Contenido del artículo
VIII SINDROMES DE VASCULITIS SISTEMICA
DR. BRIAN F. MANDELL, PH.D.
El diagnóstico de un síndrome de vasculitis primaria depende de la demostración de vasculitis y de la exclusión de enfermedades que puedan causar vasculitis secundaria. El diagnóstico de un trastorno específico se basa en el patrón de afección orgánica, los datos de patología y el tamaño de los vasos sanguíneos afectados.
Los principales determinantes del pronóstico y el tratamiento incluyen el tipo de vasculitis, la severidad de la afección a órganos vitales, la velocidad de progresión de la enfermedad y la etiología en los casos en que puede identificarse. El proceso inflamatorio suele asociarse con síntomas inespecíficos y alteraciones de laboratorio (aumento en la velocidad de sedimentación globular, anemia y fiebre) que no distinguen los procesos de vasculitis de otros procesos inflamatorios, infecciosos o neoplásicos. La naturaleza tóxica de los tratamientos para las vasculitis sistémica obliga a contar con un diagnóstico exacto.
Estudio del paciente con probable vasculitis
El médico no debe tener temor a realizar estudios invasivos como parte de la evaluación diagnóstica de los pacientes con enfermedad multisistémica, pero debe evitarse el realizar biopsias de tejidos clínicamente no afectados o realizar pruebas inespecíficas. Debe intentarse un enfoque dirigido a investigar formas específicas de vasculitis, descartando las alternativas razonables.
El primer paso en el diagnóstico de las vasculitis consiste en realizar una historia clínica y examen físico detallados del paciente para demostrar afección a órganos específicos. Debe ponerse especial atención a la piel, ojos, oídos, vías aéreas superiores, articulaciones, riñones, ganglios linfáticos, nervios periféricos y grandes vasos. En la evaluación inicial deben incluirse algunas pruebas seleccionadas [ver tabla 1]. Los estudios especializados, incluyendo la serología, se solicitarán en forma selectiva una vez que se complete el diagnóstico diferencial. Si la tira reactiva indica sangre o proteínas en la orina deberá solicitarse de inmediato el estudio del sedimento fresco. La orina que ha estado almacenada por varias horas antes del análisis no es útil para identificar cilindros celulares, que rápidamente degeneran ex vivo. Los cilindros de eritrocitos son muy sugestivos de glomerulonefritis, pero también pueden observarse cilindros leucocitarios. La glomerulonefritis suele ser asintomática. Con base en el patrón de afección orgánica podrá establecerse un diagnóstico diferencial que incluya los tipos posibles de vasculitis sistémica y otros padecimientos.
|
||||||||||||||||||||||||||||||||
ALT- alanino aminotransferasa, AF- anticuerpos antifosfolípido, SAF- síndrome antifosfolípido, AST- aspartato aminotransferasa, CID- coagulación intravascular diseminada, VSG- velocidad de sedimentación globular, PHS- púrpura de Henoch-Schönlein, MAP- poliangeítis microscópica, PAN- poliarteritis nodosa, TPT- tiempo parcial de tromboplastina, PVVR- prueba con veneno de víbora de Rusell, LEG- lupus eritematoso generalizado, PTT- púrpura trombocitopénica trombótica, GW- granulomatosis de Wegener |
Se han propuesto varios esquemas de clasificación para organizar las vasculitis sistémicas en forma consistente. Estas clasificaciones son útiles para distinguir los trastornos clínicos que tienen diferencias en pronóstico y respuesta al tratamiento.1 No existe un esquema con aceptación universal. Todos reiteran las características de la afección fulminante o clásica, poniendo énfasis en la especificidad del diagnóstico. Si se emplea el esquema de clasificación en forma muy estricta con frecuencia el paciente nuevo y sin la expresión total de la enfermedad quedará sin un diagnóstico definitivo. A pesar de ello, los sistemas de clasificación proporcionan esquemas útiles para publicaciones y para el diseño de protocolos de investigación [ver figura 1]. Los esquemas de clasificación más empleados se basan en el calibre de los vasos afectados, el patrón de afección orgánica y la presencia o ausencia de granulomas, depósito significativo de complejos inmunes e infiltrados eosinofílicos. Algunos autores han propuesto que la presencia o ausencia de anticuerpos específicos contra citoplasma de neutrófilos (ANCA) tienen utilidad diagnóstica, en especial los anticuerpos contra la proteinasa 3 y la mieloperoxidasa. En la actualidad el papel adecuado de estas pruebas consiste en apoyar un diagnóstico clínico y no definirlo. En los pacientes que no pueden clasificarse dentro de una categoría diagnóstica bien definida estas pruebas serológicas no deben sustituir los intentos por obtener un diagnóstico histológico. La presencia de ANCA no equivale a diagnóstico de vasculitis.
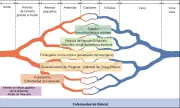
|
| Figura 1 |
| Clasificación del síndrome de vasculitis sistémica. |
Cuando los síntomas y signos principales (i.e., neuropatía y vasculitis cutánea) no sugieren un trastorno vasculítico específico aislado, puede ser útil realizar un examen físico dirigido y pruebas serológicas. El estudio más valioso para confirmar el diagnóstico será la biopsia. El valor de las pruebas indiscriminadas de anticuerpos antinucleares, ANCA, factor reumatoide y enzima convertidora de angiotensina es debatible. La infección con virus de la hepatitis B o C se asocia con un rango amplio de síndromes de vasculitis y siempre deben excluirse estas infecciones.2
Las vasculitis sistémicas pueden poner en peligro la vida y requieren tratamiento antinflamatorio e inmunosupresor potente. El diagnóstico debe hacerse con la mayor certeza posible. Sin embargo, pueden persistir dudas respecto a diagnósticos alternativos o enfermedades coexistentes. Por ello, incluso después de iniciado el tratamiento el médico debe mantener un alto grado de vigilancia para detectar problemas médicos no relacionados, complicaciones del tratamiento o ambos. Los signos y síntomas de infección pueden ocultarse por el tratamiento esteroideo.3 Con el inicio de terapia inmunosupresora potente existe una mayor ventana de riesgo para infecciones oportunistas. El mayor riesgo ocurre en pacientes con neutropenia marcada o con dosis altas de esteroides. Los médicos deben estar especialmente atentos a atribuir los nuevos problemas a las llamadas exacerbaciones del padecimiento sin primero descartar una infección nueva o recurrente. El virus varicela-zoster puede presentarse con fiebre y dolor antes de la aparición de las vesículas. Pneumocystis carinii, citomegalovirus, las infecciones micóticas sistémicas y la reactivación de infecciones por micobacterias se observan con más frecuencia que en el huésped normal. La inmunosupresión por esteroides y otros medicamentos se asocia en forma común con candidiasis mucosa, menos con molusco contagioso y rara vez con sarcoma de Kaposi.
