Contenido del artículo
VIII LEUCEMIAS Y TRASTORNOS MIELOPROLIFERATIVOS
- Biología de las leucemias
- Identificación y diagnóstico de las leucemias
- PRUEBAS CITOQUIMICAS
- MARCADORES DE SUPERFICIE CELULAR
- OTROS MARCADORES CELULARES
- MARCADORES GENETICOS
- Principios de tratamiento
- Trastornos mieloproliferativos
- Trastornos mieloproliferativos crónicos
- TROMBOCITEMIA ESENCIAL
- METAPLASIA MIELOIDE AGNOGENICA
- LEUCEMIA MIELOIDE CRONICA
- LEUCEMIA EOSINOFILICA CRONICA
- Leucemias mieloides agudas
- Síndromes mielodisplásicos
- Leucemias crónicas y subagudas originadas de células B
- LEUCEMIA LINFOCITICA CRONICA
- LEUCEMIA CRONICA DE CELULAS DE LINFOSARCOMA
- LEUCEMIA PROLINFOCITICA
- LEUCEMIA DE CELULAS PELUDAS (RETICULOENDOTELIOSIS LEUCEMICA)
- Leucemias crónicas y subagudas originadas de células T
- SINDROME DE SEZARY (FASE LEUCEMICA DE LAS MICOSIS FUNGOIDES)
- LEUCEMIA/LINFOMA DE CELULAS T DEL ADULTO
- LEUCEMIA PROLINFOCITICA DE CELULAS T
- LINFOCITOSIS DE LINFOCITOS GRANULARES GRANDES
- Leucemia de linfocitos granulares grandes-T
- Leucemia de linfocitos granulares grandes-asesinos naturales
- POSIBLES TRASTORNOS CLONALES DE CELULAS T
- Leucemia linfoblástica aguda
- Principios psicosociales generales para el tratamiento de pacientes con leucemia
DR. STANLEY L. SCHRIER
Biología de las leucemias
La leucemia puede definirse como la proliferación de un clon de células hematopoyéticas anormales que tiene las siguientes características: (1) mala respuesta a los mecanismos reguladores normales, (2) tendencia a tener menor capacidad de diferenciación celular normal, (3) capacidad de expanderse a expensas de las líneas mieloides o linfoides normales, y (4) capacidad de suprimir o deteriorar el crecimiento de células mieloides normales. Las leucemias se denominan y se clasifican de acuerdo con el tipo de célula hematopoyética que esté principalmente afectada. Las leucemias mieloides afectan a los descendientes de las células madre mieloide, mientras que las leucemias linfocíticas implican alteraciones de la línea celular linfoide. Las leucemias pueden ser agudas o crónicas, o pueden adoptar formas intermedias o variables. Sin tratamiento, incluso los trastornos crónicos pueden ser mortales.
Al caracterizar los rasgos de crecimiento de cualquier línea celular o in vitro, se usan términos específicos para definir los parámetros cinéticos celulares y tisulares. Se usa el examen microscópico para determinar el número de células en mitosis. La capacidad de incorporar timidina tritiada (3H-TdR) en el núcleo define el índice de marcación.
