Contenido del artículo
IX ENFERMEDADES DESMIELINIZANTES
- Definición
- Esclerosis múltiple
- EPIDEMIOLOGIA
- PATOLOGIA
- ETIOLOGIA
- GENETICA
- DIAGNOSTICO
- DIAGNOSTICO DIFERENCIAL
- TRATAMIENTO
- PRONOSTICO
- Neuritis óptica
- Encefalomielitis diseminada aguda
- Mielitis transversa
- Enfermedades desmielinizantes hereditarias
- Enfermedades desmielinizantes metabólicas
- Desmielinización inducida por virus
DR. J. WILLIAM LINDSEY
DR. JERRY S. WOLINSKY
Tanto en el sistema nervioso central como en el periférico, los axones de diámetro grande están mielinizandos. La mielina y se forma y mantiene por las células oligodendrogliales dentro del sistema nervioso central y por las células de Schwann en el sistema nervioso periférico. La mielina aísla los axones y organiza los constituyentes de su superficie de membrana, funciones que son cruciales para la transferencia rápida de señales necesarias para la actividad motora coordinada, la integración e interpretación apropiadas de los estímulos sensoriales y para facilitar la función cognitiva. Las enfermedades que afectan la integridad de la célula oligodendroglial y su capacidad para producir y mantener la mielina o las enfermedades que dañan en forma directa la vaina de mielina alteran la conducción en las vías de sustancia blanca mielinizada y producen muy diversas disfunciones de tipo motor, sensorial y cognitivo [ver figura 1].
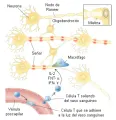
|
| Figura 1 |
| Estructura de los nervios mielinizados |
Las enfermedades desmielinizantes alteran la integridad de la mielina, respetando los axones [ver tabla 1]. Estos padecimientos afectan principalmente la supervivencia oligodendroglial (v.gr., leucoencefalopatía multifocal progresiva), el metabolismo oligodendroglial (v.gr., deficiencia de vitamina B12) y la vaina de mielina, con efectos secundarios sobre las células oligodendrogliales (v.gr., esclerosis múltiple [EM]).
|
||||||||||
|
Las enfermedades desmielinizantes con origen inmunológico incluyen
padecimientos desmielinizantes recurrentes y crónicos progresivos (EM y
sus variantes) y enfermedades desmielinizantes monofásicas (neuritis
óptica, encefalomielitis aguda diseminada y mielitis transversa). La
desmielinización monofásica puede ser el primer signo
clínico de EM.
La EM se caracteriza clínicamente por disfunción neurológica recurrente o crónica progresiva causada por lesiones en el SNC. Desde el punto de vista patológico las lesiones consisten en áreas múltiples de desmielinización que afectan el cerebro, los nervios ópticos y la médula espinal.
Alrededor de 250,000 a 350,000 personas tienen EM en los Estados Unidos, la prevalencia es de alrededor de 100 casos por 100,000 habitantes.1 La EM es más común en las mujeres que en los varones, con una relación de 2:1. Ocurre en todos los grupos raciales, pero es más frecuente en blancos, menos común en negros y rara en asiáticos. El inicio de la enfermedad suele ocurrir entre los 20 y 50 años de edad, con un pico alrededor de los 30 años. La prevalencia de la EM varía mucho según el sitio geográfico, siendo la mayor en latitudes altas en las regiones norte de Europa y América. Esta variación geográfica sugiere que la EM puede ser causada en parte por la acción de algún factor ambiental que es más común en esas latitudes. Hasta cierto grado la idea es apoyada por estudios de migración. El movimiento de un área de alto riesgo a una región de bajo riesgo a edades tempranas de la vida tiende a disminuir el riesgo de la enfermedad, y el movimiento contrario aumenta el riesgo. Sin embargo, mucha de la variación por estados en los Estados Unidos se relaciona con la ascendencia escandinava, lo que sugiere que la variación geográfica puede ser solo un reflejo de la distribución de grupos étnicos con diferente susceptibilidad.
Las placas, que consisten en cantidades variables de inflamación periventricular con células mononucleares, desmielinización sin afectar los axones, pérdida de células oligodendrogliales y proliferación de astrocitos con gliosis secundaria, son el dato característico en la EM.2 Dentro del SNC las placas pueden aparecer en cualquier sitio en que existan fibras mielinizadas. La mayoría de las placas se congregan a lo largo de las venas periventriculares de drenaje, pero también son frecuentes en la médula espinal, nervios ópticos, tallo cerebral y sustancia blanca de los hemisferios cerebrales y el cerebelo. Ocurren placas en las vías de conexión de la sustancia blanca subcortical. También pueden surgir placas intracorticales debido a que los axones de los cuerpos neuronales están mielinizados en todo su curso desde la vecindad del montecillo axonal.
La apariencia histopatológica de las placas varía con el tiempo. En las lesiones agudas y recientes predominan los infiltrados periventriculares de células T y macrófagos. Se altera la función de la barrera hematoencefálica en la región, lo que se asocia con edema vasogénico. Existen evidencias inmunocitoquímicas y citoquímicas de la activación de las células endoteliales por citocinas (mayor expresión de marcadores de superficie de las células endoteliales como el complejo principal de histocompatibilidad, la molécula de adhesión intracelular y la selectina E), activación de células T (mayor expresión de antígeno Ia, de interferón gama y secreción de linfotoxina) y activación de macrófagos (secreción de factor de necrosis tumoral-a y fagocitosis activa de axones mielinizados con ingestión y degradación temprana de la mielina).3 Los astrocitos se activan en fases tempranas de formación de las placas. Al inicio pueden existir células dendrogliales dentro o en los bordes de las nuevas lesiones.4 Es posible que exista cierto grado de remielinización, que puede ser extensa en las placas tempranas. Eventualmente el infiltrado inflamatorio puede resolverse por completo. Las lesiones más antiguas se caracterizan por una pérdida total de la mielina y las células oligodendrogliales, astrogliosis intensa, grados variables de pérdida axonal y un infiltrado residual escaso de células mononucleares, algunas de las cuales son células B que secretan inmunoglobulinas. Es común la sección axonal en muchas lesiones de EM, en especial en las que aparecen como áreas con persistente menor intensidad de señales en las imágenes de IRM de T1, y es posible que la sección axonal sea la causa de las deficiencias neurológicas permantentes.5,6
La etiología de la EM se desconoce. Sin embargo, la importancia de los factores genéticos ha sido firmemente establecida por los estudios de EM familiar y la contribución de factores no genéticos se demuestra por la falta de concordancia en la mayoría de los gemelos idénticos y por el efecto de la migración sobre el riesgo de la enfermedad.
Se desconoce el proceso patológico subyacente, aunque la mayoría de los especialistas están de acuerdo en que la EM es en parte una enfermedad autoinmune o de mediación inmunológica. La evidencia para esta conclusión incluye hallazgos patológicos (i.e., placas en las fibras mielinizadas), aumento en la síntesis de IgG en el líquido cefalorraquídeo y diversas alteraciones in vitro en la función de los leucocitos. La respuesta autoinmune puede ser la causa primaria de la enfermedad o un epifenómeno de otro proceso. El modelo popular de mimetismo molecular postula que el ataque autoinmune sobre la mielina es precipitado por un organismo infeccioso que contiene una proteína semejante a la mielina. La infección despierta una respuesta inmune intensa de los linfocitos que reconocen la proteína con reacción cruzada. En el proceso de eliminación del organismo los linfocitos activados dañan la mielina. La veracidad de esta teoría ha sido demostrada en elegantes experimentos realizados en ratones transgénicos,7 pero no se ha demostrado la relevancia de este modelo en la EM. La EM puede ser causada por otros procesos o mecanismos, como la infección persistente del SNC o un defecto bioquímico en la mielina.8,9
La sospecha de que la susceptibilidad a la EM es por lo menos en parte familiar ha sido confirmada en múltiples estudios. El riesgo de que ocurra EM en el gemelo monocigoto de un paciente con EM es del 31 por ciento, mientras que el riesgo en el gemelo dicigoto es de alrededor del 5 por ciento. El riesgo para un hermano o familiar de una persona afectada fue del 3 a 4 por ciento, comparado con el riesgo en la población general, que es de solo el 0.1 por ciento.11 Otros estudios en hermanos adoptados y medios hermanos demostró que este mayor riesgo familiar se atribuyó totalmente a factores genéticos y no ambientales.12,13 Estos datos implican que varios genes interactuantes influyen en la susceptibilidad a la EM.14
Los factores genéticos que confieren susceptibilidd a la EM se conocen solo en parte. El único locus reproducible que se ha demostrado se asocia con la EM es un antígeno leucocitario humano (HLA) de clase II. El haplotipo extendido DRB1*1501 se asocia con EM en pacientes de raza blanca.15 En un intento por identificar otros loci que contribuyan a la EM, tres grupos independientes de investigadores han realizado escrutinio genómicos extensos en grupos de familias con varios casos de EM.16-18 No se observaron regiones con una especial asociación a la EM, aunque existieron varias regiones con evidencia relativamente débil de asociación que requieren de más estudio.
