Neurología
⭳ Abrir artículo (PDF)319.2 KBEste artículo es idéntico en la Edición 2/2000.
Contenido del artículo
X ATAXIAS HEREDITARIAS
X ATAXIAS HEREDITARIAS
DR. S.H. SUBRAMONY
Las ataxias hereditarias son trastornos que causan desequilibrio progresivo como resultado de patología en el cerebelo y sus diversas vías de conección. Las personas con ataxias hereditarias suelen tener también otros signos que indican disfunción en los ganglios basales, la neurona motora superior, los núcleos oculomotores y otros núcleos del tallo cerebral, las células del asta anterior y los ganglios de la raíz dorsal. En algunos casos ocurre también pérdida de la visión y deterioro cognitivo. El cuadro clínico general de muchas ataxias hereditarias es semejante, por lo que las clasificaciones que se basan en el fenotipo clínico no son óptimas. La edad de inicio del trastorno, el patrón de herencia y la combinación particular de los signos clínicos pueden ayudar a determinar el genotipo subyacente.
Durante la última década, los estudios de genética molecular han identificado mutaciones genéticas específicas en muchas ataxias hereditarias y localizado la causa de otras en loci cromosómicos específicos, lo que permite lo siguiente:
1. Clasificación de estos trastornos con base en sus genotipos (ver tabla 1 y 2).
2. Identificación del genotipo preciso en muchos pacientes.
3. Realización de estudios diagnósticos presintomáticos y prenatales.
4. Comprensión de parte de la variabilidad fenotípica relacionada a mutaciones en el mismo gen.
5. Especulación sobre los mecanismos de muerte neuronal selectiva. La sintomatología prograsiva de estos padecimientos se relaciona con la pérdida de neuronas en muchas partes del sistema nervioso, incluyendo las células cerebelosas de Purkinje, así como neuronas de la protuberancia, olivas, sustancia negra, médula espinal y ganglios de las raíces dorsales.
Mecanismos de mutación y patrones de herencia
Las mutaciones patogénicas son alteraciones en la secuencia de nucléotidos de los genes que producen un fenotipo alterado (uno que manifiesta enfermedad). Los genes están compuestos de pares complementarios de nucleótidos que especifican la secuencia de aminoácidos que constituyen proteínas específicas. Las secuencias de nucleótidos que se transcriben a los correspondientes ARNm (ARNm) se denominan exones, las secuencias de nucleótidos que no se transcriben son los intrones. Dentro de los genes, las secuencias de tres nucleótidos (codones) especifican un aminoácido particular. Las mutaciones, incluyendo sustituciones de nucleótidos, deleciones, inserciones, y duplicación y expansión trinucléotida pueden afectar a los codones de muchas maneras.
MECANISMOS DE MUTACION
Mutaciones puntiformes En una mutación puntiforme un solo nucleótido dentro de un codón es sustituido por otro. El codón alterado puede especificar un aminoácido diferente, que puede ocasionar la creación de una proteína mutante (mutación con sentido equivocado). Un solo cambio en un nucleótido puede también producir un codón que no codifique ya para un aminoácido (v.gr., un codón de alto), causando terminación prematura de la síntesis proteica (mutación sin sentido).
Mutación de marco Una mutación de marco es causada por inserción o deleción de uno o más pares de nucleósidos en un gen. Esto desplaza el marco de lectura de todos los codones después del sitio de la mutación. La cadena polipéptida resultante puede estar compuesta de aminoácidos no codificados en el gen original y puede tener una longitud diferente.
Duplicación Una duplicación es una aberración cromosómica. Toda una secuencia de nucleótidos dentro de un segmento de un cromosoma se duplica en el genoma haploide. El genoma diploide tiene tres, en lugar de dos, copias de un segmento particular de material genético.
Expansión repetida de tres nucleótidos Muchos genes normales tienen secuencias repetidas del mismo nucleótido. En los trastornos de expansión repetida de tres nucleótidos, las personas afectadas tienen una expansión de los trinucleótidos repetidos más allá del rango normal.
PATRONES DE HERENCIA
Las enfermedades hereditarias se caracterizan por un amplio rango de edad de inicio, anticipación (i.e., la ocurrencia aparente de la enfermedad a una edad progresivamente más temprana en las generaciones sucesivas) en la edad de inicio y variabilidad del fenotipo del padecimiento dentro de la misma familia. Un patrón autosómico recesivo de herencia en una enfermedad determinada requiere que ambas copias de un gen para una proteína específica muten para producir el fenotipo de la enfermedad. La enfermedad no se manifestará en los padres que son heterocigotos para la mutación, pero aparecerá en los descendientes homocigotos. En la herencia autosómico dominante, una mutación en una copia del gen es suficiente para causar la enfermedad. Por lo tanto, las personas heterocigotas manifestarán la enfermedad y la trasmitirán a parte de su descendencia. En la herencia recesiva ligada al X, el gen mutado se encuentra en el cromosoma X, las portadoras mujeres con una copia normal del gen en el otro cromosoma X no padecen la enfermedad, pero la trasmiten a parte de su descendencia de sexo masculino. No ocurre trasmisión de la enfermedad de hombre a hombre en los padecimientos ligados al X; sin embargo, los hombres pueden trasmitir la enfermedad a sus nietos a través de sus hijas. Por último, las mutaciones genéticas en el ADN mitocondrial pueden causar alteración en la función de la cadena respiratoria, lo que puede producir la enfermedad. Debido a que todas las mitocondrias se heredan de la madre, los padecimientos secundarios a mutaciones en el ADN mitocondrial se trasmiten exclusivamente a través de la madre, pero tanto los varones como las mujeres están afectados.
Por lo tanto, las mutaciones genéticas son de diversos tipos y magnitud, causan variabilidad intrafamiliar considerable en muchas enfermedades genéticas (por ejemplo, los trastornos de expansión de trinucleótidos) así como diferencias fenotípicas entre lo que parecen ser trastornos muy semejantes (por ejemplo, distrofias musculares de Duchenne y de Becker). En esta subsección se usan las ataxias hereditarias como un modelo para discutir algunos de estos mecanismos de mutación y para mostrar la forma como los enfoques moleculares han cambiado la evaluación de estas enfermedades [ver tablas 1 y 2]. Se han logrado progresos semejantes en algunas otras enfermedades neurogenéticas [ver tabla 3].
Estudio del paciente con ataxia progresiva
Los pacientes que presentan ataxia progresiva deben ser examinados en forma cuidadosa, y deberán registrarse los detalles del fenotipo. Se realizará una historia familiar detallada, construyendo un pedigree. En algunas familias será evidente el patrón de herencia, que puede ayudar a la selección de las pruebas genpéticas adecuadas. En otras familias puede ser útil el examen directo de los familiares si la historia del paciente no es suficiente.
