.34 Estos anticuerpos también reaccionan con un antígeno similar en los pulmones, produciendo alveolitis, con hemorragia pulmonar.39 La hemoptisis suele preceder los datos clínicos de enfermedad renal, aunque algunos pacientes sólo presentan afección renal. La aparición de daño renal sin afección pulmonar indica que debe existir una lesión pulmonar simultánea que permita que los anticuerpos circulantes entren a los alveolos. Como ejemplos se encuentran la influenza, la exposición a hidrocarburos y el tabaquismo crónico.40,41
El cuadro patológico renal característico es la glomerulonefritis proliferativa, necrosante, con formación de medias lunas en más del 50 porciento de los glomérulos [ver figura 3]. La microscopía de inmunofluorescencia muestra tinción linear de IgG y C3 a lo largo de la MBG [ver figura 4]. Los datos observados por microscopía de luz y de inmunofluorescencia son diagnósticos de enfermedad de anticuerpos anti-MBG. Se puede observar una tinción inmunofluorescente similar en los alveolos de los pacientes afectados.
La manifestación renal primaria de este trastorno es la insuficiencia renal rápidamente progresiva con sedimento urinario nefrítico activo. La proteinuria suele ser leve, lo que podría reflejar el notable deterioro de la filtración glomerular, y no suele observarse edema. Los niveles de complemento son normales.
La enfermedad de anticuerpos anti-MBG generalmente evoluciona a la insuficiencia renal, aunque algunos pacientes desarrollan enfermedad focal leve y tienen un buen pronóstico. Por ejemplo, en un estudio se demostró que la insuficiencia renal terminal se desarrolló en el 80 porciento de los pacientes con el síndrome en término de un año después del diagnóstico.42 El tratamiento farmacológico aislado, incluyendo los pulsos intravenosos con dosis altas de metilprednisolona, parece ser ineficaz contra las lesiones renales.33,43
Aunque no se han llevado a cabo estudios controlados, varios pacientes parecen beneficiarse con la combinación de plasmaféresis que extrae los anticuerpos anti-MBG circulantes y fármacos inmunosupresores, que evitan la formación de nuevos anticuerpos.44,45 Un esquema consiste en realizar cambios plasmáticos de 4 L/día durante aproximadamente una semana, combinados con prednisona (60 mg/día), ciclofosfamida (3 mg/kg/día) y azatioprina (1 mg/kg/día).44 La dosis de prednisona se reduce cada semana, y en pacientes mayores de 55 años la dosis de ciclofosfamida se reduce hasta 2 mg/kg/día omitiendo la azatioprina. El tratamiento suele suspenderse después de nueve a 12 meses, o antes si desaparecen los anticuerpos circulantes anti-MBG, medidos por radioinmunoanálisis. Los fármacos citotóxicos también deben suspenderse si se desarrolla neutropenia (cuenta leucocitaria menor de 3,500/mm3) o una infección grave.
Este esquema por lo general disminuye la concentración plasmática de anticuerpos anti-MBG, pero no necesariamente se acompaña de mejoría paralela en la función renal. En particular, es poco probable que los pacientes oligúricos con concentración plasmática de creatinina mayor de 7 mg/dl recuperen la función renal.44,45 Por comparación, ocurre mejoría funcional en dos terceras partes de los pacientes no oligúricos con concentración plasmática de creatinina menor de 7 mg/dl antes de la institución del tratamiento; asimismo, el deterioro ulterior de la función renal en este grupo es poco frecuente, afectando sólo del 10 al 15 porciento de los enfermos. La plasmaféresis también resulta eficaz para el control de la hemoptisis en más del 90 porciento de los casos.45
Los investigadores que han estudiado este método terapéutico concluyen que su administración antes de que ocurra daño glomerular irreversible puede preservar y mejorar la función renal. Sin embargo, las infecciones graves en pacientes sometidos a plasmaféresis y que reciben medicamentos inmunosupresores para la glomerulonefritis rápidamente progresiva son frecuentes, por lo que es necesaria la vigilancia estrecha.46-49 Es poco probable que los pacientes que presentan concentración plasmática de creatinina mayor de 7 mg/dl respondan al tratamiento, a menos de que la lesión sea aguda. En consecuencia, es probable que el riesgo relacionado con el tratamiento exceda los beneficios posibles en esta situación, a no ser que exista hemorragia pulmonar. El riesgo de hemorragia pulmonar no debe subestimarse, ya que la muerte es común en los pacientes tratados en forma inadecuada.
Por lo general la formación de anticuerpos anti-MBG es autolimitada; estos anticuerpos desaparecen en forma espontánea en término de seis a 12 meses, y las recurrencias tardías son raras.34 Los individuos que evolucionan a insuficiencia renal en etapa terminal (como consecuencia del daño irreversible durante la fase aguda de la enfermedad) no deben recibir trasplantes hasta que la concentración de anticuerpos circulantes sea mínima o nula, para evitar la recurrencia en el trasplante.47,50
GLOMERULONEFRITIS FIBRILAR
La glomerulonefritis fibrilar, una enfermedad descrita hace poco tiempo, es más común en algunos hospitales que muchas de las glomerulonefritis mejor conocidas.51,52 La microscopía de luz muestra expansión mesangial y proliferación celular variable, y la microscopía electrónica permite observar el depósito patognomónico de fibrillas. Estas fibras son más grandes que las amiloides y no se tiñen con rojo Congo ni con tioflavina T [ver figura 5]. El origen de estas fibrillas es dudoso, la microscopía inmunofluorescente suele ser positiva para IgG, lo que indica que las inmunoglobulinas pueden estar implicadas. Sin embargo, no existen datos que apoyen la existencia de alguna paraproteína urinaria o plasmática, como sucede en forma característica en la amiloidosis primaria.
Los pacientes con glomerulonefritis fibrilar suelen manifestar
hipertensión arterial, hematura y piuria microscópicas,
insuficiencia renal y grados variables de proteinuria, que puede alcanzar el
rango nefrótico. En la mayoría de los pacientes la enfermedad es
evolutiva. No existe tratamiento para esta enfermedad. La relación entre
la glomerulonefritis fibrilar y la glomeulopatía inmunotactoide
semejante no se ha aclarado aún.53
Glomerulonefritis focal
La mayoría de los enfermos con glomerulonefritis focal presentan hematuria y proteinuria asintomáticas o episodios de hematuria macroscópica. La insuficiencia renal, la proteinuria intensa y la hipertensión arterial, tan frecuentes en la glomerulonefritis difusa, son poco comunes en esta entidad. Las manifestaciones clínicas leves correlacionan con los datos histológicos, que por lo general consisten en alteraciones proliferativas focales (con lesión de unos cuantos glomérulos) y segmentarias (con afección de una parte del penacho glomerular). Aunque estas enfermedades parecen focales, la inmunofluorescencia suele mostrar depósito difuso de inmunoglobulinas en el glomérulo, por lo que es posible que en realidad representen variedades más leves de glomerulonefritis difusa (ver antes). Dos causas comunes de glomerulonefritis focal son la nefropatía por IgA y la nefritis hereditaria.
NEFROPATIA POR IgA
En algunas partes del mundo (v.gr., Finlandia), la nefropatía por IgA (enfermedad de Berger) es la causa más común de enfermedad glomerular, sobre todo en pacientes con hematuria asintomática o episodios recurrentes de hematuria franca. Es frecuente sobre todo en varones adultos, jóvenes, de raza blanca, y tiene una tendencia familiar.54
El examen histológico se caracteriza por mostrar proliferación focal o difusa de células mensagiales [ver figura 6a], y en la microscopía electrónica se observan depósitos mesangiales densos. Sin embargo, la característica patognomónica es el depósito mesangial difuso de IgA, que se detecta por microscopía de inmunofluorescencia [ver figura 6b], a menudo acompañado por depósitos menos notables de otras inmunoglobulinas y complemento. Estos datos son indistinguibles de los observados en la púrpura de Schönlein-Henoch; no obstante, esta última también incluye afección articular, de piel y del aparato digestivo.55
La nefropatía por IgA a menudo es idiopática, pero también
coexiste con otras enfermedades, incluyendo la cirrosis hepática, en la
que puede estar alterada la capacidad de eliminación de complejos
inmunes que contienen IgA circulante por las células de Kupffer, y la
enteropatía por gluten, en la que los anticuerpos de IgA-antigliadina
desempeñan un papel crucial.22,56,57 La patogenia de la
nefropatía por IgA no se ha esclarecido por completo.22,58 La
presencia de niveles elevados de IgA, polímeros de IgA y complejos
inmunes que contienen IgA en el plasma indican una alteración en la
regulación de la síntesis de esta inmunoglobulina en las mucosas.
Una vez que se ha formado un exceso de complejos que contienen IgA,
éstos parecen depositarse de preferencia en los glomérulos porque
no activan la vía clásica del complemento y, en consecuencia, no
se unen a los receptores C3b de los eritrocitos.59 En condiciones
normales los receptores para C3b evitan el depósito de complejos en los
tejidos al transportar los depósitos al sistema
reticuloendotelial.
La mayoría de los individuos con nefropatía por IgA presentan hematuria microscópica asintomática con o sin proteinuria, o episodios de hematuria franca que aparecen después de un cuadro de faringitis aguda o de un síndrome febril menor, parecido al resfriado.55,60 La mayoría de los pacientes son menores de 35 años y ambos sexos se afectan con la misma frecuencia. La función renal suele ser normal al momento en el que aparecen los síntomas, aunque puede ocurrir insuficiencia renal aguda cuando existe hematuria importante, con oclusión de la luz tubular por eritrocitos.61 El diagnóstico puede sospecharse por la historia clínica, pero sólo puede confirmarse por biopsia renal. Aunque la IgA puede encontrarse en biopsias cutáneas de áreas expuestas al sol, este hallazgo no es lo suficiente sensible o específico para el diagnóstico. Alrededor de la mitad de los pacientes tienen niveles séricos de IgA ligeramente elevados.
La nefropatía por IgA suele tener una evolución benigna a corto plazo. La función renal se conserva, aunque puede haber episodios recurrentes de hematuria franca durante muchos años.55,60 En consecuencia, los pacientes que sólo presentan hematuria leve, cilindros hemáticos (que indican hematuria de origen glomerular), proteinuria mínima y concentración normal de creatinina plasmática, no necesitarán biopsia renal si no existen datos clínicos aparentes de progresión de la enfermedad. Cerca del 20 al 40 porciento de los pacientes con estos síntomas presentan nefropatía por IgA; el resto tiene otras enfermedades, por lo general benignas, como la enfermedad de membrana basal delgada (ver adelante).62,63
Sin embargo, aproximadamente el 50 porciento sufrirán insuficiencia renal terminal en un periodo de 20 años. Los signos pronósticos que suelen indicar una probable evolución al deterioro son: excreción de proteínas mayor de 1 g/día, hipertensión, medias lunas en la biopsia o cambios extensos tubulointersticiales y vasculares.64-66
No se ha encontrado un tratamiento eficaz para la nefropatía por IgA, aunque se han tratado esquemas que incluyen esteroides, agentes alquilantes, ciclosporina, plasmaféresis, anticoagulantes o antiplaquetarios, dietas libres en gluten, amigdalectomía o difenilhidantoína. El depósito de IgA secundario a enfermedad celíaca puede responder a la restricción de gluten, y el depósito de IgA que acompaña a la cirrosis alcohólica puede mejorar con la abstinencia de alcohol. Un subgrupo de pacientes que presentan síndrome nefrótico, hematuria mínima y lesiones glomerulares leves (en contraste con la mayoría de los casos que presentan proteinuria intensa) a menudo se comportan como si presentaran enfermedad por cambios mínimos; estos pacientes manifiestan remisión completa de la proteinuria después de la administración de prednisona por poco tiempo.67,68
Como con muchas otras enfermedades glomerulares, la nefropatía por IgA recurre con frecuencia en el riñón trasplantado a pesar del tratamiento contra el rechazo con prednisona, azatioprina y ciclosporina. Sin embargo, esta enfermedad a menudo es asintomática, por lo que la pérdida del injerto a consecuencia de la enfermedad recurrente es poco común.69
NEFRITIS HEREDITARIA
La nefritis hereditaria, o síndrome de Alport, es una enfermedad relativamente frecuente que a menudo se acompaña de pérdida de la audición y alteraciones del cristalino.70 Se han descrito tres formas de herencia: dominante ligada al cromosoma X, autosómica dominante y con menor frecuencia autosómica recesiva. El primer mecanismo parece estar presente en casi todas las familias con afección renal y sordera.71 Esta forma de herencia puede explicar el porqué la enfermedad no suele trasmitirse de los padres afectados a sus hijos, en vista de que el padre sólo podría transferir el cromosoma Y normal. El hecho de que la herencia esté ligada al cromosoma X también puede explicar la evolución por lo general más benigna en las mujeres, ya que uno de los dos cromosomas X es inactivado al azar en cada célula (hipótesis de Lyon),70 y como resultado, la mitad de las células será normal, reduciendo así al mínimo la gravedad de la enfermedad. Por otra parte, es más probable que la herencia autosómica dominante o recesiva ocurra en familias con enfermedad renal progresiva pero sin signos auditivos u oculares.71 En este caso las mujeres pueden afectarse con la misma gravedad que los hombres.
La manifestación histológica más temprana de la nefritis hereditaria es el adelgazamiento difuso de la membrana basal glomerular.72 Con el tiempo, existe cada vez mayor división longitudinal de la MGB, produciendo el aspecto laminado característico que se observa por microscopía electrónica [ver figura 7]. La microscopía de luz revela que esta división se relaciona con mayor daño glomerular, desde la expansión mesangial inicial hasta la esclerosis glomerular e infiltrado intersticial que contiene células espumosas de origen dudoso.
La patogenia de la nefritis hereditaria no se ha esclarecido por completo. Varios estudios han demostrado que la mayoría de los pacientes con esta enfermedad carecen de un antígeno que al parecer forma parte del dominio no colágeno de la colágena tipo IV.73,74 Al parecer este antígeno está relacionado con el antígeno de Goodpasture (el antígeno que reacciona con el anticuerpo anti-MBG); los glomérulos de la mayoría de los enfermos con nefritis hereditaria muestran disminución o falta de unión de los anticuerpos anti-MBG.73 Este dato es potencialmente importante para el diagnóstico.
Las manifestaciones clínicas principales de la nefritis hereditaria incluyen lo siguiente: enfermedad renal, que puede ocurrir en forma aislada; alteraciones oftálmicas, incluyendo cataratas y lenticono anterior, y sordera neurosensorial para tonos altos.70 Las alteraciones renales iniciales son parecidas a las que ocurren en otros tipos de glomerulonefritis focal: proteinuria y hematuria asintomáticas o episodios de hematuria franca. La hematuria puede aparecer antes de los cinco años de edad en los hombres, pero no ser detectada hasta la adolescencia. Algunos niños afectados se han identificado por los antecedentes familiares.
La enfermedad renal en hombres suele ser evolutiva: se relaciona con proteinuria progresiva, insuficiencia renal e hipertensión arterial. Finalmente, aparece insuficiencia renal en etapa terminal por lo general entre los 16 y 35 años de edad, aunque en ocasiones se retrasa hasta los 45 o 60 años de edad. Las mujeres sólo presentan hematuria microscópica persistente con función renal normal; sin embargo, en algunas familias puede desarrollarse insuficiencia renal antes de los 25 años de edad en las mujeres.63
El diagnóstico de nefritis hereditaria suele confirmarse por los antecedentes familiares, incluyendo el examen de orina de los miembros asintomáticos de la familia75 y, en caso necesario, por biopsia renal. Unos cuantos pacientes no presentan antecedentes familiares de la enfermedad; es probable que estos casos indiquen la existencia de una mutación fresca del gen responsable del defecto de la membrana basal. Debe resaltarse que el adelgazamiento aislado de la MBG no es diagnóstico del síndrome. Otra entidad que a menudo ocurre en familias, la enfermedad de membrana basal delgada, también puede producir hematuria persistente.62,63 Sin embargo, este trastorno por lo general tiene un pronóstico benigno, no se relaciona con división de la MBG, ni con insuficiencia renal progresiva.
Ningún procedimiento terapéutico ha demostrado ser eficaz para la nefritis hereditaria. Es posible que la MBG sea débil en esta enfermedad y que la hendidura de la MBG refleje ciclos de daño y reparación. Si esta teoría es correcta, la disminución de la presión intraglomerular con un inhibidor de la enzima convertidora de angiotensina (ECA) puede ofrecer cuando menos protección parcial a largo plazo.
Puede utilizarse el trasplante renal en los pacientes con insuficiencia renal. La enfermedad recurrente no representa un problema en vista de que la MBG del donador es normal. No obstante, algunos pacientes tienen riesgo de sufrir una enfermedad por anticuerpos anti-MBG de novo debido a que el riñón donado puede contener antígenos no observados previamente.76 Esta complicación a menudo es subclínica pero puede ocasionar la pérdida del injerto en casos aislados.
Síndrome nefrótico
Los pacientes con síndrome nefrótico presentan proteinuria intensa (> 3 g/día), hipoalbuminemia, edema, hiperlipidemia, y con menor frecuencia, fenómenos tromboembólicos. El síndrome nefrótico puede producirse tanto por una enfermedad renal primaria como por lesión glomerular causada por una enfermedad sistémica [ver tabla 2].22 Esta sección revisa los tres tipos principales de síndrome nefrótico primario, que se caracterizan por relacionarse con sedimento urinario benigno y se definen por sus manifestaciones histológicas: (1) enfermedad de cambios mínimos, (2) glomerulosclerosis focal, y (3) nefropatía membranosa. El lupus eritematoso generalizado, la diabetes mellitus y la amiloidosis también son causas relativamente frecuentes de síndrome nefrótico.
Los trastornos nefríticos difusos, como la glomerulonefritis posinfecciosa, la nefritis lúpica, la glomerulonefritis membranoproliferativa, y con menor frecuencia, las vasculitis, también pueden relacionarse con proteinuria en límites nefróticos. Sin embargo, estas enfermedades se relacionan en forma distintiva con sedimento nefrítico activo, que contiene eritrocitos, leucocitos y cilindros granulares y celulares.
En general la causa del síndrome nefrótico se establece por biopsia renal. No obstante, la evaluación antes de la biopsia debe incluir determinación de nitrógeno de urea en sangre, excreción de proteínas en orina de 24 horas, y concentraciones plasmáticas de creatinina, albúmina, glucosa y colesterol. La orina y el plasma de los pacientes mayores de 25 años también debe examinarse por inmunoelectroforesis, con el fin de detectar paraproteínas, presentes en la mayoría de los casos de amiloidosis primaria. Si uno de estos estudios resulta positivo, la presencia de amiloidosis de fondo a menudo puede confirmarse por biopsia rectal o de pliegues de tejido adiposo, procedimiento menos cruento que la biopsia renal. Aunque la búsqueda de LES está indicada en pacientes con síntomas sistémicos compatibles, por lo general no elimina la necesidad de realizar biopsia renal, porque los diferentes tipos de nefritis lúpica se tratan de manera distinta.
