Oncología
⭳ Abrir artículo (PDF)567.0 KBEste artículo fue revisado respecto a la Edición 2/2000. Ver esa versión →
Contenido del artículo
V CRECIMIENTO TUMORAL Y QUIMIOTERAPIA
- Crecimiento celular y tisular
- CONCEPTOS DE CRECIMIENTO CELULAR
- CONCEPTOS DE CRECIMIENTO TISULAR
- CURVA DE GOMPERTZ
- APOPTOSIS
- MODELO DE FRACCION DE CRECIMIENTO
- Quimioterapia
- Heterogeneidad de las células tumorales
- Estrategias en el uso de la quimioterapia
- PERFUSION SISTEMICA Y REGIONAL
- QUIMIOTERAPIA CON DOSIS INTENSIVAS Y TRASPLANTE DE MEDULA OSEA ALOGENICA O SINGENICA O DE CELULAS TRONCO DE SANGRE PERIFERICA
- ADMINISTRACION INTRACAVITARIA DE LA QUIMIOTERAPIA
- QUIMIOTERAPIA DIRIGIDA
- MODIFICADORES DE LA RESPUESTA BIOLOGICA
- VACUNAS TUMORALES
- QUlMIOTERAPIA NEOADYUVANTE
- QUIMIOPREVENCION E INDUCCION DE LA DIFERENCIACION
- Terapia génica
- Marcadores tumorales en el manejo del cáncer
- Estudio de células tumorales tronco en humanos
- Agentes anticancerosos de uso común
V CRECIMIENTO TUMORAL Y QUIMIOTERAPIA
DR. FRANK E. STOCKDALE, PH.D.
La era moderna de la quimioterapia para el cáncer empezó a principios de los años 40 con la introducción de la mostaza nitrogenada (hidroclomro de mecloretamina). Aunque actualmente ésta se usa poco, muchos medicamentos con origen, estructura y acción diferentes se emplean comúnmente para tratar una amplia variedad de cánceres en el humano. Estos agentes comparten una característica: por lo general son más eficaces para matar o dañar células malignas que células normales. Sin embargo, el hecho de que dañen células normales señala su gran potencial para causar toxicidad. La comprensión del uso de la químíoterapia requiere del entendimento tanto del mecanismo de acción del medicamento como de la fisiopatología del cáncer, que tiene su origen en un trastorno del crecimento celular y tisular. La estrategia de la quimioterapia se basa en los conocimientos existentes sobre crecimiento del tejido tumoral.
Crecimiento celular y tisular
El término cáncer describe un grupo de enfermedades caracterizadas por acúmulo inadecuado de tejido. Esta alteración es más evidente clínicamente cuando la masa de tejido tumoral compromete la función de órganos vitales. Al contrario de lo que suele pensarse, las neoplasias malignas en los humanos no suelen ser enfermedades de proliferación celular rápida; de hecho, las células de la mayor parte de las neoplasias comunes proliferan en forma más lenta que muchas células normales.1-3 Es el acúmulo relativamente lento del tejido tumoral en los órganos vitales lo que provoca la muerte en muchos de los pacientes con cáncer.
Los conceptos que describen el crecimiento de los tejidos normales son aplicables a los tejidos malignos porque los tejidos y células normales y malignos comparten características similares de crecimiento.4 Se deben distinguir los conceptos que describen el crecimiento de células individuales de los que describen el crecimiento de poblaciones de células o de tejidos [ver tabla 1].
CONCEPTOS DE CRECIMIENTO CELULAR
El tiempo del ciclo celular, o tiempo de generación de una célula normal o maligna, es el lapso requerido para que una sola célula atraviese por un ciclo celular complcto.5 El ciclo celular se divide dé manera arbitraria en cuatro o cinco fases. G1, o intervalo 1, es la fase durante la que se sintetízan ARN y proteínas para una variedad de funciones celulares diferenciadoras. Aunque de duración variable, la fase G1, es la porción más larga del ciclo celular. Durante la fase S, o de síntesis de ADN, se replica todo el genoma. La fase G2, o intervalo 2, ocurre entre el término de la síntesis de ADN y el inicio de los cambios nucleares que ocurren en preparación a la condensación de los cromosomas. Durante la fase M, o mitosis, ocurren los movimientos de los cromosomas y la división celular (citocinesis), que causan la distribución de los cromosomas en dos células hijas. G0 describe una fase prolongada de reposo durante la cual las células no responden a estímulos que normalmente iniciarían la síntesis de ADN. El tiempo del ciclo celular de la mayor parte de las células normales es de cerca de 24 a 48 horas, mientras que el de las células malignas de los tumores humanos comunes puede ser hasta de 120 horas. Sin embargo, el tiempo de generación, tanto de las células normales como de las malignas, no es fijo, y puede ser fácilmente modulado por una variedad de factores en el ambiente de la célula.
CONCEPTOS DE CRECIMIENTO TISULAR
Los cinco conceptos de crecimiento tisular no deben ser confundidos con los conceptos de crecimiento celular [ver tabla 1]. El cáncer es una enfermedad caracterizada por un trastorno en la regulación del crecimiento tisular, al igual que por un trastorno en la regulación del crecimiento celular. Los conceptos tisulares son los más importantes para el entendimiento clínico del cáncer y de su quimioterapia.
El tiempo de duplicación se refiere a la duración requerida para que un tejido o turnor duplique su tamaño o número de células. Aunque pudiera esperarse que los tiempos del ciclo celular y de duplicación tisular fueran iguales, el tiempo de duplicación de un tumor clínicamente aparente es por lo común mucho más prolongado que el del ciclo celular de las células que conforman al tumor. Cuatro parámetros del crecimiento tisular (el índice de marcado, el índice mitótico, la fracción de crecimiento y la fracción de muerte celular) pueden ser responsables de esta discrepancia.
El índice de marcado es el porcentaje de células que producen ADN en un tejido que se encuentra en la fase S del ciclo celular). En la mayor parte de los tumores humanos este porcentaje es relativamente bajo (cinco a 20 porciento).6 De manera similar, el índice mitótico es el porcentaje de células en un tejido o tumor que están en mitosis en cualquier momento.
La fracción de crecímiento es el porcentaje de la población celular total que participa en el crecimiento del tejido o tumor, y representa la fracción de células que está en el ciclo.7 La fracción de crecimiento es la mejor medida de la velocidad de crecimiento tisular. Este concepto ha influido profundamente en la práctica de la quimioterapia. La fracción de crecimiento clínicamente aparente de los cánceres humanos es por lo general pequeña (menos de la mitad de las células están en ciclo), de manera que gran parte de las células cancerosas en la mayoría de los pacientes están en la fase G0. Así, la fracción de crecimiento de muchos tejidos normales en un paciente con cáncer frecuentemente excede la del tejido tumoral. Los tejidos normales con fracciones de crecimiento mayores son los más afectados por los agentes quimioterápicos, que son tóxicos para las células en proliferación. Dichos tejidos normales (y sus respuestas a la toxicidad) incluyen la médula ósea (leucopenia, trombocitopenia, anemia), los precursores de los gametos (esterilidad), los folículos pilosos (alopecia), y el epitelio que cubre el tracto digestivo (ulceraciones orales, diarrea).
La fracción de muerte celular es el porcentaje de células hijas recién producidas que mueren. Este número es alto en el tejido tumoral de muchos de los pacientes con cáncer, lo que indica que la mayor parte de la progenie de las células tumorales muere en lugar de acumularse. Así, el grado de desequilibrio entre la producción de células tumorales nuevas (medido por el índice de marcado, el índice mitótico y la fracción de crecimiento) y la muerte celular, determina la velocidad con la que crece una masa tumoral. Cuando ésta relación se altera de manera que favorece la producción de células tumorales, el tejido tumoral se acumula en el sitio primario de un tumor o en los sitios de metástasis.
CURVA DE GOMPERTZ
A diferencia de un tumor, el hígado, el corazón o los pulmones no tienen tiempo de duplicación en un adulto sano. Estos granos se encuentran en un estado estable, las velocidades de producción y muerte celular son iguales, de modo que con el paso del tiempo no existe aumento en su tamaño o en el número de células. Sin embargo, las características de crecimiento de los tumores pueden compararse con las de los fetos humanos y de los niños, en quienes la producción de células nuevas excede a la muerte celular. En cada caso existe un tiempo de duplicación, y se puede definir un patrón de crecimiento específico para todo un tejido, órgano u organismo. La pendiente de las curvas de crecimiento para un feto humano normal y para las células de la leucemia humana son semejantes [ver figura 1]. El matemático Gompertz desarrolló una ecuación que describe este fenómeno de manera muy aproximada. Desde entonces esta curva se conoce como curva de Gompertz.1
La curva de Gompertz indica que durante el desarrollo de un feto humano normal o de un tumor, la velocidad de crecimiento es muy rápida al inicio y luego disminuye progresivamente al aumentar el tamaño. Esta observación tiene gran importancia en las limitaciones de la quimioterapia actual contra el cáncer. Los mecanismos que contribuyen a la norma de la curva de Gompertz siguen sin comprenderse en su totalidad, pero al menos dos factores importantes participan en la disminución de la velocidad de crecimiento al aumentar el tamaño de los tumores: la disminución en la fracción de crecimiento y el aumento en la muerte celular espontánea. Al acumularse las células en un tejido normal, en un tumor primario o en una metástasis, se produce disminución de la frecuencia con la que se inician nuevos ciclos de síntesis de ADN, y por lo tanto, con la que se producen nuevas células. Cuando un tumor o un feto es muy pequeño, se encuentran en la parte baja de la curva de Gompertz y tienen una gran fracción de crecimiento. Cuando un tejido es grande, se sitúa más arriba en la curva de Gompertz y tiene una fracción de crecimiento pequeña. Cuando un tejido es muy pequeño es probable que casi cada célula esté en ciclo y, por tanto, participando en el crecimiento. Aunque la ecuación de Gompertz describe el crecimiento de los tejidos y organismos en general, no todo el crecimiento tumoral o normal ocurre por un proceso continuo.8 Por ejemplo, los estudios sobre el crecimiento temprano en niños han demostrado que cuando se mide con frecuencia el crecimiento, este se presenta como un fenómeno intermitente, en el que periodos de crecimiento rápido se intercalan con periodos de poco o ningún crecimiento. En forma semejante, las medidas de los tumores humanos han demostrado que periodos de crecimiento acelerado pueden ser seguidos de periodos de crecimiento más lento o nulo. Los cálculos empleando el concepto de Gompertz sugieren que el acumulo de células cancerosas en la mayoría de los tumores humanos comienza alrededor de dos años antes de su detección. En la actualidad puede demostrarse la equivocación de los investigadores de hace dos décadas que calcularon en forma errónea, basándose en la lenta velocidad de crecimiento de tumores lo suficientemente grandes para ser observados por radiografías, que éstos se habían originado aproximadamente 10 a 20 años antes de su detección. Este error se ejemplifica mejor en el cáncer de los lactantes. Sin aplicar el concepto de crecimiento de Gompertz, podría pensarse que ciertas neoplasias de la infancia se originaron antes de la concepción.
Factores que afectan la curva de Gompertz
La disminución progresiva de la entrada de células a la síntesis de ADN ( fase S), descrita por la curva de Gompertz, no es causada por un cambio estable en las células tumorales individuales. Más bien, refleja el hecho de que las células son parte de una población celular y de que las señales responsables de iniciar el crecimiento (v.gr., hormonas, facíores de crecimiento y nutrientes) cambian al acumularse las células.Si se quitan unas cuantas células de un tumor lo suficientemente grande para estar en la parte superior de la curva de Gompertz y luego se implantan en otro sitio, estas células vuelven a entrar al ciclo celular, su fracción de crecimiento aumenta, y forman un nuevo tumor a una velocidad que se describe en forma exacta en la curva de Gompertz. La capacidad de una célula cancerosa para volver a entrar en el ciclo celular al reducir se la población de células tumorales es aprovechada por el tratamiento de modalidad combinada contra el cáncer.
Es probable que la muerte celular espontánea y la necrosis tisular sean las principales causas naturales de retraso en el crecimiento tumoral en el cáncer clínicamente aparente.9 Los informes de patología a menudo afirman que las piezas contienen grandes áreas de necrosis, y es posible que por este mecanismo se pierdan rápidamente gramos de tejido (> 109 células). Se calcula que del 70 al 90 porciento de las células tumorales recientemente producidas en los humanos mueren espontáneamente. Si este no fuera el caso, es probable que muchos pacientes murieran antes de entrar al sistema de salud, porque los tumores duplicarían su tamaño en cuestión de días.
Se entienden poco los mecanismos responsables de la muerte celular, pero quizá se relacionen con cambios en la estructura del genoma de la célula cancerosa, la apoptosis, con lo que se puede llamar en forma amplia nutrición celular, con varios mecanismos inmunológicos y con otros factores. Por ejemplo pueden surgir defectos en la estructura de los cromosomas durante su replicación y segregación. También se pueden segregar números anormales de cromosomas durante la mitosis, de manera que muchas células hijas carecen sin duda de los genes o de los mecanismos reguladores que se requieren para la supervivencia. Diversos mecanismos pueden causar apoptosis, o muerte celular iniciada en forma activa (ver adelante). Además, los tumores tienen vasculatura anormal, son comunes la ruptura de vasos, la coagulación y la hemorragia. La mala oxigenación y la carencia de nutrientes pue den matar muchas células. El papel del sistema inmunológico humano en la muerte de las células tumorales no se comprende aún, pero es probable que ciertas clases de linfocitos contribuyan también a este proceso.10 Además, genes específicos regulan la duración de la vida de muchos tipos celulares. Uno de estos genes, el bc1-2, ha mutado en algunas neoplasias humanas, en especial en los linfomas, causando pérdida del control de la supervivencia celular, con supervivencia prolongada de las células que han sufrido cambios neoplásicos.11
APOPTOSIS
La apoptosis es un proceso fisiológico normal que regula en forma específica la supervivencia celular.12,13 Es indispensable para la morfogénesis durante el proceso de desarrollo embrionario, para la selección clonal en el sistema inmunológico y quizá en la citotoxicidad mediada por células, y para la muerte de algunas células inducida por quimioterapia y radiación.14,15 Algunos cambios en la morfología nuclear y en la degradación del ADN en fragmentos son característicos de las células que sufren apoptosis. La apoptosis es un proceso activo que requiere síntesis de proteínas que puede ser desencadenada por cambios en el ambiente celular. Los aumentos o reducciones en los niveles de hormonas o de citocinas pueden iniciar la apoptosis. Por ejemplo, la eliminación de andrógenos puede iniciar la apoptosis en células epiteliales de la próstata. El gen supresor de tumores p53, cuya expresión es inducida cuando se daña el genoma de una célula y el oncogene bc1-2 tienen un papel crucial en la regulación de la apoptosis. Aunado a otros genes, el gen p53 vigila el estado del ADN de la célula. Si no se repara el daño al ADN, el producto del gen p53 inicia un proceso que termina en la apoptosis.16 El oncogen bc1-2 produce una sustancia que prohibe la muerte celular o apoptosis. Cuando el gen bc1-2 está activo se altera la apoptosis. Algunos agentes como las citocinas, los factores de crecimiento parácrino, los agonistas de los canales del calcio y los agentes anti hormonas esteroideas favorecen la apoptosis, mientras que otros agentes, como los productos del gen bcl-2, otros oncogenes, las hormonas endócrinas y quizá el virus Epstein-Barr, la retrasan.17,18 La falla de la muerte celular programada o la inhibición de los pasos intermedios que activan los mecanismos que causan la muerte celular pueden ser importantes en el inicio y progresión de los tumores desde el punto de vista clínico. Por lo tanto, la mejor comprensión de la apoptosis permitirá desarrollar nuevas estrategias para combatir el acúmulo de células neoplásicas.
MODELO DE FRACCION DE CRECIMIENTO
Se ha usado un modelo conceptual del crecimiento tumoral, comúnmente llamado modelo de la fracción de crecimiento, para desarrollar la mayor parte de las estrategias de quimioterapia [ver figura 2].20 En este modelo un tumor comprende cuatro compartimientos celulares: el compartimiento A contiene células en ciclo, que participan en la producción de células nuevas; el comportamiento B contiene células fuera de ciclo, que se encuentran detenidas temporalmente en la fase G0, pero que pueden reingresar al compartimiento de células en ciclo cuando se les aplica un estímulo apropiado; el comportamiento C contiene unas cuantas células vivas, pero detenidas, que no responden a las señales que estimulan el crecimiento ni vuelven a entrar al compartimiento de células en ciclo, y el compartimiento D representa a las células muertas, que pueden haber estado en cualquier compartimiento antes de morir. Cuando un tumor tiene una fracción de crecimiento alta, la mayor parte de sus células está en el compartimiento A; cuando un tumor tiene una fracción de crecimiento baja, la mayor parte de sus células está en los compartimientos B y C.
Casi todos los agentes quimioterápicos actualmente en uso interfieren con la síntesis de ADN, con la provisión de precursores para la síntesis de ADN y ARN o con la mitosis. Estos medicamentos son más eficaces contra las células en ciclo. Los mecanismos de muerte celular después del tratamiento con un solo agente o con la combinación de varios son complejos, y es probable que incluyan más de un proceso. Debido a que la mayoría de los tumores detectables por clínica se encuentran en la porción alta de la curva de Gompertz y están compuestos principalmente por células del compartimiento B que no están en el ciclo, no es sorprendente que la quimioterapia no siempre sea eficaz para erradicar el cáncer.
La estrategia del tratamiento del cáncer consiste en hacer que las células tumorales cambien del compartimiento B, fuera de ciclo, al compartimiento A, en ciclo. Varios métodos que promueven este cambio forman la base del tratamiento combinado. La cirugía es el método que más se emplea para reducir el tamaño del tumor y facilitar así la reentrada de células neoplásicas dentro del ciclo celular. Después de que el tumor primario se ha extirpado por completo, pueden quedar metástasis microscópicas en sitios distantes. Debido a su tamaño pequeño, las micrometástasis están compuestas principalmente de células en ciclo. El pequeño número de células que permanecen en el sitio del tumor primario pueden también reentrar al ciclo celular (i.e., cambiarse al compartimiento A). Por lo tanto, las células neoplásicas restantes son susceptibles a la quimioterapia. Puede usarse el tratamiento con radioterapia o quimioterapia solos para reducir la masa tumoral y así reclutar células dentro del compartimiento A, en ciclo. La estrategia de la quimioterapia adyuvante para el cáncer de mama se basa en estos conceptos.21,22
Quimioterapia
QUIMIOTERAPIA COMBINADA
Las investigaciones de tumores en animales y los estudios clínicos en humanos han demostrado que las combinaciones de medicamentos producen tasas de respuesta objetiva superiores y supervivencia más prolongada que los agentes únicos.23 Por lo tanto, el tratamiento combinado es la base de la mayoría de los esquemas de quimioterapia que se emplean en la actualidad. La quimioterapia combinada emplea mecanismos diferentes de acción y combina la citotoxicidad potencial de los diversos agentes. Aunque todos los medicamentos quimioterapéuticos son más eficaces sobre las células que sintetizan ADN, muchos agentes (en especial los alquilantes) pueden destruir células que no están en ciclo. Estos agentes se denominan medicamentos no dependientes de la proliferación celular. Algunos agentes, incluyendo muchos de los antimetabolitos y antibióticos, son mas eficaces contra las células durante la síntesis de ADN, por lo que se denominan medicamentos dependientes de la proliferación celular. La administración repetitiva de agentes no dependientes de la proliferación celular puede disminuir la masa tumoral al destruir células tanto de los compartimientos en ciclo como de fuera del ciclo. A continuación las células supervivientes se dirigen hacia el compartimiento de células en ciclo, donde son más sensibles a los medicamentos dependientes de la proliferación celular. El uso combinado de agentes no dependientes del ciclo celular, seguido de aquellos sí dependientes de este ciclo, es eficaz para aumentar la muerte de las células tumorales [ver figuras 2 y 3]. Cada ciclo de tratamiento destruye una fracción fija de células, de manera que se requieren ciclos repetidos para lograr la curación. Por ejemplo, una combinación de medicamento que destruya el 99.9 porciento de las células cancerosas por ciclo de tratamiento tendría que ser repetida por lo menos seis veces para eliminar una masa tumoral promedio (si no ocurriera crecimiento de células tumorales entre los ciclos).