El diagnóstico de EM se realiza con base en los signos y síntomas clínicos; la imagen de resonancia magnética y otras pruebas de laboratorio ayudan al mismo. Para diagnosticar EM se requiere evidencia de lesiones diseminadas en el tiempo y espacio y de la exclusión cuidadosa de otras causas. El paciente debe tener más de un episodio de disfunción neurológica y lesiones en la sustancia blanca en más de un sitio del SNC. Debido a que no existe un signo o síntoma patognomónico ni un resultado de laboratorio definitivo, para el diagnóstico se requerirá del juicio clínico cuidadoso de un neurólogo con experiencia.
Existen varios grupos validados de criterios diagnósticos para la EM. Los de Poser y colaboradores19 [ver tabla 2] son bien aceptados e incluyen tanto datos clínicos como de laboratorio. La categoría clínica definida requiere de la presencia de dos episodios clínicos y dos lesiones, los casos con menos certeza se clasifican como probables o posibles. Todas las categorías requieren de la exclusión de otros diagnósticos. Estos criterios se describieron originalmente para el uso de los investigadores en pacientes seleccionados que participarían en estudios clínicos. Por lo tanto, son especialmente estrictos y no todos los pacientes con EM los satisfacen. De cualquier modo, los criterios son útiles en la práctica clínica general porque ayudan a identificar a pacientes con posible o probable EM y que deben ser vigilados periódicamente.
|
||||||||||||||||||||||||
|
En la EM puede ocurrir casi cualquier deficiencia neurológica, pero existen varios signos y síntomas que son característicos. Ninguno de estos datos característicos son patognomónicos de la EM, aunque su presencia debe hacer pensar en este diagnóstico, en especial en los adultos jóvenes.
Los datos típicos incluyen neuritis óptica, oftalmoplegia internuclear, sensibilidad al calor y el signo de Lhermitte. Alrededor del 20 por ciento de los pacientes presentan neuritis óptica como síntoma inicial, y ésta se desarrolla en más de la mitad de todos los pacientes con EM. Con la neuritis óptica el paciente sufre alteraciones de la visión limitadas a un ojo. Estas alteraciones característicamente ocurren con un escotoma central. Suele existir dolor retrocular que se acentúa al mover el ojo. Los resultados del examen fundoscópico suelen ser normales, aunque puede existir papilitis o palidez del disco por episodios previos. Los síntomas suelen evolucionar en varios días antes de que se alcance la deficiencia máxima. Otro síntoma ocular típico es la diplopia causada por la oftalmoplegia internuclear. Al examen clínico existe incapacidad para la aducción en la mirada lateral, aunque se conserva la aducción con la convergencia. La oftalmoplegia internuclear es causada por una lesión en el fascículo longitudinal medio y es sugestiva de EM.
La sensibilidad al calor es una molestia característica en la EM. El ejercicio, fiebre, un baño caliente u otras actividades que aumentan la temperatura corporal pueden causar la aparición de nuevos síntomas o la recurrencia de los antiguos. Estos eventos ocurren como resultado del bloqueo en la conducción inducido por la temperatura en las fibras con desmielinización parcial. Los síntomas se resuelven al normalizarse la temperatura. El signo de Lhermitte también es característico de la EM. Se caracteriza por parestesias que irradían desde la espalda hacia las extremidades inferiores al flexionar el cuello. Este síntoma indica la presencia de una lesión en la médula cervical.
Existen otros síntomas que son muy comunes en la EM pero que también ocurren en muchos otros procesos patológicos. Entre ellos destacan debilidad, hipoestesias, ataxia y síntomas intestinales y vesicales. Todos se desarrollan en la mayoría de los pacientes con EM durante el curso del padecimiento [ver tabla 3]. La debilidad asociada con la ES es de tipo motoneurona superior y se acompaña de espasticidad e incremento en los reflejos. Puede ocurrir debilidad con cualquier patrón, incluyendo paraparesia, hemiparesia y monoparesia. En la enfermedad establecida las extremidades inferiores suelen afectarse más que las superiores. Los síntomas sensoriales también son frecuentes y pueden ocurrir en diferentes patrones. Los pacientes pueden observar parestesias o pérdida de la sensación. De nuevo, las extremidades inferiores se afectan más. La pérdida de la sensación de vibración suele ser la más significativa. La ataxia es poco común al inicio, pero ocurre en cierto grado en la mayoría de los pacientes. Son frecuentes las alteraciones en el control del esfínter vesical, estas molestias pueden ser muy incómodas para los pacientes, que sufren urgencia, polaquiuria e incontinencia de urgencia compatibles con una vejiga espástica. También pueden ocurrir goteo, retención, incontinencia por reflujo y disinergia. La constipación suele ser el síntoma intestinal más común.
|
||||||||
*Evidencia clínica significa signos presentes al examen neurológicos. Los signos demostrados en el pasado por un examinador competente se aceptan, incluso si han desaparecido. La evidencia paraclínica consiste en la demostración de lesiones por medios auxiliares (v.gr., neuroimagen o respuestas evocadas) y que no causan signos clínicos. |
Aunque casi cualquier alteración puede ocurrir en la EM, algunas son
extremadamente raras, como las crisis convulsivas, la afasia, los trastornos de
movimiento y la atrofia muscular, que reflejan principalmente afección
de la materia gris o de los nervios periféricos. Aunque estos
síntomas pueden ocurrir en la EM, su presencia no es habitual y obligan
a reconsiderar el diagnóstico.
La evolución clínica de la EM varía mucho entre los pacientes. Típicamente la enfermedad tiene un patrón de recaídas y remisiones, con exacerbaciones agudas seguidas de resolución parcial o completa. En el curso de varias horas o días aparecen nuevos síntomas neurológicos, que permanecen estables por un periodo de días a semanas y después mejoran en forma gradual. Al principio de la enfermedad los síntomas pueden resolverse casi sin secuelas. Con las exacerbaciones repetitivas tienden a desarrollarse deficiencias permanentes. Los pacientes suelen tener intervalos de meses o años libres de síntomas entre los ataques.
Los síntomas también pueden ocurrir en forma progresiva, con acúmulo constante de deficiencias neurológicas sin que existan exacerbaciones claramente definidas. Se dice que los pacientes que al inicio tienen enfermedad con recaídas y remisiones y después entran a una fase progresiva tienen enfermedad progresiva secundaria, mientras que en los que tienen síntomas progresivos desde el inicio el padecimiento se considera progresivo primario. Entre el 15 al 30 por ciento de los pacientes tienen enfermedad progresiva primaria, y de los que al inicio tienen enfermedad con recaídas y remisiones el 30 a 50 por ciento sufrirá síntomas progresivos en los primeros 10 años.20 Los estudios radiológicos y genéticos sugieren que el grupo progresivo primario puede ser diferente del grupo con recaídas y remisiones y progresión secundaria.21
Se han identificado variantes clínicas o patológicas de la EM. La neuromielitis óptica o enfermedad de Devic, se caracteriza por la afección simultánea o secuencial de los nervios ópticos y la médula espinal, y con frecuencia tiene una evolución maligna. La EM tipo Marburg o EM aguda es una variante con un curso monofásico fulminante. La enfermedad de Schilder, o esclerosis difusa, es la variante rápidamente progresiva asociada con lesiones cerebrales desmielinizantes extensas. La enfermedad de Baló es una variante patológica en la que ocurren áreas concéntricas de desmielinización en el cerebro.
Imagen por resonancia magnética La IRM es el estudio aislado más útil para el diagnóstico de la EM.22 La mayoría de los pacientes con EM tienen alteraciones que pueden observarse con esta técnica, y la excelente resolución anatómica de la prueba permite la exclusión de muchas enfermedades que podrían dar alteraciones semejantes. Las lesiones de EM son hiperintentsas en la imagen en T2 o de densidad de protones e hipo o isointensas en la imagen en T1 [ver figura 2]. Las lesiones típicas son ovoides y periventriculares, con su eje mayor perpendicular al ventrículo, aunque pueden aparecer lesiones en cualquier área en la sustancia blanca. Algunas lesiones pueden reforzar al administrar gadolinio y esto indica una lesión de la barrera hematoencefálica. Incluso en la EM temprana suelen existir varias lesiones, aún cuando no produzcan síntomas obvios.
