Endocrinología
⭳ Abrir artículo (PDF)665.6 KBEste artículo es idéntico en la Edición 2/2000.
Contenido del artículo
V HIPOFISIS
- Neuroendocrinología
- La hipofisis anterior
- SOBREPRODUCCION DE HORMONAS HIPOFISIARIAS TROFICAS
- MENOR PRODUCCION DE HORMONAS HIPOFISIARIAS TROFICAS
- TUMORES HIPOFISIARIOS Y CRECIMIENTO DE LA SILLA TURCA
- HORMONA DEL CRECIMIENTO
- PROLACTINA
- La hipófisis posterior
- Síndromes de neoplasias endócrinas múltiples
- Interacciones entre el sistema endócrino y el inmunológico
V HIPOFISIS
DR. DANIEL D. FEDERMAN
Neuroendocrinología
Los principales órganos de comunicación para la respuesta a los estímulos exógenos y endógenos estresantes, los procesos patológicos y la cirugía, son los sistemas nerviosos y endócrino. Las respuestas neurales al estrés requieren de comunicación intercelular en distancias minúsculas y usan trasmisores químicos; el tiempo de respuesta y el periodo que transcurre antes de que pueda ocurrir otra respuesta (latencia) se miden en fracciones de segundo. Por el contrario, las respuestas endócrinas son mediadas por hormonas que circulan en la sangre de un sitio a otro. La latencia se mide en minutos u horas. Además, las adaptaciones hormonales pueden continuar hasta por un día o un mes.
El eje hipotálamo-hipófisis permite que exista una convergencia entre las capacidades de adaptación neurales y hormonales. Las neuronas del hipotálamo son ricas en sinapsis de conección con el sistema nervioso central, por lo que reciben influencias aminérgicas, peptidérgicas y opioides. Además, el hipotálamo se encuentra fuera de la barrera hematoencefálica, por lo que puede percibir y responder a las concentraciones circulantes de glucosa, cortisol, sodio y otras sustancias. El hipotálamo es también el sitio de principal control homeostático, incluyendo la regulación del hambre, sed, osmolaridad, presión arterial, temperatura y respiración. Por último, es el origen de la comunicación humoral estimuladora e inhibidora con la hipófisis anterior [ver figura 1]. Los pequeños péptidos producidos en las neuronas del hipotálamo penetran a los capilares fenestrados, descendiendo a través de las venas porta-hipofisiarias y distribuyéndose por otros capilares fenestrados hasta llegar a las células de la hipófisis anterior.1
En la actualidad se han identificado ya las principales hormonas hipofisiotróficas del hipotálamo [ver tabla 1], y en este capítulo se analiza su significado clínico, su uso diagnóstico y sus aplicaciones terapéuticas. Es pertinente mencionar algunos datos generales sobre el control del eje hipotálamo-hipofisiario. Primero, la secreción hipotalámica e hipofisiaria es pulsátil, más que tónica [ver figura 2], en ciertos casos puede requerirse la toma de muestras cada 2 a 3 minutos para demostrar los pulsos. Estos pulsos dependen de ritmos biológicos, como la liberación circadiana de la hormona adrenocorticotrópica (ACTH), la liberación asociada al sueño de la hormona del crecimiento (GH) y la prolactina (PRL), y el ciclo mensual de las gonadotropinas en la mujer. Segundo, el hipotálamo es una rica fuente de las aminas biogénicas dopamina, epinefrina, norepinefrina y serotonina, y cada una de ellas modula los ritmos hipofisiarios. También los opioides endógenos son importantes en el control de la función hipofisiaria: las endorfinas promueven la liberación de GH, tirotropina (hormona estimulante del tiroides o TSH) y PRL, y suprimen la liberación de hormona foliculo estimulante (FSH), hormona luteinizante (LH) y ACTH. Las conecciones hipotalámicas con otros centros cerebrales pueden participar también en el control hipofisiario (v.gr., al parecer el hipocampo-amígdala inhibe el control hipotalámico de la ACTH y la función suprarrenal).
La fisiología y la fisiopatología de las hormonas hipofisiarias tróficas específicas (i.e., FSH, LH, ACTH y TSH) se analizan en las subsecciones de ovario, testículo, suprarrenal y tiroides. Esta subsección trata sobre alteraciones más amplias de la hipófisis anterior, padecimientos específicos de las hormonas no troficas (GH y PRL), y síndromes que afectan a la hipófisis posterior y a la función endócrina en general.
El uso de agonistas y antagonistas sintéticos de la hormona liberadora de gonadotropina (GnRH) ha revolucionado la práctica de la endocrinología reproductiva y de trastornos asociados. Estas aplicaciones se mencionan en esta sección pero, por conveniencia, se analizan en las subsecciones respectivas.3
La hipofisis anterior
SOBREPRODUCCION DE HORMONAS HIPOFISIARIAS TROFICAS
La producción excesiva de hormonas hipofisiarias tróficas suele detectarse como una enfermedad de la glándula blanco.4 Por ejemplo, la hipersecreción de ACTH por un adenoma hipofisiario es la causa más frecuente del síndrome de Cushing.5,6 El tumor suele ser un microadenoma (<10 mm de diámetro) y no causa síntomas locales; la cirugía transesfenoidal, asociada a técnicas de laser, endoscopía o radiofrecuencia, es el tratamiento de elección.7 Es raro que ocurra hipersecreción de otras hormonas hipofisiarias tróficas. Los tumores que secretan gonadotropinas o fragmentos de gonadotropinas en exceso suelen ocurrir en hombres de edad media que han tenido un desarrollo sexual normal.8 Algunos de estos pacientes presentan hipogonadismo secundario, y la mayoría tienen tumores grandes que provocan defectos de los campos visuales. La cirugía transesfenoidal inmediata puede restablecer o mejorar la visión y dar tiempo para un tratamiento definitivo. Los adenomas hipofisiarios que producen TSH causan un síndrome hipertiroideo sin las características autoinmunes de la enfermedad de Graves. Pueden asociarse síntomas mecánicos causados por el tumor hipofisiario.
MENOR PRODUCCION DE HORMONAS HIPOFISIARIAS TROFICAS
Deficiencia hipofisiaria de una hormona trófica
La deficiencia hipofisiaria de una sola hormona trófica es una causa poco frecuente, pero bien definida, de hipofunción de una glándula blanco. Los síndromes resultantes se describen en las subsecciones referentes a las gónadas, tiroides y suprarrenales, pero es conveniente hacer algunos comentarios en este capítulo.
La deficiencia aislada de gonadotropina suele implicar secreción inadecuada tanto de FSH como de LH por la hipófisis anterior, que es secundaria a una deficiencia congénita de la hormona liberadora en el hipotálamo. Los individuos afectados no tienen pubertad, todos los aspectos de la función gonadal son deficientes y las concentraciones plasmáticas de gonadotropinas son bajos o están en el límite inferior normal, lo que distingue este padecimiento de la falla gonadal primaria.9,10
La deficiencia aislada de TSH puede ser adquirida, y se presenta como un hipotiroidismo típico, excepto por la ausencia de bocio y anticuerpos antitiroideos. Los pacientes tienen concentraciones séricas de TSH bajas o normales, que pueden aumentar un poco, pero no en forma importante, después de la administración de hormona liberadora de tirotropina (TRH).
El diagnóstico de deficiencia de ACTH es muy difícil. La función de la aldosterona se afecta en etapas tardías de la evolución, si acaso, y el síndrome se caracteriza por deficiencia selectiva de esteroides sin asociarse a hiperpigmentación o deficiencia de volumen. Los pacientes suelen tener un desarrollo deficiente, caracterizado por disminución del apetito, falta de ánimo o ambición, propensión a intoxicación por agua e hiponatremia y astenia generalizada. La diferencia clínica con la depresión o una enfermedad orgánica crónica, como el cáncer o la anemia, es casi imposible. La mayor parte de los pacientes con deficiencia selectiva de ACTH se diagnostican sólo después de que se detecta la hiponatremia o la incapacidad para responder en forma normal a una situación de estrés. La concentración plasmática de cortisol es baja (i.e., <4.5 µg/dl), y no aumenta después de la hipoglucemia inducida por insulina, pero sí al infundir ACTH durante un periodo de varios días.
Panhipopituitarismo
El panhipopituitarismo es más común que cualquiera de las deficiencias aisladas de hormonas tróficas.11 La hipofunción generalizada de la hipófisis suele ser resultado de las destrucción de la glándula por varias causas. La hipófisis es la única glándula endócrina en la que la destrucción por un tumor es una causa importante de su hipofunción, y esto se debe a compresión de la glándula contra los huesos que forman la silla turca (fosa hipofisiaria). El tumor hipofisiario primario más frecuente es el adenoma cromófobo. El craneofaringioma, un tumor de estructuras vecinas, es otra causa importante de hipofunción hipofisiaria. Suele manifestarse en niños, aunque puede aparecer en la edad adulta, causando hipofunción hipofisiaria o disminución visual como síntomas principales. También los tumores metastásicos, por lo general de mama o de pulmón, pueden causar hipofunción en algunas ocasiones.
El término síndrome de Sheehan se refiere, en forma típica, al hipopituitarismo moderado a severo secundario a un estado de choque por hemorragia uterina periparto. La necrosis hipofisiaria se explica por un infarto inducido por el colapso de la arteria hipofisiaria superior. Con la mejoría de los cuidados obstétricos cada vez se observan menos casos floridos. Sin embargo, en ocasiones se encuentra una variante sutil de síndrome de Sheehan en pacientes que desarrollan anemia al final del embarazo y que no reciben transfusiones. Los síntomas habituales suelen ser incapacidad para lactar, no reaparición de las menstruaciones, fatiga y debilidad. La tomografía computada puede indicar el diagnóstico al demostrar una silla turca de tamaño normal, pero parece ser que la imagen por resonancia magnética (RM) es más sensible.
El síndrome de silla vacía sigue siendo una variante enigmática de las enfermedades hipofisiarias.13 Aunque el crecimiento de la fosa hipofisiaria demostrada por radiografías suele interpretarse como un tumor hipofisiario, en algunos pacientes este aumento aparente de tamaño corresponde a la extensión de la cisterna subaracnoidea hacia la silla turca. La patogenia de la lesión no es clara. El síndrome de silla turca vacía se refiere a los pacientes en los que el evento inicial es la proyección del espacio subaracnoideo hacia la silla. En estos pacientes la función hipofisiaria suele ser normal, aunque puede existir hipersecreción de PRL o ACTH si coexiste un microadenoma. El síndrome de silla turca vacía secundaria se refiere a un padecimiento en el que la hipofisis disminuye de tamaño por un infarto, cirugía, radiación u otro mecanismo. En estos casos es frecuente la deficiencia hipofisiaria anterior parcial o completa. Puede requerirse de un neumoencefalograma para el diagnóstico, pero la TC con metrizamida proporciona una imagen definitiva con menos morbilidad. Es probable que la RM sea superior a la TC.
La destrucción de tejido hipofisiario por un granuloma o infección es un problema poco frecuente, pero que debe tenerse en cuenta en los pacientes que tienen hipopituitarismo inexplicable o una enfermedad granulomatosa diseminada. De hecho, la combinación de hipopituitarismo parcial y defectos visuales causada por sarcoidosis que afecte los nervios ópticos puede sugerir un tumor hipofisiario. Sin embargo, en estos casos la silla turca no está aumentada de tamaño, los defectos visuales son en parches (a diferencia de los causados por compresión quiasmática) y las pruebas funcionales sugieren hipopituitarismo causado por deficiencia hipotalámica.
La insuficiencia hipofisiaria secundaria a radioterapia de las estructuras vecinas se presenta cada vez con más frecuencia.14,15 Las dosis actuales, más grandes, la mayor supervivencia de los pacientes tratados, y el uso de pruebas sutiles de reserva hipofisiaria, contribuyen a aumentar el número detectado de estos casos. Los conocimientos actuales indican que el defecto es con más frecuencia hipotalámico que hipofisiario.16 La necrosis hipofisiaria secundaria a hemorragia dentro de un tumor puede causar deficiencia hormonal. En alguna ocasión se consideró que el hipopituitarismo era un padecimiento permanente, pero la secreción hormonal puede recuperarse después de la reducción farmacológica de los macroprolactinomas17 y después de la descompresión transesfenoidal de tumores o de infartos hemorrágicos en la hipófisis.
Datos clínicos Los datos clínicos del panhipopituitarismo son muy variables, pueden incluir cambios tanto mecánicos como hormonales. Los cambios mecánicos son secundarios al crecimiento de un tumor que sobrepasa la silla turca, e incluyen cefalea y alteraciones de la visión como visión borrosa, disminución en la agudeza y defectos en los campos visuales. El primer defecto suele ser la pérdida del cuadrante superior bitemporal, que se detecta mejor por el examen con una luz roja intermitente. Debido a que la mácula no se afecta, los pacientes pueden tener grandes pérdidas de los campos visuales sin notarlas.
Las características endócrinas del panhipopituitarismo dependen del grado de deficiencia hormonal, de la velocidad con que se desarrolla la deficiencia, de la edad del individuo y de la presencia o ausencia de enfermedades asociadas. La GH y las gonadotropinas tienen una sensibilidad semejante al daño hipofisiario parcial; si el síndrome se desarrolla en forma gradual, la producción de estas hormonas es la primera que se altera. La deficiencia de hormona del crecimiento (DGH) se caracteriza por disminución del bienestar, menor masa corporal magra y densidad ósea, aumento de la grasa abdominal, piel delgada y seca y reducción en la fuerza muscular.18,19 En las mujeres en edad reproductiva la amenorrea es el primer síntoma, y en los hombres lo es la disfunción eréctil. En las mujeres posmenopáusicas la deficiencia de gonadotropina no provoca síntomas, por lo que su detección se retrasa. La apatía y falta de preocupación sobre el propio bienestar, típicos de los pacientes hipopituitarios, retrasa el diagnóstico definitivo aún más.
A diferencia de lo que ocurre con las gonadotropinas, el desarrollo de la deficiencia de TSH y de ACTH puede ser muy tardado. Cuando se producen aparecen diferentes características clínicas de hipotiroidismo e hipocortisolismo. Es frecuente que los síntomas y signos se atribuyan en forma equívoca a depresión o envejecimiento.
El diagnóstico del hipopituitarismo en los ancianos representa un reto porque los síntomas inespecíficos como fatiga, falta de ambición, debilidad y palidez, pueden ser los únicos presentes hasta que se desarrolla hipotiroidismo o hiponatremia francos.20
Diagnóstico de laboratorio La confirmación del panhipopituitarismo por laboratorio se ha simplificado mucho con la introducción de las pruebas de radioinmunoensayo para hormonas peptídicas. El enfoque general de diagnóstico consiste en demostrar primero la deficiencia de la glándula blanco y después medir la hormona hipofisiaria correspondiente. Para demostrar deficiencia de gonadotropinas en el varón se mide la testosterona plasmática. Si la concentración de testosterona es baja, se determinan las concentraciones de LH y FSH. Si éstas son bajas se confirma que el defecto tiene origen central. En la mujer en edad reproductiva, la amenorrea, una citología vaginal inmadura y la ausencia de hemorragia después de la administración de una progestina sintética reflejan deficiencia de estrógenos; una concentración normal o baja de FSH indica deficiencia hipofisiaria. La deficiencia de TSH se demuestra por un valor bajo de tiroxina y de TSH. Podría pensarse que la administración de TRH permite diferenciar entre la deficiencia hipotalámica y la hipofisiaria de TSH, pero esta prueba ha dado resultados muy poco satisfactorios.
La deficiencia de ACTH suele ser más sutil que las analizadas antes, por lo que su diagnóstico requiere de pruebas de estimulación. Un nivel de cortisol matutino en ayuno menor de 5 µg/dl sugiere deficiencia de ACTH, en especial cuando el nivel de ACTH es bajo o normal. Sin embargo, cuando la sospecha es alta y el nivel de cortisol no es bajo, se requieren las pruebas de estimulación. Puede usarse, por ejemplo, metirapona, un inhibidor de la 11 ß-hidroxilasa suprarrenal [ver figura 3]. Se administra una dosis oral única de 3 g a media noche, y se mide el cortisol plasmático y el 11-desoxicortisol a la mañana siguiente. Si ambas hormonas tienen una concentración baja, el paciente tiene deficiencia de ACTH; si el cortisol es bajo y el 11-desoxicortisol es mayor de 10 µg/dl, el paciente tiene una reserva hipofisiaria normal. Otra prueba para investigar la respuesta de ACTH consiste en administrar insulina para inducir hipoglucemia. Después de una dosis estándar de 0.1 unidad/kg, la glucemia disminuye a menos de la mitad del valor control; si el sistema hipotálamo-hipófisis-suprarrenal está intacto, la concentración plasmática de cortisol se duplicará en respuesta a la hipoglucemia. Si la concentración de cortisol no aumenta, pero sí lo hace después de una infusión prolongada de ACTH (40 unidades I.V. durante un periodo de 8 horas en 2 días consecutivos), se confirma el diagnóstico de deficiencia de ACTH. Una prueba más simple utiliza ACTH sintética, o cosyntropin. Se administra una dosis de 0.25 mg I.V., la respuesta normal consiste en aumento en el nivel de cortisol plasmático a más de 18 mg/dl. Esta prueba es útil porque la glándula suprarrenal que ha estado deprivada en forma crónica de ACTH no responde con rapidez a un sólo estímulo con ACTH.
