Contenido del artículo
VI MIOPATIAS INFLAMATORIAS IDIOPATICAS
- Clasificación
- DERMATOMIOSITIS
- POLIMIOSITIS
- MIOSITIS JUVENIL
- MIOSITIS CON NEOPLASIAS
- MIOSITIS CON OTRAS ENFERMEDADES REUMATICAS
- MIOSITIS POR CUERPOS DE INCLUSION
- Epidemiología
- Etiología y patogenia
- Diagnóstico
- MANIFESTACIONES CLINICAS
- Debilidad muscular
- Erupción cutánea
- Afección pulmonar
- Alteraciones cardiacas
- Calcinosis
- Alteraciones vasculares
- PRUEBAS DE LABORATORIO
- TECNICAS DE IMAGEN Y ESPECTROSCOPIA
- Imagen por resonancia magnética
- Ultrasonido
- Tomografía computada
- Espectroscopía con resonancia magnética
- EVALUACION DE UNA NEOPLASIA OCULTA
- Diagnóstico diferencial
- Tratamiento
DRA. NANCY J. OLSEN
Las miopatías inflamatorias idiopáticas, que incluyen a la polimiositis y la dermatomiositis, afectan principalmente el músculo esquelético. Las características comunes de estas enfermedades son debilidad y cambios inflamatorios del músculo esquelético. En general, las miopatías inflamatorias idiopáticas son trastornos serios que tienen una respuesta variable al tratamiento. La polimiositis y la dermatomiositis pueden asociarse con otras enfermedades reumáticas, especialmente esclerodermia, y con neoplasias. El pronóstico varía según el síndrome específico que se exprese.
La clasificación de estos trastornos musculares heterogéneos en subtipos es útil para determinar los enfoques diagnósticos y terapéuticos.1,2 La categoría de estas definiciones se basa en las características clínicas e histológicas, más que en pruebas de laboratorio o radiológicas [ver tabla 1].
|
||||||||||||||
|
Los pacientes con dermatomiositis suelen mostrar un patrón de debilidad simétrica proximal en todas las extremidades, acompañada de una erupción cutánea característica. Los músculos del cuello y la espalda también pueden mostrar debilidad. Las áreas de la piel más afectadas por la erupción son las superficies extensoras de las manos y las rodillas. Los subtipos de dermatomiositis incluyen la forma juvenil [ver adelante, Miositis juvenil]. Otro subtipo recientemente detectado es la dermatomiositis amiopática.3 Los pacientes con este trastorno tienen la erupción característica, pero no alteraciones musculares demostrables. Un estudio de estos pacientes ha demostrado que las técnicas de imagen por resonancia magnética pueden revelar cambios después del ejercicio que indican una alteración metabólica.4
Los pacientes con polimiositis tienen debilidad muscular proximal simétrica semejante a la que ocurre en la dermatomiositis, pero no existe erupción cutánea. El inicio de la polimiositis puede ser más difícil de determinar, en parte porque no existe una erupción cutánea como indicador de posible inflamación. La debilidad muscular y la atrofia pueden ser más severos de lo que habitualmente se observa en pacientes con dermatomiositis. Sin embargo, no se han realizado estudios formales de evolución a largo plazo que demuestren esta observación.
Los niños con edad entre menos de 5 años hasta la adolescencia pueden tener miositis juvenil. La mayoría presenta erupción cutánea y la vasculitis y las calcificaciones de los tejidos blandos son mucho más comunes en niños que en adultos. La evolución a largo plazo es variable.5
La mayoría, pero no todos los casos de miositis asociada a neoplasias se acompañan de la erupción típica de la dermatomiositis. La incidencia de neoplasia subyacente aumenta con la edad.6 El inicio de la miositis puede preceder o aparecer después de la detección de la neoplasia. Los adultos con dermatomiositis deben ser evaluados en busca de una neoplasia oculta durante los 2 primeros años después del inicio de la enfermedad.
MIOSITIS CON OTRAS ENFERMEDADES REUMATICAS
La miositis inflamatoria puede ocurrir aunada a otra enfermedad reumática establecida, principalmente esclerodermia. Otras condiciones que pueden asociarse a miositis son la artritis reumatoide, el lupus eritematoso generalizado (LEG) y el síndrome de Sjögren. Muchos pacientes con estos padecimientos tienen una forma leve de inflamación muscular que responde bien al tratamiento. Sin embargo, un pequeño subgrupo de pacientes, la mayoría de los cuales tiene también esclerodermia, pueden tener debilidad muscular severa y muy debilitante, resistente al tratamiento.7
MIOSITIS POR CUERPOS DE INCLUSION
El patrón y la severidad de la debilidad muscular en la miositis por cuerpos de inclusión (MCI), difiere del patrón de severidad observado en las otras miopatías inflamatorias idiopáticas. Además de la presencia de debilidad proximal, pueden participar los músculos distales y, en algunos casos, las alteraciones musculares son asimétricas. A diferencia de otros padecimientos musculares analizados en esta subsección, la MCI afecta más a los hombres que a las mujeres, alrededor de dos terceras partes de los pacientes son varones. La respuesta al tratamiento suele ser mala.
