Contenido del artículo
III MECANISMOS DE LA RESPUESTA INMUNE
- Respuestas de las células T a los antígenos
- Procesamiento del antígeno
- FORMACIÓN DE COMPLEJOS ANTIGENO-CPH
- COMPLEJOS ALTERNATIVOS DE PRESENTACION DEL ANTIGENO
- CELULAS PRESENTADORAS DE ANTIGENO PROFESIONALES
- MECANISMO DE PRESENTACION DEL ANTIGENO A LAS CELULAS T
- MOLECULAS COESTIMULADORAS
- Transducción de la señal de la célula T durante el reconocimiento del antígeno
- Citocinas y subtipos de células T
- Citocinas e inflamación
- CITOCINAS PROINFLAMATORIAS
- Factor de necrosis tumoral-[alpha]
- Linfotoxina
- Interleucina-1 e interleucina-6
- Interferón gama
- Factor inhibidor de la migración
- INTERLEUCINA-5
- CITOCINAS ANTINFLAMATORIAS
- Quimocinas
- Respuestas de las células B al antígeno
- Mecanismos efectores en la inmunidad mediada por células
- Información adicional
DR. JOHN DAVID
COX TERHORST, PH.D.
La respuesta inmune se define por los principios de discriminación entre lo propio y lo no propio, la especificidad y la memoria. Ante la exposición a un patógeno u otra fuente de antígenos, los macrófagos y las células dendríticas inician una respuesta que estimula la migración de células T y B hacia los sitios inflamatorios y a los ganglios linfáticos que los drenan. Los antígenos se concentran en los macrófagos y células dendríticas y en estas células son procesados a péptidos. Estas células presentadoras de antígenos (CPA) presentan los antígenos procesados en su superficie extracelular en un complejo con las moléculas del complejo principal de histocompatibilidad (CPH) de clase I o de clase II.
El primer paso crítico en la respuesta inmune específica a un antígeno es el reconocimiento y unión del complejo péptido antigénico-molécula del CPH en la superficie de las CPA al receptor de la célula T (RCT) ab en la superficie de las células T cooperadoras CD4+ y CD8+. Este evento influye en el núcleo de la célula T cooperadora por una cascada de moléculas de señal citoplásmica. En el núcleo la activación de factores específicos de transcripción estimula la expresión de los genes que codifican factores solubles o citocinas, que median la respuesta inmune.
Las células T CD4+ ab activadas secretan citocinas que inducen que las células B estimuladas por antígenos se diferencien a células plasmáticas secretoras de anticuerpos (respuesta humoral) y causa que las células T CD8+ se diferencien a células efectoras citolíticas (respuesta mediada por células) Además, las células T y B son inducidas a expandirse y controlar la infección inicial y a producir células de memoria para la inmunidad adquirida duradera.
Respuestas de las células T a los antígenos
La diversidad de las regiones variables en los RCT facilita en mucho las respuestas a los antígenos. Las células T prototipo, T ab, tienen un RCT formado por cadenas a y b, que se expresan en asociación con CD3. Un subtipo de células T periféricas, las células T gd, tienen un RCT formado por cadenas gama y delta. Estas células T gd no parecen requerir la presentación de antígenos por las moléculas del CPH.
LOS RECEPTORES DE LAS CELULAS T RECONOCEN COMPLEJOS ANTIGENO-CPH
Los productos de los genes que forman el CPH constituyen un complejo de glucoproteína de membrana que se une al péptido antigénico dentro de una CPA y los transporta a la superficie celular por interacción con las células T [ver adelante, Procesamiento del antígeno]. 1 Existe un gran polimorfismo en el CPH (i.e., un gran número de alelos por locus), dentro de la población humana; sin embargo, cada individuo expresa solo un pequeño número de moléculas diferentes del CPH. Para asegurar una respuesta inmunológica adecuada contra un gran número de antígenos no propios, cada molécula del CPH debe ser capaz de unirse a un gran número de péptidos diferentes.
Las moléculas del CPH de clase I consisten en una cadena pesada a y una cadena ligera de microglobulina-b2 (b2M), mientras que las moléculas del CPH de clase II consisten en cadenas a y b de un tamaño semejante. Las dos clases de moléculas tienen una estructura parecida: dos dominios semejantes a los de las inmunoglobulinas y un sitio de unión al péptido formado por una placa plisada b con 8 cadenas y dos regiones helicoidales a. Mientras las moléculas de clase I fijan solo péptidos pequeños de longitud definida ( 8 a 11 aminoácidos), las moléculas de clase II fijan péptidos más largos, sin restricción aparente en su longitud. Un dato interesante es que ciertos péptidos se fijan solo a alelos específicos de moléculas del CPH de clase I o de clase II. Por lo tanto, las personas que no tienen un alelo particular pueden no desarrollar una respuesta inmune al péptido asociado.
Los péptidos que se fijan a las moléculas de clase I del CPH están unidas a sus terminales amino y carboxilo por una red de uniones de hidrógeno a residuos conservados de aminoácidos en el sitio de unión del péptido de la molécula de clase I del CPH. La unión de los péptidos a las moléculas de clase II del CPH es un poco diferente porque estos péptidos tienden a ser mayores y de longitudes arbitrarias. La cadena principal del péptido se fija en la mayor parte de su longitud por medio de enlaces de hidrógeno a los residuos conservados de aminoácidos colocados a través del sitio de unión del péptido.
Las diferencias en la unión de los péptidos entre las moléculas del CPH de clase I y II se debe a pequeñas diferencias estructurales dentro del marco relativamente fijo del sitio de unión del péptido y quizá también a diferencias fundamentales en el mecanismo de procesamiento del péptido, que tiene lugar en el retículo endoplásmico (RE) para las moléculas de clase I y en los endosomas y lisosomas para las moléculas de clase II.
FORMACIÓN DE COMPLEJOS ANTIGENO-CPH
El procesamiento del antígeno asociado con las moléculas del CPH de clase II consiste en tres pasos: (1) generación de péptidos antigénicos en el citosol de la CPA, (2) transporte de los péptidos hacia el RE y (3) ensamble de los complejos péptido-CPH de clase I. Los complejos formados migran a través del aparato de Golgi hacia la superficie celular y se fusionan a la membrana plasmática para su presentación en la superficie extracelular [ver figura 1a].
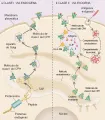
|
| Figura 1 |
| Vías de formación de los complejos antígeno-molécula del CPH |
Las proteínas antigénicas, proteínas nucleares y citosólicas endógenas del huésped, así como proteínas introducidas por patógenos intracelulares, como las proteínas virales, se fijan primero por conjugación ubicua antes de sufrir degradación proteolítica en los proteosomas. Una gran endoproteasa con múltiples subunidades, el proteosoma, es el principal mecanismo de degradación proteica no lisosómica y se encuentra en el núcleo y el citoplasma de todas las células eucariotas. Además de generar péptidos antigénicos, los proteosomas eliminan el exceso de proteínas sintetizadas por las células.
