Hematología
⭳ Abrir artículo (PDF)882.1 KBEste artículo fue revisado respecto a la Edición 2/2000. Ver esa versión →
Contenido del artículo
III ANEMIAS POR DEFECTOS EN LA PRODUCCION
- Clasificación de los defectos de producción
- Defectos de producción asociados con una médula ósea aparentemente normal
- ANEMIA DE LAS ENFERMEDADES CRONICAS
- ANEMIA EN LA ENFERMEDAD RENAL GRAVE
- DESNUTRICION
- EDAD
- MEDICAMENTOS Y OTROS AGENTES QUE AFECTAN LA PRODUCCION DE ERITROCITOS
- PADECIMIENTOS ENDOCRINOLOGICOS
- Defectos de la producción relacionados con aplasia o remplazo de la médula ósea
- Defectos en la producción relacionados con hiperplasia eritroide en la médula y eritropoyesis ineficaz
- Otros aspectos del metabolismo del folato y la cobalamina
III ANEMIAS POR DEFECTOS EN LA PRODUCCION
DR. STANLEY L. SCHRIER
Clasificación de los defectos de producción
Los defectos en la producción de eritrocitos causan anemia que se caracteriza por una cuenta absoluta baja de reticulocitos o un índice de reticulocitos bajo (cuenta de reticulocitos x [hematocrito real/hematocrito normal]). El examen de la sangre periférica y la médula ósea ayudan a clasificar estos padecimientos. La médula ósea característicamente muestra alguno de los siguientes datos:
1. Proporción normal de las células mieloides a eritroides (proporción M:E), celularidad total normal y patrón de maduración eritroide normal.
2. Ausencia virtual de elementos normales en la médula ósea debida a aplasia (ausencia de células en la médula) o sustitución de los elementos normales de la médula por fibrosis, tumores sólidos, granulomas o leucemia.
3. Hiperplasia eritroide con aumento en la celularidad. Debido a defectos en la maduración eritroide se produce eritropoyesis ineficaz o hemólisis intramedular. Los precursores mueren en la médula, y muy pocas células alcanzan la periferia.
Defectos de producción asociados con una médula ósea aparentemente normal
ANEMIA DE LAS ENFERMEDADES CRONICAS
Definición
La anemia de las enfermedades crónicas ocurre en forma secundaria a padecimientos neoplásicos, infecciosos, inflamatorios y otras enfermedades crónicas, incluyendo trastornos hepáticos, insuficiencia cardiaca congestiva y diabetes mellitus.1,2 Los valores de hematocrito suelen variar de 27 a 35 porciento, aunque el 20 porciento de los pacientes puede tener valores de hematocrito menores de 25 porciento.2
Fisiopatología
La anemia de las enfermedades crónicas puede ser causada por un ligero acortamiento en el tiempo de vida de los eritrocitos, porque el hierro es atrapado en el sistema reticuloendotelial y no está disponible para la hematopoyesis, y por una respuesta disminuida a la eritropoyetina. Característicamente, los niveles de eritropoyetina en pacientes con este tipo de anemia son bastante menores que los observados en pacientes con grados comparables de anemia por deficiencia de hierro.1,2 Los eritrocitos suelen tener apariencia normal, aunque en ocasiones son poco hipocrómicos y microcíticos. La concentración de hierro sérico y transferrina están bajas, y el porcentaje de saturación de hierro suele ser tan bajo como del 15 porciento.2 La concentración sérica de ferritina suele estar elevada, pero puede ser normal incluso en pacientes que no tienen reservas de hierro en la médula2. La concentración de protoporfirina eritrocitaria libre es alta porque el hierro es atrapado en células del sistema monocito-macrófago y es incapaz de unirse a la protoporfirina para formar el hem.
Se ha propuesto que las condiciones que producen la anemia de las enfermedades crónicas liberan citocinas (v.gr., interleucina-1 (IL-1) y factor de necrosis tumoral-alfa, interferones alfa, beta y gama y quizá factor de transformación de crecimiento-ß).3 Se ha demostrado en condiciones experimentales que las complejas interacciones entre estas citocinas recién liberadas reducen la producción de eritropoyetina, causan hipoferremia, aumentan la concentración de ferritina en suero, dañan la eritropoyesis y bloquean la liberación de hierro de las células reticuloendoteliales.1 La administración de dosis farmacológicas de eritropoyetina corrige la anemia de la enfermedad crónica en muchos pacientes.1 Por lo tanto, el defecto en la eritropoyesis puede superarse.
Diagnóstico
La anemia leve (por lo general normocítica y normocrómica), con cifras normales o elevadas de leucocitos y plaquetas en un paciente con una enfermedad crónica sugiere el diagnóstico. No es raro que esta anemia leve sea hipocrómica y microcítica, y se diagnostique como anemia por deficiencia de hierro, rasgo de talasemia o anemia sideroblástica. Si el diagnóstico está en duda después de un examen cuidadoso del frotis sanguíneo y una cuenta absoluta de reticulocitos baja, la medición del nivel de ferritina sérica y el examen de la médula ósea son las pruebas más útiles para hacer el diagnóstico diferencial [ver tabla 1 y figura 1]. En algunos casos existe más de una forma de anemia y puede requerirse un examen exhaustivo y repetitivo del paciente para establecer la causa primaria. Por ejemplo, un paciente con anemia secundaria a cáncer de colon también puede tener deficiencia de hierro por hemorragia intestinal y quimioterapia mielosupresora. La infección por VIH produce efectos hematológicos complejos, pero también causa anemia relacionada a enfermedades crónicas en la mayoría de los pacientes con SIDA. Además, los pacientes infectados por el VIH pueden tener otros tipos de anemia, incluyendo anemia hemolítica autoinmune Coombs positiva.
Tratamiento
Es importante identificar y tratar a la enfermedad primaria. La administración de hierro parenteral u oral no ayudará. Antes de comenzar el tratamiento con eritropoyetina es útil realizar una medición basal de la concentración de eritropoyetina en plasma, debido a que la respuesta a la eritropoyetina es poco frecuente en los enfermos cuyos niveles endógenos son mayores de 500 mU/ml. Se han reportado respuestas a la eritropoyetina en pacientes con artritis reumatoide,4 SIDA,5 enfermedad inflamatoria del intestino,6,7 y cáncer.8 Para obtener una respuesta óptima, el paciente debe tener reservas de hierro adecuadas (i.e., nivel de ferritina normal o elevado o adecuada tinción de hierro en la médula). Un esquema común consiste en iniciar con 100 a 150 U/kg por vía subcutánea tres veces por semana y, si no existe aumento en la hemoglobina a las seis a ocho semanas, aumentar la dosis a 300 U/kg tres veces por semana8 o cambiar a inyecciones diarias. Si aún no aumenta la hemoglobina después de 12 semanas, deberá suspenderse la eritropoyetina.
ANEMIA EN LA ENFERMEDAD RENAL GRAVE
Fisiopatología
La causa predominante de la anemia en la enfermedad renal es la deficiencia en la producción de eritropoyetina por los riñones enfermos. Si existe enfermedad renal inflamatoria subyacente, puede existir también un componente de anemia de enfermedad crónica. La anorexia y la mala ingesta de hierro, las muestras de sangre frecuentes y la pérdida de eritrocitos durante la hemodiálisis pueden producir deficiencia de hierro. la deficiencia de ácido fólico, el hiperesplenismo y el hiperpartiroidismo secundario con fibrosis de la médula3 puede promover también la anemia. Dependiendo de qué procesos causen la anemia, el frotis periférico puede mostrar fragmentación eritrocitaria o equinocitosis. La presencia de cuerpos de Heinz sugiere que ha ocurrido hemólisis oxidativa, quizá por oxidantes del líquido de hemodiálisis.
La toxicidad por aluminio también puede causar anemia en los pacientes en hemodiálisis. Este tipo de anemia se identificó inicialmente en pacientes con la llamada demencia por diálisis. Los niveles de aluminio muy altos en plasma parecen ser causados por contaminación con aluminio del líquido de diálisis o por absorción gastrointestinal de los geles de aluminio empleados para fijar el fosfato de la dieta. Experimentos in vitro han demostrado que el aluminio se une a los precursores eritroides unidad formadora de colonias eritroide (UFC-E) y unidad formadora del estallido eritroide (UFE-E).9
Tratamiento
La eritropoyetina es el tratamiento estándar para los pacientes con anemia por enfermedad renal. El tratamiento con eritropoyetina puede eliminar los requerimientos de transfusión en los pacientes con hemodiálisis y en pacientes con enfermedad renal progresiva que aún no requieren de hemodiálisis.10 El tratamiento mejora en forma sustancial su calidad de vida.11 Son poco frecuentes los efectos adversos, como la hipercalemia y la hipertensión. Es costumbre iniciar el tratamiento con 50 U/kg de eritropoyetina tres veces por semana, por vía intravenosa o subcutánea, y aumentar la dosis según sea necesario hasta alcanzar el nivel de hemoglobina deseado. En un estudio el tratamiento con desferroxamina, 30 mg/kg I.V., al final de cada sesión de diálisis, produjo mejoría importante en los pacientes con anemia causada por toxicidad por aluminio.12
DESNUTRICION
La desnutrición debida a anorexia nervosa o a deficiencias proteicas puede ocasionar anemia e incluso pancitopenia. Puede presentarse hemólisis [ver figura 2]. La biopsia de médula ósea es hipocelular, con un material gelatinoso característico que consiste en mucopolisacáridos ácidos. La anemia puede ocurrir a pesar de niveles normales de folato y cobalamina (vitamina B12) y puede corregirse con una nutrición adecuada.
EDAD
Aún es motivo de controversia si la hematopoyesis y, en especial la eritropoyesis, declinan con la edad.13,14 Sin embargo, por lo general los ancianos sanos tienen concentraciones normales de hemoglobina e índices eritrocitarios normales. Este dato fue confirmado por un estudio de pacientes de 84 y más años de edad.14 Por lo tanto, parece ser que la presencia de anemia en este grupo de pacientes amerita estudio y análisis.
MEDICAMENTOS Y OTROS AGENTES QUE AFECTAN LA PRODUCCION DE ERITROCITOS
La ingestión importante de alcohol (aguda o crónica) tiene importantes efectos hematológicos.15 La ingestión de alrededor de 80 g de alcohol (una botella de vino, seis tarros de cerveza o un tercio de una botella de whiskey) al día puede producir macrocitosis,16 vacuolización de proeritroblastos,17 trombocitopenia,18 disminución importante en los niveles séricos de folatos, y elevación en los niveles de hierro sérico. También puede afectar la respuesta de los reticulocitos a la administración de folato en pacientes con deficiencia de folato. La ingestión de alcohol por sí misma no produce un cuadro sideroblástico o megaloblástico.15 Los megaloblastos, macro-ovalocitos y neutrófilos polimorfonucleares (PMN) hipersegmentados suelen aparecer cuando existe una deficiencia concomitante de folatos. El abuso crónico de alcohol suele causar deficiencia de folato o hierro, enfermedad hepática severa, hemorragia gastrointestinal, hiperesplenismo y la anemia de la enfermedad crónica.
El cloranfenicol, además de producir anemia aplástica esporádica o idiosincrática (ver adelante), produce en forma esporádica una supresión eritroide reversible y dependiente de la dosis.19 El arsénico puede producir anemia al interferir con la producción de eritrocitos. También puede haber leucopenia y trombocitopenia, así como neuropatía periférica como resultado de la exposición a arsénico. La celularidad de la médula ósea puede estar aumentada, disminuida o normal.
La inmunoterapia para el cáncer a base de IL-2, sola o en combinación con células asesinas activadas por linfocinas (LAK, por sus siglas en inglés, n. el t.) ha producido diversos efectos hematológicos, y el más serio es la anemia dependiente de transfusión. Se piensa que la IL-2 suprime la hematopoyesis, quizá al causar la liberación de interferón gama, un conocido inhibidor de la misma. Otros efectos hematológicos producidos por la IL-2 y las células LAK incluyen trombocitopenia, linfopenia y eosinofilia.20
PADECIMIENTOS ENDOCRINOLOGICOS
En el hipotiroidismo aparece con frecuencia anemia secundaria a una menor producción de eritrocitos. La presencia de macrocitosis en un paciente hipotiroideo puede deberse a una deficiencia dietética de folatos concomitante o a una anemia perniciosa. La anemia ocasional que se presenta en el hipertiroidismo parece ser ocasionada por eritropoyesis ineficaz. La anemia leve que se relaciona con un panhipopituitarismo grave puede corregirse por medio del remplazo de hormonas suprarrenales, tiroideas y gonadales. Es bien conocido el efecto benéfico que tienen los andrógenos sobre la acción de la eritropoyetina. Tanto el hiperparatiroidismo primario como el secundario pueden ocasionar anemia; se ha observado aumento en el valor del hematocrito después de la realización de paratiroidectomía.
Defectos de la producción relacionados con aplasia o remplazo de la médula ósea
La presencia de pancitopenia o combinaciones diversas de anemia, neutropenia y trombocitopenia suelen indicar daño en la médula hematopoyética. Si la cavidad medular está infiltrada pero las células tronco pluripotenciales están intactas, suele desarrollarse hematopoyesis extramedular en los órganos de la hematopoyesis fetal (i.e., bazo, hígado y huesos distales).
Las citopenias combinadas de reciente inicio o un frotis de sangre leucoeritroblástico, que es la característica de la hematopoyesis extramedular [ver figura 2], constituyen indicación para la realización de una biopsia y aspirado de la médula ósea.
ANEMIA APLASICA
Definición
La pancitopenia (i.e., anemia, neutropenia y trombocitopenia) y una médula aplástica en el examen de la biopsia [ver figura 3] establecen la necesidad de una evaluación diagnóstica de anemia aplástica. La muestra de biopsia debe haberse tomado de un sitio no irradiado previamente. La anemia aplástica severa se define por los siguientes criterios: (1) una médula con celularidad de menos del 25 porciento del normal o con celularidad de menos del 50 porciento del normal en la que menos del 30 porciento de las células son hematopoyéticas, (2) dos de tres frotis de sangre periférica anormales (con índice corregido de reticulocitos menos del uno porciento o una cuenta absoluta de reticulocitos de menos de 40,000/µl, una cuenta de granulocitos de menos de 500 /µl o una cifra de plaquetas menor de 20,000/µl). Estos criterios han sido criticados como poco sensibles. Por ejemplo, en una serie de pacientes con diagnóstico de anemia aplástica, el índice corregido de reticulocitos menor de uno porciento se alcanzó con facilitad y no fue un criterio de severidad tan exacto como la cuenta de granulocitos menor de 500/µl.21 Otro grupo de investigadores notó que para interpretar sus datos tenían que identificar una cohorte de pacientes con anemia aplástica muy severa, con cuenta de granulocitos de menos de 200 /µl.22
Fisiopatología
Diversos medicamentos y otros agentes producen de manera regular y predecible aplasia medular [ver tabla 2]. La radiación ionizante y los medicamentos quimioterápicos empleados en el tratamiento de padecimientos malignos e inmunológicos tienen la capacidad de destruir las células tronco hematopoyéticas. Ciertos medicamentos, como el cloranfenicol, pueden producir aplasia medular de una manera no dependiente de la dosis. El tratamiento con oro y la inhalación de vapores de solventes orgánicos (v.gr., benceno o cola) también pueden inducir anemia aplástica mortal.
En el dos a 10 porciento de los casos de hepatitis ocurre aplasia severa dos a tres meses después de un caso típico de enfermedad aguda, por lo general en varones jóvenes. Con frecuencia la hepatitis no es la causa obvia, y las pruebas para hepatitis A, B y C son negativas. La enfermedad puede ser fatal a menos que se trate en forma agresiva con agentes inmunosupresores o trasplante de médula ósea.23 Existe una alta incidencia de anemia aplástica después del trasplante hepático en pacientes con hepatitis no-A no-B severa.24
La aplasia puede ser parte de los pródromos de una leucemia de células pilosas, de una leucemia linfoblástica aguda o, rara vez, de una leucemia mieloblástica aguda, o puede desarrollarse durante el curso de una mielodisplasia.
Existen varias líneas de evidencia que apoyan la posibilidad de que los padecimientos inmunológicos puedan ocasionar aplasia. La aplasia medular se presenta en la enfermedad de injerto contra huésped (EICH),25 y en la fascitis difusa.26 El precondicionamiento inmunosupresor mejora las oportunidades de que se lleve a cabo un trasplante exitoso de médula singénica en pacientes con anemia aplástica,27 y el tratamiento inmunosupresor se está empleando con éxito en el manejo de la anemia aplástica idiopática.25,27 En la sangre de algunos pacientes con anemia aplástica aparecen linfocitos T supresores que impiden el crecimiento de las células progenitoras comprometidas conocidas como unidades formadoras de colonias de granulocitos y macrófagos (CFU-GM). Las células T supresoras pueden actuar produciendo interferón gama.27 El resultado de estos complejos mecanismos inmunes que incluyen a las células T supresoras es una profunda reducción en las células hematopoyéticas primitivas medidas tanto por ensayo de cultivo a largo plazo de células iniciales (CLP-CI) como por la capacidad para formar colonias secundarias a partir de las colonias que sobreviven a cinco semanas de cultivo medular.28
Diagnóstico
El diagnóstico de anemia aplástica requiere de un aspirado y biopsia medular [ver figura 3], así como una historia cuidadosa de la exposición a fármacos, infecciones y especialmente síntomas que sugieran enfermedades virales y pruebas serológicas para hepatitis, mononucleosis infeciosa, VIH y parvovirus [ver figura 4].
El hiperesplenismo produce eliminación y secuestro esplénico rápido de las tres líneas celulares mieloides, pudiendo causar pancitopenia en padecimientos como la leucemia linfoblástica crónica, el lupus eritematoso generalizado o la esplenomegalia congestiva. Sin embargo, en tales casos la médula no está aplástica, sino que muestra hiperplasia de las líneas celulares involucradas. Debe realizarse medición de los eritrocitos CD59 para investigar hemoglobinuria paroxística nocturna.
