Contenido del artículo
VI INSUFICIENCIA RENAL AGUDA
- Generalidades sobre la insuficiencia renal aguda
- DIAGNOSTICO DIFERENCIAL: ENFERMEDAD PRERRENAL CONTRA NECROSIS TUBULAR AGUDA
- HISTORIA CLINICA Y EXAMEN FISICO
- Enfermedad prerrenal
- Azoemia prerrenal causada por depleción verdadera de volumen
- Azoemia prerrenal causada por enfermedad hepática avanzada
- Azoemia prerrenal causada por insuficiencia cardiaca congestiva
- Necrosis tubular aguda
- Necrosis tubular aguda posisquémica
- Necrosis tubular aguda causada por nefrotoxinas
DR. ROBERT M. BLACK
Generalidades sobre la insuficiencia renal aguda
La insuficiencia renal aguda (IRA) es un problema clínico frecuente caracterizado por disminución relativamente súbita de la función renal. Esta reducción en la velocidad de filtración glomerular (VFG) se manifiesta por aumento en la concentración plasmática de creatinina (Pcr) y, por lo general, por disminución del volumen urinario diario. Cuando se desarrolla IRA, el diagnóstico diferencial debe ser muy amplio, incluyendo enfermedades que afectan las arterias, glomérulos, túbulos o intersticio renal, así como padecimientos que obstruyen la salida de orina del riñón. Sin embargo, de las muchas enfermedades que causan IRA en los adultos, dos son responsables de alrededor del 70 a 75 porciento de todos los casos: la disminución en la perfusión renal (enfermedad prerrenal) y la necrosis tubular aguda (NTA). La nefritis intersticial aguda, otra causa relativamente frecuente de IRA, se analiza en otro capítulo. Las glomerulonefritis, las vasculitis y la obstrucción de las vías urinarias, padecimientos que pueden asociarse con disminución súbita de la VFG, se describen también en otra parte de la obra.
DIAGNOSTICO DIFERENCIAL: ENFERMEDAD PRERRENAL CONTRA NECROSIS TUBULAR AGUDA
La causa más frecuente de IRA es la disminución de la circulación sanguínea renal o el daño a los túbulos renales por toxinas o isquemia. En el primer caso (enfermedad prerrenal), el riñón conserva sodio en la depleción verdadera de volumen o en respuesta a la enfermedad cardiaca o hepática avanzadas. Esta respuesta es apropiada porque, en ambas circunstancias, el volumen sanguíneo arterial efectivo (que corresponde al que perfunde los órganos vitales) está disminuido.
Por el contrario, el daño renal característico de la NTA limita la capacidad normal de los túbulos renales para conservar líquido y electrolitos. Esta menor capacidad para reabsorber sodio es ocasionada por necrosis celular, así como por traslocación de la ATPasa de Na+-K+ del lado basolateral (o sanguíneo) de las células tubulares renales hacia la luz.1
Estas diferencias en la función renal son de gran importancia para distinguir entre las principales formas de insuficiencia renal aguda.
HISTORIA CLINICA Y EXAMEN FISICO
La historia clínica y el examen físico suelen brindar información diagnóstica importante, como la identificación de las posibles causas de disminución en la perfusión renal o de la NTA y el estado del volumen extracelular del paciente.
Distinción de la insuficiencia renal aguda y crónica
Al distinguir entre la insuficiencia renal aguda o crónica, el antecedente de insuficiencia renal, la observación de riñones pequeños por radiografía de abdomen o ultrasonido, y la anemia, son los datos de laboratorio que indican cronicidad en forma más confiable. Por el contrario, cuando la duración de la insuficiencia renal no se conoce, debe excluirse la posibilidad de insuficiencia renal aguda reversible. Por ejemplo, debe realizarse un estudio de ultrasonido en todos los individuos para detectar posibles lesiones obstructivas, a menos de que exista otra causa obvia de la IRA.
Existen varias pruebas de laboratorio que se usan con mucha frecuencia para distinguir entre la azoemia prerrenal y la NTA [ver tabla 1]. Estos estudios son especialmente útiles en los pacientes oligúricos (con diuresis <500 ml/día), porque los individuos con NTA no oligúrica tienen, típicamente, menos daño tubular y sus resultados de laboratorio muestran más sobreposición con los de pacientes con azoemia prerrenal.2,3
|
||||||
NTA- necrosis tubular aguda, Bun- nitrógeno de urea en sangre, Pcr -concentración plasmática de creatina Uosm- osmoralidad urinaria, UNa- concentración urinaria de sodio |
Relación entre nitrógeno de urea en sangre y creatinina
La disminución en la VFG suele aumentar el nitrógeno de urea en sangre (BUN) y la creatinina en proporciones iguales. La mayor avidez de sal y agua asociada a los estados prerrenales causa un aumento desproporcionado en la relación plasmática entre el BUN y la creatinina. Una relación mayor de 20:1 es sugestiva de azoemia prerrenal en ausencia de una fuente de mayor producción de urea, como hemorragia gastrointestinal, destrucción tisular, alta ingesta proteica o tratamiento con esteroides.
Examen general de orina
El examen general de orina suele ser normal en la azoemia prerrenal, siendo el único hallazgo frecuente la presencia de cilindros hialinos. Los cilindros están compuestos de mucoproteína de Tamm-Horsfall, que es secretada en condiciones normales por las células de la porción gruesa de la rama ascendente del asa de Henle y que se precipita en la orina concentrada y ácida, típica del estado prerrenal. Por el contrario, en la NTA la orina contiene muchos cilindros granulares café obscuro, con células libres del epitelio renal tubular y cilindros de células epiteliales [ver figura 1], aunque la orina puede ser normal hasta en el 20 a 30 porciento de los pacientes.
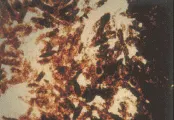
|
| Figura 1 |
| Cilindros granulares |
Osmolaridad urinaria
El riñón normal puede elaborar orina con una concentración máxima mayor de 1,200 mOsm/kg en los estados de deshidratación. La capacidad para realizar esta función depende de una función tubular intacta, en especial en el asa de Henle. Una osmolaridad urinaria (Uosm) mayor de 500 mOsm/kg es muy sugestiva de azoemia prerrenal.
En comparación, el daño tubular extenso, como el que ocurre en la NTA, disminuye la capacidad del riñón para generar una osmolaridad intersticial alta, en parte por el daño a las células del asa de Henle. Como resultado, la máxima Uosm que puede alcanzarse se reduce. Típicamente, la Uosm en la NTA es de 300 a 350 mOsm/kg, un valor muy similar a la osmolaridad plasmática (Posm).
A pesar de estas diferencias, la Uosm suele no ser útil para distinguir la causa de la insuficiencia renal aguda. Con la hipoperfusión renal prolongada, la reabsorción tubular de solutos (NaCl y urea) aumenta, y la cantidad de solutos que llega al asa de Henle y a los segmentos más distales de la nefrona disminuye en forma progresiva. Como resultado de la menor presentación de solutos al asa de Henle y la menor función de ésta, tanto la reabsorción medular de solutos como la osmolaridad intersticial disminuyen. Por lo tanto, una Uosm menor de 500 mOsm/kg no tiene utilidad diagnóstica porque puede observarse en pacientes con NTA, enfermedad prerrenal o enfermedad renal subyacente.
Concentración urinaria y fracción de excreción de sodio
La concentración urinaria de sodio (UNa) suele ser muy útil en el diagnóstico de la insuficiencia renal aguda porque la menor perfusión renal es el estímulo más potente de reabsorción tubular de sodio. En la azoemia prerrenal la UNa disminuye en forma típica a menos de 20 mEq/L, y este valor es independiente de la ingesta dietética de sal y de la concentración plasmática de sodio. En la NTA la UNa suele ser mayor. El cálculo de la fracción de excreción de sodio (FENa) puede ser especialmente útil para hacer el diagnóstico correcto, ya que esta medida refleja sólo la reabsorción tubular de sodio, y no el manejo del agua.
La FENa se define como el porcentaje del sodio filtrado (VFG x PNa) que se excreta en la orina (UNa x volumen urinario).
| FENa | = |
sodio excretado ________________ |
x | 100 |
| sodio filtrado |
| FENa | = |
UNax volumen ____________________ |
x | 100 |
| PNa x VFG |
| FENa | = |
UNax PCr ________________________ |
x | 100 |
| PNa x UCr |
en donde Ucr es la concentraciones urinarias de creatinina, Pcr y Ucr se miden en mg/dl; UNa y PNa se miden en mEq/L; y VFG = (Ucr x volumen)/Pcr. En los pacientes con función renal normal y en los que tienen insuficiencia renal aguda por azoemia prerrenal, la Fena suele ser menor de uno porciento.
Parece ser que la Fena es más específica que la Una o cualquier otra prueba aislada de laboratorio para distinguir entre azoemia prerrenal y NTA.4,5 Debe utilizarse siempre que la menor perfusión renal sea una causa probable de insuficiencia renal aguda pero la Una sea mayor de 20 mEq/L. En este caso, una Fena menor de uno porciento es muy sugestiva de enfermedad prerrenal, mientras que una Fena mayor de dos porciento indica NTA (siempre que no se estén administrando diuréticos). En el rango entre uno y dos porciento existe sobreposición entre la azoemia prerrenal y la NTA.6
Aunque la Fena es muy útil, en la actualidad parece ser que otras causas de insuficiencia renal aguda diferentes a la enfermedad prerrenal pueden, en ocasiones, asociarse con valores de Fena menores del uno porciento [ver tabla 2]. Por ejemplo, el tipo de disfunción renal que se presenta durante las primeras 48 horas de la enfermedad, en el momento en que ocurre la transición de enfermedad prerrenal a NTA, puede caracterizarse por una Fena menor de uno porciento.7
|
|
|
En resumen, aún no existe una prueba única que pueda distinguir siempre entre la azoemia prerrenal y la NTA. Por lo tanto, el diagnóstico apropiado se realiza basándose en toda la información disponible.
Enfermedad prerrenal
La disminución en la circulación sanguínea renal es la causa más frecuente de IRA. La disminución consecuente en la perfusión tisular puede ser consecuencia de depleción real de volumen o de isquemia renal selectiva (como en la estenosis bilateral de la arteria renal) [ver tabla 3]. Los factores fisiopatológicos que contribuyen al aumento en la concentración de BUN y la Pcr cuando existe hipovolemia son importantes desde el punto de vista diagnóstico y terapéutico.8
|
|
ECA - enzima convertidora de angiotensina, AINE - antinflamatorio no esteroide
|
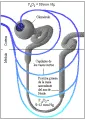
La caída en la VFG que ocurre al disminuir la circulación renal depende de dos mecanismos: dilatación de la arteriola aferente (preglomerular), lo que permite que mayor porcentaje de la presión sistémica se trasmita al glomérulo, y la constricción de la arteriola eferente (posglomerular), que aumenta más la presión de perfusión a través de los capilares glomerulares [ver figura 2]. Estudios experimentales realizados en animales sugieren que la vasodilatación aferente es mediada por una respuesta miogénica directa, por prostaglandinas vasodilatadoras y por retroalimentación túbuloglomerular. Por el contrario, la vasoconstrición eferente depende principalmente de la angiotensina II.9 La retroalimentación túbuloglomerular es un proceso por el cual la arteriola aferente se vasodilata en respuesta a un bajo aporte tubular distal de NaCl, ayudando así a restablecer el aporte distal hacia lo normal. Las observaciones realizadas en humanos han confirmado en forma indirecta la importancia de las prostaglandinas (vasodilatadores aferentes) y de la angiotensina II (vasoconstrictor eferente) en la autorregulación. Por ejemplo, si la vasodilatación preglomerular se altera por agentes que inhiben la producción de prostaglandinas (como indometacina), la vasoconstricción sin oposición de la arteriola eferente causa una disminución súbita en la circulación renal y la VFG.10 Por el contrario, en la estenosis bilateral de la arteria renal (en la que la presión intrarrenal distal a la estenosis tiende a ser menor que la presión sistémica), la administración de un inhibidor de la enzima convertidora de angiotensina (ECA) puede causar insuficiencia renal aguda por eliminación de la constricción de la arteriola eferente mediada por la angiotensina II.11 Puede ocurrir un problema semejante en los pacientes con insuficiencia cardiaca congestiva (ICC) que tienen presión arterial sistémica baja; en este caso, la velocidad de filtración glomerular puede caer a pesar de que el inhibidor de la ECA aumente el gasto cardiaco al disminuir las resistencias vasculares periféricas.12 Tanto en la estenosis bilateral de la arteria renal como en la insuficiencia cardiaca congestiva, la presión hidráulica del capilar glomerular disminuye, lo que ocasiona el principal cambio en la función renal.
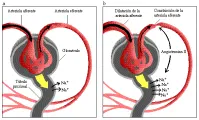
|
| Figura 2 |
| Flujo sanguíneo renal |
Las causas habituales de azoemia prerrenal son la depleción verdadera de volumen, la enfermedad hepática avanzada y la ICC. En muchos individuos existe algún medicamento (como los antiinflamatorios no esteroideos [AINE] o un inhibidor de la ECA) que actúa como un factor contribuyente al problema.
Azoemia prerrenal causada por depleción verdadera de volumen
DIAGNOSTICO
La presencia de una de las causas de depleción verdadera de volumen suele ser muy evidente desde la historia clínica. Una pérdida de líquido tan severa como para causar insuficiencia renal deberá asociarse por lo menos con algunos de los datos físicos característicos, incluyendo disminución en la turgencia de la piel, sequedad de mucosas, colapso de las venas del cuello y taquicardia postural, con o sin hipotensión. Aquellos pacientes con el síndrome clínico de choque hipovolémico también tienen signos de hipoperfusión importante (causada en parte por la vasoconstricción periférica intensa), como frío, extremidades húmedas y frías, agitación, confusión y diuresis escasa o nula.
Existe oliguria en la mayoría de los individuos. En vista de que la respuesta normal a la hipoperfusión renal es la reteción de Na+ y agua en el riñón, la oliguria es adecuada.
La reducción rápida de la hipertensión crónica grave de cualquier origen es otra causa de azoemia prerrenal aguda. En este caso existe hiperplasia arteriolar renal, que representa un cambio adaptativo porque protege a los capilares glomerulares de la presión sistémica elevada. Sin embargo, estos vasos tienen una menor capacidad para dilatarse cuando la presión sanguínea disminuye en forma brusca, lo que causa menor perfusión tisular. El control continuo de la presión arterial permite que la hiperplasia arteriolar desaparezca y la función renal mejore en un periodo de uno a tres meses.13 No es necesario suspender los medicamentos antihipertensivos durante este intervalo, a menos de que exista una disminución excesiva en la presión arterial o un aumento inaceptable en la Pcr.
TRATAMIENTO
El tratamiento de la depleción de volumen real se dirige a restituir el volumen sanguíneo circulante normal. Las principales preguntas a considerar son el tipo de líquido que va a utilizarse y la velocidad a la que deba administrarse. Excluyendo a los pacientes con hipotensión, en quienes la solución salina isotónica es la de elección, la tonicidad del líquido a administrar dependerá de la PNa.
Aunque antes se preferían las soluciones coloides a la solución salina isotónica o lactada de Ringer en el choque hipovolémico porque se creía que expandía el volumen plasmático con más eficacia (al aumentar la presión osmótica), estudios controlados no han demostrado esta ventaja en ausencia de hipoalbuminemia concomitante.14,15 Aún más, la infusión de suficiente albúmina para aumentar la concentración plasmática por arriba de lo normal se ha asociado con insuficiencia renal aguda, quizá causada por disminución de la VFG debida a una presión oncótica alta en el capilar glomerular.16
La velocidad a la que se administren los líquidos depende del estado clínico del paciente. Como regla general, la administración de líquidos a una velocidad de 75 a 100 ml/hr es adecuada para la mayoría de los adultos. La administración mprápida puede ser dañina en pacientes con enfermedad cardiaca subyacente, y es innecesaria en ausencia de pérdidas continuas, hipovolemia grave (como la que ocurre en la hiperglucemia no controlada), o estado de choque (en el que pueden requerirse 1 a 2 L de líquido durante la primera hora y hasta que mejore la perfusión).
Lo adecuado de la repleción de líquidos puede evaluarse por medio del examen físico y vigilando la función renal. Por ejemplo, el aumento de la diuresis y la excreción urinaria de sodio suelen indicar el restablecimiento a la normovolemia. Toma más tiempo revertir la azoemia y disminuir la Pcr.
Azoemia prerrenal causada por enfermedad hepática avanzada
La enfermedad hepática avanzada se asocia con dos cambios principales en la función renal: retención de sodio, que se manifiesta en un principio por ascitis, y disminución progresiva en la VFG que, en las etapas tardías, se denomina síndrome hepatorrenal (SHR). Tanto los factores humorales como los hemodinámicos tienen un papel predominante en el desarrollo de estos problemas.
