Infectología
⭳ Abrir artículo (PDF)501.9 KBEste artículo es idéntico en la Edición 2/2000.
Contenido del artículo
XXX SEPSIS
- Epidemiología
- Definición de sepsis
- Patogenia: factores microbianos
- MICRORGANISMOS CAUSALES
- PAPEL DE LA ENDOTOXINA BACTERIANA
- SUPERANTIGENOS BACTERIANOS
- OTROS MEDIADORES MICROBIANOS
- Patogenia: Mediadores derivados del huésped
- RED DE CITOCINAS
- PAPEL DEL SISTEMA DE COAGULACION
- INTERACCIONES ENTRE NEUTROFILOS Y CELULAS ENDOTELIALES
- PAPEL DEL OXIDO NITRICO
- Técnicas diagnósticas en el choque séptico
- CARACTERISTICAS GENERALES
- SINDROME DE DISFUNCION ORGANICA MULTIPLE
- TECNICAS DIAGNOSTICAS EXPERIMENTALES
- Tratamiento del choque séptico
- Tratamientos experimentales en la sepsis
XXX SEPSIS
DR. STEVEN M. OPAL
La sepsis, junto con la falla orgánica múltiple que con frecuencia acompaña al síndrome de respuesta inflamatoria sistémica (SRIS), es la principal causa de mortalidad en las unidades de cuidados intensivos.1,2 Hasta 400,000 pacientes desarrollan sepsis cada año en los Estados Unidos. Casi la mitad de estos pacientes sufren sepsis severa y choque séptico. La mortalidad del choque séptico sigue siendo alrededor de 35 a 45 porciento, a pesar de un gran esfuerzo por mejorar el tratamiento y la evolución.1-3
El choque séptico se ha convertido en un aspecto crucial de investigación en el área de los cuidados intensivos. Aunque se han logrado modestas mejoras en el pronóstico en la última década, aún se requieren inovaciones en el tratamiento del choque séptico. Este capítulo revisa algunos de los adelantos importantes logrados en el conocimiento de la fisiopatología molecular de la sepsis. Esto permite contar con una base para desarrollar estrategias de investigación que mejoren el diagnóstico y agentes terapéuticos para el manejo del choque séptico. Se analiza el tratamiento actual de la sepsis y se destacan las prioridades de investigación para el manejo futuro de los pacientes con este padecimiento.
Epidemiología
La incidencia de la sepsis ha aumentado en las últimas dos décadas.4 Es probable que esta tendencia continúe en el futuro porque la sepsis se ha convertido en una enfermedad del progreso médico. El tratamiento exitoso de diversos padecimientos médicos y quirúrgicos ha creado una gran población de pacientes con enfermedad grave y deterioro de las defensas, y estos pacientes tienen un riesgo muy alto de desarrollar sepsis. Las inovaciones en trasplante de órganos, implante de prótesis y equipos de acceso vascular de uso prolongado continúan aumentando esta población de pacientes. El envejecimiento gradual de la población en muchos países desarrollados y la mayor prevalencia de agentes microbianos resistentes a antibióticos contribuye también a la mayor incidencia del choque séptico.
Definición de sepsis
La sepsis, el choque séptico, el síndrome de respuesta inflamatoria sistémica y el síndrome de disfunción orgánica múltiple (SDOM) fueron definidos en una conferencia de consenso del American College of Chest Physicians/Society for Critical Care Medicine (ACCP/SCCM) de los EUA [ver tabla 1].5 Las definiciones toman en cuenta el dato de que la sepsis puede ser causada por múltiples agentes infecciosos y mediadores microbianos y puede o no asociarse con infección en el torrente sanguíneo. A pesar de la lógica clínica, la simplicidad intrínseca y la aceptación amplia de estas definiciones, su aplicación clínica ha sido criticada por buenos motivos.6,7 La definición de SRIS es tan amplia e inespecífica que pierde su poder de discriminación, muchos pacientes ingresados a servicios de medicina general y la mayoría de los pacientes de la UCI satisfacen la definición de SRIS. La definición actual de sepsis incluye virtualmente a cualquier persona que esté en las primeras 24 horas de un episodio de influenza porque se satisfacen los criterios de SRIS y el síndrome es causado por un agente infeccioso (un ortomixovirus). Sin embargo, los clínicos no suelen considerar la gripe como una sepsis.
Uno de los principios en los que se basan estas definiciones es que la respuesta inflamatoria en sí, y no el organismo infeccioso, es la causante del proceso séptico. La hipótesis puede ser correcta en principio, pero la naturaleza del patógeno microbiano responsable de la sepsis claramente contribuye a la evolución del paciente. Las variaciones en la susceptibilidad a las defensas del huésped, el potencial de resistencia antimicrobiana y la capacidad para generar toxinas afectan la patogenicidad del microrganismo.8 El no tomar en cuenta las diferencias intrínsecas en la virulencia microbiana limita la utilidad de las definiciones actuales de sepsis.
Existe desacuerdo sobre el nivel de reanimación con líquido necesario para asegurar que el paciente está séptico en lugar de hipovolémico. La cantidad de agente vasopresor necesario para concluir con confianza que el paciente tiene un verdadero choque séptico continúa en duda. También existe controversia por la dificultad para distinguir los padecimeintos y difunción orgánica previos de la morbilidad y disfunción orgánica inducidos por el proceso séptico en sí. Muchos pacientes con sepsis tienen disfunción orgánica severa subyacente por diversas enfermedades. El grado en el que la sepsis contribuye a una mayor alteración puede ser difícil de determinar con exactitud. Lo mismo puede decirse para el grado en que la sepsis contribuye a la mortalidad de un paciente determinado que sufre otros padecimientos graves. Todos estos factores limitan el valor discriminatorio de las definiciones de la sepsis y ponen en duda la validez de comparaciones en diferentes poblaciones de estudio, comprometiendo la capacidad para acumular datos y generalizar resultados.
Podrá ser posible definir mejor la terminología relacionada a la sepsis cuando se disponga de técnicas de diagnóstico rápido para evaluar el estado inmunológico de los pacientes sépticos. Mientras tanto, deben usarse las definiciones de consenso actuales a pesar de sus limitaciones.
Patogenia: factores microbianos
MICRORGANISMOS CAUSALES
La microbiología de la sepsis ha sufrido una transición muy importante en los últimos 25 años. Los patógenos principalmente responsables de la sepsis en los años 60 y 70 eran bacilos gram negativos y Pseudomonas aeruginosa, pero ha existido un incremento progresivo en la incidencia de sepsis causada por bacterias gram positivas9,10 y hongos oportunistas. La rápida evolución de genes resistentes a antibióticos en las bacterias gram positivas y la frecuencia de sepsis bacteriana asociada a catéteres vasculares puede ser responsable de la mayor prevalencia de patógenos gram positivos como causa de sepsis.
PAPEL DE LA ENDOTOXINA BACTERIANA
La endotoxina bacteriana, que se compone de lipopolisacáridos (LPS), es un componente intrínseco de la membrana exterior de las bacterias gram negativas y es indispensable para la viabilidad de las mismas.11 Los humanos son una de las especies más susceptibles a las potentes propiedades inmunoestimulantes de la endotoxina, que puede ser letal en dosis diminutas. Sea que la endotoxina se libere al sistema circulatorio humano en su forma libre (a partir de organismos muertos o liberada de la membrana de organismos viables) o unida a la pared celular de la bacteria intacta, se presenta una intensa respuesta inflamatoria sistémica. Parece ser que la endotoxina funciona como una molécula de alarma que alerta al huésped sobre la presencia de una bacteria gram negativa invasora.12 Es la respuesta del huésped a la endotoxina, más que ella misma, la responsable de las propiedades letales de la endotoxina.
El LPS es una macromolécula polar fosforilada que contiene elementos hidrofóbicos en los ácidos grasos de su estructura central de lípido A u elementos hidrofílicos en sus componentes polisacáridos de repetición de superficie. El LPS forma microagregados en los líquidos biológicos e interactúa en forma rápida con una variedad de proteínas lipofílicas en el suero o unidas a la membrana. En los humanos existen tres receptores conocidos para el LPS: (1) moléculas CD14 solubles o unidas a membranas, (2) moléculas CD11/CD18 (integrinas ß2) y (3) receptores depuradores de moléculas lípidas. Las moléculas CD14 solubles y unidas a la membrana representan el sistema más sensible para la activación de las células inmunes con la exposición a pequeñas cantidades de LPS.13
El mecanismo de señal más importante del LPS está mediado por interacciones con una proteína plasmática de fase aguda derivada del hígado conocida como proteína de unión al LPS (PUL).14 Esta actúa principalmente como una molécula transportadora que se une a los agregados de LPS polimérico y transfiere monómeros de LPS a la molécula CD14. CD14 es una proteína unida a glucosil fosfatidilinositol que se encuentra en las superficies celulares de células efectoras inmunológicas como los monocitos-macrófagos y los neutrófilos. Las evidencias actuales sugieren que el CD14 unido a la membrana no funciona en forma directa iniciando el mensaje intracelular de la presencia del LPS.15 En lugar de ello, después de unirse al CD14 unido a la membrana, el LPS es entregado a una molécula adyacente receptora del LPS (que debe aún identificarse y caracterizarse), y que origina una señal al espacio intracelular, activando los genes que responden al LPS.
Las vías de señal intracelular del LPS son un área de gran interés y no se han caracterizado del todo.16 La trasducción de señales se realiza por la activación de varios activadores intracelulares de transcripción, incluyendo el NFkB, la proteína cinasa, las proteínas G, RAS, RAF-1, MEK-1, ERK-1, tirosina cinasas y una serie de proteinas cinasas activadas por mitógenos. El LPS tiene semejanzas estructurales con otra molécula lípida de señales intracelulares, la ceramida. La ceramida actúa a través de la cinasa activada de ceramida. El LPS semeja a la ceramida en que también activa a esta cinasa intracelular, lo que puede contribuir a la transducción de señales del mensaje del LPS.17
El resultado final de la acción del LPS consiste en estimular la síntesis de una serie grande de genes inducibles por LPS en las células inflamatorias. Estos genes incluyen las citocinas proinflamatorias y la sintetasa de óxido nítrico. La salida de citocinas proinflamatorias y de otros mediadores inflamatorios después de la exposición a LPS contribuye al SRIS y es crucial en la patogenia del choque séptico inducido por bacterias gram negativas.12
La actividad del LPS es modulada por diversos componentes derivados del huésped. Los niveles de PUL en la circulación varían entre las personas y en diferentes estados de inflamación. La PUL es una proteína de fase aguda y sus niveles pueden aumentar siete veces durante la inflamación aguda.18 La importancia de la PUL en la fisiopatología del choque inducido por endotoxina se demuestra con claridad en ratones sin PUL que son resistentes a los efectos letales de la endotoxina bacteriana.19 Los ratones que tienen deficiencia genética de CD14 también son resistentes a la endotoxina.20 Existen ciertas evidencias de que los pacientes con niveles más altos de PUL tienen mayor riesgo de morir por choque inducido por endotoxina, comparado con los que tienen niveles bajos de PUL.20a
Otra proteína fijadora de endotoxina importante que se encuentra en el plasma humano es la proteína bactericida/aumentadora de permeabilidad (PBAP). Esta proteína tiene 456 aminoacidos, es producida por los neutrófilos humanos y se encuentra en mayor cantidad en los gránulos azurófilos (primarios).18 Tiene un 45 porciento de homología en sus aminoácidos con la PUL, pero su función con respecto al LPS es opuesta. La PUL facilita la unión del LPS al CD14, activando a las células mientras que la PBAP tiene el efecto fisiológico opuesto, inhibe la llegada del LPS a CD14. la PBAP funciona como un antagonista molecular y compite con la PUL por la unión al LPS en los líquidos biológicos .18,21 Las concentraciones relativas de estas dos proteínas fijadoras de endotoxina determinan el efecto neto de la liberación del LPS.
En el plasma humano la concentración de PUL es de dos o tres órdenes de magnitud mayor que la de PBAP. Lo opuesto parece suceder en los abscesos, en donde existe PBAP en mucho mayor cantidad que PUL.18 Esto favorece la función activadora de LPS en el plasma y la inhibitoria en los abscesos. Por lo tanto, la PBAP funciona como una molécula antiendotoxina endógena y puede ser un componente del tratamiento de la lesión inducida por endotoxina.22
Otra proteína endógena del plasma que funciona para regular la actividad del LPS en la circulación es la lipoproteína de alta densidad (LAD). El LPS se une con gran afinidad a las regiones hidrofóbicas de la LAD, y este complejo es eliminado en forma eficaz de la circulación por mecanismos de depuración hepática.23 Los ratones transgénicos que expresan grandes cantidades de apolipoproteína A-1 (la principal proteína que constituye la LAD) son menos susceptibles a la toxicidad por endotoxina que los ratones normales.24 La administración de LAD reconstituyente es un tratamiento potencial para los trastornos relacionados con la endotoxina en la sepsis.25
El fenómeno de tolerancia a endotoxina ha sido bien caracterizado en modelos animales de sepsis y es probable que también ocurra en humanos.26 La tolerancia a la endotoxina se describe como una desensibilización a la letalidad inducida por ésta sustancia al administrar primero una dosis pequeña de endotoxina y después una dosis letal. Este fenómeno parece estar mediado prncipalmente a nivel de transcripción, por regulación inhibitoria de los genes de las citocinas proinflamatorias, pero los mecanismos precisos se desconocen. La dosis de desensibilización de la endotoxina puede inducir esteroides endógenos o citocinas antinflamatorias (v.gr., interleucina-10 [IL-10]), disminuir la expresión de receptores de citocinas en la superficie celular, alterar la traslocación nuclear de moléculas de trasducción de señales o disminuir la estabilidad del ARN mensajero de citocinas.