También es importante determinar la gravedad de la enfermedad. Los casos graves tienen una tasa de remisión espontánea muy baja y una mortalidad del 70 porciento en un año. En comparación, un 80 porciento de los pacientes que tienen formas más leves de anemia aplástica sobreviven un año.21,22
Tratamiento
El tratamiento de las formas leves de la enfermedad incluye eliminar el agente agresor y proporcionar medidas de apoyo. Se espera que exista un número adecuado de células madre pluripotenciales como para repoblar la médula. En la anemia aplástica inducida por oro el dimercaprol, que facilita su excreción, puede ser de ayuda. Debido a que el oro causa la anemia aplástica por un efecto tóxico sobre las células tronco, se ha sugerido el uso de acetilcisteína intravenosa como agente quelante. Una teoría alterna afirma que el oro media una lesión inmunológica sobre las células tronco, lo que ha originado estudios sobre tratamiento inmunosupresor consistente en globulina antitimocito (GAT) y esteroides en dosis altas para pacientes con aplasia inducida por oro.29
Tratamiento de sostén Las hemorragias trombocitopénicas deberán manejarse mediante el empleo de transfusiones plaquetarias según se requiera para controlarlas. El remplazo muy liberal de plaquetas puede causar alosensibilización y complicar un futuro trasplante alogénico de médula ósea. Se administran transfusiones de eritrocitos según se requiera para controlar la anemia e hipoxia. Se han administrado factores de crecimiento hematopoyético como el factor estimulador de colonias de granulocitos (FEC-G) y el factor estimulador de colonias de granulocitos y macrófagos (FEC-GM) para aumentar la cuenta absoluta de neutrófilos y ayudar a combatir las infecciones. Sin embargo, la mayoría de las veces son ineficaces, principalmente por la grave deficiencia de células precursoras, que son el sitio blanco de su acción.30 Por lo general es preferible establecer el tratamiento definitivo, trasplante alogénico de médula ósea, de un hermano donador complatible, o tratamiento inmunosupresor.31
La elección del tratamiento adecuado en la anemia aplástica severa es crítico, y varios tratamientos se están revalorando en la actualidad. La mayoría de los expertos recomiendan el trasplante alogénico o singénico de médula ósea en pacientes menores de 20 años si existe un hermano donador compatible. Aunque existen varios riesgos, incluyendo EICH crónica y disfunción orgánica causada por el programa de acondicionamiento,30 el 50 a 80 porciento de estos pacientes pueden curarse, con una incidencia muy baja de trastornos clonales posteriores.32 El trasplante alogénico de médula ósea con programas de acondicionamiento que usan ciclofosfamida y GAT producen una supervivencia actuarial de 69 porciento a 15 años.32
En los pacientes mayores de 45 años se piensa que el impacto de la EICH es demasiado severa y que estos pacientes deben recibir mejor tratamiento inmunosupresor.30,32 Tres formas de inmunosupresión han demostrado producir remisión parcial en la anemia aplástica.30-32 La GAT produjo remisión duradera en alrededor de la mitad de los pacientes en un estudio aleatorio.31 Se ha comprobado de los esteroides en dosis altas aumentan las cuentas hemáticas en alrededor del 40 porciento de los pacientes tratados, y se ha demostrado que la ciclosporina también es benéfica.31 (Los andrógenos como la oximetolona pueden tener algún beneficio en el tratamiento de la anemia aplástica severa, pero no se administran solos.30,32) Aunque cada uno de estos agentes puede usarse en forma individual o consecutiva en el tratamiento de la anemia aplástica, un estudio controlado sugiere que los resultados son mejores cuando se usan los tres en forma simultánea.30,31 La combinación de GAT, un esteroide y ciclosporina permitió una supervivencia actuarial de 62 porciento a 36 meses, y los primeros signos de respuesta se presentaron a las 4 semanas; el tiempo promedio para la remisión fue de 60 a 82 días.31
La dosis eficaz de GAT depende del origen de la preparación (si es de caballo o conejo) y el modo de fabricación (si se produce como suspensión líquida o se administra por peso). El efecto adverso tóxico de la GAT es la enfermedad del suero, que suele controlarse con esteroides. La recomendación actual, basada en la GAT de caballo habitualmente disponible en los Estados Unidos,30,31 consiste en administrarla en una dosis de 40 mg/kg/día durante cuatro días. La GAT se diluye primero en 500 ml a 1 L de solución fisiológica y se infunde durante un periodo de cuatro a cinco horas a través de una vía intravenosa equipada con un filtro para microagregados. Se agregan 40 mg de metilprednisolona al frasco de infusión y se administra metilprednisolona por vía oral en cuatro dosis divididas hasta alcanzar una dosis de1 mg/kg/día. Se continúa el tratamiento con metilprednisolona hasta una dosis máxima de 56 mg/día en los pacientes que sufren calosfríos, fiebre o urticaria. En los enfermos en los que se desarrolla el cuadro florido de enfermedad del suero equino, la dosis de metilprednisolona se aumenta hasta un máximo de 64 mg/día. La reducción gradual de esteroides comienza al final de la segunda semana, y se suspenden en 30 días. Debido a que la GAT puede disminuir la cuenta plaquetaria, se administran transfusiones de plaquetas según se requiera para mantenerlas por arriba de 20,000/µl.
La ciclosporina se administra por vía oral en una dosis de 10 a 12 mg/kg/día en dos dosis divididas, con objeto de alcanzar niveles sanguíneos de 500 a 800 ng/ml o una concentración sérica de 100 a 200 ng/ml. Después de 29 días la dosis de ciclosporina puede disminuirse hasta una concentración sanguínea total de 200 a 500 ng/ml.30,31 La ciclosporina se continúa por lo menos durante seis meses; puede causar hipertensión, toxicidad renal, hipomagnesemia, vitiligo, temblores, hipertricosis, propensión a neumonía por Pneumocystis carinii (NPC) e hiperplasia gingival.30,31 En un estudio se administró 300 mg de pentamidina aerosolizada cada cuatro semanas como profilaxis para la NPC.31
A diferencia de los pacientes sometidos a trasplante alogénico de médula ósea, los que responden al tratamiento inmunosupresor no están realmente curados. Muchos continúan teniendo citopenias moderadas,33 el 20 a 36 porciento de estos pacientes recae con anemia aplástica,30,31,33 y hasta el 20 a 36 porciento desarrollará trastornos clonales, como hemoglobinuria paroxística nocturna, síndrome mielodisplásico e incluso leucemia aguda.30,31 Existe también mayor riesgo de desarrollar tumores sólidos después del tratamiento de la anemia aplástica, pero es el mismo para los pacientes que reciben tratamiento inmunosupresor o trasplante alogénico de médula ósea.34 De los enfermos que recaen después de responder en forma inicial al tratamiento inmunosupresor, más del 50 porciento responde por segunda ocasión.30,31
Debido a que la inmunosupresión no cura la anemia aplástica y se asocia con tasas significativas de recaída y evolución a trastornos clonales, un aspecto importante es cómo tratar a los pacientes entre 20 y 40 años de edad. Algunos autores proponen que debe considerarse el trasplante alogénico de médula ósea en este grupo de edad, en especial porque los nuevos programas de acondicionamieno parecen ser capaces de reducir la gravedad de la EICH.32,35 A falta de resultados de estudios clínicos controlados que comparen el tratamiento inmunosupresor de tres agentes con el trsplante alogénico de médula ósea de un hermano donador compatible, los médicos deben individualizar el tratamiento para los pacientes con edades entre 20 y 40 años. Una persona de 38 años de edad que realiza ejercicios en forma regular y no tiene factores de riesgo identificables puede ser un candidato para trasplante de médula ósea, mientras que alguien de la misma edad con múltiples factores de riesgo, como enfermedad cardiopulmonar por tabaquismo, no será un buen candidato. El trasplante alogénico de donador no relacionado compatible ha producido tasas de supervivencia a dos años de solo 29 porciento por EICH severa.30 Un trabajo inquietante evaluó a 10 pacientes con anemia aplástica severa que fueron tratados con dosis altas de ciclofosfamida IV (45 mg/kg/día) durante cuatro días consecutivos.36 Algunos pacientes recibieron también ciclosporina. Solo se administró un curso de ciclofosfamida. Siete de los 10 pacientes tuvo una respuesta hematológica completa y seis aún están vivos después de un seguimiento promedio de 10.8 años (rango de 7.3 a 17.8 años). Es claro que este manejo debe ser probado y evaluado en forma más profunda.
APLASIA PURA DE ERITROCITOS
Definición
En la aplasia pura de eritrocitos adquirida (APEA), la reticulocitopenia es profunda y casi no existen precursores eritroides en la médula ósea. Sin embargo, los elementos mieloides y megacariocíticos medulares están conservados y las cuentas de leucocitos y plaquetas en sangre periférica son normales. Estos datos indican que, en este desorden, se afectan los precursores eritroides, pero no las células tronco pluripotenciales.
Fisiopatología
Se piensa que en la APEA la eritropoyesis está inhibida principalmente por mecanismos inmunes, incluyendo autoanticuerpos y supresión de los progenitores eritroides mediada por células T en una fase posterior a la fase de diferenciación eritroide de la UFC-E y antes de la formación de los proeritroblastos.37 Las células T, en especial los linfocitos granulares grandes (LGG-T), pueden participar en la supresión de la eritropoyesis, y en algunos casos existió evidencia de que la supresión era causada por células clonales T.38 Se ha descrito también inhibición de la eritropoyetina por autoanticuerpos, aunque parece muy poco común.39 Es probable que existan otros dos mecanismos que causen APEA: el ataque específico a los precursores eritroides por el parvovirus B-19 (un reporte reciente indica que el 14 porciento de los casos fueron causados por este virus40) y una alteración hematopoyética clonal subyacente que puede ser un pródromo de un síndrome mielodisplásico.39 La APEA puede clasificarse con base en su etiología [ver tabla 3].
Diagnóstico
La evaluación diagnóstica de la APEA suele incluir una tomografía computada de tórax por la posibilidad de timoma, el análisis inmunofenotípico de los linfocitos de sangre circulante o de médula ósea para identificar proliferación de LGG , citogenética de la médula para estudiar la posibilidad de síndromes mielodisplásicos y pruebas de anticuerpos contra parvovirus.40
Ocurre eritroblastopenia en una pequeña proporción de pacientes con anemia hemolítica autoinmune, que puede ser causada por autoanticuerpos dirigidos contra normoblastos en maduración. El tratamiento de la infección por VIH con zidovudina (también conocida como AZT) produce anemia en virtualmente todos los pacientes, caracterizada por macrocitosis significativa.41 La hipoplasia eritroide moderada es la causa habitual de esta anemia, que puede progresar a APEA.
La APEA puede complicar al trasplante alogénico de médula ósea con incompatibilidad ABO; en estos casos el receptor del suero continúa expresando isohemaglutininas anti-A o anti-B contra el antígeno A o B del donador expresado en la superficie de los progenitores eritroides.42 En la APEA del embarazo los anticuerpos contra las UFE-E suelen desaparecer después del parto, coincidiendo con la remisión clínica.43
La infección por parvovirus es la causa de las crisis aplásticas temporales que ocurren en los pacientes que tienen trastornos hemolíticos severos. La médula de los pacientes con estos trastornos debe compensar la hemólisis periférica aumentando su preducción hasta siete veces, por lo que suele presentar una intensa hiperplasia eritroide. Aunque la infección por parvovirus puede afectar a todas las células precursoras, las de tipo eritroide son las más dañadas.40 Por lo general ocurre recuperación espontánea en una a dos semanas después del inicio de la infección. Una característica diagnóstica de la infección por parvovirus es la aparición de pronormoblastos gigantes en la médula [ver figura 4]. La asociación de la APEA con proliferación de LGG y leucemia cada vez se reconoce más. El uso rutinario de estudios de rearreglos de genes del receptor de la célula T en una serie mostró que nueve de 14 pacientes tuvo un trastorno clonal de LGG.44 Al parecer estas células median directamente la inhibición de la eritropoyesis.40,44
En quizá hasta el 20 porciento de los casos la APEA puede ser el pródromo de un síndrome mielodisplásico o incluso de leucemia mieloide aguda.40,42 La distinción en el momento del diagnóstico puede ser difícil a menos que se detecte una alteración citogenética mielodisplásica típica durante el estudio de la médula ósea.
Tratamiento
El tratamiento de la APEA depende de la causa identificada. Deben suspenderse los medicamentos agresores y, si existe un timoma, éste debe extirparse,42 lo que causa mejoría en alrededor de un tercio de los pacientes.42 Cuando la cirugía es imposible o el timoma es resistente a la quimioterapia, debe considerarse un curso de prednisona combinado con ocreótido, un análogo de somatostatina que se une a los timomas y puede inhibir la función de las células tímicas inmunes.45
En los casos de aplasia primaria de eritrocitos el tratamiento puede iniciarse con prednisona, 60 mg por vía oral diario en dosis divididas, continuando este esquema durante uno a tres meses.40 Si el paciente no responde, lo que se valora por los reticulocitos, se agregará ciclofosfamida o azatioprina, en dosis de 2 a 3 mg/kg/día por vía oral. Los pacientes con alteraciones citogenéticas de la médula ósea sugestivas de síndrome mielodisplásico responden mal.40,44 Algunos pacientes refractarios a otras formas de tratamiento han tenido buena respuesta a la infusión de IgG I.V., siendo la dosis estándar de 0.4 g/kg/día durante cinco días.46 Este tratamiento da buen resultado en los pacientes en los que la infección por parvovirus es la causa de la APEA.40 Para los pacientes con SIDA, infección por parvorvirus y APEA, puede ser necesario continuar la administración de IgG I.V. En un caso se administró IgG I.V. en dosis de 0.4 g/kg cada 2 a 3 semanas durante más de 50 meses.47 Los pacientes con proliferación de LGG como causa subyacente responden bien a la ciclofosfamida.44,48 Por lo general, las dosis bajas de ciclofosfamida (50 a 100 mg v.o.) administradas diario durante 3 a 6 meses son suficientes para producir remisión, que se asocia en ocasiones con desaparición de la proliferación de LGG.44,47 Cuando existe mala respuesta los pacientes suelen mejorar con ciclosporina oral.44,47
Se ha demostrado que la ciclosporina, en dosis de 12 mg/kg/día80 produce respuestas del orden del 65 porciento, incluso en pacientes que no responden a los esteroides, plasmaféresis, ciclofosfamida y azatioprina.42,49 Se ha administrado GAT para la APEA en pacientes refractarios, en forma semejante a como se usa en la anemia aplástica (40 mg/kg/día I.V. durante cuatro días).42
Defectos en la producción relacionados con hiperplasia eritroide en la médula y eritropoyesis ineficaz
Puede presentarse anemia con reticulocitopenia a pesar de que en la médula exista una hiperplasia eritroide intensa. Esta situación paradójica se presenta cuando existe eritropoyesis ineficaz o hemólisis intramedular. El defecto primario puede afectar en forma generalizada la línea eritroide, o bien subpoblaciones específicas de precursores eritroides en desarrollo. Algunas de estas subpoblaciones escapan a la muerte en la médula, pero su progenie ha sufrido un daño tan grave que es retirada rápidamente de la circulación, dando de esta manera la impresión de que existe hemólisis periférica. Se pueden observar otros signos de eritropoyesis ineficaz, incluyendo ictericia, un nivel de urobilinógeno fecal marcadamente elevado, un nivel muy alto de deshidrogenasa láctica en suero, una saturación de la capacidad de fijación de hierro sérico de 75 a 90 porciento y, ocasionalmente, depósito hepático de hierro en un patrón idéntico al que se ve en la hemocromatosis. El cuadro ferrocinético típico muestra depuración rápida del hierro plasmático, lo que indica intensa actividad de las células eritroides precursoras. Sin embargo, la llegada de eritrocitos marcados a la circulación periférica está muy disminuida, lo que sugiere que los precursores son destruidos por hemólisis intramedular.
Entre las enfermedades que producen eritropoyesis ineficaz importante se incluyen las anemias megaloblásticas, las anemias sideroblásticas, la talasemia, los síndromes mielodisplásicos y la metaplasia mieloide agnogénica.
ANEMIAS MEGALOBLASTICAS
Definición
Las anemias megaloblásticas se caracterizan por la presencia de macrocitosis; el frotis periférico muestra macroovalocitos, eritrocitos con forma de cola de pescado, neutrófilos hipersegmentados, y ocasionalmente eritrocitos nucleados [ver figura 2]. El encontrar un neutrófilo polimorfonuclear con seis lóbulos o que un cinco porciento de los polimorfonucleares tengan cinco lóbulos constituye una fuerte evidencia de anemia megaloblástica. En los casos graves se encuentra también granulocitopenia y trombocitopenia. La médula ósea muestra hiperplasia eritroide megaloblástica y metamielocitos gigantes.50 Si coexiste una deficiencia grave de hierro estará bloqueada la expresión morfológica completa de la megaloblastosis, aunque la presencia de metamielocitos gigantes en la médula y de polimorfonucleares hipersegmentados en la sangre periférica no se verá afectada.
Fisiopatología
Las anemias megaloblásticas son causadas por deficiencia de cobalamina o ácido fólico, por medicamentos que interfieren con la síntesis del ADN, con la absorción o el metabolismo de la cobalamina, y por trastornos genéticos que interfieren con el metabolismo del ADN o con la absorción o distribución de la cobalamina.
La eritropoyesis megaloblástica se caracteriza por síntesis de ADN defectuosa y detención en la fase G2, con alteraciones en la maduración celular y aparición de células que no sintetizan ADN que tienen un contenido anómalo del mismo. Esto ocasiona maduración asincrónica entre el núcleo y el citoplasma.50 La producción de ARN y la síntesis de proteínas continúa y, por lo tanto, se producen células más grandes, o megaloblastos. La eritropoyesis ineficaz es causada quizá por incremento de la apoptosis.51,52 Se presume que defectos semejantes en la síntesis de ADN caracterizan las alteraciones mucosas del estómago y la lengua. En la línea granulocítica la presencia de metamielocitos gigantes representa granulopoyesis ineficaz.50
Papel del ácido fólico y de la cobalamina Las interacciones entre el ácido fólico y la cobalamina son críticas en el metabolismo de las unidades de carbono único, principalmente de los análogos de metileno y de formil, que tienen un papel clave en la síntesis de ADN y las purinas [ver figura 5a]. Existen dos coenzimas principales de la cobalamina, la adenosilcobalamina y la metilcobalamina. La adenosilcobalamina es la coenzima para la metilmalonil-CoA mutasa, que cataliza un paso en el catabolismo del ácido propiónico [ver figura 5b].53 La metilcobalamina es la coenzima de la metionina sintetasa, que funciona como una metiltransferasa en la reacción que convierte 5-metiltetrahidrofolato (CH3-THF1) a tetrahidrofolato (THF1) [ver figura 5a].53 La cobalamina y el folato [ver figura 6] se combinan en la reacción de la sintetasa de metionina [ver figura 5a], en la que el grupo metil del CH3-THF1 es transferido a la cobalamina para formar metilcobalamina. La metilcobalamina transfiere entonces su grupo metil a la homocisteína para formar metionina. El monoglutamato tetrahidrofolato (THF1) que se forma por esta reacción es poliglutamado por medio de la enzima folilpoliglutamato sintetasa, y un grupo metilo se añade a él por medio de la serina-glicina metiltransferasa para formar 5,10 metileno THFn. El 5,10-metileno THFn proporciona su metileno para convertir el desoxiuridilato a timidilato, un paso clave en la síntesis del ADN. El 5-10, metileno THFn puede también convertirse en forma directa en CH3-THFn por medio de la enzima 5,10-metileno tetrahidrofolato reductasa, lo que hace que se disponga de su grupo metil para la sintetasa de metionina dependiente de cobalamina. El formil THFn (también llamado leucovorín, ácido folínico o factor citrovorum) también tiene un papel importante en la síntesis de purinas y, por lo tanto, en el metabolismo del ADN. Puede ser generado por oxidación del 5-10 metileno THFn o directamente del THFn por acción de la enzima formil THF sintetasa, en donde la metionina proporciona el grupo metil [ver figura 5a].53 Cuando existe deficiencia de cobalamina, el metileno THF1 no puede transferir su grupo metil a la cobalamina, por lo que no se libera para poliglutamarse por medio de la poliglutamato sintetasa [ver figura 5a]. Se requiere la forma poliglutamada para la síntesis de metileno THFn o de formil THFn, por lo que se bloquea la síntesis de ADN y de purinas. Esta es la hipótesis del atrapamiento del metilfolato y la apoya el encontrar incremento en los niveles de CH3-THF1 en el plasma de pacientes con deficiencia de cobalamina. Una explicación alternativa es la hipótesis de ausencia de grupos metil, en la que la deficiencia de cobalamina altera la generación de metionina, que entonces no puede proporcionar los grupos metilo necesarios para que la enzima formil THFn sintetasa produzca formil THFn.