Deben excluirse las causas no hereditarias de ataxia progresiva, como la esclerosis múltiple, los eventos cerebrovasculares, la degeneración cerebelosa atáxica, las lesiones ocupativas y las lesiones meníngeas, en especial si la historia familiar no es convincente. Los estudios de imagen, en especial de resonancia magnética, son útiles a este respecto. En casos seleccionados, en especial en los trastornos infantiles de herencia recesiva, deben descartarse enfermedades susceptibles de tratamiento, lo que con frecuencia se logra con estudios de sangre simples [ver tabla 4]. Por último, es posible sugerir un genotipo diagnóstico con base en el fenotipo específico observado y usando los análisis de mutación actualmente disponibles, muchos de los cuales pueden obtenerse a través de laboratorios comerciales [ver tablas 1 y 2]. Es posible emitir un consejo genético más adecuado si se confirma el genotipo esperado. En algunos casos puede usarse un ensayo genético para estudiar a los familiares con riesgo.
Las terapias tanto sintomáticas como neuroprotectoras para la mayoría de las ataxias hereditarias son desalentadoras. En situaciones seleccionadas puede ser útil el tratamiento específico de problemas como espasticidad y síndromes extrapiramidales con agentes como el baclofen y agonistas de dopamina, respectivamente. Puede ocurrir mejoría marginal en la función motora con el uso de buspirona o amantadina. Los pacientes requieren también programas apropiados de rehabilitación.
Ataxias autosómico recesivas
Ataxia de Friedreich La ataxia de Friedrich (AF) es el tipo más común de ataxia de herencia recesiva. El inicio de los síntomas suele ocurrir en la primera o segunda década de la vida, pero puede retrasarse hasta la tercera o después. La enfermedad se caracteriza por ataxia progresiva de la marcha, pérdida de los reflejos tendinosos profundos y perdida de la sensación propioceptiva en las extremidades, todos signos de patología temprana importante en las células ganglionares de las raíces dorsales y sus proyecciones sensoriales periféricas.1 Otros signos incluyen disartria, respuesta plantar extensora y alteraciones oculomotoras, incluyendo sacudidas en onda cuadrada. En la fase tardía ocurren atrofia muscular, debilidad y disfagia. En pocos pacientes se presentan temblor, pérdida de la visión y de la audición. Alrededor del 30 a 50 porciento de los pacientes desarrolla cardiopatía sintomática. Ocurre diabetes en el 10 porciento de los enfermos, y son frecuentes las deformidades esqueléticas, como las escoliosis.
La mutación en la AF consiste en una expansión de una repetición trinucleótida GAA intrónica en un gen denominado X-25, que se localiza en el cromosoma 9q.2 Este gen codifica para una proteína de 210 aminoácidos denominada frataxina. En el sistema nervioso central, la mayor concentración de frataxina se encuentra en la médula. En las personas normales se encuentran entre el 7 y el 22 porciento de las repeticiones GAA dentro del primer intrón. Los alelos agrandados portan 200 a 900 o más repeticiones. La mayoría de los pacientes con AF son homocigotos para esta expansión, el otro alelo porta una mutación puntiforme. La expansión es inestable durante la trasmisión meiótica del gen de los padres a la descendencia. Las evidencias preliminares sugieren que las personas afectadas tienen niveles bajos de ARNm de frataxina. Por lo tanto, la expansión de la repetición intrónica puede causar pérdida del mensaje para la frataxina, lo que ocasiona la enfermedad.
Series recientes de pacientes estudiados después de la identificación de la mutación genética han demostrado que el fenotipo asociado con la expansión GAA es más variable de lo que antes se afirmó con base en los estudios clínicos. La edad de inicio puede ser muy posterior (los pacientes pueden estar iniciando los 50) y el fenotipo puede incluir ataxia asociada a conservación de los reflejos, en ocasiones intensos.3 Debido a estos datos, el análisis de mutación para la AF está indicado no solo en pacientes con AF clásica, sino también en enfermos que presentan ataxias esporádicas y recesivas tan tarde como en la sexta década de la vida.
Ataxia con deficiencia aislada de vitamina E La ataxia con deficiencia aislada de vitamina E (ADVE) es un trastorno hereditario recesivo que recuerda a la AF, con inicio en la infancia de ataxia, arreflexia y pérdida de la sensación prospectiva. Los niveles de vitamina E son bajos y los suplementos pueden detener la progresión de la enfermedad. La mutación ocurre en el gen de la proteína de transferencia del a-tocoferol (PTT-a) en el cromosoma 8.4 La PTT-a participa en el procesamiento hep[daggerdbl]tico del tocoferol y en su incorporación a las lipoproteínas de muy baja densidad (LMBD). La absorción de vitamina E es normal.
En los pacientes que expresan un fenotipo severo la mutación consiste en una deleción de marco de una sola base de pares que remplaza los últimos 30 aminoácidos de la proteína con un péptido aberrante de 14 aminoácidos. Sin embargo, los pacientes que tuvieron inserciones o mutaciones puntiformes en el gen TPP-a expresaron un fenotipo menos severo. Por ejemplo, un paciente japonés presentó ataxia con inicio en la edad adulta y arreflexia.5 Por lo tanto, el tipo de mutación en el gen PTT-a determina el grado de deterioro funcional de la proteína e influye en el fenotipo final. Es adecuado obtener niveles de vitamina E en pacientes con cualquier tipo de ataxia esporádica, en especial si se disminuyen los reflejos tendinosos profundos. Ciertamente, debe excluirse la posibilidad de ADVA en todos los niños con sospecha de ataxia de Friedreich.
Ataxia-telangiectasia La ataxia telangiectasia típicamente se presenta pronto en la primera década de la vida como ataxia de la marcha. Los signos neurológicos que se desarrollan durante la primera década incluyen hipotonía, cierto grado de coreoatetosis, arreflexia y un trastorno oculomotor característico que se asocia con menor capacidad para generar sacadas, lo que obliga a empujar la cabeza para mover los ojos.6 Las telangiectasias típicas ocurren en niños de alrededor de cinco años de edad y pueden encontrarse en las conjuntivas, párpados y fosa cubital y poplítea. Estos niños tienen alto riesgo de neoplasias, en especial linfomas. La medición de una sensibilidad alterada a la radiación y los niveles elevados de a-fetoproteína en suero son útiles para confirmar el diagnóstico.
Las deleciones, y en ocasiones inserciones en el gen mutado de la ataxia telangiectasia (GAT) en el cromosoma 11q se asocian con la enfermedad,7 El producto proteico de este gen tiene semejanzas con las cinasas 3-fosfatidilinositol, que participan en la transducción de señales mitogénicas.
Ataxias autosómico dominantes
Las ataxias autosómico dominantes son genéticamente heterogéneas, pero comparten muchas manifestaciones clínicas [ver tabla 2]. Por lo tanto, puede ser difícil la identificación precisa del genotipo con base solo en las características clínicas. El diagnóstico clínico puede facilitarse examinando a varios miembros afectados de la familia para evaluar el grado de variabilidad dentro de la misma familia. La mayoría de las ataxias dominantes que se han definido por técnicas de genética molecular son etiquetadas como ataxia espinocerebelosa (AEC) seguido de un número para definir el locus genético específico afectado.
