Reumatología
⭳ Abrir artículo (PDF)481.6 KBEste artículo fue revisado respecto a la Edición 3/2000. Ver esa versión →
Contenido del artículo
VIII VASCULITIS SISTEMICAS
- Clasificación y diagnóstico
- Etiología de las vasculitis
- Síndromes clínicos y su tratamiento
- ARTERITIS DE TAKAYASU
- ARTERITIS DE CELULAS GIGANTES
- POLIARTERITIS NODOSA
- VASCULITIS GRANULOMATOSAS
- Granulomatosis de Wegener
- Sindrome de Churg-Strauss: Angeítis y granulomatosis alérgica
- Angeítis limitada al sistema nervioso central
- Granulomatosis linfomatoide
- ENFERMEDAD DE BEHCET
- SINDROME DE COGAN
- ENFERMEDAD DE KAWASAKI
- POLIANGEITIS MICROSCOPICA
VIII VASCULITIS SISTEMICAS
DR. EDWARD D. HARRIS. JR.
Clasificación y diagnóstico
En todos los sistemas y órganos del cuerpo existen vasos sanguíneos con calibre y funciones diferentes. Por ello no es sorprendente que los síndromes clínicos que producen inflamación en los vasos sanguíneos, o vasculitis, puedan ser tan variados en su cuadro clínico y en su evolución.
El primer reto en el tratamiento de las vasculitis sistémicas es hacer el diagnóstico correcto. La inflamación vascular puede presentarse sin signos o síntomas específicos de un lecho vascular en particular. Los hallazgos sistémicos son inespecíficos y pueden sugerir alguna de las formas de enfermedad difusa del tejido conjuntivo, una neoplasia maligna o una infección. Si el paciente presenta estos hallazgos y se puede descartar una infección sistémica o una enfermedad maligna, se debe recurrir a la toma de una biopsia a ciegas o a los estudios por imágenes para hacer el diagnóstico.
La asociación entre autoinmunidad y enzimas lisosómicas ha sido útil para diagnosticar vasculitis. En las vasculitis se han encontrado dos de los principales tipos de anticuerpos citoplásmicos antineutrófilo (ANCA, por sus siglas en inglés, n. del t.): los c-ANCA, que están dirigidos contra la proteasa serina de los gránulos de los neutrófilos (proteinasa 3) y que se detectan por inmunofluorescencia citoplásmica, y los p-ANCA, que se dirigen sobre todo contra la mieloperoxidasa (MPO) y que se detectan por tinción perinuclear.1 Los datos sugieren que cuando las citocinas inflamatorias activan a los neutrófilos, estas proteasas migran a la superficie celular,2 y esta transmigración puede facilitar la activación adicional de neutrófilos y el daño tisular subsecuente. Los resultados positivos de c-ANCA tienen una sensibilidad del 85 porciento para la granulomatosis de Wegener. Los resultados positivos a MPO de p-ANCA se asocian sobre todo con poliarteritis nodosa microscópica, poliarteritis nodosa, síndrome de Churg-Strauss o glomerulonefritis idiopática con medias lunas. Por el contrario, en la enfermedad hepática o intestinal inflamatoria, los resultados positivos de p-ANCA suelen ser negativos para MPO.3 Las pruebas c-ANCA o MPO positiva p-ANCA positivas indican un 96 porciento de probabilidad de que un paciente tenga una forma de vasculitis, mientras que los resultados negativos de estas pruebas se asocian con más de 90 porciento de probabilidad de que el paciente no tenga vasculitis.
Además, cualquier enfermedad que produzca una lesión aguda de las células endoteliales es probable que produzca aumentos en los niveles del factor VIII-factor de Von Willebrand en la sangre (FvW); estos factores se elevan en la vasculitis, especialmente en la arteritis de células gigantes (ACG) y en la polimialgia reumática (PMR).4 La especificidad de la determinación del factor VIII-FvW debe investigarse más antes de poder determinar la utilidad de esta prueba.
La primera decisión que debe tomar el médico cuando diagnostica un padecimiento primario de los vasos sanguíneos es si se trata una trombosis, de vasculitis o de ambos. Desde el punto de vista clínico, la trombosis puede semejar vasculitis y vice versa. Por ejemplo, el síndrome antifosfolípido (SAF), descrito por primera vez en 1983,5 puede enmascararse como una vasculitis. Las características del SAF incluyen trombosis venosas profundas, episodios cerebrovasculares e isquemia cerebral transitoria, lívedo reticularis, hipertensión pulmonar, abortos recurrentes y, con menos frecuencia, hipertensión lábil, trombosis de la vena renal, migraña, epilepsia, mielopatía transversa, trombocitopenia, valculopatía cardiaca (por lo general de la válvula mitral) e isquemia ocular.6 Aunque el SAF fue descrito en un principio en pacientes con lupus eritematoso sistémico (LES), se ha encontrado un síndrome primario en pacientes sin LES. Un pequeño subgrupo de pacientes con SAF desarrollan colapso multisistémico con trombocitopenia, síndrome de insuficiencia respiratoria progresiva del adulto, ictericia y muerte causada por trombosis generalizada. Los anticuerpos AF están dirigidos contra la cardiolipina, un fosfolípido con carga negativa, en presencia de ß2-glucoproteína, que puede considerarse como un cofactor.6
Un motivo importante para diferenciar el SAF de las vasculitis es el tratamiento diferente en ambas enfermedades. Los esteroides, con frecuencia útiles en las vasculitis, rara vez están indicados en el SAF. Para este síndrome son tratamientos aceptados la aspirina en dosis bajas (75 mg/día) y la anticoagulación a largo plazo.7 Sin embargo, pueden encontrarse manifestaciones tanto de vasculitis como trombosis inapropiadas en la misma enfermedad, y el LES es un ejemplo frecuente.
Se han propuesto muchas clasificaciones para las vasculitis.8 La más reciente, propuesta por una conferencia de consenso internacional,9 es de especial utilidad porque estandariza la nomenclatura de las vasculitis. (ver tabla 1 para la lista de los síndromes de vasculitis.) El cambio más notable en la nueva clasificación es que elimina el término de angeítis por hipersensibilidad, que se usaba antes para abarcar a todas las vasculitis que afectan a los vasos cutáneos. Además, esta clasificación refleja el reconocimiento de que algunas formas de vasculitis afectan vasos con diámetro muy diferente. Por ejemplo, aunque la angeítis leucocitoclástica cutánea no daña vasos mayores que la arteriola, la granulomatosis de Wegener puede afectar vasos que varían en tamaño desde capilares hasta arterias pequeñas.
Etiología de las vasculitis
Sólo en los síndromes propiamente infecciosos se puede demostrar la etiología, aunque incluso en estos casos la fisiopatología raramente está bien definida. Por ejemplo, la neuropatía vasculítica asociada con la enfermedad de Lyme puede ser producida tanto por la invasión directa de la pared arterial por el agente etiológico Borrelia burgdorferi, como por el depósito de complejos inmunes.
Se conoce poco acerca de las causas de las vasculitis que afectan los grandes vasos. Hay algunos que relacionan a la ACG con la inmunidad celular contra componentes del tejido conjuntivo. Las infecciones por micoplasmas, herpes zoster o citomegalovirus han sido algunas veces ligadas a la angeítis aislada del SNC. Los virus se han señalado frecuentemente como causa primaria de la poliarteritis nodosa. La asociación con el virus de la hepatitis B o C es la más firme.10,11 Existen evidencias de infección por un virus linfotrópico en la enfermedad de Kawasaki. Las células mononucleares de sangre periférica de los pacientes con esta enfermedad presentan actividad de polimerasa retroviral.12
Un conjunto de síndromes vasculíticos se han relacionado con el virus de la inmunodeficiencia humana (VIH).13 Los hallazgos histológicos en varios de estos síndromes son similares a los observados en los procesos inmunoproliferativos angiocéntricos, caracterizados por una densa infiltración de las paredes vasculares por células T CD8+ y células plasmáticas.
Sin embargo, en la mayoría de estos síndromes se identifica un mecanismo de tipo inmune, generado en respuesta a uno o más factores, como la causa inmediata de la inflamación vascular. Se han descrito en los últimos 20 años varios ejemplos de este fenómeno. Un estudio demostró la presencia del antígeno de superficie de la hepatitis B (AgHBs) e IgM en las paredes arteriales de un paciente con poliarteritis nodosa,10 y se calcula que el 10 porciento de los casos de poliarteritis son secundarios a una respuesta de tipo inmune al AgHBs, aunque esto bien puede ser exagerado. En la vasculitis asociada con cáncer se han demostrado complejos inmunes formados por anticuerpos y antígenos tumorales.14 Se ha implicado a anticuerpos de IgG dirigidos contra la fracción C1q del complemento en la patogenia de la vasculitis urticariana hipocomplementémica.
Se han identificado anticuerpos dirigidos contra antígenos de células endoteliales en muchos tipos de vasculitis, incluyendo granulomatosis de Wegener, poliarteritis nodosa microscópica, enfermedad de Kawasaki y vasculitis reumatoidea.16 La investigación al respecto se ha centrado en el daño a las células endoteliales como causa principal de la vasculitis. Las citocinas y los anticuerpos pueden actuar sobre estas células de muchas maneras. Por ejemplo, la interleucina-1 (IL-1) es liberada por los macrófagos, células endoteliales y otras células. Además de sus otros efectos, la IL-1 puede inducir la expresión de moléculas de adhesión en las células endoteliales, permitiendo a los neutrófilos que se adhieran a estas células. El factor-a de necrosis tumoral ejerce la misma acción. La IL-8, producida en respuesta a la inflamación cerca de los vasos, ayuda a los neutrófilos a invadir las paredes vasculares. Por último, la unión de los ANCA a los neutrófilos puede oiginar degranulación neutrofílica y toxicidad de las células endoteliales (ver antes).16
Síndromes clínicos y su tratamiento
ARTERITIS DE TAKAYASU
La arteritis de Takayasu, también conocida como enfermedad sin pulsos, afecta principalmente a mujeres orientales jóvenes. En el grupo (de cohorte) más grande estudiado en Estados Unidos (60 pacientes), el 97 porciento fueron mujeres, y la edad promedio de inicio fue 25 años.17 Los síntomas inespecíficos son frecuentes, pero los síntomas específicos se relacionan con el lecho vascular afectado. Los vasos afectados con más frecuencia fueron las arterias cubital, radial, braquial, carótida y axilar. Se encontraron soplos arteriales y reducción de los pulsos.17
En el estudio de cohorte de 60 pacientes de los Institutos Nacionales de Salud de los EUA (NIH), los datos clínicos iniciales se clasificaron como vasculares, del SNC, musculoesqueléticos, sistémicos y cardiacos.17 Los síntomas y signos vasculares más frecuentes fueron claudicación (en el 35 porciento), debilidad o ausencia de pulso (22 porciento), soplo carotídeo (20 porciento), hipertensión (18 porciento) y carotodinia (17 porciento). Los datos del SNC incluyeron sensación de cabeza ligera y mareo asociados con estenosis de la arteria vertebral (18 porciento), aberraciones en la visión (nueve porciento) y episodios cerebrovasculares (tres porciento). Las alteraciones musculoesqueléticas incluyeron dolor articular, en la pared torácica y mialgias. Entre los síntomas generales se observó malestar (20 porciento) y fiebre (18 porciento). Existió insuficiencia aórtica al inicio de la arteritis en el ocho porciento de los pacientes. La velocidad de sedimentación globular (VSG), única prueba de laboratorio útil, estuvo elevada en el 75 porciento de los pacientes. De los pacientes de la serie de los NIH, el 65 porciento tuvo lesiones aórticas en la angiografía,17 detectándose lesiones estenóticas largas en todos los vasos afectados, incluyendo los del árbol pulmonar.
