Neurología
⭳ Abrir artículo (PDF)534.7 KBEste artículo es idéntico en la Edición 2/2000.
Contenido del artículo
XII EPILEPSIA
- Definición
- Clasificación
- Evaluación diagnóstica
- Diagnóstico diferencial
- Tratamiento de las crisis convulsivas y la epilepsia
- Tratamiento del estado epiléptico
- Pronóstico del tratamiento epiléptico
- Aspectos especiales
XII EPILEPSIA
DR. L. JAMES WILLMORE
DR. JAMES A. FERRENDELLI
Definición
Las crisis convulsivas son descargas temporales, paroxísticas y sincrónicas de grupos de neuronas en el cerebro. Aunque se consideran anormales, las crisis convulsivas pueden ocurrir en tejido cerebral tanto normal como anormal. Las manifestaciones clínicas de las crisis dependen de la localización y tamaño del grupo de neuronas que participan en la descarga convulsiva, además de la duración de la descarga. Las crisis convulsivas pueden ser causadas por una alteración temporal de la función cerebral causada por diversas causas como hipoglucemia, hiponatremia y toxicidad por medicamentos. En estos casos el individuo tiene solo una o algunas pocas crisis, y el tratamiento del padecimiento de base corrige el trastorno convulsivo.
La epilepsia se define como crisis recurrentes y puede deberse a factores congénitos o adquiridos. La epilepsia adquirida es resultado de una alteración neurológica crónica, con frecuencia estática, que ha producido una cicatriz en la corteza cerebral. Alrededor del 10 porciento de la población tendrá una o varias crisis convulsivas durante su vida, y ocurre epilepsia en el uno a dos porciento de la población. Las crisis convulsivas son el trastorno neurológico más común, y puede ocurrir a cualquier edad. La incidencia de epilepsia es alta en la infancia, pero disminuye durante la niñez, es muy baja en la adolescencia y la edad adulta y aumenta mucho en los ancianos.
Clasificación
Se han usado varias clasificaciones para las crisis convulsivas, y esto creó confusión importante en relación con los tipos de crisis. En 1981 la Liga Internacional contra la Epilepsia propuso una clasificación basada en criterios clínicos y electroencefalográficos [ver tabla 1]. Esta clasificación es muy aceptada y divide a las crisis en tres categorías principales: crisis parciales, crisis generalizadas y no clasificadas.1
CRISIS PARCIALES
Las crisis parciales pueden ser simples o complejas.2 Una crisis convulsiva que comienza en una región de la neocorteza que controla el movimiento o la sensación puede causar manifestaciones motoras o inducir experiencias sensoriales según la función de la estructura en donde se origina. Si el paciente permanece consciente la crisis se clasifica como parcial simple. Sin embargo, si la descarga focal afecta regiones cerebrales relacionadas con el estado de alerta o si la crisis se generaliza lo suficiente para causar pérdida de la conciencia del paciente, la crisis se clasifica como parcial compleja.
Las crisis parciales simples pueden originarse de cualquier región neocortical . El espectro varía desde la mioclonía local de una extremidad o región de la mano o cara hasta experiencias sensoriales como ver manchas o líneas de colores, como ocurre con las descargas en la corteza visual primaria. Una crisis parcial simple puede causar síntomas que el paciente reconoce, conocidos con frecuencia como aura.3 La crisis parcial puede diseminarse para volverse compleja o incluso tónico-clónica generalizada. Los pacientes con crisis parciales complejas pierden la conciencia independiente de la experiencia que haya precedido a la crisis. Aunque el nivel de conciencia del paciente es un elemento clave en la definición, esta información puede ser difícil de obtener. Durante el estado de conciencia alterada los pacientes pueden ser incapaces de obedecer órdenes o interactuar con el medio exterior, así como de recordar los eventos que ocurrieron durante la crisis. La diferenciación entre una crisis parcial simple o compleja puede depender de un observador, con frecuencia un familiar. Aunque el paciente puede opinar, el reporte de un testigo es un aspecto importante para asegurarse que la clasificación es la adecuada. Por ejemplo, un paciente puede perder la capacidad de hablar durante una crisis que se origine en la corteza temporal, frontal o parietal dominante. Sin embargo, al observar que el paciente obedece órdenes durante la crisis o recuerda información verbal específica, puede determinarse que un paciente está manteniendo el conocimiento a pesar de la impresión obtenida durante la crisis.
Las crisis parciales complejas suelen comenzar con detención del movimiento y la mirada en blanco. Pueden ocurrir automatismos como movimientos simples de la mano o actitudes oroalimentarias como movimientos de saborear o deglutir, así como repeticiones verbales al inicio o durante la crisis. Si el paciente está realizando alguna tarea compleja al inicio de la crisis, puede continuar con ella, aunque la exactitud del comportamiento disminuirá. Son puntos clave la duración de la crisis, porque la mayoría de las crisis parciales complejas duran solo algunos minutos, y la característica estereotipada del comportamiento. Una crisis se considera estereotipada cuando los comportamientos son los mismos o semejantes en todas. Al final de la crisis el paciente puede presentar confusión momentánea, fatiga o desorientación. Esta alteración tiene gran importancia diagnóstica y debe investigarse con preguntas dirigidas. Los efectos postictales distinguen a una crisis parcial compleja de lóbulo temporal de una crisis de ausencia, porque en la segunda el paciente no tiene síntomas postictales.4
Las crisis convulsivas parciales simples y complejas pueden generalizarse en forma secundaria y causar una convulsión tónico-clónica. Es común que la descarga focal se disemine de un área local a lodo el cerebro, aunque la mayoría de las crisis parciales no se generalizan en forma secundaria. Debido a que es posible que los testigos o el paciente no noten la manifestación focal de una crisis que es secundariamente generalizada, es mejor asumir de una crisis tónico clónica recién diagnosticada se originó en un foco hasta que no se demuestre lo contrario.5 Las crisis tónico clónicas de reciente inicio en el adulto obligan a excluir una lesión focal, como un tumor, infarto o infección cerebral.
CRISIS GENERALIZADAS
Las crisis generalizadas causan un espectro de alteración que va desde el patrón no convulsivo de una simple ausencia hasta mioclonías y crisis tónico-clónicas generalizadas.6 Las crisis de ausencia, parte de un síndrome epiléptico que inicia en la infancia, son breves, suelen durar 10 segundos o menos. El paciente no tiene aura y cuando la crisis termina no existen efectos postictales. El paciente detiene su actividad y deja la vista fija. En ocasiones existe una manifestación clónica leve como parpadeo sutil de los párpados o cambios en el tono postural. Los pacientes con crisis de ausencia tienen un patrón electroencefalográfico característico de descargas generalizadas espiga onda a velocidad de 3/seg. Esta alteración del EEG se activa al forzar la respiración. Por lo demás, los pacientes tienen función neurológica e intelectual normales. Las crisis de ausencia atípicas difieren de las simples porque inician a edad más temprana, el EEG de base es anormal y las descargas son más lentas de 3/seg. Estos pacientes pueden tener también crisis atónicas y mioclónicas y sufrir retraso mental.
Los movimientos motores de las mioclonías se manifiestan como contracciones breves de un músculo o grupo de músculos específico. Las mioclonías hipnagógicas son comunes y normales. Las mioclonías focales pueden ser causadas por lesiones destructivas del tallo cerebral o la médula espinal. La enfermedad metabólica, la hipoxia, los procesos tóxicos y las infecciones pueden causar mioclonías focales o difusas. Las mioclonías asociadas con epilepsia suelen ser simétricas, y éstas son un componente de varios síndromes epilépticos y ocurren antes o como parte de las crisis tanto de ausencia como tónico-clónicas generalizadas. Un síndrome importante que debe identificarse es el de la epilepsia mioclónica juvenil porque se trata en forma específica con ácido valproico. Los pacientes con este síndrome tienen mal pronóstico si se suspende el tratamiento, por lo que deben recibirlo de por vida.7 Las crisis mioclónicas pueden ser causadas por otros trastornos además de la disfunción cerebral primaria, incluyendo enfermedades metabólicas o trastornos cerebrales genéticos como la enfermedad de Lafora.
Las crisis atónicas causa pérdida súbita del tono postural, en ocasiones con caídas y traumatismos de la cabeza o resto del cuerpo. Este tipo de crisis suelen ser refractarias a tratamiento y con frecuencia se asocian con el síndrome de Lennox-Gastaut.
Las convulsiones son el tipo más común de crisis generalizadas. Se caracterizan por pérdida de la conciencia asociada con apnea y contracturas violentas de la musculatura del tronco y las extremidades. Con frecuencia los pacientes tienen traumatismo oral e incontinencia vescial. Durante la crisis aumentan la salivación, la frecuencia cardiaca y la presión arterial. La mayoría de las convulsiones generalizadas comienzan con una fase tónica, en la que existe contracción sostenida de todos los músculos con extensión de las extremidades inferiores y flexión o extensión de las superiores. Esta fase dura varios segundos y es seguida de una fase clónica, en la que se presentan contracciones rítmicas de las extremidades que comienzan con movimientos de alta frecuencia y poca amplitud y después disminuyen gradualmente en frecuencia en un periodo de varios segundos a minutos. Algunos pacientes solo tienen crisis tónicas o clónicas. Puede observarse una secuencia de movimientos clónicos, tónicos y clónicos de nuevo en los pacientes con crisis generalizadas primarias. Después de que ceden las contracciones musculares violentas, el paciente entra en una fase postictal, en la que se restablece la respiración y la falta de respuesta es seguida de recuperación gradual del estado de conciencia. El paciente puede permanecer confuso por algunos minutos y referir después dolor muscular y cefalea. Esta secuencia es estereotipada, independientemente de la causa. Si no se encuentra una causa estructural, la convulsión se considera como idiopática o generalizada primaria. Si la convulsión es precedida por una crisis parcial se dice que es una convulsión generalizada en forma secundaria.
SINDROMES EPILEPTICOS
En 1989 la Liga Internacional contra la Epilepsia reconoció que muchos pacientes con crisis convulsivas tienen alteraciones cerebrales que afectan su calidad de vida en forma independiente a la epilepsia. Esta clasificación6 reconoce que un síndrome epiléptico incluye no solo el comportamiento durante la crisis, sino también los cambios EEG, el desarrollo mental y motor del paciente y la historia familiar [ver tabla 2]. Debido a que el repertorio de manifestaciones convulsivas es limitado o estereotipado, pueden encontrarse tipos similares de crisis como componentes de varios síndromes con pronósticos muy diversos. La definición de un síndrome específico de epilepsia con frecuencia requiere de evaluaciones repetidas, valoración del desarrollo y revisión de las respuestas al tratamiento. Los síndromes se consideran benignos o progresivos. Estos términos se aplican generalmente para referirse a la evolución final de la función intelectual y la supervivencia. Aunque algunas crisis sindromáticas son benignas en relación con su impacto sobre la función intelectual, puede requerirse tratamiento de por vida con fármacos antiepilépticos. A continuación se describen cinco síndromes epilépticos comunes.
Epilepsia benigna de la infancia con espigas centrotemporales
Este síndrome común se caracteriza por crisis motoras hemifaciales que son breves, parciales, por lo general nocturnas y en ocasiones generalizadas. Los pacientes tienen también síntomas neurosensoriales como fasciculaciones faciales y parestesias de la lengua. El desarrollo de estos pacientes es normal, sin cambios focales en los estudios contrastados de cerebro. El EEG muestra activación durante el sueño de espigas centrotemporales de alto voltaje con una configuración truncada y una onda lenta característica. Se presenta remisión completa en más del 90 porciento de los pacientes hacia la mitad de la adolescencia.8
Ausencias infantiles
Se calcula que la prevalencia de las ausencias infantiles es de dos a ocho porciento.9 Las alteraciones características del EEG de descargas espiga onda a 3/seg ocurren sobre un patrón basal normal [ver figura 1].10 Aunque se han reportado remisiones hasta en el 80 porciento de los casos, el seguimiento a largo plazo muestra que el desarrollo de crisis tónico-clónicas reduce el número de pacientes que logran una remisión sin medicamentos. Solo el 30 porciento de los pacientes con ausencia tienen resolución completa sin medicamentos antiepilépticos si presentan crisis tónico-clónicas generalizadas. La evolución tampoco es muy optimista para los pacientes que tienen más de 15 años de edad o con más de 15 años de evolución.11 La remisión es más frecuente cuando la enfermedad es más reciente, lo que implica la necesidad de identificar pronto a los pacientes e instituir el tratamiento de inmediato. Los factores asociados con un buen pronóstico son un coeficiente de inteligencia (IQ) normal e historia negativa de crisis tónico-clónicas generalizadas por desgracia, estas últimas complican la evolución de las crisis de ausencia en alrededor del 50 porciento de los pacientes.l2 Los factores de riesgo para el desarrollo de crisis tónico-clónicas incluyen edad de inicio más tardía, dificultad para controlar las crisis de ausencia con los medicamentos y actividad basal anormal en el EEG.10,13 Antes del desarrollo de medicamentos de amplio espectro algunos pacientes recibían un medicamento adicional para prevenir las crisis tónico-clónicas.
Este enfoque de tratamiento dual ha sido sustituido por el uso del valproato, un medicamento de amplio espectro con eficacia combinada contra crisis tanto de ausencia como tónico-clónicas. La discusión del tratamiento con los padres y los pacientes debe incluir el informarles sobre la posibilidad de desarrollar crisis tónico-clónicas.l4
Epilepsia mioclónica juvenil
Los pacientes con epilepsia mioclónica juvenil tienen contracciones mioclónicas características, crisis generalizadas tónico-clónicas, historia familiar de crisis convulsivas y fotosensibilidad en el EEG. Entre los precipitantes de las crisis se incluyen la deprivación de sueño, el estrés y el uso de alcohol. Las mioclonías son más importantes por la mañana y pueden afectar los músculos grandes de las extremidades inferiores. Es importante realizar un diagnóstico correcto porque las crisis suelen controlarse bien con valproato. El pronóstico para una función intelectual normal suele ser bastante bueno, sin embargo, para la remisión sin medicamentos es malo.11,15
Epilepsia parcial continua crónica progresiva
Los pacientes con crisis parciales o alteraciones cerebrales focales pueden presentar crisis focales continuas. El EEG muestra espigas focales continuas y ondas lentas. El pronóstico se relaciona con la lesión patogénica subyacente y no suele asociarse con progresión clínica. La forma infantil (encefalitis de Rassmusen) comienza con crisis focales que afectan los sistemas motores y después progresan para provocar deficiencias motoras y deterioro mental.8,16 También se han reportado casos de esta forma rara de encefalitis en adultos.
Epilepsia mioclónica severa de la infancia
Aunque las crisis mioclónicas generalizadas o focales son características de varios síndromes de la infancia, la observación de disfunción neurológica progresiva con deterioro de los sistemas visual o motor piramidal o extrapiramidal identifica a los pacientes con esta forma de epilepsia mioclónica.l7 Las causas incluyen enfermedad de Unvericht-Lundborg, Lafora, lipofuscinosis neuronal ceroide, sialidosis y encefalomielopatía mitocondrial.18 Debido a que los síndromes de la infancia benignos en ocasiones son difíciles de diferenciar de las crisis mioclónicas progresivas, es importante realizar exámenes repetidos con evaluación de la función neurológica. La evolución depende de la enfermedad subyacente, las epilepsias mioclónicas progresivas son fatales.
Evaluación diagnóstica
Aunque la historia clínica del paciente constituye la base para la caracterización de las crisis convulsivas, se requieren varios estudios de laboratorio para establecer el diagnóstico, esquema de tratamiento y pronóstico.
ELECTROENCEFALOGRAFIA
Un EEG es una imagen gráfica o electrónica de la actividad fisiológica cerebral amplificada registrada en electrodos colocados en el cráneo del paciente con una distribución ya establecida. Los patrones convulsivos focales o generalizados que interrumpen la actividad de base confirman el diagnóstico de epilepsia. La utilidad diagnóstica de un EEG mejora si el paciente es preparado con deprivación de sueño, de modo que ocurra sueño natural durante el estudio, ya que éste aumenta la actividad epileptiforme. Los pacientes con crisis parciales que se originan en estructuras de la porción media del lóbulo temporal con frecuencia tienen un EEG normal al inicio del estudio. Se requieren varios EEG para confirmar el diagnóstico. Los patrones diagnósticos incluyen espigas agudas focales, ondas o complejos espiga-onda [ver figura 2]. Las alteraciones generalizadas varían desde supresión momentánea de los ritmos de base hasta racimos de espigas rápidas múltiples, o al patrón clásico de descargas espiga onda con frecuencia de 3/seg, característico de las ausencias.