La prevalencia estimada de las miopatías inflamatorias idiopáticas es de alrededor de un caso por cada 100,000 individuos. Esta prevalencia hace a estos padecimientos alrededor de 1,000 veces menos comunes que las artritis reumatoide. Los síndromes más raros, como la MCI, pueden constituir el 20 por ciento o menos de todos los casos. En un estudio, la incidencia anual de miopatías inflamatorias idiopáticas fue de 5.5 casos por millón de población.8 Sin embargo, las tasas de incidencia pueden estar aumentando, quizá por mejores métodos de detección.
DIFERENCIAS ETNICAS, RACIALES Y DE GENERO
No se han reportado diferencias étnicas en las miopatías inflamatorias idiopáticas. Se ha sugerido que las tasas de incidencia en Estados Unidos aumentan con mayor rapidez en afromaericanos que en blancos.8 En los adultos la polimiositis es más común que la dermatomiositis, mientras que en los niños y adultos jóvenes la dermatomiositis es la forma predominante. La polimiositis y la dermatomiositis muestran una relación mujer a hombre de alrededor de 2:1. El riesgo de neoplasia aumenta en forma significativa después de los 40 años.6 El diagnóstico de miositis por cuerpos de inclusión rara vez se hace en personas menores de 50 años.
La etiología de las enfermedades musculares inflamatorias sigue sin conocerse. La hipótesis más aceptada sugiere múltiples factores. Un posible escenario consiste en una lesión inicial (por ejemplo un virus u otro agente infeccioso o toxina ambiental) que causa daño muscular en un huésped genéticamente susceptible. Este proceso desencadena una respuesta inmunológica que causa inflamación muscular crónica.
El acúmulo de casos nuevos en forma estacional y geográfica ha sugerido que los virus pueden tener algún papel en la etiología de las miopatías inflamatorias idiopáticas. Además, la infección por VIH se asocia con el desarrollo de miopatía. Sin embargo, los estudios que han buscado evidencias de material genómico en tejido muscular han dado siempre resultados negativos.9 Estos datos sugieren que los virus pueden no infectar el músculo en forma directa, sino mediar daño tisular, que a su vez causa respuestas inmunológicas que se dirigen contra el tejido muscular.10 La relativa rareza de los síndromes de miositis sugiere que si existiera un agente infeccioso común responsable, se requerirían también otros factores concomitantes. Estos factores podrían incluir un loci genético específico del huésped que controle la respuesta inmune u otros factores no infecciosos como medicamentos o toxinas ambientales.
Los agentes hipolipemiantes, como el clofibrato y las estatinas, se han asociado con aumento en los niveles de enzimas musculares en suero, con debilidad muscular en un pequeño número de pacientes. Sin embargo, la mayoría de los individuos están asintomáticos. La lista de medicamentos que se ha reportado se asocian con miopatía es muy larga. Por este motivo, deben investigarse con cuidado los medicamentos usados en cualquier paciente con debilidad muscular inexplicable.11 Tanto la infección por VIH como los medicamentos usados en su tratamiento, como la zidovudina (AZT), han sido implicados en el desarrollo de la miopatía. Es posible que toxinas ambientales no definidas participen en el proceso.
Ocurre acúmulo familiar de síndromes de miositis inflamatoria, pero la gran mayoría de los casos son esporádicos. Los casos esporádicos se han asociado al HLA-DRB1*0301, mientras que los casos familiares muestran aumento en la prevalencia de HLA-DQA1 (DQA1*0501). Se ha descrito una forma de MCI hereditaria en varios grupos étnicos, identificándose asociaciones cromosómicas con este trastorno, aunque no se han identificado los posibles genes.12 Muchos de los estudios reportados de asociación genética en las miopatías inflamatorias han agrupado varios tipos de síndromes. Es probable que estudios futuros que realizan análisis independientes de varios síndromes clínicos diferentes mostrarán asociaciones más intensas con marcadores genéticos.
La presencia de infiltrados celulares en el tejido muscular es una característica definitiva en las enfermedades inflamatorias del músculo [ver figura 1]. El examen con microscopio de luz de estos infiltrados revela un patrón característico de infiltración. En los tejidos de los pacientes con dermatomiositis los linfocitos suelen localizarse alrededor de los vasos y en la periferia de los haces musculares. Rara vez se observa invasión de las fibras musculares por células mononucleares, y existe relativa escasez de fibras musculares necróticas. Se observa daño capilar mediado por complemento con más frecuencia en las muestras de biopsia de los pacientes con dermatomiositis, en especial en los que tienen una neoplasia subyacente. Algunos estudios sugieren que los pacientes con dermatomiositis tienen un síndrome más agudo y con mejor respuesta al tratamiento inmunosupresor. En la polimiositis las fibras musculares pueden ser invadidas por infiltrados mononucleares y se observan áreas de destrucción local. El tejido de los pacientes con MCI suele mostrar cierto grado de inflamación aunado con vacuolas intracelulares.13
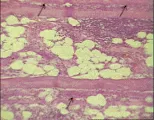
|
| Figura 1 |
| Cambios histológicos en miopatías inflamatorias |
Las diferencias más significativas entre la polimiositis y la dermatomiositis se demuestran por inmunofenotipificación de los infiltrados celulares.14 Los infiltrados de células mononucleares en la polimiositis y quizá en la MCI tienen en forma predominante un fenotipo de células T citotóxicas CD8+. Las células T CD8+ en la polimiositis muestran evidencia de expansión clonal, que es causada probablemente por antígenos específicos del músculo.15 Es probable que las células T CD8+ activadas medien una destrucción citotóxica, mediada por el sistema inmune y específica contra antígenos de la célula muscular. En la dermatomiositis predominan las células T cooperadoras-inductoras CD4+, aunadas a células B, no se observa restricción clonal.15 Estas diferencias en la histología apoyan la hipótesis de que la polimiositis y la dermatomiositis son trastornos diferentes, con etiología distinta.