En la fase activa, la arteritis de Takayasu se caracteriza por engrosamiento e inflamación de la íntima sin degeneración fibrinoide de la media. Por el contrario, la ACG que afecta la aorta u otras arterias grandes ocasiona focos de necrosis de la media rodeados con células gigantes, y la afección de la íntima es mínima.18
Alrededor del 50 porciento de todos los pacientes con arteritis de Takayasu que son tratados con 1 mg/kg/día de prednisona oral por tres meses presentan remisión. Pueden agregarse medicamentos citotóxicos (ciclofosfamida o azatioprina, 1 mg/kg/día o methotrexate, 0.15 a 0.35 mg/kg/semana) si la prednisona no induce remisión. El methotrexate tiene menos efectos adversos y la dosis puede aumentarse hasta 25 mg/semana. Aunque estos agentes pueden inducir remisión en la mayoría de los pacientes, muchos enfermos sufren por lo menos una recaída. Como en el caso de todas las vasculitis tratadas con medicamentos inmunosupresores, la infección es la complicación más común y grave del tratamiento. Aunque la muerte no es frecuente en estos pacientes jóvenes, puede ocurrir por insuficiencia cardiaca, infarto del miocardio, enfermedad cerebrovascular, ruptura aneurismática o insuficiencia renal.
Los bloqueadores de los canales del calcio o los inhibidores de la enzima convertidora de angiotensina pueden ayudar a aliviar los síntomas hipertensivos asociados con arteritis de Takayasu.10 Sin embargo, a pesar de estas medidas, el padecimiento tiende a recurrir y el pronóstico es malo.20
Para manejar la hipertensión, al igual que en otros procesos que afectan a vasos más pequeños, es importante prescribir medicamentos que disminuyan la reactividad e inflamación vascular. Debido a que el vasoespasmo y la agregación plaquetaria pueden ocasionar síntomas isquémicos, los bloqueadores de los canales del calcio y los inhibidores de la agregación plaquetaria pueden ser benéficos cuando se combinan con esteroides.
ARTERITIS DE CELULAS GIGANTES
El diagnóstico de ACG es histopatológico; los síndromes clínicos con los que se asocia con más frecuencia son la arteritis temporal y la PMR.
Arteritis temporal
En la arteritis temporal están afectadas las arterias de diferentes calibres de la cabeza y el cuello, y sólo en raras ocasiones arterias de cualquier otra localización; el paciente se presenta habitualmente con cefalea, hipersensibilidad del cuero cabelludo o claudicación mandibular [ver figura 1]. Una forma de presentación menos frecuente es la neuritis óptica isquémica, secundaria a una arteritis oclusiva de las ramas de la arteria oftálmica, que produce una pérdida súbita unilateral de la visión. Los síntomas generales son fiebre, malestar general, fatiga y pérdida de peso. Las ramas superficiales de las arterias temporales con frecuencia están doloridas, engrosadas y/o se pierde el pulso. Los estudios de laboratorio muestran una VSG de Westergren mayor de 60 mm/h y anemia normocrómica microcítica; o normocítica. Los anticuerpos antinucleares y el factor reumatoide son negativos. La enfermedad es poco frecuente en personas menores de 50 años.21
Los hallazgos histopatológicos, que suelen observarse en las biopsias de las ramas superficiales de la arteria temporal, son característicos [ver figura 2] y consisten en fragmentación de la lámina elástica interna, marcada proliferación de la íntima con infiltrado histiocítico y formación de células gigantes. La vasculitis necrosante aguda y los cambios de tipo fibrinoide son frecuentes. Aunque estos cambios se han identificado con mayor frecuencia en la arteritis temporal, se han relacionado a otras manifestaciones clínicas como demencia, hepatopatía, vasculitis coronaria e infarto intestinal o de la vesícula biliar.
La arteritis temporal es, hasta cierto punto, una enfermedad autolimitada. Los pacientes que responden al tratamiento con esteroides suelen cursar sin evidencia de enfermedad durante varios años, después de disminuir lentamente los fármacos y eventualmente suspenderlos. Otros pacientes presentan recaídas, que con frecuencia van precedidas de aumento en la velocidad de eritrosedimentación y disminución en la concentración de hemoglobina.
La biopsia de las ramas de la arteria temporal es un procedimiento simple y seguro cuando se realiza por cirujanos con experiencia. Se deben obtener segmentos largos (3 a 4 cm) de las biopsias de los segmentos arteriales y, debido a que son comunes las lesiones en parches, deben examinarse múltiples cortes.22 En la práctica, si la probabilidad de la enfermedad es alta o moderada, está indicado iniciar el tratamiento con esteroides (60 a 80 mg de prednisona al día dividido en varias dosis), incluso antes de realizar la biopsia. Esta medida tiene por objeto evitar la pérdida visual. Sin embargo, es extraordinariamente raro que un paciente con síndrome de arteritis temporal sin síntomas visuales presente pérdida de la visión, incluso si el tratamiento se retrasa.
En una serie de 535 casos consecutivos de pacientes con biopsia de la arteria temporal, el porcentaje de pacientes con resultados positivos fue semejante (31 y 35 porciento) en los pacientes no tratados y en los tratados con esteroides (15 mg/día) hasta una semana antes de la biopsia.23 Por lo tanto, si existen signos clínicos de arteritis temporal, es razonable realizar una biopsia de la arteria, incluso si ya se comenzó el tratamiento esteroideo. Por otro lado, en un estudio de la Clínica Mayo, solo el nueve porciento de todos los pacientes con resultados negativos en la biopsia que fueron seguidos por un promedio de seis años requirieron tratamiento con esteroides.22
Las dosis altas de esteroides deben disminuirse después de varias semanas de tratamiento, sobre todo si se observa una mejoría inmediata. Es necesario realizar seguimiento de estos pacientes para vigilar la aparición de síntomas y la VSG. En algunos pacientes los esteroides pueden disminuirse y suspenderse después de seis meses; otros pacientes requerirán tratamiento de sostén con dosis que aceleran la osteopenia, producen cataratas y potencian la hiperglucemia, la hipertensión y la enfermedad vascular ateromatosa.
Polimialgia reumática
La polimialgia reumática (PMR) es un síndrome clínico y no un diagnóstico patológico. En alguna ocasión se consideró que la PMR era secundaria a la arteritis de células gigantes, aunque sólo el 20 porciento de las biopsias a ciegas de arterias temporales eran positivas. Sin embargo, en la actualidad suele admitirse que la polimialgia reumática es un síndrome que puede ser una manifestación de muchos procesos patológicos, incluyendo artritis reumatoide, cáncer y ACG.21
La PMR suele afectar a personas mayores de 50 años de edad, y las mujeres la padecen con el doble de frecuencia que los varones. Puede presentarse de forma aguda o insidiosa, produciendo dolor y rigidez en la musculatura proximal. También existe fatiga, depresión, pérdida de peso y fiebre. La incidencia de artropatía periférica en la polimialgia reumática no se ha determinado,24 y es aún motivo de controversia. La PMR es una enfermedad que afecta a las arterias de grande y mediano calibre y que se asocia con otros padecimientos, como la ACG y la artritis reumatoide. La prevalencia de la PMR puede igualar a la de la artritis reumatoide en individuos mayores de 50 años. Estas dos enfermedades con frecuencia se superponen en sus hallazgos clínicos y de laboratorio, y la clasificación representa en ocasiones un problema.25
Los signos y síntomas son inespecíficos. En el diagnóstico diferencial se incluye a la artritis reumatoide, la polimiositis, la enfermedad maligna oculta, las enfermedades infecciosas, los síndromes de dolor miofascial y las alteraciones funcionales. Los datos objetivos son pocos. En un estudio se encontró que la VSG estaba muy elevada en el 99 porciento de los pacientes, los niveles de hemoglobina disminuidos en el 47 porciento, los niveles de a2-globulina estaban alterados en el 33 porciento, los niveles de aspartato aminotransferasa estaban aumentados en el 23 porciento y los niveles de fosfatasa alcalina elevados en el 10 porciento.26 También es frecuente el aumento de fibrinógeno y las cifras normales de creatina cinasa (CK) y aldolasa. Una respuesta notable al tratamiento con esteroides (en 24 a 48 horas) confirma el diagnóstico, aunque este hallazgo no es específico.
Se desconoce la patogenia de la enfermedad. Los cambios histológicos están limitados a los vasos con lámina elástica íntegra. La microscopía electrónica muestra fragmentación de la lámina con acúmulo de mononucleares. Puede existir una lesión mediada por células y la arteritis podría representar una reacción inmunológica contra la membrana elástica dañada por el efecto de la edad avanzada.21
Debido a que la PMR y la ACG parecen ser parte del espectro de una misma enfermedad, el tratamiento debe dirigirse al alivio sintomático y a evitar la catastrófica pérdida de la visión. Un régimen terapéutico inicial con dosis bajas de prednisona (6 a 10 mg/día) o el empleo de agentes no esteroides pueden controlar la rigidez matutina y el dolor. En ausencia de síntomas oculares, la polimialgia reumática puede tratarse con dosis máximas de fármacos antinflamatorios no esteroides. Se puede administrar prednisona (10 a 15 mg diarios) para lograr una respuesta terapéutica más rápida, pero la toxicidad del uso prolongado de dosis superiores a 7.5 mg/día es elevada. Normalmente puede alcanzarse una dosis de mantenimiento baja en el plazo de unas semanas. Es importante evaluar de nuevo al paciente si aparecen síntomas oculares o de otro tipo; en este caso deberá aumentarse la dosis de esteroides. Algunos pacientes requieren de dosis bajas de esteroides de por vida; en otros pueden suspenderse después de dos a cuatro años de tratamiento.26
POLIARTERITIS NODOSA
La poliarteritis nodosa es una enfermedad multisistémica caracterizada por inflamación aguda y necrosis fibrinoide de las arterias de mediano y pequeño calibre. Por definición, excluye la afección de vasos más pequeños, los denominados microscópicos, como las arteriolas, capilares y vénulas. El término periarteritis nodosa se refería a los nódulos subcutáneos y viscerales que en ocasiones se observan a lo largo de las arterias afectadas. Aunque la etiología de la poliarteritis nodosa es desconocida, sus asociaciones patogénicas incluyen la antigenemia por hepatitis B (un estudio demostró una asociación del 30 porciento27), la enfermedad del suero, reacciones alérgicas a medicamentos y a otras toxinas, abuso de anfetaminas e infección por VIH.