La hipoglucemia inducida por insulina también es el estándar de oro para detectar DGH, pero no se usa en pacientes con cardiopatía conocida o mayores de 60 años.21 Pueden usarse mediciones de GH estimulada con arginina, pero se requiere de un sofisticado laboratorio de endocrinología para distinguir la DGH hipofisiaria de la respuesta de GH lenta de los ancianos. Un enfoque aceptable consiste en calcular la probabilidad de DGH con base en la deficiencia de otras hormonas hipofisiarias. Si una hormona, por lo general gonadotropina, está deficiente, la posibilidad de DGH es de 55 porciento. Si las tres hormonas están deficientes el rango aumenta a más de 90 porciento. La disponibilidad de hormonas hipotalámicas liberadoras purificadas o sintéticas permite una prueba sensible y simple para el diagnóstico de la reserva hipofisiaria. Se administra un bolo intravenoso de una o más de las hormonas liberadoras y se mide la hormona u hormonas hipofisiarias correspondientes en los intervalos adecuados [ver tabla 2]. Las hormonas liberadoras son bien toleradas, excepto por ocasionar rubor y náusea.
Tratamiento Los pacientes con hipopituitarismo necesitan de tratamiento sustitutivo permanente, cuidadoso e individualizado. Este debe seguir lo más posible los principios fisiológicos, usando dosis y tiempos de administración que simulen los patrones naturales de secreción. Es preferible utilizar las hormonas de las glándulas blanco que las hormonas hipofisiarias porque éstas últimas se administran por vía parenteral, son incómodas, caras y antigénicas. En los pacientes con panhipopituitarismo parcial es adecuado usar dosis de hormonas tiroideas y de hidrocortisona más pequeñas que las utilizadas en el hipotiroidismo o hipoadrenalismo primarios.
La sustitución suprarrenal puede lograrse administrando hidrocortisona oral. Los pacientes que han tenido hipopituitarismo durante más de un año deben comenzar con dosis bajas, alrededor de 5 mg/día, a menos de que exista alguna urgencia. Las dosis iniciales más altas pueden en ocasiones inducir un comportamiento hipomaniaco. En pacientes con deficiencia leve, el esquema inicial lógico de tratamiento es de 10 mg de hidrocortisona dos veces al día. La dosis matutina puede aumentarse a 15 mg cuando se requiera hasta alcanzar un bienestar total. La dosis puede aumentarse en forma súbita en casos de estrés agudo, como en el paciente con insuficiencia suprarrenal primaria. Los pacientes con insuficiencia suprarrenal secundaria suelen no requerir remplazo de mineralocorticoides, pero éstos se administran cuando persiste la hiponatremia o la hipotensión postural a pesar de la administración de hidrocortisona.
La tiroxina es el tratamiento adecuado para la deficiencia tiroidea. La dosis inicial es de 0.05 mg/día, a menos de que el paciente tenga más de 60 años o padezca insuficiencia cardiaca, caso en el cual es más seguro iniciar con 0.025 mg/día. El autor aumenta la dosis a razón de 0.025 mg/día cada mes hasta que se alcanza una dosis de 0.075 a 0.150 mg/día.
Para los varones, el enantato de testosterona en dosis de 300 mg cada tres semanas proporciona una sustitución adecuada. De nuevo, en pacientes que han tenido hipogonadismo durante más de 2 años puede comenzarse con un tercio de la dosis total de sustitución. Esto evita el aumento brusco en la agresividad y la líbido y las atemorizantes fantasias sexuales que ocurren en ocasiones cuando se administra la dosis total en forma súbita.22
Para las mujeres menores de 50 años el autor utiliza estrógenos conjugados,1.25 mg/día durante los primeros 25 días de cada mes, y un progestágeno, como acetato de medroxiprogesterona, los días 16 a 25. Después de los 50 años de edad es adecuado administrar 0.625 mg/día de estrógenos conjugados y el progestágeno. Como con otras mujeres posmenopáusicas, pueden usarse dosis bajas continuas de estrógenos y progestágeno para evitar la menstruación. Para los pacientes en edad reproductiva que desean ser fértiles puede intentarse el tratamiento pulsátil con hormona liberadora de gonadotropinas (GnRH), para inducir la espermatogénesis o la ovulación si el problema básico del paciente es hipotalámico. Si el hipopituitarismo es causado por destrucción hipofisiaria, puede administrarse tratamiento secuencial con FSH y LH.
La disponibilidad ilimitada de GH recombinante ha iniciado una nueva era para los pacientes con deficiencia de esta hormona. Su administración regular aumenta la masa corporal magra, reduce la obesidad, restablece el volumen de líquido, mejora la masa muscular y la fuerza y, después de 1 a 2 años, aumenta la densidad mineral ósea. Muchos pacientes presentan también mejoría psicológica impresionante. No se sabe si el tratamiento con GH revierte la mayor mortalidad de los pacientes con hipopituitarismo, ni es claro hasta qué momento de la vida debe continuarse el tratamiento.23,24
El uso de hormona del crecimiento como tratamiento sustitutivo en ancianos con deficiencia limítrofe relacionada a la edad, en un intento por retrasar los cambios involutivos, es motivo de gran controversia. Algunos estudios han sugerido mejoría en la fuerza y bienestar, pero se requiere mucho más información sobre los beneficios y riesgos. Según los conocimientos actuales, existe poca justificación para prescribir esta hormona a individuos que no tengan una deficiencia clara de GH. Otro factor que debe considerarse es el alto costo del tratamiento con hormona del crecimiento, que tiene un promedio de 10,000 a 20,000 dolares al año para cada paciente.25
TUMORES HIPOFISIARIOS Y CRECIMIENTO DE LA SILLA TURCA
El crecimiento de la silla turca suele descubrirse en forma accidental en una radiografía de cráneo realizada por otro motivo, por ejemplo por un traumatismo craneano o durante una evaluación de los senos paranasales. En ocasiones el crecimiento es causado por una silla vacía, pero la causa más común es una neoplasia benigna.
Los tumores hipofisiarios varían en tamaño de microadenomas de 1a 2 mm a lesiones masivas. Algunas evidencias indican que estos tumores tienen un origen clonal, esto es, que se originan de una sola célula.26 Los tumores más pequeños no ocasionan síntomas por vecindad, sino que se manifiestan por síndromes hormonales, como la amenorrea hiperprolactinémica y el síndrome de Cushing. Por el contrario, los tumores que no producen hormonas funcionales y suelen ser descubiertos cuando causan síntomas locales, casi siempre son macroadenomas (> 10 mm de diámetro) que en muchos casos se extienden más allá de la silla turca. Estos tumores lesionan los campos y agudeza visual, y pueden afectar también otras áreas cerebrales. Algunos de estos tumores grandes producen fragmentos hormonales, en especial las cadenas a o ß de las hormonas glucoproteicas FSH, LH o TSH [ver figura 4]. Estos fragmentos son demostrados por tinción inmunológica del tejido tumoral, y pueden también encontrarse en la circulación. Una prueba monoclonal muy sensible ha detectado hipersecreción de la subunidad a en alrededor de la cuarta parte de los tumores clínicamente no funcionantes. Alrededor del 30 porciento de los macroadenomas no secretan hormonas ni fragmentos y son denominados tumores nulos.
El impacto endócrino de un macroadenoma debe evaluarse por los métodos descritos para detectar hipopituitarismo y es crucial medir el nivel de prolactica en suero.
Es menos claro cómo debe estudiarse un microadenoma descubierto en forma incidental por una IRM realizada por otro motivo. Alrededor del 10 a 15 porciento de las hipófisis muestran microadenomas en la autopsia, y la inmunotinción sugiere que muchos de éstos contienen prolactina. Si la persona no tiene síntomas y su función menstrual o eréctil es normal, lo más adecuado es realizar una sola medición de prolactina en suero.27 Es muy poco probable que se detecte una acromegalia o Cushing presintomáticos, ni tampoco tendrá algún beneficio.
La IRM es el método más sensible para evaluar la silla turca, la hipofisis y el hipotálamo, aunque la TC con medio de contraste también es adecuada.
El tratamiento adecuado de los tumores hipofisiarios depende de los resultados de los estudios endócrinos, radiográficos y neuroftalmológicos. Los tumores secretores requieren tratamiento, pero los microadenomas no secretores pueden ser sólo vigilados. Los macroadenomas se tratan con una combinación juiciosa de cirugía transesfenoidal (en ocasiones transfrontal) y radioterapia.28,29 El hipopituitarismo descubierto antes o después del tratamiento debe ser tratado con sustitución hormonal. En algunos pacientes el hipopituitarismo detectado antes de la cirugía mejora o se corrige por la resección transesfenoidal.30
La complicación más urgente de los tumores hipofisiarios es la apoplejía hipofisiaria. En algunos pacientes con tumores grandes puede ocurrir hemorragia y necrosis súbitas, creando una urgencia neuroquirúrgica. Los datos clínicos incluyen cefalea, obnubilación, oftalmoplejía, pérdida de la visión, y líquido cefalorraquídeo hemorrágico. La TC o la IRM han simplificado mucho el diagnóstico, y el tratamiento inmediato con hidrocortisona proporciona tiempo para la intervención neuroquirúrgica.31 La apoplejía subaguda, en la que los síntomas son mínimos o ausentes, puede ser confirmada por IRM. Puede ocurrir hipopituitarismo como una característica de la hemorragia y necrosis en los tumores. Sólo la deficiencia suprarrenal requiere de tratamiento inmediato, los otros estados hormonales pueden ser evaluados después de tratar la urgencia.32,33
HORMONA DEL CRECIMIENTO
La hormona del crecimiento, una proteína compuesta de 191 aminoácidos, es secretada por los somatotropos de la hipófisis anterior. A pesar de su nombre, la GH no promueve el crecimiento en forma directa, más bien actúa al estimular la formación hepática de somatomedina C (también conocida como factor de crecimiento semejante a insulina 1, o FCI-1), que es uno de los muchos factores de crecimiento. El FCI-1 entra a la circulación y se fija a uno de dos receptores; el receptor de membrana que fija FCI-1 es muy parecido al receptor de la insulina. En algunos tejidos la GH se fija en forma directa a un receptor de membrana.34 Aún no es claro qué efectos de la GH son causados por cuál de los eceptores.
La concentración plasmática de GH está controlada principalmente por los efectos opuestos de dos neuropéptidos que se producen en el hipotálamo y llegan a la hipófisis a través de la circulación portal.35 El neuropéptido hormona liberadora de hormona del crecimiento (GHRH) estimula la liberación de GH por la hipófisis, mientras que la somatostatina inhibe su secreción. La combinación de la secreción pulsátil, la vida media de 20 minutos y los controles opuestos sobre la secreción, causan concentraciones de GH en plasma muy variables. Aún más, tanto en niños como en adultos, se secreta GH en respuesta a la disminución en la glucemia (ver adelante), la ingestión de aminoácidos, el ejercicio, el sueño, el estrés y muchos otros factores, incluyendo estímulos adrenérgicos alfa, como la levodopa. La secreción de GH es suprimida por el aumento en la glucemia y por estímulos adrenérgicos beta.
En condiciones normales la concentración plasmática de GH en una persona varía durante el día, desde niveles no detectables hasta valores típicos de acromegalia. En vista de esta variabilidad, las mediciones aleatorias de GH no son definitivas desde el punto de vista clínico y se requieren pruebas estimuladoras y supresoras.
Acromegalia
El exceso de hormona del crecimiento produce acromegalia en los adultos y, en raras ocasiones, gigantismo en los niños.36 La causa más frecuente de exceso de GH es un adenoma hipofisiario. Para el momento en que se detecta la acromegalia por clínica, la mayoría de los pacientes tienen aumento de tamaño de la silla turca y un macroadenoma, por lo que la extensión supraselar es frecuente.
Un mecanismo de formación del adenoma, que se encuentra en el 30 a 40 porciento de los pacientes con acromegalia, es la mutación somática del gen aS, que codifica la cadena a de una proteína G reguladora unida a la membrana (GS). Esta proteína se fija a receptores de superficie de la célula para estimular segundos mensajeros intracelulares. La proteína G trimérica se mueve entre el difosfato de guanosina (GDP), que es inactivo, y el trifosfato de guanosina (GTP), que es activo. Varias mutaciones en el gen aS causan inhibición de la hidrólisis del GTP a GDP; con lo que la proteína GS se vuelve constitutivamente activa y la célula secreta GH en forma continua.
Una segunda causa de exceso de GH es la estimulación patológica por GHRH derivado del hipotálamo (eutópico) o de tumores en otra parte del organismo (ectópico). El GHRH se aisló por primera vez de un carcinoide bronquial y después de un tumor pancreático, en la actualidad se ha encontrado en muchos otros tipos de tumores. Estos tumores suelen derivar de lesiones benignas del intestino anterior y son especialmente frecuentes en los pulmones y el páncreas.
Diagnóstico Los signos más frecuentes de acromegalia son los secundarios a las manifestaciones paraselares y al exceso de GH [ver tabla 3]. Quizá la combinación más común consista en diaforesis, cefalea y debilidad o fatiga. El edema inexplicable de pies y manos, el crecimiento progresivo de las regiones acrales, el engrosamiento de labios y lengua y un olor desagradable del organismo son otras características de este síndrome. El diagnóstico suele ser tardío porque el paciente, su familia y su médico familiar suelen no notar los cambios graduales en su apariencia. Por lo tanto, por lo general es un familiar lejano o un nuevo médico quien nota el cambio en la apariencia facial del enfermo. Una vez que se ha pensado en el diagnóstico, los siguientes datos ayudan a corroborarlo: rasgos toscos, múltiples marcas en la piel (fibroma molluscum), piel gruesa y grasosa, prominencia frontal, ensanchamiento de los dientes y la mordida, antecedentes de ronquidos o apnea del sueño,37 y un tono ronco peculiar en la voz. La comparación de fotografías antiguas y recientes puede ser muy útil, al igual que en el mixedema y el síndrome de Cushing.
La característica hormonal esencial de la acromegalia consiste en la ausencia de la supresibilidad normal de la GH por la glucosa. A través de todo un día los individuos normales38,39 tienen concentraciones muy bajas de GH después de la ingesta de glucosa. El paciente con acromegalia pierde esta capacidad de respuesta a la glucosa y libera GH a una concentración que siempre es mayor que la mínima alcanzada por una persona normal. Esta diferencia es muy importante. El paciente con acromegalia no necesariamente debe producir grandes cantidades de GH; de hecho, su concentración de GH puede no ser mayor que la alcanzada por una persona normal después de realizar ejercicio o durante la hipoglucemia preprandial, pero esta concentración no disminuye después de la ingesta de glucosa. Para diagnosticar acromegalia es necesario medir la concentración de GH 1 a 2 horas después que el paciente ha ingerido 75 a 100 g de glucosa. Si la concentración de la hormona es mayor a 3 ng/ml debe sospecharse acromegalia, y se realizarán las pruebas confirmatorias definitivas. El valor posglucosa puede ser mayor de 2 ng/ml en adolescentes y en pacientes con hepatopatía crónica, uremia o anorexia nervosa, pero estos padecimientos son fáciles de diagnosticar.
La medición de la concentración de FCI-1, que correlaciona mejor que la concentración de GH con la actividad clínica de la acromegalia, es un método alterno o suplementario para confirmar el diagnóstico. En muchas instituciones esta prueba es la preferida para la evaluación inicial de escrutinio. Los valores normales varían con la edad y con diferentes métodos de laboratorio. Por lo tanto, es mandatorio interpretar el nivel de FCI-1 acorde a esto.40
Debe medirse la prolactina sérica en todos los pacientes con acromegalia. Su concentración está aumentada en alrededor de un tercio de los enfermos, principalmente por su secreción en el adenoma. Sin embargo, en el caso de algunos macroadenomas de gran tamaño con extensión supraselar, la concentración de prolactina puede estar aumentada por compresión del tallo hipofisiario, lo que ocasiona interferencia con la inhibición de la prolactina por la dopamina.