El significado de la tolerancia a la endotoxina en humanos no es claro pero se ha observado cierto grado de tolerancia en pacientes tratados con lípido A monofosforilado.26 Sin duda, la endotoxina es un mediador importante en la patogenia del choque séptico. Por lo tanto, los esfuerzos para limitar la síntesis del LPS, prevenir la activación de los elementos de respuesta inmunológica del huésped y aumentar la depuración del LPS son estrategias terapéuticas importantes para el manejo futuro del choque séptico.
SUPERANTIGENOS BACTERIANOS
Otros mediadores microbianos importantes en la patoenia del choque séptico son los superantígenos bacterianos. Los superantígenos son un grupo diverso de exotoxinas proteicas derivadas de microrganismos estreptococos, estafilococos y otros patógenos que comparten una propiedad inmunológica poco habitual, los superantígenos pueden activar gran número de células T CD4+ en un periodo breve saltándose el mecanismo habitual de procesamiento y presentación del antígeno.27
Los antígenos bacterianos convencionales son internalizados por células presentadoras de antígeno (CPA), en donde sufren proteólisis limitada y procesamiento dentro del componente endosómico del macrófago. Las secuencias peptídicas apropiadas de los antígenos microbianos (epítopes) se insertan en el surco central de las moléculas de clase II del complejo principal de histocompatibilidad (CPH) y después son expresadas en la superficie celular de las CPA. Entonces se activan células T CD4+ específicas que reconocen el epítope. La expansión clonal de este subgrupo pequeño de células T causa una respuesta inmune fisiológica al nuevo antígeno introducido. Por el contrario, los superantígenos no requieren del procesamiento intracelular por la CPA. Se unen en forma directa a los antígenos de clase II en un sitio adyacente al surco específico para el epítope, y también se unen a un número limitado de regiones Bß del receptor de la célula T en las células T CD4+. Esta unión aproxima las células T CD4+ y los macrófagos, lo que activa a ambas poblaciones celulares. Mientras que un péptido antigénico convencional estimula solo alrededor de uno en 105 linfocitos circulantes que pueden reconocer este epítope estructural único, un superantígeno (v.gr., toxina-1 del síndrome de choque tóxico de Staphylococcus aureus, que se une a la región Vß2 de las células T) puede reconocer hasta una de cada 10 linfocitos humanos circulantes. Por lo tanto, un solo superantígeno bacteriano puede activar hasta al 10 porciento de toda la población linfocitaria.28 Esto causa activación excesiva tanto de linfocitos como de macrófagos, y ocurre síntesis y liberación de citocinas proinflamatorias en forma incontrolable. La actvación inmune inducida por superantígenos puede terminar en choque séptico si el proceso no se limita. Las infecciones polimicrobianas que liberan tanto superantígenos bacterianos como endotoxina pueden ser especialmente dañinos para el huésped, la toxicidad de la endotoxina bacteriana puede aumentar mucho por los superantígenos que activan al sistema inmune para que reaccione a la endotoxina con mayor sensibilidad [ver figura 1].
OTROS MEDIADORES MICROBIANOS
El peptidoglucano de la pared celular de la bacteria, los antígenos capsulares, las toxinas microbianas y las sustancias procoagulantes producidas por patógenos microbianos pueden contribuir a la patogenia de la sepsis. Se ha observado que los peptidoglucanos, como el ácido teicoico y otros componentes de las bacterias gram positivas, pueden interactuar con moléculas CD14 y activar células inflamatorias en una forma semejante a la de la endotoxina bacteriana.29 Puede ocurrir activación dependiente de CD14 de las células mononucleares con componentes de bacterias gram positivas, aunque el nivel de activación es cuantitativamente menor que el observado con los componentes de bacterias gram negativas.30
Las bacterias gram positivas y los hongos pueden inducir hipotensión sistémica, con redistribución del flujo sanguíneo y vasoconstricción esplácnica. La isquemia y la reperfusión de los vasos sanguíneos que irrigan el tubo digestivo puede alterar la barrera mucosa intestinal contra los productos bacterianos. Ocurre entonces traslocación de patógenos microbianos intactos y de endotoxina bacteriana durante los periodos de estrés severo y de hipoperfusión de la mucosa gastrointestinal.31 La endotoxina bacteriana puede tener un papel patogénico en el proceso inflamatorio después de la hipotensión sistémica producida por lesiones infecciosas y no infecciosas. Por lo tanto, los estados de choque pueden ser potencializados y mantenidos por la liberación continua de endotoxina y otros mediadores microbianos de la luz del tubo digestivo. Estos hallazgos han iniciado el interés por fortalecer la mucosa intestinal a través de inmunonutrición, factores de crecimiento epitelial y descontaminación selectiva del tubo digestivo en enfermedades graves. Estas opciones de tratamiento pueden ser potencialmente viables y son motivo de intensa investigación dentro del manejo de la sepsis.
Patogenia: Mediadores derivados del huésped
RED DE CITOCINAS
Las citocinas proinflamatorias juegan un papel crucial en la patogenia de la sepsis. En estudios en animales, la adminitración de factor de necrosis tumoral-alfa (FNT-alfa), una proteína endógena derivada de monocitos-macrófagos, ha demostrado tener consecuencias letales, y se observaron dramáticos cambios hemodinámicos, metabólicos y hematológicos después de la administración de FNT-alfa a voluntarios humanos.32,33 También se han demostrado los efectos dañinos de los niveles sistémicos de la IL-1ß.34
Las principales citocinas proinflamatorias, FNT-alfa e IL-1ß. inducen sus efectos hemodinámicos y metabólicos en conjunto con un grupo amplio de mediadores proinflamatorios derivados del huésped que actúan en forma coordinada para producir la respuesta inflamatoria sistémica [ver figura 1 y tabla 2].35 El sistema de citocinas funciona como una red de comunicación de señales entre neutrófilos, monocitos, macrófagos y células endoteliales. La activación autócrina y parácrina causa potencialización sinérgica de la respuesta inflamatoria una vez que ésta es activada por un insulto microbiano sistémico (v. gr., endotoxemia). Mucha de la respuesta proinflamatoria es localizada y compartamentalizada en la región de la inflamación inicial (v.gr., tejido pulmonar o tubo digestivo). Si no se limita, la respuesta inflamatoria se disemina a la circulación sistémica, causando una reacción generalizada y culminando en lesión endotelial difusa y choque séptico. El efecto seudoendócrino de la liberación sistémica de citocinas y quimocinas difunde el proceso inflamatorio en todo el organismo.1-3
La gran cantidad de citocinas y quimocinas inflamatorias que se encuentran en cantidades excesivas en el torrente sanguíneo en el choque séptico es impresionante, y se equipara con un grupo igualmente enorme de mediadores antinflamatorios [ver tabla 2]. Los mediadores proinflamatorios tienden a predominar en la fase temprana de la sepsis (las primeras 12 a 24 horas), mientras que los componentes endógenos antinflamatorios suelen prevalecer en las fases tardías.36 Se ha observado que los ratones deficientes en células T y B responden al reto con endotoxina de la misma forma que los ratones normales.37 Por lo tanto, las citocinas generadas por monocitos y macrófagos son suficientes para desencadenar el proceso séptico inicial. Sin embargo, las citocinas derivadas de linfocitos e interferones son importantes en la regulación de las fases tardías de la sepsis y pueden determinar la evolución final en el choque séptico.
Papel de las células T CD4+
Existen importantes diferencias funcionales entre las células T CD4+. Las células T CD4+ activadas, pero no comprometidas (células THO) siguen dos vías principales de diferenciación funcional. Las células T expuestas a IL-12, en presencia de IL-2, desarrollan un tipo funcional TH1. Estas células producen grandes cantidades de interferón gama, FNT-alfa e IL-2 y promueven una respuesta inmune proinflamatoria mediada por células. Las células T CD4+ aún no comprometidas expuestas a IL-4 se desarrollan hacia un fenotipo TH2, que secreta IL-4, IL-10 e IL-13. Estas citocinas promueven respuestas inmunes humorales y atenúan la actividad de macrofagos y neutrófilos.
Es interesante que las citocinas elaboradas por las TH1 suprimen la expresión de las citocinas de tipo TH2; por ejemplo, el interferón gama inhibe la síntesis de IL-10. Por el contrario, la IL-10 de las TH2 es un inhibidor potente de la síntesis de FNT-alfa y de interferón gama por las células TH1. El tipo de respuesta linfocitaria inicial es crucial porque el sistema tiende a polarizarse con el tiempo hacia un predominio TH2 oTH1.38 Este proceso es importante porque la sepsis suele acompañarse de una respuesta tardía tipo TH2 después de la lesión séptica inicial. La respuesta de hormonas de estrés en el choque séptico, con expresión de hormona adrenocorticotrópica, esteroides y catecolaminas, promueve una respuesta TH2 después de la lesión sistémica. Esto puede causar una fase de refractariedad inmune relativa (parálisis inmune) en la que el paciente puede tener mayor riesgo de infección bacteriana o micótica secundaria.39 Este estado fisiopatológico se asocia con tolerancia a endotoxina, síntesis de citocinas antinflamatorias y desactivación de monocitos, macrófagos y neutrófilos. Los métodos para detectar este estado de inmunosupresión y restablecer la competencia inmune aún son motivo de investigación. Los pacientes con menor expresión de antígenos de clase II del CPH, como el HLA-DR, en la superficie de los macrófagos, pueden tener un estado de inmunosupresión funcional y beneficiarse con el tratamiento con interferón gama.40
PAPEL DEL SISTEMA DE COAGULACION
La activación de la cascada de la coagulación y la generación de una coagulopatía por consumo y de microtrombos difusos es una complicación bien reconocida de la sepsis severa. El reto con endotoxina y con FNT en voluntarios humanos normales indica que la vía extrínseca es el mecanismo principal por el que el sistema de coagulación se activa en la sepsis humana.41,42 Los factores de contacto de la vía intrínseca se activan también, y en forma secundaria inician la vasodilatación por generación de bradicinina.42 La activación de coagulación intravascular causa microtrombos y puede contribuir a la falla orgánica múltiple que se observa en la sepsis. La depleción de los factores de coagulación y la activación de plasmina, antitrombina III y proteína C activada pueden causar diátesis hemorrágica. La reducción de estos anticoagulantes endógenos provoca en forma secundaria un estado procoagulante que implica mal pronóstico.43 El interés actual en la administración de inhibidor de la vía del factor tisular, proteína C activada y antitrombina III para el tratamiento de la sepsis se basa en el posible valor terapéutico de regular el sistema de la coagulación.44
INTERACCIONES ENTRE NEUTROFILOS Y CELULAS ENDOTELIALES
El reclutamiento de neutrófilos en un área de infección local es un componente esencial en la respuesta inflamatoria del huésped. La lcoalización y erradicación de los patógenos invasores en el sitio de la infección inicial es el principal objetivo de la respuesta inmunológica contra los microrganismos patógenos. Este proceso fisiológico puede volverse dañino si ocurren interacciones difusas entre neutrófilos y células endoteliales en toda la circulación en respuesta a la inflamación sistémica.
Los mecanismos responsables de la migración de neutrófilos del espacio intravascular hacia el intersticio, en donde residen los microrganismos invasores, son complejos [ver figura 2]. Los neutrófilos activados degranulan y exponen las superficies endoteliales y estructuras circundantes para reactivar intermediarios del oxígeno, óxido nítrico y diversas proteasas. Este proceso contribuye no solo a la depuración de microrganismos, sino también a la lesión endotelial difusa en el caso de respuestas inflamatorias generalizadas. La regulación de la actividad de los neutrófilos puede representar un área nueva para la intervención terapéutica en la sepsis.45
PAPEL DEL OXIDO NITRICO
El óxido nítrico es un radical libre muy reactivo que tiene un papel esencial en la fisiopatología del choque séptico. Su vida media es muy corta (1 a 3 segundos), lo que limita su actividad a tejidos locales, en donde es generado por una de tres isoformas de la sintetasa de óxido nítrico. La regulación de la sintetasa de óxido nítrico es compleja. La expresión completa de la misma requiere de FNT-alfa, IL-1, LPS y quizá de otros elementos de control.
El óxido nítrico es el principal factor de relajación derivado del endotelio, e inicia la vasodilatación y la hipotensión sistémica observada en el choque séptico. El óxido nítrico activa a la guanilato ciclasa, que aumenta los niveles de monofosfato cíclico de guanosina dentro de las células del músculo liso vascular. Esto produce vasodilatación sistémica y reducción de la resistencia vascular. Sin embargo, en minutos después de la administración de un inhibidor de la síntesis de óxido nítrico la presión arterial de los pacientes hipotensos con choque séptico regresa a los niveles normales.46
Los otros efectos fisiológicos importantes del óxido nítrico en el choque séptido con la mayor destrucción intracelular y la regulación de la adherencia de plaquetas y neutrófilos. El óxido nítrico es un gas que difunde con facilidad y que no requiere de receptores específicos para entrar a las células eucariotas o procariotas. En presencia del anión superóxido forma peroxinitrito. El peroxinitrido se descompone en moléculas muy citotóxicas, los radicales hidroxilo y cloruro de nitrosil que, a su vez, inician la peroxidación de lípidos y provocan daño celular irreversible. El óxido nítrico inhibe diversas enzimas clave en la vía del ácido tricarboxílico, la vía glucolítica, los sistemas de reparación del ADN, las vías de transporte de electrones y las vías de intercambio de energía. Debido a su potente reactividad, el óxido nítrico altera la función de muchas metaloenzimas, proteínas transportadoras y elementos estructurales.