La poliarteritis nodosa es tres veces más frecuente en varones que en mujeres. Normalmente se presenta entre los 20 y los 60 años de edad, aunque puede hacerlo a cualquier edad. Entre sus manifestaciones clínicas se incluyen fiebre, malestar general, debilidad, anorexia, pérdida de peso, mialgias y artralgias. Otras posibles complicaciones son glomerulonefritis, hipertensión, infarto del miocardio (debido a arteritis de las coronarias), pericarditis, neuropatías periféricas (mononeuritis múltiple), convulsiones, hemiplejia, hemoptisis, asma, arteritis mesentérica y erupciones.28
La afección renal, la causa más frecuente de muerte, se presenta en alrededor de un 75 porciento de los pacientes. Puede manifestarse como una arteritis de los vasos de mediano calibre, que produce hematuria, proteinuria, hipertensión y azoemia. De nuevo, por definición, no existe glomerulonefritis en la poliarteritis nodosa.
La participación de las arterias abdominales también produce complicaciones graves. Es una serie, cinco de 16 pacientes desarrollaron crisis abdominales relacionadas con la enfermedad, que requirieron de intervención quirúrgica. Los cinco pacientes afectados tenían datos de arteritis mesentérica con segmentos isquémicos que ocasionaron infarto y perforación.29
Sólo recientemente se ha observado la vasculitis asociada con infección por VIH. En un estudio, cuatro de 14 casos de vasculitis asociada a infección por VIH pudieron describirse como similares a poliarteritis, y se observó un infiltrado inflamatorio agudo en vasos de mediano y pequeño calibre de músculos, nervios y piel.13
El diagnóstico de poliarteritis nodosa depende de la evidencia histológica de vasculitis; un músculo con dolor a la palpación u otro órgano clínicamente afectado pueden servir como sitio para la toma de biopsia. La biopsia del músculo o de nervio es la prueba más específica para observar la vasculitis necrosante,30 aunque es relativamente poco sensible. La arteriografía visceral selectiva (v. gr., de las arterias renales o celiacas) puede demostrar la presencia de los aneurismas característicos de la poliarteritis nodosa [ver figura 3]. Los cambios en las pruebas de laboratorio no son específicos. Suele haber aumento de la velocidad de sedimentación globular (VSG); también pueden encontrarse anemia, leucocitosis o eosinofilia.
La poliarteritis nodosa es relativamente rara, con una frecuencia de aproximadamente seis casos por 100,0000 habitantes en la población general.28 Por lo menos se han comunicado 13 casos de poliarteritis nodosa en pacientes con fiebre familiar del Mediterráneo, lo que sugiere una incidencia mucho mayor en este subgrupo de población.31 La edad promedio de inicio de la enfermedad en este grupo es de 20.8 años, mucho menor que en la población general (45 años). En otros aspectos, la poliarteritis nodosa en pacientes con fiebre familiar del Mediterráneo no difiere significativamente de la observada en la población general.
La actitud terapéutica ante un paciente con poliarteritis nodosa clásica o con alguna de las vasculitis alérgicas relacionadas es empírico e incluye los siguientes pasos:28 (1) identificación y eliminación del antígeno, por ejemplo, un agente infeccioso o un medicamento, (2) tratamiento de una enfermedad primaria subyacente, como una neoplasia, (3) en síndromes en los cuales no exista evidencia de enfermedad grave diseminada o sistémica o en pacientes con reacciones medicamentosas por hipersensibilidad debe administrarse prednisona (1 mg/kg/día), (4) intentos continuos para disminuir la dosis de prednisona, y (5) ciclosfamida en dosis bajas (1 a 2 mg/kg/día), con o sin esteroides, en los pacientes con vasculitis necrosante diseminada o enfermos que no respondan al tratamiento esteroideo.
VASCULITIS GRANULOMATOSAS
Granulomatosis de Wegener
La granulomatosis de Wegener se caracteriza por la formación de granulomas necrosantes en las proximidades de los vasos inflamados.28 Los órganos más afectados son la mucosa nasal, los senos paranasales, el aparato respiratorio, el parénquima pulmonar y los riñones. Los pacientes pueden presentarse con pansinusitis recurrente o una enfermedad renal caracterizada por hematuria, piuria y azoemia. Los síntomas inespecíficos de este proceso incluyen fiebre, poliartralgias y poliartritis. También pueden aparecen nódulos necróticos en la piel. El diagnóstico se sospecha por la presentación clínica y los cambios radiológicos en los senos paranasales y el tejido pulmonar, y se confirma mediante el estudio histológico.
Como se mencionó antes, las pruebas para detectar c-ANCA tienen un alto grado de especificidad y sensibilidad para la granulomatosis de Wegener.32 Prácticamente, la única fuente de falsos positivos son otras formas de vasculitis. Es importante la forma de realizar esta prueba, ya que se requiere una gran experiencia para interpretar el estudio de inmunofluorescencia indirecta. Se dispone de pruebas de inmunoabsorción ligada a enzimas que utilizan antígenos altamente purificados.32,33
En la granulomatosis de Wegener no tratada, la muerte, casi siempre por enfermedad renal, suele ocurrir semanas o meses después del inicio de la enfermedad. Sin embargo, los avances terapéuticos han mejorado notablemente las posibilidades de vida. Los esteroides prolongan la supervivencia media algunos meses. Se ha logrado una respuesta aún mejor con la ciclosfamida, que constituye el tratamiento de elección.34 En pacientes con una evolución relativamente estable (función renal anormal pero sin insuficiencia renal fulminante) se administra ciclofosfamida por vía oral (1 a 2 mg/kg/día) durante dos semanas. Si el tratamiento tiene éxito, se ajusta la dosis para mantener el recuento leucocitario por encima de 3,000/mm.3 35,36 Si no existe una respuesta favorable, debe incrementarse la dosis en 25 mg cada dos semanas, hasta que haya respuesta o se presente leucopenia. En los pacientes en los que la enfermedad ponga en peligro la vida se administra ciclofosfamida (4 mg/kg/día) durante tres días, continuando después con el régimen ya mencionado. En pacientes con enfermedad grave se añaden dosis altas de prednisona (60 mg/día), que se disminuye en forma gradual en algunas semanas hasta administrarla en días alternos. Posteriormente pueden suspenderse los esteroides y mantener al paciente sólo con el agente citotóxico. En muchos pacientes la enfermedad remite, permitiendo suspender el tratamiento en el plazo de 12 a 18 meses. El tratamiento con pulsos de ciclofosfamida es una alternativa en la enfermedad agresiva.37 También se demostrado que el trimetoprim-sulfametoxazol es útil en la granulomatosis de Wegener limitada a algunos órganos y con una severidad moderada.38
Sindrome de Churg-Strauss: Angeítis y granulomatosis alérgica
El síndrome de Churg-Strauss se caracteriza por eosinofilia y vasculitis sistémica que se presenta en pacientes con asma y rinitis alérgica. La afección pulmonar fulminante puede ser mortal en pacientes con este síndrome, aunque las lesiones iniciales pueden variar desde infiltrados parenquimatosos no segmentarios a enfermedad intersticial difusa. Los esteroides constituyen el tratamiento principal, aunque se ha demostrado que la ciclofosfamida (750 mg), administrada en pulsos por vía intravenosa, puede ser eficaz en pacientes con enfermedad que pone en peligro la vida y es resistente a los esteroides.37
Angeítis limitada al sistema nervioso central
La angeítis limitada al SNC, difícil de diagnosticar por estar limitados los cambios histopatológicos a la vasculatura intracraneal, puede ser progresiva y poner en peligro la vida del paciente. Consiste en un proceso granulomatoso vascular con cambios histopatológicos semejantes a los encontrados en la granulomatosis de Wegener y que afecta a los vasos de gran calibre que surgen de las arterias carótidas o basilar. Los síntomas y signos pueden variar desde una disfunción global, similar a una encefalitis, hasta alteraciones más localizadas que simulan un accidente cerebrovascular. Aunque la angiografía con contraste es el procedimiento que define las características de los vasos relacionados con vasculitis, los estudios actuales con imágenes por resonancia magnética sugieren que esta modalidad puede ser útil en el diagnóstico. El tratamiento se limita a la administración de esteroides en dosis altas; no se han comunicado ensayos con fármacos citotóxicos, como la ciclofosfamida.28
Granulomatosis linfomatoide
La granulomatosis linfomatoide es una forma poco frecuente de vasculitis,39 caracterizada por infiltración destructiva de varios tejidos, especialmente los pulmones, por células linfocitoides y plasmocitoides atípicas. El infiltrado es visible en la angiografía. Los granulomas y la necrosis son menos característicos que en la granulomatosis de Wegener, pero el tratamiento es el mismo para ambas enfermedades; la forma más agresiva de esta enfermedad es considerado como un linfoma angiocéntrico y debe tratarse como tal.
ENFERMEDAD DE BEHCET
Esta enfermedad se describió en 1937 como un síndrome crónico con recaídas, caracterizado por ulceración oral, úlceras genitales dolorosas y uveítis. Es una forma característica de vasculitis40 y se asocia a manifestaciones como sinovitis, vasculitis cutánea similar al eritema nosodo, meningoencefalitis, flebitis, aneurismas de grandes arterias y úlceras intestinales. En Japón, la prevalencia de la enfermedad de Behçet es de uno por 1,000 individuos; en los Estados Unidos es menor de uno por15,000. En Japón la prevalencia del antígeno de histocompatibilidad HLA-B5 es tres veces superior en los pacientes con esta enfermedad, pero esto no se ha observado en los Estados Unidos. Desde el punto de histopatológico la enfermedad se caracteriza por un importante infiltrado perivascular no necrosante de linfocitos y monocitos.
El tratamiento con esteroides tiene sólo una eficacia moderada en la enfermedad de Behçet. Los mejores resultados se han obtenido con la administración oral de clorambucil (0.1 a 0.2 mg/kg/día) y este esquema ha sido eficaz en el tratamiento tanto de la meningoencefalitis como de las manifestaciones mucosas de la enfermedad.41 Se ha recomendado el tratamiento con ciclosporina (5 mg/kg/día) para la enfermedad ocular.42
SINDROME DE COGAN
El síndrome de Cogan consiste en una queratitis intersticial no luética y enfermedad vestíbulo-auditiva. Se presenta en adultos jóvenes y se manifiesta por dolor ocular, fotofobia, vértigo e hipoacusia progresiva. El tratamiento consiste en la administración de dosis altas de esteroides (60 a 100 mg de prednisona al día); la dosis exacta se determina según la respuesta terapéutica a través de pruebas auditivas y de la visión.
ENFERMEDAD DE KAWASAKI
En 1967 Kawasaki describió un nuevo síndrome clínico en niños japoneses.43 Los rasgos cardinales de la enfermedad de Kawasaki (también llamada síndrome mucocutáneo con adenopatías) son fiebre, inyección conjuntival, labios secos y enrojecidos, eritema en palmas y plantas, edema indurado de las zonas distales de las extremidades, descamación de las puntas de los dedos, eritema cutáneo del tórax y linfoadenopatía cervical. En algunos aspectos la enfermedad se parece a la escarlatina o al síndrome de Steven-Johnson. También se pueden presentar complicaciones cardiovasculares importantes, como miocarditis, pericarditis, soplos, aneurismas arteriales coronarios, infarto del miocardio, angina de pecho, enfermedad vascular cerebral, insuficiencia vascular periférica y gangrena. Otras complicaciones son hidropesía de la vesícula, hepatitis, obstrución del intestino delgado, anemia, artritis, meningitis aséptica, uveítis, piuria y meatitis. La enfermedad suele ser autolimitada, pero en algunos casos ha ocurrido la muerte por trombosis coronaria durante la etapa aguda de la enfermedad o varios meses después de una aparente recuperación completa. La tasa de mortalidad es del uno al dos porciento.