ENFERMEDAD DE CAMBIOS MINIMOS
La enfermedad de cambios mínimos, también conocida como nefrosis lipoide, enfermedad nula, o síndrome nefrótico por lesión mínima, es la causa más frecuente de síndrome nefrótico en niños, aunque también es responsable del 20 al 25 porciento de los casos en adultos.77 El dato histológico que confirma el diagnóstico de esta enfermedad es la observación de fusión difusa de los podocitos de las células epiteliales por microscopía electrónica [ver figura 8]. La microscopía de luz muestra glomérulos normales o con hipercelularidad mesangial leve como única alteración. Los estudios de inmunoflorescencia suelen ser negativos para inmunoglobulinas y complemento, pero en algunos casos selectos se observa depósito focal de IgM y C3. Se considera que estos depósitos representan atrapamiento inespecífico en la pared capilar glomerular con permeabilidad anormal.78
La enfermedad de cambios mínimos suele ser idiopática; en algunos casos pueden participar en la etiología la alergia a los alimentos, los inhalantes u otros agentes extraños, incluyendo las picaduras de abejas.22,79 Esta enfermedad se ha relacionado con neoplasias malignas (sobre todo con enfermedad de Hodgkin),80,81 tratamiento con litio,82,83 con el uso de medicamentos antinflamatorios no esteroides (AINE) (sobre todo fenoprofen y otros derivados del ácido propiónico),84,85 y puede ocurrir al inicio de la diabetes mellitus insulinodependiente.86
Etiología
La etiología de la enfermedad de cambios mínimos no se ha esclarecido por completo. Los estudios experimentales indican que el daño celular epitelial participa de manera importante en este trastorno, ocasionando disminución secundaria en la producción de proteoglicanos cargados en forma negativa (v.gr., sulfato de heparán) y las sialoproteínas; estas moléculas están presentes en la MBG y cubren los podocitos.22,87 La producción disminuida podría ser responsable de los dos principales datos de la enfermedad de cambios mínimos: proteinuria, por pérdida de la barrera de carga aniónica en la MBG y fusión de podocitos (la separación normal de los podocitos adyacentes se mantiene en parte por la repulsión electrostática modulada por las cargas aniónicas).22,87
Varias observaciones indican que las células T desempeñan una función crucial, tal vez a través de los efectos de una linfocina, en la patogenia del daño a la célula epitelial.88 Entre éstas se encuentra la respuesta generalmente positiva al tratamiento con prednisona y las relaciones de la enfermedad de cambios mínimos con la enfermedad de Hodgkin (en la que la función de las células T es anormal)80 y con el uso de antinflamatorios no esteroides (AINE). El síndrome nefrótico relacionado con el uso de AINE a menudo se acompaña de nefritis intersticial, con infiltrado intersticial compuesto principalmente por células T.85 Las células T de estos pacientes parecen producir una linfocina que aumenta la permeabilidad glomerular.89
Los pacientes con enfermedad de cambios mínimos presentan de manera característica un inicio súbito con edema, proteinuria intensa, sedimento urinario benigno y concentración plasmática de creatinina normal.77 Sin embargo, se ha descrito insuficiencia renal aguda en algunos casos, sobre todo en mayores de 60 años de edad.77 Se desconoce el mecanismo productor de insuficiencia renal, pero es probable que contribuyan el edema intersticial, que ocasiona colapso tubular, o la pérdida del área de superficie de filtración causada por la fusión de los podocitos.90,91
Diagnóstico y tratamiento
El diagnóstico de enfermedad de cambios mínimos en adultos se confirma por biopsia renal. No obstante, en los niños suele administrarse prednisona en forma empírica por la elevada prevalencia del trastorno en este grupo de edad. Sólo los niños resistentes a los esteroides deben someterse a cirugía renal. La enfermedad de cambios mínimos es responsable del 50 porciento de los casos de resistencia a los esteroides en niños menores de seis años de edad, pero sólo del cuatro porciento en niños mayores.92
La administración de esteroides es el tratamiento inicial de elección, y produce remisión de la proteinuria en más del 90 porciento de los pacientes. La enfermedad remite en término de dos a ocho semanas en niños, pero puede tardar de 12 a 16 meses en adultos.77,92 Un esquema que se ha utilizado con frecuencia en adultos incluye la administración de prednisona en dosis diaria de 1 mg/kg durante cuatro semanas (o hasta una semana después de que se ha logrado la remisión). Para disminuir al mínimo los efectos adversos, los pacientes siguen el esquema de dosis única en días alternos con 1 mg/kg. Si la proteinuria se resuelve o se reduce a niveles muy bajos, el tratamiento en días alternos debe mantenerse hasta un mes y reducirse posteriormente en forma paulatina por un periodo de varios meses, en un intento por reducir al mínimo la incidencia de recaídas. El diagnóstico de resistencia a esteroides no debe considerarse en el adulto a menos de que hayan transcurrido 12 semanas de tratamiento sin respuesta adecuada.77
Si la remisión va seguida por recurrencia, como sucede en más de la mitad de los casos, suele administrarse otro ciclo de esteroides. Sin embargo, cuando las recaídas ocurren con frecuencia, puede requerirse tratamiento continuo en días alternos y en dosis bajas durante seis a 12 meses para mantener la remisión.77 Las recaídas suelen ocurrir en término de un año después de la suspensión del tratamiento con esteroides, aunque algunos enfermos sufren recaídas que responden a los esteroides años después de la remisión.93,94
Hay tres opciones adicionales en los pacientes que requieren tratamiento continuo con prednisona para evitar las recaídas (v.gr., dependientes de esteroides), o en los que sufren recaídas frecuentes: fármacos alquilantes (v.gr., ciclofosfamida o clorambucil), ciclosporina, o bien, no administrar tratamiento. Se ha demostrado que los medicamentos alquilantes tienen un efecto notable, reduciendo o evitando el número de recaídas en los pacientes con recurrencias frecuentes del síndrome nefrótico.77,95-98 Estos fármacos también pueden resultar benéficos en pacientes dependientes de esteroides, en los que son resistentes a los esteroides desde un principio o en los que desarrollan resistencia secundaria a los esteroides.77,97,98 Sin embargo, la frecuencia de resultados satisfactorios en estos enfermos es menor que en los pacientes sensibles a los esteroides,96 a menos que la duración del tratamiento se prolongue de ocho a 12 meses.97 Si ocurren recaídas después de suspender el tratamiento citotóxico, el enfermo a menudo responde a los esteroides.99
Los estudios realizados en niños con enfermedad de cambios mínimos indican que la ciclofosfamida debe administrarse en dosis máxima de 2.0 a 2.5 mg/kg/día durante ocho semanas en los enfermos que sufren recaídas frecuentes y tal vez durante 12 meses en los que son dependientes de los esteroides o resistentes a estos fármacos.96,97 Los efectos colaterales potenciales incluyen supresión de la médula ósea, fibrosis gonadal (probablemente relacionada con esterilidad),100 cistitis hemorrágica y supresión a largo plazo de la función linfocitaria.101 También existen datos de que la ciclofosfamida puede ocasionar neoplasias de aparición tardía, que se caracterizan por aparecer en los pacientes que reciben tratamiento prolongado.102 El clorambucil también se ha utilizado con éxito en la enfermedad de cambios mínimos,103 pero la toxicidad con este medicamento puede ser incluso más limitante que con la ciclofosfamida.104 Aunque la azatioprina suele ser mejor tolerada, no parece brindar beneficio a los pacientes con enfermedad de cambios mínimos a menos que se administre de seis a 12 meses cuando menos.105 En vista que la administración prolongada de esteroides o fármacos citotóxicos puede relacionarse con toxicidad grave, estos medicamentos deben utilizarse con precaución, sobre todo en adultos con enfermedad de cambios mínimos, porque el tratamiento inmunosupresor no ha mostrado beneficios a largo plazo.106,107 Aunque los pacientes tratados remiten con más rapidez, casi todos los adultos no tratados remiten en forma espontánea a los cuatro años,106 y la evolución a la insuficiencia renal es muy rara. Por lo tanto, debe evitarse la administración repetida de fármacos inmunosupresores en los enfermos con recurrencias frecuentes; sólo debe administrarse tratamiento sintomático, como el uso de diuréticos para el edema.
Un ejemplo del riesgo potencial del tratamiento se demuestra en el estudio de seguimiento a largo plazo de 389 niños con enfermedad de cambios mínimos.108 Diez enfermos fallecieron: uno por insuficiencia renal, con glomerulosclerosis focal en la biopsia repetida, y seis como consecuencia de infecciones relacionadas en parte con el tratamiento inmunosupresor.
Se ha sugerido el tratamiento con ciclosporina para dos grupos de pacientes con enfermedad de cambios mínimos: (1) pacientes con toxicidad por esteroides y que sufren recaídas frecuentes o pacientes dependientes de esteroides en los que dos a tres meses de tratamiento con ciclofosfamida o clorambucil no han logrado inducir una remisión prolongada y (2) pacientes resistentes a los esteroides y agentes alquilantes. En el primer grupo la ciclosporina causa remisión en el 70 a 80 porciento de los casos,109,110 pero la mayor parte de los enfermos recaen al disminuir la dosis111 o suspender el medicamento.112,113 Se ha usado una dosis de mantenimiento de 4 a 5 mg/kg/día, vigilando con cuidado los niveles sanguíneos, la presión arterial y la creatinina sérica. En el segundo grupo, que puede incluir algunos pacientes con glomeruloesclerosis focal no diagnosticada, la respuesta es menos frecuente. Después de un periodo de tratamiento de seis meses, más de la mitad de los pacientes resistentes a esteroides logran por lo menos una remisión parcial, pero las recaídas son frecuentes.114
Una posible excepción a la evolución de la enfermedad de cambios mínimos, generalmente benigna a largo plazo, ocurre en pacientes que finalmente se vuelven resistentes a los esteroides y en quienes la segunda biopsia renal demuestra evolución de la enfermedad de cambios mínimos a glomerulosclerosis focal. Aunque el tratamiento citotóxico puede ser eficaz en esta situación, algunos de estos pacientes evolucionan a insuficiencia renal en etapa terminal.115,116
Relación con otras enfermedades
La patogenia y las manifestaciones clínicas de la enfermedad de cambios mínimos parecen ser similares a la de otros trastornos glomerulares, incluyendo glomerulosclerosis focal (ver adelante), nefropatía por IgM y glomerulonefritis proliferativa mesangial idiopática. Algunos enfermos con fusión difusa de los podocitos presentan depósitos de IgM en el mesangio, lo que indica que esta lesión podría representar una enfermedad distinta.78 Sin embargo, el depósito de IgM ocurre con igual frecuencia en pacientes con enfermedad de cambios mínimos y en enfermos con glomerulosclerosis focal, con correlación pronóstica de acuerdo con el diagnóstico confirmado por microscopía de luz. Los individuos con enfermedad de cambios mínimos aparente y depósito de IgM responden al tratamiento con prednisona, mientras que la respuesta es menos predecible en los pacientes con glomerulosclerosis focal.78
Aproximadamente del tres al cinco porciento de los pacientes con síndrome nefrótico presentan proliferación mesangial difusa y fusión de podocitos, pero los estudios con inmunofluorescencia son negativos (aunque otras enfermedades como la nefritis lúpica y la nefropatía por IgA pueden producir un cuadro similar en la microscopía de luz, estos padecimientos se relacionan con un depósito importante de IgG y de IgA, respectivamente). Es probable que estos pacientes sólo presenten daño inicial más severo que los sujetos con enfermedad de cambios mínimos, con un aumento en la frecuencia de recaídas y menor incidencia de recuperación.117-119 Más de la mitad de los pacientes con esta enfermedad responden en un principio a la administración de prednisona; la mayoría de los pacientes que no responden a la prednisona muestran remisión espontánea en término de un año.117-118 No obstante, la insuficiencia renal evolutiva se desarrolla finalmente en el 10 a 30 porciento de los casos con glomerulosclerosis focal diagnosticada por biopsia.78,119 Algunos pacientes con proliferación mesangial difusa progresiva responden a la ciclofosfamida; en estos casos es razonable realizar la prueba terapéutica cuando existe síndrome nefrótico persistente y elevación de la concentración plasmática de creatinina.118,119
GLOMERULOSCLEROSIS FOCAL
La glomerulosclerosis focal es la tercera causa más común de síndrome nefrótico en adultos, después de la nefropatía membranosa y la enfermedad de cambios mínimos.22,120 Los datos histológicos, incluyendo la presencia de fusión difusa de los podocitos, son parecidos a los de la enfermedad de cambios mínimos. Sin embargo, la microscopía de luz muestra que algunos glomérulos presentan lesiones escleróticas segmentarias [ver figura 9]. Este cambio es más notable en la región yuxtaglomerular, más que en los glomérulos corticales externos, y es probable que esta distribución refleje diferencias en la hemodinámica regional.
La distinción patológica entre enfermedad de cambios mínimos y glomerulosclerosis focal no siempre puede realizarse con certeza. En las etapas tempranas de la glomerulosclerosis focal las lesiones escleróticas características sólo están presentes en una pequeña fracción de los glomérulos. Es probable que en la biopsia por punción no se obtengan glomérulos afectados, lo cual conduce al diagnóstico histológico de enfermedad de cambios mínimos.
Es importante destacar que la esclerosis segmentaria aislada es inespecífica; lesiones similares pueden ocurrir con el daño modulado en forma hemodinámica por la pérdida previa de nefronas y después de la fase de cicatrización de cualquier forma de glomerulosclerosis focal, como la glomerulonefritis posinfecciosa leve. Sin embargo, en estas situaciones la fusión de los podocitos es focal, limitada a las zonas escleróticas; además; también se observan grandes glomérulos hipertrofiados con daño secundario.
En muchos casos la glomerulosclerosis focal parece ser parte del espectro de la enfermedad de cambios mínimos, tal vez representando una lesión más grave a las células epiteliales glomerulares.121 Por ejemplo, algunos pacientes con enfermedad de cambios mínimos aparentemente evolucionan a la glomerulosclerosis focal al repetir la biopsia.115 Además, muchos pacientes con glomerulosclerosis focal siguen una evolución sensible a los esteroides, con recurrencia frecuente, similar a lo observado en la enfermedad de cambios mínimos.98,122
Varias observaciones sugieren que un factor de permeabilidad circulante puede ser responsable de la proteinuria nefrótica en la glomeruloesclerosis focal. En algunos pacientes que progresan hasta la insuficiencia renal, la enfermedad recurre en el paciente trasplantado, con retorno de la proteinuria en minutos a horas después del trasplante.123-125 Un extracto de plasma de estos pacientes puede aumentar la permeabilidad a las proteínas en glomérulos aislados de rata.126 Cuando se sometió a estos pacientes a absorción del plasma con una columna de proteína A, la proteinuria se redujo durante dos semanas, y un eluído (no una inmunoglobulina) de la columna indujo proteinuria en ratas.127 El intercambio plasmático produjo una respuesta temporal en un grupo semejante de pacientes.128 Se ha sugerido que la permeabilidad alterada de proteínas puede ser la causa de la esclerosis subsecuente y la progresión de la enfermedad.129
Aunque la glomerulosclerosis focal suele ser idiopática, también ocurre en adictos a la heroína intravenosa y hasta en el 20 porciento de los pacientes con síndrome de inmunodeficiencia adquirida (SIDA), sobre todo en los que no son homosexuales.130-133 Es probable que las toxinas virales o exógenas desempeñen un papel importante en estas situaciones.
Los enfermos con glomerulosclerosis focal tienden a presentar datos clínicos y químicos del síndrome nefrótico, aunque en algunos casos el único signo es la proteinuria no nefrótica, asintomática.120,134,135 Hasta en la mitad de los casos se presenta al inicio hipertensión arterial, hematuria leve y creatinina plasmática elevada. En los pacientes con enfermedad de cambios mínimos, el desarrollo tardío de resistencia a los esteroides es el dato principal que indica evolución a glomerulosclerosis focal.115
Los pacientes con esta enfermedad también presentan mayor incidencia de uno o más defectos tubulares, incluyendo glucosuria renal, aminoaciduria, acidosis tubular renal y pérdida de fosfato.136,137 Estas alteraciones pueden representar daño tubular secundario en nefronas con lesiones glomerulares escleróticas. En forma alternativa, el deterioro de la función tubular puede reflejar los efectos tóxicos de la reabsorción de proteínas por los túbulos proximales.
Se ha considerado que el pronóstico de la glomerulosclerosis focal es invariablemente malo, en vista de que el padecimiento es relativamente insensible al tratamiento inmunosupresor y porque evoluciona a la insuficiencia renal en etapa terminal en término de uno a 20 años. Aunque ocurre evolución rápida cuando la enfermedad se relaciona con el abuso de heroína o con el SIDA, cerca de la tercera parte de los pacientes con glomerulosclerosis focal idiopática se comportan en forma similar a los enfermos con enfermedad de cambios mínimos, presentan remisiones inducidas por esteroides y en ocasiones siguen una evolución con recaídas frecuentes.18,122,134,138 Los enfermos con recaídas frecuentes o dependientes de esteroides pueden beneficiarse con la administración de ciclofosfamida durante dos a tres meses,98,122,139 con un esquema parecido al que se prescribe en la enfermedad por cambios mínimos.98,122 También puede inducirse la remisión de la proteinuria en los pacientes resistentes a los esteroides; sin embargo, en esta situación la frecuencia de éxito es menor del 25 porciento,98,139 a menos que el tratamiento sea vigoroso y prolongado.140 Un esquema prolongado que incluyó esteroides en pulsos y por vía oral y agentes alquilantes, indujo una remisión del 60 porciento en un estudio no controlado de niños con la enfermedad.141 Sin embargo, siempre debe tenerse en cuenta la toxicidad potencial del tratamiento citotóxico.
No es soprendente que los pacientes que responden al tratamiento presenten mucho mejor pronóstico que los que no responden.134,138,139 Los pacientes con proteinuria no nefrótica también conservan mejor la función renal a largo plazo.135 El embarazo puede exacerbar la evolución de la enfermedad.142
La ciclosporina induce remisión completa en el 15 a 20 porciento de los pacientes resistentes a esteroides con glomeruloesclerosis focal,109,110,143,144 aunque el porcentaje puede ser más alto después del tratamiento durante un año.114 La insuficiencia renal por toxicidad de la ciclosporina constituye un riesgo, lo mismo que las posibles recaídas cuando se suspende el tratamiento.
En los pacientes que no responden a estos tratamientos o en los que la relación riesgo-beneficio no es favorable, los inhibidores de la ECA y en especial los AINE [ver adelante, Tratamiento general del síndrome nefrótico] han sido útiles para reducir la proteinuria.123
Los pacientes que evolucionan a la etapa terminal de la insuficiencia renal suelen ser jóvenes y, por lo tanto, candidatos excelentes para el trasplante renal. Sin embargo, se desarrollará enfermedad renal recurrente en el 20 a 30 porciento de los casos; por lo general la proteinuria intensa reaparece en términos de horas o semanas.124,125 La glomerulosclerosis focal recurrente produce pérdida del injerto en una tercera parte a una mitad de estos casos, en particular en niños pequeños,145 en pacientes en los que la enfermedad ha ocasionado insuficiencia renal en los primeros tres años después de su aparición, y en los casos en que existe proliferación mesangial.124,125,126
NEFROPATIA MEMBRANOSA
La nefropatía membranosa es la causa más frecuente de síndrome nefrótico idiopático en adultos. El nombre de esta enfermedad se deriva del cambio característico observado por microscopía de luz: engrosamiento difuso de la membrana basal glomerular y la pared capilar, con escaso o nulo aumento en la celularidad [ver figura 10a].147 En los casos leves o en etapas tempranas, el engrosamiento de la pared capilar es relativamente normal, por lo que se necesita microscopía electrónica o de inmunofluorescencia para establecer en forma definitiva el diagnóstico. Los complejos inmunes pueden observarse por medio de microscopía electrónica, que demuestra que el engrosamiento de la pared capilar glomerular está causada por depósito subepiteliales, entre la MBG y las células epiteliales [ver figura 10b]. El estudio de inmunofluorescencia revela depósitos difusos de IgG y C3 en la pared capilar con un patrón granular o lumpy-bumpy [ver figura 10c].