A la fecha, todas las mutaciones genéticas identificadas entre las ataxias dominantes (con excepción de las ataxias episódicas) han sido expansiones inestables de la secuencia de trinucléotidos CAG dentro de las regiones codificantes de los genes responsables [ver figura 1]. Estos padecimientos incluyen a la AEC1, la AEC2, la AEC3 (enfermedad de Machado-Jospeh [EMJ]), la AEC6 y la atrofia dentadorrubral-palidolusiana (ADRPL) [ver tabla 2].
Estos trastornos comparten ciertas características. La edad de inicio típicamente correlaciona en forma inversa con el número de repeticiones CAG en el alelo expandido [ver figura 2]. Además, el grado de expansión correlaciona también, hasta cierto punto, con las tasas de progresión de la enfermedad y ciertas características fenotípicas. Por lo tanto, las expansiones más importantes pueden causar pérdida más rápida de la función neuronal, así como patología neuronal diseminada. Los alelos normales en estos trastornos son estables durante la trasmisión intergeneracional. Los alelos expandidos con frecuencia son inestables, y es más probable que ocurra mayor expansión que contracción. La expansión de la repetición ocurre más con la herencia paterna que materna, y con frecuencia es de mayor magnitud con la herencia paterna. Esto explica parte de la anticipación en la edad de inicio observada.
La mayoría de los genes mutados se expresan en forma diseminada en los tejidos neurales y no neurales. En los tejidos de las personas afectadas pueden detectarse tanto el ARNm como la proteína correspondiente de los alelos normales y expandidos.8,9 La hipótesis de ganancia de la función sugiere que la degeneración neuronal en estos padecimientos puede ser resultado de alguna función tóxica novedosa del producto proteico del alelo expandido. Esta hipótesis ha sido apoyada usando modelos murinos transgénicos.10,11 El codón GAG especifica al aminoácido glutamina; por lo tanto, las proteínas mutantes tienen una tira poliglutamina más larga que la proteína nativa. Estas tiras de poliglutamina pueden ser importantes en las interacciones proteína-proteína o proteína-ácido nucleico [ver figura 3]. Las tiras de poliglutamina expandida en la proteína mutante pueden ocasionar interacciones aberrantes y pérdida neuronal. Datos recientes incluyen interacciones entre la proteína, una proteína cazadora asociada12 y la gliceraldenido-3-fosfato deshidrogenasa,13 una enzima que participa en el metabolismo energético.
Ataxia espinocerebelosa tipo 1 La AEC1 ocurre en muchos grupos étnicos y es responsable del 10 al 15 porciento de todas las ataxias con herencia autosómica dominante. Aunque la edad de inicio es variable, para la mayoría de los pacientes las dificultades de la marcha y el habla comienzan entre los 20 y los 40 años de edad. Los primeros signos neurológicos incluyen marcha atáxica, disartria, reflejos tendinosos profundos excitados y, en ocasiones, nistagmus.14 Los pacientes se vuelven no ambulatorios durante el curso de 10 años por aumento de la ataxia. Se desarrollan otros signos, incluyendo arreflexia en las extremidades inferiores, respuestas plantares extensoras, alteración de los movimientos extraoculares, disfagia y tos ineficaz. También pueden ocurrir defectos cognitivos menores. La neuropatología de la AEC1 incluye atrofia olivopontocerebelosa.
La mutación genética responsable de la enfermedad consiste en la expansión inestable de una repetición CAG dentro de un gen en el cromosoma 6p.15 La configuración de la repetición es diferente en las personas afectadas. Todos los alelos normales, con excepción de los más pequeños, tienen una a tres interrupciones CAT dentro de la secuencia de repetición CAG. Sin embargo, todos los alelos SCA1 tienen una configuración continua de la repetición CAG. Esta es una manera útil de distinguir un alelo normal más grande de un alelo afectado más pequeño. Las interrupciones CAT pueden detectarse por técnicas simples de biología molecular.
El gen afectado ocupa un ADN genómico de 450 kb y está organizado en nueve exones. La proteína que se codifica tiene 792 a 825 aminoácidos, no tiene dominios transmembrana y parece ser una proteína citoplasmática.16
Ataxia espinocerebelosa tipo 2 La ataxia espinocerebelosa tipo 2 descrita originalmente en el este de Cuba, recuerda a la AEC1 y a la EMJ, y cursa con ataxia progresiva y disartria. Los pacientes con este trastorno con frecuencia desarrollan sacadas extremadamente lentas e hiperreflexia al inicio de la enfermedad. Es raro el nistagmus. Pueden ocurrir calambres, fasciculaciones y temblor. Desde el punto de vista patológico, existe pérdida de las células de Purkinje en el cerebelo y de las neuronas pontina y olivar inferior en el tallo cerebral. Recientemente se ha identificado la mutación AEC2 como una expansión CAG dentro de un gen en el cromosoma 12q.17-19 El trastorno ha sido identificado en varios grupos étnicos, y entre las ataxias dominantes puede ser tan común como la EMJ.
Enfermedad de Machado-Joseph La mutación de la enfermedad de Machado-Joseph (ataxia espinocerebelosa tipo 3) puede corresponder al 20 a 25 porciento de todas las ataxias dominantes. Este padecimiento, descrito originalmente en emigrantes de las Azores Portuguesas, tiene muchas semejanzas clínicas con la AEC1. Los pacientes presentan marcha atáxica progresiva, reflejos tendinosos profundos exaltados y dificultades oculomotoras.20 La edad de inicio y el curso de la enfermedad también son semejantes. Sin embargo, en muchas de las familias portuguesas originales algunos de los miembros afectados tienen diferentes fenotipos, incluyendo síndromes de rigidez espástica, aquinesia, parkinsonismo y distonía. La neuropatología de la enfermedad también es característica, con alteración prominente en las neuronas pontinas, sustancia negra y neuronas en la médula espinal, y conservación relativa de las células cerebelosas de Purkinje y de las neuronas olivares inferiores.
La mutación responsable de la EMJ es una expansión inestable de la repetición CAG dentro de un gen en el cromosoma 14 [ver figura 1].21 Muchas familias que no tenían ninguna de las variaciones fenotípicas descritas antes parecen tener la misma mutación. Estas familias no pueden distinguirse clínicamente de las que tienen AEC1 y han sido etiquetadas con AEC3. Por lo tanto, la EMJ y la AEC3 no son genéticamente distintas.22
La proteína EMJ tiene un perfil hidrofílico general que no muestra una secuencia de señal o un dominio transmembrana, lo que sugiere que puede ser una proteína intracelular.
Ataxia espinocerebelosa tipo 6 La ataxia espinocerebelosa tipo 6 se ha relacionado recientemente a una repetición CAG expandida en el gen que codifica para la subunidad alfa1a del gen del canal neuronal de calcio en el cromosoma 19p.23 La enfermedad es relativamente benigna, el inicio ocurre en la vida adulta tardía y la persona afectada suele tener una supervivencia normal. El síndrome neurológico se caracteriza por una ataxia cerebelosa relativamente pura con mínima deficiencia del tallo cerebral u otro tipo de alteración neurológica.
