Contenido del artículo
XIII TRASTORNOS HEMORRAGICOS
- Estudio del paciente con trastornos hemorrágicos
- Trombocitopenia - Cuenta de plaquetas disminuida
- DEFECTOS EN LA PRODUCCION DE PLAQUETAS
- ELIMINACION ACELERADA DE PLAQUETAS POR DESTRUCCION INMUNE
- Púrpura trombocitopénica idiopática
- Púrpura trombocitopénica secundaria
- Destrucción plaquetaria inducida por fármacos
- ELIMINACIÓN ACELERADA DE PLAQUETAS POR MECANISMOS NO INMUNOLOGICOS
- Trastornos funcionales de las plaquetas
- Trombocitosis y trombocitemia
- Púrpuras vasculares
- TELANGIECTASIA HEMORRAGICA HEREDITARIA
- ESCORBUTO
- EXCESO DE ESTEROIDES
- AMILOIDOSIS
- VASCULITIS LEUCOCITOCLASTICA
- PURPURA SENIL
- DAÑO A LA MICROVASCULATURA POR EMBOLOS
- Trastornos hereditarios de la coagulación
- ENFERMEDAD DE VON WILLEBRAND
- HEMOFILIA A
- Principios de tratamiento general para la hemofilia
- Tratamiento de la hemorragia aguda
- Principios del tratamiento sustitutivo
- Cirugía electiva y extracción dental
- Tratamiento de un inhibidor
- OTROS TRASTORNOS HEMORRAGICOS HEREDITARIOS
- Trastornos hemorrágicos adquiridos
- DEFICIENCIA DE VITAMINA K
- HEMORRAGIA INDUCIDA POR FARMACOS
- DISPROTEINEMIAS
- COAGULACION INTRAVASCULAR DISEMINADA
- INHIBIDORES ADQUIRIDOS DE LA COAGULACION (ANTICOAGULANTES ADQUIRIDOS)
- HEPATOPATIA SEVERA
- FIBRINOLISIS PRIMARIA
- HEMORRAGIA DESPUES DE UNA DERIVACION CARDIOPULMONAR
- EVALUACION DE LA HEMORRAGIA POSOPERATORIA
DR. LAWRENCE L. K. LEUNG
Estudio del paciente con trastornos hemorrágicos
Muchas personas sanas consideran sus hemorragias y equímosis excesivas. Por otro lado, los pacientes con enfermedad de von Willebrand, el trastorno hemorragíparo hereditario más común, pueden no identificar sus síntomas de hemorragia.1 En consecuencia, es importante obtener información objetiva (v.gr., si el paciente tiene equímosis fáciles, el tamaño de las mismas, si el paciente ha sido sometido a cirugía o transfusiones). La historia dental puede ser especialmente útil. Si el paciente tuvo una extracción dental, en especial con algún testigo, ¿fue necesario regresar a la consulta para un taponamiento o sutura? ¿Se requirió transfusión? Las historias dentales y quirúrgicas pueden ayudar a distinguir entre los trastornos hereditarios y los adquiridos. Por ejemplo, el inicio súbito de una hemorragia sugerirá una diátesis hemorrágia adquirida, como un inhibidor del factor VIII. El paciente debe ser interrogado sobre el uso de medicamentos (incluyendo drogas intravenosas) y patrones de actividad sexual, así como la historia de transfusión, anemia, infección recurrente, enfermedad del tejido conectivo, linfoma y estados de inmunosupresión. También es muy importante contar con la historia familiar.
El tipo de hemorragia da información El sangrado activo puede ser causado por una lesión anatómica local o una diátesis hemorrágica subyacente. La hemorragia mucosa, con epistaxis recurrente, gingivorragia, equímosis y menorragia, es sugestiva de enfermedad de von Willebrand u otros trastornos plaquetarios, mientras que las hemorragias de tejidos profundos (v.gr., hemartrosis y hematomas musculares dolorosos) se observan con más frecuencia en la hemofilia y las deficiencias de los factores de la coagulación. Los pacientes con deficiencias en los factores de la coagulación pueden tener sangrado tardío, quizá porque el trombo plaquetario inicial proporciona la hemostasia inmediata, pero no es estabilizado en forma adecuada por un coágulo de fibrina. Como ya se mencionó, es importante obtener una historia farmacológica detallada. La aspirina puede alterar en parte la función plaquetaria y desencadenar síntomas hemorrágicos en un paciente con enfermedad de von Willebrand subyacente leve. Debido a que varios cientos de medicamentos contienen aspirina (con frecuencia sin indicación de su contenido en el nombre del producto), puede ser difícil identificar a la aspirina como causa del trastorno hemorragíparo.
Trombocitopenia - Cuenta de plaquetas disminuida
La trombocitopenia puede ser causada por producción anormal de plaquetas, eliminación acelerada resultado de mecanismos inmunológicos o no inmunológicos, secuestro de plaquetas en el bazo o combinación de estos mecanismos.
La principal característica de la trombocitopenia son las petequias, que reflejan sangrado a nivel de los capilares o vénulas poscapilares. Las petequias suelen ocurrir en sitios de mayor presión intravascular (v.gr., sobre las extremidades, en la mucosa oral y en los sitios que sufren presión por la ropa, como por las tiras del brassiere). Las petequias por trombocitopenia en general no se palpan. La púrpura palpable indica un componente adicional de inflamación vascular y sugiere una vasculitis sistémica subyacente, como la crioglobulinemia.
La trombocitopenia causa púrpura y hemorragia mucosa, los sangrados de tejidos blandos son poco comunes. No existe un umbral exacto en el nivel de plaquetas con el cual pueda considerarse que el paciente está seguro. El riesgo de hemorragia intracraneal suele dirigir el tratamiento. En general, una cuenta de plaquetas mayor de 20,000/µl se considera tolerable, las cuentas de 10,000/µl o menos pueden tolerarse en pacientes no quirúrgicos. En general, los pacientes con púrpura trombocitopénica idiopática sangran menos con una cuenta determinada de plaquetas que los pacientes con anemia aplástica. Al parecer, mientras más grandes y jóvenes sean las plaquetas son más eficaces en la hemostasia porque son más activas desde el punto de vista metabólico.
Los ancianos y los pacientes con enfermedades concomitantes sangran más que los pacientes jóvenes y los enfermos que solo tienen trombocitopenia. Un trastorno asociado, como la disfunción hepática o una enfermedad de tejido conectivo, aumenta el riesgo de hemorragia grave.
En la evaluación inicial de laboratorio la biometría hemática completa establecerá si la trombocitopenia es un dato aislado o se asocia con anemia o leucopenia. Si la trombocitopenia es aislada el médico debe confirmar la cuenta plaquetaria repitiendo la prueba. La agregación de plquetas in vitro causada por la presencia de aglutininas frías o dependientes de ácido etilendiaminotetracético puede causar una trombocitopenia ficticia. El examen del frotis de sangre y la repetición de la cuenta de plaquetas en una muestra anticoagulada con heparina o con citrato resolverá este problema.2
El frotis de sangre periférica revelará alteraciones morfológicas e indica la presencia de policromatofilia, neutropenia, linfopenia, esferocitosis, blastos o eritrocitos microangiopáticos fragmentados. El volumen plaquetario promedio, determinado por una citología hemática automatizada, proporciona información adicional sobre la causa de la trombocitopenia. Los volúmenes de plaquetas bajos (<6.4 femtolitros) sugieren mala producción, mientras que los volúmenes altos indican regeneración rápida o producción displástica.
DEFECTOS EN LA PRODUCCION DE PLAQUETAS
Es crucial realizar un aspirado de médula ósea para diagnosticar la trombocitopenia. El encontrar una médula hipoplásica en la que la celularidad total está disminuida con reducción concomitante en los megacariocitos implica anemia aplástica o hipoplástica. La primera sospecha en estos casos debe ser toxicidad por fármacos. Una médula fibrosada o infiltrada con células leucémicas o neoplásicas de otro tipo representa el síndrome de pancitopenia por infiltración de la médula ósea.
El aspirado y biopsia de médula que muestra celularidad normal y maduración normal de los precursores eritroides y mieloides con un número menor de megacariocitos aparentemente normales sugiere que el paciente ha ingerido un medicamento, como etanol, que afecta en forma específica a las células progenitoras megacariocíticas.3 El etanol produce también megacariopoyesis ineficaz. En la deficiencia de vitamina B12 y de folato se afectan las tres líneas de la médula. El frotis de médula muestra muchos megacariocitos grandes e hiperlobulados. Algunos trastornos mieloproliferativos se caracterizan por megacariopoyesis ineficaz con megacariocitos binucleados bizarros.
Si se sospecha un medicamento como causa de la trombocitopenia, éste debe suspenderse. Se requiere sustitución específica para las deficiencias de vitamina B12 y folato. Cuando la trombocitopenia está causando hemorragia significativa pueden requerirse transfusiones de plaquetas hasta resolver la situación.
En la actualidad se realizan estudios clínicos con trombopoyetina recombinante. Se ha demostrado que la IL-11 (IL-11), que tiene un papel importante en la megacariopoyesis, es eficaz para disminuir la necesidad de transfusiones de plaquetas después de la quimioterapia4 y ha sido autorizada por la Food and Drug Administration de los EUA para la profilaxis secundaria contra la trombocitopenia después de quimioterapia.
ELIMINACION ACELERADA DE PLAQUETAS POR DESTRUCCION INMUNE
Cuando la destrucción acelerada de plaquetas parece ser la causa de la trombocitopenia, debe hacerse un diagnóstico diferencial rápido [ver tabla 1]. Será muy útil un aspirado y biopsia de médula ósea. Por lo general, la trombocitopenia con megacariocitos normales y abundantes en la médula se debe a eliminación acelerada de plaquetas.5 En condiciones normales las plaquetas sobreviven 10 días y tienen una vida media de alrededor de 4 días: en los estados de remoción acelerada, como la púrpura trombocitopénica idiopática, la vida media de la plaqueta puede ser tan breve como de 30 a 60 minutos. La cuenta de plaquetas reflejará el equilibrio entre eliminación acelerada y megacariopoyesis compensadora.
|
|||||||||||||||||||
HELLP – hemólisis, elevación de enzimas hepáticas, trombocitopenia |
Los estudios de supervivencia de plaquetas no están fácilmente
disponibles y no suelen ser necesarios para determinar si está
ocurriendo eliminación acelerada de las mismas. La infusión de
plaquetas de donador aleatorio puede usarse como un procedimiento tanto
diagnóstico como terapéutico. Cuando la remoción acelerada
es la responsable de la trombocitopenia, la transfusión con 6 paquetes
plaquetarios elevará la cuenta solo un poco, con retorno al nivel basal
en menos de 24 horas. Sin embargo, esta prueba terapéutica pierde
confiabilidad si el paciente ha sido aloinmunizado antes con transfusiones de
sangre o plaquetas o por múltiples embarazos.
Púrpura trombocitopénica idiopática
Fisiopatología La púrpura trombocitopénica idiopática (PTI) es un trastorno autoinmune caracterizado por destrucción acelerada de plaquetas. En este padecimiento existen anticuerpos contra las plaquetas del paciente. Estos autoanticuerpos se unen a proteínas específicas en la superficie de la plaqueta y las plaquetas cubiertas con anticuerpo son eliminadas por el sistema reticuloendotelial, en especial en el bazo. La inmunoglobulina sobre la membrana de la plaqueta suele ser IgG (principalmente de la subclase IgG1). Esta inmunoglobulina con frecuencia va dirigida contra la glucoproteína plaquetaria (GP) IIb-IIIa, el complejo receptor que media la unión del fibrinógeno y la agregación plaquetaria. Con menos frecuencia va dirigida contra el complejo GPIb.6 La médula ósea responde a la trombocitopenia aumentando la producción de plaquetas. En algunos casos la respuesta medular es subóptima, quizá porque los anticuerpos antiplaqueta también reaccionan con antígenos de la superficie celular de los megacariocitos. Las plaquetas producidas en la PTI suelen ser grandes y funcionales, lo que puede explicar la observación clínica de que la mayoría de los pacientes con PTI no sufren hemorragias significativas.
Manifestaciones clínicas La PTI típicamente aparece en mujeres jóvenes. En ciertas comunidades la prevalencia de PTI en mujeres jóvenes ha sido superada por su ocurrencia en varones seropositivos para infección por VIH. Entre las enfermedades y factores que pueden contribuir a la PTI se incluyen la mononucleosis infecciosa y otras enfermedades virales agudas, la enfermedad de Graves, la tiroiditis de Hashimoto,7 así como el síndrome de anticuerpos anticardiolipina y el síndrome de anticuerpos antifosfolípido.8 En los pacientes con anticuerpos antifosfolípido la evolución, pronóstico y respuesta al tratamiento no son diferentes a los de la PTI.
El inicio de la PTI suele ser insidioso. La historia y el examen físico son negativos excepto por la presencia de petequias, principalmente en las extremidades inferiores. Las hemorragias clínicas son leves y consisten en púrpura, epistaxis, gingivorragia y menorragia. Las ampollas con sangre (púrpura húmeda) en la boca indican la presencia de trombocitopenia severa. Son raras las hemorragias retinianas. El bazo no suele ser palpable. La presencia de bazo palpable sugiere la posibilidad de lupus eritematoso generalizado (LEG), linfoma, mononucleosis infecciosa o hiperesplenismo por hepatopatía crónica.
Evaluación de laboratorio El frotis periférico suele ser normal, las pocas plaquetas presentes son grandes y poseen gránulos. La presencia de hipocromía sugiere deficiencia de hierro por pérdida crónica de sangre, los esferocitos aumentan la posibilidad de hemólisis autoinmune asociada (síndrome de Evans) y los fragmentos de eritrocitos (esquistocitos) sugieren otra enfermedad, como coagulación intravascular diseminada (CID), púrpura trombocitopénica trombótica (PTT) o síndrome urémico-hemolítico (SUH) (ver adelante). La médula muestra megacariocitos abundantes, muchos de los cuales son pequeños, mientras que los precursores eritroides y mieloides son normales. Los resultados de las pruebas para LEG son negativas. Los niveles de IgG asociada a las plaquetas (IgG-AP) están aumentados; sin embargo, debido a que en condiciones normales las plaquetas contienen IgG en sus gránulos a, la IgG-AP no distingue entre los anticuerpos antiplaqueta, los complejos inmunes depositados en la superficie de la plaqueta y los anticuerpos liberados de los gránulos de las plaquetas y fijos en su superficie. Por lo tanto, las pruebas para IgG-AP no son útiles en el diagnóstico de PTI.9
Diagnóstico diferencial El diagnóstico diferencial de la PTI incluye una cuenta de plaquetas falsamente baja por aglutininas dependientes de ácido etilenodiaminotetracético o frías que causan agregación in vitro de las plaquetas (y que se diagnostica revisando la cuenta de plaquetas en una muestra de sangre anticoagulada con heparina o con citrato), la trombocitopenia gestacional del embarazo (por lo general un problema leve que no se asocia con mayor riesgo de sangrado), el síndrome mielodisplásico (que suele asociarse con anemia y leucopenia) y las enfermedades linfoproliferativas subyacentes.
Evolución y pronóstico La PTI es un trastorno relativamente benigno que tiene una mortalidad de alrededor del 1 al 5 por ciento, la mayoría de las muertes en adultos se deben a hemorragia cerebral. La PTI aguda suele limitarse a niños y adultos jóvenes y con frecuencia es precedida por una infección viral. En menos de 3 meses ocurre remisión espontánea y permanente. La PTI crónica, la variante habitual en el adulto, se define como un proceso que persiste por más de 3 meses. Aunque ocurren remisiones y recaídas espontáneas en la PTI crónica, las remisiones espontáneas duraderas son poco comunes. Por otro lado, el pronóstico a largo plazo de la PTI es benigno si se maneja a los pacientes en forma apropiada, incluso en los casos refractarios.10
Tratamiento El tratamiento depende de la edad y la severidad de la enfermedad, de si sólo existen petequias u ocurre hemrragia moderada a severa en mucosas o el sistema nervioso central, o si la paciente está embarazada.11
La Sociedad Americana de Hematología ha elaborado una guía basada en evidencias para el tratamiento de la PTI,9 que puede resumirse de la siguiente manera:
(1) Los pacientes con cuentas de plaquetas mayores a 50,000/µl no suelen requerir tratamiento.
(2) Está indicado el tratamiento en pacientes con cuentas de plaquetas menores de 20,000 a 30,000/µl y en pacientes con cuentas de plaquetas de menos de 50,000/µl y que tienen hemorragia mucosa significativa o factores de riesgo de sangrado (v.gr., hipertensión, enfermedad ulcerosa péptica o un estilo de vida vigoroso).
(3) Los pacientes con cuentas de plaquetas de menos de 20,000/µl no necesitan hospitalizarse si están asintomáticos o si sólo tienen púrpura leve.
Los pacientes con trombocitopenia asintomática leve o moderada (i.e., cuenta de plaquetas > 50,000/ml) no requieren tratamiento activo. Pueden ser vigilados y deberán reportar cualquier sangrado de las mucosas o la aparición de nuevas petequias. Debe indicarse evitar aspirina y otros antinflamatorios no esteroides (AINE).
Para pacientes con hemorragia mucosa moderada se aconseja el tratamiento con prednisona en dosis de 60 a 100 mg/día en dosis divididas. Los esteroides interfieren con el ataque de los macrófagos sobre las plaquetas y eventualmente reducen la cantidad de anticuerpos antiplaquetarios producidos por las células esplénicas y linfoides de la médula ósea. A menos que la hemorragia sea severa el paciente no requiere hospitalización. Debe evitarse la actividad física intensa, en especial cualquiera que incluya una maniobra de Valsalva, para no aumentar la presión intracraneal. Deben evitarse la aspirina y otros AINE. Si se requiere pueden administrarse transfusiones de eritrocitos; sin embargo, rara vez es necesario transfundir plaquetas en estos pacientes.