El mecanismo por el que estos complejos son capaces de pasar a través de
la MBG y se alojan en el espacio subepitelial aún no se esclarece del
todo. Estudios experimentales indican un mecanismo alternativo: desarrollo de
agregados inmunes por un mecanismo in situ en el que los antígenos y
anticuerpos libres se depositan por separado.148,149 De acuerdo con
esta teoría, el antígeno ya se encuentra presente (v.gr.,
producido por células epiteliales glomerulares) o se deposita en el
espacio subepitelial y a continuación reacciona con anticuerpos libres
circulantes. La adición constante de antígenos y anticuerpos
libres conduce al desarrollo de la red inmune hasta el tamaño en el que
pueden ser observados por microscopía electrónica.
Los modelos animales indican que el antígeno estimulador debe ser catiónico, lo que permitiría que cruce la MBG cargada negativamente, o estar presente en condiciones normales en el espacio subepitelial, como es el caso de los antígenos epiteliales tubulares renales.149 La activación subsecuente del sistema del complemento, en particular de los componentes terminales del complejo de ataque a la membrana (C5b-C9), parece ser responsable del daño asociado a la pared capilar,150 que produce el aumento en el tamaño de los poros necesario para que ocurra la proteinuria.151
La mayoría de los casos de nefropatía membranosa son idiopáticos. Sin embargo, diversos padecimientos sistémicos se han relacionado con la enfermedad.22 De éstos, los más comunes son los tumores sólidos (con depósito de antígenos tumorales en los glomérulos),81 el lupus eritematoso sistémico, los medicamentos como el oro y la penicilamina,152,153 y las infecciones por el virus de la hepatitis B.154 Es posible que la hepatitis B sea la causa de la nefropatía membranosa en niños y adultos jóvenes en áreas endémicas155 y puede acompañarse de antigenemia por el hepatitis Be.156 La remisión espontánea es frecuente, en especial cuando la antigenemia desaparece. No se ha demostrado que el tratamiento con interferón sea eficaz.157 En raras ocasiones se encuentra asociación entre la nefropatía membranosa y el virus de la hepatitis C,158 la artritis reumatoide no tratada, la sarcoidosis o la sífilis.22 Se ha calculado que ocurren neoplasias malignas ocultas, generalmente tumores sólidos, hasta en el10 porciento de los casos nefropatía membranosa en adultos.81 En la mayoría de estos casos el tumor ya se ha diagnosticado o es evidente desde el punto de vista clínico al momento del comienzo del síndrome nefrótico. Es probable que las neoplasias malignas ocultas sean responsables de menos del dos porciento de los casos.159 En consecuencia, no está justificada la evaluación excesiva del paciente con nefropatía membranosa en busca de neoplasias ocultas, a menos que existan datos compatibles como pérdida de peso, anemia de causa desconocida o búsqueda de sangre en heces positiva.
La nefropatía membranosa también puede ocurrir en el uno al tres porciento de los pacientes con artritis reumatoide tratados con oro parenteral (el oro por vía oral es menos nefrotóxico) y en el siete por ciento de los casos tratados con penicilamina,152,153 y se ha notificado en asociación con la exposición a formaldehido.160
Los pacientes con nefropatía membranosa de manera característica presentan edema y proteinuria en rangos nefróticos.147,161 No obstante, cerca del 20 porciento de los casos presentan proteinuria asintomática, no nefrótica, que se detecta en los exámenes de rutina. Puede observarse hematuria leve o hipertensión arterial como manifestación clínica inicial. El diagnóstico correcto sólo se confirma por medio de biopsia renal.
La evolución de la nefropatía membranosa es variable. La enfermedad inducida por medicamentos tiende a remitir después de la suspensión del fármaco causante, aunque pueden trascurrir de uno a dos años antes de que desaparezca la proteinuria.153 Este retraso en la recuperación puede relacionarse en parte con la lentitud con que son eliminados los complejos del espacio subepitelial, separado de la circulación sistémica por la MBG.162
La historia natural de la enfermedad idiopática es variable: en el lapso de cinco a 10 años los pacientes pueden clasificarse, en proporciones iguales, en cuatro grupos: remisión espontánea, proteinuria no nefrótica persistente, síndrome nefrótico persistente y evolución a la insuficiencia renal.161,163-166 Esta variabilidad se atribuye en parte a la presencia de subgrupos con diferente pronóstico. El pronóstico es mejor en mujeres, niños y en pacientes con proteinuria no nefrótica; menos del 10 porciento de estos pacientes evolucionarán hacia el deterioro en cinco a 10 años.161,163,167,168 Por comparación, los hombres con síndrome nefrótico tienen peor pronóstico, sobre todo cuando la concentración de creatinina plasmática se encuentra elevada desde el principio de la enfermedad.161,163,165,169 La enfermedad tubulointersticial indica también mal pronóstico. Casi siempre ocurre enfermedad progresiva en término de dos a tres años. Por lo tanto, el mantenimiento de la concentración normal de creatinina plasmática a los tres años es un signo de buen pronóstico.161
La evolución de la nefropatía membranosa, más que la de otros estados nefróticos, puede complicarse también por el desarrollo de trombosis venosa renal.170,171 La función renal no suele estar afectada; la embolia pulmonar es el indicador principal para sospechar la presencia de trombosis venosa renal. Aunque en el 10 a 15 porciento de los pacientes puede demostrarse la trombosis en la venografía renal, la posibilidad de embolia pulmonar secundaria es incierta, de ahí que exista controversa sobre si se requiere un estudio rutinario con el fin de anticoagular al paciente en ausencia de signos clínicos de trombosis. Un análisis reciente concluyó que es adecuado administrar tratamiento anticoagulante con warfarina para la mayoría de los pacientes con nefropatía membranosa.172
No se ha establecido el tratamiento óptimo de la nefropatía membranosa.173 Varios estudios controlados y no controlados indican que la administración de prednisona durante dos o tres meses (125 mg cada tercer día) retarda la evolución a la insuficiencia renal.174-175Sin embargo, el grado de proteinuria no parece afectarse por el tratamiento con prednisona a corto plazo,175 y no se han encontrado efectos benéficos en otros estudios.164, 176
Se han revisado varios programas de tratamiento que emplean ciclofosfamida o clorambucil, por lo general asociados a esteroides.177,178 Sin embargo, estos programas se acompañan de riesgos, que en algunos pacientes pueden superar los beneficios. Los pacientes que pueden sufrir una recuperación espontánea eventual (v.gr., pacientes jóvenes, mujeres y los que tienen función renal normal, presión arterial normal y proteinuria leve) deben mantenerse en vigilancia estrecha, poniendo atención a los elementos del manejo general (ver adelante). Por otro lado, los que tienen mal pronóstico (v.gr., pacientes de edad avanzada, varones, pacientes con hipertensión severa, hiperlipidemia resistente, proteinuria en el rango de 6 a 10 g/día o mayor, o evidencia de aumento en la creatinina sérica) pueden beneficiarse del tratamiento.179 En los pacientes con enfermedad avanzada y un nivel de creatinina sérica mayor de 3.0 mg/dl, el beneficio potencial puede ser limitado por la cicatrización renal irreversible.
El programa con agentes alquilantes mejor revisado es el de Ponticelli y colaboradores,180 en el que se administra un ciclo de un mes de metilprednisolona en pulsos (1 g/día por tres días) y después prednisona o prednisolona oral (0.5 mg/kg/día o 0.4 mg/kg/día, respectivamente, por lo siguientes 27 días) alternando con un ciclo de un mes de clorambucil por vía oral (0.2 mg/kg/día), cada uno durante tres ciclos por un periodo total de seis meses. Los beneficios del programa son significativos, incluso para los pacientes con función renal en deterioro,181 pero parecen disminuir después de cuatro años, en especial si se compara con el tratamiento que usa solo prednisolona en pulsos.182 La incidencia de efectos colaterales, en especial leucopenia,183 es alta, lo que ha originado que algunos investigadores recomienden una dosis de clorambucil menor (0.15 mg/kg/día).
También se ha estudiado el uso de ciclofosfamida oral combinada con prednisona en pacientes que tienen progresión de la insuficiencia renal,184-186 con aparente beneficio, en especial cuando se administra por tiempo prolongado. La administración de ciclofosfamida intravenosa en pulsos185-187 y los cursos breves de ciclofosfamida por vía oral188 parecen ser menos efectivos.
Al parecer la ciclosporina (4 a 5 mg/kg/día) produce remisión completa o parcial en algunos pacientes con nefropatía membranosa y puede mantenerse con seguridad razonable si se vigilan en suero su concentración y la creatinina.189,190 Sin embargo, la ciclosporina es muy costosa, las recaídas son frecuentes después de suspender el medicamento y su efecto en la evolución no se ha demostrado. Se ha propuesto el tratamiento con g-globulina intravenosa en dosis altas,191 pero no se han realizado estudios controlados al respecto.
MANEJO GENERAL DEL SINDROME NEFROTICO
Algunas medidas generales pueden ser adecuadas para lospacientes con síndrome nefrótico, independientemente de la patología subyacente [ver tabla 3].
El aumento en la proteína dietética en los pacientes nefróticos no aumenta la concentración de albúmina en suero e incluso puede ser dañino.192,193 Al parecer es adecuada una ingesta de 1 g/kg/día de proteína, sobre todo de proteína de alta calidad biológica y la reducción de las grasas animales. Al parecer, limitar la ingesta proteína detiene la progresión a insuficiencia renal en pacientes con enfermedades glomerulares proteinúricas.194 También parece ser que la hiperlipidemia del síndrome nefrótico predispone a ateroesclerosis temprana y severa, por lo que amerita tratamiento.195,196 La dieta sola suele ser insuficiente, aunque puede ser eficaz si se suplemente con tratamiento farmacológico con inhibidores de la 3-hidroxi-3-metilglutaril coenzima A reductasa (HMG-CoA), como el lovastatin y el simvastatin.197,198 Los pacientes nefróticos pueden ser susceptibles a rabdomiolisis inducida por medicamentos, por lo que deben vigilarse los niveles de creatina fosfoquinasa sérica. Los derivados del ácido fíbrico se toleran menos.
La reducción de la presión arterial, en especial con el uso de inhibidores de la ECA en combinación con diuréticos (para estimular la producción de angiotensina) suele causar una reducción en la proteinuria nefrótica199-201 y retrasar la progresión a insuficiencia renal en los pacientes con enfermedades glomerulares proteinúricas.194 Esta reducción puede tardar varias semanas en ocurrir, lo que sugiere un mecanismo diferente de la reducción inmediata de la presión intraglomerular.202 Pueden usarse AINE como indometacina y meclofenamato con un objetivo semejante.123,203 Cuando ocurre una reducción importante en la proteinuria con estas medidas, suele mejorar el control del edema y la hiperlipidemia, y el pronóstico a largo plazo es relativamente bueno.
En ocasiones un paciente con síndrome nefrótico muy severo y que no responde puede presentar proteinuria masiva, trombosis e hiperlipidemia, progresando a la insuficiencia renal terminal. En estos pacientes puede estar indicada una nefrectomía médica por medio de la embolización bilateral de las arterias renales para producir un infarto terapéutico.204
DR. CECIL H. COGGINS
DR. HELMUT G. RENNKE
DR. BURTON D. ROSE
En este capítulo se discuten las principales características clínicas de las enfermedades glomerulares individuales, que se catalogan de acuerdo con su forma de aparición en: glomerulonefritis difusa, glomerulonefritis focal y síndrome nefrótico. Las manifestaciones renales de las enfermedades sistémicas, como la diabetes mellitus, el lupus eritematoso generalizado (LEG), las vasculitis necrosantes y la amiloidosis no se incluyen en esta subsección.
Glomerulonefritis difusa
Los trastornos relacionados con la glomerulonefritis difusa suelen manifestarse por datos de síndrome nefrítico: sedimento urinario activo que contiene eritrocitos, leucocitos y cilindros granulares y celulares, grados variables de proteinuria, que pueden alcanzar el rango nefrótico (> 3 g/día), concentración elevada de creatinina plasmática y, con frecuencia, hipertensión arterial y edema.
GLOMERULONEFRITIS POSINFECCIOSA
Glomerulonefritis posestreptocócica
La glomerulonefritis posestreptocócica es la enfermedad típica relacionada con el síndrome nefrítico agudo (ver adelante).1 Las manifestaciones clínicas se caracterizan por aparecer después de una infección por estreptococos ß-hemolíticos del grupo A, de los cuales algunas cepas son especialmente nefritogénicas en particular, como el tipo 12 (relacionado con faringitis) y el tipo 49 (relacionado con impétigo).2,3
En la microscopía de luz la glomerulonefritis posinfecciosa se caracteriza por aumento focal o difuso en la celularidad glomerular, que produce cierre de la luz capilar. En este trastorno se observa infiltración de neutrófilos circulantes y de células mononucleares y proliferación de células endoteliales y mensagiales glomerulares, que puede complicarse por la formación de medias lunas en los casos graves [ver figura 1a]. La microscopía por inmunofluorescencia muestra de manera característica un patrón granular con IgG y complemento, que indica depósito de complejos inmunes. Sin embargo, el cambio más distintivo lo constituye el depósito de medias lunas subepiteliales, o en forma de giba, observado por microscopía electrónica [ver figura 1b]. Estos depósitos inician una reacción mediada por complemente que origina daño glomerular. Se supone que los complejos inmunes incluyen un antígeno relacionado a los estreptococos, aunque no se ha definido el antígeno específico.
|
|

Las manifestaciones clínicas de la glomerulonefritis posestreptocócica varían desde la hematuria asintomática y proteinuria leve hasta el síndrome nefrítico típico con hematuria franca, proteinuria, oliguria, edema, hipertensión arterial e insuficiencia renal. Los niveles de complemento sérico por lo general están bajos; los títulos de antiestreptolisina O y sobre todo de anti-DNasa B se encuentran elevados. El diagnóstico diferencial para los pacientes que presentan enfermedad posestreptocócica incluye otras causas de síndrome nefrítico.
No existe tratamiento específico para la glomerulonefritis posestreptocócica, que se caracteriza por ser una enfermedad autolimitada. La proteinuria leve y la hematuria microscópica pueden persistir durante meses o incluso años. La proteinuria persistente en rango nefrótico es muy poco común, pero puede ocurrir, en especial en pacientes con gran cantidades de gibas subepiteliales.5 Por lo tanto, debe sospecharse uno de los trastornos nefróticos, como nefropatía membranosa, en los casos en que el paciente es revisado por primera vez en esta etapa tardía.
En la mayoría de los casos ocurre la recuperación completa, en especial en niños y cuando la enfermedad se adquiere durante una epidemia de infección estreptocócica.4,6 En algunos casos, sobretodo esporádicos y en adultos, ocurre solo recuperación parcial, aunque esto no es frecuente. Cuando la enfermedad aguda es severa, con oliguria prolongada, la insuficiencia renal puede ser permanente. Existen controversias acerca de la persistencia de las alteraciones renales a largo plazo. Se han presentado algunos casos de hipertensión, disminución de la velocidad de filtración glomerular (VFG) sin insuficiencia renal clínica y alteraciones histológicas, incluyendo esclerosis mesangial, que ocurren varios años después de la enfermedad aguda (sin embargo, es difícil establecer en estos casos si el diagnóstico original en realidad fue glomerulonefritis posestreptocócica).
Otras formas de glomerulonefritis posinfecciosa
El síndrome nefrítico agudo también puede ocurrir después de infecciones bacterianas (estafilocócicas, neumocócicas o estreptocócicas del grupo C), virales o parasitarias.9-12 Un ejemplo es la nefritis relacionada con endocarditis bacteriana o con derivaciones auriculoventriculares infectadas.13-15 Las manifestaciones renales de estos dos trastornos pueden ser parecidas, histológica y clínicamente, a las que se observan en la glomerulonefritis posestreptocócica (con gibas subepiteliales) o en la glomerulonefritis membranoproliferativa tipo I (con depósitos mensangiales y subendoteliales). La gravedad de las manifestaciones clínicas se relaciona sobre todo con la duración de la infección antes de iniciar el tratamiento antimicrobiano adecuado. En general, el control de la infección, incluyendo la extracción del cortocircuito infectado, conduce a la resolución rápida de la enfermedad, con recuperación de la función renal a los límites basales. Sin embargo, puede ocurrir insuficiencia renal irreversible, sobre todo cuando el tratamiento se ha retrasado.14
En pacientes con endocarditis la afección glomerular debe distinguirse de las embolias renales y de la nefritis intersticial aguda inducida por antimicrobianos. Las embolias renales se sospechan por dolor unilateral en los flancos y por otros síntomas generales; el diagnóstico puede confirmarse por la presencia de defectos de perfusión local o por gamagrafía con radioisótopos. La nefritis intersticial debe sospecharse por la aparición tardía de la enfermedad (por lo general más de 10 días después de la infección). Las manifestaciones de la nefritis intersticial incluyen eosinofilia, exantema y eosinofiluria. En comparación, la glomerulonefritis suele encontrarse en su grado máximo de gravedad antes de iniciar el tratamiento.
GLOMERULONEFRITIS MEMBRANOPROLIFERATIVA
La glomerulonefritis membranoproliferativa (GNMP), también llamada glomerulonefritis hipocomplementémica, glomerulonefritis lobular y glomerulonefritis mesangiocapilar, puede manifestarse como nefritis aguda o con una evolución más indolente relacionada con el síndrome nefrótico.16-18 La mayoría de los pacientes son niños o adultos jóvenes, y la GNMP puede causar el cinco a 10 porciento de los casos de síndrome nefrótico en este grupo. Durante su evolución, la mayoría de los pacientes puede tener cifras bajas de la fracción C3 del complemento.
La microscopía de luz revela paredes capilares glomerulares engrosadas y proliferación celular mesangial [ver figura 2a]. Por microscopía electrónica se han identificado dos subgrupos de GNMP. La enfermedad tipo I se define por dos características: (1) división de la membrana basal por la interposición de matriz mesangial, que produce una apariencia en vías de ferrocarril cuando la muestra de biopsia se tiñe con el reactivo ácido periódico de Schiff (PAS) o se tiñe con plata, y (2) depósitos de complejos inmunes subendoteliales y mesangiales notables. En la enfermedad tipo II la interposición de la matriz mesangial es menos obvia, y se observan depósitos prominentes, densos, en forma de listón (en lugar de complejos aislados) en la membrana basal; de aquí el nombre alternativo de enfermedad de depósitos densos [ver figura 2b]. Se desconoce el origen de estos depósitos.
|
|

|
||||||||||||||||||
|
Aunque la GNMP suele ser idiopática, puede relacionarse con algunos trastornos generalizados. Es común que los pacientes con lipodistrofia parcial presenten el tipo II de la GNMP y FC3NeF.20 El patrón membranoproliferativo también se ha observado en otros trastornos relacionados con la formación de complejos inmunes, incluyendo LEG, crioglobulinemia mixta, infecciones en derivaciones, endocarditis bacteriana, linfoma linfocítico crónico y esquistosomiasis.22
El diagnóstico de GNMP sólo puede confirmarse por biopsia renal. El dato clínico principal que indica la presencia de este trastorno es hipocomplementemia sin datos clínicos o serológicos de LEG o de infección estreptocócica reciente.