Atrofia dentadorrubral-palidolusiana La clave para el diagnóstico de la atrofia dentadorrubral-palidolusiana, un trastorno que se ha reportado principalmente en Japón, es la presencia de una variabilidd fenotípica aoen mayor dentro de la misma familia.24 la edad de inicio varía de seis meses a 60 años o más, y la anticipación en la edad de inicio puede ser considerable. Cuando inicia en la niñez, el trastorno se presenta como una epilepsia mioclónica progresiva, ataxia y demencia. La enfermedad que inicia en el adulto se caracteriza por ataxia, coreoatetosis y demencia, y se parece en algo a la enfermedad de Huntington y a la EMJ. Otros signos clínicos incluyen psicosis, sacadas lentas, discinesia e hiperreflexia. La anticipación significativa en la edad de inicio puede causar casos aparentemente esporádicos en niños hijos de padres que portan la mutación y que aún están asintomáticos. El daño patológico incluye al núcleo dentado, el núcleo rojo, el núcleo subtalámico y el globo pálido.
La mutación en la ADRPL incluye la expansión de una secuencia repetida de CAG dentro de la región que codifica un gen en el cromosoma 12.24,25 Aunque la mayoría de los pacientes han sido descritos en Japón, el análisis de la mutación ha identificado el mismo defecto genético en pacientes de otros orígenes étnicos, como los de Carolina del Norte que tienen el síndrome de Haw River. El gen de la ADRPL tiene alrededor de 4,300 pares de bases que codifican una proteína 1,185 aminoácidos mayor, con un peso molecular de 125 kd.
En los niños con epilepsia mioclónica progresiva y ataxia debe realizarse análisis en busca de la mutación de la ADRPL, incluso en ausencia de una historia familiar positiva, lo mismo que en personas que tienen el diagnóstico clínico de enfermedad de Huntington y en los que el análisis para esta mutación es negativo.
Ataxia espinocerebelosa tipo 4 La ataxia espinocerebelosa tipo 4 es una ataxia de herencia dominante que fue descrita en una familia de Utah. La mutación genética se localizó en el cromosoma 16.26 La enfermedad se caracteriza por pérdida sensorial prominente que se relaciona con una neuropatía periférica de tipo axonal.
Ataxia espinocerebelosa tipo 5 El defecto genético que produjo ataxia dominante, la ataxia espinocerebelosa tipo 5, en una familia que era descendiente de los abuelos de Abraham Lincoln, recientemente se localizó en el cromosoma 11.27 Los pacientes en esta familia comenzaron en la tercera y cuarta década. Los datos clínicos eran puramente cerebelosos, con mínimos defectos de motilidad ocular, deficiencias bulbares o alteraciones de las vías piramidales.
Ataxia dominante con degeneración retiniana La ataxia dominante con degeneración retiniana (AEC7) tiene un rango muy amplio de edad de inicio, y los niños pueden presentar síntomas antes que sus padres que portan la mutación. Los pacientes sufren varias combinaciones de ataxia progresiva y deficiencia visual debida a maculopatía. El fenotipo dentro de una familia puede variar desde pacientes que tienen tanto retinopatía como ataxia, a los que solo tienen dificultad visual y a los que solo tienen ataxia. La enfermedad subclínica de la retina puede detectarse por pruebas de visión con color y electrorretinografía. Otros signos neurológicos incluyen hiperreflexia, sacadas lentas y respuestas plantares extensoras; los niños con la enfermedad pueden presentar también crisis convulsivas, mioclonías e hiporreflexia. Recientemente el locus de la enfermedad ha sido mapeado al cromosoma 3p.28,29
Ataxias paroxísticas dominantes Los pacientes que tienen ataxias paroxísticas dominantes tienen episodios autolimitados de ataxia. Una variedad, causada por una mutación puntiforme en el gen del canal de potasio en el cromosoma 12p, se caracteriza por episodios breves de ataxia, con resolución completa de los síntomas y descargas mioclónicas asociadas.30 Otra variedad, asociada a marcadores del cromosoma 19 y relacionada con mutaciones puntiformes en la subunidad alfa del canal del calcio, se asocia con episodios más duraderos y signos cerebelosos interictales leves.31
Bibliografía
DR. S.H. SUBRAMONY
Las ataxias hereditarias son trastornos que causan desequilibrio progresivo como resultado de patología en el cerebelo y sus diversas vías de conección. Las personas con ataxias hereditarias suelen tener también otros signos que indican disfunción en los ganglios basales, la neurona motora superior, los núcleos oculomotores y otros núcleos del tallo cerebral, las células del asta anterior y los ganglios de la raíz dorsal. En algunos casos ocurre también pérdida de la visión y deterioro cognitivo. El cuadro clínico general de muchas ataxias hereditarias es semejante, por lo que las clasificaciones que se basan en el fenotipo clínico no son óptimas. La edad de inicio del trastorno, el patrón de herencia y la combinación particular de los signos clínicos pueden ayudar a determinar el genotipo subyacente.
Durante la última década, los estudios de genética molecular han identificado mutaciones genéticas específicas en muchas ataxias hereditarias y localizado la causa de otras en loci cromosómicos específicos, lo que permite lo siguiente:
1. Clasificación de estos trastornos con base en sus genotipos (ver tabla 1 y 2).
2. Identificación del genotipo preciso en muchos pacientes.
3. Realización de estudios diagnósticos presintomáticos y prenatales.
4. Comprensión de parte de la variabilidad fenotípica relacionada a mutaciones en el mismo gen.
5. Especulación sobre los mecanismos de muerte neuronal selectiva. La sintomatología prograsiva de estos padecimientos se relaciona con la pérdida de neuronas en muchas partes del sistema nervioso, incluyendo las células cerebelosas de Purkinje, así como neuronas de la protuberancia, olivas, sustancia negra, médula espinal y ganglios de las raíces dorsales.
|
||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||
|
Mecanismos de mutación y patrones de herencia
Las mutaciones patogénicas son alteraciones en la secuencia de nucléotidos de los genes que producen un fenotipo alterado (uno que manifiesta enfermedad). Los genes están compuestos de pares complementarios de nucleótidos que especifican la secuencia de aminoácidos que constituyen proteínas específicas. Las secuencias de nucleótidos que se transcriben a los correspondientes ARNm (ARNm) se denominan exones, las secuencias de nucleótidos que no se transcriben son los intrones. Dentro de los genes, las secuencias de tres nucleótidos (codones) especifican un aminoácido particular. Las mutaciones, incluyendo sustituciones de nucleótidos, deleciones, inserciones, y duplicación y expansión trinucléotida pueden afectar a los codones de muchas maneras.