Estos modelos son necesariamente simplistas y sin duda minimizan factores importantes que pudieran reforzar la quimioterapia contra el cáncer. Sin embargo, cuando en los modelos fueron desarrollados por Skipper y colaboradores, proporcionaron la base teórica que se utilizó para curar la leucemia en niños.20
Varios principios guían la selección de medicamentos que se usan en combinación.24 Los medicamentos que son eficaces en forma individual se deben combinar en dosis tan altas y administrarse con tanta frecuencia como lo permita la toxicidad. Por lo tanto, son preferibles las combinaciones en donde no se superponen los tipos de toxicidad. Los medicamentos seleccionados deben también tener mecanismos diferentes de acción. Esta medida aumenta la destrucción celular, reduce la posibilidad de que surjan poblaciones de células resistentes al medicamento e interrumpen las funciones de las células neoplásicas al atacar múltiples vías metabólicas.
FACTORES QUE INFLUYEN EN LA EFICACIA DE LA QUIMIOTERAPIA
Para que la quimioterapia sea eficaz, el paciente debe estar en buenas condiciones fisiológicas, el tumor debe tener características de crecimiento favorables y los medicamentos deben ser eficaces contra ese tumor en particular. Entre las características relacionadas con el desarrollo tumoral que limitan la eficacia de la quimioterapia se encuentran la existencia de una gran masa tumoral en el momento del diagnóstico, una fracción de crecimiento tumoral baja, resistencia de las células tumorales a los medicamentos quimioterápicos, y la existencia de santuarios dentro del organismo que limiten el acceso de las drogas a las células tumorales [ver tabla 2].
Una gran masa tumoral y la resistencia a varios medicamentos son los dos factores más importantes que limitan la eficacia de la quimioterapia. Las neoplasias malignas graves más comunes en humanos (cáncer de pulmón, mama, colon y próstata) rara vez se detectan antes de que el tumor primario haya crecido por lo menos hasta 1 cm y contenga aproximadamente 109 células. En el momento del diagnóstico muchos pacientes con cáncer tienen más de 1010 células neoplásicas (10 billones de células, o alrededor de 10 g de tumor) y con frecuencia pueden tener más de 100 billones de células. (La mayoría de los pacientes con cáncer mueren cuando sus masas celulares tumorales se acercan a las 1012 células, o 1 kg. de tumor). Por lo tanto, aunque existiera un medicamento que destruyera con eficacia el 99.9 porciento de las células tumorales, quedarían de 106 a 107 (de un millón a 10 millones) de células en el paciente típico con cáncer después del tratamiento inicial.
La eficacia de la quimioterapia puede alterarse por la cinética de crecimiento tumoral, la masa de células tumorales en todo el organismo, los efectos tóxicos de la quimioterapia sobre las células y otros tejidos diferentes al tumor y el deterioro de la función renal y hepática. Cuando las neoplasias son lo suficientemente grandes para ser detectadas, se encuentran en la parte alta de la curva de Gompertz (donde la fracción de crecimiento es baja) y ya ha ocurrido la mayor parte del crecimiento. La mayoría de estas células tumorales no están proliferando, por lo que es menos probable que cualquier agente quimioterápico las destruya. En forma semejante, a mayor tamaño del tumor primario, mayor probabilidad de que un número importante de células (resistentes y sensibles a los medicamentos) hayan metastatizado antes del diagnóstico y sean resistentes a la quimioterapia. Debido a que la mayoría de los agentes quimioterapéuticos dependen de la función renal y hepática para la excreción o modificación de los medicamentos, cuando existe daño en la función renal o hepática será necesario modificar las dosis para evitar toxicidad severa.
En el momento del diagnóstico muchos tumores humanos ya contienen células cancerosas que son resistentes a la quimioterapia estándar [ver adelante, Heterogeneidad de las células tumorales, Resistencia a los medicamentos].25,26 Se calcula en forma conservadora que existen mutaciones espontáneas que confieren resistencia a los medicamentos en una de cada l06-107 células neoplásicas. Esta velocidad de mutación parece ser independiente de cualquier presión selectiva por parte del tratamiento medicamentos, aunque la radioterapia y la quimioterapia pueden originar mutaciones adicionales. Las mutaciones, la pérdida de genes supresores de tumores y la activación de oncogenes aumentan también la velocidad de mutación y contribuyen a la progresión del tumor dentro de la población de células cancerosas.27-30 De ahí que la masa de células cancerosas en el momento del diagnóstico es de gran importancia porque incluso tumores de 1 cm (109 células) podrían contener hasta 100 a 1,000 células resistentes a medicamentos antes de iniciar el tratamiento.
Algunas metástasis se originan en ciertos sitios del organismo en donde son resistentes a la quimioterapia por la poca distribución tisular de los agentes administrados en dosis estándar. Estos sitios actúan como santuarios que protegen a las células neoplásicas de los medicamentos que circulan en sangre. Por ejemplo, existe este tipo de barrera en el cerebro y los testículos, que impiden la difusión de los medicamentos de los capilares hacia el tejido. Por lo tanto, estos sitios requieren modalidades especiales de tratamiento.
Heterogeneidad de las células tumorales
Han ocurrido grandes adelantos en el conocimiento de la composición celular de las neoplasias. Antes se pensaba que los tumores malignos estaban compuestos por poblaciones celulares uniformes u homogéneas, y que la caracterización de una sola célula podía ayudar a entender el funcionamiento de toda la población celular. Ahora está claro que un tumor maligno se encuentra compuesto por células que son heterogéneas para casi todas las características estudiadas.31,32 Se han observado las principales divergencias en el tamaño y forma de las células (morfología e histología), cariotipo (i.e., composición cromosómica), antígenos de superficie celular, respuesta a medicamentos (resistencia), capacidad de metastatizar y proliferar y concentración intracélular de enzimas y marcadores de células cancerosas (v.gr., receptores para estrógenos).
La mayor parte de los tumores malignos, aunque no todos, se originan de una sola célula o clona, y la población celular descendiente se diversifica debido a la constante aparición y selección de nuevas clonas,29,33,34 Los procesos que determinan la evolución clonal en los cánceres humanos son poco conocidos. Es posible que participen los procesos ocasionados por la inestabilidad genética (quizá provocados por genes que aumentan las mutaciones), como las rupturas cromosómicas, las traslocaciones, la activación de oncogenes y la pérdida de genes supresores de tumores. También pueden participar cambios en el ambiente del huésped, como las alteraciones en la regulación inmunológica Algunos linfomas parecen originarse de más de una clona y continúan cambiando respecto a la estructura celular y la respuesta al tratamiento, lo mismo que los cánceres que se originan de una sola clona.35 La comprensión de los mecanismos responsables de la diversificación continua de las poblaciones tumorales celulares que originan la progresión del tumor es el principal reto para poder desarrollar nuevos tratamientos contra el cáncer.31-33,36
RESISTENCIA A LOS MEDICAMENTOS
La resistencia de las células tumorales a los efectos destructores de la quimioterapia es uno de los problemas centrales en el tratamiento del cáncer. La destrucción selectiva de las células tumorales sensibles a los medicamentos causa una mayor proliferación de las células tumorales que son resistentes a la quimioterapia. Entre los mecanismos de resistencia a los medicamentos se incluyen el menor acumulo de medicamento (en especial la resistencia a medicamentos múltiples), el aumento en los mecanismos de transferencia de glutatión y de detoxificación, los niveles bajos de ADN topoisomerasa II, las alteraciones de enzimas o metabolismo de medicamentos, y el aumento en la capacidad de las células neoplásicas para reparar el daño inducido por medicamentos.37 Las células que crecen en exceso en la población tumoral no solo son resistentes a los agentes empleados, sino tienden a ser resistentes también a otros medicamentos, muchos de los cuales tienen mecanismos de acción distintos. Este fenómeno, denominado resistencia pleiotrópica a medicamentos o resistencia a múltiples fármacos, puede ser responsable de gran parte de la resistencia a los medicamentos que ocurre en los pacientes con cáncer tratados en forma previa.
La amplificación de los genes (i.e., la producción de copias extra de genes dentro de una célula) es uno de los mecanismos que puede originar resistencia a los medicamentos. Este mecanismo se ha investigado sobre todo en tumores que son resistentes a metotrexate tanto in vitro como in vivo.38 El metotrexate actúa al inhibir la acción de la enzima tetrahidrofolato reductasa. La amplificación del gen que codifica la tetrahidrofolato reductasa causa producción excesiva de la enzima, originando resistencia al medicamento. Además de la amplificación genética, los cambios en la velocidad del transporte del metotrexate hacia las células cancerosas y en la estructura de la dihidrofolato reductasa puede contribuir a la resistencia clínica a este fármaco.
La amplificacíón de genes puede influir también en el fenómeno de resistencia a múltiples fármacos. Esta parece estar asociada a la mayor expresión de una glucoproteína de la membrana celular denominada glucoproteína P, que se encuentra en la superficie de las células neoplásicas.39-4l Esta glucoproteína de membrana sirve como un transportador para el flujo de sustancias nocivas como los agentes quimioterápicos, y su mayor expresión se asocia con menor acumulo de múltiples agentes quimioterápicos dentro de las células neoplásicas resistentes. Se ha aislado e identificado la secuencia de un gen resistente a múltiples fármacos, el mdr-1, que codifica la glucoproteína P. Cuando este gen se transfiere, confiere resistencia a medicamentos en células antes sensibles a los mismos.42 El gen mdr 1 se expresa en varios tejidos normales, como el riñón, la glándula suprarrenal, el hígado y el colon, y su expresión puede estar aumentada en las células cancerosas de pacientes con leucemia, linfoma, mieloma múltiple y otras neoplasias que son resistentes a la quimioterapia.43 Aunque la amplificación de los genes puede causar expresión excesiva de la glucoproteína P, la rnayoría de los casos clínicos de resistencia a múltiples medicamentos es resultado de una mayor velocidad de transcripción del gen mdr 1, más que de amplificación del gen. Sin embargo, es posible que no toda la resistencia a múltiples fármacos se relacione con la expresión de la glucoproteína-P. Evidencias circunstanciales indican que en algunos casos ésta se relaciona con otras proteínas.44,45
En un esfuerzo por minimizar el efecto tóxico de la quimioterapia sobre las células tronco de la médula ósea, se ha transrectado el gen mdr-1 a células tronco normales de la médula ósea de ratones y de humanos en protocolos de investigación.46 En estos modelos, las células normales de la médula ósea son protegidas del efecto tóxico de la quimioterapia, por lo que estas células aumentan, en lugar de disminuir, con el tratamiento.47,48
La resistencia a múltiples fármacos en los cánceres humanos puede combatirse con medicamentos no citotóxicos que interactúan y evitan la función de la glucoproteína P. Entre los fármacos no citotóxicos que se usan aunados a la quimioterapia para prevenir la salida de agentes quimioterápicos de las células neoplásicas se incluyen los bloqueadores del calcio (v.gr., verapamil) los esteroides y antagonistas de esteroides (v.gr., tamoxifén), los antagonistas de la calmodulina (v.gr., trifluopcrazina), la ciclosporina y agentes cardiacos (v.gr., quinidina y amiodarona).49-5l Estos agentes compiten por la acción del transportador glucoproteína P o interfieren con su función. Esta medida inhibe la función normal de la glucoproteína P, causando menor excreción del agente citotóxico y, quizá, mayor destrucción de células.51 Aunque cuando se usa esta estrategia se encuentra regresión de los tumores, las respuestas son limitadas.
Se ha demostrado que muchos tumores expresan el gen mdr-1, pero debido a que muchos pacientes con tratamientos intensivos que recurren tienen tumores que no tienen expresión aumentada de este gen, es probable que otros mecanismos contribuyan a la resistencia a la quimioterapia.44,45,52,53 En muchos casos la resistencia a múltiples fármacos puede estar mediada por mutaciones o disminución de concentración en las topoisomerasas de ADN (enzimas que participan en la síntesis del ADN, en la separación de los cromosomas durante la mitosis, en la recombinación, y quizá en la transcripción genética). Debido a que las antraciclinas y algunas epipodofilotoxinas (v.gr., etopósido) pueden unirse al ADN y a su vez bloquear la acción de las topoisomerasas de ADN, los mecanismos de resistencia a estos medicamentos pueden incluir alteraciones en las topoisormerasas de ADN. Independientemente de los cambios en el gen mdr-1, la resistencia a múltiples fármacos puede ser causada también por mutaciones en el gen p53, que evita que un agente quimioterápico inicie la apoptosis, permitiendo que la célula se divida y se produzcan nuevas células aberrantes.
Estos hallazgos enfatizan la necesidad de una detección temprana del cáncer y de la atención cuidadosa a los esquemas y dosis de tratamiento.54-56 Los pacientes deben ser tratados con dosis altas de agentes quimioterápicos cuando la población neoplásica es pequeña y la posibilidad de que exista un número grande de células resistentes o metastásicas es escasa. Este concepto se refiere a la intensidad de dosis, esto es, la cantidad de medicamento que se administra por ciclo o unidad de tiempo. Es importante administrar varios medicamentos en forma simultánea o en días secuenciales y usar las dosis máximas toleradas.57 Debido a que los niveles de resistencia a fármacos baja o intermedia pueden sobrepasarse usando quimioterapia en dosis altas, los esfuerzos actuales se enfocan a determinar las dosis óptimas de los medicamentos y a usar programas de tratamiento con dosis intensivas asociados a trasplante de médula ósea . La resistencia causada por la amplificación genética es menos probable cuando se emplean esquemas de dosis altas porque esta amplificación puede desarrollarse con más facilidad cuando se inhibe la replicación del ADN solo en forma temporal. Un área activa de estudio es el uso de agentes que interfieren en forma directa con los mecanismos de resistencia a múltiples fármacos.
Estrategias en el uso de la quimioterapia
La quimioterapia combinada ha sido muy eficaz para aumentar el porcentaje de respuesta de una gran variedad de neoplasias. También ha permitido la curación de otros [ver tabla 2]. El 50 a 80 porciento de los pacientes con enfermedad de Hodgkin, linfoma histiocítico difuso, linfoma nodular mixto, carcinoma del testículo y coriocarcioma gestacional pueden curarse en la actualidad por medio de la quimioterapia combinada. Alrededor del 10 porciento de las pacientes con cáncer de ovario y el 15 a 25 porciento de los enfermos con leucemia mielógena pueden también ser curados. Aún más, la quimioterapia combinada con trasplante de la médula ósea puede aumentar estos porcentajes (ver adelante). La quimioterapia combinada ha causado resultados importantes en las neoplasias de la infancia: el 60 a 80 porciento de los niños con leucemia linfocítica aguda, linfoma de Burkitt o tumor de Wilms puede curarse en la actualidad.
Se requieren nuevas estrategias para tratar aquellas enfermedades malignas que son responsables de más del 50 porciento de muertes debidas al cáncer en adultos (cáncer del pulmón, colon, mama y próstata). Estos cánceres carecen de las características comunes a la mayoría de las enfermedades malignas que son curables con quimioterapia. Los tumores curables crecen rápidamente y tienden a presentar fracciones de crecimiento elevadas. Sin embargo, con excepción del cáncer de células pequeñas del pulmón, los tumores menos curables tienen fracciones de crecimiento bajas. Muchos de los tumores curables son mucho más sensibles a los agentes quimioterápicos que los tejidos normales en crecimiento, como las células de la médula ósea y el epitelio intestinal; el índice terapéutico, que expresa la relación entre las dosis terapéuticas y tóxicas de un medicamento, es alto en tales casos. Los agentes que son eficaces contra las enfermedades malignas comunes por lo general son limitados en número o tienen índices terapéuticos bajos. La mayor parte de los tumores actualmente curables ocurren en personas jóvenes, mientras que las enfermedades malignas comunes que escapan a la curación por quimioterapia afectan a personas mayores. Se considera que la resistencia a los medicamentos es el principal impedimento para el tratamiento útil de estas neoplasias, pero están emergiendo nuevos métodos terapéuticos diseñados para combatir los frecuentes problemas de baja fracción de crecimiento y resistencia a fármacos.
Algunos de los métodos descritos en esta sección están en etapa experimental y no deben iniciarse en la práctica clínica hasta que se demuestre su validez en estudios clínicos controlados. Estos estudios son necesarios para asegurarse de que se logra un avance significativo en el tratamiento y para proteger al paciente de la exposición a tratamientos tóxicos de valor limitado.
PERFUSION SISTEMICA Y REGIONAL
Se han usado las infusiones venosa y arterial de medicamentos quimioterápicos en lugar de las inyecciones en bolo para minimizar la toxicidad y lograr concentraciones altas de medicamento en los tejidos. Este método asegura que las células de proliferación más lenta comiencen la síntesis de ADN en presencia de concentraciones altas del fármaco.57 Pueden usarse diversos equipos para administrar estas infusiones, y todos ellos permiten tener un acceso permanente a la vena subclavia.58 Entre ellos se incluyen las bombas implantables subcutáneas o bombas externas unidas a sitios de acceso o catéteres subcutáneos que protruyen en forma permanente a través de la piel. En el procedimiento que parece ser más frecuente, se colocan sitios de acceso en forma subcutánea en pacientes que recibirán ciclos múltiples de quimioterapia (incluso cuando no la van a recibir en forma de infusión continua) para tener una vía venosa permeable adecuada y disminuir el efecto esclerosante que pueden tener algunas formas de quimioterapia sobre las venas periféricas. La infusión arterial y la infusión a la vena porta, aunque no se realizan con frecuencia, son otros métodos que sirven para administrar concentraciones altas de medicamentos a las metástasis .