Los criterios diagnósticos que fueron propuestos por Bohan y Peter en 1975 1,2 siguen siendo útiles para definir a la mayoría de los síndromes de miositis. Sin embargo, la MCI, que no se conocía en el momento en que se escribieron estos criterios, difiere un poco de la polimiositis y la dermatomiositis. Aunque en la actualidad se cuenta con pruebas diagnósticas sofisticadas, incluyendo perfiles de autoanticuerpos y técnicas de imagen, la historia clínica cuidadosa y el examen físico siguen siendo indispensables para hacer el diagnóstico inicial y evaluar las respuestas al tratamiento.
En la polimiositis y la dermatomiositis la debilidad es principalmente proximal. Suele conservarse la fuerza distal. En la MCI la debilidad puede ser asimétrica y con frecuencia se observa reducción en la fuerza distal. La fuerza muscular puede evaluarse en el consultorio o al lado de la cama del paciente empleando una escala semicuantitativa de 1 a 5+. Pueden hacerse mediciones más parecidas con equipos para aplicar fuerza, como los que existen en los servicios de terapia física.
La erupción de la dermatomiositis es eritematosa, de color rojo oscuro y con o sin descamación y atrofia. Ocurre en la cara, cuello, parte superior del tronco y superficies extensoras de las articulaciones como las de los codos y las manos. Puede presentarse edema periorbitario, lo mismo que eritema en heliotropo, que se caracteriza por coloración volácea o lila, en especial en los párpados. Ocasionalmente la erupción es más diseminada o adopta formas diferentes [ver figura 2]. Puede presentarse eritema y telangiectasias en las áreas periungueales. En los adultos la vasculitis suele confinarse a la piel y toma la forma de urticaria, nódulos subcutáneos, infartos periungueales y ulceraciones digitales. La vasculitis cutánea se ha asociado con neoplasias.
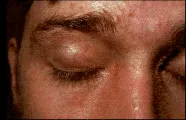
|
| Figura 2 |
| Alteraciones cutáneas en dermatomiositis |
Afección pulmonar
En casi el 50 por ciento de los pacientes que tienen miositis ocurre afección pulmonar, siendo la neumonía la alteración más común. La neumonía por aspiración, que suele ser recurrente, es prevalente en los pacientes con debilidad de los músculos faríngeos. También ocurre tos ineficaz por la debilidad de los músculos ventilatorios, pero que es mucho menos común que los problemas de deglución. En general, el paciente con broncoaspiración recurrente tiene mal pronóstico, porque esto indica disfunción de muchos grupos musculares. La neumonía bacteriana causada por la aspiración es la principal causa de muerte en los pacientes ancianos.6 Pueden ocurrir infecciones oportunistas en los pacientes sometidos a tratamiento inmunosupresor. Además, algunos de estos medicamentos, principalmente el metotrexate, puede causar neumonitis, que suele ser reversible pero potencialmente fatal.
Ocurre enfermedad pulmonar intersticial (EPI) en hasta el 30 por ciento de los pacientes con miositis y en el 60 por ciento de los que tienen anticuerpos dirigidos contra la aminoacil-ARN de transferencia (ARNt) sintetasas. La disponibilidad y aplicación de técnicas diagnósticas nuevas y sensibles como la tomografía computada de alta resolución, puede aumentar la detección de alteraciones pulmonares. La manifestación clínica más común es la disnea progresiva, que puede asociarse con tos no productiva. Al examen físico suelen detectarse estertores crepitantes basales. La progresión puede ser lenta y pueden ocurrir síntomas en los pacientes con enfermedad ya establecida, o la evolución puede ser rápida y ocurrir al mismo tiempo que la debilidad muscular.16 En ocasiones existen hipoxemia y alcalosis respiratoria. En algunos pacientes estas alteraciones se detectan solo después del ejercicio. La TC de alta resolución es útil para detectar la fibrosis intersticial, que puede no detectarse en la radiografía de tórax de rutina. Las pruebas de función respiratoria pueden revelar reducción del volumen pulmonar y menor capacidad de difusión. Suele encontrarse una de tres formas histológicas de EPI: neumonía intersticial, daño alveolar difusa o bronquiolitis obliterante con o sin neumonía organizada. La EPI ocurre con o sin afección cutánea. No existe correlación entre el desarrollo de EPI y la severidad de la afección pulmonar, y la EPI puede preceder o ser posterior al inicio de la debilidad muscular. La EPI se asocia con gran mortalidad. En algunos pacientes se ha reportado que el tratamiento con ciclofosfamida o azatioprina ha sido útil.16 Un pequeño número de pacientes con neumonitis aguda pueden responder solo al tratamiento esteroideo.