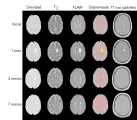
|
| Figura 2 |
| IRM en la esclerosis múltiple |
Aunque la IRM es muy sensible para detectar lesiones en la sustancia blanca de pacientes con EM, no es muy específica. Muchos otros padecimientos producen lesiones múltiples en la sustancia blanca, de modo que la IRM nunca debe emplearse como base única para el diagnóstico. Se han propuesto varios grupos de criterios para aumentar la especificidad de los hallazgos en la IRM, aunque todos los incrementos en especificidad se asocian con reducción en la sensibilidad. Las características útiles para aumentar el valor predictivo de la IRM para el diagnóstico de EM incluyen la presencia de tres o más lesiones en la sustancia blanca, lesiones confinadas al cuerpo de los ventrículos laterales, lesiones infratentoriales, lesiones mayores de 5 mm y lesiones que muestren reforzamiento con gadolinio.23
En IRM seriadas se han investigado los cambios que ocurren en las lesiones de EM. El tamaño y número de las lesiones hiperintensas en T2 fluctúa, pero tienden a acumularse nuevas lesiones y la masa total de lesión aumenta.24 En estudios que incluyen a pacientes con recaídas y remisiones tratados solo con esquemas breves de esteroides para los ataques clínicos, la carga de lesión total en T2 aumentó en un 6 a 8 por ciento por año.25,26 El reforzamiento con gadolinio es un fenómeno transitorio que suele durar menos de 8 semanas y que por lo general ocurre cuando aparece una lesión por vez primera. Las lesiones que refuerzan con gadolinio corresponden a zonas de inflamación aguda en el examen patológico, mientras que las lesiones que no refuerzan corresponden a un padecimiento más crónico.27 Las lesiones hipointensas y estables en la imagen en T1 que no refuerzan con gadolinio parecen reflejar lesión tisular extensa, incluyendo pérdida axonal. Estas lesiones correlacionan en forma estrecha con la evaluación clínica de disfunción neurológica.28
El uso de la IRM seriada para estudiar la EM ha provocado cambios fundamentales en el concepto del proceso patológico. La actividad de la enfermedad medida por la aparición de nuevas lesiones hiperintensas en las imágenes de T2 o la aparición de lesiones que incrementan con contraste en T1 excede con mucho la severidad de la enfermedad según la evidencian los síntomas clínicos. La actividad de la enfermedad evaluada por IRM también es episódica, pero ocurre 5 a 10 veces más frecuente que los síntomas clínicos, lo que sugiere que la EM es un proceso activo y no el proceso intermitente que sugiere la actividad clínica.
Los estudios actuales sobre nuevos tratamientos para la EM incluyen medidas de actividad de la enfermedad por IRM como uno de los puntos finales. Para facilitar el uso de la IRM en estudios extensos varios grupos ha desarrollado métodos automatizados para medir la cantidad de lesión y otros parámetros de interés.29 Se han desarrollado nuevas secuencias, como la imagen por transferencia de magnetización, la recuperación inversa atenuada por líquido (FLAIR, por sus siglas en inglés, n. del t.) y la combinación de ambas,30 para mostrar cambios más sutiles y mayor patología [ver figura 2]. En forma semejante, la imagen por resonancia magnética espectroscópica [ver figura 3] proporciona información adicional sobre la magnitud de pérdida axonal dentro de las lesiones y medidas más directas sobre la liberación de lípidos durante la desmielinización activa.31-33 La IRM puede también aumentar el conocimiento sobre la patogenia de la lesión por EM. Estudios seriados de IRM demuestran una reducción en la relación de transferencia de magnetización en la IRM o la presencia de picos de lípidos en la imagen de resonancia magnética espectroscópica en sustancia blanca de apariencia normal que pueden preceder al desarrollo de lesiones que refuerzan, lo que sugeriría que la respuesta inflamatoria es un factor secundario en el desarrollo de una nueva lesión.34,35
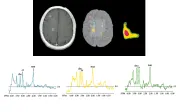
|
| Figura 3 |
| Resonancia magnética de protones con imagen espectroscópica en esclerosis múltiple |
Líquido cefalorraquídeo La mayoría de los constituyentes del LCR se afectan muy poco en la EM. Puede observarse pleocitosis leve a base de mononucleares durante las recaídas, pero la cuenta total de células rara vez es mayor a 50 células/mm3. El nivel de proteínas puede estar un poco elevado, pero rara vez excede 100 mg/dl. Durante los ataques agudos, en especial los que afectan la médula y el tallo cerebral, el LCR puede contener cantidades mesurables de proteína básica de mielina. La alteración más característica en la EM es la síntesis intratecal de inmunoglobulinas de heterogeneidad limitada. La presencia de esta alteración se determina mejor por electroforesis comparativa de suero y LCR concentrado, lo que muestra bandas oligoclonales de inmunoglobulina específicas del LCR. Las mediciones cuantitativas del contenido de inmunoglobulinas en el LCR, como el índice de IgG y la velocidad de síntesis de IgG, también son muy sensibles y útiles en la práctica clínica. Las bandas oligoclonales o la síntesis anormal de inmunoglobulinas se encuentran en alrededor del 90 por ciento de los pacientes con EM clínicamente definida y, aunque no son específicas de EM, estos datos apoyan el diagnóstico en los casos dudosos. Las infecciones del SNC o los padecimientos que causan inflamación crónica del SNC pueden también estimular síntesis anormal de inmunoglobulinas.
El retraso en la conducción en los segmentos desmielinizados de los axones o en las vías con remielinización incompleta proporciona un marcador útil para identificar lesiones subclínicas adicionales en las vías sensoriales. La conducción puede medirse en las vías visuales, auditivas y somatosensoriales por medio de potenciales corticales evocados. En estas pruebas se obtiene un registro cronometrado del electroencefalograma sobre la corteza aferente de interés después de un estímulo visual, auditivo o sensorial repetido. Si la desmielinización es significativa la conducción en las vías centrales estará retrasada. Las respuestas evocadas pueden usarse en la EM para evidenciar desmielinización en una vía sensorial en particular si no existen datos clínicos o para confirmar la afección clínica cuando no existen datos convincentes. En general las respuestas evocadas visuales y somatosensoriales proporcionan información más útil que las respuestas auditivas.
El diagnóstico diferencial de la EM depende del cuadro clínico. Para el caso clásico de síntomas con exacerbaciones y remisiones en un adulto joven con datos típicos en la IRM el diagnóstico diferencial es limitado. En el paciente mayor que presenta mielopatía progresiva el diagnóstico diferencial es extenso. No existe una lista estándar de diagnósticos alternativos que deban considerarse en cada paciente con sospecha de EM. En lugar de ello, el médico tratante debe analizar en forma cuidadosa las características clínicas y de laboratorio de cada caso para generar la lista de posibles alternativas. Entre las consideraciones diagnósticas frecuentes se incluyen lesiones estructurales, enfermedades desmielinizantes hereditarias o degenerativas, vasculitis, afección vascular, infecciones crónicas (v.gr., sífilis, enfermedad de Lyme y virus linfotrópico de células T humanas tipo I), deficiencia de vitamina B12 y neurosarcoidosis.
El tratamiento de la EM puede analizarse considerando el manejo de los episodios agudos, la prevención de las recaídas intentando modificar el proceso patológico y el manejo de los síntomas y las deficiencias neurológicas.
El tratamiento de las recaídas varía con la severidad de los signos y síntomas de presentación. Los episodios leven que no alteran en forma significativa la capacidad del paciente para funcionar no requieren más que una relación médico-paciente de apoyo. Está indicado el tratamiento con dosis altas de esteroides para las exacerbaciones que afectan de modo adverso la función del paciente. El uso intravenoso de metilprednisolona en dosis diarias de 0.5 a 1.0 g por 3 a 7 días reduce la duración de los signos y síntomas neurológicos y suele revertir con rapidez la fatiga que con frecuencia acompaña a los episodios agudos.36,37 Después puede administrarse un esquema corto y de reducción de esteroides por vía oral. Este tratamiento se asocia con supresión rápida de la captación de gadolinio en la IRM, quizá por menor inflamación en las lesiones agudas, que ocurre cuando se normaliza la barrera hematoencefálica.38,39 Sin embargo, sin duda los efectos benéficos del tratamiento son multifactoriales y no pueden explicarse simplemente por este efecto evidente pero temporal de los esteroides sobre el reforzamiento de la imagen. Las dosis equivalentes de esteroides por vía oral pueden tener un efecto semejante,40 pero el manejo con dosis menores (v.gr., 1 mg/kg/día) es motivo de controversia. Los beneficios iniciales pueden ser similares, aunque los esteroides en dosis bajas pueden acortar el intervalo entre los episodios.41,42 No deben ignorarse el papel de la menor actividad física durante la exacerbación y de la rehabilitación temprana. Las contraindicaciones relativas para el uso de esteroides incluyen diabetes mellitus tipo 1 (insulinodependiente), hipertensión no controlada y depresión o psicosis previas inducidas por esteroides. Aunque los esteroides tienen un efecto benéfico a corto plazo cuando se usan para las exacerbaciones agudas, su efecto a largo plazo sobre la evolución de la EM se desconoce.