PATOGENIA DE LA DISMINUCION EN LA FUNCION RENAL
El aumento en la norepinefrina y angiotensina II circulantes, que promueve la retención de sodio en la enfermedad hepática, también causa vasoconstricción renal, un cambio que contribuye a la caída progresiva de la VFG.17 La disminución relativamente temprana de la filtración glomerular, suele pasar desapercibida porque existe poco o ningún cambio en el BUN o en la Pcr. La menor producción hepática de urea limita el aumento en el BUN, mientras que la desnutrición (que disminuye la masa muscular) y quizá disminuye la producción hepática de creatinina (precursora de la creatinina), limita el aumento de la Pcr.18 Como resultado, hasta la mitad de los pacientes cirróticos con concentraciones plasmáticas de creatinina dentro del rango normal (1.0 a 1.2 mg/dl) tienen menor VFG (que se mide por depuración de insulina), que puede ser tan baja como 15 ml/min.18
La disminución progresiva en la función renal que ocurre en la cirrosis hepática parece depender de alteraciones hemodinámicas porque la función tubular está intacta (según lo evidencía una menor UNa y el examen general de orina normal)19 y los riñones no muestran anomalías histológicas características (como lo demuestra la utilidad de estos riñones como órganos de trasplante).20
Una posible explicación del deterioro de la función renal en la cirrosis hepática es el desequilibrio neurohumoral en el que el nivel de vosoconstrictores renales aumenta a la vez que la actividad vasodilatadoa renal disminuye.8 Como se describió antes, la hipertensión intrahepática y el menor llenado arterial causa un aumento progresivo en las actividades del sistema nervioso simpático y en los sistemas renina-angiotensina. Esta tendencia a la isquemia renal es, en un principio, contrarrestada en forma parcial por la mayor producción de prostaglandinas y cininas vasodilatadoras.21-23 Sin embargo, estas respuestas protectoras importantes se alteran cuando la enfermedad hepática se agrava y las concentraciones tanto de prostaglandinas como de cininas disminuyen.21,22,24 Estos cambios pueden reflejar la importancia de una función hepática normal en la producción de la precalicreína (que, cuando se activa la calicreína, divide el cininógeno para formar el potente vasodilatador lisil-bradicinina) en la conversión de ácido linoleico a ácido araquidónico (el precursor de las protaglandinas).21
SINDROME HEPATORRENAL
El síndrome hepatorrenal (SHR) se refiere al desarrollo de IRA en un paciente con enfermedad hepática avanzada, casi siempre por cirrosis, y menos por un tumor metastásico o hepatitis alcohólica severa.25
La vasodilatación esplácnica parece tener un papel importante en la patogenia del SHR.25 Al empeorar la función hepática, el gasto cardiaco aumenta y las resistencias vasculares sistémicas disminuyen. Ocurre reducción en la resistencia vascular sistémica a pesar de los incrementos locales en la resistencia vascular renal y femoral causada, en parte, por activación del sistema renina-angiotensina y del sistema nervioso simpático (ambos por la hipotensión).23
La reducción en la perfusión renal en este caso se asocia con disminución en la VFG, en la excreción de sodio (por lo general a < 10 mEq/día) y en la presión arterial media, a pesar de la intensa vasoconstricción renal.17,26 El papel patogénico de la vasodilatación esplácnica puede ilustrarse en forma indirecta por la respuesta de los pacientes con cirrosis hepática avanzada a la ornipresina, un análogo de la hormona antidiurética (HAD, también conocida como arginina vasopresina) que es un vasoconstrictor esplácnico:27 la administración de ornipresina corrige en forma parcial muchas de las alteraciones hemodinámicas sistémicas y renales, causando elevación en la presión arterial media, reducción en la actividad de renina plasmática y en la concentración de norepinefrina, y aumento en el flujo sanguíneo renal, la VFG, la excreción urinaria de sodio y el volumen urinario.
Independientemente del mecanismo por el que la VFG se reduce en pacientes con enfermedad hepática, esta reducción suele estar clínicamente enmascarada. Tanto la producción de urea como de creatinina pueden estar muy reducidas en estos pacientes por la enfermedad hepática, la menor masa muscular y la menor ingesta de proteínas vegetales y animales. Como resultado, la Pcr puede estar en un rango normal (1.0 a 1.3 mg/dl), dependiendo de la masa muscular, y asociarse con una VFG en rangos desde 20 ml/min hasta normales, por arriba de 100 ml/min.18,28
Presentación clínica
El SHR se caracteriza por oliguria, sedimento urinario que suele contener pocas células o cilindros, una excreción de sodio muy baja e incremento discreto en la Pcr.25,26 La Pcr puede aumentar tan solo 0.1 mg/dl/día, con periodos intermitentes de estabilización e incluso con discreta mejoría. El inicio de la insuficiencia renal suele ser insidioso, pero puede ser precipitado por una lesión aguda, como hemorragia gastrointestinal, infección o diuresis demasiado rápida.26
Un estudio prospectivo de 234 pacientes no azoémicos con cirrosis y ascitis encontró que se desarrolló SHR en 18 porciento de los pacientes en un año y 39 porciento en cinco años.26 Los pacientes con hiponatremia y nivel elevado de renina en plasma tuvieron el riesgo más alto de SHR. Estos signos de activación neurohumoral parecen reflejar una reducción más severa en la perfusión tisular eficaz.23
Aunque los cambios en la perfusión renal y en la función típicos del SHR pueden observarse en muchas formas de enfermedad hepática severa, los pacientes con cirrosis biliar primaria parecen estar relativamente protegidos.29 La retención de sodio, la formación de ascitis y la presencia de SHR no suelen presentarse en este padecimiento, lo que quizá refleje las acciones natriuréticas y de vasodilatación renal de las sales biliares retenidas.
Diagnóstico diferencial de la insuficiencia renal aguda en la enfermedad hepática diferente al síndrome hepatorrenal
Enfermedad hepática veno-oclusiva Recientemente se ha descrito la enfermedad hepática veno-oclusiva, un padecimiento que semeja al SHR y que se desarrolla sobre todo después del trasplante de médula ósea.30 Alrededor del 90 porciento de los pacientes en los que se desarrolla IRA pronto después del trasplante de médula desarrollan enfermedad hepática veno-oclusiva.30 La patogenia de este trastorno es incierta.
Las características clínicas de la enfermedad veno-oclusiva se vuelven aparentes en un lapso de una semana después de la infusión del trasplante, desarrollándose un estado de retención de sodio que causa edema y en ocasiones ascitis. Casi siempre existe disfunción hepática concomitante, que se manifiesta por hiperbilirrubinemia progresiva. Alrededor de siete a 14 días después se desarrolla azoemia, precedida por retención urinaria de sodio (el UNa suele ser < 20 mEq/L). El aumento en las concentraciones plasmáticas de creatinina y urea suele presentarse después de alguna lesión coadyuvante, como sepsis.
Se observa mayor riesgo de enfermedad veno-oclusiva en pacientes con hepatopatía previa, en quienes sufren fiebre durante el tratamiento citorreductivo y en los que reciben ciertos medicamentos (v.gr., estrógenos, progestágenos, anfotericina B y metotrexate).30 Aunque la enfermedad veno-oclusiva puede ser severa y progresar al estado de coma y muerte, también ocurren formas más leves.
Necrosis tubular aguda Los pacientes con cirrosis hepática pueden desarrollar NTA después del tratamiento con aminoglucósidos, la administración de medios de contraste o un episodio de sepsis o hemorragia. Casi siempre puede sospecharse NTA por la historia clínica y por el incremento rápido en la Pcr.
Algunos métodos de laboratorio que se usan en forma tradicional para distinguir la enfermedad prerrenal de la NTA pueden no ser útiles en el paciente con enfermedad hepática. La FENa puede ser menor de uno porciento en pacientes con cirrosis por la isquemia renal persistente inducida por la hepatopatía. Los resultados del examen de orina también pueden ser dudosos porque pueden observarse cilindros granulares en pacientes que solo tienen hiperbilirrubinemia importante, por lo que no son específicos de NTA.31
Enfermedad prerrenal El SHR es una forma de enfermedad prerrenal porque en ambas condiciones los riñones son histológicamente normales y pueden usarse con éxito para el trasplante renal.20 Sin embargo, la enfermedad prerrenal también puede ser inducida por pérdidas de líquidos gastrointestinales, hemorragia o tratamiento con diuréticos o AINE (en vista de que las prostaglandinas vasodilatadoras renales mantienen en parte la perfusión renal en estas circunstancias).32 Por lo tanto, para hacer el diagnóstico de SHR debe demostrarse la ausencia de mejoría de la función renal después de suspender agentes potencialmente nefrotóxicos y de realizar una prueba de hidratación.
Es importante distinguir el SHR de otras formas de enfermedad prerrenal porque el pronóstico es muy diferente. La NTA y las otras causas de enfermedad prerrenal suelen ser reversibles. Por el contrario, el pronóstico de los pacientes con SHR es malo: la mayoría de los enfermos fallece en semanas después del inicio de la insuficiencia renal.29,33 La encefalopatía hepática concomitante es frecuente, y la muerte casi siempre se debe a una complicación de la hepatopatía, como hemorragia gastrointestinal.
Tratamiento
En los pacientes con cirrosis hepática la VFG disminuye al progresar la enfermedad17 (ver adelante). En los estadios iniciales de la enfermedad, la administración intravenosa del agente simpaticolítico clonidina disminuye en forma aguda el tono simpático renal y la resistencia vascular, lo que puede aumentar la VFG hasta en un 25 porciento. Sin embargo, a pesar de la reducción persistente en la actividad simpática, el tratamiento a largo plazo por vía oral no parece aumentar en forma sostenida la VFG.34
Puede ocurrir mejoría de la insuficiencia renal si la función hepática mejora como resultado de la resolución parcial de la enfermedad primaria o del trasplante hepático exitoso.26,35 Por el contrario, la supervivencia en diálisis suele ser limitada por la gravedad de la insuficiencia.36 No existen evidencias hasta el momento de que ningún medicamento aumente en forma persistente y significativa la función renal en pacientes con SHR.25
Azoemia prerrenal causada por insuficiencia cardiaca congestiva
La insuficiencia cardiaca congestiva (ICC) se asocia con dos alteraciones principales en la función renal: retención de sodio en etapas tempranas de la enfermedad y disminución en la VFG al empeorar la función cardiaca. Los factores neurohumorales y ciertos tratamientos pueden contribuir a estos problemas.37
INSUFICIENCIA RENAL
Los pacientes con ICC pueden también desarrollar IRA o crónica como resultado de la menor perfusión tisular. La disminución en la función renal se asocia con los datos característicos de la enfermedad prerrenal: aumento en la relación entre el BUN y la creatinina plasmática, examen general de orina normal y UNa bajo (< 20 mEq/L), a menos que se hayan administrado diuréticos.
Patogenia
Diversos mecanismos pueden causar insuficiencia renal en los pacientes con ICC, cada uno de los cuales requiere un tratamiento diferente [ver tabla 4].
|
||||||||
|
Uso excesivo de diuréticos La administración de diuréticos causa pérdida de líquido y disminución de la presión intravascular. Estos efectos son benéficos porque permiten movilizar el edema pulmonar y periférico, mejorando los síntomas. Sin embargo, la disminución en la presión de llenado ventricular izquierdo causa con frecuencia disminución en el gasto cardiaco.38
Aunque la disminución resultante en la perfusión tisular suele no ser importante desde el punto de vista clínico, el BUN y la Pcr aumentan en algunos pacientes. El tratamiento adecuado en ese momento consiste en evitar por un tiempo el uso de diuréticos y, si el paciente ha tenido una diuresis excesiva, intentar una repleción cuidadosa de líquido. Sin embargo, si persiste el edema, estará indicado el tratamiento vasodilatador para mejorar la función cardiaca.
Antinflamatorios no esteroides Aunque las prostaglandinas renales son principalmente vasodilatadoras, no tienen un papel importante en la regulación de la hemodinámica renal en los individuos normales porque su síntesis basal es relativamente escasa. Sin embargo, la liberación de estas hormonas (en especial prostaciclina y prostaglandina E2) aumenta por los vasoconstrictores angiotensina II y norepinefrina; la secreción de estos vasoconstrictores se incrementa en condiciones de depleción efectiva de volumen, como en la falla cardiaca.8 En estas circunstancias, las prostaglandinas vasodilatadoras actúan conservando la circulación sanguínea renal y la VFG al antagonizar los efectos vasoconstrictores de la angiotensina II y la norepinefrina, en especial en la arteriola aferente.
En estos pacientes, la menor producción de prostaglandinas vasodilatadoras inducidas por los AINE pueden ocasionar falla renal por dos mecanismos: vasoconstricción renal sin oposición causada por la angiotensina II y la norepinefrina, y disminución en el gasto cardiaco secundario al aumento asociado en la resistencia sistémica vascular (un efecto que se opone a la disminución benéfica de la poscarga cardiaca inducida por los vasodilatadores).39 Por lo tanto, la inhibición de la síntesis de prostaglandinas por un AINE puede causar isquemia renal reversible, disminución en la presión hidráulica glomerular (la principal fuerza para la filtración glomerular), e IRA.10,40 El aumento en la Pcr ocurre, en forma típica, entre los primeros tres a siete días de tratamiento, que es el tiempo requerido para lograr concentraciones estables del fármaco y, en consecuencia, máxima inhibición de la síntesis de prostaglandinas.41
Inhibidores de la enzima convertidora de angiotensina Los inhibidores de la enzima convertidora de angiotensina (ECA) se usan mucho en el tratamiento de la ICC. Estos agentes disminuyen la formación de angiotensina II, disminuyendo la resistencia arteriolar y venosa.
Al mejorar el gasto cardiaco y la circulación renal, puede suponerse que la VFG también aumentará. Sin embargo, esto sucede en menos del 10 porciento de los pacientes, y la Pcr incluso aumenta en alrededor del 30 porciento de los mismos.12 Lo más probable es que esta complicación ocurra en los casos en que el mantenimiento de la VFG depende de altas concentraciones ambientales de angiotensina II.12,42 Entre estos casos se incluyen los pacientes en tratamiento con diuréticos a dosis altas y que han tenido una respuesta excesiva a los mismos, los enfermos con hipotensión relativa, que tienen una presión arterial media menor de 65 mm Hg, y los pacientes con concentración plasmática de sodio previa al tratamiento menor a 137 mEq/L, porque este hallazgo es indicativo de activación neurohumoral importante.
Por tanto, la IRA que puede ocurrir en los pacientes con insuficiencia cardiaca se desarrolla por un mecanismo semejante al observado en algunos pacientes con estenosis bilateral de la arteria renal: dilatación preferencial de la arteriola eferente con caída subsecuente de la presión interaglomerular. El nivel basal de función renal puede restablecerse al diminuir la dosis de diuréticos. En algunos individuos, el mantenimiento de la VFG puede ser tan dependiente de la angiotensina II que se requerirá combinar hidralacina y dinitrato de isosorbide (que no relajan en forma preferente la arteriola eferente).43
Es importante mencionar que muchos pacientes con insuficiencia cardiaca avanzada tienen aumento del BUN yla Pcr antes de que se instituya el tratamiento vasodilatador. Esta observación tiene implicaciones terapéuticas importantes porque la posibilidad de lograr una respuesta cardiovascular benéfica es mucho menor en estos casos (< 25 porciento si la Pcr es > 2.8 mg/dl, comparada con 75 porciento cuando la Pcr es < 1.4 mg/dl).44 No se conoce cual es el motivo por el que los pacientes con concentraciones elevadas de creatinina responden menos al tratamiento vasodilatador.
Los enfermos con insuficiencia renal subyacente tienen mayor riesgo de desarrollar hipercalemia después de la administración de un inhibidor de la ECA.45
Empeoramiento de la función cardiaca El empeoramiento de la función cardiaca es la causa final de la enfermedad prerrenal progresiva en la ICC. En muchos pacientes, el diagnóstico será aparente por aumento de los síntomas cardiacos o un infarto al miocardio reciente.
En por lo menos algunos pacientes con ICC, la eliminación extracorpórea de líquido por medio de ultrafiltración aislada puede ser útil. En un estudio, la eliminación de alrededor de 1,880 ml de líquido en tres horas causó mejoría clínica y de laboratorio.46 El mecanismo por el que se logra este beneficio no es claro, pero la persistencia de la mejoría durante varias semanas o más en algunos pacientes sugiere que el procedimiento depleta un cardiodepresor circulante. Debido a que no es posible identificar a los pacientes con ICC que tienen más posibilidad de responder a la ultrafiltración, la aplicación diseminada de esta técnica sería un tanto prematura.
Necrosis tubular aguda
La necrosis tubular aguda es una de las dos causas más frecuentes de IRA, siendo la otra la enfermedad prerrenal. El daño tubular característico de este padecimiento representa una respuesta inespecífica que puede observarse con diversas lesiones renales, incluyendo isquemia renal y exposición a nefrotoxinas endógenas y exógenas. El efecto neto consiste en una disminución rápida de la función renal que, en muchos pacientes , requiere de un periodo variable de diálisis antes de que ocurra la resolución espontánea.
Los cambios histológicos principales de la NTA son dos: (1) necrosis tubular con desprendimiento de células epiteliales y (2) oclusión de la luz tubular con cilindros y detritos celulares (incluyendo el borde en cepillo de las células tubulares proximales y la mucoproteína de Tamm-Horsfall liberada de las células dañadas en la porción gruesa de la rama ascendente del asa de Henle) [ver figura 3].47 Estos cambios suelen ser en parches, y parecer relativamente leves en relación con la gravedad de la insuficencia renal. Sin embargo, es importante recordar que muchas nefronas drenan en un sólo túbulo colector cortical, por lo que la obstrucción de un pequeño número de ellos puede causar disfunción renal grave.
|
|
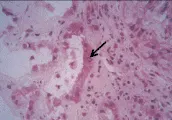
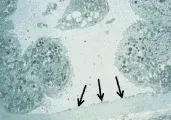
Necrosis tubular aguda posisquémica
ETIOLOGIA
La IRA posisquemia, generalmente causada por NTA, es una complicación no poco frecuente de la isquemia grave o la cirugía mayor. Los pacientes posoperados tienen mayor riesgo de IRA posisquémica por la depleción preoperatoria de líquidos, la anestesia y las pérdidas transoperatorias,48 aunque la mayoría de los pacientes tolera bien estos cambios. La posibilidad de daño tubular aumenta si existe una lesión adicional, como hemólisis o hipotensión.