QUIMIOTERAPIA CON DOSIS INTENSIVAS Y TRASPLANTE DE MEDULA OSEA ALOGENICA O SINGENICA O DE CELULAS TRONCO DE SANGRE PERIFERICA
Los esquemas de quirnioterapia con dosis altas pueden emplearse si se rescata al paciente con un trasplante autólogo o alogénico de células hematopoyéticas tronco o por medio de la administración de agentes que actúan como antídotos ante la acción tóxica del medicamento. La estrategia base de todos los esquemas con dosis altas consiste en vencer los mecanismos de resistencia a fármacos por medio de un efecto de acción en masa que produce concentraciones intracelulares muy altas de los quimioterápicos.59 La quimioterapia con dosis intensivas combate el problema frecuente de menor acumulo del medicamento al exponer a todas las células, incluyendo las que proliferan con lentitud, a una concentración consistentemente alta del fármaco.57
El trasplante de médula ósea, ya sea singénico (i.e., con genotipo idéntico) o alogénico (i.e., genéticamente distinto), después de la administración de dosis tumoricidas de quimioterapia, radioterapia o ambas, se usa cada vez más para tratar a pacientes con leucemias y algunos tumores sólidos.60 En la actualidad el trasplante de médula ósea es útil para tratar la leucemia linfocítica aguda, la leucemia mielógena aguda y crónica y el mieloma.6l El trasplante autólogo de médula ósea es un tratamiento eficaz en pacientes con leucemias agudas y en enfermos con recurrencia de linfomas de grado alto, enfermedad de Hodgkin o cáncer testicular.60,62,63 En varias instituciones se emplea también el trasplante autólogo en mujeres con cáncer de mama con recidivas, con un cáncer de mama primario con riesgo alto de recidiva (i.e., cáncer de mama que afecta 10 o más ganglios linfáticos) y cáncer de ovario recurrente64-68
Cada vez se usa más la reinfusión de células tronco de sangre periférica, en lugar del trasplante de médula ósea, en pacientes con cáncer sometidos a tratamiento intensivo para cánceres no hematológicos.69 Las células hema-topoyéticas tronco circulan dentro de la población de células nucleadas de la sangre periférica tanto de individuos normales como de pacientes con cáncer. Las células nucleadas son recolectadas por leucoféresis después de varios ciclos en dosis convencionales de quimioterapia y de estimulación con factores estimuladores de colonias [ver adelante, Modificadores de la respuesta biológica]. Después de la quimioterapia y de la administración de los factores estimuladores de crecimiento, el número de células hematopoyéticas tronco en la circulación periférica aumenta. Estas células tronco se asocian con las células nuceladas en la circulación periférica y pueden ser enriquecidas seleccionando las células CD-34 positivas. Cuando las células tronco periféricas se reinfunden, suele administrarse factor estimulador de colonias de granulocitos (FEC-G) o factor estimulador de colonias de granulocitos y macrófagos (FEC-GM) para favorecer el implante de célulares periféricas.70 Cuando va a realizarse un autoinjerto con células tronco periféricas el paciente es sometido a leucoféresis en varias ocasiones y se colectan y almacenan alrededor de 108 células nucleadas para su reinfusión posterior. Después de la quimioterapia en dosis intensivas que abate las células tronco de la médula ósea, la médula ósea del paciente se reconstituye por medio de la reinfusión de esta población de células nucleadas. La cuenta de plaquetas y granulocitos puede normalizarse con mayor rapidez después de la rehinfusión de las células tronco periféricas que después de la reinfusión de células autólogas de la médula ósea.
Los peligros potenciales con estos programas de tratamiento con quimioterapia en dosis altas seguido de rescate con células hematopoyéticas tronco incluyen enfermedad venoclusiva del hígado, neurnonitis intersticial, trombocitopenia persistente y enfermedad injerto contra huésped (cuando se usa un injerto alogénico). Algunos de estos riesgos pueden disminuir en forma significativa por medio del uso de FEC-G y FEC-GM en estos programas [ver adelante, Modificadores de la respuesta biológica]. Estos factores de crecimiento, al administrarse poco después de la quimioterapia intensiva, provocan una reaparición más rápida de granulocitos y plaquetas en sangre periférica. Sin embargo, es menos fácil de manejar la toxicidad al hígado, pulmones y corazón que puede asociarse con algunos agentes quimioterápicos administrados en dosis mayores de las convencionales. La reacción tóxica más significativa es la enfermedad venoclusiva del hígado: el uso de algunos agentes a dosis altas puede causar deterioro en la microcirculación del hígado, con falla hepática subsecuente.71
ADMINISTRACION INTRACAVITARIA DE LA QUIMIOTERAPIA
La administración directa de agentes en las cavidades peritoneal y pleural, el espacio cerebroespinal o la vejiga ha sido empleada para tratar tumores que están confinados a estos espacios, porque este método expone a las células tumorales a dosis altas de los agentes citotóxicos durante periodos prolongados y suele causar toxicidad sistémica leve.72,73 La quimioterapia intracavitaria ha sido útil sobre todo en el tratamiento del cáncer de ovario y en el cáncer superficial de la vejiga.
QUIMIOTERAPIA DIRIGIDA
Otro método para vencer la resistencia a los fármacos incluye técnicas de adminis-tración innovadoras diseñadas para lograr concentraciones locales altas de fármacos citotóxicos o toxinas en el sitio de la célula tumoral.74-76 Estos métodos incluyen el uso de agentes citotóxicos incorporados a los liposomas o unidos a lipoproteínas de baja densidad a ADN o a microesferas. Estos medicamentos han sido unidos también a anticuerpos monoclonales específicos dirigidos contra las células neoplásicas77-81 Estas técnicas, que aún están en evolución, intentan distribuir en forma específica o dirigir la acción de los medicamentos (el llamado método de proyectil mágico), provocando así el máximo daño a las células tumorales y minimizando la lesión para las células normales.
Los anticuerpos monoclonales, administrados solos o en combinación con otros agentes como el interferón alfa, parecen ser promisorios en el tratamiento de un número limitado de tipos de cáncer.82-84 Se ha observado regresión de linfomas B después del tratamiento con anticuerpos monoclonales dirigidos contra componentes de superficie específicos de las células de linfoma de un paciente en particular.79,85-87 El tratamiento con anticuerpos monoclonales contra el HER-2/neu, un receptor de factores de crecimiento, puede causar regresión del cáncer en el 25 porciento de las pacientes con cáncer de mama en las que esta molécula se expresa en forma exagerada. Es posible que los anticuerpos monoclonales demuestren ser útiles para la observación de las metástasis [ver adelante, Marcadores tumorales en el tratamiento del cáncer].81
MODIFICADORES DE LA RESPUESTA BIOLOGICA
Los modificadores de la respuesta biológica, que en condiciones normales son producidos por los humanos y otros mamíferos para aumentar la respuesta inmunológica contra los tumores y la infección, han sido empleados solos o en combinación con otros agentes para el tratamiento experimental de las neoplasias humanas.89,90 Los interferones, una gran familia de glucoproteínas, son ejemplos de estos modificadores de la respuesta biológica. Hasta el 90 porciento de los pacientes con leucemia de células peludas responde en forma favorable al tratamiento con interferón alfa.90 Los interferones también han demostrado cierta eficacia en los linfomas no Hodgkin de grado bajo, el linfomacutáneo de células T, la leucemia mielógena crónica, el mieloma múltiple, el sarcoma de Kaposi asociado con síndrome de inmunodeficiencia adquirida (SIDA), el cáncer superficial de vejiga y quizá el cáncer de células renales.91,92 Sin embargo, el tratamiento con interferón para cánceres corno el de mama, colon y prostata no ha producido resultados alentadores. La mayoría de los estudios de tratamiento con interferón en el manejo del cáncer se refieren al uso del interferón alfa, pero en la actualidad se sintetizan otros miembros de la familia de los interferones por medio de técnicas de recombinación de ADN, lo que facilitará el estudio de estos compuestos y ayudará a resolver las dudas en relación con los esquemas de dosificación.
La inmunoterapia adoptiva empleando linfocinas, como la interleucina-2 (IL-2) ha recibido atención considerable porque aprovecha un mecanismo endógeno de destrucción de células tumorales.10,93-95 La IL-2 parece activar a las células T citotóxicas, a las células asesinas naturales y quizá a otras células linfoides que a su vez atacan a las células neoplásicas. El tratamiento solo con dosis altas de IL-2 o con células linfoides de sangre periférica activadas con IL-2 autóloga ha producido regresión de metástasis en algunos pacientes refractarios al tratamiento convencional o para los que no existe un tratamiento sistémico establecido. El melanona y el cáncer de células renales son de los tumores que mejor han respondido a esta modalidad. Aunque se ha autorizado ya el uso de IL-2 para el tratamiento del cáncer de células renales, su toxicidad puede ser importante.
Quizá el tratamiento biológico que más se emplea en la actualidad consiste en el uso de FEC-G y FEC-GM. Estas citocinas, que en condiciones normales son producidas por varios tipos celulares en la médula ósea (y quizá en otros sitios), pueden producirse en grandes cantidades por medio de biotecnología recombinante. El FEC-G y el FEC-GM son factores de crecimiento hematopoyético que están autorizados para la prevención de la melosupresión inducida por quimioterapia y que en la actualidad se emplean en otras situacioncs además del trasplante de células hematopoyéticas tronco.96
El FEC-G se encuentra en el suero de los individuos normales y su nivel aumenta en respuesta a las infecciones bacterianas. Por el contrario, no se detecta FEC-GM en suero, y se piensa que esta citocina actúa en forma parácrina, más que en forma endócrina. El FEC-GM actúa sobre muchas células precursoras y sobre células maduras de diversas líneas dentro de la médula ósea. Puede aumentar el número de neutrófilos, monocitos y eosinófilos en la circulación periférica y puede prolongar la vida media circulante de los neutrófilos. El FEC-G apoya la expresión, supervivencia y maduración de los precursores de neutrófilos, disminuyendo el tiempo para su maduración a granulocitos maduros. Estudios aleatorios que incluyeron pacientes con cáncer pulmonar de células pequeñas mostraron que el FEC-G asociado a un esquema de quimioterapia intensiva redujo el número de hospitalizaciones y los días de estancia hospitalaria, así como el número de episodios febriles, de infecciones confirmadas por cultivo y de ciclos de quimioterapia en los que tuvieran que reducirse las dosis por la leucopenia97 El FEC-G se emplea también después de la quimioterapia ablativa y de la reinfusión de médula osea autóloga para la enfermedad de Hodgkin, el linfoma no Hodgkin, el cáncer de mama y otros tumores sólidos y leucemias agudas. En estos estudios, el tiempo de injerto y el número de días de fiebre se redujeron en forma significativa.70,98,99 El uso de FEC-G no parece aumentar el riesgo de recurrencia de la leucemia en pacientes que son sometidos a trasplante de médula ósea por leucemia aguda.100
VACUNAS TUMORALES
Una estrategia de tratamiento en vías de investigación es cl desarrollo de vacunas tumorales, que intentan cumplir con dos objetivos: inducir una respuesta inmunológica contra antígenos específicos del tumor e inducir una respuesta inmunológica contra las metástasis tumorales. En un primer enfoque se han inyectado células tumorales completas o lisados celulares después de resecar un melanoma o un cáncer de colon y recto primarios para inmunizar a los pacientes que tienen riesgo alto de recurrencia.101,102 En una segunda aplicación, se han empleado inmunoglobulinas idiotípicas expresadas en la superficie de células de linfoma B como antígenos específicos del tumor para estimular las respuestas inmunológicas en entermos con linfomas clínicamente activos,103 y se han administrado antígenos autólogos de melanoma a pacientes con melanoma avanzado.104 Debido a que los linfomas de células B son clonales, todas las células de un linfoma expresan una misma inmunoglobulina de superficie (i.e., inmunoglobulina idiotípica). Las inmunoglobulinas idiolípicas pueden producirse en grandes cantidades en el laboratorio. Los pacientes que reciben inmunoglobulinas idiotípicas de sus propios linfomas montan respuestas inmunológicas humorales y mediadas por células contra este idiotipo. Sin embargo, las vacunas para tratar o prevenir el crecimiento de los tumores humanos aún están en etapas muy tempranas de investigación.
QUlMIOTERAPIA NEOADYUVANTE
La quimioterapia neoadyuvante se refiere a la administración de agentes quimioterápicos antes del tratamiento estándar con cirugía, radioterapia o ambos.105 El objetivo de la misma es reducir el tamaño del tumor lo suficiente como para que los pacientes sean mejores candidatos a la cirugía o radioterapia. La quimioterapia neoadyuvante ha sido empleada sobre todo en el tratamiento del cáncer de cabeza y cuello, el sarcoma osteogénico y el cáncer de mama localmente avanzado.106-110 Los medicamentos como los radiosensibilizadores y los radioprotectores pueden usarse en esta forma para aumentar los efectos de la radioterapia sobre las células tumorales o para proteger a las células normales del daño por radiación.
QUIMIOPREVENCION E INDUCCION DE LA DIFERENCIACION
Las células diferenciadas sintetizan proteínas específicas y tienden a proliferar poco o no proliferar. Con frecuencia se considera a las células neoplásicas como no diferenciadas porque no sintetizan productos proteicos que las distingan de otras células y porque son capaces de proliferar casi sin límite. Un enfoque de tratamiento (y quizá de prevención) para el cáncer que no requiere de la destrucción celular consiste en alterar la velocidad de crecimiento de las células dentro de un tumor aumentando la diferenciación de las mismas.111,112 El uso de medicamentos que promueven la diferenciación celular puede ser también importante en el manejo de condiciones premalignas como la leucoplasia. Por ejemplo, la administración sistémica de isotretinoín (ácido I3-cis-retinoico) causa que la mucosa leucoplásica oral revierta hacia un epitelio normal diferenciado. Los estudios aleatorios con adyuvantes demostraron que la administración de isotretinoín después del tratamiento principal redujo también la incidencia de cánceres secundarios en pacientes con cáncer de cabeza y cuello113,114 tenerse cuidado con cualquier tratamiento quimopreventivo para el cáncer; por ejemplo, los estudios controlados demuestran que se desarrolla cáncer de pulmón con más frecuencia en los fumadores que reciben b caroteno que en los que no reciben este medicamento.l15
Terapia génica
La introducción de genes a las células tumorales o a células normales se encuentra en las fases iniciales de aplicación clínica para el tratamiento del cáncer.46,l16 Aun que los adelantos recientes en biología molecular han tenido un gran impacto en el diagnóstico, su impacto sobre el tratamiento de tumores humanos ha sido mucho menor. Sin embargo), se están desarrollando enfoques sofisticados para bloquear los efectos de los productos de los oncogenes, identificar a las células neoplásicas y destruirlas.46,117 Los enfoques que se han examinado incluyen lo siguiente: (1) aumentar la inmullogenicidad de los tumores insertando genes de citocinas, (2) insertar genes cuyos productos, al ser procesados, sean tóxicos para las células, (3) codificar ARN antisentido, que bloquee los efectos de los oncogenes y los genes de resistencia a fármacos, y (4) marcar células de modo que puedan detectarse después.46,117,118 Las limitaciones actuales de estas medidas se relacionan con los métodos para administrar los genes en forma específica a las células tumorales y para restringir la expresión de los genes de las células normales cuando éstas son el blanco. Es necesario solucionar problemas de selectividad, especificidad y seguridad antes de poder aplicar en forma amplia estos enfoques de tratamiento génico.
INHIBICION DE LA ANGIOGENESIS
La angiogéllesis (i.e., la formación de arteriolas que promueven el aporte de sangre a los tumores en crecimiento y a las metástasis) ocurre en respuesta a los factores de crecimiento parácrino producidos por el tumor y por los tejidos normales en respuesta al tumor.119 Un área de gran interés es el papel de la vascularización en el desarrollo del tumor. Durante la progresión del tumor los factores de crecimiento parácrino sirven para reclutar tejido del estroma y ampliar el aporte sanguíneo al tumor en expansión.l20 Los tumores producen estímulos que incitan o inhiben la formación de nuevos vasos sanguíneos, que son indispensables para el crecimiento y remodelación del tumor y sus metástasis.121 Al teñir los tejidos (en especial los cánceres primarios de la mama o la próstata) en busca de capilares y contar el número de vasos en un área definida es posible predecir la evolución.122,123 Varios estudios han demostrado un peor pronóstico para los pacientes cuyo tumores tienen gran número de capilares pequeños. En la actualidad se estudian estrategias para interferir con la angiogénesis como un método para tratar el cáncer. Algunos agentes, como el angiostatin, parecen bloquear la formación de los vasos nuevos.124 Aún no se ha definido el papel de la inhibición de la angiogénesis en el tratamiento de los canceres primariosmetastásicos, así como de otras enfermedades.125
Marcadores tumorales en el manejo del cáncer
El manejo del cáncer incluye el desarrollo de métodos inmunológicos que pueden detectar neoplasias cuando su tamaño es pequeño y que pasan desapercibidas desde el punto de vista clínico.l26,127 Pueden detectarse en el suero de pacientes con cáncer testicular, coriocarcinoma gestacional, hepatoma, cáncer de próstata o cáncer ovárico, antígenos circulantes que pueden ser de utilidad para detectar o confirmar la presencia de un cáncer o su recurrencia.128 Cuando existe la posibilidad de curación para un cáncer diseminado (en especial en el caso del cáncer testicular y el coriocarcinoma gestacional), la medición de estos antígenos tiene especial importancia. Por ejemplo, los inmunoensayos que miden gonadotropinas en circulantes en las mujeres con corlocarcinoma gestacional pueden detectar la presencia de micrometástasis, lo que orientará a la continuación del tratamiento hasta que estas metástasis hayan desaparecido. Existen también marcadores tumorales circulantes para el cáncer de testículo (gonadotropinascoriónicas), el hepatoma(a-fetoproteína), el cáncer de próstata (antígeno prostático específico) y algunos otros carcinomas. Los ensayos de anticuerpos monoclonales pueden detectar un antígeno del cáncer de ovario humano conocido como CA 125 en el suero del 80 porciento de las pacientes con cáncer de ovario demostrado por biopsia.l29-131 Aunque este estudio es útil para vigilar a las pacientes con cáncer de ovario y se ha demostrado que puede detectarlo, el antígeno CA 125 es también un indicador inespecífico de afección peritoneal, y su concentración se eleva en otros padecimientos incluyendo la cirrosis hepática con ascitis.l32 Sin embargo, los valores muy altos suelen asociarse con cáncer de ovario. Aún no es muy clara la manera como pueden emplearse los marcadores tumorales en el manejo inicial de cánceres diseminados cuando no existe la posibilidad de tratamientos curativos. Por ejemplo, la detección temprana de la recidiva del cáncer de mama no ofrece en la actualidad ningún beneficio clínico.133 Los marcadores como el antígeno carcinoembriónico (ACE), que se encuentran en cánceres frecuentes, no han sido tan útiles como se esperaba. El ACE no es específico para un carcinoma, ni siempre se eleva en presencia de neoplasia.