Al igual que con otras glomerulopatías, la gravedad de la enfermedad en el momento de aparición es un dato pronóstico importante. Los siguientes datos se relacionan con un riesgo mayor de evolución a insuficiencia renal terminal: síndrome nefrótico persistente, hipertensión arterial, insuficiencia renal, presencia de medias lunas (ver adelante) o de esclerosis en la biopsia, y la presencia de FC3NeF.18 En los pacientes con hematuria o proteinuria asintomática que presentan lesiones focales y segmentarias en la biopsia renal el pronóstico suele ser mejor.23
El tratamiento óptimo de la GNMP no se ha establecido. En un estudio retrospectivo, no controlado, de niños con enfermedad tipo I, el posible beneficio de la prednisona a dosis bajas para mantener la VFG fue cotrarrestado por un empeoramiento de la hipertensión.24,25 No existen en la actualidad estudios controlados de esteroides en adultos o en pacientes con enfermedad de tipo II. El tratamiento antiplaquetario, con aspirina o dipiridamol, puede ser benéfico a corto plazo (un año) para mantener la VFG, pero está en duda el beneficio a largo plazo.26 Los estudios controlados de ciclofosfamida, dipiridamol y warfarina no han demostrado beneficio a largo plazo.27,28
En resumen, ninguno de los tratamientos probados tienen un efecto benéfico demostrado a largo plazo. En los casos de síndrome nefrótico, algunos nefrólogos recomiendan, además de las medidas generales, administrar tratamiento a largo plazo en dosis moderadas y días alternos de esteroides en los pacientes en los que no es difícil controlar la hipertensión.
Las lesiones histológicas de la GNMP tipo I, y sobre todo del tipo II, con frecuencia recurren en los riñones trasplantados; sin embargo, esta afección a menudo es silenciosa, con mínimas manifestaciones clínicas o asintomática.29.30 Por lo tanto, el riesgo de recurrencia no impide que el trasplante tenga éxito en estos pacientes.31 La pérdida del injerto por GNMP recurrente ocurre en menos del 10 porciento de los casos, la mayoría de los cuales presentaron evolución relativamente rápida de su enfermedad inicial.
GLOMERULONEFRITIS EN MEDIAS LUNAS, O RAPIDAMENTE PROGRESIVA
A diferencia del inicio súbito del síndrome nefrítico que se observa en la glomerulonefritis aguda, con resolución en varias semanas, algunos pacientes sufren deterioro progresivo en semanas a meses que llega hasta insuficiencia renal. El examen general de orina revela proteinuria, en ocasiones en rango nefrótico, y sedimento activo con eritrocitos dismórficos y cilindros eritrocitarios. En los pacientes con glomerulonefritis rápidamente progresiva (GNRP), la biopsia suele mostrar medias luns celulares que cubren la capa parietal de la cápsula de Bowman en más del 50 porciento del glomérulo [ver figura 3]. Las medias lunas constituyen una respuesta inespecífica al daño glomerular y pueden ocurrir en cualquier enfermedad glomerular grave. El evento inicial en la formación de medias lunas es el daño a la pared capilar glomerular que permite al fibrinógeno o a la fibrina entrar al espacio de Bowman.32 La entrada de macrófagos y la proliferación secundaria de células epiteliales acompañan estas lesiones, y estos son los componentes celulares de la media luna. La insuficiencia renal se produce sobre todo por compresión del penacho capilar por la media luna expandida.
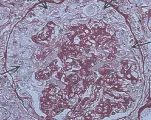
|
| Figura 3 |
| Glomerulonefritis de medias lunas |
El examen de muestras de biopsia renal por microscopía de luz es insuficiente para confirmar el diagnóstico. Por ejemplo, la identificación de depósitos mensagiales prominentes de IgA en la microscopía de inmunofluorescencia es diagnóstica de nefropatía por IgA de fondo, y la identificación de gibas subepiteliales por microscopía electrónica es diagnóstica de glomerulonefritis posinfecciosa [ver figura 1b].
Los pacientes con evolución clínica de GNRP y biopsias que muestran medias lunas celulares abundantes por microscopía de luz pueden subdividirse en tres grupos patogénicos con base en la inmunofluorescencia.33-36 Alrededor del 50 porciento tendrá evidencia mínima o nula de depósito de inmunoglobulinas en el glomérulo (GNRP pauci-inmune), el 40 porciento mostrará depósitos de complejos inmunes, y alrededor del 10 porciento presentará un patrón de anticuerpos dirigidos contra un componente de la membrana basal del capilar glomerular (enfermedad antimembrana basal glomerular o anti-MBG). La presencia de cilindros hemáticos y de leucocitos en la orina ayuda a distinguir la GNRP de otras causas de insuficiencia renal aguda, como la nefritis intersticial aguda, la obstrucción de las vías urinarias o la enfermedad ateroembólica.
GNRP sin anticuerpos o pauci-inmune
Los pacientes con GNRP sin anticuerpos o pauci-inmune suelen tener anticuerpos anticitoplasma de neutrófilo (ANCA, por sus siglas en inglés), lo que indica que la GNRP es parte de un síndrome de vasculitis. Los que tienen ANCA dirigidos contra la proteinasa 3 leucocitaria suelen tener granulomatosis de Wegener, mientras que los que tienen anticuerpos antimieloperoxidasa con más frecuencia tienen poliarteritis microscópica. El tratamiento inmediato e intensivo con esteroides y alquilantes, en ocasiones aunado a plasmaféresis, suele conservar la función e interrumpir lo que de otro modo sería una evolución progresiva hacia el deterioro.
GNRP por complejos inmunes
La mayoría de los pacientes con GNRP mediada por complejos inmunes tiene evidencia de una enfermedad sistémica, incluyendo LES, púrpura de Schönlein-Henoch, endocarditis bacteriana subaguda o infección de derivaciones, y crioglobulinemia mixta. Algunos parecen tener medias lunas sobrepuestas a otras enfermedades renales primarias, incluyendo glomerulonefritis membranoproliferativa, nefropatía por IgA y nefropatía membranosa. Algunos tienen un padecimiento que parece idiopático, aunque se han implicado a la penicilamina, la sífilis y las neoplasias en casos aislados.22 También es posible que exista cierta predisposición genética, el trastorno se ha asociado con el fenotipo BfF de la properdina y con el haplotipo HLA-DR2 o MT3.37 El pronóstico y tratamiento del padecimiento es semejante al de la GNRP sin anticuerpos.
Enfermedad por anticuerpos anti-MBG
El Dr. Ernest W. Goodpasture describió por primera vez la combinación de glomerulonefritis y hemorragia pulmonar. En la actualidad se ha demostrado que este grupo de síntomas puede producirse por diversos trastornos;38 por lo tanto, el término síndrome de Goodpasture debe limitarse a la entidad patológica específica, observada con más frecuencia en hombres jóvenes, en la cual la presencia de anticuerpos circulantes contra la MBG explica la glomerulonefritis.34 Estos anticuerpos también reaccionan con un antígeno similar en los pulmones, produciendo alveolitis, con hemorragia pulmonar.39 La hemoptisis suele preceder los datos clínicos de enfermedad renal, aunque algunos pacientes sólo presentan afección renal. La aparición de daño renal sin afección pulmonar indica que debe existir una lesión pulmonar simultánea que permita que los anticuerpos circulantes entren a los alveolos. Como ejemplos se encuentran la influenza, la exposición a hidrocarburos y el tabaquismo crónico.40,41
El cuadro patológico renal característico es la glomerulonefritis proliferativa, necrosante, con formación de medias lunas en más del 50 porciento de los glomérulos [ver figura 3]. La microscopía de inmunofluorescencia muestra tinción linear de IgG y C3 a lo largo de la MBG [ver figura 4]. Los datos observados por microscopía de luz y de inmunofluorescencia son diagnósticos de enfermedad de anticuerpos anti-MBG. Se puede observar una tinción inmunofluorescente similar en los alveolos de los pacientes afectados.

|
| Figura 4 |
| Glomerulonefritis de medias lunas: inmunofluorescencia |
La manifestación renal primaria de este trastorno es la insuficiencia renal rápidamente progresiva con sedimento urinario nefrítico activo. La proteinuria suele ser leve, lo que podría reflejar el notable deterioro de la filtración glomerular, y no suele observarse edema. Los niveles de complemento son normales.
La enfermedad de anticuerpos anti-MBG generalmente evoluciona a la insuficiencia renal, aunque algunos pacientes desarrollan enfermedad focal leve y tienen un buen pronóstico. Por ejemplo, en un estudio se demostró que la insuficiencia renal terminal se desarrolló en el 80 porciento de los pacientes con el síndrome en término de un año después del diagnóstico.42 El tratamiento farmacológico aislado, incluyendo los pulsos intravenosos con dosis altas de metilprednisolona, parece ser ineficaz contra las lesiones renales.33,43
Aunque no se han llevado a cabo estudios controlados, varios pacientes parecen beneficiarse con la combinación de plasmaféresis que extrae los anticuerpos anti-MBG circulantes y fármacos inmunosupresores, que evitan la formación de nuevos anticuerpos.44,45 Un esquema consiste en realizar cambios plasmáticos de 4 L/día durante aproximadamente una semana, combinados con prednisona (60 mg/día), ciclofosfamida (3 mg/kg/día) y azatioprina (1 mg/kg/día).44 La dosis de prednisona se reduce cada semana, y en pacientes mayores de 55 años la dosis de ciclofosfamida se reduce hasta 2 mg/kg/día omitiendo la azatioprina. El tratamiento suele suspenderse después de nueve a 12 meses, o antes si desaparecen los anticuerpos circulantes anti-MBG, medidos por radioinmunoanálisis. Los fármacos citotóxicos también deben suspenderse si se desarrolla neutropenia (cuenta leucocitaria menor de 3,500/mm3) o una infección grave.
Este esquema por lo general disminuye la concentración plasmática de anticuerpos anti-MBG, pero no necesariamente se acompaña de mejoría paralela en la función renal. En particular, es poco probable que los pacientes oligúricos con concentración plasmática de creatinina mayor de 7 mg/dl recuperen la función renal.44,45 Por comparación, ocurre mejoría funcional en dos terceras partes de los pacientes no oligúricos con concentración plasmática de creatinina menor de 7 mg/dl antes de la institución del tratamiento; asimismo, el deterioro ulterior de la función renal en este grupo es poco frecuente, afectando sólo del 10 al 15 porciento de los enfermos. La plasmaféresis también resulta eficaz para el control de la hemoptisis en más del 90 porciento de los casos.45
Los investigadores que han estudiado este método terapéutico concluyen que su administración antes de que ocurra daño glomerular irreversible puede preservar y mejorar la función renal. Sin embargo, las infecciones graves en pacientes sometidos a plasmaféresis y que reciben medicamentos inmunosupresores para la glomerulonefritis rápidamente progresiva son frecuentes, por lo que es necesaria la vigilancia estrecha.46-49 Es poco probable que los pacientes que presentan concentración plasmática de creatinina mayor de 7 mg/dl respondan al tratamiento, a menos de que la lesión sea aguda. En consecuencia, es probable que el riesgo relacionado con el tratamiento exceda los beneficios posibles en esta situación, a no ser que exista hemorragia pulmonar. El riesgo de hemorragia pulmonar no debe subestimarse, ya que la muerte es común en los pacientes tratados en forma inadecuada.
Por lo general la formación de anticuerpos anti-MBG es autolimitada; estos anticuerpos desaparecen en forma espontánea en término de seis a 12 meses, y las recurrencias tardías son raras.34 Los individuos que evolucionan a insuficiencia renal en etapa terminal (como consecuencia del daño irreversible durante la fase aguda de la enfermedad) no deben recibir trasplantes hasta que la concentración de anticuerpos circulantes sea mínima o nula, para evitar la recurrencia en el trasplante.47,50
GLOMERULONEFRITIS FIBRILAR
La glomerulonefritis fibrilar, una enfermedad descrita hace poco tiempo, es más común en algunos hospitales que muchas de las glomerulonefritis mejor conocidas.51,52 La microscopía de luz muestra expansión mesangial y proliferación celular variable, y la microscopía electrónica permite observar el depósito patognomónico de fibrillas. Estas fibras son más grandes que las amiloides y no se tiñen con rojo Congo ni con tioflavina T [ver figura 5]. El origen de estas fibrillas es dudoso, la microscopía inmunofluorescente suele ser positiva para IgG, lo que indica que las inmunoglobulinas pueden estar implicadas. Sin embargo, no existen datos que apoyen la existencia de alguna paraproteína urinaria o plasmática, como sucede en forma característica en la amiloidosis primaria.
|
|
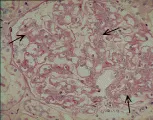
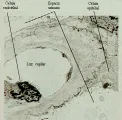
Glomerulonefritis focal
La mayoría de los enfermos con glomerulonefritis focal presentan hematuria y proteinuria asintomáticas o episodios de hematuria macroscópica. La insuficiencia renal, la proteinuria intensa y la hipertensión arterial, tan frecuentes en la glomerulonefritis difusa, son poco comunes en esta entidad. Las manifestaciones clínicas leves correlacionan con los datos histológicos, que por lo general consisten en alteraciones proliferativas focales (con lesión de unos cuantos glomérulos) y segmentarias (con afección de una parte del penacho glomerular). Aunque estas enfermedades parecen focales, la inmunofluorescencia suele mostrar depósito difuso de inmunoglobulinas en el glomérulo, por lo que es posible que en realidad representen variedades más leves de glomerulonefritis difusa (ver antes). Dos causas comunes de glomerulonefritis focal son la nefropatía por IgA y la nefritis hereditaria.
NEFROPATIA POR IgA
En algunas partes del mundo (v.gr., Finlandia), la nefropatía por IgA (enfermedad de Berger) es la causa más común de enfermedad glomerular, sobre todo en pacientes con hematuria asintomática o episodios recurrentes de hematuria franca. Es frecuente sobre todo en varones adultos, jóvenes, de raza blanca, y tiene una tendencia familiar.54
El examen histológico se caracteriza por mostrar proliferación focal o difusa de células mensagiales [ver figura 6a], y en la microscopía electrónica se observan depósitos mesangiales densos. Sin embargo, la característica patognomónica es el depósito mesangial difuso de IgA, que se detecta por microscopía de inmunofluorescencia [ver figura 6b], a menudo acompañado por depósitos menos notables de otras inmunoglobulinas y complemento. Estos datos son indistinguibles de los observados en la púrpura de Schönlein-Henoch; no obstante, esta última también incluye afección articular, de piel y del aparato digestivo.55
|
|
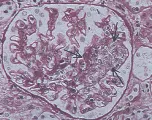

La mayoría de los individuos con nefropatía por IgA presentan hematuria microscópica asintomática con o sin proteinuria, o episodios de hematuria franca que aparecen después de un cuadro de faringitis aguda o de un síndrome febril menor, parecido al resfriado.55,60 La mayoría de los pacientes son menores de 35 años y ambos sexos se afectan con la misma frecuencia. La función renal suele ser normal al momento en el que aparecen los síntomas, aunque puede ocurrir insuficiencia renal aguda cuando existe hematuria importante, con oclusión de la luz tubular por eritrocitos.61 El diagnóstico puede sospecharse por la historia clínica, pero sólo puede confirmarse por biopsia renal. Aunque la IgA puede encontrarse en biopsias cutáneas de áreas expuestas al sol, este hallazgo no es lo suficiente sensible o específico para el diagnóstico. Alrededor de la mitad de los pacientes tienen niveles séricos de IgA ligeramente elevados.
La nefropatía por IgA suele tener una evolución benigna a corto plazo. La función renal se conserva, aunque puede haber episodios recurrentes de hematuria franca durante muchos años.55,60 En consecuencia, los pacientes que sólo presentan hematuria leve, cilindros hemáticos (que indican hematuria de origen glomerular), proteinuria mínima y concentración normal de creatinina plasmática, no necesitarán biopsia renal si no existen datos clínicos aparentes de progresión de la enfermedad. Cerca del 20 al 40 porciento de los pacientes con estos síntomas presentan nefropatía por IgA; el resto tiene otras enfermedades, por lo general benignas, como la enfermedad de membrana basal delgada (ver adelante).62,63
Sin embargo, aproximadamente el 50 porciento sufrirán insuficiencia renal terminal en un periodo de 20 años. Los signos pronósticos que suelen indicar una probable evolución al deterioro son: excreción de proteínas mayor de 1 g/día, hipertensión, medias lunas en la biopsia o cambios extensos tubulointersticiales y vasculares.64-66
No se ha encontrado un tratamiento eficaz para la nefropatía por IgA, aunque se han tratado esquemas que incluyen esteroides, agentes alquilantes, ciclosporina, plasmaféresis, anticoagulantes o antiplaquetarios, dietas libres en gluten, amigdalectomía o difenilhidantoína. El depósito de IgA secundario a enfermedad celíaca puede responder a la restricción de gluten, y el depósito de IgA que acompaña a la cirrosis alcohólica puede mejorar con la abstinencia de alcohol. Un subgrupo de pacientes que presentan síndrome nefrótico, hematuria mínima y lesiones glomerulares leves (en contraste con la mayoría de los casos que presentan proteinuria intensa) a menudo se comportan como si presentaran enfermedad por cambios mínimos; estos pacientes manifiestan remisión completa de la proteinuria después de la administración de prednisona por poco tiempo.67,68
Como con muchas otras enfermedades glomerulares, la nefropatía por IgA recurre con frecuencia en el riñón trasplantado a pesar del tratamiento contra el rechazo con prednisona, azatioprina y ciclosporina. Sin embargo, esta enfermedad a menudo es asintomática, por lo que la pérdida del injerto a consecuencia de la enfermedad recurrente es poco común.69
NEFRITIS HEREDITARIA
La nefritis hereditaria, o síndrome de Alport, es una enfermedad relativamente frecuente que a menudo se acompaña de pérdida de la audición y alteraciones del cristalino.70 Se han descrito tres formas de herencia: dominante ligada al cromosoma X, autosómica dominante y con menor frecuencia autosómica recesiva. El primer mecanismo parece estar presente en casi todas las familias con afección renal y sordera.71 Esta forma de herencia puede explicar el porqué la enfermedad no suele trasmitirse de los padres afectados a sus hijos, en vista de que el padre sólo podría transferir el cromosoma Y normal. El hecho de que la herencia esté ligada al cromosoma X también puede explicar la evolución por lo general más benigna en las mujeres, ya que uno de los dos cromosomas X es inactivado al azar en cada célula (hipótesis de Lyon),70 y como resultado, la mitad de las células será normal, reduciendo así al mínimo la gravedad de la enfermedad. Por otra parte, es más probable que la herencia autosómica dominante o recesiva ocurra en familias con enfermedad renal progresiva pero sin signos auditivos u oculares.71 En este caso las mujeres pueden afectarse con la misma gravedad que los hombres.