IMAGEN
Debido a que la epilepsia es un síntoma, en la evaluación diagnóstica debe buscarse alguna enfermedad susceptible de tratamiento. Los estudios de la estructura cerebral, incluyendo la tomografía computada y la resonancia magnética, tienen utilidad y aplicaciones diferentes, dependiendo de la edad del paciente al inicio de la epilepsia. La mayoría de los pacientes con epilepsia que tienen un IQ normal y patrones basales normales en el EEG tendrán imágenes estructurales normales en la IRM cerebral. Sin embargo, se encuentran alteraciones en la IRM en alrededor de la mitad de los pacientes con crisis parciales complejas originadas en el lóbulo temporal. En los adultos con epilepsia de reciente inicio y crisis focales o generalizadas debe usarse la IRM para tratar de definir la causa. Por ejemplo, el 19 porciento de los pacientes entre 18 y 50 años de edad pueden tener tumores cerebrales, con crisis convulsivas como manifestación clínica inicial.19 Otras lesiones que causan crisis convulsivas en este grupo de edad incluyen malformaciones arteriovenosas, infección intracraneal e infarto cerebral. El volumen del hipocampo puede evaluarse con la IRM. Aunque se encuentra esclerosis media temporal en alrededor del 15 porciento de los pacientes con crisis parciales complejas originadas en este lóbulo, se observa atrofia focal de la formación del hipocampo en por lo menos el 60 porciento de los casos.20
La tomografía computada por emisión de fotón único (SPECT, por sus siglas en inglés, n. del t.) demuestra reducción del flujo sanguíneo en la región en que se origina la crisis en algunos pacientes. La certeza diagnóstica mejora al inyectar el isótopo del SPECT durante una crisis parcial compleja. La tomografía de emisión de positrones es un estudio adyuvante importante en pacientes seleccionados y revela hipometabolismo ictal en el 70 a 79 porciento de los pacientes con crisis parciales complejas originadas en el lóbulo temporal.21
Diagnóstico diferencial
Aunque las alteraciones súbitas en la función neurológica son características de las crisis convulsivas, también ocurren cuando las estructuras craneales dejan de recibir el aporte adecuado de glucosa u oxígeno. La ausencia de perfusión cerebral adecuada provoca pérdida de la conciencia. La disfunción cardiaca por falla de la pared miocárdica, bloqueo de rama, bloqueo auriculoventricular y taquicardia paroxística, disminuirá el gasto cardíaco y causarán síncope. Si el paciente queda en posición horizontal, con el corazón en el mismo plano que el cerebro, la perfusión cerebral se restablece. Sin embargo, algunos observadores mantienen al paciente en posición erecta, lo que prolonga la interrupción de la circulación cerebral. Pueden ocurrir movimientos convulsivos o incluso tónico clónicos durante la interrupción prolongada de la perfusión cerebral.
La alteración para mantener la resistencia vascular periférica suele ser de tipo insidioso, y por lo general se debe a neuropatía autonómica que complica a la neuropatía periférica, la diabetes mellitus o el síndrome de Shy-Drager. El espectro de síntomas de la hipotensión ortostática varía de vértigo a síncope. Algunos medicamentos causan alteraciones en el tono vascular periférico, provocando síntomas de tipo ortostático como alteración de la conciencia. En cualquier paciente con alteración de la conciencia se requiere revisar el tratamiento farmacológico y la historia clínica para detectar la inducción postural de los síntomas.
La disfunción sensorial o motora puede ser causada por eventos isquémicos temporales. Estos pueden deberse a enfermedad vascular embólica cerebral, lesión extracraneal de la arteria carótida o basilar, o incluso migraña. La enfermedad metabólica, en especial la alteración del metabolismo de la glucosa que amerita tratamiento con insulina, puede causar hipoglucemia.
Como en otros aspectos del diagnóstico de epilepsia, la historia clínica es la clave para el diagnóstico diferencial. Todos los pacientes que inician con una crisis convulsiva deben someterse a estudios de escrutinio bioquímico y celular. Estos deben incluir un perfil químico de glucosa, nitrógeno de urea, electrolitos y niveles circulantes de enzimas hepáticas. La biometría hemática puede sugerir una infección, alteración plaquetaria o anemia. Debe realizarse punción lumbar con estudios de líquido cefalorraquídeo si está indicado o en caso de duda clínica. Está indicado realizar punción lumbar si el paciente tiene fiebre o presenta alteraciones en la función cognitiva, por ejemplo cambios de comportamiento que puedan atribuirse a un proceso encefalopático. Este estudio se realizará después de excluir la presencia de una masa intracraneana o de aumento en la presión intracraneal.
Un problema común es la ocurrencia de seudocrisis. Alrededor del 20 porciento de los pacientes que ingresan a unidades de vigilancia de epilepsia para evaluación diagnóstica tienen alteraciones episódicas en el comportamiento que no son causadas por disfunción fisiológica del cerebro. Aunque el término seudocrisis es apropiado para la codificación diagnóstica y la comunicación entre médicos, los eufemismos como crisis o eventos no epilépticos son más adecuados al hablar con los pacientes. El uso de estos términos ayuda a los pacientes a comprender su problema y facilita su referencia para una terapia de comportamiento. Los pacientes reaccionan con enojo ante el término seudo, y es menos probable que crean que el médico se interesa por su problema. Las claves para el diagnóstico incluyen eventos periódicos que tienden a no ser estereotipados. Tanto los pacientes como los observadores reportan un comportamiento diferente en cada evento. Otra clave es la duración prolongada. Las seudocrisis pueden durar 30 minutos a varias horas. La mayoría de las crisis, tanto parciales como generalizadas, rara vez persisten por más de varios minutos. Los pacientes que tienen tanto seudocrisis como epilepsia real constituyen un problema clínico especial, y esta coexistencia existe en alrededor del 25 porciento de los pacientes sometidos a unidades de vigilancia de epilepsia. El tratamiento de las seudocrisis requiere de intervención en el comportamiento. Si se encuentran ambos problemas se requerirá tratamiento antiepiléptico en paralelo a la terapia de comportamiento.
Tratamiento de las crisis convulsivas y la epilepsia
TRATAMIENTO FARMACOLOGICO
El tratamiento principal de las crisis convulsivas es el farmacológico. Los medicamentos anticonvulsivantes, conocidos también como antiepilépticos (AE), están disponibles para uso clínico desde hace más de un siglo. Sin embargo, la era moderna del tratamiento AE comenzó a principios del siglo XX con la introducción del fenobarbital. En la actualidad se dispone en los Estados Unidos de más de una docena de agentes clasificados como AE. La mayoría comenzaron a usarse antes de 1980, aunque existen nuevos medicamentos y se espera que se introduzcan algunos más en los próximos años.
El tratamiento debe dirigirse tanto a controlar las crisis como, si es posible, a corregir la enfermedad o trastorno subyacente. Para los pacientes que han tenido una sola crisis o muy pocas por un trastorno temporal, como una intoxicación farmacológica, supresión alcohólica o de sedantes-hipnóticos, hiponatremia o hipoglucemia, pueden no requerirse AE o usarse por muy poco tiempo. Los pacientes que tienen crisis recurrentes secundarias a una enfermedad neurológica trabable, como un tumor cerebral o una infección intracraneal, deben recibir AE y también el tratamiento de la enfermedad de base. En muchos pacientes la causa de las crisis no se identifica o es un proceso estático, como un infarto cerebral establecido o una contusión secundaria a un traumatismo craneal. En estos pacientes el tratamiento con EA es el único posible.
Los pacientes con crisis recurrentes crónicas, independientemente de la etiología, deben tratarse con AE. Sin embargo, existe controversia respecto a si el tratamiento debe iniciarse después de la primera crisis. Muchos especialistas afirman que debido a que muchos pacientes no tienen una segunda crisis durante varios años de seguimiento, no parece existir justificación para comenzar el tratamiento y someter al paciente a sus posibles efectos adversos.22 Esta idea es apoyada por un estudio retrospectivo que reportó que solo el 60 porciento de los pacientes con una primera convulsión tuvieron una segunda en un periodo de cinco años.23 Sin embargo, este punto de vista no es aceptado por todos los clínicos porque muchos pacientes tienen crisis recurrentes. Los autores recomiendan no tratar al paciente después de la primera crisis si el examen neurológico, EEG y estudio de imagen del cerebro son normales, y el enfermo acepta someterse al riesgo asociado con otra crisis convulsiva. Sin embargo, debe iniciarse tratamiento anticonvulsivo para la mayoría de los pacientes después de una primera crisis cuando la evaluación revela cualquier alteración cerebral estructural o funcional. También, el riesgo de recurrencia de las crisis disminuye en forma progresiva mientras más tiempo permanezca el paciente sin sufrir crisis.
El tratamiento con AE debe seguir ciertos principios básicos. El tratamiento debe iniciarse con un solo agente adecuado. Debe lograrse el control de las convulsiones aumentando la dosis de este medicamento en lugar de agregando un segundo. Si no se controlan las crisis con el primer fármaco, debe considerarse un segundo como alternativa. Siempre que sea posible debe evitarse administrar dos o más AE en combinación, aunque las combinaciones racionales pueden ser útiles cuando falla la monoterapia. Los cambios en la dosis deben ser guiados por la respuesta clínica del paciente y no por las concentraciones séricas, y el control inadecuado indica la necesidad de elevar la dosis, mientras que la presencia de toxicidad sugiere que ésta debe disminuirse. Por lo general no es necesario vigilar los niveles séricos del medicamento en los pacientes con buen control de las crisis que toman un medicamento bien tolerado. Sin embargo, esto puede ser útil en ciertas circunstancias, como para establecer el cumplimiento o investigar cambios inexplicables en el control de las crisis o la toxicidad del fármaco.
Mecanismos de acción de los AE
Esencialmente todos los AE disponibles fueron identificados al investigar su actividad anticonvulsivantes en crisis inducidas en forma experimental en ratas y ratones de laboratorio. Todos bloquean las crisis por electrochoques, las crisis inducidas por químicos o ambas. Se han realizado gran cantidad de investigaciones para definir los mecanismos de acción de los AE. Cada AE parece tener varias acciones moleculares y celulares, pero es probable que solo algunas de estas acciones individuales sean responsables de los efectos anticonvulsivantes y antiepilépticos. En general, los AE parecen disminuir la excitabilidad o aumentar la inhibición neuronal. Esto se logra alterando las corrientes intrínsecas de la membrana, como la conductancia de sodio, potasio y calcio, o afectando la actividad de varios neurotransmisores, como el ácido 8-aminobutírico (GABA), el glutamato u otros neurotrasmisores que pueden participar en la actividad convulsiva. Aunque varios AE comparten mecanismos en común, cada uno parece tener acciones diferentes.
Los medicamentos que actúan sobre las corrientes intrínsecas de la membrana pueden dividirse en forma burda en los que afectan principalmente el canal de sodio con puerta dependiente de voltaje y los que alteran las corrientes de calcio. Los medicamentos que modifican los canales de sodio en concentraciones terapéuticas (fenitoína, carbamacepina, primidona, ácido valproico y lamotrigina) inhiben las descargas neuronales de alta frecuencia.24-27 Debido a que son dependientes tanto de uso como de voltaje, tienen poco efecto sobre la actividad neuronal normal. Sin embargo, durante las descargas de alta frecuencia retrasan la reactivación del canal de sodio y producen un mayor efecto inhibitorio sobre el potencial de acción hasta que la descarga está completamente bloqueada. La actividad antiepiléptica de la etosuximida es resultado de la acción en los canales de calcio.28,29 La etosuximida bloquea en forma selectiva la corriente de calcio tipo T, que no se afecta mucho por la mayoría de los otros AE. Se considera que esta corriente actúa como un marcapaso en las neuronas talámicas y puede ser importante en la actividad de la epilepsia por ausencias.30 El ácido valproico y el lamotrigine son también eficaces contra las crisis de ausencia, aunque aún no se determina si su utilidad es resultado de acción sobre la corriente de calcio tipo T o por otros mecanismos aún no definidos. El bloqueo de otras corrientes de calcio, como la tipo N, principalmente por el fenobarbital y la fenitoína, puede tener un efecto menos específico que interfiere principalmente con la liberación de trasmisores.
Los medicamentos que alteran la función sináptica actúan principalmente aumentando la inhibición neuronal mediada por GABA, el principal mecanismo inhibitorio cerebral. Estos medicamentos incluyen fenobarbital, benzodiacepinas y quizá otros. Cada uno de estos fármacos actúa por un mecanismo diferente para aumentar la influencia del GABA en el sistema nervioso central.31 Las benzodiacepinas aumentan la frecuencia de apertura en los canales receptores de GABAA,32,33 y los barbitúricos aumentan la duración de apertura de los mismos.34,35
Es claro que los mecanismos anticonvulsivantes de los principales medicamentos usados para los trastornos convulsivos aún no se conocen del todo. Algunos AE pueden afectar la conductancia de iones diferentes a los analizados, y es posible que algunos afecten en forma directa o indirecta procesos de neurotrasmisores diferentes al sistema GABA. Estas acciones pueden ser importantes por sus efectos clínicos. Se requieren más investigaciones para definir por completo los mecanismos moleculares y celulares, además de los sitios de acción, de los fármacos AE.
Selección de AE
Los AE más empleados en Estados Unidos para tratar la epilepsia son la carbamacepina, la etosuximida, el gabapentin, el lamotrigine, el fenobarbital, la fenitoína, la primidona y el valproato [ver tabla 3]. Algunas benzodiacepinas, incluyendo el clonacepam, el diacepam y el loracepam, se usan también para tratar las crisis convulsivas. Con excepción del clonacepam, las benzodiacepinas se usan para tratamiento a corto plazo de las crisis agudas o del estado epiléptico y por lo general se administran por vía parenteral. El clonacepam puede usarse para tratar la epilepsia, aunque esto no es recomendable porque la mayoría de los pacientes desarrollan tolerancia a su efecto antiepiléptico. El felbemato, un AE de reciente introducción, se ha prescrito en algunos pacientes con crisis incontrolables, pero su uso está limitado por una alta incidencia de efectos adversos. Se han usado otros medicamentos adicionales, incluyendo metosuximida trimetadiona y acetazolamida, pero no se analizarán en esta subsección.
La mayoría de los especialistas en epilepsia están de acuerdo en que los medicamentos de elección para las crisis parciales son la carbamacepina y la fenitoína, que son muy eficaces y tienen efectos adversos aceptables.36 También se ha demostrado que el valproato es eficaz en el tratamiento de las crisis parciales.37 Es probable que tanto el fenobarbital como la primidona sean tan eficaces como la carbamacepina o la fenitoína, pero ambos barbitúricos se asocian con una incidencia mucho mayor de efectos adversos, en especial sedación y deterioro cognoscitivo.36 Dos de los medicamentos más nuevos, gabapentin y lamotrigine, son eficaces contra las crisis parciales, y en la actualidad cada uno se usa en combinación con alguno de los fármacos mencionados antes. Las crisis convulsivas generalizadas pueden controlarse con carbamacepina, fenitoína, valproato, barbitúricos, gabapentin o lamotrigine. La carbamacepina, la fenitoína y el valproato parecen tener una eficacia semejante para las crisis convulsivas generalizadas. La carbamacepina, la fenitoína, el gabapentin y el lamotrigine se usan en pacientes con crisis generalizadas secundarias porque muchos de estos pacientes tienen también crisis parciales. Por otro lado, el valproato y el lamotrigine se usan con más frecuencia en pacientes con crisis generalizadas primarias porque algunos de estos pacientes tienen también crisis de ausencias, que pueden controlarse con estos medicamentos. Los barbitúricos no son de elección en el tratamiento de las crisis generalizadas principalmente por sus efectos sedantes. Las crisis generalizadas no convulsivas, en especial las de ausencia, pueden tratarse con etosuximida o valproato.38 Para los pacientes que solo tienen crisis de ausencia la etosuximida es adecuada; sin embargo, para los enfermos que tienen ausencias junto con otros tipos de crisis, como convulsiones generalizadas o crisis mioclónicas, el valproato es el medicamento de elección y se ha sugerido que también el lamotrigine es eficaz.
Farmacocinética de los AE
La mayoría de los pacientes tratados con AE requieren manejo durante varios años. Todos los AE se administran por vía oral una vez al día o con más frecuencia [ver tabla 3]. La absorción de la mayoría de los AE suele ocurrir en forma lenta durante varias horas y puede ser incompleta, en especial en el caso del gabapentin. La unión a proteínas varía mucho entre los medicamentos, variando desde cero para el gabapentin a 90 porciento o más para la fenitoína. Excepto por el gabapetin, todos los AE se metabolizan en el hígado antes de su excreción renal. La depuración y vida media de los AE varía de horas a días. Las vidas medias de la carbamacepina, valproato, primidona y gabapentin son relativamente cortas, variando de cuatro a ocho horas, lo que obliga a administrar estos medicamentos por lo menos tres veces al día. La fenitoína, el lamotrigine y el fenobarbital tienen vidas medias de un día o más y pueden administrarse una o dos veces al día. La fenitoína, la carbamacepina, los barbitúricos y quizá el lamotrigine, causan inducción enzimática, por lo que el tratamiento prolongado con estos medicamentos puede afectar su propia velocidad de metabolismo, así como el de otros medicamentos concomitantes. Todos los AE que se metabolizan pueden competir con otros fármacos que también se metabolizan en el hígado, retrasando su eliminación. Debido a su capacidad para inducir o bloquear el metabolismo farmacológico, todos los AE excepto el gabapentin pueden asociarse con interacciones farmacocinéticas en la que se afectan las concentraciones y efectos de los medicamentos administrados en forma simultánea. Este tipo de interacción puede anticiparse en todos los pacientes. Pueden vigilarse los síntomas clínicos y los niveles sanguíneos de los AE, en especial los niveles del fármaco libre, y ajustarse la dosis en caso necesario. También puede ocurrir interacción farmacodinámica. En esta situación, la combinación de dos o más medicamentos con mecanismos semejantes o antagónicos causa que sus efectos clínicos aumenten o disminuyan, y estos cambios pueden requerir de ajuste en las dosis. Las interacciones farmacodinámicas pueden anticiparse si se conocen los mecanismos de acción del medicamento.