Ciertos procedimientos quirúrgicos parecen asociarse a un riesgo mucho mayor de NTA. La cirugía de un aneurisma de la aorta abdominal puede provocar NTA porque ocurren periodos de isquemia renal total cuando se requiere pinzar la arteria a nivel suprarrenal.48 La cirugía para corregir la ictericia obstructiva se asocia con una reducción en la VFG del doble comparado con otras formas de cirugía abdominal, y es posible que la absorción de endotoxina del intestino tenga algún papel al respecto. La cirugía cardiaca también predispone a la NTA. En este caso, la combinación de cardiopatía previa, hipotensión y hemólisis durante la derivación cardiopulmonar pueden ser todos factores agravantes.
Además de poscirugía, puede ocurrir NTA posisquémica en pacientes con sepsis o hipovolemia grave. Incluso los grados moderados de depleción de volumen pueden aumentar el riesgo de falla renal en presencia de una nefrotoxina, como medio de contraste. Por el contrario, la NTA parece ser una complicación poco frecuente en pacientes con ICC, incluso en los que tienen grados moderados de hipotensión sistémica.49
PATOGENIA
El daño isquémico en la NTA suele ser más severo en el principio del túbulo proximal y en los segmentos medulares externos del riñón, incluyendo la porción gruesa de la rama ascendente del asa de Henle.50,51 Las células tubulares distales casi no sufren daño importante.52
Las células tubulares proximales parecen ser especialmente susceptibles a la isquemia, también la médula renal es susceptible porque en general se encuentra en medio hipóxico (PaO2 < 15 mm Hg). Esta hipoxia basal en la médula renal es causada por la combinación de un bajo flujo sanguíneo medular y el mecanismo de contracorriente de los vasos rectos, que proporcionan sustratos y oxígeno al asa de Henle50,53 [ver figura 4].
|
| Figura 4 |
| Difusión tubular de oxígeno |
La mala oxigenación causa diversos factores secundarios que promueven el desarrollo de lesión a las células tubulares y menor filtración glomerular. Los eventos más importantes causados por la isquemia o hipoxia son menor energía en las células tubulares, acúmulo de calcio intracelular, generación de radicales de oxígeno, aumento en la actividad de la fosfolipasa intracelular, desprendimiento y adhesión celular, y alteraciones en la hemodinámica glomerular.54,55
Ocurre depleción de la energía de las células tubulares durante casi todos los tipos de insuficiencia renal experimental55 lo que causa reducción en las reservas de trifosfato de adenosina (ATP) celular. Es posible que la pérdida de potasio intracelular inducida por la hipoxia contribuya a este proceso.56 Como consecuencia de la depleción de ATP se inhibe la Ca2+-ATPasa y la Na+-K+-ATPasa, lo que ocasiona acúmulo intracelular de calcio.55 Sin embargo, no se sabe con certeza si el acúmulo de calcio contribuye o solo es consecuencia de la lesión celular.
Los radicales libres de oxígeno, metabolitos reactivos producidos después de la isquemia o hipoxia cuando se reinicia el aporte de oxígeno,55 causan lesión celular al dañar las membranas lípidas, alterando la permeabilidad de la membrana, los procesos enzimáticos y la bomba de iones. Otro factor que causa disfunción celular es la mayor actividad de la fosfolipasa intracelular. Las fosfolipasas son enzimas que convierten los fosfolípidos en ácidos grasos libres, que tienen acción detergente tóxica55 y pueden alterar las membranas celulares.
El hallazgo de células tubulares renales desprendidas y viables en los diferentes modelos de IRA ha originado el estudio del papel de la adhesión de las células epiteliales a la membrana basal en la fisiopatología de la NTA.55 Este proceso puede, en parte, afectar la redistribución inducida por estrés de los receptores de integrina de la membrana basolateral, sitio donde ocurre la adhesión a la matriz en condiciones normales, a la membrana luminal. La obstrucción intratubular por las células desprendidas y detritos celulares es un resultado importante de estos procesos fisiopatológicos. La obstrucción de solo algunos túbulos colectores puede asociarse con NTA severa porque muchos túbulos de diferentes nefronas pueden vaciarse en cada túbulo colector.
Entre los diferentes factores que pueden participar en la reducción de la VFG en la NTA se encuentran la obstrucción tubular (ver antes) y el escape del filtrado a través de las membranas tubulares dañadas. Además, son importantes los procesos que alteran la hemodinámica glomerular. Por ejemplo, la reabsorción defectuosa de NaCl en cualquier sitio proximal a la mácula densa (en donde el túbulo distal se encuentra con la arteriola aferente) puede desencadenar una reducción de la VFG por retroalimentación para conservar sal y agua. Este proceso se ha demostrado en varias formas de IRA.55 Las sustancias vasoactivas como la adenosina, las prostaglandinas, el óxido nítrico y la endotelina, parecen tener también un papel importante en la alteración de la hemodinámica glomerular.
PREVENCION
Se ha intentado conservar la función renal en pacientes con riesgo de NTA por dos mecanismos generales: conservando la viabilidad celular y previniendo o revirtiendo la obstrucción intratubular. Aunque varias estrategias farmacológicas preventivas han demostrado ser eficaces en modelos animales experimentales, la aplicación de estas estrategias a los humanos no se ha demostrado aún.
Estudios no controlados han demostrado que los pacientes que responden al manitol o al furosemide y dopamina con un aumento en la diuresis tienen mejor evolución que los que no responden a este tratamiento.57-59 Sin embargo, los que respondieron quizá solo tuvieron una enfermedad menos grave, manifestada por menor duración de la oliguria (menos de 24 horas), mayor diuresis y mayor Uosm (sugiriendo una mayor conservación de la función tubular). Aún más, algunos estudios han puesto en duda el beneficio de la dopamina sola en estos casos60 o han sugerido que la administración de este agente tiene un efecto dañino sobre el lecho esplácnico.61
Ni el péptido auricular natriurético (PAN) ni los bloqueadores de los canales del calcio han sido aún evaluados en humanos en estudios controlados. Sin embargo, el tratamiento con bloqueadores del calcio es más eficaz en los modelos en animales si se administra antes o durante el episodio isquémico, más que después de que ha iniciado la insuficiencia renal. Por lo tanto, su principal utilidad en los humanos puede ser conservar la función en los aloinjertos renales, mas que minimizar la gravedad de la NTA posisquémica.
Los únicos estudios controlados en humanos se han realizado durante la fase de NTA establecida, no en la fase isquémica temprana. En este momento la perfusión tisular se ha restablecido, y ya existe la mayor parte del daño tubular. Por lo tanto, no es sorprendente que aumentar la diuresis con un diurético de asa no tenga efecto sobre la gravedad o duración de la insuficiencia renal.58 Al parecer, el diurético de asa aumenta la diursis en las pocas nefronas que aún funcionan, pero existe muy poco reclutamiento de las nefronas no funcionantes previamente. Aún más, las altas dosis de diuréticos de asa que se requieren para inducir la diuresis pueden causar sordera, que puede ser permanente.58
Sin embargo, las evidencias experimentales sugieren que la duración de la enfermedad establecida puede acortarse por medio de tratamiento farmacológico específico. Por ejemplo, los factores de crecimiento o la combinación de PAN y dopamina pueden ser benéficos [ver adelante, Necrosis tubular aguda posisquémica, Tratamiento].
PRESENTACION CLINICA Y EVOLUCION
La disminución de la función renal en la NTA tiene un inicio variable. Suele comenzar en forma súbita después de un episodio de hipotensión, rabdomiolisis, o de la administración de medios de contraste. En cambio, cuando la causa es nefrotoxicidad por aminoglucósidos, el inicio es más insidioso, aumentando la concentración plasmática de creatinina en forma lenta después de siete o más días de tratamiento. Algunos pacientes con enfermedad prerrenal importante pueden también tener una evolución diferente, y los hallazgos urinarios muestran una transición gradual entre los índices típicos de enfermedad prerrenal (UNa baja, FENa baja y examen general de orina normal) y los característicos de NTA (FENa alta y cilindros granulares y epiteliales en la orina).
Una vez que comienza la insuficiencia renal, el BUN y la Pcr aumentan alrededor de 10 a 25 mg/dl y 0.5 a 2.5 mg/dl cada día respectivamente. Sin embargo, el aumento en el BUN puede alcanzar 50 mg/dl al día o más en los pacientes hipercatabólicos (en especial en los que reciben soluciones parenterales de aminoácidos), la hipercalemia grave también es más frecuente en estos casos.
NTA oligúrica contra no oligúrica
Aunque la VFG puede disminuir hasta niveles muy bajos en los pacientes con NTA, este cambio no siempre se asocia con una reducción paralela en la diuresis, que puede variar desde niveles oligúricos (< 400 a 500 ml/día) hasta valores relativamente normales.3,62 Esta diferencia en la diuresis puede ser causada por uno de dos factores: los pacientes no oligúricos pueden tener una VFG más alta que los oligúricos, o los pacientes no oligúricos pueden tener la misma VFG pero reabsorber menos líquido en los túbulos. Por ejemplo, si la VFG cae a 7 L/día (5 ml/min), la diuresis puede ser relativamente normal, de 1 a 2 L, si se reabsorben solo 5 a 6 L de líquido.
Estudios en modelos animales han demostrado que ocurre menos daño morfológico y funcional en la IRA no oligúrica que en la oligúrica.63 También existen evidencias en humanos que sugieren que la ausencia de oliguria en la NTA refleja por lo general enfermedad menos severa.3,62 Por ejemplo, en un estudio que comparó a pacientes no oligúricos con oligúricos, los pacientes no oligúricos tuvieron una Pcr máxima más baja (6 y 9 mg/dl, respectivamente) y menor necesidad de diálisis durante la fase aguda de la enfermedad (28 y 84 porciento, respectivamente).62
Un aspecto que aún no se resuelve es si es posible mejorar el pronóstico renal en la NTA establecida corrigiendo la oliguria con un diurético de asa o manitol. Aunque estos agentes pueden prevenir la lesión tubular si se administran durante o poco después de que ocurre la lesión isquémica, las dosis altas de diuréticos de asa no acortan la duración de la insuficiencia renal o disminuyen la necesidad de diálisis en pacientes tratados comparando con pacientes no tratados que permanecen oligúricos.58 En esta fase tardía de la enfermedad, la lesión isquémica está casi completa, las nefronas dañadas en forma seria son no funcionantes y el aumento en la diuresis es causado por disminución en la reabsorción tubular en las nefronas funcionantes restantes, y no por el reclutamiento de las nefronas que antes no funcionaban (ver antes).
Duración de la necrosis tubular aguda
La fase de insuficiencia renal en la NTA isquémica suele durar de siete a 21 días,51 y la mayoría de los enfermos recuperan su función renal previa. Sin embargo, la duración es muy variable, y depende de la duración y gravedad del episodio isquémico inicial y de la presencia o ausencia de isquemia recurrente.51
Como un ejemplo, los pacientes sometidos a pinzamiento aórtico suprarrenal para cirugía de aneurismas aórticos sufren 20 a 80 minutos de isquemia renal total. Aunque es frecuente que exista evidencia clínica de NTA en el posoperatorio, la función renal suele retornar a la normalidad en dos a tres días, porque la isquemia es breve y no recurrente.48,51 En forma similar, la evolución típica de la insuficiencia renal aguda inducida por medios de contraste es de tres a cinco días, al parecer porque el daño renal es muy breve.
TRATAMIENTO
Además de la corrección del problema subyacente (como la suspensión de un aminoglucósido), el tratamiento de la NTA establecida es sobre todo de sostén. En especial, debe ponerse atención a conservar el balance hídrico y electrolítico, y a mantener una nutrición adecuada. Sin embargo, a pesar de que el manejo sea óptimo, muchos pacientes requieren un periodo temporal de diálisis.
Dopamina en dosis bajas
La dopamina es una hormona natriurética que aumenta la excreción de sodio al disminuir la reabsorción, principalmente en el túbulo proximal.64 Cuando se infunde en dosis bajas (0.5 a 3.0 µg/kg/min), también es un vasodilatador renal que dilata las arterias interlobulares y las arteriolas aferentes (preglomerulares) y eferentes (posglomerulares).59 El efecto neto es un incremento relativamente grande en el flujo renal con menos o ninguna elevación en la VFG.65 La ausencia relativa de aumento en la VFG es causada por dilatación eferente, que minimiza el aumento en la presión intraglomerular. En dosis más altas (> 5 µg/kg/min), la dopamina induce vasoconstricción renal, una respuesta que es mediada por la activación de receptores alfa-adrenérgicos.59
Estos datos han originado el uso frecuente de dopamina en dosis bajas (la llamada dosis renal) (0.5 a 3.0 µg/kg/min) tanto para aumentar la diuresis como para preservar la función renal en pacientes oligúricos con riesgo de NTA posisquémica.59 Aunque la dopamina puede aumentar la diuresis y el flujo sanguíneo renal, en la actualidad existen pocas evidencias de que la dopamina induce un efecto protector renal cuando se usa sola.
Además, existen pocos estudios controlados sobre el efecto de la dopamina en humanos.59 En un estudio pequeño no controlado, se administró dopamina en dosis bajas (3 µg/kg/min) con diurético de asa (furosemide) a pacientes oligúridos en las primeras 48 horas después de una lesión isquémica.57 Los pacientes que respondieron con diuresis, comparado con los que no respondieron, tuvieron una recuperación más rápida de la función renal (10 contra 41 días), menor posibilidad de requerir diálisis aguda y fueron tratados antes (17 contra 33 horas). Sin embargo, los que respondieron tuvieron también evidencia de menos lesión tubular (diuresis más alta y menor UNa) y quizá hubieran tenido también mejor evolución sin tratamiento.
En un estudio controlado de 37 pacientes sometidos a reparación aórtica electiva o injerto aortobifemoral, los pacientes recibieron dopamina en dosis bajas o placebo.66 En esta pequeña población, la dopamina no confirió mayor protección renal que el placebo.
En resumen, en la actualidad no existen datos clínicos que apoyen la hipótesis de que la dopamina tiene un efecto protector renal, en especial después de que se establece la NTA. Algunos médicos administran dosis bajas de dopamina con un diurético de asa si el tratamiento se comienza temprano y si el paciente no ha tenido respuesta a la administración previa de un diurético de asa, manitol o ambos. Sin embargo, este esquema no debe continuarse si la diuresis no aumenta en forma significativa.59
Manitol
Con frecuencia se administra manitol para prevenir la NTA y en ocasiones para aumentar la diuresis en la IRA establecida. Aunque el tratamiento con manitol suele ser bien tolerado, pueden ocurrir diversas complicaciones hídricas, electrolíticas y renales si el paciente no es vigilado con cuidado. Estas complicaciones incluyen depleción de volumen, hipernatremia, expansión de volumen, hiponatremia, hipercalemia y acidosis metabólica.
Además, aunque el manitol se usa como un intento para prevenir la NTA posisquémica, una concentración de manitol muy alta en plasma (> 1,050 mg/dl) puede inducir IRA. Debido a que no se realizan mediciones de niveles de manitol en la mayoría de los hospitales, la concentración puede calcularse usando la brecha aniónica. Este valor puede determinarse restando la Posm calculada de la osmolaridad medida por el laboratorio clínico. Los pacientes con acúmulo importante de manitol (una brecha osmolar > 60 a 75 mOsm/kg) parecen tener mayor riesgo de IRA reversible.67 La IRA está causada principalmente por vasoconstricción renal, aunque también la vacuolización tubular puede influir.
Diuréticos
Los diuréticos (por lo general de asa, como el furosemide) se usan con frecuencia en los pacientes con IRA, en especial en los oligúricos (i.e., con diuresis de < 400 a 500 ml/día). A pesar de que los pacientes con NTA no oligúrica suelen tener un mejor pronóstico que los oligúricos, este dato ha sido mejor estudiado en los pacientes con NTA oligúrica que espontáneamente se vuelven no oligúricos. Por el contrario, existen pocas evidencias de que el pronóstico de recuperación en la NTA establecida mejore por medio de la administración de diuréticos. Aunque estos agentes pueden facilitar el manejo de líquidos, cualquier aumento en la diuresis puede ser causado por mayor excreción de sal y agua en el cinco a 10 porciento de las nefronas que permanecen intactas, y no por el reclutamiento de nuevas nefronas.
Hemodiálisis en la insuficiencia renal aguda
La mortalidad en la IRA suele asociarse con infección o hemorragia. Debido a que estas compliaciones pueden ser exacerbadas por la uremia, se ha sugerido que la evolucón del paciente puede mejorar por medio de la diálisis temprana o más intensiva para mantener el BUN en menos de 80 a 100 mg/dl. Sin embargo, en la mayoría de los estudios la diálisis profiláctica no ha producido ningún beneficio.68 Algunas evidencias sugieren que la diálisis puede en realidad exacerbar la lesión renal y retrasar la recuperación de la función.
Por lo tanto, por lo general la diálisis se inicia para ayudar a resolver los síntomas urémicos o las alteraciones severas de líquidos y electrolidos, más que para lograr una concentración específica de BUN o Pcr. Las indicaciones de diálisis incluyen sobrecarga de líquidos, que puede ser tratada por hemofiltración, hipercalemia, y signos de uremia, como pericarditis, neuropatía o deterioro del estado mental no explicable por otras causas.