La tecnología con ADN recombinante puede ser de utilidad para el diagnóstico del cáncer y para definir los mecanismos por los que las células normales se transforman en células neoplásicas . Las técnicas de ADN recombinante también se han empleado con éxito para diagnosticar linfomas,35,l34,l35 y parecen ser útiles para identificar algunos pacientes con riesgo alto de recidiva de cáncer de mama y neuroblastoma.136,137 Esta tecnología parece tener un papel cada vez más importante en el diagnóstico del cáncer y para evaluar el pronóstico. Por ejemplo, pueden usarse técnicas de ADN recombinante para realizar escrutinio de carcinoma de colon.l38
Estudio de células tumorales tronco en humanos
El estudio de células tumorales tronco en humanos, también llamado ensayo clonogénico, se ha empleado para medir la sensibilidad de las células tumorales a la quimioterapia.139 Este estudio compara el número de colonias formadas en el cultivo celular en presencia y ausencia de varias diluciones de medicamentos aislados. Sin embargo, varios factores limitan su aplicación clínica: (1) es frecuente que los tumores no tengan suficientes células clonables, (2) la prueba predice mejor a qué medicamentos no responderán las células tumorales y no a cuáles fármacos son sensibles, y (3) predice los efectos de los agentes aislados, más que de la combinación de los mismos (que es la manera como suelen administrarse estos fármacos).~4~) ~4l
Aunque la esperanza inicial de que esta técnica pudiera servir como una guía general de tratamiento para el paciente individual parece no haberse cumplido, el ensayo proporciona información sobre la heterogeneidad de las células tumorales y la resistencia a los medicamentos. Por ejemplo, el ensayo reveló que incluso en ausencia de medicamentos, solo una pequeña proporción de células tumorales (0.01 a 0.10 porciento) de la muestra de biopsia es capaz de crecer en forma clonal. Aunque las dificultades técnicas podrían explicar esta baja eficiencia de clonación, parece ser que la mayoría de las células tumorales tienen un potencial limitado de crecimiento y no pueden proliferar lo suficiente para producir una nueva masa tumoral. Este hallazgo tiene implicaciones importantes para la formación de metástasis, porque indica que la mayoría de las células tumorales pueden proliferar solo durante varias generaciones y que solo una pequeña proporción de las células lo hacen en forma indefinida. Estas células capaces de proliferar de modo casi ilimitado, quizá solo una de cada 1,000 a 10,000 células tumorales, se conocen como células clonogénicas o tronco. Las células tumorales que se comportan como células tronco tienen gran importancia clínica porque una pequeña fracción de las misulas son resistentes a los medicamentos quimioterápicos, incluso en pacientes que no han sido tratados previamente con estos agentes.
En la actualidad el ensayo clonogénico tiene poca utilidad en el manejo clínico del cáncer.
Agentes anticancerosos de uso común
Los agentes quimioterápicos comúnmente usados se enlistan en la tabla 3. Están clasificados por un modo de acción, origen o estructura, aunque algunos de los agentes no se ajustan de manera clara a un solo grupo. Las categorías incluyen agentes alquilantes, antimetabólitos, antibióticos, alcaloides y agentes diversos (incluyendo hormonas). La figura 4 resume los sitios de acción de muchos de estos medicamentos.
Los agentes alquilantes forman enlaces convalentes con los ácidos nucleicos. Estos agentes alteran la integridad del ADN transfiriendo un grupo alquilo a los ácidos nucleicos. Algunos tienen dos sitios reactivos, lo que les permite formar enlaces cruzados con el ADN. Los agentes de esta clase son tanto citotóxicos como carcinógenos. La ciclofosfamida es el agente alquilante utilzado con mayor frecuencia
Los antimetabolitos son estructuralmente similares a los sustratos metabólicos normales. Alteran las funciones celulares sustituyendo los precursores normales en reacciones fisiológicas vitales, o bloqueando estas reacciones. Los antimetabolitos que se emplean más a menudo son el metotrexate y el 5-fluorouracilo, los cuales interfieren con la síntesis de los ácidos nucleicos.
Los antibióticos son productos biológicos de bacterias u hongos y tienen gran importancia en el tratamiento del cáncer. No comparten un solo mecanismo de acción. La bleomicina y las antraciclinas daunorrubucina y doxorrubicina son algunos de los antibióticos comúnmente usados. La bleomicina produce su citotoxicidad rompiendo físicamente el ADN. Las antraciclinas logran su efecto citotóxico por diversos mecanismos, incluyendo intercalación entre las hélices de ADN, interfiriendo de esta manera con la síntesis de ADN y ARN y la acción de las ADN topoisomerasas, producción de radicales libres que reaccionan con las proteínas y ácidos nucleicos intercelulares dañándolos, quelación de cationes divalentes, y reacción con las membranas celulares. La amplia variedad de sitios potenciales de acción puede ser responsable tanto del grado de eficacia como de la toxicidad de las antraciclinas.142
Los alcaloides, como la vincristina, la vinblastina, la vindesina y el paclitaxel (Taxol), se unen a la tubulina, proteína citoplasmática estructural, y evitan el ensamble o desensamble de los microtúbulos. Como el aparato en huso responsable del movimiento cromosómico durante la mitosis consta de microtúbulos, los alcaloides son especialmente activos durante la mitosis. Muchos otros procesos celulares, como el mantenimiento de la forma y el transporte intracelular de nutrientes en el sistema nervioso periférieo, dependen también de la formación de microtúbulos y son afectados por los alcaloides. Es probable que la neuropatía que se asocia al uso de estos medicamentos resulte de su acción sobre los microtúbulos de los axones largos de los nervios. El etopósido (VP- 16), una epipodofilotoxina, es un nuevo alcaloide que detiene a las células en fase G2 y en fase S temprana al actuar sobre las ADN topoisomerasas.
Los fármacos en la categoría de diversos tienen diversas acciones. Por ejemplo, la dacarbazina y la procarbazina son similares en su modo de acción a los agentes alquilantes, mientras que la L-asparaginasa actúa enzimáticamente. La L-asparaginasa es una de las pocas proteínas usadas en la quimioterapia contra el cáncer. No interfiere directamente con la síntesis de ADN, sino que inhibe la síntesis proteica. La L-asparagina es un aminoácido no esencial. En la mayor parte de los tejidos humanos normales, la síntesis de L asparagina a partir del ácido aspártico es catalizada por la enzima L-asparagina sintetasa. Sin embargo, muchos tumores derivados de linfocitos T carecen de esta enzima y por lo tanto dependen del plasma para su aporte de L asparagina. El tratamiento con L-asparaginasa agota el fondo circulante de L-asparagina y así deteriora la síntesis proteica en dichas células tumorales, destruyéndolas finalmente.
Las hormonas, en especial las homonas esteroideas prednisona, progesterona, estrógeno y androgeno, se usan con frecuencia en el tratamiento contra el cáncer. Sin embargo, estas cuatro hormonas se han ido sustituyendo por nuevos agentes que interfieren en forma selectiva o no selectiva con la acción hormonal. Estos agentes incluyen tamoxifen, toremifine y droloxifene, que son antiestrógenos usados en el tratamiento del cáncer de mama, leuprolide, un análogo de la hormona liberadora de gonadotropinas que evita la liberación de gonadotropinas y se usa en el tratamiento del cáncer de mama y próstata, y anastrozole, que bloquea la acción de la arimidasa, una enzima que convierte los andrógenos en esírógenos en el tejido adiposo y otros tejidos. Los agentes que son semejantes en acción a las hormonas naturales pueden ser eficaces también en el tratamiento de los cánceres. Un ejemplo es el megestrol, un agente progestágeno que es útil contra el cáncer de mama cuando se emplea en dosis altas. Aunque no se conocen del todo los mecanismos exactos por los que varias hormonas reducen el tamaño tumoral, parece ser que alteran la cinética de crecimiento tisular del tumor y favorecen la muerte celular, como la apoptosis. Por ejemplo, la prednisona puede ser eficaz en la leucemia linfocítica aguda porque induce apoptosis en células linfoides inmaduras. Otras hormonas inician la producción de factores de crecimiento parácrino a partir de las células del estroma normal de un tumor, lo que puede alterar el crecimiento tumoral.
Los agentes quimioterápicos, que casi siempre se administran en forma combinada, ofrecen paliaciónexitosa para la mayoría de los pacientes con cáncer y pueden proporcionar curación para algunos. La mayoría de los esquemas causan toxicidad considerable, por lo que se requiere tener experiencia y estar familiarizado con la farmacología de cada combinación que vaya a emplearse.
Bibliografía
DR. FRANK E. STOCKDALE, PH.D.
La era moderna de la quimioterapia para el cáncer empezó a principios de los años 40 con la introducción de la mostaza nitrogenada (hidroclomro de mecloretamina). Aunque actualmente ésta se usa poco, muchos medicamentos con origen, estructura y acción diferentes se emplean comúnmente para tratar una amplia variedad de cánceres en el humano. Estos agentes comparten una característica: por lo general son más eficaces para matar o dañar células malignas que células normales. Sin embargo, el hecho de que dañen células normales señala su gran potencial para causar toxicidad. La comprensión del uso de la químíoterapia requiere del entendimento tanto del mecanismo de acción del medicamento como de la fisiopatología del cáncer, que tiene su origen en un trastorno del crecimento celular y tisular. La estrategia de la quimioterapia se basa en los conocimientos existentes sobre crecimiento del tejido tumoral.
Crecimiento celular y tisular
El término cáncer describe un grupo de enfermedades caracterizadas por acúmulo inadecuado de tejido. Esta alteración es más evidente clínicamente cuando la masa de tejido tumoral compromete la función de órganos vitales. Al contrario de lo que suele pensarse, las neoplasias malignas en los humanos no suelen ser enfermedades de proliferación celular rápida; de hecho, las células de la mayor parte de las neoplasias comunes proliferan en forma más lenta que muchas células normales.1-3 Es el acúmulo relativamente lento del tejido tumoral en los órganos vitales lo que provoca la muerte en muchos de los pacientes con cáncer.
Los conceptos que describen el crecimiento de los tejidos normales son aplicables a los tejidos malignos porque los tejidos y células normales y malignos comparten características similares de crecimiento.4 Se deben distinguir los conceptos que describen el crecimiento de células individuales de los que describen el crecimiento de poblaciones de células o de tejidos [ver tabla 1].
|
||||
|
CONCEPTOS DE CRECIMIENTO CELULAR
El tiempo del ciclo celular, o tiempo de generación de una célula normal o maligna, es el lapso requerido para que una sola célula atraviese por un ciclo celular complcto.5 El ciclo celular se divide dé manera arbitraria en cuatro o cinco fases. G1, o intervalo 1, es la fase durante la que se sintetízan ARN y proteínas para una variedad de funciones celulares diferenciadoras. Aunque de duración variable, la fase G1, es la porción más larga del ciclo celular. Durante la fase S, o de síntesis de ADN, se replica todo el genoma. La fase G2, o intervalo 2, ocurre entre el término de la síntesis de ADN y el inicio de los cambios nucleares que ocurren en preparación a la condensación de los cromosomas. Durante la fase M, o mitosis, ocurren los movimientos de los cromosomas y la división celular (citocinesis), que causan la distribución de los cromosomas en dos células hijas. G0 describe una fase prolongada de reposo durante la cual las células no responden a estímulos que normalmente iniciarían la síntesis de ADN. El tiempo del ciclo celular de la mayor parte de las células normales es de cerca de 24 a 48 horas, mientras que el de las células malignas de los tumores humanos comunes puede ser hasta de 120 horas. Sin embargo, el tiempo de generación, tanto de las células normales como de las malignas, no es fijo, y puede ser fácilmente modulado por una variedad de factores en el ambiente de la célula.
CONCEPTOS DE CRECIMIENTO TISULAR
Los cinco conceptos de crecimiento tisular no deben ser confundidos con los conceptos de crecimiento celular [ver tabla 1]. El cáncer es una enfermedad caracterizada por un trastorno en la regulación del crecimiento tisular, al igual que por un trastorno en la regulación del crecimiento celular. Los conceptos tisulares son los más importantes para el entendimiento clínico del cáncer y de su quimioterapia.
El tiempo de duplicación se refiere a la duración requerida para que un tejido o turnor duplique su tamaño o número de células. Aunque pudiera esperarse que los tiempos del ciclo celular y de duplicación tisular fueran iguales, el tiempo de duplicación de un tumor clínicamente aparente es por lo común mucho más prolongado que el del ciclo celular de las células que conforman al tumor. Cuatro parámetros del crecimiento tisular (el índice de marcado, el índice mitótico, la fracción de crecimiento y la fracción de muerte celular) pueden ser responsables de esta discrepancia.
El índice de marcado es el porcentaje de células que producen ADN en un tejido que se encuentra en la fase S del ciclo celular). En la mayor parte de los tumores humanos este porcentaje es relativamente bajo (cinco a 20 porciento).6 De manera similar, el índice mitótico es el porcentaje de células en un tejido o tumor que están en mitosis en cualquier momento.
La fracción de crecímiento es el porcentaje de la población celular total que participa en el crecimiento del tejido o tumor, y representa la fracción de células que está en el ciclo.7 La fracción de crecimiento es la mejor medida de la velocidad de crecimiento tisular. Este concepto ha influido profundamente en la práctica de la quimioterapia. La fracción de crecimiento clínicamente aparente de los cánceres humanos es por lo general pequeña (menos de la mitad de las células están en ciclo), de manera que gran parte de las células cancerosas en la mayoría de los pacientes están en la fase G0. Así, la fracción de crecimiento de muchos tejidos normales en un paciente con cáncer frecuentemente excede la del tejido tumoral. Los tejidos normales con fracciones de crecimiento mayores son los más afectados por los agentes quimioterápicos, que son tóxicos para las células en proliferación. Dichos tejidos normales (y sus respuestas a la toxicidad) incluyen la médula ósea (leucopenia, trombocitopenia, anemia), los precursores de los gametos (esterilidad), los folículos pilosos (alopecia), y el epitelio que cubre el tracto digestivo (ulceraciones orales, diarrea).
La fracción de muerte celular es el porcentaje de células hijas recién producidas que mueren. Este número es alto en el tejido tumoral de muchos de los pacientes con cáncer, lo que indica que la mayor parte de la progenie de las células tumorales muere en lugar de acumularse. Así, el grado de desequilibrio entre la producción de células tumorales nuevas (medido por el índice de marcado, el índice mitótico y la fracción de crecimiento) y la muerte celular, determina la velocidad con la que crece una masa tumoral. Cuando ésta relación se altera de manera que favorece la producción de células tumorales, el tejido tumoral se acumula en el sitio primario de un tumor o en los sitios de metástasis.
CURVA DE GOMPERTZ
A diferencia de un tumor, el hígado, el corazón o los pulmones no tienen tiempo de duplicación en un adulto sano. Estos granos se encuentran en un estado estable, las velocidades de producción y muerte celular son iguales, de modo que con el paso del tiempo no existe aumento en su tamaño o en el número de células. Sin embargo, las características de crecimiento de los tumores pueden compararse con las de los fetos humanos y de los niños, en quienes la producción de células nuevas excede a la muerte celular. En cada caso existe un tiempo de duplicación, y se puede definir un patrón de crecimiento específico para todo un tejido, órgano u organismo. La pendiente de las curvas de crecimiento para un feto humano normal y para las células de la leucemia humana son semejantes [ver figura 1]. El matemático Gompertz desarrolló una ecuación que describe este fenómeno de manera muy aproximada. Desde entonces esta curva se conoce como curva de Gompertz.1

|
| Figura 1 |
| Crecimiento fetal contra tumoral |
La curva de Gompertz indica que durante el desarrollo de un feto humano normal o de un tumor, la velocidad de crecimiento es muy rápida al inicio y luego disminuye progresivamente al aumentar el tamaño. Esta observación tiene gran importancia en las limitaciones de la quimioterapia actual contra el cáncer. Los mecanismos que contribuyen a la norma de la curva de Gompertz siguen sin comprenderse en su totalidad, pero al menos dos factores importantes participan en la disminución de la velocidad de crecimiento al aumentar el tamaño de los tumores: la disminución en la fracción de crecimiento y el aumento en la muerte celular espontánea. Al acumularse las células en un tejido normal, en un tumor primario o en una metástasis, se produce disminución de la frecuencia con la que se inician nuevos ciclos de síntesis de ADN, y por lo tanto, con la que se producen nuevas células. Cuando un tumor o un feto es muy pequeño, se encuentran en la parte baja de la curva de Gompertz y tienen una gran fracción de crecimiento. Cuando un tejido es grande, se sitúa más arriba en la curva de Gompertz y tiene una fracción de crecimiento pequeña. Cuando un tejido es muy pequeño es probable que casi cada célula esté en ciclo y, por tanto, participando en el crecimiento. Aunque la ecuación de Gompertz describe el crecimiento de los tejidos y organismos en general, no todo el crecimiento tumoral o normal ocurre por un proceso continuo.8 Por ejemplo, los estudios sobre el crecimiento temprano en niños han demostrado que cuando se mide con frecuencia el crecimiento, este se presenta como un fenómeno intermitente, en el que periodos de crecimiento rápido se intercalan con periodos de poco o ningún crecimiento. En forma semejante, las medidas de los tumores humanos han demostrado que periodos de crecimiento acelerado pueden ser seguidos de periodos de crecimiento más lento o nulo. Los cálculos empleando el concepto de Gompertz sugieren que el acumulo de células cancerosas en la mayoría de los tumores humanos comienza alrededor de dos años antes de su detección. En la actualidad puede demostrarse la equivocación de los investigadores de hace dos décadas que calcularon en forma errónea, basándose en la lenta velocidad de crecimiento de tumores lo suficientemente grandes para ser observados por radiografías, que éstos se habían originado aproximadamente 10 a 20 años antes de su detección. Este error se ejemplifica mejor en el cáncer de los lactantes. Sin aplicar el concepto de crecimiento de Gompertz, podría pensarse que ciertas neoplasias de la infancia se originaron antes de la concepción.
Factores que afectan la curva de Gompertz
La disminución progresiva de la entrada de células a la síntesis de ADN ( fase S), descrita por la curva de Gompertz, no es causada por un cambio estable en las células tumorales individuales. Más bien, refleja el hecho de que las células son parte de una población celular y de que las señales responsables de iniciar el crecimiento (v.gr., hormonas, facíores de crecimiento y nutrientes) cambian al acumularse las células.Si se quitan unas cuantas células de un tumor lo suficientemente grande para estar en la parte superior de la curva de Gompertz y luego se implantan en otro sitio, estas células vuelven a entrar al ciclo celular, su fracción de crecimiento aumenta, y forman un nuevo tumor a una velocidad que se describe en forma exacta en la curva de Gompertz. La capacidad de una célula cancerosa para volver a entrar en el ciclo celular al reducir se la población de células tumorales es aprovechada por el tratamiento de modalidad combinada contra el cáncer.
Es probable que la muerte celular espontánea y la necrosis tisular sean las principales causas naturales de retraso en el crecimiento tumoral en el cáncer clínicamente aparente.9 Los informes de patología a menudo afirman que las piezas contienen grandes áreas de necrosis, y es posible que por este mecanismo se pierdan rápidamente gramos de tejido (> 109 células). Se calcula que del 70 al 90 porciento de las células tumorales recientemente producidas en los humanos mueren espontáneamente. Si este no fuera el caso, es probable que muchos pacientes murieran antes de entrar al sistema de salud, porque los tumores duplicarían su tamaño en cuestión de días.