La cuenta de plaquetas suele aumentar en varios días a 2 a 3 semanas después de iniciado el tratamiento. Cuando la cuenta de plaquetas alcanza niveles normales la dosis de prednisona puede disminuirse en un periodo de 3 a 4 semanas. Aunque se han reportado remisiones prolongadas a largo plazo solo con la prednisona, es poco frecuente la remisión completa y duradera, que ocurre en menos del 10 por ciento de los pacientes.
Está indicada la esplenectomía si las cuentas de plaquetas permanecen por debajo de 30,000/µl después de 4 a 6 semanas de tratamiento esteroide o cuando la cuenta de plaquetas comienza a disminuir de nuevo después de disminuir el esteroide. El procedimiento produce remisión prolongada en alrededor del 65 por ciento de los pacientes con PTI. Es mejor reanudar los esteroides orales antes de la esplenectomía, de modo que el paciente tenga una cuenta plaquetaria de por lo menos 30,000 a 50,000/µl en el momento de la cirugía. En forma alternativa, si el paciente no responde al tratamiento esteroideo puede administrarse inmunoglobulina intravenosa (IGIV) en dosis de 1 g/kg/día por 2 días o 0.4 g/kg/día por 5 días algunos días antes de la cirugía (ver adelante). La IGIV producirá un incremento temporal en la cuenta de plaquetas en la mayoría de los pacientes, pero es un tratamiento muy costoso. La cuenta de plaquetas suele comenzar a aumentar el primer día posoperatorio, alcanzando niveles normales para la segunda semana. Deben administrarse vacunas contra neumococo, Haemophilus influenzae y meningococo 1 a 2 semanas antes de la cirugía.
Si el paciente es anciano y débil y por tanto es difícil que tolere la esplenectomía, la enfermedad puede controlarse administrando la cantidad mínima necesaria de esteroides para aumentar la cuenta plaquetaria a 30,000 a 50,000/µl, un nivel con el que rara vez ocurren sangrados. Debido a que los pacientes con PTI que se clasifican como fracasos terapéuticos en general tienen buena evolución clínica, el papel de agentes potencialmente peligrosos, como ciclofosfamida y azatioprina, para el tratamiento de estos casos, requiere de individualización.
La hemorragia mucosa severa o del SNC es una verdadera urgencia médica y requiere de hospitalización. Se transfunden eritrocitos según se requiera y se administra prednisona de inmediato, comenzando con una dosis de 100 mg y continuando con 25 mg cada 6 horas. Debe administrarse IGIV en dosis de 0.4 g/kg/5 días o 1g/kg/2 días y se transfundirán 8 a 10 unidades de concentrados plaquetarios al terminar la primera infusión de IGIV, que suele administrarse en aproximadamente 60 minutos. La transfusión de plaquetas después de la infusión de IGIV produce un incremento mayor y más duradero en la cuenta plaquetaria. Sus efectos adversos incluyen dolor generalizado, cefalea, rubor, fiebre y calosfríos. Cuando ocurre hemorragia uterina severa puede administrarse una sola dosis de 25 mg de estrógenos conjugados por vía intravenosa para controlar la hemorragia. Debe enfatizarse que el beneficio de la IGIV suele ser temporal, durando solo algunos días, por lo que deberán realizarse planes para la esplenectomía.
El mecanismo de acción de la IGIV no se conoce del todo. Puede producir bloqueo reticuloendotelial al ocupar los sitios para la fracción Fc de la IgG en los monocitos y macrófagos. También puede contener anticuerpos anti-idiotipo muy específicos que bloquean la unión de los anticuerpos antiplaqueta al antígeno plaquetario GPIIb-IIIa.12 La IGIV causa la liberación de IL-6, IL-8, factor de necrosis tumoral (FNT), receptor del FNT y antagonista del receptor de IL-1. Estudios más recientes indican que la velocidad catabólica de la IgG está mediada por un nuevo receptor para el componente Fc de la IgG, denominado FcRn (receptor Fc neonatal, llamado así porque se identificó inicialmente en el epitelio intestinal de neonatos), sobre el endotelio de las células vasculares. En condiciones normales la IgG, pero no la IgM, que entra en la célula a través de un proceso de pinocitosis, es protegida de su ruptura catabólica al unirse al FcRn. Después de la administración de IGIV en dosis altas este receptor se satura, permitiendo la degradación del anticuerpo patogénico, que ocurre en proporción con su concentración en el plasma.13
Se considera que el paciente que después de la esplenectomía y el tratamiento esteroideo permanece con trombocitopenia severa o presenta remisión pero después recae y no responde a dosis altas de prednisona, tiene PTI refractaria. Debido a que las hemorragias serias no son frecuentes cuando la cuenta de plaquetas es mayor de 30,000/µl, suele ser prudente aceptar una respuesta incompleta y no administrar formas más tóxicas de tratamiento. Por lo general los inmunosupresores son la base del tratamiento en esta etapa. Sin embargo, debe enfatizarse que no existen estudios aleatorizados a largo plazo que analicen este difícil problema y que por lo general los pacientes deben ser enviados a un hematólogo.
Existen diversas alternativas de tratamiento. Una de ellas consiste en el tratamiento inmunosupresor a largo plazo con azatioprina (100 a 150 mg/día por vía oral) más prednisona (40 a 60 mg/día vía oral) con vigilancia semanal de la citología hemática con cuenta de plaquetas. Pueden pasar hasta 4 meses para que ocurra una respuesta, durante este periodo la prednisona puede disminuirse en forma gradual y la azatioprina ajustarse para evitar la leucopenia. Una equivocación frecuente consiste en suspender la prueba terapéutica en forma prematura. Otra alternativa es emplear ciclofosfamida, por lo general 100 a 150 mg/día por vía oral. Tanto la azatioprina como la ciclofosfamida son mielosupresoras y ambas se han asociado con el desarrollo subsecuente de síndrome mielodisplásico y leucemia mieloide aguda. Si se requiere administración de prednisona por tiempo prolongado es mejor emplearla en días alternos para disminuir los efectos adversos.
Otra alternativa es el tratamiento con dosis intermitentes de IGIV en las dosis ya mencionadas. Los costos y las respuestas, que generalmente son breves, hacen que esta sea una opción poco atractiva. Se han administrado anticuerpos anti-D a pacientes Rh+ (D+) con PTI por la teoría de que los eritrocitos cubiertos con anticuerpos pueden bloquear los receptores Fc en los macrófagos y evitar la eliminación acelerada de las plaquetas. Esto ha producido un incremento moderado y breve en la vida de las plaquetas, en especial en pacientes no esplenectomizados, con toxicidad mínima y una reducción de solo 1 a 2 g/dl en la hemoglobina (causada por hemólisis inmune).14
Otras opciones terapéuticas incluyen vincristina, vinblastina,15 danazol16 y dexametasona en dosis altas. Puede administrarse dexametasona (40 mg/día por vía oral por 4 días consecutivos), repitiendo los ciclos cada 28 días por seis ciclos.17 También se han empleado combinaciones de ciclofosfamida, vincristina y prednisolona en esquemas semejantes a los usados para tratar los linfomas.18 Existen también reportes anecdóticos que apoyan el uso de ciclosporina, interferón alfa y plasmaféresis en pacientes con PTI refractaria.
En el paciente esplenectomizado y refractario es importante investigar la presencia de cuerpos de Howell-Jolly y la posibilidad de un bazo accesorio. La desaparición de los cuerpos de Howell-Joly sugiere la presencia de un bazo accesorio o regenerado.
Los pacientes con sangrado por trombocitopenia clínicamente significativo pueden beneficiarse con el inhibidor de la fibrinolisis ácido e-aminocaproico (EACA). Un esquema comienza con una dosis de impregnación de 5 g, administrada por vía intravenosa u oral, seguido de 1 g cada 4 horas hasta que la hemorragia se detenga o disminuya. La dosis se reduce a 1 g cada 6 horas y se administra en esa dosis mientras sea necesario para controlar el episodio hemorrágico.19
Se ha identificado un péptido linear en la GPIIIa de la membrana plaquetaria como el principal determinante antigénico del anticuerpo antiplaqueta anti-GPIIIa en la PTI relacionada al VIH-1.20 En los pacientes con infección por VIH los plaquetas contienen también cantidades aumentadas de IgG, IgM, complemento y complejos inmunes. La supevivencia de las plaquetas es moderadamente corta y su producción está disminuida, en especial en la fase tardía de la enfermedad.21
El uso de agentes inmunosupresores en los pacientes infectados por VIH es riesgoso. Si la reducción en la cuenta de plaquetas es moderada no se requiere tratamiento. Cuando la trombocitopenia es severa puede administrarse un curso breve de prednisona, seguido de esplenectomía.
La hemorragia trombocitopénica aguda en la PTI asociada al VIH puede tratarse administrando IGIV en dosis altas, en forma semejante al tratamiento en otras PTI. La PTI crónica asociada al VIH puede responder a la zidovudina por vía oral (AZT) o a otros tratamientos antivirales. El anticuerpo anti-D (ver antes), la dapsona y el intererón pueden usarse también con cierto éxito.22-24 Los pacientes que rechazan la esplenectomía o que se piensa que son malos candidatos quirúrgicos pueden responder a radiación esplénica en dosis bajas.25
En el 5 por ciento de las mujeres embarazadas ocurren cuentas de plaquetas tan bajas como de 70,000/µl. Cuando la trombocitopenia se detecta por primera vez durante el embarazo el diagnóstico diferencial debe incluir preclampsia [ver tabla 1]. Al excluir otros diagnósticos se establece el de trombocitopenia gestacional o trombocitopenia incidental del embarazo y no se requiere tratamiento.26 Sin embargo, si se realiza diagnóstico de PTI las opciones son limitadas porque la esplenectomía puede causar un aborto espontáneo y los agentes inmunosupresores tienen riesgo de dañar los órganos fetales en desarrollo. En estos casos el tratamiento suele limitarse a esteroides o IGIV. Los esteroides aumentan el riesgo de preclampsia y diabetes gestacional. En los casos de hemorragia trombocitoénica severa deben usarse todas las opciones disponibles para proteger la vida y bienestar de la madre.
Debido a que los autoanticuerpos antiplaqueta en la PTI tienen una especificidad amplia y casi siempre son de tipo IgG, pueden cruzar la placenta y producir trombocitopenia en el feto. Durante el parto vaginal la presión aplicada a la cabeza del feto trombocitopénico puede inducir una hemorragia intracraneal. La preocupación a este respecto ha orillado a muchos expertos a recomendar la cesárea temprana en las mujeres con historia de PTI o enfermedad activa. Aunque la cesárea puede ayudar a minimizar la morbilidad fetal, puede producir hemorragia significativa en la madre trombocitopénica. Ningún tema dentro de la hematología ha producido mayor divergencia de opinión que el tratamiento de la PTI durante el parto por la posibilidad de dañar tanto al feto como a la madre.
Un estudio prospectivo extenso de vigilancia ha demostrado que la medición de los anticuerpos antiplaqueta en la madre no tienen utilidad clínica. Una madre con historia de PTI que tiene cuenta de plaquetas normales puede dar a luz un neonato trombocitopénico (2 de 15 nacimientos, o 13 por ciento). La incidencia global de trombocitopenia en los neonatos en mujeres con PTI es bastante baja (4 de 46 nacimientos, o 9 por ciento).26 No se observaron hemorragias intracraneales durante el estudio. En los 6 neonatos con trombocitopenia más severa (cuentas de plaquetas menores de 20,000/µl), el trastorno no fue causado por la PTI durante el embarazo, sino por el síndrome de trombocitopenia aloinmune neonatal.27 A pesar de ello, este tema sigue siendo motivo de controversia y algunos investigadores expertos recomiendan tomar muestras percutáneas de sangre del cordón umbilical en mujeres con cuentas de plaquetas menores de 70,000/µl y realizar cesárea si la cuenta de plaquetas del feto es menor de 50,000/µl.
Púrpura trombocitopénica secundaria
Los pacientes con LEG, enfermedad de Hodgkin o linfoma no Hodgkin pueden presentar un cuadro clínico idéntico al observado en la PTI. El enfoque diagnóstico y terapéutico es el mismo que el de la PTI. Deben excluirse la presencia de esplenomegalia con secuestro esplénico, la infiltración medular con células malignas y el uso reciente de antineoplásicos o inmunosupresores. Los pacientes con LEG o linfoma pueden tener síndrome de Evans, la asociación de PTI con anemia hemolítica autoinmune. El tratamiento del síndrome de Evans es el mismo que el de la PTI y anemia hemolítica autoinmune.
Púrpura postransfusión La púrpura postransfusión (PPT) se caracteriza por inicio agudo de trombocitopenia severa, con frecuencia menor de 10,000/µl aunado con hemorragia clínica. Puede ocurrir 2 a 10 días después de una transfusión de sangre total, paquete de eritrocitos o concentrado plaquetario. Casi todas las pacientes afectadas son mujeres multíparas. En el diagnóstico diferencial deben considerarse padecimientos como trombocitopenia séptica, CID y trombocitopenia inducida por heparina (ver adelante). La trombocitopenia suele durar alrededor de 4 semanas. Debido a que por lo general las transfusiones de plaquetas son inútiles y en ocasiones precipitan respuestas sistémicas severas, siempre que puedan deben evitarse.
La fisiopatología de la PPT no se conoce del todo. En la mayoría de los casos el paciente ha estado expuesto a aloantígenos plaquetarios durante el embarazo o como resultado de una transfusión. La mayoría de los pacientes con este trastorno tienen anticuerpos contra el antígeno plaquetario PLA-1 (también denominado HPA-1), un antígeno presente en la GPIIIa de la superficie plaquetaria. Alrededor del 98 por ciento de la población caucásica y el 99 por ciento de la población afroamericana y asiática son homocigotos para el PLA-1. Los pacientes en los que se desarrolla PPT suelen ser PLA-1 negativos y PLA-2 (HPA-1b) positivos. El paciente ha sido sensibilizado al antígeno PLA-1, por lo general durante un embarazo, y la reexposición a plaquetas PLA-1 durante la transfusión de eritrocitos causa una respuesta anamnésica y la destrucción de las plaquetas extrañas. Parece una pradoja el que los aloanticuerpos dirigidos contra un antígeno presente en plaquetas extrañas causen destrucción de las plaquetas autólogas del paciente, que no expresan el antígeno PLA-1. Existen evidencias que sugieren que el antígeno PLA-1 se vuelve soluble y se fija a las plaquetas PLA-1 negativas. En forma alternativa, la exposición a plaquetas extrañas puede inducir la formación de un verdadero anticuerpo contra las plaquetas endógenas. El polimorfismo PLA-1/PLA-2 causa el 80 a 90 por ciento de los casos de PPT. La confirmación del diagnóstico requiere de estudios serológicos que demuestren la presencia de anticuerpos anti-PLA-1 y un genotipo PLA-2 homocigoto. Se han desarrollado varias técnicas rápidas para determinar el genotipo de las plaquetas con base en la reacción en cadena de la polimerasa.
No existen estudios controlados que evalúen el tratamiento para la PPT por el número limitado de casos. Reportes recientes indican que la IGIV, usada en dosis similares a las empleadas en el tratamiento de la PTI, es eficaz en alrededor del 80 por ciento de los casos. Otra opción es la plasmaféresis, que parece ser tan útil como la IGIV pero más complicada de realizar. Las dosis altas de esteroides también son eficaces, aunque el resultado de este tratamiento no es tan constante como con la IGIV.28
Destrucción plaquetaria inducida por fármacos
La destrucción inmune de las plaquetas por fármacos es indistinguible de la PTI. La médula ósea muestra megacariocitos abundantes y en laboratorios especiales puede detectarse la presencia de anticuerpos antifármacos.
Púrpura por quinidina y quinina Los anticuerpos patogénicos en casos de la púrpura por quinidina y quinina se desarrollan tan pronto como 12 días después de la exposición al agente agresor. En la mayoría de los casos se han identificado en el suero de los pacientes anticuerpos dependientes del medicamento contra la GPIb-IX de la superficie de la plaqueta.29 Los anticuerpos son dependientes del fármaco porque se unen a las plaquetas sólo en presencia de quinina o quinidina. Al parecer, la unión de estos fármacos a las glucoproteínas de superficie de la plaqueta inducen nuevos sitios antigénicos en las proteínas que son reconocidos por los anticuerpos.
Es obvio que en estos casos debe suspenderse el agente causal (quinidina o quinina). Ni el tratamiento con esteroides ni la esplenectomía de urgencia han demostrado ser útiles en la púrpura inducida por estos agentes. La plasmaféresis para eliminar el medicamento y los anticuerpos parece una medida lógica, pero no existen estudios sistemáticos sobre su eficacia. Las plaquetas transfundidas se eliminan tan rápidamente como las propias del paciente. Sin embargo, puede intentarse la transfusión plaquetaria para controlar las hemorragias graves. No se cuenta con estudios controlados al respecto del manejo. Se recomienda emplear prednisona e IGIV en dosis similares a las empleadas en la PTI.
Se ha detectado una trombocitopenia inducida por quinina que va seguida pronto por un SUH. En el suero de los pacientes se han detectado anticuerpos contra plaquetas dependientes de quinina, así como contra células endoteliales.30 Incluso la pequeña cantidad de quinina en el agua de quina puede ser suficiente para desencadenar recurrencias del síndrome. Otros medicamentos que en ocasiones pueden producir trombocitopenia dependiente del fármaco incluyen al dipiridamol y al trimetoprim-sulfametoxazol.31
Trombocitopenia inducida por heparina La trombocitopenia inducida por heparina (TIH) es una causa frecuente de trombocitopenia inducida por fármacos en los pacientes hosptalizados. A pesar de la presencia de trombocitopenia leve o moderada, ésta rara vez se asocia con hemorragia, sino con trombosis que puede incluso ser mortal.