Efectos adversos de los AE
La mayoría de los efectos adversos de los AE pueden ser tolerables. Sin embargo, los efectos idiosincráticos raros pueden ser graves. Para realizar una buena práctica médica es necesario obtener algunos estudios de escrutinio antes de iniciar los AE. Los estudios son los siguientes: biometría hemática completa con diferencial y cuenta de plaquetas, química sérica con glucosa, nitrógeno de urea, electrolitos, calcio, fósforo, magnesio, ácido úrico, hierro libre, colesterol, bilirrubinas, fosfatasa alcalina, aspartato y alanino aminotransferasas, proteínas totales, albúmina y globulina, y pruebas de coagulación. Sin embargo, el valor y necesidad de obtener estas pruebas de laboratorio de rutina durante el tratamiento no se ha demostrado. En los pacientes estables la vigilancia de rutina parece no estar justificada. Por otro lado, la vigilancia clínica por medio de revisión y reporte de síntomas u observación por medio de un examen físico tiene mejor relación costo-beneficio y es más práctica. De hecho, la vigilancia de rutina como se practica en la actualidad no permite anticipar los efectos graves asociados con la mayoría de los AE, incluyendo carbamacepina, fenitoína y fenobarbital. Por ejemplo, estudios prospectivos de evaluación rutinaria de sangre y orina en pacientes que recibieron AE por tiempo prolongado no demostraron ninguna utilidad en pacientes asintomáticos.39 Mucho más importante que la vigilancia rutinaria de la sangre es la comunicación continua con el paciente y la familia. Ellos deben estar al tanto de las posibles complicaciones y de los síntomas que pueden indicar un evento adverso. Los autores consideran que la participación informada en la vigilancia clínica tiene buena relación costo beneficio, es segura y adecuada. El uso de la experiencia clínica aunada con la educación del paciente y el conocimiento de los riesgos y beneficios del tratamiento por parte de las personas que le atienden se ha convertido en el manejo estándar.40
Todos los AE pueden producir efectos adversos, que son numerosos y varían mucho de paciente a paciente. Algunos de estos efectos indeseables están claramente relacionados con la dosis, como la neurotoxicidad, mientras que otros parecen ser idiosincráticos, como algunas formas de hepatotoxicidad o anemia aplástica, y aún otros parecen tener mecanismos distintos. Consideramos que la forma mas eficaz de comprender los efectos adversos de los AE consiste en definirlos de acuerdo con el sistema orgánico afectado y este esquema de clasificación es el que se usa en este capítulo.
Efectos sobre el sistema nervioso central La neurotoxicidad dependiente de la dosis es el efecto adverso más frecuente de los AE, y puede limitar la cantidad de medicamento que puede emplearse. Los síntomas del SNC se usan para calcular la dosis aceptable en forma independiente de los niveles de AE en sangre. Por ejemplo, la carbamacepina causa diplopia o sedación, y estos síntomas sirven como indicadores de que el paciente ha llegado a la dosis máxima tolerada. El medicamento puede administrarse en dosis progresivas hasta alcanzar el límite superior terapéutico en sangre según las estadísticas. Sin embargo, una vez que se alcance éste, la dosis se aumenta hasta que el paciente presenta neurotoxicidad. Esta define la dosis máxima tolerada. Todos los AE provocan depresión de la función cortical, con síntomas de sedación y letargo. Aunque los efectos relacionados con la dosis de la fenitoína sobre el cerebelo son indicadores clásicos de toxicidad, los hallazgos típicos con frecuencia están bloqueados o no son aparentes por las respuestas individuales de cada paciente.41 No siempre ocurre la progresión esperada de los síntomas de nistagmus a ataxia y confusión o la sensación de intoxicación. Aunque este aspecto no es del todo claro, puede ocurrir ataxia residual después de la intoxicación prolongada con fenitoína, en especial en pacientes vulnerables.42
La ataxia persistente o fluctuante parece relacionarse con la duración de la epilepsia, el uso de múltiples drogas y una nivel persistentemente alto del medicamento en suero. Se han reportado cambios en la función intelectual y alteraciones en la personalidad o incluso efectos psiquiátricos con varios AE. La alteración del IQ en niños tratados con fenitoína y primidona parece relacionarse con niveles altos en sangre de estos fármacos.
La evaluación de la función cognitiva en pacientes tratados en forma aleatoria no reveló diferencias entre los principales AE, aunque el fenobarbital pareció tener un efecto relativamente mayor que otros medicamentos.43 Los medicamentos administrados a los niños pueden tener efectos impredecibles, y los barbitúricos son una causa bien conocida de hiperactividad en niños. Los AE pueden provocar también depresión y psicosis. La mayoría de los efectos sobre el comportamiento parecen relacionarse con la dosis. Se ha reportado psicosis después del control de las crisis, y este proceso denominado normalización forzada parece relacionarse con normalización del EEG.44
La fenitoína puede causar movimientos anormales relacionados con la dosis, la carbamacepina rara vez causa movimientos involuntarios. Las disquinesias se manifiestan como movimientos asociados al uso crónico de neurolépticos, incluyendo disquinesias de la cara, extrernidades y lengua.45
Se ha reportado neuropatía sensorial leve en ocho a 15 porciento de los pacientes tratados con AE46 y al parecer requiere exposición prolongada. La exposición prolongada a niveles altos en plasma causa pérdida de las fibras mielinizadas grandes y distribución no aleatoria en racimos de desmielinización segmentaria que sugiere un patrón de neuropatía axonal y desmielinización segmentaria.47 El uso de múltiples medicamentos también se asocia con neuropatía crónica. La alteración en la función de los nervios periféricos parece deberse al efecto de los AE sobre el metabolismo del folato, con alteración leve secundaria en la función periférica. El efecto clínico no suele ser severo, pero puede ocurrir pérdida de las respuestas tendinosas profundas o de la sensibilidad vibratoria a nivel de los tobillos.
Además de estos efectos dependientes de la dosis, los AE pueden inducir una encefalopatía reversible y deterioro mental progresivo o delirio.48 En raras ocasiones la exposición inicial del paciente a valproato causa estado de coma, el mecanismo puede relacionarse con el metabolismo mitocondrial.49
Efectos gastrointestinales y hepáticos Todos los AE pueden causar daño hepático, pero la incidencia es poco frecuente, con algunas excepciones. Se ha reportado que el valproato produce falla hepática en alrededor de uno de cada 10,000 pacientes. La tasa más alta se observa en niños menores de dos años que están siendo tratados con múltiples fármacos. Los pacientes de mayor edad con un solo medicamento tienen una tasa mucho más baja: uno en 45,000. El mecanismo de la hepatotoxicidad inducida por valproato se desconoce, pero puede incluir a metabolitos tóxicos del ácido valproico. La ocurrencia poco frecuente de daño hepático asociada con la fenitoína, la carbamacepina o el fenobarbital sugiere que el mecanismo es una reacción de hipersensibilidad idiosincrática.
Se ha observado pancreatitis durante el tratamiento con valproato, y han ocurrido muertes tanto en niños como en adultos.50 Aunque el mecanismo de este efecto adverso no se ha confirmado, la reexposición de los pacientes afectados ha causado recurrencia de la pancreatitis.51
Efectos hematológicos Las reacciones hematológicas adversas asociadas con respuestas idiosincráticas de los AE varían de una reducción leve en el número de células sanguíneas hasta anemia aplástica. Por fortuna los problemas serios son poco frecuentes. La fenitoína puede alterar la función linfocitaria porque el 21 a 25 porciento de los pacientes con tratamiento prolongado tienen disminución de los niveles circulantes de IgA con depresión de la transformación linfocitaria con fitohemaglutinina.52,53 La hipersensibilidad por fenitoína puede causar linfadenopatía y en casos raros puede asociarse con linfoma.54,55 El tratamiento con carbamacepina causa con frecuencia cambios hematológicos. Se ha reportado que ocurre leucopenia relacionada con la dosis en el 12 porciento de los pacientes tratados.56,57 Este cálculo es bajo según la experiencia de los autores. Los mecanismos que provocan este efecto sobre los granulocitos se desconocen, pero el efecto parece relacionarse con la dosis y no presagia reacciones más serias. Al parecer no hay necesidad de preocuparse hasta que la cuenta total de leucocitos sea menor a 2,500/mm3 o la cuenta total de granulocitos sea de menos de 750/mm3.
Se ha reportado anemia aplástica con todos los AE, pero es poco frecuente y ningún medicamento en particular parece asociarse con más riesgo de causar esta complicación seria. La anemia aplástica relacionada al tratamiento con carbamacepina parece relacionarse con la dosis y la edad avanzada de los pacientes.56 De los 65 pacientes que fallecieron por anemia aplástica asociada con carbamacepina, solo cuatro eran niños.58 El valproato causa una reducción dependiente de la dosis en el nivel de plaquetas circulantes que solo en ocasiones es sintomático. La disminución en los niveles de plaquetas en los pacientes sin síntomas varía de 149,000 a 35,000.59 La ocurrencia de púrpura o hemorragias petequiales obliga a suspender el medicamento. El mecanismo parece ser un cambio en la adhesividad plaquetaria, con aceleración en la segunda fase de consumo de plaquetas que causa una mayor pérdida de las plaquetas circulantes. En ocasiones se detecta un autoanticuerpo IgM contra plaquetas.60 Se ha reportado macrocitosis leve y reducción en los niveles de folato en los eritrocitos asociados al uso de AE, en especial fenitoína.6l La anemia megaloblástica es un efecto ocasional del tratamiento AE que parece relacionarse con alteraciones en el metabolismo del folato. Se requieren suplementos de folato en algunos casos.
Reacciones dermatológicas Las reacciones dermatológicas de los AE no son raras; sin embargo, las respuestas serias sí lo son.62 Las erupciones inducidas por fármacos varían desde eritema leve, con o sin prurito, hasta reacciones exfoliativas serias o presencia de bulas. El exantema relacionado con medicamentos es la reacción cutánea más común. La erupción puede ser pruriginosa, maculopapular o morbiliforme, y alrededor del 50 porciento de los pacientes pueden tener síntomas prodrómicos que incluyen malestar y fiebre. Esta forma de erupción puede responder a la reducción del medicamento.62 Las erupciones farmacológicas que comienzan con una reacción pruriginosa con eritemas morbiliformes o escarlatiniformes pueden progresar hasta dermatitis exfoliativa severa. En este caso el medicamento agresor debe suspenderse pronto, sustituyéndose por otro AE. El mecanismo de la reacción cutánea puede estar determinado por factores farmacogenéticos relacionados con la capacidad de metabolizar los compuestos formados durante el metabolismo del medicamento.63,64 Las dermatitis exfoliativas producidas por los AE incluyen eritema multiforme, síndrome de Stevens-Johnson y síndrome de Lyell.65 La dermatitis exfoliativa suele ocurrir entre la primera y cuatro semanas después de iniciado el tratamiento y puede ser fatal. El medicamento que inició la reacción debe suspenderse, instituyéndose tratamiento con esteroides sistémicos.60 El eritema multiforme, como reacción a fármacos, tiene un inicio rápido, con lesiones eritematosas que pueden variar de un patrón macular con formas variadas hasta el desarrollo de vesículas o bulas. Las lesiones mucosas no son una manifestación universal, a menos que la reacción consista en un síndrome de Stevens-Johnson. En cualquiera de los casos es imperativa la consulta dermatológica. El acné es un problema cutáneo menor asociado con la fenitoína. La hipertricosis se asocia tanto con fenitoína como con carbamacepina. Ocurre alopecia durante las primeras semanas de tratamiento con valproato. La administración de multivitamínicos que contienen zinc parece prevenir este cambio en la fuerza del pelo.
Enfermedades del tejido conjuntivo Los AE pueden causar lupus eritematoso generalizado, esclerodermia, síndrome de Sjogren y fascitis eosinofílica. Los criterios de diagnóstico incluyen eritema malar con cambios discoides en la piel, fotosensibilidad, úlceras orales, artritis no erosiva, serositis y nefropatía. Los cambios hematológicos e inmunológicos incluyen preparaciones positivas de células de lupus eritematoso y anticuerpos antinucleares positivos, incluyendo anti-ADN de doble cadena.67 Los medicamentos pueden precipitar la presentación de lupus, causar exacerbación del existente o asociarse con una forma aislada relacionada al medicamento. El lupus inducido por medicamentos suele desaparecer al suspender el medicamento responsable. Además, el patrón de afección orgánica suele respetar la piel, los riñones y el SNC. El patrón inmunológico no suele incluir la inducción de anticuerpos contra ADN de doble cadena. El uso de fenitoína a largo plazo ha causado un síndrome de equimosis, hemorragia gingival y detección de anticuerpos anticoagulante lúpico asociado con deficiencia de protrombina.68 También la carbamacepina se ha relacionado con el desarrollo de lupus inducido por medicamentos.69
Efectos metabólicos y endócrinos La hiponatremia es un efecto de la carbamacepina relacionado con la dosis y que parece presentarse solo en adultos.70 Se ha considerado que el mecanismo podría incluir efectos centrales sobre la hormona antidiurética y efectos periféricos o renales, pero la patogenia exacta se desconoce. La hiponatremia asociada al tratamiento con carbamacepina rara vez causa un problema clínico. Los efectos de la fenitoína sobre la función hipófisis-suprarrenal se relacionan con los efectos periféricos de la inducción de enzimas hepáticas del citocromo P-450, con acentuación resultante del metabolismo de la hormona endógena. El metabolismo acelerado de las hormonas esteroideas administradas en forma exógena, como las pastillas anticonceptivas, puede causar fracaso del sistema anticonceptivo. La función hipotalámica puede afectarse por la fenitoína. Los cambios incluyen liberación alterada de la hormona antidiurética, bloqueo del efecto de la hormona estimulante del tiroides y mayor secreción de la hormona folículo estimulante y de la hormona luteinizante en mujeres.7l,72 Los estudios de función tiroidea se alteran por la fenitoína, pero por lo general no se encuentran efectos clínicamente significativos. La triyodotironina total (T3) y la tiroxina (T4) disminuyen porque la fenitoína causa su desplazamiento de la globulina fijadora de tiroxina.73
TRATAMIENTO QUIRURGICO
El tratamiento quirúrgico es una opción para los pacientes que no responden al tratamiento convencional con AE o que tienen efectos adversos intolerables, cuyas crisis tienen un origen focal y que se originan en tejido que puede ser extirpado sin causar incapacidad.74 La epilepsia puede asociarse con una lesión que tiene un cambio estructural que correlacione con la región de inicio de la crisis. La presencia de un foco epiléptico en el hemisferio dominante para el lenguaje y la ocurrencia de crisis parciales complejas que se originen del teido extratemporal requiere de una evaluación especial. La detección de descargas epileptiformes bilaterales, el desarollo de crisis secundariamente generalizadas y el inicio ocasional de una crisis del tejido contralateral tienden a aumentar la complejidad de la evaluación preoperatoria.75,76
La técnica estándar de cirugía requiere la localización del foco epileptógeno de varias maneras. Aunque se han usado el EEG ictal e interictal de superficie y la electrocorticografía intraoperatoría, estos métodos no proporcionan una información adecuada sobre localización o lateralización. Por lo tanto, la mayoría de los centros de epilepsia emplean la combinación de un EEG de superficie y de un video para grabar por lo menos tres de las crisis típicas del paciente. Se obtiene información adicional por medio de IRM, neuropsicología y estudios de isótopos de circulación sanguínea y metabolismo. La localización de la función de memoria y la lateralización del lenguaje también son importantes. Es necesario insertar electrodos intracraneales si el EEG de superficie no proporciona información adecuada sobre la información o la lateralización.
El procedimiento quirúrgico realizado con más frecuencia para la epilepsia es la lobectomía temporal. En alrededor del 70 porciento de los pacientes desaparecen las convulsiones y un 20 porciento adicional mejora mucho después de la cirugía. La evaluación histopatológica muestra que en por lo menos el 60 porciento de los pacientes ocurren cambios escleróticos en el hipocampo seccionado. En algunos centros de epilepsia se realiza resección de focos extratemporales, sección del cuerpo calloso y hemisferectomía funcional. La corpuscallosotomía es un procedimiento paliativo para los pacientes que tienen lesiones durante las crisis.77 Las complicaciones asociadas con la cirugía de resección son bajas y en general aceptables. Puede ocurrir un defecto en el campo superior del cuadrante en hasta el 75 porciento de los pacientes sometidos a lobectomía temporal clásica. Se reporta hemiparesia permanente en hasta el 2.4 porciento de los pacientes, y la mortalidad varía de cero a 1.7 porciento de los pacientes.75
Tratamiento del estado epiléptico
El estado epiléptico constituye un peligro para el paciente y un reto para el médico. La característica cardinal del estado epiléptico serio es la presencia de crisis convulsivas continuas o dos o más crisis que ocurren en secuencia sin recuperación de la conciencia entre ellas. El estado epiléptico tiene varias formas de presentación clínica, que incluyen crisis convulsivas generalizadas de repetición, con coma entre las mismas, crisis no convulsivas, causando un cambio en la función cognitiva y crisis focales secuenciales, incluyendo crisis motoras focales o molestias sensoriales focales. El tratamiento se divide en manejo agudo y administración lógica de los medicamentos.
El estado epiléptico con crisis generalizadas es el más común y el más difícil de manejar. El paciente tiene movimientos convulsivos y está inconsciente. Las manifestaciones motoras del estado epiléptico convulsivo pueden ser simétricas, con actividad tónica y después clónica. Si las crisis generalizadas se originan en un foco, ocurren movimientos lateralizados al inicio o durante la crisis.
TRATAMlENTO AGUDO
El tratamiento inicia con apoyo vital, incluyendo permeabilidad de las vías aéreas y apoyo ventilatorio, mantenimiento de la presión arterial y colocación de un acceso intravenoso. La vigilancia fisiológica debe incluir electrocardiografía, presión arterial, gases en sangre, evaluación bioquímica y temperatura corporal. La vía aérea debe protegerse y asegurarse una oxigenación adecuada. Aunque la intubación puede ser necesaria, la decisión de usarla debe ir en paralelo con la selección y administración del medicamento anticonvulsivante. Aunque algunas definiciones del estado epiléptico son útiles para la investigación, cualquier paciente que tiene una crisis convulsiva cuando llega al servicio de urgencia o en quien se observa una crisis durante 10 minutos debe tratarse con el supuesto de que está en estado epiléptico.
Debe excluirse o corregirse la hipoglucemia. Los niveles de glucemia se miden de inmediato y, si esto no es posible, el paciente debe recibir glucosa intravenosa. Los adultos deben recibir primero 100 mg de tiamina intravenosa y la dosis de glucosa para adultos es de un bolo de 50 ml al 50 porciento. Para niños la dosis es de 2 ml/kg de solución de glucosa al 25 porciento.