El metotrexate, la azatioprina y la ciclofosfamida pueden causar leucopenia y, con menos frecuencia, otras citopenias. El metotrexate debe usarse con precaución, si es que se emplea, en los pacientes con menor velocidad de filtración glomerular (VFG).
La vasculitis de capilares y vénulas es la forma más común y casi de modo invariable afecta la piel. Puede ocurrir a cualquier edad y afecta a hombres y mujeres con igual frecuencia.
La vasculitis de vasos pequeños ocurre como un trastorno idiopático o secundaria a alergia a medicamentos, endocarditis bacteriana e infecciones virales, como las causadas por los virus de hepatitis B o C, Neisseria diseminada y ricketsias. Puede también ser parte de un trastorno autoinmune definido como síndrome de Sjögren, lupus eritematoso generalizado (LEG) o artritis reumatoide, o puede ocurrir en asociación con neoplasias hematológicas, linfoides y de órganos sólidos [ver figura 2].
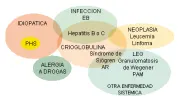
|
| Figura 2 |
| Causas de vasculitis de vasos pequeños |
DIAGNOSTICO
Puede ocurrir afección cutánea en muchos de los síndromes de vasculitis primaria o secundaria. La oclusión de los vasos grandes, medianos o pequeños puede ocasionar livedo, fenómeno de Raynaud o necrosis. La púrpura es la manifestación más común de la vasculitis de vasos pequeños. Este tipo de vasculitis con frecuencia se asocia con depósito de complejos inmunes y las vasculitis que afectan principalmente a las vénulas poscapilares se denominan también vasculitis por hipersensibilidad.4 También puede ocurrir vasculitis de vasos pequeños en las vasculitis sistémicas de vasos grandes (v.gr., granulomatosis de Wegener y poliangeítis microscópica). La vasculitis de vasos pequeños puede limitarse a la piel o asociarse con daño visceral, incluyendo hemorragia alveolar, isquemia o hemorragia intestinal y glomerulonefritis.
La púrpura tiende a ocurrir en brotes de lesiones de edad semejante, y es más evidente en las áreas sometidas a gravedad [ver figura 3]. Cuando la púrpura no se limita a estas zonas deben excluirse enfermedad por crioaglutininas, crioglobulinemia (que puede asociarse con infecciones como hepatitis C o con linfoma), embolia y enfermedades infiltrativas. Las vasculitis cutáneas de cualquier etiología pueden asociarse con edema de declive importante.
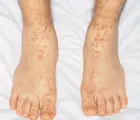
|
| Figura 3 |
| Púrpura vascular |
En una serie de casos de vasculitis cutánea de vasos pequeños,4 casi el 100 por ciento de los pacientes menores de 20 años tuvo enfermedad limitada a la piel, mientras que alrededor del 40 por ciento de los 172 pacientes mayores de 20 años tuvieron un padecimiento sistémico o asociado subyacente. Diecisiete adultos tuvieron vasculitis necrosante sistémica, 4 neoplasias, 4 infección bacteriana que causó la vasculitis, 11 crioglobulinemia y 59 púrpura de Henoch-Schönlein. No se investigó la posibilidad de infección con virus de la hepatitis C, que es la causa más probable de la crioglobulinemia mixta.2
La biopsia es útil para excluir causas de púrpura no vasculítica como amiloidosis, leucemia cutis, sarcoma de Kaposi, linfomas de células T y embolias de colesterol y mixomatosas. La tinción del tejido con inmunofluorescencia apoya el diagnóstico de púrpura de Henoch-Schönlein o LEG. Las células infiltrantes y que quizá destruyen la pared vascular pueden ser neutrófilos o linfocitos, dependiendo de la etiología. La patología en la mayoría de los casos de vasculitis de vasos pequeños corresponde a una angeítis leucocitoclásica.
La púrpura de Henoch-Schönlein es un síndrome vasculítico de vasos pequeños en el que las manifestaciones cutáneas suelen ser importantes y es común la afección visceral. Este padecimiento es menos común en adultos que en niños,5 y suele asociarse con depósito vascular y renal de complejos inmunes que contienen IgA. Las manifestaciones comunes incluyen púrpura, urticaria, dolor abdominal, hemorragia gastrointestinal o intususcepción (principalmente en niños), artralgias o artritis y glomerulonefritis. Los síntomas viscerales pueden preceder a las lesiones de la piel. La púrpura de Henoch-Schönlein puede ser desencadenada por medicamentos o infecciones estreptocócicas o virales. Suele ser un padecimiento autolimitado, pero la glomerulonefitis progresa en raros casos (sobre todo en adultos) a insuficiencia renal.
La vasculitis urticariana representa un subtipo peculiar de vasculitis de vasos pequeños.6 El cuadro clínico consiste en ampollas o pápulas, en ocasiones con angioedema circundante o topográficamente independiente. Las lesiones individuales se resuelven en forma muy lenta, durando varios días. Suele existir molestia ardorosa y disestésica en las lesiones. Como la púrpura, las lesiones de vasculitis urticariana con frecuencia se localizan en áreas declive y suelen cicatrizar con hiperpigmentación de la piel o un área con equimosis. La mayoría de los casos son idiopáticos, aunque puede existir asociación con un padecimiento autoinmune sistémico subyacente, como LEG, paraproteinemia de IgM o una infección viral. En casos raros la vasculitis urticariana se ha asociado con hipocomplementemia y enfermedad pulmonar intersticial.
El tratamiento de la vasculitis cutánea se dirige primero a eliminar cualquier factor precipitante. Deben buscarse y tratarse las causas infecciosas, y se suspenderá cualquier medicamento sospechoso. También debe considerarse la posibilidad de asociación con mielodisplasia y enfermedad mieloproliferativa, en especial si existen alteraciones hematológicas. Si no se encuentran factores precipitantes aparentes puede intentarse un tratamiento no riesgoso con antinflamatorios no esteroides, colchicina, pentoxifilina, dapsona o esteroides en dosis bajas y por tiempo breve. Debe evitarse hasta donde sea posible el tratamiento esteroideo prolongado. Las medias elásticas pueden ser útiles para disminuir el edema que con frecuencia acompaña a la vasculitis cutánea.