La afección clínica del músculo cardiaco es poco común y parece asociarse con mal pronóstico. Las alteraciones cardiacas pueden ser muy diversas, desde trastornos del ritmo o de la conducción hasta inflamación miocárdica o fibrosis. Las alteraciones del músculo cardiaco pueden detectarse por estudios de gamagrafía con radionúclidos. Sin embargo, muchas alteraciones histológicas y eléctricas no son clínicamente significativas.17 Por lo tanto, rara vez está indicado realizar más que los estudios diagnósticos de rutina.
La calcificación de los tejidos blandos ocurre principalmente en los niños. Los depósitos cálcicos pueden ocurrir a lo largo de los planos de las fascias profundas o en la dermis superficial, en ocasiones con ulceración en la piel. Los tratamientos se basan mucho en reportes anecdóticos y no se han realizado estudios sistematizados al respecto.18 Entre los agentes que se han observado fueron útiles en algunos casos se incluyen probenecid, diltiacem y warfarina. Algunos pacientes tienen regresión espontánea de la calcinosis sin un tratamiento específico.
El fenómeno de Raynaud es frecuente en los pacientes con miositis asociada a otras enfermedades reumatológicas (v.gr., esclerodermia). La vasculitis clínicamente significativa es poco habitual en los adultos, aunque los pacientes con dermatomiositis pueden mostrar cambios vasculares al examen histológico.
Se requiere la confirmación histológica de la inflamación muscular en muchos, pero no todos, los casos de enfermedad muscular inflamatoria. Los pacientes con erupción cutánea característica de dermatomiositis y con elevación de enzimas musculares en suero pueden ser tratados sin biopsia muscular, porque estos dos indicadores pueden usarse para vigilar la evolución de la enfermedad. En ausencia de afección cutánea o elevación enzimática el diagnóstico es más difícil y la mayoría de los pacientes requerirán de la biopsia para confirmar la presencia de inflamación muscular. Se usan dos tipos de método de biopsia, abierta o cerrada por punción. La biopsia por punción ofrece la ventaja de menor morbilidad y menor costo porque no se requiere de un quirófano. Las muestras de tejido obtenidas por punción pueden proporcionar información diagnóstica suficiente para que las interprete el patólogo. Sin embargo, la calidad de la muestra obtenida depende de la destreza y la experiencia de quien la realiza. En ausencia de este recurso o en casos especiales en los que se desea obtener tejido extra es preferible el método abierto. Pueden emplearse estudios de imagen, como la IRM o la TC, para establecer el sitio óptimo de la biopsia.
Todas las muestras necesitan ser manejadas de inmediato por un equipo con experiencia que trabaje en el laboratorio de patología para asegurar un resultado óptimo. El análisis con microscopía de luz suele dar suficiente información en la mayoría de los casos. Debido a que el tratamiento de la polimiositis y la dermatomiositis es el mismo, no está indicado realizar inmunofenotipificación de los infiltrados celulares para distinguir entre estos dos trastornos. Puede requerirse de microscopía electrónica para demostrar los cuerpos de inclusión que definen a la MCI.
La mayoría, pero no todos los pacientes, con miopatía inflamatoria tienen elevación de las enzimas musculares en algún momento durante la evolución de la miositis activa.19 La presencia de enzimas musculares intracelulares en el suero refleja principalmente el daño a la membrana de las células musculares. La enzima muscular más usada para la evaluación es la creatina cinasa (CK), y se observa elevación de varias veces el límite normal. La isoenzima MB de la CK puede aumentar por la presencia de esta isoforma en el músculo esquelético que se regenera. La medición de la CK puede confundirse por la presencia de inhibidores de la enzima que ocurren en forma natural. Además, existen variaciones de raza y género en los niveles normales de CK, ya que los varones de raza negra suelen tener valores más altos.20 La aldolasa es otra enzima muscular que puede medirse en el suero y quizá tiene menos variabilidad. Sin embargo, existe en otros tejidos además del músculo, por lo que no es específica de daño muscular. Los estudios de IRM han demostrado que puede existir inflamación muscular activa en pacientes con niveles persistentemente elevados de CK en suero.21 No se conocen los motivos de esta discordancia pero los datos sugieren que las estrategias de tratamiento deben basarse en el estado clínico del paciente, más que en los niveles de enzimas musculares.22
Se encuentran autoanticuerpos contra antígenos nucleares y citoplásmicos en hasta el 90 por ciento de los pacientes con una miopatía inflamatoria. Estos autoanticuerpos con frecuencia son útiles para distinguir las miopatías inflamatorias de enfermedades que no son padecimientos autoinmunes. Algunos de estos autoanticuerpos son inespecíficos y se observan en varios trastornos autoinmunes. Otros son relativamente específicos para los síndromes de miositis inflamatorias en general o para categorías específicas de diagnóstico. Se observan anticuerpos antinucleares positivos en alrededor del 25 por ciento de los pacientes con miositis inflamatoria, con un porcentaje más alto en el subgrupo de pacientes con síndromes de sobreposición. Los anticuerpos antinucleares no suelen ser útiles para establecer el diagnóstico de miositis o alguno de sus subgrupos. Los autoanticuerpos que están dirigidos principalmente contra ribonucleoproteínas citoplásmicas han sido designados como autoanticuerpos específicos de miositis (AEM). Alrededor del 30 por ciento de los pacientes con miositis tienen uno o más de estos autoanticuerpos. Por lo tanto, son relativamente específicos, pero no sensibles, para la presencia de miositis y, como tales, no pueden usarse como prueba de escrutinio para detectar la enfermedad.