Para alcanzar el sitio de unión del péptido de la molécula de clase I del CPH en la luz del RE, los péptidos atraviesan la bicapa lípida del RE por medio de un sistema de bomba dependiente de trifosfato de adenosina (ATP) que consiste en las proteínas TAP1 y TAP2 (TAP significa proteína asociada al transportador). Un tercer componente, denominado tapasina (TAP-A) influye en la asociación de la molécula de clase I con la TAP y en el ensamble del complejo péptido-molécula de clase I del CPH. 2 El ensamble de la cadena pesada a de clase I y de la cadena ligera b2M en el RE requiere la presencia del péptido. Si no se forma el complejo péptido-ab2M, las moléculas de clase I del CPH no viajan a la superficie celular y son degradadas en la luz del RE.
El ensamble del complejo péptido antigénico-molécula de clase II del CPH requiere de cuatro pasos: (1) captación de antígenos exógenos por las vesículas proteolíticas (endosomas, lisosomas y quizá subcompartimientos endosómicos no definidos) en la CPA, (2) degradación proteolítica de las proteínas en el endosoma, (3 ensamble de las moléculas de clase II del CPH en el RE y migración de estas moléculas a través del aparato de Golgi a los endosomas, y (4) ensamble de los complejos péptido-molécula clase II del CPH en el endosoma. Después de que los péptidos se unen a las moléculas de clase II del CPH, el endosoma que contiene estos complejos migra a la superficie celular y se fusiona a la membrana plasmática [ver figura 1b]. Es importante que las proteínas antigénicas parcialmente desdobladas pueden fijarse a las moléculas de clase II antes de sufrir degradación proteolítica. Esto puede explicar porqué la longitud de los péptidos unidos a las moléculas de clase II es muy variable.
Dos proteínas críticas controlan las interacciones péptido-molécula de clase II del CPH: la cadena invariante (Ii) y el producto genético del HLA-DM. La Ii tiene un papel importante en el ensamble y transporte intracelular de las moléculas de clase II del CPH y la selección de los péptidos. La Ii bloquea el sitio de unión al péptido durante el ensamble de las subunidades a y b de las moléculas de clase II en el RE, evitando así la unión de péptidos procesados en el RE. La Ii se degrada en forma parcial en un compartimiento endosómico, dejando un pequeño fragmento que se denomina péptido Ii asociado a la clase Ii (CLIP) en el sitio de unión al péptido de la molécula de clase II del CPH. La eliminación del CLIP cataliza la unión de los péptidos antigénicos. 3,4 El HLA-DM facilita la eliminación del CLIP.
COMPLEJOS ALTERNATIVOS DE PRESENTACION DEL ANTIGENO
Un tercer grupo de moléculas presentadoras de antígeno son las proteínas no polimórficas de clase I del CPH parecidas a la b2M, que incluyen los productos de los genes CD1 no polimórficos. Las moléculas CD1 fijan antígenos lípidos y glucolípidos para presentarlos a una gran variedad de células T. Por ejemplo, las moléculas CD1 presentan glucolípidos de Mycobacterium tuberculosis a células T ab que no portan CD4 o CD8. 5 Este ejemplo de reconocimiento de un antígeno microbiano no proteico sugiere que las células T ab reconocen un rango mucho mayor de antígenos de lo que se pensaba. Las células T con restricción de CD1 con frecuencia son autorreactivas y han sido implicadas en padecimeintos autoinmunes como la diabetes mellitus tipo 1 (insulinodependiente) y el lupus eritematoso. Se ha sugerido que participan en la fase innata temprana de estas respuestas inmunes. 6
Otro método de presentación de antígeno incluye a las moléculas de clase II del CPH y los superantígenos, una clase de proteínas inmunoestimuladoras derivadas de agentes microbianos (v.gr., proteínas virales y toxinas producidas por Staphylococcus aureus que causan síndrome de choque tóxico e intoxicación alimentaria). Los superantígenos se unen a las moléculas de clase II del CPH fuera del sitio convencional de unión de antígeno y estimulan a las células T. 7 Por ejemplo, el superantígeno bacteriano intacto enterotoxina B de S. aureus se une al dominio a1 de la subunidad a de la molécula humana de clase II del CPH HLA-DR1, creando un sitio de unión al RCT novedoso adyacente al sitio convencional de unión antígeno-CPH. Los diferentes alelos de clase II del CPH tienen diferentes constantes de unión para los superantígenos, por lo que éstos pueden activar segmentos diferentes del repertorio de células T.
CELULAS PRESENTADORAS DE ANTIGENO PROFESIONALES
Mientras que las moléculas de clase I del CPH se expresan en la superficie de todas las células eucarióticas, las moléculas de clase II tienen una distribución tisular restringida. De hecho, ciertas células, incluyendo células B, células dendríticas, macrófagos, células de Langherhans y células endoteliales, se denominan CPA profesionales porque presentan al péptido antigénico con las moléculas de clase II del CPH en forma más eficiente que otras CPA. 8 Esta eficacia se atribuye a su capacidad para procesar antígenos endocitados. Las CPA profesionales también interactúan con células T en forma más eficaz porque tienen varios marcadores de superficie celular que se fijan a moléculas coestimuladoras en la superficie de las células T (ver adelante). Las CPA más potentes son las células dendríticas, que presentan el antígeno solo al madurar y migrar a los ganglios linfáticos. Este proceso es desencadenado por la captación del antígeno.
MECANISMO DE PRESENTACION DEL ANTIGENO A LAS CELULAS T
El reconocimiento del antígeno por la célula tiene dos fases diferentes: un paso inespecífico de adhesión intercelular entre una célula T de una clona que expresa un tipo de RCT ab y una CPA, seguido de la interacción específica entre el RCT y el complejo antígeno-CPH. Este proceso destaca dos propiedades fundamentales de las células T: su capacidad para migrar por todo el organismo a muchos tipos de células y su gran especificidad por antígenos particulares.
Las CPA procesan en forma simultánea muchos péptidos antigénicos, por lo que expresan en sus superficies celulares un gran número de diferentes complejos antigénicos. Solo un número pequeño de estos complejos puede ser reconocido por una clona específica de células T ab. A través de la adhesión intercelular, las células T buscan CPA, la adhesión se pierde en ausencia del reconocimiento específico del antígeno por el RCT y se intensifica cuando se realiza el contacto correcto RCT-péptido-molécula CPH. Al adherirse los complejos RCT-CD3 se agregan en la superficie de la célula T y se unen a los complejos antígeno-CPH que se han colocado en la superficie de la CPA. La regulación inhibitoria de las moléculas de adhesión permite la separación celular. En ausencia de reconocimiento del antígeno los complejos RCT-CD3 son incapaces de acumularse y la separación ocurre de inmediato.