No hay datos de que la enfermedad se trasmita de persona a persona. En las células mononucleares de sangre periférica se ha encontrado actividad de transcriptasa inversa, que se ha podido trasmitir a células T en cultivos.13 Estos hallazgos, junto con la linfocitopenia de células T y la marcada activación de las células B, apoyan la posibilidad de que el síndrome sea causado por un retrovirus.44 Se ha relacionado la fase aguda del síndrome de Kawasaki con la presencia de anticuerpos citotóxicos contra las células endoteliales.45
Un estudio multicéntrico aleatorio ha mostrado que un esquema de gamaglobulina administrada por vía intravenosa en dosis elevadas, junto con ácido acetilsalicílico, es eficaz para reducir la prevalencia de las alteraciones en la circulación coronaria y la severidad de las manifestaciones inflamatorias sistémicas.46 Se administran 400 mg/kg/día de gamaglobulina durante dos horas por cuatro días consecutivos y 100 mg/kg de ácido acetilsalicílico diariamente, divididos en cuatro dosis, durante 14 días de la enfermedad, posteriormente se reduce la dosis a 3 a 5 mg/kg/día en una sola dosis. Este tratamiento debe administrarse en las etapas tempranas de la enfermedad y parece ser el tratamiento más eficaz. Se recomienda a los médicos que informen los casos de este síndrome a sus departamentos de salud locales y estales.
POLIANGEITIS MICROSCOPICA
La manifestación más frecuente de la este tipo de angeítis es la púrpura palpable de las extremidades inferiores y las áreas en declive, que se relaciona con reacciones idiosincráticas por fármacos. Sin embargo, muchas y diferentes situaciones pueden precipitar los mismos cambios patológicos tanto en la piel como en múltiples órganos. Las lesiones papulares, no blanquecinas, aparecen en grupos, suelen abrirse al cuarto día y al evolucionar cambian de color rojo-púrpura a café, aplanándose y diseminándose. La afección glomerular puede producir insuficiencia renal, las lesiones intestinales causan hemorragias gastrointestinales. Cuando se afecta el parénquima pulmonar pueden observarse infiltrados difusos o nodulares, y la afección neurológica se manifiesta por cefalea, diplopia y síntomas orgánicos cerebrales.
En ocasiones se han observado pacientes que tienen una forma crónica y recurrente de poliangeítis microscópica en ausencia de enfermedad generalizada o de un suceso precipitante obvio. Los pacientes con esta enfermedad no muestran progresión hacia una afección generalizada durante el seguimiento a largo plazo. Sin embargo, este proceso no suele responder a los esteroides ni a la ciclofosfamida y, por lo tanto, deberá evitarse el uso de regímenes inmunosupresores tóxicos en estos pacientes.47 El tratamiento de las lesiones de las extremidades inferiores es difícil.
Púrpura de Henoch-Schönlein
La púrpura de Henoch-Schönlein afecta primariamente a las arteriolas pequeñas y a las capilares en los niños. Los órganos más afectados son la piel, las grandes articulaciones, los riñones y el aparato gastrointestinal. Se ha observado un depósito simultáneo de inmunoglobulinas (predominantemente IgA), componentes del complemento y fibrina en las paredes de los vasos cutáneos afectados, en el mesangio glomerular y en el tubo digestivo.48 La púrpura de Henoch-Schönlein con frecuencia es benigna, pero en pacientes con participación renal puede desarrollarse insuficiencia renal. Aunque suele ser suficiente el tratamiento sintomático, la enfermedad renal asociada se trata generalmente con esteroides (40 a 60 mg/día en adultos).
Síndrome de Goodpasture
Este síndrome se caracteriza por una marcada capilaritis pulmonar, púrpura palpable y glomerulitis. Se asocia con la presencia de anticuerpos circulantes contra los componentes de la colágena tipo IV de las membranas basales vasculares. Las pruebas de estos anticuerpos y para los ANCA pueden ayudar a diferenciar esta alteración de la granulomatosis de Wegener.1,3
Vasculitis urticariana hipocomplementémica
La vasculitis urticariana hipocomplementémica (VUH) es un síndrome poco frecuente que se caracteriza por urticaria recurrente y angioedema. Las lesiones urticarianas muestran una vasculitis leucocitoclástica. También puede presentarse conjuntivitis, uveítis, glomerulonefritis, dolor abdominal, artritis o artralgias. La enfermedad pulmonar obstructiva crónica parece acentuarse en los fumadores con este proceso, pero se desconoce la causa.49 Hay una considerable superposición clínica con el LES, pero los estudios inmunológicos recientes han establecido importantes diferencias entre estos dos síndromes. La IgG purificada de pacientes con vasculitis urticariana hipocomplementémica tiene una actividad de unión para el primer componente del complemento, C1q.15 Se ha postulado que la activación de C1 por anticuerpos anti-C1q puede conducir a la activación de la fase fluida de la cascada del complemento, y de esta forma generar los cambios patológicos que se observan clínicamente.
Las lesiones urticarianas se presentan principalmente en la cara, extremidades superiores y tórax. La urticaria puede durar más de 48 horas y desvanecerse sin dejar huellas, a diferencia de la púrpura palpable. En comparación con el LES, no se encuentran anticuerpos antinucleares en el plasma. Los esteroides suelen ser eficaces en el alivio de los signos y síntomas.
Vasculitis crioglobulinémica
Las crioglobulinas son inmunoglobulinas y complejos inmunes que se precipitan con el frío (4°C) y se disuelven al volver a calentarse. Pueden estar relacionadas con diversas enfermedades y síntomas reumatológicos, como fenómeno de Raynaud, vasculitis, púrpura, artralgias, fiebre y enfermedad renal. Los complejos inmunes pueden en parte ser responsable de la vasculitis asociada. El principal síndrome clínico asociado con crioprecipitados de IgG e IgM se caracteriza por púrpura, artralgias y debilidad, y suele cursar con glomerulonefritis rápidamente progresiva. Dos terceras partes de estos pacientes son positivos para AgHBs o para los anti-HBs cuando se analiza el plasma y los crioprecipitados.50 Se ha usado la plasmaféresis en el tratamiento de los casos graves resistentes a los esteroides.
Bibliografía
DR. EDWARD D. HARRIS. JR.
Clasificación y diagnóstico
En todos los sistemas y órganos del cuerpo existen vasos sanguíneos con calibre y funciones diferentes. Por ello no es sorprendente que los síndromes clínicos que producen inflamación en los vasos sanguíneos, o vasculitis, puedan ser tan variados en su cuadro clínico y en su evolución.
El primer reto en el tratamiento de las vasculitis sistémicas es hacer el diagnóstico correcto. La inflamación vascular puede presentarse sin signos o síntomas específicos de un lecho vascular en particular. Los hallazgos sistémicos son inespecíficos y pueden sugerir alguna de las formas de enfermedad difusa del tejido conjuntivo, una neoplasia maligna o una infección. Si el paciente presenta estos hallazgos y se puede descartar una infección sistémica o una enfermedad maligna, se debe recurrir a la toma de una biopsia a ciegas o a los estudios por imágenes para hacer el diagnóstico.
La asociación entre autoinmunidad y enzimas lisosómicas ha sido útil para diagnosticar vasculitis. En las vasculitis se han encontrado dos de los principales tipos de anticuerpos citoplásmicos antineutrófilo (ANCA, por sus siglas en inglés, n. del t.): los c-ANCA, que están dirigidos contra la proteasa serina de los gránulos de los neutrófilos (proteinasa 3) y que se detectan por inmunofluorescencia citoplásmica, y los p-ANCA, que se dirigen sobre todo contra la mieloperoxidasa (MPO) y que se detectan por tinción perinuclear.1 Los datos sugieren que cuando las citocinas inflamatorias activan a los neutrófilos, estas proteasas migran a la superficie celular,2 y esta transmigración puede facilitar la activación adicional de neutrófilos y el daño tisular subsecuente. Los resultados positivos de c-ANCA tienen una sensibilidad del 85 porciento para la granulomatosis de Wegener. Los resultados positivos a MPO de p-ANCA se asocian sobre todo con poliarteritis nodosa microscópica, poliarteritis nodosa, síndrome de Churg-Strauss o glomerulonefritis idiopática con medias lunas. Por el contrario, en la enfermedad hepática o intestinal inflamatoria, los resultados positivos de p-ANCA suelen ser negativos para MPO.3 Las pruebas c-ANCA o MPO positiva p-ANCA positivas indican un 96 porciento de probabilidad de que un paciente tenga una forma de vasculitis, mientras que los resultados negativos de estas pruebas se asocian con más de 90 porciento de probabilidad de que el paciente no tenga vasculitis.
Además, cualquier enfermedad que produzca una lesión aguda de las células endoteliales es probable que produzca aumentos en los niveles del factor VIII-factor de Von Willebrand en la sangre (FvW); estos factores se elevan en la vasculitis, especialmente en la arteritis de células gigantes (ACG) y en la polimialgia reumática (PMR).4 La especificidad de la determinación del factor VIII-FvW debe investigarse más antes de poder determinar la utilidad de esta prueba.
La primera decisión que debe tomar el médico cuando diagnostica un padecimiento primario de los vasos sanguíneos es si se trata una trombosis, de vasculitis o de ambos. Desde el punto de vista clínico, la trombosis puede semejar vasculitis y vice versa. Por ejemplo, el síndrome antifosfolípido (SAF), descrito por primera vez en 1983,5 puede enmascararse como una vasculitis. Las características del SAF incluyen trombosis venosas profundas, episodios cerebrovasculares e isquemia cerebral transitoria, lívedo reticularis, hipertensión pulmonar, abortos recurrentes y, con menos frecuencia, hipertensión lábil, trombosis de la vena renal, migraña, epilepsia, mielopatía transversa, trombocitopenia, valculopatía cardiaca (por lo general de la válvula mitral) e isquemia ocular.6 Aunque el SAF fue descrito en un principio en pacientes con lupus eritematoso sistémico (LES), se ha encontrado un síndrome primario en pacientes sin LES. Un pequeño subgrupo de pacientes con SAF desarrollan colapso multisistémico con trombocitopenia, síndrome de insuficiencia respiratoria progresiva del adulto, ictericia y muerte causada por trombosis generalizada. Los anticuerpos AF están dirigidos contra la cardiolipina, un fosfolípido con carga negativa, en presencia de ß2-glucoproteína, que puede considerarse como un cofactor.6
Un motivo importante para diferenciar el SAF de las vasculitis es el tratamiento diferente en ambas enfermedades. Los esteroides, con frecuencia útiles en las vasculitis, rara vez están indicados en el SAF. Para este síndrome son tratamientos aceptados la aspirina en dosis bajas (75 mg/día) y la anticoagulación a largo plazo.7 Sin embargo, pueden encontrarse manifestaciones tanto de vasculitis como trombosis inapropiadas en la misma enfermedad, y el LES es un ejemplo frecuente.