La exposición a anestesia con óxido nitroso (N2O) por
un tiempo tan corto como seis horas produce megaloblastosis en la
médula; la exposición crónica a N2O puede
ocasionar una neuropatía similar a la que se ve en la deficiencia grave
de cobalamina. Aparecen promielocitos gigantes y metamielocitos en la
médula en forma rápida y pueden encontrarse neutrófilos
hipersegmentados en sangre periférica una semana después. La
concentración de folato en suero aumenta con rapidez y los niveles de
metionina caen porque el N2O inactiva la metionina
sintetasa. Los niveles aparentemente normales de cobalamina pueden
ser explicados por la incapacidad que tiene el ensayo en el plasma para
distinguir los análogos inactivos de la metilcobalamina
activa.54 Esta observación apoya la importancia del
metabolismo de la metionina en la megaloblastosis de la deficiencia de folatos
y cobalamina.
Otros aspectos del metabolismo del folato y la cobalamina
Debido a que ni el ácido fólico ni la cobalamina se producen en cantidades adecuadas en los humanos, ambos deben ser absorbidos a partir de los alimentos. En particular, la cobalamina deriva de fuentes microbianas y se ingiere a través de la carne o los huevos.
La mayor parte del ácido fólico presente en la dieta se encuentra en la forma de poliglutamato y es absorbido en la mucosa intestinal. La absorción del ácido fólico marcado radiactivamente se acerca al 80 porciento de una dosis de 200 µg.50,53 En el suero una pequeña cantidad de folato se fija a una proteína específica, y aún se desconoce el papel de esta proteína fijadora en el metabolismo del folato. Los niveles séricos de folato parecen mantenerse gracias al folato que se absorbe de los alimentos. Se ha observado circulación enterohepática del folato, en la que el ácido fólico que pasa hacia la bilis y al intestino delgado se absorbe de manera cuantitativa. En modelos animales la administración de etanol bloquea la entrada de folato hacia la bilis. Este efecto podría, en parte, ser la causa de la rápida caída en los niveles de folato sérico que se observa ocho horas después del consumo de alcohol. Una disminución similar en los niveles séricos de folato se observa después de la ingestión de fenitoína. Los requerimientos diarios de cobalamina son de alrededor de 1 µg, y la cantidad proporcionada por la dieta occidental habitual, que es rica en productos animales, varía de 5 a 15 µg.55
Las proteínas R son un tipo de glucoproteínas fijadoras de cobalamina producidas por granulocitos y otros tejidos. El factor intrínseco (FI) es una glucoproteína de 45 kd secretada por las células parietales gástricas que es muy específica para la cobalamina íntegra. El complejo proteína R-cobalamina no se une a los receptores ileales, por lo que no se absorbe. En el estómago la cobalamina se une preferentemente a las proteínas R más que al FI,50,53,55 por lo tanto, es el complejo proteína R-cobalamina fisiológicamente inactivo el que se libera al duodeno. Sin embargo, en el duodeno y el intestino delgado las proteasas pancreáticas, junto con la pepsina, degradan las proteínas R, liberando la cobalamina y permitiendo su unión al FI. Por lo tanto, la atrofia gástrica y la insuficiencia pancreática contribuyen a la malabsorción de la cobalamina.53,55 El complejo FI-cobalamina, en presencia de Ca2+ y un pH mayor de 5.4, se une en forma específica a un número limitado de sitios en las microvellosidades de las células de la mucosa en la porción terminal del íleo, en donde tiene lugar la absorción [ver figura 7].53,55
En el plasma la mayor parte de la cobalamina se une a las proteínas R transcobalaminas I y III (TC-I y TC-III), que no son muy importantes desde el punto de vista fisiológico, que se sintetizan en forma parcial en los precursores de granulocitos y que pueden servir como almacén de cobalamina. Estas se saturan en alrededor del 70 porciento con cobalamina y sus análogos.56 La proteína transportadora de importancia fisiológica es la transcobalamina-II (TC-II), que tiene considerable especificidad para la cobalamina y que se satura en solo el cinco a 10 porciento. Existen receptores para el complejo TC-II-cobalamina en muchas membranas celulares. La TC-II fija alrededor del 90 porciento de una dosis recién inyectada de cobalamina, y el complejo es depurado con rapidez con una vida media de seis a nueve minutos.55,56 En la deficiencia congénita de TC-II, que causa anemia megaloblástica severa, tanto el transporte de cobalamina en plasma como su absorción están deteriorados. La menor absorción de cobalamina en esos casos implica que la TC-II tiene alguna función dentro del enterocito ileal, en donde la cobalamina es transferida del FI a la TC-II.
La elevación de los niveles de cobalamina que se observa en pacientes con leucemia granulocítica crónica o granulocitosis significativa es causada por aumentos en la TC-I y, en menor grado, en la TC-III.
DEFICIENCIA DE COBALAMINA
Además de la anemia macrocítica megaloblástica, el paciente con deficiencia de cobalamina puede tener atrofia de las papilas linguales y glositis. La neuropatía es la característica inicial en alrededor del 12 porciento de los pacientes con anemia perniciosa sin anemia concomitante.57 Los pacientes con deficiencia grave de cobalamina se quejan al inicio de parestesias. El sentido del tacto y el de la temperatura pueden estar afectados en un grado mínimo, con disminución en los reflejos y en la percepción de las vibraciones de alta frecuencia. La enfermedad puede progresar y afectar las porciones dorsales de la médula, lo que ocasiona ataxia y debilidad, y en la exploración física se encuentra una marcha con una base amplia, signo de Romberg, y una pérdida del sentido de la posición y de la vibración (en especial cuando se explora este último con un diapasón de 256 Hz). Si no se detecta y se trata el padecimiento, se extenderá hasta afectar las columnas laterales. Esto ocasionará incapacidad para caminar debido a la presencia de debilidad y espasticidad, las cuales pueden confirmarse por los datos de clonus sostenido, hiperreflexia y signo de Babinsky. Debido a que tanto los nervios periféricos como las columnas laterales y dorsales se encuentran afectadas, en algunas ocasiones estas manifestaciones neurológicas son conocidas como degeneración subaguda combinada o enfermedad sistémica combinada subaguda. La afección de la memoria y la depresión pueden ser manifestaciones prominentes.57 La deficiencia de cobalamina parece ser la causa de varios trastornos neuropsiquiátricos, manifestados por síntomas como parestesias, ataxia, debilidad de las extremidades, alteraciones de la marcha y defectos de la memoria. Otros síntomas reportados como manifestaciones de deficiencia de cobalamina incluyen alucinaciones y cambios en la personalidad y el estado de ánimo.56 Sin embargo, es difícil relacionar estos síntomas con las lesiones de la médula espinal que ocurren en los estados de deficiencia de cobalamina.
Los investigadores han tratado de determinar si un defecto en la síntesis de metionina o alguna alteración en el metabolismo del ácido propiónico son la causa de la neuropatía asociada con la deficiencia de cobalamina [ver figura 5b], pero el mecanismo exacto sigue sin aclararse. Sin embargo, dos observaciones apuntan hacia la alteración en la sintetasa de metionina como causa de la neuropatía. Primero, la administración de N2O a monos produce una neuropatía que es bloqueada por el tratamiento simultáneo con metionina. Segundo, un paciente con alteración congénita en la actividad de la sintetasa de metionina tuvo una neuropatía caracterizada por alteraciones en la marcha, torpeza y parestesias.50,57
Diagnóstico
El cuadro clínico que en el pasado sugería el diagnóstico de deficiencia de cobalamina ha cambiado en forma significativa. El cuadro clínico clásico de la deficiencia de cobalamina consiste en anemia macrocítica con glositis o neuropatía. El aspirado y biopsia de la médula ósea revelan típicamente hiperplasia eritroide megaloblástica con metamielocitos gigantes.50 La hipercelularidad puede ser tan importante y los megaloblastos tan inmaduros que los clínicos en ocasiones hacen el diagnóstico equívoco de leucemia.50,58 La introducción de los contadores electrónicos de células que pueden medir con exactitud el volumen corpuscular promedio (VCM) ha permitido la identificación de grados más sutiles de macrocitosis (VCM > 100 fl) que podrían no ser aparentes en el examen del frotis de sangre periférica. El dato de macrocitosis justifica la medición de los niveles de cobalamina y folato, pero es importante recordar que la macrocitosis puede ser causada por otras condiciones, incluyendo deficiencia de folatos, enfermedad hepática, abuso de alcohol, reticulocitosis e ingestión de medicamentos como los antimetabolitos, agentes alquilantes y zidovudina.41,59 Por otro lado, la macrocitosis de la anemia megaloblástica puede ser enmascarada por condiciones que pueden producir hipocromia y eritrocitos microcíticos, como la talasemia. Esta posibilidad debe considerarse en especial en pacientes de raza negra, entre los cuales existe una frecuencia alta de talasemia-alfa (alrededor del 30 porciento). La talasemia-alfa concomitante puede minimizar la macrocitosis de la anemia perniciosa.59 La anemia de la enfermedad crónica o la que es ocasionada por pérdida de sangre y deficiencia de hierro puede disminuir el grado de macrocitosis, pero no afecta la hipersegmentación de los neutrófilos. En un estudio se descubrió deficiencia de hierro en el 20 porciento de 121 pacientes con anemia perniciosa.60 En otro estudio, el 19 porciento de los pacientes con anemia perniciosa no tuvieron anemia, y el 33 porciento no tuvieron macrocitosis.61
La incidencia de cáncer gástrico, tumores carcinoides gástricos, cáncer esofágico,62 y quizá cáncer de colon y recto63 es mayor en pacientes con anemia perniciosa que en los sujetos controles. La anemia perniciosa ocurre en asociación con otros padecimientos autoimunes. Se observaron trastornos tiroideos autoinmunes en el 24 porciento de 162 pacientes con anemia perniciosa.64 Por lo tanto, la evaluación clínica del estado tiroideo debe ser parte del estudio diagnóstico en la anemia perniciosa.
Debe considerarse el diagnóstico de deficiencia de cobalamina en los pacientes que tengan trastornos neuropsiquiátricos, incluso en ausencia de anemia o macrocitosis. Los peligros de no pensar en el diagnóstico correcto son muchos. Pueden no existir anemia, macrocitosis y alteraciones en el frotis periférico, incluso en pacientes con concentraciones de cobalamina en suero entre 100 y 199 pg/ml. Por lo tanto, las indicaciones para considerar un diagnóstico de deficiencia de cobalamina han cambiado desde un cuadro clínico más o menos especifico hasta uno más general que incluye neuropatía y ciertas formas poco descritas aún de disfunción cerebral.57 Es posible que estas formas de presentación ocasionen mayor frecuencia de solicitudes para medir la concentración de cobalamina en suero. Las pruebas disponibles en la actualidad en los laboratorios clínicos proporcionan mediciones mucho más exactas que antes. Debe hacerse en forma simultánea la medición del nivel de folatos en los eritrocitos, ya que el metabolismo de la cobalamina se relaciona con el del folato [ver figura 5a]. Debido a que la deficiencia de cobalamina causa aumento en los niveles de homocisteína y ácido metilmalónico, se ha propuesto que la medición de estos produtos ayuda a definir el estado de deficiencia.59,65,66
Durante el embarazo y en los estados de deficiencia de folatos ocurren concentraciones de cobalamina bajas en forma falsa.59 Hasta hace poco la reducción en la cobalamina sérica no se consideraba significativa sino hasta que el valor era muy bajo (i.e., < 150 pg/ml). Sin embargo, en la actualidad es claro que los pacientes con concentraciones de cobalamina tan altas como de 250 pg/ml, y quizá incluso mayores, pueden tener deficiencia de cobalamina.59,61,65 Por fortuna, el encontrar macro-ovalocitos o PMN hipersegmentados en el frotis de sangre periférica sigue siendo un indicador sensible de la presencia de deficiencia de cobalamina.
Los pacientes que tienen una concentración sérica de cobalamina muy baja y anemia macrocítica deben someterse a un ensayo de tratamiento con cobalamina parenteral. El diagnóstico de deficiencia de cobalamina se confirma si el tratamiento produce reticulocitosis en tres o cuatro días y se asocia con aumento en la concentración de hemoglobina y reducción del VCM. La prueba de supresión con desoxiuridina (dU) es muy confiable para identificar a los pacientes con deficiencia de cobalamina. Esta prueba, que no está disponible en todos los laboratorios, se ha usado para investigar la anemia megaloblástica y ha proporcionado mucha de la información actual sobre las funciones de la cobalamina y el folato. Cuando las células de la médula ósea normal se preincuban con desoxiuridina fría, la incorporación subsecuente de la timidina-H3 al ADN se reduce mucho. Esto se debe a que las células normales de la médula osea convierten la dU a desoxiuridilato, que después es metilado a timidilato [ver figura 5a], que a su vez se convierte en trifosfato de desoxitimidina (dTTP), un sustrato para la síntesis de ADN. Por lo tanto, la preincubación con dU aumenta mucho el reservorio celular de dTPP, de modo que la conversión de timidina-H3 en ADN se diluye y disminuye. Sin embargo, en las células de la médula deficientes en cobalamina y folato la deficiencia interfiere con la metilación de la dU a timidilato [ver figura 5a], de modo que la preincubación con dU no suprime la incorporación de timidina-H3 al ADN.50,66
Una vez que se hace el diagnóstico de deficiencia de cobalamina deberá buscarse la causa de la misma [ver tabla 4]. La fisiopatología de la anemia perniciosa parece incluir un fenómeno autoinmune, principalmente gastritis autoinmune y dos tipos de anticuerpos contra el factor intrínseco. Uno de estos anticuerpos anti-FI bloquea la unión de la cobalamina al FI, y el otro bloquea la fijación del complejo FI-cobalamina a los receptores ileales.67 Clínicamente, se encuentran anticuerpos específicos anti-FI en el 70 porciento de los pacientes con anemia perniciosa.
Existen otras causas de deficiencia de cobalamina, En los vegetarianos se desarrolla una anemia megaloblástica severa como resultado de la ingesta muy baja de cobalamina. También se han observado deficiencias de folato y hierro. Los infantes de madres vegetarianas pueden tener deficiencia severa de cobalamina, en especial si reciben leche materna.56 La deficiencia de cobalamina es sorprendentemente común en países en vías de desarrollo, en donde las personas no son vegetarianos estrictos.56 La incidencia es especialmente alta en mujeres embarazadas y niños prescolares.56 La cirugía gástrica, en la que se eliminan el FI, la pepsina y componentes secretores de ácido, suele causar deficiencia de cobalamina (en el 31 porciento de los pacientes según un estudio69). Los pacientes que han sido sometidos a cirugía gastrica deben ser vigilados en forma rutinaria midiendo los niveles de cobalamina u homocisteína en plasma, recibiendo suplementos con cobalamina de por vida si los niveles están bajos.69
El método general de estudio para el diagnóstico diferencial de la deficienfia de cobalamina se apoya en la prueba de Schilling, la cual mide la absorción de cobalamina marcada con cobalto 57 (57Co). Después de que se administra una dosis de 1 µg de cobalamina marcada por vía oral , se administra una dosis de 1,000 µg de cobalamina no marcada por vía parenteral. La dosis parenteral satura a las transcobalaminas I, II y III, de tal manera que una porción importante del material absorbido es excretado por la orina. Si la cantidad de cobalamina marcada con 57Co que se mide en una muestra de orina de 24 horas debidamente recolectada es menor del 10 porciento de la cantidad administrada por vía oral, existe una absorción deficiente de cobalamina.
Se puede repetir la prueba de Schilling, agregando factor intrínseco oral suplementario. En la anemia perniciosa la adición de FI deberá corregir los resultados anormales de la prueba a menos que el FI administrado no sea completamente activo, el paciente secrete anticuerpos contra el FI o esté tomando medicamentos que interfieran con la absorción de cobalamina. La deficiencia prolongada de cobalamina afecta la captación a nivel del íleon, lo que altera los resultados de las pruebas. Por lo tanto, algunas personas no realizan la prueba de Schilling con FI hasta que el paciente ha recibido tratamiento con cobalamina por varias semanas.
Existen cada vez más reportes de pacientes con anemia perniciosa demostrada que tienen concentraciones séricas de cobalamina bajas o limítrofes pero prueba de Schilling normal. Parece ser que al progresar la lesión de atrofia gástrica de la anemia perniciosa, la capacidad de producir ácido-pepsina se pierde antes de que desaparezca toda la actividad del FI. Por lo tanto, en pacientes que se encuentran en una fase temprana de la enfermedad, puede existir suficiente FI para fijar la cobalamina libre administrada por vía oral en la prueba de Schilling y dar un resultado normal. Puede demostrarse malabsorción de cobalamina en estos pacientes por medio de una prueba denominada de Schilling con alimento, que se realiza con huevos de gallinas que han sido inyectados con cobalamina radioactiva.70 Esta prueba revela si no existe suficiente ácido-pepsina para separar el complejo cobalamina-enzima y liberar la cobalamina libre para que se una al FI. Sin embargo, esta prueba de Schilling con alimento no está disponible con facilidad, por lo que si se sospecha anemia perniciosa en un paciente que aparentemente tiene una prueba de Schilling normal, deben realizarse otros estudios para confirmar el diagnóstico, incluyendo examinar la morfología de las células sanguíneas, medir anticuerpos anti-FI o los niveles séricos de homocisteína o ácido metilmalónico, o realizar un estudio terapéutico con administración parenteral de cobalamina.
Si las pruebas de Schilling con factor intrínseco no demuestran mejoría en los resultados en forma repetida, probablemente deberán investigarse causas de malabsorción de cobalamina no relacionadas con la deficiencia del FI. Por ejemplo, la insuficiencia pancreática puede originar malabsorción de la cobalamina si el páncreas dañado no produce tripsina o quimiotripsina en cantidades suficientes para digerir el complejo proteína R-cobalamina y de esta manera liberar a la vitamina para que forme el complejo con el FI [ver figura 7].