La manifestación histológica más temprana de la nefritis hereditaria es el adelgazamiento difuso de la membrana basal glomerular.72 Con el tiempo, existe cada vez mayor división longitudinal de la MGB, produciendo el aspecto laminado característico que se observa por microscopía electrónica [ver figura 7]. La microscopía de luz revela que esta división se relaciona con mayor daño glomerular, desde la expansión mesangial inicial hasta la esclerosis glomerular e infiltrado intersticial que contiene células espumosas de origen dudoso.

|
| Figura 7 |
| Nefritis hereditaria |
La patogenia de la nefritis hereditaria no se ha esclarecido por completo. Varios estudios han demostrado que la mayoría de los pacientes con esta enfermedad carecen de un antígeno que al parecer forma parte del dominio no colágeno de la colágena tipo IV.73,74 Al parecer este antígeno está relacionado con el antígeno de Goodpasture (el antígeno que reacciona con el anticuerpo anti-MBG); los glomérulos de la mayoría de los enfermos con nefritis hereditaria muestran disminución o falta de unión de los anticuerpos anti-MBG.73 Este dato es potencialmente importante para el diagnóstico.
Las manifestaciones clínicas principales de la nefritis hereditaria incluyen lo siguiente: enfermedad renal, que puede ocurrir en forma aislada; alteraciones oftálmicas, incluyendo cataratas y lenticono anterior, y sordera neurosensorial para tonos altos.70 Las alteraciones renales iniciales son parecidas a las que ocurren en otros tipos de glomerulonefritis focal: proteinuria y hematuria asintomáticas o episodios de hematuria franca. La hematuria puede aparecer antes de los cinco años de edad en los hombres, pero no ser detectada hasta la adolescencia. Algunos niños afectados se han identificado por los antecedentes familiares.
La enfermedad renal en hombres suele ser evolutiva: se relaciona con proteinuria progresiva, insuficiencia renal e hipertensión arterial. Finalmente, aparece insuficiencia renal en etapa terminal por lo general entre los 16 y 35 años de edad, aunque en ocasiones se retrasa hasta los 45 o 60 años de edad. Las mujeres sólo presentan hematuria microscópica persistente con función renal normal; sin embargo, en algunas familias puede desarrollarse insuficiencia renal antes de los 25 años de edad en las mujeres.63
El diagnóstico de nefritis hereditaria suele confirmarse por los antecedentes familiares, incluyendo el examen de orina de los miembros asintomáticos de la familia75 y, en caso necesario, por biopsia renal. Unos cuantos pacientes no presentan antecedentes familiares de la enfermedad; es probable que estos casos indiquen la existencia de una mutación fresca del gen responsable del defecto de la membrana basal. Debe resaltarse que el adelgazamiento aislado de la MBG no es diagnóstico del síndrome. Otra entidad que a menudo ocurre en familias, la enfermedad de membrana basal delgada, también puede producir hematuria persistente.62,63 Sin embargo, este trastorno por lo general tiene un pronóstico benigno, no se relaciona con división de la MBG, ni con insuficiencia renal progresiva.
Ningún procedimiento terapéutico ha demostrado ser eficaz para la nefritis hereditaria. Es posible que la MBG sea débil en esta enfermedad y que la hendidura de la MBG refleje ciclos de daño y reparación. Si esta teoría es correcta, la disminución de la presión intraglomerular con un inhibidor de la enzima convertidora de angiotensina (ECA) puede ofrecer cuando menos protección parcial a largo plazo.
Puede utilizarse el trasplante renal en los pacientes con insuficiencia renal. La enfermedad recurrente no representa un problema en vista de que la MBG del donador es normal. No obstante, algunos pacientes tienen riesgo de sufrir una enfermedad por anticuerpos anti-MBG de novo debido a que el riñón donado puede contener antígenos no observados previamente.76 Esta complicación a menudo es subclínica pero puede ocasionar la pérdida del injerto en casos aislados.
Síndrome nefrótico
Los pacientes con síndrome nefrótico presentan proteinuria intensa (> 3 g/día), hipoalbuminemia, edema, hiperlipidemia, y con menor frecuencia, fenómenos tromboembólicos. El síndrome nefrótico puede producirse tanto por una enfermedad renal primaria como por lesión glomerular causada por una enfermedad sistémica [ver tabla 2].22 Esta sección revisa los tres tipos principales de síndrome nefrótico primario, que se caracterizan por relacionarse con sedimento urinario benigno y se definen por sus manifestaciones histológicas: (1) enfermedad de cambios mínimos, (2) glomerulosclerosis focal, y (3) nefropatía membranosa. El lupus eritematoso generalizado, la diabetes mellitus y la amiloidosis también son causas relativamente frecuentes de síndrome nefrótico.
|
||||||
|
Los trastornos nefríticos difusos, como la glomerulonefritis posinfecciosa, la nefritis lúpica, la glomerulonefritis membranoproliferativa, y con menor frecuencia, las vasculitis, también pueden relacionarse con proteinuria en límites nefróticos. Sin embargo, estas enfermedades se relacionan en forma distintiva con sedimento nefrítico activo, que contiene eritrocitos, leucocitos y cilindros granulares y celulares.
En general la causa del síndrome nefrótico se establece por biopsia renal. No obstante, la evaluación antes de la biopsia debe incluir determinación de nitrógeno de urea en sangre, excreción de proteínas en orina de 24 horas, y concentraciones plasmáticas de creatinina, albúmina, glucosa y colesterol. La orina y el plasma de los pacientes mayores de 25 años también debe examinarse por inmunoelectroforesis, con el fin de detectar paraproteínas, presentes en la mayoría de los casos de amiloidosis primaria. Si uno de estos estudios resulta positivo, la presencia de amiloidosis de fondo a menudo puede confirmarse por biopsia rectal o de pliegues de tejido adiposo, procedimiento menos cruento que la biopsia renal. Aunque la búsqueda de LES está indicada en pacientes con síntomas sistémicos compatibles, por lo general no elimina la necesidad de realizar biopsia renal, porque los diferentes tipos de nefritis lúpica se tratan de manera distinta.
ENFERMEDAD DE CAMBIOS MINIMOS
La enfermedad de cambios mínimos, también conocida como nefrosis lipoide, enfermedad nula, o síndrome nefrótico por lesión mínima, es la causa más frecuente de síndrome nefrótico en niños, aunque también es responsable del 20 al 25 porciento de los casos en adultos.77 El dato histológico que confirma el diagnóstico de esta enfermedad es la observación de fusión difusa de los podocitos de las células epiteliales por microscopía electrónica [ver figura 8]. La microscopía de luz muestra glomérulos normales o con hipercelularidad mesangial leve como única alteración. Los estudios de inmunoflorescencia suelen ser negativos para inmunoglobulinas y complemento, pero en algunos casos selectos se observa depósito focal de IgM y C3. Se considera que estos depósitos representan atrapamiento inespecífico en la pared capilar glomerular con permeabilidad anormal.78
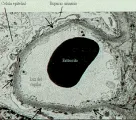
|
| Figura 8 |
| Enfermedad de cambios mínimos |
La enfermedad de cambios mínimos suele ser idiopática; en algunos casos pueden participar en la etiología la alergia a los alimentos, los inhalantes u otros agentes extraños, incluyendo las picaduras de abejas.22,79 Esta enfermedad se ha relacionado con neoplasias malignas (sobre todo con enfermedad de Hodgkin),80,81 tratamiento con litio,82,83 con el uso de medicamentos antinflamatorios no esteroides (AINE) (sobre todo fenoprofen y otros derivados del ácido propiónico),84,85 y puede ocurrir al inicio de la diabetes mellitus insulinodependiente.86
Etiología
La etiología de la enfermedad de cambios mínimos no se ha esclarecido por completo. Los estudios experimentales indican que el daño celular epitelial participa de manera importante en este trastorno, ocasionando disminución secundaria en la producción de proteoglicanos cargados en forma negativa (v.gr., sulfato de heparán) y las sialoproteínas; estas moléculas están presentes en la MBG y cubren los podocitos.22,87 La producción disminuida podría ser responsable de los dos principales datos de la enfermedad de cambios mínimos: proteinuria, por pérdida de la barrera de carga aniónica en la MBG y fusión de podocitos (la separación normal de los podocitos adyacentes se mantiene en parte por la repulsión electrostática modulada por las cargas aniónicas).22,87
Varias observaciones indican que las células T desempeñan una función crucial, tal vez a través de los efectos de una linfocina, en la patogenia del daño a la célula epitelial.88 Entre éstas se encuentra la respuesta generalmente positiva al tratamiento con prednisona y las relaciones de la enfermedad de cambios mínimos con la enfermedad de Hodgkin (en la que la función de las células T es anormal)80 y con el uso de antinflamatorios no esteroides (AINE). El síndrome nefrótico relacionado con el uso de AINE a menudo se acompaña de nefritis intersticial, con infiltrado intersticial compuesto principalmente por células T.85 Las células T de estos pacientes parecen producir una linfocina que aumenta la permeabilidad glomerular.89
Los pacientes con enfermedad de cambios mínimos presentan de manera característica un inicio súbito con edema, proteinuria intensa, sedimento urinario benigno y concentración plasmática de creatinina normal.77 Sin embargo, se ha descrito insuficiencia renal aguda en algunos casos, sobre todo en mayores de 60 años de edad.77 Se desconoce el mecanismo productor de insuficiencia renal, pero es probable que contribuyan el edema intersticial, que ocasiona colapso tubular, o la pérdida del área de superficie de filtración causada por la fusión de los podocitos.90,91
Diagnóstico y tratamiento
El diagnóstico de enfermedad de cambios mínimos en adultos se confirma por biopsia renal. No obstante, en los niños suele administrarse prednisona en forma empírica por la elevada prevalencia del trastorno en este grupo de edad. Sólo los niños resistentes a los esteroides deben someterse a cirugía renal. La enfermedad de cambios mínimos es responsable del 50 porciento de los casos de resistencia a los esteroides en niños menores de seis años de edad, pero sólo del cuatro porciento en niños mayores.92
La administración de esteroides es el tratamiento inicial de elección, y produce remisión de la proteinuria en más del 90 porciento de los pacientes. La enfermedad remite en término de dos a ocho semanas en niños, pero puede tardar de 12 a 16 meses en adultos.77,92 Un esquema que se ha utilizado con frecuencia en adultos incluye la administración de prednisona en dosis diaria de 1 mg/kg durante cuatro semanas (o hasta una semana después de que se ha logrado la remisión). Para disminuir al mínimo los efectos adversos, los pacientes siguen el esquema de dosis única en días alternos con 1 mg/kg. Si la proteinuria se resuelve o se reduce a niveles muy bajos, el tratamiento en días alternos debe mantenerse hasta un mes y reducirse posteriormente en forma paulatina por un periodo de varios meses, en un intento por reducir al mínimo la incidencia de recaídas. El diagnóstico de resistencia a esteroides no debe considerarse en el adulto a menos de que hayan transcurrido 12 semanas de tratamiento sin respuesta adecuada.77
Si la remisión va seguida por recurrencia, como sucede en más de la mitad de los casos, suele administrarse otro ciclo de esteroides. Sin embargo, cuando las recaídas ocurren con frecuencia, puede requerirse tratamiento continuo en días alternos y en dosis bajas durante seis a 12 meses para mantener la remisión.77 Las recaídas suelen ocurrir en término de un año después de la suspensión del tratamiento con esteroides, aunque algunos enfermos sufren recaídas que responden a los esteroides años después de la remisión.93,94
Hay tres opciones adicionales en los pacientes que requieren tratamiento continuo con prednisona para evitar las recaídas (v.gr., dependientes de esteroides), o en los que sufren recaídas frecuentes: fármacos alquilantes (v.gr., ciclofosfamida o clorambucil), ciclosporina, o bien, no administrar tratamiento. Se ha demostrado que los medicamentos alquilantes tienen un efecto notable, reduciendo o evitando el número de recaídas en los pacientes con recurrencias frecuentes del síndrome nefrótico.77,95-98 Estos fármacos también pueden resultar benéficos en pacientes dependientes de esteroides, en los que son resistentes a los esteroides desde un principio o en los que desarrollan resistencia secundaria a los esteroides.77,97,98 Sin embargo, la frecuencia de resultados satisfactorios en estos enfermos es menor que en los pacientes sensibles a los esteroides,96 a menos que la duración del tratamiento se prolongue de ocho a 12 meses.97 Si ocurren recaídas después de suspender el tratamiento citotóxico, el enfermo a menudo responde a los esteroides.99
Los estudios realizados en niños con enfermedad de cambios mínimos indican que la ciclofosfamida debe administrarse en dosis máxima de 2.0 a 2.5 mg/kg/día durante ocho semanas en los enfermos que sufren recaídas frecuentes y tal vez durante 12 meses en los que son dependientes de los esteroides o resistentes a estos fármacos.96,97 Los efectos colaterales potenciales incluyen supresión de la médula ósea, fibrosis gonadal (probablemente relacionada con esterilidad),100 cistitis hemorrágica y supresión a largo plazo de la función linfocitaria.101 También existen datos de que la ciclofosfamida puede ocasionar neoplasias de aparición tardía, que se caracterizan por aparecer en los pacientes que reciben tratamiento prolongado.102 El clorambucil también se ha utilizado con éxito en la enfermedad de cambios mínimos,103 pero la toxicidad con este medicamento puede ser incluso más limitante que con la ciclofosfamida.104 Aunque la azatioprina suele ser mejor tolerada, no parece brindar beneficio a los pacientes con enfermedad de cambios mínimos a menos que se administre de seis a 12 meses cuando menos.105 En vista que la administración prolongada de esteroides o fármacos citotóxicos puede relacionarse con toxicidad grave, estos medicamentos deben utilizarse con precaución, sobre todo en adultos con enfermedad de cambios mínimos, porque el tratamiento inmunosupresor no ha mostrado beneficios a largo plazo.106,107 Aunque los pacientes tratados remiten con más rapidez, casi todos los adultos no tratados remiten en forma espontánea a los cuatro años,106 y la evolución a la insuficiencia renal es muy rara. Por lo tanto, debe evitarse la administración repetida de fármacos inmunosupresores en los enfermos con recurrencias frecuentes; sólo debe administrarse tratamiento sintomático, como el uso de diuréticos para el edema.
Un ejemplo del riesgo potencial del tratamiento se demuestra en el estudio de seguimiento a largo plazo de 389 niños con enfermedad de cambios mínimos.108 Diez enfermos fallecieron: uno por insuficiencia renal, con glomerulosclerosis focal en la biopsia repetida, y seis como consecuencia de infecciones relacionadas en parte con el tratamiento inmunosupresor.
Se ha sugerido el tratamiento con ciclosporina para dos grupos de pacientes con enfermedad de cambios mínimos: (1) pacientes con toxicidad por esteroides y que sufren recaídas frecuentes o pacientes dependientes de esteroides en los que dos a tres meses de tratamiento con ciclofosfamida o clorambucil no han logrado inducir una remisión prolongada y (2) pacientes resistentes a los esteroides y agentes alquilantes. En el primer grupo la ciclosporina causa remisión en el 70 a 80 porciento de los casos,109,110 pero la mayor parte de los enfermos recaen al disminuir la dosis111 o suspender el medicamento.112,113 Se ha usado una dosis de mantenimiento de 4 a 5 mg/kg/día, vigilando con cuidado los niveles sanguíneos, la presión arterial y la creatinina sérica. En el segundo grupo, que puede incluir algunos pacientes con glomeruloesclerosis focal no diagnosticada, la respuesta es menos frecuente. Después de un periodo de tratamiento de seis meses, más de la mitad de los pacientes resistentes a esteroides logran por lo menos una remisión parcial, pero las recaídas son frecuentes.114
Una posible excepción a la evolución de la enfermedad de cambios mínimos, generalmente benigna a largo plazo, ocurre en pacientes que finalmente se vuelven resistentes a los esteroides y en quienes la segunda biopsia renal demuestra evolución de la enfermedad de cambios mínimos a glomerulosclerosis focal. Aunque el tratamiento citotóxico puede ser eficaz en esta situación, algunos de estos pacientes evolucionan a insuficiencia renal en etapa terminal.115,116
Relación con otras enfermedades
La patogenia y las manifestaciones clínicas de la enfermedad de cambios mínimos parecen ser similares a la de otros trastornos glomerulares, incluyendo glomerulosclerosis focal (ver adelante), nefropatía por IgM y glomerulonefritis proliferativa mesangial idiopática. Algunos enfermos con fusión difusa de los podocitos presentan depósitos de IgM en el mesangio, lo que indica que esta lesión podría representar una enfermedad distinta.78 Sin embargo, el depósito de IgM ocurre con igual frecuencia en pacientes con enfermedad de cambios mínimos y en enfermos con glomerulosclerosis focal, con correlación pronóstica de acuerdo con el diagnóstico confirmado por microscopía de luz. Los individuos con enfermedad de cambios mínimos aparente y depósito de IgM responden al tratamiento con prednisona, mientras que la respuesta es menos predecible en los pacientes con glomerulosclerosis focal.78
Aproximadamente del tres al cinco porciento de los pacientes con síndrome nefrótico presentan proliferación mesangial difusa y fusión de podocitos, pero los estudios con inmunofluorescencia son negativos (aunque otras enfermedades como la nefritis lúpica y la nefropatía por IgA pueden producir un cuadro similar en la microscopía de luz, estos padecimientos se relacionan con un depósito importante de IgG y de IgA, respectivamente). Es probable que estos pacientes sólo presenten daño inicial más severo que los sujetos con enfermedad de cambios mínimos, con un aumento en la frecuencia de recaídas y menor incidencia de recuperación.117-119 Más de la mitad de los pacientes con esta enfermedad responden en un principio a la administración de prednisona; la mayoría de los pacientes que no responden a la prednisona muestran remisión espontánea en término de un año.117-118 No obstante, la insuficiencia renal evolutiva se desarrolla finalmente en el 10 a 30 porciento de los casos con glomerulosclerosis focal diagnosticada por biopsia.78,119 Algunos pacientes con proliferación mesangial difusa progresiva responden a la ciclofosfamida; en estos casos es razonable realizar la prueba terapéutica cuando existe síndrome nefrótico persistente y elevación de la concentración plasmática de creatinina.118,119
GLOMERULOSCLEROSIS FOCAL
La glomerulosclerosis focal es la tercera causa más común de síndrome nefrótico en adultos, después de la nefropatía membranosa y la enfermedad de cambios mínimos.22,120 Los datos histológicos, incluyendo la presencia de fusión difusa de los podocitos, son parecidos a los de la enfermedad de cambios mínimos. Sin embargo, la microscopía de luz muestra que algunos glomérulos presentan lesiones escleróticas segmentarias [ver figura 9]. Este cambio es más notable en la región yuxtaglomerular, más que en los glomérulos corticales externos, y es probable que esta distribución refleje diferencias en la hemodinámica regional.
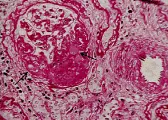
|
| Figura 9 |
| Glomeruloesclerosis focal idiopática |
La distinción patológica entre enfermedad de cambios mínimos y glomerulosclerosis focal no siempre puede realizarse con certeza. En las etapas tempranas de la glomerulosclerosis focal las lesiones escleróticas características sólo están presentes en una pequeña fracción de los glomérulos. Es probable que en la biopsia por punción no se obtengan glomérulos afectados, lo cual conduce al diagnóstico histológico de enfermedad de cambios mínimos.