Ha sido aparente que el óxido nítrico, como muchos otros componentes de la respuesta inflamatoria del huésped, puede tener propiedades tanto útiles como dañinas en la sepsis. Regula la microcirculación a los órganos vitales y contribuye a la destrucción intracelular de microrganismos patógenos. Sin embargo, la liberación excesiva y prolongada de óxido nítrico causa la vasodilatación generalizada e hipotensión sistémica en el choque séptico. El óxido nítrico se ha vuelto un blanco importante en las estrategias terapéuticas de la sepsis, y en la actualidad se realizan estudios clínicos para determinar la eficacia protectora de los inhibidores de esta sustancia.46 Por ejemplo, se ha demostrado que los inhibidores no selectivos de la sintetasa de óxido nítrico mejoran la hemodinamia de los pacientes sépticos.47
Técnicas diagnósticas en el choque séptico
CARACTERISTICAS GENERALES
En su tratado clásico de naturaleza humana (The Prince, cerca de 1505), Maquiavelo establece, "La fiebre éctica [i.e., sepsis según las definiciones actuales] es difícil de reconocer en su inicio pero fácil de tratar; si no se atiende se vuelve fácil de reconocer pero difícil de tratar." Esta afirmación es tan cierta ahora como lo era hace 500 años. El choque séptico ya establecido es un síndrome clínico muy aparente que rara vez se confunde con otros estados patológicos. Sin embargo, las fases iniciales pueden ser muy sutiles, incluso en pacientes vigilados con cuidado. Los signos y síntomas tempranos pueden incluir confusión, aprehensión o deterioro del estado de alerta. Aunque la fiebre es característica, puede observarse hipotermia, que implica un mal pronóstico. La reducción inexplicable en la diuresis, el inicio súbito de ictericia colestática, la alcalosis metabólica inexplicable, la hemorragia excesiva en sitios de venopunción o incluso la hipotensión súbita inexplicable pueden ser el dato inicial del choque séptico. Es indispensable que el clínico detecte estos signos y síntomas tempranos porque el tratamiento exitoso del choque séptico depende de su detección temprana e intervención apropiada.
En el choque séptico se detectan diversas alteraciones clínicas, de laboratorio y hemodinámicas [ver tablas 3 y 4]. Por desgracia, no existe una sola prueba clínica o de laboratorio que sea suficientemente sensible y específica para confirmar con certeza el diagnóstico. Los hemocultivos pueden o no ser positivos, se observa leucocitosis o neutropenia, hiperglucemia o hipoglucemia y alcalosis respiratoria o acidosis metabólica. Es el conjunto de signos y síntomas lo que origina el diagnóstico de choque séptico.
Los datos hemodinámicos más comunes en el choque séptico temprano son un gasto cardiaco alto con resistencias vasculares sistémicas bajas, con mantenimiento inicial de la presión arterial sistólica al intentar el corazón compensar por la pérdida de tono vascular sistémico. La función miocárdica está disminuida incluso en las fases tempranas del choque séptico.48 Sin una intervención adecuada el volumen de sangre circulante se pierde en forma continua hacia el espacio intersticial y los sitios intracelulares. El corazón no puede compensar suficiente y ocurre hipotensión sistólica. El deterioro de la función miocárdica asociada con vasoconstricción difusa marca el estado refractario tardío del choque séptico.
El choque séptico puede asociarse con pérdida de la autorregulación normal entre el aporte de oxígeno y su consumo en los tejidos. La disoxia dependiente del aporte puede contribuir a la lesión tisular en la falla multiorgánica de la sepsis.49 Los intentos por aumentar el aporte de oxígeno a niveles supranormales, aunque factibles en teoría, no han aumentado la supervivencia en estudios clínicos.50 Quizá el uso más juicioso de aporte de oxígeno a los tejidos podrá beneficiar a los pacientes en choque séptico.
SINDROME DE DISFUNCION ORGANICA MULTIPLE
Una de las características del choque séptico es el desarrollo de disfunción orgánica múltiple. El desarrollo de falla orgánica al inicio o durante la evolución de la sepsis es un factor de mal pronóstico y es el principal determinante de la evolución [ver tabla 5].51 La lesión endotelial difusa que acompaña al choque séptico causa disfunción orgánica distante al sitio original de la lesión. Se supone que la señal que provoca el daño endovascular difuso depende de factores del plasma (v.gr., citocinas proinflamatorias, complemento, cininas y otros mediadores inflamatorios derivados del huésped) o por un elemento celular encontrado en uno o más de las células inmunes efectoras.
El aporte sanguíneo inadecuado a los tejidos vitales produce SDOM. La falla de la microcirculación para mantener a los tejidos puede ser resultado de hipoperfusión de los lechos capilares, redistribución del flujo sanguíneo dentro de los lechos vasculares, derivaciones arteriovenosas funcionales, obstrucción del flujo sanguíneo por microtrombos, agregados de plaquetas o leucocitos o deformación anormal de los eritrocitos. El óxido nítrico, los intermediarios reactivos del oxígeno, las citocinas proinflamatorias y los inductores de apoptosis pueden dañar en forma directa las superficies del endotelio. También el edema endotelial por el movimiento del líquido intravascular hacia los espacios extravascular e intracelulares puede obstruir en forma mecánica la luz de los lechos capilares.
Aunque el origen de la falla orgánica múltiple en la sepsis se relaciona principalmente con los efectos microvasculares, la función miocárdica y pulmonar también disminuyen durante el curso del choque séptico y pueden contribuir en forma significativa al desarrollo de la falla multiorgánica. La contractilidad miocárdica disminuye en respuesta a diversos factores depresores del miocardio encontrados en el plasma de pacientes sépticos. El FNT-alfa es una causa prominente de disfunción miocárdica, la IL-1, el óxido nítrico y otros mediadores inflamatorios derivados del huésped pueden también ser factores que contribuyen.52
La lesión pulmonar aguda ocurre como resultado del daño a la circulación vascular pulmonar y las membranas alveolocapilares. El pulmón es susceptible de lesionarse en varios estados inflamatorios sistémicos. El síndrome de insuficiencia respiratoria progresiva aguda (SIRPA) sigue siendo una causa importante de morbimortalidad en el choque séptico. Los esfuerzos por prevenir la lesión pulmonar incluyen inovaciones en el apoyo respiratorio, evitar el barotrauma y volutrauma a las unidades alveolocapilares, evitar la lesión pulmonar inducida por oxidantes, salvar las unidades alveolocapilares funcionales por medio de cambio de posición (posición prona) y manejo juicioso de líquidos.53 Han ocurrido mejoras mesurables en la evolución del SIRPA, pero aún puede protegerse mucho más la función pulmonar en el caso del choque séptico.
La falla orgánica múltiple es potencialmente revesible si se aplican medidas rápidas para corregir las alteraciones hemodinámicas e inflamatorias.
TECNICAS DIAGNOSTICAS EXPERIMENTALES
Los ensayos actuales usados para diagnosticar el choque séptico son incómodos y no útiles para el diagnóstico rápido. En la actualidad se investigan métodos mejores. La medición de niveles de endotoxina en plasma, por ejemplo, puede ser útil para el diagnóstico de sepsis y se ha demostrado que tiene cierto valor predictivo para determinar los pacientes con riesgo de desarrollar estado de choque.54 Por desgracia, los niveles de endotoxina no aumentan de modo uniforme en el choque séptico y pueden elevarse en forma falsa en infecciones por gram positivos y otros trastornos que cursan con hipotensión. La respuesta del huésped a la endotoxina es variable y no puede determinarse con base en los niveles de endotoxina circulante.
Se han medido con éxito los niveles circulantes de superantígenos bacterianos en pacientes seleccionados con síndrome de choque tóxico.55 Estas mediciones pueden ser útiles en situaciones clínicas específicas.
La IL-6 es una citocina multifuncional que tiene gran cantidad de actividades biológicas, algunas de las cuales son proinflamatorias y otras antinflamatorias. La IL-6 se ha considerado como un indicador de activación de citocinas porque se presenta en forma constante después de la activación del FNT-alfa y de la IL-1ß.56 Los pacientes con niveles elevados de IL-6 pueden responder en forma favorable a los tratamientos anticitocina.57 En varios estudios,57-60 la elevación de IL-6, así como la no disminución en el nivel de IL-6 después de iniciar el tratamiento para la sepsis, se asociaron con mala evolución. Por desgracia, los niveles de IL-6 no son específicos de sepsis y pueden aumentar en diversos estados inflamatorios e infecciosos. La falta de especificidad limita la confiabilidad de la medición de IL-6 como método diagnóstico de choque séptico.
La procalcitonina es el propéptido de la calcitonina que en condiciones fisiológicas es producido por las células C del tiroides. Una proteasa específica convierte la procalcitonina en calcitonina, katalcina y un péptido amino-terminal.61 La procalcitonina tiene atributos que le hacen un marcador potencial de sepsis. Tiene una vida media larga (alrededor de 24 horas) y aumenta desde niveles no detectables hasta más de 100 ng/ml durante el choque séptico. El origen preciso de la procalcitonina en la sepsis no es claro, pero es extratiroideo.61,62 Los niveles de procalcitocina no aumentan tan pronto como los de la IL-6 o la IL-8, sino que se observan hasta 4 a 6 horas después del reto sistémico con endotoxina u otro estímulo séptico.62
Es interesante que los niveles de procalcitonina se elevan en la sepsis severa, pero no en las infecciones localizadas.62 Esto indica que la procalcitonina puede ser un marcador para la respuesta inflamatoria sistémica. También se ha observado que en pacientes que han sido sometidos a trasplante de órganos los niveles de calcitonina pueden permitir la diferenciación entre la fiebre asociada con rechazo y la asociada con sepsis.63 Los niveles de procalcitonina no parecen elevarse en varias infecciones virales o condiciones inflamatorias de origen no infeccioso. Aunque no se ha definido el papel fisiológico preciso de la procalcitonina en la sepsis, ésta parecer ser el indicador más sensible y razonablemente específico de la sepsis severa hasta el momento.62,64
La proteína C reactiva y el lactato del plasma se han usado como marcadores potenciales de sepsis, pero la falta de especificidad y sensibilidad de ambos indicadores limitan su valor diagnóstico. La IL-8 puede ser útil, con o sin IL-6, como indicador de sepsis.65 La fosfolipasa A2 o su precursor, el propéptido tipo I de fosfolipasa A2, puede convertirse en un marcador potencial de sepsis, pero el valor diagnóstico de medir esta enzima requiere ser confirmado en estudios clínicos.66 La fosfolipasa A2 es esencial para la generación de factor activador de plaquetas y derivados del ácido araquidónico, incluyendo tromboxano, prostaciclina, prostaglandinas y leucotrienos.
Tratamiento del choque séptico
Existen cuatro metas en el manejo del choque séptico: (1) detección temprana y reanimación, (2) restablecimiento de la perfusión tisular y de la presión arterial, (3) manejo de sostén óptimo y (4) inicio oportuno de tratamiento para erradicar el foco séptico causal. El enfoque de tratamiento estándar no ha cambiado en forma importante en las dos últimas décadas. Sin embargo, se han logrado modestas mejoras en la evolución como resultado de una mejor nutrición y tratamiento de sostén además del uso de agentes vasopresores. El determinante clave para la supervivencia es la detección temprana de la sepsis y el inicio del tratamiento mientras el proceso es aún reversible. Esto requiere de vigilancia constante por el clínico que atiende pacientes con diversas enfermedades médicas y quirúrgicas.
La reanimación con líquidos es un primer paso obligado en el manejo del choque séptico. La fuga vascular difusa que ocurre en el choque séptico requiere de la reposición de un volumen sanguíneo adecuado para mantener la perfusión tisular. Continúa el debate sobre si deben administrarse coloides o cristaloides. La falta de evidencia clara de beneficios con los agentes coloides (v.gr., albúmina, dextrán y expansores del plasma) y su alto costo determinan que se usen más soluciones salinas para expander el volumen. La cantidad óptima de líquido para pacientes en choque séptico también es motivo de debate. Se requiere un delicado balance entre el mantenimiento de la perfusión tisular y la prevención de sobrecarga, con el riesgo asociado de lesión pulmonar. La menor función miocárdica en la sepsis puede requerir una mayor presión de llenado para lograr un gasto cardiaco adecuado; sin embargo, el exudado de líquidos hacia el espacio alveolar en el tejido pulmonar y hacia el intersticio en otros órganos vitales continúa siendo un problema importante. El mantenimiento de una presión de oclusión de la arteria pulmonar de alrededor de 12 mm Hg se considera como un punto de inicio razonable en pacientes que tienen monitores hemodinámicos.67
Cuando el estado hemodinámico de los pacientes no mejora solo con líquidos, suelen usarse agentes vasopresores para restablecer la presión arterial sistémica. La dopamina ha sido el agente vasopresor de elección durante las últimas dos décadas por sus efectos favorables de vasodilatación renal y su modesto inotropismo. Sin embargo, la validez de esta elección ha sido puesta en duda.68 Los efectos de la dopamina se complican por el hecho de que esta catecolamina tiene sus propios receptores (D1 y D2) así como afinidades variables para los receptores adrenérgicos alfa y beta. Los efectos de la dopamina dependen de la densidad de receptores en los lechos vasculares específicos, el volumen de sangre y la dosis usada. Las dosis mayores de dopamina aumentan la resistencia vascular sistémica por sus efectos sobre los receptores adrenérgicos alfa de la circulación periférica. La dopamina puede tener efectos adversos sobre el flujo sanguíneo esplácnico,68,69 y nunca se ha demostrado con claridad en estudios clínicos controlados que sea benéfica en pacientes sépticos.