Las interacciones entre una clona de células T cooperadoras y una CPA o entre una célula T citotóxica y su célula blanco siguen el mismo patrón general. Debido a que la constante de unión entre el RCT y el complejo antígeno-CPH es baja, solo un pequeño número de complejos inicia la unión con RCT específicos en un área del contacto intercelular establecido por la adhesión. En consecuencia, los complejos RCT-CD3 y las moléculas del CPH que portan el péptido antigénico correcto migran hacia la región de contacto célula T-CPA. Esto establece una alta densidad local de RCT, que promueve la unión al antígeno y la activación de las células T. La existencia de estos RCT acumulados se ha demostrado usando microscopía confocal y anticuerpos monoclonales dirigidos en estudios de la interfase entre una clona de células T cooperadora y una CPA específica de antígeno. 9 Además, los acúmulos de RCT pueden observarse usando tetrámeros fluorescentes de complejos antígeno-CPH. 10 Esta técnica novedosa ha permitido estudios incisivos sobre las respuestas de las células T específicas contra epítopes en infecciones con virus Epstein-Barr o VIH. 11
La activación aberrante del RCT puede iniciar una respuesta inmune desastrosa, como la observada en las enfermedades autoinmunes y otras inmunopatologías. Por lo tanto, el reconocimiento del antígeno por el RCT y la transducción subsecuente de señales son controlados en forma positiva o negativa por otras moléculas de superficie de la célula T [ver figura 2]. 12 Las moléculas coestimuladoras participan tanto en la adhesión como en la transducción de señales de las células T y tienen un papel importante en la coordinación y regulación cinética de la activación de la célula T.
|
|
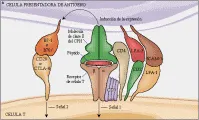
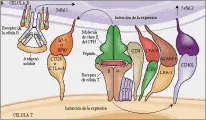
Las moléculas CD4 y CD8 son correceptores del RCT que interactúan con moléculas del CPH en la superficie de las CPA. Esta interacción puede influir en la generación de células T CD4+ y CD8+ en el timo. Debido a que tanto el CD4 como el CD8 son parte de los complejos RCT-CPH y se asocian con la proteína tirosina cinasa Lck, influyen en forma directa en la transducción de señales iniciada por el RCT (ver adelante). Otros coestimuladores clave son las moléculas de adhesión CD2 y CD11/CD18. El ligando natural en la superficie de la CPA para CD2 es CD58, mientras que los ligandos para CD11/CD18 son la molécula de adhesión intercelular-1 (ICAM-1, también denominado CD54), el componente del complemento iC3b y las proteínas de la matriz extracelular.
CD28 y CTLA-4 proporcionan la segunda señal más importante, iniciando una vía de transducción de señales que es diferente y con frecuencia independiente de la vía mediada por el complejo RCT-CD3. CD28 es una glucoproteína de superficie celular que se expresa esencialmente en todas las células T CD4 + y en la mayoría de las CD8 +. CD28 se fija a B7-1 (CD-80), que se expresa en baja densidad en las CPA (aunque su expresión puede aumentar). La unión de CD28 a B7-1 o a una segunda contraestructura, B7-2 (CD86), causa una señal de transducción coestimuladora y activación y proliferación consecuente de las células T. Por el contrario, la expresión de CTLA-4, que es homóloga a CD28, aumenta después de la activación de las células T. La unión de CTLA-4 a B7-1 o B7-2 brinda una señal inhibitoria que causa regulación inhibitoria de la proliferación de las células T. Debe enfatizarse que las investigaciones que consisten en manipulación de las interacciones de CD28/CTLA-4 con sus ligandos naturales han producido resultados novedosos en trasplantes y tratamiento tumoral y pueden constituir un tratamiento potencial para enfermedades como la artritis, la esclerosis múltiple y el asma, así como para proteger contra la infección por VIH. 13
La molécula coestimuladora CD27 se expresa durante la activación de las células T y actúa en forma sinérgica con la activación asociada al RCT para inducir proliferación de linfocitos T. La CD27 en las células T constituye un miembro de la familia del receptor del factor de necrosis tumoral (RFNT), mientras que su ligando, CD70, que se encuentra en las células activadas T y B, es miembro de la familia del FNT.
Otro miembro de la familia del RFNT, Fas (también denominado APO-1 y CD95) ha sido implicado en eventos de señales tanto positivos como negativos por su interacción con el ligando de Fas (CD95L) que se expresa en efectores citolíticos. La unión de Fas en las células T a CD95L típicamente causa su muerte celular programada (i.e., apoptosis, ver adelante). Sin embargo, el ligando de Fas a un anticuerpo monoclonal anti-Fas ha demostrado que Fas puede funcionar como una molécula coestimuladora para la activación RCT-CD3. Por lo tanto, una sola molécula puede provocar diferentes resultados en las diferentes fases del desarrollo de la célula T.
Transducción de la señal de la célula T durante el reconocimiento del antígeno
En respuesta al reconocimiento del antígeno las células T en reposo sufren una serie compleja de eventos conocidos como activación de las células T. 14,15 El evento extracelular, reconocimiento y unión del RCT a los complejos antígeno-CPH y de las moléculas coestimuladoras a sus ligandos apropiados, es seguida de una cascada intracelular de eventos bioquímicos (i.e., transducción de señales) que al final alcanza genes específicos en el núcleo. La producción específica de citocinas, quimocinas y otras moléculas inmunomoduladoras causa proliferación y diferenciación celular, así como expresión de funciones efectoras únicas.
La mayoría de los eventos bioquímicos que ocurren inmediatamente después del contacto del RCT con el complejo antígeno-CPH ya se han definido [ver figura 3]. El primer paso es el reclutamiento de una de dos proteínas tirosina cinasas de la familia Srk, Lck y Fyn, a los elementos del CD3 (CD3-g, CD3-d, CD3-e y CD3-s) del complejo RCT-CD3. Estas enzimas fosforilan residuos de tirosina específicos en las secuencias llamadas motivo de activación basado en el inmunorreceptor de tirosina (ITAM) que se encuentra en las colas citoplásmicas del CD3. El nivel de fosforilación de CD3-s depende de la avidez entre el complejo antígeno-CPH y el RCT ab.16,17
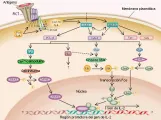
|
| Figura 3 |
| Vías de señales después de la estimulación del RCT |
Un ciclo de fosforilación y defosforilación de tirosina regula la activación de las células T inducida por antígeno. La defosforilación y prevención de la fosforilación de la tirosina son llevadas a cabo por fosfatasas de tirosina (v.gr., CD45) y por el inactivador-Src Csk, respectivamente. Se ha demostrado que Csk inhibe la activación genética del RCT inducida por citocinas por fosforilación de los sitios de regulación inhbitoria en Lck y Fyn, causando la inactivación de estas dos enzimas.
Después de la fosforilación en los sitios de anclaje de las colas citoplásmicas del CD3, una de dos proteínas tirosina cinasa de membrana, Syk o ZAP-70, se agregan al complejo. Estas cinasas activan las proteínas de señales citoplásmicas, que inducen la activación de factores de transcripción que controlan la expresión de genes que median la función efectora de la célula T (v.gr., el gen para la citocina interleucina-2 [IL-2]) [ver figura 3]. Por lo tanto, la interacción exquisita clona célula T-RCT específico conecta, a través de diversas moléculas intermediarias, con vías universales de transducción de señales.