Tratamiento El autor considera que todos los pacientes con pruebas de escrutinio sugestivas de acromegalia deben ser referidos al endocrinólogo. El tratamiento inicial de la mayoría de los pacientes consiste en cirugía transesfenoidal de la hipósifis, un procedimiento muy especializado pero con frecuencia no curativo. El tratamiento subsecuente puede incluir radioterapia, bromocriptina o el análogo de somatostatina, ocreótido. La mayoría de los clínicos intentan la normalización total de los niveles de GH a menos de 2 ng/ml para minimizar la morbimortalidad por exceso de GH. Sin embargo, el tratamiento adecuado, la vigilancia de los efectos adversos e incluso la confirmación por laboratorio del control completo son temas de gran discrepancia.41-43
Hormona del crecimiento y envejecimiento
La disponibilidad de GH humana recombinante ha iniciado una nueva área en la endocrinología, la posibilidad de tratar a los adultos sin deficiencia de GH. Los niveles de FCI-1 son menores en los ancianos que en los jóvenes, y también lo es la liberación de GH inducida por insulina,44 lo que ha originado varias preguntas. ¿Cuándo deben considerarse los niveles descendientes de GH en los ancianos como deficiencia?45 ¿Si existe o no una deficiencia patológica, los menores niveles contribuyen a la reducción de la masa corporal magra, del músculo y del hueso en el anciano? ¿Podría el tratamiento con GH detener estos cambios sin causar efectos adversos dañinos?46,47 Varios estudios a corto plazo han demostrado un efecto anabólico significativo del tratamiento con GH, pero existen pocos datos sobre los efectos a largo plazo en el sistema cardiovascular o la predisposición al cáncer, que son una grave preocupación en el anciano.
PROLACTINA
La prolactina es la segunda hormona producida en la hipófisis anterior que actúa en forma directa sobre tejidos blancos, más que como una hormona trófica. Aunque tiene muchas funciones en otros animales, su única función conocida en el humano es la estimulación posparto de la producción de leche. Sin embargo, existe en varones y en mujeres no embarazadas. Además, ocurren pulsos de liberación de prolactina en respuesta al sueño, estrés y otros estímulos. Sin embargo, a diferencia de otras hormonas hipofisiarias, la prolactina está controlada, principalmente, por inhibición tónica, más que por estimulación intermitente. Su principal inhibidor es la dopamina. La prolactina aumenta la secreción de dopamina, por lo que inhibe su propia secreción. Otros inhibidores fisiológicos conocidos son la somatostatina y la triyodotironina (T3). La liberación de prolactina es estimulada por serotonina, acetilcolina, opiáceos, estrógenos, TRH y angiotensina II, entre otros, pero no se sabe con exactitud cuales de estas sustancias, si es que alguna, son fisiológicamente importantes.
La secreción de prolactina aumenta durante el embarazo, alcanzando al término del mismo concentraciones hasta de 20 a 30 veces la normal. Cuando la concentración de estrógeno, que también aumenta en forma dramática durante el embarazo, comienza a disminuir, se vuelve posible la lactancia; al continuar la lactancia los niveles basales de prolactina disminuyen hasta niveles semejantes a los de las mujeres no embarazadas. Aunque la succión estimula la liberación de prolactina, la lactación puede continuar con niveles de prolactina normales en reposo. En forma semejante, las concentraciones basales de prolactina son normales en algunos pacientes con galactorrea, no se sabe si las concentraciones estaban aumentadas antes en esos pacientes.
Alteraciones en la secreción de prolactina
Ocurre hipoprolactinemia en el síndrome de Sheehan y en otros tipos de panhipopituitarismo. Las mediciones basales de prolactina no diferencían en forma confiable entre los valores bajos y los limítrofes, pero la prolactina sérica no aumenta después de la estimulación con TRH en los pacientes afectados.
La hiperprolactinemia es mucho más frecuente que la deficiencia de prolactina.48 El principal síntoma directo del exceso de prolactina es la galactorrea, o lactación no puerperal. La lactación puede ser espontánea o detectarse por compresión suave del pezón. La correlación entre concentraciones elevadas de prolactina y lactación es escasa: muchos pacientes tienen concentraciones elevadas de prolactina sin lactación, y puede persistir una lactación significativa sin hiperprolactinemia.
Las consecuencias indirectas de la hiperprolactinemia son mucho más frecuentes que la galactorrea. La impotencia en el varón o las alteraciones menstruales en la mujer son quejas frecuentes. El defecto menstrual puede consistir en una fase lútea corta, periodos anovulatorios, oligomenorrea o amenorrea. Por lo tanto, debe determinarse la concentración de prolactina en todas las pacientes con alteraciones reproductivas.
Puede ocurrir hiperprolactinemia después del uso de fenotiacinas, inhibidores de la monoaminoxidasa u otros fármacos que influyen en la función hipotalámica o adrenérgica. También se ha implicado a los anticonceptivos orales en los síndromes de lactación; puede ocurrir hiperprolactinemia durante o poco después de la suspensión de los anticonceptivos orales.
El hipotiroidismo causa hiperprolactinemia, pero los valores de prolactina muy pocas veces son mayores a 50 ng/ml. Debe realizarse medición de TSH para diferenciar entre el hipotiroidismo primario (que causa elevación secundaria de prolactina) y la enfermedad hipofisiaria que provoca tanto hipotiroidismo como hiperprolactinemia [ver figura 5]. La insuficiencia renal puede también ocasionar aumento de prolactina.
Diagnóstico En la evaluación de los pacientes con hiperprolactinemia, una historia clínica y examen físico cuidadosos, junto con la evaluación de la función tiroidea y renal, suelen ser suficientes para detectar causas no hipofisiarias.49 Se han usado diversas pruebas de supresión y estimulación para distinguir entre las causas hipotalámicas e hipofisiarias de la hiperprolactinemia, pero ninguna es muy confiable. Por lo tanto, es indispensable realizar una TC de cráneo de alta resolución con contraste o una IRM.
El grado de aumento de la prolactina y los hallazgos radiográficos deben relacionarse en forma cuidadosa. Las concentraciones séricas de prolactina superiores a 300 ng/ml suelen reflejar un tumor hipofisiario, ya sea un microadenoma (< 10 mm de diámetro) o un macroadenoma (> 10 mm). Pueden encontrarse concentraciones muy altas (> 600 ng/ml) en los tumores grandes. Una concentración de sólo 50 a 300 ng/ml asociada a un tumor grande puede significar compresión del tallo hipofisiario por el tumor, con bloqueo consecuente de la inhibición de la dopamina sobre la liberación de prolactina, y no la presencia de un prolactinoma. En el nivel inferior de la escala, pueden existir microadenomas con concentraciones de prolactina tan bajas como de 30 ng/ml. En estos casos el radiólogo debe distinguir en forma cuidadosa entre las variaciones normales de los tumores verdaderos, realizando correlaciones clínicas para descubrir o descartar causas no tumorales del aumento de la prolatina. Si no se encuentra otra explicación, puede realizarse un diagnóstico provisional de hiperprolactinemia idiopática.
Tratamiento La mayor parte de los casos de hiperprolactinemia causados por un tumor o funcionales pueden ser controlados con el agonista dopaminérgico bromocriptina.50 El medicamento se administra en una dosis inicial de 1.25 mg/día, con los alimentos y en el momento de acostarse para minimizar los efectos colaterales de náusea e hipotensión postural. Después de una semana la dosis puede aumentarse a 1.25 mg dos veces al día, que suele ser bien tolerada. A las dos semanas debe medirse la concentración sérica de prolactina, antes de considerar un nuevo aumento en la dosis. La dosis de bromocriptina puede aumentarse 2.5 mg cada dos semanas hasta que la concentración de prolactina disminuya, aparezcan síntomas colaterales o se alcance una dosis máxima de 7.5 mg/día. La concentración de prolactina puede normalizarse varios días después de iniciado el tratamiento, y las menstruaciones o la potencia se recuperan después de varias semanas. Un agonista dopaminérgico alterno, el mesilato de pergolide, parece ser tan eficaz como la bromocriptina y en ocasiones es mejor tolerado. De hecho, el esquema de una dosis al día del pergolide es mejor en los pacientes que tienen dificultad para tolerar la bromocriptina. Otro agonista de dopamina, el cabergoline, también tiene algunas ventajas sobre la bromocriptina.51 El tratamiento con cualquiera de estos dos fármacos suele suspenderse durante el embarazo. Sin embargo, las pacientes embarazadas deben ser vigiladas en forma estrecha porque en alrededor del 1 a 2 porciento de los casos (casi siempre en pacientes con macroadenomas), las altas concentraciones de estrógenos inducen crecimiento significativo de los prolactinomas. Los pacientes con tumores en ocasiones son resistentes a la bromocriptina, y esta resistencia parece relacionarse con concentraciones bajas de receptores de dopamina D2 en las membranas de las células del prolactinoma.52 Para estos y otros pacientes que prefieren evitar los medicamentos la adenomectomía transesfenoidal ofrece una alternativa más conveniente.
Entre los pacientes con microadenomas y concentraciones de prolactina menores de 100 ng/ml, el porcentaje de curación con la cirugía alcanza un 88 porciento. Sin embargo, con el tiempo se observa una frecuencia significativa de recurrencias. Por ello, a los enfermos que se han sometido a una cirugía aparentemente exitosa se les medirá la prolactina sérica en forma periódica, en especial si los síntomás recurren. El seguimiento deberá continuar durante 5 años. Los macroadenomas representan un mayor reto quirúrgico tanto por el riesgo de la operación como por el menor porcentaje de éxito. Por este motivo se usa mucho la bromocriptina como tratamiento inicial,15 incluso cuando al final se requerirá de tratamiento quirúrgico. El tratamiento preoperatorio con bromocriptina puede mejorar la exploración quirúrgica en esos casos; de hecho, la disminución de tamaño de los prolactinomas puede ser tan rápida como para considerarse una opción para el tratamiento urgente de pacientes con cefalea y defectos de los campos visuales severos. En raras ocasiones están indicadas la radioterapia y la bromocriptina posoperatorias. La radioterapia aislada se emplea en casos seleccionados, pero parece ser menos eficaz que la bromocriptina o que la cirugía transesfenoidal.
¿Deben tratarse todos los pacientes con hiperprolactinemia? Los resultados de estudios sobre la historia natural de la hiperprolactinemia son variables pero varias series han notificado lo siguiente: (1) los microadenomas progresan muy poco, si es que lo hacen, (2) en algunos pacientes las concentraciones de prolactina se normalizan en forma espontánea, y (3) la vigilancia de la hipófisis por medio de imágenes es una manera segura de seguir a los pacientes que no desean tratamiento. El autor considera que es razonable vigilar a las mujeres con menstruaciones normales y que tienen concentraciones de prolactina menores a 100 ng/ml. Debe utilizarse tratamiento con progestágenos para inducir menstruaciones regulares en las pacientes con oligomenorrea. En las enfermas que no responden a los progestágenos deberá medirse la concentración plasmática de estradiol y realizar estudios de densitometría ósea. Es mejor indicar a estas pacientes que no responden que deben iniciar tratamiento con bromocriptina .
La hipófisis posterior
La hipófisis posterior está compuesta por porciones terminales de neuronas que se originan en los núcleos supraópticos y paraventriculares del hipotálamo. La vasopresina (AVP u hormona antidiurética HAD) y la oxitocina son sintetizadas en el hipotálamo como parte de un gran complejo molecular precursor que incluye proteínas asociadas llamadas neurofisinas. La molécula precursora es procesada durante su movimiento a lo largo del axón de la célula nerviosa. Después la vasopresina y la oxitocina son almacenadas en la hipófisis posterior y liberadas en respuesta a los estímulos apropiados. El primer ensayo satisfactorio de AVP [ver figura 6] ha demostrado lo siguiente:53
1. El adulto normal con una osmolaridad sérica de 287 ± 5 mOsm/kg tiene una concentración plasmática de AVP entre 1 y 2 pg/ml.
2. Un incremento de uno porciento en la osmolaridad provoca un cambio de 1 pg/ml en la concentración plasmática de AVP. La sensibilidad y, en menor grado, el umbral de la respuesta de AVP a un cambio en la tonicidad muestra una variabilidad considerable entre un individuo y otro, por lo menos parte de esta variación es hereditaria.54
3.El cambio de 1 pg/ml en la AVP aumenta la concentración urinaria alrededor de 200 mOsm/kg; por lo tanto, un cambio de 3 a 4 porciento en la osmolaridad sérica aumentará la concentración urinaria 600 a 800 mOsm/kg, o de muy diluida a con máxima concentración.
4. En la persona normal la tonicidad está balanceada en el punto medio, con cambios en cualquier dirección que alteran en forma directa la liberación de AVP y la concentración urinaria.
5. La insuficiencia cardiaca congestiva disminuye el umbral osmótico para la liberación de AVP, mientras que el envejecimiento y otros factores disminuyen la sensibilidad de la liberación de AVP (i.e., la velocidad de liberación de AVP por unidad de cambio en la osmolaridad).
6. Los cambios leves en el volumen y presión sanguíneos (i.e., de menos del 10 porciento) no afectan la liberación de AVP en forma significativa, pero cambios mayores sí. La hipotensión y la hipovolemia estimulan la liberación de AVP al disminuir el umbral osmótico, la hipertensión y la hipovolemia inhiben la liberación al aumentar el umbral. Estas influencias son mediadas por barorreceptores que tienen sus terminales aferentes en la aurícula izquierda.
7. La náusea, pero no el vómito, es un estímulo potente para la liberación de AVP, y aumenta la concentración plasmática hasta 1,000 veces el valor requerido para una antidiuresis máxima. Sin embargo, el dolor no es un estímulo importante para la liberación de AVP.
8. Muchas vías neurales influyen en la liberación de AVP en respuesta a estímulos no osmóticos. Por lo general, las vías adrenérgicas alfa estimulan y las vías adrenérgicas beta inhiben la liberación de AVP.
Las principales alteraciones en la secreción de AVP consisten en la deficiencia parcial o completa (diabetes insípida) y el síndrome de secreción excesiva inapropiada de hormona antidiurética (SIHAD). En este capítulo se analizan los síndromes de deficiencia de vasopresina.
DIABETES INSIPIDA
La poliuria es un problema clínico frecuente.55 Un paciente que tiene un gran volumen de diuresis suele tener una de tres alteraciones: una diuresis osmótica, resistencia a la vasopresina o secreción deficiente de HAD.56 La causa más común de diuresis osmótica es la glucosuria, síntoma característico de la diabetes mellitus. La resistencia a la vasopresina (i.e., diabetes insípida nefrogénica) ocurre en ciertas formas de daño renal crónico, después de la liberación de una obstrucción urinaria y como un padecimiento hereditario. Se han identificado defectos genéticos en el receptor renal de AVP.57,58 La deficiencia de HAD (i.e., diabetes insípida neurogénica) refleja una alteración funcional o estructural de las neuronas supraópticas del hipotálamo que secretan la hormona. De nuevo, en la actualidad se conocen defectos genéticos en la secreción de la AVP.59,60 Dos datos clave (inicio súbito de la poliuria y preferencia por bebidas heladas) sugieren deficiencia de HAD. Sin embargo, la diabetes insípida neurogénica debe diferenciarse de la polidipsia primaria porque la ingesta excesiva de agua también causa poliuria y supresión de la secreción de vasopresina.
Diagnóstico
Por lo general la diabetes insípida neurógenica y nefrogénica pueden distinguirse por medio de pruebas clínicas [ver figura 7]. El paciente es deprivado de agua hasta que se pierde el 3 a 5 porciento del peso corporal y la tonicidad sérica es mayor de 295 mOsm/kg. Si la poliuria desaparece y la concentración urinaria es mayor de 500 mOsm/kg, la secreción de HAD es adecuada. Si persisten la poliuria y la orina diluida (< 300 mOsm/kg), se administran 20 µg de acetato de desmopresina (DDAVP) por vía inhalada, en forma alterna, pueden administrarse 300 µU de AVP por vía intravenosa. Si la diuresis disminuye y su concentración aumenta puede inferirse que existe deficiencia de HAD. Sin embargo, si el plasma se concentra y la orina permanece diluida a pesar de la administración de DDAVP, se diagnostica diabetes insípida nefrogénica.
Deben tenerse en cuenta algunas precauciones cuando se realicen pruebas de deshidratación. Primero, el término de diabetes insípida parcial describe a un paciente que, al estar sediento, alcanza una concentración urinaria mayor que la osmolaridad del plasma pero menor que la obtenida después de la administración de hormona antidiurética. Las pruebas funcionales pueden ser confusas en pacientes con diabetes insípida neurogénica o nefrogénica parciales. En estos pacientes, que tienen concentraciones urinarias entre 300 y 500 mOsm/kg, la medición de la concentración plasmática de AVP puede ser muy útil. Una concentración alta de AVP en presencia de plasma concentrado y orina relativamente diluida indica diabetes insípida nefrogénica; un valor bajo indica deficiencia de la hormona. Por el contrario, la resistencia parcial a la vasopresina puede ser causada por polidipsia crónica, con dilución secundaria de la concentración medular en el riñón. Si estos pacientes controlan su ingesta excesiva de agua, recuperarán la concentración medular renal normal y, al mismo tiempo, una respuesta normal a la vasopresina. Por último, la deprivación de agua produce menos sed en ancianos que en jóvenes.61 Los hombres mayores de 80 años deben ser vigilados con cuidado después de la prueba para asegurarse de que ingieran cantidades adecuadas de agua.