La norepinefrina es un vasoconstrictor potente que se ha usado con frecuencia para tratar los efectos hemodinámicos del choque séptico. Las preocupaciones iniciales sobre las consecuencias adversas de la norepinefrina sobre el flujo sanguíneo renal han sido superadas, los estudios sugieren que la norepinefrina incluso aumenta la diuresis y la depuración de creatinina en pacientes sépticos.70 La norepinefrina puede restablecer la presión de perfusión en el glomérulo y mejorar la filtración glomerular en pacientes con reposición adecuada de líquido.
La dobutamina, un agonista beta, puede mejorar el gasto cardiaco y el aporte de oxígeno en algunos pacientes en choque séptico que tienen bajo gasto cardiaco. Sin embargo, puede causar vasodilatación periférica, lo que es dañino en pacientes con choque séptico. Además, la dobutamina aumenta el consumo miocárdico de oxígeno por sus efectos inotrópicos positivos, lo cual tampoco es conveniente. El uso de cualquier agente vasopresor en el choque séptico se asocia con ciertos riesgos y estos fármacos deben emplearse solamente cuando el tratamiento solo con líquidos sea insuficiente para restablecer la estabilidad hemodinámica.
Se desconoce cual es el indicador más confiable para evaluar lo adecuado de la perfusión tisular en el choque séptico. Parecería lógico proporcionar un aporte adecuado de oxígeno a los tejidos para satisfacer las demandas metabólicas del paciente séptico. Shoemaker y colaboradores han desarrollado el concepto de consumo de oxígeno dependiente del aporte,49 y esta idea aún debe probarse como guía clínica en el tratamiento del choque séptico. Se han desarrollado numerosos métodos de medición de oxigenación tisular usando tonometría gátrica, mediciones de oxígeno en venas hepáticas, mediciones directas de oxígeno tisular y otros con objeto de medir mejor y comprender los requerimientos críticos de aporte de oxígeno a los tejidos de los pacientes sépticos. Sin embargo, el valor práctico de estas mediciones en el ámbito clínico de la sepsis aún está en duda.
La necesidad de mejorar la capacidad transportadora de oxígeno de la sangre por medio de transfusiones ha sido motivo de gran debate.71 Se ha demostrado que los humanos son muy resistentes a los efectos adversos de la reducción isovolumétrica en los valores de hemoglobina.72 El límite inferior para indicar una transfusión en el choque séptico no se ha definido, pero parece ser bastante menor que el umbral tradicional de menos de 10 g/dl.71
El tratamiento experto de la insuficiencia renal aguda, del SIRPA, de la descompensación hepática, la coagulopatía, las alteraciones ácido-base y los trastornos hemodinámicos son de gran importancia en el manejo de la sepsis.
El apoyo nutricional en el choque séptico ha cambiado mucho durante las últimas dos décadas. El uso de la nutrición parenteral total ha cambiado hacia un uso más temprano y frecuente de hiperalimentación enteral. Se ha demostrado que la alimentación enteral en los pacientes con sepsis beneficia la función de los enterocitos, ayuda a mantener la permeabilidad de la barrera intestinal y a evitar la presencia de endotoxina derivada del intestino y la generación de citocinas.73 El suplemento nutricional con glutamina, arginina y ácidos grasos omega-3 tiene bases experimentales y se usa cada vez más en pacientes sépticos.74 El valor de las fórmulas enterales enriquecidas en comparación con las estándar no ha sido confirmado en estudios clínicos extensos.
La fiebre es común en la sepsis severa y parece constituir una respuesta positiva.75 Las proteínas de calor de choque funcionan como chaperones intracelulares para estabilizar y prevenir la desnaturalización de las proteínas del huésped. La inducción de proteínas de calor de choque puede en realidad disminuir la mortalidad asociada a la administración experimental de endotoxina.76 Los esfuerzos por reducir la temperatura corporal con mantas frías han sido ineficaces y no benefician al paciente en choque séptico. Esta estretegia debe evitarse a menos que exista verdadera hipertermia.77
El tratamiento antimicrobiano más apropiado en la sepsis depende de la fuente de infección, los patrones de sensibilidd de los microrganismos patógenos en una determinada institución, la exposición previa a antimicrobianos, la presencia o ausencia de embarazo, la función hepática y renal y la historia de alergia a medicamentos [ver tabla 6]. En el choque séptico suelen administrarse combinaciones de antimicrobianos bactericidas en forma empírica. Las combinaciones disminuyen el riesgo de que un patógeno resistente a múltiples fármacos no sea cubierto y aumenta la probabilidad de que todos los patógenos importantes sean inhibidos por lo menos por uno de los agentes antimicrobianos. Aún existe preocupación sobre la liberación de endotoxina inducida por antibióticos en la sepsis, pero la importancia clínica de este efecto no se ha demostrado y no debe afectar la elección de antibióticos en el paciente séptico.78
Tratamientos experimentales en la sepsis
Durante los últimos 10 años han existido más de 20 estudios doble ciego, controlados con placebo, multicéntricos, en fase II o III que han evaluado la eficacia de nuevos agentes experimentales en el tratamiento del choque séptico.2,6 Ninguno de estos agentes ha demostrado en forma convincente ser útil en el ámbito clínico. A pesar de ello, continúan los trabajos por mejorar el tratamiento de la sepsis [ver tabla 7]. Es posible que en algunos años contemos con mejores estrategias para inhibir los efectos de la endotoxina, modular la síntesis del óxido nítrico, atenuar la coagulopatía y regular la respuesta inmunológica contra la sepsis. Se espera que las mejoras en el tratamiento de sostén, manejo de vasopresores y líquidos y estrategias de tratamiento para alterar la fisiopatología de la sepsis sean benéficas para los pacientes en un futuro cercano. El choque séptico sigue siendo un gran reto en el manejo actual de los pacientes graves. Mejorar la evolución del síndrome requiere de un esfuerzo concertado de científicos, investigadores clínicos y clínicos dedicados.
Reconocimientos
Figuras 1 y 2 Seward Hung.
Bibliografía
DR. STEVEN M. OPAL
La sepsis, junto con la falla orgánica múltiple que con frecuencia acompaña al síndrome de respuesta inflamatoria sistémica (SRIS), es la principal causa de mortalidad en las unidades de cuidados intensivos.1,2 Hasta 400,000 pacientes desarrollan sepsis cada año en los Estados Unidos. Casi la mitad de estos pacientes sufren sepsis severa y choque séptico. La mortalidad del choque séptico sigue siendo alrededor de 35 a 45 porciento, a pesar de un gran esfuerzo por mejorar el tratamiento y la evolución.1-3
El choque séptico se ha convertido en un aspecto crucial de investigación en el área de los cuidados intensivos. Aunque se han logrado modestas mejoras en el pronóstico en la última década, aún se requieren inovaciones en el tratamiento del choque séptico. Este capítulo revisa algunos de los adelantos importantes logrados en el conocimiento de la fisiopatología molecular de la sepsis. Esto permite contar con una base para desarrollar estrategias de investigación que mejoren el diagnóstico y agentes terapéuticos para el manejo del choque séptico. Se analiza el tratamiento actual de la sepsis y se destacan las prioridades de investigación para el manejo futuro de los pacientes con este padecimiento.
Epidemiología
La incidencia de la sepsis ha aumentado en las últimas dos décadas.4 Es probable que esta tendencia continúe en el futuro porque la sepsis se ha convertido en una enfermedad del progreso médico. El tratamiento exitoso de diversos padecimientos médicos y quirúrgicos ha creado una gran población de pacientes con enfermedad grave y deterioro de las defensas, y estos pacientes tienen un riesgo muy alto de desarrollar sepsis. Las inovaciones en trasplante de órganos, implante de prótesis y equipos de acceso vascular de uso prolongado continúan aumentando esta población de pacientes. El envejecimiento gradual de la población en muchos países desarrollados y la mayor prevalencia de agentes microbianos resistentes a antibióticos contribuye también a la mayor incidencia del choque séptico.
Definición de sepsis
La sepsis, el choque séptico, el síndrome de respuesta inflamatoria sistémica y el síndrome de disfunción orgánica múltiple (SDOM) fueron definidos en una conferencia de consenso del American College of Chest Physicians/Society for Critical Care Medicine (ACCP/SCCM) de los EUA [ver tabla 1].5 Las definiciones toman en cuenta el dato de que la sepsis puede ser causada por múltiples agentes infecciosos y mediadores microbianos y puede o no asociarse con infección en el torrente sanguíneo. A pesar de la lógica clínica, la simplicidad intrínseca y la aceptación amplia de estas definiciones, su aplicación clínica ha sido criticada por buenos motivos.6,7 La definición de SRIS es tan amplia e inespecífica que pierde su poder de discriminación, muchos pacientes ingresados a servicios de medicina general y la mayoría de los pacientes de la UCI satisfacen la definición de SRIS. La definición actual de sepsis incluye virtualmente a cualquier persona que esté en las primeras 24 horas de un episodio de influenza porque se satisfacen los criterios de SRIS y el síndrome es causado por un agente infeccioso (un ortomixovirus). Sin embargo, los clínicos no suelen considerar la gripe como una sepsis.
|
|||||||||||||||||||||
|
Uno de los principios en los que se basan estas definiciones es que la respuesta inflamatoria en sí, y no el organismo infeccioso, es la causante del proceso séptico. La hipótesis puede ser correcta en principio, pero la naturaleza del patógeno microbiano responsable de la sepsis claramente contribuye a la evolución del paciente. Las variaciones en la susceptibilidad a las defensas del huésped, el potencial de resistencia antimicrobiana y la capacidad para generar toxinas afectan la patogenicidad del microrganismo.8 El no tomar en cuenta las diferencias intrínsecas en la virulencia microbiana limita la utilidad de las definiciones actuales de sepsis.
Existe desacuerdo sobre el nivel de reanimación con líquido necesario para asegurar que el paciente está séptico en lugar de hipovolémico. La cantidad de agente vasopresor necesario para concluir con confianza que el paciente tiene un verdadero choque séptico continúa en duda. También existe controversia por la dificultad para distinguir los padecimeintos y difunción orgánica previos de la morbilidad y disfunción orgánica inducidos por el proceso séptico en sí. Muchos pacientes con sepsis tienen disfunción orgánica severa subyacente por diversas enfermedades. El grado en el que la sepsis contribuye a una mayor alteración puede ser difícil de determinar con exactitud. Lo mismo puede decirse para el grado en que la sepsis contribuye a la mortalidad de un paciente determinado que sufre otros padecimientos graves. Todos estos factores limitan el valor discriminatorio de las definiciones de la sepsis y ponen en duda la validez de comparaciones en diferentes poblaciones de estudio, comprometiendo la capacidad para acumular datos y generalizar resultados.
Podrá ser posible definir mejor la terminología relacionada a la sepsis cuando se disponga de técnicas de diagnóstico rápido para evaluar el estado inmunológico de los pacientes sépticos. Mientras tanto, deben usarse las definiciones de consenso actuales a pesar de sus limitaciones.
Patogenia: factores microbianos
MICRORGANISMOS CAUSALES
La microbiología de la sepsis ha sufrido una transición muy importante en los últimos 25 años. Los patógenos principalmente responsables de la sepsis en los años 60 y 70 eran bacilos gram negativos y Pseudomonas aeruginosa, pero ha existido un incremento progresivo en la incidencia de sepsis causada por bacterias gram positivas9,10 y hongos oportunistas. La rápida evolución de genes resistentes a antibióticos en las bacterias gram positivas y la frecuencia de sepsis bacteriana asociada a catéteres vasculares puede ser responsable de la mayor prevalencia de patógenos gram positivos como causa de sepsis.
PAPEL DE LA ENDOTOXINA BACTERIANA
La endotoxina bacteriana, que se compone de lipopolisacáridos (LPS), es un componente intrínseco de la membrana exterior de las bacterias gram negativas y es indispensable para la viabilidad de las mismas.11 Los humanos son una de las especies más susceptibles a las potentes propiedades inmunoestimulantes de la endotoxina, que puede ser letal en dosis diminutas. Sea que la endotoxina se libere al sistema circulatorio humano en su forma libre (a partir de organismos muertos o liberada de la membrana de organismos viables) o unida a la pared celular de la bacteria intacta, se presenta una intensa respuesta inflamatoria sistémica. Parece ser que la endotoxina funciona como una molécula de alarma que alerta al huésped sobre la presencia de una bacteria gram negativa invasora.12 Es la respuesta del huésped a la endotoxina, más que ella misma, la responsable de las propiedades letales de la endotoxina.