La afección visceral con disfunción orgánica puede necesitar un enfoque más agresivo que el empleado en la vasculitis cutánea limitada. Suelen ser eficaces las dosis moderadas de esteroides. En el caso de complicaciones por el uso crónico de esteroides o si existe afección visceral severa son útiles el metotrexate, la azatioprina, la ciclofosfamida y otros agentes inmunosupresores. Cuando se trata la afección de pequeños vasos crónica y refractaria debe ponerse especial atención a la relación riesgo-beneficio de los diferentes tratamientos.
La granulomatosis de Wegener (GW) es un padecimiento relativamente poco común y potencialmente letal, que se caracteriza por inflamación granulomatosa necrosante y vasculitis de vasos pequeños y medianos.7,8 Pueden afectarse hombres y mujeres de todas las edades.
En la granulomatosis de Wegener existe afección orgánica múltiple, con predilección por la vía respiratoria superior e inferior, los ojos y los riñones.
Afección de las vías respiratorias superiores La afección de las vías aéreas superiores puede ser muy evidente, pero suele atribuirse por muchos meses o incluso años a una sinusitis, hasta que se detectan otras manifestaciones de la GW. Incluso después de hacer el diagnóstico y administrar el tratamiento inmunosupresor, la afección sinusal puede ser resistente al tratamiento. Esta resistencia puede ser causada en parte por sobreinfección del tejido dañado por Staphylococcus aureus. El daño anatómico puede incluir perforaciones septales y deformidades en silla de montar. La afección traqueal causa estenosis subglótica. Es común el daño ótico, en especial la otitis media, que puede producir pérdida de la audición conductiva. Los seudotumores orbitarios pueden producir proptosis con dolor intratable y pérdida de la visión, estas masas inflamatorias y fibrosas en ocasiones son refractarias al tratamiento antinflamatorio, al tratamiento inmunosupresor e incluso a la radioterapia. Con frecuencia ocurren conjuntivitis, uveítis y afección de la retina, solas o en combinación.
Afección de las vías respiratorias inferiores Puede no existir daño pulmonar al inicio de la enfermedad, o presentarse en forma dramática como una hemorragia alveolar difusa. La tercera parte de las lesiones pulmonares que se observan en los estudios de imagen [ver figura 4] son asintomáticas (la TC es más sensible que la radiografía simple). Los nódulos suelen sufrir necrosis, causando formación de cavidades. El broncoespasmo no es característico de la GW. Si se sospecha obstrucción de las vías respiratorias debe considerarse realizar una broncoscopía para excluir estenosis endobronquiales o subglóticas. Con frecuencia es necesario descartar procesos infecciosos como causa de los infiltrados pulmonares y la broncoscopía es útil para esto. Sin embargo, el material obtenido de biopsias transbronquiales suele ser insuficiente para hacer el diagnóstico histológico de GW.
|
|
|
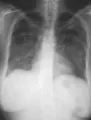
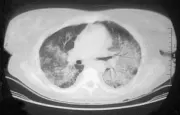
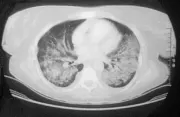
La biopsia abierta de pulmón puede proporcionar la mejor oportunidad para demostrar los datos patológicos típicos de la GW y excluir neoplasias e infecciones atípicas. Las biopsias típicas a cielo abierto9 contienen áreas de necrosis, con frecuencia con un patrón amplio, células gigantes en el tejido parenquimatoso y vasculitis. Pueden no coexistir todos los componentes histológicos en el mismo corte. Debido a que estos hallazgos también ocurren en las infecciones crónicas por micobacterias u hongos, es indispensable realizar tinciones especiales y cultivos para estos agentes.
Glomerulonefritis La glomerulonefritis es una causa común de morbimortalidad en la GW. Su presencia o ausencia define las formas generalizadas o limitadas de la enfermedad. La glomerulonefritis puede ser relativamente indolente o agresiva, generalmente esto último. Es clínica y patológicamente indistinguible de la glomerulonefritis idiopática rápidamente progresiva con medias lunas. En semanas puede ocurrir evolución de afección renal subclínica a enfermedad dependiente de diálisis. La glomerulonefritis puede presentarse al inicio de la enfermedad o desarrollarse después de que el paciente ha padecido una forma aparentemente limitada del padecimiento. Nunca podrá subestimarse la importancia de los exámenes microscópicos frecuentes de orina al inicio y durante el seguimiento en los pacientes con GW. En especial en los pacientes ancianos o debilitados puede obtenerse información valiosa realizando estudios periódicos en orina de 24 horas, que permiten establecer con más exactitud la VFG que la medición de creatinina en suero. La biopsia renal puede revelar glomerulonefritis focal y segmentaria con cambios proliferativos glomerulares diversos, formación de medias lunas y necrosis, en ausencia de depósito significativo de complejos inmunes. Aunque apoyan el diagnóstico de GW, estos datos no son diagnósticos de la enfermedad.
Otras manifestaciones clínicas Puede ocurrir afección musculoesquelética en casi la mitad de los pacientes con GW. Los síntomas pueden incluir artralgias o artritis migratorias, aditivas o fijas. Con frecuencia existe factor reumatoide positivo, que puede causar confusión diagnóstica con artritis reumatoide cuando las manifestaciones articulares son importantes. La afección articular en la GW rara vez causa erosiones óseas. Ocurren síntomas y signos neurológicos en menos del 50 por ciento de los pacientes, neuropatía periférica en menos del 20 por ciento y afección del sistema nervioso central en menos del 10 por ciento. Pueden presentarse defectos oculomotores por compresión por una masa retrorbitaria. La isquemia y las úlceras gastrointestinales no son comunes, pero pueden confundirse con enfermedad inflamatoria del intestino, en especial porque esta última puede asociarse con ANCA (por lo general con patrón perinuclear, o p-ANCA). En hasta el 50 por ciento de los pacientes se presentan lesiones cutáneas, caracterizadas por púrpura, paniculitis o ulceraciones. La actividad de la afección cutánea suele ser paralela a la de la enfermedad sistémica.
La inflamación inexplicable de las vías respiratorias, de los ojos, o la presencia de glomerulonefritis, es compatible con el diagnóstico de GW. La probabilidad aumenta cuando existe afección orgánica múltiple, si la afección de las vías respiratorias superiores es destructiva o si se demuestran nódulos pulmonares (en especial cavitados) en la radiografía. Cualquier combinación de afección orgánica es posible, pero la mayoría de los pacientes tienen afección de las vías respiratorias superiores en el momento del diagnóstico.