En los años previos se han autorizado tres medicamentos diferentes que influyen sobre la evolución clínica a largo plazo de la EM: el inteferón beta-1b, el interferón beta-1a y el acetato de glatiramero (antes conocido como copolímero-1). En los pacientes con EM con recaídas y remisiones estos medicamentos reducen la frecuencia de los episodios, la velocidad de acúmulo de la lesión en la IRM o el incremento en la discapacidad.
Los beta interferones disminuyen la expresión del complejo principal de histocompatibilidad (CPH) de clase II, alteran el patrón de secreción de citocinas, inhiben la actividad de las metaloproteinasas de la matriz y aumentan los mecanismos supresores o específicos de antígeno en los modelos experimentales. En los estudios clínicos disminuyen la frecuencia de ataques clínicos, el reforzamiento de lesiones, limitan el acúmulo de lesiones fijas en la IRM y quizá retrasan la progresión de la incapacidad. El interferón beta-1b y el interferón beta-1a recombinantes humanos varían en sus vías de administración, perfil de efectos adversos y magnitud aparente de los efectos. Estas diferencias se explican solo en parte por las propiedades estructurales de las dos moléculas creadas por ingeniería y los diseños de los estudios clínicos piloto. Ambas preparaciones inducen anticuerpos que pueden limitar sus efectos clínicos benéficos.25,43,44
En los pacientes con enfermedad recurrente de severidad leve a moderada se administran 2.5 mg (8 millones de UI) de interferón beta-1b por vía subcutánea cada tercer día, lo que reduce la tasa de exacerbación anual en 30 por ciento. Esta reducción se mantuvo durante 5 años en un estudio controlado amplio. La frecuencia de lesiones con reforzamiento en la IRM se redujo en forma franca y la carga total de enfermedad, medida por IRM, aumentó con el tiempo en el grupo control pero se estabilizó en los que recibieron tratamiento con dosis altas. Los beneficios del tratamiento con interferón-1b fueron dependientes de la dosis, observándose los mejores resultados con las dosis mayores. En el 38 por ciento de los pacientes tratados se desarrollaron anticuerpos, que pueden limitar el efecto del fármaco.25,43,45
La mayoría de los pacientes sufren síntomas de tipo catarral de severidad variable al iniciar el tratamiento con interferón beta-1b. En la mayor parte de los pacientes estos síntomas pueden controlarse con administración previa de un antinflamatorio no esteroide. Los síntomas se vuelven menos severos y pueden disminuir con el tiempo. Las reacciones locales en el sitio de la inyección suelen tener solo importancia cosmética, pero puede ocurrir necrosis cutánea franca. Alrededor del 20 por ciento de los pacientes suspenderá el tratamiento por los efectos adversos locales o sistémicos de otros tejidos. De los pacientes que toleran el tratamiento, el beneficio puede perderse en aquellos que desarrollan anticuerpos neutralizantes. A pesar de ello, los pacientes que superan estas desventajas pueden beneficiarse en forma importante, independientemente del estado de incapacidad al iniciar el tratamiento.46,47
El interferón beta-1a produce beneficios semejantes. En los pacientes con recaídas e incapacidad leve se administra una dosis de 30 µg (6 millones de UI) una vez por semana por vía intramusuclar, que reduce la tasa de recaídas en 18 por ciento y la proporción de pacientes con progresión de la incapacidad neurológica. Cuando se administró por vía subcutánea en dosis mayores de 44 µg tres veces por semana a los pacientes con incapacidad leve a moderada, el inteferón beta-1a disminuyó la tasa de recaídas en 33 por ciento y retrasó la progresión de la incapacidad. El número de lesiones reforzadas en la IRM y la carga total de enfermedad también mejoró en los pacientes que recibieron interferón beta-1a. Durante el tratamiento se desarrollan anticuerpos que neutralizan la actividad biológica. Para ambos tipos de interferón beta los efectos benéficos pueden ser mayores con dosis más altas.26,44
El glatiramero es un polímero sintético aleatorio de cuatro aminoácidos. Su mecanismo de acción no se ha establecido en definitiva, pero se une en forma promiscua a los antígenos de clase II del CPH e induce respuestas de células T cooperadoras tipo 2 órgano específicas. El glatiramero reduce la frecuencia de recaídas en la EM, puede disminuir la progresión de la incapacidad y no parece inducir respuestas del huésped que limiten su beneficio con el tiempo. Este medicamento reduce la tasa anual de episodios en 29 por ciento, observándose el mayor efecto en los pacientes con el menor deterioro neurológico. También se ha retrasa el aumento en la incapacidad. El número de lesiones reforzadas en la IRM disminuye en 35 por ciento, lo que correlaciona bien con el efecto en la tasa de recaídas. Los efectos adversos son mínimos, pero ocurre una reacción sistémica temporal en alrededor del 15 por ciento de los pacientes después de una o más inyecciones del fármaco. Esencialmente todos los pacientes tratados con glatiramero desarrollan anticuerpos que se fijan al medicamento, pero esto no parece limitar su actividad.48-50
En resumen, el inicio del tratamiento profiláctico con beta interferon o glatiramero es apropiado para los pacientes con enfermedad recurrente e incapacidad leve a moderada. La elección del agente a emplear depende de cada paciente. Los pacientes que se mantienen con estos tratamientos pueden esperar una reducción del 18 al 50 por ciento en la frecuencia de ataques. Por desgracia, los dos interferones disponibles en los Estados Unidos vienen en una sola dosis. A pesar de las evidencias sustanciales de que las dosis mayores pueden producir mayor beneficio clínico, es difícil ajustar la dosis. Al parecer con ambos medicamentos se obtienen mejores respuestas si se inician en etapas relativamente tempranas de la enfermedad. El papel de estos medicamentos en pacientes con afección más severa (o en los que tienen un padecimiento progresivo desde el inicio) aún es motivo de investigación. El tratamiento combinado con glatiramero e interferón es atractivo, pero no ha producido beneficios adicionales en estudios en animales. En la actualidad se realiza un estudio cuidadoso y controlado sobre la combinación de interferón y glatiramero. También se están estudiando otros fármacos inmunomoduladores, así como varias intervenciones específicas contra el antígeno y la enfermedad, y pronto se contará con nuevas alternativas terapéuticas.
El tratamiento para la EM progresiva no está tan bien establecido como para la enfermedad recurrente. Como ya se mencionó (ver antes), la enfermedad progresiva desde el inicio se denomina progresiva primaria, mientras que el padecimiento que al inicio tiene recaídas y remisiones y después entra en una fase progresiva se llama progresiva secundaria. Al parecer el interferón beta es genérico en la EM progresiva secundaria. En un estudio clínico el interferón beta-1b retrasó el tiempo en que se confirmó la progresión. Durante el estudio la enfermedad progresó en el 50 por ciento de los pacientes que recibieron placebo, comparando con 30 por ciento de los tratados con interferón beta-1b.51 Un estudio no publicado de interferón beta-1a en enfermedad progresiva secundaria mostró un efecto convincente en las tasas de recaídas y en varias medidas de IRM, pero no alcanzó un beneficio estadístico en el punto de medición final del estudio que fue el momento en el que existió progresión clínica sostenida. El glatiramero causó un beneficio limitado o nulo en un pequeño estudio de enfermedad progresiva, probablemente de ambos tipos.52 En la actualidad se realiza un estudio sobre este medicamento en la enfermedad progresiva primaria. Además del interferón y el glatiramero, se han intentado varios medicamentos inmunosupresores agresivos. La mayoría se han usado poco por la falta de eficacia, efectos adversos excesivos o ambos. Se ha sugerido el uso de ciclofosfamida, cladribina, metotrexate, azatioprina y tratamiento mensual en dosis altas de esteroides, pero es necesario balancear con cuidado los riesgos contra los pocos beneficios demostrados. Varios estudios controlados y no controlados con mitoxantrone sugieren que este agente intercalante puede tener cierta utilidad en grupos muy seleccionados de pacientes con EM, habiendo demostrado beneficios en evaluaciones clínicas y de IRM. En la actualidad el medicamento está sometido a revisiones regulatorias. Sin embargo, su potencial cardiotóxico limita la exposición total al fármaco y su uso general en esta enfermedad crónica.