Se han propuesto muchas clasificaciones para las vasculitis.8 La más reciente, propuesta por una conferencia de consenso internacional,9 es de especial utilidad porque estandariza la nomenclatura de las vasculitis. (ver tabla 1 para la lista de los síndromes de vasculitis.) El cambio más notable en la nueva clasificación es que elimina el término de angeítis por hipersensibilidad, que se usaba antes para abarcar a todas las vasculitis que afectan a los vasos cutáneos. Además, esta clasificación refleja el reconocimiento de que algunas formas de vasculitis afectan vasos con diámetro muy diferente. Por ejemplo, aunque la angeítis leucocitoclástica cutánea no daña vasos mayores que la arteriola, la granulomatosis de Wegener puede afectar vasos que varían en tamaño desde capilares hasta arterias pequeñas.
|
||
* También puede clasificarse como vasculitis granulomatosa. |
Etiología de las vasculitis
Sólo en los síndromes propiamente infecciosos se puede demostrar la etiología, aunque incluso en estos casos la fisiopatología raramente está bien definida. Por ejemplo, la neuropatía vasculítica asociada con la enfermedad de Lyme puede ser producida tanto por la invasión directa de la pared arterial por el agente etiológico Borrelia burgdorferi, como por el depósito de complejos inmunes.
Se conoce poco acerca de las causas de las vasculitis que afectan los grandes vasos. Hay algunos que relacionan a la ACG con la inmunidad celular contra componentes del tejido conjuntivo. Las infecciones por micoplasmas, herpes zoster o citomegalovirus han sido algunas veces ligadas a la angeítis aislada del SNC. Los virus se han señalado frecuentemente como causa primaria de la poliarteritis nodosa. La asociación con el virus de la hepatitis B o C es la más firme.10,11 Existen evidencias de infección por un virus linfotrópico en la enfermedad de Kawasaki. Las células mononucleares de sangre periférica de los pacientes con esta enfermedad presentan actividad de polimerasa retroviral.12
Un conjunto de síndromes vasculíticos se han relacionado con el virus de la inmunodeficiencia humana (VIH).13 Los hallazgos histológicos en varios de estos síndromes son similares a los observados en los procesos inmunoproliferativos angiocéntricos, caracterizados por una densa infiltración de las paredes vasculares por células T CD8+ y células plasmáticas.
Sin embargo, en la mayoría de estos síndromes se identifica un mecanismo de tipo inmune, generado en respuesta a uno o más factores, como la causa inmediata de la inflamación vascular. Se han descrito en los últimos 20 años varios ejemplos de este fenómeno. Un estudio demostró la presencia del antígeno de superficie de la hepatitis B (AgHBs) e IgM en las paredes arteriales de un paciente con poliarteritis nodosa,10 y se calcula que el 10 porciento de los casos de poliarteritis son secundarios a una respuesta de tipo inmune al AgHBs, aunque esto bien puede ser exagerado. En la vasculitis asociada con cáncer se han demostrado complejos inmunes formados por anticuerpos y antígenos tumorales.14 Se ha implicado a anticuerpos de IgG dirigidos contra la fracción C1q del complemento en la patogenia de la vasculitis urticariana hipocomplementémica.
Se han identificado anticuerpos dirigidos contra antígenos de células endoteliales en muchos tipos de vasculitis, incluyendo granulomatosis de Wegener, poliarteritis nodosa microscópica, enfermedad de Kawasaki y vasculitis reumatoidea.16 La investigación al respecto se ha centrado en el daño a las células endoteliales como causa principal de la vasculitis. Las citocinas y los anticuerpos pueden actuar sobre estas células de muchas maneras. Por ejemplo, la interleucina-1 (IL-1) es liberada por los macrófagos, células endoteliales y otras células. Además de sus otros efectos, la IL-1 puede inducir la expresión de moléculas de adhesión en las células endoteliales, permitiendo a los neutrófilos que se adhieran a estas células. El factor-a de necrosis tumoral ejerce la misma acción. La IL-8, producida en respuesta a la inflamación cerca de los vasos, ayuda a los neutrófilos a invadir las paredes vasculares. Por último, la unión de los ANCA a los neutrófilos puede oiginar degranulación neutrofílica y toxicidad de las células endoteliales (ver antes).16
Síndromes clínicos y su tratamiento
ARTERITIS DE TAKAYASU
La arteritis de Takayasu, también conocida como enfermedad sin pulsos, afecta principalmente a mujeres orientales jóvenes. En el grupo (de cohorte) más grande estudiado en Estados Unidos (60 pacientes), el 97 porciento fueron mujeres, y la edad promedio de inicio fue 25 años.17 Los síntomas inespecíficos son frecuentes, pero los síntomas específicos se relacionan con el lecho vascular afectado. Los vasos afectados con más frecuencia fueron las arterias cubital, radial, braquial, carótida y axilar. Se encontraron soplos arteriales y reducción de los pulsos.17
En el estudio de cohorte de 60 pacientes de los Institutos Nacionales de Salud de los EUA (NIH), los datos clínicos iniciales se clasificaron como vasculares, del SNC, musculoesqueléticos, sistémicos y cardiacos.17 Los síntomas y signos vasculares más frecuentes fueron claudicación (en el 35 porciento), debilidad o ausencia de pulso (22 porciento), soplo carotídeo (20 porciento), hipertensión (18 porciento) y carotodinia (17 porciento). Los datos del SNC incluyeron sensación de cabeza ligera y mareo asociados con estenosis de la arteria vertebral (18 porciento), aberraciones en la visión (nueve porciento) y episodios cerebrovasculares (tres porciento). Las alteraciones musculoesqueléticas incluyeron dolor articular, en la pared torácica y mialgias. Entre los síntomas generales se observó malestar (20 porciento) y fiebre (18 porciento). Existió insuficiencia aórtica al inicio de la arteritis en el ocho porciento de los pacientes. La velocidad de sedimentación globular (VSG), única prueba de laboratorio útil, estuvo elevada en el 75 porciento de los pacientes. De los pacientes de la serie de los NIH, el 65 porciento tuvo lesiones aórticas en la angiografía,17 detectándose lesiones estenóticas largas en todos los vasos afectados, incluyendo los del árbol pulmonar.
En la fase activa, la arteritis de Takayasu se caracteriza por engrosamiento e inflamación de la íntima sin degeneración fibrinoide de la media. Por el contrario, la ACG que afecta la aorta u otras arterias grandes ocasiona focos de necrosis de la media rodeados con células gigantes, y la afección de la íntima es mínima.18
Alrededor del 50 porciento de todos los pacientes con arteritis de Takayasu que son tratados con 1 mg/kg/día de prednisona oral por tres meses presentan remisión. Pueden agregarse medicamentos citotóxicos (ciclofosfamida o azatioprina, 1 mg/kg/día o methotrexate, 0.15 a 0.35 mg/kg/semana) si la prednisona no induce remisión. El methotrexate tiene menos efectos adversos y la dosis puede aumentarse hasta 25 mg/semana. Aunque estos agentes pueden inducir remisión en la mayoría de los pacientes, muchos enfermos sufren por lo menos una recaída. Como en el caso de todas las vasculitis tratadas con medicamentos inmunosupresores, la infección es la complicación más común y grave del tratamiento. Aunque la muerte no es frecuente en estos pacientes jóvenes, puede ocurrir por insuficiencia cardiaca, infarto del miocardio, enfermedad cerebrovascular, ruptura aneurismática o insuficiencia renal.
Los bloqueadores de los canales del calcio o los inhibidores de la enzima convertidora de angiotensina pueden ayudar a aliviar los síntomas hipertensivos asociados con arteritis de Takayasu.10 Sin embargo, a pesar de estas medidas, el padecimiento tiende a recurrir y el pronóstico es malo.20
Para manejar la hipertensión, al igual que en otros procesos que afectan a vasos más pequeños, es importante prescribir medicamentos que disminuyan la reactividad e inflamación vascular. Debido a que el vasoespasmo y la agregación plaquetaria pueden ocasionar síntomas isquémicos, los bloqueadores de los canales del calcio y los inhibidores de la agregación plaquetaria pueden ser benéficos cuando se combinan con esteroides.
ARTERITIS DE CELULAS GIGANTES
El diagnóstico de ACG es histopatológico; los síndromes clínicos con los que se asocia con más frecuencia son la arteritis temporal y la PMR.
Arteritis temporal
En la arteritis temporal están afectadas las arterias de diferentes calibres de la cabeza y el cuello, y sólo en raras ocasiones arterias de cualquier otra localización; el paciente se presenta habitualmente con cefalea, hipersensibilidad del cuero cabelludo o claudicación mandibular [ver figura 1]. Una forma de presentación menos frecuente es la neuritis óptica isquémica, secundaria a una arteritis oclusiva de las ramas de la arteria oftálmica, que produce una pérdida súbita unilateral de la visión. Los síntomas generales son fiebre, malestar general, fatiga y pérdida de peso. Las ramas superficiales de las arterias temporales con frecuencia están doloridas, engrosadas y/o se pierde el pulso. Los estudios de laboratorio muestran una VSG de Westergren mayor de 60 mm/h y anemia normocrómica microcítica; o normocítica. Los anticuerpos antinucleares y el factor reumatoide son negativos. La enfermedad es poco frecuente en personas menores de 50 años.21
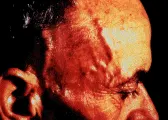
|
| Figura 1 |
| Arteritis de la temporal |
Los hallazgos histopatológicos, que suelen observarse en las biopsias de las ramas superficiales de la arteria temporal, son característicos [ver figura 2] y consisten en fragmentación de la lámina elástica interna, marcada proliferación de la íntima con infiltrado histiocítico y formación de células gigantes. La vasculitis necrosante aguda y los cambios de tipo fibrinoide son frecuentes. Aunque estos cambios se han identificado con mayor frecuencia en la arteritis temporal, se han relacionado a otras manifestaciones clínicas como demencia, hepatopatía, vasculitis coronaria e infarto intestinal o de la vesícula biliar.

|
| Figura 2 |
| Biopsia de la arteria temporal |
La arteritis temporal es, hasta cierto punto, una enfermedad autolimitada. Los pacientes que responden al tratamiento con esteroides suelen cursar sin evidencia de enfermedad durante varios años, después de disminuir lentamente los fármacos y eventualmente suspenderlos. Otros pacientes presentan recaídas, que con frecuencia van precedidas de aumento en la velocidad de eritrosedimentación y disminución en la concentración de hemoglobina.
La biopsia de las ramas de la arteria temporal es un procedimiento simple y seguro cuando se realiza por cirujanos con experiencia. Se deben obtener segmentos largos (3 a 4 cm) de las biopsias de los segmentos arteriales y, debido a que son comunes las lesiones en parches, deben examinarse múltiples cortes.22 En la práctica, si la probabilidad de la enfermedad es alta o moderada, está indicado iniciar el tratamiento con esteroides (60 a 80 mg de prednisona al día dividido en varias dosis), incluso antes de realizar la biopsia. Esta medida tiene por objeto evitar la pérdida visual. Sin embargo, es extraordinariamente raro que un paciente con síndrome de arteritis temporal sin síntomas visuales presente pérdida de la visión, incluso si el tratamiento se retrasa.