Se entienden poco los mecanismos responsables de la muerte celular, pero quizá se relacionen con cambios en la estructura del genoma de la célula cancerosa, la apoptosis, con lo que se puede llamar en forma amplia nutrición celular, con varios mecanismos inmunológicos y con otros factores. Por ejemplo pueden surgir defectos en la estructura de los cromosomas durante su replicación y segregación. También se pueden segregar números anormales de cromosomas durante la mitosis, de manera que muchas células hijas carecen sin duda de los genes o de los mecanismos reguladores que se requieren para la supervivencia. Diversos mecanismos pueden causar apoptosis, o muerte celular iniciada en forma activa (ver adelante). Además, los tumores tienen vasculatura anormal, son comunes la ruptura de vasos, la coagulación y la hemorragia. La mala oxigenación y la carencia de nutrientes pue den matar muchas células. El papel del sistema inmunológico humano en la muerte de las células tumorales no se comprende aún, pero es probable que ciertas clases de linfocitos contribuyan también a este proceso.10 Además, genes específicos regulan la duración de la vida de muchos tipos celulares. Uno de estos genes, el bc1-2, ha mutado en algunas neoplasias humanas, en especial en los linfomas, causando pérdida del control de la supervivencia celular, con supervivencia prolongada de las células que han sufrido cambios neoplásicos.11
APOPTOSIS
La apoptosis es un proceso fisiológico normal que regula en forma específica la supervivencia celular.12,13 Es indispensable para la morfogénesis durante el proceso de desarrollo embrionario, para la selección clonal en el sistema inmunológico y quizá en la citotoxicidad mediada por células, y para la muerte de algunas células inducida por quimioterapia y radiación.14,15 Algunos cambios en la morfología nuclear y en la degradación del ADN en fragmentos son característicos de las células que sufren apoptosis. La apoptosis es un proceso activo que requiere síntesis de proteínas que puede ser desencadenada por cambios en el ambiente celular. Los aumentos o reducciones en los niveles de hormonas o de citocinas pueden iniciar la apoptosis. Por ejemplo, la eliminación de andrógenos puede iniciar la apoptosis en células epiteliales de la próstata. El gen supresor de tumores p53, cuya expresión es inducida cuando se daña el genoma de una célula y el oncogene bc1-2 tienen un papel crucial en la regulación de la apoptosis. Aunado a otros genes, el gen p53 vigila el estado del ADN de la célula. Si no se repara el daño al ADN, el producto del gen p53 inicia un proceso que termina en la apoptosis.16 El oncogen bc1-2 produce una sustancia que prohibe la muerte celular o apoptosis. Cuando el gen bc1-2 está activo se altera la apoptosis. Algunos agentes como las citocinas, los factores de crecimiento parácrino, los agonistas de los canales del calcio y los agentes anti hormonas esteroideas favorecen la apoptosis, mientras que otros agentes, como los productos del gen bcl-2, otros oncogenes, las hormonas endócrinas y quizá el virus Epstein-Barr, la retrasan.17,18 La falla de la muerte celular programada o la inhibición de los pasos intermedios que activan los mecanismos que causan la muerte celular pueden ser importantes en el inicio y progresión de los tumores desde el punto de vista clínico. Por lo tanto, la mejor comprensión de la apoptosis permitirá desarrollar nuevas estrategias para combatir el acúmulo de células neoplásicas.
MODELO DE FRACCION DE CRECIMIENTO
Se ha usado un modelo conceptual del crecimiento tumoral, comúnmente llamado modelo de la fracción de crecimiento, para desarrollar la mayor parte de las estrategias de quimioterapia [ver figura 2].20 En este modelo un tumor comprende cuatro compartimientos celulares: el compartimiento A contiene células en ciclo, que participan en la producción de células nuevas; el comportamiento B contiene células fuera de ciclo, que se encuentran detenidas temporalmente en la fase G0, pero que pueden reingresar al compartimiento de células en ciclo cuando se les aplica un estímulo apropiado; el comportamiento C contiene unas cuantas células vivas, pero detenidas, que no responden a las señales que estimulan el crecimiento ni vuelven a entrar al compartimiento de células en ciclo, y el compartimiento D representa a las células muertas, que pueden haber estado en cualquier compartimiento antes de morir. Cuando un tumor tiene una fracción de crecimiento alta, la mayor parte de sus células está en el compartimiento A; cuando un tumor tiene una fracción de crecimiento baja, la mayor parte de sus células está en los compartimientos B y C.
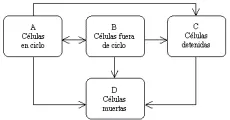
|
| Figura 2 |
| Modelo de crecimiento tumoral |
Casi todos los agentes quimioterápicos actualmente en uso interfieren con la síntesis de ADN, con la provisión de precursores para la síntesis de ADN y ARN o con la mitosis. Estos medicamentos son más eficaces contra las células en ciclo. Los mecanismos de muerte celular después del tratamiento con un solo agente o con la combinación de varios son complejos, y es probable que incluyan más de un proceso. Debido a que la mayoría de los tumores detectables por clínica se encuentran en la porción alta de la curva de Gompertz y están compuestos principalmente por células del compartimiento B que no están en el ciclo, no es sorprendente que la quimioterapia no siempre sea eficaz para erradicar el cáncer.
La estrategia del tratamiento del cáncer consiste en hacer que las células tumorales cambien del compartimiento B, fuera de ciclo, al compartimiento A, en ciclo. Varios métodos que promueven este cambio forman la base del tratamiento combinado. La cirugía es el método que más se emplea para reducir el tamaño del tumor y facilitar así la reentrada de células neoplásicas dentro del ciclo celular. Después de que el tumor primario se ha extirpado por completo, pueden quedar metástasis microscópicas en sitios distantes. Debido a su tamaño pequeño, las micrometástasis están compuestas principalmente de células en ciclo. El pequeño número de células que permanecen en el sitio del tumor primario pueden también reentrar al ciclo celular (i.e., cambiarse al compartimiento A). Por lo tanto, las células neoplásicas restantes son susceptibles a la quimioterapia. Puede usarse el tratamiento con radioterapia o quimioterapia solos para reducir la masa tumoral y así reclutar células dentro del compartimiento A, en ciclo. La estrategia de la quimioterapia adyuvante para el cáncer de mama se basa en estos conceptos.21,22
Quimioterapia
QUIMIOTERAPIA COMBINADA
Las investigaciones de tumores en animales y los estudios clínicos en humanos han demostrado que las combinaciones de medicamentos producen tasas de respuesta objetiva superiores y supervivencia más prolongada que los agentes únicos.23 Por lo tanto, el tratamiento combinado es la base de la mayoría de los esquemas de quimioterapia que se emplean en la actualidad. La quimioterapia combinada emplea mecanismos diferentes de acción y combina la citotoxicidad potencial de los diversos agentes. Aunque todos los medicamentos quimioterapéuticos son más eficaces sobre las células que sintetizan ADN, muchos agentes (en especial los alquilantes) pueden destruir células que no están en ciclo. Estos agentes se denominan medicamentos no dependientes de la proliferación celular. Algunos agentes, incluyendo muchos de los antimetabolitos y antibióticos, son mas eficaces contra las células durante la síntesis de ADN, por lo que se denominan medicamentos dependientes de la proliferación celular. La administración repetitiva de agentes no dependientes de la proliferación celular puede disminuir la masa tumoral al destruir células tanto de los compartimientos en ciclo como de fuera del ciclo. A continuación las células supervivientes se dirigen hacia el compartimiento de células en ciclo, donde son más sensibles a los medicamentos dependientes de la proliferación celular. El uso combinado de agentes no dependientes del ciclo celular, seguido de aquellos sí dependientes de este ciclo, es eficaz para aumentar la muerte de las células tumorales [ver figuras 2 y 3]. Cada ciclo de tratamiento destruye una fracción fija de células, de manera que se requieren ciclos repetidos para lograr la curación. Por ejemplo, una combinación de medicamento que destruya el 99.9 porciento de las células cancerosas por ciclo de tratamiento tendría que ser repetida por lo menos seis veces para eliminar una masa tumoral promedio (si no ocurriera crecimiento de células tumorales entre los ciclos).

|
| Figura 3 |
| Respuesta a la quimioterapia cíclica |
Estos modelos son necesariamente simplistas y sin duda minimizan factores importantes que pudieran reforzar la quimioterapia contra el cáncer. Sin embargo, cuando en los modelos fueron desarrollados por Skipper y colaboradores, proporcionaron la base teórica que se utilizó para curar la leucemia en niños.20
Varios principios guían la selección de medicamentos que se usan en combinación.24 Los medicamentos que son eficaces en forma individual se deben combinar en dosis tan altas y administrarse con tanta frecuencia como lo permita la toxicidad. Por lo tanto, son preferibles las combinaciones en donde no se superponen los tipos de toxicidad. Los medicamentos seleccionados deben también tener mecanismos diferentes de acción. Esta medida aumenta la destrucción celular, reduce la posibilidad de que surjan poblaciones de células resistentes al medicamento e interrumpen las funciones de las células neoplásicas al atacar múltiples vías metabólicas.
FACTORES QUE INFLUYEN EN LA EFICACIA DE LA QUIMIOTERAPIA
Para que la quimioterapia sea eficaz, el paciente debe estar en buenas condiciones fisiológicas, el tumor debe tener características de crecimiento favorables y los medicamentos deben ser eficaces contra ese tumor en particular. Entre las características relacionadas con el desarrollo tumoral que limitan la eficacia de la quimioterapia se encuentran la existencia de una gran masa tumoral en el momento del diagnóstico, una fracción de crecimiento tumoral baja, resistencia de las células tumorales a los medicamentos quimioterápicos, y la existencia de santuarios dentro del organismo que limiten el acceso de las drogas a las células tumorales [ver tabla 2].
|
|||||||||||||||||||||||||||
Nota: curable indica que más del 50% se curan, subcurable indica que menos del 50% se curan y precurable que los pacientes rara vez o nunca se curan con los esquemas actuales de quimioterapia. |
Una gran masa tumoral y la resistencia a varios medicamentos son los dos factores más importantes que limitan la eficacia de la quimioterapia. Las neoplasias malignas graves más comunes en humanos (cáncer de pulmón, mama, colon y próstata) rara vez se detectan antes de que el tumor primario haya crecido por lo menos hasta 1 cm y contenga aproximadamente 109 células. En el momento del diagnóstico muchos pacientes con cáncer tienen más de 1010 células neoplásicas (10 billones de células, o alrededor de 10 g de tumor) y con frecuencia pueden tener más de 100 billones de células. (La mayoría de los pacientes con cáncer mueren cuando sus masas celulares tumorales se acercan a las 1012 células, o 1 kg. de tumor). Por lo tanto, aunque existiera un medicamento que destruyera con eficacia el 99.9 porciento de las células tumorales, quedarían de 106 a 107 (de un millón a 10 millones) de células en el paciente típico con cáncer después del tratamiento inicial.
La eficacia de la quimioterapia puede alterarse por la cinética de crecimiento tumoral, la masa de células tumorales en todo el organismo, los efectos tóxicos de la quimioterapia sobre las células y otros tejidos diferentes al tumor y el deterioro de la función renal y hepática. Cuando las neoplasias son lo suficientemente grandes para ser detectadas, se encuentran en la parte alta de la curva de Gompertz (donde la fracción de crecimiento es baja) y ya ha ocurrido la mayor parte del crecimiento. La mayoría de estas células tumorales no están proliferando, por lo que es menos probable que cualquier agente quimioterápico las destruya. En forma semejante, a mayor tamaño del tumor primario, mayor probabilidad de que un número importante de células (resistentes y sensibles a los medicamentos) hayan metastatizado antes del diagnóstico y sean resistentes a la quimioterapia. Debido a que la mayoría de los agentes quimioterapéuticos dependen de la función renal y hepática para la excreción o modificación de los medicamentos, cuando existe daño en la función renal o hepática será necesario modificar las dosis para evitar toxicidad severa.
En el momento del diagnóstico muchos tumores humanos ya contienen células cancerosas que son resistentes a la quimioterapia estándar [ver adelante, Heterogeneidad de las células tumorales, Resistencia a los medicamentos].25,26 Se calcula en forma conservadora que existen mutaciones espontáneas que confieren resistencia a los medicamentos en una de cada l06-107 células neoplásicas. Esta velocidad de mutación parece ser independiente de cualquier presión selectiva por parte del tratamiento medicamentos, aunque la radioterapia y la quimioterapia pueden originar mutaciones adicionales. Las mutaciones, la pérdida de genes supresores de tumores y la activación de oncogenes aumentan también la velocidad de mutación y contribuyen a la progresión del tumor dentro de la población de células cancerosas.27-30 De ahí que la masa de células cancerosas en el momento del diagnóstico es de gran importancia porque incluso tumores de 1 cm (109 células) podrían contener hasta 100 a 1,000 células resistentes a medicamentos antes de iniciar el tratamiento.
Algunas metástasis se originan en ciertos sitios del organismo en donde son resistentes a la quimioterapia por la poca distribución tisular de los agentes administrados en dosis estándar. Estos sitios actúan como santuarios que protegen a las células neoplásicas de los medicamentos que circulan en sangre. Por ejemplo, existe este tipo de barrera en el cerebro y los testículos, que impiden la difusión de los medicamentos de los capilares hacia el tejido. Por lo tanto, estos sitios requieren modalidades especiales de tratamiento.
Heterogeneidad de las células tumorales
Han ocurrido grandes adelantos en el conocimiento de la composición celular de las neoplasias. Antes se pensaba que los tumores malignos estaban compuestos por poblaciones celulares uniformes u homogéneas, y que la caracterización de una sola célula podía ayudar a entender el funcionamiento de toda la población celular. Ahora está claro que un tumor maligno se encuentra compuesto por células que son heterogéneas para casi todas las características estudiadas.31,32 Se han observado las principales divergencias en el tamaño y forma de las células (morfología e histología), cariotipo (i.e., composición cromosómica), antígenos de superficie celular, respuesta a medicamentos (resistencia), capacidad de metastatizar y proliferar y concentración intracélular de enzimas y marcadores de células cancerosas (v.gr., receptores para estrógenos).
La mayor parte de los tumores malignos, aunque no todos, se originan de una sola célula o clona, y la población celular descendiente se diversifica debido a la constante aparición y selección de nuevas clonas,29,33,34 Los procesos que determinan la evolución clonal en los cánceres humanos son poco conocidos. Es posible que participen los procesos ocasionados por la inestabilidad genética (quizá provocados por genes que aumentan las mutaciones), como las rupturas cromosómicas, las traslocaciones, la activación de oncogenes y la pérdida de genes supresores de tumores. También pueden participar cambios en el ambiente del huésped, como las alteraciones en la regulación inmunológica Algunos linfomas parecen originarse de más de una clona y continúan cambiando respecto a la estructura celular y la respuesta al tratamiento, lo mismo que los cánceres que se originan de una sola clona.35 La comprensión de los mecanismos responsables de la diversificación continua de las poblaciones tumorales celulares que originan la progresión del tumor es el principal reto para poder desarrollar nuevos tratamientos contra el cáncer.31-33,36
RESISTENCIA A LOS MEDICAMENTOS
La resistencia de las células tumorales a los efectos destructores de la quimioterapia es uno de los problemas centrales en el tratamiento del cáncer. La destrucción selectiva de las células tumorales sensibles a los medicamentos causa una mayor proliferación de las células tumorales que son resistentes a la quimioterapia. Entre los mecanismos de resistencia a los medicamentos se incluyen el menor acumulo de medicamento (en especial la resistencia a medicamentos múltiples), el aumento en los mecanismos de transferencia de glutatión y de detoxificación, los niveles bajos de ADN topoisomerasa II, las alteraciones de enzimas o metabolismo de medicamentos, y el aumento en la capacidad de las células neoplásicas para reparar el daño inducido por medicamentos.37 Las células que crecen en exceso en la población tumoral no solo son resistentes a los agentes empleados, sino tienden a ser resistentes también a otros medicamentos, muchos de los cuales tienen mecanismos de acción distintos. Este fenómeno, denominado resistencia pleiotrópica a medicamentos o resistencia a múltiples fármacos, puede ser responsable de gran parte de la resistencia a los medicamentos que ocurre en los pacientes con cáncer tratados en forma previa.
La amplificación de los genes (i.e., la producción de copias extra de genes dentro de una célula) es uno de los mecanismos que puede originar resistencia a los medicamentos. Este mecanismo se ha investigado sobre todo en tumores que son resistentes a metotrexate tanto in vitro como in vivo.38 El metotrexate actúa al inhibir la acción de la enzima tetrahidrofolato reductasa. La amplificación del gen que codifica la tetrahidrofolato reductasa causa producción excesiva de la enzima, originando resistencia al medicamento. Además de la amplificación genética, los cambios en la velocidad del transporte del metotrexate hacia las células cancerosas y en la estructura de la dihidrofolato reductasa puede contribuir a la resistencia clínica a este fármaco.
La amplificacíón de genes puede influir también en el fenómeno de resistencia a múltiples fármacos. Esta parece estar asociada a la mayor expresión de una glucoproteína de la membrana celular denominada glucoproteína P, que se encuentra en la superficie de las células neoplásicas.39-4l Esta glucoproteína de membrana sirve como un transportador para el flujo de sustancias nocivas como los agentes quimioterápicos, y su mayor expresión se asocia con menor acumulo de múltiples agentes quimioterápicos dentro de las células neoplásicas resistentes. Se ha aislado e identificado la secuencia de un gen resistente a múltiples fármacos, el mdr-1, que codifica la glucoproteína P. Cuando este gen se transfiere, confiere resistencia a medicamentos en células antes sensibles a los mismos.42 El gen mdr 1 se expresa en varios tejidos normales, como el riñón, la glándula suprarrenal, el hígado y el colon, y su expresión puede estar aumentada en las células cancerosas de pacientes con leucemia, linfoma, mieloma múltiple y otras neoplasias que son resistentes a la quimioterapia.43 Aunque la amplificación de los genes puede causar expresión excesiva de la glucoproteína P, la rnayoría de los casos clínicos de resistencia a múltiples medicamentos es resultado de una mayor velocidad de transcripción del gen mdr 1, más que de amplificación del gen. Sin embargo, es posible que no toda la resistencia a múltiples fármacos se relacione con la expresión de la glucoproteína-P. Evidencias circunstanciales indican que en algunos casos ésta se relaciona con otras proteínas.44,45
En un esfuerzo por minimizar el efecto tóxico de la quimioterapia sobre las células tronco de la médula ósea, se ha transrectado el gen mdr-1 a células tronco normales de la médula ósea de ratones y de humanos en protocolos de investigación.46 En estos modelos, las células normales de la médula ósea son protegidas del efecto tóxico de la quimioterapia, por lo que estas células aumentan, en lugar de disminuir, con el tratamiento.47,48
La resistencia a múltiples fármacos en los cánceres humanos puede combatirse con medicamentos no citotóxicos que interactúan y evitan la función de la glucoproteína P. Entre los fármacos no citotóxicos que se usan aunados a la quimioterapia para prevenir la salida de agentes quimioterápicos de las células neoplásicas se incluyen los bloqueadores del calcio (v.gr., verapamil) los esteroides y antagonistas de esteroides (v.gr., tamoxifén), los antagonistas de la calmodulina (v.gr., trifluopcrazina), la ciclosporina y agentes cardiacos (v.gr., quinidina y amiodarona).49-5l Estos agentes compiten por la acción del transportador glucoproteína P o interfieren con su función. Esta medida inhibe la función normal de la glucoproteína P, causando menor excreción del agente citotóxico y, quizá, mayor destrucción de células.51 Aunque cuando se usa esta estrategia se encuentra regresión de los tumores, las respuestas son limitadas.