Varios de los síntomas comunes de la EM responden al tratamiento farmacológico [ver tabla 4]. Entre los síntomas más importantes de la EM se encuentran la depresión y la fatiga.
|
|||||||||||||||
* Para los síndromes de EM suelen usarse las dosis habituales en el adulto. Ver las referencias apropiadas sobre la información para prescribir completa, incluyendo las contraindicaciones, advertencias, los efectos adversos y el inicio y fin del tratamiento. |
Depresión y fatiga Ocurre depresión en alrededor del 20
por ciento de los pacientes con EM, probablemente con más frecuencia que
en otras enfermedades crónicas. El tratamiento de la depresión en
los pacientes con EM no difiere de la de otros enfermos con depresión.
La fatiga ocurre en el 50 por ciento de los casos de EM y no suele ser
proporcional al compromiso neurológico. Se asocia con las
recaídas, aunque puede existir también entre los ataques. Se
distingue de la fatiga normal por su severidad y su frecuente falta de
relación con la actividad. Alrededor del 50 por ciento de los pacientes
con EM tienen un alivio por lo menos parcial con el tratamiento con amantadina,
aunque deben evitarse las dosis al atardecer porque inducen insomnio. Cuando la
amantadina no alivia la fatiga puede ser útil el tratamiento con
pemoline, hidrocloruro de metilfenidato o inhibidores selectivos de la
recaptura de serotonina.
Espasticidad La espasticidad es otro síntoma común susceptible de tratamiento. El aumento en el tono extensor en las extremidades inferiores puede compensar en cierto grado la debilidad asociada, pero la espasticidad excesiva es contraproducente para una marcha fluida y puede ocasionar espasmos dolorosos o contractura articular. Los agonistas del ácido g-aminobutírico (GABA) de acción central, como el baclofen, suelen disminuir la espasticidad, pero conservan la marcha y fuerza en las extremidades inferiores necesaria para trasladarse. Las dosis eficaces de baclofen varían desde 5 hasta 240 mg al día en dosis divididas. El tratamiento debe iniciarse en dosis bajas y aumentarse en forma gradual hasta el nivel que proporcione el máximo beneficio. Otros medicamentos que pueden ser útiles incluyen diacepam, clonacepam y los agonistas adrenérgicos alfa clonidina y tizanidina. Aunque no se ha investigado en forma específica como un agente espasmolítico, la gabapentina puede ser útil para disminuir la espasticidad. Algunos pacientes mejoran con dantrolene.
Disfunción vesical La disfunción vesical es común en la EM. La fisiopatología es compleja. Pueden ocurrir síntomas de disinergia del esfínter del detrusor, función hiperactiva del detrusor y vejiga flácida, en forma individual o en diversas combinaciones, que suelen variar con el tiempo. Son indispensables los estudios urodinámicos funcionales para definir los diversos patrones y determinar un tratamiento lógico. La mayoría de los pacientes tienen síntomas de urgencia y polaquiuria, que pueden aliviarse con relajantes del músculo liso y anticolinérgicos. La determinación periódica de los volúmenes urinarios residuales posmicción permite identificar a los pacientes que se beneficiarán de los programas de autosondeo.
Es importante detectar las infecciones asociadas, que son comunes en las mujeres con EM. Debido a la pérdida frecuente de sensación perineal, las infecciones vesicales agudas pueden no causar disuria, sino ser evidentes como deterioro global en la función neurológica, lo que puede confundirse con una recaída aguda. La evaluación de los pacientes en las recaídas debe incluir la búsqueda de piuria. La baciluria asintomática puede demandar el tratamiento concomitante de las recaídas agudas con una combinación de metilprednisolona y los antibióticos apropiados. La retención urinaria significativa con incontinencia por rebosamiento puede asociarse con exageración del reflejo de espasticidad en los miembros inferiores. La institución de un programa de autosondeo en los pacientes afectados suele disminuir la espasticidad en forma más eficaz que la farmacoterapia.
Dolor El dolor no es raro en la EM. Con frecuencia es secundario a estrés mecánico por debilidad asimétrica o espasticidad. Son útiles las ortosis, espasmolíticos y rutinas de ejercicio, suplementadas con analgésicos simples. Los síndromes de dolor paroxístico, como la neuralgia del trigémino, suelen mejorar con dosis bajas de carbamacepina u otros medicamentos antiepilépticos, en especial gabapentina. Las sensaciones disestésicas distales también pueden responder con estos fármacos. Pueden ser útiles los antidepresivos tricíclicos en dosis bajas. La ataxia y el temblor de intención pueden ser especialmente difíciles de manejar. Los aditamentos para aumentar el equilibrio pueden ayudar a algunos pacientes. Las intervenciones farmacológicas suelen ser desalentadoras, pero en ocasiones el clonacepam o la gabapentina en la dosis máxima tolerada pueden proporcionar alivio sintomático para los pacientes con ataxia de las extremidades superiores.
Aunque el curso de la EM varía de paciente a paciente, se ha determinado el efecto de la enfermedad en cohortes grandes de pacientes. El tiempo promedio desde el inicio de la enfermedad hasta una incapacidad suficientemente severa para que el paciente requiera ayuda para la deambulación es de 15 años. La EM tiene un efecto mínimo en la expectancia de vida.20 El 15 a 20 por ciento de los pacientes con EM pueden tener una evolución relativamente benigna, y algunos enfermos tienen una incapacidad mínima o nula después de 20 años de evolución. En los pacientes con enfermedad caracterizada por recaídas y remisiones la frecuencia promedio de recaídas es de una cada 2 años. No existen factores conocidos que predigan el curso clínico en un paciente individual, aunque el sexo femenino, la menor edad al inicio y la presencia de neuritis óptica o síntomas sensoriales como manifestaciones de inicio tienden a asociarse con un pronóstico más favorable.
La neuritis óptica es una neuropatía óptica inflamatoria aguda. Los síntomas cardinales son pérdida unilatral de la visión y dolor retrobulbar al mover el ojo. El diagnóstico diferencial incluye neuropatía óptica isquémica anterior, que suele ser indolora y se observa en pacientes mayores de 50 años, padecimientos hereditarios como la neuropatía óptica hereditaria de Leber y las neuropatías ópticas tóxicas o nutricionales.53 El tratamiento con metilprednisolona intravenosa en dosis de 1 g/día por 3 días seguido de prednisona oral por 11 días acelera la recuperación de la visión, aunque proporciona muy poco beneficio residual al año de volución.54,55 Un estudio mostró que la administración de prednisona en dosis de 1 mg/kg/día por 14 días no tuvo beneficio y se asoció con más recurrencias.54 Incluso sin tratamiento, casi todos los pacientes comienzan a recuperar la visión en 4 semanas.56
La relación entre la neuritis óptica y la EM es motivo de controversia. Algunos expertos consideran que la neuritis óptica es una entidad diferente, pero otros piensan que es parte del espectro clínico de la EM. Más de la mitad de todos los pacientes con EM tienen neuritis óptica en algún momento durante la enfermedad. De los pacientes que presentan neuritis óptica sin otra deficiencia neurológica, casi el 40 por ciento tiene una o más lesiones ovoides o periventriculares en la IRM de cerebro, y eventualmente el 60 por ciento desarrolla EM definitiva.42,57
Encefalomielitis diseminada aguda
La encefalomielitis diseminada aguda (EMDA) es un síntoma monofásico que suele ir precedido de un exantema viral, una infección de vías respiratorias altas o la aplicación de una vacuna. Los virus asociados con más frecuencia son sarampión, paramixovirus, varicela, rubéola y virus Epstein-Barr. El inicio suele ser rápido y se caracteriza por signos meníngeos, cefalea, crisis convulsivas y alteraciones del estado mental. Los signos neurológicos asociados son variables y pueden incluir hemiplegia, paraplegia, pérdida sensorial, pérdida de la visión y mielitis transversa. La EMDA puede ser fatal, pero la mayoría de los pacientes comienzan a recuperarse en 2 a 4 semanas. La encefalomielitis hemorrágica aguda probablemente es una variante fulminnate de EMDA. La principal característica patológica consiste en múltiples áreas de inflamación y desmielinización perivascular, sin evidencia de infección viral activa. La EMDA puede ser causada por una respuesta autoinmune contra antígenos de mielina debido a reacción cruzada contra proteínas virales. Por lo general existen múltiples lesiones de sustancia blanca en la IRM y la mayoría de estas refuerza con el medio de contraste.58 Con frecuencia se emplea tratamiento esteroideo, aunque su eficacia no ha sido demostrada en estudios clínicos. También la plasmaféresis ha sido útil.59 El pronóstico varía según el virus desencadenante, pero la mortalidad puede ser tan alta como 30 por ciento y los sobrevivientes pueden sufrir síntomas residuales.60
La mielitis transversa aguda es un síndrome de disfunción de la médula espinal de inicio rápido. Al igual que la EMDA, puede ocurrir después de una infección o vacuna, pero puede no tener un evento precipitante identificable. También puede ser el síntoma inicial de la EM. Los síntomas incluyen paraparesia, que al inicio es flácida y después espástica, pérdida de la sensación con un nivel sensorial en el tronco y disfunción intestinal y vesical. El dolor lumbar precede los síntomas neurológicos y los síntomas sensoriales pueden comenzar en la región distal y ascender. La médula torácica es la más afectada. El diagnóstico diferencial incluye otras causas de mielopatía aguda, como compresión de la médula por una lesión estructural extradural, neoplasias de la médula espinal, isquemia y lupus eritematoso generalizado. La IRM es muy útil para excluir lesiones estructurales y para confirmar la presencia de una lesión intramedular en el nivel de la médula compatible con los síntomas. Las lesiones de mielitis transversa aguda suelen ser hiperintensas en la imagen en T2 y afectan la mayoría del corte transversal de la médula en varios segmentos, pudiendo reforzar con contraste. Las lesiones pueden causar inflamación de la médula espinal.61,62 Ningún tratamiento ha demostrado ser benéfico, pero con frecuencia se emplean esteroides. El pronóstico es variable, un tercio de los pacientes tienen buena evolución, un tercio evolución regular y un tercio no se recuperan.63,64 El choque espinal, la lumbalgia y el inicio catastrófico se asocian con mal pronóstico.