Por medio de la especificidad de los AEM pueden definirse tres grupos de pacientes. Estos subgrupos difieren en la presentación clínica y el pronóstico.23 El primer grupo se define por la presencia de anticuerpos dirigidos contra las aminoacil-ARNt sintetasas. Estos pacientes suelen tener afección muscular de inicio agudo, con alta incidencia de enfermedad pulmonar intersticial asociada. También pueden tener artritis y una erupción hiperqueratósica en las manos, conocida como manos del mecánico. La mayoría de los pacientes en este grupo tienen HLA-DR3. Las respuestas al tratamiento son variables y la mortalidad es significativa. El segundo grupo incluye pacientes con anticuerpos contra la partícula de reconocimiento de señales (SRP, por sus siglas en inglés, n. del t.). Este complejo proteico facilita la traslocación de polipéptidos recién sintetizados a través del retículo endoplásmico. Los pacientes con anti-SRP tienen debilidad muscular de inicio súbito y pueden sufrir afección asociada del músculo cardiaco. La mayoría de los pacientes son mujeres afroamericanas. La respuesta al tratamiento no es buena y el pronóstico es malo. En una serie, la mortalidad a 5 años para los pacientes anti-PRS fue de 75 por ciento. Un tercer grupo se identifica por autoanticuerpos contra Mi-2, que es una proteína nuclear con función desconocida. Estos pacientes pueden tener un síndrome clínico de dermatomiositis, con una erupción conocida como de chal en el tronco y con proliferación cuticular. La respuesta al tratamiento suele ser buena y la mortalidad es menor que en los otros grupos.
En la mayoría de los pacientes la electromiografía revela potenciales de unidad motora polifásicos, de baja amplitud, que indican una falta de contractura sincrónica entre las fibras musculares de las unidades motoras. Este dato correlaciona con la distribución no homogénea de degeneración muscular en el examen histológico. Las fibrilaciones y la irritabilidad a la inserción son evidencia de alteraciones en la membrana. Los datos son característicos, pero no específicos de miositis.
TECNICAS DE IMAGEN Y ESPECTROSCOPIA
Las radiografías convencionales tienen poco valor para evaluar el músculo esquelético. Sin embargo, otras técnicas, incluyendo ultrasonido, TC e IRM pueden mejorar los enfoques diagnósticos de muchas miopatías.24 De estas modalidades, la IRM ha sido la más útil para evaluar y vigilar los síndromes musculares inflamatorios. Sin embargo, puede no ser el método de elección en todas las circunstancias y, en algunos de estos casos, el ultrasonido y la TC pueden proporcionar también información útil. Las ventajas de las tres técnicas son que son no invasivas y permiten examinar un mayor volumen de músculo que el obtenido con la biopsia. En los pacientes para los que la biopsia puede ser difícil o traumática, como los niños pequeños, los estudios de imagen pueden proporcionar suficiente información como para establecer el tratamiento.
Imagen por resonancia magnética
La IRM es un método muy exacto de imagen muscular que ha sido muy útil en el diagnóstico y tratamiento de los pacientes con enfermedades musculares inflamatorias de muchos tipos. La evaluación completa requiere obtener imágenes en T1 y T2. La imagen en T1 es más útil para definir la anatomía muscular porque detecta cambios en la masa muscular causados por atrofia o infiltración grasa. La inflamación se detecta mejor en la imagen en T2, en donde las áreas anormales se observan brillantes, contrastando con el fondo oscuro del músculo normal [ver figura 3]. Los estudios que emplean IRM han demostrado con claridad la naturaleza en parches de la inflamación muscular, que quizá explica porqué algunos pacientes con debilidad muscular significativa tienen biopsias normales. En la dermatomiositis se observa inflamación en los músculos del muslo principalmente en los compartimientos anteriores y la masa muscular suele conservarse. Los pacientes con polimiositis y miositis por cuerpos de inclusión pueden tener infiltración grasa y atrofia muscular más extensa, que puede incluir todos los grupos musculares. Pueden usarse estudios longitudinales de IRM para demostrar la eficacia del tratamiento inmunosupresor. Los pacientes pueden ser estudiados en la cámara habitual, lo que permite observar ambas piernas.25 Otros estudios han empleado una cámara de rodilla colocada en la cara anterior del cuadríceps, con lo que se obtiene mayor detalle.21 Como en todos los estudios de IRM, los pacientes deben ser interrogados en forma cuidadosa para detectar la presencia de metales antes de colocar el magneto.
|
|
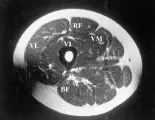
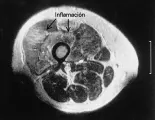
Ultrasonido
El ultrasonido es una técnica accesible y relativamente económica que se ha empleado para examinar muy diversos trastornos musculares.26,27 La inflamación dentro del tejido muscular se observa por ultrasonido como áreas de menor ecogenicidad. Además, los cambios de flujo sanguíneo pueden medirse con técnicas relacionadas como la imagen Doppler en color. El ultrasonido puede ser útil para guiar el sitio de biopsia por punción o abierta.