Citocinas y subtipos de células T
Las citocinas, un grupo diverso de proteínas producido por diferentes tipos celulares, son cruciales en la regulación de las respuestas inmunes. También son importantes en la diferenciación celular. En general, las citocinas no se almacenan en las células, sino que se sintetizan con el estímulo apropiado. El ARN mensajero (ARNm) resultante tiene vida media corta. En algunos casos la citocina activa se libera a partir de un precursor inactivo por proteólisis. Después de su secreción, las citocinas suelen actuar en forma local uniéndose a receptores en las superficies celulares. Las citocinas pueden actuar en muchas células diferentes y las diversas citocinas pueden tener actividades semejantes. Los receptores de citocinas con frecuencia están compuestos de las mismas cadenas proteicas. Por ejemplo, los receptores de citocinas IL-2R, IL-4R. IL-7R, IL-9R, IL-13R e IL-15R tienen en común una cadena g. La mayoría de las citocinas forma una red que regula la activación de las células y la producción de otras citocinas. 18 A través de la unión a receptores en las superficies celulares las citocinas ejercen sus efectos al activar mecanismos intracelulares de señales que originan la expresión de genes particulares (v.gr., genes para sí mismas u otras citocinas). Los principales transductores intracelulares de las señales de las citocinas son la proteína tirosina cinasa Jak (cinasa Janus) y la familia de factores de transcripción STAT (transductores de señales y activadores de la transcripción).
Las citocinas tienen un papel muy importante en el desarrollo y regulación de las células T. De hecho, dos subtipos de células T cooperadoras, Th1 y Th2, se definen por las citocinas que producen. La Th1 y la Th2 tienen papeles clave para determinar el equilibrio entre dos resultados inmunológicos: la resistencia del huésped y la inmunopatología. 19 Las respuestas Th1 son potencialmente eficaces para erradicar agentes infecciosos, pero cuando una respuesta dominada por Th1 es poco eficaz o demasiado prolongada puede producirse daño al huésped. Las respuestasTh2 participan principalmente en las reacciones alérgicas, la producción de anticuerpos y el cambio de clase de anticuerpos. Pueden limitar las respuestas mediadas por Th1 potencialmente dañinas y ser parte de los mecanismos supresores para respuestas Th1 inapropiadas o exageradas. 20
Cada uno de los dos subtipos de células T cooperadoras inhibe el desarrollo y función del otro. El interferón gama producido por las células Th1 inhibe el desarrollo y función de las células Th2. Por el contrario, la IL-4 y la IL-10 producidas por las células Th2 inhiben el desarrollo y función de las células Th1. La IL-4 actúa en parte inhibiendo la expresión del receptor de IL-12, IL-12Rb, cuya expresión aumenta por el interferón gama [ver figura 4]. La IL-12 aumenta la función celular porque es un potente factor de crecimiento para las células asesinas naturales (NK), que también producen interferón fama. El hecho de que las células Th2 inhiban las respuestas inmunes Th1 y que las células TH1 inhiban las respuestas de anticuerpos mediadas por Th2 puede atribuirse al hecho de que ambos subtipos producen inflamación, que debe ser controlada [ver adelante, Citocinas e inflamación].
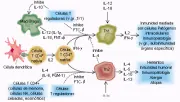
|
| Figura 4 |
| Regulación de las respuestas de la célula T cooperadora |
Los estudios sobre las respuestas inmunes a parásitos Schistosoma han demostrado que un carbohidrato de superficie, la lacto-N-fucopentaosa-III (LNFP-III) puede cambiar una respuesta protectora Th1 mediada por células en una respuesta Th2 mediada principalmente por anticuerpos. 21 La LNFP-III estimula la expresión de células B1 y causa que secreten grandes cantidades de IL-10. Al actuar a través de los macrófagos, la IL-10 inhibe las células Th1 y favorece la respuesta Th2. La IL-12 bloquea la expansión de las células B1 por los carbohidratos. Este fenómeno tiene implicaciones importantes para la inmunidad de muchos organismos porque este carbohidrato, que contiene Lewisx, existe en otros parásitos y en las células neoplásicas. El microrganismo o la célula cancerosa podría usar este carbohidrato para inhibir la respuesta inmune protectora mediada por células del huésped. Los carbohidratos podrían también inhibir las enfermedades autoinmunes controladas por Th1.
Al exponerse nuevamente al antígeno, las células de memoria median una respuesta inmunológica más rápida e intensa que las células T vírgenes o nativas. Comparado con las células T vírgenes, las células de memoria responden de modo diferente y tienen otro patrón de marcadores de superficie. CD45RO es el marcador encontrado en las células de memoria. Estas células secretan gran variedad de citocinas y pueden mostrar la misma polarización que las células Th1 y Th2. 12 Los requerimientos de proliferación y producción de citocinas en las células de memoria no son tan estrictos como para las células T nativas, pero la respuesta óptima requiere de coestimulación. 22 Las células T de memoria migran en forma selectiva a tejidos no linfoides específicos como el intestino, la piel y el pulmón. Las que llegan al intestino tienen integrinas especializadas (moléculas de adhesión) en su superficie denominadas a4b7 que median esta migración. Las células de memoria parecen persistir en ausencia de estimulación antigénica como células que no se dividen. El reencuentro con un antígeno puede expandir la población a un nivel más alto y estable, la competencia por otro antígeno puede disminuir la población. 22
La activación crónica de las células T CD4 + humanas en presencia de IL-10 da origen a un subtipo de clonas de células T CD4+ antígeno específicas con baja capacidad de proliferación y que producen niveles altos de IL-10, niveles bajos de IL-2 y nada de IL-4. Estas células T, llamadas células T reguladoras (Tr1), suprimen la proliferación de las respuestas inmunes antígeno-anticuerpo y ejercen regulación inhibitoria activa sobre la respuesta inmune patológica. 23
Las citocinas son cruciales en el proceso inflamatorio. El FNT-a, una citocina proinflamatoria prominente, es una de las sustancias más abundantes producidas por los macrófagos después de la estimulación con interferón gama, factor inhibitorio de migración (FIM) o lipopolisacárido bacteriano (LPS). El FNT-a también es producido por células T activadas, células NK y células cebadas.
La respuesta inflamatoria comienza con la expresión de las moléculas de adhesión ICAM-1, molécula de adhesión de células vasculares-1 (VCAM-1) y E-selectina (ELAM-1) en las células del endotelio vascular. Estas moléculas promueven la adhesión de neutrófilos, monocitos y linfocitos a la pared del vaso, del que se extravasan hacia los tejidos. En concentraciones bajas el FNT-a aumenta la respuesta inflamatoria protectora, activando y aumentando la función de varios leucocitos, incluyendo neutrófilos, macrófagos y eosinófilos. Esto puede estimular la producción de citocinas por los macrófagos, incluyendo el FNT-a per se, la IL-1, la IL-6 y diversas citocinas quimiotácticas [ver adelante, Quimocinas]. El FNT-a aumenta la expresión de moléculas de clase I del CPH, potencia la lisis celular inducida por células T citotóxicas, funciona como un pirógeno endógeno (i.e., el FNT-a induce fiebre, junto con IL-1 e IL-6, por medio de acciones directas e indirectas en el cerebro), activa el sistema de coagulación y la producción de proteínas de fase aguda por el hígado y puede causar inmunodeficiencia por la supresión de la médula ósea. El FNT-a causa también caquexia cuando está presente por periodos prolongados.