La diálisis puede afectar de modo adverso la función renal por tres de los efectos del procedimiento: reducción de la diuresis, inducción de hipotensión y activación del complemento como resultado de la interacción entre la sangre y la membrana de diálisis.69 Los estudios recientes sugieren que este último problema, que puede ocasionar acúmulo de neutrófilos en varios órganos, incluyendo los pulmones y riñones, puede ser de gran importancia. Por ejemplo, el uso de cartuchos de diálisis con membranas celulósicas bioincompatibles que activan el complemento, causó peor evolución que el uso de cartuchos con membranas no celulósicas biocompatibles que no tienen este efecto.70,71 La recuperación de la IRA ocurre en forma más temprana y más completa cuando se usan membranas no celulósicas biocompatibles, y la mortalidad general puede ser menor. En consecuencia, se recomienda el uso de estas membranas cuando los pacientes con IRA requieren hemodiálisis.
Hemofiltración continua
Debido a la velocidad más lenta de remoción de solutos, la hemofiltración continua (también conocida como tratamiento de remplazo renal continuo) puede ser preferible a la hemodiálisis convencional en pacientes hemodinámicamente inestables con IRA severa, en especial en casos de sobrecarga severa de volumen con edema masivo. Una vez que se toma la decisión de usar hemofiltración continua, se requiere un acceso arteriovenoso o venovenoso. Para establecer el primero se coloca un catéter arterial de modo que la presión arterial sistémica dirija la sangre a través del hemofiltro y se coloca un catéter venoso para el retorno de la misma. En el acceso venoso se emplean dos catéteres venosos o una de doble luz. Debido a que la presión venosa es menor, se requiere una bomba sanguínea extracorpórea (de diálisis) para hacer circular la sangre a través del hemofiltro.
Con la hemofiltración continua, a diferencia de la diálisis, no ocurre eliminación de solutos por difusión. Por lo tanto, los solutos pequeños (como la urea) se eliminan por ultrafiltración en la misma concentración en la que existen en el plasma. Esta relación se mantiene hasta que se alcanza el limite de poros de la membrana. La convección durante la hemofiltración causa una mayor eliminación de moléculas grandes que la difusión durante la diálisis. Sin embargo, no es claro si esta mayor eliminación de compuestos de mayor tamaño y potencialmente más toxicos es benéfica para el paciente. Aunque el control metabólico (v.gr., reducción del BUN) puede mejorar con la hemofiltración continua,72 la mayoría de los estudios sugieren que los pacientes que se someten a hemofiltración o a diálisis tienen evoluciones semejantes.73,74
Tratamientos experimentales: Factores de crecimiento y péptido auricular natriurético
Los estudios experimentales sugieren que las tasas de regeneración tubular y recuperación funcional en la NTA pueden acelerarse por la administración de factores de crecimiento o de la combinación de PAN y dopamina. La regeneración tubular después de la lesión isquémica se asocia con la activación de ciertos genes y la liberación de factores de crecimiento que son necesarios para el crecimiento celular.75 Estas observaciones tienen utilidad clínica potencial: aunque la expresión del factor de crecimiento semejante a insulina (FCI-1) y del factor de crecimiento epidérmico (FCE) no parece estar aumentada en el riñón durante la recuperación de la NTA experimental inducida por aminoglucósidos,76 la administración exógena de estos factores de crecimiento puede acelerar tanto la regeneración tubular como la recuperación de la función renal en animales de experimentación con IRA posisquémica o nefrotóxica.77,78 La aplicación de estos datos a la enfermedad en humanos aún no es un hecho.
En animales de experimentación, la combinación de PAN y dopamina administrada para prevenir la hipotensión reduce el grado de insuficiencia renal cuando se administra en forma concomitante a la lesión isquémica. Esta combinación se ha evaluado también en la NTA establecida cuando se administra dos días después del episodio isquémico.79 En este estudio, la Pcr en animales controles aumentó del valor basal de 0.7 mg/dl a 1.7 mg/dl el segundo día, para el día 4 había aumentado a 2.8 mg/dl. En los animales tratados con PAN y dopamina, la Pcr tuvo una evolución semejante al segundo día, pero disminuyó a 1.2 mg/dl para el día 4. El aumento en la VFG se debió principalmente al aumento de la presión intraglomerular inducido por dilatación arteriolar. Los animales tratados tuvieron también menos necrosis tubular y cilindros tubulares, lo que sugiere que se facilitó la recuperación tubular. No se conoce aún el mecanismo de recuperación ni su aplicabilidad en humanos. En la actualidad se realizan estudios para evaluar la eficacia del PAN y la NTA establecida en humanos, pero no se han obtenido aún resultados positivos constantes.
Necrosis tubular aguda causada por nefrotoxinas
AMINOGLUCOSIDOS
La IRA causada por necrosis tubular es una complicación relativamente frecuente del tratamiento con aminoglucósidos, asociándose con un aumento en la Pcr mayor de 0.5 a 1 mg/dl en el 10 al 20 porciento de los pacientes.80,81 Los aminoglucósidos se filtran en forma libre, y casi todo el fármaco se excreta, captándose y almacenándose una pequeña fracción en las células tubulares, lo que ocasiona el daño, en especial en el túbulo proximal. Puede ocurrir IRA incluso si se vigilan con cuidado las concentraciones del fármaco.82
Parece ser que el número de grupos amino catiónicos (NH3+) por molécula es un determinante importante de nefrotoxicidad.80,83Por lo tanto, entre los aminoglucósidos, la neomicina (seis grupos amino por molécula) causa el mayor daño renal y la estreptomicina (tres grupos por molécula) el menor. La gentamicina (cinco grupos), tobramicina (cinco grupos), netilmicina (cuatro grupos) y amikacina (cuatro grupos) tienen toxicidad intermedia.81 Aunque las publicaciones iniciales sugerían que la gentamicina era más nefrotóxica que la tobramicina, los enfermos con sepsis o hipotensión fueron excluidos de estos estudios en un intento por eliminar otras causas potenciales de insuficiencia renal aguda. En esta población seleccionada, la gentamicina era más nefrotóxica, pero el daño renal era leve y la concentración plasmática máxima de creatinina era menor de 2 mg/dl en casi todos los pacientes.82 Cuando se incluyeron a todos los grupos de pacientes, parece que no existió diferencia en la nefrotoxicidad de la gentamicina y la tobramicina.84,85
Manifestaciones clínicas y diagnóstico
El daño renal inducido por aminoglucósidos se asocia con acúmulo preferencial en la corteza renal. Los aminoglucósidos se filtran y después son captados en forma parcial y almacenados, induciendo daño a las células tubulares proximales. El almacén prolongado de los aminoglucósicos (hasta 28 días para una sola dosis) explica la observación de que la insuficiencia renal puede volverse clínicamente aparente hasta varios días después de que se han eliminado estos medicamentos.
También pueden afectarse los segmentos más distales de la nefrona. Las dos principales manifestaciones de disfunción distal son la poliuria, causada por disminución en la capacidad de concentración y la hipomagnesemia, causada por aumento en las pérdidas urinarias.80 La depleción de magnesio puede causar hipocalemia e hipocalcemia secundarias. El tratamiento de la pérdida de magnesio inducida por aminoglucósidos consiste en la administración de suplementos orales de magnesio. Sin embargo, la eficacia de este tratamiento está limitado por la excreción urinaria de la mayoría del magnesio que se administre.
La mayoría de los enfermos con NTA inducida por aminoglucósidos son no oligúricos, quizá por la alteración concomitante en la capacidad de concentración.86 El diagnóstico de este padecimiento se realiza con base en el antecedente de insuficiencia renal aguda que comenzó más de cinco días después de la institución del tratamiento aminoglucósido, un sedimento urinario normal o que muestra cilindros celulares granulares y epiteliales, y una FENa mayor de uno porciento.
Factores predisponentes
Se han identificado diversos factores que aumentan el riesgo de nefrotoxicidad por aminoglucósidos.80
Duración del tratamiento e isquemia renal El desarrollo de insuficiencia renal suele requerir por lo menos siete días de tratamiento con aminoglucósidos. Sin embargo, en presencia de isquemia renal concomitante, el daño combinado puede causar insuficiencia renal en uno a dos días, incluso cuando ninguno de los factores aislados sea suficiente para causar daño tubular.87,88
Frecuencia de las dosis y niveles en plasma La vigilancia cuidadosa de las concentraciones plasmáticas es un componente importante del tratamiento con aminoglucósidos, aunque puede ocurrir deterioro de la función renal incluso cuando las concentraciones plasmáticas del fármaco se encuentran dentro del rango normal.50 Las concentraciones máximas basales (> 2 a 3 µg/dl) suelen ser una guía útil de la función renal porque son la primera evidencia clínica de una reducción en la VFG. Por otro lado, la eficacia terapéutica se relaciona con la concentración máxima. Cuando se administran dosis divididas existe una diferencia estrecha entre el rango terapéutico y el rango tóxico.90
En comparación, estudios en animales de experimentación han demostrado que la incidencia de IRA disminuye cuando se administra el medicamento en una dosis alta una vez al día.89,91,92 Las concentraciones máximas más altas logradas con el tratamiento en bolo parecen ser suficientes para promover la destrucción bacteriana, a pesar de que los niveles basales circulantes sean relativamente bajos durante la mayor parte del día.92
Varios estudios sugieren que estos datos pueden aplicarse a los humanos.89,93,94 En un estudio, 67 pacientes con infecciones serias fueron distribuidos para recibir gentamicina en dosis de 1.33 mg/kg tres veces al día o a 4 mg/kg una vez al día.93 Se encontró que el esquema de un día fue por lo menos tan eficaz como la dosificación múltiple para controlar la infección: se observó una buena respuesta clínica en el 91 porciento de los pacientes en este grupo y en el 78 porciento de los que recibieron dosis múltiples. La incidencia de nefrotoxicidad (definida como un aumento en la Pcr de por lo menos 0.5 mg/dl) fue mucho menor en los que recibieron una sola dosificación, ocurriendo en el cinco porciento de estos pacientes contra el 24 porciento de los que recibieron dosis múltiples. No existió diferencia en la incidencia de ototoxicidad.
Un estudio mucho más extenso evaluó el tratamiento de la infección en 667 pacientes con cáncer y granulocitopenia.94 De nuevo, el tratamiento con amikacina en una dosis (20 mg/kg) fue tan eficaz como las dosis diarias múltiples (6.5 mg/kg cada ocho horas). La incidencia de nefropatía fue semejante en los dos grupos (tres porciento y dos porciento en los grupos de una y múltiples dosis, respectivamente). Sin embargo, el diseño del estudio minimizó la posibilidad de lesión renal porque los pacientes eran jóvenes (en promedio 28 años de edad) y la duración del tratamiento corta (en promedio ocho días) A pesar de estas limitaciones, existieron indicios de que la nefrotoxicidad producida por el tratamietno con una dosis fue menos severo que la causada por el esquema múltiple. La enfermedad renal ocurrió después (10 contra siete días) y se asoció con un incremento promedio menor en la Pcr (0.9 contra 1.4 mg/dl) en los pacientes que recibieron el esquema de una sola dosis.
Además de disminuir la gravedad de la nefrotoxicidad, la administración de una sola dosis al día (v.gr., gentamicina en dosis de 4 a 5 mg/kg) garantiza los niveles máximos, también requiere menos pruebas de sangre porque solo es necesario vigilar los niveles mínimos.95 Los estudios sobre una sola dosis al día aún son limitados, pero ninguno ha dado resultados peores a los del uso de aminoglucósidos en dosis divididas.89
Sepsis y otras nefrotoxinas Además de causar depleción de volumen, la endotoxina aumenta la nefrotoxicidad de los aminoglucósidos. Aunque la menor perfusión renal causada por vasoconstricción puede contribuir al desarrollo de nefrotoxicidad inducida por aminoglucósidos en pacientes sépticos, la endotoxina parece también asociarse con mayor acúmulo del medicamento en los túbulos. No se sabe como ocurre este fenómeno.
Ocurren efectos semejantes cuando se administra otro agente nefrotóxico con el aminoglucósido. Por ejemplo, puede ocurrir reducción aguda en la función renal casi con el doble de frecuencia cuando se administran un aminoglucósido y vancomicina que cuando se administra un aminoglucósido solo [ver adelante, Vancomicina].
Enfermedad hepática La enfermedad hepática, en especial la ictericia obstructiva con concentración plasmática mayor de 5 mg/dl, predispone a la insuficiencia renal aguda asociada con aminoglucósidos.81,96 No se conoce el mecanismos de esta interacción, pero la bilirrubina o la endotoxina (que se absorbe por la menor entrada de sales biliares a la luz intestinal) puede aumentar la sensibilidad de las células tubulares renales a la toxicidad por los aminoglucósidos.96
Posible papel de las cefalosporinas y penicilinas Los estudios iniciales sugerían que la cefalotina aumentaba la nefrotoxicidad por aminoglucósidos.97 Sin embargo, en estos estudios se excluyó a los pacientes con sepsis o hipotensión. En esta población limitada la cefalotina sí tuvo un efecto potencializador, pero el daño renal fue leve y la Pcr permaneció menor de 2 mg/dl en casi todos los pacientes.97 Además, parece ser que no existe un efecto tóxico aditivo si se incluye a todos los pacientes, lo que simula con más exactitud la práctica clínica.80 Por último, no es claro si los resultados iniciales con cefalotina pueden extrapolarse a otras cefalosporinas. Las evidencias preliminares sugieren que la administración concomitante de una cefalosporina no afecta la nefrotoxicidad por aminoglucósidos y que los dos tipos de medicamentos pueden administrarse juntos son seguridad.84
Las evidencias sugieren que la administración concomitante de una penicilina puede tener incluso un efecto protector.83 Sin embargo, debe mencionarse que las penicilinas, en especial la carbenicilina, piperacilina y ticarcilina, pueden formar complejos físicos con los aminoglucósidos, y que estos agentes no pueden administrarse juntos en la misma infusión intravenosa.
Evolución y tratamiento
El tratamiento inicial de la nefrotoxicidad por aminoglucósidos es de sostén y consiste en suspender el aminoglucósido y mantener el equilibrio hidroelectrolítico. La recuperación, con retorno de la función renal al nivel previo, suele ocurrir antes de 21 días después de que se suspende el tratamiento con aminoglucósidos. Sin embargo, ésta puede retrasarse si el paciente permanece hipovolémico, séptico o catabólico, y en este caso no puede ocurrir regeneración tubular.
Como en otras formas de IRA reversible, la normalización de la Pcr e incluso de la VFG pueden no indicar una recuperación completa. La pérdida irreversible de las nefronas puede estar enmascarada por hiperfiltración compensatoria en los glomérulos normales restantes. Además, aunque el daño túbulointersticial es poco común en los pacientes con nefrotoxicidad aguda por aminoglucósidos, puede ocurrir después del tratamiento a largo plazo, incluso si el medicamento se administra en dosis bajas.98
MEDIO DE CONTRASTE
La administración de medio de contraste puede causar una forma reversible de IRA que comienza pronto después de que se administra el agente.99-103 El mecanismo del daño no se conoce bien, pero quizá participen la vasoconstricción renal y la generación de radicales libres del oxígeno.104-106 Como en la mayor parte de los casos de NTA, la FENa suele ser mayor de uno porciento. Sin embargo, no es raro encontrar valores menores (sugestivos de una función intacta) en este padecimiento.101,104
Factores predisponentes
Los estudios prospectivos han demostrado que es frecuente que se presente un pequeño aumento en la Pcr (en promedio de 0.2 mg/dl) después de un estudio por medio de contraste.101 Sin embargo, puede ocurrir deterioro más importante en la función renal, sobre todo si los pacientes tienen uno o más de los siguientes factores de riesgo:99-104 (1) insuficiencia renal subyacente, con concentración plasmática de creatinina mayor de 1.5 mg/dl, (2) nefropatía diabética asociada con insuficiencia renal, (3) insuficiencia cardiaca grave, (4) dosis total de medio de contraste alta, o (5) mieloma múltiple.
Por lo general, la incidencia de un aumento en la concentración plasmática de creatinina de más del 50 porciento sobre el nivel previo, o de más de 1 mg/dl es negligible en pacientes con función renal normal, incluso si son diabéticos.99 La incidencia es de alrededor de cuatro a once porciento en enfermos con insuficiencia renal leve o moderada (con concentración plasmática de creatinina entre 1.5 y 4.0 mg/dl).99,100,102 Sin embargo, este riesgo puede aumentar hasta el 40 porciento si la disfunción renal es más grave, o si se asocian depleción marcada de volumen, insuficiencia cardiaca grave o la realización de múltiples estudios con medio de contraste en un periodo de 72 horas.100 La posibilidad de un aumento significativo en la creatinina plasmática aumenta entre nueve y 38 porciento en enfermos con insuficiencia renal leve a moderada y diabetes mellitus,99,103 y es mayor del 50 porciento cuando la creatinina plasmática basal es mayor de 4 a 5 mg/dl.104, 107
Algunos estudios han demostrado un riesgo de insuficiencia renal dependiente de la dosis. Cuando se compara una dosis baja en pacientes con función renal normal, que se define en forma variable como menor de 125 ml o de menos de 5 ml/kg (hasta un máximo de 300 ml) de medio de contraste con dosis mayores, la incidencia de insuficiencia renal aguda puede ser de dos contra 20 porciento en pacientes solo con insuficiencia renal100,103 y de seis contra 38 porciento en pacientes con insuficiencia renal y diabetes mellitus.103
En casi todos estos pacientes, la disminución en la función renal es leve, temporal y de poca importancia clínica. Sin embargo, algunos pacientes tienen un aumento máximo en la Pcr que puede ser mayor de 5 mg/dl, lo que ocasionalmente requiere de diálisis aguda. Este aumento ocurre con más frecuencia cuando la Pcr basal es mayor de 4 mg/dl.100,104 La insuficiencia renal persistente es rara, y ha sido descrita pricipalmente en pacientes con enfermedad subyacente avanzada (i.e., con creatinina plasmática basal mayor de 8 mg/dl).104,107
Los enfermos con mieloma múltiple también pueden tener mayor riesgo cuando se someten a un estudio con medio de contraste. La deshidratación previa (que promueve la precipitación intratubular de las cadenas ligeras filtradas) y una posible interacción entre las cadenas ligeras y el agente de contraste pueden ser importantes en estas circunstancias.108
Tratamiento y prevención
El mejor tratamiento para la insuficiencia renal inducida por medio de contraste es la prevención. Si es posible, es mejor utilizar ultrasonido o TC sin medio de contraste, sobre todo en los pacientes con alto riesgo, y si no, es conveniente utilizar el medio de contraste a bajas dosis,100,103,104 evitando los estudios repetidos muy seguidos. Cuando sea posible se corregirá la depleción de volumen y se suspenderán los antinflamatorios no esteroideos antes de iniciar los estudios con medio de constraste, porque ambos factores pueden aumentar la vasoconstrición renal.