Enfermedades desmielinizantes hereditarias
La adrenoleucodistrofia es un trastorno hereditario que se asocia con desmielinización progresiva y disfunción de la corteza suprarrenal.65,66 El patrón de herencia puede ser autosómico recesivo o recesivo ligado al X. La forma ligada al X es causada por la mutación de un gen que codifica una proteína integral de la membrana que se encuentra en el peroxisoma. Los defectos en este gen causan el acúmulo de ácidos grasos de cadena muy larga (AGCML). Los fenotipos pueden variar en forma considerable, incluso dentro de la misma familia. En la fomra infantil el paciente presenta deficiencias cognitivas y después ocurre deterioro neurológico, presentándose la muerte en 2 a 5 años. La forma adulta, denominada adrenomieloneuropatía, se presenta alrededor de los 28 años de edad en forma de disfunción progresiva de la médula espinal con paraparesia espástica, pérdida sensorial y síntomas intestinales y vesicales. La afección cerebral puede ser mínima. Solo la mitad de los pacientes con enfermedad de inicio en el adulto tienen alteraciones cerebrales en la IRM, que se encuentran con más frecuencia en las vías corticoespinales.67 La mayoría de los pacientes tienen atrofia difusa de la médula espinal. El diagnóstico se realiza con base en la combinación de afección neurológica y suprarrenal, historia familiar y niveles elevados en suero de AGCML. El tratamiento dietético con ácidos grasos no saturados disminuye los niveles de AGCML, pero no afecta en forma significativa la progresión de los síntomas.68 El trasplante de médula ósea puede ser eficaz si se realiza antes de que se desarrollen síntomas severos. El pronóstico es malo para los pacientes con la forma infantil de la enfermedad. Los pacientes con la enfermedad de inicio en el adulto suelen requerir asistencia para la deambulación después de 10 a 15 años y se desarrollan lesiones cerebrales rápidamente progresivas en un gran porcentaje de los pacientes 5 a 10 años después del inicio de los síntomas de tipo medular.66
La leucodistrofia metacromática es un padecimiento autosómico recesivo que ocasiona desmielinización de los axones en los sistemas nerviosos central y periférico. Es causada por mutaciones en el gen de la arilsulfatasa A que ocasiona acúmulo de sulfátidos con tinción metacromática.69 El inicio suele ser en la infancia o la niñez, es raro en la edad adulta. Los síntomas de la enfermedad del adulto consisten en alteraciones progresivas en el comportamiento, demencia, ataxia y neuropatía.70 La IRM o la TC cerebral muestra atrofia y alteraciones difusas en la sustancia blanca, en especial en los lóbulos frontales. El diagnóstico se confirma por medición de la actividad de la arilsulfatasa A en los leucocitos de sangre periférica, la orina o los fibroblastos de la piel. La deficiencia verdadera de arilsulfatasa debe distinguirse de un estado común de seudodeficiencia que es causado por un alelo con actividad enzimática baja.69 Los síntomas de leucodistrofia metacromática son progresivos y el inicio temprano se asocia con progresión más rápida. La supervivencia promedio en la enfermedad del adulto es de 12 años. No existe un tratamiento eficaz, aunque en la actualidad se investiga la utilidad del trasplante alogénico de médula ósea y la terapia génica.
Enfermedades desmielinizantes metabólicas
La mielinolisis pontina central (MPC) es un síndrome en el que ocurren deficiencias neurológicas después de la corrección rápida de la hiponatremia.71 La MPC suele ocurrir en adultos jóvenes o de edad media y se asocia con abuso de alcohol y desnutrición. Los signos y síntomas comienzan 3 días después del inicio de la sustitución de sodio y consisten en cambios del estado mental, disartria y otros signos de disfunción corticobulbar, y cuadriplegia espástica. La mejoría comienza alrededor de 2 semanas después del comienzo de los síntomas, pero el grado de recuperación es variable. El dato más característico al examen patológico es la presencia de lesiones desmielinizantes simétricas en la porción central del puente. Pueden ocurrir también lesiones desmielinizantes con un patrón relativamente simétrico en los ganglios basales, el tálamo, la cápsula interna, la sustancia blanca subcortical y el cerebelo. La IRM en T2 suele demostrar la presencia de lesiones hiperintensas. Estas lesiones no suelen reforzar con medio de contraste. También después del trasplante hepático ocurre MPC. No existe un tratamiento específico una vez aparecidos los síntomas. La mayor duración y la corrección rápida de la hiponatremia aumentan el riesgo de MPC, por lo que se recomienda una velocidad de corrección no mayor de 10 a 12 mEq en 24 horas.
La deficiencia de vitamina B12 causa desmielinización de los axones en los sistemas nerviosos central y periférico. Las zonas más afectadas son la sustancia blanca de las vías dorsal y lateral de la médula espinal, una característica que le ha dado el nombre de degeneración combinada subaguda de la médula espinal. Los síntomas más comunes consisten en parestesias, pérdida sensorial que comienza en los pies y progresa en dirección proximal y ataxia sensorial.72 La debilidad casi siempre inicia después de la pérdida sensorial. En una minoría de pacientes ocurren trastornos de la memoria, irritabilidad y confusión. Al examen físico los pacientes suelen tener menor sentido de la vibración y posición, que es peor en los pies que en las manos y pueden tener paraparesia espástica. El examen patológico revela pérdida simétrica de la mielina en las columnas posteriores y laterales de la médula espinal y en ocasiones desmielinización en parches en la sustancia blanca cerebral. La IRM de la médula espinal suele demostrar lesiones en la sustancia blanca, que se resuelven con el tratamiento. El diagnóstico se realiza con base en los datos clínicos y un nivel sérico disminuido de cobalamina. En la mayoría de los pacientes existe macrocitosis o anemia, pero este dato no puede usarse en lugar del nivel de cobalamina como diagnóstico.73 En los pacientes que tienen síntomas y cobalamina baja, la demostración de niveles elevados de ácido metilmalónico y homocisteína total en suero confirman la presencia de una deficiencia funcionalmente significativa de cobalamina. Si existe deficiencia de cobalamina debe investigarse la etiología subyacente. Alrededor del 80 por ciento de los pacientes con deficiencia de cobalamina tienen anemia perniciosa. La administración de cobalamina evita la progresión de los síntomas y produce mejoría clínica en la mayoría de los pacientes.72 El óxido nitroso evita el metabolismo de la cobalamina y puede causar síntomas semejantes después de la exposición prolongada. Después de una sola exposición puede desenmascarar una deficiencia subclínica de cobalamina.