Trombocitopenia inducida por oro El tratamiento con sales de oro para la artritis reumatoide produce trombocitopenia en el 1 a 3 por ciento de los pacientes. No existen evidencias de una reacción de anticuerpos fármaco-antifármaco como la que ocurre con la trombocitopenia por quinidina y quinina. El proceso se caracteriza por aumento en los megacariocitos de la médula ósea, acortamiento en la vida de las plaquetas y, con poca frecuencia, anticuerpos antiplaqueta. La mayoría de los pacientes responden al tratamiento con 60 mg de prednisona al día. No se ha establecido la utilidad del dimercaprol como agente quelante. Los pacientes que no responden al tratamiento esteroideo parecen beneficiarse con la esplenectomía.
Trombocitopenia asociada con cocaína Se ha reportado un síndrome semejante a la PTI en personas que emplean cocaína intravenosa. Estos individuos mejoran con un tratamiento parecido al de la PTI.32
Trombocitopenia causada por antagonistas del receptor de la glucoproteína plaquetaria IIb-IIIa Se han autorizado tres antagonistas parenterales de la GPIIb-IIIa, abciximab (ReoPro), eptifibatide (Integrilin) y tirofiban (Aggrastat), para su uso en el tratamiento del síndrome coronario agudo y como manejo adyuvante en la angioplastía coronaria. Diversos antagonistas activos por vía oral de la GPIIb-IIIa también son motivo actual de estudios clínicos. Los meta-análisis de los estudios clínicos con antagonistas de la GPIIb-IIIa sugieren que la administración parenteral de estos compuestos aumenta la posibilidad de trombocitopenia (cuenta plaquetaria menor de 100,000/µl) en alrededor del 50 por ciento (riesgo global, 1.48), comparado con el placebo, con una incidencia de alrededor de 1 a 2 casos por 100 pacientes tratados.33 La inclusión de heparina no tiene un efecto aditivo aparente. El desarrollo de la trombocitopenia puede ser agudo (i.e., en 24 horas después de la exposición al fármaco) o tardía (i.e., hasta 14 días después del inicio del tratamiento crónico). Se ha observado trombocitopenia aguda y grave, con cuentas de plaquetas menores de 20,000/µl, en el 0.3 a 0.7 por ciento de los pacientes que reciben abciximab.34
El mecanismo de la trombocitopenia parece ser autoinmune. Es probable que en estos pacientes existan autoanticuerpos previos anti-GPIIb-IIIa, y que después de la administración del antagonista anti-GPIIb-IIIa la unión del medicamento a esta molécula induzca un cambio conformacional en la GPIIb-IIIa de modo que se expongan nuevos epítopes que son reconocidos por los autoanticuerpos. Esto explicaría el inicio agudo de trombocitopenia grave. La incidencia de anticuerpos anti-GPIIb-IIIa que se unen a las plaquetas autólogas se ha calculado en alrededor del 1 por ciento en poblaciones seleccionadas de pacientes.35 No existe correlación entre el anticuerpo humano antiquimérico y el desarrollo de trombocitopenia en estos casos.
Cuando se desarrolla trombocitopenia con cuentas de plaquetas menores a 100,000/µl deben suspenderse de inmediato el antagonista de la GPIIb-IIIa y cualquier otro medicamento potencialmente agresor (v.gr., heparina). Dependiendo de la cuenta de plaquetas, puede no ser necesario suspender los agentes antiplaquetarios como la aspirina, la ticlopidina o e clopidogrel (Plavix), porque los pacientes con este trastorno tienen alto riesgo de una trombosis aguda coronaria o de la derivación. Si la cuenta de plaquetas es menor de 11,000/µl, debe pensarse seriamente en infundir plaquetas. En general, es suficiente con una sola transfusión plaquetaria. Existen reportes anecdóticos de trombosis coronaria aguda asociada con transfusión de plaquetas en pacientes que tenían cifras mayores a 50,000/µl y en los que se habían retirado los agentes antiplaquetarios. Por lo tanto, en ocasiones es necesario reiniciar los antiplaquetarios. En la mayoría de los casos la cuenta de plaquetas se normaliza en 4 días, aunque puede tardar hasta 2 semanas en el caso del abciximab. Se recomienda que se revise la cuenta de plaquetas en todos los pacientes 2 a 4 horas después del inicio de un antagonista intravenoso de la GPIIb-IIIa y a las 24 horas después de la ingestión de un inhibidor oral.
ELIMINACIÓN ACELERADA DE PLAQUETAS POR MECANISMOS NO INMUNOLOGICOS
Existen varias causas de trombocitopenia no inmunológica. En varias de estas condiciones ocurre lesión de la pared de los vasos sanguíneos con aumento en la generación de trombina, lo que produce mayor activación plaquetaria y su consumo.
Púrpura trombocitopénica trombotica y síndrome urémico-hemolítico del adulto
Características clínicas y diagnóstico La PTT incluye a un grupo de síndromes clínicos en los que un evento inicial daña el endotelio de los pequeños vasos, causando depósito diseminado de trombos de plaquetas y fibrina en las arterias pequeñas y arteriolas. Las cinco principales manifestaciones de la PTT son (1) anemia hemolítica microangiopática severa asociada con deshidrogenasa láctica (DHL) muy alta y un frotis que muestra los esquistocitos y células en casco característicos, (2) trombocitopenia moderada a severa con aumento en los megacariocitos de la médula ósea, lo que indica activación y consumo intravascular de las plaquetas, (3) fiebre, que en ocasiones es alta, (4) signos y síntomas del SNC que pueden ser leves al inicio, con inquietud, cefalea y desorientación, pero que en ocasiones progresan en forma explosiva a hemiparesia, afasia, crisis convulsivas, deficiencias focales, coma y muerte, y (5) afección renal, que suele ser leve y produce elevaciones moderadas de la creatinina sérica y las proteínas en la orina. Debe enfatizarse que muchos pacientes no tienen todas estas manifestaciones.
La forma adulta del SUH es semejante. Las características comunes de la PTT y el SUH incluyen anemia hemolítica microangiopática, trombocitopenia y la presencia de trombos de fibrina y plaquetas en los vasos pequeños. La afección renal siempre es severa en el SUH, mientras que la afección del SNC es menos importante que en la PTT. Existe una forma característica de SUH y que ocurre principalmente en niños pequeños, quienes sufren diarrea sanguinolenta y colitis hemorrágica, insuficiencia renal aguda y trombocitopenia causada por Escherichia coli, por lo general del serotipo 0157:H7, que libera una verotoxina que daña las células endoteliales.36
La PTT/SUH ocurre en forma espontánea y se asocia también con embarazo, cáncer, trasplante de médula ósea, enfermedades autoinmunes y varios fármacos. Durante el embarazo recuerda a la preclampsia severa. En el periodo posparto las manifestaciones del SNC pueden confundirse al inicio con depresión posparto, con resultados trágicos. Se han reportado casos después de un alumbramiento normal, o asociados a desprendimiento de placenta y preclampsia.
Varios fármacos parecen causar PTT/SUH. Entre estos se incluyen medicamentos quimioterápicos (v.gr., mitomicina C, bleomicina y cisplatino), agentes inmunosupresores (v.gr., ciclosporina y FK506), el agente antiplaquetario ticlopidina, anticonceptivos orales y quinina. Para la ticlopidina la incidencia es muy baja (0.02 por ciento) y la mayoría de los casos ocurren después de 2 a 4 semanas bajo tratamiento. Sin embargo, el trastorno es difícil de predecir y puede ocurrir después de menos de 2 semanas de iniciado el tratamiento con ticlopidina. En un estudio, la mortalidad global por PTT/SUH inducido por ticlopidina fue de 21 por ciento, y todas las muertes ocurrieron en pacientes que no fueron tratados con plasmaféresis.37 Estos datos destacan la importancia de un alto grado de sospecha y de la institución inmediata de la plasmaféresis.
Tanto la PTT como el SUH deben distinguirse del LEG y el síndrome de Evans. La hemólisis microangiopática, la leucocitosis neutrofílica y una prueba directa de Coombs negativa (prueba de antiglobulina directa) sugieren en forma intensa PTT o SUH. Las pruebas de coagulación no suelen revelar alteraciones significativas (i.e., no hay evidencia de CIH) y la DHL suele estar alta. Por lo general no se requiere biopsia de médula ósea, que puede mostrar trombos hialinos de plaquetas y fibrina característicos, pero no patognomónicos, en las pequeñas arterias y las arteriolas.
Fisiopatología Se han propuesto dos mecanismos responsables de la PTT/SUH. Una hipótesis postula que la presencia de un factor activador de plaquetas circulante estimula a las plaquetas, causando su agregación intravascular. La segunda hipótesis sugiere que la lesión inicial es el daño extenso al endotelio arteriolar. La activación plaquetaria, su adherencia, oclusión y la formación de tiras de fibrina causan la trombocitopenia y la hemólisis microangiopática. Existen datos que apoyan ambas hipótesis.
Muchos investigadores han observado que existen multímeros anormalmente grandes de factor de von Willebrand (vWF) en los pacientes con PTT crónica recurrente.38,39 Estos múltímeros anormales tienen mayor afinidad por las plaquetas y pueden contribuir al desarrollo de trombosis microvascular. Se acumulan por un defecto en el procesamiento normal de los multímeros de vWF, predisponiendo al paciente a sufrir trombosis. En apoyo a esto se ha descrito una enzima que contiene metales (metaloproteasa) y que normalmente rompe los multímeros grandes de vWF.40,41 Además, dos grupos reportaron en forma reciente el aislamiento de anticuerpos en el plasma de pacientes con PTT crónica recurrente que inhiben la actividad de esta enzima.42,43 Estos anticuerpos inhibitorios se encontraron específicamente en el plasma de casos de PTT y no en pacientes con trombocitopenia por otras causas. Existían durante el episodio agudo y estaban ausentes cuando los pacientes permanecían en remisión. Además, un grupo encontró lo que parece ser una deficiencia constitutiva de la enzima de escinde el vWF en pacientes con PTT familiar. La plasmaféresis podría eliminar los multímeros y los anticuerpos inhibitorios. Los beneficios ocasionales descritos después de la infusión de plasma normal pueden deberse a la administración de la enzima proteolítica.
Otros autores sostienen que estos multímeros de vWF anormales no son suficientes para explicar la patogenia de la PTT. Pueden detectarse grandes multímeros en el plasma de pacientes con PTT recurrente cuando están en remisión clínica.43 En caso muy bien estudiado de PTT crónica recurrente, la normalización de los multímeros de vWF no aumentó la cuenta plaquetaria ni indujo remisión clínica.44 El daño extenso o la activación de las células endoteliales, quizá en el caso de infecciones, embarazo o exposición a ciertos fármacos, causa que se liberen los multímeros más grandes que, como resultado de un procesamiento defectuoso, pueden causar agregación plaquetaria extensa con trombosis microvascular.
Tratamiento El inicio pronto de intercambio plasmático con plasma fresco congelado es el manejo de elección para la PTT/SUH. En un estudio extenso y aleatorizado realizado por el Grupo Canadiense de Aféresis, el intercambio extenso de plasma fue más eficaz que las infusiones e plasma en términos de supervivencia del paciente (78 contra 63 por ciento).45 En ese estudio, se removió 1.5 veces el volumen calculado de plasma, sustituyéndolo con plasma fresco congelado durante cada uno de los 3 primeros días de tratamiento y después un volumen de plasma por día durante un mínimo de 7 días. Algunos investigadores obtuvieron buenos resultados con un intercambio diario de un volumen en lugar de 1.5 veces el volumen.46 Es lógico iniciar con un intercambio de un volumen al día si el paciente está relativamente estable, con trombocitopenia moderada y sin deterioro neurológico significativo. Sin embargo, si la situación clínica empeora está indicado un intercambio de plasma más intenso y con un volumen doble (5,000 a 6,000 ml/día o alrededor de 80 ml/kg/día). Debido a que existen multímeros de vWF en el crioprecipitado, puede sustituirse el criosupernadante (i.e., plasma fresco congelado del que se ha retirado el crioprecipitado) como líquido de remplazo cuando un paciente no responde al intercambio plasmático de rutina. Un estudio no controlado, mostró mayor beneficio con esta preparación al comparar con el plasma fresco congelado.47 Una vez que se ha logrado beneficio terapéutico (que se mide por restablecimiento de la función del SNC, aumento en la cuenta de plaquetas y reducción de la DHL), la intensidad y frecuencia del recambio plasmático puede disminuirse, alejando los intercambios de plasma de un volumen, primero a 3 por semana y después a 2 semanales.
Aunque se conoce la importancia del intercambio inmediato de plasma, no se ha probado en estudios clínicos prospectivos el uso de esteroides,48 aspirina y dipiridamol. Debido a que la aféresis tiende a disminuir la cuenta de plaquetas en los pacientes ya de sí trombocitopénicos, persiste el problema de si deben transfundirse plaquetas. Algunos investigadores han observado que la infusión de plaquetas puede exacerbar la PTT,49 mientras que otros usan transfusiones según se requiera.
La microangiopatía puede persistir por semanas o meses después de que han desaparecido las demás evidencias de la enfermedad. En un estudio extenso de seguimiento de pacientes con PTT, alrededor de la tercera parte de los que entraron en remisión recayeron durante un periodo de vigilancia de 10 años y uno de cada 10 presentó otros problemas médicos serios. Por lo tanto, se requiere un seguimiento cuidadoso.50
Algunos clínicos experimentados recomiendan la esplenectomía,51 y el autor ha tenido cierto éxito con esta medida. Sin embargo, también han ocurrido fracasos que se complicaron por los efectos de la esplenectomía en una situación clínica ya de sí difícil. Por lo tanto, el autor no la realiza de rutina en pacientes con PTT. Las mismas modalidades empleadas para la PTT se usan en el SUH, junto con hemodiálisis para la insuficiencia renal y tratamiento médico para la hipertensión.
Trombocitopenia inducida por infección
Varias infecciones serias virales, bacterianas, micóticas y parasitarias pueden producir CID y en consecuencia trombocitopenia. Sin embargo, también existen otros mecanismos de trombocitopenia inducida por infección independientes de la CID.
Infecciones virales Las infecciones virales como el dengue y la rubéola congénita pueden dañar en forma directa a los megacariocitos. La varicela puede causar una forma de trombocitopenia que tiene características de una reacción inmune: número aumentado de megacariocitos, no evidencia de CID y presencia de AP de IgG o IgM. Por lo general no se requiere tratamiento. La trombocitopenia aguda en la mononucleosis infecciosa parece tener un mecanismo inmune, como lo demuestra el aumento en los megacariocitos de la médula, en los AP de IgG y la respuesta favorable a esteroides. En ocasiones la trombocitopenia es severa, pero como se mencionó, responde a esteroides.
Septicemia bacteriana Los pacientes que tienen septicemia severa por gram negativos y cuentas de plaquetas menores de 50,000/µl suelen tener evidencia de CID. Sin embargo, muchos pacientes que tienen septicemia tanto por gram negativos como positivos y cuentas de plaquetas entre 50,000 y 150,000/µl no tienen signos de CID. Es posible que un mecanismo inmunológico participe en estos casos porque suelen existir AP-IgG elevados y el grado de elevación correlaciona con la severidad de la trombocitopenia. El aumento en los AP-IgG puede representar complejos inmunes depositados en la superficie plaquetaria en lugar de autoanticuerpos antiplaqueta. La clave para controlar la trombocitopenia consiste en establecer el tratamiento adecuado para la infección. Si existe CID debe manejarse con control cuidadoso de la hipotensión y del volumen sanguíneo (ver adelante).
Si existe trombocitopenia clínicamente significativa no relacionada con CID deben administrarse concentrados plaquetarios para evitar hemorragias.
Infección por protozoarios La trombocitopenia es común en el paludismo, aunque la CID es rara. La supervivencia de las plaquetas es corta y se han encontrado AP-IgG elevados. El anticuerpo IgG parece unirse a los antígenos del paludismo adsorbidos en la superficie de la plaqueta.52
Trombocitopenia durante el embarazo y el periodo periparto
En el 5 al 8 por ciento de las mujeres embarazadas ocurre trombocitopenia leve con cuentas de plaquetas tan bajas como 70,000/µl (trombocitopenia gestacional). Este trastorno no tiene significado clínico. Si ocurre trombocitopenia aguda durante el embarazo, debe distinguirse de la PTI (ver antes), la PTT asociada al embarazo y la preclampsia.
Además de hipertensión, proteinuria y evidencia de cambios patológicos en el riñón, hígado, SNC y placenta, alrededor del 15 por ciento de los pacientes con preclampsia tienen trombocitopenia moderada. En solo una minoría de los pacientes con preclampsia y trombocitopenia existen evidencias por laboratorio de CID. El número de megacariocitos está aumentado y la supervivencia de las plaquetas un poco disminuida. Algunos pacientes con preclampsia y trombocitopenia tienen también hemólisis microangiopática, lo que sugiere que los vasos dañados que contienen bandas de fibrina destruyen tanto a los eritrocitos como a las plaquetas. El vasoespasmo intenso, que causa daño endotelial con activación, adherencia y destrucción plaquetarias, puede tener también algún papel. El cuadro clínico puede ser indistinguible de la PTT, y en estos casos debe manejarse como tal. En caso contrario, el tratamiento consiste en el manejo prenatal de la preclampsia y en un esfuerzo por detectar la trombocitopenia lo más pronto posible.