Tratamiento
El tratamiento con ácido fólico puede producir remisiones hematológicas parciales en los pacientes con deficiencia de cobalamina, pero no mejora la sintomatología neurológica. Debe iniciarse tratamiento específico inmediatamente después de que se haya hecho el diagnóstico de megaloblastosis y de que se hayan tomado muestras para determinar los niveles de cobalamina. Si el paciente se encuentra sintomático por la presencia de una anemia grave, puede transfundirse en forma muy lenta un paquete globular para evitar el precipitar o el agravar una insuficiencia cardiaca congestiva. Esta constituye una de las pocas circunstancias en las cuales puede estar justificada la transfusión de una sola unidad, ya que ésta puede producir un incremento de un 25 porciento en la capacidad de transporte de oxígeno. Debe administrarse una dosis grande de cobalamina debido a que la retención de la cobalamina parenteral es poca, pero variable, y la vitamina no es cara ni tiene efectos colaterales peligrosos. La respuesta de reticulocitos se inicia en cuatro a seis días, y si la cuenta de los granulocitos es baja, aumenta al mismo tiempo. La hipersegmentación de los polimorfonucleares desaparece después de 10 a 14 días, lo que sugiere que en las anemias megaloblasticas la granulopoyesis está afectada por la deficiencia de la cobalamina a dos niveles diferentes: el paso en el cual se determina el número de lóbulos de los polimorfonucleares, y otro nivel en el que los granulocitos maduran y abandonan la médula.50 Las dosis semanales de 1,000 µg de cobalamina parenteral por seis semanas deben continuarse con dosis parenterales de 1,000 µg por mes de por vida. La preparación parenteral estándar es la cianobalamina. En el caso de insuficiencia pancreática puede administrarse cobalamina por vía parenteral o enzimas pancreáticas por vía oral. Debe diseñarse un tratamiento específico para los pacientes con formas intestinales de malabsorción.
Debido a que una pequeña cantidad de cobalamina se absorbe incluso en ausencia de FI y ya que solo se requiere 1 µg/día, se ha demostrado que son adecuadas las dosis orales de cobalamina de 300 a 1,000 µg/día en pacientes con anemia perniciosa, y esta forma de tratamiento evita las inyecciones mensuales (se recomiendan 1,000 µg/día).71
DEFICIENCIA DE ACIDO FOLICO
Los pacientes con anemia megaloblástica que no tienen glositis, antecedentes familiares de anemia perniciosa, o el cuadro neurológico de una enfermedad sistémica combinada probablemente tienen deficiencia de ácido fólico. Es importante el interrogatorio meticuloso acerca de los hábitos debido a que los malos hábitos alimenticios, la ingesta dietética insuficiencia y el alcoholismo pueden ocasionar deficiencia grave de ácido fólico. Es factible que la anemia megaloblástica que se presenta durante la administración de medicamentos o durante el embarazo sea debida a deficiencia de ácido fólico. Debido a que la combinación de deficiencia de folato y hierro es común, con frecuencia se bloquea la expresión completa de la megaloblastosis y el paciente presenta una anemia dimórfica, más que la macro-ovalocitos fácilmente identificable. Sin embargo, persiste la hipersegmentación de los polimorfonucleares.50,72 Recientemente se ha descubierto una variable termolábil de la enzima 5,10-metileno tetrahidrofolato reductasa, C677T que causa una deficiencia enzimática relativa. Alrededor del cinco a 10 porciento de la población general es homocigota para esta variante. La deficiencia enzimática interfiere con el reciclamiento del 5,10-metileno THFn a metil THFn, que a su vez proporciona el grupo metil para la conversión de homocisteína a metionina. No es sorprendente que el nivel de homocisteína en los pacientes afectados aumente, y éstos pueden desarrollar un estado de hipercoagulabilidad . Las mujeres embarazadas y no embarazadas que son homocigotas para la mutación C677T tienen niveles de folato en eritrocitos significativamente bajos,73 pueden ser susceptible a enfermedad cardiovascular y cerebrovascular y pueden tener hijos con defectos del tubo neural.73,74
Fisiopatología
Se han identificado numerosas causas de deficiencia de ácido fólico [ver tabla 5]. La ingestión de etanol en voluntarios bien nutridos no produce megaloblastosis, pero en pacientes con reservas de folato en límites el etanol puede disminuir la concentración sérica de folatos y bloquear la respuesta de reticulocitos a la administración de folato. El alcohol puede bloquear también la liberación de ácido fólico de los tejidos al suero.
Diagnóstico
Los niveles séricos de folato disminuyen súbitamente después de la ingestión de etanol; los niveles disminuyen en un plazo de dos semanas después de que se suspende la ingestión de folato en la dieta. Por lo tanto muchos pacientes hospitalizados tienen niveles séricos de folato disminuidos sin que exista una deprivación verdadera de folatos en los tejidos. En la evaluación de los pacientes con deficiencia de folato deben obtenerse los valores de folato sérico, cobalamina sérica y valores de folato en eritrocitos. El valor de folato en los eritrocitos refleja el depósito tisular,72 pero puede estar reducido en la deficiencia grave de cobalamina. En los estados de deficiencia aislada de cobalamina el valor sérico del folato suele ser normal o elevado (la llamada trampa de metilfolato). Es importante recordar que la deficiencia grave y de larga duración de la cobalamina ocasiona anorexia y alteraciones gastrointestinales, que pueden ocasionar deficiencia dietética de folatos. Como resultado, tanto los niveles de cobalamina como los de folatos serán bajos, lo que indica un estado de doble deficiencia. Cuando se ha diagnosticado deficiencia de folato deben establecerse otras medidas para esclarecer con precisión las causas de la deficiencia. En los casos en que es difícil pero necesario distinguir la megaloblastosis de la deficiencia de cobalamina de la de deficiencia de folato, son útiles las concentraciones séricas de ácido metilmalónico y de homocisteína, que en la deficiencia de cobalamina estarán elevadas, mientras que en la deficiencia de folatos solo aumenta la concentración de homocisteína [ver figura 5].72
En la mayoría de los pacientes (i.e., que no requieren una gran cantidad de folato por situaciones como hemólisis o embarazo), se presenta una respuesta hemotológica después de la administración de 200 µg de folato por día. La mayor demanda de folato durante el embarazo es de alrededor de 200 a 300 µg/día.75 Aún más, los suplementos de folato parecen prevenir los defectos del tubo neural fetal.76 Estos defectos del tubo neural pueden ocurrir en el embrión en etapas muy tempranas de la gestación (incluso antes de que se confirme el embarazo).73,74 Por lo tanto, se recomienda que las mujeres en edad reproductiva o que estén planeando embarazarse reciban alrededor de 400 µg de folato al día. Las mujeres homocigotas para la mutación C677T deben también tomar suplementos de folato. Algunos alimentos como la harina y los cereales pueden estar enriquecidos con ácido fólico. Sin embargo, se ha expresado la preocupación de que los suplementos de folato enmascaren la megaloblastosis de la anemia perniciosa, causando neuropatía severa en lugar de anemia.77
Tratamiento
El tratamiento estándar para la deficiencia de ácido fólico consiste en la administración de 1 mg/día por vía oral. La respuesta se manifiesta por la presencia de reticulocitosis en cuatro a seis días, ausencia de magaloblastosis y normalización de los valores hematológicos, lo que confirma el diagnóstico de deficiencia de folato. Sin embargo, la hipersegmentación de los neutrófilos desaparece solamente después de que han pasado 10 a 14 días.50 La megaloblastosis y la depresión medular grave secundaria a medicamentos que bloquean a la dihidrofolato reductasa, como la pirimetamina y el metotrexate, pueden tratarse con ácido folínico. En el caso de toxicidad después de la administración de dosis únicas elevadas de metotrexate será suficiente la administración de una dosis única equivalente (i.e., miligramo por miligramo) de ácido folínico IM. En los casos de toxicidad después del tratamiento crónico con pirimetamina, pueden administrarse de 1 a 5 mg de ácido folínico por día sin bloquear los efectos antipalúdicos de la pirimetamina. La megaloblastosis debida a tratamiento anticonvulsivante puede tratarse con 1 mg de ácido fólico por día. Se recomienda la profilaxis durante el embarazo, y también puede ser de utilidad en los pacientes que tienen hemólisis crónica grave.
Si la causa de la megaloblastosis no está claramente relacionada con anormalidades del metabolismo de la cobalamina o del ácido fólico, deberán buscarse otras causas de alteración en la síntesis del ADN. Muchos de los medicamentos antineoplásicos e inmunosupresores que interfieren con la síntesis de ADN producen también megaloblastosis. Entre los medicamentos se incluye el fluoruracilo, la hidroxiurea, la mercaptopurina, la tioguanina, la citarabina y la azatioprina.
ANEMIAS SIDEROBLASTICAS
Las anemias sideroblásticas son un grupo heterogéneo de padecimientos que se caracterizan por la presencia de anemia y eritropoyesis ineficaz.
Fisiopatología
Las anemias sideroblásticas no tienen una etiología única. En estos padecimientos aparecen subpoblaciones de células eritroides en la médula y grupos distintos de células se ven afectados en grados diferentes. Esto puede explicar la apariencia dimórfica de los eritrocitos en el frotis de sangre periférica. Las alteraciones en la síntesis del hem pueden ser la causa más frecuente, y se han descrito defectos moleculares de la enzima 5-aminolevulinato sintetasa.78,79 Esta enzima inicia la vía de síntesis del hem y su alteración afecta en forma importante la producción de esta molécula. En otros casos existen deleciones importantes en el ADN mitocondrial. El hierro penetra en los precursores eritroides pero, debido a que la síntesis del hem está alterada, no puede incorporarse al hem y se acumula en la cresta de la mitocondria.
Diagnóstico
Los pacientes pueden tener anemia refractaria o hemocromatosis. El diagnóstico de anemia sideroblástica se establece por reticulocitopenia, presencia de sideroblastos en anillo (normoblastos en la médula ósea con grandes incrustaciones de hierro no ferritina en la mitocondria [ver figura 8], capacidad de fijación de hierro saturada (por lo general de 80 porciento), elevación de deshidrogenasa láctica (DHL) en suero, depósito de hierro en el hígado en un patrón indistinguible de la hemocromatosis y un frotis de sangre periférica bizarro, con hipocromía, eritrocitos distorsionados y puntilleo basófilo.78,79 El estudio citogenético de la médula ósea puede revelar uno de los patrones típicos observados en los síndromes mielodisplásicos. Las anemias sideroblásticas se clasifican en cuatro grupos diferentes [ver tabla 6].
Tratamiento
Para fines pronósticos es importante decidir si el paciente tiene una forma benigna o maligna de la enfermedad. Los indicadores de la forma maligna son granulocitopenia, trombocitopenia, granulopoyesis displásica en la médula, megacariocitos bilobulados [ver figura 2], y cuerpos de Auer. En forma esporádica, algunos pacientes con la variedad benigna pueden tener una respuesta de los reticulocitos y la hemoglobina a la administración de piridoxina en dosis de 200 mg/día, con o sin ácido fólico.80 Los pacientes con signos y síntomas de hemocromatosis tienen serios problemas debido a que su anemia no suele permitir la extracción de hierro por medio de flebotomías repetidas. En tales circunstancias pudiera ser de utilidad el empleo de infusiones intravenosas o subcutáneas de desferroxamina para favorecer la salida de hierro. El beber té con los alimentos puede ayudar a bloquear el aumento en la absorción de hierro. La anemia puede ser lo bastante grave como para necesitar la transfusión de paquetes globulares, pero esto sólo intensificará los problemas relacionados con el metabolismo del hierro. En el manejo de la anemia es de importancia crítica el suspender la exposición a los agentes agresores, particularmente el alcohol.
Reconocimientos
Figuras 1 y 6 Talar Agasyan.
Figura 5 Alan D. Iselin.
Figura 7 Tom Moore.
Blibliografía
DR. STANLEY L. SCHRIER
Clasificación de los defectos de producción
Los defectos en la producción de eritrocitos causan anemia que se caracteriza por una cuenta absoluta baja de reticulocitos o un índice de reticulocitos bajo (cuenta de reticulocitos x [hematocrito real/hematocrito normal]). El examen de la sangre periférica y la médula ósea ayudan a clasificar estos padecimientos. La médula ósea característicamente muestra alguno de los siguientes datos:
1. Proporción normal de las células mieloides a eritroides (proporción M:E), celularidad total normal y patrón de maduración eritroide normal.
2. Ausencia virtual de elementos normales en la médula ósea debida a aplasia (ausencia de células en la médula) o sustitución de los elementos normales de la médula por fibrosis, tumores sólidos, granulomas o leucemia.
3. Hiperplasia eritroide con aumento en la celularidad. Debido a defectos en la maduración eritroide se produce eritropoyesis ineficaz o hemólisis intramedular. Los precursores mueren en la médula, y muy pocas células alcanzan la periferia.
Defectos de producción asociados con una médula ósea aparentemente normal
ANEMIA DE LAS ENFERMEDADES CRONICAS
Definición
La anemia de las enfermedades crónicas ocurre en forma secundaria a padecimientos neoplásicos, infecciosos, inflamatorios y otras enfermedades crónicas, incluyendo trastornos hepáticos, insuficiencia cardiaca congestiva y diabetes mellitus.1,2 Los valores de hematocrito suelen variar de 27 a 35 porciento, aunque el 20 porciento de los pacientes puede tener valores de hematocrito menores de 25 porciento.2
Fisiopatología
La anemia de las enfermedades crónicas puede ser causada por un ligero acortamiento en el tiempo de vida de los eritrocitos, porque el hierro es atrapado en el sistema reticuloendotelial y no está disponible para la hematopoyesis, y por una respuesta disminuida a la eritropoyetina. Característicamente, los niveles de eritropoyetina en pacientes con este tipo de anemia son bastante menores que los observados en pacientes con grados comparables de anemia por deficiencia de hierro.1,2 Los eritrocitos suelen tener apariencia normal, aunque en ocasiones son poco hipocrómicos y microcíticos. La concentración de hierro sérico y transferrina están bajas, y el porcentaje de saturación de hierro suele ser tan bajo como del 15 porciento.2 La concentración sérica de ferritina suele estar elevada, pero puede ser normal incluso en pacientes que no tienen reservas de hierro en la médula2. La concentración de protoporfirina eritrocitaria libre es alta porque el hierro es atrapado en células del sistema monocito-macrófago y es incapaz de unirse a la protoporfirina para formar el hem.
Se ha propuesto que las condiciones que producen la anemia de las enfermedades crónicas liberan citocinas (v.gr., interleucina-1 (IL-1) y factor de necrosis tumoral-alfa, interferones alfa, beta y gama y quizá factor de transformación de crecimiento-ß).3 Se ha demostrado en condiciones experimentales que las complejas interacciones entre estas citocinas recién liberadas reducen la producción de eritropoyetina, causan hipoferremia, aumentan la concentración de ferritina en suero, dañan la eritropoyesis y bloquean la liberación de hierro de las células reticuloendoteliales.1 La administración de dosis farmacológicas de eritropoyetina corrige la anemia de la enfermedad crónica en muchos pacientes.1 Por lo tanto, el defecto en la eritropoyesis puede superarse.
Diagnóstico
La anemia leve (por lo general normocítica y normocrómica), con cifras normales o elevadas de leucocitos y plaquetas en un paciente con una enfermedad crónica sugiere el diagnóstico. No es raro que esta anemia leve sea hipocrómica y microcítica, y se diagnostique como anemia por deficiencia de hierro, rasgo de talasemia o anemia sideroblástica. Si el diagnóstico está en duda después de un examen cuidadoso del frotis sanguíneo y una cuenta absoluta de reticulocitos baja, la medición del nivel de ferritina sérica y el examen de la médula ósea son las pruebas más útiles para hacer el diagnóstico diferencial [ver tabla 1 y figura 1]. En algunos casos existe más de una forma de anemia y puede requerirse un examen exhaustivo y repetitivo del paciente para establecer la causa primaria. Por ejemplo, un paciente con anemia secundaria a cáncer de colon también puede tener deficiencia de hierro por hemorragia intestinal y quimioterapia mielosupresora. La infección por VIH produce efectos hematológicos complejos, pero también causa anemia relacionada a enfermedades crónicas en la mayoría de los pacientes con SIDA. Además, los pacientes infectados por el VIH pueden tener otros tipos de anemia, incluyendo anemia hemolítica autoinmune Coombs positiva.
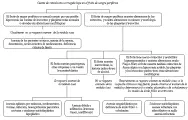
|
| Figura 1 |
| Diagnóstico de anemia por defecto de producción |
|
|||||||||||||||||||||||||||||||||||
|
Tratamiento
Es importante identificar y tratar a la enfermedad primaria. La administración de hierro parenteral u oral no ayudará. Antes de comenzar el tratamiento con eritropoyetina es útil realizar una medición basal de la concentración de eritropoyetina en plasma, debido a que la respuesta a la eritropoyetina es poco frecuente en los enfermos cuyos niveles endógenos son mayores de 500 mU/ml. Se han reportado respuestas a la eritropoyetina en pacientes con artritis reumatoide,4 SIDA,5 enfermedad inflamatoria del intestino,6,7 y cáncer.8 Para obtener una respuesta óptima, el paciente debe tener reservas de hierro adecuadas (i.e., nivel de ferritina normal o elevado o adecuada tinción de hierro en la médula). Un esquema común consiste en iniciar con 100 a 150 U/kg por vía subcutánea tres veces por semana y, si no existe aumento en la hemoglobina a las seis a ocho semanas, aumentar la dosis a 300 U/kg tres veces por semana8 o cambiar a inyecciones diarias. Si aún no aumenta la hemoglobina después de 12 semanas, deberá suspenderse la eritropoyetina.
ANEMIA EN LA ENFERMEDAD RENAL GRAVE
Fisiopatología
La causa predominante de la anemia en la enfermedad renal es la deficiencia en la producción de eritropoyetina por los riñones enfermos. Si existe enfermedad renal inflamatoria subyacente, puede existir también un componente de anemia de enfermedad crónica. La anorexia y la mala ingesta de hierro, las muestras de sangre frecuentes y la pérdida de eritrocitos durante la hemodiálisis pueden producir deficiencia de hierro. la deficiencia de ácido fólico, el hiperesplenismo y el hiperpartiroidismo secundario con fibrosis de la médula3 puede promover también la anemia. Dependiendo de qué procesos causen la anemia, el frotis periférico puede mostrar fragmentación eritrocitaria o equinocitosis. La presencia de cuerpos de Heinz sugiere que ha ocurrido hemólisis oxidativa, quizá por oxidantes del líquido de hemodiálisis.