El FNT-a tiene un papel importante en la respuesta del huésped ante bacterias gram negativas, la fuente de la endotoxina, y al LPS. El LPS causa la liberación de FIM, que a su vez aumenta la producción de FNT-a por los macrófagos. Ante concentraciones bajas de LPS, el FNT-a media una respuesta protectora. Sin embargo, en concentraciones altas el FNT-a media la coagulación intravascular diseminada (parte de la cual se conoce como reacción de Shwartzman) y la muere por choque.
La inmunidad protectora contra ciertos organismos intracelulares, como Leishmania, está aumentada por el FNT-a y éste tiene potente actividad antiviral también. Sin embargo, muchos de los síntomas del paludismo, en especial del cerebral y algunos síntomas de la infección por VIH pueden estar mediados por FNT-a. Se ha autorizado ya el uso de anticuerpos contra FNT-a para el tratamiento de la enfermedad de Crohn. 24
Producida en forma casi exclusiva por células T activadas (células Th1), la linfotoxina tiene muchas de las mismas propiedades biológicas del FNT-a y se le conoce también como FNT-b. Al igual que el FNT-a, la linfotoxina lisa células tumorales pero no células normales, activa neutrófilos y aumenta la adhesión vascular y la extravasación de los leucocitos. Los efectos de la linfotoxina dependen de la unión de esta proteína con otra, la linfotoxina-b para formar un complejo. Este utiliza el mismo receptor celular que el FNT-a. Además, la linfotoxina es importante en el desarrollo del tejido linfoide.
Interleucina-1 e interleucina-6
La IL-1 es producida principalmente por monocitos y macrófagos, pero también por otras células, como las células epiteliales y endoteliales. Es un pirógeno endógeno y muchas de sus funciones son semejantes a las del FNT-a. Induce la producción de IL-1 adicional y de IL-6 a partir de los macrófagos, así como síntesis de glucocorticoides y liberación de prostaglandinas, colagenasa y proteínas de fase aguda. La IL-1 aumenta la expresión de moléculas de superficie en las células endoteliales, causando adhesión de leucococitos y coagulación, y estimula la producción de quimocinas de macrófagos que a su vez activan a los neutrófilos. La IL-1 difiere del FNT-a en que no produce necrosis de tumores o lesión tumoral, aumenta la expresión del CPH o, por si misma, media la reacción de Shwartzman.
Los macrófagos producen un antagonista del receptor de IL-1 (IL-1ra) que es estructuralmente semejante a la IL-1 y se fija al receptor, pero es inactivo. El IL-1ra, junto con el receptor de IL-1 se liberan de las células activadas, inhiben a la IL-1 y actúan de esa forma como un regulador. Estos inhibidores naturales de la IL-1 han sido investigados como agentes potenciales para su uso clínico con objeto de contrarrestar ciertos procesos inflamatorios.
La IL-6 es inducida por la IL-1 y el FNT-a a partir de los macrófagos y a su vez inhibe la producción por estas células de IL-1 y FNT-a. La IL-6 actúa sobre las células hepáticas para producir proteínas de fase aguda como fibrinógeno, a2-macroglobulina y proteína de amiloide sérico A. Esta citocina puede inhibir también la activación de los macrófagos.
El interferón gama, producido por las células T las NK, es el principal factor activador de macrófagos (FAM) y por lo tanto una citocina potente en la inmunidad mediada por células. Los macrófagos activados producen muchas citocinas y quimocinas que participan en forma íntima en la inflamación, incluyendo FNT-a, IL-1, IL-6 y FIM. Otros FAM incluyen el factor estimulador de colonias de granulocitos y macrófagos (FEC-GM) y el FIM. La IL-1 y el FNT-a tienen actividad débil de FAM. La IL-12 estimula a las células NK para producir mayores cantidades de interferón fama, aumentando las acciones dependientes de estas sustancias. Per se y por aumentar los efectos del FNT-a, el interferón gama causa la expresión de moléculas de adhesión en la superficie de las células del endotelio vascular, causando adhesión y extravasación de células T.
Los efectos proinflamatorios del interferón fama son contrarrestados por el factor de transformación de crecimiento-b (FTC-b) y la IL-10, que inhibe la activación de los macrófagos. El interferón gama se ha usado con éxito para tratar la enfermedad granulomatosa crónica y la leishmaniasis visceral resistente a fármacos. 25
Factor inhibidor de la migración
El FIM fue la primera citocina descubierta que derivaba de células T producida después de la estimulación de estas células sensibilizadas con el antígeno específico. Su nombre deriva del hecho de que inhibe la migración aleatoria de los macrófagos. El FIM actúa como una hormona endógena que contrarresta la acción de los esteroides. 26 Los macrófagos y las células T liberan FIM en respuesta a los esteroides y otros estímulos proinflamatorios. El FIM supera los efectos inmunosupresores de los esteroides sobre los macrófagos y la producción de citocinas por las células T. 26
El gen del FIM es un gen temprano retardado que se expresa en muchos tejidos diferentes. 27 Se encuentran grandes cantidades del mismo en los macrófagos y células de la hipófisis. De hecho, el LPS estimula la liberación de FIM por la hipófisis. Cuando se administra a ratones, el FIM aumenta mucho la mortalidad por LPS, por el contrario, los anticuerpos anti-FIM revierten por completo la letalidad del LPS. 28 Se ha demostrado que el FIM recombinante activa a los macrófagos para que destruyan a Leishmania y los estimulan para producir FNT-a y óxido nítrico. Los ratones que no tienen el gen para FIM muestran mayor resistencia a los efectos letales de dosis altas de LPS y enterotoxina de S. aureus, sí como Pseudomonas aeruginosa, pero son susceptibles a Leishmania. 29
Es importante mencionar que en la actualidad se desarrollan tratamientos anti-FIM. El objetivo es incrementar las propiedades inmunosupresoras y antinflamatorias de los esteroides de liberación endógena, disminuyendo así la necesidad de tratamiento esteroideo en diversos padecimientos autoinmunes e inflamatorios. 26
La IL-5 afecta principalmente el reclutamiento y activación de los eosinófilos. La IL-5 es producida por las células Th2 y células cebadas activadas y estimula el crecimiento y diferenciación de los eosinófilos. Los eosinófilos son activados por la IL-5, el FNT-a y el factor incrementador de citotoxicidad de eosinófilos derivado de los monocitos. Los eosinófilos activados producen daño tisular en los estados alérgicos y destruyen parásitos helmínticos.
Interleucina-4 e interleucina-13
La IL-4 estimula la expresión de VCAM en las células endoteliales, causando unión de eosinófilos, linfocitos, neutrófilos y monocitos con su subsecuente extravasación. Sin embargo, también actúa como una citocina antinflamatoria, inhibiendo los macrófagos activados y disminuyendo la producción de FNT-a y óxido nítrico.