Es importante destacar que la esclerosis segmentaria aislada es inespecífica; lesiones similares pueden ocurrir con el daño modulado en forma hemodinámica por la pérdida previa de nefronas y después de la fase de cicatrización de cualquier forma de glomerulosclerosis focal, como la glomerulonefritis posinfecciosa leve. Sin embargo, en estas situaciones la fusión de los podocitos es focal, limitada a las zonas escleróticas; además; también se observan grandes glomérulos hipertrofiados con daño secundario.
En muchos casos la glomerulosclerosis focal parece ser parte del espectro de la enfermedad de cambios mínimos, tal vez representando una lesión más grave a las células epiteliales glomerulares.121 Por ejemplo, algunos pacientes con enfermedad de cambios mínimos aparentemente evolucionan a la glomerulosclerosis focal al repetir la biopsia.115 Además, muchos pacientes con glomerulosclerosis focal siguen una evolución sensible a los esteroides, con recurrencia frecuente, similar a lo observado en la enfermedad de cambios mínimos.98,122
Varias observaciones sugieren que un factor de permeabilidad circulante puede ser responsable de la proteinuria nefrótica en la glomeruloesclerosis focal. En algunos pacientes que progresan hasta la insuficiencia renal, la enfermedad recurre en el paciente trasplantado, con retorno de la proteinuria en minutos a horas después del trasplante.123-125 Un extracto de plasma de estos pacientes puede aumentar la permeabilidad a las proteínas en glomérulos aislados de rata.126 Cuando se sometió a estos pacientes a absorción del plasma con una columna de proteína A, la proteinuria se redujo durante dos semanas, y un eluído (no una inmunoglobulina) de la columna indujo proteinuria en ratas.127 El intercambio plasmático produjo una respuesta temporal en un grupo semejante de pacientes.128 Se ha sugerido que la permeabilidad alterada de proteínas puede ser la causa de la esclerosis subsecuente y la progresión de la enfermedad.129
Aunque la glomerulosclerosis focal suele ser idiopática, también ocurre en adictos a la heroína intravenosa y hasta en el 20 porciento de los pacientes con síndrome de inmunodeficiencia adquirida (SIDA), sobre todo en los que no son homosexuales.130-133 Es probable que las toxinas virales o exógenas desempeñen un papel importante en estas situaciones.
Los enfermos con glomerulosclerosis focal tienden a presentar datos clínicos y químicos del síndrome nefrótico, aunque en algunos casos el único signo es la proteinuria no nefrótica, asintomática.120,134,135 Hasta en la mitad de los casos se presenta al inicio hipertensión arterial, hematuria leve y creatinina plasmática elevada. En los pacientes con enfermedad de cambios mínimos, el desarrollo tardío de resistencia a los esteroides es el dato principal que indica evolución a glomerulosclerosis focal.115
Los pacientes con esta enfermedad también presentan mayor incidencia de uno o más defectos tubulares, incluyendo glucosuria renal, aminoaciduria, acidosis tubular renal y pérdida de fosfato.136,137 Estas alteraciones pueden representar daño tubular secundario en nefronas con lesiones glomerulares escleróticas. En forma alternativa, el deterioro de la función tubular puede reflejar los efectos tóxicos de la reabsorción de proteínas por los túbulos proximales.
Se ha considerado que el pronóstico de la glomerulosclerosis focal es invariablemente malo, en vista de que el padecimiento es relativamente insensible al tratamiento inmunosupresor y porque evoluciona a la insuficiencia renal en etapa terminal en término de uno a 20 años. Aunque ocurre evolución rápida cuando la enfermedad se relaciona con el abuso de heroína o con el SIDA, cerca de la tercera parte de los pacientes con glomerulosclerosis focal idiopática se comportan en forma similar a los enfermos con enfermedad de cambios mínimos, presentan remisiones inducidas por esteroides y en ocasiones siguen una evolución con recaídas frecuentes.18,122,134,138 Los enfermos con recaídas frecuentes o dependientes de esteroides pueden beneficiarse con la administración de ciclofosfamida durante dos a tres meses,98,122,139 con un esquema parecido al que se prescribe en la enfermedad por cambios mínimos.98,122 También puede inducirse la remisión de la proteinuria en los pacientes resistentes a los esteroides; sin embargo, en esta situación la frecuencia de éxito es menor del 25 porciento,98,139 a menos que el tratamiento sea vigoroso y prolongado.140 Un esquema prolongado que incluyó esteroides en pulsos y por vía oral y agentes alquilantes, indujo una remisión del 60 porciento en un estudio no controlado de niños con la enfermedad.141 Sin embargo, siempre debe tenerse en cuenta la toxicidad potencial del tratamiento citotóxico.
No es soprendente que los pacientes que responden al tratamiento presenten mucho mejor pronóstico que los que no responden.134,138,139 Los pacientes con proteinuria no nefrótica también conservan mejor la función renal a largo plazo.135 El embarazo puede exacerbar la evolución de la enfermedad.142
La ciclosporina induce remisión completa en el 15 a 20 porciento de los pacientes resistentes a esteroides con glomeruloesclerosis focal,109,110,143,144 aunque el porcentaje puede ser más alto después del tratamiento durante un año.114 La insuficiencia renal por toxicidad de la ciclosporina constituye un riesgo, lo mismo que las posibles recaídas cuando se suspende el tratamiento.
En los pacientes que no responden a estos tratamientos o en los que la relación riesgo-beneficio no es favorable, los inhibidores de la ECA y en especial los AINE [ver adelante, Tratamiento general del síndrome nefrótico] han sido útiles para reducir la proteinuria.123
Los pacientes que evolucionan a la etapa terminal de la insuficiencia renal suelen ser jóvenes y, por lo tanto, candidatos excelentes para el trasplante renal. Sin embargo, se desarrollará enfermedad renal recurrente en el 20 a 30 porciento de los casos; por lo general la proteinuria intensa reaparece en términos de horas o semanas.124,125 La glomerulosclerosis focal recurrente produce pérdida del injerto en una tercera parte a una mitad de estos casos, en particular en niños pequeños,145 en pacientes en los que la enfermedad ha ocasionado insuficiencia renal en los primeros tres años después de su aparición, y en los casos en que existe proliferación mesangial.124,125,126
NEFROPATIA MEMBRANOSA
La nefropatía membranosa es la causa más frecuente de síndrome nefrótico idiopático en adultos. El nombre de esta enfermedad se deriva del cambio característico observado por microscopía de luz: engrosamiento difuso de la membrana basal glomerular y la pared capilar, con escaso o nulo aumento en la celularidad [ver figura 10a].147 En los casos leves o en etapas tempranas, el engrosamiento de la pared capilar es relativamente normal, por lo que se necesita microscopía electrónica o de inmunofluorescencia para establecer en forma definitiva el diagnóstico. Los complejos inmunes pueden observarse por medio de microscopía electrónica, que demuestra que el engrosamiento de la pared capilar glomerular está causada por depósito subepiteliales, entre la MBG y las células epiteliales [ver figura 10b]. El estudio de inmunofluorescencia revela depósitos difusos de IgG y C3 en la pared capilar con un patrón granular o lumpy-bumpy [ver figura 10c].
|
|
|
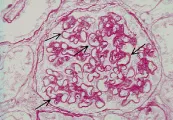

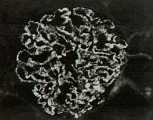
Los modelos animales indican que el antígeno estimulador debe ser catiónico, lo que permitiría que cruce la MBG cargada negativamente, o estar presente en condiciones normales en el espacio subepitelial, como es el caso de los antígenos epiteliales tubulares renales.149 La activación subsecuente del sistema del complemento, en particular de los componentes terminales del complejo de ataque a la membrana (C5b-C9), parece ser responsable del daño asociado a la pared capilar,150 que produce el aumento en el tamaño de los poros necesario para que ocurra la proteinuria.151
La mayoría de los casos de nefropatía membranosa son idiopáticos. Sin embargo, diversos padecimientos sistémicos se han relacionado con la enfermedad.22 De éstos, los más comunes son los tumores sólidos (con depósito de antígenos tumorales en los glomérulos),81 el lupus eritematoso sistémico, los medicamentos como el oro y la penicilamina,152,153 y las infecciones por el virus de la hepatitis B.154 Es posible que la hepatitis B sea la causa de la nefropatía membranosa en niños y adultos jóvenes en áreas endémicas155 y puede acompañarse de antigenemia por el hepatitis Be.156 La remisión espontánea es frecuente, en especial cuando la antigenemia desaparece. No se ha demostrado que el tratamiento con interferón sea eficaz.157 En raras ocasiones se encuentra asociación entre la nefropatía membranosa y el virus de la hepatitis C,158 la artritis reumatoide no tratada, la sarcoidosis o la sífilis.22 Se ha calculado que ocurren neoplasias malignas ocultas, generalmente tumores sólidos, hasta en el10 porciento de los casos nefropatía membranosa en adultos.81 En la mayoría de estos casos el tumor ya se ha diagnosticado o es evidente desde el punto de vista clínico al momento del comienzo del síndrome nefrótico. Es probable que las neoplasias malignas ocultas sean responsables de menos del dos porciento de los casos.159 En consecuencia, no está justificada la evaluación excesiva del paciente con nefropatía membranosa en busca de neoplasias ocultas, a menos que existan datos compatibles como pérdida de peso, anemia de causa desconocida o búsqueda de sangre en heces positiva.
La nefropatía membranosa también puede ocurrir en el uno al tres porciento de los pacientes con artritis reumatoide tratados con oro parenteral (el oro por vía oral es menos nefrotóxico) y en el siete por ciento de los casos tratados con penicilamina,152,153 y se ha notificado en asociación con la exposición a formaldehido.160
Los pacientes con nefropatía membranosa de manera característica presentan edema y proteinuria en rangos nefróticos.147,161 No obstante, cerca del 20 porciento de los casos presentan proteinuria asintomática, no nefrótica, que se detecta en los exámenes de rutina. Puede observarse hematuria leve o hipertensión arterial como manifestación clínica inicial. El diagnóstico correcto sólo se confirma por medio de biopsia renal.
La evolución de la nefropatía membranosa es variable. La enfermedad inducida por medicamentos tiende a remitir después de la suspensión del fármaco causante, aunque pueden trascurrir de uno a dos años antes de que desaparezca la proteinuria.153 Este retraso en la recuperación puede relacionarse en parte con la lentitud con que son eliminados los complejos del espacio subepitelial, separado de la circulación sistémica por la MBG.162
La historia natural de la enfermedad idiopática es variable: en el lapso de cinco a 10 años los pacientes pueden clasificarse, en proporciones iguales, en cuatro grupos: remisión espontánea, proteinuria no nefrótica persistente, síndrome nefrótico persistente y evolución a la insuficiencia renal.161,163-166 Esta variabilidad se atribuye en parte a la presencia de subgrupos con diferente pronóstico. El pronóstico es mejor en mujeres, niños y en pacientes con proteinuria no nefrótica; menos del 10 porciento de estos pacientes evolucionarán hacia el deterioro en cinco a 10 años.161,163,167,168 Por comparación, los hombres con síndrome nefrótico tienen peor pronóstico, sobre todo cuando la concentración de creatinina plasmática se encuentra elevada desde el principio de la enfermedad.161,163,165,169 La enfermedad tubulointersticial indica también mal pronóstico. Casi siempre ocurre enfermedad progresiva en término de dos a tres años. Por lo tanto, el mantenimiento de la concentración normal de creatinina plasmática a los tres años es un signo de buen pronóstico.161
La evolución de la nefropatía membranosa, más que la de otros estados nefróticos, puede complicarse también por el desarrollo de trombosis venosa renal.170,171 La función renal no suele estar afectada; la embolia pulmonar es el indicador principal para sospechar la presencia de trombosis venosa renal. Aunque en el 10 a 15 porciento de los pacientes puede demostrarse la trombosis en la venografía renal, la posibilidad de embolia pulmonar secundaria es incierta, de ahí que exista controversa sobre si se requiere un estudio rutinario con el fin de anticoagular al paciente en ausencia de signos clínicos de trombosis. Un análisis reciente concluyó que es adecuado administrar tratamiento anticoagulante con warfarina para la mayoría de los pacientes con nefropatía membranosa.172
No se ha establecido el tratamiento óptimo de la nefropatía membranosa.173 Varios estudios controlados y no controlados indican que la administración de prednisona durante dos o tres meses (125 mg cada tercer día) retarda la evolución a la insuficiencia renal.174-175Sin embargo, el grado de proteinuria no parece afectarse por el tratamiento con prednisona a corto plazo,175 y no se han encontrado efectos benéficos en otros estudios.164, 176
Se han revisado varios programas de tratamiento que emplean ciclofosfamida o clorambucil, por lo general asociados a esteroides.177,178 Sin embargo, estos programas se acompañan de riesgos, que en algunos pacientes pueden superar los beneficios. Los pacientes que pueden sufrir una recuperación espontánea eventual (v.gr., pacientes jóvenes, mujeres y los que tienen función renal normal, presión arterial normal y proteinuria leve) deben mantenerse en vigilancia estrecha, poniendo atención a los elementos del manejo general (ver adelante). Por otro lado, los que tienen mal pronóstico (v.gr., pacientes de edad avanzada, varones, pacientes con hipertensión severa, hiperlipidemia resistente, proteinuria en el rango de 6 a 10 g/día o mayor, o evidencia de aumento en la creatinina sérica) pueden beneficiarse del tratamiento.179 En los pacientes con enfermedad avanzada y un nivel de creatinina sérica mayor de 3.0 mg/dl, el beneficio potencial puede ser limitado por la cicatrización renal irreversible.
El programa con agentes alquilantes mejor revisado es el de Ponticelli y colaboradores,180 en el que se administra un ciclo de un mes de metilprednisolona en pulsos (1 g/día por tres días) y después prednisona o prednisolona oral (0.5 mg/kg/día o 0.4 mg/kg/día, respectivamente, por lo siguientes 27 días) alternando con un ciclo de un mes de clorambucil por vía oral (0.2 mg/kg/día), cada uno durante tres ciclos por un periodo total de seis meses. Los beneficios del programa son significativos, incluso para los pacientes con función renal en deterioro,181 pero parecen disminuir después de cuatro años, en especial si se compara con el tratamiento que usa solo prednisolona en pulsos.182 La incidencia de efectos colaterales, en especial leucopenia,183 es alta, lo que ha originado que algunos investigadores recomienden una dosis de clorambucil menor (0.15 mg/kg/día).
También se ha estudiado el uso de ciclofosfamida oral combinada con prednisona en pacientes que tienen progresión de la insuficiencia renal,184-186 con aparente beneficio, en especial cuando se administra por tiempo prolongado. La administración de ciclofosfamida intravenosa en pulsos185-187 y los cursos breves de ciclofosfamida por vía oral188 parecen ser menos efectivos.
Al parecer la ciclosporina (4 a 5 mg/kg/día) produce remisión completa o parcial en algunos pacientes con nefropatía membranosa y puede mantenerse con seguridad razonable si se vigilan en suero su concentración y la creatinina.189,190 Sin embargo, la ciclosporina es muy costosa, las recaídas son frecuentes después de suspender el medicamento y su efecto en la evolución no se ha demostrado. Se ha propuesto el tratamiento con g-globulina intravenosa en dosis altas,191 pero no se han realizado estudios controlados al respecto.
MANEJO GENERAL DEL SINDROME NEFROTICO
Algunas medidas generales pueden ser adecuadas para lospacientes con síndrome nefrótico, independientemente de la patología subyacente [ver tabla 3].
|
|
|
El aumento en la proteína dietética en los pacientes nefróticos no aumenta la concentración de albúmina en suero e incluso puede ser dañino.192,193 Al parecer es adecuada una ingesta de 1 g/kg/día de proteína, sobre todo de proteína de alta calidad biológica y la reducción de las grasas animales. Al parecer, limitar la ingesta proteína detiene la progresión a insuficiencia renal en pacientes con enfermedades glomerulares proteinúricas.194 También parece ser que la hiperlipidemia del síndrome nefrótico predispone a ateroesclerosis temprana y severa, por lo que amerita tratamiento.195,196 La dieta sola suele ser insuficiente, aunque puede ser eficaz si se suplemente con tratamiento farmacológico con inhibidores de la 3-hidroxi-3-metilglutaril coenzima A reductasa (HMG-CoA), como el lovastatin y el simvastatin.197,198 Los pacientes nefróticos pueden ser susceptibles a rabdomiolisis inducida por medicamentos, por lo que deben vigilarse los niveles de creatina fosfoquinasa sérica. Los derivados del ácido fíbrico se toleran menos.
La reducción de la presión arterial, en especial con el uso de inhibidores de la ECA en combinación con diuréticos (para estimular la producción de angiotensina) suele causar una reducción en la proteinuria nefrótica199-201 y retrasar la progresión a insuficiencia renal en los pacientes con enfermedades glomerulares proteinúricas.194 Esta reducción puede tardar varias semanas en ocurrir, lo que sugiere un mecanismo diferente de la reducción inmediata de la presión intraglomerular.202 Pueden usarse AINE como indometacina y meclofenamato con un objetivo semejante.123,203 Cuando ocurre una reducción importante en la proteinuria con estas medidas, suele mejorar el control del edema y la hiperlipidemia, y el pronóstico a largo plazo es relativamente bueno.
En ocasiones un paciente con síndrome nefrótico muy severo y que no responde puede presentar proteinuria masiva, trombosis e hiperlipidemia, progresando a la insuficiencia renal terminal. En estos pacientes puede estar indicada una nefrectomía médica por medio de la embolización bilateral de las arterias renales para producir un infarto terapéutico.204
Bibliografía
-
Nissenson AR, Baraff LJ, Fine RN, et al: Poststreptococcal
acute glomerulonephritis: fact and controversy. Ann Intern Med 91:76, 1979
[PMID
380430]
-
Stetson CA, Rammelkamp CH Jr, Krause RM, et al: Epidemic
acute nephritis: studies on etiology, natural history, and prevention.
Medicine (Baltimore) 34:431, 1955
-
Anthony BF, Kaplan EL, Wannamaker LW, et al: Attack rates
of acute nephritis after type 49 streptococcal infection of the skin and
of the respiratory tract. J Clin Invest 48:1697, 1969 [PMID
5822578]
-
Rodriguez-Iturbe B: Epidemic poststreptococcal glomerulonephritis.