TRATAMIENTO FARMACOLOGICO
Las crisis clínicas y eléctricas deben yugularse lo más rápido posible, ya que la duración del estado epiléptico correlaciona con la respuesta al tratamiento y con la evolución.78 Los médicos que traten estados epilépticos deben estar familiarizados con los medicamentos disponibles, incluyendo métodos de administración, dosis y efectos agudos adversos. Se presenta una mejor evolución si se tiene un plan, se usan medicamentos eficaces administrados por vías y en dosis apropiadas y se evita la apnea. Debe seguirse un protocolo y los medicamentos deben administrarse por vía intravenosa. La naturaleza del medicamento seleccionado debe ser guiado por la situación clínica. Si un paciente tiene una crisis convulsiva en el momento de la evaluación se requiere una benzodiaepina. Si el paciente tiene historia de crisis seriadas pero éstas se han abatido es mejor elegir un AE de acción duradera.
Benzodiacepinas
Las benzodiacepinas son eficaces y muy potentes. Es común contar con diacepam y loracepam. El diacepam es muy liposoluble y penetra con rapidez al tejido cerebral. Sin embargo, la redistribución en los tejidos grasos no neurales causa rápida disminución de su concentración tanto en sangre como en cerebro. Si se usa el diacepam para terminar con las crisis, debe administrarse también un AE de larga acción, como la fenitoína. La acción del loracepam es más larga, pero se asocia con un tiempo prolongado para la recuperación completa.79 Ambos medicamentos causan depresión de la respiración e incluso apnea. Debe administrarse apoyo a la ventilación, y en ocasiones es necesario intubar al paciente.
Fenitoína
La impregnación del paciente con fenitoína por vía intravenosa terminará con el estado epiléptico.80 La dosis inicial para el adulto es dc 18 mg/kg administrados a través de una vena periférica. La velocidad de infusión no debe ser mayor de 40 mg/min o 1 mg/kg/min en niños. Los pacientes con enfermedad cardiaca o dificultad para mantener la presión arterial requieren vigilancia cuidadosa. Los que sufren hipotensión requieren una velocidad de infusión más lenta. La vigilancia electrocardiográfica puede mostrar prolongación del intervalo QT o incluso inducción de arritmias. Estos cambios indican la necesidad de disminuir la velocidad de la infusión aún más.
Fenobarbital
El fenobarbital es muy eficaz y muchos médicos están familiarizados con su uso. La dosis para el adulto es de 10 a 20 mg/kg. Puede provocar sedación y la apnea es un riesgo, en especial si el paciente ha recibido benzodiacepinas. La vigilancia de la presión arterial es crítica y la hipotensión responde a la disminución de la velocidad de administración.
TRATAMIENTO DEL ESTADO EPILEPTICO REFRACTARIO
Si el paciente no recupera la conciencia o continua con crisis convulsivas después del tratamiento de primera línea, se requiere entonces consultar a un neurólogo. Se realizará un EEG de urgencia y considerará la administración de anestesia. El mejor anestésico en estos casos es el pentobarbital.81,82 El paciente debe ser intubado y se establece vigilancia de cuidados intensivos adecuada. Los pacientes se mantienen en coma con pentobarbital por periodos variables. El tratamiento continuo requiere ajuste de la dosis en forma gradual hasta la de mantenimiento a las cuatro horas, de nuevo a las ocho horas y en forma regular después, observando para determinar si las crisis convulsivas se han abatido y el EEG no muestra descargas convulsivas. Pueden requerirse vasopresores durante el coma con pentobarbital. En ocasiones se desarrolla estado convulsivo sutil, y se requiere vigilar con EEG, en especial si no son obvias las descargas motoras.
Después de realizar los estudios diagnósticos iniciales y controlar las crisis debe buscarse la causa del estado epiléptico. Es importante realizar una evaluación médica y neurológica. Si el paciente es conocido como epiléptico y se dispone de los antecedentes para revisarlos, entonces no se requiere mayor evaluación. Es adecuado realizar estudios de imagen después de que se controlen las crisis. Si existe antecedente de traumatismo craneal, crisis focales o signos de enfermedad sistémica se realizará la evaluación adecuada. Si el paciente tiene fiebre es necesario realizar una punción lumbar con examen del líquido cefalorraquídeo, pero solo después de excluir lesiones ocupativas y obstrucción ventricular por tomografía computada o IRM. La situación clínica determinará la decisión.
Pronóstico del tratamiento epiléptico
El pronóstico está determinado por la evolución de la enfermedad y la perspectiva futura según el trastorno de base. En relación con la epilepsia, control implica ausencia de crisis convulsivas con uso de medicamentos. Remisión indica ausencia de crisis sin medicamentos.
El pronóstico para el control médico de las crisis generalizadas y parciales ha mejorado por los métodos más exactos de evaluación, la introducción de nuevos medicamentos y el uso más racional de los agentes antiguos.36,83 El EEG y la vigilancia con video simultáneos han mejorado la exactitud diagnóstica.
Se logra una buena evolución en el 60 a 65 porciento de los pacientes con crisis de reciente inicio tratadas con un solo medicamento.84 Es más difícil lograr el control en los pacientes que tienen epilepsia de larga duración, crisis parciales, más crisis antes de iniciar el tratamiento, crisis con una causa conocida y patrones epileptiformes en el EEG.85,86 El mal control de las crisis se asocia también con menor adaptación social, múltiples tipos de crisis con alteraciones en el EEG, retraso en el inicio del tratamiento de más de un año, y crisis frecuentes.87 Algunos estudios muestran remisión a cinco años en por lo menos el 70 porciento de los pacientes que fueron vigilados durante 20 años. De estos pacientes, el 50 porciento estaba en remisión verdadera y no tomaba ningún medicamento.88
Los pacientes con crisis parciales complejas tienen un pronóstico menos favorable para el control que los pacientes con crisis generalizadas, pero la buena evolución de los primeros se asocia con estado mental normal, duración breve de la enfermedad y baja frecuencia de las crisis.89 Sin embargo, si las crisis parciales complejas se complican con crisis generalizadas tónico-clónicas, es difícil lograr un control completo. Los pacientes con riesgo alto para desarrollar crisis parciales complejas intratables son los que tienen crisis en racimos, más de una crisis al día, aura al inicio de la crisis y enfermedad psiquiátrica.90 En estudios farmacológicas que emplearon carbamacepina, renitoina y barbitúricos en dosis altas hasta alcanzar la toxicidad clínica en pacientes con crisis intratables, el tratamiento en dosis altas causó control completo en el 22 porciento de los pacientes, el 38 porciento tuvo aumento o ningún cambio en la frecuencia de las crisis y alrededor del 30 porciento no se afectó por el medicamento.91,92
La epilepsia puede afectar la esperanza de vida. La revisión de casos revela factores en común en pacientes con epilepsia que fallecieron por muerte súbita.93 La mayoría de las muertes ocurrieron mientras los pacientes estaban en cama, con seis a 30 porciento durante el sueño. En la autopsia, pocos pacientes tenían niveles terapéuticos en sangre de los AE prescritos, el 50 porciento no tenía niveles detectables. Las crisis convulsivas no se asocian con muerte inmediata,94 y se han implicado a las arritmias cardiacas.94,95 La prevalencia de muerte súbita va de uno en 2,000 a uno en 900 pacientes con epilepsia.
Aspectos especiales
INCAPACIDAD PSICOSOCIAL
Las condiciones médicas crónicas que requieren de tratamiento médico continuo afectan la autoimagen y autoestima. La epilepsia se asocia además con el temor a la pérdida del control y presenta presiones situacionales como falta de empleo, incapacidad para obtener un seguro y restricciones para manejar. Todos estos problemas exacerban el aislamiento social. Los recursos locales, como el capítulo de la Fundación Americana contra la Epilepsia, en los EUA, proporciona una oportunidad a los pacientes para recibir ayuda para estos problemas importantes.
Debe indicarse a cada paciente que debe cumplir con las leyes locales sobre el manejo de vehículos de motor, y esta advertencia debe anotarse en su expediente.
ANTICONCEPTIVOS ORALES
Los AE inductores de enzimas, como la carbamacepina aceleran el metabolismo de los anticonceptivos orales.96 Las pastillas anticonceptivas pueden fallar y puede ser necesario ajustar la dosis de hormona, además de avisar a la paciente de esta interacción farmacológica. La discusión debe anotarse en el expediente.
EMBARAZO
Aunque los ginecólogos suelen instruir a las pacientes para evitar los medicamentos durante el embarazo, las mujeres con epilepsia requieren tratamiento AE a largo plazo para controlar las convulsiones. Los médicos deben revisar los aspectos sobre teratogénesis con todas las mujeres en edad reproductiva, y esta discusión se registrará en el expediente.97 La epilepsia materna se asocia con riesgo de malformaciones fetales, y el tratamiento con AE aumenta el riesgo de dos a tres porciento a cuatro a seis porciento.98 Todos los AE de uso común causan malformaciones mayores, incluyendo defectos del tabique ventricular y labio y paladar hendido. El valproato y la carbamacepina se asocian con espina bífida, la incidencia es de uno a dos porciento, que es 10 a 20 veces mayor que la de la población general.99,100 Sin embargo, son comunes las malformaciones morfológicas menores. Se han observado alteraciones de la porción central de la cara, hipoplasia digital e hipoplasia de los lechos ungueales en el cinco a 30 porciento de los hijos de madres tratadas con AE. Las mujeres que reciben valproato y carbamacepina necesitan orientación especial antes de la concepción.
Se desconocen los mecanismos de teratogénesis pero se han desarrollado recomendaciones con base en estudios de población. Las madres epilépticas deben ser tratadas con la dosis eficaz más baja de un medicamento, y la frecuencia de las dosis se diseñará para evitar las concentraciones séricas altas. Cuando es posible, es mejor emplear un solo fármaco. Se requiere administrar suplementos de folato antes y después de la concepción. Todas las mujeres epilépticas que estén planeando un embarazo deben recibir por lo menos 1 mg de folato antes del embarazo y durante el mismo. Debe administrarse vitamina K1 durante las últimas cuatro semanas de la gestación para reducir la ocurrencia de coagulopatía en el recién nacido. Si la madre considera la opción del aborto terapéutico en caso de que el feto tenga un defecto del tuho neural, se requerirá evaluación cuidadosa con ultrasonido e incluso amniocentesis.
PACIENTES ANCIANOS
La incidencia de epilepsia entre los ancianos aumenta al avanzar la edad de la población. Las causas incluyen infarto cerebral, neoplasias y enfermedad cerebral degenerativa. La elección de los medicamentos es semejante a la de los pacientes más jóvenes, pero los cambios en la función corporal asociados con el envejecimiento complican el tratamiento. La reducción en la función hepática y renal alteran la unión de los medicamentos a las proteínas y su eliminación. Estos cambios obligan a los médicos a usar aspectos clínicos, como la frecuencia de las crisis, y la vigilancia estrecha para detectar síntomas de efectos adversos, en lugar de medir las concentraciones séricas del fármaco. Además, por los padecimientos concomitantes los pacientes suelen tomar múltiples fármacos, lo que causa problemas de interacción. La medición de los niveles libres del medicamento es más útil que medir los niveles totales en plasma.101
Bibliografía
DR. L. JAMES WILLMORE
DR. JAMES A. FERRENDELLI
Definición
Las crisis convulsivas son descargas temporales, paroxísticas y sincrónicas de grupos de neuronas en el cerebro. Aunque se consideran anormales, las crisis convulsivas pueden ocurrir en tejido cerebral tanto normal como anormal. Las manifestaciones clínicas de las crisis dependen de la localización y tamaño del grupo de neuronas que participan en la descarga convulsiva, además de la duración de la descarga. Las crisis convulsivas pueden ser causadas por una alteración temporal de la función cerebral causada por diversas causas como hipoglucemia, hiponatremia y toxicidad por medicamentos. En estos casos el individuo tiene solo una o algunas pocas crisis, y el tratamiento del padecimiento de base corrige el trastorno convulsivo.
La epilepsia se define como crisis recurrentes y puede deberse a factores congénitos o adquiridos. La epilepsia adquirida es resultado de una alteración neurológica crónica, con frecuencia estática, que ha producido una cicatriz en la corteza cerebral. Alrededor del 10 porciento de la población tendrá una o varias crisis convulsivas durante su vida, y ocurre epilepsia en el uno a dos porciento de la población. Las crisis convulsivas son el trastorno neurológico más común, y puede ocurrir a cualquier edad. La incidencia de epilepsia es alta en la infancia, pero disminuye durante la niñez, es muy baja en la adolescencia y la edad adulta y aumenta mucho en los ancianos.
Clasificación
Se han usado varias clasificaciones para las crisis convulsivas, y esto creó confusión importante en relación con los tipos de crisis. En 1981 la Liga Internacional contra la Epilepsia propuso una clasificación basada en criterios clínicos y electroencefalográficos [ver tabla 1]. Esta clasificación es muy aceptada y divide a las crisis en tres categorías principales: crisis parciales, crisis generalizadas y no clasificadas.1
|
|
* De la Commission on Classification and Terminology of the International League Against Epilepsy.1 |
CRISIS PARCIALES
Las crisis parciales pueden ser simples o complejas.2 Una crisis convulsiva que comienza en una región de la neocorteza que controla el movimiento o la sensación puede causar manifestaciones motoras o inducir experiencias sensoriales según la función de la estructura en donde se origina. Si el paciente permanece consciente la crisis se clasifica como parcial simple. Sin embargo, si la descarga focal afecta regiones cerebrales relacionadas con el estado de alerta o si la crisis se generaliza lo suficiente para causar pérdida de la conciencia del paciente, la crisis se clasifica como parcial compleja.
Las crisis parciales simples pueden originarse de cualquier región neocortical . El espectro varía desde la mioclonía local de una extremidad o región de la mano o cara hasta experiencias sensoriales como ver manchas o líneas de colores, como ocurre con las descargas en la corteza visual primaria. Una crisis parcial simple puede causar síntomas que el paciente reconoce, conocidos con frecuencia como aura.3 La crisis parcial puede diseminarse para volverse compleja o incluso tónico-clónica generalizada. Los pacientes con crisis parciales complejas pierden la conciencia independiente de la experiencia que haya precedido a la crisis. Aunque el nivel de conciencia del paciente es un elemento clave en la definición, esta información puede ser difícil de obtener. Durante el estado de conciencia alterada los pacientes pueden ser incapaces de obedecer órdenes o interactuar con el medio exterior, así como de recordar los eventos que ocurrieron durante la crisis. La diferenciación entre una crisis parcial simple o compleja puede depender de un observador, con frecuencia un familiar. Aunque el paciente puede opinar, el reporte de un testigo es un aspecto importante para asegurarse que la clasificación es la adecuada. Por ejemplo, un paciente puede perder la capacidad de hablar durante una crisis que se origine en la corteza temporal, frontal o parietal dominante. Sin embargo, al observar que el paciente obedece órdenes durante la crisis o recuerda información verbal específica, puede determinarse que un paciente está manteniendo el conocimiento a pesar de la impresión obtenida durante la crisis.
Las crisis parciales complejas suelen comenzar con detención del movimiento y la mirada en blanco. Pueden ocurrir automatismos como movimientos simples de la mano o actitudes oroalimentarias como movimientos de saborear o deglutir, así como repeticiones verbales al inicio o durante la crisis. Si el paciente está realizando alguna tarea compleja al inicio de la crisis, puede continuar con ella, aunque la exactitud del comportamiento disminuirá. Son puntos clave la duración de la crisis, porque la mayoría de las crisis parciales complejas duran solo algunos minutos, y la característica estereotipada del comportamiento. Una crisis se considera estereotipada cuando los comportamientos son los mismos o semejantes en todas. Al final de la crisis el paciente puede presentar confusión momentánea, fatiga o desorientación. Esta alteración tiene gran importancia diagnóstica y debe investigarse con preguntas dirigidas. Los efectos postictales distinguen a una crisis parcial compleja de lóbulo temporal de una crisis de ausencia, porque en la segunda el paciente no tiene síntomas postictales.4
Las crisis convulsivas parciales simples y complejas pueden generalizarse en forma secundaria y causar una convulsión tónico-clónica. Es común que la descarga focal se disemine de un área local a lodo el cerebro, aunque la mayoría de las crisis parciales no se generalizan en forma secundaria. Debido a que es posible que los testigos o el paciente no noten la manifestación focal de una crisis que es secundariamente generalizada, es mejor asumir de una crisis tónico clónica recién diagnosticada se originó en un foco hasta que no se demuestre lo contrario.5 Las crisis tónico clónicas de reciente inicio en el adulto obligan a excluir una lesión focal, como un tumor, infarto o infección cerebral.
CRISIS GENERALIZADAS
Las crisis generalizadas causan un espectro de alteración que va desde el patrón no convulsivo de una simple ausencia hasta mioclonías y crisis tónico-clónicas generalizadas.6 Las crisis de ausencia, parte de un síndrome epiléptico que inicia en la infancia, son breves, suelen durar 10 segundos o menos. El paciente no tiene aura y cuando la crisis termina no existen efectos postictales. El paciente detiene su actividad y deja la vista fija. En ocasiones existe una manifestación clónica leve como parpadeo sutil de los párpados o cambios en el tono postural. Los pacientes con crisis de ausencia tienen un patrón electroencefalográfico característico de descargas generalizadas espiga onda a velocidad de 3/seg. Esta alteración del EEG se activa al forzar la respiración. Por lo demás, los pacientes tienen función neurológica e intelectual normales. Las crisis de ausencia atípicas difieren de las simples porque inician a edad más temprana, el EEG de base es anormal y las descargas son más lentas de 3/seg. Estos pacientes pueden tener también crisis atónicas y mioclónicas y sufrir retraso mental.