La TC no es útil para detectar los cambios inflamatorios en el músculo. Sin embargo, las áreas de atrofia o infiltración grasa causan menor densidad muscular, que se detecta con facilidad con la TC. Las calcificaciones de los tejidos blandos, como las que ocurren en la dermatomiositis juvenil, se observan mejor con la TC. En ocasiones estas calcificaciones ocurren en áreas profundas y no pueden apreciarse al examen físico.
Espectroscopía con resonancia magnética
La espectroscopía es una herramienta principalmente de investigación. Sin embargo, los estudios publicados21,28 han demostrado la utilidad de esta técnica no invasiva para evaluar la función muscular, y en un futuro cercano tendrá aplicaciones clínicas. En los pacientes con dermatomiositis se ha demostrado con espectroscopía con resonancia magnética P-31 pérdida de los compuestos de fosfato de alta energía que se requieren para la contracción muscular eficaz.28 Los estudios longitudinales han demostrado que la corrección de estas alteraciones metabólicas puede persistir a pesar de la mejoría en la inflamación muscular.
EVALUACION DE UNA NEOPLASIA OCULTA
Los pacientes con dermatomiositis y polimiositis tienen mayor riesgo de una neoplasia subyacente. La magnitud de este riesgo es difícil de determinar y varía mucho entre los reportes. Un estudio en una población de cohortes calculó el riesgo relativo de cáncer como 1.8 en varones y 1.7 en mujeres.29 En general, el riesgo es mayor en pacientes con dermatomiositis que en pacientes con polimiositis y en todos los pacientes mayores de 40 años. Existe el consenso general de que el escrutinio de rutina para neoplasias debe incluir radiografía de tórax, mamografía (en mujeres), sangre oculta en heces, examen ginecológico completo y antígeno prostático específico (en varones). Las alteraciones observadas en las pruebas de escrutinio pueden sugerir la necesidad de estudios adicionales, como endoscopía, colonoscopía y biopsia tisular. Las neoplasias más difíciles de detectar son las que surgen del ovario. Debe hacerse ultrasonido uterino transvaginal o TC de la pelvis en las mujeres mayores de 40 años, pero algunas neoplasias ováricas ocultas no son detectadas incluso por estas pruebas. Algunos investigadores sugieren realizar estudios del tubo digestivo bajo para detectar cáncer de colon en los pacientes mayores de 65 años.6 Por lo general no se recomiendan estudios más profundos para realizar escrutinio de neoplasias ocultas.
El diagnóstico de dermatomiositis se basa en la presencia de la erupción característica. Sin embargo, debido a que las manifestaciones cutáneas son parecidas a las del LEG, el diagnóstico puede confundirse, en especial si existen anticuerpos antinucleares positivos. Los pacientes con polimiositis pueden ser difíciles de distinguir de los pacientes con otros trastornos miopáticos [ver tabla 2]. Estos otros padecimientos incluyen miopatías metabólicas, disfunción endócrina, trastornos inducidos por medicamentos, infecciones y síndromes diversos como sarcoidosis. Debe pensarse en algunos tipos de distrofias en pacientes con debilidad muscular y elevación de las enzimas musculares. Los síndromes caracterizados por mialgias, como la polimialgia reumática, en la que la molestia predominante puede ser la rigidez, pueden confundirse en algunos pacientes. La fibromialgia se asocia más con fatiga que con debilidad muscular, y se caracteriza por la presencia de puntos dolorosos que no suelen existir en los pacientes con miositis.
|
||||||||||||||
|
Las guías de tratamiento de las miopatías inflamatorias idiopáticas no se han establecido por diversos motivos. Estos padecimientos son poco comunes, lo que hace difícil acumular un número suficiente de pacientes para realizar estudios aleatorios y controlados. Además, algunas formas tienen una evolución lenta y prolongada, requiriendo largos periodos de observación. Por último, no existe un esquema de clasificación totalmente aceptado, por lo que la comparación de tratamientos administrados a diferentes grupos de pacientes en diferentes momentos y lugares puede no ser válida. Como ejemplos, en el pasado la polimiositis y la dermatomiositis se incluían en la misma categoría y la MCI no se había detectado. En la actualidad es claro que las diferentes formas de estas enfermedades tienen diferente pronóstico y respuesta al tratamiento.
Los esteroides son la base del tratamiento inicial. La mayoría de los pacientes con inflamación muscular demostrada deben inicial con estos fármacos en dosis relativamente altas (1 mg/kg/día), administrados en dosis divididas. Un enfoque estándar ha sido mantener esta dosis por hasta 3 meses o hasta que ocurra mejoría clínica. Después de este periodo inicial la dosis puede administrarse una vez al día por la mañana y después disminuir en forma gradual en 20 a 25 por ciento por mes. Con este esquema debe alcanzarse una dosis de mantenimiento de 5 a 10 mg al día en alrededor de 6 a 8 meses. Se considera que si no hay mejoría después de los primeros 3 meses el tratamiento con prednisona ha fracasado. Con el mayor uso de los inmunosupresores se recomienda la adición más temprana de medicamentos de segunda línea. Los pacientes de edad avanzada con trastornos concomitantes como diabetes y osteoporosis tienen especial riesgo por los efectos adversos de los esteroides. Los efectos adversos pueden incluir una apariencia cushinoide, fracturas por compresión, necrosis avascular, cataratas e infecciones. Un estudio ha sugerido que los efectos adversos de los esteroides pueden contribuir en forma significativa a la morbilidad de la polimiositis y dermatomiositis.30 Por este motivo, cualquier paciente con debilidad muscular severa, estado funcional limitado o condiciones que hagan que los esteroides impliquen mayor riesgo (v.gr., diabetes mellitus u osteoporosis) deben iniciar un fármaco inmunosupresor desde el inicio.