En una serie de 535 casos consecutivos de pacientes con biopsia de la arteria temporal, el porcentaje de pacientes con resultados positivos fue semejante (31 y 35 porciento) en los pacientes no tratados y en los tratados con esteroides (15 mg/día) hasta una semana antes de la biopsia.23 Por lo tanto, si existen signos clínicos de arteritis temporal, es razonable realizar una biopsia de la arteria, incluso si ya se comenzó el tratamiento esteroideo. Por otro lado, en un estudio de la Clínica Mayo, solo el nueve porciento de todos los pacientes con resultados negativos en la biopsia que fueron seguidos por un promedio de seis años requirieron tratamiento con esteroides.22
Las dosis altas de esteroides deben disminuirse después de varias semanas de tratamiento, sobre todo si se observa una mejoría inmediata. Es necesario realizar seguimiento de estos pacientes para vigilar la aparición de síntomas y la VSG. En algunos pacientes los esteroides pueden disminuirse y suspenderse después de seis meses; otros pacientes requerirán tratamiento de sostén con dosis que aceleran la osteopenia, producen cataratas y potencian la hiperglucemia, la hipertensión y la enfermedad vascular ateromatosa.
Polimialgia reumática
La polimialgia reumática (PMR) es un síndrome clínico y no un diagnóstico patológico. En alguna ocasión se consideró que la PMR era secundaria a la arteritis de células gigantes, aunque sólo el 20 porciento de las biopsias a ciegas de arterias temporales eran positivas. Sin embargo, en la actualidad suele admitirse que la polimialgia reumática es un síndrome que puede ser una manifestación de muchos procesos patológicos, incluyendo artritis reumatoide, cáncer y ACG.21
La PMR suele afectar a personas mayores de 50 años de edad, y las mujeres la padecen con el doble de frecuencia que los varones. Puede presentarse de forma aguda o insidiosa, produciendo dolor y rigidez en la musculatura proximal. También existe fatiga, depresión, pérdida de peso y fiebre. La incidencia de artropatía periférica en la polimialgia reumática no se ha determinado,24 y es aún motivo de controversia. La PMR es una enfermedad que afecta a las arterias de grande y mediano calibre y que se asocia con otros padecimientos, como la ACG y la artritis reumatoide. La prevalencia de la PMR puede igualar a la de la artritis reumatoide en individuos mayores de 50 años. Estas dos enfermedades con frecuencia se superponen en sus hallazgos clínicos y de laboratorio, y la clasificación representa en ocasiones un problema.25
Los signos y síntomas son inespecíficos. En el diagnóstico diferencial se incluye a la artritis reumatoide, la polimiositis, la enfermedad maligna oculta, las enfermedades infecciosas, los síndromes de dolor miofascial y las alteraciones funcionales. Los datos objetivos son pocos. En un estudio se encontró que la VSG estaba muy elevada en el 99 porciento de los pacientes, los niveles de hemoglobina disminuidos en el 47 porciento, los niveles de a2-globulina estaban alterados en el 33 porciento, los niveles de aspartato aminotransferasa estaban aumentados en el 23 porciento y los niveles de fosfatasa alcalina elevados en el 10 porciento.26 También es frecuente el aumento de fibrinógeno y las cifras normales de creatina cinasa (CK) y aldolasa. Una respuesta notable al tratamiento con esteroides (en 24 a 48 horas) confirma el diagnóstico, aunque este hallazgo no es específico.
Se desconoce la patogenia de la enfermedad. Los cambios histológicos están limitados a los vasos con lámina elástica íntegra. La microscopía electrónica muestra fragmentación de la lámina con acúmulo de mononucleares. Puede existir una lesión mediada por células y la arteritis podría representar una reacción inmunológica contra la membrana elástica dañada por el efecto de la edad avanzada.21
Debido a que la PMR y la ACG parecen ser parte del espectro de una misma enfermedad, el tratamiento debe dirigirse al alivio sintomático y a evitar la catastrófica pérdida de la visión. Un régimen terapéutico inicial con dosis bajas de prednisona (6 a 10 mg/día) o el empleo de agentes no esteroides pueden controlar la rigidez matutina y el dolor. En ausencia de síntomas oculares, la polimialgia reumática puede tratarse con dosis máximas de fármacos antinflamatorios no esteroides. Se puede administrar prednisona (10 a 15 mg diarios) para lograr una respuesta terapéutica más rápida, pero la toxicidad del uso prolongado de dosis superiores a 7.5 mg/día es elevada. Normalmente puede alcanzarse una dosis de mantenimiento baja en el plazo de unas semanas. Es importante evaluar de nuevo al paciente si aparecen síntomas oculares o de otro tipo; en este caso deberá aumentarse la dosis de esteroides. Algunos pacientes requieren de dosis bajas de esteroides de por vida; en otros pueden suspenderse después de dos a cuatro años de tratamiento.26
POLIARTERITIS NODOSA
La poliarteritis nodosa es una enfermedad multisistémica caracterizada por inflamación aguda y necrosis fibrinoide de las arterias de mediano y pequeño calibre. Por definición, excluye la afección de vasos más pequeños, los denominados microscópicos, como las arteriolas, capilares y vénulas. El término periarteritis nodosa se refería a los nódulos subcutáneos y viscerales que en ocasiones se observan a lo largo de las arterias afectadas. Aunque la etiología de la poliarteritis nodosa es desconocida, sus asociaciones patogénicas incluyen la antigenemia por hepatitis B (un estudio demostró una asociación del 30 porciento27), la enfermedad del suero, reacciones alérgicas a medicamentos y a otras toxinas, abuso de anfetaminas e infección por VIH.
La poliarteritis nodosa es tres veces más frecuente en varones que en mujeres. Normalmente se presenta entre los 20 y los 60 años de edad, aunque puede hacerlo a cualquier edad. Entre sus manifestaciones clínicas se incluyen fiebre, malestar general, debilidad, anorexia, pérdida de peso, mialgias y artralgias. Otras posibles complicaciones son glomerulonefritis, hipertensión, infarto del miocardio (debido a arteritis de las coronarias), pericarditis, neuropatías periféricas (mononeuritis múltiple), convulsiones, hemiplejia, hemoptisis, asma, arteritis mesentérica y erupciones.28
La afección renal, la causa más frecuente de muerte, se presenta en alrededor de un 75 porciento de los pacientes. Puede manifestarse como una arteritis de los vasos de mediano calibre, que produce hematuria, proteinuria, hipertensión y azoemia. De nuevo, por definición, no existe glomerulonefritis en la poliarteritis nodosa.
La participación de las arterias abdominales también produce complicaciones graves. Es una serie, cinco de 16 pacientes desarrollaron crisis abdominales relacionadas con la enfermedad, que requirieron de intervención quirúrgica. Los cinco pacientes afectados tenían datos de arteritis mesentérica con segmentos isquémicos que ocasionaron infarto y perforación.29
Sólo recientemente se ha observado la vasculitis asociada con infección por VIH. En un estudio, cuatro de 14 casos de vasculitis asociada a infección por VIH pudieron describirse como similares a poliarteritis, y se observó un infiltrado inflamatorio agudo en vasos de mediano y pequeño calibre de músculos, nervios y piel.13
El diagnóstico de poliarteritis nodosa depende de la evidencia histológica de vasculitis; un músculo con dolor a la palpación u otro órgano clínicamente afectado pueden servir como sitio para la toma de biopsia. La biopsia del músculo o de nervio es la prueba más específica para observar la vasculitis necrosante,30 aunque es relativamente poco sensible. La arteriografía visceral selectiva (v. gr., de las arterias renales o celiacas) puede demostrar la presencia de los aneurismas característicos de la poliarteritis nodosa [ver figura 3]. Los cambios en las pruebas de laboratorio no son específicos. Suele haber aumento de la velocidad de sedimentación globular (VSG); también pueden encontrarse anemia, leucocitosis o eosinofilia.
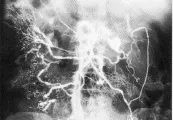
|
| Figura 3 |
| Poliarteritis nodosa: arteriografía |
La poliarteritis nodosa es relativamente rara, con una frecuencia de aproximadamente seis casos por 100,0000 habitantes en la población general.28 Por lo menos se han comunicado 13 casos de poliarteritis nodosa en pacientes con fiebre familiar del Mediterráneo, lo que sugiere una incidencia mucho mayor en este subgrupo de población.31 La edad promedio de inicio de la enfermedad en este grupo es de 20.8 años, mucho menor que en la población general (45 años). En otros aspectos, la poliarteritis nodosa en pacientes con fiebre familiar del Mediterráneo no difiere significativamente de la observada en la población general.
La actitud terapéutica ante un paciente con poliarteritis nodosa clásica o con alguna de las vasculitis alérgicas relacionadas es empírico e incluye los siguientes pasos:28 (1) identificación y eliminación del antígeno, por ejemplo, un agente infeccioso o un medicamento, (2) tratamiento de una enfermedad primaria subyacente, como una neoplasia, (3) en síndromes en los cuales no exista evidencia de enfermedad grave diseminada o sistémica o en pacientes con reacciones medicamentosas por hipersensibilidad debe administrarse prednisona (1 mg/kg/día), (4) intentos continuos para disminuir la dosis de prednisona, y (5) ciclosfamida en dosis bajas (1 a 2 mg/kg/día), con o sin esteroides, en los pacientes con vasculitis necrosante diseminada o enfermos que no respondan al tratamiento esteroideo.
VASCULITIS GRANULOMATOSAS
Granulomatosis de Wegener
La granulomatosis de Wegener se caracteriza por la formación de granulomas necrosantes en las proximidades de los vasos inflamados.28 Los órganos más afectados son la mucosa nasal, los senos paranasales, el aparato respiratorio, el parénquima pulmonar y los riñones. Los pacientes pueden presentarse con pansinusitis recurrente o una enfermedad renal caracterizada por hematuria, piuria y azoemia. Los síntomas inespecíficos de este proceso incluyen fiebre, poliartralgias y poliartritis. También pueden aparecen nódulos necróticos en la piel. El diagnóstico se sospecha por la presentación clínica y los cambios radiológicos en los senos paranasales y el tejido pulmonar, y se confirma mediante el estudio histológico.