El LPS es una macromolécula polar fosforilada que contiene elementos hidrofóbicos en los ácidos grasos de su estructura central de lípido A u elementos hidrofílicos en sus componentes polisacáridos de repetición de superficie. El LPS forma microagregados en los líquidos biológicos e interactúa en forma rápida con una variedad de proteínas lipofílicas en el suero o unidas a la membrana. En los humanos existen tres receptores conocidos para el LPS: (1) moléculas CD14 solubles o unidas a membranas, (2) moléculas CD11/CD18 (integrinas ß2) y (3) receptores depuradores de moléculas lípidas. Las moléculas CD14 solubles y unidas a la membrana representan el sistema más sensible para la activación de las células inmunes con la exposición a pequeñas cantidades de LPS.13
El mecanismo de señal más importante del LPS está mediado por interacciones con una proteína plasmática de fase aguda derivada del hígado conocida como proteína de unión al LPS (PUL).14 Esta actúa principalmente como una molécula transportadora que se une a los agregados de LPS polimérico y transfiere monómeros de LPS a la molécula CD14. CD14 es una proteína unida a glucosil fosfatidilinositol que se encuentra en las superficies celulares de células efectoras inmunológicas como los monocitos-macrófagos y los neutrófilos. Las evidencias actuales sugieren que el CD14 unido a la membrana no funciona en forma directa iniciando el mensaje intracelular de la presencia del LPS.15 En lugar de ello, después de unirse al CD14 unido a la membrana, el LPS es entregado a una molécula adyacente receptora del LPS (que debe aún identificarse y caracterizarse), y que origina una señal al espacio intracelular, activando los genes que responden al LPS.
Las vías de señal intracelular del LPS son un área de gran interés y no se han caracterizado del todo.16 La trasducción de señales se realiza por la activación de varios activadores intracelulares de transcripción, incluyendo el NFkB, la proteína cinasa, las proteínas G, RAS, RAF-1, MEK-1, ERK-1, tirosina cinasas y una serie de proteinas cinasas activadas por mitógenos. El LPS tiene semejanzas estructurales con otra molécula lípida de señales intracelulares, la ceramida. La ceramida actúa a través de la cinasa activada de ceramida. El LPS semeja a la ceramida en que también activa a esta cinasa intracelular, lo que puede contribuir a la transducción de señales del mensaje del LPS.17
El resultado final de la acción del LPS consiste en estimular la síntesis de una serie grande de genes inducibles por LPS en las células inflamatorias. Estos genes incluyen las citocinas proinflamatorias y la sintetasa de óxido nítrico. La salida de citocinas proinflamatorias y de otros mediadores inflamatorios después de la exposición a LPS contribuye al SRIS y es crucial en la patogenia del choque séptico inducido por bacterias gram negativas.12
La actividad del LPS es modulada por diversos componentes derivados del huésped. Los niveles de PUL en la circulación varían entre las personas y en diferentes estados de inflamación. La PUL es una proteína de fase aguda y sus niveles pueden aumentar siete veces durante la inflamación aguda.18 La importancia de la PUL en la fisiopatología del choque inducido por endotoxina se demuestra con claridad en ratones sin PUL que son resistentes a los efectos letales de la endotoxina bacteriana.19 Los ratones que tienen deficiencia genética de CD14 también son resistentes a la endotoxina.20 Existen ciertas evidencias de que los pacientes con niveles más altos de PUL tienen mayor riesgo de morir por choque inducido por endotoxina, comparado con los que tienen niveles bajos de PUL.20a
Otra proteína fijadora de endotoxina importante que se encuentra en el plasma humano es la proteína bactericida/aumentadora de permeabilidad (PBAP). Esta proteína tiene 456 aminoacidos, es producida por los neutrófilos humanos y se encuentra en mayor cantidad en los gránulos azurófilos (primarios).18 Tiene un 45 porciento de homología en sus aminoácidos con la PUL, pero su función con respecto al LPS es opuesta. La PUL facilita la unión del LPS al CD14, activando a las células mientras que la PBAP tiene el efecto fisiológico opuesto, inhibe la llegada del LPS a CD14. la PBAP funciona como un antagonista molecular y compite con la PUL por la unión al LPS en los líquidos biológicos .18,21 Las concentraciones relativas de estas dos proteínas fijadoras de endotoxina determinan el efecto neto de la liberación del LPS.
En el plasma humano la concentración de PUL es de dos o tres órdenes de magnitud mayor que la de PBAP. Lo opuesto parece suceder en los abscesos, en donde existe PBAP en mucho mayor cantidad que PUL.18 Esto favorece la función activadora de LPS en el plasma y la inhibitoria en los abscesos. Por lo tanto, la PBAP funciona como una molécula antiendotoxina endógena y puede ser un componente del tratamiento de la lesión inducida por endotoxina.22
Otra proteína endógena del plasma que funciona para regular la actividad del LPS en la circulación es la lipoproteína de alta densidad (LAD). El LPS se une con gran afinidad a las regiones hidrofóbicas de la LAD, y este complejo es eliminado en forma eficaz de la circulación por mecanismos de depuración hepática.23 Los ratones transgénicos que expresan grandes cantidades de apolipoproteína A-1 (la principal proteína que constituye la LAD) son menos susceptibles a la toxicidad por endotoxina que los ratones normales.24 La administración de LAD reconstituyente es un tratamiento potencial para los trastornos relacionados con la endotoxina en la sepsis.25
El fenómeno de tolerancia a endotoxina ha sido bien caracterizado en modelos animales de sepsis y es probable que también ocurra en humanos.26 La tolerancia a la endotoxina se describe como una desensibilización a la letalidad inducida por ésta sustancia al administrar primero una dosis pequeña de endotoxina y después una dosis letal. Este fenómeno parece estar mediado prncipalmente a nivel de transcripción, por regulación inhibitoria de los genes de las citocinas proinflamatorias, pero los mecanismos precisos se desconocen. La dosis de desensibilización de la endotoxina puede inducir esteroides endógenos o citocinas antinflamatorias (v.gr., interleucina-10 [IL-10]), disminuir la expresión de receptores de citocinas en la superficie celular, alterar la traslocación nuclear de moléculas de trasducción de señales o disminuir la estabilidad del ARN mensajero de citocinas.
El significado de la tolerancia a la endotoxina en humanos no es claro pero se ha observado cierto grado de tolerancia en pacientes tratados con lípido A monofosforilado.26 Sin duda, la endotoxina es un mediador importante en la patogenia del choque séptico. Por lo tanto, los esfuerzos para limitar la síntesis del LPS, prevenir la activación de los elementos de respuesta inmunológica del huésped y aumentar la depuración del LPS son estrategias terapéuticas importantes para el manejo futuro del choque séptico.
SUPERANTIGENOS BACTERIANOS
Otros mediadores microbianos importantes en la patoenia del choque séptico son los superantígenos bacterianos. Los superantígenos son un grupo diverso de exotoxinas proteicas derivadas de microrganismos estreptococos, estafilococos y otros patógenos que comparten una propiedad inmunológica poco habitual, los superantígenos pueden activar gran número de células T CD4+ en un periodo breve saltándose el mecanismo habitual de procesamiento y presentación del antígeno.27
Los antígenos bacterianos convencionales son internalizados por células presentadoras de antígeno (CPA), en donde sufren proteólisis limitada y procesamiento dentro del componente endosómico del macrófago. Las secuencias peptídicas apropiadas de los antígenos microbianos (epítopes) se insertan en el surco central de las moléculas de clase II del complejo principal de histocompatibilidad (CPH) y después son expresadas en la superficie celular de las CPA. Entonces se activan células T CD4+ específicas que reconocen el epítope. La expansión clonal de este subgrupo pequeño de células T causa una respuesta inmune fisiológica al nuevo antígeno introducido. Por el contrario, los superantígenos no requieren del procesamiento intracelular por la CPA. Se unen en forma directa a los antígenos de clase II en un sitio adyacente al surco específico para el epítope, y también se unen a un número limitado de regiones Bß del receptor de la célula T en las células T CD4+. Esta unión aproxima las células T CD4+ y los macrófagos, lo que activa a ambas poblaciones celulares. Mientras que un péptido antigénico convencional estimula solo alrededor de uno en 105 linfocitos circulantes que pueden reconocer este epítope estructural único, un superantígeno (v.gr., toxina-1 del síndrome de choque tóxico de Staphylococcus aureus, que se une a la región Vß2 de las células T) puede reconocer hasta una de cada 10 linfocitos humanos circulantes. Por lo tanto, un solo superantígeno bacteriano puede activar hasta al 10 porciento de toda la población linfocitaria.28 Esto causa activación excesiva tanto de linfocitos como de macrófagos, y ocurre síntesis y liberación de citocinas proinflamatorias en forma incontrolable. La actvación inmune inducida por superantígenos puede terminar en choque séptico si el proceso no se limita. Las infecciones polimicrobianas que liberan tanto superantígenos bacterianos como endotoxina pueden ser especialmente dañinos para el huésped, la toxicidad de la endotoxina bacteriana puede aumentar mucho por los superantígenos que activan al sistema inmune para que reaccione a la endotoxina con mayor sensibilidad [ver figura 1].
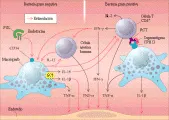
|
| Figura 1 |
| Papel de la endotoxina y los superantígenos en la sepsis |
OTROS MEDIADORES MICROBIANOS
El peptidoglucano de la pared celular de la bacteria, los antígenos capsulares, las toxinas microbianas y las sustancias procoagulantes producidas por patógenos microbianos pueden contribuir a la patogenia de la sepsis. Se ha observado que los peptidoglucanos, como el ácido teicoico y otros componentes de las bacterias gram positivas, pueden interactuar con moléculas CD14 y activar células inflamatorias en una forma semejante a la de la endotoxina bacteriana.29 Puede ocurrir activación dependiente de CD14 de las células mononucleares con componentes de bacterias gram positivas, aunque el nivel de activación es cuantitativamente menor que el observado con los componentes de bacterias gram negativas.30
Las bacterias gram positivas y los hongos pueden inducir hipotensión sistémica, con redistribución del flujo sanguíneo y vasoconstricción esplácnica. La isquemia y la reperfusión de los vasos sanguíneos que irrigan el tubo digestivo puede alterar la barrera mucosa intestinal contra los productos bacterianos. Ocurre entonces traslocación de patógenos microbianos intactos y de endotoxina bacteriana durante los periodos de estrés severo y de hipoperfusión de la mucosa gastrointestinal.31 La endotoxina bacteriana puede tener un papel patogénico en el proceso inflamatorio después de la hipotensión sistémica producida por lesiones infecciosas y no infecciosas. Por lo tanto, los estados de choque pueden ser potencializados y mantenidos por la liberación continua de endotoxina y otros mediadores microbianos de la luz del tubo digestivo. Estos hallazgos han iniciado el interés por fortalecer la mucosa intestinal a través de inmunonutrición, factores de crecimiento epitelial y descontaminación selectiva del tubo digestivo en enfermedades graves. Estas opciones de tratamiento pueden ser potencialmente viables y son motivo de intensa investigación dentro del manejo de la sepsis.
Patogenia: Mediadores derivados del huésped
RED DE CITOCINAS
Las citocinas proinflamatorias juegan un papel crucial en la patogenia de la sepsis. En estudios en animales, la adminitración de factor de necrosis tumoral-alfa (FNT-alfa), una proteína endógena derivada de monocitos-macrófagos, ha demostrado tener consecuencias letales, y se observaron dramáticos cambios hemodinámicos, metabólicos y hematológicos después de la administración de FNT-alfa a voluntarios humanos.32,33 También se han demostrado los efectos dañinos de los niveles sistémicos de la IL-1ß.34
Las principales citocinas proinflamatorias, FNT-alfa e IL-1ß. inducen sus efectos hemodinámicos y metabólicos en conjunto con un grupo amplio de mediadores proinflamatorios derivados del huésped que actúan en forma coordinada para producir la respuesta inflamatoria sistémica [ver figura 1 y tabla 2].35 El sistema de citocinas funciona como una red de comunicación de señales entre neutrófilos, monocitos, macrófagos y células endoteliales. La activación autócrina y parácrina causa potencialización sinérgica de la respuesta inflamatoria una vez que ésta es activada por un insulto microbiano sistémico (v. gr., endotoxemia). Mucha de la respuesta proinflamatoria es localizada y compartamentalizada en la región de la inflamación inicial (v.gr., tejido pulmonar o tubo digestivo). Si no se limita, la respuesta inflamatoria se disemina a la circulación sistémica, causando una reacción generalizada y culminando en lesión endotelial difusa y choque séptico. El efecto seudoendócrino de la liberación sistémica de citocinas y quimocinas difunde el proceso inflamatorio en todo el organismo.1-3
|
||||||||||||||||||||||||||||||||||
|
La gran cantidad de citocinas y quimocinas inflamatorias que se encuentran en cantidades excesivas en el torrente sanguíneo en el choque séptico es impresionante, y se equipara con un grupo igualmente enorme de mediadores antinflamatorios [ver tabla 2]. Los mediadores proinflamatorios tienden a predominar en la fase temprana de la sepsis (las primeras 12 a 24 horas), mientras que los componentes endógenos antinflamatorios suelen prevalecer en las fases tardías.36 Se ha observado que los ratones deficientes en células T y B responden al reto con endotoxina de la misma forma que los ratones normales.37 Por lo tanto, las citocinas generadas por monocitos y macrófagos son suficientes para desencadenar el proceso séptico inicial. Sin embargo, las citocinas derivadas de linfocitos e interferones son importantes en la regulación de las fases tardías de la sepsis y pueden determinar la evolución final en el choque séptico.