Si el cuadro clínico es compatible con el diagnóstico de GW y se han descartado en forma adecuada otros diagnósticos alternativos, el encontrar ANCA citoplasmáticos, o c-ANCA, con especificidad contra la proteinasa 3 es suficiente para hacer el diagnóstico provisional e iniciar el tratamiento. Alrededor del 20 por ciento de los pacientes con GW pueden tener p-ANCA con especificidad a mieloperoxidasa. Si existen manifestaciones atípicas o preocupación por el inicio del tratamiento inmunosupresor, o si el paciente no responde al tratamiento adecuado, será obligado obtener la confirmación histológica. La presencia de ANCA no es equivalente a vasculitis, y pueden encontrarse estos anticuerpos en otras enfermedades. El nivel de ANCA no es confiable para vigilar la actividad de la enfermedad.10,11 Debido a que la GW suele requerir de tratamiento esteroideo más citotóxico, debe distinguirse de otros padecimientos inflamatorios, incluyendo otros síndromes vasculíticos [ver tabla 2], que pueden tratarse en forma eficaz con un esquema menos tóxico.
|
|||||||||||||||||||||||||
*La presencia de depósitos inmunes sugiere posible infección
por virus de hepatitis B o C.
|
El tratamiento inicial de la GW generalizada se basa en un esquema dual de inmunosupresión. Los esteroides pueden producir mejoría sintomática en las vías respiratorias, pulmones, piel y sistema musculoesquelético, pero su reducción suele causar recaída de la enfermedad. La afección seria y aguda, en especial el daño renal progresivo, se trata al inicio con esteroides y ciclofosfamida diaria, con reducción subsecuente de los esteroides. Existen ciertas contraindicaciones relativas pero importantes para el uso de la ciclofosfamida, incluyendo la alteración vesical y la leucopenia. En la GW más leve o limitada puede sustituirse la ciclofosfamida con metotrexate semanal (0.20 a 0.30 mg/kg, según la función renal) con ácido fólico o leucovorin.12 Los pacientes tratados con metotrexate o ciclofosamida deben ser vigilados en forma constante en busca de recaídas de la enfermedad, infecciones oportunistas y efectos adversos. Los efectos adversos incluyen citopenias y neumonitis inducida por medicamentos. El metotrexate puede causar hepatitis y, en raras ocasiones, cirrosis. Debe evitarse si existe insuficiencia renal significativa. La ciclofosfamida puede producir cistitis y cáncer de vejiga. Algunos autores han sugerido el uso de trimetoprim-sulfametoxazol como tratamiento adyuvante para el manejo de la GW y para la prevención de infecciones bacterianas que puedan promover recaídas de la enfermedad de las vías respiratorias superiores. Este aspecto sigue siendo motivo de controversia. Sin embargo, la administración de trimetoprim-sulfametoxazol tres veces por semana es útil para proteger al paciente contra la neumonía por P. carinii mientras recibe el tratamiento inmunosupresor intenso. La higiene local nasal y sinusal, así como las evaluaciones otolaringoscópicas, son parte rutinaria de la atención de los pacientes con afección de las vías respiratorias superiores.
El síndrome de Churg-Strauss (SCS), o angeítis alérgica granulomatosa, es un síndrome raro que afecta arterias y venas de pequeño a mediano calibre.
El SCS tiene manifestaciones clínicas semejantes a las de la GW en términos de afección orgánica y patología, en especial en pacientes con afección de las vías respiratorias superiores o inferiores o glomerulonefritis. El SCS difiere principalmente de la GW en que el primero ocurre en pacientes con historia de atopia, asma o rinitis alérgica, que con frecuencia está activa. En la fase atópica prevasculítica, así como durante la fase sistémica de la enfermedad, la eosinofilia es característica y con frecuencia muy marcada (> 1,000 eosinófilos/mm3). Cuando existe eosinofilia en la GW está suele ser más moderada (< 500 eosinófilos/mm3).
Las manifestaciones sistémicas del SCS incluyen cierta combinación de infiltrados pulmonares, cardiomiopatía, arteritis coronaria, pericarditis, polineuropatía (simétrica o mononeuritis múltiple), enfermedad intestinal isquémica, gastroenteritis eosinofílica, inflamación ocular, perforaciones nasales, glomerulonefritis, nódulos cutáneos y púrpura.13,14
Los infiltrados pulmonares en parches del SCS suelen ser temporales y asociarse con hemorragia alveolar. Los nódulos pulmonares son muy poco comunes y rara vez sufren cavitación. Son frecuentes los derrames pleurales y con frecuencia contienen eosinófilos abundantes. En ocasiones es difícil la diferencia clínica entre neumonitis por hipersensibilidad, aspergilosis alérgica y linfoma pulmonar. Se han reportado varios casos de SCS después del tratamiento con zafirlukast y la supensión de los esteroides en pacientes con asma bronquial crónica.
La afección cardiaca puede ser severa y es la principal causa de mortalidad. El daño vascular no es tan común o característico como en el síndrome hipereosinofílico idiopático. Ocurre afección neurológica en más del 60 por ciento de los pacientes, puede ser severa y suele atribuirse a arteritis. Ocurren púrpura, urticaria, erupciones eritematosas polimórficas y nódulos en la piel. El daño gastrointestinal se debe a vasculitis isquémica, gastroenteritis eosinofílica o ambos, y puede causar dolor tipo cólico y diarrea.
La histopatología típicamente muestra inflamación granulomatosa extravascular, con infiltrado eosinofílico prominente y vasculitis. La vasculitis en un corte determinado de tejido puede ser granulomatosa o no granulomatosa. Pueden encontrarse granulomas en áreas tisulares independientes de la vasculitis demostrable. Los infiltrados eosinofílicos son más importantes que en la GW. Ni en la poliarteritis nodosa (PAN) clásica ni en la poliangeítis microscópica (PAM) se encuentran eosinófilos abundantes, granulomas o células gigantes. La patología de los nódulos no es suficiente para hacer el diagnóstico de SCS porque pueden encontrase datos similares en el linfoma o la sarcoidosis. La glomerulonefritis no es tan común o severa como en la GW, pero cuando existe suele ser focal y segmentaria, indistinguible de otras formas de la llamada glomerulonefritis pauci-inmune (sin depósito significativo de complejos inmunes en el tejido).