Anteriormente se pensaba que la división rápida y descontrolada explicaba el aumento rápido de la masa tumoral en los pacientes con leucemia. Sin embargo, los mieloblastos normales tienen un tiempo de generación de 24 a 48 horas, mientras que los mieloblastos leucémicos tienen un tiempo de generación 15 a 60 horas. Aún más, la fracción de crecimiento de los mieloblastos leucémicos en el momento del diagnóstico es con frecuencia tan baja como del cinco al siete porciento. La diferencia clave parece estar en el hecho que las células mieloides normales siguen una secuencia fija de desarrollo y pueden ser sometidas a división celular sólo hasta la etapa de mielocito, después de la cual las células se diferencían, circulan y mueren.1 En las células normales, la apoptosis (en ocasiones denominada muerte celular programada) ocasiona condensación nuclear y degradación del ADN, produciendo patrones en escalera característicos en los geles de ADN.2 En las células malignas, incluyendo las leucémicas, los programas de apoptosis parecen estar suprimidos en grado variable.3 Al parecer varios agentes antileucémicos actúan, en parte, al recuperar la apoptosis. Muchas células leucémicas se salen del ciclo celular normal y, en grados variables, son capaces de sufrir divisiones celulares en forma casi indefinida. El resultado es una masa de células en expansión que no mueren con una secuencia ordenada, y que retienen la capacidad de crecer aún más. Estas células se encuentran en fase G1 prolongada, algunas veces llamada G0. Los mieloblastos leucémicos pueden dejar la médula, circular, establecerse en los órganos, dividirse y entrar de nuevo a la circulación.4
La fisiopatología de las leucemias se relaciona casi en forma directa con el impacto del número de células en expansión. La población celular en crecimiento infiltra la médula ósea y hace que el paciente esté funcionalmente aplásico, lo que causa la muerte por infección o hemorragia. De 1 a 2 kg de células leucémicas que equivalen a 1-2 x 1012 células, parecen ser suficientes para causar la muerte. El volumen ocupado por todas estas células será del orden de 1.7 L, que es el volumen medular total de un adulto medio. Las células leucémicas pueden también infiltrar otros órganos, principalmente hígado, bazo, ganglios linfáticos y meninges, y causar disfunción orgánica.
Debido a que el diagnóstico de leucemia no suele hacerse por métodos clínicos estándar a menos de que la masa de células tumorales sea de 109 células, es evidente que solo 10 duplicaciones de células tumorales (210 es igual a un aumento de 1,000 veces) separan el número más pequeño detectable de células del número potencialmente letal. Las técnicas de reacción en cadena de la polimerasa (RCP), que pueden detectar una célula neoplásica en 100,000 células, son 100 a 1,000 veces más sensibles que los métodos morfológicos o citogenéticos estandar y mejoran mucho la detección de la enfermedad residual mínima [ver adelante, Leucemia mieloide crónica y Leucemias mieloides agudas]. Si no se usan técnicas de RCP, un paciente que se afirma que está en remisión completa de una leucemia aguda puede tener células leucémicas que no son detectables por clínica, y el número de estas células será de menos de 109.
ONCOGENESIS
La etiología de las leucemias se está dilucidando gracias a los avances en la citogenética y biología molecular que comienzan a integrar piezas de información discordantes en apariencia: algunas leucemias en animales y al menos una leucemia humana (leucemia de células T del adulto) son causadas por virus tumorales ARN.5 Las leucemias en el hombre pueden ocurrir después de la exposición a radiaciones ionizantes.6, 7 Agentes como el cloranfenicol, la fenilbutazona y el benceno llegan a producir hipoplasia medular, y un número importante de los pacientes que sobreviven al episodio pancitopénico8-10 desarrollan posteriormente leucemia; la terapia con agentes alquilantes se relaciona en ciertas situaciones clínicas con una incidencia de leucemia superior al 10 porciento.11-14 El tratamiento con agentes como el etopósido, que ataca la topoisomerasa II del ADN, puede causar rearreglo del gen MLL (leucemia mieloide-linfoide), que se localiza en la banda cromosómica 11q23. Se observan rearreglos del 11q23 en la leucemia mieloide aguda, leucemia linfoblástica aguda (LLA) y en los síndromes mielodisplásicos (SMD).15 En la actualidad es evidente que existen alteraciones citogenéticas específicas en muchas, si no es que en todas, las leucemias [ver tabla 1].16,17
|
||||||||||||||||||||||||||||||||||||||||||||||||
Nota: ver referencia 95. |
La relación aparente de estas observaciones parece residir en los oncogenes, genes que pueden mediar la carcinogénesis. Los oncogenes están presentes en el genoma normal y es probable que participen en el crecimiento y/o diferenciación celulares. Parece ser que, en su sitio habitual dentro del cromosoma, los oncogenes están flanqueados por regiones que controlan su acción. Sin embargo, después de la exposición a eventos que causan aberraciones cromosómicas o a la acción de los retrovirus ARN que alteran al oncogén o su contexto, el oncogén es liberado de su restricción. Puede replicarse muchas veces e inducir el crecimiento no controlado de una clona de células que descienda de la célula que sufrió la alteración inicial. El cambio en oncogenes como el bcl-2 puede suprimir la apoptosis, contribuyendo a la expansión de una clona.3 Es posible que se encuentren muchos factores adicionales involucrados en la etiología de las leucemias, como la predisposición genética o la resistencia y respuesta inmunológica a una clona maligna que haya surgido.