MECANISMOS DE MUTACION
Mutaciones puntiformes En una mutación puntiforme un solo nucleótido dentro de un codón es sustituido por otro. El codón alterado puede especificar un aminoácido diferente, que puede ocasionar la creación de una proteína mutante (mutación con sentido equivocado). Un solo cambio en un nucleótido puede también producir un codón que no codifique ya para un aminoácido (v.gr., un codón de alto), causando terminación prematura de la síntesis proteica (mutación sin sentido).
Mutación de marco Una mutación de marco es causada por inserción o deleción de uno o más pares de nucleósidos en un gen. Esto desplaza el marco de lectura de todos los codones después del sitio de la mutación. La cadena polipéptida resultante puede estar compuesta de aminoácidos no codificados en el gen original y puede tener una longitud diferente.
Duplicación Una duplicación es una aberración cromosómica. Toda una secuencia de nucleótidos dentro de un segmento de un cromosoma se duplica en el genoma haploide. El genoma diploide tiene tres, en lugar de dos, copias de un segmento particular de material genético.
Expansión repetida de tres nucleótidos Muchos genes normales tienen secuencias repetidas del mismo nucleótido. En los trastornos de expansión repetida de tres nucleótidos, las personas afectadas tienen una expansión de los trinucleótidos repetidos más allá del rango normal.
PATRONES DE HERENCIA
Las enfermedades hereditarias se caracterizan por un amplio rango de edad de inicio, anticipación (i.e., la ocurrencia aparente de la enfermedad a una edad progresivamente más temprana en las generaciones sucesivas) en la edad de inicio y variabilidad del fenotipo del padecimiento dentro de la misma familia. Un patrón autosómico recesivo de herencia en una enfermedad determinada requiere que ambas copias de un gen para una proteína específica muten para producir el fenotipo de la enfermedad. La enfermedad no se manifestará en los padres que son heterocigotos para la mutación, pero aparecerá en los descendientes homocigotos. En la herencia autosómico dominante, una mutación en una copia del gen es suficiente para causar la enfermedad. Por lo tanto, las personas heterocigotas manifestarán la enfermedad y la trasmitirán a parte de su descendencia. En la herencia recesiva ligada al X, el gen mutado se encuentra en el cromosoma X, las portadoras mujeres con una copia normal del gen en el otro cromosoma X no padecen la enfermedad, pero la trasmiten a parte de su descendencia de sexo masculino. No ocurre trasmisión de la enfermedad de hombre a hombre en los padecimientos ligados al X; sin embargo, los hombres pueden trasmitir la enfermedad a sus nietos a través de sus hijas. Por último, las mutaciones genéticas en el ADN mitocondrial pueden causar alteración en la función de la cadena respiratoria, lo que puede producir la enfermedad. Debido a que todas las mitocondrias se heredan de la madre, los padecimientos secundarios a mutaciones en el ADN mitocondrial se trasmiten exclusivamente a través de la madre, pero tanto los varones como las mujeres están afectados.
Por lo tanto, las mutaciones genéticas son de diversos tipos y magnitud, causan variabilidad intrafamiliar considerable en muchas enfermedades genéticas (por ejemplo, los trastornos de expansión de trinucleótidos) así como diferencias fenotípicas entre lo que parecen ser trastornos muy semejantes (por ejemplo, distrofias musculares de Duchenne y de Becker). En esta subsección se usan las ataxias hereditarias como un modelo para discutir algunos de estos mecanismos de mutación y para mostrar la forma como los enfoques moleculares han cambiado la evaluación de estas enfermedades [ver tablas 1 y 2]. Se han logrado progresos semejantes en algunas otras enfermedades neurogenéticas [ver tabla 3].
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Estudio del paciente con ataxia progresiva
Los pacientes que presentan ataxia progresiva deben ser examinados en forma cuidadosa, y deberán registrarse los detalles del fenotipo. Se realizará una historia familiar detallada, construyendo un pedigree. En algunas familias será evidente el patrón de herencia, que puede ayudar a la selección de las pruebas genpéticas adecuadas. En otras familias puede ser útil el examen directo de los familiares si la historia del paciente no es suficiente.
Deben excluirse las causas no hereditarias de ataxia progresiva, como la esclerosis múltiple, los eventos cerebrovasculares, la degeneración cerebelosa atáxica, las lesiones ocupativas y las lesiones meníngeas, en especial si la historia familiar no es convincente. Los estudios de imagen, en especial de resonancia magnética, son útiles a este respecto. En casos seleccionados, en especial en los trastornos infantiles de herencia recesiva, deben descartarse enfermedades susceptibles de tratamiento, lo que con frecuencia se logra con estudios de sangre simples [ver tabla 4]. Por último, es posible sugerir un genotipo diagnóstico con base en el fenotipo específico observado y usando los análisis de mutación actualmente disponibles, muchos de los cuales pueden obtenerse a través de laboratorios comerciales [ver tablas 1 y 2]. Es posible emitir un consejo genético más adecuado si se confirma el genotipo esperado. En algunos casos puede usarse un ensayo genético para estudiar a los familiares con riesgo.
|
||||||||||||||||||||||||||||||||
|
Las terapias tanto sintomáticas como neuroprotectoras para la mayoría de las ataxias hereditarias son desalentadoras. En situaciones seleccionadas puede ser útil el tratamiento específico de problemas como espasticidad y síndromes extrapiramidales con agentes como el baclofen y agonistas de dopamina, respectivamente. Puede ocurrir mejoría marginal en la función motora con el uso de buspirona o amantadina. Los pacientes requieren también programas apropiados de rehabilitación.
Ataxias autosómico recesivas
Ataxia de Friedreich La ataxia de Friedrich (AF) es el tipo más común de ataxia de herencia recesiva. El inicio de los síntomas suele ocurrir en la primera o segunda década de la vida, pero puede retrasarse hasta la tercera o después. La enfermedad se caracteriza por ataxia progresiva de la marcha, pérdida de los reflejos tendinosos profundos y perdida de la sensación propioceptiva en las extremidades, todos signos de patología temprana importante en las células ganglionares de las raíces dorsales y sus proyecciones sensoriales periféricas.1 Otros signos incluyen disartria, respuesta plantar extensora y alteraciones oculomotoras, incluyendo sacudidas en onda cuadrada. En la fase tardía ocurren atrofia muscular, debilidad y disfagia. En pocos pacientes se presentan temblor, pérdida de la visión y de la audición. Alrededor del 30 a 50 porciento de los pacientes desarrolla cardiopatía sintomática. Ocurre diabetes en el 10 porciento de los enfermos, y son frecuentes las deformidades esqueléticas, como las escoliosis.
La mutación en la AF consiste en una expansión de una repetición trinucleótida GAA intrónica en un gen denominado X-25, que se localiza en el cromosoma 9q.2 Este gen codifica para una proteína de 210 aminoácidos denominada frataxina. En el sistema nervioso central, la mayor concentración de frataxina se encuentra en la médula. En las personas normales se encuentran entre el 7 y el 22 porciento de las repeticiones GAA dentro del primer intrón. Los alelos agrandados portan 200 a 900 o más repeticiones. La mayoría de los pacientes con AF son homocigotos para esta expansión, el otro alelo porta una mutación puntiforme. La expansión es inestable durante la trasmisión meiótica del gen de los padres a la descendencia. Las evidencias preliminares sugieren que las personas afectadas tienen niveles bajos de ARNm de frataxina. Por lo tanto, la expansión de la repetición intrónica puede causar pérdida del mensaje para la frataxina, lo que ocasiona la enfermedad.