El SCS suele ser una enfermedad que responde a esteroides. Algunos pacientes incluso pueden llegar a suspenderlos. Sin embargo, el asma bronquial y la afección sinusal pueden requerir de tratamiento continuo, incluso si remite el componente vasculítico de la enfermedad. Los pacientes con afección orgánica severa o refractaria visceral se tratan en forma empírica con agentes como ciclofosfamida, metotrexate o azatioprina.
Poliarteritis nodosa y poliangeítis microscópica
Los intentos por separar la PAN y la PAM, dos formas de vasculitis necrosantes de arterias pequeñas a medianas, no han sido aceptadas en forma universal. Una conferencia de consenso internacional reciente propuso que el diagnóstico de estos padecimientos se base en la ausencia de inflamación granulomatosa en ambas y la alta de afección de arteriolas, capilares, vénulas y capilares glomerulares en la PAN. Estudios más antiguos de pacientes con PAN no hicieron esta distinción en forma uniforme. Incluso más importante, los pacientes con hepatitis viral B o C no pudieron ser excluidos de los estudios previos. La detección de hepatitis viral es de gran importancia porque la hepatitis crónica B o C15 puede causar un síndrome de vasculitis secundaria indistinguible de la PAN o la PAM desde el punto de vista clínico, pero con diferente respuesta al tratamiento.16 La PAM afecta a los vasos que van desde capilares y vénulas hasta arterias de mediano tamaño [ver figura 1].17
La glomerulonefritis, en especial la glomerulonefritis rápidamente progresiva, y la hemorragia alveolar, son comunes en la PAM y están ausentes, por definición, en la PAN clásica.
La PAN afecta arterias musculares de tamaño mediano y, como la PAM, se asocia con neuropatía periférica e isquemia intestinal.18-20 Pueden ocurrir azoemia e hipertensión en la PAN por arteritis de las arterias renales pero no por glomerulonefritis. La formación de microaneurismas en las arterias de vasos medianos puede ser importante, con ruptura ocasional.
Los síntomas constitucionales como fiebre, astenia y mialgias son comunes tanto en la PAN como en la PAM. Son frecuentes los reactantes de fase aguda elevados, la trombocitosis, la leucocitosis y la anemia de la enfermedad inflamatoria, aunque no siempre existen.
Cuando se sospecha clínicamente PAN o PAM, debe excluirse una infección bacteriana (v.gr. endocarditis ) o viral (v.gr., hepatitis B y C). La asociación con infección por virus de la hepatitis B o C puede no alterar en forma dramática la presentación del síndrome de PAN o PAM, excepto porque la glomerulonefritis membranosa, la crioglobulinemia, la glomerulonefritis asociada a complejos inmunes, la falla hepática y la trombocitopenia son más probables en la vasculitis asociada a hepatitis viral.
El síndrome de anticuerpos antifosfolípido (SAF) puede simular PAN al causar isquemia mesentérica o insuficiencia renal por oclusión trombótica de los vasos mesentéricos y renales.21 Las características del SAF y la PAN incluyen livedo reticularis [ver figura 5]. No se espera que ocurran glomerulonefritis crioglobulinémica, glomerulonefritis asociada a complejos inmunes, y neuropatía periférica en el SAF a menos que el paciente tenga también LEG. La trombocitopenia puede ocurrir en el SAF, pero no es común en la PAN. También debe pensarse en émbolos de colesterol22 como causa de livedo, insuficiencia renal y síntomas constitucionales.
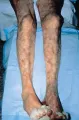
|
| Figura 5 |
| Livedo reticularis |
Pruebas de laboratorio
El diagnóstico de PAM y de PAN idealmente debe basarse en la demostración histológica de arteritis y en el patrón clínico de la enfermedad. La presencia de p-ANCA en suero con especificidad antimieloperoxidasa (en el 60 por ciento de los pacientes con PAM) apoya el diagnóstico clínico de PAM, pero los p-ANCA no son específicos de esta enfermedad. Los ANCA no son característicos de la PAN. La biopsia renal en la PAM, como en la GW y en el SCS, no contiene gran cantidad de complejos inmunes en la tinción de inmunofluorescencia o la microscopía electrónica, constituyendo una glomerulonefritis pauci-inmune. Cuando existen infiltrados pulmonares o hemorragia la biopsia pulmonar revela capilaritis, un patrón histológico que puede observarse también en GW, LEG y enfermedad anti-membrana basal glomerular. La biopsia es útil principalmente para descartar diagnósticos pulmonares alternativos, y la biopsia abierta o por toracoscopía demuestra vasculitis con más frecuencia que la biopsia transbronquial. La PAN clásica no causa glomerulonefritis o afección pulmonar parenquimatosa.
Puede ser difícil la demostración de la arteritis en la PAN, en especial cuando existen síntomas constitucionales y ausencia de un tejido afectado fácilmente accesible. Las biopsias iniciales deben tomarse de tejidos con evidencia de afección según los síntomas o signos. La biopsia del nervio sural se ha vuelto una opción popular cuando se intenta diagnosticar una arteritis que afecta vasos musculares de tamaño mediano. El nervio sural es un nervio puramente sensorial accesible y su vasa nervorum contiene arterias musculares de tamaño tanto pequeño como mediano. Los estudios de conducción nerviosa permiten identificar un nervio isquémico antes de la aparición de los síntomas clíniocs.23 Múltiples reportes han enfatizado la poca certeza diagnóstica por la biopsia de nervios asintomáticos y normales desde el punto de vista eléctrico. Incluso los nervios con conducción anormal no muestran patología diagnóstica en el 46 por ciento de los casos.24 Este procedimiento causa una morbilidad notable, 13 de 60 pacientes sufrieron infecciones de las heridas o retraso en la cicatrización, y 3 pacientes tuvieron dolor de reciente inicio en la distribución del nervio sural biopsiado.24 La biopsia de tejido clínicamente no afectado (i.e., músculo asintomático) tiene una utilidad diagnóstica menor al 30 por ciento.