Síndrome de HELLP El síndrome de HELLP se refiere a un trastorno que ocurre durante el embarazo y se caracteriza por hemólisis, aumento en las enzimas hepáticas y reducción en la cuenta de plaquetas. No es claro si este trastorno tan grave es una forma de preclampsia, CID o ninguna de las dos. En algún momento entre la 23 y 39 semanas de embarazo las pacientes afectadas presentan trombocitopenia marcada, con una cuenta de plaquetas menor de 100,000/µl, hemólisis microangiopática, pruebas anormales del hígado y en ocasiones hipertensión.53 Los resultados de las pruebas estándar de coagulación para CID son normales, aunque puede haber cierta elevación en el nivel de productos de degradación de la fibrina y disminución en la concentración de antitrombina-III (AT-III). Las pacientes con el síndrome de HELLP suelen estar muy graves, con falla circulatoria, respiratoria y renal, hemorragia posparto, hemorragia intrahepática y crisis convulsivas. El padecimiento se trata suspendiendo el embarazo, por lo general por parto, y proporcionando manejo cuidadoso de sostén. En una serie grande de pacientes con HELLP el nadir de la trombocitopenia ocurrió 1 a 2 días después del parto.54 La trombocitopenia persistente con microangiopatía o la presencia de falla orgánica sugiere PTT/SUH posparto y debe considerarse el intercambio del plasma.
Trombocitopenia por hipotermia
En ocasiones puede ocurrir trombocitopenia durante la hipotermia inducida durante la cirugía cardiaca. La hipotermia en los ancianos también parece causar trombocitopenia, y se han reportado cifras de plaquetas hasta de 30,000/µl.55 Los mecanismos propuestos como responsables de la trombocitopenia incluyen CID y secuestro hepático y esplénico. Después de que la temperatura del paciente se ha normalizado las plaquetas aumentan en forma espontánea en un periodo de 1 a 2 semanas.
Lavado de plaquetas y alteraciones del lecho vascular
El lavado perioperatorio de plaquetas era antes una causa frecuente de trombocitopenia no inmune. Los pacientes que tienen sangrado importante durante una cirugía y después se transfunden con más de 10 unidades de sangre total almacenada desarrollan trombocitopenia. En efecto, en este caso los pacientes son sometidos a transfusión de intercambio con sangre que contiene plaquetas no viables. La cuenta de plaquetas es baja, el tiempo de protrombina (TP), el tiempo parcial de tromboplastina (TPT) y el tiempo de trombina (TT) son normales. Por lo tanto, debe vigilarse la cuenta de plaquetas en los pacientes que reciben transfusiones masivas (v.gr., 10 unidades de eritrocitos o sangre total). Si la cuenta disminuye a menos de 100,000/µl y el paciente va a someterse a cirugía u otro reto hemostático deben transfundirse concentrados plaquetarios.
Las plaquetas también pueden ser eliminadas por un lecho vascular anormal. En los hemangiomas gigantes existe enlentecimiento del lecho sanguíneo por canales mal endotelizados. Estas superficies pueden producir CID de grado bajo.
El tercer mecanismo importante de trombocitopenia es el secuestro. Son comunes las cuentas de plaquetas entre 40,000 y 80,000/µl en los pacientes con esplenomegalia importante. Rara vez ocurre hemorragia clínicamente significativa a menos que exista un padecimiento hemorrágico concomitante. El tratamiento se dirige hacia la enfermedad primaria. Rara vez se requiere esplenectomía.
Trastornos funcionales de las plaquetas
La clave para detectar la existencia de un defecto funcional de plaquetas consiste en encontrar hemorragia clínica en presencia de un tiempo de sangrado prolongado y una cuenta plaquetaria mayor de 100,000/µl. Son raras las petequias. La morfología de las plaquetas y las pruebas funcionales pueden ser anormales [ver tabla 2].
|
|||||||||||||||
|
Trastornos de la membrana de las plaquetas
El síndrome de Bernard-Soulier es un padecimiento autosómico recesivo poco frecuente que se caracteriza por plaquetas gigantes, un tiempo de sangrado prolongado, trombocitopenia moderada y riesgo de hemorragia fatal. El defecto, que consiste en ausencia del complejo GPIb-IX-V (el principal sitio de unión de la plaqueta del vWF) causa menor adhesión plaquetaria a las superficies lesionadas. La aglutinación plaquetaria inducida por ristocetina es anormal y no se corrige por la adición de plasma normal que contiene vWF. La hemorragia aguda se trata con transfusiones de plaquetas.
La trombastenia de Glanzmann es un padecimiento autosómico recesivo raro en el que la morfología y la cuenta de las plaquetas son normales pero el tiempo de sangrado está prolongado. Debido a que el complejo crucial GPIIb-IIIa que forma el sitio de unión de la plaqueta al fibrinógeno está ausente, las plaquetas no sufren agregación inducida por difosfato de adenosina (ADP) o colágena. Sin embargo, la aglutinación inducida por ristocetina es normal. Cuando se requiere, el tratamiento consiste en transfusiones plaquetarias.
Trastornos de los gránulos plaquetarios
Síndromes de plaquetas grises y deficiencia de gránulos densos Los pacientes con el síndrome de plaquetas grises, un trastorno raro, tienen hemorragias mucosas, equimosis y petequias. Existe trombocitopenia moderada y el tiempo de sangrado está prolongado. Las plaquetas son mayores de lo normal y se observan agranulares por la ausencia de gránulos a. Debido a que el contenido de los gránulos a está muy reducido, la adhesión plaquetaria y coagulación apoyada por plaquetas están deficientes. La agregación plaquetaria con colágena es anormal. Los episodios de sangrado deben tratarse con infusión de plaquetas normales.
Otro trastorno poco común, el síndrome por deficiencia de gránulos densos, se caracteriza por hemorragia mucosa asociada con cuenta normal de plaquetas, que tienen morfología normal y prolongación del tiempo de sangrado. La agregación plaquetaria con ADP y colágena es anormal. La reducción en el contenido de ADP de los gránulos densos altera los eventos mediados por ADP. La hemorragia se trata con transfusión de plaquetas.
La administración de 1-deamino-8-D-arginina vasopresina (DDAVP o desmopresina) es una alternativa para los pacientes con trastornos plaquetarios primarios que requieren cirugía.
Padecimientos mieloproliferativos
Ocurren alteraciones en la función plaquetaria en los padecimientos mieloproliferativos como la leucemia mieloide crónica, la policitemia vera, la trombocitemia esencial y la leucemia aguda. La cuenta de plaquetas en los trastornos mieloproliferativos crónicos suele ser muy alta, pero el tiempo de sangrado puede estar prolongado y aparecer hemorragia clínica en forma de hemorragia de mucosas y hematomas. Las alteraciones recuerdan un defecto adquirido de almacenamiento. Los megacariocitos suelen ser anormales, con núcleos separados; las plaquetas de sangre periférica son grandes y pueden no tener gránulos. El tratamiento de la hemorragia aguda consiste en transfusión de plaquetas normales para aumentar la cuenta a más de 50,000/µl. Deben evitarse la aspirina y otros AINE.
Se ha demostrado en la uremia la ocurrencia de tiempo de sangrado prolongado con hemorragia clínica a pesar de cuentas de plaquetas normales. La identidad de la sustancia inhibitoria en el plasma urémico que es responsable de la trombocitopenia no se ha identificado aún. La DDAVP (en dosis de 0.3 µg/kg en 50 ml de solución salina durante un periodo de 30 minutos) es eficaz para controlar la hemorragia urémica en alrededor de 4 a 6 horas. La infusión de DDAVP produce aumento en la actividad plasmática de vWF y en especial en los multímeros grandes de vWF, que pueden aumentar la adhesión plaquetaria.
El hematocrito debe mantenerse por arriba del 30 por ciento en los pacientes urémicos que sangran porque el tiempo de sangrado se prolonga cuando éste disminuye por debajo de 26 por ciento.56 La hemorragia también puede controlarse con el uso de estrógenos conjugados. Los estrógenos conjugados (Premarin) administrados por vía oral (50 µg/día) o intravenosa (0.6 µg/kg/día) por 4 a 5 días acorta el tiempo de sangrado en alrededor del 50 por ciento por alrededor de 2 semanas.57 La ventaja de los estrógenos conjugados sobre la DDAVP es la mayor duración de su efecto benéfico sobre la función plaquetaria, aunque el inicio de su acción es más lento. Ambos medicamentos pueden usarse en forma concomitante.
Además del hiperesplenismo y la síntesis defectuosa de procoagulantes, existe evidencia de que ocurre CID de bajo grado en forma continua en la enfermedad hepática severa. La menor eliminación de productos de degradación de la fibrina puede contribuir también a los niveles plasmáticos altos de productos de degradación de fibrina-fibrinógeno. Estos productos interfieren con la función plaquetaria y la polimerización de la fibrina, y su nivel correlaciona con la hemorragia clínica en la cirrosis hepática severa. El tratamiento para esta condición debe dirigirse a la enfermedad primaria.
Macroglobulinemia y otras disproteinemias
La presencia de concentraciones altas de proteínas viscosas produce efectos complejos sobre el mecanismo hemostático. Las proteínas parecen cubrir a las plaquetas e interfieren con la adhesión y quizá con la agregación. El tratamiento se dirige a la enfermedad primaria, pero si la hiperviscosidad y la hemorragia son significativas puede requerirse plasmaféresis pronta para disminuir el nivel de proteínas anormales y corregir el trastorno hemorrágico.
Trastornos inducidos por fármacos
Aspirina y otros antinflamatorios no esteroides En las personas normales la ingestión de 0.6 g de aspirina prolonga el tiempo de sangrado templado en 2 a 3 minutos. Las plaquetas se afectan en forma irreversible. El tromboxano A2 (TXA2) es un inductor potente de la liberación y agregación plaquetaria. La aspirina acetila e inhibe en forma irreversible a la cicloxigenasa-1 (COX-1) y bloquea la generación subsecuente de tromboxano. Algunas personas aparentemente normales tienen especial sensibilidad a la acción de la aspirina, de modo que sus tiempos de sangrado se prolongan mucho más y pueden tener hemorragia clínicamente significativa, en especial durante o después de una cirugía o traumatismo. Estos pacientes pueden tener una forma leve de enfermedad de von Willebrand o trastorno del almacenamiento y su diátesis hemorrágica leve se exacerba por el efecto antiplaquetario de la aspirina.
Los pacientes urémicos son especialmente sensibles al sangrado inducido por la aspirina. Una dosis pequeña de este medicamento no prolonga el tiempo de sangrado en las personas normales, pero sí, en ocasiones hasta 15 minutos, en los pacientes urémicos. La combinación de alcohol y aspirina también es peligrosa por su capacidad para prolongar el tiempo de sangrado.
La hemorragia inducida por aspirina se diagnostica primero determinando la existencia de un defecto adquirido en la función plaquetaria (cuenta de plaquetas mayor de 100,000/µl, prueba de agregación plaquetaria anormal y ausencia de historia de sangrado) y encontrando evidencia de ingestión de aspirina. Debido a que cerca de 300 compuestos en el mercado contienen aspirina, la historia negativa debe complementarse midiendo la concentración sérica de salicilatos o detectando una alteración en el patrón de agregación con colágena que se normaliza en 7 días (el patrón típico por ingestión de aspirina).
Si la hemorragia es significativa puede manejarse por transfusión de plaquetas. Debido a que la inhibición de la COX-1 es irreversible, la afección hemostática puede durar 4 a 5 días después de que se suspende la aspirina. Si el paciente requiere analgesia puede emplearse acetaminofén o codeína porque ninguno de éstos afecta la función plaquetaria. Si el paciente requiere antinflamatorios no esteroides para tratamiento, por ejemplo, de una artritis reumatoide, pueden usarse inhibidores de la cicloxigenasa-2 (COX-2), que no afectan la función plaquetaria.
Alcohol Además de producir trombocitopenia por suprimir la producción de plaquetas, el consumo de alcohol puede causar también defectos en la función plaquetaria.58 Los estudios in vitro han demostrado que el alcohol altera la agregación plaquetaria y la liberación de TXA2. La función plaquetaria se normaliza después de 2 a 3 semanas de abstinencia.
Dextranes El dextrán de peso molecular de 40,000 se excreta con facilidad, pero el de 70,000 puede persistir en la circulación por 3 días e interferir con la superficie plaquetaria. El tratamiento es de sostén hasta que se excreta el dextrán. Las plaquetas transfundidas son afectadas por el dextrán en el plasma.
Antibióticos La carbenicilina y la ticarcilina pueden inhibir la agregación plaquetaria y contribuir a un trastorno hemorrágico, lo mismo que las dosis masivas de penicilina. Estas últimas alteran la agregación plaquetaria inducida por colágena y ristocetina. El moxalactam, una cefalosporina de tercera generación, causa también un trastorno en la función plaquetaria. La situación clínica es más importante cuando se desarrolla un defecto adquirido en la función plaquetaria en un paciente pancitopénico tratado por septicemia. El cambio de antibióticos suele corregir el problema.
Agentes diversos Muchos otros medicamentos pueden modificar la función plaquetaria [ver tabla 3].59
|
|||||
*Se usa como agente terapéutico antitrombótico
|
La cuenta de plaquetas mayor de 500,000/µl se conoce como trombocitosis reactiva. En la trombocitosis reactiva las pruebas de función plaquetaria (incluyendo los estudios de agregación) suelen ser normales, los pacientes no sufren mayor incidencia de hemorragia o tromboembolia incluso si la cuenta de plaquetas excede 1 millón/µl.
Las cuentas elevadas de plaquetas (con frecuencia hasta 1 a 3 millones/µl o más) ocurren también en la leucemia mieloide crónica, la metaplasia mieloide agnogénica con mielofibrosis, la policitemia vera y la trombocitemia esencial. En el diagnóstico de trombocitemia esencial la cuenta de plaquetas es mayor de 600,000/µl y se han excluido otras causas de trombocitosis, como otros padecimientos mieloproliferativos o trombosis reactiva.
En los trastornos mieloproliferativos las pruebas de función plaquetaria suelen ser anormales [ver adelante, Trastornos de la función plaquetaria]. Algunos pacientes con trastornos mieloproliferativos parecen tener mayor propensión a sufrir hemorragia y tromboembolismo. Ni el número de plaquetas ni la medición de su función predicen el grado de trombosis o hemorragia.
Desde el punto de vista clínico, los signos de hemorragia incluyen sangrado de mucosas (en especial gastrointestinal), hematomas y equimosis. Puede existir trombosis de la vena esplénica, trombosis portal o mesentérica y trombosis recurrente de venas profundas con o sin embolia pulmonar. La trombosis arterial es menos común.
Los pacientes con trombocitemia esencial y policitemia vera pueden tener eritromelalgia debilitante (ardor y comezón en los dedos de manos y pies) que pueden progresar a acrocianosis isquémica.60 Este complejo sintomático parece ser causado por oclusión e inflamación de arteriolas por los agregados plaquetarios. La aspirina o la indometacina producen alivio en cuestión de horas. La aspirina, en dosis de 0.3 a 0.6 g cada tercer día, puede producir un beneficio duradero.
La hemorragia y la trombosis son eventos poco comunes incluso cuando la cuenta de plaquetas es de 1 millón/µl. En un paciente con trombocitemia esencial que tiene hemorragia clínicamente significativa o trombosis puede lograrse un buen control de la cuenta plaquetaria por medio de hidroxiurea oral (15 mg/kg/día) con ajustes en la dosis según se requiera para disminuir la cuenta plaquetaria. El tratamiento con hidroxiurea requiere de vigilancia cuidadosa de la citología hemática, y este tratamiento no parece hasta ahora aumentar el riesgo de un segundo trastorno maligno. Los nuevos tratamientos para la trombocitemia incluyen el uso de anagrelide, un agente poderoso que disminuye el número de plaquetas.61
Las púrpuras vasculares son un grupo heterogéneo de trastornos [ver tabla 4] que se caracterizan por hemorragia cutánea, en ocasiones asociada con hemorragia mucosa. La fuga ocurre de las arteriolas terminales, los capilares y las vénulas poscapilares. Los resultados de las pruebas en el número y función plaquetaria, así como las pruebas de función procoagulante son normales.
|
||||||||||
|
TELANGIECTASIA HEMORRAGICA HEREDITARIA
La telangiectasia hemorrágica hereditaria (THH) se trasmite como un rasgo autosómico dominante y se manifiesta principalmente por epistaxis o hemorragia gastrointestinal.62 Análisis de asociación recientes han identificado por lo menos tres loci de la THH, incluyendo tres genes para la endoglina y una cinasa semejante al receptor de activina. Ambas proteínas se expresan en las células endoteliales vasculares y pueden funcionar como receptores para el factor de transformación del crecimiento-b (FTC-b). El FTC-b tiene un papel complejo para coordinar la respuesta entre las células endoteliales y la matriz extracelular, y las mutaciones en los genes para la endoglina y la cinasa semejante al receptor de activina causan el desarrollo de vasos sanguíneos y canales arterioovenosos anormales.63 El examen físico revela telangiectasias en las yemas de los dedos, mucosa oral, la lengua y los labios. Las pruebas de coagulación suelen ser normales. Las malformaciones arteriovenosas pulmonares que ocurren en algunos pacientes pueden producir disnea, hemoptisis, tensión arterial de oxígeno (PaO2) baja y eritremia secundaria. El diagnóstico puede confirmarse por angiografía pulmonar. Si los cortocircuitos son grandes y clínicamente significativos, pueden tratarse por emboloterapia con balón.64 Puede ocurrir embolia paradójica con eventos cerebrovasculares en los pacientes con THH que tienen derivaciones y malformaciones arteriovenosas pulmonares. El tratamiento de la epistaxis recurrente incluye los diversos métodos de taponamiento nasal. La hemorragia gastrointestinal se maneja usando preparados con hierro siempre que sea posible.