Desmielinización inducida por virus
La encefalopatía multifocal progresiva es una enfermedad desmielinizante lenta causada por una infección viral oportunista de los oligodendrocitos en pacientes inmunosuprimidos. El agente causal es el virus JC, un papovavirus ubicuo que infecta a la mayoría de la población antes de la edad adulta y establece una infección latente en el riñón. En los huéspedes inmunosuprimidos el virus puede reactivarse e infectar a los oligodendrocitos en forma activa. Esta condición, antes poco frecuente, es más común ahora porque ocurre en el 4 por ciento de los pacientes con SIDA. Los enfermos suelen sufrir alteraciones neurológicas focales progresivas, como hemiparesia o deficiencias en los campos visuales o alteraciones del estado mental. En la IRM cerebral existen una o más lesiones de la sustancia blanca, que son hiperintensas en las imágenes en T2 e hipointensas en las imágenes en T1. No existe efecto de masa y el reforzamiento con contraste es raro. El diagnóstico puede confirmarse por biopsia cerebral, con demostración del virus por hibridización in situ o inmunocitoquímica. La reacción de amplificación por reacción en cadena de la polimerasa de las secuencias del virus JC en el LCR puede confirmar el diagnóstico sin necesidad de biopsia.74 En la actualidad no existe un tratamiento eficaz. La supervivencia después del diagnóstico es de alrededor de 3 a 5 meses en los pacientes con SIDA.
La panencefalitis esclerosante subaguda (PEES) es una complicación tardía rara de la infección por virus del sarampión. Ocurre con más frecuencia en pacientes que tuvieron la infección inicial con este virus antes de los 2 años de edad y el tiempo promedio entre la infección inicial y el síndrome neurológico es de 7 años. El uso de la vacuna contra el sarampión ha reducido en mucho la incidencia de esta complicación en países desarrollados. El síntoma inicial suele ser deterioro cognitivo progresivo, que es seguido de disfunción motora y mioclonus asociados con alteraciones electroencefalográficas características. El examen patológico revela infección viral activa en el cerebro, con proteínas y ARN del virus del sarampión que pueden detectarse tanto en los oligodendrocitos como en las neuronas y una respuesta inflamatoria vigorosa. La evolución es progresiva, con remisiones temporales ocasionales. No existe un tratamiento satisfactorio.
Figura 1 Seward Hung.
Bibliografía
- Anderson DW, Ellenberg JH, Leventhal CM, et al: Revised estimate of the prevalence of multiple sclerosis in the United States. Ann Neurol 31:333, 1992 [PMID 1637140 ]
- Brueck W, Schmied M, Suchanek G, et al: Oligodendrocytes in the early course of multiple sclerosis. Ann Neurol 35:65, 1994
- Raine CS: Multiple sclerosis: TNF revisited, with promise. Nat Med 1:211, 1995
- Prineas JW, Barnard RO, Kwon EE, et al: Multiple sclerosis: remyelination of nascent lesions. Ann Neurol 33:137, 1993 [PMID 8434875 ]
- Trapp BD, Peterson J, Ransohoff RM, et al: Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338:278, 1998 [PMID 9445407 ]
- van Walderveen MA, Kamphorst W, Scheltens P, et al: Histopathologic correlate of hypointense lesions on T1-weighted spin-echo MRI in multiple sclerosis. Neurology 50:1282, 1998 [PMID 9595975 ]
- Evans CF, Horwitz MS, Hobbs MV, et al: Viral infection of transgenic mice expressing a viral protein in oligodendrocytes leads to chronic central nervous system autoimmune disease. J Exp Med 184:2371, 1996 [PMID 8976191 ]
- Challoner PB, Smith KT, Parker JD, et al: Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci USA 92:7440, 1995 [PMID 7638210 ]
- Moscarello MA, Wood DD, Ackerley C, et al: Myelin in multiple sclerosis is developmentally immature. J Clin Invest 94:146, 1994 [PMID 7518827 ]
- Sadovnick AD, Armstrong H, Rice GPA, et al: A population-based study of multiple sclerosis in twins: update. Ann Neurol 33:281, 1993 [PMID 8498811 ]
- Sadovnick AD, Baird PA, Ward RH: Multiple sclerosis: updated risks for relatives. Am J Med Genet 29:533, 1988 [PMID 3376997 ]
- Ebers GC, Sadovnick AD, Risch NJ, et al: A genetic basis for familial aggregation in multiple sclerosis. Nature 377:150, 1995 [PMID 7675080 ]
- Sadovnick AD, Ebers GC, Dyment DA, et al: The Canadian Collaborative Study Group: evidence for genetic basis of multiple sclerosis. Lancet 347:1728, 1996 [PMID 8656905 ]
- Ebers GC, Sadovnick AD: The role of genetic factors in multiple sclerosis susceptibility. J Neuroimmunol 54:1, 1994 [PMID 7929798 ]
- Olerup O, Hillert J: HLA class II-associated genetic susceptibility in multiple sclerosis: a critical evaluation. Tissue Antigens 38:1, 1991 [PMID 1926129 ]
- Sawcer S, Jones HB, Feakes R, et al: A genome screen in multiple sclerosis reveals susceptibility loci on chromosome 6p21 and 17q22. Nat Genet 13:464, 1996 [PMID 8696343 ]
- A complete genomic screen for multiple sclerosis underscores a role for the major histocompatibility complex. The Multiple Sclerosis Genetics Group. Nat Genet 13:469, 1996
- Ebers GC, Kukay K, Bulman DE, et al: A full genome search in multiple sclerosis. Nat Genet 13:472, 1996 [PMID 8696345 ]
- Poser CM, Paty DW, Scheinberg L, et al: New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol 13:227, 1983 [PMID 6847134 ]
- Runmarker B, Andersen O: Prognostic factors in a multiple sclerosis incidence cohort with twenty-five years of follow-up. Brain 116:117, 1993 [PMID 8453453 ]
- Thompson AJ, Kermode AG, Wicks D, et al: Major differences in the dynamics of primary and secondary progressive multiple sclerosis. Ann Neurol 29:53, 1991 [PMID 1996879 ]
- Fazekas F, Barkhof F, Filippi M, et al: The contribution of magnetic resonance imaging to the diagnosis of multiple sclerosis. Neurology (in press)
- Offenbacher H, Fazekas F, Schmidt R, et al: Assessment of MRI criteria for a diagnosis of MS. Neurology 43:905, 1993 [PMID 8274173 ]
- Stone LA, Albert PS, Smith ME, et al: Changes in the amount of diseased white matter over time in patients with relapsing-remitting multiple sclerosis. Neurology 45:1808, 1995 [PMID 7477973 ]
- Interferon beta-1b in the treatment of multiple sclerosis: final outcome of the randomized controlled trial. The IFNB Multiple Sclerosis Study Group, University of British Columbia MS/MRI Analysis Group. Neurology 45:1277, 1995
- Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS Study Group. Lancet 352:1498, 1998
- Katz D, Taubenberger JK, Cannella B, et al: Correlation between magnetic resonance imaging findings and lesion development in chronic, active multiple sclerosis. Ann Neurol 34:661, 1993 [PMID 8239560 ]
- Truyen L, van Waesberghe JHTM, van Walderveen MAA, et al: Accumulation of hypointense lesions ("black holes") on T1 spin-echo MRI correlates with disease progression in multiple sclerosis. Neurology 47:1469, 1996 [PMID 8960729 ]
- Filippi M, Horsfield MA, Ader HJ, et al: Guidelines for using quantitative measures of brain magnetic resonance imaging abnormalities in monitoring the treatment of multiple sclerosis. Ann Neurol 43:499, 1998 [PMID 9546332 ]
- Bedell BJ, Narayana PA, Wolinsky JS: A dual approach for minimizing false lesion classifications on magnetic resonance images. Magn Reson Med 37:94, 1997 [PMID 8978637 ]
- Narayana PA, Wolinsky JS, Jackson EF, et al: Proton MR spectroscopy of gadolinium-enhanced multiple sclerosis plaques. J Magn Reson Imaging 2:263, 1992 [PMID 1627860 ]
- Davie CA, Hawkins CP, Barker GJ, et al: Serial proton magnetic resonance spectroscopy in acute multiple sclerosis lesions. Brain 117:49, 1994 [PMID 8149214 ]