Como se mencionó antes, las pruebas para detectar c-ANCA tienen un alto grado de especificidad y sensibilidad para la granulomatosis de Wegener.32 Prácticamente, la única fuente de falsos positivos son otras formas de vasculitis. Es importante la forma de realizar esta prueba, ya que se requiere una gran experiencia para interpretar el estudio de inmunofluorescencia indirecta. Se dispone de pruebas de inmunoabsorción ligada a enzimas que utilizan antígenos altamente purificados.32,33
En la granulomatosis de Wegener no tratada, la muerte, casi siempre por enfermedad renal, suele ocurrir semanas o meses después del inicio de la enfermedad. Sin embargo, los avances terapéuticos han mejorado notablemente las posibilidades de vida. Los esteroides prolongan la supervivencia media algunos meses. Se ha logrado una respuesta aún mejor con la ciclosfamida, que constituye el tratamiento de elección.34 En pacientes con una evolución relativamente estable (función renal anormal pero sin insuficiencia renal fulminante) se administra ciclofosfamida por vía oral (1 a 2 mg/kg/día) durante dos semanas. Si el tratamiento tiene éxito, se ajusta la dosis para mantener el recuento leucocitario por encima de 3,000/mm.3 35,36 Si no existe una respuesta favorable, debe incrementarse la dosis en 25 mg cada dos semanas, hasta que haya respuesta o se presente leucopenia. En los pacientes en los que la enfermedad ponga en peligro la vida se administra ciclofosfamida (4 mg/kg/día) durante tres días, continuando después con el régimen ya mencionado. En pacientes con enfermedad grave se añaden dosis altas de prednisona (60 mg/día), que se disminuye en forma gradual en algunas semanas hasta administrarla en días alternos. Posteriormente pueden suspenderse los esteroides y mantener al paciente sólo con el agente citotóxico. En muchos pacientes la enfermedad remite, permitiendo suspender el tratamiento en el plazo de 12 a 18 meses. El tratamiento con pulsos de ciclofosfamida es una alternativa en la enfermedad agresiva.37 También se demostrado que el trimetoprim-sulfametoxazol es útil en la granulomatosis de Wegener limitada a algunos órganos y con una severidad moderada.38
Sindrome de Churg-Strauss: Angeítis y granulomatosis alérgica
El síndrome de Churg-Strauss se caracteriza por eosinofilia y vasculitis sistémica que se presenta en pacientes con asma y rinitis alérgica. La afección pulmonar fulminante puede ser mortal en pacientes con este síndrome, aunque las lesiones iniciales pueden variar desde infiltrados parenquimatosos no segmentarios a enfermedad intersticial difusa. Los esteroides constituyen el tratamiento principal, aunque se ha demostrado que la ciclofosfamida (750 mg), administrada en pulsos por vía intravenosa, puede ser eficaz en pacientes con enfermedad que pone en peligro la vida y es resistente a los esteroides.37
Angeítis limitada al sistema nervioso central
La angeítis limitada al SNC, difícil de diagnosticar por estar limitados los cambios histopatológicos a la vasculatura intracraneal, puede ser progresiva y poner en peligro la vida del paciente. Consiste en un proceso granulomatoso vascular con cambios histopatológicos semejantes a los encontrados en la granulomatosis de Wegener y que afecta a los vasos de gran calibre que surgen de las arterias carótidas o basilar. Los síntomas y signos pueden variar desde una disfunción global, similar a una encefalitis, hasta alteraciones más localizadas que simulan un accidente cerebrovascular. Aunque la angiografía con contraste es el procedimiento que define las características de los vasos relacionados con vasculitis, los estudios actuales con imágenes por resonancia magnética sugieren que esta modalidad puede ser útil en el diagnóstico. El tratamiento se limita a la administración de esteroides en dosis altas; no se han comunicado ensayos con fármacos citotóxicos, como la ciclofosfamida.28
Granulomatosis linfomatoide
La granulomatosis linfomatoide es una forma poco frecuente de vasculitis,39 caracterizada por infiltración destructiva de varios tejidos, especialmente los pulmones, por células linfocitoides y plasmocitoides atípicas. El infiltrado es visible en la angiografía. Los granulomas y la necrosis son menos característicos que en la granulomatosis de Wegener, pero el tratamiento es el mismo para ambas enfermedades; la forma más agresiva de esta enfermedad es considerado como un linfoma angiocéntrico y debe tratarse como tal.
ENFERMEDAD DE BEHCET
Esta enfermedad se describió en 1937 como un síndrome crónico con recaídas, caracterizado por ulceración oral, úlceras genitales dolorosas y uveítis. Es una forma característica de vasculitis40 y se asocia a manifestaciones como sinovitis, vasculitis cutánea similar al eritema nosodo, meningoencefalitis, flebitis, aneurismas de grandes arterias y úlceras intestinales. En Japón, la prevalencia de la enfermedad de Behçet es de uno por 1,000 individuos; en los Estados Unidos es menor de uno por15,000. En Japón la prevalencia del antígeno de histocompatibilidad HLA-B5 es tres veces superior en los pacientes con esta enfermedad, pero esto no se ha observado en los Estados Unidos. Desde el punto de histopatológico la enfermedad se caracteriza por un importante infiltrado perivascular no necrosante de linfocitos y monocitos.
El tratamiento con esteroides tiene sólo una eficacia moderada en la enfermedad de Behçet. Los mejores resultados se han obtenido con la administración oral de clorambucil (0.1 a 0.2 mg/kg/día) y este esquema ha sido eficaz en el tratamiento tanto de la meningoencefalitis como de las manifestaciones mucosas de la enfermedad.41 Se ha recomendado el tratamiento con ciclosporina (5 mg/kg/día) para la enfermedad ocular.42
SINDROME DE COGAN
El síndrome de Cogan consiste en una queratitis intersticial no luética y enfermedad vestíbulo-auditiva. Se presenta en adultos jóvenes y se manifiesta por dolor ocular, fotofobia, vértigo e hipoacusia progresiva. El tratamiento consiste en la administración de dosis altas de esteroides (60 a 100 mg de prednisona al día); la dosis exacta se determina según la respuesta terapéutica a través de pruebas auditivas y de la visión.
ENFERMEDAD DE KAWASAKI
En 1967 Kawasaki describió un nuevo síndrome clínico en niños japoneses.43 Los rasgos cardinales de la enfermedad de Kawasaki (también llamada síndrome mucocutáneo con adenopatías) son fiebre, inyección conjuntival, labios secos y enrojecidos, eritema en palmas y plantas, edema indurado de las zonas distales de las extremidades, descamación de las puntas de los dedos, eritema cutáneo del tórax y linfoadenopatía cervical. En algunos aspectos la enfermedad se parece a la escarlatina o al síndrome de Steven-Johnson. También se pueden presentar complicaciones cardiovasculares importantes, como miocarditis, pericarditis, soplos, aneurismas arteriales coronarios, infarto del miocardio, angina de pecho, enfermedad vascular cerebral, insuficiencia vascular periférica y gangrena. Otras complicaciones son hidropesía de la vesícula, hepatitis, obstrución del intestino delgado, anemia, artritis, meningitis aséptica, uveítis, piuria y meatitis. La enfermedad suele ser autolimitada, pero en algunos casos ha ocurrido la muerte por trombosis coronaria durante la etapa aguda de la enfermedad o varios meses después de una aparente recuperación completa. La tasa de mortalidad es del uno al dos porciento.
No hay datos de que la enfermedad se trasmita de persona a persona. En las células mononucleares de sangre periférica se ha encontrado actividad de transcriptasa inversa, que se ha podido trasmitir a células T en cultivos.13 Estos hallazgos, junto con la linfocitopenia de células T y la marcada activación de las células B, apoyan la posibilidad de que el síndrome sea causado por un retrovirus.44 Se ha relacionado la fase aguda del síndrome de Kawasaki con la presencia de anticuerpos citotóxicos contra las células endoteliales.45
Un estudio multicéntrico aleatorio ha mostrado que un esquema de gamaglobulina administrada por vía intravenosa en dosis elevadas, junto con ácido acetilsalicílico, es eficaz para reducir la prevalencia de las alteraciones en la circulación coronaria y la severidad de las manifestaciones inflamatorias sistémicas.46 Se administran 400 mg/kg/día de gamaglobulina durante dos horas por cuatro días consecutivos y 100 mg/kg de ácido acetilsalicílico diariamente, divididos en cuatro dosis, durante 14 días de la enfermedad, posteriormente se reduce la dosis a 3 a 5 mg/kg/día en una sola dosis. Este tratamiento debe administrarse en las etapas tempranas de la enfermedad y parece ser el tratamiento más eficaz. Se recomienda a los médicos que informen los casos de este síndrome a sus departamentos de salud locales y estales.
POLIANGEITIS MICROSCOPICA
La manifestación más frecuente de la este tipo de angeítis es la púrpura palpable de las extremidades inferiores y las áreas en declive, que se relaciona con reacciones idiosincráticas por fármacos. Sin embargo, muchas y diferentes situaciones pueden precipitar los mismos cambios patológicos tanto en la piel como en múltiples órganos. Las lesiones papulares, no blanquecinas, aparecen en grupos, suelen abrirse al cuarto día y al evolucionar cambian de color rojo-púrpura a café, aplanándose y diseminándose. La afección glomerular puede producir insuficiencia renal, las lesiones intestinales causan hemorragias gastrointestinales. Cuando se afecta el parénquima pulmonar pueden observarse infiltrados difusos o nodulares, y la afección neurológica se manifiesta por cefalea, diplopia y síntomas orgánicos cerebrales.
En ocasiones se han observado pacientes que tienen una forma crónica y recurrente de poliangeítis microscópica en ausencia de enfermedad generalizada o de un suceso precipitante obvio. Los pacientes con esta enfermedad no muestran progresión hacia una afección generalizada durante el seguimiento a largo plazo. Sin embargo, este proceso no suele responder a los esteroides ni a la ciclofosfamida y, por lo tanto, deberá evitarse el uso de regímenes inmunosupresores tóxicos en estos pacientes.47 El tratamiento de las lesiones de las extremidades inferiores es difícil.
Púrpura de Henoch-Schönlein
La púrpura de Henoch-Schönlein afecta primariamente a las arteriolas pequeñas y a las capilares en los niños. Los órganos más afectados son la piel, las grandes articulaciones, los riñones y el aparato gastrointestinal. Se ha observado un depósito simultáneo de inmunoglobulinas (predominantemente IgA), componentes del complemento y fibrina en las paredes de los vasos cutáneos afectados, en el mesangio glomerular y en el tubo digestivo.48 La púrpura de Henoch-Schönlein con frecuencia es benigna, pero en pacientes con participación renal puede desarrollarse insuficiencia renal. Aunque suele ser suficiente el tratamiento sintomático, la enfermedad renal asociada se trata generalmente con esteroides (40 a 60 mg/día en adultos).
Síndrome de Goodpasture
Este síndrome se caracteriza por una marcada capilaritis pulmonar, púrpura palpable y glomerulitis. Se asocia con la presencia de anticuerpos circulantes contra los componentes de la colágena tipo IV de las membranas basales vasculares. Las pruebas de estos anticuerpos y para los ANCA pueden ayudar a diferenciar esta alteración de la granulomatosis de Wegener.1,3
Vasculitis urticariana hipocomplementémica
La vasculitis urticariana hipocomplementémica (VUH) es un síndrome poco frecuente que se caracteriza por urticaria recurrente y angioedema. Las lesiones urticarianas muestran una vasculitis leucocitoclástica. También puede presentarse conjuntivitis, uveítis, glomerulonefritis, dolor abdominal, artritis o artralgias. La enfermedad pulmonar obstructiva crónica parece acentuarse en los fumadores con este proceso, pero se desconoce la causa.49 Hay una considerable superposición clínica con el LES, pero los estudios inmunológicos recientes han establecido importantes diferencias entre estos dos síndromes. La IgG purificada de pacientes con vasculitis urticariana hipocomplementémica tiene una actividad de unión para el primer componente del complemento, C1q.15 Se ha postulado que la activación de C1 por anticuerpos anti-C1q puede conducir a la activación de la fase fluida de la cascada del complemento, y de esta forma generar los cambios patológicos que se observan clínicamente.