Los agentes de segunda línea de uso más común para el tratamiento de las miopatías inflamatorias son el metotrexate y la azatioprina. El metotrexate puede administrarse por vía oral o subcutánea en dosis inicial de 7.5 a 10 mg por semana, aumentando en forma gradual hasta 25 mg por semana. Al aumentar la dosis de metotrexate suele disminuirse la dosis de prednisona. En general, el metotrexate es bien tolerado por los pacientes, aunque se ha reportado toxicidad semejante a la de los pacientes que usan este medicamento por artritis reumatoide. Se requiere vigilancia regular de la función hepática. Es necesario vigilar otras enzimas hepáticas diferentes a las aminotranferasas para evitar interferencia por la inflamación muscular. Una buena alternativa es la gama-glutamiltranspeptidasa. El metotrexate puede ser útil en el tratamiento de la enfermedad pulmonar intersticial asociada con miositis, pero debido a que este medicamento puede causar toxicidad pulmonar en algunos casos, está relativamente contraindicado en pacientes con problemas pulmonares significativos.
La azatioprina ha demostrado ser eficaz en pacientes con miositis en un estudio doble ciego, prospectivo y controlado, pero se requiere tratamiento por lo menos durante 6 meses para que ocurra mejoría. El tratamiento se inicia con 50 a 100 mg/día y la dosis se aumenta en forma gradual hasta un máximo de 150 a 200 mg/día. Los efectos adversos incluyen supresión de la médula ósea, desarrollo de infecciones y posiblemente, de neoplasias. Los pacientes en los que ha fracasado el tratamiento con esteroides y metotrexate o azatioprina pueden responder a una combinación de metotrexate y azatioprina.31,32
Otros agentes inmunosupresores
Se ha administrado ciclofosfamida tanto en pulsos intravenosos como en forma diaria. Algunos datos sugieren que puede ser útil en adultos con síndromes antisintetasa y en niños con dermatomiositis y complicaciones por vasculitis. La ciclofosfamida también puede ser útil en el tratamiento de la afección pulmonar intersticial.16 Otros medicamentos que pueden ser valiosos en los pacientes que no responden al manejo habitual son la ciclosporina A, el FK506 (tacrolimus) y el clorambucil. El análogo de adenina fludarabina mostró cierto beneficio en un estudio de pacientes refractarios.33
La inmunoglobulina intravenosa (IGIV) parece tener cierto beneficio en ciertos pacientes con poli o dermatomiositis. Un estudio controlado de inmunoglobulina en pacientes con dermatomiositis demostró que esta era eficaz cuando se administró en dosis de 1 g/kg/día por 2 días, repitiendo cada mes durante 3 meses.34 No existen estudios controlados en pacientes con polimiositis o miositis por cuerpos de inclusión, pero algunos reportes han descrito beneficio en pacientes con polimiositis o dermatomiositis juvenil. El tratamiento con IGIV está limitado por su escasez y alto costo.
La erupción de la dermatomiositis suele ser fotosensible. Por lo tanto, es importante evitar la exposición al sol lo más posible. Las pantallas solares, la ropa con protección para sol y las micas en las ventanas suelen ser útiles. Algunos dermatólogos recomiendan el uso de b-caroteno, 25 a 30 mg, dos veces al día al inicio y después aumentar a no más de 5 veces al día.
La terapia física tiene un papel muy importante en la
rehabilitación de los pacientes con miositis. Durante la fase de
enfermedad inflamatoria activa se requieren ejercicios pasivos con rangos de
movimiento para evitar las contracturas. Una vez que se controla el componente
inflamatorio de la enfermedad serán útiles los movimientos
activos contra resistencia para recuperar la fuerza muscular.