La toxicidad por aluminio también puede causar anemia en los pacientes en hemodiálisis. Este tipo de anemia se identificó inicialmente en pacientes con la llamada demencia por diálisis. Los niveles de aluminio muy altos en plasma parecen ser causados por contaminación con aluminio del líquido de diálisis o por absorción gastrointestinal de los geles de aluminio empleados para fijar el fosfato de la dieta. Experimentos in vitro han demostrado que el aluminio se une a los precursores eritroides unidad formadora de colonias eritroide (UFC-E) y unidad formadora del estallido eritroide (UFE-E).9
Tratamiento
La eritropoyetina es el tratamiento estándar para los pacientes con anemia por enfermedad renal. El tratamiento con eritropoyetina puede eliminar los requerimientos de transfusión en los pacientes con hemodiálisis y en pacientes con enfermedad renal progresiva que aún no requieren de hemodiálisis.10 El tratamiento mejora en forma sustancial su calidad de vida.11 Son poco frecuentes los efectos adversos, como la hipercalemia y la hipertensión. Es costumbre iniciar el tratamiento con 50 U/kg de eritropoyetina tres veces por semana, por vía intravenosa o subcutánea, y aumentar la dosis según sea necesario hasta alcanzar el nivel de hemoglobina deseado. En un estudio el tratamiento con desferroxamina, 30 mg/kg I.V., al final de cada sesión de diálisis, produjo mejoría importante en los pacientes con anemia causada por toxicidad por aluminio.12
DESNUTRICION
La desnutrición debida a anorexia nervosa o a deficiencias proteicas puede ocasionar anemia e incluso pancitopenia. Puede presentarse hemólisis [ver figura 2]. La biopsia de médula ósea es hipocelular, con un material gelatinoso característico que consiste en mucopolisacáridos ácidos. La anemia puede ocurrir a pesar de niveles normales de folato y cobalamina (vitamina B12) y puede corregirse con una nutrición adecuada.
|
|
|

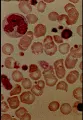

Aún es motivo de controversia si la hematopoyesis y, en especial la eritropoyesis, declinan con la edad.13,14 Sin embargo, por lo general los ancianos sanos tienen concentraciones normales de hemoglobina e índices eritrocitarios normales. Este dato fue confirmado por un estudio de pacientes de 84 y más años de edad.14 Por lo tanto, parece ser que la presencia de anemia en este grupo de pacientes amerita estudio y análisis.
MEDICAMENTOS Y OTROS AGENTES QUE AFECTAN LA PRODUCCION DE ERITROCITOS
La ingestión importante de alcohol (aguda o crónica) tiene importantes efectos hematológicos.15 La ingestión de alrededor de 80 g de alcohol (una botella de vino, seis tarros de cerveza o un tercio de una botella de whiskey) al día puede producir macrocitosis,16 vacuolización de proeritroblastos,17 trombocitopenia,18 disminución importante en los niveles séricos de folatos, y elevación en los niveles de hierro sérico. También puede afectar la respuesta de los reticulocitos a la administración de folato en pacientes con deficiencia de folato. La ingestión de alcohol por sí misma no produce un cuadro sideroblástico o megaloblástico.15 Los megaloblastos, macro-ovalocitos y neutrófilos polimorfonucleares (PMN) hipersegmentados suelen aparecer cuando existe una deficiencia concomitante de folatos. El abuso crónico de alcohol suele causar deficiencia de folato o hierro, enfermedad hepática severa, hemorragia gastrointestinal, hiperesplenismo y la anemia de la enfermedad crónica.
El cloranfenicol, además de producir anemia aplástica esporádica o idiosincrática (ver adelante), produce en forma esporádica una supresión eritroide reversible y dependiente de la dosis.19 El arsénico puede producir anemia al interferir con la producción de eritrocitos. También puede haber leucopenia y trombocitopenia, así como neuropatía periférica como resultado de la exposición a arsénico. La celularidad de la médula ósea puede estar aumentada, disminuida o normal.
La inmunoterapia para el cáncer a base de IL-2, sola o en combinación con células asesinas activadas por linfocinas (LAK, por sus siglas en inglés, n. el t.) ha producido diversos efectos hematológicos, y el más serio es la anemia dependiente de transfusión. Se piensa que la IL-2 suprime la hematopoyesis, quizá al causar la liberación de interferón gama, un conocido inhibidor de la misma. Otros efectos hematológicos producidos por la IL-2 y las células LAK incluyen trombocitopenia, linfopenia y eosinofilia.20
PADECIMIENTOS ENDOCRINOLOGICOS
En el hipotiroidismo aparece con frecuencia anemia secundaria a una menor producción de eritrocitos. La presencia de macrocitosis en un paciente hipotiroideo puede deberse a una deficiencia dietética de folatos concomitante o a una anemia perniciosa. La anemia ocasional que se presenta en el hipertiroidismo parece ser ocasionada por eritropoyesis ineficaz. La anemia leve que se relaciona con un panhipopituitarismo grave puede corregirse por medio del remplazo de hormonas suprarrenales, tiroideas y gonadales. Es bien conocido el efecto benéfico que tienen los andrógenos sobre la acción de la eritropoyetina. Tanto el hiperparatiroidismo primario como el secundario pueden ocasionar anemia; se ha observado aumento en el valor del hematocrito después de la realización de paratiroidectomía.
Defectos de la producción relacionados con aplasia o remplazo de la médula ósea
La presencia de pancitopenia o combinaciones diversas de anemia, neutropenia y trombocitopenia suelen indicar daño en la médula hematopoyética. Si la cavidad medular está infiltrada pero las células tronco pluripotenciales están intactas, suele desarrollarse hematopoyesis extramedular en los órganos de la hematopoyesis fetal (i.e., bazo, hígado y huesos distales).
Las citopenias combinadas de reciente inicio o un frotis de sangre leucoeritroblástico, que es la característica de la hematopoyesis extramedular [ver figura 2], constituyen indicación para la realización de una biopsia y aspirado de la médula ósea.
ANEMIA APLASICA
Definición
La pancitopenia (i.e., anemia, neutropenia y trombocitopenia) y una médula aplástica en el examen de la biopsia [ver figura 3] establecen la necesidad de una evaluación diagnóstica de anemia aplástica. La muestra de biopsia debe haberse tomado de un sitio no irradiado previamente. La anemia aplástica severa se define por los siguientes criterios: (1) una médula con celularidad de menos del 25 porciento del normal o con celularidad de menos del 50 porciento del normal en la que menos del 30 porciento de las células son hematopoyéticas, (2) dos de tres frotis de sangre periférica anormales (con índice corregido de reticulocitos menos del uno porciento o una cuenta absoluta de reticulocitos de menos de 40,000/µl, una cuenta de granulocitos de menos de 500 /µl o una cifra de plaquetas menor de 20,000/µl). Estos criterios han sido criticados como poco sensibles. Por ejemplo, en una serie de pacientes con diagnóstico de anemia aplástica, el índice corregido de reticulocitos menor de uno porciento se alcanzó con facilitad y no fue un criterio de severidad tan exacto como la cuenta de granulocitos menor de 500/µl.21 Otro grupo de investigadores notó que para interpretar sus datos tenían que identificar una cohorte de pacientes con anemia aplástica muy severa, con cuenta de granulocitos de menos de 200 /µl.22
|
|
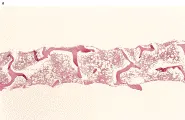
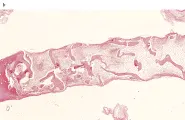
Diversos medicamentos y otros agentes producen de manera regular y predecible aplasia medular [ver tabla 2]. La radiación ionizante y los medicamentos quimioterápicos empleados en el tratamiento de padecimientos malignos e inmunológicos tienen la capacidad de destruir las células tronco hematopoyéticas. Ciertos medicamentos, como el cloranfenicol, pueden producir aplasia medular de una manera no dependiente de la dosis. El tratamiento con oro y la inhalación de vapores de solventes orgánicos (v.gr., benceno o cola) también pueden inducir anemia aplástica mortal.
|
|
|
En el dos a 10 porciento de los casos de hepatitis ocurre aplasia severa dos a tres meses después de un caso típico de enfermedad aguda, por lo general en varones jóvenes. Con frecuencia la hepatitis no es la causa obvia, y las pruebas para hepatitis A, B y C son negativas. La enfermedad puede ser fatal a menos que se trate en forma agresiva con agentes inmunosupresores o trasplante de médula ósea.23 Existe una alta incidencia de anemia aplástica después del trasplante hepático en pacientes con hepatitis no-A no-B severa.24
La aplasia puede ser parte de los pródromos de una leucemia de células pilosas, de una leucemia linfoblástica aguda o, rara vez, de una leucemia mieloblástica aguda, o puede desarrollarse durante el curso de una mielodisplasia.
Existen varias líneas de evidencia que apoyan la posibilidad de que los padecimientos inmunológicos puedan ocasionar aplasia. La aplasia medular se presenta en la enfermedad de injerto contra huésped (EICH),25 y en la fascitis difusa.26 El precondicionamiento inmunosupresor mejora las oportunidades de que se lleve a cabo un trasplante exitoso de médula singénica en pacientes con anemia aplástica,27 y el tratamiento inmunosupresor se está empleando con éxito en el manejo de la anemia aplástica idiopática.25,27 En la sangre de algunos pacientes con anemia aplástica aparecen linfocitos T supresores que impiden el crecimiento de las células progenitoras comprometidas conocidas como unidades formadoras de colonias de granulocitos y macrófagos (CFU-GM). Las células T supresoras pueden actuar produciendo interferón gama.27 El resultado de estos complejos mecanismos inmunes que incluyen a las células T supresoras es una profunda reducción en las células hematopoyéticas primitivas medidas tanto por ensayo de cultivo a largo plazo de células iniciales (CLP-CI) como por la capacidad para formar colonias secundarias a partir de las colonias que sobreviven a cinco semanas de cultivo medular.28
Diagnóstico
El diagnóstico de anemia aplástica requiere de un aspirado y biopsia medular [ver figura 3], así como una historia cuidadosa de la exposición a fármacos, infecciones y especialmente síntomas que sugieran enfermedades virales y pruebas serológicas para hepatitis, mononucleosis infeciosa, VIH y parvovirus [ver figura 4].
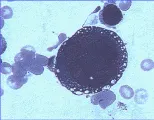
|
| Figura 4 |
| Pronormoblasto gigante |
El hiperesplenismo produce eliminación y secuestro esplénico rápido de las tres líneas celulares mieloides, pudiendo causar pancitopenia en padecimientos como la leucemia linfoblástica crónica, el lupus eritematoso generalizado o la esplenomegalia congestiva. Sin embargo, en tales casos la médula no está aplástica, sino que muestra hiperplasia de las líneas celulares involucradas. Debe realizarse medición de los eritrocitos CD59 para investigar hemoglobinuria paroxística nocturna.
También es importante determinar la gravedad de la enfermedad. Los casos graves tienen una tasa de remisión espontánea muy baja y una mortalidad del 70 porciento en un año. En comparación, un 80 porciento de los pacientes que tienen formas más leves de anemia aplástica sobreviven un año.21,22
Tratamiento
El tratamiento de las formas leves de la enfermedad incluye eliminar el agente agresor y proporcionar medidas de apoyo. Se espera que exista un número adecuado de células madre pluripotenciales como para repoblar la médula. En la anemia aplástica inducida por oro el dimercaprol, que facilita su excreción, puede ser de ayuda. Debido a que el oro causa la anemia aplástica por un efecto tóxico sobre las células tronco, se ha sugerido el uso de acetilcisteína intravenosa como agente quelante. Una teoría alterna afirma que el oro media una lesión inmunológica sobre las células tronco, lo que ha originado estudios sobre tratamiento inmunosupresor consistente en globulina antitimocito (GAT) y esteroides en dosis altas para pacientes con aplasia inducida por oro.29
Tratamiento de sostén Las hemorragias trombocitopénicas deberán manejarse mediante el empleo de transfusiones plaquetarias según se requiera para controlarlas. El remplazo muy liberal de plaquetas puede causar alosensibilización y complicar un futuro trasplante alogénico de médula ósea. Se administran transfusiones de eritrocitos según se requiera para controlar la anemia e hipoxia. Se han administrado factores de crecimiento hematopoyético como el factor estimulador de colonias de granulocitos (FEC-G) y el factor estimulador de colonias de granulocitos y macrófagos (FEC-GM) para aumentar la cuenta absoluta de neutrófilos y ayudar a combatir las infecciones. Sin embargo, la mayoría de las veces son ineficaces, principalmente por la grave deficiencia de células precursoras, que son el sitio blanco de su acción.30 Por lo general es preferible establecer el tratamiento definitivo, trasplante alogénico de médula ósea, de un hermano donador complatible, o tratamiento inmunosupresor.31
La elección del tratamiento adecuado en la anemia aplástica severa es crítico, y varios tratamientos se están revalorando en la actualidad. La mayoría de los expertos recomiendan el trasplante alogénico o singénico de médula ósea en pacientes menores de 20 años si existe un hermano donador compatible. Aunque existen varios riesgos, incluyendo EICH crónica y disfunción orgánica causada por el programa de acondicionamiento,30 el 50 a 80 porciento de estos pacientes pueden curarse, con una incidencia muy baja de trastornos clonales posteriores.32 El trasplante alogénico de médula ósea con programas de acondicionamiento que usan ciclofosfamida y GAT producen una supervivencia actuarial de 69 porciento a 15 años.32
En los pacientes mayores de 45 años se piensa que el impacto de la EICH es demasiado severa y que estos pacientes deben recibir mejor tratamiento inmunosupresor.30,32 Tres formas de inmunosupresión han demostrado producir remisión parcial en la anemia aplástica.30-32 La GAT produjo remisión duradera en alrededor de la mitad de los pacientes en un estudio aleatorio.31 Se ha comprobado de los esteroides en dosis altas aumentan las cuentas hemáticas en alrededor del 40 porciento de los pacientes tratados, y se ha demostrado que la ciclosporina también es benéfica.31 (Los andrógenos como la oximetolona pueden tener algún beneficio en el tratamiento de la anemia aplástica severa, pero no se administran solos.30,32) Aunque cada uno de estos agentes puede usarse en forma individual o consecutiva en el tratamiento de la anemia aplástica, un estudio controlado sugiere que los resultados son mejores cuando se usan los tres en forma simultánea.30,31 La combinación de GAT, un esteroide y ciclosporina permitió una supervivencia actuarial de 62 porciento a 36 meses, y los primeros signos de respuesta se presentaron a las 4 semanas; el tiempo promedio para la remisión fue de 60 a 82 días.31
La dosis eficaz de GAT depende del origen de la preparación (si es de caballo o conejo) y el modo de fabricación (si se produce como suspensión líquida o se administra por peso). El efecto adverso tóxico de la GAT es la enfermedad del suero, que suele controlarse con esteroides. La recomendación actual, basada en la GAT de caballo habitualmente disponible en los Estados Unidos,30,31 consiste en administrarla en una dosis de 40 mg/kg/día durante cuatro días. La GAT se diluye primero en 500 ml a 1 L de solución fisiológica y se infunde durante un periodo de cuatro a cinco horas a través de una vía intravenosa equipada con un filtro para microagregados. Se agregan 40 mg de metilprednisolona al frasco de infusión y se administra metilprednisolona por vía oral en cuatro dosis divididas hasta alcanzar una dosis de1 mg/kg/día. Se continúa el tratamiento con metilprednisolona hasta una dosis máxima de 56 mg/día en los pacientes que sufren calosfríos, fiebre o urticaria. En los enfermos en los que se desarrolla el cuadro florido de enfermedad del suero equino, la dosis de metilprednisolona se aumenta hasta un máximo de 64 mg/día. La reducción gradual de esteroides comienza al final de la segunda semana, y se suspenden en 30 días. Debido a que la GAT puede disminuir la cuenta plaquetaria, se administran transfusiones de plaquetas según se requiera para mantenerlas por arriba de 20,000/µl.
La ciclosporina se administra por vía oral en una dosis de 10 a 12 mg/kg/día en dos dosis divididas, con objeto de alcanzar niveles sanguíneos de 500 a 800 ng/ml o una concentración sérica de 100 a 200 ng/ml. Después de 29 días la dosis de ciclosporina puede disminuirse hasta una concentración sanguínea total de 200 a 500 ng/ml.30,31 La ciclosporina se continúa por lo menos durante seis meses; puede causar hipertensión, toxicidad renal, hipomagnesemia, vitiligo, temblores, hipertricosis, propensión a neumonía por Pneumocystis carinii (NPC) e hiperplasia gingival.30,31 En un estudio se administró 300 mg de pentamidina aerosolizada cada cuatro semanas como profilaxis para la NPC.31
A diferencia de los pacientes sometidos a trasplante alogénico de médula ósea, los que responden al tratamiento inmunosupresor no están realmente curados. Muchos continúan teniendo citopenias moderadas,33 el 20 a 36 porciento de estos pacientes recae con anemia aplástica,30,31,33 y hasta el 20 a 36 porciento desarrollará trastornos clonales, como hemoglobinuria paroxística nocturna, síndrome mielodisplásico e incluso leucemia aguda.30,31 Existe también mayor riesgo de desarrollar tumores sólidos después del tratamiento de la anemia aplástica, pero es el mismo para los pacientes que reciben tratamiento inmunosupresor o trasplante alogénico de médula ósea.34 De los enfermos que recaen después de responder en forma inicial al tratamiento inmunosupresor, más del 50 porciento responde por segunda ocasión.30,31
Debido a que la inmunosupresión no cura la anemia aplástica y se asocia con tasas significativas de recaída y evolución a trastornos clonales, un aspecto importante es cómo tratar a los pacientes entre 20 y 40 años de edad. Algunos autores proponen que debe considerarse el trasplante alogénico de médula ósea en este grupo de edad, en especial porque los nuevos programas de acondicionamieno parecen ser capaces de reducir la gravedad de la EICH.32,35 A falta de resultados de estudios clínicos controlados que comparen el tratamiento inmunosupresor de tres agentes con el trsplante alogénico de médula ósea de un hermano donador compatible, los médicos deben individualizar el tratamiento para los pacientes con edades entre 20 y 40 años. Una persona de 38 años de edad que realiza ejercicios en forma regular y no tiene factores de riesgo identificables puede ser un candidato para trasplante de médula ósea, mientras que alguien de la misma edad con múltiples factores de riesgo, como enfermedad cardiopulmonar por tabaquismo, no será un buen candidato. El trasplante alogénico de donador no relacionado compatible ha producido tasas de supervivencia a dos años de solo 29 porciento por EICH severa.30 Un trabajo inquietante evaluó a 10 pacientes con anemia aplástica severa que fueron tratados con dosis altas de ciclofosfamida IV (45 mg/kg/día) durante cuatro días consecutivos.36 Algunos pacientes recibieron también ciclosporina. Solo se administró un curso de ciclofosfamida. Siete de los 10 pacientes tuvo una respuesta hematológica completa y seis aún están vivos después de un seguimiento promedio de 10.8 años (rango de 7.3 a 17.8 años). Es claro que este manejo debe ser probado y evaluado en forma más profunda.