Con frecuencia se utiliza la angiografía abdominal en la evaluación de pacientes que pueden tener arteritis en vasos de calibre mediano cuando la biopsia no da un buen resultado o no es una opción adecuada. Las arterias afectadas por la poliarteritis nodosa y otros padecimientos que afectan arterias musculares de tamaño mediano pueden causar microaneurismas o estenosis que pueden observarse por angiografía. Cuando se usa la angiografía en un esfuerzo por diagnosticar vasculitis sistémica necrosante en ausencia de evidencia patológica de la enfermedad, deben considerarse varias dificultades. La angiografía tiene una capacidad de resolución espacial limitada, por lo que los vasos pequeños no se observan bien. Los pacientes con afección principalmente de vasos pequeños es poco probable que tengan una angiografía diagnóstica. En un estudio, solo 4 de 30 pacientes con PAM, una enfermedad que afecta arterias tanto pequeñas como medianas, tuvo angiografías diagnósticas.17 Diferentes investigadores han reportado aneurismas en el 60 a 90 por ciento de los pacientes con PAN. Los aneurismas tardan en desarrollarse y pueden no existir al inicio de la enfermedad. Además de asociarse con aneurismas, la arteritis puede asociarse con estenosis, que pueden ser más largas y graduales que las lesiones u oclusiones ateroescleróticas típicas. Para maximizar la utilidad del procedimiento el estudio debe incluir los vasos celiaco, renal y mesentéricos. La falta de afección clínica de un órgano (i.e., la ausencia de isquemia intestinal) no excluye la posibilidad de encontrar vasos anormales en la angiografía. Se ha sugerido que la observación de aneurismas en la PAN denota afección más severa. No es claro si su presencia puede relacionarse en forma alternativa con la duración de la enfermedad. Los aneurismas pueden resolverse con el tratamiento exitoso de la afección primaria o asociada a hepatitis viral. La presencia de microaneurismas viscerales no es diagnóstica de PAN. Estos se han descrito en forma anecdótica en pacientes con GW y PAM y es probable que representen afección de arterias musculares de tamaño mediano en esos padecimientos. También ocurren microaneurismas en trastornos no vasculíticos. Se han descrito casos aislados de aneurismas en pacientes con mixoma auricular, endocarditis bacteriana, carcinomatosis peritoneal e hipertensión arterial severa, así como después del abuso de metanfetaminas. No existen datos adecuados para evaluar la sensibilidad y especificidad o el valor predictivo de la angiografía abdominal en el diagnóstico de la arteritis necrosante. Como en el caso en que se interpreta el resultado de una biopsia en una probable vasculitis, los estudios de imagen deben analizarse en conjunto con el cuadro clínico. En general debe evitarse la angiografía si existe insuficiencia renal progresiva o significativa.
El tratamiento tanto de la PAN como de la PAM es empírico.25 Los esteroides en dosis altas siguen siendo la base del tratamiento para ambos padecimientos. Los esteroides son suficientes en los pacientes que no tienen afección a órganos vitales, definida como insuficiencia renal, isquemia gastrointestinal, cardiomiopatía o neuropatía periférica. El tratamiento con esteroides solos puede fracasar con más frecuencia en la PAM que en la PAN por la tendencia de la primera a que existan recaídas.17 Los pacientes con marcadores de enfermedad severa suelen tratarse con esteroides y un agente inmunosupresor adicional, como ciclofosfamida. Aunque este enfoque es el habitual, no se han estudiado en forma adeucada las indicaciones para iniciar con tratamiento combinado.
Cuando existe infección activa por virus de la hepatitis B o C debe considerarse un esquema relativamente breve de esteroides según la severidad de la enfermedad, junto con el tratamiento antiviral.
La enfermedad de Kawasaki (EK) fue descrita por primera vez en 1967 como síndrome mucocutáneo ganglionar.26 Típicamente afecta lactantes y niños pequeños, causando principalmente manifestaciones cutáneas, muy raras veces afecta adultos.
La presencia de características clínicas definidas en la EK ha permitido establecer criterios diagnósticos [ver tabla 3]. Esta vasculitis afecta vasos que varían en tamaño desde vénulas hasta la aorta. Se observa inflamación importante en las arterias coronarias, lo que provoca la formación de aneurismas en alrededor del 25 por ciento de los pacientes no tratados. Las complicaciones cardiacas inmediatas y tardías que ponen en peligro la vida, aunadas con el tratamiento específico que se requiere (aspirina y gamaglobulina intravenosa) obligan a realizar un diagnóstico clínico pronto.
|
||
|
Sin tratamiento puede existir fiebre alta en picos durante 1 a 2 semanas. Al
iniciar la terapia es característica la defervescencia rápida. Es
frecuente que junto con la fiebre exista conjuntivitis no exudativa, y la
meningitis aséptica (linfocítica) es común. Las
manifestaciones orales incluyen eritema, sequedad y fisuras en los labios,
faringitis no exudativa y eritema en la lengua con papilas muy prominentes. Las
ulceras mucosas no son características de la enfermedad. Puede ocurrir
inflamación distal en las extremidades días después de la
fiebre, con eritema y dolor que no se limitan a las articulaciones. La
descamación, con frecuencia es placas, puede comenzar días a
semanas después de iniciada la fiebre. Cuando la descamación es
temprana puede aparecer en forma concomitante una erupción en el tronco
y cambios en los ojos y labios, simulando una reacción medicamentosa o
un síndrome de Stevens-Johnson. La erupción suele ser difusa y
polimorfa, con componentes urticarianos, morbiliformes, anulares o en placa,
pero no forma vesículas. La adenopatía, que existe en el 75 por
ciento de los pacientes, es más evidente en la región cervical.
La morbimortalidad de la EK (< 3 por ciento) se asocia principalmente con el desarrollo de aneurismas inflamatorios en las arterias coronarias, la mayoría asintomáticos en el momento de su formación. Los aneurismas pueden detectarse por ecocardiografía. Ocurren trombos en los aneurismas, provocando oclusión coronaria directa o por embolia. Los eventos coronarios pueden ocurrir semanas o incluso mucho años después del proceso febril. Debe obtenerse un ecocardiografma basal en el momento del padecimiento agudo y repetirse 2 y 6 semanas después. La detección temprana del padecimiento y el tratamiento con inmunoglobulina intravenosa y aspirina ha disminuido en forma significativa la frecuencia de formación de aneurismas y los eventos coronarios trombóticos.
El tratamiento debe iniciarse con inmunoglobulina intravenosa (2 g/kg en dosis única) y aspirina (80 a 100 mg/kg/día cada 6 horas) en cuanto la sospecha diagnóstica sea seria.27 La aspirina es más eficaz que los esteroides para prevenir los aneurismas. Por lo general no se requiere tratamiento esteroideo y algunos autores consideran que está relativamente contraindicado. La fiebre, la conjuntivitis y la erupción tienden a resolverse en varios días después de iniciados la aspirina y la inmunoglobulina.