La vitamina C es necesaria para el metabolismo normal de la colágena, el folato y quizá el hierro. El paciente con escorbuto sufre principalmente de alteración en la síntesis de colágena. La falta de un apoyo de colágena apropiado para la microvasculatura causa hemorragias perifoliculares, gingivorragia e incluso hematomas en tejidos profundos. Al parecer, defectos semejantes en la colágena causan el cabello denominado de espantapájaros y la hiperqueratosis asociados a este trastorno.65 El cuadro clínico característico en un paciente desnutrido sugiere el diagnóstico. Los niveles de ácido ascórbico en plasma son bajos y suelen existir también otras deficiencias vitamínicas. El tratamiento eficaz consiste en 1 g de ácido ascórbico al día en dosis divididas.
El exceso de esteroides, por causa endógena o exógena, produce hemorragias cutáneas, quizá por catabolismo de las proteínas en los tejidos de apoyo vascular inducido por los esteroides.
La amiloidosis puede causar equímosis subcutáneas que tienen predilección por el cuello y la porción superior del tórax. La biopsia muestra amiloide, que por infiltración puede debilitar las paredes vasculares o interferir con la activación de superficie de las plaquetas, procoagulantes o ambos. En los pacientes con amiloidosis sistémica primaria, en especial cuando se acompaña de esplenomegalia masiva, el amiloide puede en casos muy raros adsorber suficiente factor X como para producir deficiencia severa de este factor y hemorragia clínica.
Las lesiones purpúricas en pacientes con vasculitis leucocitoclástica pueden ser elevadas (púrpura palpable). En la biopsia estas lesiones pueden mostrar degranulación de células cebadas y, cuando se tiñen en forma apropiada, depósito de complejos inmunes. Al parecer los complejos inmunes proporcionan el estímulo quimiotáctico que causa el acúmulo de neutrófilos. El daño a la microvasculatura es causado por el complejo de ataque del complemento y por liberación del contenido de los gránulos de los neutrófilos. Este componente inflamatorio produce la púrpura palpable.
Los pacientes con púrpura senil tienen hemorragias cutáneas sobre el dorso de las manos, muñecas, la porción superior de brazos y en ocasiones las pantorrillas. No ocurre hemorragia seria ni se requiere de tratamiento. Al parecer este proceso representa un deterioro dependiente de la edad del tejido de apoyo vascular.
DAÑO A LA MICROVASCULATURA POR EMBOLOS
La CID y la PTT pueden causar vaso-oclusiones localizadas que causan daño microvascular y fuga de eritrocitos. Puede ocasionarse un daño semejante por las embolias que se originan de válvulas cardiacas infectadas. La embolia grasa puede complicar las fracturas de los huesos largos y la pelvis. El síndrome consiste en fiebre, confusión y petequias, púrpura o ambos, sobre el cuello, tórax, cara y axilas. Las embolias de colesterol pueden causar también petequias, por lo general sobre las extremidades inferiores. Ocurren típicamente en un paciente con ateroesclerosis severa que es sometido a un procedimiento invasivo en la aorta abdominal o arterias renales. La biopisa de la púrpura muestra los cristales de colesterol cuando se usa la tinción apropiada.
Trastornos hereditarios de la coagulación
Los trastornos de la coagulación aparecen clínicamente como hemorragias espontáneas o excesivas después de un traumatismo o cirugía. La historia suele indicar si el padecimiento es congénito o adquirido. Los trastornos hereditarios se caracterizan por aparición en etapas tempranas de la vida y por la presencia de una sola alteración que puede explicar todo el cuadro clínico.
La enfermedad de Von Willebrand, el trastorno hemorrágico hereditario más común, es causado por un defecto o deficiencia en el vWF en el plasma. El gen que codifica al vWF se encuentra en el cromosoma 12. El vFW tiene dominios específicos para unirse con el factor de coagulación VIII, heparina, colágena, GPIb y GPIIb-IIIa plaquetarias. Estos dominios se relacionan en forma directa con las siguientes funciones del vWF: (1) su acción como molécula transportadora para el factor VIII:C, por la que protege el factor de la coagulación de la proteolisis y prolonga en forma sustancial su vida media en plasma, (2) el aumento de la adhesión plaquetaria primaria cuando la fuerza de cizalla es alta al unir a las plaquetas a través del receptor GPIb-IX-V a los tejidos subendoteliales en el sitio de la lesión, y (3) su apoyo a la agregación plaquetaria al unir plaquetas a través de sus receptores GPIIb-IIIa.66 El vWF circula como multímeros que varían en tamaño de 0.5 millones de daltons (el dímero) a 20 millones de daltons. Incluso existen multímeros no circulantes más grandes en las células endoteliales, en donde se almacenan en los cuerpos de Weibel-Palade. El vWF se libera a la circulación o al lado de la luz, en donde se fija a la colágena subendotelial. Los gránulos a de las plaquetas también contienen vWF, que se libera cuando las plaquetas se activan. Los multímeros de vWF que son de 12 millones de daltons o mayores probablemente son los más eficaces para apoyar la adhesión plaquetaria.
Existen muchas formas variantes de la enfermedad de von Willebrand que diferen en sus manifestaciones clínicas, alteraciones de laboratorio y tratamiento. Por lo tanto, se requieren pruebas específicas para identificar el tipo de enfermedad y su severidad. Las pruebas comienzan con un TPT activado (TPTa) y una cuenta de plaquetas. Debido a que el vWF es una proteína transportadora para el factor VIII, el TPTa se prolonga cuando la concentración de vWF es baja. La cuenta de plaquetas suele ser, aunque no siempre, normal. El tiempo de sangrado por lo general está prolongado, pero no es suficientemente confiable como para emplearse para el diagnóstico. El diagnóstico depende, por tanto, de los niveles de factor VIII y vWF. Existen dos desventajas respecto a las pruebas para la enfermedad de von Willebrand: (1) los resultados de las pruebas de laboratorio en este padecimiento son muy variables y (2) el grupo sanguíneo del paciente afecta la concentración del vWF, esto es, los pacientes con grupo sanguíneo tipo O tienen niveles más bajos que los pacientes con sangre tipo A, B o AB, hasta en un 30 por ciento.67
El nivel de vWF se mide por métodos inmunológicos. El resultado suele reportarse como un porcentaje del antígeno vWF normal (actor VIIIR:Ag). Debido a que el vWF circula en formas multiméricas importantes desde el punto de vista fisiológico, en ocasiones es útil para determinar la composición multimérica del vWF en el plasma del paciente. Las capacidades funcionales del vWF se evalúan por medio de la prueba de agregación plaquetaria inducida por ristocetina. La ristocetina se agrega al plasma del paciente rico en paquetas, en donde causa que el vWF se una a las plaquetas a través del receptor GPIb-IX-V, causando activación y agregación de las plaquetas. En algunos laboratorios se usan plaquetas fijas en formalina y se mide la aglutinación de estas plaquetas fijas después de medir la adición de ristocetina. El diagnóstico genético preciso para algunos subtipos de enfermedad de von Willebrand está disponible solo en algunos laboratorios de investigación.
En el esquema de clasificación actual para las variantes de la enfermedad de von Willebrand existen tres grupos principales: el tipo 1 es una deficiencia cuantitativa parcial del vWF, el tipo 2 una alteración cualitativa del factor y el tipo 3 una deficiencia severa y prácticamente total desde el punto de vista cuantitativo [ver tabla 5].68
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
N- normal ¯-disminuido -aumentado vWF- factor de von Willebrand |
La enfermedad de von Willebran tipo 1 es la forma más común
(causa el 75 por ciento de los casos). Generalmente es de tipo
autosómico dominante y suele aparecer en su forma heterocigota. Los
pacientes con enfermedad de von Willebrand tipo 1 clásica tienen
historia de hemorragias leves a moderadas durante la vida, típicamente
en las superficies mucosas. Pueden no conocer del trastorno
hemorragíparo hasta que se someten a cirugía o sufren un
traumatismo, casos en los que sangran en forma severa. Los niveles del
antígeno vWF, el factor VIII y el cofactor ristocetina están
todos disminuidos. La rara forma homocigota o doble heterocigota (enfermedad de
von Willebrand tipo 3) se caracteriza por hemorragias severas,
prolontación del TPT y una concentración de factor VIII menor del
5 por ciento.
La enfermedad de von Willebrand tipo 2 se caracteriza por alteraciones cualitativas del vWF y una reducción variable en el antígeno vWF, el factor VIII y el cofactor ristocetina. En el tipo 2a los multímeros de mayor tamaño están ausentes. En el tipo 2B los multímeros se unen en forma excesiva a las plaquetas por una mutación que incrementa la función. En el tipo 2M el vWF anormal no se une al GPIb-X-V, y en el tipo 2N el sitio de unión del vWF para el factor VIII ha sufrido una mutación.
Se ha descrito una forma plaquetaria para la enfermedad de von Willebrand, que se denomina enfermedad de seudo-von Willebrand. En ella existe una GPIb en las plaquetas, causando unión excesiva del vWF normal en plasma a plaquetas no estimuladas.
La enfermedad de von Willebrand suele tratarse con infusión de crioprecipitados que contengan vWF, factor VIII y fibrinógeno. Cada bolsa de crioprecipitado contiene alrededor de 100 unidades de factor VIII y 40 a 70 por ciento del vWF originalmente presente en el plasma. En general, la administración de 10 bolsas de crioprecipitado cada 12 a 24 horas proporciona hemostasia adecuada. Debe evitarse la aspirina. El humate-P, un concentrado de factor VIII pasteurizado y de pureza intermedia, tiene una cantidad importante de multímeros grandes de vWF y se ha usado para tratar a los pacientes con enfermedad de von Willebrand con éxito. Típicamente se infunden dosis de 20 a 40 unidades de factor VIII por kilogramo cada 12 a 24 horas. Para controlar la hemorragia grave que no responde a tratamiento con crioprecipitado o concentrado de factor VIII puede intentarse una transfusión de plaquetas normales con la idea de que el vWF plaquetario puede ser hemostáticamente eficaz.69
Se ha demostrado que la DDAVP es eficaz en el tratamiento del sangrado traumático y antes de la cirugía en algunos pacientes con enfermedad de von Willebrand. La administración intravenosa de DDAVP en dosis de 0.3 mg/kg en un periodo de 15 a 30 minutos causa la liberación de vWF a partir de las reservas de las células endoteliales. La respuesta pico suele ocurrir en 30 a 60 minutos y persiste por hasta 4 a 6 horas. La administración repetida de DDAVP durante un periodo de 24 horas es ineficaz porque ocurre taquifilaxia después de la depleción de la reserva de vWF endotelial. Puede usarse el DDAVP en aerosol nasal para el manejo ambulatorio de los pacientes con enfermedad de von Willebrand, tanto para el tratamiento de los episodios hemorrágicos como para preparación para cirugía menor.70 Los efectos adversos del DDAVP intravenosos suelen ser leves, incluyendo retención significativa de agua y, rara vez, trombosis. Se ha reportado infarto al miocardio.
La hemofilia A afecta a uno de cada 10,000 varones y se caracteriza por un factor de coagulación VIII deficiente o defectuoso. El gen del factor VIII, que se localiza en el cromosoma X en Xq28, es uno de los genes humanos más grandes, abarcando 186 kb y conteniendo 26 exones. Codifica una proteína de alrededor de 300,000 daltons, que circula en el plasma en concentraciones muy bajas y normalmente se une y es protegido por el vWF. Se desconoce cual es la principal fuente de producción del factor VIII, pero el hígado debe ser importante porque la hemofilia A puede corregirse con el trasplante de hígado. Debido a que el gen para la actividad coagulante del factor VIII se encuentra en el cromosoma X, la enfermedad se manifiesta en los varones hemicigotos. Todas las hijas de un varón con hemofilia serán portadoras, la mitad de los hijos de una madre que porte el gen serán hemofílicos y la mitad de las hijas serán portadoras. Las familias parecen afectarse en diversos grados, dependiendo de la naturaleza específica del defecto genético.
La severidad clínica de la hemofilia A correlaciona bien con los niveles medidos de actividad coagulante del factor VIII. En general, los niveles de factor VIII por debajo del 1 por ciento se asocian con síntomas hemorrágicos severos, los niveles entre 1 y 5 por ciento con hemofilia moderada y los niveles entre 5 y 25 por ciento con hemofilia leve [ver tabla 6].
|
|||||||||||
|
Alrededor de la tercera parte de los pacientes con hemofilia A tienen nuevas
mutaciones e historia familiar negativa. En la actualidad se han encontrado
más de 300 genes del factor VIII anormales. Las alteraciones, que
incluyen mutaciones puntiformes, inserciones y deleciones de genes, causan
producción deficiente del factor VIII o la generación de un
factor funcionalmente ineficaz. Es interesante que en el 45 por ciento de todos
los pacientes con hemofilia A severa (factor VIII menor del 1 por ciento) se
encuentra inversión dentro del intrón 22 del gen del factor VIII,
que ocasiona una proteína trunca e inestable.71
El diagnóstico se hace con el cuadro clínico, la historia familiar (positiva en dos terceras partes de los casos) y el nivel de actividad coagulante del factor VIII. En la mayoría de los casos el tipo de hemorragias y la historia familiar clásica descartan enfermedad de von Willebrand (que, a diferencia de la hemofilia A, se trasmite en forma autosómica). En la actualidad se dispone de análisis de ADN para detectar la inversión del intrón 22. Esta prueba proporciona el diagnóstico molecular en el 45 por ciento de los pacientes con hemofilia severa. Sin embargo, no debe solicitarse en pacientes con hemofilia leve a moderada.
Principios de tratamiento general para la hemofilia
Los aspectos psicosociales de la hemofilia son complejos. Los niños se ausentan con frecuencia de la escuela, tienen riesgo de deformidades y de adicción a fármacos por el dolor severo. Los padres se preocupan mucho y en ocasiones se sienten culpables. El tratamiento debe enfocar estos aspectos, lo mismo que los problemas específicos de la coagulación.
Los concentrados de factor VIII son eficaces para controlar las hemorragias espontáneas y traumáticas en la hemofilia severa. El uso de crioprecipitados, evaluados o tratados para prevenir los virus de la hepatitis y el VIH, siguen siendo la base del tratamiento. La posibilidad de contaminación ha aumentado los esfuerzos para desarrollar preparaciones libres de virus. En la actualidad existen varias de éstas disponibles y el problema consiste en equilibrar el costo contra el probable beneficio. Las preparaciones de factor VIII muy purificadas (v.gr., Monoclate-P y Hemofil M) se elaboran por medio de anticuerpos monoclonales y cromatografía de alta afinidad. Se ha clonado ya el gen del factor VIII y en el mercado existen desde hace varios años dos formas de factor VIII recombinante completo (Recombinate y Kogenate). Los estudios formales, así como la amplia experiencia clínica indica que estos preparados son seguros y eficaces.72,73 Se ha desarrollado también un factor VIII recombinante de segunda generación con deleción del dominio B, que se ha empleado en estudios clínicos desde 1993. El nuevo factor VIII recombinante tiene la ventaja de tener actividad específica considerablemente mayor y la fórmula final es estable sin la adición de albúmina sérica humana, lo que disminuye más el riesgo potencial de trasmisión de agentes infecciosos humanos.74
La profilaxis dental es crucial para disminuir la necesidad de cirugía dental. Debe evitarse la aspirina y considerarse también la revacunación contra el virus de la hepatitis B.
El consejo genético debe ser parte del programa de tratamiento. Debido a la difícil vida de los pacientes con hemofilia severa, una mujer puede optar por terminar con el embarazo si está cierta de su estado de portadora o de que el feto está afectado. Existen varias estrategias para detectar a los portadores. En las mujeres portadoras los niveles de factor VIII son típicamente de la mitad de lo normal, mientras que los de vWF son normales. La relación entre factor VIII y vWF para los portadores es de 0.5;75 sin embargo, la tasa de error para esta prueba va de 10 a 17 por ciento. Puede hacerse un diagnóstico genético más exacto para los portadores por medio de un enfoque de asociación. Este se basa en fragmentos de restricción de longitud polimórfica (FRLP) dentro del gen del factor VIII. El análisis del varón afectado establecerá el patrón para el cromosoma X que porta el alelo de la hemofilia, aún sin conocer la mutación precisa. Existe un gran número de polimorfismos intragénicos que permiten distinguir las dos copias de los genes del factor VIII en una mujer potencialmente portadora, identificando su estado de portadora con gran exactitud.
Estas sondas moleculares para los FRLP se emplean en la actualidad para determinar el estado del feto. El tejido puede obtenerse por amniocentesis o muestra de las vellosidades coriónicas.
Tratamiento de la hemorragia aguda
La hemorragia en los tejidos profundos, la hemartrosis y la hematuria son las formas más comunes de hemorragia clínica en la hemofilia A. La hemorragia retroperitoneal, la hemorragia en la boca, lengua o cuello que obstruyen la vía aérea y la hemorragia intracraneal pueden poner en peligro la vida. Tanto el ultrasonido como la tomografía computada pueden usarse para identificar los hematomas retroperitoneales e intramusculares.