Los granulomas, los traumatismos, las infecciones y otros procesos infiltrativos pueden producir diabetes insípida. Es raro que los tumores metastásicos causen insuficiencia en otras glándulas endócrinas, pero los tumores que se originan del pulmón, mama y otros órganos pueden producir insuficiencia en la hipófisis posterior. La sensibilidad de la IRM ha afinado en forma considerable el esquema diagnóstico de la diabetes insípida.62
La diabetes insípida puede desarrollarse en forma súbita después de un tramatismo externo o una neurocirugía. En este caso la poliuria diluida con un nivel de sodio sérico mayor de 145 mEq/L permite el diagnóstico presuntivo y debe administrarse de inmediato DDAVP parenteral.63 Por el contrario, debe esperarse hiponatremia por aumento en la secreción de AVP después de la cirugía transesfenoidal, por lo que se vigilará el sodio sérico.64
Se han notificado casos de diabetes mellitus central y de diabetes insípida súbita y fatal en mujeres jóvenes con hiponatremia posoperatoria que no fue tratada en forma agresiva. La patogenia de este padecimiento no se conoce, pero la secuencia patológica incluyó edema cerebral y herniación, compresión del tercer par craneal, infarto hipóxico de la hipófisis e hipotálamo, paro respiratorio y coma. La rapidez del deterioro en estas pacientes indica que la hiponatremia en estos casos debe ser corregida de inmediato, aunque la dilatación pupilar fija, secundaria a la compresión del nervio oculomotor, puede sugerir muerte cerebral.65
Tratamiento
Existen varios esquemas de tratamiento para la diabetes insípida.66 Si la poliuria es leve y no interfiere con el sueño, no se requiere tratamiento. La cloropropamida potencializa el efecto de la vasopresina sobre la capacidad de concentración renal y puede ser utilizada para tratar la diabetes insípida parcial. Se administra en una dosis de 250 a 375 mg una vez al día y suele no producir hipoglucemia en las personas normales. Sin embargo, si los pacientes no comen en forma regular o si tienen insuficiencia hipofisiaria anterior no sospechada, la cloropropamida puede ser peligrosa.
Para los pacientes con diabetes insípida severa, el tanato de vasopresina en aceite, 5 unidades cada 2 a 4 días por vía intramuscular, proporciona un control excelente de la poliuria y la polidipsia. Es conveniente permitir que el efecto antidiurético desaparezca y se presente poliuria antes de administrar la siguiente dosis, si no la hiponatremia es una complicación frecuente. Esta precaución es importante, en especial después de una neurocirugía, en la que puede ocurrir una secuencia trifásica de deficiencia de AVP, exceso de AVP y nuevamente deficiencia. El tratamiento estándar previo de la diabetes insípida ha sido remplazado por la administración intranasal u oral de DDAVP. La administración intranasal de DDAVP en el tratamiento de la diabetes insípida es eficaz, no tóxica y no irritativa. Las tabletas de DDAVP, en dosis de 0.1 a 0.2 mg una a tres veces al día, es el tratamiento más novedoso para la diabetes insípida. Todos los pacientes con diabetes insípida deben ser advertidos de que en circunstancias de pérdida extrema de agua o en momentos de inconciencia están expuestos a un riesgo importante, a menos de que sean atendidos por un médico que sepa que padecen esta enfermedad.
Síndromes de neoplasias endócrinas múltiples
Varios síndromes hereditarios afectan a múltiples glándulas endócrinas en distintos patrones.67 La neoplasia endócrina múltiple tipo I (NEM I) incluye diversas alteraciones de la hipófisis anterior, las paratiroides y los islotes pancreáticos. Las familias afectadas muestran una alta incidencia de úlcera péptica y una frecuencia irregular y baja de otras lesiones somáticas y endócrinas. Las siguientes lesiones son las principales alteraciones endócrinas:
1. Hipófisis: adenomas o hiperplasia de las células acidófilas o cromófobas, con acromegalia, hiperprolactinemia o hipopituitarismo. La función endócrina puede ser normal.
2. Paratiroides: se han notificado adenomas, adenomas múltiples, hiperplasia y carcinoma de las paratiroides, el hiperparatiroidismo es la alteración endócrina más frecuente en los pacientes con NEM I. La afección paratiroidea suele frustrar los esfuerzos del patólogo y del cirujano para distinguir entre los adenomas y la hiperplasia, porque ambos padecimientos pueden ocurrir en la misma familia.
3. Páncreas: pueden ocurrir adenomas, hiperplasia y carcinomas en cualquiera de las células de los islotes pancreáticos. Los síndromes endócrinos resultantes incluyen hiperinsulinismo, hipergastrinemia (se produce un síndrome de Zollinger-Ellison), cólera pancreática y otras alteraciones menos bien definidas.
La NEM I se hereda en forma autosómica dominante, pero sólo en raras ocasiones se manifiesta antes de la pubertad. El hiperparatiroidismo es la manifestación endócrina espontánea más frecuente, aunque algunas series indican que el escrutinio prospectivo de los familiares de primer orden permite detectar lesiones pancreáticas o hipofisiarias antes del comienzo del hiperparatiroidismo.68,69 Sin embargo, no se sabe si estas series son típicas.
La genética y las bases moleculares de la NEM I son especialmente interesantes. La expresión del rasgo varía mucho entre los individuos y familias. De acuerdo con la teoría de dos etapas de la carcinogénesis de Knudson,70 los cánceres hereditarios son ocasionados por una mutación en una célula germinal que crea la predisposición, y una mutación en una célula somática que origina la manifestación clínica. La comparación entre las secuencias de ADN de los tumores paratiroideos y pancreáticos con las de los leucocitos de sangre periférica y otros tejidos de pacientes con NEM I demuestra pérdida de un segmento de la banda q13 del cromosoma 11 en los tumores.71-73 Se considera (aunque no ha podido demostrarse con las técnicas actuales) que la lesión hereditaria es una mutación en el locus q13 en un miembro del par cromosómico 11 y que este defecto existe en todos los tejidos del organismo [ver figura 8]. Al parecer, el gen en el locus q13 del cromosoma 11 codifica un producto supresor de tumores. Cuando se pierde el locus q13 del segundo miembro del par cromosómico 11, el cambio produce pérdida de la heterocigocidad, y la deficiencia homocigota de la proteína supresora de tumores permite que se desarrollen cánceres. A diferencia de los tumores de la NEM II, los tumores en los pacientes con NEM I no requieren de una hiperplasia subyacente para su aparición. Sin embargo, la presencia de hiperplasia paratiroidea y de mitógenos paratiroideos en algunos pacientes con NEM I parecen sugerir que, como en la NEM II, la hiperplasia precede a la neoplasia. También es de interés que algunos adenomas paratiroideos aislados muestran el mismo defecto clonal en el segmento q13 del cromosoma 11.74
La aparición tardía y las manifestaciones inespecíficas de la NEM I originan una pregunta clínica muy importante: ¿Debe considerarse la búsqueda de otros signos en un paciente que tenga alguna de las características clínicas de la NEM I? Esta investigación debe ser mínima si el índice de sospecha no es alto. Por ejemplo, en un paciente con un adenoma paratiroideo curado por cirugía existen pocos motivos para realizar evaluaciones adicionales. Sin embargo, si se encuentra hiperplasia paratiroidea, existe afección de otra glándula endócrina o la historia familiar incluye un segundo individuo afectado, deberá realizarse un estudio formal de otras glándulas y de los otros miembros de la familia. Los familiares de primer orden tienen un 50 porciento de riesgo de la enfermedad. Deberá realizarse un pedigree detallado, informando a todos los familiares del riesgo. Los enfoques de investigación varían, pero el autor sugiere medir por lo menos las concentraciones séricas de calcio, prolactina y glucosa. Estas pruebas deberán repetirse en intervalos de una a dos veces por año, comenzando desde los 15 años de edad. La detección temprana permite realizar un tratamiento más eficaz y disminuir la morbilidad de la familia.
Aunque el gen de la NEM I no ha sido clonado, la asociación genética con secuencias en el cromosoma 11 permite realizar estudios predictivos en las personas afectadas.75-77 Este enfoque es tanto más importante como presionante en la NEM II, pero en las familias con NEM I los familiares afectados pueden detectarse con el seguimiento adecuado.
La ausencia de historia familiar no obvia la investigación en busca de una NEM I. Las mutaciones de novo son responsables de casi el 50 porciento de los casos de este padecimiento. Cuando ocurren casos espontáneos, solo los hijos de los pacientes, y no los padres o hermanos, tienen mayor riesgo de NEM I.
Por último, puede ocurrir NEM I en asociación con el síndrome de Zollinger-Ellison, ya sea en un individuo o en una familia afectada. Si la historia familiar sugiere síndrome de Zollinger-Ellison, deberá incluirse la gastrina sérica en el escrutinio. La hipercalcemia del hiperparatiroidismo puede exagerar los síntomas y las alteraciones secretoras del síndrome de Zollinger-Ellison.
La neoplasia endócrina múltiple de tipo II, o NEM II ( que incluye feocromocitomas, carcinomas medulares de tiroides y adenomas o hiperplasia paratiroidea), se analiza en otra parte de la obra.
Interacciones entre el sistema endócrino y el inmunológico
Existen muchas interacciones entre el sistema inmunológico y el endócrino. El cortisol es una sustancia antinflamatoria importante y su producción constituye una de las principales respuestas del organismo al estrés. Tanto la hormona liberadora de corticotropina como la AVP tienen efectos proinflamatorios, pero ambas causan mayor producción de cortisol. Por lo menos cuatro citocinas influyen en la inflamación aguda: la interleucina-1 (IL-1), la IL-6, el factor de necrosis tumoral-a y el antagonista del receptor de IL-1 (IL-1ra). Las primeras tres son proinflamatorias, pero el IL-1ra es un freno a la respuesta inflamatoria. El efecto neto de las citocinas y las respuestas relacionadas consiste en inducir un estado del 'todo enfermo', una condición de alteración eutiroidea, hipogonadal, diabetogénica que se manifiesta más por alteraciones de laboratorio que por un reto terapéutico clínicamente significativo. La importancia de estos cambios y de las interacciones inmuno-endócrinas se estudia con gran interés.78,79
Reconocimientos
Figura 1 Carol Donner.
Figuras 2 y 3 Alan Iselin.
Figuras 4 y 8 Tom Moore.
Figura 5 Al Miller.
Figura 6 Hank Iken.
DR. DANIEL D. FEDERMAN
Neuroendocrinología
Los principales órganos de comunicación para la respuesta a los estímulos exógenos y endógenos estresantes, los procesos patológicos y la cirugía, son los sistemas nerviosos y endócrino. Las respuestas neurales al estrés requieren de comunicación intercelular en distancias minúsculas y usan trasmisores químicos; el tiempo de respuesta y el periodo que transcurre antes de que pueda ocurrir otra respuesta (latencia) se miden en fracciones de segundo. Por el contrario, las respuestas endócrinas son mediadas por hormonas que circulan en la sangre de un sitio a otro. La latencia se mide en minutos u horas. Además, las adaptaciones hormonales pueden continuar hasta por un día o un mes.
El eje hipotálamo-hipófisis permite que exista una convergencia entre las capacidades de adaptación neurales y hormonales. Las neuronas del hipotálamo son ricas en sinapsis de conección con el sistema nervioso central, por lo que reciben influencias aminérgicas, peptidérgicas y opioides. Además, el hipotálamo se encuentra fuera de la barrera hematoencefálica, por lo que puede percibir y responder a las concentraciones circulantes de glucosa, cortisol, sodio y otras sustancias. El hipotálamo es también el sitio de principal control homeostático, incluyendo la regulación del hambre, sed, osmolaridad, presión arterial, temperatura y respiración. Por último, es el origen de la comunicación humoral estimuladora e inhibidora con la hipófisis anterior [ver figura 1]. Los pequeños péptidos producidos en las neuronas del hipotálamo penetran a los capilares fenestrados, descendiendo a través de las venas porta-hipofisiarias y distribuyéndose por otros capilares fenestrados hasta llegar a las células de la hipófisis anterior.1
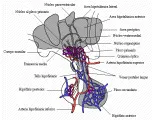
|
| Figura 1 |
| Anatomía hipofisiaria |
En la actualidad se han identificado ya las principales hormonas hipofisiotróficas del hipotálamo [ver tabla 1], y en este capítulo se analiza su significado clínico, su uso diagnóstico y sus aplicaciones terapéuticas. Es pertinente mencionar algunos datos generales sobre el control del eje hipotálamo-hipofisiario. Primero, la secreción hipotalámica e hipofisiaria es pulsátil, más que tónica [ver figura 2], en ciertos casos puede requerirse la toma de muestras cada 2 a 3 minutos para demostrar los pulsos. Estos pulsos dependen de ritmos biológicos, como la liberación circadiana de la hormona adrenocorticotrópica (ACTH), la liberación asociada al sueño de la hormona del crecimiento (GH) y la prolactina (PRL), y el ciclo mensual de las gonadotropinas en la mujer. Segundo, el hipotálamo es una rica fuente de las aminas biogénicas dopamina, epinefrina, norepinefrina y serotonina, y cada una de ellas modula los ritmos hipofisiarios. También los opioides endógenos son importantes en el control de la función hipofisiaria: las endorfinas promueven la liberación de GH, tirotropina (hormona estimulante del tiroides o TSH) y PRL, y suprimen la liberación de hormona foliculo estimulante (FSH), hormona luteinizante (LH) y ACTH. Las conecciones hipotalámicas con otros centros cerebrales pueden participar también en el control hipofisiario (v.gr., al parecer el hipocampo-amígdala inhibe el control hipotalámico de la ACTH y la función suprarrenal).

|
| Figura 2 |
| Secreción de hormonas hipofisiarias |
|
||||||||||||||||
|
La fisiología y la fisiopatología de las hormonas hipofisiarias tróficas específicas (i.e., FSH, LH, ACTH y TSH) se analizan en las subsecciones de ovario, testículo, suprarrenal y tiroides. Esta subsección trata sobre alteraciones más amplias de la hipófisis anterior, padecimientos específicos de las hormonas no troficas (GH y PRL), y síndromes que afectan a la hipófisis posterior y a la función endócrina en general.
El uso de agonistas y antagonistas sintéticos de la hormona liberadora de gonadotropina (GnRH) ha revolucionado la práctica de la endocrinología reproductiva y de trastornos asociados. Estas aplicaciones se mencionan en esta sección pero, por conveniencia, se analizan en las subsecciones respectivas.3
La hipofisis anterior
SOBREPRODUCCION DE HORMONAS HIPOFISIARIAS TROFICAS
La producción excesiva de hormonas hipofisiarias tróficas suele detectarse como una enfermedad de la glándula blanco.4 Por ejemplo, la hipersecreción de ACTH por un adenoma hipofisiario es la causa más frecuente del síndrome de Cushing.5,6 El tumor suele ser un microadenoma (<10 mm de diámetro) y no causa síntomas locales; la cirugía transesfenoidal, asociada a técnicas de laser, endoscopía o radiofrecuencia, es el tratamiento de elección.7 Es raro que ocurra hipersecreción de otras hormonas hipofisiarias tróficas. Los tumores que secretan gonadotropinas o fragmentos de gonadotropinas en exceso suelen ocurrir en hombres de edad media que han tenido un desarrollo sexual normal.8 Algunos de estos pacientes presentan hipogonadismo secundario, y la mayoría tienen tumores grandes que provocan defectos de los campos visuales. La cirugía transesfenoidal inmediata puede restablecer o mejorar la visión y dar tiempo para un tratamiento definitivo. Los adenomas hipofisiarios que producen TSH causan un síndrome hipertiroideo sin las características autoinmunes de la enfermedad de Graves. Pueden asociarse síntomas mecánicos causados por el tumor hipofisiario.
MENOR PRODUCCION DE HORMONAS HIPOFISIARIAS TROFICAS
Deficiencia hipofisiaria de una hormona trófica
La deficiencia hipofisiaria de una sola hormona trófica es una causa poco frecuente, pero bien definida, de hipofunción de una glándula blanco. Los síndromes resultantes se describen en las subsecciones referentes a las gónadas, tiroides y suprarrenales, pero es conveniente hacer algunos comentarios en este capítulo.
La deficiencia aislada de gonadotropina suele implicar secreción inadecuada tanto de FSH como de LH por la hipófisis anterior, que es secundaria a una deficiencia congénita de la hormona liberadora en el hipotálamo. Los individuos afectados no tienen pubertad, todos los aspectos de la función gonadal son deficientes y las concentraciones plasmáticas de gonadotropinas son bajos o están en el límite inferior normal, lo que distingue este padecimiento de la falla gonadal primaria.9,10
La deficiencia aislada de TSH puede ser adquirida, y se presenta como un hipotiroidismo típico, excepto por la ausencia de bocio y anticuerpos antitiroideos. Los pacientes tienen concentraciones séricas de TSH bajas o normales, que pueden aumentar un poco, pero no en forma importante, después de la administración de hormona liberadora de tirotropina (TRH).
El diagnóstico de deficiencia de ACTH es muy difícil. La función de la aldosterona se afecta en etapas tardías de la evolución, si acaso, y el síndrome se caracteriza por deficiencia selectiva de esteroides sin asociarse a hiperpigmentación o deficiencia de volumen. Los pacientes suelen tener un desarrollo deficiente, caracterizado por disminución del apetito, falta de ánimo o ambición, propensión a intoxicación por agua e hiponatremia y astenia generalizada. La diferencia clínica con la depresión o una enfermedad orgánica crónica, como el cáncer o la anemia, es casi imposible. La mayor parte de los pacientes con deficiencia selectiva de ACTH se diagnostican sólo después de que se detecta la hiponatremia o la incapacidad para responder en forma normal a una situación de estrés. La concentración plasmática de cortisol es baja (i.e., <4.5 µg/dl), y no aumenta después de la hipoglucemia inducida por insulina, pero sí al infundir ACTH durante un periodo de varios días.