Series recientes de pacientes estudiados después de la identificación de la mutación genética han demostrado que el fenotipo asociado con la expansión GAA es más variable de lo que antes se afirmó con base en los estudios clínicos. La edad de inicio puede ser muy posterior (los pacientes pueden estar iniciando los 50) y el fenotipo puede incluir ataxia asociada a conservación de los reflejos, en ocasiones intensos.3 Debido a estos datos, el análisis de mutación para la AF está indicado no solo en pacientes con AF clásica, sino también en enfermos que presentan ataxias esporádicas y recesivas tan tarde como en la sexta década de la vida.
Ataxia con deficiencia aislada de vitamina E La ataxia con deficiencia aislada de vitamina E (ADVE) es un trastorno hereditario recesivo que recuerda a la AF, con inicio en la infancia de ataxia, arreflexia y pérdida de la sensación prospectiva. Los niveles de vitamina E son bajos y los suplementos pueden detener la progresión de la enfermedad. La mutación ocurre en el gen de la proteína de transferencia del a-tocoferol (PTT-a) en el cromosoma 8.4 La PTT-a participa en el procesamiento hep[daggerdbl]tico del tocoferol y en su incorporación a las lipoproteínas de muy baja densidad (LMBD). La absorción de vitamina E es normal.
En los pacientes que expresan un fenotipo severo la mutación consiste en una deleción de marco de una sola base de pares que remplaza los últimos 30 aminoácidos de la proteína con un péptido aberrante de 14 aminoácidos. Sin embargo, los pacientes que tuvieron inserciones o mutaciones puntiformes en el gen TPP-a expresaron un fenotipo menos severo. Por ejemplo, un paciente japonés presentó ataxia con inicio en la edad adulta y arreflexia.5 Por lo tanto, el tipo de mutación en el gen PTT-a determina el grado de deterioro funcional de la proteína e influye en el fenotipo final. Es adecuado obtener niveles de vitamina E en pacientes con cualquier tipo de ataxia esporádica, en especial si se disminuyen los reflejos tendinosos profundos. Ciertamente, debe excluirse la posibilidad de ADVA en todos los niños con sospecha de ataxia de Friedreich.
Ataxia-telangiectasia La ataxia telangiectasia típicamente se presenta pronto en la primera década de la vida como ataxia de la marcha. Los signos neurológicos que se desarrollan durante la primera década incluyen hipotonía, cierto grado de coreoatetosis, arreflexia y un trastorno oculomotor característico que se asocia con menor capacidad para generar sacadas, lo que obliga a empujar la cabeza para mover los ojos.6 Las telangiectasias típicas ocurren en niños de alrededor de cinco años de edad y pueden encontrarse en las conjuntivas, párpados y fosa cubital y poplítea. Estos niños tienen alto riesgo de neoplasias, en especial linfomas. La medición de una sensibilidad alterada a la radiación y los niveles elevados de a-fetoproteína en suero son útiles para confirmar el diagnóstico.
Las deleciones, y en ocasiones inserciones en el gen mutado de la ataxia telangiectasia (GAT) en el cromosoma 11q se asocian con la enfermedad,7 El producto proteico de este gen tiene semejanzas con las cinasas 3-fosfatidilinositol, que participan en la transducción de señales mitogénicas.
Ataxias autosómico dominantes
Las ataxias autosómico dominantes son genéticamente heterogéneas, pero comparten muchas manifestaciones clínicas [ver tabla 2]. Por lo tanto, puede ser difícil la identificación precisa del genotipo con base solo en las características clínicas. El diagnóstico clínico puede facilitarse examinando a varios miembros afectados de la familia para evaluar el grado de variabilidad dentro de la misma familia. La mayoría de las ataxias dominantes que se han definido por técnicas de genética molecular son etiquetadas como ataxia espinocerebelosa (AEC) seguido de un número para definir el locus genético específico afectado.
A la fecha, todas las mutaciones genéticas identificadas entre las ataxias dominantes (con excepción de las ataxias episódicas) han sido expansiones inestables de la secuencia de trinucléotidos CAG dentro de las regiones codificantes de los genes responsables [ver figura 1]. Estos padecimientos incluyen a la AEC1, la AEC2, la AEC3 (enfermedad de Machado-Jospeh [EMJ]), la AEC6 y la atrofia dentadorrubral-palidolusiana (ADRPL) [ver tabla 2].

|
| Figura 1 |
| Ataxia espinocerebelosa tipo 3 |
Estos trastornos comparten ciertas características. La edad de inicio típicamente correlaciona en forma inversa con el número de repeticiones CAG en el alelo expandido [ver figura 2]. Además, el grado de expansión correlaciona también, hasta cierto punto, con las tasas de progresión de la enfermedad y ciertas características fenotípicas. Por lo tanto, las expansiones más importantes pueden causar pérdida más rápida de la función neuronal, así como patología neuronal diseminada. Los alelos normales en estos trastornos son estables durante la trasmisión intergeneracional. Los alelos expandidos con frecuencia son inestables, y es más probable que ocurra mayor expansión que contracción. La expansión de la repetición ocurre más con la herencia paterna que materna, y con frecuencia es de mayor magnitud con la herencia paterna. Esto explica parte de la anticipación en la edad de inicio observada.
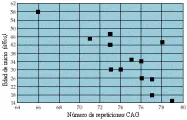
|
| Figura 2 |
| Ataxia espinocerebelosa tipo 3: edad de inicio y número de repeticiones |
La mayoría de los genes mutados se expresan en forma diseminada en los tejidos neurales y no neurales. En los tejidos de las personas afectadas pueden detectarse tanto el ARNm como la proteína correspondiente de los alelos normales y expandidos.8,9 La hipótesis de ganancia de la función sugiere que la degeneración neuronal en estos padecimientos puede ser resultado de alguna función tóxica novedosa del producto proteico del alelo expandido. Esta hipótesis ha sido apoyada usando modelos murinos transgénicos.10,11 El codón GAG especifica al aminoácido glutamina; por lo tanto, las proteínas mutantes tienen una tira poliglutamina más larga que la proteína nativa. Estas tiras de poliglutamina pueden ser importantes en las interacciones proteína-proteína o proteína-ácido nucleico [ver figura 3]. Las tiras de poliglutamina expandida en la proteína mutante pueden ocasionar interacciones aberrantes y pérdida neuronal. Datos recientes incluyen interacciones entre la proteína, una proteína cazadora asociada12 y la gliceraldenido-3-fosfato deshidrogenasa,13 una enzima que participa en el metabolismo energético.