Papel de las células T CD4+
Existen importantes diferencias funcionales entre las células T CD4+. Las células T CD4+ activadas, pero no comprometidas (células THO) siguen dos vías principales de diferenciación funcional. Las células T expuestas a IL-12, en presencia de IL-2, desarrollan un tipo funcional TH1. Estas células producen grandes cantidades de interferón gama, FNT-alfa e IL-2 y promueven una respuesta inmune proinflamatoria mediada por células. Las células T CD4+ aún no comprometidas expuestas a IL-4 se desarrollan hacia un fenotipo TH2, que secreta IL-4, IL-10 e IL-13. Estas citocinas promueven respuestas inmunes humorales y atenúan la actividad de macrofagos y neutrófilos.
Es interesante que las citocinas elaboradas por las TH1 suprimen la expresión de las citocinas de tipo TH2; por ejemplo, el interferón gama inhibe la síntesis de IL-10. Por el contrario, la IL-10 de las TH2 es un inhibidor potente de la síntesis de FNT-alfa y de interferón gama por las células TH1. El tipo de respuesta linfocitaria inicial es crucial porque el sistema tiende a polarizarse con el tiempo hacia un predominio TH2 oTH1.38 Este proceso es importante porque la sepsis suele acompañarse de una respuesta tardía tipo TH2 después de la lesión séptica inicial. La respuesta de hormonas de estrés en el choque séptico, con expresión de hormona adrenocorticotrópica, esteroides y catecolaminas, promueve una respuesta TH2 después de la lesión sistémica. Esto puede causar una fase de refractariedad inmune relativa (parálisis inmune) en la que el paciente puede tener mayor riesgo de infección bacteriana o micótica secundaria.39 Este estado fisiopatológico se asocia con tolerancia a endotoxina, síntesis de citocinas antinflamatorias y desactivación de monocitos, macrófagos y neutrófilos. Los métodos para detectar este estado de inmunosupresión y restablecer la competencia inmune aún son motivo de investigación. Los pacientes con menor expresión de antígenos de clase II del CPH, como el HLA-DR, en la superficie de los macrófagos, pueden tener un estado de inmunosupresión funcional y beneficiarse con el tratamiento con interferón gama.40
PAPEL DEL SISTEMA DE COAGULACION
La activación de la cascada de la coagulación y la generación de una coagulopatía por consumo y de microtrombos difusos es una complicación bien reconocida de la sepsis severa. El reto con endotoxina y con FNT en voluntarios humanos normales indica que la vía extrínseca es el mecanismo principal por el que el sistema de coagulación se activa en la sepsis humana.41,42 Los factores de contacto de la vía intrínseca se activan también, y en forma secundaria inician la vasodilatación por generación de bradicinina.42 La activación de coagulación intravascular causa microtrombos y puede contribuir a la falla orgánica múltiple que se observa en la sepsis. La depleción de los factores de coagulación y la activación de plasmina, antitrombina III y proteína C activada pueden causar diátesis hemorrágica. La reducción de estos anticoagulantes endógenos provoca en forma secundaria un estado procoagulante que implica mal pronóstico.43 El interés actual en la administración de inhibidor de la vía del factor tisular, proteína C activada y antitrombina III para el tratamiento de la sepsis se basa en el posible valor terapéutico de regular el sistema de la coagulación.44
INTERACCIONES ENTRE NEUTROFILOS Y CELULAS ENDOTELIALES
El reclutamiento de neutrófilos en un área de infección local es un componente esencial en la respuesta inflamatoria del huésped. La lcoalización y erradicación de los patógenos invasores en el sitio de la infección inicial es el principal objetivo de la respuesta inmunológica contra los microrganismos patógenos. Este proceso fisiológico puede volverse dañino si ocurren interacciones difusas entre neutrófilos y células endoteliales en toda la circulación en respuesta a la inflamación sistémica.
Los mecanismos responsables de la migración de neutrófilos del espacio intravascular hacia el intersticio, en donde residen los microrganismos invasores, son complejos [ver figura 2]. Los neutrófilos activados degranulan y exponen las superficies endoteliales y estructuras circundantes para reactivar intermediarios del oxígeno, óxido nítrico y diversas proteasas. Este proceso contribuye no solo a la depuración de microrganismos, sino también a la lesión endotelial difusa en el caso de respuestas inflamatorias generalizadas. La regulación de la actividad de los neutrófilos puede representar un área nueva para la intervención terapéutica en la sepsis.45
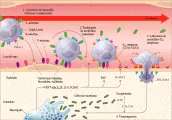
|
| Figura 2 |
| Interacciones neutrófilo-célula endotelial en la sepsis. |
PAPEL DEL OXIDO NITRICO
El óxido nítrico es un radical libre muy reactivo que tiene un papel esencial en la fisiopatología del choque séptico. Su vida media es muy corta (1 a 3 segundos), lo que limita su actividad a tejidos locales, en donde es generado por una de tres isoformas de la sintetasa de óxido nítrico. La regulación de la sintetasa de óxido nítrico es compleja. La expresión completa de la misma requiere de FNT-alfa, IL-1, LPS y quizá de otros elementos de control.
El óxido nítrico es el principal factor de relajación derivado del endotelio, e inicia la vasodilatación y la hipotensión sistémica observada en el choque séptico. El óxido nítrico activa a la guanilato ciclasa, que aumenta los niveles de monofosfato cíclico de guanosina dentro de las células del músculo liso vascular. Esto produce vasodilatación sistémica y reducción de la resistencia vascular. Sin embargo, en minutos después de la administración de un inhibidor de la síntesis de óxido nítrico la presión arterial de los pacientes hipotensos con choque séptico regresa a los niveles normales.46
Los otros efectos fisiológicos importantes del óxido nítrico en el choque séptido con la mayor destrucción intracelular y la regulación de la adherencia de plaquetas y neutrófilos. El óxido nítrico es un gas que difunde con facilidad y que no requiere de receptores específicos para entrar a las células eucariotas o procariotas. En presencia del anión superóxido forma peroxinitrito. El peroxinitrido se descompone en moléculas muy citotóxicas, los radicales hidroxilo y cloruro de nitrosil que, a su vez, inician la peroxidación de lípidos y provocan daño celular irreversible. El óxido nítrico inhibe diversas enzimas clave en la vía del ácido tricarboxílico, la vía glucolítica, los sistemas de reparación del ADN, las vías de transporte de electrones y las vías de intercambio de energía. Debido a su potente reactividad, el óxido nítrico altera la función de muchas metaloenzimas, proteínas transportadoras y elementos estructurales.
Ha sido aparente que el óxido nítrico, como muchos otros componentes de la respuesta inflamatoria del huésped, puede tener propiedades tanto útiles como dañinas en la sepsis. Regula la microcirculación a los órganos vitales y contribuye a la destrucción intracelular de microrganismos patógenos. Sin embargo, la liberación excesiva y prolongada de óxido nítrico causa la vasodilatación generalizada e hipotensión sistémica en el choque séptico. El óxido nítrico se ha vuelto un blanco importante en las estrategias terapéuticas de la sepsis, y en la actualidad se realizan estudios clínicos para determinar la eficacia protectora de los inhibidores de esta sustancia.46 Por ejemplo, se ha demostrado que los inhibidores no selectivos de la sintetasa de óxido nítrico mejoran la hemodinamia de los pacientes sépticos.47
Técnicas diagnósticas en el choque séptico
CARACTERISTICAS GENERALES
En su tratado clásico de naturaleza humana (The Prince, cerca de 1505), Maquiavelo establece, "La fiebre éctica [i.e., sepsis según las definiciones actuales] es difícil de reconocer en su inicio pero fácil de tratar; si no se atiende se vuelve fácil de reconocer pero difícil de tratar." Esta afirmación es tan cierta ahora como lo era hace 500 años. El choque séptico ya establecido es un síndrome clínico muy aparente que rara vez se confunde con otros estados patológicos. Sin embargo, las fases iniciales pueden ser muy sutiles, incluso en pacientes vigilados con cuidado. Los signos y síntomas tempranos pueden incluir confusión, aprehensión o deterioro del estado de alerta. Aunque la fiebre es característica, puede observarse hipotermia, que implica un mal pronóstico. La reducción inexplicable en la diuresis, el inicio súbito de ictericia colestática, la alcalosis metabólica inexplicable, la hemorragia excesiva en sitios de venopunción o incluso la hipotensión súbita inexplicable pueden ser el dato inicial del choque séptico. Es indispensable que el clínico detecte estos signos y síntomas tempranos porque el tratamiento exitoso del choque séptico depende de su detección temprana e intervención apropiada.
En el choque séptico se detectan diversas alteraciones clínicas, de laboratorio y hemodinámicas [ver tablas 3 y 4]. Por desgracia, no existe una sola prueba clínica o de laboratorio que sea suficientemente sensible y específica para confirmar con certeza el diagnóstico. Los hemocultivos pueden o no ser positivos, se observa leucocitosis o neutropenia, hiperglucemia o hipoglucemia y alcalosis respiratoria o acidosis metabólica. Es el conjunto de signos y síntomas lo que origina el diagnóstico de choque séptico.
|
||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||
|
Los datos hemodinámicos más comunes en el choque séptico temprano son un gasto cardiaco alto con resistencias vasculares sistémicas bajas, con mantenimiento inicial de la presión arterial sistólica al intentar el corazón compensar por la pérdida de tono vascular sistémico. La función miocárdica está disminuida incluso en las fases tempranas del choque séptico.48 Sin una intervención adecuada el volumen de sangre circulante se pierde en forma continua hacia el espacio intersticial y los sitios intracelulares. El corazón no puede compensar suficiente y ocurre hipotensión sistólica. El deterioro de la función miocárdica asociada con vasoconstricción difusa marca el estado refractario tardío del choque séptico.
El choque séptico puede asociarse con pérdida de la autorregulación normal entre el aporte de oxígeno y su consumo en los tejidos. La disoxia dependiente del aporte puede contribuir a la lesión tisular en la falla multiorgánica de la sepsis.49 Los intentos por aumentar el aporte de oxígeno a niveles supranormales, aunque factibles en teoría, no han aumentado la supervivencia en estudios clínicos.50 Quizá el uso más juicioso de aporte de oxígeno a los tejidos podrá beneficiar a los pacientes en choque séptico.
SINDROME DE DISFUNCION ORGANICA MULTIPLE
Una de las características del choque séptico es el desarrollo de disfunción orgánica múltiple. El desarrollo de falla orgánica al inicio o durante la evolución de la sepsis es un factor de mal pronóstico y es el principal determinante de la evolución [ver tabla 5].51 La lesión endotelial difusa que acompaña al choque séptico causa disfunción orgánica distante al sitio original de la lesión. Se supone que la señal que provoca el daño endovascular difuso depende de factores del plasma (v.gr., citocinas proinflamatorias, complemento, cininas y otros mediadores inflamatorios derivados del huésped) o por un elemento celular encontrado en uno o más de las células inmunes efectoras.
|
||||||||||||||||||||||||
|
El aporte sanguíneo inadecuado a los tejidos vitales produce SDOM. La falla de la microcirculación para mantener a los tejidos puede ser resultado de hipoperfusión de los lechos capilares, redistribución del flujo sanguíneo dentro de los lechos vasculares, derivaciones arteriovenosas funcionales, obstrucción del flujo sanguíneo por microtrombos, agregados de plaquetas o leucocitos o deformación anormal de los eritrocitos. El óxido nítrico, los intermediarios reactivos del oxígeno, las citocinas proinflamatorias y los inductores de apoptosis pueden dañar en forma directa las superficies del endotelio. También el edema endotelial por el movimiento del líquido intravascular hacia los espacios extravascular e intracelulares puede obstruir en forma mecánica la luz de los lechos capilares.
Aunque el origen de la falla orgánica múltiple en la sepsis se relaciona principalmente con los efectos microvasculares, la función miocárdica y pulmonar también disminuyen durante el curso del choque séptico y pueden contribuir en forma significativa al desarrollo de la falla multiorgánica. La contractilidad miocárdica disminuye en respuesta a diversos factores depresores del miocardio encontrados en el plasma de pacientes sépticos. El FNT-alfa es una causa prominente de disfunción miocárdica, la IL-1, el óxido nítrico y otros mediadores inflamatorios derivados del huésped pueden también ser factores que contribuyen.52
La lesión pulmonar aguda ocurre como resultado del daño a la circulación vascular pulmonar y las membranas alveolocapilares. El pulmón es susceptible de lesionarse en varios estados inflamatorios sistémicos. El síndrome de insuficiencia respiratoria progresiva aguda (SIRPA) sigue siendo una causa importante de morbimortalidad en el choque séptico. Los esfuerzos por prevenir la lesión pulmonar incluyen inovaciones en el apoyo respiratorio, evitar el barotrauma y volutrauma a las unidades alveolocapilares, evitar la lesión pulmonar inducida por oxidantes, salvar las unidades alveolocapilares funcionales por medio de cambio de posición (posición prona) y manejo juicioso de líquidos.53 Han ocurrido mejoras mesurables en la evolución del SIRPA, pero aún puede protegerse mucho más la función pulmonar en el caso del choque séptico.
La falla orgánica múltiple es potencialmente revesible si se aplican medidas rápidas para corregir las alteraciones hemodinámicas e inflamatorias.