- De Stefano N, Matthews PM, Antel JP, et al: Chemical pathology of acute demyelinating lesions and its correlation with disability. Ann Neurol 38:901, 1995 [PMID 8526462 ]
- Filippi M, Rocca MA, Martino G, et al: Magnetization transfer changes in the normal appearing white matter precede the appearance of enhancing lesions in patients with multiple sclerosis. Ann Neurol 43:809, 1998 [PMID 9629851 ]
- Narayana PA, Doyle TJ, Lai DJ, et al: Serial proton magnetic resonance spectroscopic imaging, contrast-enhanced magnetic resonance imaging, and quantitative lesion volumetry in multiple sclerosis. Ann Neurol 43:56, 1998 [PMID 9450769 ]
- Durelli L, Cocito D, Ficcio A, et al: High-dose intravenous methylprednisolone in the treatment of multiple sclerosis: clinical-immunologic correlations. Neurology 36:238, 1986 [PMID 3511404 ]
- Milligan NM, Newcombe R, Compston DAS: A double-blind controlled trial of high dose methylprednisolone in patients with multiple sclerosis: 1. Clinical effects. J Neurol Neurosurg Psychiatry 50:511, 1987
- Barkhof F, Hommes OR, Scheltens P, et al: Quantitative MRI changes in gadolinium-DTPA enhancement after high-dose intravenous methylprednisolone in multiple sclerosis. Neurology 41:1219, 1991 [PMID 1866009 ]
- Burnham JA, Wright RR, Dreisbach J, et al: The effect of high-dose steroids on MRI gadolinium enhancement in acute demyelinating lesions. Neurology 41:1349, 1991 [PMID 1891079 ]
- Alam SM, Kyriakides T, Lawden M, et al: Methylprednisolone in multiple sclerosis: a comparison of oral with intravenous therapy at equivalent high dose. J Neurol Neurosurg Psychiatry 56:1219, 1993 [PMID 8229035 ]
- Barnes D, Hughes RAC, Morris RW, et al: Randomised trial of oral and intravenous methylprednisolone in acute relapses of multiple sclerosis. Lancet 349:902, 1997 [PMID 9093250 ]
- Beck RW, Cleary PA, Trobe JD, et al: The effect of corticosteroids for acute optic neuritis on the subsequent development of multiple sclerosis. N Engl J Med 329:1764, 1993 [PMID 8232485 ]
- Interferon beta-1b is effective in relapsing-remitting multiple sclerosis: I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology 43:665, 1993
- Jacobs LD, Cookfair DL, Rudick RA, et al: Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol 39:285, 1996 [PMID 8602746 ]
- Stone LA, Frank JA, Albert PS, et al: The effect of interferon-beta on blood-brain barrier disruptions demonstrated by contrast-enhanced magnetic resonance imaging in relapsing-remitting multiple sclerosis. Ann Neurol 37:611, 1995 [PMID 7755356 ]
- Lublin FD, Whitaker JN, Eidelman BH, et al: Management of patients receiving interferon beta-1b for multiple sclerosis: report of a consensus conference. Neurology 46:12, 1996 [PMID 8559358 ]
- Neilley LK, Goodin DS, Goodkin DE, et al: Side effect profile of interferon beta-1b in MS: results of an open label trial. Neurology 46:552, 1996 [PMID 8614531 ]
- Teitelbaum D, Fridkis-Hareli M, Arnon R, et al: Copolymer 1 inhibits chronic relapsing experimental allergic encephalomyelitis induced by proteolipid protein (PLP) peptides in mice and interferes with PLP-specific T cell responses. J Neuroimmunol 64:209, 1996 [PMID 8632064 ]
- Bornstein MB, Miller A, Slagle S, et al: A pilot trial of Cop 1 in exacerbating-remitting multiple sclerosis. N Engl J Med 317:408, 1987 [PMID 3302705 ]
- Johnson KP, Brooks BR, Cohen JA, et al: Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. Neurology 45:1268, 1995 [PMID 7617181 ]
- Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. European Study Group on Interferon Beta-1b in Secondary Progressive MS. Lancet 352:1491, 1998
- Bornstein MB, Miller A, Slagle S, et al: A placebo-controlled, double-blind, randomized, two-center, pilot trial of Cop 1 in chronic progressive multiple sclerosis. Neurology 41:533, 1991 [PMID 2011253 ]
- Newman NJ: Optic neuropathy. Neurology 46:315, 1996
- Beck RW, Cleary PA, Anderson MM, et al: A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. N Engl J Med 326:581, 1992 [PMID 1734247 ]
- Beck RW, Cleary PA: Optic neuritis treatment trial: one-year follow-up results. Arch Ophthalmol 111:773, 1993 [PMID 8512477 ]
- Beck RW, Cleary PA, Backlund JC: The course of visual recovery after optic neuritis: experience of the Optic Neuritis Treatment Trial. Ophthalmology 101:1771, 1994 [PMID 7800355 ]
- Rodriguez M, Siva A, Cross SA, et al: Optic neuritis: a population-based study in Olmsted County, Minnesota. Neurology 45:244, 1995 [PMID 7854520 ]
- Mader I, Stock KW, Ettlin T, et al: Acute disseminated encephalomyelitis: MR and CT features. AJNR Am J Neuroradiol 17:104, 1996 [PMID 8770258 ]
- Kanter DS, Horensky D, Sperling RA, et al: Plasmapheresis in fulminant acute disseminated encephalomyelitis. Neurology 45:824, 1995 [PMID 7723979 ]
- Johnson RT, Griffin DE, Gendelman HE: Postinfectious encephalomyelitis. Semin Neurol 5:180, 1985
- Choi KH, Lee KS, Chung SO, et al: Idiopathic transverse myelitis: MR characteristics. AJNR Am J Neuroradiol 17:1151, 1996 [PMID 8791931 ]
- Tartaglino LM, Croul SE, Flanders AE, et al: Idiopathic acute transverse myelitis: MR imaging findings. Radiology 201:661, 1996 [PMID 8939212 ]
- Ropper AH, Poskanzer DC: The prognosis of acute and subacute transverse myelopathy based on early signs and symptoms. Ann Neurol 4:51, 1978 [PMID 697326 ]
- Christensen PB, Wermuth L, Hinge HH, et al: Clinical course and long-term prognosis of acute transverse myelopathy. Acta Neurol Scand 81:431, 1990 [PMID 2375246 ]
- Moser HW: Clinical and therapeutic aspects of adrenoleukodystrophy and adrenomyeloneuropathy. J Neuropathol Exp Neurol 54:740, 1995
- Aubourg P, Mandel JL: X-linked adrenoleukodystrophy. Ann NY Acad Sci 804:461, 1996 [PMID 8993565 ]
- Kumar AJ, Kohler W, Kruse B, et al: MR findings in adult-onset adrenoleukodystrophy. AJNR Am J Neuroradiol 16:1227, 1995 [PMID 7677014 ]
- Aubourg P, Adamsbaum C, Lavallard-Rousseau MC, et al: A two-year trial of oleic and erucic acids ("Lorenzo's oil") as treatment for adrenomyeloneuropathy. N Engl J Med 329:745, 1993 [PMID 8350883 ]
- Gieselmann V, Polten A, Kreysing J, et al: Molecular genetics of metachromatic leukodystrophy. J Inherit Metab Dis 17:500, 1994 [PMID 7967499 ]
- Hageman AT, Gabreels FJ, de Jong JG, et al: Clinical symptoms of adult metachromatic leukodystrophy and arylsulfatase A pseudodeficiency. Arch Neurol 52:408, 1995 [PMID 7710377 ]
- Karp BI, Laureno R: Pontine and extrapontine myelinolysis: a neurologic disorder following rapid correction of hyponatremia. Medicine (Baltimore) 72:359, 1993 [PMID 8231786 ]
- Healton EB, Savage DG, Brust JC, et al: Neurologic aspects of cobalamin deficiency. Medicine (Baltimore) 70:229, 1991 [PMID 1648656 ]
- Lindenbaum J, Healton EB, Savage DG, et al: Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med 318:1720, 1988 [PMID 3374544 ]
- Antinori A, Ammassari A, DeLuca A, et al: Diagnosis of AIDS-related focal brain lesions: a decision-making analysis based on clinical and neuroradiologic characteristics combined with polymerase chain reaction assays in CSF. Neurology 48:687, 1997 [PMID 9065549 ]
- Waxman SG, Ritchie JM: Molecular dissection of the myelinated axon. Ann Neurol 33:121, 1993 [PMID 7679565]
- Shepherd DI: Clinical features of multiple sclerosis in north-east Scotland. Acta Neurol Scand 60:218, 1979
- Posner S, Wikstrom J, Bauer HJ: Clinical data and the identification of special forms of multiple sclerosis in 1271 cases studied with a standardized documentation system. J Neurol Sci 40:159, 1979