Panhipopituitarismo
El panhipopituitarismo es más común que cualquiera de las deficiencias aisladas de hormonas tróficas.11 La hipofunción generalizada de la hipófisis suele ser resultado de las destrucción de la glándula por varias causas. La hipófisis es la única glándula endócrina en la que la destrucción por un tumor es una causa importante de su hipofunción, y esto se debe a compresión de la glándula contra los huesos que forman la silla turca (fosa hipofisiaria). El tumor hipofisiario primario más frecuente es el adenoma cromófobo. El craneofaringioma, un tumor de estructuras vecinas, es otra causa importante de hipofunción hipofisiaria. Suele manifestarse en niños, aunque puede aparecer en la edad adulta, causando hipofunción hipofisiaria o disminución visual como síntomas principales. También los tumores metastásicos, por lo general de mama o de pulmón, pueden causar hipofunción en algunas ocasiones.
El término síndrome de Sheehan se refiere, en forma típica, al hipopituitarismo moderado a severo secundario a un estado de choque por hemorragia uterina periparto. La necrosis hipofisiaria se explica por un infarto inducido por el colapso de la arteria hipofisiaria superior. Con la mejoría de los cuidados obstétricos cada vez se observan menos casos floridos. Sin embargo, en ocasiones se encuentra una variante sutil de síndrome de Sheehan en pacientes que desarrollan anemia al final del embarazo y que no reciben transfusiones. Los síntomas habituales suelen ser incapacidad para lactar, no reaparición de las menstruaciones, fatiga y debilidad. La tomografía computada puede indicar el diagnóstico al demostrar una silla turca de tamaño normal, pero parece ser que la imagen por resonancia magnética (RM) es más sensible.
El síndrome de silla vacía sigue siendo una variante enigmática de las enfermedades hipofisiarias.13 Aunque el crecimiento de la fosa hipofisiaria demostrada por radiografías suele interpretarse como un tumor hipofisiario, en algunos pacientes este aumento aparente de tamaño corresponde a la extensión de la cisterna subaracnoidea hacia la silla turca. La patogenia de la lesión no es clara. El síndrome de silla turca vacía se refiere a los pacientes en los que el evento inicial es la proyección del espacio subaracnoideo hacia la silla. En estos pacientes la función hipofisiaria suele ser normal, aunque puede existir hipersecreción de PRL o ACTH si coexiste un microadenoma. El síndrome de silla turca vacía secundaria se refiere a un padecimiento en el que la hipofisis disminuye de tamaño por un infarto, cirugía, radiación u otro mecanismo. En estos casos es frecuente la deficiencia hipofisiaria anterior parcial o completa. Puede requerirse de un neumoencefalograma para el diagnóstico, pero la TC con metrizamida proporciona una imagen definitiva con menos morbilidad. Es probable que la RM sea superior a la TC.
La destrucción de tejido hipofisiario por un granuloma o infección es un problema poco frecuente, pero que debe tenerse en cuenta en los pacientes que tienen hipopituitarismo inexplicable o una enfermedad granulomatosa diseminada. De hecho, la combinación de hipopituitarismo parcial y defectos visuales causada por sarcoidosis que afecte los nervios ópticos puede sugerir un tumor hipofisiario. Sin embargo, en estos casos la silla turca no está aumentada de tamaño, los defectos visuales son en parches (a diferencia de los causados por compresión quiasmática) y las pruebas funcionales sugieren hipopituitarismo causado por deficiencia hipotalámica.
La insuficiencia hipofisiaria secundaria a radioterapia de las estructuras vecinas se presenta cada vez con más frecuencia.14,15 Las dosis actuales, más grandes, la mayor supervivencia de los pacientes tratados, y el uso de pruebas sutiles de reserva hipofisiaria, contribuyen a aumentar el número detectado de estos casos. Los conocimientos actuales indican que el defecto es con más frecuencia hipotalámico que hipofisiario.16 La necrosis hipofisiaria secundaria a hemorragia dentro de un tumor puede causar deficiencia hormonal. En alguna ocasión se consideró que el hipopituitarismo era un padecimiento permanente, pero la secreción hormonal puede recuperarse después de la reducción farmacológica de los macroprolactinomas17 y después de la descompresión transesfenoidal de tumores o de infartos hemorrágicos en la hipófisis.
Datos clínicos Los datos clínicos del panhipopituitarismo son muy variables, pueden incluir cambios tanto mecánicos como hormonales. Los cambios mecánicos son secundarios al crecimiento de un tumor que sobrepasa la silla turca, e incluyen cefalea y alteraciones de la visión como visión borrosa, disminución en la agudeza y defectos en los campos visuales. El primer defecto suele ser la pérdida del cuadrante superior bitemporal, que se detecta mejor por el examen con una luz roja intermitente. Debido a que la mácula no se afecta, los pacientes pueden tener grandes pérdidas de los campos visuales sin notarlas.
Las características endócrinas del panhipopituitarismo dependen del grado de deficiencia hormonal, de la velocidad con que se desarrolla la deficiencia, de la edad del individuo y de la presencia o ausencia de enfermedades asociadas. La GH y las gonadotropinas tienen una sensibilidad semejante al daño hipofisiario parcial; si el síndrome se desarrolla en forma gradual, la producción de estas hormonas es la primera que se altera. La deficiencia de hormona del crecimiento (DGH) se caracteriza por disminución del bienestar, menor masa corporal magra y densidad ósea, aumento de la grasa abdominal, piel delgada y seca y reducción en la fuerza muscular.18,19 En las mujeres en edad reproductiva la amenorrea es el primer síntoma, y en los hombres lo es la disfunción eréctil. En las mujeres posmenopáusicas la deficiencia de gonadotropina no provoca síntomas, por lo que su detección se retrasa. La apatía y falta de preocupación sobre el propio bienestar, típicos de los pacientes hipopituitarios, retrasa el diagnóstico definitivo aún más.
A diferencia de lo que ocurre con las gonadotropinas, el desarrollo de la deficiencia de TSH y de ACTH puede ser muy tardado. Cuando se producen aparecen diferentes características clínicas de hipotiroidismo e hipocortisolismo. Es frecuente que los síntomas y signos se atribuyan en forma equívoca a depresión o envejecimiento.
El diagnóstico del hipopituitarismo en los ancianos representa un reto porque los síntomas inespecíficos como fatiga, falta de ambición, debilidad y palidez, pueden ser los únicos presentes hasta que se desarrolla hipotiroidismo o hiponatremia francos.20
Diagnóstico de laboratorio La confirmación del panhipopituitarismo por laboratorio se ha simplificado mucho con la introducción de las pruebas de radioinmunoensayo para hormonas peptídicas. El enfoque general de diagnóstico consiste en demostrar primero la deficiencia de la glándula blanco y después medir la hormona hipofisiaria correspondiente. Para demostrar deficiencia de gonadotropinas en el varón se mide la testosterona plasmática. Si la concentración de testosterona es baja, se determinan las concentraciones de LH y FSH. Si éstas son bajas se confirma que el defecto tiene origen central. En la mujer en edad reproductiva, la amenorrea, una citología vaginal inmadura y la ausencia de hemorragia después de la administración de una progestina sintética reflejan deficiencia de estrógenos; una concentración normal o baja de FSH indica deficiencia hipofisiaria. La deficiencia de TSH se demuestra por un valor bajo de tiroxina y de TSH. Podría pensarse que la administración de TRH permite diferenciar entre la deficiencia hipotalámica y la hipofisiaria de TSH, pero esta prueba ha dado resultados muy poco satisfactorios.
La deficiencia de ACTH suele ser más sutil que las analizadas antes, por lo que su diagnóstico requiere de pruebas de estimulación. Un nivel de cortisol matutino en ayuno menor de 5 µg/dl sugiere deficiencia de ACTH, en especial cuando el nivel de ACTH es bajo o normal. Sin embargo, cuando la sospecha es alta y el nivel de cortisol no es bajo, se requieren las pruebas de estimulación. Puede usarse, por ejemplo, metirapona, un inhibidor de la 11 ß-hidroxilasa suprarrenal [ver figura 3]. Se administra una dosis oral única de 3 g a media noche, y se mide el cortisol plasmático y el 11-desoxicortisol a la mañana siguiente. Si ambas hormonas tienen una concentración baja, el paciente tiene deficiencia de ACTH; si el cortisol es bajo y el 11-desoxicortisol es mayor de 10 µg/dl, el paciente tiene una reserva hipofisiaria normal. Otra prueba para investigar la respuesta de ACTH consiste en administrar insulina para inducir hipoglucemia. Después de una dosis estándar de 0.1 unidad/kg, la glucemia disminuye a menos de la mitad del valor control; si el sistema hipotálamo-hipófisis-suprarrenal está intacto, la concentración plasmática de cortisol se duplicará en respuesta a la hipoglucemia. Si la concentración de cortisol no aumenta, pero sí lo hace después de una infusión prolongada de ACTH (40 unidades I.V. durante un periodo de 8 horas en 2 días consecutivos), se confirma el diagnóstico de deficiencia de ACTH. Una prueba más simple utiliza ACTH sintética, o cosyntropin. Se administra una dosis de 0.25 mg I.V., la respuesta normal consiste en aumento en el nivel de cortisol plasmático a más de 18 mg/dl. Esta prueba es útil porque la glándula suprarrenal que ha estado deprivada en forma crónica de ACTH no responde con rapidez a un sólo estímulo con ACTH.
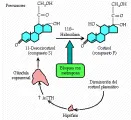
|
| Figura 3 |
| Bloqueo hormonal con metirapona |
La hipoglucemia inducida por insulina también es el estándar de oro para detectar DGH, pero no se usa en pacientes con cardiopatía conocida o mayores de 60 años.21 Pueden usarse mediciones de GH estimulada con arginina, pero se requiere de un sofisticado laboratorio de endocrinología para distinguir la DGH hipofisiaria de la respuesta de GH lenta de los ancianos. Un enfoque aceptable consiste en calcular la probabilidad de DGH con base en la deficiencia de otras hormonas hipofisiarias. Si una hormona, por lo general gonadotropina, está deficiente, la posibilidad de DGH es de 55 porciento. Si las tres hormonas están deficientes el rango aumenta a más de 90 porciento. La disponibilidad de hormonas hipotalámicas liberadoras purificadas o sintéticas permite una prueba sensible y simple para el diagnóstico de la reserva hipofisiaria. Se administra un bolo intravenoso de una o más de las hormonas liberadoras y se mide la hormona u hormonas hipofisiarias correspondientes en los intervalos adecuados [ver tabla 2]. Las hormonas liberadoras son bien toleradas, excepto por ocasionar rubor y náusea.
|
||||||||||
|
Tratamiento Los pacientes con hipopituitarismo necesitan de tratamiento sustitutivo permanente, cuidadoso e individualizado. Este debe seguir lo más posible los principios fisiológicos, usando dosis y tiempos de administración que simulen los patrones naturales de secreción. Es preferible utilizar las hormonas de las glándulas blanco que las hormonas hipofisiarias porque éstas últimas se administran por vía parenteral, son incómodas, caras y antigénicas. En los pacientes con panhipopituitarismo parcial es adecuado usar dosis de hormonas tiroideas y de hidrocortisona más pequeñas que las utilizadas en el hipotiroidismo o hipoadrenalismo primarios.
La sustitución suprarrenal puede lograrse administrando hidrocortisona oral. Los pacientes que han tenido hipopituitarismo durante más de un año deben comenzar con dosis bajas, alrededor de 5 mg/día, a menos de que exista alguna urgencia. Las dosis iniciales más altas pueden en ocasiones inducir un comportamiento hipomaniaco. En pacientes con deficiencia leve, el esquema inicial lógico de tratamiento es de 10 mg de hidrocortisona dos veces al día. La dosis matutina puede aumentarse a 15 mg cuando se requiera hasta alcanzar un bienestar total. La dosis puede aumentarse en forma súbita en casos de estrés agudo, como en el paciente con insuficiencia suprarrenal primaria. Los pacientes con insuficiencia suprarrenal secundaria suelen no requerir remplazo de mineralocorticoides, pero éstos se administran cuando persiste la hiponatremia o la hipotensión postural a pesar de la administración de hidrocortisona.
La tiroxina es el tratamiento adecuado para la deficiencia tiroidea. La dosis inicial es de 0.05 mg/día, a menos de que el paciente tenga más de 60 años o padezca insuficiencia cardiaca, caso en el cual es más seguro iniciar con 0.025 mg/día. El autor aumenta la dosis a razón de 0.025 mg/día cada mes hasta que se alcanza una dosis de 0.075 a 0.150 mg/día.
Para los varones, el enantato de testosterona en dosis de 300 mg cada tres semanas proporciona una sustitución adecuada. De nuevo, en pacientes que han tenido hipogonadismo durante más de 2 años puede comenzarse con un tercio de la dosis total de sustitución. Esto evita el aumento brusco en la agresividad y la líbido y las atemorizantes fantasias sexuales que ocurren en ocasiones cuando se administra la dosis total en forma súbita.22
Para las mujeres menores de 50 años el autor utiliza estrógenos conjugados,1.25 mg/día durante los primeros 25 días de cada mes, y un progestágeno, como acetato de medroxiprogesterona, los días 16 a 25. Después de los 50 años de edad es adecuado administrar 0.625 mg/día de estrógenos conjugados y el progestágeno. Como con otras mujeres posmenopáusicas, pueden usarse dosis bajas continuas de estrógenos y progestágeno para evitar la menstruación. Para los pacientes en edad reproductiva que desean ser fértiles puede intentarse el tratamiento pulsátil con hormona liberadora de gonadotropinas (GnRH), para inducir la espermatogénesis o la ovulación si el problema básico del paciente es hipotalámico. Si el hipopituitarismo es causado por destrucción hipofisiaria, puede administrarse tratamiento secuencial con FSH y LH.
La disponibilidad ilimitada de GH recombinante ha iniciado una nueva era para los pacientes con deficiencia de esta hormona. Su administración regular aumenta la masa corporal magra, reduce la obesidad, restablece el volumen de líquido, mejora la masa muscular y la fuerza y, después de 1 a 2 años, aumenta la densidad mineral ósea. Muchos pacientes presentan también mejoría psicológica impresionante. No se sabe si el tratamiento con GH revierte la mayor mortalidad de los pacientes con hipopituitarismo, ni es claro hasta qué momento de la vida debe continuarse el tratamiento.23,24
El uso de hormona del crecimiento como tratamiento sustitutivo en ancianos con deficiencia limítrofe relacionada a la edad, en un intento por retrasar los cambios involutivos, es motivo de gran controversia. Algunos estudios han sugerido mejoría en la fuerza y bienestar, pero se requiere mucho más información sobre los beneficios y riesgos. Según los conocimientos actuales, existe poca justificación para prescribir esta hormona a individuos que no tengan una deficiencia clara de GH. Otro factor que debe considerarse es el alto costo del tratamiento con hormona del crecimiento, que tiene un promedio de 10,000 a 20,000 dolares al año para cada paciente.25
TUMORES HIPOFISIARIOS Y CRECIMIENTO DE LA SILLA TURCA
El crecimiento de la silla turca suele descubrirse en forma accidental en una radiografía de cráneo realizada por otro motivo, por ejemplo por un traumatismo craneano o durante una evaluación de los senos paranasales. En ocasiones el crecimiento es causado por una silla vacía, pero la causa más común es una neoplasia benigna.
Los tumores hipofisiarios varían en tamaño de microadenomas de 1a 2 mm a lesiones masivas. Algunas evidencias indican que estos tumores tienen un origen clonal, esto es, que se originan de una sola célula.26 Los tumores más pequeños no ocasionan síntomas por vecindad, sino que se manifiestan por síndromes hormonales, como la amenorrea hiperprolactinémica y el síndrome de Cushing. Por el contrario, los tumores que no producen hormonas funcionales y suelen ser descubiertos cuando causan síntomas locales, casi siempre son macroadenomas (> 10 mm de diámetro) que en muchos casos se extienden más allá de la silla turca. Estos tumores lesionan los campos y agudeza visual, y pueden afectar también otras áreas cerebrales. Algunos de estos tumores grandes producen fragmentos hormonales, en especial las cadenas a o ß de las hormonas glucoproteicas FSH, LH o TSH [ver figura 4]. Estos fragmentos son demostrados por tinción inmunológica del tejido tumoral, y pueden también encontrarse en la circulación. Una prueba monoclonal muy sensible ha detectado hipersecreción de la subunidad a en alrededor de la cuarta parte de los tumores clínicamente no funcionantes. Alrededor del 30 porciento de los macroadenomas no secretan hormonas ni fragmentos y son denominados tumores nulos.