TECNICAS DIAGNOSTICAS EXPERIMENTALES
Los ensayos actuales usados para diagnosticar el choque séptico son incómodos y no útiles para el diagnóstico rápido. En la actualidad se investigan métodos mejores. La medición de niveles de endotoxina en plasma, por ejemplo, puede ser útil para el diagnóstico de sepsis y se ha demostrado que tiene cierto valor predictivo para determinar los pacientes con riesgo de desarrollar estado de choque.54 Por desgracia, los niveles de endotoxina no aumentan de modo uniforme en el choque séptico y pueden elevarse en forma falsa en infecciones por gram positivos y otros trastornos que cursan con hipotensión. La respuesta del huésped a la endotoxina es variable y no puede determinarse con base en los niveles de endotoxina circulante.
Se han medido con éxito los niveles circulantes de superantígenos bacterianos en pacientes seleccionados con síndrome de choque tóxico.55 Estas mediciones pueden ser útiles en situaciones clínicas específicas.
La IL-6 es una citocina multifuncional que tiene gran cantidad de actividades biológicas, algunas de las cuales son proinflamatorias y otras antinflamatorias. La IL-6 se ha considerado como un indicador de activación de citocinas porque se presenta en forma constante después de la activación del FNT-alfa y de la IL-1ß.56 Los pacientes con niveles elevados de IL-6 pueden responder en forma favorable a los tratamientos anticitocina.57 En varios estudios,57-60 la elevación de IL-6, así como la no disminución en el nivel de IL-6 después de iniciar el tratamiento para la sepsis, se asociaron con mala evolución. Por desgracia, los niveles de IL-6 no son específicos de sepsis y pueden aumentar en diversos estados inflamatorios e infecciosos. La falta de especificidad limita la confiabilidad de la medición de IL-6 como método diagnóstico de choque séptico.
La procalcitonina es el propéptido de la calcitonina que en condiciones fisiológicas es producido por las células C del tiroides. Una proteasa específica convierte la procalcitonina en calcitonina, katalcina y un péptido amino-terminal.61 La procalcitonina tiene atributos que le hacen un marcador potencial de sepsis. Tiene una vida media larga (alrededor de 24 horas) y aumenta desde niveles no detectables hasta más de 100 ng/ml durante el choque séptico. El origen preciso de la procalcitonina en la sepsis no es claro, pero es extratiroideo.61,62 Los niveles de procalcitocina no aumentan tan pronto como los de la IL-6 o la IL-8, sino que se observan hasta 4 a 6 horas después del reto sistémico con endotoxina u otro estímulo séptico.62
Es interesante que los niveles de procalcitonina se elevan en la sepsis severa, pero no en las infecciones localizadas.62 Esto indica que la procalcitonina puede ser un marcador para la respuesta inflamatoria sistémica. También se ha observado que en pacientes que han sido sometidos a trasplante de órganos los niveles de calcitonina pueden permitir la diferenciación entre la fiebre asociada con rechazo y la asociada con sepsis.63 Los niveles de procalcitonina no parecen elevarse en varias infecciones virales o condiciones inflamatorias de origen no infeccioso. Aunque no se ha definido el papel fisiológico preciso de la procalcitonina en la sepsis, ésta parecer ser el indicador más sensible y razonablemente específico de la sepsis severa hasta el momento.62,64
La proteína C reactiva y el lactato del plasma se han usado como marcadores potenciales de sepsis, pero la falta de especificidad y sensibilidad de ambos indicadores limitan su valor diagnóstico. La IL-8 puede ser útil, con o sin IL-6, como indicador de sepsis.65 La fosfolipasa A2 o su precursor, el propéptido tipo I de fosfolipasa A2, puede convertirse en un marcador potencial de sepsis, pero el valor diagnóstico de medir esta enzima requiere ser confirmado en estudios clínicos.66 La fosfolipasa A2 es esencial para la generación de factor activador de plaquetas y derivados del ácido araquidónico, incluyendo tromboxano, prostaciclina, prostaglandinas y leucotrienos.
Tratamiento del choque séptico
Existen cuatro metas en el manejo del choque séptico: (1) detección temprana y reanimación, (2) restablecimiento de la perfusión tisular y de la presión arterial, (3) manejo de sostén óptimo y (4) inicio oportuno de tratamiento para erradicar el foco séptico causal. El enfoque de tratamiento estándar no ha cambiado en forma importante en las dos últimas décadas. Sin embargo, se han logrado modestas mejoras en la evolución como resultado de una mejor nutrición y tratamiento de sostén además del uso de agentes vasopresores. El determinante clave para la supervivencia es la detección temprana de la sepsis y el inicio del tratamiento mientras el proceso es aún reversible. Esto requiere de vigilancia constante por el clínico que atiende pacientes con diversas enfermedades médicas y quirúrgicas.
La reanimación con líquidos es un primer paso obligado en el manejo del choque séptico. La fuga vascular difusa que ocurre en el choque séptico requiere de la reposición de un volumen sanguíneo adecuado para mantener la perfusión tisular. Continúa el debate sobre si deben administrarse coloides o cristaloides. La falta de evidencia clara de beneficios con los agentes coloides (v.gr., albúmina, dextrán y expansores del plasma) y su alto costo determinan que se usen más soluciones salinas para expander el volumen. La cantidad óptima de líquido para pacientes en choque séptico también es motivo de debate. Se requiere un delicado balance entre el mantenimiento de la perfusión tisular y la prevención de sobrecarga, con el riesgo asociado de lesión pulmonar. La menor función miocárdica en la sepsis puede requerir una mayor presión de llenado para lograr un gasto cardiaco adecuado; sin embargo, el exudado de líquidos hacia el espacio alveolar en el tejido pulmonar y hacia el intersticio en otros órganos vitales continúa siendo un problema importante. El mantenimiento de una presión de oclusión de la arteria pulmonar de alrededor de 12 mm Hg se considera como un punto de inicio razonable en pacientes que tienen monitores hemodinámicos.67
Cuando el estado hemodinámico de los pacientes no mejora solo con líquidos, suelen usarse agentes vasopresores para restablecer la presión arterial sistémica. La dopamina ha sido el agente vasopresor de elección durante las últimas dos décadas por sus efectos favorables de vasodilatación renal y su modesto inotropismo. Sin embargo, la validez de esta elección ha sido puesta en duda.68 Los efectos de la dopamina se complican por el hecho de que esta catecolamina tiene sus propios receptores (D1 y D2) así como afinidades variables para los receptores adrenérgicos alfa y beta. Los efectos de la dopamina dependen de la densidad de receptores en los lechos vasculares específicos, el volumen de sangre y la dosis usada. Las dosis mayores de dopamina aumentan la resistencia vascular sistémica por sus efectos sobre los receptores adrenérgicos alfa de la circulación periférica. La dopamina puede tener efectos adversos sobre el flujo sanguíneo esplácnico,68,69 y nunca se ha demostrado con claridad en estudios clínicos controlados que sea benéfica en pacientes sépticos.
La norepinefrina es un vasoconstrictor potente que se ha usado con frecuencia para tratar los efectos hemodinámicos del choque séptico. Las preocupaciones iniciales sobre las consecuencias adversas de la norepinefrina sobre el flujo sanguíneo renal han sido superadas, los estudios sugieren que la norepinefrina incluso aumenta la diuresis y la depuración de creatinina en pacientes sépticos.70 La norepinefrina puede restablecer la presión de perfusión en el glomérulo y mejorar la filtración glomerular en pacientes con reposición adecuada de líquido.
La dobutamina, un agonista beta, puede mejorar el gasto cardiaco y el aporte de oxígeno en algunos pacientes en choque séptico que tienen bajo gasto cardiaco. Sin embargo, puede causar vasodilatación periférica, lo que es dañino en pacientes con choque séptico. Además, la dobutamina aumenta el consumo miocárdico de oxígeno por sus efectos inotrópicos positivos, lo cual tampoco es conveniente. El uso de cualquier agente vasopresor en el choque séptico se asocia con ciertos riesgos y estos fármacos deben emplearse solamente cuando el tratamiento solo con líquidos sea insuficiente para restablecer la estabilidad hemodinámica.
Se desconoce cual es el indicador más confiable para evaluar lo adecuado de la perfusión tisular en el choque séptico. Parecería lógico proporcionar un aporte adecuado de oxígeno a los tejidos para satisfacer las demandas metabólicas del paciente séptico. Shoemaker y colaboradores han desarrollado el concepto de consumo de oxígeno dependiente del aporte,49 y esta idea aún debe probarse como guía clínica en el tratamiento del choque séptico. Se han desarrollado numerosos métodos de medición de oxigenación tisular usando tonometría gátrica, mediciones de oxígeno en venas hepáticas, mediciones directas de oxígeno tisular y otros con objeto de medir mejor y comprender los requerimientos críticos de aporte de oxígeno a los tejidos de los pacientes sépticos. Sin embargo, el valor práctico de estas mediciones en el ámbito clínico de la sepsis aún está en duda.
La necesidad de mejorar la capacidad transportadora de oxígeno de la sangre por medio de transfusiones ha sido motivo de gran debate.71 Se ha demostrado que los humanos son muy resistentes a los efectos adversos de la reducción isovolumétrica en los valores de hemoglobina.72 El límite inferior para indicar una transfusión en el choque séptico no se ha definido, pero parece ser bastante menor que el umbral tradicional de menos de 10 g/dl.71
El tratamiento experto de la insuficiencia renal aguda, del SIRPA, de la descompensación hepática, la coagulopatía, las alteraciones ácido-base y los trastornos hemodinámicos son de gran importancia en el manejo de la sepsis.
El apoyo nutricional en el choque séptico ha cambiado mucho durante las últimas dos décadas. El uso de la nutrición parenteral total ha cambiado hacia un uso más temprano y frecuente de hiperalimentación enteral. Se ha demostrado que la alimentación enteral en los pacientes con sepsis beneficia la función de los enterocitos, ayuda a mantener la permeabilidad de la barrera intestinal y a evitar la presencia de endotoxina derivada del intestino y la generación de citocinas.73 El suplemento nutricional con glutamina, arginina y ácidos grasos omega-3 tiene bases experimentales y se usa cada vez más en pacientes sépticos.74 El valor de las fórmulas enterales enriquecidas en comparación con las estándar no ha sido confirmado en estudios clínicos extensos.
La fiebre es común en la sepsis severa y parece constituir una respuesta positiva.75 Las proteínas de calor de choque funcionan como chaperones intracelulares para estabilizar y prevenir la desnaturalización de las proteínas del huésped. La inducción de proteínas de calor de choque puede en realidad disminuir la mortalidad asociada a la administración experimental de endotoxina.76 Los esfuerzos por reducir la temperatura corporal con mantas frías han sido ineficaces y no benefician al paciente en choque séptico. Esta estretegia debe evitarse a menos que exista verdadera hipertermia.77
El tratamiento antimicrobiano más apropiado en la sepsis depende de la fuente de infección, los patrones de sensibilidd de los microrganismos patógenos en una determinada institución, la exposición previa a antimicrobianos, la presencia o ausencia de embarazo, la función hepática y renal y la historia de alergia a medicamentos [ver tabla 6]. En el choque séptico suelen administrarse combinaciones de antimicrobianos bactericidas en forma empírica. Las combinaciones disminuyen el riesgo de que un patógeno resistente a múltiples fármacos no sea cubierto y aumenta la probabilidad de que todos los patógenos importantes sean inhibidos por lo menos por uno de los agentes antimicrobianos. Aún existe preocupación sobre la liberación de endotoxina inducida por antibióticos en la sepsis, pero la importancia clínica de este efecto no se ha demostrado y no debe afectar la elección de antibióticos en el paciente séptico.78
|
||||||||||||||
|
Tratamientos experimentales en la sepsis
Durante los últimos 10 años han existido más de 20 estudios doble ciego, controlados con placebo, multicéntricos, en fase II o III que han evaluado la eficacia de nuevos agentes experimentales en el tratamiento del choque séptico.2,6 Ninguno de estos agentes ha demostrado en forma convincente ser útil en el ámbito clínico. A pesar de ello, continúan los trabajos por mejorar el tratamiento de la sepsis [ver tabla 7]. Es posible que en algunos años contemos con mejores estrategias para inhibir los efectos de la endotoxina, modular la síntesis del óxido nítrico, atenuar la coagulopatía y regular la respuesta inmunológica contra la sepsis. Se espera que las mejoras en el tratamiento de sostén, manejo de vasopresores y líquidos y estrategias de tratamiento para alterar la fisiopatología de la sepsis sean benéficas para los pacientes en un futuro cercano. El choque séptico sigue siendo un gran reto en el manejo actual de los pacientes graves. Mejorar la evolución del síndrome requiere de un esfuerzo concertado de científicos, investigadores clínicos y clínicos dedicados.
|
||||||||||||||||||||||||||||||
|
Figuras 1 y 2 Seward Hung.