Aún no se ha definido el papel de los medios de contraste de baja osmolaridad, nuevos y mucho más costosos. Parece que existen pocas ventajas de usar estos agentes comparando con los agentes iónicos en pacientes con función renal normal,102,109 incluso en los que tienen diabetes mellitus.110 En estudios de pacientes con insuficiencia renal moderada (una Pcr de 1.4 a 2.4 mg/dl) los agentes no iónicos de baja osmolaridad se asociaron con baja incidencia de reducción leve a moderada en la función renal, aunque la reducción no fue siempre estadísticamente significativa.109-111 Sin embargo, el beneficio clínico fue pequeño porque ningún paciente tuvo empeoramiento de la función renal. Por lo tanto, el uso de estos agentes puede producir beneficios significativos solo en los pacientes de alto riesgo como diabéticos con una Pcr mayor de 2.0 a 2.5 mg/dl.110-112
Además de estas medidas preventivas, existen evidencias que sugieren que la hidratación inicial puede ser benéfica.113 Por el contrario, no se ha demostrado la utilidad de agregar manitol o diuréticos al esquema de hidratación. Un estudio reciente de diabéticos de alto riesgo mostró que la adición de furosemide o manitol al esquema de hidratación no produjo mejores resultados que los causados por la hidratación con solución salina al 0.45 porciento.113 No se sabe si puede generalizarse esta falta de protección a los no diabéticos.
Por último, un estudio reciente de 39 pacientes sugiere que la caída en la VFG inducida por el medio de radiocontraste puede prevenirse por medio del pretratamiento con teofilina.114 Debido a que la teofilina es un antagonista potente de la adenosina, este efecto puede ser causado por bloqueo de la vasoconstricción que induce la adenosina. Sin embargo, aún no es claro que estos datos puedan aplicarse en general porque los pacientes que se incluyeron en este estudio tuvieron bajo riesgo de IRA (solo 15 porciento tenía diabetes con una concentración plasmática de creatinina de 1.2 mg/dl).
Aunque algunos agentes pueden proteger a los pacientes de la IRA inducida por el medio de contraste, el dextrán de bajo peso molecular (que puede administrarse para prevenir la coagulación después de la aplicación de férulas o injertos coronarios) puede tener el efecto opuesto. Algunos reportes sugieren que el uso de dextrán puede en realizar promover la IRA en estos casos.115
PLATINO
Patogenia y prevalencia
Los compuestos con base de platino son agentes quimioterapéuticos eficaces, pero con potencial nefrotóxico.116 Por ejemplo, el cisplatino provoca disminución progresiva de la función renal. Alrededor del 25 a 35 porciento de los pacientes desarrollan disminución leve y parcialmente reversible en la VFG después del primer episodio de tratamiento. La incidencia y gravedad de la insuficiencia renal aumenta con los tratamientos subsecuentes, hasta volverse irreversible. Por lo tanto, suele estar indicado suspender el tratamiento en los pacientes que experimentan aumento progresivo en la Pcr. Los estudios preliminares sugieren que el carboplatino puede ser menos tóxico para el riñón que el cisplatino; sin embargo, el carboplatino es más tóxico para la médula ósea, sobre todo por la trombocitopenia que produce.116, 117
La patogenia de la nefrotoxicidad por cisplatino incluye daño a las células tubulares. Dentro del riñón, se afecta sobre todo el último segmento S3 del túbulo proximal, aunque también la porción distal de la nefrona. El cisplatino es una toxina tubular potente, en especial en los ambientes con poca concentración de cloruro, como el interior de la célula. Los grupos cloruro en la posición cis de la molécula del cisplatino son remplazados por moléculas de agua, esta reacción es seguida por la formación de radicales hidroxilo muy reactivos, que al parecer producen daño al unirse a sitios nucleofílicos del ADN.116
Además del aumento en la Pcr, en la mitad de los pacientes tratados cisplatino ocurre hipomagnesemia potencialmente irreversible, secundaria a la perdida urinaria de magnesio.118 Los sujetos con función renal normal son capaces de no excretar magnesio en la orina cuando la concentración plasmática de magnesio (PMg) cae por debajo de 1.5 mg/dl (la concentración normal es de 1.7 a 2.1 mg/dl).119 Por lo tanto, una fracción de excreción de magnesio (FEMg) mayor de 2.5 porciento es indicativa de cierta pérdida del mismo.120 El valor de la FEMg puede ser calculado con una fórmula semejante a la de la FENa:
| FEMg | = |
UMg x Pcr ______________________ |
x | 100 |
| (0.7 x PMg) x Ucr |
en donde UMg se refiere a la concentración urinaria (en una muestra aleatoria) de magnesio. La PMg se multiplica por 0.7 porque sólo alredor del 70 porciento del magnesio circulante es libre (i.e., no unido a albúmina) y capaz de filtrarse a través del glomérulo.
La diuresis suele ser de más de 1,000 ml/día en estos pacientes (a menos de que la insuficiencia renal esté muy avanzada), con una Uosm semejante a la Posm. Es probable que este defecto de concentración refleje el daño inducido por el platino en el asa de Henle (en donde se requiere un gradiente de contracorriente para establecer la concentración urinaria) o en los túbulos colectores (el sitio de acción de la HAD).
El cisplatino puede producir una forma diferente de IRA cuando se administra junto con bleomicina: una microangiopatía trombótica con características del síndrome urémico-hemolítico o de púrpura trombocitopénica. Esa complicación parece reflejar una lesión vascular directa, con activación secundaria de plaquetas. El inicio de la insuficiencia renal aguda puede ser súbito o insidioso, en este último caso puede desarrollarse varios meses después de que se ha suspendido el tratamiento. El diagnóstico de esta forma de nefrotoxicidad es sugerido por la presencia de anemia hemolítica microangiopática y trombocitopenia. En los animales de experimentación, las infusiones I.V. de glicina protegen contra la nefrotoxicidad por cisplatino, pero este tratamiento no se ha probado en humanos.120
Tratamiento y prevención
Existe evidencia de que la nefrotoxicidad asociada al tratamiento con cisplatino o carboplatino puede disminuirse con una hidratación vigorosa (250 ml de solución salina por hora) y quizá administrando el fármaco en una solución hipertónica (como 250 ml de solución salina al tres porciento).116,121 La gran concentración de cloro que proporciona la solución salina hipertónica puede disminuir la formación de los compuestos reactivos del platino que produce daño tubular (ver antes), mientras que la solución isotónica o hipertónica puede disminuir la captación de cisplatino por las células tubulares renales (al aumentar el flujo del filtrado).
Los pacientes con tumores intraperitoneales pueden ser tratados con cisplatino o carboplatino intraperitoneal para alcanzar concentraciones locales altas. Se ha administrado tiosulfato intravenoso en forma concomitante para que reaccione en forma covalente y se fije al platino que entra a la circulación sistémica. El complejo resultante no tiene toxicidad sistémica o renal, ni efecto antitumoral;116,122 por tanto, sólo el fármaco intraperitoneal es activo.
GRUPOS HEM
La insuficiencia renal aguda puede ser inducida por la liberación de pigmentos hem, como ocurre en la mioglobinuria secundaria a rabdomiolisis y la hemoglobina ocasionada por hemólisis intravascular.123-125 La insuficiencia renal aguda en estos casos es causada por varios factores, incluyendo obstrucción intratubular por cilindros pigmentados de hem, toxicidad celular inducida por pigmento y hierro (que puede depletar las reservas de ATP126 y generar radicales libres127), constricción y dilatación de las arteriolas aferente y eferente, respectivamente,128 y depleción de volumen causada por secuestro de líquido en el músculo dañado.
Las causas más frecuentes de rabdomiolisis son los traumatismos (incluyendo el daño isquémico al músculo después de una sobredosis de drogas), el alcoholismo, las crisis convulsivas y el golpe de calor por ejercicio, en especial en sujetos sin la capacidad física necesaria o que tienen rasgo de células falciformes.123, 129-131 Causas menos frecuentes de rabdomiolisis son hipocalemia, hipofosfatemia, intoxicación por cocaína, infección por virus de inmunodeficiencia humana (VIH) o administración de zidovudina (también conocida como AZT), síndrome neuroléptico maligno, y uso de un inhibidor de la 3-hidroxi-3-metilglutaril coenzima A (HMG-CoA) reductasa (como el lovastatin) para el tratamiento de la hipercolesterolemia.129-136 El riesgo de rabdomiolisis es bajo cuando se usa lovastatin en forma aislada, pero puede ser hasta del 30 porciento cuando se administra con ciclosporina, y del cinco al ocho porciento si se administra con gemfibrozil.135,136
La sobreproducción importante de hemoglobina o mioglobina causa, en forma típica, coloración roja o café de la orina, a menos de que la excreción del pigmento sea menor por una baja velocidad de filtración glomerular o porque el pigmento sea eliminado del plasma por mecanismos extrarrenales.129 Sin embargo, estos compuestos tienen efectos diferentes, con importancia diagnóstica, sobre el color del plasma. La hemoglobina se filtra muy poco por su gran tamaño (peso molecular del tetrámero de 69 kd y del dímero de 34 kd) y por la unión de proteínas a la haptoglobina. En consecuencia, la hemoglobina alcanza una concentración total en plasma relativamente alta antes de excretarse en la orina, dando un color rojo o café en la cinta reactiva de coloración del plasma. En comparación, la mioglobina es un monómero (con peso molecular de 17 kd) y no se fija a las proteínas, por lo que se filtra y excreta con rapidez. Es por esto que el plasma conserva su color normal en la sobreproducción de mioglobina, a menos de que la insuficiencia renal limite su excreción.
Los pacientes con rabdomiolisis suelen presentar la triada característica de cilindros granulares pigmentados en la orina, coloración rojo o café en el sobrenadante de la orina y aumento marcado en la concentración plasmática de creatina cinasa. También pueden liberarse otros elementos celulares, causando hipercalemia, hiperfosfatemia, hipocalcemia (causada tanto por precipitación de fosfato de calcio en el músculo dañado, como por entrada de calcio a las células musculares isquémicas), hiperuricemia y aumento rápido en la Pcr.123,129 En contraste con los hallazgos de otras formas de NTA, la FENa suele ser menor de uno porciento, un dato que puede reflejar predominio de la obstrucción tubular, más que necrosis tubular.137
La liberación de fosfato de las células musculares dañadas causa con frecuencia confusión en los pacientes que tienen rabdomiolisis inducida por hipofosfatemia porque la hipofosfatemia subyacente puede enmascararse.138 En estos casos, la demostración de una concentración baja de fosfato en plasma antes o después de la máxima destrucción muscular puede ser la única clave para la presencia de depleción de fosfato.
Se ha sugerido que la Pcr aumenta con más rapidez en los pacientes con rabdomiolisis (hasta 2.5 mg/día) que en los que tienen otras causas de IRA. La liberación de creatinina preformada del músculo lesionado, la liberación de creatina que se convierte a creatinina en el líquido extracelular, o ambos factores, pueden explicar este hallazgo. Sin embargo, una explicación alternativa es que la rabdomiolisis afecta con frecuencia a varones con masa muscular importante, y que se ha observado una mayor producción de creatinina que la que se presenta en pacientes enfermos con otras formas de IRA.139
Tratamiento y prevención
El tratamiento es más eficaz si se inicia pronto; por ejemplo, con la rabdomiolisis causada por lesión por aplastamiento, el tratamiento puede iniciarse antes de aliviar la obstrucción y, por lo tanto, antes de que el pigmento hem sea liberado a la circulación.123,140 Existen dos medidas que pueden prevenir o minimizar la gravedad de la insuficiencia renal aguda en estas circunstancias:
1.Hidratación con solución salina isotónica, tanto para aumentar la perfusión renal (minimizando la lesión por isquemia) como para aumentar la diuresis y eliminar los cilindros que obstruyen.
2.La diuresis forzada con alcalinos y manitol (aumentando el pH de la orina a más de 6.5) puede disminuir en forma importante la toxicidad renal por mioglobina y hemoglobina. La alcalinización de la orina puede aumentar la solubilidad del pigmento hem y disminuir la conversión de hemoglobina al compuesto más tóxico, metahemoglobina.124,125
Existe cierto riesgo con la alcalinización porque puede facilitar la precipitación de fosfato de calcio y disminuir la concentración de calcio ionizado. Sin embargo, parece que el efecto neto es benéfico si el tratamiento se inicia de inmediato.123
Estudios experimentales sugieren que el efecto protector conferido por el manitol es causado principalmente por la diuresis asociada, que minimiza el depósito intratubular de pigmento hem y la formación de cilindros. También se ha propuesto que el manitol actúa como un depurador de radicales libres, disminuyendo así la lesión celular. Sin embargo, estudios cuidadosos han demostrado que el manitol no reduce la necrosis tubular proximal.107
El objeto del tratamiento consiste en mantener una diuresis de alrededor de 300 ml/hr hasta que cese la pigmenturia. En las primeras seis a 12 horas después de que se inicia la hidratación por una lesión por aplastamiento, la mayoría de los pacientes con rabdomiolisis tendrán un balance hídrico muy positivo por el secuestro de líquido hacia el músculo lesionado.123 Este esquema de hidratación es menos eficaz y puede causar sobrecarga hídrica sintomática si se inicia después del periodo inicial, cuando la insuficiencia renal se ha establecido.123
Un último problema que es único de la insuficiencia renal aguda inducida por rabdomiolisis es el desarrollo de hipercalcemia durante la fase de recuperación en alrededor del 20 a 30 porciento de los pacientes.141,142 En parte este problema es causado por la movilización de calcio que se ha depositado en el músculo dañado.143 Tanto la correción de la hiperfosfatemia (secundaria al aumento en la velocidad de filtración glomerular) como un aumento inexplicable en la 1,25-dehidroxivitamina D3 contribuyen a esta respuesta.141 Para disminuir esta complicación, debe evitarse la administración de calcio durante la fase de insuficiencia renal aguda, a menos de que el paciente tenga hipocalcemia sintomática o hipercalemia grave.
ACIDO URICO
Existen tres tipos diferentes de enfermedad renal por ácido úrico: la nefrolitiasis de ácido úrico, la nefropatía crónica por uratos, y la nefropatía aguda por ácido úrico. La nefropatía crónica por uratos es una forma de insuficiencia renal aguda inducida por el depósito de cristales de urato de sodio en el intersticio medular. Este depósito causa una respuesta inflamatoria secundaria (semejante a la que se observa con la formación de microtofos en otras partes del organismo), llegando a producir fibrosis intersticial e insuficiencia renal crónica. Aunque la nefropatía por uratos se observó en el pasado en pacientes con gota tofácea, tanto la formación de tofos como la nefropatía por uratos son poco frecuentes en la actualidad.144 Existen pacientes con insuficiencia renal crónica, sedimento urinario casi normal e hiperuricemia desproporcionada en relación con el grado de insuficiencia renal, datos que son compatibles con nefropatía por uratos. Sin embargo, en la gran mayoría de estos enfermos parece ser que la intoxicación por plomo es la causa primaria de la nefropatía.145-147
En cambio, la nefropatía aguda por ácido úrico se manifiesta como insuficiencia renal aguda oligúrica o anúrica causada por precipitación de ácido úrico dentro de los túbulos.148 Con más frecuencia es resultado de la sobreproducción y excreción excesiva de ácido úrico en pacientes con linfoma, leucemia u otros padecimientos mieloproliferativos (como policitemia vera), en especial después de que la quimio o radioterapia han inducido una lisis celular masiva. Son causas menos frecuentes de nefropatía por ácido úrico el catabolismo tisular secundario a crisis convulsivas o al tratamiento de tumores sólidos, la sobreproducción primaria de ácido úrico (como la que ocurre en el raro síndrome de deficiencia de hipoxantina-guanina fosforribosiltransferasa) o la hiperuricosuria secundaria a disminución en la reabsorción de ácido úrico en el túbulo proximal (como la que se presenta en un síndrome similar al de Fanconi).148,149
En forma típica, la nefropatía por ácido úrico no se asocia con síntomas relacionados con las vías urinarias, aunque puede ocurrir dolor en el flanco si existe obstrucción de la pelvis renal o del urétero. El diagnóstico debe sospecharse cuando se desarrolla un cuadro inexplicable de insuficiencia renal aguda en un paciente con alguno de los padecimientos ya mencionados y que tiene hiperuricemia importante (con concentración plasmática de ácido úrico mayor de 15 mg/dl). En la mayoría de las otras formas de insuficiencia renal aguda, la concentración plasmática de ácido úrico es menor de 12 mg/dl, excepto en la enfermedad prerrenal, en la que puede haber aumento en la reabsorción de sodio y urato en el túbulo proximal.