Principios del tratamiento sustitutivo
Un nivel de 100 por ciento en el procoagulante del plasma indica que existe una unidad de procoagulante por cada mililitro de plasma. La mayoría de las personas tienen 40 ml de plasma por kilogramo de peso corporal. Por lo tanto, al determinar el volumen plasmático de un paciente y su nivel procoagulante puede calcularse la cantidad requerida de factor VIII a sustituir. Por ejemplo, en el caso de un niño de 60 kg que tiene hemartrosis no complicada de la rodilla y un factor VIII basal de menos del 1 por ciento, será suficiente aumentar el nivel de factor VIII a 25 por ciento (0.25 U/ml) por 2 a 3 días. Este paciente tiene un volumen plasmático de 60 kg por 40 ml/kg, o 2,400 ml, y necesitará 0.25 U/ml x 2,400 ml, o 600 unidades de factor VIII como bolo inicial. Otro método para calcular la dosis se basa en el siguiente efecto: la infusión de una unidad de factor VIII por kilogramo aumenta los niveles de factor VIII en 2 por ciento. Por lo tanto, al dividir la concentración de factor VIII deseado entre 2 se obtendrá el número de unidades/kg requeridas. En el ejemplo citado, para lograr un 25 por ciento de factor VIII se requieren 12.5 unidades/kg, o 750 unidades.
La cantidad deseada de factor VIII puede administrarse como concentrado purificado o como factor recombinante por medio de 6 a 8 bolsas de crioprecipitado (cada bolsa contiene alrededor de 100 unidades). El número de unidades está indicado en la etiqueta. La vida media biológica del factor VIII es de alrededor de 12 horas, la dosis puede repetirse cada 12 a 24 horas mientras se requiera para controlar la hemorragia. En los pacientes con hemartrosis el nivel de factor VIII debe mantenerse por 2 a 3 días.
Cirugía electiva y extracción dental
La atención dental debe realizarla un dentista con experiencia en el tratamiento de hemofílicos. Antes de una extracción dental se administra factor VIII para aumentar el nivel a alrededor del 50 por ciento. El inhibidor de la fibrinolisis AEAC se inicia la noche antes de la cirugía en dosis de impregnación de 3 g por vía oral y se continúa en dosis de 2 a 3 g tres a cuatro veces al día por 7 a 10 días después de terminado el trabajo dental. Por lo general no se requiere mayor administración de factor VIII.
Antes de la cirugía electiva el nivel de factor VIII debe aumentarse a 50 a 100 por ciento (0.5 a 1.0 U/ml) y después mantenerse por arriba del 50 por ciento durante los siguientes 10 a 14 días. El mantener una concentración mayor de factor VIII no reduce la frecuencia de hemorragias.76
Puede usarse DDAVP para tratar las hemorragias traumáticas agudas en los pacientes con hemofilia leve a moderada e incluso para preparar a estos pacientes para cirugía menor. El DDAVP, que causa la liberación de vWF de las reservas de las células endoteliales, no puede usarse en forma repetida durante muchos días porque las reservas se depletan. El DDAVP se infunde en dosis de 0.3 µg/kg en 50 ml de solución salina durante 15 a 30 minutos y produce un incremento rápido en el factor VIII. La vida media biológica del factor VIII liberado es de 11 a 12 horas.
Los inhibidores tienden a ocurrir en pacientes con afección severa, que tienden a recibir el mayor número de concentrados de factor VIII. En un estudio reciente de un solo centro de 431 pacientes durante 3 décadas, alrededor del 10 por ciento de los pacientes con hemofilia A severa tuvieron un inhibidor (alrededor de la tercera parte eran niños menores de 10 años).77 No todos los inhibidores producen problemas clínicos. Deben realizarse estudios en busca de inhibidores del factor VIII a intervalos regulares en todos los pacientes que tienen hemofilia severa.
Hemorragia en pacientes con un inhibidor La hemorragia en un paciente con un inhibidor puede ser grave. En un enfermo que tiene un título de inhibidor menor de 5 unidades Bethesda y que no es un respondedor de anticuerpos vigoroso, deben administrarse cantidades grandes de concentrado de factor VIII en un intento por superar al anticuerpo. Debe medirse el nivel posinfusión de factor VIII para determinar si se logra el punto final esperado, 0.3 U/ml de plasma.78
Para los pacientes con hemorragias más serias o niveles más altos de inhibidor contra el factor VIII humano, el factor VIII porcino (Hyate:C) es el tratamiento de elección. La medición del título del inhibidor contra el factor VIII porcino proporciona información útil.
Un estudio controlado ha establecido la eficacia de los concentrados de complejo de protrombina (v.gr., Konyne y Proplex) para superar la deficiencia de factor VIII en el tratamiento de la hemartrosis.79 Se han usado también concentrados activados del complejo de protrombina, como Autoplex y FEIBA, con cierto éxito. Recientemente se ha desarrollado y encontrado que el factor VII activado recombinante (rFVIIa) fue seguro y eficaz en el 70 a 85 por ciento de más de 1,500 episodios de sangrado en pacientes hemofílicos con inhibidores,80 aunque aún no está autorizado por la Food and Drug Administration. Se han empleado dosis altas de IgG intravenosa para tratar a no hemofílicos con inhibidores adquiridos del factor VIII, pero ésta medida no suele ser útil en pacientes hemofílicos.
OTROS TRASTORNOS HEMORRAGICOS HEREDITARIOS
Deficiencia de factor IX (hemofilia B)
La deficiencia de factor IX (hemofilia B o enfermedad de Christmas) es un trastorno ligado al X que clínicamente es indistinguible de la hemofilia A. El gen del factor IX se encuentra en el cromosoma X y produce una proteína de 56 kd que, como otros factores vitamina K dependientes, tiene una región rica en ácidos glutámicos g-carboxilados. Al parecer los iones calcio establecen puentes entre esta región y la superficie activada de la plaqueta, en donde el factor IXa interactúa con el VIIIa para formar un complejo asociado a la membrana que convierte en forma eficaz el factor X a factor Xa (tenaza intrínseca). Se han detectado en el gen del factor IX un gran número de inserciones rearreglos y deleciones, y el síndrome de la hemofilia B es muy heterogéneo.81
El diagnóstico requiere de una prueba para factor IX. Los principios de tratamiento son los mismos que para la hemofilia A (ver antes). El factor IX es sustituido con plasma fresco congelado o con concentrados del complejo de protrombina. Se ha esterilizado ya uno de los nuevos concentrados del factor IX (Mononine), que tiene excelente actividad específica y una vida media biológica muy conveniente de 18 a 34 horas. También se cuenta con factor IX recombinante comercial.
El nivel de factor IX necesario para controlar la hemostasia en pacientes con hemofilia B es un poco menor que el requerido para el tratamiento de la hemofilia A, alrededor de 0.15 a 0.20 U/ml para el primero y 0.30 a 0.50 U /ml para el segundo. El factor IX es una molécula más pequeña que el factor VIII y se distribuye en el espacio de la albúmina. Al hacer los cálculos para sustituir, se supone que una unidad de factor IX por kilogramo aumentará el nivel en plasma en 1 por ciento o 0.01 U/ml. El factor IX tiene una vida media bifásica y los niveles en plasma de este factor pueden mantenerse al infundir el concentrado cada 24 horas durante un episodio de hemorragia aguda en el paciente con hemofilia B. En la actualidad las técnicas de clonación de genes pueden detectar el estado de portador de deficiencia del factor IX y permitir un consejo genético exacto.
La terapia génica ha permitido la corrección sostenida del trastorno hemorragíparo en ratones con hemofilia B,82 y en la actualidad se inician ya estudios clínicos al respecto.
En ocasiones las pruebas de escrutinio preoperatorio revelan que un paciente tiene un TP con prolongación leve en ausencia de hepatopatía, desnutrición o administración de antibióticos. Algunos de estos pacientes pueden ser heterocigotos para un estado de deficiencia de factor VII, como lo confirman los estudios familiares y la medición del nivel de antígeno de este factor en el plasma. No se requiere tratamiento a menos que se piense realizar una cirugía mayor, caso en el que el factor VII puede suplementarse en la forma de plasma fresco congelado. La hemorragia clínica en estos pacientes es muy variable, variando de inexistente a severa. Con menos frecuencia, se ha reportado deficiencia de factor VII asociada con trombosis.83
Dos trastornos hemorrágicos congénitos poco comunes se han atribuido a alteraciones en la fibrinolisis. La deficiencia de a2-antiplasmina, el principal inhibidor de la plasmina, causa actividad no controlada de la plasmina con hemorragias secundarias. El incremento en la actividad fibrinolítica con hemorragia clínica ocasional se ha asociado también con deficiencia de activador del plasminógeno-1 (PAI-1), del inhibidor fisiológico del activador de plasminógeno tisular (t-PA) y de urocinasa.84 El tratamiento de ambos tipos de alteraciones fibrinolóticas consiste en los agentes antifibrinolíticos ácido tranexámico y AEAC, que bloquean la unión del plasminógeno y la plasmina con la fibrina.
Trastornos hemorrágicos adquiridos
Además de los trastornos hereditarios de la coagulación, se han identificado varios trastornos adquiridos que pueden causar hemorragia generalizada.
Una carboxilasa dependiente de vitamina K en el hígado sintetiza ácido g-carboxigutámico, que se requiere para la función biológica de la protrombina y de los factores VII, IX y X. En ausencia de vitamina K se sintetiza una protrombina anormal que no tiene los residuos g-carboxiglutámicos. Los inmunoensayos específicos realizados en pacientes con deficiencia de vitamina K revelan una reducción profunda en la protrombina normal y un incremento concomitante en la des-g-carboxiprotrombina anormal. La misma alteración molecular ocurre con los factores VII, IX y X.85
La deficiencia de vitamina K, que disminuye la protrombina y los factores VII, IX y X, ocurre en casos de desnutrición severa, malabsorción intestinal e ictericia obstructiva. En la ictericia obstructiva las sales biliares, que son necesarias para la emulsificación y absorción de vitaminas liposolubles (A, D, E y K), no pueden llegar al intestino. La ingestión crónica de antibióticos orales suprime la producción de vitamina K por los organismos intestinales. El efecto es especialmente importante en los pacientes que, por su enfermedad, son incapaces de consumir una dieta adecuada. Ocurren hemorragia mucosa y equímosis si los niveles de los procoagulantes disminuyen por debajo del 10 a 15 por ciento de lo normal.
El tratamiento con fitonadiona (10 a 25 mg/día por vía oral) por 2 a 3 días, o fitonadiona parenteral en el caso de ictericia obstructiva, suele revertir la alteración en alrededor de 6 a 24 horas. Si existe hemorragia severa la administración de plasma fresco congelado (alrededor de 3 unidades) restablecerá los niveles de los procoagulantes con rapidez [ver antes, Principios del tratamiento sustitutivo].
HEMORRAGIA INDUCIDA POR FARMACOS
La sobredosis de warfarina o la potenciación de su acción por otros medicamentos puede causar hemorragias muy severas. El TP se prolonga y ocurren hemorragias mucosas, gastrointestinales o equímosis. Si la hemorragia es importante debe iniciarse tratamiento para restablecer los niveles de procoagulantes al 30 por ciento de lo normal por medio de plasma fresco congelado. Si no existe urgencia puede administrarse fitonadiona por vía oral. El uso a escondidas de warfarina puede identificarse por una medición de la misma, disponible en laboratorios especializados. Debe mencionarse que algunos de los antagonistas de acción prolongada de vitamina K que se usan como raticidas (superwarfarinas) pueden causar hemorragias prolongadas después e su ingestión accidental o intencionada. La síntesis de factores de la coagulación dependientes de vitamina K puede alterarse durante meses después de la exposición inicial. Puede requerirse de la administración repetida de plasma fresco congelado, suplementado con dosis masivas de vitamina K1 por vía oral (100 a 150 mg/día) para controlar la hemorragia.
La sobredosis de heparina puede no ser obvia. Suele causar hemorragias subcutáneas y de tejidos profundos. Los TPT, TP y TT se prolongan, pero el tiempo de reptilasa (TR) es normal. La administración intravenosa de protamina en dosis de 1 mg/100 U de heparina administrada corrige el trastorno. Debido a que la vida media de la protamina es más corta que la de la heparina, puede ocurrir un rebote en la acción de esta última, necesitándose una segunda administración de protamina. Las preparaciones de heparina de bajo peso molecular (HBPM) causan tanta hemorragia como la heparina estándar no fraccionada. La capacidad de la protamina para revertir las acciones de estas preparaciones de HBPM es muy específica para cada compuesto. Por ejemplo, la protamina no revierte por completo las acciones de la enoxaparina.
En la actualidad se emplea tratamiento trombolítico para el infarto agudo del miocardio y para algunos casos de embolia pulmonar. Las complicaciones del tratamiento trombolítico son esencialmente hemorrágicas. En general, las hemorragias se limitan a un rezumamiento leve en los sitios de punción vascular, pero pueden ocurrir hematomas subdurales, infarto cerebral y hemorragia intracraneana. Los agentes trombolíticos, incluso los diseñados para ser relativamente específicos de fibrina, en ocasiones producen un estado lítico sistémico significativo, con reducción en los niveles de fibrinógeno, factor V y factor VIII. Además, la generación de productos de degradación de fibrinógeno interfiere a su vez con la formación de un coágulo firme y con la función plaquetaria. Si se sospecha que el tratamiento trombolítico es la causa de la hemorragia en un paciente debe obtenerse sangre de inmediato para valuar el TPT, TT, TR y nivel de fibrinógeno. Si el tratamiento trombolítico es la causa, el TPTa estará prolongado, la concentración de fibrinógeno será menor de 50 mg/dl y tanto el TT como el TR estarán prolongados (como resultado de los productos de degradación de la fibrina). El trastorno se trata con crioprecipitados (para aumentar la concentración de fibrinógeno a alrededor de 10 mg/dl), alrededor de dos unidades de plasma fresco congelado (que pueden aumentarse hasta 6 unidades según se requiera para sustituir el factor V y otros procoagulantes) y alrededor de 6 unidades de concentrados plaquetarios. Si estas medidas no detienen el sangrado deberá considerarse el uso de un agente antifibrinolítico específico, como el AEAC. Este se administra en un bolo de 5 g IV durante 30 a 60 minutos y después en dosis de 1 g/h por infusión IV continua.86
Las proteínas anormales asociadas con el mieloma y la macroglobulinemia pueden interferir con la función plaquetaria y causar hemorragia clínica. Estas proteínas también producen alteraciones en las pruebas de coagulación. Tanto las proteínas IgG como IgA del mieloma provocan TT prolongados al interferir con el proceso de polimerización de la fibrina. Con menos frecuencia pueden interactuar con factores específicos de la coagulación. El tratamiento se dirige a la enfermedad primaria. En general estas paraproteínas no causan hemorragia clínica significativa. Si ocurre hemorragia la plasmaféresis corrige con rapidez los defectos al reducir en forma súbita el nivel de la proteína anormal.
COAGULACION INTRAVASCULAR DISEMINADA
Muchas circunstancias diferentes pueden provocar CID [ver tabla 7]. En cada caso, la activación masiva de la cascada de la coagulación supera los mecanismos antitrombóticos naturales, provocando generación no controlada de trombina. Esto causa trombosis en los lechos arteriales y venosos, con infartos isquémicos y necrosis que intensifican el daño, liberan factor tisular y activan aún más la cascada de la coagulación. La coagulación masiva depleta procoagulantes y plaquetas, con una coagulopatía por consumo y hemorragia. El daño tisular y el depósito de fibrina libera y activa activadores del plasminógeno y provoca la generación de plasmina en cantidades que superan a su inhibidor a2-antiplasmina. La plasmina degrada al fibrinógeno, la protrombina y los factores V y VIII de la coagulación, y forma productos de degradación de fibrina-fibrinógeno. Estas sustancias interfieren con la polimerización normal de la fibrina y alteran la función plaquetaria al unirse al receptor del fibrinógeno plaquetario, la GPIIb-IIIa de superficie. Los productos de degradación de fibrina-fibrinógeno funcionan como anticoagulantes circulantes y agentes antiplaquetarios, aumentando la coagulopatía por consumo y participando en forma importante en la diátesis hemorrágica [ver figura 1].
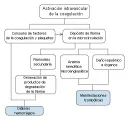
|
| Figura 1 |
| Coagulación intravascular diseminada |
|
|
|
La endotoxina liberada durante la septicemia por gram negativos aumenta la
expresión de factor tisular, acelerando la activación
procoagulante y suprimiendo la expresión de trombomodulina. Estos
efectos disminuyen el sistema de proteína S/proteína C,
promoviendo la tendencia a la CID.87 En los pacientes con
hemangiomas solitarios o múltiples asociados con trombocitopenia
(síndrome de Kasabach-Merritt) la CID parece iniciar por el contacto
prolongado de la superficie endotelial anormal con sangre en las áreas
de estasis vascular. Las plaquetas y el fibrinógeno se consumen dentro
de los hemangiomas y la fibrinolisis parece aumentar,88 este consumo
puede ocasionar hemorragia. El veneno de ciertas serpientes también
puede producir CID y se han identificado diversos mecanismos. Por ejemplo, el
veneno de víbora de Russell contiene una proteasa que activa en forma
directa al factor X y produce desfibrinación casi instantánea.
Las consecuencias de la CID dependen de su causa y de la rapidez con la que se propague el evento desencadenante. Si la activación ocurre en forma lenta se produce un exceso de procoagulantes que predispone a trombosis. Al mismo tiempo, mientras el hígado pueda compensar el consumo de factores de coagulación y la médula ósea mantenga una producción adecuada de plaquetas no habrá una diátesis hemorrágica aparente. El cuadro clínico en este momento consiste principalmente en manifestaciones trombóticas que pueden ser tanto arteriales como venosas. Las trombosis venosas con frecuencia son trombosis de venas profundas en las extremidades o tromboflebitis migratoria superficial. Los pacientes pueden desarrollar también trombosis arterial que provoca isquemia digital, infarto renal o un evento cerebrovascular. Parte de la isquemia arterial puede deberse a émbolos que se originan de coágulos de fibrina en la válvula mitral, una condición denominada endocarditis trombótica no bacteriana o endocarditis marántica. Este proceso se conoce en ocasiones como CID compensada o crónica y causa el síndrome de Trousseau89 (una CID crónica causada por una neoplasia maligna, casi siempre un cáncer de páncreas o gastrointestinal de otro tipo). Es posible que las células neoplásicas produzcan un factor tisular u otro procoagulante que active el sistema de coagulación.