- Bone RC. Toward a theory regarding the pathogenesis of the systemic inflammatory response syndrome: what we do and do not know about cytokine regulation. Crit Care Med 24.163, 1996
- Dellinger RP, Zimmerman J, Opal SM, et al. From the bench to the bedside: the future of sepsis research-Executive summary of an American College of Chest Physicians. National lnstitute of AlIergy and lnfectious Diseases, and National Heart, Lung, and Blood Institute Workshop, Chest 111744, 1997
- Opal SM: Lessons learned from clinical trials of sepsis, J Endotoxin Res 2:1.1995
- Increase in national hospital discharge survey rates for septicemia-United States, 1979-1987. MMWR 39:3l, 1990
- Bone RC, Balk RA, Cerra FB, et al: Definitions for sepsis in organ failure and guidelines for the use of innovative therapies in sepsis. Chest 101:1644, 1992
- Zeni F, Freeman B, Natanson C: Anti-inflammatory therapies to treat sepsis and septic shock: a reassessment, Crit Care Med 25:1097, 1997
- Bone RC, Fisher CJ Jr, Clemmer TP, et al: Sepsis syndrome: a valid clinical entity. Crit Care Med 17:339, 1989
- Cross AS, Opal SM, Sadoff JC, et al: Choice of bacteria in animal models of sepsis, Infect Immun 61:274l, 1993
- Sands KE, Bates DW, Lanken PN, et al: Epidemiology of sepsis syndrome in eight academic medical centers. JAMA 278:234, 1997
- Bone RC: Gram-positive organisms in sepsis. Arch Intern Med 154:26, 1994
- Rietschel ET, Brade H, Holst O, et al: Bacterial endotoxin: chemical composition, biological recognition, host response, and immunological detoxification, Curr Top Microbiol ImmunoI 216:39, 1996
- Horn DL, Opal SM, Lomastro E: Antibiotics, cytokines, and endotoxin: a complex and evolving relationship in gram-negative sepsis. Scand J Infect Dis 101:9, 1996
- Ulevitch RJ, Tobias PS: Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annu Rev ImmunoI 13:437, 1995
- Schumann RR, Leong SR, Flaggs GW: Structure and function of lipopolysaccharidebinding protein. Science 49:143l.1992
- Wright SD: CD14 and innate recognition of bacteria, J ImmunoI155:6, 1995
- Ulevitch RJ, Tobias PS. Recognition of endotoxin by cells leading to transmembrane signaling. Curr Opin ImmunoI 6:l25, 1994
- Barber SA, Perera P-Y, Vogel SN: Defective ceramide response in C3H/HeJ (Lpsd) macrophages. J ImmunoI 155:2303, 1995
- Opal SM, Palardy JE, Marra MN, et al: Relative concentrations of endotoxin-binding proteins in body fluids during infection. Lancet 344:429, 1994
- Jack RS, Fan X, Bemheiden M, et al: Lipopolysaccharide-binding protein is required to combat a murine gram-negative bacterial infection. Nature 389:742, 1997
- Haziot A, Ferrero E, Kontgen F, et al: Resistance to endotoxin shock and reduced dissemination of gram-negative bacteria in CD14-deficient mice. Immunity 4:407, 1996
| 20a | Scannon P: Personal communication, March 1998
|
- Marra MN, Wilde CG, Collins MS, et al: The role of bactericidal / permeability-increasing protein as a natural inhibitor of bacterial endotoxin. J lmmunoI 148:532, 1992
- von der Mohlen MAM, Kimmings AN, Wedel NI, et al: Inhibition of endotoxin-induced cytokine release and neutrophil activation in humans using recombinant bactericidal / permeability-increasing protein (rBPI23). J Infect Dis 172:144, 1995
- Flegel WA, Baumstark MW, Weinstock C, et al: Prevention of endotoxin induced monokine release by human low- and high-density lipoproteins and by human apolipoprotein A-1. Infect Immun 61:514, 1993
- Levine DM, Parker TS, Donnelly TM, et al: In vivo protection against endotoxin by plasma high density lipoprotein- Proc Natl Acad Sci USA 90:12040, 1993
- Pajkrt D, Doran JE, Coster F, et al: Anti-inflammatory effects of reconstituted highdensity lipoprotein during human endotoxemia. J Exp Med 184:1601, 1996
- Astiz ME, Rackow EC, Still JG, et al: Pretreatment of normal humans with monophosphoryl lipid A induces tolerance to endotoxin: a prospective, double-blind, randomized, controlled trial Crit Care Med 23:9, 1995
- Florquine S, Goldman M: Immunoregulatory mechanisms of T-cell-dependent shock induced by bacterial superantigen in mice. Infect Immun 64:3443, 1996
- Choi Y, Lafferty JA, Clements JRL: Selective expansion of T cells expressing V[beta]2 in toxic shock syndrome. J Exp Med 172:981, 1990
- Kusunoki T, Hailman E, Juan TS-C, et al: Molecules from Staphylococcus aureus that bind CD14 and stimulate innate immune responses. J Exp Med 182:1673, 1995
- Cleveland MG, Gorham JD, Murphy TL, et al: Lipoteichoic acid preparations from gram-positive bacteria induce interleukin-12 through a CD14-dependent pathway. lnfect Immun 64:1906, 1996
- Casey LC, BaIk RA, Bone RC: Plasma cytokine and endotoxin levels correlate with survival in patients with sepsis syndrome. Ann Intem Med 119:771, 1993
- Dinarello CA: The proinflammatory cytokines interleukin-1 and tumor necrosis factor and treatment of the septic shock syndrome. J Infect Dis 63:1177, 1991
- Dinarello CA, Gelfand JA, Wolff SM: Anticytokine strategies in the treatment of the systemic inflammatory response syndrome: JAMA 269:1825, 1993
- Fantuzzi G, Dinarello CA: The inflammatory response in interleukin-1 beta-deficient mice: comparison with other cytokine-related knockout mice- J Leukoc BioI 59:489, 1996
- Bone RC: Sir lsaac Newton, sepsis, SIRS and CARS. Crit Care Med 24:1125, 1996
- Abraham E: Physiologic stress and cellular ischemia: relationship to immunosuppression and susceptibility to infection. Crit Care Med 19:613, 1991
- Falk LA, McNally R, Perera PY, et al: LPS-inducible responses in severe combined immunodeficiency (SCID) mice. J Endotoxin Res 2:273, 1995
- Szabo SJ, Dinghe AS, Gubler U, et al: Regulation of the interleukin (lL)-12R[beta]2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med 185-817, 1997
- Williams MA, White SA, Miller JJ, et al: Granulocyte-macrophage colony-stimulating factor induces activation and restores respiratory burst activity in monocytes from septic patients. J Infect Dis 177:107, 1998
- Kox WJ, Bone RC, Krausch D, et al: Interferon gamma-1b in the treatment of compensatory anti-inflammatory response syndrome: new approach: proof of principle. Arch Intern Med 157:389, 1997
- van Deventer SJH, Buller HR, ten Cate JW , et al: Experimental endotoxemia in humans: analysis of cytokine release and coagulation, fibrinolytic and complement pathways- Blood 76:2520, 1990
- Bauer KA, ten Cate H, Barzegar S, et al: Tumor necrosis factor infusions have a procoagulant effect on the hemostatic mechanism of humans. Blood 74:165, 1989
- Levi M, ten Cate H, van der Poll T, et al: Pathogenesis of disseminated intravascular coagulation in sepsis. JAMA 270:975, 1993
- Smith OP, White B, Vaughn D, et al: Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. Lancet 350:1590, 1997
- Luster AD: Chemokines: chemotactic cytokines that mediate inflammation. N Engl J Med 338:436, 1998
- Shenep JL, Toumanen E: Prospective: targeting nitric oxide in the adjuvant therapy of sepsis in meningitis. J Infect Dis 177:766, 1998
- Petros A, Bennett D, Vallance P: Effect of nitric oxide synthase inhibitors on hypotension in patients with septic shock. Lancet 338:1557, 1991
- Parker MN, Suffredini AF, Natanson C, et al: Responses of left ventricular function in survivors and non-survivors of septic shock. J Crit Care 4:19, 1989
- Shoemaker WC, Montgomery ES, Kaplan E, et al: Physiologic patterns in surviving and non-surviving shock patients. Arch Surg 106:630, 1973
- Hayes MA, Timmins AC, Yau EHS, et al: Elevation of systemic oxygen delivery in thetreatment of critically ill patients. N Engl J Med 330:17l7, 1994
- Pittet D, Thiévent B, Wenzel RP , et al: Importance of pre-existing co-morbidities for prognosis of septicemia in critically ill patients. Intensive Care Med 19:265, 1993
- Finkel MS, Oddis CV, Jacob TD, et al: Negative inotropic effect of cytokines on the heart mediated by nitric oxide. Science 257:387, 1992
- Milberg JA, Davis DR, Steinberg KP, et al: Improved survival of patients with acute respiratory distress syndrome (ARDS): 1983-1993. JAMA 273:306, 1995
- Opal SM, Palardy JE, Parejo N, et al: Endotoxemia in sepsis: therapeutic and prognostic implications [abstr # G-88]. 31st Interscience Conference in Antimicrobial Agents and Chemotherapy 1995, San Francisco
- Sriskandan S, Moyes D, Cohen J: Detection of circulating bacterial superantigen and lymphotoxin alpha in patients with streptococcal toxic-shock syndrome Lancet 348: 1315, 1986
- Damas P, Ledoux D, Nys M, et al: Cytokine serum levels during severe sepsis in humans: IL-6 as a marker of severity. Ann Surg 215:356, 1992
- Reinhart K, Wiegand-Löhnert C, Grimminger F, et al: Assessment of the safety and efficacy of the monoclonal anti-tumor necrosis factor antibody-fragment, MAK-l95F, in patients with sepsis and septic shock: a multicenter, randomized, placebo-controlled, dose-ranging study. Crit Care Med 24:733, 1996
- Opal SM, Fisher CJ, Pribble JP, et al: The confirmatory interleukin-1 receptor antagonist trial in sepsis: a phase III randomized, double-blind, placebo-controlled, multicenter triaI. Crit Care Med 25:1115, 1997
- Fisher CJ, Dhainaut J-F, Opal SM, et al: Recombinant human interleukin-1 receptor antagonist in the treatment of patients with sepsis syndrome: results from a randomized, double-blind, placebo-controlled triaI, JAMA 271:1836, 1994
- Fisher CJ, Opal SM, Dhainaut J-F, et al: Influence of a murine anti-TNF monoclonal antibody (CB0006) on cytokine levels in patients with sepsis. Crit Care Med 2:318, 1993
- Gendrel D, Assicot M, Raymond J, et al: Procalcitonin as a marker for the early diagnosis of neonatal infection. J Pediatr l28:570, 1996
- deWerra I, Jacchard C, Corradin SB, et al: Cytokines, nitrite / nitrate, soluble tumor necrosis factor receptors, and procalcitonin concentration: comparisons in patients with septic shock, cardiogenic shock, and bacterial pneumonia. Crit Care Med 25:607, 1997
- Staehler M, Hammer C, Meiser B, et al: Procalcitonin: a new marker for differential diagnosis of acute rejection and bacterial infection in heart transplantation. Transplant Proc 29:584, 1997
- Karzai W, Oberhoffer M, Meier-HeImann A, et al: Procaldtonin a new inclicator of systemic response to severe infections. Infection 6:629, 1997
- Hack CA, Hart M, van SchIjndel RJ, et al: Interleukin-8 in sepsis: relation to shock and inflammatory mediators. Infect lmmun 60:2835, 1992
- Rae D, Porter J, Beechey-Newman N, et al: Type 1-prophospholipase A2 propeptide in acute lung injury. Lancet 344:1472, 1994
- Rackow EC, Kaufman Bs, Falk JL, et al: Hemodynamic response to fluid resuscitation in patients with septic shock: evidence for early depression of carcliac performance. Circulatory Shock 22:1l, 1987
- Martin C, Papazian L, Perrin G, et al: Norepinephrine or dopamine for the treatment of hyperdynamic septic shock? Chest 103:1826, 1993
- Germann R, Haiskackl M, Hasibeder W, et al: Dopamine and mucosal oxygenation in porcine jejunum. J Appl Physiol 77:2845, 1994
- Marin C, Eon B, Saux P, et al: Renal effects of norepinephrine used to treat septic shock patients. Crit Care Med 18:282, 1990
- Hébert PC, Wells G, Marshall J, et al: Transfusion requirements in critical care. JAMA 273:1439, 1995
- Weiskopf RB, Viely MK, Feiner J, et al: Human cardiovascular and metabolic response to acute, severe isovolemic anemia. JAMA 279:217, 1998
- Kudsk KA, Croce MA, Fabian TC, et al: Enteral vs parenteral feeding: effects on septic morbidity after blunt and penetrating abdominal trauma. Ann surg 215:503, 1992
- Bower RH, Cerra FB, Bershadshy B, et al: Early enteral administration of a formula (Impact) supplemented with arginine, nucleotides, and fish oil in intensive care unit patients: results of a multicenter prospective, randomized, clinical trial. Crit Care Med 23:436, 1995
- Kluger MJ, Kozak W, Conn C, et al: The adaptive value of fever. Infect Dis Clin North Am 10:1, 1996
- Ribeiro SP, Chu E, Slutsky As: Heat stress after injection of LPS in rats decreases mortality. Am J Respir Crit Care Med 153:A252, 1996
- O'Donnell J, Axelrod R, Fisher C, et al: Use and effectiveness of hypothermia blankets for febrile patients in the intensive care unit. Clin Infect Dis 24:1208, 1997
- Prins JM, van Deventer SJH, Kuijper EJ, et al: Clinical relevance of antibiotic-induced endotoxin release. Antimicrob Agents Chemother 38:1211, 1994
- Carlos TN, Harlan JM: Leukocyte-endothelial adhesion molecules. Blood 84:2068, 1994