El examen general de orina en la nefropatía por ácido úrico puede mostrar cristales múltiples de ácido úrico, pero puede ser relativamente normal cuando no sale orina de las nefronas obstruidas. Puede demostrarse excreción excesiva de ácido úrico en muchos pacientes al encontrar una relación entre ácido úrico y creatinina (mg/mg) mayor de 1.0 en una muestra aleatoria de orina; en comparación, se encuentra un valor menor de 0.60 a 0.75 en la mayoría de las otras formas de IRA.150
También se liberan otros componentes celulares cuando existe ruptura tisular importante, lo que puede causar hipercalemia, hiperfosfatemia e hipocalcemia (causada por el depósito de fosfato de calcio en los tejidos). En algunos individuos, la precipitación de calcio y fosfato en los túbulos renales puede ocasionar IRA, independientemente de la precipitación de ácido úrico.151
Tratamiento y prevención
El tratamiento ideal para la nefropatía por ácido úrico es la prevención. Los pacientes que van a recibir quimioterapia o radiación por un cáncer con recambio celular rápido deben recibir antes alopurinol (al principio en dosis más altas que las habituales, 600 a 900 mg/día) más una carga de líquido para mantener una diuresis abundante (más de 2.5 L/día), con o sin bicarbonato de sodio para alcalinizar la orina (lograr un pH urinario de alrededor de 6.5). En la orina alcalina, el ácido úrico se convierte en sales de urato, que son más solubles, lo que disminuye la tendencia a la precipitación. Sin embargo, incluso con una profilaxis óptima, el grado de lisis celular es tan grande que puede desarrollarse IRA en algunos pacientes.148
El tratamiento de la insuficiencia renal temprana consiste en administrar alopurinol (si no se ha administrado antes) e intentar expulsar los cristales de ácido úrico que obstruyen por medio de un diurético de asa e hidratación. Es indispensable la vigilancia estrecha porque la retención del líquido administrado puede causar sobrecarga de volumen, provocando edema pulmonar. En algunos pacientes en quienes no puede inducirse diuresis deberá utilizarse hemodiálisis para eliminar el ácido úrico circulante. El pronóstico para una recuperación completa es excelente si el tratamiento se inicia en forma rápida.
El papel de la alcalinización de la orina con acetazolamida y bicarbonato de sodio es menos claro. Los estudios experimentales sugieren que la hidratación con solución salina sola es tan eficaz como la alcalinización para disminuir la precipitación del ácido úrico.252 Incluso si se administra bicarbonato, puede ser difícil elevar el pH urinario en un paciente con IRA. La alcalinización también tiene la desventaja potencial de facilitar el depósito de fosfato de calcio en los enfermos con hiperfosfatemia importante.
ANFOTERICINA B
La anfotericina B se utiliza en el tratamiento de las infecciones micóticas graves. Sin embargo, su administración se asocia con frecuencia con deterioro funcional renal que se manifiesta por un aumento en la Pcr, pérdida urinaria de potasio e hipocalemia, pérdida urinaria de magnesio e hipomagnesemia, acidosis metabólica secundaria a acidosis tubular renal tipo 1 (o distal), y poliuria causada por diabetes insípida nefrogénica.153-155 Sin embargo, no todos los pacientes desarrollan insuficiencia renal, y el tratamiento a largo plazo (i.e., mayor de un año) puede no disminuir la función renal.156
La disminución de la VFG inducida por la anfotericina B puede exceder el 50 porciento, y se asocia con vasoconstricción renal concomitante. Estos cambios se manifiestan clínicamente por aumento (sobre el nivel basal) en la Pcr por arriba de 2.5 mg/dl.153,157 La disminución en la VFG suele comenzar a las dos semanas de iniciado el tratamiento. La insuficiencia renal más grave, causada sólo por la administración de anfotericina B, es poco frecuente, incluso si se continúa el tratamiento; aunque puede ocurrir si se asocia la depleción de volumen inducida por diuréticos con la administración de otra nefrotoxina, como los antibióticos aminoglucósidos.154 La posibilidad de nefrotoxicidad también es dependiente de la dosis, y el riesgo de disfunción renal es relativamente bajo si se administran dosis menores de 0.5 mg/kg/día.158
El mecanismo por el que ocurren estos cambios no está bien comprendido aún. Se ha propuesto que la anfotericina B ocasiona cambios tubulares importantes, quizá al activar el sistema de retroalimentación túbuloglomerular. En la mayoría de los casos, la mayor reabsorción de sodio por las células de la mácula densa al principio del túbulo distal causa reducción en la VFG.159 Este sistema de retroalimentación es fisiológicamente adecuado porque el aumento en la entrada de NaCl suele reflejar mayor aporte de NaCl al túbulo proximal; al reducir la VFG se disminuye el aporte distal hacia lo normal, evitando así pérdidas excesivas de NaCl.
La anfotericina B interactúa con los esteroles de la membrana, aumentando así la permeabilidad de la misma, un proceso que puede activar el sistema de retroalimentación túbuloglomerular.153,160 Dentro de las células de la mácula densa en el túbulo distal, la mayor entrada de moléculas puede activar el sistema de retroalimentación, causando disminución en la VFG.153,157
Se piensa también que el aumento en la permeabilidad de la membrana inducido por la anfotericina B es responsable de las alteraciones electrolíticas que pueden observarse en pacientes que reciben el fármaco. La mayor permeabilidad puede favorecer la secreción distal de potasio por un gradiente favorable de concentración entre la célula y la luz tubular,161 también puede disminuir la secreción neta de hidrógeno. Por ejemplo, si la orina tiene un pH de 5.0, la concentración de iones hidrógeno es 200 veces mayor que la del líquido extracelular. Cualquier aumento en la permeabilidad tubular favorecerá la difusión retrógrada de los iones de hidrógeno secretados, limitando así la excreción de ácido.161,162
El efecto neto de estos cambios es que son frecuentes la hipocalemia y la acidosis metabólica, causadas por la pérdida de potasio y la retención de hidrógeno, respectivamente.161-163 Puede ocurrir resistencia a la HAD y disminución en la reabsorción neta de magnesio, lo que puede ocasionar poliuria, polidipsia e hipomagnesemia.155 A diferencia de la disminución en la VFG, estos signos de disminución tubular no parecen disminuir por la expansión de volumen.164
La nefrotoxicidad asociada con anfotericina B suele ser reversible si se suspende el tratamiento.157,165 Sin embargo, la disfunción renal puede recurrir cuando éste se reinicia.165
Tratamiento y prevención
La hipótesis de que la retroalimentación túbuloglomerular puede activarse por la anfotericina B ha provocado el uso de cargas de solución salina, porque la expansión de volumen reduce la sensibilidad de este sistema. Los estudios realizados tanto en humanos como en animales han demostrado que la administración de solución salina puede proteger o reducir la disminución en la VFG causada por este medicamento.153,157,164 Sin embargo, no se ha demostrado que este efecto benéfico se relacione con la retroalimentación túbuloglomerular.
Los estudios sugieren que la incidencia y gravedad de estas alteraciones renales pueden disminuirse administrando anfotericina B en vesículas fosfolípidas (liposomas).166 Los liposomas se distribuyen en forma preferencial en el sistema reticuloendotelial, de donde la anfotericina B puede transferirse en forma directa a los hongos atrapados, disminuyendo así su entrada a otras células que contienen colesterol, como las de los túbulos renales. A pesar de estos hallazgos iniciales favorables, se requiere más observación en mayor número de pacientes para establecer en forma definitiva la utilidad de este tipo de tratamiento.167
Además, observaciones en animales sugieren que la administración de un bloqueador de los canales de calcio poco antes y después de la administración de la anfotericina B, protege contra la nefrotoxicidad.168 El mecanismo preciso de este efecto y su aplicación potencial en los humanos no es clara.
PENTAMIDINA
La neumonía causada por Pneumocystis carinii en los pacientes con síndrome de inmunodeficiencia adquirida (SIDA) se trata con dosis relativamente altas de pentamidina intravenosa. En estos casos, la insuficiencia renal aguda reversible es una complicación relativamente frecuente, que ocurre en más del 25 porciento de los pacientes, según algunas series.169,170 Aunque no es frecuente, también puede ocurrir disminución en la función renal con el tratamiento de pentamidina en aerosol.171
El mecanismo por el que la pentamidina produce insuficiencia renal aguda no es claro, pero quizá participe un efecto nefrotóxico directo, que causa necrosis tubular aguda. Como en el caso de otras toxinas tubulares (como los aminoglucósidos, el cisplatino y la anfotericina B), la pentamidina también puede inducir pérdida urinaria de magnesio, causando hipomagnesemia e hipocalcemia.172 A diferencia del efecto de estas otras toxinas tubulares (con las que es frecuente la hipocalemia), la pentamidina produce hipercalemia, efecto que puede explicarse por la semejanza estructural de la pentamidina con los agentes ahorradores de potasio (como el amiloride).173
La suspensión de la pentamidina permite la recuperación de la función renal, aunque ésta puede no presentarse en semanas.169,172 Es probable que la persistencia de la pentamidina en el parénquima renal después de la suspensión del tratamiento contribuya a un retraso en la mejoría.169
Los pacientes que desarrollan nefrotoxicidad por pentamidina deben ser tratados, cuando es necesario, con agentes alternativos como el trimetoprim-sulfametoxazol. Debe evitarse, si es posible, el retratamiento con pentamidina porque la insuficiencia renal puede recurrir.169
ACICLOVIR
La IRA no es una complicación rara del tratamiento intravenoso con aciclovir, que suele administrarse en dosis de 500 mg/m2. Este medicamento se excreta principalmente por la orina y tiene una solubilidad relativamente baja. Por lo tanto, el tratamiento con bolos intravenosos, en especial si el paciente no está bien hidratado, puede causar el depósito de cristales de aciclovir en los túbulos, con obstrución intratubular y focos de inflamación intersticial.174
La función renal comienza a deteriorarse pronto después de iniciado el tratamiento. Los enfermos pueden referir náusea y dolor en el flanco o abdomen, quizá inducidos por la obstrucción de las vías urinarias.174 En algunos casos pueden observarse cristales de aciclovir,birrefringentes y en aguja, en la muestra de orina que se observa bajo luz polarizada.
La disminución en la función renal puede ser grave, y en ocasiones la Pcr es mayor de 8 mg/dl. Sin embargo, es típico que se presente recuperación completa en cuatro a nueve días después de suspendido el medicamento.
Es probable que la mayor parte de los casos de nefrotoxicidad por aciclovir puedan prevenirse con hidratación previa (manteniendo la diuresis a niveles mayores de 75 ml/hr) e infundiendo el fármaco en forma lenta en una a dos horas. Los pacientes que desarrollan insuficiencia renal aguda pueden ser tratados de nuevo, si es necesario, disminuyendo la dosis de aciclovir intravenoso a 250 mg/m2 o menos.126 También el tratamiento por vía oral es bien tolerado, quizá porque disminuye la velocidad de excreción del medicamento. Sin embargo, en ocasiones ocurre IRA.175
SULFONAMIDAS
Algunos antibióticos sulfonamidas son relativamente insolubles en la orina ácida. La sulfadiacina y el sulfametoxazol, que son especialmente insolubles, se usan en la actualidad en dosis mayores a las habituales para tratar la toxoplasmosis, la infección del sistema nervioso central más común en pacientes con SIDA. Hasta el siete porciento de los pacientes tratados con sulfadiacina pueden tener riesgo de IRA porque este medicamento es muy insoluble en orina con un pH de 5.5 o menor.176 La precipitación intrarrenal, que puede también causar la formación de cálculos en el riñón,177 con frecuencia puede ser prevenida manteniendo la ingesta de líquidos en más de 3 L/día, vigilando la orina en busca de cristales que tengan apariencia de `choque de trigo' [ver figura 5] y, si se observan cristales, alcalinizado la orina hasta un pH mayor de 7.15.
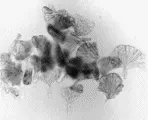
|
| Figura 5 |
| Nefrotoxicidad por sulfadiacina |
En algunos pacientes, el polvo de sulfadiacina o pequeños cálculos que se localizan en los cálices pueden detectarse como acúmulos bilaterales de material ecogénico en capas en el ultrasonido renal. En otros los cristales de acetilsulfadiacina puede coalescer para formar cálculos más grandes que pueden obstruir el uretero, ocasionando hidronefrosis.
METOTREXATE
El metotrexate en dosis altas administrado por vía intravenosa puede precipitarse en los túbulos e inducir lesión tubular porque alrededor del 90 porciento del medicamento se excreta en condiciones normales en la orina. El riesgo de IRA puede minimizarse por la hidratación previa (tanto para mantener un flujo urinario alto como para disminuir la concentración del fármaco en el líquido tubular) y alcalinizando la orina a un pH mayor de 7.0, lo que aumenta la solubilidad del metotrexate hasta en 10 veces.178
Cuando ocurre IRA el tratamiento se dirige a lavar los túbulos administrando un diurético de asa. Aunque puede intentarse la administración de alcalinos, su eficacia para elevar el pH urinario en presencia de falla renal es muy dudosa. La eliminación del medicamento por diálisis, la hemoperfusión con carbón o el intercambio plasmático suelen ser de valor limitado porque el metotrexate se une a proteínas (por lo que no es dializable) y tiene un volumen de distribución extravascular muy grande.
En la mayoría de los casos la enfermedad renal inducida por metotrexate es reversible. La Pcr suele alcanzar un máximo en la primera semana y regesa a la basal en una a tres semanas. Aunque muchos pacientes tienen una evolución benigna, la excreción urinaria reducida causa niveles elevados de medicamento en plasma que pueden durar hasta dos a tres semanas.
VANCOMICINA
Con frecuencia se administra vancomicina a los pacientes con infecciones sistémicas. En vista de la frecuencia de IRA en estos casos (principalmente por NTA), se ha pensado en la posibilidad de que la vancomicina sea responsable de la nefrotoxicidad. En estudios iniciales sobre lesión renal inducida por vancomicina, el daño parecía ser causado por preparaciones que contenían cantidades relativamente grandes de impurezas. Los avances posteriores en la tecnología han permitido mejorar mucho la pureza del antibiótico.
No se conoce con certeza la incidencia de la nefrotoxicidad por vancomicina porque otros factores que pueden alterar la función renal (v.gr., infección, hipotensión y administración de medio de contraste o aminoglucósidos) coexisten con frecuencia. La mayoría de los estudios indican que el cinco a 15 porciento de los pacientes tratados solo con vancomicina desarrollan deterioro agudo en la función renal (definido como un aumento en la Pcr de por lo menos 0.5 mg/dl). En la mayoría de los pacientes el grado de insuficiencia renal es relativamente leve a menos que se administren dosis excesivas.
Por lo tanto, la inducción de daño renal importante por la vancomicina es poco frecuente. Sin embargo, este antibiótico puede potencializar la nefrotoxicidad de un aminoglucósido administrado en forma concomitante. La incidencia de IRA en este caso puede ser hasta de 20 a 30 porciento.
ACETAMINOFEN
La ingestión prolongada de acetaminofén puede asociarse con un mayor riesgo de insuficiencia renal crónica.180 Sin embargo, está un poco mejor documentada la IRA inducida por este medicamento.181 La IRA casi siempre ocurre en personas que han tomados una sobredosis de acetaminofén, pero algunos alcohólicos tienen riesgo de IRA incluso con dosis terapéuticas.
Aunque puede ocurrir daño renal aislado, la mayoría de los pacientes tienen lesión hepática concomitante de gravedad variable. La lesión casi siempre se manifiesta por elevación importante en la concentración plasmática de aminotransferaa y, en algunos casos, por insuficiencia hepática, que puede ser fatal. Los mecanismos que ocasionan toxicidad en el riñón y el hígado son semejantes. Un tóxico intermediario, el N-acetilimidoquinone, se forma localmente por medio de la vía del citocromo P450 y después se une en forma covalente a macromoléculas celulares, causando lesión o muerte celular. El tiempo requerido para la formación de este metabolito, que comienza a acumularse solo después de que se han depletado las concentraciones de glutatión, y para que ocurra el daño celular explica el retraso habitual de 72 a 96 horas antes de que se haga aparente la falla hepática y renal.
La incidencia de disfunción renal se asocia con la dosis de acetaminofén que se ingiere y con la gravedad de la lesión hepática concomitante. El daño renal puede ser más frecuente en pacientes con insuficiencia hepática fulminante. Es posible que en estos individuos un síndrome semejante al SHR, aunado a la toxicidad directa, contribuya al desarrollo de insuficiencia renal.
Se ha estudiado el tejido renal de un pequeño número de pacientes con IRA inducida por acetaminofén, revelándose por lo general NTA.181 Estos datos patológicos son compatibles con los resultados del examen de orina, que por lo general demuestra cilindros granulares y epiteliales. La función renal retorna en forma espontánea a la basal en una a cuatro semanas, aunque puede requerirse diálisis durante el epidosio agudo. No se sabe si la acetilcisteina, que se administra para minimizar la hepatotoxicidad, tiene efecto protector sobre el riñón, en especial si se ha desarrollado IRA.
FOSCARNET
El foscarnet (ácido fosfonofórmico) es un agente antiviral empleado sobre todo en el tratamiento de las infecciones serias por citomegalovirus en pacientes con SIDA. La IRA es el principal efecto tóxico del foscarnet.182 Se ha calculado que hasta dos terceras partes de los pacientes con IRA inducida por foscarnet tienen aumento en la Pcr y que hasta el 10 a 15 porciento requiere diálisis temporal.182 Ocurre recuperación después de suspender el tratamiento, pero la Pcr puede no normalizarse en varios meses.
Cuando ocurre nefrotoxicidad, la disminución en la función renal suele comenzar después de seis a 15 días de tratamiento. Aunque los resultados del examen de orina permanecen relativamente normales, la biopsia renal revela la presencia de NTA. Por lo tanto, es probable que el foscarnet tenga un efecto tóxico directo en los túbulos. El riesgo de IRA parece aumentarse por la depleción de volumen efectivo, y existen ciertas evidencias que sugieren que la hidratación agresiva es protectora.