APLASIA PURA DE ERITROCITOS
Definición
En la aplasia pura de eritrocitos adquirida (APEA), la reticulocitopenia es profunda y casi no existen precursores eritroides en la médula ósea. Sin embargo, los elementos mieloides y megacariocíticos medulares están conservados y las cuentas de leucocitos y plaquetas en sangre periférica son normales. Estos datos indican que, en este desorden, se afectan los precursores eritroides, pero no las células tronco pluripotenciales.
Fisiopatología
Se piensa que en la APEA la eritropoyesis está inhibida principalmente por mecanismos inmunes, incluyendo autoanticuerpos y supresión de los progenitores eritroides mediada por células T en una fase posterior a la fase de diferenciación eritroide de la UFC-E y antes de la formación de los proeritroblastos.37 Las células T, en especial los linfocitos granulares grandes (LGG-T), pueden participar en la supresión de la eritropoyesis, y en algunos casos existió evidencia de que la supresión era causada por células clonales T.38 Se ha descrito también inhibición de la eritropoyetina por autoanticuerpos, aunque parece muy poco común.39 Es probable que existan otros dos mecanismos que causen APEA: el ataque específico a los precursores eritroides por el parvovirus B-19 (un reporte reciente indica que el 14 porciento de los casos fueron causados por este virus40) y una alteración hematopoyética clonal subyacente que puede ser un pródromo de un síndrome mielodisplásico.39 La APEA puede clasificarse con base en su etiología [ver tabla 3].
|
|
|
Diagnóstico
La evaluación diagnóstica de la APEA suele incluir una tomografía computada de tórax por la posibilidad de timoma, el análisis inmunofenotípico de los linfocitos de sangre circulante o de médula ósea para identificar proliferación de LGG , citogenética de la médula para estudiar la posibilidad de síndromes mielodisplásicos y pruebas de anticuerpos contra parvovirus.40
Ocurre eritroblastopenia en una pequeña proporción de pacientes con anemia hemolítica autoinmune, que puede ser causada por autoanticuerpos dirigidos contra normoblastos en maduración. El tratamiento de la infección por VIH con zidovudina (también conocida como AZT) produce anemia en virtualmente todos los pacientes, caracterizada por macrocitosis significativa.41 La hipoplasia eritroide moderada es la causa habitual de esta anemia, que puede progresar a APEA.
La APEA puede complicar al trasplante alogénico de médula ósea con incompatibilidad ABO; en estos casos el receptor del suero continúa expresando isohemaglutininas anti-A o anti-B contra el antígeno A o B del donador expresado en la superficie de los progenitores eritroides.42 En la APEA del embarazo los anticuerpos contra las UFE-E suelen desaparecer después del parto, coincidiendo con la remisión clínica.43
La infección por parvovirus es la causa de las crisis aplásticas temporales que ocurren en los pacientes que tienen trastornos hemolíticos severos. La médula de los pacientes con estos trastornos debe compensar la hemólisis periférica aumentando su preducción hasta siete veces, por lo que suele presentar una intensa hiperplasia eritroide. Aunque la infección por parvovirus puede afectar a todas las células precursoras, las de tipo eritroide son las más dañadas.40 Por lo general ocurre recuperación espontánea en una a dos semanas después del inicio de la infección. Una característica diagnóstica de la infección por parvovirus es la aparición de pronormoblastos gigantes en la médula [ver figura 4]. La asociación de la APEA con proliferación de LGG y leucemia cada vez se reconoce más. El uso rutinario de estudios de rearreglos de genes del receptor de la célula T en una serie mostró que nueve de 14 pacientes tuvo un trastorno clonal de LGG.44 Al parecer estas células median directamente la inhibición de la eritropoyesis.40,44
En quizá hasta el 20 porciento de los casos la APEA puede ser el pródromo de un síndrome mielodisplásico o incluso de leucemia mieloide aguda.40,42 La distinción en el momento del diagnóstico puede ser difícil a menos que se detecte una alteración citogenética mielodisplásica típica durante el estudio de la médula ósea.
Tratamiento
El tratamiento de la APEA depende de la causa identificada. Deben suspenderse los medicamentos agresores y, si existe un timoma, éste debe extirparse,42 lo que causa mejoría en alrededor de un tercio de los pacientes.42 Cuando la cirugía es imposible o el timoma es resistente a la quimioterapia, debe considerarse un curso de prednisona combinado con ocreótido, un análogo de somatostatina que se une a los timomas y puede inhibir la función de las células tímicas inmunes.45
En los casos de aplasia primaria de eritrocitos el tratamiento puede iniciarse con prednisona, 60 mg por vía oral diario en dosis divididas, continuando este esquema durante uno a tres meses.40 Si el paciente no responde, lo que se valora por los reticulocitos, se agregará ciclofosfamida o azatioprina, en dosis de 2 a 3 mg/kg/día por vía oral. Los pacientes con alteraciones citogenéticas de la médula ósea sugestivas de síndrome mielodisplásico responden mal.40,44 Algunos pacientes refractarios a otras formas de tratamiento han tenido buena respuesta a la infusión de IgG I.V., siendo la dosis estándar de 0.4 g/kg/día durante cinco días.46 Este tratamiento da buen resultado en los pacientes en los que la infección por parvovirus es la causa de la APEA.40 Para los pacientes con SIDA, infección por parvorvirus y APEA, puede ser necesario continuar la administración de IgG I.V. En un caso se administró IgG I.V. en dosis de 0.4 g/kg cada 2 a 3 semanas durante más de 50 meses.47 Los pacientes con proliferación de LGG como causa subyacente responden bien a la ciclofosfamida.44,48 Por lo general, las dosis bajas de ciclofosfamida (50 a 100 mg v.o.) administradas diario durante 3 a 6 meses son suficientes para producir remisión, que se asocia en ocasiones con desaparición de la proliferación de LGG.44,47 Cuando existe mala respuesta los pacientes suelen mejorar con ciclosporina oral.44,47
Se ha demostrado que la ciclosporina, en dosis de 12 mg/kg/día80 produce respuestas del orden del 65 porciento, incluso en pacientes que no responden a los esteroides, plasmaféresis, ciclofosfamida y azatioprina.42,49 Se ha administrado GAT para la APEA en pacientes refractarios, en forma semejante a como se usa en la anemia aplástica (40 mg/kg/día I.V. durante cuatro días).42
Defectos en la producción relacionados con hiperplasia eritroide en la médula y eritropoyesis ineficaz
Puede presentarse anemia con reticulocitopenia a pesar de que en la médula exista una hiperplasia eritroide intensa. Esta situación paradójica se presenta cuando existe eritropoyesis ineficaz o hemólisis intramedular. El defecto primario puede afectar en forma generalizada la línea eritroide, o bien subpoblaciones específicas de precursores eritroides en desarrollo. Algunas de estas subpoblaciones escapan a la muerte en la médula, pero su progenie ha sufrido un daño tan grave que es retirada rápidamente de la circulación, dando de esta manera la impresión de que existe hemólisis periférica. Se pueden observar otros signos de eritropoyesis ineficaz, incluyendo ictericia, un nivel de urobilinógeno fecal marcadamente elevado, un nivel muy alto de deshidrogenasa láctica en suero, una saturación de la capacidad de fijación de hierro sérico de 75 a 90 porciento y, ocasionalmente, depósito hepático de hierro en un patrón idéntico al que se ve en la hemocromatosis. El cuadro ferrocinético típico muestra depuración rápida del hierro plasmático, lo que indica intensa actividad de las células eritroides precursoras. Sin embargo, la llegada de eritrocitos marcados a la circulación periférica está muy disminuida, lo que sugiere que los precursores son destruidos por hemólisis intramedular.
Entre las enfermedades que producen eritropoyesis ineficaz importante se incluyen las anemias megaloblásticas, las anemias sideroblásticas, la talasemia, los síndromes mielodisplásicos y la metaplasia mieloide agnogénica.
ANEMIAS MEGALOBLASTICAS
Definición
Las anemias megaloblásticas se caracterizan por la presencia de macrocitosis; el frotis periférico muestra macroovalocitos, eritrocitos con forma de cola de pescado, neutrófilos hipersegmentados, y ocasionalmente eritrocitos nucleados [ver figura 2]. El encontrar un neutrófilo polimorfonuclear con seis lóbulos o que un cinco porciento de los polimorfonucleares tengan cinco lóbulos constituye una fuerte evidencia de anemia megaloblástica. En los casos graves se encuentra también granulocitopenia y trombocitopenia. La médula ósea muestra hiperplasia eritroide megaloblástica y metamielocitos gigantes.50 Si coexiste una deficiencia grave de hierro estará bloqueada la expresión morfológica completa de la megaloblastosis, aunque la presencia de metamielocitos gigantes en la médula y de polimorfonucleares hipersegmentados en la sangre periférica no se verá afectada.
Fisiopatología
Las anemias megaloblásticas son causadas por deficiencia de cobalamina o ácido fólico, por medicamentos que interfieren con la síntesis del ADN, con la absorción o el metabolismo de la cobalamina, y por trastornos genéticos que interfieren con el metabolismo del ADN o con la absorción o distribución de la cobalamina.
La eritropoyesis megaloblástica se caracteriza por síntesis de ADN defectuosa y detención en la fase G2, con alteraciones en la maduración celular y aparición de células que no sintetizan ADN que tienen un contenido anómalo del mismo. Esto ocasiona maduración asincrónica entre el núcleo y el citoplasma.50 La producción de ARN y la síntesis de proteínas continúa y, por lo tanto, se producen células más grandes, o megaloblastos. La eritropoyesis ineficaz es causada quizá por incremento de la apoptosis.51,52 Se presume que defectos semejantes en la síntesis de ADN caracterizan las alteraciones mucosas del estómago y la lengua. En la línea granulocítica la presencia de metamielocitos gigantes representa granulopoyesis ineficaz.50
Papel del ácido fólico y de la cobalamina Las interacciones entre el ácido fólico y la cobalamina son críticas en el metabolismo de las unidades de carbono único, principalmente de los análogos de metileno y de formil, que tienen un papel clave en la síntesis de ADN y las purinas [ver figura 5a]. Existen dos coenzimas principales de la cobalamina, la adenosilcobalamina y la metilcobalamina. La adenosilcobalamina es la coenzima para la metilmalonil-CoA mutasa, que cataliza un paso en el catabolismo del ácido propiónico [ver figura 5b].53 La metilcobalamina es la coenzima de la metionina sintetasa, que funciona como una metiltransferasa en la reacción que convierte 5-metiltetrahidrofolato (CH3-THF1) a tetrahidrofolato (THF1) [ver figura 5a].53 La cobalamina y el folato [ver figura 6] se combinan en la reacción de la sintetasa de metionina [ver figura 5a], en la que el grupo metil del CH3-THF1 es transferido a la cobalamina para formar metilcobalamina. La metilcobalamina transfiere entonces su grupo metil a la homocisteína para formar metionina. El monoglutamato tetrahidrofolato (THF1) que se forma por esta reacción es poliglutamado por medio de la enzima folilpoliglutamato sintetasa, y un grupo metilo se añade a él por medio de la serina-glicina metiltransferasa para formar 5,10 metileno THFn. El 5,10-metileno THFn proporciona su metileno para convertir el desoxiuridilato a timidilato, un paso clave en la síntesis del ADN. El 5-10, metileno THFn puede también convertirse en forma directa en CH3-THFn por medio de la enzima 5,10-metileno tetrahidrofolato reductasa, lo que hace que se disponga de su grupo metil para la sintetasa de metionina dependiente de cobalamina. El formil THFn (también llamado leucovorín, ácido folínico o factor citrovorum) también tiene un papel importante en la síntesis de purinas y, por lo tanto, en el metabolismo del ADN. Puede ser generado por oxidación del 5-10 metileno THFn o directamente del THFn por acción de la enzima formil THF sintetasa, en donde la metionina proporciona el grupo metil [ver figura 5a].53 Cuando existe deficiencia de cobalamina, el metileno THF1 no puede transferir su grupo metil a la cobalamina, por lo que no se libera para poliglutamarse por medio de la poliglutamato sintetasa [ver figura 5a]. Se requiere la forma poliglutamada para la síntesis de metileno THFn o de formil THFn, por lo que se bloquea la síntesis de ADN y de purinas. Esta es la hipótesis del atrapamiento del metilfolato y la apoya el encontrar incremento en los niveles de CH3-THF1 en el plasma de pacientes con deficiencia de cobalamina. Una explicación alternativa es la hipótesis de ausencia de grupos metil, en la que la deficiencia de cobalamina altera la generación de metionina, que entonces no puede proporcionar los grupos metilo necesarios para que la enzima formil THFn sintetasa produzca formil THFn.
|
|
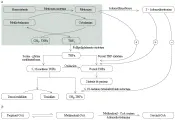
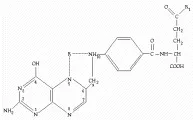
Otros aspectos del metabolismo del folato y la cobalamina
Debido a que ni el ácido fólico ni la cobalamina se producen en cantidades adecuadas en los humanos, ambos deben ser absorbidos a partir de los alimentos. En particular, la cobalamina deriva de fuentes microbianas y se ingiere a través de la carne o los huevos.
La mayor parte del ácido fólico presente en la dieta se encuentra en la forma de poliglutamato y es absorbido en la mucosa intestinal. La absorción del ácido fólico marcado radiactivamente se acerca al 80 porciento de una dosis de 200 µg.50,53 En el suero una pequeña cantidad de folato se fija a una proteína específica, y aún se desconoce el papel de esta proteína fijadora en el metabolismo del folato. Los niveles séricos de folato parecen mantenerse gracias al folato que se absorbe de los alimentos. Se ha observado circulación enterohepática del folato, en la que el ácido fólico que pasa hacia la bilis y al intestino delgado se absorbe de manera cuantitativa. En modelos animales la administración de etanol bloquea la entrada de folato hacia la bilis. Este efecto podría, en parte, ser la causa de la rápida caída en los niveles de folato sérico que se observa ocho horas después del consumo de alcohol. Una disminución similar en los niveles séricos de folato se observa después de la ingestión de fenitoína. Los requerimientos diarios de cobalamina son de alrededor de 1 µg, y la cantidad proporcionada por la dieta occidental habitual, que es rica en productos animales, varía de 5 a 15 µg.55
Las proteínas R son un tipo de glucoproteínas fijadoras de cobalamina producidas por granulocitos y otros tejidos. El factor intrínseco (FI) es una glucoproteína de 45 kd secretada por las células parietales gástricas que es muy específica para la cobalamina íntegra. El complejo proteína R-cobalamina no se une a los receptores ileales, por lo que no se absorbe. En el estómago la cobalamina se une preferentemente a las proteínas R más que al FI,50,53,55 por lo tanto, es el complejo proteína R-cobalamina fisiológicamente inactivo el que se libera al duodeno. Sin embargo, en el duodeno y el intestino delgado las proteasas pancreáticas, junto con la pepsina, degradan las proteínas R, liberando la cobalamina y permitiendo su unión al FI. Por lo tanto, la atrofia gástrica y la insuficiencia pancreática contribuyen a la malabsorción de la cobalamina.53,55 El complejo FI-cobalamina, en presencia de Ca2+ y un pH mayor de 5.4, se une en forma específica a un número limitado de sitios en las microvellosidades de las células de la mucosa en la porción terminal del íleo, en donde tiene lugar la absorción [ver figura 7].53,55
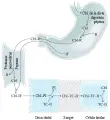
|
| Figura 7 |
| Asimilación de la cobalamina. |
En el plasma la mayor parte de la cobalamina se une a las proteínas R transcobalaminas I y III (TC-I y TC-III), que no son muy importantes desde el punto de vista fisiológico, que se sintetizan en forma parcial en los precursores de granulocitos y que pueden servir como almacén de cobalamina. Estas se saturan en alrededor del 70 porciento con cobalamina y sus análogos.56 La proteína transportadora de importancia fisiológica es la transcobalamina-II (TC-II), que tiene considerable especificidad para la cobalamina y que se satura en solo el cinco a 10 porciento. Existen receptores para el complejo TC-II-cobalamina en muchas membranas celulares. La TC-II fija alrededor del 90 porciento de una dosis recién inyectada de cobalamina, y el complejo es depurado con rapidez con una vida media de seis a nueve minutos.55,56 En la deficiencia congénita de TC-II, que causa anemia megaloblástica severa, tanto el transporte de cobalamina en plasma como su absorción están deteriorados. La menor absorción de cobalamina en esos casos implica que la TC-II tiene alguna función dentro del enterocito ileal, en donde la cobalamina es transferida del FI a la TC-II.
La elevación de los niveles de cobalamina que se observa en pacientes con leucemia granulocítica crónica o granulocitosis significativa es causada por aumentos en la TC-I y, en menor grado, en la TC-III.
DEFICIENCIA DE COBALAMINA
Además de la anemia macrocítica megaloblástica, el paciente con deficiencia de cobalamina puede tener atrofia de las papilas linguales y glositis. La neuropatía es la característica inicial en alrededor del 12 porciento de los pacientes con anemia perniciosa sin anemia concomitante.57 Los pacientes con deficiencia grave de cobalamina se quejan al inicio de parestesias. El sentido del tacto y el de la temperatura pueden estar afectados en un grado mínimo, con disminución en los reflejos y en la percepción de las vibraciones de alta frecuencia. La enfermedad puede progresar y afectar las porciones dorsales de la médula, lo que ocasiona ataxia y debilidad, y en la exploración física se encuentra una marcha con una base amplia, signo de Romberg, y una pérdida del sentido de la posición y de la vibración (en especial cuando se explora este último con un diapasón de 256 Hz). Si no se detecta y se trata el padecimiento, se extenderá hasta afectar las columnas laterales. Esto ocasionará incapacidad para caminar debido a la presencia de debilidad y espasticidad, las cuales pueden confirmarse por los datos de clonus sostenido, hiperreflexia y signo de Babinsky. Debido a que tanto los nervios periféricos como las columnas laterales y dorsales se encuentran afectadas, en algunas ocasiones estas manifestaciones neurológicas son conocidas como degeneración subaguda combinada o enfermedad sistémica combinada subaguda. La afección de la memoria y la depresión pueden ser manifestaciones prominentes.57 La deficiencia de cobalamina parece ser la causa de varios trastornos neuropsiquiátricos, manifestados por síntomas como parestesias, ataxia, debilidad de las extremidades, alteraciones de la marcha y defectos de la memoria. Otros síntomas reportados como manifestaciones de deficiencia de cobalamina incluyen alucinaciones y cambios en la personalidad y el estado de ánimo.56 Sin embargo, es difícil relacionar estos síntomas con las lesiones de la médula espinal que ocurren en los estados de deficiencia de cobalamina.