Los movimientos motores de las mioclonías se manifiestan como contracciones breves de un músculo o grupo de músculos específico. Las mioclonías hipnagógicas son comunes y normales. Las mioclonías focales pueden ser causadas por lesiones destructivas del tallo cerebral o la médula espinal. La enfermedad metabólica, la hipoxia, los procesos tóxicos y las infecciones pueden causar mioclonías focales o difusas. Las mioclonías asociadas con epilepsia suelen ser simétricas, y éstas son un componente de varios síndromes epilépticos y ocurren antes o como parte de las crisis tanto de ausencia como tónico-clónicas generalizadas. Un síndrome importante que debe identificarse es el de la epilepsia mioclónica juvenil porque se trata en forma específica con ácido valproico. Los pacientes con este síndrome tienen mal pronóstico si se suspende el tratamiento, por lo que deben recibirlo de por vida.7 Las crisis mioclónicas pueden ser causadas por otros trastornos además de la disfunción cerebral primaria, incluyendo enfermedades metabólicas o trastornos cerebrales genéticos como la enfermedad de Lafora.
Las crisis atónicas causa pérdida súbita del tono postural, en ocasiones con caídas y traumatismos de la cabeza o resto del cuerpo. Este tipo de crisis suelen ser refractarias a tratamiento y con frecuencia se asocian con el síndrome de Lennox-Gastaut.
Las convulsiones son el tipo más común de crisis generalizadas. Se caracterizan por pérdida de la conciencia asociada con apnea y contracturas violentas de la musculatura del tronco y las extremidades. Con frecuencia los pacientes tienen traumatismo oral e incontinencia vescial. Durante la crisis aumentan la salivación, la frecuencia cardiaca y la presión arterial. La mayoría de las convulsiones generalizadas comienzan con una fase tónica, en la que existe contracción sostenida de todos los músculos con extensión de las extremidades inferiores y flexión o extensión de las superiores. Esta fase dura varios segundos y es seguida de una fase clónica, en la que se presentan contracciones rítmicas de las extremidades que comienzan con movimientos de alta frecuencia y poca amplitud y después disminuyen gradualmente en frecuencia en un periodo de varios segundos a minutos. Algunos pacientes solo tienen crisis tónicas o clónicas. Puede observarse una secuencia de movimientos clónicos, tónicos y clónicos de nuevo en los pacientes con crisis generalizadas primarias. Después de que ceden las contracciones musculares violentas, el paciente entra en una fase postictal, en la que se restablece la respiración y la falta de respuesta es seguida de recuperación gradual del estado de conciencia. El paciente puede permanecer confuso por algunos minutos y referir después dolor muscular y cefalea. Esta secuencia es estereotipada, independientemente de la causa. Si no se encuentra una causa estructural, la convulsión se considera como idiopática o generalizada primaria. Si la convulsión es precedida por una crisis parcial se dice que es una convulsión generalizada en forma secundaria.
SINDROMES EPILEPTICOS
En 1989 la Liga Internacional contra la Epilepsia reconoció que muchos pacientes con crisis convulsivas tienen alteraciones cerebrales que afectan su calidad de vida en forma independiente a la epilepsia. Esta clasificación6 reconoce que un síndrome epiléptico incluye no solo el comportamiento durante la crisis, sino también los cambios EEG, el desarrollo mental y motor del paciente y la historia familiar [ver tabla 2]. Debido a que el repertorio de manifestaciones convulsivas es limitado o estereotipado, pueden encontrarse tipos similares de crisis como componentes de varios síndromes con pronósticos muy diversos. La definición de un síndrome específico de epilepsia con frecuencia requiere de evaluaciones repetidas, valoración del desarrollo y revisión de las respuestas al tratamiento. Los síndromes se consideran benignos o progresivos. Estos términos se aplican generalmente para referirse a la evolución final de la función intelectual y la supervivencia. Aunque algunas crisis sindromáticas son benignas en relación con su impacto sobre la función intelectual, puede requerirse tratamiento de por vida con fármacos antiepilépticos. A continuación se describen cinco síndromes epilépticos comunes.
|
||||||||
* De la Commission on Classification and Terminology of the
International League Against Epilepsy.6
|
Epilepsia benigna de la infancia con espigas centrotemporales
Este síndrome común se caracteriza por crisis motoras hemifaciales que son breves, parciales, por lo general nocturnas y en ocasiones generalizadas. Los pacientes tienen también síntomas neurosensoriales como fasciculaciones faciales y parestesias de la lengua. El desarrollo de estos pacientes es normal, sin cambios focales en los estudios contrastados de cerebro. El EEG muestra activación durante el sueño de espigas centrotemporales de alto voltaje con una configuración truncada y una onda lenta característica. Se presenta remisión completa en más del 90 porciento de los pacientes hacia la mitad de la adolescencia.8
Ausencias infantiles
Se calcula que la prevalencia de las ausencias infantiles es de dos a ocho porciento.9 Las alteraciones características del EEG de descargas espiga onda a 3/seg ocurren sobre un patrón basal normal [ver figura 1].10 Aunque se han reportado remisiones hasta en el 80 porciento de los casos, el seguimiento a largo plazo muestra que el desarrollo de crisis tónico-clónicas reduce el número de pacientes que logran una remisión sin medicamentos. Solo el 30 porciento de los pacientes con ausencia tienen resolución completa sin medicamentos antiepilépticos si presentan crisis tónico-clónicas generalizadas. La evolución tampoco es muy optimista para los pacientes que tienen más de 15 años de edad o con más de 15 años de evolución.11 La remisión es más frecuente cuando la enfermedad es más reciente, lo que implica la necesidad de identificar pronto a los pacientes e instituir el tratamiento de inmediato. Los factores asociados con un buen pronóstico son un coeficiente de inteligencia (IQ) normal e historia negativa de crisis tónico-clónicas generalizadas por desgracia, estas últimas complican la evolución de las crisis de ausencia en alrededor del 50 porciento de los pacientes.l2 Los factores de riesgo para el desarrollo de crisis tónico-clónicas incluyen edad de inicio más tardía, dificultad para controlar las crisis de ausencia con los medicamentos y actividad basal anormal en el EEG.10,13 Antes del desarrollo de medicamentos de amplio espectro algunos pacientes recibían un medicamento adicional para prevenir las crisis tónico-clónicas.
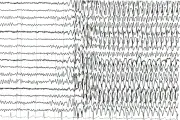
|
| Figura 1 |
| Ausencias de la infancia: electroencefalograma |
Este enfoque de tratamiento dual ha sido sustituido por el uso del valproato, un medicamento de amplio espectro con eficacia combinada contra crisis tanto de ausencia como tónico-clónicas. La discusión del tratamiento con los padres y los pacientes debe incluir el informarles sobre la posibilidad de desarrollar crisis tónico-clónicas.l4
Epilepsia mioclónica juvenil
Los pacientes con epilepsia mioclónica juvenil tienen contracciones mioclónicas características, crisis generalizadas tónico-clónicas, historia familiar de crisis convulsivas y fotosensibilidad en el EEG. Entre los precipitantes de las crisis se incluyen la deprivación de sueño, el estrés y el uso de alcohol. Las mioclonías son más importantes por la mañana y pueden afectar los músculos grandes de las extremidades inferiores. Es importante realizar un diagnóstico correcto porque las crisis suelen controlarse bien con valproato. El pronóstico para una función intelectual normal suele ser bastante bueno, sin embargo, para la remisión sin medicamentos es malo.11,15
Epilepsia parcial continua crónica progresiva
Los pacientes con crisis parciales o alteraciones cerebrales focales pueden presentar crisis focales continuas. El EEG muestra espigas focales continuas y ondas lentas. El pronóstico se relaciona con la lesión patogénica subyacente y no suele asociarse con progresión clínica. La forma infantil (encefalitis de Rassmusen) comienza con crisis focales que afectan los sistemas motores y después progresan para provocar deficiencias motoras y deterioro mental.8,16 También se han reportado casos de esta forma rara de encefalitis en adultos.
Epilepsia mioclónica severa de la infancia
Aunque las crisis mioclónicas generalizadas o focales son características de varios síndromes de la infancia, la observación de disfunción neurológica progresiva con deterioro de los sistemas visual o motor piramidal o extrapiramidal identifica a los pacientes con esta forma de epilepsia mioclónica.l7 Las causas incluyen enfermedad de Unvericht-Lundborg, Lafora, lipofuscinosis neuronal ceroide, sialidosis y encefalomielopatía mitocondrial.18 Debido a que los síndromes de la infancia benignos en ocasiones son difíciles de diferenciar de las crisis mioclónicas progresivas, es importante realizar exámenes repetidos con evaluación de la función neurológica. La evolución depende de la enfermedad subyacente, las epilepsias mioclónicas progresivas son fatales.
Evaluación diagnóstica
Aunque la historia clínica del paciente constituye la base para la caracterización de las crisis convulsivas, se requieren varios estudios de laboratorio para establecer el diagnóstico, esquema de tratamiento y pronóstico.
ELECTROENCEFALOGRAFIA
Un EEG es una imagen gráfica o electrónica de la actividad fisiológica cerebral amplificada registrada en electrodos colocados en el cráneo del paciente con una distribución ya establecida. Los patrones convulsivos focales o generalizados que interrumpen la actividad de base confirman el diagnóstico de epilepsia. La utilidad diagnóstica de un EEG mejora si el paciente es preparado con deprivación de sueño, de modo que ocurra sueño natural durante el estudio, ya que éste aumenta la actividad epileptiforme. Los pacientes con crisis parciales que se originan en estructuras de la porción media del lóbulo temporal con frecuencia tienen un EEG normal al inicio del estudio. Se requieren varios EEG para confirmar el diagnóstico. Los patrones diagnósticos incluyen espigas agudas focales, ondas o complejos espiga-onda [ver figura 2]. Las alteraciones generalizadas varían desde supresión momentánea de los ritmos de base hasta racimos de espigas rápidas múltiples, o al patrón clásico de descargas espiga onda con frecuencia de 3/seg, característico de las ausencias.
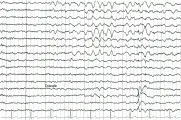
|
| Figura 2 |
| Crisis parciales complejas: electroencefalograma |
IMAGEN
Debido a que la epilepsia es un síntoma, en la evaluación diagnóstica debe buscarse alguna enfermedad susceptible de tratamiento. Los estudios de la estructura cerebral, incluyendo la tomografía computada y la resonancia magnética, tienen utilidad y aplicaciones diferentes, dependiendo de la edad del paciente al inicio de la epilepsia. La mayoría de los pacientes con epilepsia que tienen un IQ normal y patrones basales normales en el EEG tendrán imágenes estructurales normales en la IRM cerebral. Sin embargo, se encuentran alteraciones en la IRM en alrededor de la mitad de los pacientes con crisis parciales complejas originadas en el lóbulo temporal. En los adultos con epilepsia de reciente inicio y crisis focales o generalizadas debe usarse la IRM para tratar de definir la causa. Por ejemplo, el 19 porciento de los pacientes entre 18 y 50 años de edad pueden tener tumores cerebrales, con crisis convulsivas como manifestación clínica inicial.19 Otras lesiones que causan crisis convulsivas en este grupo de edad incluyen malformaciones arteriovenosas, infección intracraneal e infarto cerebral. El volumen del hipocampo puede evaluarse con la IRM. Aunque se encuentra esclerosis media temporal en alrededor del 15 porciento de los pacientes con crisis parciales complejas originadas en este lóbulo, se observa atrofia focal de la formación del hipocampo en por lo menos el 60 porciento de los casos.20
La tomografía computada por emisión de fotón único (SPECT, por sus siglas en inglés, n. del t.) demuestra reducción del flujo sanguíneo en la región en que se origina la crisis en algunos pacientes. La certeza diagnóstica mejora al inyectar el isótopo del SPECT durante una crisis parcial compleja. La tomografía de emisión de positrones es un estudio adyuvante importante en pacientes seleccionados y revela hipometabolismo ictal en el 70 a 79 porciento de los pacientes con crisis parciales complejas originadas en el lóbulo temporal.21
Diagnóstico diferencial
Aunque las alteraciones súbitas en la función neurológica son características de las crisis convulsivas, también ocurren cuando las estructuras craneales dejan de recibir el aporte adecuado de glucosa u oxígeno. La ausencia de perfusión cerebral adecuada provoca pérdida de la conciencia. La disfunción cardiaca por falla de la pared miocárdica, bloqueo de rama, bloqueo auriculoventricular y taquicardia paroxística, disminuirá el gasto cardíaco y causarán síncope. Si el paciente queda en posición horizontal, con el corazón en el mismo plano que el cerebro, la perfusión cerebral se restablece. Sin embargo, algunos observadores mantienen al paciente en posición erecta, lo que prolonga la interrupción de la circulación cerebral. Pueden ocurrir movimientos convulsivos o incluso tónico clónicos durante la interrupción prolongada de la perfusión cerebral.
La alteración para mantener la resistencia vascular periférica suele ser de tipo insidioso, y por lo general se debe a neuropatía autonómica que complica a la neuropatía periférica, la diabetes mellitus o el síndrome de Shy-Drager. El espectro de síntomas de la hipotensión ortostática varía de vértigo a síncope. Algunos medicamentos causan alteraciones en el tono vascular periférico, provocando síntomas de tipo ortostático como alteración de la conciencia. En cualquier paciente con alteración de la conciencia se requiere revisar el tratamiento farmacológico y la historia clínica para detectar la inducción postural de los síntomas.
La disfunción sensorial o motora puede ser causada por eventos isquémicos temporales. Estos pueden deberse a enfermedad vascular embólica cerebral, lesión extracraneal de la arteria carótida o basilar, o incluso migraña. La enfermedad metabólica, en especial la alteración del metabolismo de la glucosa que amerita tratamiento con insulina, puede causar hipoglucemia.
Como en otros aspectos del diagnóstico de epilepsia, la historia clínica es la clave para el diagnóstico diferencial. Todos los pacientes que inician con una crisis convulsiva deben someterse a estudios de escrutinio bioquímico y celular. Estos deben incluir un perfil químico de glucosa, nitrógeno de urea, electrolitos y niveles circulantes de enzimas hepáticas. La biometría hemática puede sugerir una infección, alteración plaquetaria o anemia. Debe realizarse punción lumbar con estudios de líquido cefalorraquídeo si está indicado o en caso de duda clínica. Está indicado realizar punción lumbar si el paciente tiene fiebre o presenta alteraciones en la función cognitiva, por ejemplo cambios de comportamiento que puedan atribuirse a un proceso encefalopático. Este estudio se realizará después de excluir la presencia de una masa intracraneana o de aumento en la presión intracraneal.
Un problema común es la ocurrencia de seudocrisis. Alrededor del 20 porciento de los pacientes que ingresan a unidades de vigilancia de epilepsia para evaluación diagnóstica tienen alteraciones episódicas en el comportamiento que no son causadas por disfunción fisiológica del cerebro. Aunque el término seudocrisis es apropiado para la codificación diagnóstica y la comunicación entre médicos, los eufemismos como crisis o eventos no epilépticos son más adecuados al hablar con los pacientes. El uso de estos términos ayuda a los pacientes a comprender su problema y facilita su referencia para una terapia de comportamiento. Los pacientes reaccionan con enojo ante el término seudo, y es menos probable que crean que el médico se interesa por su problema. Las claves para el diagnóstico incluyen eventos periódicos que tienden a no ser estereotipados. Tanto los pacientes como los observadores reportan un comportamiento diferente en cada evento. Otra clave es la duración prolongada. Las seudocrisis pueden durar 30 minutos a varias horas. La mayoría de las crisis, tanto parciales como generalizadas, rara vez persisten por más de varios minutos. Los pacientes que tienen tanto seudocrisis como epilepsia real constituyen un problema clínico especial, y esta coexistencia existe en alrededor del 25 porciento de los pacientes sometidos a unidades de vigilancia de epilepsia. El tratamiento de las seudocrisis requiere de intervención en el comportamiento. Si se encuentran ambos problemas se requerirá tratamiento antiepiléptico en paralelo a la terapia de comportamiento.
Tratamiento de las crisis convulsivas y la epilepsia
TRATAMIENTO FARMACOLOGICO
El tratamiento principal de las crisis convulsivas es el farmacológico. Los medicamentos anticonvulsivantes, conocidos también como antiepilépticos (AE), están disponibles para uso clínico desde hace más de un siglo. Sin embargo, la era moderna del tratamiento AE comenzó a principios del siglo XX con la introducción del fenobarbital. En la actualidad se dispone en los Estados Unidos de más de una docena de agentes clasificados como AE. La mayoría comenzaron a usarse antes de 1980, aunque existen nuevos medicamentos y se espera que se introduzcan algunos más en los próximos años.
El tratamiento debe dirigirse tanto a controlar las crisis como, si es posible, a corregir la enfermedad o trastorno subyacente. Para los pacientes que han tenido una sola crisis o muy pocas por un trastorno temporal, como una intoxicación farmacológica, supresión alcohólica o de sedantes-hipnóticos, hiponatremia o hipoglucemia, pueden no requerirse AE o usarse por muy poco tiempo. Los pacientes que tienen crisis recurrentes secundarias a una enfermedad neurológica trabable, como un tumor cerebral o una infección intracraneal, deben recibir AE y también el tratamiento de la enfermedad de base. En muchos pacientes la causa de las crisis no se identifica o es un proceso estático, como un infarto cerebral establecido o una contusión secundaria a un traumatismo craneal. En estos pacientes el tratamiento con EA es el único posible.
Los pacientes con crisis recurrentes crónicas, independientemente de la etiología, deben tratarse con AE. Sin embargo, existe controversia respecto a si el tratamiento debe iniciarse después de la primera crisis. Muchos especialistas afirman que debido a que muchos pacientes no tienen una segunda crisis durante varios años de seguimiento, no parece existir justificación para comenzar el tratamiento y someter al paciente a sus posibles efectos adversos.22 Esta idea es apoyada por un estudio retrospectivo que reportó que solo el 60 porciento de los pacientes con una primera convulsión tuvieron una segunda en un periodo de cinco años.23 Sin embargo, este punto de vista no es aceptado por todos los clínicos porque muchos pacientes tienen crisis recurrentes. Los autores recomiendan no tratar al paciente después de la primera crisis si el examen neurológico, EEG y estudio de imagen del cerebro son normales, y el enfermo acepta someterse al riesgo asociado con otra crisis convulsiva. Sin embargo, debe iniciarse tratamiento anticonvulsivo para la mayoría de los pacientes después de una primera crisis cuando la evaluación revela cualquier alteración cerebral estructural o funcional. También, el riesgo de recurrencia de las crisis disminuye en forma progresiva mientras más tiempo permanezca el paciente sin sufrir crisis.