Las lesiones urticarianas se presentan principalmente en la cara, extremidades superiores y tórax. La urticaria puede durar más de 48 horas y desvanecerse sin dejar huellas, a diferencia de la púrpura palpable. En comparación con el LES, no se encuentran anticuerpos antinucleares en el plasma. Los esteroides suelen ser eficaces en el alivio de los signos y síntomas.
Vasculitis crioglobulinémica
Las crioglobulinas son inmunoglobulinas y complejos inmunes que se precipitan con el frío (4°C) y se disuelven al volver a calentarse. Pueden estar relacionadas con diversas enfermedades y síntomas reumatológicos, como fenómeno de Raynaud, vasculitis, púrpura, artralgias, fiebre y enfermedad renal. Los complejos inmunes pueden en parte ser responsable de la vasculitis asociada. El principal síndrome clínico asociado con crioprecipitados de IgG e IgM se caracteriza por púrpura, artralgias y debilidad, y suele cursar con glomerulonefritis rápidamente progresiva. Dos terceras partes de estos pacientes son positivos para AgHBs o para los anti-HBs cuando se analiza el plasma y los crioprecipitados.50 Se ha usado la plasmaféresis en el tratamiento de los casos graves resistentes a los esteroides.
Bibliografía
-
Lesavre P: Antineutrophil cytoplasmic autoantibodies antigen
specificity. Am J Kidney Dis 18:159, 1991
-
Falk RJ, Terrell RS, Charles LA, et al: Anti-neutrophil cytoplasmic
autoantibodies induce neutrophils to degranulate and produce oxygen radicals
in vitro. Proc Natl Acad Sci USA 87:4115, 1990 [PMID
2161532]
-
Kallenberg CG, Mulder AH, Tervaert JW: Antineutrophil cytoplasmic
antibodies: a still-growing class of autoantibodies in inflammatory disorders.
Am J Med 93:675, 1992 [PMID
1466365]
-
Persellin ST, Daniels TM, Rings LJ, et al: Factor VIII-von
Willebrand factor in giant cell arteritis and polymyalgia rheumatica. Mayo
Clin Proc 60:457, 1985 [PMID
3925247]
-
Hughes GR: Thrombosis, abortion, cerebral disease, and the
lupus anticoagulant (editorial). BMJ 287:1088, 1983
-
Hughes GR: The antiphospholipid syndrome: ten years on. Lancet
342:341, 1993
-
Asherson RA, Khamashta MA, Ordi-Ros J, et al: The "primary"
antiphospholipid syndrome: major clinical and serological features. Medicine
(Baltimore) 68:366, 1989 [PMID
2509856]
-
Lie JT: Nomenclature and classification of vasculitis: plus
ca change, plus c'est la meme chose (editorial). Arthritis Rheum 37:181,
1994
-
Jennette JC, Falk RJ, Andrassy K, et al: Nomenclature of
systemic vasculitides: proposal of an international consensus conference.
Arthritis Rheum 37:187, 1994 [PMID
8129773]
-
Gocke DJ, Hsu K, Morgan C, et al: Association between polyarteritis
and Australian antigen. Lancet 2:1149, 1970 [PMID
4098431]
-
Quint L, Deny P, Guillevin L, et al: Hepatitis C virus in
patients with polyarteritis nodosa: prevalence in 38 patients. Clin Exp
Rheumatol 9:253, 1991 [PMID
1715249]
-
Burns JC, Geha RS, Schneeberger EE, et al: Polymerase activity
in lymphocyte culture supernatants from patients with Kawasaki disease.
Nature 323:814, 1986 [PMID
2430187]
-
Calabrese LH, Estes M, Yen-Lieberman B, et al: Systemic vasculitis
in association with human immunodeficiency virus infection. Arthritis Rheum
32:569, 1989 [PMID
2655605]
-
Andrasch RH, Bardana EJ Jr, Porter JM, et al: Digital ischemia
and gangrene preceding renal neoplasm: an association with sarcomatoid
adenocarcinoma of the kidney. Arch Intern Med 136:486, 1976 [PMID
1267558]
-
Wisnieski JJ, Naff GB: Serum IgG antibodies to C1q in hypocomplementemic
urticarial vasculitis syndrome. Arthritis Rheum 32:1119, 1989 [PMID
2528353]
-
Brasile L, Kremer JM, Clarke JL, et al: Identification of
an autoantibody to vascular endothelial cell-specific antigens in patients
with systemic vasculitis. Am J Med 87:74, 1989 [PMID
2741984]
-
Kerr GS, Hallahan CW, Giordano J, et al: Takayasu arteritis.
Ann Intern Med 120:919, 1994
-
Daggett WM Jr, Fallan JT: A 55-year-old man with intermittent
claudication of the arms. N Engl J Med 305:1519, 1981
-
Grossman E, Morag B, Nussinovitch N, et al: Clinical use
of captopril in Takayasu's disease. Arch Intern Med 144:95, 1984 [PMID
6140907]
-
Ishikawa K: Natural history and classification of occlusive
thromboaortopathy (Takayasu's disease). Circulation 57:27, 1978
-
Hunder GG: Giant cell arteritis and polymyalgia rheumatica.
Textbook of Rheumatology, 3rd ed. Kelley WN, Harris ED Jr, Ruddy S, et
al, Eds. WB Saunders Co, Philadelphia, 1989, p 1200
-
Hall S, Persellin S, Lie JT, et al: The therapeutic impact
of temporal artery biopsy. Lancet 2:1217, 1983 [PMID
6139569]
-
Achkar AA, Lie JT, Hunder GG, et al: How does previous corticosteroid
treatment affect the biopsy findings in giant cell (temporal) arteritis?
Ann Intern Med 120:987, 1994
-
Healey LA: Long-term follow-up of polymyalgia rheumatica:
evidence for synovitis. Semin Arthritis Rheum 13:322, 1984
-
Healey LA: Polymyalgia rheumatica and the American Rheumatism
Association criteria for rheumatoid arthritis. Arthritis Rheum 26:1417,
1983
-
Chuang T-Y, Hunder GG, Ilstrup DM, et al: Polymyalgia rheumatica:
a 10-year epidemiologic and clinical study. Ann Intern Med 97:672, 1982
-
Sergent JS, Lockshin MD, Christian CL, et al: Vasculitis
with hepatitis B antigenemia: long-term observations in nine patients.
Medicine (Baltimore) 55:1, 1976 [PMID
1628]
-
Conn DL, Hunder GG: Vasculitis and related disorders. Textbook
of Rheumatology, 3rd ed. Kelley WN, Harris ED Jr, Ruddy S, et al, Eds.
WB Saunders Co, Philadelphia, 1989, p 1167
-
Zizic TM, Classen JN, Stevens MB: Acute abdominal complications
of systemic lupus erythematosus and polyarteritis nodosa. Am J Med 73:525,
1982 [PMID
6127033]
-
Dahlberg PJ, Lockhart JM, Overholt EL: Diagnostic studies
for systemic necrotizing vasculitis. Arch Intern Med 149:161, 1989 [PMID
2912404]
-
Glikson M, Galun E, Schlesinger M, et al: Polyarteritis nodosa
and familial Mediterranean fever: a report of 2 cases and review of the
literature. J Rheumatol 16:536, 1989 [PMID
2568489]
-
Nolle B, Specks U, Ludemann J, et al: Anticytoplasmic autoantibodies:
their immunodiagnostic value in Wegener granulomatosis. Ann Intern Med
111:28, 1989 [PMID
2660645]
-
Specks U, Wheatley CL, McDonald TJ, et al: Anticytoplasmic
autoantibodies in the diagnosis and follow-up of Wegener's granulomatosis.
Mayo Clin Proc 64:28, 1989 [PMID
2642995]
-
Haynes BF: Treatment of the granulomatous vasculitides: the
spectrum of vasculitis: clinical, pathologic, immunologic, and therapeutic
considerations. Fauci AS, moderator. Ann Intern Med 89:671, 1978
-
Fauci AS, Haynes BF, Katz P, et al: Wegener's granulomatosis:
prospective clinical and therapeutic experience with 85 patients for 21
years. Ann Intern Med 98:76, 1983 [PMID
6336643]
-
Fan PT, Davis JA, Somer T, et al: A clinical approach to
systemic vasculitis. Semin Arthritis Rheum 9:248, 1980 [PMID
6105711]
-
Chow CC, Li EK, Lai FM: Allergic granulomatosis and angiitis
(Churg-Strauss syndrome): response to 'pulse' intravenous cyclophosphamide.
Ann Rheum Dis 48:605, 1989 [PMID
2774702]
-
DeRemee RA, McDonald TJ, Weiland LH: Wegener's granulomatosis:
observations on treatment with antimicrobial agents. Mayo Clin Proc 60:27,
1985 [PMID
3871238]
-
Lie JT: Classification and immunodiagnosis of vasculitis:
a new solution or promises unfulfilled? J Rheumatol 15:728, 1988
-
O'Duffy JD: Behcet's disease. Textbook of Rheumatology, 3rd
ed. Kelley WN, Harris ED Jr, Ruddy S, et al, Eds. WB Saunders Co, Philadelphia,
1989, p 1209
-
O'Duffy JD, Robertson DM, Goldstein NP: Chlorambucil in the
treatment of uveitis and meningoencephalitis of Behcet's disease. Am J
Med 76:75, 1984 [PMID
6691363]
-
Masuda K, Nakajima A, Urayama A, et al: Double-masked trial
of cyclosporin versus colchicine and long-term open study of cyclosporin
in Behcet's disease. Lancet 1:1093, 1989 [PMID
2566048]
-
Kawasaki T: Acute febrile mucocutaneous syndrome with lymphoid
involvement with specific desquamation of the fingers and toes in children.
Arerugi 16:178, 1967
-
Katz P: Treatment of the polyarteritis nodosa group of systemic
necrotizing vasculitis: the spectrum of vasculitis: clinical, pathologic,
immunologic, and therapeutic considerations. Fauci AS, moderator. Ann Intern
Med 89:673, 1978
-
Leung DY, Geha RS, Newburger JW, et al: Two monokines, interleukin
1 and tumor necrosis factor, render cultured vascular endothelial cells
susceptible to lysis by antibodies circulating during Kawasaki syndrome.
J Exp Med 164:1958, 1986 [PMID
3491174]
-
Newburger JW, Takahashi M, Burns JC, et al: The treatment
of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med 315:341,
1986 [PMID
2426590]
-
Cupps TR, Springer RM, Fauci AS: Chronic, recurrent small-vessel
cutaneous vasculitis: clinical experience in 13 patients. JAMA 247:1994,
1982 [PMID
7062504]
-
Touchard G, Maire P, Beauchant M, et al: Vascular IgA and
C3 deposition in gastrointestinal tract of patients with Henoch-Schonlein
purpura (letter). Lancet 1:771, 1983 [PMID
6132119]
-
Schwartz HR, McDuffie FC, Black LF, et al: Hypocomplementemic
urticarial vasculitis: association with chronic obstructive pulmonary disease.
Mayo Clin Proc 57: 231, 1982 [PMID
7040825]
-
Levo Y, Gorevic PD, Kassab HJ, et al: Association between
hepatitis B virus and essential mixed cryoglobulinemia. N Engl J Med 296:1501,
1977 [PMID
865530]