Bibliografía
- Bohan A, Peter JB: Polymyositis and dermatomyositis (pt I). N Engl J Med 292:344, 1975 [PMID 1090839 ]
- Bohan A, Peter JB: Polymyositis and dermatomyositis (pt II). N Engl J Med 292:403, 1975 [PMID 1089199 ]
- Euwer RL, Sontheimer RD: Amyopathic dermatomyositis (dermatomyositis sine myositis). J Am Acad Dermatol 24:959, 1991 [PMID 1869684 ]
- Park JH, Olsen NJ, King LE, et al: MRI and P-31 magnetic resonance spectroscopy detect and quantify muscle dysfunction in the amyopathic and myopathic variants of dermatomyositis. Arthritis Rheum 38:68, 1995 [PMID 7818575 ]
- Pachman LM, Hayford JR, Chung A, et al: Juvenile dermatomyositis at diagnosis: clinical characteristics of 79 children. J Rheumatol 25:1198, 1998 [PMID 9632086 ]
- Marie I, Hatron PY, Levesque H, et al: Influence of age on characteristics of polymyositis and dermatomyositis in adults. Medicine (Baltimore) 78:139, 1999 [PMID 10352646 ]
- Olsen NJ, King LE Jr, Park JH: Muscle abnormalities in scleroderma. Rheum Dis Clin North Am 22:783, 1996 [PMID 8923596 ]
- Oddis CV, Conte CG, Steen VD, et al: Incidence of polymyositis-dermatomyositis: a 20-year study of hospital diagnosed cases in Allegheny County, PA 1963-1982. J Rheumatol 17:1329, 1990 [PMID 2254890 ]
- Pachman LM, Litt DL, Rowley AH, et al: Lack of detection of enteroviral RNA or bacterial DNA in magnetic resonance imaging-directed muscle biopsies from twenty children with active untreated juvenile dermatomyositis. Arthritis Rheum 38:1513, 1995 [PMID 7575702 ]
- Ytterberg SR: Infectious agents associated with myopathies. Curr Opin Rheumatol 8:507, 1996
- Pascuzzi RM: Drugs and toxins associated with myopathies. Curr Opin Rheumatol 10:511, 1998
- Dalakas MC: Molecular immunology and genetics of inflammatory muscle diseases. Arch Neurol 55:1509, 1998
- Vogel H: Inclusion body myositis-a review. Adv Anat Pathol 5:164, 1998
- Engel AG, Arahata K: Mononuclear cells in myopathies: quantitation of functionally distinct subsets, recognition of antigen-specific cell-mediated cytotoxicity in some diseases, and implications for the pathogenesis of the different inflammatory myopathies. Hum Pathol 17:704, 1986 [PMID 3459704 ]
- Mantegazza R, Andreetta F, Bernasconi P, et al: Analysis of T cell receptor repertoire of muscle-infiltrating T lymphocytes in polymyositis: Restricted Va/b rearrangements may indicate antigen-driven selection. J Clin Invest 91:2880, 1993 [PMID 8514895 ]
- Schwarz MI: The lung in polymyositis. Clin Chest Med 19:701, 1998
- Gonzalez-Lopez L, Gamez-Nava JI, Sanchez L, et al: Cardiac manifestations in dermato-polymyositis. Clin Exp Rheumatol 14:373, 1996 [PMID 8871835 ]
- Spiera R, Kagen L: Extramuscular manifestations in idiopathic inflammatory myopathies. Curr Opin Rheumatol 10:556, 1998 [PMID 9812216 ]
- Hochberg MC, Feldman D, Stevens MB: Adult onset polymyositis/dermatomyositis: an analysis of clinical and laboratory features and survival in 76 patients with a review of the literature. Semin Arthritis Rheum 15:168, 1986 [PMID 3515559 ]
- Worrall JG, Phongsathorn V, Hooper RJL, et al: Racial variation in serum creatinine kinase unrelated to lean body mass. Br J Rheumatol 29:371, 1990 [PMID 2224407 ]
- Park JH, Vital T, Ryder N, et al: MR imaging and P-31 MR spectroscopy provide unique quantitative data for longitudinal management of patients with dermatomyositis. Arthritis Rheum 37:736, 1994 [PMID 8185702 ]
- Dalakas MC: Polymyositis, dermatomyositis, and inclusion-body myositis. N Engl J Med 325:1487, 1991
- Love LA, Leff RL, Fraser DD, et al: A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 70:360, 1991 [PMID 1659647 ]
- Olsen NJ, Park J: Skeletal muscle imaging for the evaluation of myopathies. Diseases of Skeletal Muscle. Wortmann R, Ed. Lippincott, Williams & Wilkins, New York, 1999, p 293
- Fraser DD, Frank JA, Dalakas M, et al: Magnetic resonance imaging in the idiopathic inflammatory myopathies. J Rheumatol 18:1693, 1991 [PMID 1787491 ]
- Reimers CD, Fleckenstein JL, Witt TN, et al: Muscular ultrasound in idiopathic inflammatory myopathies of adults. J Neurol Sci 116:82, 1993 [PMID 8509807 ]
- Fleckenstein JL, Reimers CD: Inflammatory myopathies: radiologic evaluation. Radiol Clin North Am 34:427, 1996 [PMID 8633124 ]
- Newman ED, Kurland RJ: P31 magnetic resonance spectroscopy in polymyositis and dermatomyositis: altered energy utilization during exercise. Arthritis Rheum 35:199, 1992 [PMID 1734909 ]
- Sigurgeirsson B, Lindelof B, Edhag O, et al: Risk of cancer in patients with dermatomyositis or polymyositis. N Engl J Med 326:363, 1992 [PMID 1729618 ]
- Clarke AE, Bloch DA, Medsger TA, et al: A longitudinal study of functional disability in a national cohort of patients with polymyositis/dermatomyositis. Arthritis Rheum 38: 1218, 1995 [PMID 7575715 ]
- Joffe MM, Love, LA, Leff RL, et al: Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 94:379, 1993 [PMID 8386437 ]
- Villalba ML, Hicks JE, Thornton B, et al: A combination of oral methotrexate and azathioprine is more effective than high dose intravenous MTX with leucovorin rescue in treatment-resistant myositis. Arthritis Rheum 38:S307, 1995
- Adams EM, Pucino F, Yarboro C, et al: A pilot study: use of fludarabine for refractory dermatomyositis and polymyositis, and examination of endpoint measures. J Rheumatol 26:352, 1999 [PMID 9972969 ]
- Dalakas MC, Illa I, Dambrosia JM, et al: A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med 329:1993, 1993 [PMID 8247075 ]