La IL-13 tiene la capacidad de realizar algunas de las funciones de la IL-4. Ambas citocinas inducen síntesis de IgE en las células B y diferencian células T a células Th2, pudiendo suprimir el proceso inflamatorio inducido por las células Th1. La IL-4 y la IL-13 realizan sus efectos a través de la unión a receptores sobre células blanco (IL-4R e IL-13R, respectivamente) y activan la transducción intracelular de señales a través de las cinasas Jak y el factor de transcripción STAT6, que media la transcripción de genes que responden a IL-4. Estas citocinas actúan también sobre los macrófagos para suprimir diversas citocinas proinflamatorias, quimocinas y factores de crecimiento hematopoyético, incluyendo IL-1, IL-8, FNT-a, proteína inflamatoria de macrófagos-1a (PIM-1a) y FEC-GM. Este provoca la inhibición de los receptores Fc sobre los macrófagos y la inhibición de la citotoxicidad mediada por células dependiente de anticuerpos (CCDA) y la producción de óxido nítrico.
Las células cebadas y basófilos activados producen más IL-4. Es importante que la IL4 mutada puede inhibir la síntesis de IgE por IL-4 e IL-13 y ser útil para el tratamiento de algunos estados alérgicos.
Factor de transformación de crecimiento-b
El FTC-b es producido por diversas células, incluyendo plaquetas, linfocitos, macrófagos activados y células de la placenta. Es una citocina antinflamatoria importante porque inhibe la activación de los macrófagos y la maduración de las células T citotóxicas, controlando los efectos de muchas citocinas.
La IL-10 es producida por las células T CD4+ y CD8+, las células B, los macrófagos, las células cebadas activadas y los queratinocitos. Aunque suele asociarse con actividad de las células Th2, la IL-10 puede también ser producida por células Th1. La IL-10 suprime las respuestas de linfocitos al regular en forma inhibitoria las citocinas de macrófagos, incluyendo IL-1, FNT-a, IL-6, IL-8, FEC-GM y factor estimulador de colonias de granulocitos (FEC-G), e inhibir la producción de óxido nítrico.
Las quimocinas son una superfamilia de citocinas de bajo peso molecular que median la migración direccional de los leucocitos en el estado normal y el proceso inflamatorio.30 Tienen un papel importante para atraer granulocitos hacia los sitios de inflamación. Existen cuatro familias diferentes de quimocinas que se distinguen con base en la posición de sus dos primeros residuos conservados de cisteína: CXC (las primeras dos cisteínas están separadas por un aminoácido), CC, C y CX3C. Los receptores para quimocinas son receptores proteicos acoplados a la membrana G y constituyen una de las clases más grandes de moléculas de señal.
Las quimocinas CXC activan principalmente neutrófilos.31 Esta familia incluye a la IL-8, la tromboglobulina-b (TG-b), gro-a, gro-b, gro-g y factor plaquetario 4. Suelen ser producidas por monocitos, algunas son producidas por otras células, incluyendo células T, células endoteliales y plaquetas. La IL-8 induce la expresión de integrinas de unión a enutrófilos en las células endoteliales, provocando la acumulación rápida de este tipo de células en los tejidos. La quimocina gro (producto génico relacionado con el crecimiento) estimula también el acúmulo de neutrófilos, así como la liberación de enzima lisosómicas que contribuyen a la respuesta inflamatoria local. El factor plaquetario 4 y la TG-b se liberan de plaquetas agregadas y fibroblastos estimulados, necesarios en sitios de reparación de hemorragia o trombosis.
Las quimocinas CC activan a células T, monocitos y eosinófilos. Esta familia incluye a las RANTES (factor regulado, expresado y secretado en las células T normales activadas), el factor quimiotáctico y activador de macrófagos (FQAM), PIM-1a y PIM-1b. Las quimocinas CC son producidas por células T y monocitos activados. RANTES es un potente quimiotáctico para las células T de memoria (pero no para las nativas) y también atrae monocitos. El FQAM actúa solo sobre los monocitos, atrayéndolos, activándolos y regulando la expresión de integrinas en su superficie. Los PIM-1a y 1b atraen solo monocitos. Las quimocinas CC, eotaxina, eotaxina-2 y proteína quimioatrayente de monocitos-4 (PQM-4) activan en forma predominante eosinófilos.31
Los receptores de quimocinas tienen un papel importante como correceptores para el VIH. El virus actúa primero sobre el CD4 en las células T, pero requiere un correceptor para penetrar la membrana celular. El receptor-5 de la quimocina CC (CCR5) que media la activación de las células T y los macrófagos es el principal correceptor para algunas cepas de VIH-1. Los ligandos naturales para el CCR5 incluyen al RANTES, PIM-1a y PIM-1b.32 El receptor-4 de quimocina CXC (CXCR4) parece ser importante en la fase tardía d de la infección por VIH.33
Respuestas de las células B al antígeno
Cuando un receptor de célula B (RCB) se une al antígeno soluble ocurre uno de dos eventos: apoptosis o proliferación y maduración posterior de la célula. Las señales para estos procesos son generadas intracelularmente por Iga e Igb. Estas proteínas se asocian con el RCB del mismo modo como las proteínas CD3 se asocian con el RCT. La Iga y la Igb reclutan moléculas de señales de transducción en forma semejante a la del complejo RCT-CD3, principalmente dos cascadas de tirosina cinasas, la primera que consiste en Lyn, Fyn y Btk y la segunda que consiste en Syk. Algunas de las mismas proteínas que participan en la activación de la célula T son reclutadas (v.gr., PI-3 cinasa, Vav, Ras, Raf-1, MEK, PLC-b y PLC-g). La vía de activación de las células B es semejante, pero no idéntica a la de las células T.
Se sabe relativamente poco sobre las señales que tienen lugar en las células B cuando éstas presentan el antígeno a una célula T cooperadora que lo reconoce. La interacción célula T-célula B puede también causar apoptosis o proliferación de las células B. Sin embargo, con la ayuda de la célula T la proliferación provoca la generación de varias clases diferentes de células B.
Las células del manto folicular expresan IgD (IgD +, CD38-) en su superficie y pueden madurar a células plasmáticas que producen solo anticuerpos de IgM. Estas células producen IgM que muestran gran heterogeneidad, pero las células no sufren mutación somática para generar números más grandes de moléculas heterogéneas de anticuerpo.
El proceso que causa la mutación somática ocurre en los centrocitos, que son células B IgD-, CD38+ que han sufrido cambio de isotipo para producir IgG. El paso de maduración de célula B IgD +, CD38- a IgD -, CD38+ depende del contacto intercelular con células T específicas de antígeno y citocinas. En especial la IL-4 es de gran importancia para los eventos que provocan cambio de clase.
La célula B más madura, la célula de memoria, no expresa IgD o CD38, pero se distingue por otros marcadores de superficie y por su localización dentro del centro germinal. Las células de memoria pueden también evolucionar a células plasmáticas, produciendo IgM, IgG, IgA e IgE. En general, la generación de células plasmáticas a partir de las células B de memoria es independiente de la ayuda de las células T.