Kidney Int 25:129, 1984
-
Sorger K, Gessler M, Hubner FK, et al: Follow-up studies
of three subtypes of acute postinfectious glomerulonephritis ascertained
by renal biopsy. Clin Nephrol 27:111, 1987
-
Potter EV, Lipschultz SA, Abidh S, et al: Twelve to seventeen-year
follow-up of patients with poststreptococcal acute glomerulonephritis in
Trinidad. N Engl J Med 307:725, 1982
-
Baldwin DS: Poststreptococcal glomerulonephritis: a progressive
disease? Am J Med 62:1, 1977
-
Baldwin DS, Gluck MC, Schacht RG, et al: The long-term course
of poststreptococcal glomerulonephritis. Ann Intern Med 80:342, 1974 [PMID
4816174]
-
Barnham M, Thornton TJ, Lange K: Nephritis caused by
Streptococcus
zooepidemicus (Lancefield group C). Lancet 1:945, 1983 [PMID
6132267]
-
Kim Y, Michael AF: Chronic bacteremia and nephritis. Annu
Rev Med 29:319, 1978 [PMID
348039]
-
Smith MC, Cooke JH, Zimmerman DM, et al: Asymptomatic glomerulonephritis
after nonstreptococcal upper respiratory infections. Ann Intern Med 91:697,
1979 [PMID
227300]
-
Levy M, Kleinknecht C, Droz D, et al: Glomerular nephropathies
and hepatitis B virus infection. Adv Nephrol 11:341, 1982
-
Gutman RA, Striker GE, Gilliland BC, et al: The immune complex
glomerulonephritis of bacterial endocarditis. Medicine (Baltimore) 51:1,
1972 [PMID
4550571]
-
Neugarten J, Baldwin DS: Glomerulonephritis in bacterial
endocarditis. Am J Med 77:297, 1984
-
Schoeneman M, Bennett B, Greifer I: Shunt nephritis progressing
to chronic renal failure. Am J Kidney Dis 2:375, 1982 [PMID
6756132]
-
West CD: Childhood membranoproliferative glomerulonephritis:
an approach to management. Kidney Int 29:1077, 1986
-
Donadio JV Jr, Anderson CF, Mitchell JC III, et al: Membranoproliferative
glomerulonephritis: a prospective clinical trial of platelet-inhibitor
therapy. N Engl J Med 310:1421, 1984
-
Cameron JS, Turner DR, Heaton J, et al: Idiopathic mesangiocapillary
glomerulonephritis: comparison of types I and II in children and adults
and long-term prognosis. Am J Med 74:175, 1983 [PMID
6337487]
-
Daha MR, Austen KF, Fearon DT: Heterogeneity, polypeptide
chain composition and antigenic reactivity of C3 nephritic factor. J Immunol
120:1389, 1978 [PMID
76663]
-
Sissons JGP, West RJ, Fallows J, et al: The complement abnormalities
of lipodystrophy. N Engl J Med 294:461, 1976
-
Coleman TH, Forristal J, Kosaka T, et al: Inherited complement
component deficiencies in membranoproliferative glomerulonephritis. Kidney
Int 24:681, 1983 [PMID
6663990]
-
Rose BD: Pathophysiology of Renal Disease, 2nd ed. McGraw-Hill,
New York, 1987, chapter 6
-
Strife CF, McAdams AJ, West CD: Membranoproliferative glomerulonephritis
characterized by focal, segmental proliferative lesions. Clin Nephrol 18:9,
1982 [PMID
6749363]
-
A Report of the International Study of Kidney Disease in
Children: Alternate day steroid therapy in membranoproliferative glomerulonephritis:
a randomized controlled clinical trial (abstr). Kidney Int 21:150, 1982
-
Edelman CM: Long-term low-dose prednisone ameliorates the
course of membranoproliferative glomerulonephritis (MPGN): a report of
the International Study of Kidney Disease in Children (abstr). Pediatr
Res 21:474A, 1987
-
Donadio JV, Offord KP: Reassessment of treatment results
in membranoproliferative glomerulonephritis, with emphasis on life-table
analysis. Am J Kidney Dis 14:445, 1989
-
Cattran DC, Cardella CJ, Roscoe JM, et al: Results of a controlled
drug trial in membranoproliferative glomerulonephritis. Kidney Int 27:436,
1985 [PMID
3886998]
-
Tiller DJ, Clarkson AR, Mathew T, et al: A prospective randomized
trial in the use of cyclophosphamide, dipyridamole and warfarin in membranous
and mesangiocapillary glomerulonephritis. Proceedings of the 8th International
Congress of Nephrology: advances in Basic and Clinical Nephrology. Zurukzoglu
W, Papadimitriou M, Pyrpasopoulos M, et al, Eds. Karger, Basel, 1981, p
345
-
McLean RH, Geiger H, Burke B, et al: Recurrence of membranoproliferative
glomerulonephritis following kidney transplantation: serum complement component
studies. Am J Med 60:60, 1976 [PMID
766620]
-
Droz D, Nabarra B, Noel L-H, et al: Recurrence of dense deposits
in transplanted kidneys: I. Sequential survey of the lesions. Kidney Int
15:386, 1979
-
Curtis JJ, Wyatt RJ, Bhathena D, et al: Renal transplantation
for patients with type I and type II membranoproliferative glomerulonephritis:
serial complement and nephritic factor measurements and the problem of
recurrence of disease. Am J Med 66:216, 1979
-
Salant DJ: Immunopathogenesis of crescentic glomerulonephritis
and lung purpura. Kidney Int 32:408, 1987
-
Couser WG: Rapidly progressive glomerulonephritis: classification,
pathogenetic mechanisms, and therapy. Am J Kidney Dis 11:449, 1988
-
Briggs WA, Johnson JP, Teichman S, et al: Anti-glomerular
basement membrane antibody mediated glomerulonephritis and Goodpasture's
syndrome. Medicine (Baltimore) 58:348, 1979
-
Beirne GJ, Wagnild JP, Zimmerman SW, et al: Idiopathic crescentic
glomerulonephritis. Medicine (Baltimore) 56:349, 1977 [PMID
329051]
-
Lewis EJ, Schwartz MN: Idiopathic crescentic glomerulonephritis.
Semin Nephrol 2:193, 1982
-
Muller GA, Gebhardt M, Komph J, et al: Association between
rapidly progressive glomerulonephritis and the properdin factor BfF and
different HLA-D region products. Kidney Int 25:115, 1984 [PMID
6233449]
-
Thysell H, Oxelius VA, Norlin M: Successful treatment of
hemolytic uremic syndrome and thrombotic thrombocytopenic purpura with
fresh frozen plasma and plasma exchange. Acta Med Scand 212:285, 1982 [PMID
6891171]
-
Kefalides NA: The Goodpasture antigen and basement membranes:
the search must go on (editorial). Lab Invest 56:1, 1987
-
Keogh AM, Ibels LS, Allen DH, et al: Exacerbation of Goodpasture's
syndrome after inadvertent exposure to hydrocarbon fumes. Br Med J 288:188,
1984
-
Donaghy M, Rees AJ: Cigarette smoking and lung haemorrhage
in glomerulonephritis caused by autoantibodies to glomerular basement membrane.
Lancet 2:1390, 1983
-
Wilson CB, Dixon FJ: Anti-glomerular basement membrane antibody-induced
glomerulonephritis. Kidney Int 3:74, 1973 [PMID
4571918]
-
Bolton WK, Couser WG: Intravenous pulse methylprednisolone
therapy of acute crescentic rapidly progressive glomerulonephritis. Am
J Med 66:495, 1979 [PMID
433955]
-
Savage CO, Pusey CD, Bowman C, et al. Antiglomerular basement
membrane antibody mediated disease in the British Isles 1980-4. Br Med
J 292:301, 1986
-
Pusey CD, Lockwood CM, Peters DK: Plasma exchange and immunosuppressive
drugs in the treatment of glomerulonephritis due to antibodies to the glomerular
basement membrane. Int J Artif Organs 6(suppl):15, 1983 [PMID
6642729]
-
Wing EJ, Bruns FJ, Fraley DS, et al: Infectious complications
with plasmapheresis in rapidly progressive glomerulonephritis. JAMA 244:2423,
1980 [PMID
7431570]
-
Hazards of apheresis (editorial). Lancet 2:1025, 1982
-
Huestis DW: Mortality in therapeutic haemapheresis (letter).
Lancet 1:1043, 1983
-
Kleit SA: Apheresis. Am J Kidney Dis 2:573, 1983
-
Bergrem H, Jervell J, Brodwall EK, et al: Goodpasture's syndrome:
a report of seven patients including long-term follow-up of three who received
a kidney transplant. Am J Med 68:54, 1980 [PMID
6985766]
-
Alpers CE, Rennke HG, Hopper J Jr, et al: Fibrillary glomerulonephritis:
a new morphological entity with unusual immunofluorescence features. Kidney
Int 31:781, 1987 [PMID
3106698]
-
Korbet SM, Schwartz MM, Rosenberg BF, et al: Immunotactoid
glomerulopathy. Medicine (Baltimore) 64:228, 1985 [PMID
4010500]
-
Fogo A, Qureshi N, Horn RG: Morphologic and clinical features
of fibril lary glomerulonephritis versus immunotactoid glomerulopathy.
Am J Kidney Dis 22:367, 1993 [PMID
8372831]
-
Julian BA, Quiggins PA, Thompson JS, et al: Familial IgA
nephropathy: evidence of an inherited mechanism of disease. N Engl J Med
312:202, 1985 [PMID
3855328]
-
Nakamoto Y, Asano K, Dohi K, et al: Primary IgA glomerulonephritis
and Schonlein-Henoch purpura nephritis: clinicopathological and immunohistological
characteristics. Q J Med 47:495, 1978 [PMID
375278]
-
Newell GC: Cirrhotic glomerulonephritis: incidence, morphology,
clinical features, and pathogenesis. Am J Kidney Dis 9:183, 1987
-
Fornasieri A, Sinico RA, Maldifassi P, et al: IgA-antigliadin
antibodies in IgA mesangial nephropathy (Berger's disease). Br Med J 295:78,
1987
-
Rifai A: Experimental models for IgA-associated glomerulonephritis.
Kidney Int 31:1, 1987
-
Waxman FJ, Hebert LA, Cosio FG, et al: Differential binding
of immunoglobulin A and immunoglobulin G1 immune complexes to primate erythrocytes
in vitro: immunoglobulin A immune complexes bind less well to erythrocytes
and are preferentially deposited in glomeruli. J Clin Invest 77:82, 1986
[PMID
3944261]
-
Nicholls KM, Fairley KF, Dowling JP, et al: The clinical
course of mesangial IgA associated nephropathy in adults. Q J Med 53:227,
1984 [PMID
6463197]
-
Praga M, Gutierrez-Millet V, Navas JJ, et al: Acute worsening
of renal function during episodes of macroscopic hematuria in IgA nephropathy.
Kidney Int 28:69, 1985 [PMID
4046327]
-
Trachtman H, Weiss RA, Bennett B, et al: Isolated hematuria
in children: indications for a renal biopsy. Kidney Int 25:94, 1984 [PMID
6727131]
-
Tiebosch ATMG, Frederik PM, van Breda Vriesman PJC, et al:
Thin-basement-membrane nephropathy in adults with persistent hematuria.
N Engl J Med 320:14, 1989
-
Cattran DC, Greenwood C, Ritchie S: Long-term benefits of
angiotensin-converting enzyme inhibitor therapy in patients with severe
immunoglobulin A nephropathy: a comparison to patients receiving treatment
with other antihypertensive agents and to patients receiving no therapy.
Am J Kidney Dis 23:247, 1994 [PMID
8311083]
-
Hogg RJ, Silva FG, Wyatt RJ, et al: Prognostic indicators
in children with IgA nephropathy: report of the Southwest Pediatric Nephrology
Study Group. Pediatr Nephrol 8:15, 1994
-
Donadio JV, Bergstralh EJ, Offord KP, et al: Clinical and
histopathologic associations with impaired renal function in IgA nephropathy.
Clin Nephrol 41:65, 1994 [PMID
8004831]
-
Mustonen J, Pasternack A, Rantala I: The nephrotic syndrome
in IgA glomerulonephritis: response to corticosteroid therapy. Clin Nephrol
20:172, 1983 [PMID
6641024]
-
Lai KN, Lai FM, Ho CP, et al: Corticosteroid therapy in IgA
nephropathy with nephrotic syndrome: a long-term controlled trial. Clin
Nephrol 26:174, 1986 [PMID
3536231]
-
Bachman U, Biava C, Amend W, et al: The clinical course of
IgA nephropathy and Henoch-Schonlein purpura following renal transplantation.
Transplantation 42:511, 1980
-
Grunfeld JP: The clinical spectrum of hereditary nephritis.
Kidney Int 27:83, 1985
-
Flinter FA, Cameron JS, Chantler C, et al: Genetics of classic
Alport's syndrome. Lancet 2:1005, 1988 [PMID
2902439]
-
Rumpelt HJ: Hereditary nephropathy (Alport syndrome): correlation
of clinical data with glomerular basement membrane alterations. Clin Nephrol
13:203, 1980
-
Savage CO, Pusey CD, Kershaw MJ, et al: The Goodpasture antigen
in Alport's syndrome: studies with a monoclonal antibody. Kidney Int 30:107,
1986 [PMID
3528615]
-
Kashtan C, Fish AJ, Kleppel M, et al: Nephritogenic antigen
determinants in epidermal and renal basement membranes of kindreds with
Alport-type familial nephritis. J Clin Invest 78:1035, 1986 [PMID
2428839]
-
Blumenthal SS, Fritsche C, Lemann J Jr: Establishing the
diagnosis of benign familial hematuria: the importance of examining the
urine sediment of family members. JAMA 259:2263, 1988 [PMID
3352118]
-
Querin S, Noel LH, Grunfeld JP, et al: Linear glomerular
IgG fixation in renal allografts: incidence and significance in Alport's
syndrome. Clin Nephrol 25:134, 1986 [PMID
3514017]
-
Nolasco F, Cameron JS, Heywood EF, et al: Adult-onset minimal
change nephrotic syndrome: a long-term follow-up. Kidney Int 29:1215, 1986
[PMID
3747335]
-
Ji-Yun Y, Melvin T, Sibley R, et al: No evidence for a specific
role of IgM in mesangial proliferation of idiopathic nephrotic syndrome.
Kidney Int 25:100, 1984 [PMID
6727121]
-
Olivero JJ, Ayus JC, Eknoyan G: Nephrotic syndrome developing
after bee stings. South Med J 74:82, 1981 [PMID
7455752]
-
Moorthy AV, Zimmerman SW, Burkholder PM: Nephrotic syndrome
in Hodgkin's disease: evidence for pathogenesis alternative to immune complex
deposition. Am J Med 61:471, 1976
-
Alpers CE, Cotran RS: Neoplasia and glomerular injury. Kidney
Int 30:465, 1986 [PMID
3537450]
-
Moskovitz R, Springer P, Urquhart M: Lithium-induced nephrotic
syndrome. Am J Psychiatry 138:382, 1981
-
Kalina KM, Burnett GB: Lithium and the nephrotic syndrome.
J Clin Psychopharmacol 4:148, 1984
-
Feinfeld DA, Olesnicky L, Pirani CL, et al: Nephrotic syndrome
associated with use of the nonsteroidal anti-inflammatory drugs: case report
and review of the literature. Nephron 37:174, 1984 [PMID
6738768]
-
Finkelstein A, Fraley DS, Stachura I, et al: Fenoprofen nephropathy:
lipoid nephrosis and interstitial nephritis: a possible T lymphocyte disorder.
Am J Med 72:81, 1982 [PMID
6977270]
-
Dornan TL, Jenkins S, Cotton RE, et al: The nephrotic syndrome
at presentation of insulin-dependent diabetes mellitus: cause or coincidence?
Diabetic Med 5:387, 1988
-
Carrie BJ, Salyer WR, Myers BD: Minimal change nephropathy:
an electrochemical disorder of the glomerular membrane. Am J Med 70:262,
1981 [PMID
6162382]
-
Schnaper HW, Aune TM: Steroid-sensitive mechanism of soluble
immune response suppressor production in steroid-responsive nephrotic syndrome.
J Clin Invest 79:257, 1987
-
Koyama A, Fujisaki M, Kobayashi M, et al: A glomerular permeability
factor produced by human T cell hybridomas. Kidney Int 40:453, 1991 [PMID
1787645]
-
Lowenstein J, Schacht RG, Baldwin DS: Renal failure in minimal
change nephrotic syndrome. Am J Med 70:227, 1981 [PMID
7468609]
-
Bohman SO, Jaremko G, Bohlin AB, et al: Foot process fusion
and glomerular filtration rate in minimal change nephrotic syndrome. Kidney
Int 25:696, 1984 [PMID
6482174]
-
A Report of the International Study of Kidney Disease in
Children: The primary nephrotic syndrome in children: identification of
patients with minimal change nephrotic syndrome from initial response to
prednisone. J Pediatr 98:561, 1981
-
Kida H, Iida H, Dohi K, et al: Period of freedom from relapse
as an indication of cure in minimal change nephrotic syndrome in adults.
Nephron 19:153, 1977 [PMID
895965]
-
Pru C, Kjellstrand CM, Cohn RA, et al: Late recurrence of
minimal lesion nephrotic syndrome. Ann Intern Med 100:69, 1984 [PMID
6691659]
-
Berns JS, Gaudio KM, Krassner LS, et al: Steroid-responsive
nephrotic syndrome of childhood: a long-term study of clinical course,
histopathology, efficacy of cyclophosphamide therapy, and effects on growth.
Am J Kidney Dis 9:108, 1987 [PMID
3826059]
-
Arbeitsgemeinschaft fur Padiatrische Nephrologie: Effect
of cytotoxic drugs in frequently relapsing nephrotic syndrome with and
without steroid dependence. N Engl J Med 306:451, 1982
-
Cyclophosphamide treatment of steroid dependent nephrotic
syndrome: comparison of eight week with 12 week course: report of Arbeitsgemeinschaft
fur Padiatrische Nephrologie. Arch Dis Child 62:1102, 1987
-
Schulman SL, Kaiser BA, Polinsky MS, et al: Predicting the
response to cytotoxic therapy for childhood nephrotic syndrome: superiority
of response to corticosteroid therapy over histopathologic patterns. J
Pediatr 113:996, 1988 [PMID
3193322]
-
Trainin EB, Boichis H, Spitzer A, et al: Late nonresponsiveness
to steroids in children with the nephrotic syndrome. J Pediatr 87:519,
1975 [PMID
1159578]
-
Trompeter RS, Evans PR, Barratt TM: Gonadal function in boys
with steroid-responsive nephrotic syndrome treated with cyclophosphamide
for short periods. Lancet 1:1177, 1981 [PMID
6112527]
-
Taube D, Brown Z, Williams DG: Long-term impairment of suppressor-cell
function by cyclophosphamide in minimal-change nephropathy and its association
with therapeutic response. Lancet 1:235, 1981 [PMID
6109898]
-
Baker GL, Kahl LE, Zee BC, et al: Malignancy following treatment
of rheumatoid arthritis with cyclophosphamide: long-term case-control follow-up
study. Am J Med 83:1, 1987 [PMID
3605161]
-
Williams SA, Makker SP, Ingelfinger JR, et al: Long-term
evaluation of chlorambucil plus prednisone in the idiopathic nephrotic
syndrome of childhood. N Engl J Med 302:929, 1980
-
Kleinknecht C, Guesry P, Lenoir G, et al: High-cost benefit
of chlorambucil in frequently relapsing nephrosis. N Engl J Med 296:48,
1977 [PMID
830272]
-
Cade R, Mars D, Privette M, et al: Effect of long-term azathioprine
administration in adults with minimal-change glomerulonephritis and nephrotic
syndrome resistant to corticosteroids. Arch Intern Med 146:737, 1986 [PMID
3963956]
-
Black DAK, Rose G, Brewer DB: Controlled trial of prednisone
in adult patients with the nephrotic syndrome. Br Med J 3:421, 1970
-
Coggins CH: Adult minimal change nephropathy: experience
of the collaborative study of glomerular disease. Trans Am Clin Climatol
Assoc 97:18, 1985
-
A Report of the International Study of Kidney Disease in
Children: Minimal change nephrotic syndrome in children: deaths during
the first 5 to 15 years' observation. Pediatrics 73:497, 1984
-
Ponticelli C: Treatment of the nephrotic syndrome with cyclosporin
A. J Autoimmun 5(suppl A):315, 1992
-
Meyrier A, Condamin MC, Broneer D: Treatment of adult idiopathic
nephrotic syndrome with cyclosporin A: minimal-change disease and focal-segmental
glomerulosclerosis: Collaborative Group of the French Society of Nephrology.