El tratamiento con AE debe seguir ciertos principios básicos. El tratamiento debe iniciarse con un solo agente adecuado. Debe lograrse el control de las convulsiones aumentando la dosis de este medicamento en lugar de agregando un segundo. Si no se controlan las crisis con el primer fármaco, debe considerarse un segundo como alternativa. Siempre que sea posible debe evitarse administrar dos o más AE en combinación, aunque las combinaciones racionales pueden ser útiles cuando falla la monoterapia. Los cambios en la dosis deben ser guiados por la respuesta clínica del paciente y no por las concentraciones séricas, y el control inadecuado indica la necesidad de elevar la dosis, mientras que la presencia de toxicidad sugiere que ésta debe disminuirse. Por lo general no es necesario vigilar los niveles séricos del medicamento en los pacientes con buen control de las crisis que toman un medicamento bien tolerado. Sin embargo, esto puede ser útil en ciertas circunstancias, como para establecer el cumplimiento o investigar cambios inexplicables en el control de las crisis o la toxicidad del fármaco.
Mecanismos de acción de los AE
Esencialmente todos los AE disponibles fueron identificados al investigar su actividad anticonvulsivantes en crisis inducidas en forma experimental en ratas y ratones de laboratorio. Todos bloquean las crisis por electrochoques, las crisis inducidas por químicos o ambas. Se han realizado gran cantidad de investigaciones para definir los mecanismos de acción de los AE. Cada AE parece tener varias acciones moleculares y celulares, pero es probable que solo algunas de estas acciones individuales sean responsables de los efectos anticonvulsivantes y antiepilépticos. En general, los AE parecen disminuir la excitabilidad o aumentar la inhibición neuronal. Esto se logra alterando las corrientes intrínsecas de la membrana, como la conductancia de sodio, potasio y calcio, o afectando la actividad de varios neurotransmisores, como el ácido 8-aminobutírico (GABA), el glutamato u otros neurotrasmisores que pueden participar en la actividad convulsiva. Aunque varios AE comparten mecanismos en común, cada uno parece tener acciones diferentes.
Los medicamentos que actúan sobre las corrientes intrínsecas de la membrana pueden dividirse en forma burda en los que afectan principalmente el canal de sodio con puerta dependiente de voltaje y los que alteran las corrientes de calcio. Los medicamentos que modifican los canales de sodio en concentraciones terapéuticas (fenitoína, carbamacepina, primidona, ácido valproico y lamotrigina) inhiben las descargas neuronales de alta frecuencia.24-27 Debido a que son dependientes tanto de uso como de voltaje, tienen poco efecto sobre la actividad neuronal normal. Sin embargo, durante las descargas de alta frecuencia retrasan la reactivación del canal de sodio y producen un mayor efecto inhibitorio sobre el potencial de acción hasta que la descarga está completamente bloqueada. La actividad antiepiléptica de la etosuximida es resultado de la acción en los canales de calcio.28,29 La etosuximida bloquea en forma selectiva la corriente de calcio tipo T, que no se afecta mucho por la mayoría de los otros AE. Se considera que esta corriente actúa como un marcapaso en las neuronas talámicas y puede ser importante en la actividad de la epilepsia por ausencias.30 El ácido valproico y el lamotrigine son también eficaces contra las crisis de ausencia, aunque aún no se determina si su utilidad es resultado de acción sobre la corriente de calcio tipo T o por otros mecanismos aún no definidos. El bloqueo de otras corrientes de calcio, como la tipo N, principalmente por el fenobarbital y la fenitoína, puede tener un efecto menos específico que interfiere principalmente con la liberación de trasmisores.
Los medicamentos que alteran la función sináptica actúan principalmente aumentando la inhibición neuronal mediada por GABA, el principal mecanismo inhibitorio cerebral. Estos medicamentos incluyen fenobarbital, benzodiacepinas y quizá otros. Cada uno de estos fármacos actúa por un mecanismo diferente para aumentar la influencia del GABA en el sistema nervioso central.31 Las benzodiacepinas aumentan la frecuencia de apertura en los canales receptores de GABAA,32,33 y los barbitúricos aumentan la duración de apertura de los mismos.34,35
Es claro que los mecanismos anticonvulsivantes de los principales medicamentos usados para los trastornos convulsivos aún no se conocen del todo. Algunos AE pueden afectar la conductancia de iones diferentes a los analizados, y es posible que algunos afecten en forma directa o indirecta procesos de neurotrasmisores diferentes al sistema GABA. Estas acciones pueden ser importantes por sus efectos clínicos. Se requieren más investigaciones para definir por completo los mecanismos moleculares y celulares, además de los sitios de acción, de los fármacos AE.
Selección de AE
Los AE más empleados en Estados Unidos para tratar la epilepsia son la carbamacepina, la etosuximida, el gabapentin, el lamotrigine, el fenobarbital, la fenitoína, la primidona y el valproato [ver tabla 3]. Algunas benzodiacepinas, incluyendo el clonacepam, el diacepam y el loracepam, se usan también para tratar las crisis convulsivas. Con excepción del clonacepam, las benzodiacepinas se usan para tratamiento a corto plazo de las crisis agudas o del estado epiléptico y por lo general se administran por vía parenteral. El clonacepam puede usarse para tratar la epilepsia, aunque esto no es recomendable porque la mayoría de los pacientes desarrollan tolerancia a su efecto antiepiléptico. El felbemato, un AE de reciente introducción, se ha prescrito en algunos pacientes con crisis incontrolables, pero su uso está limitado por una alta incidencia de efectos adversos. Se han usado otros medicamentos adicionales, incluyendo metosuximida trimetadiona y acetazolamida, pero no se analizarán en esta subsección.
|
||||||||||||
* Dosis total |
||||||||||||
La mayoría de los especialistas en epilepsia están de acuerdo en que los medicamentos de elección para las crisis parciales son la carbamacepina y la fenitoína, que son muy eficaces y tienen efectos adversos aceptables.36 También se ha demostrado que el valproato es eficaz en el tratamiento de las crisis parciales.37 Es probable que tanto el fenobarbital como la primidona sean tan eficaces como la carbamacepina o la fenitoína, pero ambos barbitúricos se asocian con una incidencia mucho mayor de efectos adversos, en especial sedación y deterioro cognoscitivo.36 Dos de los medicamentos más nuevos, gabapentin y lamotrigine, son eficaces contra las crisis parciales, y en la actualidad cada uno se usa en combinación con alguno de los fármacos mencionados antes. Las crisis convulsivas generalizadas pueden controlarse con carbamacepina, fenitoína, valproato, barbitúricos, gabapentin o lamotrigine. La carbamacepina, la fenitoína y el valproato parecen tener una eficacia semejante para las crisis convulsivas generalizadas. La carbamacepina, la fenitoína, el gabapentin y el lamotrigine se usan en pacientes con crisis generalizadas secundarias porque muchos de estos pacientes tienen también crisis parciales. Por otro lado, el valproato y el lamotrigine se usan con más frecuencia en pacientes con crisis generalizadas primarias porque algunos de estos pacientes tienen también crisis de ausencias, que pueden controlarse con estos medicamentos. Los barbitúricos no son de elección en el tratamiento de las crisis generalizadas principalmente por sus efectos sedantes. Las crisis generalizadas no convulsivas, en especial las de ausencia, pueden tratarse con etosuximida o valproato.38 Para los pacientes que solo tienen crisis de ausencia la etosuximida es adecuada; sin embargo, para los enfermos que tienen ausencias junto con otros tipos de crisis, como convulsiones generalizadas o crisis mioclónicas, el valproato es el medicamento de elección y se ha sugerido que también el lamotrigine es eficaz.
Farmacocinética de los AE
La mayoría de los pacientes tratados con AE requieren manejo durante varios años. Todos los AE se administran por vía oral una vez al día o con más frecuencia [ver tabla 3]. La absorción de la mayoría de los AE suele ocurrir en forma lenta durante varias horas y puede ser incompleta, en especial en el caso del gabapentin. La unión a proteínas varía mucho entre los medicamentos, variando desde cero para el gabapentin a 90 porciento o más para la fenitoína. Excepto por el gabapetin, todos los AE se metabolizan en el hígado antes de su excreción renal. La depuración y vida media de los AE varía de horas a días. Las vidas medias de la carbamacepina, valproato, primidona y gabapentin son relativamente cortas, variando de cuatro a ocho horas, lo que obliga a administrar estos medicamentos por lo menos tres veces al día. La fenitoína, el lamotrigine y el fenobarbital tienen vidas medias de un día o más y pueden administrarse una o dos veces al día. La fenitoína, la carbamacepina, los barbitúricos y quizá el lamotrigine, causan inducción enzimática, por lo que el tratamiento prolongado con estos medicamentos puede afectar su propia velocidad de metabolismo, así como el de otros medicamentos concomitantes. Todos los AE que se metabolizan pueden competir con otros fármacos que también se metabolizan en el hígado, retrasando su eliminación. Debido a su capacidad para inducir o bloquear el metabolismo farmacológico, todos los AE excepto el gabapentin pueden asociarse con interacciones farmacocinéticas en la que se afectan las concentraciones y efectos de los medicamentos administrados en forma simultánea. Este tipo de interacción puede anticiparse en todos los pacientes. Pueden vigilarse los síntomas clínicos y los niveles sanguíneos de los AE, en especial los niveles del fármaco libre, y ajustarse la dosis en caso necesario. También puede ocurrir interacción farmacodinámica. En esta situación, la combinación de dos o más medicamentos con mecanismos semejantes o antagónicos causa que sus efectos clínicos aumenten o disminuyan, y estos cambios pueden requerir de ajuste en las dosis. Las interacciones farmacodinámicas pueden anticiparse si se conocen los mecanismos de acción del medicamento.
Efectos adversos de los AE
La mayoría de los efectos adversos de los AE pueden ser tolerables. Sin embargo, los efectos idiosincráticos raros pueden ser graves. Para realizar una buena práctica médica es necesario obtener algunos estudios de escrutinio antes de iniciar los AE. Los estudios son los siguientes: biometría hemática completa con diferencial y cuenta de plaquetas, química sérica con glucosa, nitrógeno de urea, electrolitos, calcio, fósforo, magnesio, ácido úrico, hierro libre, colesterol, bilirrubinas, fosfatasa alcalina, aspartato y alanino aminotransferasas, proteínas totales, albúmina y globulina, y pruebas de coagulación. Sin embargo, el valor y necesidad de obtener estas pruebas de laboratorio de rutina durante el tratamiento no se ha demostrado. En los pacientes estables la vigilancia de rutina parece no estar justificada. Por otro lado, la vigilancia clínica por medio de revisión y reporte de síntomas u observación por medio de un examen físico tiene mejor relación costo-beneficio y es más práctica. De hecho, la vigilancia de rutina como se practica en la actualidad no permite anticipar los efectos graves asociados con la mayoría de los AE, incluyendo carbamacepina, fenitoína y fenobarbital. Por ejemplo, estudios prospectivos de evaluación rutinaria de sangre y orina en pacientes que recibieron AE por tiempo prolongado no demostraron ninguna utilidad en pacientes asintomáticos.39 Mucho más importante que la vigilancia rutinaria de la sangre es la comunicación continua con el paciente y la familia. Ellos deben estar al tanto de las posibles complicaciones y de los síntomas que pueden indicar un evento adverso. Los autores consideran que la participación informada en la vigilancia clínica tiene buena relación costo beneficio, es segura y adecuada. El uso de la experiencia clínica aunada con la educación del paciente y el conocimiento de los riesgos y beneficios del tratamiento por parte de las personas que le atienden se ha convertido en el manejo estándar.40
Todos los AE pueden producir efectos adversos, que son numerosos y varían mucho de paciente a paciente. Algunos de estos efectos indeseables están claramente relacionados con la dosis, como la neurotoxicidad, mientras que otros parecen ser idiosincráticos, como algunas formas de hepatotoxicidad o anemia aplástica, y aún otros parecen tener mecanismos distintos. Consideramos que la forma mas eficaz de comprender los efectos adversos de los AE consiste en definirlos de acuerdo con el sistema orgánico afectado y este esquema de clasificación es el que se usa en este capítulo.
Efectos sobre el sistema nervioso central La neurotoxicidad dependiente de la dosis es el efecto adverso más frecuente de los AE, y puede limitar la cantidad de medicamento que puede emplearse. Los síntomas del SNC se usan para calcular la dosis aceptable en forma independiente de los niveles de AE en sangre. Por ejemplo, la carbamacepina causa diplopia o sedación, y estos síntomas sirven como indicadores de que el paciente ha llegado a la dosis máxima tolerada. El medicamento puede administrarse en dosis progresivas hasta alcanzar el límite superior terapéutico en sangre según las estadísticas. Sin embargo, una vez que se alcance éste, la dosis se aumenta hasta que el paciente presenta neurotoxicidad. Esta define la dosis máxima tolerada. Todos los AE provocan depresión de la función cortical, con síntomas de sedación y letargo. Aunque los efectos relacionados con la dosis de la fenitoína sobre el cerebelo son indicadores clásicos de toxicidad, los hallazgos típicos con frecuencia están bloqueados o no son aparentes por las respuestas individuales de cada paciente.41 No siempre ocurre la progresión esperada de los síntomas de nistagmus a ataxia y confusión o la sensación de intoxicación. Aunque este aspecto no es del todo claro, puede ocurrir ataxia residual después de la intoxicación prolongada con fenitoína, en especial en pacientes vulnerables.42
La ataxia persistente o fluctuante parece relacionarse con la duración de la epilepsia, el uso de múltiples drogas y una nivel persistentemente alto del medicamento en suero. Se han reportado cambios en la función intelectual y alteraciones en la personalidad o incluso efectos psiquiátricos con varios AE. La alteración del IQ en niños tratados con fenitoína y primidona parece relacionarse con niveles altos en sangre de estos fármacos.
La evaluación de la función cognitiva en pacientes tratados en forma aleatoria no reveló diferencias entre los principales AE, aunque el fenobarbital pareció tener un efecto relativamente mayor que otros medicamentos.43 Los medicamentos administrados a los niños pueden tener efectos impredecibles, y los barbitúricos son una causa bien conocida de hiperactividad en niños. Los AE pueden provocar también depresión y psicosis. La mayoría de los efectos sobre el comportamiento parecen relacionarse con la dosis. Se ha reportado psicosis después del control de las crisis, y este proceso denominado normalización forzada parece relacionarse con normalización del EEG.44
La fenitoína puede causar movimientos anormales relacionados con la dosis, la carbamacepina rara vez causa movimientos involuntarios. Las disquinesias se manifiestan como movimientos asociados al uso crónico de neurolépticos, incluyendo disquinesias de la cara, extrernidades y lengua.45
Se ha reportado neuropatía sensorial leve en ocho a 15 porciento de los pacientes tratados con AE46 y al parecer requiere exposición prolongada. La exposición prolongada a niveles altos en plasma causa pérdida de las fibras mielinizadas grandes y distribución no aleatoria en racimos de desmielinización segmentaria que sugiere un patrón de neuropatía axonal y desmielinización segmentaria.47 El uso de múltiples medicamentos también se asocia con neuropatía crónica. La alteración en la función de los nervios periféricos parece deberse al efecto de los AE sobre el metabolismo del folato, con alteración leve secundaria en la función periférica. El efecto clínico no suele ser severo, pero puede ocurrir pérdida de las respuestas tendinosas profundas o de la sensibilidad vibratoria a nivel de los tobillos.
Además de estos efectos dependientes de la dosis, los AE pueden inducir una encefalopatía reversible y deterioro mental progresivo o delirio.48 En raras ocasiones la exposición inicial del paciente a valproato causa estado de coma, el mecanismo puede relacionarse con el metabolismo mitocondrial.49
Efectos gastrointestinales y hepáticos Todos los AE pueden causar daño hepático, pero la incidencia es poco frecuente, con algunas excepciones. Se ha reportado que el valproato produce falla hepática en alrededor de uno de cada 10,000 pacientes. La tasa más alta se observa en niños menores de dos años que están siendo tratados con múltiples fármacos. Los pacientes de mayor edad con un solo medicamento tienen una tasa mucho más baja: uno en 45,000. El mecanismo de la hepatotoxicidad inducida por valproato se desconoce, pero puede incluir a metabolitos tóxicos del ácido valproico. La ocurrencia poco frecuente de daño hepático asociada con la fenitoína, la carbamacepina o el fenobarbital sugiere que el mecanismo es una reacción de hipersensibilidad idiosincrática.
Se ha observado pancreatitis durante el tratamiento con valproato, y han ocurrido muertes tanto en niños como en adultos.50 Aunque el mecanismo de este efecto adverso no se ha confirmado, la reexposición de los pacientes afectados ha causado recurrencia de la pancreatitis.51
Efectos hematológicos Las reacciones hematológicas adversas asociadas con respuestas idiosincráticas de los AE varían de una reducción leve en el número de células sanguíneas hasta anemia aplástica. Por fortuna los problemas serios son poco frecuentes. La fenitoína puede alterar la función linfocitaria porque el 21 a 25 porciento de los pacientes con tratamiento prolongado tienen disminución de los niveles circulantes de IgA con depresión de la transformación linfocitaria con fitohemaglutinina.52,53 La hipersensibilidad por fenitoína puede causar linfadenopatía y en casos raros puede asociarse con linfoma.54,55 El tratamiento con carbamacepina causa con frecuencia cambios hematológicos. Se ha reportado que ocurre leucopenia relacionada con la dosis en el 12 porciento de los pacientes tratados.56,57 Este cálculo es bajo según la experiencia de los autores. Los mecanismos que provocan este efecto sobre los granulocitos se desconocen, pero el efecto parece relacionarse con la dosis y no presagia reacciones más serias. Al parecer no hay necesidad de preocuparse hasta que la cuenta total de leucocitos sea menor a 2,500/mm3 o la cuenta total de granulocitos sea de menos de 750/mm3.