INTERACCIONES CELULA T- CELULA B
CD40 es un receptor de superficie celular que pertenece a la familia del RFNT y que fue identificado por primera vez en las células B. El ligando natural de CD40, CD40L (también llamado CD154) es una glucoproteína de tipo 2 en la superficie de la célula T relacionada al FNT. La expresión temporal de CD40L en la superficie de las células T es inducida por la unión del RCT al complejo antígeno-CPH y por la unión de B7 a CD28 o CTLA-4. La unión CD40-CD40L estimula el cambio de isotipo de inmunoglobulina en la célula B. Esto se demostró mejor por la detección de un defecto molecular en una inmunodeficiencia genética denominada síndrome de hiper-IgM. Una mutación en el gen que codifica CD40L es responsable del cambio defectuoso de isotipo de anticuerpo en este síndrome. Las respuestas de la célula B independientes de la célula T y las respuestas inducidas por un anti-CD40 no se afectan en esta mutación. Por lo tanto, la ayuda de la célula T para la activación de la célula B está mediada principalmente por una vía coestimuladora CD40-CD40L. Las personas con síndrome de hiper-IgM son susceptibles a patógenos que producen infecciones oportunistas como las que ocurren en el SIDA y que normalmente dependen de células T, lo que resalta el papel de la coestimulación de CD40L en la activación normal de las células T. la ausencia de CD40L tiene un efecto más dramático sobre el cambio de clase de Ig que la ausencia de células T. Por lo tanto, parece ser que CD40L puede expresarse en otras células además de las T, que pudieran inducir el cambio de clase de inmunoglobulinas en las células B a través de la unión de CD40.
Mecanismos efectores en la inmunidad mediada por células
La inmunidad mediada por células provoca la destrucción de microrganismos invasores como bacterias, virus, hongos y parásitos, la destrucción de células tumorales, el rechazo de injertos tisulares y la lesión de tejidos en diversos estados patológicos, incluyendo autoinmunidad. Las reacciones inmunes mediadas por células pueden ser inducidas también por contacto con antígenos, como las encontradas en la intoxicación con hiedra venenosa y numerosos fármacos. Los fármacos provocan reacciones mediadas por células con más frecuencia cuando se aplican tópicamente y no por vía sistémica.
La mayoría de las reacciones inmunes mediadas por células incluyen una interacción inicial entre células T sensibilizadas y antígenos en las células presentadoras. Esta reacción puede despertar varias vías efectoras, incluyendo la activación de células T citotóxicas, la estimulación de la producción de citocinas por la célula T que activan macrófagos y promueven la proliferación de células NK y la producción de anticuerpos que participan en la citotoxicidad mediada por células dependiente de anticuerpos por las células NK y otros tipos celulares. Aunque las reacciones inmunes mediadas por células diferentes a la CCDA no requieren la presencia de anticuerpo o complemento, pueden modificarse por estos factores humorales. Los eventos subsecuentes requieren de la cooperación entre diferentes subtipos de células, las reacciones que participan son controladas por diversas citocinas.
Los mecanismos de inmunidad mediada por células incluyen interacciones célula T-macrófago que pueden ser tanto protectoras (causando destrucción de microrganismos invasores) como dañinas (causando inflamación y destrucción tisular). En ocasiones ambas van de la mano, por ejemplo, en la tuberculosis tanto la destrucción del bacilo como la producción de cavidades en los pulmones son consecuencia de interacciones célula T-macrófago.
Las células T citotóxicas y las células NK son la principal defensa del organismo contra las células infectadas por virus y tumorigénicas. Estas células asesinas inducen muerte celular programada por dos mecanismos. El mecanismo clásico es la apoptosis mediada por gránulos. Un segundo mecanismo es la apoptosis mediada por receptores. En respuesta a la unión del ligando expresado en la célula asesina, los receptores de superficie celular se agregan en la célula blanco, lo que causa reclutamiento de las proteínas citoplásmicas a los receptores y transducción de una señal de muerte a la célula blanco. 34
Las células T citotóxicas son células efectoras específicas de antígeno que son importantes para atacar agentes infecciosos, en especial los virus presentes en células diferentes a los macrófagos, destruir células tumorales y rechazar aloinjertos. La mayoría de las células T citotóxicas son CD8 + que reconocen antígenos presentados por moléculas de clase I del CPH, aunque un número considerable de células T CD4+ son capaces de destruir células blanco.
Una fase de adhesión célula T citotóxica-célula blanco, dependiente de Mg2+ es seguida de una fase dependiente de Ca2+ que causa la llegada de químicos citotóxicos a la célula blanco. La célula T citotóxica se disocia entonces de la célula blanco y ocurre la muerte en ausencia de la célula T citotóxica, que se recicla para atacar otra célula blanco. Si la célula T citotóxica se adhiere a una célula que no porta la combinación correcta péptido antigénico-molécula del CPH, no se liberan químicos citotóxicos y las células se disocian con más rapidez.
Las células T citotóxicas desarrollan gránulos que contienen moléculas citotóxicas, incluyendo perforinas (proteínas que producen orificios o poros en la superficie de la membrana celular), proteasas de serina (granzima A y granzima B) y estereasas de serina. De estas, las perforinas son las más importantes, como se ha demostrado en ratones en los que se ha eliminado el gen que codifica la perforina. Un segundo mecanismo de destrucción incluye la interacción ligando de Fas en la célula T citotóxica y Fas en la célula blanco. Este es el único mecanismo de destrucción disponible para el ratón nulo a perforina y se usa preferencialmente (aunque no en forma exclusiva) en las células T CD4+ citotóxicas.
En respuesta a los virus se produce un gran número de células T citotóxicas específicas. Esto se demuestra en forma evidente durante las respuestas iniciales a las células B infectadas con virus Epstein-Barr. Se ha encontrando que las clonas de células T citotóxicas específicas para algún complejo antígeno-CPH son muy abundantes, constituyendo alrededor del 50 por ciento de todas las células T citotóxicas. 11 Cuando la respuesta de la célula T citotóxica disminuye estas clonas abundantes de células T parecen eliminarse por mecanismos de apoptosis.
Las células NK son los efectores citolíticos del sistema inmunológico innato. La IL-12 es un inductor potente de la activación y proliferación de las células NK; el interferón gama y la IL-2 también activan estas células. La IL-2 aumenta la capacidad tumoricida de las células NK, lo que causa desarrollo de células asesinas activadas por linfocinas (LAK). Las células NK pueden secretar también interferón gama, que actúa en forma autoactiva para incrementar la actividad de las células NK. El FNT-a puede contrarrestar el desarrollo de las células NK inducido por IL-12.
La fijación mediada por receptor de las células NK a la célula blanco ocurre en forma Ca2+ independiente. Sin embargo, una vez que las células NK se activan requieren de iones Ca2+ para la lisis. La citólisis se inicia cuando las células NK orientan sus gránulos hacia la célula blanco y liberan perforinas y enzimas proteolíticas. El mecanismo de acción es semejante al de las células T citotóxicas. La destrucción de las células NK, a diferencia de la destrucción por células T citotóxicas, no es bloqueada por anticuerpos monoclonales anti-CD3 o anti-CD8, pero sí por anticuerpos contra la molécula de adhesión LFA-1.