IFOSFAMIDA
La ifosfamida es un análogo sintético de la ciclofosfamida que se ha autorizado para el uso con otros medicamentos en el tratamiento de ciertos tipos de cáncer. A diferencia de los efectos de la ciclofosfamida, los efectos de la ifosfamida incluyen nefrotoxicidad causada por lesión tubular directa, que puede constituir una complicación significativa. Aunque puede ocurrir IRA, la disminución en la VFG suele ser leve a menos que la ifosfamida se administre en combinación con otra nefrotoxina (como cisplatino).183 Con más frecuencia, la lesión renal se manifiesta clínicamente por uno o más de los siguientes signos de disfunción tubular: acidosis tubular renal tipo 1 (distal) o tipo 2 (proximal), hipofosfatemia, diabetes insípida nefrogénica o hipocalemia.
Estudios preliminares in vitro sugieren que es un metabolito, el cloracetaldehido, más que la ifosfamida, el que es tóxico para las células tubulares proximales.184 Se ha propuesto que el grado de disfunción renal puede minimizarse por la administración concomitante de mesna, un compuesto sulfihidrilo sintético que actúa en la orina para detoxificar el cloracetaldehido y quizá otros metabolitos. Sin embargo, no se ha aclarado porqué algunos pacientes han desarrollado toxicidad por ifosfamida a pesar de recibir el medicamento en combinación con mesna.
La evolución a largo plazo de la lesión renal inducida por ifosfamida es poco clara. Puede ocurrir mejoría si se suspende el tratamiento, aunque en casos graves puede ser solo parcial.
TRIAMTIRENO
El triamtireno es un diurético ahorrador de potasio que se usa con frecuencia en combinación con una tiacida para tratar la hipertensión. Es una nefrotoxina potencial que con frecuencia produce cristaluria y formación de cilindros, y en casos raros formación de cálculos o IRA reversible.185
En hasta el 50 porciento de los pacientes tratados con triamtireno ocurre excreción de cristales del medicamento y cilindros granulares. Este hallazgo no se observa con el amiloride, otro diurético ahorrador de potasio. Se han propuesto dos mecanismos etiológicos de la nefrotoxicidad por triamtireno: obstrucción intratubular por los cristales y tratamiento concomitante con un AINE.185
Bibliografía
- Spiegel DM, Wilson PD, Molitoris BA: Epithelial polarity following ischemia: a requirement for normal cell function. Am J Physiol 265:F430, 1989
- Miller TR, Anderson RJ, Linas SL, et al: Urinary diagnostic indices in acute renal failure: a prospective study. Ann Intern Med 89:47, 1978 [PMID 666184]
- Dixon BS, Anderson RJ: Nonoliguric acute renal failure. Am J Kidney Dis 6:71, 1985
- Oken DE: On the differential diagnosis of acute renal failure. Am J Med 71:916, 1981
- Espinel CH, Gregory AW: Differential diagnosis of acute renal failure. Clin Nephrol 13:73, 1980 [PMID 7363517]
- Steiner RW: Interpreting the fractional excretion of sodium. Am J Med 77: 699, 1984
- Brosius FC, Lau K: Low fractional excretion of sodium in acute renal failure: role of timing of the test and ischemia. Am J Nephrol 6:450, 1986 [PMID 3565502]
- Schrier RW: Body fluid volume regulation in health and disease: a unifying hypothesis. Ann Intern Med 113:155, 1990
- Schnermann J, Briggs JP, Weber PC: Tubuloglomerular feedback, prostaglandins, and angiotensin in the autoregulation of glomerular filtration rate. Kidney Int 25:53, 1984 [PMID 6587164]
- Patrono C, Dunn MJ: The clinical significance of inhibition of renal prostaglandin synthesis. Kidney Int 32:1, 1987 [PMID 3306093]
- Jackson B, Matthews PG, McGrath BP, et al: Angiotensin converting enzyme inhibition in renovascular hypertension: frequency of reversible renal failure. Lancet 1: 225, 1984 [PMID 6198567]
- Packer M, Lee WH, Medina N, et al: Functional renal insufficiency during long-term therapy with captopril and enalapril in severe chronic heart failure. Ann Intern Med 106:346, 1987
- Mroczek WJ, Davidov M, Gavrilovich L, et al: The value of aggressive therapy in the hypertensive patient with azotemia. Circulation 40:893, 1969
- Moss GS, Lowe RJ, Jilek J, et al: Colloid or crystalloid in the resuscitation of hemorrhagic shock: a controlled clinical trial. Surgery 89:434, 1981 [PMID 7010650]
- Monafo W: Expensive salt water. Surgery 89:525, 1981
- Rozich JD, Paul RV: Acute renal failure precipitated by elevated colloid osmotic pressure. Am J Med 87:359, 1989 [PMID 2773975]
- Epstein M, Berk DP, Hollenberg NK, et al: Renal failure in the patient with cirrhosis: the role of active vasoconstriction. Am J Med 49:175, 1970 [PMID 5452940]
- Papadakis MA, Arieff AI: Unpredictability of clinical evaluation of renal function in cirrhosis: prospective study. Am J Med 82:945, 1987 [PMID 3578363]
- Arroyo V, Bosch J, Gaya-Beltran J, et al: Plasma renin activity and urinary sodium excretion as prognostic indicators in nonazotemic cirrhosis with ascites. Ann Intern Med 94:198, 1981
- Koppel MH, Coburn JW, Mims MM, et al: Transplantation of cadaveric kidneys from patients with hepatorenal syndrome: evidence for the functional nature of renal failure in advanced liver disease. N Engl J Med 280:1367, 1969 [PMID 4890476]
- Perez-Ayuso RM, Arroyo V, Camps J, et al: Evidence that renal prostaglandins are involved in renal water metabolism in cirrhosis. Kidney Int 26:72, 1984 [PMID 6434791]
- Laffi G, La Villa G, Pinzani M, et al: Altered renal and platelet arachidonic acid metabolism in cirrhosis. Gastroenterology 90:274, 1986 [PMID 3079716]
- Fernandez-Seara J, Prieto J, Quiroga J, et al: Systemic and regional hemodynamics in patients with liver cirrhosis and ascites with and without functional renal failure. Gastroenterology 97:1304, 1989 [PMID 2676683]
- Wong PY, Talamo RC, Williams GH: Kallikrein-kinin and renin-angiotensin systems in functional renal failure of cirrhosis of the liver. Gastroenterology 73:1114, 1977 [PMID 332579]
- Levy M: Hepatorenal syndrome. Kidney Int 43:737, 1993
- Gines A, Escorsell A, Gines P, et al: Incidence, predictive factors, and prognosis of the hepatorenal syndrome in cirrhosis with ascites. Gastroenterology 105: 229, 1993 [PMID 8514039]
- Lenz K, Hortnagl H, Druml W, et al: Ornipressin in the treatment of functional renal failure in decompensated liver cirrhosis: effects on renal hemodynamics and atrial natriuretic factor. Gastroenterology 101:1060, 1991 [PMID 1832407]
- Caregaro L, Menon F, Angeli P, et al: Limitations of serum creatinine level and creatinine clearance as filtration markers in cirrhosis. Arch Intern Med 154:201, 1994 [PMID 8285815]
- Better OS: Renal and cardiovascular dysfunction in liver disease. Kidney Int 29:598, 1986
- Zager RA: Acute renal failure in the setting of bone marrow transplantation. Kidney Int 46:1443, 1994
- Baldus WP, Feichter RN, Summerskill WHJ: The kidney in cirrhosis: I. Clinical and biochemical features of azotemia in hepatic failure. Ann Intern Med 60:353, 1994
- Laffi G, Daskalopoulos G, Kronborg I, et al: Effects of sulindac and ibuprofen in patients with cirrhosis and ascites: an explanation for the renal-sparing effect of sulindac. Gastroenterology 90:182, 1986 [PMID 3079594]
- Linas SL, Schaefer JW, Moore EE, et al: Peritoneovenous shunt in the management of the hepatorenal syndrome. Kidney Int 30:736, 1986 [PMID 3537462]
- Roulot D, Moreau R, Gaudin C, et al: Long-term sympathetic and hemodynamic responses to clonidine in patients with cirrhosis and ascites. Gastroenterology 102:1309, 1992 [PMID 1551536]
- Gonwa TA, Morris CA, Goldstein RM, et al: Long-term survival and renal function following liver transplantation in patients with and without hepatorenal syndrome-experience in 300 patients. Transplantation 51:428, 1991 [PMID 1994538]
- Wilkinson SP, Weston MJ, Parsons V, et al: Dialysis in the treatment of renal failure in patients with liver disease. Clin Nephrol 8:287, 1977 [PMID 884909]
- Dzau VJ: Renal and circulatory mechanisms in congestive heart failure. Kidney Int 3:1402, 1987
- Stampfer M, Epstein SE, Beiser GD, et al: Hemodynamic effects of diuresis at rest and during intense upright exercise in patients with impaired cardiac function. Circulation 37:900, 1968 [PMID 5653053]
- Dzau VJ, Packer M, Lilly LS, et al: Prostaglandins in severe congestive heart failure. N Engl J Med 310:347, 1984 [PMID 6361570]
- Oates JA, FitzGerald GA, Branch RA, et al: Clinical implications of prostaglandin and thromboxane A2 formation. N Engl J Med 319:761, 1988 [PMID 3045551]
- Whelton A, Stout RL, Spilman PS, et al: Renal effects of ibuprofen, piroxicam, and sulindac in patients with asymptomatic renal failure: a prospective, randomized, crossover comparison. Ann Intern Med 112:568, 1990 [PMID 2183665]
- Packer M, Lee WH, Kessler PD, et al: Identification of hyponatremia as a risk factor for the development of functional renal insufficiency during converting enzyme inhibition in severe chronic heart failure. J Am Coll Cardiol 10:837, 1987 [PMID 2821091]
- Packer M, Lee WH, Yushak M, et al: Comparison of captopril and enalapril in patients with severe chronic heart failure. N Engl J Med 315:847, 1986 [PMID 3018566]
- Packer M, Lee WH, Medina N, et al: Influence of renal function on the hemodynamic and clinical responses to long-term captopril therapy in severe chronic heart failure. Ann Intern Med 104:147, 1986 [PMID 2936291]
- Textor SC, Bravo EL, Fouad FM, et al: Hyperkalemia in azotemic patients during angiotensin-converting enzyme inhibition and aldosterone reduction with captopril. Am J Med 73:719, 1982 [PMID 6291388]
- Agostoni PG, Marenzi GC, Pepi M, et al: Isolated ultrafiltration in moderate congestive heart failure. J Am Coll Cardiol 21:424, 1993 [PMID 8426008]
- Solez K, Morel-Maroger L, Sraer J-D: The morphology of "acute tubular necrosis" in man: analysis of 57 renal biopsies and a comparison with the glycerol model. Medicine (Baltimore) 58:362, 1979 [PMID 481195]
- Myers BD, Miller DC, Mehigan JT, et al: Nature of the renal injury following total renal ischemia in man. J Clin Invest 73:329, 1984 [PMID 6421876]
- Schrier RW: Acute renal failure. Kidney Int 15:205, 1979
- Brezis M, Rosen S, Silva P, et al: Renal ischemia: a new perspective. Kidney Int 26:375, 1984 [PMID 6396435]
- Myers BD, Moran SM: Hemodynamically mediated acute renal failure. N Engl J Med 314:97, 1986
- Kiyama S, Yoshioka T, Burr IM, et al: Strategic locus for the activation of the superoxide dismutase gene in the nephron. Kidney Int 47:536, 1995 [PMID 7536859]
- Brezis M, Rosen SN, Epstein FH: The pathophysiological implications of medullary hypoxia. Am J Kidney Dis 13:253, 1989 [PMID 2493191]
- Bonventre JV: Mechanisms of ischemic acute renal failure. Kidney Int 43: 1160, 1993
- Iaina A, Schwartz D: Renal tubular cellular and molecular events in acute renal failure. Nephron 68:413, 1994 [PMID 7870224]
- Reeves WB, Shah SV: Activation of potassium channels contributes to hypoxic injury in proximal tubules. J Clin Invest 94:2289, 1994 [PMID 7989584]
- Graziani G, Cantaluppi A, Casati S, et al: Dopamine and frusemide in oliguric acute renal failure. Nephron 37:39, 1984 [PMID 6717704]
- Brown CB, Ogg CS, Cameron JS: High dose frusemide in acute renal failure: a controlled trial. Clin Nephrol 15:90, 1981 [PMID 7011622]
- Szerlip HM: Renal-dose dopamine: fact and fiction. Ann Intern Med 115:153, 1991
- Duke GJ, Bersten AD: Dopamine and renal salvage in the critically ill patient. Anaesth Intensive Care 20:277, 1992 [PMID 1524166]
- Marik PE, Mohedin M: The contrasting effects of dopamine and norepinephrine on systemic and splanchnic oxygen utilization in hyperdynamic sepsis. JAMA 272:1354, 1994 [PMID 7933396]
- Anderson RJ, Linas SL, Berns AS, et al: Nonoliguric acute renal failure. N Engl J Med 296:1134, 1977
- Honda N, Hishida A: Pathophysiology of experimental nonoliguric acute renal failure. Kidney Int 43:513, 1993 [PMID 8455351]
- Lee MR: Dopamine and the kidney: ten years on. Clin Sci 84:357, 1993
- Olsen NV, Hansen JM, Ladefoged SD, et al: Renal tubular reabsorption of sodium and water during infusion of low-dose dopamine in normal man. Clin Sci 78:503, 1990
- Baldwin L, Henderson A, Hickman P: Effect of postoperative low-dose dopamine on renal function after elective major vascular surgery. Ann Intern Med 120:744, 1994 [PMID 8147547]
- Dorman HR, Sondheimer JH, Cadnapaphornchai P: Mannitol-induced acute renal failure. Medicine (Baltimore) 69:153, 1990 [PMID 2111870]
- Gillum DM, Dixon BS, Yanover MJ, et al: The role of intensive dialysis in acute renal failure. Clin Nephrol 25:249, 1986 [PMID 3720035]
- Conger JD: Does hemodialysis delay recovery from acute renal failure? Seminars in Dialysis 3:146, 1990
- Hakim RM, Wingard RL, Parker RA: Effect of the dialysis membrane in the treatment of patients with acute renal failure. N Engl J Med 331:1338, 1994 [PMID 7935703]
- Schiffl H, Lang SM, Konig A, et al: Biocompatible membranes in acute renal failure: prospective case-controlled study. Lancet 344:570, 1994 [PMID 7914959]
- Clark WR, Mueller BA, Alaka KJ, et al: A comparison of metabolic control by continuous and intermittent therapies in acute renal failure. J Am Soc Nephrol 4:1413, 1994 [PMID 8161723]
- Barton IK, Hilton PJ, Taub NA, et al: Acute renal failure treated by haemofiltration: factors affecting outcome. Q J Med 86:81, 1993 [PMID 8464996]
- Bellomo R, Parkin G, Love J, et al: Use of continuous haemodiafiltration: an approach to the management of acute renal failure in the critically ill. Am J Nephrol 12:240, 1992
- Safirstein R: Gene expression in nephrotoxic and ischemic acute renal failure. J Am Soc Nephrol 4:1387, 1994
- Verstrepen WA, Nouwen EJ, Yue XS, et al: Altered growth factor expression during toxic proximal tubular necrosis and regeneration. Kidney Int 43:1267, 1993 [PMID 8315941]
- Coimbra TM, Cieslinski DA, Humes HD: Epidermal growth factor accelerates renal repair in mercuric chloride nephrotoxicity. Am J Physiol 259:F438, 1990 [PMID 2396670]
- Ding H, Kopple JD, Cohen A, et al: Recombinant human insulin-like growth factor-I accelerates recovery and reduces catabolism in rats with ischemic acute renal failure. J Clin Invest 91:2281, 1993 [PMID 8486787]
- Conger JD, Falk SA, Hammond WS: Atrial natriuretic peptide and dopamine in established acute renal failure in the rat. Kidney Int 40:21, 1991 [PMID 1833582]
- Humes HD: Aminoglycoside nephrotoxicity. Kidney Int 33:900, 1988
- Moore RD, Smith CR, Lipsky JJ, et al: Risk factors for nephrotoxicity in patients treated with aminoglycosides. Ann Intern Med 100:352, 1984 [PMID 6364908]
- Smith CR, Lipsky JJ, Laskin OL, et al: Double-blind comparison of the nephrotoxicity and auditory toxicity of gentamicin and tobramycin. N Engl J Med 302:1106, 1980 [PMID 6988713]
- Bennett WM, Wood CA, Houghton DC, et al: Modification of experimental aminoglycoside nephrotoxicity. Am J Kidney Dis 8:292, 1986 [PMID 3788967]
- Meyer RD: Risk factors and comparisons of clinical nephrotoxicity of aminoglycosides. Am J Med 80(suppl 6B):119, 1986
- Matzke GR, Lucarotti RL, Shapiro HS: Controlled comparison of genta micin and tobramycin nephrotoxicity. Am J Nephrol 3:11, 1983 [PMID 6340504]