Se ha reportado anemia aplástica con todos los AE, pero es poco frecuente y ningún medicamento en particular parece asociarse con más riesgo de causar esta complicación seria. La anemia aplástica relacionada al tratamiento con carbamacepina parece relacionarse con la dosis y la edad avanzada de los pacientes.56 De los 65 pacientes que fallecieron por anemia aplástica asociada con carbamacepina, solo cuatro eran niños.58 El valproato causa una reducción dependiente de la dosis en el nivel de plaquetas circulantes que solo en ocasiones es sintomático. La disminución en los niveles de plaquetas en los pacientes sin síntomas varía de 149,000 a 35,000.59 La ocurrencia de púrpura o hemorragias petequiales obliga a suspender el medicamento. El mecanismo parece ser un cambio en la adhesividad plaquetaria, con aceleración en la segunda fase de consumo de plaquetas que causa una mayor pérdida de las plaquetas circulantes. En ocasiones se detecta un autoanticuerpo IgM contra plaquetas.60 Se ha reportado macrocitosis leve y reducción en los niveles de folato en los eritrocitos asociados al uso de AE, en especial fenitoína.6l La anemia megaloblástica es un efecto ocasional del tratamiento AE que parece relacionarse con alteraciones en el metabolismo del folato. Se requieren suplementos de folato en algunos casos.
Reacciones dermatológicas Las reacciones dermatológicas de los AE no son raras; sin embargo, las respuestas serias sí lo son.62 Las erupciones inducidas por fármacos varían desde eritema leve, con o sin prurito, hasta reacciones exfoliativas serias o presencia de bulas. El exantema relacionado con medicamentos es la reacción cutánea más común. La erupción puede ser pruriginosa, maculopapular o morbiliforme, y alrededor del 50 porciento de los pacientes pueden tener síntomas prodrómicos que incluyen malestar y fiebre. Esta forma de erupción puede responder a la reducción del medicamento.62 Las erupciones farmacológicas que comienzan con una reacción pruriginosa con eritemas morbiliformes o escarlatiniformes pueden progresar hasta dermatitis exfoliativa severa. En este caso el medicamento agresor debe suspenderse pronto, sustituyéndose por otro AE. El mecanismo de la reacción cutánea puede estar determinado por factores farmacogenéticos relacionados con la capacidad de metabolizar los compuestos formados durante el metabolismo del medicamento.63,64 Las dermatitis exfoliativas producidas por los AE incluyen eritema multiforme, síndrome de Stevens-Johnson y síndrome de Lyell.65 La dermatitis exfoliativa suele ocurrir entre la primera y cuatro semanas después de iniciado el tratamiento y puede ser fatal. El medicamento que inició la reacción debe suspenderse, instituyéndose tratamiento con esteroides sistémicos.60 El eritema multiforme, como reacción a fármacos, tiene un inicio rápido, con lesiones eritematosas que pueden variar de un patrón macular con formas variadas hasta el desarrollo de vesículas o bulas. Las lesiones mucosas no son una manifestación universal, a menos que la reacción consista en un síndrome de Stevens-Johnson. En cualquiera de los casos es imperativa la consulta dermatológica. El acné es un problema cutáneo menor asociado con la fenitoína. La hipertricosis se asocia tanto con fenitoína como con carbamacepina. Ocurre alopecia durante las primeras semanas de tratamiento con valproato. La administración de multivitamínicos que contienen zinc parece prevenir este cambio en la fuerza del pelo.
Enfermedades del tejido conjuntivo Los AE pueden causar lupus eritematoso generalizado, esclerodermia, síndrome de Sjogren y fascitis eosinofílica. Los criterios de diagnóstico incluyen eritema malar con cambios discoides en la piel, fotosensibilidad, úlceras orales, artritis no erosiva, serositis y nefropatía. Los cambios hematológicos e inmunológicos incluyen preparaciones positivas de células de lupus eritematoso y anticuerpos antinucleares positivos, incluyendo anti-ADN de doble cadena.67 Los medicamentos pueden precipitar la presentación de lupus, causar exacerbación del existente o asociarse con una forma aislada relacionada al medicamento. El lupus inducido por medicamentos suele desaparecer al suspender el medicamento responsable. Además, el patrón de afección orgánica suele respetar la piel, los riñones y el SNC. El patrón inmunológico no suele incluir la inducción de anticuerpos contra ADN de doble cadena. El uso de fenitoína a largo plazo ha causado un síndrome de equimosis, hemorragia gingival y detección de anticuerpos anticoagulante lúpico asociado con deficiencia de protrombina.68 También la carbamacepina se ha relacionado con el desarrollo de lupus inducido por medicamentos.69
Efectos metabólicos y endócrinos La hiponatremia es un efecto de la carbamacepina relacionado con la dosis y que parece presentarse solo en adultos.70 Se ha considerado que el mecanismo podría incluir efectos centrales sobre la hormona antidiurética y efectos periféricos o renales, pero la patogenia exacta se desconoce. La hiponatremia asociada al tratamiento con carbamacepina rara vez causa un problema clínico. Los efectos de la fenitoína sobre la función hipófisis-suprarrenal se relacionan con los efectos periféricos de la inducción de enzimas hepáticas del citocromo P-450, con acentuación resultante del metabolismo de la hormona endógena. El metabolismo acelerado de las hormonas esteroideas administradas en forma exógena, como las pastillas anticonceptivas, puede causar fracaso del sistema anticonceptivo. La función hipotalámica puede afectarse por la fenitoína. Los cambios incluyen liberación alterada de la hormona antidiurética, bloqueo del efecto de la hormona estimulante del tiroides y mayor secreción de la hormona folículo estimulante y de la hormona luteinizante en mujeres.7l,72 Los estudios de función tiroidea se alteran por la fenitoína, pero por lo general no se encuentran efectos clínicamente significativos. La triyodotironina total (T3) y la tiroxina (T4) disminuyen porque la fenitoína causa su desplazamiento de la globulina fijadora de tiroxina.73
TRATAMIENTO QUIRURGICO
El tratamiento quirúrgico es una opción para los pacientes que no responden al tratamiento convencional con AE o que tienen efectos adversos intolerables, cuyas crisis tienen un origen focal y que se originan en tejido que puede ser extirpado sin causar incapacidad.74 La epilepsia puede asociarse con una lesión que tiene un cambio estructural que correlacione con la región de inicio de la crisis. La presencia de un foco epiléptico en el hemisferio dominante para el lenguaje y la ocurrencia de crisis parciales complejas que se originen del teido extratemporal requiere de una evaluación especial. La detección de descargas epileptiformes bilaterales, el desarollo de crisis secundariamente generalizadas y el inicio ocasional de una crisis del tejido contralateral tienden a aumentar la complejidad de la evaluación preoperatoria.75,76
La técnica estándar de cirugía requiere la localización del foco epileptógeno de varias maneras. Aunque se han usado el EEG ictal e interictal de superficie y la electrocorticografía intraoperatoría, estos métodos no proporcionan una información adecuada sobre localización o lateralización. Por lo tanto, la mayoría de los centros de epilepsia emplean la combinación de un EEG de superficie y de un video para grabar por lo menos tres de las crisis típicas del paciente. Se obtiene información adicional por medio de IRM, neuropsicología y estudios de isótopos de circulación sanguínea y metabolismo. La localización de la función de memoria y la lateralización del lenguaje también son importantes. Es necesario insertar electrodos intracraneales si el EEG de superficie no proporciona información adecuada sobre la información o la lateralización.
El procedimiento quirúrgico realizado con más frecuencia para la epilepsia es la lobectomía temporal. En alrededor del 70 porciento de los pacientes desaparecen las convulsiones y un 20 porciento adicional mejora mucho después de la cirugía. La evaluación histopatológica muestra que en por lo menos el 60 porciento de los pacientes ocurren cambios escleróticos en el hipocampo seccionado. En algunos centros de epilepsia se realiza resección de focos extratemporales, sección del cuerpo calloso y hemisferectomía funcional. La corpuscallosotomía es un procedimiento paliativo para los pacientes que tienen lesiones durante las crisis.77 Las complicaciones asociadas con la cirugía de resección son bajas y en general aceptables. Puede ocurrir un defecto en el campo superior del cuadrante en hasta el 75 porciento de los pacientes sometidos a lobectomía temporal clásica. Se reporta hemiparesia permanente en hasta el 2.4 porciento de los pacientes, y la mortalidad varía de cero a 1.7 porciento de los pacientes.75
Tratamiento del estado epiléptico
El estado epiléptico constituye un peligro para el paciente y un reto para el médico. La característica cardinal del estado epiléptico serio es la presencia de crisis convulsivas continuas o dos o más crisis que ocurren en secuencia sin recuperación de la conciencia entre ellas. El estado epiléptico tiene varias formas de presentación clínica, que incluyen crisis convulsivas generalizadas de repetición, con coma entre las mismas, crisis no convulsivas, causando un cambio en la función cognitiva y crisis focales secuenciales, incluyendo crisis motoras focales o molestias sensoriales focales. El tratamiento se divide en manejo agudo y administración lógica de los medicamentos.
El estado epiléptico con crisis generalizadas es el más común y el más difícil de manejar. El paciente tiene movimientos convulsivos y está inconsciente. Las manifestaciones motoras del estado epiléptico convulsivo pueden ser simétricas, con actividad tónica y después clónica. Si las crisis generalizadas se originan en un foco, ocurren movimientos lateralizados al inicio o durante la crisis.
TRATAMlENTO AGUDO
El tratamiento inicia con apoyo vital, incluyendo permeabilidad de las vías aéreas y apoyo ventilatorio, mantenimiento de la presión arterial y colocación de un acceso intravenoso. La vigilancia fisiológica debe incluir electrocardiografía, presión arterial, gases en sangre, evaluación bioquímica y temperatura corporal. La vía aérea debe protegerse y asegurarse una oxigenación adecuada. Aunque la intubación puede ser necesaria, la decisión de usarla debe ir en paralelo con la selección y administración del medicamento anticonvulsivante. Aunque algunas definiciones del estado epiléptico son útiles para la investigación, cualquier paciente que tiene una crisis convulsiva cuando llega al servicio de urgencia o en quien se observa una crisis durante 10 minutos debe tratarse con el supuesto de que está en estado epiléptico.
Debe excluirse o corregirse la hipoglucemia. Los niveles de glucemia se miden de inmediato y, si esto no es posible, el paciente debe recibir glucosa intravenosa. Los adultos deben recibir primero 100 mg de tiamina intravenosa y la dosis de glucosa para adultos es de un bolo de 50 ml al 50 porciento. Para niños la dosis es de 2 ml/kg de solución de glucosa al 25 porciento.
TRATAMIENTO FARMACOLOGICO
Las crisis clínicas y eléctricas deben yugularse lo más rápido posible, ya que la duración del estado epiléptico correlaciona con la respuesta al tratamiento y con la evolución.78 Los médicos que traten estados epilépticos deben estar familiarizados con los medicamentos disponibles, incluyendo métodos de administración, dosis y efectos agudos adversos. Se presenta una mejor evolución si se tiene un plan, se usan medicamentos eficaces administrados por vías y en dosis apropiadas y se evita la apnea. Debe seguirse un protocolo y los medicamentos deben administrarse por vía intravenosa. La naturaleza del medicamento seleccionado debe ser guiado por la situación clínica. Si un paciente tiene una crisis convulsiva en el momento de la evaluación se requiere una benzodiaepina. Si el paciente tiene historia de crisis seriadas pero éstas se han abatido es mejor elegir un AE de acción duradera.
Benzodiacepinas
Las benzodiacepinas son eficaces y muy potentes. Es común contar con diacepam y loracepam. El diacepam es muy liposoluble y penetra con rapidez al tejido cerebral. Sin embargo, la redistribución en los tejidos grasos no neurales causa rápida disminución de su concentración tanto en sangre como en cerebro. Si se usa el diacepam para terminar con las crisis, debe administrarse también un AE de larga acción, como la fenitoína. La acción del loracepam es más larga, pero se asocia con un tiempo prolongado para la recuperación completa.79 Ambos medicamentos causan depresión de la respiración e incluso apnea. Debe administrarse apoyo a la ventilación, y en ocasiones es necesario intubar al paciente.
Fenitoína
La impregnación del paciente con fenitoína por vía intravenosa terminará con el estado epiléptico.80 La dosis inicial para el adulto es dc 18 mg/kg administrados a través de una vena periférica. La velocidad de infusión no debe ser mayor de 40 mg/min o 1 mg/kg/min en niños. Los pacientes con enfermedad cardiaca o dificultad para mantener la presión arterial requieren vigilancia cuidadosa. Los que sufren hipotensión requieren una velocidad de infusión más lenta. La vigilancia electrocardiográfica puede mostrar prolongación del intervalo QT o incluso inducción de arritmias. Estos cambios indican la necesidad de disminuir la velocidad de la infusión aún más.
Fenobarbital
El fenobarbital es muy eficaz y muchos médicos están familiarizados con su uso. La dosis para el adulto es de 10 a 20 mg/kg. Puede provocar sedación y la apnea es un riesgo, en especial si el paciente ha recibido benzodiacepinas. La vigilancia de la presión arterial es crítica y la hipotensión responde a la disminución de la velocidad de administración.
TRATAMIENTO DEL ESTADO EPILEPTICO REFRACTARIO
Si el paciente no recupera la conciencia o continua con crisis convulsivas después del tratamiento de primera línea, se requiere entonces consultar a un neurólogo. Se realizará un EEG de urgencia y considerará la administración de anestesia. El mejor anestésico en estos casos es el pentobarbital.81,82 El paciente debe ser intubado y se establece vigilancia de cuidados intensivos adecuada. Los pacientes se mantienen en coma con pentobarbital por periodos variables. El tratamiento continuo requiere ajuste de la dosis en forma gradual hasta la de mantenimiento a las cuatro horas, de nuevo a las ocho horas y en forma regular después, observando para determinar si las crisis convulsivas se han abatido y el EEG no muestra descargas convulsivas. Pueden requerirse vasopresores durante el coma con pentobarbital. En ocasiones se desarrolla estado convulsivo sutil, y se requiere vigilar con EEG, en especial si no son obvias las descargas motoras.
Después de realizar los estudios diagnósticos iniciales y controlar las crisis debe buscarse la causa del estado epiléptico. Es importante realizar una evaluación médica y neurológica. Si el paciente es conocido como epiléptico y se dispone de los antecedentes para revisarlos, entonces no se requiere mayor evaluación. Es adecuado realizar estudios de imagen después de que se controlen las crisis. Si existe antecedente de traumatismo craneal, crisis focales o signos de enfermedad sistémica se realizará la evaluación adecuada. Si el paciente tiene fiebre es necesario realizar una punción lumbar con examen del líquido cefalorraquídeo, pero solo después de excluir lesiones ocupativas y obstrucción ventricular por tomografía computada o IRM. La situación clínica determinará la decisión.
Pronóstico del tratamiento epiléptico
El pronóstico está determinado por la evolución de la enfermedad y la perspectiva futura según el trastorno de base. En relación con la epilepsia, control implica ausencia de crisis convulsivas con uso de medicamentos. Remisión indica ausencia de crisis sin medicamentos.
El pronóstico para el control médico de las crisis generalizadas y parciales ha mejorado por los métodos más exactos de evaluación, la introducción de nuevos medicamentos y el uso más racional de los agentes antiguos.36,83 El EEG y la vigilancia con video simultáneos han mejorado la exactitud diagnóstica.
Se logra una buena evolución en el 60 a 65 porciento de los pacientes con crisis de reciente inicio tratadas con un solo medicamento.84 Es más difícil lograr el control en los pacientes que tienen epilepsia de larga duración, crisis parciales, más crisis antes de iniciar el tratamiento, crisis con una causa conocida y patrones epileptiformes en el EEG.85,86 El mal control de las crisis se asocia también con menor adaptación social, múltiples tipos de crisis con alteraciones en el EEG, retraso en el inicio del tratamiento de más de un año, y crisis frecuentes.87 Algunos estudios muestran remisión a cinco años en por lo menos el 70 porciento de los pacientes que fueron vigilados durante 20 años. De estos pacientes, el 50 porciento estaba en remisión verdadera y no tomaba ningún medicamento.88
Los pacientes con crisis parciales complejas tienen un pronóstico menos favorable para el control que los pacientes con crisis generalizadas, pero la buena evolución de los primeros se asocia con estado mental normal, duración breve de la enfermedad y baja frecuencia de las crisis.89 Sin embargo, si las crisis parciales complejas se complican con crisis generalizadas tónico-clónicas, es difícil lograr un control completo. Los pacientes con riesgo alto para desarrollar crisis parciales complejas intratables son los que tienen crisis en racimos, más de una crisis al día, aura al inicio de la crisis y enfermedad psiquiátrica.90 En estudios farmacológicas que emplearon carbamacepina, renitoina y barbitúricos en dosis altas hasta alcanzar la toxicidad clínica en pacientes con crisis intratables, el tratamiento en dosis altas causó control completo en el 22 porciento de los pacientes, el 38 porciento tuvo aumento o ningún cambio en la frecuencia de las crisis y alrededor del 30 porciento no se afectó por el medicamento.91,92
La epilepsia puede afectar la esperanza de vida. La revisión de casos revela factores en común en pacientes con epilepsia que fallecieron por muerte súbita.93 La mayoría de las muertes ocurrieron mientras los pacientes estaban en cama, con seis a 30 porciento durante el sueño. En la autopsia, pocos pacientes tenían niveles terapéuticos en sangre de los AE prescritos, el 50 porciento no tenía niveles detectables. Las crisis convulsivas no se asocian con muerte inmediata,94 y se han implicado a las arritmias cardiacas.94,95 La prevalencia de muerte súbita va de uno en 2,000 a uno en 900 pacientes con epilepsia.