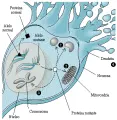
|
| Figura 3 |
| Mecanismos de muerte neuronal en los trastornos de expansión de CAG |
Ataxia espinocerebelosa tipo 1 La AEC1 ocurre en muchos grupos étnicos y es responsable del 10 al 15 porciento de todas las ataxias con herencia autosómica dominante. Aunque la edad de inicio es variable, para la mayoría de los pacientes las dificultades de la marcha y el habla comienzan entre los 20 y los 40 años de edad. Los primeros signos neurológicos incluyen marcha atáxica, disartria, reflejos tendinosos profundos excitados y, en ocasiones, nistagmus.14 Los pacientes se vuelven no ambulatorios durante el curso de 10 años por aumento de la ataxia. Se desarrollan otros signos, incluyendo arreflexia en las extremidades inferiores, respuestas plantares extensoras, alteración de los movimientos extraoculares, disfagia y tos ineficaz. También pueden ocurrir defectos cognitivos menores. La neuropatología de la AEC1 incluye atrofia olivopontocerebelosa.
La mutación genética responsable de la enfermedad consiste en la expansión inestable de una repetición CAG dentro de un gen en el cromosoma 6p.15 La configuración de la repetición es diferente en las personas afectadas. Todos los alelos normales, con excepción de los más pequeños, tienen una a tres interrupciones CAT dentro de la secuencia de repetición CAG. Sin embargo, todos los alelos SCA1 tienen una configuración continua de la repetición CAG. Esta es una manera útil de distinguir un alelo normal más grande de un alelo afectado más pequeño. Las interrupciones CAT pueden detectarse por técnicas simples de biología molecular.
El gen afectado ocupa un ADN genómico de 450 kb y está organizado en nueve exones. La proteína que se codifica tiene 792 a 825 aminoácidos, no tiene dominios transmembrana y parece ser una proteína citoplasmática.16
Ataxia espinocerebelosa tipo 2 La ataxia espinocerebelosa tipo 2 descrita originalmente en el este de Cuba, recuerda a la AEC1 y a la EMJ, y cursa con ataxia progresiva y disartria. Los pacientes con este trastorno con frecuencia desarrollan sacadas extremadamente lentas e hiperreflexia al inicio de la enfermedad. Es raro el nistagmus. Pueden ocurrir calambres, fasciculaciones y temblor. Desde el punto de vista patológico, existe pérdida de las células de Purkinje en el cerebelo y de las neuronas pontina y olivar inferior en el tallo cerebral. Recientemente se ha identificado la mutación AEC2 como una expansión CAG dentro de un gen en el cromosoma 12q.17-19 El trastorno ha sido identificado en varios grupos étnicos, y entre las ataxias dominantes puede ser tan común como la EMJ.
Enfermedad de Machado-Joseph La mutación de la enfermedad de Machado-Joseph (ataxia espinocerebelosa tipo 3) puede corresponder al 20 a 25 porciento de todas las ataxias dominantes. Este padecimiento, descrito originalmente en emigrantes de las Azores Portuguesas, tiene muchas semejanzas clínicas con la AEC1. Los pacientes presentan marcha atáxica progresiva, reflejos tendinosos profundos exaltados y dificultades oculomotoras.20 La edad de inicio y el curso de la enfermedad también son semejantes. Sin embargo, en muchas de las familias portuguesas originales algunos de los miembros afectados tienen diferentes fenotipos, incluyendo síndromes de rigidez espástica, aquinesia, parkinsonismo y distonía. La neuropatología de la enfermedad también es característica, con alteración prominente en las neuronas pontinas, sustancia negra y neuronas en la médula espinal, y conservación relativa de las células cerebelosas de Purkinje y de las neuronas olivares inferiores.
La mutación responsable de la EMJ es una expansión inestable de la repetición CAG dentro de un gen en el cromosoma 14 [ver figura 1].21 Muchas familias que no tenían ninguna de las variaciones fenotípicas descritas antes parecen tener la misma mutación. Estas familias no pueden distinguirse clínicamente de las que tienen AEC1 y han sido etiquetadas con AEC3. Por lo tanto, la EMJ y la AEC3 no son genéticamente distintas.22
La proteína EMJ tiene un perfil hidrofílico general que no muestra una secuencia de señal o un dominio transmembrana, lo que sugiere que puede ser una proteína intracelular.
Ataxia espinocerebelosa tipo 6 La ataxia espinocerebelosa tipo 6 se ha relacionado recientemente a una repetición CAG expandida en el gen que codifica para la subunidad alfa1a del gen del canal neuronal de calcio en el cromosoma 19p.23 La enfermedad es relativamente benigna, el inicio ocurre en la vida adulta tardía y la persona afectada suele tener una supervivencia normal. El síndrome neurológico se caracteriza por una ataxia cerebelosa relativamente pura con mínima deficiencia del tallo cerebral u otro tipo de alteración neurológica.
Atrofia dentadorrubral-palidolusiana La clave para el diagnóstico de la atrofia dentadorrubral-palidolusiana, un trastorno que se ha reportado principalmente en Japón, es la presencia de una variabilidd fenotípica aoen mayor dentro de la misma familia.24 la edad de inicio varía de seis meses a 60 años o más, y la anticipación en la edad de inicio puede ser considerable. Cuando inicia en la niñez, el trastorno se presenta como una epilepsia mioclónica progresiva, ataxia y demencia. La enfermedad que inicia en el adulto se caracteriza por ataxia, coreoatetosis y demencia, y se parece en algo a la enfermedad de Huntington y a la EMJ. Otros signos clínicos incluyen psicosis, sacadas lentas, discinesia e hiperreflexia. La anticipación significativa en la edad de inicio puede causar casos aparentemente esporádicos en niños hijos de padres que portan la mutación y que aún están asintomáticos. El daño patológico incluye al núcleo dentado, el núcleo rojo, el núcleo subtalámico y el globo pálido.
La mutación en la ADRPL incluye la expansión de una secuencia repetida de CAG dentro de la región que codifica un gen en el cromosoma 12.24,25 Aunque la mayoría de los pacientes han sido descritos en Japón, el análisis de la mutación ha identificado el mismo defecto genético en pacientes de otros orígenes étnicos, como los de Carolina del Norte que tienen el síndrome de Haw River. El gen de la ADRPL tiene alrededor de 4,300 pares de bases que codifican una proteína 1,185 aminoácidos mayor, con un peso molecular de 125 kd.
En los niños con epilepsia mioclónica progresiva y ataxia debe realizarse análisis en busca de la mutación de la ADRPL, incluso en ausencia de una historia familiar positiva, lo mismo que en personas que tienen el diagnóstico clínico de enfermedad de Huntington y en los que el análisis para esta mutación es negativo.
Ataxia espinocerebelosa tipo 4 La ataxia espinocerebelosa tipo 4 es una ataxia de herencia dominante que fue descrita en una familia de Utah. La mutación genética se localizó en el cromosoma 16.26 La enfermedad se caracteriza por pérdida sensorial prominente que se relaciona con una neuropatía periférica de tipo axonal.