La arteritis de la temporal, o de células gigantes (ACG) del anciano y la arteritis de Takayasu (AT) son los trastornos inflamatorios más comunes de la aorta y de sus ramas principales. Puede ocurrir afección vascular semejante en la enfermedad de Behcet, el síndrome de Cogan y la sarcoidosis. Existe incertidumbre respecto a si la AT y la ACG son padecimientos diferentes o son el mismo trastorno con una expresión modificada en diferentes grupos de edad.
ARTERITIS DE LA TEMPORAL O DE CELULAS GIGANTES
La ACG generalmente afecta individuos mayores de 50 años.28,29 Se asocia en muchos pacientes con el síndrome de polimialgia reumática (PR). La PR se caracteriza por dolor muscular proximal que empeora por la noche y temprano en la mañana. Puede existir una sensación subjetiva de debilidad, sin debilidad real a la exploración o elevación de las enzimas musculares séricas.
La ACG se asocia en forma variable con fiebre, dolor en el cuero cabelludo, cefalea, claudicación de los músculos masticatorios, enfermedad vascular periférica, aneurismas aórticos inflamatorios y síndromes isquémicos de la retina. Puede ocurrir artritis oligoarticular, con frecuencia en las extremidades superiores, así como síndrome del túnel del carpo. Los síntomas y signos isquémicos pueden ser clínicamente indistinguibles de los que ocurren en la enfermedad obliterativa ateroesclerótica.
Como parte de las visitas de seguimiento de los pacientes con ACG debe incluirse el examen de la presión arterial en las cuatro extremidades y la búsqueda de soplos y de aneurismas abdominales. Pueden datos patológicos de ACG en las arterias temporales superficiales de los pacientes con PR, incluso sin síntomas de ACG. Sin embargo, en los pacientes con PR y sin otros síntomas de ACG no está justificada la biopsia de rutina de las arterias temporales superficiales.
Los reactantes de fase aguda se elevan en más del 80 por ciento de los pacientes. El diagnóstico definitivo se hace por biopsia de la arteria temporal superficical. Los datos histológicos en la ACG suelen consistir en infiltrados mononucleares crónicos, destrucción de la lámina elástica interna y células gigantes. La presencia de células gigantes no es requisito para el diagnóstico. La existencia de manifestaciones características, como cefalea de reciente inicio y claudicación de la mandíbula, en especial con PR concomitante, puede permitir el diagnóstico presuntivo en ausencia de una biopsia o incluso cuando la biopsia de la arteria temporal superficial es negativa. Sin embargo, debido a que otras condiciones pueden simular ACG, incluyendo la ateroesclerosis, está justificado intentar diagnosticar la ACG realizando la biopsia en la mayoría de los pacientes.30 El tratamiento esteroideo no afecta de inmediato el resultado de la biopsia, por lo que no debe detenerse en un paciente con sospecha diagnóstica y en quien se espera realizar el procedimiento.
La arteritis de Takayasu (enfermedad sin pulsos) es una enfermedad inflamatoria crónica que afecta la aorta y sus ramas principales.31 Suele diagnosticarse principalmente en mujeres jóvenes, durante la época reproductiva de la vida, aunque puede ocurrir también en niños y pacientes de mayor edad de cualquier sexo. La AT se asocia más con estenosis y aneurismas de la aorta y sus ramas que la ACG.
El síndrome clínico de presentación puede incluir una enfermedad prolongada de tipo gripal, incluyendo un patrón de dolor muscular semejante a la polimialgia reumática. Otros pacientes cursan con síntomas de isquemia en las extremidades, cerebral o cardiaca. Las características de la enfermedad reflejan la isquemia producida por las estenosis inflamatorias de la aorta y sus ramas principales. La isquemia renal puede causar hipertensión con renina alta. Los sitios principales de estenosis son los vasos del arco aórtico, en especial la arteria subclavia. Son comunes la claudicación de los brazos y la presencia de soplos. Puede encontrarse dolor superficial y a la palpación en las arterias (v.gr., carotidinia), pero este dato no es diagnóstico de AT. La hipertensión central severa causada por estenosis de la arteria renal puede no detectarse por la coexistencia de estenosis en las arterias de los brazos, pro lo que al inicio debe evaluarse la presión arterial en las cuatro extremidades, vigilándola con frecuencia. En ocasiones pueden existir estenosis en todas las arterias principales de las extremidades, lo que impide vigilar la presión aórtica central. No son raros los eventos cerebrovasculares, que suelen relacionarse con hipertensión no detectada. Es muy difícil evaluar la actividad de la AT, la presencia o ausencia de manifestaciones constitucionales o el aumento en los reactantes de fase aguda son medidas poco exactas de la actividad de la enfermedad. Esta impresión ha sido corroborada por la histopatología vascular obtenida durante la cirugía reconstructiva. Más del 40 por ciento de las muestras vasculares de pacientes que se pensaba estaban en remisión revelaron inflamación activa.
El diagnóstico de AT suele hacerse por demostración angiográfica de lesiones estenóticas, con menos frecuencia se observan aneurismas. Debe evaluarse todo el arco aórtico, así como la aorta abdominal y los vasos renales. Es de gran importancia que se obtenga la presión arterial central en el momento de la angiografía y se compare con una medición simultánea obtenida con manguito en el brazo y la pierna. En la actualidad se investiga la utilidad de la resonancia magnética en la evaluación y seguimiento de estos pacientes.32 Esta técnica puede indicar cambios en el grosor de la pared vascular y edema, así como cambios en el tamaño de la luz. En la AT es difícil obtener la corroboración histológica, que suele obtenerse en el momento de la cirugía de derivación; los datos son semejantes a los de la ACG.