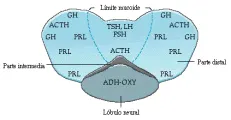
|
| Figura 4 |
| Glándula hipófisis |
El impacto endócrino de un macroadenoma debe evaluarse por los métodos descritos para detectar hipopituitarismo y es crucial medir el nivel de prolactica en suero.
Es menos claro cómo debe estudiarse un microadenoma descubierto en forma incidental por una IRM realizada por otro motivo. Alrededor del 10 a 15 porciento de las hipófisis muestran microadenomas en la autopsia, y la inmunotinción sugiere que muchos de éstos contienen prolactina. Si la persona no tiene síntomas y su función menstrual o eréctil es normal, lo más adecuado es realizar una sola medición de prolactina en suero.27 Es muy poco probable que se detecte una acromegalia o Cushing presintomáticos, ni tampoco tendrá algún beneficio.
La IRM es el método más sensible para evaluar la silla turca, la hipofisis y el hipotálamo, aunque la TC con medio de contraste también es adecuada.
El tratamiento adecuado de los tumores hipofisiarios depende de los resultados de los estudios endócrinos, radiográficos y neuroftalmológicos. Los tumores secretores requieren tratamiento, pero los microadenomas no secretores pueden ser sólo vigilados. Los macroadenomas se tratan con una combinación juiciosa de cirugía transesfenoidal (en ocasiones transfrontal) y radioterapia.28,29 El hipopituitarismo descubierto antes o después del tratamiento debe ser tratado con sustitución hormonal. En algunos pacientes el hipopituitarismo detectado antes de la cirugía mejora o se corrige por la resección transesfenoidal.30
La complicación más urgente de los tumores hipofisiarios es la apoplejía hipofisiaria. En algunos pacientes con tumores grandes puede ocurrir hemorragia y necrosis súbitas, creando una urgencia neuroquirúrgica. Los datos clínicos incluyen cefalea, obnubilación, oftalmoplejía, pérdida de la visión, y líquido cefalorraquídeo hemorrágico. La TC o la IRM han simplificado mucho el diagnóstico, y el tratamiento inmediato con hidrocortisona proporciona tiempo para la intervención neuroquirúrgica.31 La apoplejía subaguda, en la que los síntomas son mínimos o ausentes, puede ser confirmada por IRM. Puede ocurrir hipopituitarismo como una característica de la hemorragia y necrosis en los tumores. Sólo la deficiencia suprarrenal requiere de tratamiento inmediato, los otros estados hormonales pueden ser evaluados después de tratar la urgencia.32,33
HORMONA DEL CRECIMIENTO
La hormona del crecimiento, una proteína compuesta de 191 aminoácidos, es secretada por los somatotropos de la hipófisis anterior. A pesar de su nombre, la GH no promueve el crecimiento en forma directa, más bien actúa al estimular la formación hepática de somatomedina C (también conocida como factor de crecimiento semejante a insulina 1, o FCI-1), que es uno de los muchos factores de crecimiento. El FCI-1 entra a la circulación y se fija a uno de dos receptores; el receptor de membrana que fija FCI-1 es muy parecido al receptor de la insulina. En algunos tejidos la GH se fija en forma directa a un receptor de membrana.34 Aún no es claro qué efectos de la GH son causados por cuál de los eceptores.
La concentración plasmática de GH está controlada principalmente por los efectos opuestos de dos neuropéptidos que se producen en el hipotálamo y llegan a la hipófisis a través de la circulación portal.35 El neuropéptido hormona liberadora de hormona del crecimiento (GHRH) estimula la liberación de GH por la hipófisis, mientras que la somatostatina inhibe su secreción. La combinación de la secreción pulsátil, la vida media de 20 minutos y los controles opuestos sobre la secreción, causan concentraciones de GH en plasma muy variables. Aún más, tanto en niños como en adultos, se secreta GH en respuesta a la disminución en la glucemia (ver adelante), la ingestión de aminoácidos, el ejercicio, el sueño, el estrés y muchos otros factores, incluyendo estímulos adrenérgicos alfa, como la levodopa. La secreción de GH es suprimida por el aumento en la glucemia y por estímulos adrenérgicos beta.
En condiciones normales la concentración plasmática de GH en una persona varía durante el día, desde niveles no detectables hasta valores típicos de acromegalia. En vista de esta variabilidad, las mediciones aleatorias de GH no son definitivas desde el punto de vista clínico y se requieren pruebas estimuladoras y supresoras.
Acromegalia
El exceso de hormona del crecimiento produce acromegalia en los adultos y, en raras ocasiones, gigantismo en los niños.36 La causa más frecuente de exceso de GH es un adenoma hipofisiario. Para el momento en que se detecta la acromegalia por clínica, la mayoría de los pacientes tienen aumento de tamaño de la silla turca y un macroadenoma, por lo que la extensión supraselar es frecuente.
Un mecanismo de formación del adenoma, que se encuentra en el 30 a 40 porciento de los pacientes con acromegalia, es la mutación somática del gen aS, que codifica la cadena a de una proteína G reguladora unida a la membrana (GS). Esta proteína se fija a receptores de superficie de la célula para estimular segundos mensajeros intracelulares. La proteína G trimérica se mueve entre el difosfato de guanosina (GDP), que es inactivo, y el trifosfato de guanosina (GTP), que es activo. Varias mutaciones en el gen aS causan inhibición de la hidrólisis del GTP a GDP; con lo que la proteína GS se vuelve constitutivamente activa y la célula secreta GH en forma continua.
Una segunda causa de exceso de GH es la estimulación patológica por GHRH derivado del hipotálamo (eutópico) o de tumores en otra parte del organismo (ectópico). El GHRH se aisló por primera vez de un carcinoide bronquial y después de un tumor pancreático, en la actualidad se ha encontrado en muchos otros tipos de tumores. Estos tumores suelen derivar de lesiones benignas del intestino anterior y son especialmente frecuentes en los pulmones y el páncreas.
Diagnóstico Los signos más frecuentes de acromegalia son los secundarios a las manifestaciones paraselares y al exceso de GH [ver tabla 3]. Quizá la combinación más común consista en diaforesis, cefalea y debilidad o fatiga. El edema inexplicable de pies y manos, el crecimiento progresivo de las regiones acrales, el engrosamiento de labios y lengua y un olor desagradable del organismo son otras características de este síndrome. El diagnóstico suele ser tardío porque el paciente, su familia y su médico familiar suelen no notar los cambios graduales en su apariencia. Por lo tanto, por lo general es un familiar lejano o un nuevo médico quien nota el cambio en la apariencia facial del enfermo. Una vez que se ha pensado en el diagnóstico, los siguientes datos ayudan a corroborarlo: rasgos toscos, múltiples marcas en la piel (fibroma molluscum), piel gruesa y grasosa, prominencia frontal, ensanchamiento de los dientes y la mordida, antecedentes de ronquidos o apnea del sueño,37 y un tono ronco peculiar en la voz. La comparación de fotografías antiguas y recientes puede ser muy útil, al igual que en el mixedema y el síndrome de Cushing.
|
|||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||
La característica hormonal esencial de la acromegalia consiste en la ausencia de la supresibilidad normal de la GH por la glucosa. A través de todo un día los individuos normales38,39 tienen concentraciones muy bajas de GH después de la ingesta de glucosa. El paciente con acromegalia pierde esta capacidad de respuesta a la glucosa y libera GH a una concentración que siempre es mayor que la mínima alcanzada por una persona normal. Esta diferencia es muy importante. El paciente con acromegalia no necesariamente debe producir grandes cantidades de GH; de hecho, su concentración de GH puede no ser mayor que la alcanzada por una persona normal después de realizar ejercicio o durante la hipoglucemia preprandial, pero esta concentración no disminuye después de la ingesta de glucosa. Para diagnosticar acromegalia es necesario medir la concentración de GH 1 a 2 horas después que el paciente ha ingerido 75 a 100 g de glucosa. Si la concentración de la hormona es mayor a 3 ng/ml debe sospecharse acromegalia, y se realizarán las pruebas confirmatorias definitivas. El valor posglucosa puede ser mayor de 2 ng/ml en adolescentes y en pacientes con hepatopatía crónica, uremia o anorexia nervosa, pero estos padecimientos son fáciles de diagnosticar.
La medición de la concentración de FCI-1, que correlaciona mejor que la concentración de GH con la actividad clínica de la acromegalia, es un método alterno o suplementario para confirmar el diagnóstico. En muchas instituciones esta prueba es la preferida para la evaluación inicial de escrutinio. Los valores normales varían con la edad y con diferentes métodos de laboratorio. Por lo tanto, es mandatorio interpretar el nivel de FCI-1 acorde a esto.40
Debe medirse la prolactina sérica en todos los pacientes con acromegalia. Su concentración está aumentada en alrededor de un tercio de los enfermos, principalmente por su secreción en el adenoma. Sin embargo, en el caso de algunos macroadenomas de gran tamaño con extensión supraselar, la concentración de prolactina puede estar aumentada por compresión del tallo hipofisiario, lo que ocasiona interferencia con la inhibición de la prolactina por la dopamina.
Tratamiento El autor considera que todos los pacientes con pruebas de escrutinio sugestivas de acromegalia deben ser referidos al endocrinólogo. El tratamiento inicial de la mayoría de los pacientes consiste en cirugía transesfenoidal de la hipósifis, un procedimiento muy especializado pero con frecuencia no curativo. El tratamiento subsecuente puede incluir radioterapia, bromocriptina o el análogo de somatostatina, ocreótido. La mayoría de los clínicos intentan la normalización total de los niveles de GH a menos de 2 ng/ml para minimizar la morbimortalidad por exceso de GH. Sin embargo, el tratamiento adecuado, la vigilancia de los efectos adversos e incluso la confirmación por laboratorio del control completo son temas de gran discrepancia.41-43
Hormona del crecimiento y envejecimiento
La disponibilidad de GH humana recombinante ha iniciado una nueva área en la endocrinología, la posibilidad de tratar a los adultos sin deficiencia de GH. Los niveles de FCI-1 son menores en los ancianos que en los jóvenes, y también lo es la liberación de GH inducida por insulina,44 lo que ha originado varias preguntas. ¿Cuándo deben considerarse los niveles descendientes de GH en los ancianos como deficiencia?45 ¿Si existe o no una deficiencia patológica, los menores niveles contribuyen a la reducción de la masa corporal magra, del músculo y del hueso en el anciano? ¿Podría el tratamiento con GH detener estos cambios sin causar efectos adversos dañinos?46,47 Varios estudios a corto plazo han demostrado un efecto anabólico significativo del tratamiento con GH, pero existen pocos datos sobre los efectos a largo plazo en el sistema cardiovascular o la predisposición al cáncer, que son una grave preocupación en el anciano.
PROLACTINA
La prolactina es la segunda hormona producida en la hipófisis anterior que actúa en forma directa sobre tejidos blancos, más que como una hormona trófica. Aunque tiene muchas funciones en otros animales, su única función conocida en el humano es la estimulación posparto de la producción de leche. Sin embargo, existe en varones y en mujeres no embarazadas. Además, ocurren pulsos de liberación de prolactina en respuesta al sueño, estrés y otros estímulos. Sin embargo, a diferencia de otras hormonas hipofisiarias, la prolactina está controlada, principalmente, por inhibición tónica, más que por estimulación intermitente. Su principal inhibidor es la dopamina. La prolactina aumenta la secreción de dopamina, por lo que inhibe su propia secreción. Otros inhibidores fisiológicos conocidos son la somatostatina y la triyodotironina (T3). La liberación de prolactina es estimulada por serotonina, acetilcolina, opiáceos, estrógenos, TRH y angiotensina II, entre otros, pero no se sabe con exactitud cuales de estas sustancias, si es que alguna, son fisiológicamente importantes.
La secreción de prolactina aumenta durante el embarazo, alcanzando al término del mismo concentraciones hasta de 20 a 30 veces la normal. Cuando la concentración de estrógeno, que también aumenta en forma dramática durante el embarazo, comienza a disminuir, se vuelve posible la lactancia; al continuar la lactancia los niveles basales de prolactina disminuyen hasta niveles semejantes a los de las mujeres no embarazadas. Aunque la succión estimula la liberación de prolactina, la lactación puede continuar con niveles de prolactina normales en reposo. En forma semejante, las concentraciones basales de prolactina son normales en algunos pacientes con galactorrea, no se sabe si las concentraciones estaban aumentadas antes en esos pacientes.
Alteraciones en la secreción de prolactina
Ocurre hipoprolactinemia en el síndrome de Sheehan y en otros tipos de panhipopituitarismo. Las mediciones basales de prolactina no diferencían en forma confiable entre los valores bajos y los limítrofes, pero la prolactina sérica no aumenta después de la estimulación con TRH en los pacientes afectados.
La hiperprolactinemia es mucho más frecuente que la deficiencia de prolactina.48 El principal síntoma directo del exceso de prolactina es la galactorrea, o lactación no puerperal. La lactación puede ser espontánea o detectarse por compresión suave del pezón. La correlación entre concentraciones elevadas de prolactina y lactación es escasa: muchos pacientes tienen concentraciones elevadas de prolactina sin lactación, y puede persistir una lactación significativa sin hiperprolactinemia.
Las consecuencias indirectas de la hiperprolactinemia son mucho más frecuentes que la galactorrea. La impotencia en el varón o las alteraciones menstruales en la mujer son quejas frecuentes. El defecto menstrual puede consistir en una fase lútea corta, periodos anovulatorios, oligomenorrea o amenorrea. Por lo tanto, debe determinarse la concentración de prolactina en todas las pacientes con alteraciones reproductivas.
Puede ocurrir hiperprolactinemia después del uso de fenotiacinas, inhibidores de la monoaminoxidasa u otros fármacos que influyen en la función hipotalámica o adrenérgica. También se ha implicado a los anticonceptivos orales en los síndromes de lactación; puede ocurrir hiperprolactinemia durante o poco después de la suspensión de los anticonceptivos orales.
El hipotiroidismo causa hiperprolactinemia, pero los valores de prolactina muy pocas veces son mayores a 50 ng/ml. Debe realizarse medición de TSH para diferenciar entre el hipotiroidismo primario (que causa elevación secundaria de prolactina) y la enfermedad hipofisiaria que provoca tanto hipotiroidismo como hiperprolactinemia [ver figura 5]. La insuficiencia renal puede también ocasionar aumento de prolactina.

|
| Figura 5 |
| Niveles de prolactina en varias circunstancias |
Diagnóstico En la evaluación de los pacientes con hiperprolactinemia, una historia clínica y examen físico cuidadosos, junto con la evaluación de la función tiroidea y renal, suelen ser suficientes para detectar causas no hipofisiarias.49 Se han usado diversas pruebas de supresión y estimulación para distinguir entre las causas hipotalámicas e hipofisiarias de la hiperprolactinemia, pero ninguna es muy confiable. Por lo tanto, es indispensable realizar una TC de cráneo de alta resolución con contraste o una IRM.
El grado de aumento de la prolactina y los hallazgos radiográficos deben relacionarse en forma cuidadosa. Las concentraciones séricas de prolactina superiores a 300 ng/ml suelen reflejar un tumor hipofisiario, ya sea un microadenoma (< 10 mm de diámetro) o un macroadenoma (> 10 mm). Pueden encontrarse concentraciones muy altas (> 600 ng/ml) en los tumores grandes. Una concentración de sólo 50 a 300 ng/ml asociada a un tumor grande puede significar compresión del tallo hipofisiario por el tumor, con bloqueo consecuente de la inhibición de la dopamina sobre la liberación de prolactina, y no la presencia de un prolactinoma. En el nivel inferior de la escala, pueden existir microadenomas con concentraciones de prolactina tan bajas como de 30 ng/ml. En estos casos el radiólogo debe distinguir en forma cuidadosa entre las variaciones normales de los tumores verdaderos, realizando correlaciones clínicas para descubrir o descartar causas no tumorales del aumento de la prolatina. Si no se encuentra otra explicación, puede realizarse un diagnóstico provisional de hiperprolactinemia idiopática.
Tratamiento La mayor parte de los casos de hiperprolactinemia causados por un tumor o funcionales pueden ser controlados con el agonista dopaminérgico bromocriptina.50 El medicamento se administra en una dosis inicial de 1.25 mg/día, con los alimentos y en el momento de acostarse para minimizar los efectos colaterales de náusea e hipotensión postural. Después de una semana la dosis puede aumentarse a 1.25 mg dos veces al día, que suele ser bien tolerada. A las dos semanas debe medirse la concentración sérica de prolactina, antes de considerar un nuevo aumento en la dosis. La dosis de bromocriptina puede aumentarse 2.5 mg cada dos semanas hasta que la concentración de prolactina disminuya, aparezcan síntomas colaterales o se alcance una dosis máxima de 7.5 mg/día. La concentración de prolactina puede normalizarse varios días después de iniciado el tratamiento, y las menstruaciones o la potencia se recuperan después de varias semanas. Un agonista dopaminérgico alterno, el mesilato de pergolide, parece ser tan eficaz como la bromocriptina y en ocasiones es mejor tolerado. De hecho, el esquema de una dosis al día del pergolide es mejor en los pacientes que tienen dificultad para tolerar la bromocriptina. Otro agonista de dopamina, el cabergoline, también tiene algunas ventajas sobre la bromocriptina.51 El tratamiento con cualquiera de estos dos fármacos suele suspenderse durante el embarazo. Sin embargo, las pacientes embarazadas deben ser vigiladas en forma estrecha porque en alrededor del 1 a 2 porciento de los casos (casi siempre en pacientes con macroadenomas), las altas concentraciones de estrógenos inducen crecimiento significativo de los prolactinomas. Los pacientes con tumores en ocasiones son resistentes a la bromocriptina, y esta resistencia parece relacionarse con concentraciones bajas de receptores de dopamina D2 en las membranas de las células del prolactinoma.52 Para estos y otros pacientes que prefieren evitar los medicamentos la adenomectomía transesfenoidal ofrece una alternativa más conveniente.