Si la reacción es brusca y explosiva el cuadro clínico estará dominado por coagulación intravascular, depleción de plaquetas, fibrinógeno, protrombina y factores V y VIII, y la formación de productos de degradación de la fibrina por acción de la plasmina interferirá aún más con la hemostasia. La consecuencia clínica es una diátesis hemorrágica generalizada, con salida de sangre de los sitios de punción venosa, las líneas intravenosas y catéteres, así como en los tejidos profundos. Las tiras de fibrina intravascular producen anemia hemolítica microangiopática.
Los eritrocitos microangiopáticos en el frotis y la trombocitopenia moderada a severa sugieren el diagnóstico. En la CID ocurren varias alteraciones de laboratorio, dependiendo de la fase de la CID. Debido a la depleción de factores de la coagulación, el TPT y el TP están prolongados y el nivel de fibrinógeno es bajo. Ya que los productos de degradación de la fibrina interfieren con la polimerización de la misma, el TT y el TR también se prolongan. La concentración de productos de degradación de la fibrina, medida por el nivel de dímero D, está aumentada. El plasminógeno, la proteína C y la a2-antiplasmina en plasma también están bajas por consumo. Sin embargo, por lo general no es necesario medir estos factores. En el caso de la CID compensada, la mayoría de estos parámetros pueden ser normales, excepto por la elevación del nivel de dímero D, que indica la presencia de depósito de haces entrecruzados de fibrina intravascular y fibrinolisis. En ocasiones el nivel de fibrinógeno puede estar incluso alto porque este compuesto es un reactante de fase aguda. Cuando la CID se descompensa predomina la coagulopatía por consumo y existen las otras alteraciones de laboratorio ya mencionadas. Es muy importante la repetición a intervalos regulares de las pruebas de coagulación, en especial de la cuenta de plaquetas, el fibrinógeno y el nivel de dímero D. Estas pruebas proporcionan un parámetro cinético que ayuda en mucho en la evaluación de la severidad de la CID y en la elección del tratamiento apropiado.
El tratamiento debe dirigirse a la enfermedad principal para eliminar el evento desencadenante. Esto puede incluir el tratamiento quimioterápico de un tumor, la administración de antibióticos y el drenaje quirúrgico de un absceso, o el vaciamiento del útero cuando un embarazo complicado es la causa. Es indispensable el apoyo hemodinámico. Está contraindicado el uso de agentes antifibrinolíticos como AEAC o aprotinina. A pesar de las complicaciones hemorrágicas, la CID es un estado de hipercoagulabilidad severa y estos agentes bloquean el sistema fibrinolítico, pudiendo exacerbar las complicaciones trombóticas. La administración de productos sanguíneos, como plaquetas, plasma fresco congelado o crioprecipitados puede agregar combustible al fuego y empeorar la coagulopatía por consumo. Sin embargo, si la hemorragia clínica es significativa es prudente administrar apoyo vigoroso con productos sanguíneos.
El uso de la heparina en la CID aguda aún es motivo de controversia. Aunque la heparina, al activar a la AT-III es eficaz para inhibir la trombina y debe ser eficaz en el tratamiento de la CID, su uso suele limitarse a situaciones de CID crónica o compensada. La heparina, administrada por vía subcutánea, es eficaz en el tratamiento de las trombosis venosas en pacientes con síndrome de Trousseau. Otra situación en la que la heparina debe considerarse es al iniciar quimioterapia de inducción, cuando se espera una liberación explosiva de materiales tromboplásticos asociados con la lisis de células tumorales. Por ejemplo, el uso juicioso de heparina puede disminuir la exacerbación evidente de CID que se asocia con frecuencia con el uso de quimioterapia en el tratamiento de la leucemia promielocítica aguda. En esta situación, si el paciente sufre CID significativa incluso antes del tratamiento, la heparina en dosis bajas (en el rango de 300 U/h por infusión continua) puede disminuir la CID, lo que se demuestra por reducción del dímero D y aumento en el nivel de fibrinógeno. Sin embargo, con el advenimiento del ácido transretinoico total para el tratamiento inicial de la leucemia promielocítica aguda, por lo general no se requiere de heparina. En el caso de la CID descompensada, en la que la hemorragia es la principal manifestación clínica, la heparina puede exacerbar en forma significativa la hemorragia y no suele estar indicada. Recientemente se ha recomendado la infusión de AT-III en dosis altas en esta situación, pero su eficacia no se ha demostrado en estudios aleatorios.90,91
El tratamiento de la CID asociada con hemangiomas solitarios o múltiples representa un problema especial. Cuando los hemangiomas están localizados pueden extirparse, o en ocasiones tienen buena respuesta con la radioterapia local. Los intentos por controlar la CID por medio de heparina, esteroides, aspirina, sulfinpirazona, estrógenos y dipiridamol no han sido útiles. La clave para el tratamiento exitoso de la CID asociada con ciertos venenos de serpiente es la identificación de la serpiente y la administración pronta del antiveneno adecuado.
INHIBIDORES ADQUIRIDOS DE LA COAGULACION (ANTICOAGULANTES ADQUIRIDOS)
Además de los aloanticuerpos inhibidores circulantes que se observan en las hemofilias A y B severas, en ocasiones la hemorragia clínica es causada por inhibidores circulantes dirigidos contra factores específicos de la coagulación que pueden aparecer en forma espontánea. Debido a que el anticuerpo adquirido contra el factor VIII, que origina un cuadro clínico de hemofilia adquirida, es el más común de estos inhibidores circulantes, se le describirá con cierto detalle, pero mucho de lo descrito se aplica también a otros inhibidores.
Los autoanticuerpos contra el factor VIII suelen ser de tipo IgGa, principalmente IgG4 y no fijan complemento. Suelen dirigirse contra epítopes en el sitio activo del factor VIII. Alrededor de la mitad de los pacientes con un inhibidor adquirido del factor VIII no tienen un trastorno asociado identificable, pero muchos procesos patológicos se han identificado en el resto. Entre ellos se incluyen LEG, trastornos linfoproliferativos, neoplasias de células plasmáticas, reacciones farmacológicas (v.gr., reacción contra la penicilina), el estado posparto y trastornos de la piel.92
Los pacientes con un inhibidor adquirido del factor VIII con frecuencia presentan hemorragias mucosas de reciente inicio, hematomas y equímosis, junto con una historia negativa para sangrados en el pasado. Típicamente esto ocurre en un paciente mayor o una mujer joven durante un embarazo o en el periodo posparto. La característica de laboratorio de un inhibidor adquirido contra un factor de la coagulación es un tiempo de coagulación prolongado que no corrige al mezclar partes iguales del plasma del paciente con plasma normal. En el caso del inhibidor del factor VIII se prolonga el TPT y el TP y el TT son normales. El anticuerpo se une al factor VIII con una cinética compleja, de modo que el efecto inhibitorio se vuelve aparente solo después de una incubación prolongada. Por lo tanto, si se sospecha un inhibidor adquirido los estudios de mezcla deben realizarse después de incubar 5 y 60 minutos. El diagnóstico puede confirmarse demostrando niveles de factor VIII muy bajos con otros niveles de factores de la coagulación normales.
Las hemorragias pueden ser graves. Los intentos de sustitución no son útiles porque el inhibidor inactiva al factor VIII exógeno. En ocasiones, si el inhibidor se encuentra en títulos bajos, la sustitución masiva de factor VIII puede superar el efecto del inhibidor. Sin embargo, esto puede desencadenar una respuesta anamnésica en el nivel de anticuerpo, que complica el tratamiento posterior. En la mayoría de los casos puede administrarse terapia inmunosupresora con una combinación de ciclofosfamida (en forma de pulsos intravenosos mensuales o por vía oral en forma diaria) y prednisona.93,94 El inhibidor suele volverse no detectable después de tres o cuatro ciclos mensuales de quimioterapia. En el caso de hemorragias graves o que ponen en peligro la vida y en las que no existe tiempo suficiente para disminuir el nivel del inhibidor, puede administrarse factor VIII porcino porque el autoanticuerpo suele mostrar poca reacción cruzada.
El otro tratamiento alternativo para la hemorragia aguda es la administración de complejos procoagulantes, que pueden superar el inhibidor al proporcionar grandes cantidades de factor X y factor VII.95 Otras opciones terapéuticas incluyen plasmaféresis y dosis altas de IgG intravenosa, aunque la respuesta para la IGIV parece ser muy pobre.96 En pacientes con este trastorno ha sido útil el factor VII activado recombinante.
Los pacientes con hepatopatía severa pueden sufrir hemorragias graves. Las más frecuentes son las esofágicas y gastrointestinales relacionadas con várices, gastritis o úlcera péptica. También puede ocurrir sangrado en sitios de biopsias y durante o después de cirugías. Se presentan hemorragias mucosas y de tejidos blandos, pero en general no son el problema dominante.
La coagulopatía de la enfermedad hepática severa es compleja y no está bien definida. Debido a que el hígado es el principal sitio de síntesis de todos los factores de la coagulación, se observa disminución en los niveles de muchos de éstos, incluyendo fibrinógeno, protrombina, factor V y VII; exceptuando el factor VIII, quizá porque es un reactante de fase aguda. También se ha visto aumento en el nivel de fibrinógeno anormal con menor capacidad de coagulación en los pacientes con cirrosis.97 Además, existe menor eliminación de factores de la coagulación activados. Al parecer con frecuencia ocurre CID en los pacientes con cirrosis98 (quizá por la activación de la cascada de la coagulación por el daño al tejido hepático), pero su papel preciso tanto en la hepatitis fulminante aguda como en la hepatopatía crónica no se ha establecido. La vida de las plaquetas está acortada y aumenta su secuestro en el bazo, aunque la función suele mantenerse. También hay evidencias de hiperfibrinolisis, pero la contribución de este proceso en el defecto hemostático global es incierta. El hígado sintetiza también la mayoría de las proteínas anticoagulantes naturales, por lo que disminuyen la AT-III, la proteína C y la proteína S. Las mejores pruebas de escrutinio para este trastorno incluyen el TP, el TPT, la cuenta de plaquetas, el nivel de fibrinógeno y el nivel de dímero D. Pruebas específicas que pueden dirigir el tratamiento incluyen al factor V, factor VII y AT-III. La sustitución en la hemorragia activa se logra administrando plasma fresco congelado, crioprecipitados y plaquetas según se requiera. No se recomiendan los complejos de protrombina porque no sustituyen todos los factores deficientes y pueden exacerbar la CID. En general, aunque muchos defectos hemostáticos contribuyen a la diátesis hemorrágica en la hepatopatía severa, los factores hemodinámicos y anatómicos son los principales determinantes de esta situación.
Los casos de fibrinolisis primaria son raros. Muchos de los reportes iniciales sobre fibrinolisis primaria probablemente representaban fibrinolisis secundaria asociada con CID. La hematuria posprostatectomía puede constituir un ejemplo real de hemorragia causada por fibrinolisis localizada. La alta concentración de urocinasa en la orina en esta condición causa que el plasminógeno se convierta en plasmina, con lisis secundaria del coágulo. Si se logran descartar otras causas de hematuria posoperatoria persistente la condición puede tratarse con AEAC oral o intravenoso. También es eficaz la instilación local de AEAC por medio de un catéter uretral.
HEMORRAGIA DESPUES DE UNA DERIVACION CARDIOPULMONAR
Los pacientes sometidos a cirugía cardiaca con una derivación cardiopulmonar en ocasiones desarrollan hemorragia trans y posoperatoria en ausencia de consumo significativo de procoagulantes o sobredosis de heparina.99 Puede ocurrir trombocitopenia por heparina o consumo durante la derivación. Algunos pacientes desarrollan un trastorno adquirido en la función plaquetaria, quizá por contacto entre las plaquetas y el equipo oxigenador, pero la naturaleza exacta del defecto sigue sin conocerse.100 Además de la liberación del contenido de los gránulos a, puede ocurrir fibrinolisis junto con una depleción moderada de factores coagulantes.101 La hemorragia en estos casos suele corresponder a transfusiones de plaquetas. Se ha reportado que el uso de DDAVP reduce la hemorragia posoperatoria; sin embargo, un meta-análisis de 17 estudios clínicos mostró un efecto benéfico apenas modesto.102
También se ha probado la proteasa inhibidora aprotinina en pacientes sometidos a derivación cardiopulmonar con la suposición de que parte de la hemorragia es causada por incremento en la proteolisis de factores de coagulación y proteínas de la membrana plaquetaria desencadenada por el procedimiento y el oxigenador. La aprotonina parece ser superior al AEAC y a la DDAVP para disminuir la pérdida de sangre y los requerimientos de transfusión.103 La aprotonina debe reservarse para los pacientes que pueden requerir hemotransfusiones, en especial los sometidos a segundas cirugías y los que tienen defectos hemostáticos previos. Las pruebas preoperatorias de hemostasia no parecen ser útiles.
Durante la cirugía de derivación los pacientes se exponen en ocasiones a trombina tópica (goma de fibrina), que se usa para hemostasia local. En general en estas preparaciones se emplean trombina bovina y otros factores en cantidades mínimas a las que los pacientes pueden crear anticuerpos. Los anticuerpos contra la trombina bovina son inocuos per se, pero pueden ocasionar prolongación del TT, con complicaciones potenciales serias, cuando tienen reacción cruzada con la trombina humana. Algunos pacientes desarrollan anticuerpos contra el factor V bovino que tienen reacción cruzada con el factor humano y que pueden causar hemorragia clínica.104,105 Los estudios de mezcla con plasma del paciente y plasma normal revelarán la presencia de inhibidores, y la medición del nivel del factor permitirá confirmar el diagnóstico. En ocasiones se requiere de plasmaféresis para controlar la hemorragia.
EVALUACION DE LA HEMORRAGIA POSOPERATORIA
La hemorragia seria durante o después de la cirugía es un problema clínico complicado y que requiere de un diagnóstico rápido con intervención inmediata. La primera pregunta a hacerse es si la hemorragia se debe a una causa anatómica local (v.gr., un vaso no ligado) o a una falla hemostática sistémica. Si el paciente sangra solo en el área operada podrá pensarse en una causa anatómica, como un vaso no ligado. Es muy útil contar con una historia clínica previa, en especial con los resultados de procedimientos quirúrgicos anteriores, aunque quizá ésta pudiera ser inadecuada o incompleta. Un dato clínico importante para un trastorno generalizado es el sangrado en múltiples sitios, en especial en zonas no quirúrgicas. La presencia de sangre alrededor de un catéter, sitios de venopunción y venodisecciones es muy indicativa de un trastorno hemorragíparo.
Es imperativa la evaluación clínica global rápida. ¿Tiene el paciente afección renal, hepática o una neoplasia subyacente? ¿La cirugía requirió técnicas de derivación o la inducción de hipotermia, o el paciente ha estado en choque o hipotérmico? ¿Cuántas unidades de sangre y sus derivados se han administrado y en qué periodo de tiempo? ¿Se obtuvieron pruebas de coagulación de escrutinio antes de la cirugía y se cuenta aún con plasma congelado del paciente? El diagnóstico diferencial de la hemorragia posoperatoria debe incluir varios padecimientos [ver tabla 8].
|
|
|
La evaluación completa del problema requiere de un panel de pruebas de
coagulación (incluyendo TPT, TP, TT, fibrinógeno y dímero
D), cuenta plaquetaria y un frotis bien teñido para investigar la
morfología de las plaquetas. Esta batería de pruebas debe
realizarse de inmediato. Después podrán hacerse estudios
más especializados si existe evidencia de algún trastorno
específico.