TRATAMIENTO DE LA ACG Y DE LA AT
Los esteroides constituyen el tratamiento inicial tanto para la AT como para la ACG. La ACG suele responder a los esteroides, aunque la dosis inicial más adecuada es motivo de controversia. Se han sugerido dosis iniciales diarias entre 20 mg y 1 mg/kg, con reducción grdual en 8 a 12 meses. Algunos pacientes con ACG requieren de varios años de tratamiento. La medición de los reactantes de fase aguda proporciona un índice imperfecto de la actividad de la enfermedad y no debe ser la única guía para ajustar la dosis de esteroide. Si ocurren efectos adversos significativos por lo esteroides o si los pacientes refieren recaídas al disminuir estos, puede agregarse un agente de segunda línea, como metotrexate, en forma empírica. Sin embargo, hasta el momento no se ha demostrado el valor real de estos agentes ahorradores de esteroides en la ACG. La cirugía vascular reconstructiva, la angioplastía y la colocación de férulas constituyen opciones terapéuticas adyuvantes para estos pacientes. La afección tan frecuente de los vasos subclavios en la AT debe tomarse en cuenta para elegir el sitio del implante en los procedimientos de derivación coronaria. El tratamiento esteroideo en dosis altas, en especial en los pacientes de edad avanzada, tiene efectos adversos potencialmente peligrosos. Debe ponerse especial atención a prevenir infecciones oportunistas, osteoporosis, glaucoma, hiperglucemia e hiperlipidemia.
Figuras 1 y 2 Seward Hung
Bibliografía
- Jennette C, Falk RJ, Andrassy K, et al: Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum 37:187, 1994
- Agnello V, Romain PL: Mixed cryoglobulinemia secondary to hepatitis C virus infection. Rheum Dis Clin North Am 22:1, 1996 [PMID 8907062 ]
- Lawrence EC, Mills J: Bacterial endocarditis mimicking vasculitis with steroid-induced remission. West J Med 124:333, 1976 [PMID 1266218 ]
- Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, et al: Cutaneous vasculitis in children and adults: associated disease and etiologic factors in 303 patients. Medicine (Baltimore) 77:403, 1998 [PMID 9854604 ]
- Szer IS: Henoch-Schönlein purpura: when and how to treat. J Rheumatol 23:1661, 1996
- O'Donnell B, Black AK: Urticarial vasculitis. Int Angiol 14:166, 1995 [PMID 8609443 ]
- Hoffman GS, Kerr GS, Leavitt RY, et al: Wegener's granulomatosis: an analysis of 158 patients. Ann Intern Med 116:488, 1992 [PMID 1739240 ]
- Duna G, Galperin C, Hoffman GS: Wegener's granulomatosis. Rheum Dis Clin North Am 21:949, 1995 [çPMID 8592744 ]
- Travis WD, Hoffman GS, Leavitt RY, et al: Surgical pathology of the lung in Wegener's granulomatosis. Am J Surg 15:315, 1991
- Hoffman GS: Classification of the systemic vasculitides: antineutrophil cytoplasmic antibodies, consensus and controversy. Clin Exp Rheumatol 16:111, 1998
- Hoffman GS, Specks U: Antineutrophil cytoplasmic antibodies: diagnostic value in systemic vasculitis. Arthritis Rheum 41:1521, 1998 [PMID 9751084 ]
- Sneller MC, Hoffman GS, Talar-Williams C, et al: An analysis of 42 Wegener's granulomatosis patients treated with methotrexate and prednisone. Arthritis Rheum 38:608, 1995 [PMID 7748215 ]
- Guillevin L, Cohen P, Gayraud M, et al: Churg-Strauss syndrome: clinical study and long-term follow up of 96 patients. Medicine (Baltimore) 78:26, 1999 [PMID 9990352 ]
- Reid AJC, Harrison BDW, Watts RA, et al: Churg-Strauss syndrome in a district hospital. Q J Med 91:219, 1998
- Hadziyannis SJ: The spectrum of extrahepatic manifestations in hepatitis C virus infection. J Viral Hepat 4:9, 1997
- Guillevin L, Lhote F, Cohen P, et al: Polyarteritis nodosa related to hepatitis B virus: a prospective study with long-term observation of 41 patients. Medicine (Baltimore) 74:238, 1995 [PMID 7565065 ]
- Guillevin L, Durand-Gasselin B, Cevallos R, et al: Microscopic polyangiitis-clinical and laboratory findings in 85 patients. Arthritis Rheum 42:421, 1999 [PMID 10088763 ]
- Travers RL, Allison DJ, Brettle RP, et al: Polyarteritis nodosa: a clinical and angiographic analysis of 17 cases. Semin Arthritis Rheum 8:184, 1979 [PMID 34221 ]
- Lhote F, Cohen P, Guillevin L: Polyarteritis nodosa, microscopic polyangiitis and Churg-Strauss syndrome. Rheum Dis Clin North Am 21:911, 1995 [PMID 8592743 ]
- Mandell BF, Hoffman GS: Differentiating the vasculitides. Rheum Dis Clin North Am 20:409, 1994 [PMID 8016419 ]
- Triplett DA: Protean clinical presentation of antiphospholipid-protein antibodies. Thromb Haemost 74:329, 1995
- Om A, Ellahham S, DiScascio G: Cholesterol embolism: an underdiagnosed clinical entity. Am Heart J 124:1321, 1992 [PMID 1442502 ]
- Wees SJ, Sunwoo IN, Oh SJ: Sural nerve biopsy in systemic necrotizing vasculitis. Am J Med 71:525, 1981 [PMID 6116431 ]
- Rappaport WD, Valente J, Hunter GC, et al: Clinical utilization and complications of sural nerve biopsy. Am J Surg 166:252, 1993 [PMID 8396357 ]
- Guillevin L, Lhote F: Treatment of polyarteritis nodosa and microscopic polyangiitis. Arthritis Rheum 41:2100, 1998 [PMID 9870866 ]
- Schulman ST, Inocencio JD, Hirsch R, et al: Kawasaki disease. Pediatr Clin North Am 21:1013, 1995
- Leung DY, Schlievert PM, Meissner HC: The immunopathogenesis and management of Kawasaki syndrome. Arthritis Rheum 41:1538, 1998 [PMID 9751085 ]
- Evans JM, Hunder GG: Polyangiitis rheumatica and giant cell arteritis. Clin Geriatr Med 14:455, 1998 [PMID 9664102 ]
- Hunder GG: Giant cell arteritis and polymyalgia rheumatica. Med Clin North Am 81:195, 1997
- Ponge T, Barrier JH, Grolleau JY, et al: The efficacy of selective unilateral temporal artery biopsy versus bilateral biopsies for diagnosis of giant cell arteritis. J Rheumatol 15:997, 1988 [PMID 2971112 ]
- Kerr GS, Hallahan CW, Giordano J, et al: Takayasu's arteritis. Ann Intern Med 120:919, 1994 [PMID 7909656 ]
- Flamm SD, White RD, Hoffman GS: The clinical application of "edema-weighted" magnetic resonance imaging in assessment of Takayasu's arteritis. Int J Cardiol 66(suppl 1):S151, 1998