- Gary NE, Buzzeo L, Salaki J, et al: Gentamicin-associated acute renal failure. Arch Intern Med 136:1101, 1976 [PMID 788666]
- Zager RA: Gentamicin effects on renal ischemia/reperfusion injury. Circ Res 70:20, 1992
- Spiegel DM, Shanley PF, Molitoris BA: Mild ischemia predisposes the S3 segment to gentamicin toxicity. Kidney Int 38:459, 1990 [PMID 2232488]
- Gilbert DN: Once-daily aminoglycoside therapy. Antimicrob Agents Chemother 35:399, 1991
- Aronson JK, Reynolds DJM: Aminoglycoside antibiotics. BMJ 305:1421, 1992 [PMID 1486311]
- Parker SE, Davey PG: Practicalities of once-daily aminoglycoside dosing. J Antimicrob Chemother 31:4, 1993
- Levison ME: New dosing regimens for aminoglycoside antibiotics. Ann Intern Med 117:693, 1992
- Prins JM, Buller HR, Kuijper EJ, et al: Once versus thrice daily gentamicin in patients with serious infections. Lancet 341:335, 1993 [PMID 8094114]
- The International Antimicrobial Therapy Cooperative Group of the European Organization for Research and Treatment of Cancer: Efficacy and toxicity of single daily doses of amikacin and ceftriaxone versus multiple daily doses of amikacin and ceftazidime for infection in patients with cancer and granulocytopenia. Ann Intern Med 119:584, 1993
- Baines AD, Shaikh N, Ho P: Mechanisms of perfused kidney cytoprotection by alanine and glycine. Am J Physiol 259:F80, 1990 [PMID 2375394]
- Desai TK, Tsang T-K: Aminoglycoside nephrotoxicity in obstructive jaundice. Am J Med 85:47, 1988 [PMID 3389381]
- Wade JC, Smith CR, Petty BG, et al: Cephalothin plus an aminoglycoside is more nephrotoxic than methicillin plus an aminoglycoside. Lancet 2:604, 1978 [PMID 80528]
- Houghton DC, English J, Bennett WM: Chronic tubulointerstitial nephritis and renal insufficiency associated with long-term "subtherapeutic" gentamicin. J Lab Clin Med 112:694, 1988
- Parfrey PS, Griffiths SM, Barrett BJ: Contrast material-induced renal failure in patients with diabetes mellitus, renal insufficiency, or both: a prospective controlled study. N Engl J Med 320:143, 1989 [PMID 2643041]
- Taliercio CP, Vlietstra RE, Fisher LD, et al: Risks for renal dysfunction with cardiac angiography. Ann Intern Med 104:501, 1986 [PMID 3954277]
- Davidson CJ, Hlatky M, Morris KG, et al: Cardiovascular and renal toxicity of a nonionic radiographic contrast agent after cardiac catheterization: a prospective trial. Ann Intern Med 110:119, 1989 [PMID 2909204]
- Schwab SJ, Hlatky MA, Pieper KS, et al: Contrast nephrotoxicity: a randomized controlled trial of a nonionic and an ionic radiographic contrast agent. N Engl J Med 320:149, 1989
- Cigarroa RG, Lange RA, Williams RH, et al: Dosing of contrast material to prevent contrast nephropathy in patients with renal disease. Am J Med 86:649, 1989 [PMID 2729314]
- Berns AS: Nephrotoxicity of contrast media. Kidney Int 36:730, 1989
- Bakris GL, Lass N, Gaber AO, et al: Radiocontrast medium-induced declines in renal function: a role for oxygen free radicals. Am J Physiol 258:F115, 1990 [PMID 2301588]
- Heyman SN, Spokes K, Epstein FH, et al: Early functional and morphological changes in radiocontrast nephropathy (abstr). J Am Soc Nephrol 1:596, 1990
- Weinrauch LA, Healy RW, Leland OS, et al: Coronary angiography and acute renal failure in diabetic azotemic nephropathy. Ann Intern Med 86:56, 1977 [PMID 835928]
- Holland MD, Galla JH, Sanders PW, et al: Effect of urinary pH and diatrizoate on Bence Jones protein nephrotoxicity in the rat. Kidney Int 27:46, 1985 [PMID 2984452]
- Moore RD, Steinberg EP, Rowe NR, et al: Nephrotoxicity of high-osmolality versus low-osmolality contrast media: randomized clinical trial. Radiology 182:649, 1992 [PMID 1535876]
- Rudnick MR, Goldfarb S, Wexler L, et al: Nephrotoxicity of ionic and nonionic contrast media in 1196 patients: a randomized trial. Kidney Int 47:254, 1995 [PMID 7731155]
- Barrett BJ, Carlisle EJ: Metaanalysis of the relative nephrotoxicity of high- and low-osmolality iodinated contrast media. Radiology 188:171, 1993 [PMID 8511292]
- Harris KG, Smith TP, Cragg AH, et al: Nephrotoxicity from contrast material in renal insufficiency: ionic versus nonionic agents. Radiology 179:849, 1991 [PMID 2028004]
- Solomon R, Werner C, Mann D, et al: Effects of saline, mannitol, and furosemide on acute decreases in renal function induced by radiocontrast agents. N Engl J Med 331:1416, 1994
- Erley CM, Duda SH, Schlepckow S, et al: Adenosine antagonist theophylline prevents the reduction of glomerular filtration rate after contrast media application. Kidney Int 45:1425, 1994 [PMID 8072255]
- Kurnik BRC, Singer F, Groh WC: Case report: dextran-induced acute anuric renal failure. Am J Med Sci 302:28, 1991
- Ries F, Klastersky J: Nephrotoxicity induced by cancer chemotherapy with special emphasis on cisplatin toxicity. Am J Kidney Dis 8:368, 1986 [PMID 3538860]
- Reed E, Jacob J: Carboplatin and renal dysfunction (letter). Ann Intern Med 110:409, 1989
- Schilsky RL, Anderson T: Hypomagnesemia and renal magnesium wasting in patients receiving cisplatin. Ann Intern Med 90:929, 1979 [PMID 375794]
- Lam M, Adelstein DJ: Hypomagnesemia and renal magnesium wasting in patients treated with cisplatin. Am J Kidney Dis 8:164, 1986 [PMID 3752072]
- Heyman SN, Rosen S, Silva P, et al: Protective action of glycine in cisplatin nephrotoxicity. Kidney Int 40:273, 1991 [PMID 1942775]
- Ozols RF, Corden BJ, Jacob J, et al: High-dose cisplatin in hypertonic saline. Ann Intern Med 100:19, 1984 [PMID 6197916]
- Howell SB, Pfeifle CL, Wung WE, et al: Intraperitoneal cisplatin with systemic thiosulfate protection. Ann Intern Med 97:845, 1982 [PMID 6890785]
- Better OS, Stein JH: Early management of shock and prophylaxis of acute renal failure in traumatic rhabdomyolysis. N Engl J Med 322:825, 1990 [PMID 2407958]
- Zager RA, Gamelin LM: Pathogenetic mechanisms in experimental hemoglobinuric acute renal failure. Am J Physiol 256:F446, 1989 [PMID 2923223]
- Zager RA: Studies of mechanisms and protective maneuvers in myoglobinuric acute renal injury. Lab Invest 60:619, 1989
- Zager RA: Myoglobin depletes renal adenine nucleotide pools in the presence and absence of shock. Kidney Int 39:111, 1991
- Nath KA, Balla J, Croatt AJ, et al: Heme protein-mediated renal injury: a protective role for 21-aminosteroids in vitro and in vivo. Kidney Int 47:592, 1995 [PMID 7723246]
- Wolfert AI, Oken DE: Glomerular hemodynamics in established glycerol-induced acute renal failure in the rat. J Clin Invest 84:1967, 1989 [PMID 2592568]
- Gabow PA, Kaehny WD, Kelleher SP: The spectrum of rhabdomyolysis. Medicine (Baltimore) 61:141, 1982
- Honda N: Acute renal failure and rhabdomyolysis. Kidney Int 23:888, 1983
- Valeriano-Marcet J, Kerr LD: Myonecrosis and myofibrosis as complications of sickle cell anemia. Ann Intern Med 115:99, 1991 [PMID 2058869]
- Guze BH, Baxter LR Jr: Neuroleptic malignant syndrome. N Engl J Med 313:163, 1985 [PMID 3892294]
- Roth D, Alarcon FJ, Fernandez JA, et al: Acute rhabdomyolysis associated with cocaine intoxication. N Engl J Med 319:673, 1988 [PMID 3412385]
- Dalakas MC, Illa I, Pezeshkpour GH, et al: Mitochondrial myopathy caused by long-term zidovudine therapy. N Engl J Med 322:1098, 1990 [PMID 2320079]
- East C, Alivizatos PA, Grundy SM, et al: Rhabdomyolysis in patients receiving lovastatin after cardiac transplantation (letter). N Engl J Med 318:47, 1988 [PMID 3275892]
- Pierce LR, Wysowski DK, Gross TP: Myopathy and rhabdomyolysis associated with lovastatin-gemfibrozil combination therapy. JAMA 264:71, 1990 [PMID 2355431]
- Corwin HL, Schreiber MJ, Fang LST: Low fractional excretion of sodium: occurrence with hemoglobinuric- and myoglobinuric-induced acute renal failure. Arch Intern Med 144:981, 1984
- Knochel JP: Hypophosphatemia and rhabdomyolysis (editorial). Am J Med 92:455, 1992
- Oh MS: Does serum creatinine rise faster in rhabdomyolysis? Nephron 63:255, 1993
- Ron D, Taitelman U, Michaelson M, et al: Prevention of acute renal failure in traumatic rhabdomyolysis. Arch Intern Med 144:277, 1984 [PMID 6696564]
- Akmal M, Bishop JE, Telfer N, et al: Hypocalcemia and hypercalcemia in patients with rhabdomyolysis with and without acute renal failure. J Clin Endocrinol Metab 63:137, 1986
- Llach F, Felsenfeld AJ, Haussler MR: The pathophysiology of altered calcium metabolism in rhabdomyolysis-induced acute renal failure: interactions of parathyroid hormone, 25-hydroxycholecalciferol, and 1,25-dihydroxycholecalciferol. N Engl J Med 305:117, 1981 [PMID 6894630]
- Lane JT, Boudreau RJ, Kinlaw WB: Disappearance of muscular calcium deposits during resolution of prolonged rhabdomyolysis-induced hypercalcemia. Am J Med 89:523, 1990 [PMID 2220885]
- Beck LH: Requiem for gouty nephropathy. Kidney Int 30:280, 1986
- Batuman V, Maesaka JK, Haddad B, et al: The role of lead in gout nephropathy. N Engl J Med 304:520, 1981 [PMID 6779160]
- Craswell PW, Price J, Boyle PD, et al: Chronic renal failure with gout: a marker of chronic lead poisoning. Kidney Int 26:319, 1984 [PMID 6439940]
- Batuman V, Landy E, Maesaka JK, et al: Contribution of lead to hypertension with renal impairment. N Engl J Med 309:17, 1983 [PMID 6406892]
- Kjellstrand CM, Campbell DC II, von Hartitzsch B, et al: Hyperuricemic acute renal failure. Arch Intern Med 133:349, 1974 [PMID 4283735]
- Hricik DE, Goldsmith GH: Uric acid nephrolithiasis and acute renal failure secondary to streptozotocin nephrotoxicity. Am J Med 84:153, 1988 [PMID 2827466]
- Kelton J, Kelley WN, Holmes EW: A rapid method for the diagnosis of acute uric acid nephropathy. Arch Intern Med 138:612, 1978 [PMID 637642]
- Monballyu J, Zachee P, Verberckmoes R, et al: Transient acute renal failure due to tumor-lysis-induced severe phosphate load in a patient with Burkitt's lymphoma. Clin Nephrol 22:47, 1984 [PMID 6478662]
- Conger JD, Falk SA, Guggenheim SJ, et al: A micropuncture study of the early phase of acute urate nephropathy. J Clin Invest 58:681, 1976 [PMID 956394]
- Branch RA: Prevention of amphotericin B-induced renal impairment: a review on the use of sodium supplementation. Arch Intern Med 148:2389, 1988
- Butler WT, Bennet JE, Alling DW, et al: Nephrotoxicity of amphotericin B: early and late effects in 81 patients. Ann Intern Med 61:175, 1964
- Barton CH, Pahl M, Vaziri ND, et al: Renal magnesium wasting associated with amphotericin B therapy. Am J Med 77:471, 1984 [PMID 6475987]
- Reines E, Gross PA: Cryptococcal meningitis: seven years of maintenance amphotericin therapy without progressive renal failure. Am J Med 85:591, 1988 [PMID 3177423]
- Heidemann HTH, Gerkens JF, Spickard WA, et al: Amphotericin B nephrotoxicity in humans decreased by salt repletion. Am J Med 75:476, 1983
- Fisher MA, Talbot GH, Maislin G, et al: Risk factors for amphotericin B-associated nephrotoxicity. Am J Med 87:547, 1989 [PMID 2816970]
- Blantz RC, Thomson SC, Peterson OW, et al: Physiologic adaptations of the tubuloglomerular feedback system. Kidney Int 38:577, 1990 [PMID 2232499]
- Cheng J-T, Witty RT, Robinson RR, et al: Amphotericin B nephrotoxicity: increased renal resistance and tubule permeability. Kidney Int 22:626, 1982
- Douglas JB, Healy JK: Nephrotoxic effects of amphotericin B, including renal tubular acidosis. Am J Med 46:154, 1969 [PMID 4951424]
- Gil FZ, Malnic G: Effect of amphotericin B on renal tubular acidification in the rat. Pflugers Arch 413:280, 1989 [PMID 2717375]
- Battle DC: Segmental characterization of defects in collecting tubule acidification. Kidney Int 30:546, 1986
- Tolins JP, Raij L: Chronic amphotericin B nephrotoxicity in the rat, protective effect of prophylactic salt loading. Am J Kidney Dis 11:313, 1988 [PMID 3354567]
- Sacks P, Fellner SK: Recurrent reversible acute renal failure from amphotericin. Arch Intern Med 147:593, 1987 [PMID 3827439]
- Lopez-Berestein G, Bodey GP, Fainstein V, et al: Treatment of systemic fungal infections with liposomal amphotericin B. Arch Intern Med 149:2533, 1989 [PMID 2818111]
- Sarosi GA: Nosocomial amphotericin B: current status (editorial). Arch Intern Med 149:2402, 1989
- Tolins JP, Raij L: Chronic amphotericin B nephrotoxicity in the rat: protective effect of calcium channel blockade. J Am Soc Nephrol 2:98, 1991 [PMID 1912413]
- Lachaal M, Venuto RC: Nephrotoxicity and hyperkalemia in patients with acquired immunodeficiency syndrome treated with pentamidine. Am J Med 87:260, 1989 [PMID 2773964]
- Sattler FR, Cowan R, Nielsen DM, et al: Trimethoprim-sulfamethoxazole compared with pentamidine for treatment of Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome: a prospective, noncrossover study. Ann Intern Med 109:280, 1988 [PMID 3260759]
- Miller RF, Delany S, Semple SJ: Acute renal failure after nebulised pent amidine (letter). Lancet 1:1271, 1989 [PMID 2566818]
-
Shah GM, Alvarado P, Kirschenbaum MA: Symptomatic hypocalcemia
and hypomagnesemia with renal magnesium wasting associated with pentamidine
therapy in a patient with AIDS. Am J Med 89:380, 1990 [PMID
2393042]
-
Kleyman TR, Roberts C, Ling BN: A mechanism for pentamidine-induced
hyperkalemia: inhibition of distal nephron sodium transport. Ann Intern
Med 122:103, 1995 [PMID
7992983]
-
Sawyer MH, Webb DE, Balow JE, et al: Acyclovir-induced
renal failure: clinical course and histology. Am J Med 84:1067, 1988 [PMID
3376977]
-
Eck P, Silver SM, Clark EC: Acute renal failure and
coma after a high dose of oral acyclovir (letter). N Engl J Med 325:1178,
1991 [PMID
1891032]
-
Sasson JP, Dratch PL, Shortsleeve MJ: Renal US findings
in sulfadiazine-induced crystalluria. Radiology 185:739, 1992 [PMID
1438755]
-
Hein R, Brunkhorst R, Thon WF, et al: Symptomatic sulfadiazine
crystalluria in AIDS patients: a report of two cases. Clin Nephrol 39:254,
1993 [PMID
8513601]
-
Pitman SW, Frei EI III: Weekly methotrexate-calcium
leucovorin rescue: effect of alkalinization on nephrotoxicity; pharmacokinetics
in the CNS; and use in CNS non-Hodgkin's lymphoma. Cancer Treat Rep 61:695,
1977 [PMID
18282]
-
Rybak MJ, Albrecht LM, Boike SC, et al: Nephrotoxicity
of vancomycin, alone and with an aminoglycoside. J Antimicrob Chemother
25:679, 1990 [PMID
2351627]
-
Perneger TV, Whelton PK, Klag MJ: Risk of kidney failure
associated with the use of acetaminophen, aspirin, and nonsteroidal antiiflammatory
drugs. N Engl J Med 331: 1675, 1994 [PMID
7969358]
-
Bjorck S, Svalander CT, Aurell M: Acute renal failure
after analgesic drugs including paracetamol (acetaminophen). Nephron 49:45,
1988 [PMID
3380218]
-
Berns JS, Cohen RM, Stumacher RJ, et al: Renal aspects
of therapy for human immunodeficiency virus and associated opportunistic
infections. J Am Soc Nephrol 1:1061, 1991 [PMID
1912406]
-
Skinner R, Pearson ADJ, Price L, et al: Nephrotoxicity
after ifosfamide. Arch Dis Child 65:732, 1990
-
Zamlauski-Tucker MJ, Morris M, Springate J: Ifosfamide
metabolite chloroacetaldehyde induces Fanconi syndrome in the perfused
rat kidney (abstr). J Am Soc Nephrol 4:762, 1993
- Sica DA, Gehr TWB: Triamterene and the kidney. Nephron 51:454, 1989 [PMID 2662034]