Los investigadores han tratado de determinar si un defecto en la síntesis de metionina o alguna alteración en el metabolismo del ácido propiónico son la causa de la neuropatía asociada con la deficiencia de cobalamina [ver figura 5b], pero el mecanismo exacto sigue sin aclararse. Sin embargo, dos observaciones apuntan hacia la alteración en la sintetasa de metionina como causa de la neuropatía. Primero, la administración de N2O a monos produce una neuropatía que es bloqueada por el tratamiento simultáneo con metionina. Segundo, un paciente con alteración congénita en la actividad de la sintetasa de metionina tuvo una neuropatía caracterizada por alteraciones en la marcha, torpeza y parestesias.50,57
Diagnóstico
El cuadro clínico que en el pasado sugería el diagnóstico de deficiencia de cobalamina ha cambiado en forma significativa. El cuadro clínico clásico de la deficiencia de cobalamina consiste en anemia macrocítica con glositis o neuropatía. El aspirado y biopsia de la médula ósea revelan típicamente hiperplasia eritroide megaloblástica con metamielocitos gigantes.50 La hipercelularidad puede ser tan importante y los megaloblastos tan inmaduros que los clínicos en ocasiones hacen el diagnóstico equívoco de leucemia.50,58 La introducción de los contadores electrónicos de células que pueden medir con exactitud el volumen corpuscular promedio (VCM) ha permitido la identificación de grados más sutiles de macrocitosis (VCM > 100 fl) que podrían no ser aparentes en el examen del frotis de sangre periférica. El dato de macrocitosis justifica la medición de los niveles de cobalamina y folato, pero es importante recordar que la macrocitosis puede ser causada por otras condiciones, incluyendo deficiencia de folatos, enfermedad hepática, abuso de alcohol, reticulocitosis e ingestión de medicamentos como los antimetabolitos, agentes alquilantes y zidovudina.41,59 Por otro lado, la macrocitosis de la anemia megaloblástica puede ser enmascarada por condiciones que pueden producir hipocromia y eritrocitos microcíticos, como la talasemia. Esta posibilidad debe considerarse en especial en pacientes de raza negra, entre los cuales existe una frecuencia alta de talasemia-alfa (alrededor del 30 porciento). La talasemia-alfa concomitante puede minimizar la macrocitosis de la anemia perniciosa.59 La anemia de la enfermedad crónica o la que es ocasionada por pérdida de sangre y deficiencia de hierro puede disminuir el grado de macrocitosis, pero no afecta la hipersegmentación de los neutrófilos. En un estudio se descubrió deficiencia de hierro en el 20 porciento de 121 pacientes con anemia perniciosa.60 En otro estudio, el 19 porciento de los pacientes con anemia perniciosa no tuvieron anemia, y el 33 porciento no tuvieron macrocitosis.61
La incidencia de cáncer gástrico, tumores carcinoides gástricos, cáncer esofágico,62 y quizá cáncer de colon y recto63 es mayor en pacientes con anemia perniciosa que en los sujetos controles. La anemia perniciosa ocurre en asociación con otros padecimientos autoimunes. Se observaron trastornos tiroideos autoinmunes en el 24 porciento de 162 pacientes con anemia perniciosa.64 Por lo tanto, la evaluación clínica del estado tiroideo debe ser parte del estudio diagnóstico en la anemia perniciosa.
Debe considerarse el diagnóstico de deficiencia de cobalamina en los pacientes que tengan trastornos neuropsiquiátricos, incluso en ausencia de anemia o macrocitosis. Los peligros de no pensar en el diagnóstico correcto son muchos. Pueden no existir anemia, macrocitosis y alteraciones en el frotis periférico, incluso en pacientes con concentraciones de cobalamina en suero entre 100 y 199 pg/ml. Por lo tanto, las indicaciones para considerar un diagnóstico de deficiencia de cobalamina han cambiado desde un cuadro clínico más o menos especifico hasta uno más general que incluye neuropatía y ciertas formas poco descritas aún de disfunción cerebral.57 Es posible que estas formas de presentación ocasionen mayor frecuencia de solicitudes para medir la concentración de cobalamina en suero. Las pruebas disponibles en la actualidad en los laboratorios clínicos proporcionan mediciones mucho más exactas que antes. Debe hacerse en forma simultánea la medición del nivel de folatos en los eritrocitos, ya que el metabolismo de la cobalamina se relaciona con el del folato [ver figura 5a]. Debido a que la deficiencia de cobalamina causa aumento en los niveles de homocisteína y ácido metilmalónico, se ha propuesto que la medición de estos produtos ayuda a definir el estado de deficiencia.59,65,66
Durante el embarazo y en los estados de deficiencia de folatos ocurren concentraciones de cobalamina bajas en forma falsa.59 Hasta hace poco la reducción en la cobalamina sérica no se consideraba significativa sino hasta que el valor era muy bajo (i.e., < 150 pg/ml). Sin embargo, en la actualidad es claro que los pacientes con concentraciones de cobalamina tan altas como de 250 pg/ml, y quizá incluso mayores, pueden tener deficiencia de cobalamina.59,61,65 Por fortuna, el encontrar macro-ovalocitos o PMN hipersegmentados en el frotis de sangre periférica sigue siendo un indicador sensible de la presencia de deficiencia de cobalamina.
Los pacientes que tienen una concentración sérica de cobalamina muy baja y anemia macrocítica deben someterse a un ensayo de tratamiento con cobalamina parenteral. El diagnóstico de deficiencia de cobalamina se confirma si el tratamiento produce reticulocitosis en tres o cuatro días y se asocia con aumento en la concentración de hemoglobina y reducción del VCM. La prueba de supresión con desoxiuridina (dU) es muy confiable para identificar a los pacientes con deficiencia de cobalamina. Esta prueba, que no está disponible en todos los laboratorios, se ha usado para investigar la anemia megaloblástica y ha proporcionado mucha de la información actual sobre las funciones de la cobalamina y el folato. Cuando las células de la médula ósea normal se preincuban con desoxiuridina fría, la incorporación subsecuente de la timidina-H3 al ADN se reduce mucho. Esto se debe a que las células normales de la médula osea convierten la dU a desoxiuridilato, que después es metilado a timidilato [ver figura 5a], que a su vez se convierte en trifosfato de desoxitimidina (dTTP), un sustrato para la síntesis de ADN. Por lo tanto, la preincubación con dU aumenta mucho el reservorio celular de dTPP, de modo que la conversión de timidina-H3 en ADN se diluye y disminuye. Sin embargo, en las células de la médula deficientes en cobalamina y folato la deficiencia interfiere con la metilación de la dU a timidilato [ver figura 5a], de modo que la preincubación con dU no suprime la incorporación de timidina-H3 al ADN.50,66
Una vez que se hace el diagnóstico de deficiencia de cobalamina deberá buscarse la causa de la misma [ver tabla 4]. La fisiopatología de la anemia perniciosa parece incluir un fenómeno autoinmune, principalmente gastritis autoinmune y dos tipos de anticuerpos contra el factor intrínseco. Uno de estos anticuerpos anti-FI bloquea la unión de la cobalamina al FI, y el otro bloquea la fijación del complejo FI-cobalamina a los receptores ileales.67 Clínicamente, se encuentran anticuerpos específicos anti-FI en el 70 porciento de los pacientes con anemia perniciosa.
|
|
|
Existen otras causas de deficiencia de cobalamina, En los vegetarianos se desarrolla una anemia megaloblástica severa como resultado de la ingesta muy baja de cobalamina. También se han observado deficiencias de folato y hierro. Los infantes de madres vegetarianas pueden tener deficiencia severa de cobalamina, en especial si reciben leche materna.56 La deficiencia de cobalamina es sorprendentemente común en países en vías de desarrollo, en donde las personas no son vegetarianos estrictos.56 La incidencia es especialmente alta en mujeres embarazadas y niños prescolares.56 La cirugía gástrica, en la que se eliminan el FI, la pepsina y componentes secretores de ácido, suele causar deficiencia de cobalamina (en el 31 porciento de los pacientes según un estudio69). Los pacientes que han sido sometidos a cirugía gastrica deben ser vigilados en forma rutinaria midiendo los niveles de cobalamina u homocisteína en plasma, recibiendo suplementos con cobalamina de por vida si los niveles están bajos.69
El método general de estudio para el diagnóstico diferencial de la deficienfia de cobalamina se apoya en la prueba de Schilling, la cual mide la absorción de cobalamina marcada con cobalto 57 (57Co). Después de que se administra una dosis de 1 µg de cobalamina marcada por vía oral , se administra una dosis de 1,000 µg de cobalamina no marcada por vía parenteral. La dosis parenteral satura a las transcobalaminas I, II y III, de tal manera que una porción importante del material absorbido es excretado por la orina. Si la cantidad de cobalamina marcada con 57Co que se mide en una muestra de orina de 24 horas debidamente recolectada es menor del 10 porciento de la cantidad administrada por vía oral, existe una absorción deficiente de cobalamina.
Se puede repetir la prueba de Schilling, agregando factor intrínseco oral suplementario. En la anemia perniciosa la adición de FI deberá corregir los resultados anormales de la prueba a menos que el FI administrado no sea completamente activo, el paciente secrete anticuerpos contra el FI o esté tomando medicamentos que interfieran con la absorción de cobalamina. La deficiencia prolongada de cobalamina afecta la captación a nivel del íleon, lo que altera los resultados de las pruebas. Por lo tanto, algunas personas no realizan la prueba de Schilling con FI hasta que el paciente ha recibido tratamiento con cobalamina por varias semanas.
Existen cada vez más reportes de pacientes con anemia perniciosa demostrada que tienen concentraciones séricas de cobalamina bajas o limítrofes pero prueba de Schilling normal. Parece ser que al progresar la lesión de atrofia gástrica de la anemia perniciosa, la capacidad de producir ácido-pepsina se pierde antes de que desaparezca toda la actividad del FI. Por lo tanto, en pacientes que se encuentran en una fase temprana de la enfermedad, puede existir suficiente FI para fijar la cobalamina libre administrada por vía oral en la prueba de Schilling y dar un resultado normal. Puede demostrarse malabsorción de cobalamina en estos pacientes por medio de una prueba denominada de Schilling con alimento, que se realiza con huevos de gallinas que han sido inyectados con cobalamina radioactiva.70 Esta prueba revela si no existe suficiente ácido-pepsina para separar el complejo cobalamina-enzima y liberar la cobalamina libre para que se una al FI. Sin embargo, esta prueba de Schilling con alimento no está disponible con facilidad, por lo que si se sospecha anemia perniciosa en un paciente que aparentemente tiene una prueba de Schilling normal, deben realizarse otros estudios para confirmar el diagnóstico, incluyendo examinar la morfología de las células sanguíneas, medir anticuerpos anti-FI o los niveles séricos de homocisteína o ácido metilmalónico, o realizar un estudio terapéutico con administración parenteral de cobalamina.
Si las pruebas de Schilling con factor intrínseco no demuestran mejoría en los resultados en forma repetida, probablemente deberán investigarse causas de malabsorción de cobalamina no relacionadas con la deficiencia del FI. Por ejemplo, la insuficiencia pancreática puede originar malabsorción de la cobalamina si el páncreas dañado no produce tripsina o quimiotripsina en cantidades suficientes para digerir el complejo proteína R-cobalamina y de esta manera liberar a la vitamina para que forme el complejo con el FI [ver figura 7].
Tratamiento
El tratamiento con ácido fólico puede producir remisiones hematológicas parciales en los pacientes con deficiencia de cobalamina, pero no mejora la sintomatología neurológica. Debe iniciarse tratamiento específico inmediatamente después de que se haya hecho el diagnóstico de megaloblastosis y de que se hayan tomado muestras para determinar los niveles de cobalamina. Si el paciente se encuentra sintomático por la presencia de una anemia grave, puede transfundirse en forma muy lenta un paquete globular para evitar el precipitar o el agravar una insuficiencia cardiaca congestiva. Esta constituye una de las pocas circunstancias en las cuales puede estar justificada la transfusión de una sola unidad, ya que ésta puede producir un incremento de un 25 porciento en la capacidad de transporte de oxígeno. Debe administrarse una dosis grande de cobalamina debido a que la retención de la cobalamina parenteral es poca, pero variable, y la vitamina no es cara ni tiene efectos colaterales peligrosos. La respuesta de reticulocitos se inicia en cuatro a seis días, y si la cuenta de los granulocitos es baja, aumenta al mismo tiempo. La hipersegmentación de los polimorfonucleares desaparece después de 10 a 14 días, lo que sugiere que en las anemias megaloblasticas la granulopoyesis está afectada por la deficiencia de la cobalamina a dos niveles diferentes: el paso en el cual se determina el número de lóbulos de los polimorfonucleares, y otro nivel en el que los granulocitos maduran y abandonan la médula.50 Las dosis semanales de 1,000 µg de cobalamina parenteral por seis semanas deben continuarse con dosis parenterales de 1,000 µg por mes de por vida. La preparación parenteral estándar es la cianobalamina. En el caso de insuficiencia pancreática puede administrarse cobalamina por vía parenteral o enzimas pancreáticas por vía oral. Debe diseñarse un tratamiento específico para los pacientes con formas intestinales de malabsorción.
Debido a que una pequeña cantidad de cobalamina se absorbe incluso en ausencia de FI y ya que solo se requiere 1 µg/día, se ha demostrado que son adecuadas las dosis orales de cobalamina de 300 a 1,000 µg/día en pacientes con anemia perniciosa, y esta forma de tratamiento evita las inyecciones mensuales (se recomiendan 1,000 µg/día).71
DEFICIENCIA DE ACIDO FOLICO
Los pacientes con anemia megaloblástica que no tienen glositis, antecedentes familiares de anemia perniciosa, o el cuadro neurológico de una enfermedad sistémica combinada probablemente tienen deficiencia de ácido fólico. Es importante el interrogatorio meticuloso acerca de los hábitos debido a que los malos hábitos alimenticios, la ingesta dietética insuficiencia y el alcoholismo pueden ocasionar deficiencia grave de ácido fólico. Es factible que la anemia megaloblástica que se presenta durante la administración de medicamentos o durante el embarazo sea debida a deficiencia de ácido fólico. Debido a que la combinación de deficiencia de folato y hierro es común, con frecuencia se bloquea la expresión completa de la megaloblastosis y el paciente presenta una anemia dimórfica, más que la macro-ovalocitos fácilmente identificable. Sin embargo, persiste la hipersegmentación de los polimorfonucleares.50,72 Recientemente se ha descubierto una variable termolábil de la enzima 5,10-metileno tetrahidrofolato reductasa, C677T que causa una deficiencia enzimática relativa. Alrededor del cinco a 10 porciento de la población general es homocigota para esta variante. La deficiencia enzimática interfiere con el reciclamiento del 5,10-metileno THFn a metil THFn, que a su vez proporciona el grupo metil para la conversión de homocisteína a metionina. No es sorprendente que el nivel de homocisteína en los pacientes afectados aumente, y éstos pueden desarrollar un estado de hipercoagulabilidad . Las mujeres embarazadas y no embarazadas que son homocigotas para la mutación C677T tienen niveles de folato en eritrocitos significativamente bajos,73 pueden ser susceptible a enfermedad cardiovascular y cerebrovascular y pueden tener hijos con defectos del tubo neural.73,74
Fisiopatología
Se han identificado numerosas causas de deficiencia de ácido fólico [ver tabla 5]. La ingestión de etanol en voluntarios bien nutridos no produce megaloblastosis, pero en pacientes con reservas de folato en límites el etanol puede disminuir la concentración sérica de folatos y bloquear la respuesta de reticulocitos a la administración de folato. El alcohol puede bloquear también la liberación de ácido fólico de los tejidos al suero.
|
||||||||||
|
Diagnóstico
Los niveles séricos de folato disminuyen súbitamente después de la ingestión de etanol; los niveles disminuyen en un plazo de dos semanas después de que se suspende la ingestión de folato en la dieta. Por lo tanto muchos pacientes hospitalizados tienen niveles séricos de folato disminuidos sin que exista una deprivación verdadera de folatos en los tejidos. En la evaluación de los pacientes con deficiencia de folato deben obtenerse los valores de folato sérico, cobalamina sérica y valores de folato en eritrocitos. El valor de folato en los eritrocitos refleja el depósito tisular,72 pero puede estar reducido en la deficiencia grave de cobalamina. En los estados de deficiencia aislada de cobalamina el valor sérico del folato suele ser normal o elevado (la llamada trampa de metilfolato). Es importante recordar que la deficiencia grave y de larga duración de la cobalamina ocasiona anorexia y alteraciones gastrointestinales, que pueden ocasionar deficiencia dietética de folatos. Como resultado, tanto los niveles de cobalamina como los de folatos serán bajos, lo que indica un estado de doble deficiencia. Cuando se ha diagnosticado deficiencia de folato deben establecerse otras medidas para esclarecer con precisión las causas de la deficiencia. En los casos en que es difícil pero necesario distinguir la megaloblastosis de la deficiencia de cobalamina de la de deficiencia de folato, son útiles las concentraciones séricas de ácido metilmalónico y de homocisteína, que en la deficiencia de cobalamina estarán elevadas, mientras que en la deficiencia de folatos solo aumenta la concentración de homocisteína [ver figura 5].72
En la mayoría de los pacientes (i.e., que no requieren una gran cantidad de folato por situaciones como hemólisis o embarazo), se presenta una respuesta hemotológica después de la administración de 200 µg de folato por día. La mayor demanda de folato durante el embarazo es de alrededor de 200 a 300 µg/día.75 Aún más, los suplementos de folato parecen prevenir los defectos del tubo neural fetal.76 Estos defectos del tubo neural pueden ocurrir en el embrión en etapas muy tempranas de la gestación (incluso antes de que se confirme el embarazo).73,74 Por lo tanto, se recomienda que las mujeres en edad reproductiva o que estén planeando embarazarse reciban alrededor de 400 µg de folato al día. Las mujeres homocigotas para la mutación C677T deben también tomar suplementos de folato. Algunos alimentos como la harina y los cereales pueden estar enriquecidos con ácido fólico. Sin embargo, se ha expresado la preocupación de que los suplementos de folato enmascaren la megaloblastosis de la anemia perniciosa, causando neuropatía severa en lugar de anemia.77
Tratamiento
El tratamiento estándar para la deficiencia de ácido fólico consiste en la administración de 1 mg/día por vía oral. La respuesta se manifiesta por la presencia de reticulocitosis en cuatro a seis días, ausencia de magaloblastosis y normalización de los valores hematológicos, lo que confirma el diagnóstico de deficiencia de folato. Sin embargo, la hipersegmentación de los neutrófilos desaparece solamente después de que han pasado 10 a 14 días.50 La megaloblastosis y la depresión medular grave secundaria a medicamentos que bloquean a la dihidrofolato reductasa, como la pirimetamina y el metotrexate, pueden tratarse con ácido folínico. En el caso de toxicidad después de la administración de dosis únicas elevadas de metotrexate será suficiente la administración de una dosis única equivalente (i.e., miligramo por miligramo) de ácido folínico IM. En los casos de toxicidad después del tratamiento crónico con pirimetamina, pueden administrarse de 1 a 5 mg de ácido folínico por día sin bloquear los efectos antipalúdicos de la pirimetamina. La megaloblastosis debida a tratamiento anticonvulsivante puede tratarse con 1 mg de ácido fólico por día. Se recomienda la profilaxis durante el embarazo, y también puede ser de utilidad en los pacientes que tienen hemólisis crónica grave.