Aspectos especiales
INCAPACIDAD PSICOSOCIAL
Las condiciones médicas crónicas que requieren de tratamiento médico continuo afectan la autoimagen y autoestima. La epilepsia se asocia además con el temor a la pérdida del control y presenta presiones situacionales como falta de empleo, incapacidad para obtener un seguro y restricciones para manejar. Todos estos problemas exacerban el aislamiento social. Los recursos locales, como el capítulo de la Fundación Americana contra la Epilepsia, en los EUA, proporciona una oportunidad a los pacientes para recibir ayuda para estos problemas importantes.
Debe indicarse a cada paciente que debe cumplir con las leyes locales sobre el manejo de vehículos de motor, y esta advertencia debe anotarse en su expediente.
ANTICONCEPTIVOS ORALES
Los AE inductores de enzimas, como la carbamacepina aceleran el metabolismo de los anticonceptivos orales.96 Las pastillas anticonceptivas pueden fallar y puede ser necesario ajustar la dosis de hormona, además de avisar a la paciente de esta interacción farmacológica. La discusión debe anotarse en el expediente.
EMBARAZO
Aunque los ginecólogos suelen instruir a las pacientes para evitar los medicamentos durante el embarazo, las mujeres con epilepsia requieren tratamiento AE a largo plazo para controlar las convulsiones. Los médicos deben revisar los aspectos sobre teratogénesis con todas las mujeres en edad reproductiva, y esta discusión se registrará en el expediente.97 La epilepsia materna se asocia con riesgo de malformaciones fetales, y el tratamiento con AE aumenta el riesgo de dos a tres porciento a cuatro a seis porciento.98 Todos los AE de uso común causan malformaciones mayores, incluyendo defectos del tabique ventricular y labio y paladar hendido. El valproato y la carbamacepina se asocian con espina bífida, la incidencia es de uno a dos porciento, que es 10 a 20 veces mayor que la de la población general.99,100 Sin embargo, son comunes las malformaciones morfológicas menores. Se han observado alteraciones de la porción central de la cara, hipoplasia digital e hipoplasia de los lechos ungueales en el cinco a 30 porciento de los hijos de madres tratadas con AE. Las mujeres que reciben valproato y carbamacepina necesitan orientación especial antes de la concepción.
Se desconocen los mecanismos de teratogénesis pero se han desarrollado recomendaciones con base en estudios de población. Las madres epilépticas deben ser tratadas con la dosis eficaz más baja de un medicamento, y la frecuencia de las dosis se diseñará para evitar las concentraciones séricas altas. Cuando es posible, es mejor emplear un solo fármaco. Se requiere administrar suplementos de folato antes y después de la concepción. Todas las mujeres epilépticas que estén planeando un embarazo deben recibir por lo menos 1 mg de folato antes del embarazo y durante el mismo. Debe administrarse vitamina K1 durante las últimas cuatro semanas de la gestación para reducir la ocurrencia de coagulopatía en el recién nacido. Si la madre considera la opción del aborto terapéutico en caso de que el feto tenga un defecto del tuho neural, se requerirá evaluación cuidadosa con ultrasonido e incluso amniocentesis.
PACIENTES ANCIANOS
La incidencia de epilepsia entre los ancianos aumenta al avanzar la edad de la población. Las causas incluyen infarto cerebral, neoplasias y enfermedad cerebral degenerativa. La elección de los medicamentos es semejante a la de los pacientes más jóvenes, pero los cambios en la función corporal asociados con el envejecimiento complican el tratamiento. La reducción en la función hepática y renal alteran la unión de los medicamentos a las proteínas y su eliminación. Estos cambios obligan a los médicos a usar aspectos clínicos, como la frecuencia de las crisis, y la vigilancia estrecha para detectar síntomas de efectos adversos, en lugar de medir las concentraciones séricas del fármaco. Además, por los padecimientos concomitantes los pacientes suelen tomar múltiples fármacos, lo que causa problemas de interacción. La medición de los niveles libres del medicamento es más útil que medir los niveles totales en plasma.101
Bibliografía
-
Hauser WA: Seizure disorders: the changes with age.
Epilepsia 33(suppl 4):S6, 1992
-
Proposal for revised classification of epilepsies and
epileptic syndromes. Commission on Classification and Terminology of the
International League Against Epilepsy. Epilepsia 30:389, 1989
-
Theodore WH, Porter RJ, Penry JK: Complex partial seizures:
clinical characteristics and differential diagnosis. Neurology 33:1115,
1983 [PMID
6684245]
-
Rocca WA, Sharbrough FW, Hauser WA, et al: Risk factors
for complex partial seizures: a population-based case-control study. Ann
Neurol 21:22, 1987 [PMID
3827212]
-
Keranen T, Sillanpaa M, Riekkinen PR: Distribution
of seizure types in an epileptic population. Epilepsia 29:1, 1988 [PMID
3123210]
-
Delgado-Escueta AV, Enrile-Bascal F: Juvenile myoclonic
epilepsy of Janz. Neurology 34:285, 1984 [PMID
6422321]
-
Dreifuss F: Pediatric epilepsy syndromes: an overview.
Cleve Clin J Med 56(suppl):S166, 1989
-
Penry JK, Porter RJ, Dreifuss FE: Simultaneous recording
of absence seizures with videotape and electroencephalography: a study
of 374 seizures in 48 patients. Brain 98:427, 1975 [PMID
1182486]
-
Sato S, Dreifuss FE, Penry JK, et al: Long-term follow-up
of absence seizures. Neurology 33:1590, 1983 [PMID
6417557]
-
Wirrell EC, Camfield CS, Camfield PR, et al: Long-term
prognosis of typical childhood absence epilepsy: remission or progression
to juvenile myoclonic epilepsy. Neurology 47:912, 1996 [PMID
8857718]
-
Rasmussen TE, Olszewsik J, Lloyd-Smith D: Focal cortical
seizures due to chronic localized encephalitides. Neurology 8:435, 1958
-
Berkovic SF, Cochius J, Andermann E, et al: Progressive
myoclonus epilepsies: clinical and genetic aspects. Epilepsia 34(suppl
3):S19, 1993
-
Radhakrishnan K, So EL, Silbert PL, et al: Predictors
of outcome of anterior temporal lobectomy for intractable epilepsy. Neurology
51:465, 1998 [PMID
9710020]
-
Cascino GD, Jack CR Jr, Parisi JE, et al: Magnetic
resonance imaging-based volume studies in temporal lobe epilepsy: pathological
correlations. Ann Neurol 30:31, 1991 [PMID
1929226]
-
Cascino GD: Structural brain imaging. Epilepsy: A Comprehensive
Textbook. Engel J Jr, Pedley TA, Eds. Lippincott-Raven, Philadelphia, 1997,
p 937
-
Laxer KD: Clinical applications of magnetic resonance
spectroscopy. Epilepsia 38(suppl):S13, 1997
-
Kuzniecky R: Magnetic resonance and functional magnetic
resonance imaging: tools for the study of human epilepsy. Curr Opin Neurol
10:88, 1997
-
Ryvlin P, Philippon B, Cinotti L, et al: Functional
neuroimaging strategy in temporal lobe epilepsy: a comparative study of
18FDG-PET and 99mTc-HMPAO-SPECT. Ann Neurol 31:650, 1993
-
Breier JI, Mullani NA, Thomas AB, et al: Effects of
duration of epilepsy on the uncoupling of metabolism and blood flow in
complex partial seizures. Neurology 48:1047, 1997
-
Meierkord H, Will B, Fish D, et al: The clinical features
and prognosis of pseudoseizures diagnosed using video-EEG telemetry. Neurology
41:1643, 1991 [PMID
1922808]
-
Shen W, Bowman ES, Markand ON: Presenting the diagnosis
of pseudoseizure. Neurology 40:756, 1990 [PMID
2330101]
-
Hauser WA, Rich SS, Lee JR-J, et al: Rise of recurrent
seizures after two unprovoked seizures. N Engl J Med 338:429, 1998 [PMID
9459646]
-
Hauser WA, Hesdorffer DC: Epilepsy: Frequency, Causes
and Consequences. Demos, New York, 1990
-
McLean MJ, Macdonald RL: Carbamazepine and 10,11-epoxycarbamazepine
produce use- and voltage-dependent limitation of rapidly firing action
potentials of mouse central neurons in cell culture. J Pharmacol Exp Ther
238:727, 1986 [PMID
2874218]
-
Macdonald RL: Antiepileptic drug actions. Epilepsia
30(suppl):S19, 1989
-
Coulter DA, Hugenard JR, Prince DA: Specific petit
mal antiepileptics reduce calcium currents in thalamic neurons. Neurosci
Lett 98:74, 1989 [PMID
2710401]
-
Coulter DA, Hugenard JR, Prince DA: Characterization
of ethosuximide reduction of low-threshold calcium current in thalamic
neurons. Ann Neurol 25:582, 1989 [PMID
2545161]
-
Coulter DA, Hugenard JR, Prince DA: Calcium currents
in rat thalamocortical relay neurones: kinetic properties of the transient
low-threshold current. J Physiol 414:587, 1989
-
Olsen RW, Avoli M: GABA and epileptogenesis. Epilepsia
38:399, 1997 [PMID
9118844]
-
Macdonald RL, Rogers CJ, Twyman RE: Barbiturate regulation
of kinetic properties of the GABAA receptor channel of mouse
spinal neurons in culture. J Physiol 417:483, 1989 [PMID
2482885]
-
Twyman RE, Rogers CJ, Macdonald RL: Differential regulation
of gamma-aminobutyric acid receptor channels by diazepam and phenobarbital.
Ann Neurol 25:213, 1989
-
Suzdak PD, Jansen JA: A review of the preclinical pharmacology
of tiagabine: a potent and selective anticonvulsant GABA uptake inhibitor.
Epilepsia 36:612, 1995 [PMID
7555976]
-
Stables JP, Bialer M, Johannessen SI, et al: Progress
report on new antiepileptic drugs a summary of the second Eilat conference.
Epilepsy Res 22:235, 1995 [PMID
8991791]
-
Pellock JM, Brodie MJ: Felbamate: 1997 update. Epilepsia
38:1261, 1997 [PMID
9578519]
-
Mattson RH, Cramer JA, Collins JF, et al: Comparison
of carbamazepine, phenobarbital, phenytoin, and primidone in partial and
secondarily generalized tonic-clonic seizures. N Engl J Med 313:145, 1985
[PMID
3925335]
-
Willmore LJ, Shu V, Wallin B, et al: Efficacy and safety
of add-on divalproex sodium in the treatment of complex partial seizures.
M88-194 Study Group. Neurology 46:49, 1996
-
Mattson RH, Cramer JA, Collins JF, et al: A comparison
of valproate with carbamazepine for the treatment of complex partial seizures
and secondarily generalized tonic-clonic seizures in adults. Department
of Veterans Affairs Epilepsy Cooperative Study, No. 264 Group. N Engl J
Med 327:765, 1992
-
Pellock JM, Willmore LJ: A rational guide to routine
blood monitoring in patients receiving antiepileptic drugs. Neurology 41:961,
1991 [PMID
2067658]
-
Plaa GI, Willmore LJ: General Principles: Toxicology.
Antiepileptic Drugs, 4th ed. Levy RH, Mattson RH, Meldrum BS, Eds. Raven
Press, New York, 1998, p 51
-
Meador KJ, Loring DW, Huh K, et al: Comparative cognitive
effects of anticonvulsants. Neurology 40:391, 1990 [PMID
2314578]
-
Reynolds EH, Trimble MR: Adverse neuropsychiatric effects
of anticonvulsant drugs. Drugs 29:570, 1985 [PMID
2861075]
-
Chadwick DW, Reynolds EH, Marsden CD: Anticonvulsant-induced
dyskinesias: a comparison with dyskinesias induced by neuroleptics. J Neurol
Neurosurg Psychiatry 39:1210, 1976
-
Shorvon SD, Reynolds EH: Anticonvulsant peripheral
neuropathy: a clinical and electrophysiological study of patients on single
drug treatment with phenytoin, carbamazepine or barbiturates. J Neurol
Neurosurg Psychiatry 45:620, 1982 [PMID
6288881]
-
Triggs WJ, Bohan TP, Lin S, et al: Valproate-induced
coma with ketosis and carnitine insufficiency. Arch Neurol 47:1131, 1990
[PMID
2121122]
-
Bryant AE III, Dreifuss FE: Valproic acid hepatic fatalities:
III. U.S. experience since 1986. Neurology 46:465, 1996
-
Camfield PR, Bagnell P, Camfield CS, et al: Pancreatitis
due to valproic acid. Lancet 1:1198, 1979 [PMID
86928]
-
Sorrell TC, Forbes IJ: Depression of immune competence
by phenytoin and carbamazepine. Clin Exp Immunol 20:273, 1975 [PMID
1212810]
-
Kahn HD, Faguet GB, Agee JF, et al: Drug-induced liver
injury: in vitro demonstration of hypersensitivity to both phenytoin and
phenobarbital. Arch Intern Med 144:1677, 1984
-
Brown M, Schubert T: Phenytoin hypersensitivity hepatitis
and mononucleosis syndrome. J Clin Gastroenterol 8:469, 1986 [PMID
3093562]
-
Barr RD, Copeland SA, Stockwell MC, et al: Valproic
acid and immune thrombocytopenia. Arch Dis Child 57:681, 1982 [PMID
6812506]
-
. Sandler RM, Emberson C, Roberts GE, et al: IgM platelet
autoantibody due to sodium valproate. Br Med J 2:1683, 1978 [PMID
367512]
-
Roujeau J, Kelly JP, Naldi L, et al: Medication use
and the risk of Stevens-Johnson syndrome or toxic epidermal necrolysis.
N Engl J Med 333:1600, 1995 [PMID
7477195]
-
Klinman DM, Steinberg AD: Systemic lupus erythematosus
and overlap syndromes. Immunological Diseases, 4th ed. Samter M, Talmage
DW, Frank MM, et al, Eds. Little, Brown & Co, Boston, 1988, p 1335
-
Lahr MB: Hyponatremia during carbamazepine therapy.
Clin Pharmacol Ther 37:693, 1985
-
Surks MI, Ordene KW, Manu DN, et al: Diphenylhydantoin
inhibits the thyrotropin response to thyrotropin-releasing hormone in man
and rat. J Clin Endocrinol Metab 56:940, 1983
-
Toone BK, Wheller M, Fenwick PBC: Sex hormone changes
in male epileptics. Clin Endocrinol 12:391, 1980
-
Haidukewych D, Rodin EA: Chronic antiepileptic drug
therapy: classification by medication regimen and incidence of decreases
in serum thyroxine and free thyroxine index. Ther Drug Monit 9:392, 1987
[PMID
3424405]
-
Engel JJ: Surgical Treatment of the Epilepsies, 2nd
ed. Raven Press, New York, 1993
-
Salanova V, Markand ON, Worth R: Clinical characteristics
and predictive factors in 98 patients with complex partial seizures treated
with temporal resection. Ann Neurol 51:1008, 1994
-
Sorenson JM, Wheless JW, Baumgartner JE, et al: Corpus
callosotomy for medically intractable seizures. Pediatr Neurosurg 27:260,
1997 [PMID
9620004]
-
Towne AR, Pellock JM, Ko D, et al: Determinants of
mortality in status epilepticus. Epilepsia 35:27, 1994 [PMID
8112254]
-
Lowenstein DH, Alldredge BK: Status epilepticus. N
Engl J Med 338:970, 1998 [PMID
9521986]
-
Wilder BJ, Ramsay RE, Willmore LJ, et al: Efficacy
of intravenous phenytoin in the treatment of status epilepticus. Ann Neurol
1:511, 1977 [PMID
883764]
-
DeLorenzo RJ, Waterhouse EJ, Towne AR, et al: Persistent
nonconvulsive status epilepticus after control of convulsive status epilepticus.
Epilepsia 39:833, 1998 [PMID
9701373]
-
Bleck TP: Advances in the management of refractory
status epilepticus. Crit Care Med 21:955, 1993
-
Willmore LJ: Prognosis of epilepsy. Prognosis in Neurological
Disease. Evans RW, Baskin DS, Yatsu FM, Eds. Oxford University Press, New
York, 1989, p 483
-
Beghi E, Togoni G: Prognosis of epilepsy in newly referred
patients: a multicenter prospective study. Epilepsia 29:236, 1988 [PMID
3371280]
-
Kotagal P, Rothner AD, Erenberg G, et al: Complex partial
seizures of childhood onset: a five-year follow-up study. Arch Neurol 44:1177,
1987 [PMID
3314811]
-
Leestma JE, Annegers JF, Brodie MJ, et al: Sudden unexplained
death in epilepsy: observations from a large clinical development program.
Epilepsia 38:47, 1997 [PMID
9024183]
-
Annegers JF, Coan SP, Hauser WA, et al: Epilepsy, vagal
nerve stimulation by the NCP system, mortality, and sudden, unexpected,
unexplained death. Epilepsia 39:206, 1998 [PMID
9578002]
-
Mattson RH, Cramer JA, Darney PD, et al: Use of oral
contraceptives by women with epilepsy. JAMA 256:238, 1986 [PMID
3723710]
-
Delgado-Escueta AV, Janz D: Consensus guidelines: preconception
counseling, management, and care of the pregnant woman with epilepsy. Neurology
42(suppl 5):149, 1992
-
Yerby MS: Pregnancy, teratogenesis, and epilepsy. Neurol
Clin 12:749, 1994
-
Lindhout D, Meinardi H: Spina bifida and in-utero exposure
to valproate. Lancet 2:396, 1984 [PMID
6147468]
-
Rosa FW: Spina bifida in infants of women treated with
carbamazepine during pregnancy. N Engl J Med 324:674, 1991
- Willmore LJ: The effect of age on pharmacokinetics of antiepileptic drugs. Epilepsia 36(suppl):S14, 1995