Las células NK también son sujeto de inhibición de la función efectora mediada por receptores. 35 Las señales inhibitorias se transducen por receptores en las células NK que detectan moléculas de clase I del CPH en las células blanco. Al unirse el ligando al receptor inhibitorio ocurre una secuencia de foforilación del motivo de inhibición basado en el inmunorreceptor tirosina (ITIM), lo que proporciona un sustrato para la unión de las proteínas fosfatasas de tirosina SHP-1 y SHP-2. Estas fosfatasas median la transducción citoplásmica de una señal negativa que causa la inhibición de la citotoxicidad y la expresión de citocinas. También se encuentran receptores inhibitorios en la superficie de ciertas células T citotóxicas, lo que indica que las respuestas de estas células también pueden afectarse por miembros de la superfamilia de receptores inhibitorios. 35.36
En la Red Mundial de Internet se ha colocado una extensa lista de designaciones para los antígenos CD. La URL para esta lista es www.cx.unibe.ch/dkf6/immunology/info/cd_table.htm. Las ilustraciones de las estructuras conocidas de las citocinas, determinadas por cristalografía de rayos x o espectroscopía magnética nuclear multidimensional pueden también conocerse en la red y la URL de este sitio es www.psynix.co.uk/cytweb/cyt_strucs/struc_class.html.
Figuras 1 a 4 Dimitry Schidlovsky.
Bibliografía
- Davis MM, Boniface JJ, Reich Z, et al: Ligand recognition by alpha beta T cell receptors. Annu Rev Immunol 16:523, 1998 [PMID 9597140 ]
- Li S, Paulsson KM, Sjogren HO, et al: Peptide-bound major histocompatibility complex class I molecules with tapasin before dissociation from transporter associated with peptide processing. J Biol Chem 274:8649, 1999 [PMID 10085102 ]
- Ploegh HL: Viral strategies of immune evasion. Science 280:248, 1998
- Barrera CA, Almanza RJ, Ogra PL, et al: The role of the invariant chain in mucosal immunity. Int Arch Allergy Immunol 117:85, 1998 [PMID 9784651 ]
- Beckman EM, Porcelli SA, Morita CT, et al: Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature 372:691, 1994 [PMID 7527500 ]
- Park SH, Chiu YH, Jayawardena J, et al: Innate and adaptive functions of the CD1 pathway of antigen presentation. Semin Immunol 10:391, 1998 [PMID 9799714 ]
- Jardetzky TS, Brown JH, Gorga JC, et al.: Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature 368:711, 1994
- Banchereau J, Steinman RM: Dendritic cells and the control of immunity. Nature 392: 245, 1998 [PMID 9521319 ]
- Viola A, Schroeder S, Sakakibara Y, et al: T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science 283:680, 1999 [PMID 9924026 ]
- Altman JD, Moss PAH, Goulder PJR, et al: Phenotypic analysis of antigen-specific T lymphocytes. Science 274:94, 1996 [PMID 8810254 ]
- McMichael AJ, O'Callaghan CA: A new look at T cells. J Exp Med 187:1367, 1998 [PMID 9565629 ]
- Van Parijs L, Abbas AK: Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science 280:243, 1998 [PMID 9535647 ]
- Ward SG: The complexities of CD28 and CTLA-4 signalling: PI3K and beyond. Arch Immunol Ther Exp (Warsz) 47:69, 1999
- Bolen JB, Brugge JS: Leukocyte protein tyrosine kinases: potential targets for drug discovery. Annu Rev Immunol 15:371, 1997 [PMID 9143693 ]
- Rao A, Luo C, Hogan PG: Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15:707, 1997 [PMID 9143705 ]
- Kersh GJ, Kersh EN, Fremont DH, et al: High- and low-potency ligands with similar affinities for the TCR: the importance of kinetics in TCR signaling. Immunity 9:817, 1998 [PMID 9881972 ]
- Itoh Y, Hemmer B, Martin R, et al: Serial TCR engagement and down-modulation by peptide: MHC molecule ligands: relationship to the quality of individual TCR signaling events. J Immunol 162:2073, 1999 [PMID 9973480 ]
- O'Garra A: Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity 8:275, 1998
- Sher A, Gazinelli RT, Jankovic D, et al: Cytokines as determinants of disease and disease interactions. Braz J Med Biol Res 31:85, 1998 [PMID 9686183 ]
- Granger DN: Cell adhesion and migration: II. Leukocyte-endothelial cell adhesion in the digestive system. Am J Physiol 275:G982, 1997
- Velupillai P, Harn DA: Oligosaccharide-specific induction of interleukin 10 production by B220+ cells from schistosome-infected mice: a mechanism for regulation of CD4+ T-cell subsets. Proc Natl Acad Sci USA 91:18, 1994 [PMID 7904066 ]
- Dutton RW, Bradley LM, Swain SL: T cell memory. Annu Rev Immunol 16:201, 1998 [PMID 9597129 ]
- Groux H, O'Garra A, Bigler M, et al: A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 389:737, 1997 [PMID 9338786 ]
- Baert FJ, Rutgeerts PR: Anti-TNF strategies in Crohn's disease: mechanisms, clinical effects, indications. Int J Colorectal Dis 14:47, 1999 [PMID 10207730 ]
- Locksley RM, Fowell DJ, Shinkai K, et al: Development of CD4+ effector T cells and susceptibility to infectious diseases. Adv Exp Med Biol 452:45, 1998 [PMID 9889958 ]
- Bucala R: Neuroimmunomodulation by macrophage migration inhibitory factor (MIF). Ann NY Acad Sci 840:74, 1998
- Lanahan A, Williams JB, Sanders LK, et al: Growth factor-induced delayed early response genes. Mol Cell Biol 12:3919, 1992 [PMID 1508193 ]
- Bernhagen J, Calandra T, Mitchell RA, et al: MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature 365:756, 1993 [PMID 8413654 ]
- Bozza M, Satoskar AR, Lin G, et al: Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med 189:341, 1999 [PMID 9892616 ]
- Baggiolini M, Dewald B, Moser B: Human chemokines: an update. Annu Rev Immunol 15:675, 1997 [PMID 9143704 ]
- Petering H, Gotze O, Kimmig D, et al: The biologic role of interleukin-8: functional analysis and expression of CXCR1 and CXCR2 on human eosinophils. Blood 93:694, 1999 [PMID 9885232 ]
- Olbrich H, Proudfoot AE, Opperman M: Chemokine-induced phosphorylation of CC chemokine receptor 5 (CCR5). J Leukoc Biol 65:281, 1999 [PMID 10080528 ]
- Choe H: Chemokine receptors in HIV-1 and SIV infection. Arch Pharm Res 21:634, 1998
- Darmon AJ, Bleackley RC: Proteases and cell-mediated cytotoxicity. Crit Rev Immunol 18:255, 1998 [PMID 9637413 ]
- Lanier LL: NK cell receptors. Annu Rev Immunol 16:359, 1998
- Bruhns P, Marchetti P, Fridman WH, et al: Differential roles of N- and C-terminal immunoreceptor tyrosine-based inhibition motifs during inhibition of cell activation by killer cell inhibitory receptors. J Immunol 162:3168, 1999 [PMID 10092767 ]