Entre los pacientes con microadenomas y concentraciones de prolactina menores de 100 ng/ml, el porcentaje de curación con la cirugía alcanza un 88 porciento. Sin embargo, con el tiempo se observa una frecuencia significativa de recurrencias. Por ello, a los enfermos que se han sometido a una cirugía aparentemente exitosa se les medirá la prolactina sérica en forma periódica, en especial si los síntomás recurren. El seguimiento deberá continuar durante 5 años. Los macroadenomas representan un mayor reto quirúrgico tanto por el riesgo de la operación como por el menor porcentaje de éxito. Por este motivo se usa mucho la bromocriptina como tratamiento inicial,15 incluso cuando al final se requerirá de tratamiento quirúrgico. El tratamiento preoperatorio con bromocriptina puede mejorar la exploración quirúrgica en esos casos; de hecho, la disminución de tamaño de los prolactinomas puede ser tan rápida como para considerarse una opción para el tratamiento urgente de pacientes con cefalea y defectos de los campos visuales severos. En raras ocasiones están indicadas la radioterapia y la bromocriptina posoperatorias. La radioterapia aislada se emplea en casos seleccionados, pero parece ser menos eficaz que la bromocriptina o que la cirugía transesfenoidal.
¿Deben tratarse todos los pacientes con hiperprolactinemia? Los resultados de estudios sobre la historia natural de la hiperprolactinemia son variables pero varias series han notificado lo siguiente: (1) los microadenomas progresan muy poco, si es que lo hacen, (2) en algunos pacientes las concentraciones de prolactina se normalizan en forma espontánea, y (3) la vigilancia de la hipófisis por medio de imágenes es una manera segura de seguir a los pacientes que no desean tratamiento. El autor considera que es razonable vigilar a las mujeres con menstruaciones normales y que tienen concentraciones de prolactina menores a 100 ng/ml. Debe utilizarse tratamiento con progestágenos para inducir menstruaciones regulares en las pacientes con oligomenorrea. En las enfermas que no responden a los progestágenos deberá medirse la concentración plasmática de estradiol y realizar estudios de densitometría ósea. Es mejor indicar a estas pacientes que no responden que deben iniciar tratamiento con bromocriptina .
La hipófisis posterior
La hipófisis posterior está compuesta por porciones terminales de neuronas que se originan en los núcleos supraópticos y paraventriculares del hipotálamo. La vasopresina (AVP u hormona antidiurética HAD) y la oxitocina son sintetizadas en el hipotálamo como parte de un gran complejo molecular precursor que incluye proteínas asociadas llamadas neurofisinas. La molécula precursora es procesada durante su movimiento a lo largo del axón de la célula nerviosa. Después la vasopresina y la oxitocina son almacenadas en la hipófisis posterior y liberadas en respuesta a los estímulos apropiados. El primer ensayo satisfactorio de AVP [ver figura 6] ha demostrado lo siguiente:53
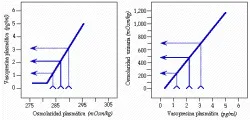
|
| Figura 6 |
| Osmolaridad y vasopresina |
1. El adulto normal con una osmolaridad sérica de 287 ± 5 mOsm/kg tiene una concentración plasmática de AVP entre 1 y 2 pg/ml.
2. Un incremento de uno porciento en la osmolaridad provoca un cambio de 1 pg/ml en la concentración plasmática de AVP. La sensibilidad y, en menor grado, el umbral de la respuesta de AVP a un cambio en la tonicidad muestra una variabilidad considerable entre un individuo y otro, por lo menos parte de esta variación es hereditaria.54
3.El cambio de 1 pg/ml en la AVP aumenta la concentración urinaria alrededor de 200 mOsm/kg; por lo tanto, un cambio de 3 a 4 porciento en la osmolaridad sérica aumentará la concentración urinaria 600 a 800 mOsm/kg, o de muy diluida a con máxima concentración.
4. En la persona normal la tonicidad está balanceada en el punto medio, con cambios en cualquier dirección que alteran en forma directa la liberación de AVP y la concentración urinaria.
5. La insuficiencia cardiaca congestiva disminuye el umbral osmótico para la liberación de AVP, mientras que el envejecimiento y otros factores disminuyen la sensibilidad de la liberación de AVP (i.e., la velocidad de liberación de AVP por unidad de cambio en la osmolaridad).
6. Los cambios leves en el volumen y presión sanguíneos (i.e., de menos del 10 porciento) no afectan la liberación de AVP en forma significativa, pero cambios mayores sí. La hipotensión y la hipovolemia estimulan la liberación de AVP al disminuir el umbral osmótico, la hipertensión y la hipovolemia inhiben la liberación al aumentar el umbral. Estas influencias son mediadas por barorreceptores que tienen sus terminales aferentes en la aurícula izquierda.
7. La náusea, pero no el vómito, es un estímulo potente para la liberación de AVP, y aumenta la concentración plasmática hasta 1,000 veces el valor requerido para una antidiuresis máxima. Sin embargo, el dolor no es un estímulo importante para la liberación de AVP.
8. Muchas vías neurales influyen en la liberación de AVP en respuesta a estímulos no osmóticos. Por lo general, las vías adrenérgicas alfa estimulan y las vías adrenérgicas beta inhiben la liberación de AVP.
Las principales alteraciones en la secreción de AVP consisten en la deficiencia parcial o completa (diabetes insípida) y el síndrome de secreción excesiva inapropiada de hormona antidiurética (SIHAD). En este capítulo se analizan los síndromes de deficiencia de vasopresina.
DIABETES INSIPIDA
La poliuria es un problema clínico frecuente.55 Un paciente que tiene un gran volumen de diuresis suele tener una de tres alteraciones: una diuresis osmótica, resistencia a la vasopresina o secreción deficiente de HAD.56 La causa más común de diuresis osmótica es la glucosuria, síntoma característico de la diabetes mellitus. La resistencia a la vasopresina (i.e., diabetes insípida nefrogénica) ocurre en ciertas formas de daño renal crónico, después de la liberación de una obstrucción urinaria y como un padecimiento hereditario. Se han identificado defectos genéticos en el receptor renal de AVP.57,58 La deficiencia de HAD (i.e., diabetes insípida neurogénica) refleja una alteración funcional o estructural de las neuronas supraópticas del hipotálamo que secretan la hormona. De nuevo, en la actualidad se conocen defectos genéticos en la secreción de la AVP.59,60 Dos datos clave (inicio súbito de la poliuria y preferencia por bebidas heladas) sugieren deficiencia de HAD. Sin embargo, la diabetes insípida neurogénica debe diferenciarse de la polidipsia primaria porque la ingesta excesiva de agua también causa poliuria y supresión de la secreción de vasopresina.
Diagnóstico
Por lo general la diabetes insípida neurógenica y nefrogénica pueden distinguirse por medio de pruebas clínicas [ver figura 7]. El paciente es deprivado de agua hasta que se pierde el 3 a 5 porciento del peso corporal y la tonicidad sérica es mayor de 295 mOsm/kg. Si la poliuria desaparece y la concentración urinaria es mayor de 500 mOsm/kg, la secreción de HAD es adecuada. Si persisten la poliuria y la orina diluida (< 300 mOsm/kg), se administran 20 µg de acetato de desmopresina (DDAVP) por vía inhalada, en forma alterna, pueden administrarse 300 µU de AVP por vía intravenosa. Si la diuresis disminuye y su concentración aumenta puede inferirse que existe deficiencia de HAD. Sin embargo, si el plasma se concentra y la orina permanece diluida a pesar de la administración de DDAVP, se diagnostica diabetes insípida nefrogénica.

|
| Figura 7 |
| Diagnóstico de poliuria y polidipsia |
Deben tenerse en cuenta algunas precauciones cuando se realicen pruebas de deshidratación. Primero, el término de diabetes insípida parcial describe a un paciente que, al estar sediento, alcanza una concentración urinaria mayor que la osmolaridad del plasma pero menor que la obtenida después de la administración de hormona antidiurética. Las pruebas funcionales pueden ser confusas en pacientes con diabetes insípida neurogénica o nefrogénica parciales. En estos pacientes, que tienen concentraciones urinarias entre 300 y 500 mOsm/kg, la medición de la concentración plasmática de AVP puede ser muy útil. Una concentración alta de AVP en presencia de plasma concentrado y orina relativamente diluida indica diabetes insípida nefrogénica; un valor bajo indica deficiencia de la hormona. Por el contrario, la resistencia parcial a la vasopresina puede ser causada por polidipsia crónica, con dilución secundaria de la concentración medular en el riñón. Si estos pacientes controlan su ingesta excesiva de agua, recuperarán la concentración medular renal normal y, al mismo tiempo, una respuesta normal a la vasopresina. Por último, la deprivación de agua produce menos sed en ancianos que en jóvenes.61 Los hombres mayores de 80 años deben ser vigilados con cuidado después de la prueba para asegurarse de que ingieran cantidades adecuadas de agua.
Los granulomas, los traumatismos, las infecciones y otros procesos infiltrativos pueden producir diabetes insípida. Es raro que los tumores metastásicos causen insuficiencia en otras glándulas endócrinas, pero los tumores que se originan del pulmón, mama y otros órganos pueden producir insuficiencia en la hipófisis posterior. La sensibilidad de la IRM ha afinado en forma considerable el esquema diagnóstico de la diabetes insípida.62
La diabetes insípida puede desarrollarse en forma súbita después de un tramatismo externo o una neurocirugía. En este caso la poliuria diluida con un nivel de sodio sérico mayor de 145 mEq/L permite el diagnóstico presuntivo y debe administrarse de inmediato DDAVP parenteral.63 Por el contrario, debe esperarse hiponatremia por aumento en la secreción de AVP después de la cirugía transesfenoidal, por lo que se vigilará el sodio sérico.64
Se han notificado casos de diabetes mellitus central y de diabetes insípida súbita y fatal en mujeres jóvenes con hiponatremia posoperatoria que no fue tratada en forma agresiva. La patogenia de este padecimiento no se conoce, pero la secuencia patológica incluyó edema cerebral y herniación, compresión del tercer par craneal, infarto hipóxico de la hipófisis e hipotálamo, paro respiratorio y coma. La rapidez del deterioro en estas pacientes indica que la hiponatremia en estos casos debe ser corregida de inmediato, aunque la dilatación pupilar fija, secundaria a la compresión del nervio oculomotor, puede sugerir muerte cerebral.65
Tratamiento
Existen varios esquemas de tratamiento para la diabetes insípida.66 Si la poliuria es leve y no interfiere con el sueño, no se requiere tratamiento. La cloropropamida potencializa el efecto de la vasopresina sobre la capacidad de concentración renal y puede ser utilizada para tratar la diabetes insípida parcial. Se administra en una dosis de 250 a 375 mg una vez al día y suele no producir hipoglucemia en las personas normales. Sin embargo, si los pacientes no comen en forma regular o si tienen insuficiencia hipofisiaria anterior no sospechada, la cloropropamida puede ser peligrosa.
Para los pacientes con diabetes insípida severa, el tanato de vasopresina en aceite, 5 unidades cada 2 a 4 días por vía intramuscular, proporciona un control excelente de la poliuria y la polidipsia. Es conveniente permitir que el efecto antidiurético desaparezca y se presente poliuria antes de administrar la siguiente dosis, si no la hiponatremia es una complicación frecuente. Esta precaución es importante, en especial después de una neurocirugía, en la que puede ocurrir una secuencia trifásica de deficiencia de AVP, exceso de AVP y nuevamente deficiencia. El tratamiento estándar previo de la diabetes insípida ha sido remplazado por la administración intranasal u oral de DDAVP. La administración intranasal de DDAVP en el tratamiento de la diabetes insípida es eficaz, no tóxica y no irritativa. Las tabletas de DDAVP, en dosis de 0.1 a 0.2 mg una a tres veces al día, es el tratamiento más novedoso para la diabetes insípida. Todos los pacientes con diabetes insípida deben ser advertidos de que en circunstancias de pérdida extrema de agua o en momentos de inconciencia están expuestos a un riesgo importante, a menos de que sean atendidos por un médico que sepa que padecen esta enfermedad.
Síndromes de neoplasias endócrinas múltiples
Varios síndromes hereditarios afectan a múltiples glándulas endócrinas en distintos patrones.67 La neoplasia endócrina múltiple tipo I (NEM I) incluye diversas alteraciones de la hipófisis anterior, las paratiroides y los islotes pancreáticos. Las familias afectadas muestran una alta incidencia de úlcera péptica y una frecuencia irregular y baja de otras lesiones somáticas y endócrinas. Las siguientes lesiones son las principales alteraciones endócrinas:
1. Hipófisis: adenomas o hiperplasia de las células acidófilas o cromófobas, con acromegalia, hiperprolactinemia o hipopituitarismo. La función endócrina puede ser normal.
2. Paratiroides: se han notificado adenomas, adenomas múltiples, hiperplasia y carcinoma de las paratiroides, el hiperparatiroidismo es la alteración endócrina más frecuente en los pacientes con NEM I. La afección paratiroidea suele frustrar los esfuerzos del patólogo y del cirujano para distinguir entre los adenomas y la hiperplasia, porque ambos padecimientos pueden ocurrir en la misma familia.
3. Páncreas: pueden ocurrir adenomas, hiperplasia y carcinomas en cualquiera de las células de los islotes pancreáticos. Los síndromes endócrinos resultantes incluyen hiperinsulinismo, hipergastrinemia (se produce un síndrome de Zollinger-Ellison), cólera pancreática y otras alteraciones menos bien definidas.
La NEM I se hereda en forma autosómica dominante, pero sólo en raras ocasiones se manifiesta antes de la pubertad. El hiperparatiroidismo es la manifestación endócrina espontánea más frecuente, aunque algunas series indican que el escrutinio prospectivo de los familiares de primer orden permite detectar lesiones pancreáticas o hipofisiarias antes del comienzo del hiperparatiroidismo.68,69 Sin embargo, no se sabe si estas series son típicas.
La genética y las bases moleculares de la NEM I son especialmente interesantes. La expresión del rasgo varía mucho entre los individuos y familias. De acuerdo con la teoría de dos etapas de la carcinogénesis de Knudson,70 los cánceres hereditarios son ocasionados por una mutación en una célula germinal que crea la predisposición, y una mutación en una célula somática que origina la manifestación clínica. La comparación entre las secuencias de ADN de los tumores paratiroideos y pancreáticos con las de los leucocitos de sangre periférica y otros tejidos de pacientes con NEM I demuestra pérdida de un segmento de la banda q13 del cromosoma 11 en los tumores.71-73 Se considera (aunque no ha podido demostrarse con las técnicas actuales) que la lesión hereditaria es una mutación en el locus q13 en un miembro del par cromosómico 11 y que este defecto existe en todos los tejidos del organismo [ver figura 8]. Al parecer, el gen en el locus q13 del cromosoma 11 codifica un producto supresor de tumores. Cuando se pierde el locus q13 del segundo miembro del par cromosómico 11, el cambio produce pérdida de la heterocigocidad, y la deficiencia homocigota de la proteína supresora de tumores permite que se desarrollen cánceres. A diferencia de los tumores de la NEM II, los tumores en los pacientes con NEM I no requieren de una hiperplasia subyacente para su aparición. Sin embargo, la presencia de hiperplasia paratiroidea y de mitógenos paratiroideos en algunos pacientes con NEM I parecen sugerir que, como en la NEM II, la hiperplasia precede a la neoplasia. También es de interés que algunos adenomas paratiroideos aislados muestran el mismo defecto clonal en el segmento q13 del cromosoma 11.74

|
| Figura 8 |
| NEM I: mutación de línea germinal y mutación somática |
La aparición tardía y las manifestaciones inespecíficas de la NEM I originan una pregunta clínica muy importante: ¿Debe considerarse la búsqueda de otros signos en un paciente que tenga alguna de las características clínicas de la NEM I? Esta investigación debe ser mínima si el índice de sospecha no es alto. Por ejemplo, en un paciente con un adenoma paratiroideo curado por cirugía existen pocos motivos para realizar evaluaciones adicionales. Sin embargo, si se encuentra hiperplasia paratiroidea, existe afección de otra glándula endócrina o la historia familiar incluye un segundo individuo afectado, deberá realizarse un estudio formal de otras glándulas y de los otros miembros de la familia. Los familiares de primer orden tienen un 50 porciento de riesgo de la enfermedad. Deberá realizarse un pedigree detallado, informando a todos los familiares del riesgo. Los enfoques de investigación varían, pero el autor sugiere medir por lo menos las concentraciones séricas de calcio, prolactina y glucosa. Estas pruebas deberán repetirse en intervalos de una a dos veces por año, comenzando desde los 15 años de edad. La detección temprana permite realizar un tratamiento más eficaz y disminuir la morbilidad de la familia.