Si la causa de la megaloblastosis no está claramente relacionada con anormalidades del metabolismo de la cobalamina o del ácido fólico, deberán buscarse otras causas de alteración en la síntesis del ADN. Muchos de los medicamentos antineoplásicos e inmunosupresores que interfieren con la síntesis de ADN producen también megaloblastosis. Entre los medicamentos se incluye el fluoruracilo, la hidroxiurea, la mercaptopurina, la tioguanina, la citarabina y la azatioprina.
ANEMIAS SIDEROBLASTICAS
Las anemias sideroblásticas son un grupo heterogéneo de padecimientos que se caracterizan por la presencia de anemia y eritropoyesis ineficaz.
Fisiopatología
Las anemias sideroblásticas no tienen una etiología única. En estos padecimientos aparecen subpoblaciones de células eritroides en la médula y grupos distintos de células se ven afectados en grados diferentes. Esto puede explicar la apariencia dimórfica de los eritrocitos en el frotis de sangre periférica. Las alteraciones en la síntesis del hem pueden ser la causa más frecuente, y se han descrito defectos moleculares de la enzima 5-aminolevulinato sintetasa.78,79 Esta enzima inicia la vía de síntesis del hem y su alteración afecta en forma importante la producción de esta molécula. En otros casos existen deleciones importantes en el ADN mitocondrial. El hierro penetra en los precursores eritroides pero, debido a que la síntesis del hem está alterada, no puede incorporarse al hem y se acumula en la cresta de la mitocondria.
Diagnóstico
Los pacientes pueden tener anemia refractaria o hemocromatosis. El diagnóstico de anemia sideroblástica se establece por reticulocitopenia, presencia de sideroblastos en anillo (normoblastos en la médula ósea con grandes incrustaciones de hierro no ferritina en la mitocondria [ver figura 8], capacidad de fijación de hierro saturada (por lo general de 80 porciento), elevación de deshidrogenasa láctica (DHL) en suero, depósito de hierro en el hígado en un patrón indistinguible de la hemocromatosis y un frotis de sangre periférica bizarro, con hipocromía, eritrocitos distorsionados y puntilleo basófilo.78,79 El estudio citogenético de la médula ósea puede revelar uno de los patrones típicos observados en los síndromes mielodisplásicos. Las anemias sideroblásticas se clasifican en cuatro grupos diferentes [ver tabla 6].

|
| Figura 8 |
| Anemia sideroblástica |
|
||||||||||
|
Tratamiento
Para fines pronósticos es importante decidir si el paciente tiene una forma benigna o maligna de la enfermedad. Los indicadores de la forma maligna son granulocitopenia, trombocitopenia, granulopoyesis displásica en la médula, megacariocitos bilobulados [ver figura 2], y cuerpos de Auer. En forma esporádica, algunos pacientes con la variedad benigna pueden tener una respuesta de los reticulocitos y la hemoglobina a la administración de piridoxina en dosis de 200 mg/día, con o sin ácido fólico.80 Los pacientes con signos y síntomas de hemocromatosis tienen serios problemas debido a que su anemia no suele permitir la extracción de hierro por medio de flebotomías repetidas. En tales circunstancias pudiera ser de utilidad el empleo de infusiones intravenosas o subcutáneas de desferroxamina para favorecer la salida de hierro. El beber té con los alimentos puede ayudar a bloquear el aumento en la absorción de hierro. La anemia puede ser lo bastante grave como para necesitar la transfusión de paquetes globulares, pero esto sólo intensificará los problemas relacionados con el metabolismo del hierro. En el manejo de la anemia es de importancia crítica el suspender la exposición a los agentes agresores, particularmente el alcohol.
Figuras 1 y 6 Talar Agasyan.
Figura 5 Alan D. Iselin.
Figura 7 Tom Moore.
Blibliografía
- Means RT Jr, Krantz SB: Progress in understanding the pathogenesis of the anemia of chronic disease. Blood 80:1639, 1992 [PMID 1391934]
- Cash JM, Sears DA: The anemia of chronic disease: spectrum of associated diseases in a series of unselected hospitalized patients. Am J Med 87:638, 1989 [PMID 2589399]
- Lacombe C: Resistance to erythropoietin. N Engl J Med 334:660, 1996
- Pincus T, Olsen NJ, Russell IJ, et al: Multicenter study of recombinant human erythropoietin in correction of anemia in rheumatoid arthritis. Am J Med 89:161, 1990
- Henry DH, Beall GN, Benson CA, et al: Recombinant human erythropoietin in the treatment of anemia associated with human immunodeficiency virus (HIV) infection and zidovudine therapy. Ann Intern Med 117:739, 1992 [PMID 1416576]
- Dowlati A, R'Zik S, Fillet G, et al: Anaemia of lung cancer is due to impaired erythroid marrow response to erythropoietin stimulation as well as relative inadequacy of erythropoietin production. Br J Haematol 97:297, 1997 [PMID 9163591]
- Schreiber S, Howaldt S, Schnoor M, et al: Recombinant erythropoietin for the treatment of anemia in inflammatory bowel disease. N Engl J Med 334:619, 1996 [PMID 8592524]
- Ludwig H, Fritz E, Leitgeb C, et al: Prediction of response to erythropoietin treatment in chronic anemia of cancer. Blood 84:1056, 1994 [PMID 7741835]
- Mladenovic J: Aluminum inhibits erythropoiesis in vitro. J Clin Invest 81:1661, 1988
- Adamson JW, Kelly MR, Haley NR, et al: Correction of the anemia of progressive renal failure with recombinant human erythropoietin (rHuEpo). Blood 72 (suppl):35a, 1988
- Eschbach JW, Egrie JC, Downing MR, et al: Correction of the anemia of end-stage renal disease with recombinant human erythropoietin: results of a combined phase I and II clinical trial. N Engl J Med 316:73, 1987 [PMID 3537801]
- Altmann P, Plowman D, Marsh F, et al: Aluminum chelation therapy in dialysis patients: evidence for inhibition of haemoglobin synthesis by low levels of aluminum. Lancet 1:1012, 1988 [PMID 2896867]
- Zauber NP, Zauber AG: Hematologic data of healthy very old people. JAMA 257: 2181, 1987 [PMID 3560399]
- Baldwin JG Jr: Hematopoietic function in the elderly. Arch Intern Med 148: 2544, 1988
- Coleman N, Herbert V: Hematologic complications of alcoholism: overview. Semin Hematol 17:164, 1980
- Lindenbaum J: Folate and vitamin B12 deficiencies in alcoholism. Semin Hematol 17:119, 1980
- Straus DJ: Hematologic aspects of alcoholism. Semin Hematol 10:183, 1973
- Conrad ME, Barton JC: Anemia and iron kinetics in alcoholism. Semin Hematol 17:149, 1980
- Yunis AA: Chloramphenicol-induced bone marrow suppression. Semin Hematol 10:225, 1973
- Ettinghausen SE, Moore JG, White DE, et al: Hematologic effects of immunotherapy with lymphokine-activated killer cells and recombinant interleukin-2 in cancer patients. Blood 69:1654, 1987 [PMID 3495302]
- Rozman C, Marin P, Nomdedeu B, et al: Criteria for severe aplastic anaemia. Lancet 2:955, 1987 [PMID 2889870]
- Bacigalupo A, Hows J, Gluckman E, et al: Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol 70:177, 1988 [PMID 3056497]
- Brown KE, Tisdale J, Barrett AJ, et al: Hepatitis-associated aplastic anemia. N Engl J Med 336:1059, 1997
- Tzakis AG, Arditi M, Whitington PF, et al: Aplastic anemia complicating orthotopic liver transplantation for non-A, non-B hepatitis. N Engl J Med 319:393, 1988 [PMID 3135496]
- Young NS: Autoimmunity and its treatment in aplastic anemia. Ann Intern Med 126:166, 1997
- Hoffman R, Young N, Ershler WB, et al: Diffuse fasciitis and aplastic anemia: a report of four cases revealing an unusual association
- Young NS, Maciejewski J: The pathophysiology of acquired aplastic anemia. N Engl J Med 336:1365, 1997
- Maciejewski JP, Selleri C, Sato T, et al: A severe and consistent deficit in marrow and circulating primitive hematopoietic cells (long-term culture-initiating cells) in acquired aplastic anemia. Blood 88:1983, 1996 [PMID 8822917]
- Doney K, Storb R, Buckner CD, et al: Treatment of gold-induced aplastic anaemia with immunosuppressive therapy. Br J Haematol 68:469, 1988 [PMID 3259892]
- Young NS, Barrett AJ: The treatment of severe acquired aplastic anemia. Blood 85:3367, 1996
- Rosenfeld SJ, Kimball J, Vining D, et al: Intensive immunosuppression with antithymocyte globulin and cyclosporine as treatment for severe acquired aplastic anemia. Blood 85:3058, 1995 [PMID 7756640]
- Doney K, Leisenring W, Storb R, et al: Primary treatment of acquired aplastic anemia: outcomes with bone marrow transplantation and immunosuppressive therapy. Ann Intern Med 126:107, 1997
- Frickhoffen N, Kaltwasser JP, Schrezenmeier H, et al: Treatment of aplastic anemia with antilymphocyte globulin and methylprednisolone with or without cyclosporine. N Engl J Med 324:1297, 1991
- Soci?? G, Henry-Amar M, Bacigalupo A, et al: Malignant tumors occurring after treatment of aplastic anemia. N Engl J Med 329:1152, 1993
- Paquette RL, Tebyani N, Frane M, et al: Long-term outcome of aplastic anemia in adults treated with antithymocyte globulin: comparison with bone marrow transplantation. Blood 85:283, 1995 [PMID 7803802]
- Brodsky RW, Sensenbrenner LL, Jones RJ: Complete remission in severe aplastic anemia after high-dose cyclophosphamide without bone marrow transplantation. Blood 87:491, 1996
- Dessypris EN: The biology of pure red cell aplasia. Semin Hematol 28:275, 1991
- Maung ZT, Norden J, Middleton PG, et al: Pure red cell aplasia: further evidence of T cell clonal disorder. Br J Haematol 87:189, 1994 [PMID 7947244]
- Casadevall N, Dufuy E, Molho-Sabatier P, et al: Brief report: autoantibodies against erythropoietin in a patient with pure red-cell aplasia. N Engl J Med 334:630, 1996 [PMID 8592526]
- Charles RJ, Sabo KM, Kidd PG, et al: The pathophysiology of pure red cell aplasia: implications for therapy. Blood 87:4831, 1996 [PMID 8639856]
- Walker RE, Parker RI, Kovacs JA, et al: Anemia and erythropoiesis in patients with the acquired immunodeficiency syndrome (AIDS) and Kaposi sarcoma treated with zidovudine. Ann Intern Med 108:372, 1988 [PMID 3422548]
- Marmont AM: Therapy of pure red cell aplasia. Semin Hematol 28:285, 1991
- Baker RI, Manoharan A, De Luca E, et al: Pure red cell aplasia of pregnancy: a distinct clinical entity. Br J Haematol 85:619, 1993 [PMID 8136286]
- Lacy MQ, Kurtin PJ, Tefferi A: Pure red cell aplasia: association with large granular lymphocyte leukemia and the prognostic value of cytogenetic abnormalities. Blood 87:3000, 1996 [PMID 8639922]
- Palmieri G, Lastoria S, Colao A, et al: Successful treatment of a patient with thymoma and pure red-cell aplasia with octreotide and prednisone. N Engl J Med 336:263, 1997 [PMID 8995089]
- McGuire WA, Yang HH, Bruno E, et al: Treatment of antibody-mediated pure red-cell aplasia with high-dose intravenous gamma globulin. N Engl J Med 317:1004, 1987 [PMID 3116428]
- Ramratnam B, Gollerkeri A, Schiffman FJ, et al: Management of persistent B19 parvovirus infection in AIDS. Br J Haematol 91:90, 1995 [PMID 7577659]
- Yamada O, Mizoguchi H, Oshimi K: Cyclophosphamide therapy for pure red cell aplasia associated with granular lymphocyte-proliferative disorders. Br J Haematol 97:392, 1997
- Means RT Jr, Dessypris EN, Krantz SB, et al: Treatment of refractory pure red cell aplasia with cyclosporine A: disappearance of IgG inhibitor associated with clinical response. Br J Haematol 78:114, 1991 [PMID 1904268]
- Wickramasinghe S: Morphology biology and biochemistry of cobalamin- and folate-deficient bone marrow cells. Bailliere's Clin Haematol 8:441, 1995
- Koury MJ, Horne DW: Apoptosis mediates and thymidine prevents erythroblast destruction in folate deficiency anemia. Proc Natl Acad Sci USA 91:4067, 1994 [PMID 8171036]
- Igram CF, Davidoff AN, Marais E, et al: Evaluation of DNA analysis for evidence of apoptosis in megaloblastic anaemia. Br J Haematol 96:576, 1997
- Tefferi A, Pruthi RK: The biochemical basis of cobalamin deficiency. Mayo Clin Proc 69:181, 1994
- Skacel PO, Hewlett AM, Lewis JD, et al: Studies on the haemopoietic toxicity of nitrous oxide in man. Br J Haematol 53:189, 1983 [PMID 6821648]
- Pruthi RK, Tefferi A: Pernicious anemia revisited. Mayo Clin Proc 69:144, 1994 [PMID 8309266]
- Allen LH: Vitamin B12 metabolism and status during pregnancy, lactation and infancy. Adv Exp Med Biol 352:173, 1996
- Weir DG, Scott JM: The biochemical basis of the neuropathy in cobalamin deficiency. Baillire's Clin Haematol 8:479, 1995
- Dokal IS, Cox TM, Galton DAG: Vitamin B-12 and folate deficiency presenting as leukaemia. BMJ 300:1263, 1990 [PMID 2354298]
- Chanarin I, Metz J: Diagnosis of cobalamin deficiency: the old and the new. Br J Haematol 97:695, 1997
- Carmel R, Weiner JM, Johnson CS: Iron deficiency occurs frequently in patients with pernicious anemia. JAMA 257:1081, 1987 [PMID 3806900]
- Carmel R: Pernicious anemia: the expected findings of very low serum cobalamin levels, anemia, and macrocytosis are often lacking. Arch Intern Med 148:1712, 1988
- Hsing AW, Hansson L-E, McLaughlin JK, et al: Pernicious anemia and subsequent cancer. Cancer 71:745, 1993 [PMID 8431855]
- Talley NJ, Chute CG, Larson DE, et al: Risk for colorectal adenocarcinoma in pernicious anemia: a population-based cohort study. Ann Intern Med 111:738, 1989 [PMID 2802432]
- Carmel R, Spencer CA: Clinical and subclinical thyroid disorders associated with pernicious anemia: observations on abnormal thyroid-stimulating hormone levels and on a possible association of blood group O with hyperthyroidism. Arch Intern Med 142:1465, 1982
- Green R: Screening for vitamin B-12 deficiency: caveat emptor. Ann Intern Med 124:509, 1996
- Carmel R, Rasmussen K, Jacobsen DW, et al: Comparison of the deoxyuridine suppression test with serum levels of methylmalonic acid and homocysteine in mild cobalamin deficiency. Br J Haematol 93:311, 1996 [PMID 8639422]
- Gueant JL, Safi A, Aimone-Gastin I, et al: Autoantibodies in pernicious anemia type I patients recognize sequence 251-256 in human intrinsic factor. Proc Assoc Am Physicians 109:462, 1997
- Pippard MJ: Megaloblastic anaemia: geography and diagnosis. Lancet 334:6, 1994
- Sumner AE, Chin MM, Abrahm JL, et al: Elevated methylmalonic acid and total homocysteine levels show high prevalence of vitamin B12 deficiency after gastric surgery. Ann Intern Med 124:469, 1996 [PMID 8602704]
- Carmel R, Sinow RM, Siegel ME, et al: Food cobalamin malabsorption occurs frequently in patients with unexplained low serum cobalamin levels. Arch Intern Med 148:1715, 1988
- Lederle FA: Oral cobalamin for pernicious anemia: medicine's best kept secret? JAMA 265:94, 1991
- Amose RJ, Dawson DW, Fish DI, et al: Guidelines on the investigation and diagnosis of cobalamin and folate deficiencies. Clin Lab Haematol 16:101, 1994
- Molloy AM, Daly S, Mills JL, et al: Thermolabile variant of 5,10-methylenetetrahydrofolate reductase associated with low red-cell folates: implications for folate intake recommendations. Lancet 349:1591, 1997 [PMID 9174561]
- Wilcken DEL: MTHFR ® 677c mutation, folate intake, neural-tube defect, and risk of cardiovascular disease. Lancet 350:603, 1997
- McPartlin J, Halligan A, Scott JM, et al: Accelerated folate breakdown in pregnancy. Lancet 341:148, 1993 [PMID 8093747]
- Wald NJ, Bower C: Folic acid, pernicious anaemia, and prevention of neural tube defects. Lancet 343:307, 1994 [PMID 7905138]
- Carmel R: Subtle cobalamin deficiency. Ann Intern Med 124:338, 1996
-
Bottomley SS, Healy HM, Brandenburg MA, et al: 5-aminolevulinate
synthase in sideroblastic anemias: mRNA and enzyme activity levels in bone
marrow cells. Am J Hematol 41:76, 1992
-
Bottomley SS, May BK, Cox TC, et al: Molecular defects
of erythroid 5-aminolevulinate synthase in X-linked sideroblastic anemia.
J Bioenerg Biomembr 27:161, 1995 [PMID
7592563]
-
Cotter PD, May A, Fitzsimons EJ, et al: Late-onset
X-linked sideroblastic anemia: missense mutations in the erythroid-aminolevulinate
synthase (ALAS2) gene in two pyridoxine-responsive patients initially diagnosed
with acquired refractory anemia and ringed sideroblasts. J Clin Invest
96:2090, 1995 [PMID
7560104]
-
Brodie MJ, Pellock JM: Taming the brain storms: felbamate
updated. Lancet 346:918, 1995
-
Ammus SS, Yunis AA: Acquired pure red cell aplasia. Am J
Hematol 24:311, 1987 [PMID
3103428]
-
Clark DA, Dessypris EN, Krantz SB: Studies on pure red cell
aplasia: XI. Results of immunosuppressive treatment of 37 patients. Blood
63:277, 1984
between rheumatologic and hematologic disorders. Medicine (Baltimore) 61:373, 1982 [PMID 7144530]