Bibliografía
- Miller CH, Graham JB, Goldin LR, Elston RC: Genetics of classic von Willebrand's disease: II. Optimal assignment of the heterozygous phenotype (diagnosis) by discriminant analysis. Blood 54:137, 1979
- George JN, El-Harake MA, Raskob GE: Chronic idiopathic thrombocytopenic purpura. N Engl J Med 331:1207, 1994
- Gewirtz AM, Hoffman R: Transitory hypomegakaryocytic thrombocytopenia: aetiological association with ethanol abuse and implications regarding regulation of human megakaryocytopoiesis. Br J Haematol 62:333, 1986
- Tepler I, Elias L, Smith JW II, et al: A randomized placebo-controlled trial of recombinant human interleukin-11 in cancer patients with severe thrombocytopenia due to chemotherapy. Blood 87:3607, 1996
- Hirsh J, Dalen JE, Anderson DR, et al: Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 114:445S, 1998
- He R, Reid DM, Jones CE, et al: Spectrum of Ig classes, specificities, and titers of serum antiglycoproteins in chronic idiopathic thrombocytopenic purpura. Blood 83:1024, 1994
- Hymes K, Blaum M, Lackner H, et al: Easy bruising, thrombocytopenia, and elevated immunoglobulin G in Graves' disease and Hashimoto's thyroiditis. Ann Intern Med 94:27, 1981
- Stasi R, Stipa E, Masi M, et al: Prevalence and clinical significance of elevated antiphospholipid antibodies in patients with idiopathic thrombocytopenic purpura. Blood 84:4203, 1994
- George JN, Woolf SH, Raskob GE, et al: Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood 88:3, 1996
- Stasi R, Stipa E, Masi M, et al: Long term observation of 208 adults with chronic idiopathic thrombocytopenic purpura. Am J Med 98:436, 1995 [PMID 7733121 ]
- Cortelazzo S, Finazzi G, Buelli M, et al: High risk of severe bleeding in aged patients with chronic idiopathic thrombocytopenic purpura. Blood 77:31, 1991 [PMID 1984800 ]
- Berchtold P, Dale GL, Tani P, et al: Inhibition of autoantibody binding to platelet glycoprotein IIb/IIIa by anti-idiotypic antibodies in intravenous gammaglobulin. Blood 74:2414, 1989 [PMID 2478230 ]
- Yu Z, Lennon VA: Mechanism of intravenous immune globulin therapy in antibody-mediated autoimmune diseases. N Engl J Med 340:227, 1999 [PMID 9895405 ]
- Boughton BJ, Chakraverty R, Baglin TP, et al: The treatment of chronic idiopathic thrombocytopenic purpura with anti-D (Rho) immunoglobulin: its effectiveness, safety and mechanism of action. Clin Lab Haematol 10:275, 1988 [PMID 2846228 ]
- Facon T, Caulier MT, Wattel E, et al: A randomized trial comparing vinblastine in slow infusion and by bolus I.V. injection in idiopathic thrombocytopenic purpura: a report on 42 patients. Br J Haematol 86:678, 1994
- Laveder F, Marcolongo R, Zamboni S: Thrombocytopenic purpura following treatment with danazol. Br J Haematol 90:970, 1995 [PMID 7669685 ]
- Anderson JC: Response of resistant idiopathic thrombocytopenic purpura to pulsed high-dose dexamethasone therapy. N Engl J Med 330:1560, 1994
- Figueroa M, Gehlsen J, Hammond D, et al: Combination chemotherapy in refractory immune thrombocytopenic purpura. N Engl J Med 328:1226, 1993 [PMID 8464433 ]
- Bartholomew JR, Salgia R, Bell WR: Control of bleeding in patients with immune and nonimmune thrombocytopenia with aminocaproic acid. Arch Intern Med 149:1959, 1989 [PMID 2774776 ]
- Nardi MA, Liu L-X, Karpatkin S: GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci USA 94:7589, 1997 [PMID 9207136 ]
- Najean Y, Rabin JD: The mechanism of thrombocytopenia in patients with HIV infection. J Lab Clin Med 123:415, 1994 [PMID 8133154 ]
- Scaradavou A, Woo B, Woloski BMR, et al: Intravenous anti-D treatment of immune thrombocytopenic purpura: experience in 272 patients. Blood 89:2689, 1997 [PMID 9108386 ]
- Durand JM, Lefevre P, Hovette P, et al: Dapsone for thrombocytopenic purpura related to human immunodeficiency virus infection. Am J Med 90:675, 1991 [PMID 2042682 ]
- Marroni M, Gresele P, Landonio G, et al: Interferon-a is effective in the treatment of HIV-1-related, severe, zidovudine-resistant thrombocytopenia. Ann Intern Med 121:423, 1994 [PMID 8053616 ]
- Needleman SW, Sorace J, Poussin-Rosillo H, et al: Low-dose splenic irradiation in the treatment of autoimmune thrombocytopenia in HIV-infected patients. Ann Intern Med 116:310, 1992 [PMID 1733386 ]
- Burrows RF, Kelton JG: Fetal thrombocytopenia and its relation to maternal thrombocytopenia. N Engl J Med 329:1463, 1993 [PMID 8413457 ]
- Flug F, Karpatkin M, Karpatkin S: Should all pregnant women be tested for their PLA (Zw, HPA-1) phenotype? Br J Haematol 86:1, 1994
- 28. McCrae KR, Herman JH: Posttransfusion purpura: two unusual cases and a literature review. Am J Hematol 52:205, 1996 [PMID 8756089 ]
- Burgess JK, Lopez JA, Berndt MC, et al: Quinine-dependent antibodies bind a restricted set of epitopes on the glycoprotein Ib-IX complex: characterization of the epitopes. Blood 92:2366, 1998 [PMID 9746776 ]
- Glynne P, Salama A, Chaudhry A, et al: Quinine-induced immune thrombocytopenic purpura followed by hemolytic uremic syndrome. Am J Kidney Dis 33:133, 1999 [PMID 9915279 ]
- Kaufman DW, Kelly JP, Johannes CB, et al: Acute thrombocytopenic purpura in relation to the use of drugs. Blood 82:2714, 1993 [PMID 8219224 ]
- Burday MJ, Martin SE: Cocaine-associated thrombocytopenia. Am J Med 91:656, 1992
- Giugliano RP, Hyatt RR Jr: Thrombocytopenia with GPIIb/IIIa inhibitors: A metaanalysis. JACC 31:185A, 1998
- Berkowitz SD, Harrington RA, Rund MM, et al: Acute profound thrombocytopenia after c7E3 Fab (abciximab) therapy. Circulation 95:809, 1997 [PMID 9054735 ]
- Bednar B, Bednar RA, Cook JJ, et al: Drug-dependent antibodies against glycoprotein IIb/IIIa induce thrombocytopenia. Circulation 94(suppl):99, 1996
- Boyce TG, Swerdlow DL, Griffin PM: Escherichia coli 0157:H7 and the hemolytic-uremic syndrome. N Engl J Med 333:364, 1995 [PMID 7609755 ]
- Steinhubl SR, Tan WA, Foody JM, et al: Incidence and clinical course of thrombotic thrombocytopenic purpura due to ticlopidine following coronary stenting. EPISTENT investigators: evaluation of platelet IIb/IIIa inhibitor for stenting. JAMA 281:806, 1999
- Moake JL: Moschcowitz, multimers, and metalloprotease. N Engl J Med 339:1629, 1998
- Rock G, Kelton JG, Shumak KH, et al: Laboratory abnormalities in thrombotic thrombocytopenic purpura. Canadian apheresis group. Br J Haematol 103:1031, 1998
- Tsai HM: Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood 87:4235, 1996
- Furlan M, Robles R, Lammle B: Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood 87:4223, 1996 [PMID 8639781 ]
- Tsai HM, Lian ECY: Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med 339:1585, 1999
- Furlan M, Robles R, Galbusera M, et al: von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med 339:1578, 1998 [PMID 9828245 ]
- Galbusera M, Ruggenenti P, Noris M, et al: a1-Antitrypsin therapy in a case of thrombotic thrombocytopenic purpura. Lancet 345:224, 1995 [PMID 7823715 ]
- Rock G, Shumak KH, Buskard NA, et al: Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian apheresis study group. N Engl J Med 325:393, 1991
- George JN, Gilcher RO, Smith JW, et al: Thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: diagnosis and management. J Clin Apheresis 13:120, 1998 [PMID 9828022 ]
- Rock G, Shumak KH, Sutton DM, et al: Cryosupernatant as replacement fluid for plasma exchange in thrombotic thrombocytopenic purpura. Members of the Canadian Apheresis Group. Br J Haematol 94:383, 1996
- Bell WR, Braine HG, Ness PM, et al: Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: clinical experience in 108 patients. N Engl J Med 325:398, 1991 [PMID 2062331 ]
- Harkness DR, Byrnes JJ, Lian EC-Y: Hazard of platelet transfusion in thrombotic thrombocytopenic purpura. JAMA 246:1931, 1981 [PMID 7197306 ]
- Shumak KH, Rock G, Nair RC, et al: Late relapses in patients successfully treated for thrombotic thrombocytopenic purpura. Ann Intern Med 122:569, 1995 [PMID 7887549 ]
- Onundarson PT, Rowe JM, Heal JM, et al: Response to plasma exchange and splenectomy in thrombotic thrombocytopenic purpura: a 10-year experience at a single institution. Arch Intern Med 152:791, 1992 [PMID 1558437 ]
- Kelton JG, Keystone J, Moore J, et al: Immune-mediated thrombocytopenia of malaria. J Clin Invest 71:832, 1983 [PMID 6220030 ]
- Van Dam PA, Renier M, Baekelandt M, et al: Disseminated intravascular coagulation and the syndrome of hemolysis, elevated liver enzymes, and low platelets in severe preeclampsia. Obstet Gynecol 73:97, 1989 [PMID 2909047 ]
- Martin JN Jr, Blake PG, Perry KG Jr, et al: The natural history of HELLP syndrome: patterns of disease progression and regression. Am J Obstet Gynecol 164:1500, 1991 [PMID 2048596 ]
- Easterbrook PJ, Davis HP: Thrombocytopenia in hypothermia: a common but poorly recognized complication. Br Med J 291:23, 1985
- Fernandez F, Goudable C, Sie P, et al: Low haematocrit and prolonged bleeding time in uraemic patients: effect of red cell transfusions. Br J Haematol 59:139, 1985 [PMID 3918557 ]
- Mannucci PM: Hemostatic drugs. N Engl J Med 339:245, 1998
- Mikhailidis DP, Jenkins WJ, Barradas MA, et al: Platelet function defects in chronic alcoholism. Br Med J 293:715, 1986
- George JN, Shattil SJ: The clinical importance of acquired abnormalities of platelet function. N Engl J Med 324:27, 1991 [PMID 1984161 ]
- Michiels JJ, Abels J, Steketee J, et al: Erythromelalgia caused by platelet-mediated arteriolar inflammation and thrombosis in thrombocythemia. Ann Intern Med 102:466, 1985 [PMID 3977194 ]
- Anagrelide, a therapy for thrombocythemic states: experience in 577 patients. Anagrelide Study Group. Am J Med 92:69, 1992
- Haitjema T, Westermann CJJ, Overtoom TTC, et al: Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): new insights into pathogenesis, complications, and treatment. Arch Intern Med 156:714, 1996 [PMID 8615703 ]
- Shovlin CL: Molecular defects in rare bleeding disorders: hereditary haemorrhagic telangiectasia. Thromb Haemost 78:145, 1997
- White RI Jr, Lynch-Nyhan A, Terry P, et al: Pulmonary arteriovenous malformation: techniques and long-term outcome of embolotherapy. Radiology 169:663, 1988 [PMID 3186989 ]
- Vilter RW: Nutritional aspects of ascorbic acid: uses and abuses. West J Med 133:485, 1980
- Clemetson KJ: Platelet GPIb-V-IX complex. Thromb Haemost 78:266, 1997
- Triplett DA: Laboratory diagnosis of von Willebrand's disease. Mayo Clin Proc 66:832, 1991
- Sadler JE, Gralnick HR: A new classification for von Willebrand's disease. Blood 84:676, 1994 [PMID 8043857 ]
- Castillo R, Monteagudo J, Escolar G, et al: Hemostatic effects of normal platelet transfusion in severe von Willebrand's disease patients. Blood 77:1901, 1991 [PMID 1902120 ]
- Rose EH, Aledort LM: Nasal spray desmopressin (DDAVP) for mild hemophilia A and von Willebrand disease. Ann Intern Med 114:563, 1991 [PMID 1900403 ]
- Antonarakis SE, Rossiter JP, Young M, et al: Factor VIII inversions in severe hemophilia A: results of an international consortium study. Blood 86:2206, 1995 [PMID 7662970 ]
- Bray GL, Gomperts ED, Courter S, et al: The Recombinate Study Group: a multicenter study of recombinant factor VIII (Recombinate): safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. Blood 83:2428, 1994 [PMID 8167332 ]
- Seremetis S, Lusher JM, Abildgaard CF, et al: Human recombinant DNA-derived antihemophilic factor (factor VIII) in the treatment of hemophilia A: conclusions of a 5-year study of home therapy. The KOGENATE Study Group. Haemophilia 5:9, 1999
- Berntop E: Second generation, B-domain deleted recombinant factor VIII. Thromb Haemost 78:256, 1997
- Green PP, Mannucci PM, Briet E, et al: Carrier detection in hemophilia A: a cooperative international study: II. The efficacy of a universal discriminant. Blood 67:1560, 1986
- Kasper CK, Boylen AL, Ewing NP, et al: Hematologic management of hemophilia A for surgery. JAMA 253:1279, 1985 [PMID 3918191 ]
- Yee TT, Pasi KJ, Lilley PA, et al: Factor VIII inhibitors in haemophiliacs: a single-center experience over 34 years, 1964-97. Br J Haematol 104:909, 1999 [PMID 10192458 ]
- Kasper CK: Prothrombin complex concentrates and inhibitors: a reappraisal. JAMA 251:68, 1984
- Lusher JM: Controlled clinical trials with prothrombin complex concentrates. Prog Clin Biol Res 150:277, 1984
- Hedner U, Glazer S, Falch J: Recombinant activated factor VII in the treatment of bleeding episodes in patients with inherited and acquired bleeding disorders. Transfusion Med Rev 7:78, 1993
- Roberts HR, Eberst ME: Current management of hemophilia B. Hematol Oncol Clin North Am 7:1269, 1993 [PMID 8294316 ]
- Wang L, Takabe K, Bidlingmaier SM, et al: Sustained correction of bleeding disorder in hemophilia B mice by gene therapy. Proc Natl Acad Sci USA 30:3906, 1999
- Cooper DN, Millar DS, Wacey A, et al: Inherited factor VII deficiency: molecular genetics and pathophysiology. Thromb Haemost 78:151, 1997 [PMID 9198146 ]
- Fay WP, Parker AC, Condrey LR, et al: Human plasminogen activator inhibitor-1 (PAI-I) deficiency: characterization of a large kindred with a null mutation in the PAI-1 gene. Blood 90:204, 1997 [PMID 9207454 ]
- Furie B, Furie BC: Molecular and cellular biology of blood coagulation. N Engl J Med 326:800, 1992 [PMID 1538724 ]
- Sane DC, Califf RM, Topol EJ, et al: Bleeding during thrombolytic therapy for acute myocardial infarction: mechanisms and management. Ann Intern Med 111:1010, 1989 [PMID 2688504 ]
- Esmon CT, Fukudome K, Mather T, et al: Inflammation, sepsis, and coagulation. Haematologica 84:254, 1999 [PMID 10189392 ]
- Warrell RP, Kempin SJ: Treatment of severe coagulopathy in the Kasabach-Merritt syndrome with aminocaproic acid and cryoprecipitate. N Engl J Med 313:309, 1985 [PMID 3925341 ]
- Rickles FR, Levine MN, Edwards RL: Hemostatic alterations in cancer patients. Cancer Metastasis Rev 11:237, 1992 [PMID 1423816 ]
- Jochum M: Influence of high-dose antithrombin concentrate therapy on the release of cellular proteases, cytokines, and soluble adhesion molecules in acute inflammation. Semin Hematol 32:14, 1995
- Fourrier F, Chopin C, Huart JJ, et al: Double-blind, placebo-controlled trial of antithrombin III concentrates in septic shock with disseminated intravascular coagulation. Chest 104:882, 1993 [PMID 8365305 ]
- Ludlam CA, Morrison AE, Kessler C: Treatment of acquired hemophilia. Sem Hematol 31:16, 1994
- Lian C-Y, Larcada AF, Chiu AY-Z: Combination immunosuppressive therapy after factor VIII infusion for acquired factor VIII inhibitor. Ann Intern Med 110:774, 1989
- Shaffer LG, Phillips MD: Successful treatment of acquired hemophilia with oral immunosuppressive therapy. Ann Intern Med 127:206, 1997 [PMID 9245226 ]
- Morrison AE, Ludlam CA, Kessler C: Use of porcine factor VIII in the treatment of patients with acquired hemophilia. Blood 81:1513, 1993 [PMID 8453098 ]
- Crenier L, Ducobu J, des Grottes JM, et al: Low response to high-dose intravenous immunoglobulin in the treatment of acquired factor VIII inhibitor. Br J Haematol 95:750, 1996 [PMID 8982056 ]
- Francis JL, Armstrong DJ: Acquired dysfibrinogenemia in liver disease. J Clin Pathol 35:667, 1982 [PMID 7085917 ]
- Stein SF, Harker LA: Kinetic and functional studies of platelets, fibrinogen, and plasminogen in patients with hepatic cirrhosis. J Lab Clin Med 99:217, 1986
- Harker LA: Bleeding after cardiopulmonary bypass. N Engl J Med 314:1446, 1986
- Kestin AS, Valeri CR, Khuri SF, et al: The platelet function defect of cardiopulmonary bypass. Blood 82:107, 1993 [PMID 7686785 ]
- Bolan CD, Alving BM: Pharmacologic agents in the management of bleeding disorders. Transfusion 30:541, 1990 [PMID 2198685 ]
- Cattaneo M, Harris AS, Stromberg U, et al: The effect of desmopressin on reducing blood loss in cardiac surgery: a meta-analysis of double-blind, placebo-controlled trials. Thromb Haemost 74:1064, 1995 [PMID 8560415 ]
- Fremes SE, Wong BI, Lee E, et al: Metaanalysis of prophylactic drug treatment in the prevention of postoperative bleeding. Ann Thorac Surg 58:1580, 1994 [PMID 7526811 ]
- Zehnder JL, Leung LL: Development of antibodies to thrombin and factor V with recurrent bleeding in a patient exposed to topical bovine thrombin. Blood 76:2011, 1990 [PMID 2242423 ]
- Berruyer M, Amiral J, French P, et al: Immunization by bovine thrombin used with fibrin glue during cardiovascular operations. J Thorac Cardiovasc Surg 105:892, 1993 [PMID 8487567 ]
- George J: Hemostasis and thrombosis. Hematology: Basic Principles and Practice, 3rd ed. Hoffman R, Banz EJ Jr, Shattil SJ, Eds. Churchill Livingstone, Philadelphia, 1999, p 1928