Aunque el gen de la NEM I no ha sido clonado, la asociación genética con secuencias en el cromosoma 11 permite realizar estudios predictivos en las personas afectadas.75-77 Este enfoque es tanto más importante como presionante en la NEM II, pero en las familias con NEM I los familiares afectados pueden detectarse con el seguimiento adecuado.
La ausencia de historia familiar no obvia la investigación en busca de una NEM I. Las mutaciones de novo son responsables de casi el 50 porciento de los casos de este padecimiento. Cuando ocurren casos espontáneos, solo los hijos de los pacientes, y no los padres o hermanos, tienen mayor riesgo de NEM I.
Por último, puede ocurrir NEM I en asociación con el síndrome de Zollinger-Ellison, ya sea en un individuo o en una familia afectada. Si la historia familiar sugiere síndrome de Zollinger-Ellison, deberá incluirse la gastrina sérica en el escrutinio. La hipercalcemia del hiperparatiroidismo puede exagerar los síntomas y las alteraciones secretoras del síndrome de Zollinger-Ellison.
La neoplasia endócrina múltiple de tipo II, o NEM II ( que incluye feocromocitomas, carcinomas medulares de tiroides y adenomas o hiperplasia paratiroidea), se analiza en otra parte de la obra.
Interacciones entre el sistema endócrino y el inmunológico
Existen muchas interacciones entre el sistema inmunológico y el endócrino. El cortisol es una sustancia antinflamatoria importante y su producción constituye una de las principales respuestas del organismo al estrés. Tanto la hormona liberadora de corticotropina como la AVP tienen efectos proinflamatorios, pero ambas causan mayor producción de cortisol. Por lo menos cuatro citocinas influyen en la inflamación aguda: la interleucina-1 (IL-1), la IL-6, el factor de necrosis tumoral-a y el antagonista del receptor de IL-1 (IL-1ra). Las primeras tres son proinflamatorias, pero el IL-1ra es un freno a la respuesta inflamatoria. El efecto neto de las citocinas y las respuestas relacionadas consiste en inducir un estado del 'todo enfermo', una condición de alteración eutiroidea, hipogonadal, diabetogénica que se manifiesta más por alteraciones de laboratorio que por un reto terapéutico clínicamente significativo. La importancia de estos cambios y de las interacciones inmuno-endócrinas se estudia con gran interés.78,79
Figura 1 Carol Donner.
Figuras 2 y 3 Alan Iselin.
Figuras 4 y 8 Tom Moore.
Figura 5 Al Miller.
Figura 6 Hank Iken.
Bibliografía
- Molitch ME: Neuroendocrinology. Endocrinology and Metabolism, 3rd ed. Felig P, Baxter JD, Frohman LA, Eds. McGraw-Hill, New York, 1995, p 221
- Veldhuis JD: Pulsatile hormone release as a window into the brain's control of the anterior pituitary gland in health and disease: implications and consequences of pulsatile luteinizing hormone secretion. Endocrinologist 4:454, 1994
- Conn PM, Crowley WF Jr: Gonadotropin-releasing hormone and its analogs. Annu Rev Med 45:391, 1994 [PMID 8198390]
- Frohman LA: Diseases of the anterior pituitary. Endocrinology and Metabolism, 3rd ed. Felig P, Baxter JD, Frohman LA, Eds. McGraw-Hill, New York, 1995, p 289
- Corticotropin-releasing hormone (CRH) in the differential diagnosis of Cushing's syndrome. Endocrinologist 7(suppl):1S, 1997
- Orth DN: Cushing's syndrome. N Engl J Med 332:791, 1995
- Blevins LS Jr, Christy JH, Khajavi M, et al: Outcomes of therapy for Cushing's disease due to adrenocorticotropin-secreting pituitary macroadenomas. J Clin Endocrinol Metab 83:63, 1998
- Young WF Jr, Scheithauer BW, Kovacs KT, et al: Gonadotroph adenoma of the pituitary gland: a clinicopathologic analysis of 100 cases. Mayo Clin Proc 71:649, 1996 [PMID 8656706]
- Yen SSC: Female hypogonadotropic hypogonadism: hypothalamic amenorrhea syndrome. Endocrinol Metab Clin North Am 22:29, 1993
- Waldstreicher J, Seminara SB, Jameson JL, et al: The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. J Clin Endocrinol Metab 81:4388, 1996
- Vance ML: Hypopituitarism. N Engl J Med 330:1651, 1994
- Dash RJ, Gupta V, Suri S: Sheehan's syndrome: clinical profile, pituitary hormone responses and computed sellar tomography. Aust NZ J Med 23:26, 1993
- Vance ML: The empty sella. Curr Ther Endocrinol Metab 6:38, 1997
- . Constine LS, Woolf PD, Cann D, et al: Hypothalamic-pituitary dysfunction after radiation for brain tumors. N Engl J Med 328:87, 1993 [PMID 8416438]
- Shalet SM: Radiation and pituitary dysfunction (editorial). N Engl J Med 328:131, 1993
- Sklar CA, Constine LS: Chronic neuroendocrinological sequelae of radiation therapy. Int J Radiat Oncol Biol Phys 31:1113, 1995 [PMID 7713777]
- Reversible hypopituitarism (editorial). Lancet 337:276, 1991
- Consensus guidelines for the diagnosis and treatment of adults with growth hormone deficiency: summary statement of the Growth Hormone Research Society workshop on adult growth hormone deficiency. J Clin Endocrinol Metab 83:379, 1998
- Carroll PV, Christ ER, Bengtsson BA, et al: Growth hormone deficiency in adulthood and the effects of growth hormone replacement: a review. J Clin Endocrinol Metab 83:382, 1998
- Mannakkara JV, Datta-Chaudhuri M: Recognizing pituitary insufficiency in the elderly. J Am Geriatr Soc 39:273, 1991 [PMID 2005342]
- Shalet SM, Toogood A, Rahim A, et al: The diagnosis of growth hormone deficiency in children and adults. Endocr Rev 19:203, 1998 [PMID 9570037]
- Bardin CW, Swerdloff RS, Santen RJ: Androgens: risks and benefits. J Clin Endocrinol Metab 73:4, 1991
- Riedel M, Brabant G, Rieger K, et al: Growth hormone therapy in adults: rationales, results, and perspectives. Exp Clin Endocrinol 102:273, 1994 [PMID 7813598]
- Bates AS, Van't Hoff W, Jones PJ, et al: The effect of hypopituitarism on life expectancy. J Clin Endocrinol Metab 81:1169, 1996 [PMID 8772595]
-
Bouillanne O, Rainfray M, Tissandier O, et al: Growth
hormone therapy in elderly people: an age delaying drug? Fund Clin Pharm
10:416, 1996
-
Levy A, Lightman SL: The pathogenesis of pituitary adenomas.
Clin Endocrinol (Oxf) 38:559, 1993 [PMID
8334742]
-
King JT Jr, Justice AC, Aron DC: Management of incidental
pituitary microadenomas: a cost-effective analysis. J Clin Endocrinol Metab
82:3625, 1997 [PMID
9360517]
-
Wilson CB: Surgical management of pituitary tumors. J Clin
Endocrinol Metab 82:2381, 1997
-
Tran LM, Blount L, Horton D, et al: Radiation therapy of
pituitary tumors: results in 95 cases. Am J Clin Oncol 14:25, 1991 [PMID
1987733]
-
Arafah BM, Kailani SH, Nekl KE, et al: Immediate recovery
of pituitary function after transsphenoidal resection of pituitary macroadenomas.
J Clin Endocrinol Metab 79:348, 1994
-
Bills DC, Meyer FB, Laws ER Jr, et al: A retrospective
analysis of pituitary apoplexy. Neurosurgery 33:602, 1993 [PMID
8232799]
-
Fraioli B, Esposito V, Palma L, et al: Hemorrhagic pituitary
adenomas: clinicopathological features and surgical treatment. Neurosurgery
27:741, 1990 [PMID
2259404]
-
Glick RP, Tiesi JA: Subacute pituitary apoplexy: clinical
and magnetic resonance imaging characteristics. Neurosurgery 27:214, 1990
[PMID
2385339]
-
Matthews LS: Molecular biology of growth hormone receptors.
Trends in Endo crinology and Metabolism 2:176, 1991
-
Scanlon MF, Issa BG, Dieguez C: Regulation of growth
hormone secretion. Horm Res 46:149, 1996
-
Jaffe CA, Friberg RD, Barkan AL: Suppression of growth
hormone (GH) secretion by a selective GH-releasing hormone (GHRH) antagonist:
direct evidence for involvement of endogenous GHRH in the generation of
GH pulses. J Clin Invest 92:695, 1993 [PMID
8349808]
-
Grunstein RR, Ho KY, Sullivan CE: Sleep apnea in acromegaly.
Ann Intern Med 115:527, 1991
-
Hartman ML, Pincus SM, Johnson ML, et al: Enhanced
basal and disorderly growth hormone secretion distinguish acromegalic from
normal pulsatile growth hormone release. J Clin Invest 94:1277, 1994 [PMID
8083369]
-
Ho KKY, Weissberger AJ: Characterization of 24-hour
growth hormone secretion in acromegaly: implications for diagnosis and
therapy. Clin Endocrinol (Oxf) 41:75, 1994
-
Barkan A: Controversies in the diagnosis and therapy
of acromegaly. Endocrinologist 7:300, 1997
-
Colao A, Merola B, Ferone D, et al: Acromegaly. J Clin
Endocrinol Metab 82:2777, 1997
-
Frohman LA: Acromegaly: what constitutes optimal therapy?
(editorial). J Clin Endocrinol Metab 81:443, 1996
-
Clayton RN, Wass JAH: Pituitary tumours: recommendations
for service provision and guidelines for management of patients. J R Coll
Physicians Lond 31:628, 1997 [PMID
9409495]
-
Rudman D, Mattson DE: Serum insulin-like growth factor
I in healthy older men in relation to physical activity. J Am Geriatr Soc
42:71, 1994 [PMID
8277119]
-
Hodes RJ: Frailty and disability: can growth hormone
or other trophic agents make a difference? J Am Geriatr Soc 42:1208, 1994
-
Corpas E, Harman SM, Blackman MR: Human growth hormone
and human aging. Endocr Rev 14:20, 1993 [PMID
8491152]
-
Toogood AA, O'Neill PA, Shalet SM: Beyond the somatopause:
growth hormone deficiency in adults over the age of 60 years. J Clin Endocrinol
Metab 81:460, 1996 [PMID
8636250]
-
Yazigi RA, Quintero CH, Salameh WA: Prolactin disorders.
Fertil Steril 67:215, 1997
-
Sarapura V, Sclaff WD: Recent advances in the understanding
of the pathophysiology and treatment of hyperprolactinemia. Curr Opin Obstet
Gynecol 5:360, 1993 [PMID
8329652]
-
Molitch ME, Thorner MO, Wilson C: Management of prolactinomas.
J Clin Endocrinol Metab 82:996, 1997
-
Webster J, Piscitelli G, Polli A, et al: A comparison
of cabergoline and bromocriptine in the treatment of hyperprolactinemic
amenorrhea. N Engl J Med 331:904, 1994 [PMID
7915824]
-
Brue T, Pellegrini I, Priou A, et al: Prolactinomas
and resistance to dopamine agonists. Horm Res 38:84, 1992 [PMID
1306523]
-
Robertson GL: Physiology of ADH secretion. Kidney Int
Suppl 21:S20, 1987
-
Zerbe RL, Miller JZ, Robertson GL: The reproducibility
and heritability of individual differences in osmoregulatory function in
normal human subjects. J Lab Clin Med 117: 51, 1991
-
Singer I, Oster JR, Fishman LM: The management of diabetes
insipidus in adults. Arch Intern Med 157:1293, 1997 [PMID
9201003]
-
Robertson GL: Posterior pituitary. Endocrinology and
Metabolism, 3rd ed. Felig P, Baxter JD, Frohman LA, Eds. McGraw-Hill, New
York, 1995, p 385
-
Holtzman EJ, Harris HW Jr, Kolakowski LF Jr, et al:
Brief report: a molecular defect in the vasopressin V2-receptor gene causing
nephrogenic diabetes insipidus. N Engl J Med 328:1534, 1993 [PMID
8479490]
-
Merendino JJ Jr, Spiegel AM, Crawford JD, et al: Brief
report: a mutation in the vasopressin V2-receptor gene in a kindred with
X-linked nephrogenic diabetes insipidus. N Engl J Med 328:1538, 1993 [PMID
8479491]
-
McLeod JF, Kovacs L, Gaskill MB, et al: Familial neurohypophyseal
diabetes insipidus associated with a signal peptide mutation. J Clin Endocrinol
Metab 77:599A, 1993
-
Miller WL: Molecular genetics of familial central diabetes
insipidus (editorial). J Clin Endocrinol Metab 77:592, 1993
-
Phillips PA, Rolls BJ, Ledingham JG, et al: Reduced
thirst after water deprivation in healthy elderly men. N Engl J Med 311:753,
1984 [PMID
6472364]
-
Tien R, Kucharczyk J, Kucharczyk W: MR imaging of the
brain in patients with diabetes insipidus. AJNR Am J Neuroradiol 12:533,
1991 [PMID
2058510]
-
Buonocore CM, Robinson AG: The diagnosis and management
of diabetes insipidus during medical emergencies. Endocrinol Metab Clin
North Am 22:411, 1993 [PMID
8325295]
-
Sane T, Rantakari K, Poranen A, et al: Hyponatremia
after transsphenoidal surgery for pituitary tumors. J Clin Endocrinol Metab
79:1395, 1994 [PMID
7962334]
-
Fraser CL, Arieff AI: Fatal central diabetes mellitus
and insipidus resulting from untreated hyponatremia: a new syndrome. Ann
Intern Med 112:113, 1990 [PMID
2294815]
-
Robinson AG, Verbalis JG: Diabetes insipidus. Curr
Ther Endocrinol Metab 6:1, 1997 [PMID
9174688]
-
Schimke RN: Multiple endocrine neoplasia: how many
syndromes? Am J Med Genet 37:375, 1990
-
Skogseid B, Eriksson B, Lundqvist G, et al: Multiple
endocrine neoplasia type 1: a 10-year prospective screening study in four
kindreds. J Clin Endocrinol Metab 73:281, 1991
-
Shepherd JJ: The natural history of multiple endocrine
neoplasia type 1: highly uncommon or highly unrecognized? Arch Surg 126:935,
1991
-
Knudson AG Jr, Strong LC, Anderson DE: Heredity and
cancer in man. Progress in Medical Genetics 9:113, 1973
-
Friedman E, Sakaguchi K, Bale AE, et al: Clonality
of parathyroid tumors in familial multiple endocrine neoplasia type 1.
N Engl J Med 321:213, 1989 [PMID
2568586]
-
Brandi ML: Multiple endocrine neoplasia type 1: general
features and new insights into etiology. J Endocrinol Invest 14:61, 1991
-
Beckers A, Abs R, Reyniers E, et al: Variable regions
of chromosome 11 loss in different pathological tissues of a patient with
multiple endocrine neoplasia type I syndrome. J Clin Endocrinol Metab 79:1498,
1994 [PMID
7962349]
-
Bale AE, Norton JA, Wong EL, et al: Allelic loss on
chromosome 11 in hereditary and sporadic tumors related to familial multiple
endocrine neoplasia type 1. Cancer Res 51: 1154, 1991 [PMID
1671755]
-
Thakker RV: The role of molecular genetics in screening
for multiple endocrine neoplasia type I. Endocrinol Metab Clin North Am
23:117, 1994
-
Larsson C, Nordenskjold M: Family screening in multiple
endocrine neoplasia type 1 (MEN 1). Ann Med 26:191, 1994 [PMID
7915525]
-
Skogseid B, Rastad J, Oberg K: Multiple endocrine neoplasia
type 1: clinical features and screening. Endocrinol Metab Clin North Am
23:1, 1994 [PMID
7913018]
-
Chrousos GP: The hypothalamic-pituitary-adrenal axis
and immune-mediated inflammation. N Engl J Med 332:1351, 1995
-
Reichlin S: Neuroendocrine-immune interactions. N Engl
J Med 329:1246, 1993
-
Weitzman ED: Circadian rhythms and episodic hormone
secretion in man. Annu Rev Med 27:225, 1976
-
Robertson GL, Shelton RL, Athar S: The osmoregulation
of vasopressin. Kidney Int 10:27, 1976
- Daughaday WH: The adenohypophysis. Textbook of Endocrinology, 5th ed. Williams RH, Ed. WB Saunders Co, Philadelphia, 1974