Clin Nephrol 35:S37, 1991
-
Tanaka R, Yoshikawa N, Kitano Y, et al: Cyclosporin treatment
in nephrotic children. Pediatr Nephrol 7:249, 1993 [PMID
8518091]
-
Niaudet P: Comparison of cyclosporin and chlorambucil in
the treatment of steroid-dependent idiopathic nephrotic syndrome: a multicentre
randomized control trial: French Society of Pediatric Nephrology. Pediatr
Nephrol 6:1, 1992
-
Melocoton TL, Kamil ES, Cohen AH, et al: Long-term cyclosporin
A treatment of steroid-resistant and steroid-dependent nephrotic syndrome.
Am J Kidney Dis 18:583, 1991 [PMID
1951339]
-
Ponticelli C, Rizzoni G, Edefonti A, et al: CsA in steroid-resistant
nephrotic syndrome: a randomized trial of cyclosporine in steroid-resistant
idiopathic nephrotic syndrome. Kidney Int 43:1377, 1993 [PMID
8315953]
-
Hayslett JP, Krassner LS, Bensch KG, et al: Progression of
"lipoid nephrosis" to renal insufficiency. N Engl J Med 281:181, 1969 [PMID
5790494]
-
A Report of the Southwest Pediatric Nephrology Study Group:
Focal segmental glomerulosclerosis in children with idiopathic nephrotic
syndrome. Kidney Int 27:442, 1985
-
A Report of the International Study of Kidney Disease in
Children: Primary nephrotic syndrome in children: clinical significance
of histopathologic variants of minimal change and of diffuse mesangial
hypercellularity. Kidney Int 20:765, 1981
-
Brown EA, Upadhyaya K, Hayslett JP, et al: The clinical course
of mesangial proliferative glomerulonephritis. Medicine (Baltimore) 58:295,
1979 [PMID
449664]
-
A Report of the Southwest Pediatric Nephrology Study Group:
Childhood nephrotic syndrome associated with diffuse mesangial hypercellularity.
Kidney Int 24:87, 1983
-
Beaufils H, Alphonse JC, Guedon J, et al: Focal glomerulosclerosis:
natural history and treatment: a report of 70 cases. Nephron 21:75, 1978
[PMID
673092]
-
Schwartz MM, Lewis EJ: Focal glomerulosclerosis: the cellular
lesion. Kidney Int 28:968, 1985
-
Siegel NJ, Gaudio KM, Krassner LS, et al: Steroid-dependent
nephrotic syndrome in children: histopathology and relapses after cyclophosphamide
treatment. Kidney Int 19:454, 1981
-
Velosa JA, Torres VE: Benefits and risks of nonsteroidal
antiinflammatory drugs in steroid-resistant nephrotic syndrome. Am J Kidney
Dis 8:345, 1986 [PMID
3788972
-
Lewis EJ: Recurrent focal sclerosis after renal transplantation.
Kidney Int 22:315, 1982
-
Striegel JE, Sibley RK, Fryd DS, et al: Recurrence of focal
segmental sclerosis in children following renal transplantation. Kidney
Int 30(suppl):S44, 1986
-
Savin VJ, Artero M, Sharma R, et al: Risk of recurrence of
focal segmental glomerular sclerosis (FSGS) after transplantation can be
assessed using an in vitro assay of serum. J Am Soc Nephrol 4:960, 1993
-
Dantal J, Bigot E, Bogers W, et al: Effect of plasma protein
adsorption on protein excretion in kidney-transplant recipients with recurrent
nephrotic syndrome. N Engl J Med 330:7, 1994
-
Dantal J, Baatard R, Hourmant M, et al: Recurrent nephrotic
syndrome following renal transplantation in patients with focal glomerulosclerosis:
a one-center study of plasma exchange effects. Transplantation 52:827,
1991 [PMID
1949168]
-
Remuzzi G, Bertani T: Is glomerulosclerosis a consequence
of altered glomerular permeability to macromolecules? Kidney Int 38:384,
1990
-
Cunningham EE, Zielezny MA, Venuto RC: Heroin-associated
nephropathy: a nationwide problem. JAMA 250:2935, 1983 [PMID
6644972]
-
Dubrow A, Mittman N, Ghali V, et al: The changing spectrum
of heroin-associated nephropathy. Am J Kidney Dis 5:36, 1985 [PMID
3966467]
-
Rao TKS, Filippone EJ, Nicastri AD, et al: Associated focal
and segmental glomerulosclerosis in the acquired immune deficiency syndrome.
N Engl J Med 310:669, 1984
-
Pardo V, Meneses R, Ossa L, et al: AIDS-related glomerulopathy:
occurrence in specific risk groups. Kidney Int 31:1167, 1987 [PMID
3599656]
-
Korbet SM, Schwartz MM, Lewis EJ: The prognosis of focal
segmental glomerulosclerosis of adulthood. Medicine (Baltimore) 65:304,
1986 [PMID
3747827]
-
Velosa JA, Holley KE, Torres VE, et al: Significance of proteinuria
on the outcome of renal function in patients with focal segmental glomerulosclerosis.
Mayo Clin Proc 58:568, 1983
-
McVicar M, Exeni R, Susin M: Nephrotic syndrome and multiple
tubular defects in children: an early sign of focal segmental glomerulosclerosis.
J Pediatr 97:918, 1980 [PMID
7441420]
-
Bouissou F, Barthe P, Pierragi MT: Severe idiopathic nephrotic
syndrome with tubular dysfunction (report of nine pediatric cases). Clin
Nephrol 14:135, 1980 [PMID
7418280]
-
Pei Y, Cattran D, Delmore T, et al: Evidence suggesting under-treatment
in adults with idiopathic focal segmental glomerulosclerosis: Regional
Glomerulonephritis Registry Study. Am J Med 82:938, 1987 [PMID
3578362]
-
Geary DF, Farine M, Thorner P, et al: Response to cyclophosphamide
in steroid-resistant focal segmental glomerulosclerosis: a reappraisal.
Clin Nephrol 22:109, 1984 [PMID
6488594]
-
Banfi G, Moriggi M, Sabadini E, et al: The impact of prolonged
immunosuppression on the outcome of idiopathic focal-segmental glomerulosclerosis
with nephrotic syndrome in adults: a collaborative retrospective study.
Clin Nephrol 36:53, 1991 [PMID
1934660]
-
Mendoza SA, Tune BM: Treatment of childhood nephrotic syndrome.
J Am Soc Nephrol 3:889, 1992
-
Taylor J, Novak R, Christiansen R, et al: Focal sclerosing
glomerulopathy with adverse effects during pregnancy. Arch Intern Med 138:1695,
1978 [PMID
718321]
-
Walker RG, Kincaid-Smith P: The effect of treatment of corticosteroid-resistant
idiopathic (primary) focal and segmental hyalinosis and sclerosis (focal
glomerulosclerosis) with ciclosporin. Nephron 54:117, 1990 [PMID
2179754]
-
Jordan SC, Querfeld U, Toyoda M, et al: Serum interleukin-2
levels in a patient with focal segmental glomerulosclerosis: relationship
to clinical course and cyclosporin A therapy. Pediatr Nephrol 4:166, 1990
[PMID
2397185]
-
Ingulli E, Tejani A: Incidence, treatment and outcome of
recurrent focal segmental glomerulosclerosis posttransplantation in 42
allografts in children: a single-center experience. Transplantation 51:401,
1991 [PMID
1994534]
-
Senggutuvan P, Cameron JS, Hartley RB, et al: Recurrence
of focal segmental glomerulosclerosis in transplanted kidneys: analysis
of incidence and risk factors in 59 allografts. Pediatr Nephrol 4:21, 1990
[PMID
2206875]
-
Row PG, Cameron JS, Turner DR, et al: Membranous nephropathy:
long-term follow-up and association with neoplasia. Q J Med 44:207, 1975
[PMID
1178811]
-
Couser WG, Salant DJ: In situ immune complex formation and
glomerular injury. Kidney Int 17:1, 1980 [PMID
6990087]
-
Couser WG: Mechanisms of glomerular injury in immune-complex
disease. Kidney Int 28:569, 1985
-
Couser WG, Baker PJ, Adler S: Complement and the direct mediation
of glomerular injury: a new perspective. Kidney Int 28:879, 1985 [PMID
2935674]
-
Shemesh O, Ross JC, Deen WM, et al: Nature of the glomerular
capillary injury in human membranous glomerulopathy. J Clin Invest 77:868,
1986 [PMID
2419362]
-
Katz WA, Blodgett RC Jr, Pietrusko RG: Proteinuria in gold-treated
rheumatoid arthritis. Ann Intern Med 101:176, 1984 [PMID
6430141]
-
Hall CL, Jawad S, Harrison PR, et al: Natural course of penicillamine
nephropathy: a long term study of 33 patients. Br Med J 296:1083, 1988
-
Hirose H, Udo K, Kojima M, et al: Deposition of hepatitis
B e antigen in membranous glomerulonephritis: identification by F (ab¢)2
fragments of monoclonal antibody. Kidney Int 26:338, 1984 [PMID
6513277]
-
Wrzolkowa T, Zurowska A, Uszycka-Karcz N, et al: Hepatitis
B virus-associated glomerulonephritis: electron microscopic studies in
98 children. Am J Kidney Dis 18:306, 1991 [PMID
1882821]
-
Lai KN, Li PKT, Lui SF, et al: Membranous nephropathy related
to hepatitis B virus in adults. N Engl J Med 324:1457, 1991 [PMID
2023605]
-
Gilbert RD, Wiggelinkhuizen J: The clinical course of hepatitis
B virus-associated nephropathy. Pediatr Nephrol 8:11, 1994 [PMID
8142208]
-
Darda R, Peterson J, Weiner R, et al: Membranous glomerulonephritis
in association with hepatitis C virus infection. Am J Kidney Dis 22:452,
1993
-
Burstein DM, Korbet SM, Schwartz MM: Membranous glomerulonephritis
and malignancy. Am J Kidney Dis 22:5, 1993 [PMID
8322793]
-
Breysse MS, Couser WG, Alpers CE, et al: Membranous nephropathy
and formaldehyde exposure. Ann Intern Med 120:396, 1994
-
Davidson AM, Cameron JS, Kerr DN, et al: The natural history
of renal function in untreated idiopathic membranous glomerulonephritis
in adults. Clin Nephrol 22:61, 1984
-
Fries JWU, Mendrick DL, Rennke HG: Determinants of immune
complex-mediated glomerulonephritis. Kidney Int 34:333, 1988
-
Mallick NP, Short CD, Manos J: Clinical membranous nephropathy.
Nephron 34:209, 1983 [PMID
6224094]
-
Donadio JV Jr, Torres VE, Velosa JA, et al: Idiopathic membranous
nephropathy: the natural history of untreated patients. Kidney Int 33:708,
1988 [PMID
3367560]
-
Schieppati A, Mosconi L, Perna A, et al: Prognosis of untreated
patients with idiopathic membranous nephropathy. N Engl J Med 329:85, 1993
[PMID
8510707]
-
Falk RJ, Hogan SL, Muller KE, et al: Treatment of progressive
membranous glomerulopathy: a randomized trial comparing cyclophosphamide
and corticosteroids with corticosteroids alone: for the Glomerular Disease
Collaborative Network. Ann Intern Med 116:438, 1992
-
Hopper J, Trew PA, Biava CG: Membranous nephropathy:
its relative benignity
-
Passerini P, Pasquali S, Cesana B, et al: Long-term outcome
of patients with membranous nephropathy after complete remission of proteinuria.
Nephrol Dial Transplant 4:525, 1989
-
D'Amico G: Influence of clinical and histological features
on actuarial renal survival in adult patients with idiopathic IgA nephropathy,
membranous nephropathy, and membranoproliferative glomerulonephritis: survey
of the recent literature. Am J Kidney Dis 20:315, 1992
-
Llach F: Hypercoagulability, renal vein thrombosis,
and other thrombotic complications of nephrotic syndrome. Kidney Int 28:429,
1985
-
Wagoner RD, Stanson AW, Holley KE, et al: Renal vein thrombosis
in idiopathic membranous glomerulopathy and nephrotic syndrome: incidence
and significance. Kidney Int 23:368, 1983
-
Sarasin FP, Schifferli JA: Prophylactic oral anticoagulation
in nephrotic patients with idiopathic membranous nephropathy. Kidney Int
45:578, 1994 [PMID
8164448]
-
Schena FP, Cameron JS: Treatment of proteinuric idiopathic
glomerulonephritides in adults: a retrospective survey. Am J Med 85:315,
1988 [PMID
3046352]
-
Ehrenreich T, Porush JG, Churg J, et al: Treatment of idiopathic
membranous nephropathy. N Engl J Med 295:741, 1976 [PMID
958260]
-
Collaborative Study of the Adult Nephrotic Syndrome: A controlled
study of short-term prednisone treatment in adults with membranous nephropathy.
N Engl J Med 301:1301, 1979
-
Cattran DC, Delmore T, Roscoe J, et al: A randomized controlled
trial of prednisone in patients with idiopathic membranous nephropathy.
N Engl J Med 320:210, 1989 [PMID
2643046]
-
Ponticelli C, Passerini P: Treatment of the nephrotic syndrome
associated with primary glomerulonephritis. Kidney Int 46:595, 1994 [PMID
7996782]
-
Rostoker G: Idiopathic membranous nephropathy: new therapeutic
trends. European Journal of Medicine 2:106, 1993
-
Piccoli A, Pillon L, Passerini P, et al: Therapy for idiopathic
membranous nephropathy: tailoring the choice by decision analysis. Kidney
Int 45:1193, 1994 [PMID
8007591]
-
Ponticelli C, Zucchelli P, Passerini P, et al: A randomized
trial of methylprednisolone and chlorambucil in idiopathic membranous nephropathy.
N Engl J Med 320:8, 1989
-
Mathieson PW, Turner AN, Maidment CGH, et al: Prednisolone
and chlorambucil treatment in idiopathic membranous nephropathy with deteriorating
renal function. Lancet 2:869, 1988
-
Ponticelli C, Zucchelli P, Passerini P, et al: Methylprednisolone
plus chlorambucil as compared with methylprednisolone alone for the treatment
of idiopathic membranous nephropathy. N Engl J Med 327:599, 1992 [PMID
1640953]
-
Warwick GL, Geddes CG, Boulton-Jones JM: Prednisolone and
chlorambucil therapy for idiopathic membranous nephropathy with progressive
renal failure. Q J Med 87:223, 1994 [PMID
8208913]
-
West ML, Jindal KK, Bear RA, et al: Controlled trial of cyclophosphamide
in patients with membranous glomerulonephritis. Kidney Int 32:579, 1987
[PMID
3323596]
-
Jindal K, West M, Bear R, et al: Long-term benefits of therapy
with cyclophosphamide and prednisone in patients with membranous glomerulonephritis
and impaired renal function. Am J Kidney Dis 19:61, 1992 [PMID
1739084]
-
Bruns FJ, Adler S, Fraley DS, et al: Sustained remission
of membranous glomerulonephritis after cyclophosphamide and prednisone.
Ann Intern Med 114:725, 1991 [PMID
2012353]
-
Reichert LJM, Huysmans FTM, Assmann K, et al: Chlorambucil
but not pulse cyclophosphamide preserves renal function in patients with
membranous nephropathy and deteriorating renal function. Ann Intern Med
121:328, 1994
-
Alexopoulos E, Sakellariou G, Memmos D, et al: Cyclophosphamide
provides no additional benefit to steroid therapy in the treatment of idiopathic
membranous nephropathy. Am J Kidney Dis 21:497, 1993 [PMID
8488817]
-
Rostoker G, Belghiti D, Maadi AB, et al: Long-term cyclosporin
A therapy for severe idiopathic membranous nephropathy. Nephron 63:335,
1993 [PMID
8446273]
-
Guasch A, Suranyi M, Newton L, et al: Short-term responsiveness
of membranous glomerulopathy to cyclosporine. Am J Kidney Dis 20:472, 1992
[PMID
1442759]
-
Palla R, Cirami C, Panichi V, et al: Treatment of idiopathic
membranous nephropathy (MGN) with high dose intravenous gammaglobulin:
results of a multicenter non-randomized and non-controlled trial. J Nephrol
4:211, 1991
-
Kaysen GA, Gambertoglio J, Jiminez I, et al: Effect of dietary
protein intake on albumin homeostasis in nephrotic patients. Kidney Int
29:572, 1986 [PMID
3702214]
-
Kaysen GA: Albumin metabolism in the nephrotic syndrome:
the effect of dietary protein intake. Am J Kidney Dis 12:461, 1988
-
Klahr S, Levey AS, Beck GJ, et al: The effects of dietary
protein restriction and blood-pressure control on the progression of chronic
renal disease. N Engl J Med 330:877, 1994
-
Wheeler DC, Bernard DB: Lipid abnormalities in the nephrotic
syndrome: causes, consequences, and treatment. Am J Kidney Dis 23:331,
1994 [PMID
8128933]
-
Harris RC, Ismail N: Extrarenal complications of the nephrotic
syndrome. Am J Kidney Dis 23:477, 1994
-
Kasiske BL, Velosa JA, Halstenson CE, et al: The effects
of lovastatin in hyperlipidemic patients with the nephrotic syndrome. Am
J Kidney Dis 15:8, 1990 [PMID
2294737]
-
Wanner C, Bohler J, Eckardt HG, et al: Effects of simvastatin
on lipoprotein (a) and lipoprotein composition in patients with nephrotic
syndrome. Clin Nephrol 41:138, 1994 [PMID
8187355]
-
Rosenberg ME, Hostetter TH: Comparative effects of antihypertensives
on proteinuria: angiotensin-converting enzyme inhibitor versus a1-antagonist.
Am J Kidney Dis 18:472, 1991 [PMID
1656730]
-
Praga M, Hernandez E, Montoyo C, et al: Long-term beneficial
effects of angiotensin-converting enzyme inhibition in patients with nephrotic
proteinuria. Am J Kidney Dis 20:240, 1992
-
Ishimitsu T, Yagi H, Ohkubo M, et al: Different effects between
antihypertensive drugs on nephrotic-range proteinuria in renovascular hypertension.
Am J Nephrol 14:60, 1994
-
Gansevoort RT, deZeeuw D, deJong PE: Dissociation between
the course of the hemodynamic and antiproteinuric effects of angiotensin
I converting enzyme inhibition. Kidney Int 44:579, 1993 [PMID
8231031]
-
Gansevoort RT, Heeg JE, Vriesendorp R, et al: Antiproteinuric
drugs in patients with idiopathic membranous glomerulopathy. Nephrol Dial
Transplant 7:91, 1992 [PMID
1337189]
-
Olivero JJ, Frommer JP, Gonzalez JM: Medical nephrectomy:
the last resort for intractable complications of the nephrotic syndrome.
Am J Kidney Dis 21:260, 1993 [PMID
8447301]