Ataxia espinocerebelosa tipo 5 El defecto genético que produjo ataxia dominante, la ataxia espinocerebelosa tipo 5, en una familia que era descendiente de los abuelos de Abraham Lincoln, recientemente se localizó en el cromosoma 11.27 Los pacientes en esta familia comenzaron en la tercera y cuarta década. Los datos clínicos eran puramente cerebelosos, con mínimos defectos de motilidad ocular, deficiencias bulbares o alteraciones de las vías piramidales.
Ataxia dominante con degeneración retiniana La ataxia dominante con degeneración retiniana (AEC7) tiene un rango muy amplio de edad de inicio, y los niños pueden presentar síntomas antes que sus padres que portan la mutación. Los pacientes sufren varias combinaciones de ataxia progresiva y deficiencia visual debida a maculopatía. El fenotipo dentro de una familia puede variar desde pacientes que tienen tanto retinopatía como ataxia, a los que solo tienen dificultad visual y a los que solo tienen ataxia. La enfermedad subclínica de la retina puede detectarse por pruebas de visión con color y electrorretinografía. Otros signos neurológicos incluyen hiperreflexia, sacadas lentas y respuestas plantares extensoras; los niños con la enfermedad pueden presentar también crisis convulsivas, mioclonías e hiporreflexia. Recientemente el locus de la enfermedad ha sido mapeado al cromosoma 3p.28,29
Ataxias paroxísticas dominantes Los pacientes que tienen ataxias paroxísticas dominantes tienen episodios autolimitados de ataxia. Una variedad, causada por una mutación puntiforme en el gen del canal de potasio en el cromosoma 12p, se caracteriza por episodios breves de ataxia, con resolución completa de los síntomas y descargas mioclónicas asociadas.30 Otra variedad, asociada a marcadores del cromosoma 19 y relacionada con mutaciones puntiformes en la subunidad alfa del canal del calcio, se asocia con episodios más duraderos y signos cerebelosos interictales leves.31
- Harding AE. Friedreich's ataxia: a clinical and genelic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 104:589, 1981
- Campuzano V, Montermini L, Molto MD, et al Friedreich's ataxia autosomal recessive diseasecaused by intronic GAA triplet repeatexpansion. Science 271:1423,1996
- Durr A, Cossee M, Agid Y, et al. Clinical and genetic abnormalities in patients with Friedreich's ataxia. N Engl J Med 335:1169, 1996
- Ouahchi K, Arita M, Kayden H, et al. Ataxia with isolated vitamin E deficiency is caused by mutations in the a-tocopherol transfer protein. Nat Genet 9:14l, 1995
- Gotoda T, Arita M, Arai H, et al. Adult-onset spinocerebellar dysfunction caused by a mutation in the gene for the a-tocopherol-transfer protein N Engl J Med 333:1313, 1995
- Swift MS, Heim RA, Lench NJ Genetic aspects of ataxia telangiectasia. Adv Neurol 61:115, 1993
- Savitsky K, Bar-shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase Science 268:1749, 1995
- Servido S, Koshy B, Annstrong D, et al: Expression and analysis of the ataxin I protein tissues from normal and spinocerebellar ataxia type I individuals. Nat Genet 10:94, 1995
- Yazawa I, Nukina N, Hashida H, et al: Abnormal gene product identified in hereditary dentatornbral pallidoluysian atrophy (DRPLA), Nat Genet 10:99, 1995
- Burright EN, Clark BH, Servido A, et al: SCA-1 transgenic mice:a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 82:937, 1995
- lkeda H, Yamaguchi M, Sugai S, et al: Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat Genet 13:196; 1996
- U XJ, U SH, Sharp AH, et al: A huntingtin-associated protein enriched in brain with implications for pathology .Nature 378:398, 1995
- Burke JR, Enghi1d JJ, Martin ME, et al: Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat Med 2:347, 1996
- Zoghbi HY, Pollack MS, Lyons LA, et al: Spinocerebellar ataxia: variable age of onset and linkage to hurnan leukocyte antigen in a large kindred. Ann Neurol 23:580, 1988
- Orr HT, Chung M, Banfi S, et al: Expansion of an unstable trinucleotide CAG repeat in spinocerebelIar ataxia type I. Nat Genet 4:221,1993
- Banfi S, Servadio A, Chung M, et al: Identification and characterization of the gene causing type 1 spinocerebellar ataxia, Nat Genet 7:513,1994
- ImbertG,Saudou F, YvertG, et al: Ooningof the gene for spinocerebellar ataxia 2 revealsa locus with high sensitivity to expanded CAG/ glutamine repeats, Nat Genet 14:285,1996
- Pulst SM, Nechiporuk A, Nechiporuk T, et al: Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 14:269, 1996
- Sanpei K, Takano H, Igarashi S, et al: Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT, Nat Genet 14:277, 1996
- Sequiros J, Coutinho p. Epidemiology and clinical aspects of Machado-Joseph disease. Adv neuroI 61:139, 1993
- Kawaguchi Y, Okarnoto T, Taniwaki M, et al: CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 8:221,1994
- Matilla T, McCall A, Subramony S, et al: Molecular and clinical correlations in spinocerebellar ataxia type 3 and Machado-Joseph disease, Ann Neurol 38:68, 1995
- Zhuchenko O, Bailey J, Bonnen P, et al' Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansion in the alpha lA.voltage-dependent calcium channel, Nat Genet 15:62,1997
- Koide R, Ikeuchi T, Onodera O,et al: Unstable expansion of CAG repeat in hereditary dentatornbral pallidoluysian atrophy (DRPLA). Nat Genet 6:9, 1994
- Nagafuchi S, Yanagisawa H, Sato K, et al: Dentatorubral and pallidoluysian atrophy expansion of an unstable CAG lrinucleotide on chromosome 12p. Nat Genet 6:14, 1994
- Flanigan K, Gardner K, Alderson K, et al: Autosomal dominant spinocerebellar ataxia with sensory axonal neuropathy (SCA4): clinical descríption and genetic localization to chromosome 16q22.1.Am J HumGenet 59:392, 1996
- Ranum LPW , Schut LJ, Lundgren JK, et al: Spinocerebellar ataxia type 5 in a family descended from the grandparents of President Lincoln maps to chromosome 11, Nat Genet 8:280, 1994
- Gouw LG, Kaplan CD, Haines JH, et al. Retinal degeneration characterizes a spinocerebellar ataxia mapping tochromosome 3p. Nat Genet 10:89,1995
- Benomar A, Krols L, Stevanin G, et al. The gene for autosomal dominant cerebellar ataxia with pigmentary macular dystrophy maps to chromosome 3p12-p21.1. NatGenet 10:84,1995
- Browne DL, Gancher ST , Nutt JG, et al. Episodic ataxia/ myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 8:136,1994
- Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2. channel gene CACNL1A4. CeIl 87:543, 1996