Se ha demostrado que muchos tumores expresan el gen mdr-1, pero debido a que muchos pacientes con tratamientos intensivos que recurren tienen tumores que no tienen expresión aumentada de este gen, es probable que otros mecanismos contribuyan a la resistencia a la quimioterapia.44,45,52,53 En muchos casos la resistencia a múltiples fármacos puede estar mediada por mutaciones o disminución de concentración en las topoisomerasas de ADN (enzimas que participan en la síntesis del ADN, en la separación de los cromosomas durante la mitosis, en la recombinación, y quizá en la transcripción genética). Debido a que las antraciclinas y algunas epipodofilotoxinas (v.gr., etopósido) pueden unirse al ADN y a su vez bloquear la acción de las topoisomerasas de ADN, los mecanismos de resistencia a estos medicamentos pueden incluir alteraciones en las topoisormerasas de ADN. Independientemente de los cambios en el gen mdr-1, la resistencia a múltiples fármacos puede ser causada también por mutaciones en el gen p53, que evita que un agente quimioterápico inicie la apoptosis, permitiendo que la célula se divida y se produzcan nuevas células aberrantes.
Estos hallazgos enfatizan la necesidad de una detección temprana del cáncer y de la atención cuidadosa a los esquemas y dosis de tratamiento.54-56 Los pacientes deben ser tratados con dosis altas de agentes quimioterápicos cuando la población neoplásica es pequeña y la posibilidad de que exista un número grande de células resistentes o metastásicas es escasa. Este concepto se refiere a la intensidad de dosis, esto es, la cantidad de medicamento que se administra por ciclo o unidad de tiempo. Es importante administrar varios medicamentos en forma simultánea o en días secuenciales y usar las dosis máximas toleradas.57 Debido a que los niveles de resistencia a fármacos baja o intermedia pueden sobrepasarse usando quimioterapia en dosis altas, los esfuerzos actuales se enfocan a determinar las dosis óptimas de los medicamentos y a usar programas de tratamiento con dosis intensivas asociados a trasplante de médula ósea . La resistencia causada por la amplificación genética es menos probable cuando se emplean esquemas de dosis altas porque esta amplificación puede desarrollarse con más facilidad cuando se inhibe la replicación del ADN solo en forma temporal. Un área activa de estudio es el uso de agentes que interfieren en forma directa con los mecanismos de resistencia a múltiples fármacos.
Estrategias en el uso de la quimioterapia
La quimioterapia combinada ha sido muy eficaz para aumentar el porcentaje de respuesta de una gran variedad de neoplasias. También ha permitido la curación de otros [ver tabla 2]. El 50 a 80 porciento de los pacientes con enfermedad de Hodgkin, linfoma histiocítico difuso, linfoma nodular mixto, carcinoma del testículo y coriocarcioma gestacional pueden curarse en la actualidad por medio de la quimioterapia combinada. Alrededor del 10 porciento de las pacientes con cáncer de ovario y el 15 a 25 porciento de los enfermos con leucemia mielógena pueden también ser curados. Aún más, la quimioterapia combinada con trasplante de la médula ósea puede aumentar estos porcentajes (ver adelante). La quimioterapia combinada ha causado resultados importantes en las neoplasias de la infancia: el 60 a 80 porciento de los niños con leucemia linfocítica aguda, linfoma de Burkitt o tumor de Wilms puede curarse en la actualidad.
Se requieren nuevas estrategias para tratar aquellas enfermedades malignas que son responsables de más del 50 porciento de muertes debidas al cáncer en adultos (cáncer del pulmón, colon, mama y próstata). Estos cánceres carecen de las características comunes a la mayoría de las enfermedades malignas que son curables con quimioterapia. Los tumores curables crecen rápidamente y tienden a presentar fracciones de crecimiento elevadas. Sin embargo, con excepción del cáncer de células pequeñas del pulmón, los tumores menos curables tienen fracciones de crecimiento bajas. Muchos de los tumores curables son mucho más sensibles a los agentes quimioterápicos que los tejidos normales en crecimiento, como las células de la médula ósea y el epitelio intestinal; el índice terapéutico, que expresa la relación entre las dosis terapéuticas y tóxicas de un medicamento, es alto en tales casos. Los agentes que son eficaces contra las enfermedades malignas comunes por lo general son limitados en número o tienen índices terapéuticos bajos. La mayor parte de los tumores actualmente curables ocurren en personas jóvenes, mientras que las enfermedades malignas comunes que escapan a la curación por quimioterapia afectan a personas mayores. Se considera que la resistencia a los medicamentos es el principal impedimento para el tratamiento útil de estas neoplasias, pero están emergiendo nuevos métodos terapéuticos diseñados para combatir los frecuentes problemas de baja fracción de crecimiento y resistencia a fármacos.
Algunos de los métodos descritos en esta sección están en etapa experimental y no deben iniciarse en la práctica clínica hasta que se demuestre su validez en estudios clínicos controlados. Estos estudios son necesarios para asegurarse de que se logra un avance significativo en el tratamiento y para proteger al paciente de la exposición a tratamientos tóxicos de valor limitado.
PERFUSION SISTEMICA Y REGIONAL
Se han usado las infusiones venosa y arterial de medicamentos quimioterápicos en lugar de las inyecciones en bolo para minimizar la toxicidad y lograr concentraciones altas de medicamento en los tejidos. Este método asegura que las células de proliferación más lenta comiencen la síntesis de ADN en presencia de concentraciones altas del fármaco.57 Pueden usarse diversos equipos para administrar estas infusiones, y todos ellos permiten tener un acceso permanente a la vena subclavia.58 Entre ellos se incluyen las bombas implantables subcutáneas o bombas externas unidas a sitios de acceso o catéteres subcutáneos que protruyen en forma permanente a través de la piel. En el procedimiento que parece ser más frecuente, se colocan sitios de acceso en forma subcutánea en pacientes que recibirán ciclos múltiples de quimioterapia (incluso cuando no la van a recibir en forma de infusión continua) para tener una vía venosa permeable adecuada y disminuir el efecto esclerosante que pueden tener algunas formas de quimioterapia sobre las venas periféricas. La infusión arterial y la infusión a la vena porta, aunque no se realizan con frecuencia, son otros métodos que sirven para administrar concentraciones altas de medicamentos a las metástasis .
QUIMIOTERAPIA CON DOSIS INTENSIVAS Y TRASPLANTE DE MEDULA OSEA ALOGENICA O SINGENICA O DE CELULAS TRONCO DE SANGRE PERIFERICA
Los esquemas de quirnioterapia con dosis altas pueden emplearse si se rescata al paciente con un trasplante autólogo o alogénico de células hematopoyéticas tronco o por medio de la administración de agentes que actúan como antídotos ante la acción tóxica del medicamento. La estrategia base de todos los esquemas con dosis altas consiste en vencer los mecanismos de resistencia a fármacos por medio de un efecto de acción en masa que produce concentraciones intracelulares muy altas de los quimioterápicos.59 La quimioterapia con dosis intensivas combate el problema frecuente de menor acumulo del medicamento al exponer a todas las células, incluyendo las que proliferan con lentitud, a una concentración consistentemente alta del fármaco.57
El trasplante de médula ósea, ya sea singénico (i.e., con genotipo idéntico) o alogénico (i.e., genéticamente distinto), después de la administración de dosis tumoricidas de quimioterapia, radioterapia o ambas, se usa cada vez más para tratar a pacientes con leucemias y algunos tumores sólidos.60 En la actualidad el trasplante de médula ósea es útil para tratar la leucemia linfocítica aguda, la leucemia mielógena aguda y crónica y el mieloma.6l El trasplante autólogo de médula ósea es un tratamiento eficaz en pacientes con leucemias agudas y en enfermos con recurrencia de linfomas de grado alto, enfermedad de Hodgkin o cáncer testicular.60,62,63 En varias instituciones se emplea también el trasplante autólogo en mujeres con cáncer de mama con recidivas, con un cáncer de mama primario con riesgo alto de recidiva (i.e., cáncer de mama que afecta 10 o más ganglios linfáticos) y cáncer de ovario recurrente64-68
Cada vez se usa más la reinfusión de células tronco de sangre periférica, en lugar del trasplante de médula ósea, en pacientes con cáncer sometidos a tratamiento intensivo para cánceres no hematológicos.69 Las células hema-topoyéticas tronco circulan dentro de la población de células nucleadas de la sangre periférica tanto de individuos normales como de pacientes con cáncer. Las células nucleadas son recolectadas por leucoféresis después de varios ciclos en dosis convencionales de quimioterapia y de estimulación con factores estimuladores de colonias [ver adelante, Modificadores de la respuesta biológica]. Después de la quimioterapia y de la administración de los factores estimuladores de crecimiento, el número de células hematopoyéticas tronco en la circulación periférica aumenta. Estas células tronco se asocian con las células nuceladas en la circulación periférica y pueden ser enriquecidas seleccionando las células CD-34 positivas. Cuando las células tronco periféricas se reinfunden, suele administrarse factor estimulador de colonias de granulocitos (FEC-G) o factor estimulador de colonias de granulocitos y macrófagos (FEC-GM) para favorecer el implante de célulares periféricas.70 Cuando va a realizarse un autoinjerto con células tronco periféricas el paciente es sometido a leucoféresis en varias ocasiones y se colectan y almacenan alrededor de 108 células nucleadas para su reinfusión posterior. Después de la quimioterapia en dosis intensivas que abate las células tronco de la médula ósea, la médula ósea del paciente se reconstituye por medio de la reinfusión de esta población de células nucleadas. La cuenta de plaquetas y granulocitos puede normalizarse con mayor rapidez después de la rehinfusión de las células tronco periféricas que después de la reinfusión de células autólogas de la médula ósea.
Los peligros potenciales con estos programas de tratamiento con quimioterapia en dosis altas seguido de rescate con células hematopoyéticas tronco incluyen enfermedad venoclusiva del hígado, neurnonitis intersticial, trombocitopenia persistente y enfermedad injerto contra huésped (cuando se usa un injerto alogénico). Algunos de estos riesgos pueden disminuir en forma significativa por medio del uso de FEC-G y FEC-GM en estos programas [ver adelante, Modificadores de la respuesta biológica]. Estos factores de crecimiento, al administrarse poco después de la quimioterapia intensiva, provocan una reaparición más rápida de granulocitos y plaquetas en sangre periférica. Sin embargo, es menos fácil de manejar la toxicidad al hígado, pulmones y corazón que puede asociarse con algunos agentes quimioterápicos administrados en dosis mayores de las convencionales. La reacción tóxica más significativa es la enfermedad venoclusiva del hígado: el uso de algunos agentes a dosis altas puede causar deterioro en la microcirculación del hígado, con falla hepática subsecuente.71
ADMINISTRACION INTRACAVITARIA DE LA QUIMIOTERAPIA
La administración directa de agentes en las cavidades peritoneal y pleural, el espacio cerebroespinal o la vejiga ha sido empleada para tratar tumores que están confinados a estos espacios, porque este método expone a las células tumorales a dosis altas de los agentes citotóxicos durante periodos prolongados y suele causar toxicidad sistémica leve.72,73 La quimioterapia intracavitaria ha sido útil sobre todo en el tratamiento del cáncer de ovario y en el cáncer superficial de la vejiga.
QUIMIOTERAPIA DIRIGIDA
Otro método para vencer la resistencia a los fármacos incluye técnicas de adminis-tración innovadoras diseñadas para lograr concentraciones locales altas de fármacos citotóxicos o toxinas en el sitio de la célula tumoral.74-76 Estos métodos incluyen el uso de agentes citotóxicos incorporados a los liposomas o unidos a lipoproteínas de baja densidad a ADN o a microesferas. Estos medicamentos han sido unidos también a anticuerpos monoclonales específicos dirigidos contra las células neoplásicas77-81 Estas técnicas, que aún están en evolución, intentan distribuir en forma específica o dirigir la acción de los medicamentos (el llamado método de proyectil mágico), provocando así el máximo daño a las células tumorales y minimizando la lesión para las células normales.
Los anticuerpos monoclonales, administrados solos o en combinación con otros agentes como el interferón alfa, parecen ser promisorios en el tratamiento de un número limitado de tipos de cáncer.82-84 Se ha observado regresión de linfomas B después del tratamiento con anticuerpos monoclonales dirigidos contra componentes de superficie específicos de las células de linfoma de un paciente en particular.79,85-87 El tratamiento con anticuerpos monoclonales contra el HER-2/neu, un receptor de factores de crecimiento, puede causar regresión del cáncer en el 25 porciento de las pacientes con cáncer de mama en las que esta molécula se expresa en forma exagerada. Es posible que los anticuerpos monoclonales demuestren ser útiles para la observación de las metástasis [ver adelante, Marcadores tumorales en el tratamiento del cáncer].81
MODIFICADORES DE LA RESPUESTA BIOLOGICA
Los modificadores de la respuesta biológica, que en condiciones normales son producidos por los humanos y otros mamíferos para aumentar la respuesta inmunológica contra los tumores y la infección, han sido empleados solos o en combinación con otros agentes para el tratamiento experimental de las neoplasias humanas.89,90 Los interferones, una gran familia de glucoproteínas, son ejemplos de estos modificadores de la respuesta biológica. Hasta el 90 porciento de los pacientes con leucemia de células peludas responde en forma favorable al tratamiento con interferón alfa.90 Los interferones también han demostrado cierta eficacia en los linfomas no Hodgkin de grado bajo, el linfomacutáneo de células T, la leucemia mielógena crónica, el mieloma múltiple, el sarcoma de Kaposi asociado con síndrome de inmunodeficiencia adquirida (SIDA), el cáncer superficial de vejiga y quizá el cáncer de células renales.91,92 Sin embargo, el tratamiento con interferón para cánceres corno el de mama, colon y prostata no ha producido resultados alentadores. La mayoría de los estudios de tratamiento con interferón en el manejo del cáncer se refieren al uso del interferón alfa, pero en la actualidad se sintetizan otros miembros de la familia de los interferones por medio de técnicas de recombinación de ADN, lo que facilitará el estudio de estos compuestos y ayudará a resolver las dudas en relación con los esquemas de dosificación.
La inmunoterapia adoptiva empleando linfocinas, como la interleucina-2 (IL-2) ha recibido atención considerable porque aprovecha un mecanismo endógeno de destrucción de células tumorales.10,93-95 La IL-2 parece activar a las células T citotóxicas, a las células asesinas naturales y quizá a otras células linfoides que a su vez atacan a las células neoplásicas. El tratamiento solo con dosis altas de IL-2 o con células linfoides de sangre periférica activadas con IL-2 autóloga ha producido regresión de metástasis en algunos pacientes refractarios al tratamiento convencional o para los que no existe un tratamiento sistémico establecido. El melanona y el cáncer de células renales son de los tumores que mejor han respondido a esta modalidad. Aunque se ha autorizado ya el uso de IL-2 para el tratamiento del cáncer de células renales, su toxicidad puede ser importante.
Quizá el tratamiento biológico que más se emplea en la actualidad consiste en el uso de FEC-G y FEC-GM. Estas citocinas, que en condiciones normales son producidas por varios tipos celulares en la médula ósea (y quizá en otros sitios), pueden producirse en grandes cantidades por medio de biotecnología recombinante. El FEC-G y el FEC-GM son factores de crecimiento hematopoyético que están autorizados para la prevención de la melosupresión inducida por quimioterapia y que en la actualidad se emplean en otras situacioncs además del trasplante de células hematopoyéticas tronco.96
El FEC-G se encuentra en el suero de los individuos normales y su nivel aumenta en respuesta a las infecciones bacterianas. Por el contrario, no se detecta FEC-GM en suero, y se piensa que esta citocina actúa en forma parácrina, más que en forma endócrina. El FEC-GM actúa sobre muchas células precursoras y sobre células maduras de diversas líneas dentro de la médula ósea. Puede aumentar el número de neutrófilos, monocitos y eosinófilos en la circulación periférica y puede prolongar la vida media circulante de los neutrófilos. El FEC-G apoya la expresión, supervivencia y maduración de los precursores de neutrófilos, disminuyendo el tiempo para su maduración a granulocitos maduros. Estudios aleatorios que incluyeron pacientes con cáncer pulmonar de células pequeñas mostraron que el FEC-G asociado a un esquema de quimioterapia intensiva redujo el número de hospitalizaciones y los días de estancia hospitalaria, así como el número de episodios febriles, de infecciones confirmadas por cultivo y de ciclos de quimioterapia en los que tuvieran que reducirse las dosis por la leucopenia97 El FEC-G se emplea también después de la quimioterapia ablativa y de la reinfusión de médula osea autóloga para la enfermedad de Hodgkin, el linfoma no Hodgkin, el cáncer de mama y otros tumores sólidos y leucemias agudas. En estos estudios, el tiempo de injerto y el número de días de fiebre se redujeron en forma significativa.70,98,99 El uso de FEC-G no parece aumentar el riesgo de recurrencia de la leucemia en pacientes que son sometidos a trasplante de médula ósea por leucemia aguda.100
VACUNAS TUMORALES
Una estrategia de tratamiento en vías de investigación es cl desarrollo de vacunas tumorales, que intentan cumplir con dos objetivos: inducir una respuesta inmunológica contra antígenos específicos del tumor e inducir una respuesta inmunológica contra las metástasis tumorales. En un primer enfoque se han inyectado células tumorales completas o lisados celulares después de resecar un melanoma o un cáncer de colon y recto primarios para inmunizar a los pacientes que tienen riesgo alto de recurrencia.101,102 En una segunda aplicación, se han empleado inmunoglobulinas idiotípicas expresadas en la superficie de células de linfoma B como antígenos específicos del tumor para estimular las respuestas inmunológicas en entermos con linfomas clínicamente activos,103 y se han administrado antígenos autólogos de melanoma a pacientes con melanoma avanzado.104 Debido a que los linfomas de células B son clonales, todas las células de un linfoma expresan una misma inmunoglobulina de superficie (i.e., inmunoglobulina idiotípica). Las inmunoglobulinas idiolípicas pueden producirse en grandes cantidades en el laboratorio. Los pacientes que reciben inmunoglobulinas idiotípicas de sus propios linfomas montan respuestas inmunológicas humorales y mediadas por células contra este idiotipo. Sin embargo, las vacunas para tratar o prevenir el crecimiento de los tumores humanos aún están en etapas muy tempranas de investigación.
QUlMIOTERAPIA NEOADYUVANTE
La quimioterapia neoadyuvante se refiere a la administración de agentes quimioterápicos antes del tratamiento estándar con cirugía, radioterapia o ambos.105 El objetivo de la misma es reducir el tamaño del tumor lo suficiente como para que los pacientes sean mejores candidatos a la cirugía o radioterapia. La quimioterapia neoadyuvante ha sido empleada sobre todo en el tratamiento del cáncer de cabeza y cuello, el sarcoma osteogénico y el cáncer de mama localmente avanzado.106-110 Los medicamentos como los radiosensibilizadores y los radioprotectores pueden usarse en esta forma para aumentar los efectos de la radioterapia sobre las células tumorales o para proteger a las células normales del daño por radiación.
QUIMIOPREVENCION E INDUCCION DE LA DIFERENCIACION
Las células diferenciadas sintetizan proteínas específicas y tienden a proliferar poco o no proliferar. Con frecuencia se considera a las células neoplásicas como no diferenciadas porque no sintetizan productos proteicos que las distingan de otras células y porque son capaces de proliferar casi sin límite. Un enfoque de tratamiento (y quizá de prevención) para el cáncer que no requiere de la destrucción celular consiste en alterar la velocidad de crecimiento de las células dentro de un tumor aumentando la diferenciación de las mismas.111,112 El uso de medicamentos que promueven la diferenciación celular puede ser también importante en el manejo de condiciones premalignas como la leucoplasia. Por ejemplo, la administración sistémica de isotretinoín (ácido I3-cis-retinoico) causa que la mucosa leucoplásica oral revierta hacia un epitelio normal diferenciado. Los estudios aleatorios con adyuvantes demostraron que la administración de isotretinoín después del tratamiento principal redujo también la incidencia de cánceres secundarios en pacientes con cáncer de cabeza y cuello113,114 tenerse cuidado con cualquier tratamiento quimopreventivo para el cáncer; por ejemplo, los estudios controlados demuestran que se desarrolla cáncer de pulmón con más frecuencia en los fumadores que reciben b caroteno que en los que no reciben este medicamento.l15
Terapia génica
La introducción de genes a las células tumorales o a células normales se encuentra en las fases iniciales de aplicación clínica para el tratamiento del cáncer.46,l16 Aun que los adelantos recientes en biología molecular han tenido un gran impacto en el diagnóstico, su impacto sobre el tratamiento de tumores humanos ha sido mucho menor. Sin embargo), se están desarrollando enfoques sofisticados para bloquear los efectos de los productos de los oncogenes, identificar a las células neoplásicas y destruirlas.46,117 Los enfoques que se han examinado incluyen lo siguiente: (1) aumentar la inmullogenicidad de los tumores insertando genes de citocinas, (2) insertar genes cuyos productos, al ser procesados, sean tóxicos para las células, (3) codificar ARN antisentido, que bloquee los efectos de los oncogenes y los genes de resistencia a fármacos, y (4) marcar células de modo que puedan detectarse después.46,117,118 Las limitaciones actuales de estas medidas se relacionan con los métodos para administrar los genes en forma específica a las células tumorales y para restringir la expresión de los genes de las células normales cuando éstas son el blanco. Es necesario solucionar problemas de selectividad, especificidad y seguridad antes de poder aplicar en forma amplia estos enfoques de tratamiento génico.
INHIBICION DE LA ANGIOGENESIS
La angiogéllesis (i.e., la formación de arteriolas que promueven el aporte de sangre a los tumores en crecimiento y a las metástasis) ocurre en respuesta a los factores de crecimiento parácrino producidos por el tumor y por los tejidos normales en respuesta al tumor.119 Un área de gran interés es el papel de la vascularización en el desarrollo del tumor. Durante la progresión del tumor los factores de crecimiento parácrino sirven para reclutar tejido del estroma y ampliar el aporte sanguíneo al tumor en expansión.l20 Los tumores producen estímulos que incitan o inhiben la formación de nuevos vasos sanguíneos, que son indispensables para el crecimiento y remodelación del tumor y sus metástasis.121 Al teñir los tejidos (en especial los cánceres primarios de la mama o la próstata) en busca de capilares y contar el número de vasos en un área definida es posible predecir la evolución.122,123 Varios estudios han demostrado un peor pronóstico para los pacientes cuyo tumores tienen gran número de capilares pequeños. En la actualidad se estudian estrategias para interferir con la angiogénesis como un método para tratar el cáncer. Algunos agentes, como el angiostatin, parecen bloquear la formación de los vasos nuevos.124 Aún no se ha definido el papel de la inhibición de la angiogénesis en el tratamiento de los canceres primariosmetastásicos, así como de otras enfermedades.125
Marcadores tumorales en el manejo del cáncer
El manejo del cáncer incluye el desarrollo de métodos inmunológicos que pueden detectar neoplasias cuando su tamaño es pequeño y que pasan desapercibidas desde el punto de vista clínico.l26,127 Pueden detectarse en el suero de pacientes con cáncer testicular, coriocarcinoma gestacional, hepatoma, cáncer de próstata o cáncer ovárico, antígenos circulantes que pueden ser de utilidad para detectar o confirmar la presencia de un cáncer o su recurrencia.128 Cuando existe la posibilidad de curación para un cáncer diseminado (en especial en el caso del cáncer testicular y el coriocarcinoma gestacional), la medición de estos antígenos tiene especial importancia. Por ejemplo, los inmunoensayos que miden gonadotropinas en circulantes en las mujeres con corlocarcinoma gestacional pueden detectar la presencia de micrometástasis, lo que orientará a la continuación del tratamiento hasta que estas metástasis hayan desaparecido. Existen también marcadores tumorales circulantes para el cáncer de testículo (gonadotropinascoriónicas), el hepatoma(a-fetoproteína), el cáncer de próstata (antígeno prostático específico) y algunos otros carcinomas. Los ensayos de anticuerpos monoclonales pueden detectar un antígeno del cáncer de ovario humano conocido como CA 125 en el suero del 80 porciento de las pacientes con cáncer de ovario demostrado por biopsia.l29-131 Aunque este estudio es útil para vigilar a las pacientes con cáncer de ovario y se ha demostrado que puede detectarlo, el antígeno CA 125 es también un indicador inespecífico de afección peritoneal, y su concentración se eleva en otros padecimientos incluyendo la cirrosis hepática con ascitis.l32 Sin embargo, los valores muy altos suelen asociarse con cáncer de ovario. Aún no es muy clara la manera como pueden emplearse los marcadores tumorales en el manejo inicial de cánceres diseminados cuando no existe la posibilidad de tratamientos curativos. Por ejemplo, la detección temprana de la recidiva del cáncer de mama no ofrece en la actualidad ningún beneficio clínico.133 Los marcadores como el antígeno carcinoembriónico (ACE), que se encuentran en cánceres frecuentes, no han sido tan útiles como se esperaba. El ACE no es específico para un carcinoma, ni siempre se eleva en presencia de neoplasia.
La tecnología con ADN recombinante puede ser de utilidad para el diagnóstico del cáncer y para definir los mecanismos por los que las células normales se transforman en células neoplásicas . Las técnicas de ADN recombinante también se han empleado con éxito para diagnosticar linfomas,35,l34,l35 y parecen ser útiles para identificar algunos pacientes con riesgo alto de recidiva de cáncer de mama y neuroblastoma.136,137 Esta tecnología parece tener un papel cada vez más importante en el diagnóstico del cáncer y para evaluar el pronóstico. Por ejemplo, pueden usarse técnicas de ADN recombinante para realizar escrutinio de carcinoma de colon.l38
Estudio de células tumorales tronco en humanos
El estudio de células tumorales tronco en humanos, también llamado ensayo clonogénico, se ha empleado para medir la sensibilidad de las células tumorales a la quimioterapia.139 Este estudio compara el número de colonias formadas en el cultivo celular en presencia y ausencia de varias diluciones de medicamentos aislados. Sin embargo, varios factores limitan su aplicación clínica: (1) es frecuente que los tumores no tengan suficientes células clonables, (2) la prueba predice mejor a qué medicamentos no responderán las células tumorales y no a cuáles fármacos son sensibles, y (3) predice los efectos de los agentes aislados, más que de la combinación de los mismos (que es la manera como suelen administrarse estos fármacos).~4~) ~4l
Aunque la esperanza inicial de que esta técnica pudiera servir como una guía general de tratamiento para el paciente individual parece no haberse cumplido, el ensayo proporciona información sobre la heterogeneidad de las células tumorales y la resistencia a los medicamentos. Por ejemplo, el ensayo reveló que incluso en ausencia de medicamentos, solo una pequeña proporción de células tumorales (0.01 a 0.10 porciento) de la muestra de biopsia es capaz de crecer en forma clonal. Aunque las dificultades técnicas podrían explicar esta baja eficiencia de clonación, parece ser que la mayoría de las células tumorales tienen un potencial limitado de crecimiento y no pueden proliferar lo suficiente para producir una nueva masa tumoral. Este hallazgo tiene implicaciones importantes para la formación de metástasis, porque indica que la mayoría de las células tumorales pueden proliferar solo durante varias generaciones y que solo una pequeña proporción de las células lo hacen en forma indefinida. Estas células capaces de proliferar de modo casi ilimitado, quizá solo una de cada 1,000 a 10,000 células tumorales, se conocen como células clonogénicas o tronco. Las células tumorales que se comportan como células tronco tienen gran importancia clínica porque una pequeña fracción de las misulas son resistentes a los medicamentos quimioterápicos, incluso en pacientes que no han sido tratados previamente con estos agentes.
En la actualidad el ensayo clonogénico tiene poca utilidad en el manejo clínico del cáncer.
Agentes anticancerosos de uso común
Los agentes quimioterápicos comúnmente usados se enlistan en la tabla 3. Están clasificados por un modo de acción, origen o estructura, aunque algunos de los agentes no se ajustan de manera clara a un solo grupo. Las categorías incluyen agentes alquilantes, antimetabólitos, antibióticos, alcaloides y agentes diversos (incluyendo hormonas). La figura 4 resume los sitios de acción de muchos de estos medicamentos.
|
| Figura 4 |
| Mecanismo de la quimioterapia |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
* En E.U.A. |
Los agentes alquilantes forman enlaces convalentes con los ácidos nucleicos. Estos agentes alteran la integridad del ADN transfiriendo un grupo alquilo a los ácidos nucleicos. Algunos tienen dos sitios reactivos, lo que les permite formar enlaces cruzados con el ADN. Los agentes de esta clase son tanto citotóxicos como carcinógenos. La ciclofosfamida es el agente alquilante utilzado con mayor frecuencia
Los antimetabolitos son estructuralmente similares a los sustratos metabólicos normales. Alteran las funciones celulares sustituyendo los precursores normales en reacciones fisiológicas vitales, o bloqueando estas reacciones. Los antimetabolitos que se emplean más a menudo son el metotrexate y el 5-fluorouracilo, los cuales interfieren con la síntesis de los ácidos nucleicos.
Los antibióticos son productos biológicos de bacterias u hongos y tienen gran importancia en el tratamiento del cáncer. No comparten un solo mecanismo de acción. La bleomicina y las antraciclinas daunorrubucina y doxorrubicina son algunos de los antibióticos comúnmente usados. La bleomicina produce su citotoxicidad rompiendo físicamente el ADN. Las antraciclinas logran su efecto citotóxico por diversos mecanismos, incluyendo intercalación entre las hélices de ADN, interfiriendo de esta manera con la síntesis de ADN y ARN y la acción de las ADN topoisomerasas, producción de radicales libres que reaccionan con las proteínas y ácidos nucleicos intercelulares dañándolos, quelación de cationes divalentes, y reacción con las membranas celulares. La amplia variedad de sitios potenciales de acción puede ser responsable tanto del grado de eficacia como de la toxicidad de las antraciclinas.142
Los alcaloides, como la vincristina, la vinblastina, la vindesina y el paclitaxel (Taxol), se unen a la tubulina, proteína citoplasmática estructural, y evitan el ensamble o desensamble de los microtúbulos. Como el aparato en huso responsable del movimiento cromosómico durante la mitosis consta de microtúbulos, los alcaloides son especialmente activos durante la mitosis. Muchos otros procesos celulares, como el mantenimiento de la forma y el transporte intracelular de nutrientes en el sistema nervioso periférieo, dependen también de la formación de microtúbulos y son afectados por los alcaloides. Es probable que la neuropatía que se asocia al uso de estos medicamentos resulte de su acción sobre los microtúbulos de los axones largos de los nervios. El etopósido (VP- 16), una epipodofilotoxina, es un nuevo alcaloide que detiene a las células en fase G2 y en fase S temprana al actuar sobre las ADN topoisomerasas.
Los fármacos en la categoría de diversos tienen diversas acciones. Por ejemplo, la dacarbazina y la procarbazina son similares en su modo de acción a los agentes alquilantes, mientras que la L-asparaginasa actúa enzimáticamente. La L-asparaginasa es una de las pocas proteínas usadas en la quimioterapia contra el cáncer. No interfiere directamente con la síntesis de ADN, sino que inhibe la síntesis proteica. La L-asparagina es un aminoácido no esencial. En la mayor parte de los tejidos humanos normales, la síntesis de L asparagina a partir del ácido aspártico es catalizada por la enzima L-asparagina sintetasa. Sin embargo, muchos tumores derivados de linfocitos T carecen de esta enzima y por lo tanto dependen del plasma para su aporte de L asparagina. El tratamiento con L-asparaginasa agota el fondo circulante de L-asparagina y así deteriora la síntesis proteica en dichas células tumorales, destruyéndolas finalmente.
Las hormonas, en especial las homonas esteroideas prednisona, progesterona, estrógeno y androgeno, se usan con frecuencia en el tratamiento contra el cáncer. Sin embargo, estas cuatro hormonas se han ido sustituyendo por nuevos agentes que interfieren en forma selectiva o no selectiva con la acción hormonal. Estos agentes incluyen tamoxifen, toremifine y droloxifene, que son antiestrógenos usados en el tratamiento del cáncer de mama, leuprolide, un análogo de la hormona liberadora de gonadotropinas que evita la liberación de gonadotropinas y se usa en el tratamiento del cáncer de mama y próstata, y anastrozole, que bloquea la acción de la arimidasa, una enzima que convierte los andrógenos en esírógenos en el tejido adiposo y otros tejidos. Los agentes que son semejantes en acción a las hormonas naturales pueden ser eficaces también en el tratamiento de los cánceres. Un ejemplo es el megestrol, un agente progestágeno que es útil contra el cáncer de mama cuando se emplea en dosis altas. Aunque no se conocen del todo los mecanismos exactos por los que varias hormonas reducen el tamaño tumoral, parece ser que alteran la cinética de crecimiento tisular del tumor y favorecen la muerte celular, como la apoptosis. Por ejemplo, la prednisona puede ser eficaz en la leucemia linfocítica aguda porque induce apoptosis en células linfoides inmaduras. Otras hormonas inician la producción de factores de crecimiento parácrino a partir de las células del estroma normal de un tumor, lo que puede alterar el crecimiento tumoral.
Los agentes quimioterápicos, que casi siempre se administran en forma combinada, ofrecen paliaciónexitosa para la mayoría de los pacientes con cáncer y pueden proporcionar curación para algunos. La mayoría de los esquemas causan toxicidad considerable, por lo que se requiere tener experiencia y estar familiarizado con la farmacología de cada combinación que vaya a emplearse.
Bibliografía
-
Steel GG: Growth Kinetics of Tumours. Oxford University Press,
New York, 1977, p 40
-
Spratt JS, Meyer JS, Spratt JA: Rates of growth of human
solid neoplasms (pt 1). J Surg Oncol 60:137, 1995 [PMID
7564383]
-
Spratt JS, Meyer JS, Spratt JA: Rates of growth of human
neoplasms (pt 2). J Surg Oncol 61:68, 1996 [PMID
8544465]
- Fridovich KJ, Hansen LJ, Keyomarsi K, et al: Progression through the cell cycle: an overview. Am Rev Respir Dis 142:S3, 1990
-
Hartwell L, Kastan M: Cell cycle and cancer. Science 266:1821,
1994 [PMID
7997877]
-
Steel GG: Growth Kinetics of Tumours. Oxford University Press,
New York, 1977, p 190
-
Mendelsohn ML: The growth fraction: a new concept applied
to tumors. Science 132:1496, 1960
-
Tubiana M: Tumor cell proliferation kinetics and tumor growth
rate. Acta Oncol 28:113, 1989
-
Steel GG: Cell loss as a factor in the growth rate of human
tumors. Eur J Cancer 3:381, 1967
-
Lotze MT, Custer MC, Bolton ES, et al: Mechanisms of immunologic
antitumor therapy: lessons from the laboratory and clinical applications.
Hum Immunol 28:198, 1990 [PMID
2190952]
-
Brada M: BCL2 gene: current relevance to clinical oncology.
Eur J Cancer 28: 270, 1992
-
Marx J: Cell death studies yield cancer clues. Science 259:760,
1993
-
Orrenius S: Apoptosis: molecular mechanisms and implications
for human disease. J Intern Med 237:529, 1995
-
Williams GT: Programmed cell death: apoptosis and oncogenesis.
Cell 65:1097, 1991
-
Wyllie AH: Apoptosis and the regulation of cell numbers in
normal and neoplastic tissues: an overview. Cancer Metastasis Rev 11:95,
1992
-
Chang F, Syrjanen S, Syrjanen K: Implications of the p53
tumor-suppressor gene in clinical oncology. J Clin Oncol 13:1009, 1995
[PMID
7707100]
-
Oren M: The involvement of oncogenes and tumor suppressor
genes in the control of apoptosis. Cancer Metastasis Rev 11:141, 1992
-
Owens GP, Cohen JJ: Identification of genes involved in programmed
cell death. Cancer Metastasis Rev 11:149, 1992 [PMID
1394794]
-
Lowe SW: Cancer therapy and p53. Curr Opin Oncol 7:547, 1995
-
Skipper HE, Perry S: Kinetics of normal and leukemic leukocyte
populations and relevance to chemotherapy. Cancer Res 30:1883, 1970 [PMID
4917694]
-
Weiss RB, DeVita VT Jr: Multimodal primary cancer treatment
(adjuvant chemotherapy): current results and future prospects. Ann Intern
Med 91:251, 1979 [PMID
380437]
-
Bonadonna G, Valagussa P: Adjuvant chemotherapy for breast
cancer. Semin Surg Oncol 4:250, 1988
-
Frei E III: Combination cancer therapy: presidential address.
Cancer Res 32: 2593, 1972
-
DeVita VT, Young RC, Canellos GP: Combination versus single
agent chemotherapy: a review of the basis for selection of drug treatment
of cancer. Cancer 35: 98, 1975 [PMID
162854]
-
Goldstein LJ, Pastan I, Gottesman MM: Multidrug resistance
in human cancer. Crit Rev Oncol Hematol 12:243, 1992 [PMID
1353966]
-
Moscow JA, Morrow CS, Cowan KH: Multidrug resistance. Cancer
Chemother Biol Response Modif 13:91, 1992
-
Goldie JH, Coldman AJ: A mathematic model for relating the
drug sensitivity of tumors to their spontaneous mutation rate. Cancer Treat
Rep 63:1727, 1979 [PMID
526911]
-
Goldie JH, Coldman AJ: The genetic origin of drug resistance
in neoplasm: implications for systemic therapy. Cancer Res 44:3643, 1984
[PMID
6744284]
-
Nowell PC: Mechanisms of tumor progression. Cancer Res 46:2203,
1986
-
Goldie JH: Modelling the process of drug resistance. Lung
Cancer 10:S91, 1994
-
Coleman WB, Tsongalis GJ: Multiple mechanisms account for
genomic instability and molecular mutation in neoplastic transformation.
Clin Chem 41:644, 1995 [PMID
7729041]
-
Shackney SE, Shankey TV: Genetic and phenotypic heterogeneity
of human malignancies: finding order in chaos. Cytometry 21:2, 1995 [PMID
8529466]
-
Nowell PC: The clonal evolution of tumor cell populations.
Science 194:23, 1976
-
Nowell PC: Molecular events in tumor development. N Engl
J Med 319:575, 1988
-
Sklar J, Cleary ML, Thielemans K, et al: Biclonal B-cell
lymphoma. N Engl J Med 311:20, 1984
-
Schnipper LE: Clinical implications of tumor-cell heterogeneity.
N Engl J Med 314:1423, 1986
-
Harris AL, Hochhauser D: Mechanisms of multidrug resistance
in cancer treatment. Acta Oncol 31:205, 1992 [PMID
1352455]
-
Schimke RT: Gene amplification: what are we learning? Mutat
Res 276:145, 1992
-
Biedler JL: Genetic aspects of multidrug resistance. Cancer
70:1799, 1992
-
Roninson IB: The role of the MDR1 (P-glycoprotein) gene in
multidrug resistance in vitro and in vivo. Biochem Pharmacol 43:95, 1992
-
Kaye S: Clinical drug resistance: the role of factors other
than P-glycoprotein. Am J Med 99:40, 1995
-
Gros P, Neriah YB, Croop JM, et al: Isolation and expression
of a complementary DNA that confers multidrug resistance. Nature 323:728,
1986 [PMID
3022150]
-
Fojo AT, Ueda K, Slamon DJ, et al: Expression of a multidrug-resistance
gene in human tumors and tissues. Proc Natl Acad Sci USA 84:265, 1987 [PMID
2432605]
-
Woodhouse JR, Ferry DR: The genetic basis of resistance to
cancer chemotherapy. Ann Med 27:157, 1995 [PMID
7632408]
-
Scheffer G, Wijngaard P, Flens M, et al: The drug resistance-related
protein LPP is the human major vault protein. Nature Medicine 1:527, 1995
-
Hanania EG, Kavanagh J, Hortobagyi G, et al: Recent advances
in the application of gene therapy to human disease. Am J Med 99:537, 1995
[PMID
7485213]
-
Podda S, Ward M, Himelstein A, et al: Transfer and
expression of the human multiple drug resistance gene into live mice. Proc
Natl Acad Sci USA 89:9676, 1992 [PMID
1357667]
-
Sorrentino BP, Brandt SJ, Bodine D, et al: Selection of drug-resistant
bone marrow cells in vivo after retroviral transfer of human MDR1. Science
257:99, 1992 [PMID
1352414]
-
Sonneveld P, Durie BG, Lokhorst HM, et al: Modulation of
multidrug-resistant multiple myeloma by cyclosporin. The Leukaemia Group
of the EORTC and the HOVON. Lancet 340:255, 1992
-
Yahanda AM, Alder KM, Fisher GA, et al: Phase I trial of
etoposide with cyclosporine as a modulator of multidrug resistance. J Clin
Oncol 10:1624, 1992 [PMID
1403040]
-
Lum BL, Kaubisch S, Yahanda AM, et al: Alteration of etoposide
pharmacokinetics and pharmacodynamics by cyclosporine in a phase I trial
to modulate multidrug resistance. J Clin Oncol 10:1635, 1992 [PMID
1403041]
-
Chabner BA, Fojo A: Multidrug resistance: P-glycoprotein
and its allies-the elusive foes. J Natl Cancer Inst 81:910, 1989 [PMID
2567356]
-
Beck WT: Unknotting the complexities of multidrug resistance:
the involvement of DNA topoisomerases in drug action and resistance. J
Natl Cancer Inst 81: 1683, 1989
-
DeVita VT: Dose-response is alive and well. J Clin Oncol
4:1157, 1986
-
Goldie JH: Genetic and kinetic factors in the drug resistance
of micrometastatic disease. Prog Clin Biol Res 354A:99, 1990
-
Goldie JH: Mathematical models of drug resistance and chemotherapy
effects. Cancer Treat Res 48:13, 1989
-
Frei E III, Canellos GP: Dose: a critical factor in cancer
chemotherapy. Am J Med 69:585, 1980
-
Busch E, Kemeny MM: Colorectal cancer: hepatic-directed therapy:
the role of surgery, regional chemotherapy, and novel modalities. Semin
Oncol 22:494, 1995 [PMID
7570060]
-
van der Wall E, Beijnen JH, Rodenhuis S: High-dose chemotherapy
regimens for solid tumors. Cancer Treat Rev 21:105, 1995 [PMID
7758003]
-
Santos GW: Bone marrow transplantation in hematologic malignancies:
current status. Cancer 65:786, 1990
-
Canellos GP, Nadler L, Takvorian T: Autologous bone marrow
transplantation in the treatment of malignant lymphoma and Hodgkin's disease.
Semin Hematol 25:58, 1988 [PMID
3041601]
-
Nichols CR, Tricot G, Williams SD, et al: Dose-intensive
chemotherapy in refractory germ cell cancer-a phase I/II trial of high-dose
carboplatin and etoposide with autologous bone marrow transplantation.
J Clin Oncol 7:932, 1989 [PMID
2544687]
-
Meehan KR, Pritchard RS, Leichter JW, et al: Autologous bone
marrow transplantation versus chemotherapy in relapsed/refractory non-Hodgkin's
lymphoma: estimates of long-term survival from the recent literature. Am
J Hematol 50:116, 1995 [PMID
7572990]
-
Marks LB, Halperin EC, Prosnitz LR, et al: Post-mastectomy
radiotherapy following adjuvant chemotherapy and autologous bone marrow
transplantation for breast cancer patients with greater than or equal to
10 positive axillary lymph nodes. Cancer and Leukemia Group B. Int J Radiat
Oncol Biol Phys 23:1021, 1992
-
Shpall EJ, Clarke PD, Soper JT, et al: High-dose alkylating
agent chemotherapy with autologous bone marrow support in patients with
stage III/IV epithelial ovarian cancer. Gynecol Oncol 38:386, 1990 [PMID
2121627]
-
Santos GW, Yeager AM, Jones RJ: Autologous bone marrow transplantation.
Annu Rev Med 40:99, 1989 [PMID
2658765]
-
Peters WP: High-dose chemotherapy with autologous bone marrow
transplantation for the treatment of breast cancer: yes. Important Adv
Oncol 215, 1995
-
Kotz KW, Schilder RJ: High-dose chemotherapy and hematopoietic
progenitor cell support for patients with epithelial ovarian cancer. Semin
Oncol 22:250, 1995 [PMID
7777869]
-
Demuynck H, Delforge M, Zachee P, et al: An update on peripheral
blood progenitor cell transplantation. Ann Hematol 71:29, 1995 [PMID
7632815]
-
Taylor KM, Jagannath S, Spitzer G, et al: Recombinant human
granulocyte colony-stimulating factor hastens granulocyte recovery after
high-dose chemotherapy and autologous bone marrow transplantation in Hodgkin's
disease. J Clin Oncol 7:1791, 1989 [PMID
2479719]
-
Shulman HM, Hinterberger W: Hepatic veno-occlusive disease-liver
toxicity syndrome after bone marrow transplantation. Bone Marrow Transplant
10:197, 1992 [PMID
1422475]
-
Markman M: Intraperitoneal chemotherapy. Semin Oncol 18:248,
1991
-
Williams RD: Intravesical interferon alfa in the treatment
of superficial bladder cancer. Semin Oncol 15:10, 1988
-
Pastan I, Chaudhary V, FitzGerald DJ: Recombinant toxins
as novel therapeutic agents. Annu Rev Biochem 61:331, 1992 [PMID
1497314]
-
Rosenberg SA: Karnofsky Memorial Lecture. The immunotherapy
and gene therapy of cancer. J Clin Oncol 10:180, 1992
-
Press OW, Eary JF, Badger CC, et al: Radiolabeled antibody
therapy of human B cell lymphomas. Adv Exp Med Biol 303:91, 1991 [PMID
1805577]
-
Pai L, Witter R, Setser A, et al: Treatment of advanced solid
tumors with immunotoxin LMB-1: an antibody linked to Pseudomonas
exotoxin. Nature Medicine 2: 350, 1996 [PMID
8612238]
-
Sugarman SM, Perez-Soler R: Liposomes in the treatment of
malignancy: a clinical perspective. Crit Rev Oncol Hematol 12:231, 1992
[PMID
1497823]
-
Pai L, Pastan I: Immunotoxin therapy for cancer. JAMA 269:78,
1993 [PMID
8416411]
-
French R, Hamlin J, Bell A, et al: Treatment of B-cell lymphomas
with combination of bispecific antibodies and saponin. Lancet 346:223,
1995 [PMID
7542357]
-
International symposium on labeled and unlabeled antibody
in cancer diagnosis and therapy. Order SE, Ed. Natl Cancer Inst Monogr
No 3, 1987
-
Oldham RK: Custom-tailored drug immunoconjugates in cancer
therapy. Mol Biother 3:148, 1991
-
Brown SL, Miller RA, Horning SJ, et al: Treatment of B-cell
lymphomas with anti-idiotype antibodies alone and in combination with alpha
interferon. Blood 73:651, 1989 [PMID
2465039]
-
Baselga J, Tripathy D, Mendelsohn J, et al: Phase II study
of weekly intravenous recombinant humanized anti-p185HER2 monoclonal
antibody in patients with HER2/neu-overexpressing metastatic breast
cancer. J Clin Oncol 14: 737, 1996 [PMID
8622019]
-
Miller RA, Maloney DG, Warnke R, et al: Treatment of B-cell
lymphoma with monoclonal anti-idiotype antibody. N Engl J Med 306:517,
1982 [PMID
6173751]
-
Levy R, Miller RA: Therapy of lymphoma directed at idiotypes.
Monogr Natl Cancer Inst 10:61, 1990
-
Maloney DG, Brown S, Czerwinski DK, et al: Monoclonal anti-idiotype
antibody therapy of B-cell lymphoma: the addition of a short course of
chemotherapy does not interfere with the antitumor effect nor prevent the
emergence of idiotype-negative variant cells. Blood 80:1502, 1992 [PMID
1520877]
-
Parkinson D: Present status of biological response modifiers
in cancer. Am J Med 99:54, 1995
-
Tursz T: Combination of BRMs with chemotherapy. Am J Med
99:56, 1995
-
Golomb HM, Ratain MJ, Mick R, et al: Interferon treatment
for hairy cell leukemia: an update on a cohort of 69 patients treated from
1983-1986. Leukemia 6:1177, 1992 [PMID
1434800]
-
Muss HB, Costanzi JJ, Leavitt R, et al: Recombinant alfa
interferon in renal cell carcinoma: a randomized trial of two routes of
administration. J Clin Oncol 5: 286, 1987 [PMID
3543247]
-
Marshall ME, Wolf M, Crawford ED, et al: Evaluation of low
dose alpha-interferon (Roferon-A) in patients with advanced renal cell
carcinoma: a Southwest Oncology Group study. Cancer Biother 10:205, 1995
[PMID
8547959]
-
Rosenberg SA, Lotze MT, Muul LM, et al: A progress report
on the treatment of 157 patients with advanced cancer using lymphokine-activated
killer cells and interleukin-2 or high dose interleukin-2 alone. N Engl
J Med 316:889, 1987 [PMID
3493432]
-
Ettinghausen SE, Rosenberg SA: Immunotherapy and gene therapy
of cancer. Adv Surg 28:223, 1995 [PMID
7879680]
-
Lummen G, Goepel M, Mollhoff S, et al: Phase II study of
interferon-gamma versus interleukin-2 and interferon-alpha 2b in metastatic
renal cell carcinoma. J Urol 155:455, 1996
-
Lieschke GJ, Burgess AW: Granulocyte colony-stimulating factor
and granulocyte-macrophage colony-stimulating factor (pt 1). N Engl J Med
327:28, 1992 [PMID
1375975]
-
Crawford J, Ozer H, Stoller R, et al: Reduction by granulocyte
colony-stimulating factor of fever and neutropenia induced by chemotherapy
in patients with small-cell lung cancer. N Engl J Med 325:164, 1991 [PMID
1711156]
-
Sheridan WP, Morstyn G, Wolf M, et al: Granulocyte colony-stimulating
factor and neutrophil recovery after high-dose chemotherapy and autologous
bone marrow transplantation. Lancet 2:891, 1989 [PMID
2477656]
-
Ohno R, Tomonaga M, Kobayashi T, et al: Effect of granulocyte
colony-stimulating factor after intensive induction therapy in relapsed
or refractory acute leukemia. N Engl J Med 323:871, 1990 [PMID
1697646]
-
Masaoka T, Shibata H, Ohno R, et al: Double-blind test of
human urinary macrophage colony-stimulating factor for allogeneic and syngeneic
bone marrow transplantation: effectiveness of treatment and 2-year follow-up
for relapse of leukaemia. Br J Haematol 76:501, 1990 [PMID
2265113]
-
Bystryn JC, Oratz R, Roses D, et al: Relationship between
immune response to melanoma vaccine immunization and clinical outcome in
stage II malignant melanoma. Cancer 69:1157, 1992 [PMID
1739915]
-
Barth A, Morton DL: The role of adjuvant therapy in melanoma
management. Cancer 75:726, 1995
-
Kwak LW, Campbell MJ, Czerwinski DK, et al: Induction of
immune responses in patients with B-cell lymphoma against the surface-immunoglobulin
idiotype expressed by their tumors. N Engl J Med 327:1209, 1992 [PMID
1406793]
-
Morton DL, Foshag LJ, Hoon DS, et al: Prolongation of survival
in metastatic melanoma after active specific immunotherapy with a new polyvalent
melanoma vaccine. Ann Surg 216:463, 1992
-
Frei E III: What's in a name-neoadjuvant (editorial). J Natl
Cancer Inst 80: 1088, 1988
-
Goffinet DR, Stockdale FE: Inflammatory breast cancer. Current
Therapy in Oncology. Niederhuber JE, Ed. BC Decker, Philadelphia, 1992,
p 361
-
Friedland DM, Comis RL: Perioperative therapy of non-small
cell lung cancer: a review of adjuvant and neoadjuvant approaches. Semin
Oncol 22:571, 1995 [PMID
8539633]
-
Freedman GM, Coia LR: Adjuvant and neoadjuvant treatment
of rectal cancer. Semin Oncol 22:611, 1995 [PMID
8539636]
-
Smith MR, Kantoff PW: Neoadjuvant and adjuvant chemotherapy
for invasive bladder cancer. Semin Oncol 22:625, 1995 [PMID
8539637]
-
Shenoy MA, Singh BB: Chemical radiosensitizers in cancer
therapy. Cancer Invest 10:533, 1992 [PMID
1422891]
-
Hong WK, Lippman SM: Cancer chemoprevention. Monogr Natl
Cancer Inst (17):49, 1995 [PMID
8573453]
-
Lippman SM, Heyman RA, Kurie JM, et al: Retinoids and chemoprevention:
clinical and basic studies. J Cell Biochem Suppl 22:1, 1995 [PMID
8538183]
-
Hong WK, Lippman SM, Itri LM, et al: Prevention of second
primary tumors with isotretinoin in squamous-cell carcinoma of the head
and neck. N Engl J Med 323:795, 1990 [PMID
2202902]
-
Lippman SM, Batsakis JG, Toth BB, et al: Comparison of low-dose
isotretinoin with beta carotene to prevent oral carcinogenesis. N Engl
J Med 328:15, 1993 [PMID
8416267]
-
The effect of vitamin E and beta carotene on the incidence
of lung cancer and other cancers in male smokers. The Alpha-Tocopherol,
Beta Carotene Cancer Prevention Study Group. N Engl J Med 330:1029, 1994
-
Herrmann F: Cancer gene therapy: principles, problems, and
perspectives. J Mol Med 73:157, 1995
-
Mercola D, Cohen JS: Antisense approaches to cancer gene
therapy. Cancer Gene Therapy 2:47, 1995
-
Rosenberg S: Gene therapy in cancer. JAMA 268:2416, 1992
-
Folkman J: Tumor angiogenesis and tissue factor (comment).
Nature Medicine 2: 167, 1996
-
Bikfalvi A: Significance of angiogenesis in tumour progression
and metastasis. Eur J Cancer 31A:1101, 1995
-
Folkman J: Angiogenesis inhibitors generated by tumors. Molecular
Medicine 1: 120, 1995
-
Gasparini G, Harris AL: Clinical importance of the determination
of tumor angiogenesis in breast carcinoma: much more than a new prognostic
tool. J Clin Oncol 13: 765, 1995 [PMID
7533829]
-
Folkman J: Angiogenesis and breast cancer. J Clin Oncol 12:441,
1994
-
O'Reilly MS, Holmgren L, Shing Y, et al: Angiostatin: a novel
angiogenesis inhibitor that mediates the suppression of metastases by a
Lewis lung carcinoma. Cell 79: 315, 1994 [PMID
7525077]
-
Folkman J: Angiogenesis in cancer, vascular, rheumatoid and
other disease. Nature Medicine 1:27, 1995
-
Stahel RA, Mabry M, Skarin AT, et al: Detection of bone marrow
metastasis in small-cell lung cancer by monoclonal antibody. J Clin Oncol
3:455, 1985 [PMID
2984342]
-
Duffy MJ: Can molecular markers now be used for early diagnosis
of malignancy? Clin Chem 41:1410, 1995
-
Seleznick MJ: Tumor markers. Prim Care 19:715, 1992
-
Bast RC, Klug TL, St. John E, et al: A radioimmunoassay using
a monoclonal antibody to monitor the course of epithelial ovarian cancer.
N Engl J Med 309:883, 1983
-
Einhorn N, Sjovall K, Knapp RC, et al: Prospective evaluation
of serum CA 125 levels for early detection of ovarian cancer. Obstet Gynecol
80:14, 1992 [PMID
1603484]
-
Jacobs IJ, Oram DH, Bast RC Jr: Strategies for improving
the specificity of screening for ovarian cancer with tumor-associated antigens
CA 125, CA 15-3, and TAG 72. 3. Obstet Gynecol 80:396, 1992
-
Hunter VJ, Weinberg JB, Haney AF, et al: CA 125 in peritoneal
fluid and serum from patients with benign gynecologic conditions and ovarian
cancer. Gynecol Oncol 36: 161, 1990 [PMID
2404835]
-
Moertel C, Fleming T, MacDonald J: An elevation of carcinoembryonic
antigen (CEA) test for monitoring patients with resected colon cancer.
JAMA 270:943, 1993 [PMID
8141873]
-
Sklar J, Weiss LM: Applications of antigen receptor gene
rearrangements to the diagnosis and characterization of lymphoid neoplasms.
Annu Rev Med 39:315, 1988 [PMID
3285779]
-
Sklar J: What can DNA rearrangements tell us about solid
hematolymphoid neoplasms? Am J Surg Pathol 14(suppl 1):16, 1990
-
Slamon DJ, Clark GM, Wong SG, et al: Human breast cancer:
correlation of relapse and survival with amplification of the HER-2/neuoncogene.
Science 235:177, 1987 [PMID
3798106]
-
Seeger RC, Brodeur GM, Sather H, et al: Association of multiple
copies of the N-myc oncogene with rapid progression of neuroblastomas.
N Engl J Med 313:1111, 1985 [PMID
4047115]
-
Sidransky D, Tokino T, Hamilton SR, et al: Identification
of ras oncogene mutations in the stool of patients with curable
colorectal tumors. Science 256: 102, 1992 [PMID
1566048]
-
Salmon SE: Human tumor colony assay and chemosensitivity
testing. Cancer Treat Rep 68:117, 1984
-
Phillips RM, Bibby MC, Double JA: A critical appraisal of
the predictive value of in vitro chemosensitivity assays. J Natl Cancer
Inst 82:1457, 1990
-
Von Hoff DD, Clark GM, Weiss GR, et al: Use of in vitro dose
response effects to select antineoplastics for high-dose or regional administration
regimens. J Clin Oncol 4:1827, 1986
-
Myers CE: Anthracyclines. Cancer Chemother Biol Response
Modif 13:45, 1992