Identificación y diagnóstico de las leucemias
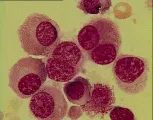
El diagnóstico de leucemia se basa en los hallazgos citológicos y morfológicos. El hallazgo de células anormales en la sangre periférica y la presencia en la médula de un infiltrado de células anormales que sustituyen a los elementos medulares normales establecen el diagnóstico. Una característica patognomónica en algunos casos de leucemia mieloblástica aguda (LMA) es el hallazgo de cuerpos de Auer (gránulos primarios anormales) en los mieloblastos. La biopsia de lesiones hepáticas, esplénicas, ganglios linfáticos o en piel también puede indicar el diagnóstico.
La aparición de un patrón leucémico es evidencia de leucemia mieloide aguda o de progresión a otra forma agresiva de leucemia. El frotis periférico no muestra una progresión ordenada de precursores granulocíticos en maduración, como lo muestra en la leucemia mielocítica crónica (LMC) estándar (i.e., 1 porciento de mieloblastos, 3 porciento de promielocitos, 10 porciento de mielocitos, 20 porciento de metamielocitos y 67 porciento de neutrófilos y bandas). En lugar de eso, el frotis puede mostrar el patrón leucémico, a base de mieloblastos y neutrófilos con pocas formas intermedias. Una interpretación es que en la médula existe una nueva clona que es capaz de sufrir maduración ulterior.
Aunque las técnicas morfológicas estándar basadas en tinciones de Romanowski o tinciones con hematoxilina-eosina tienen uso limitado para clasificar las diversas formas de leucemia, las distinciones obtenidas por estas técnicas pueden servir desde el punto de vista clínico para elegir el tratamiento apropiado. Se están usando otras técnicas, como la inmunofenotipificación con anticuerpos monoclonales, el análisis citogénico y el análisis de rearreglos de genes, para suplementar el análisis citológico estándar en el diagnóstico de las leucemias y en la detección de pequeños números de células leucémicas residuales en pacientes que en apariencia han experimentado remisión completa. Con frecuencia la célula maligna en las leucemias agudas es una célula indiferenciada primitiva y el marcador leucémico puede no ser mas que un indicador de regresión a un patrón de crecimiento más primitivo en lugar de la derivación de la línea leucémica.
PRUEBAS CITOQUIMICAS
El grupo Franco-Americano-Británico (FAB) analizó pacientes con leucemia aguda usando frotis de aspirados de médula ósea con tinción de Romanowski para hacer la diferenciación morfológica general, tinciones con mieloperoxidasa o Sudan negro B para detectar células mieloides, y la reacción de anftol ASD cloroacetato esterasa (NASDA) con inhibición por fluoruro de sodio para identificar monocitos.18, 19 Las células de leucemias agudas de origen mieloide son en general mieloperoxidasa positivas. Se han identificado siete subgrupos de leucemia mieloide aguda, de M1 a M7. Para que un trastorno se clasifique como leucemia mieloide aguda en el sistema FAB, los blastomas deben constituir más del 30 porciento y los precursores eritroides menos del 50 porciento para todas las células nucleadas de la médula. La excepción a esta regla general es la variante M6, en la que hay extensa proliferación de los precursores eritroides inmaduros. El subgrupo M7 identifica pacientes con leucemia megacarioblástica aguda. Por lo general, la médula presenta fibrosis y no puede aspirarse en tales casos. Por lo tanto, las tinciones citoquímicas de las células de la médula no pueden utilizarse para definir a este subgrupo. La identificación de M7 requiere estudios de sangre periférica mediante microscopía eletrónica para demostrar la actividad de peroxidasa plaquetaria característica o análisis de anticuerpos monoclonales o policlonales para identificar la reactividad antigénica relacionada con el Factor VIII (factor plaquetario de von Willebrand) o glucoproteína IIb de la plaqueta.20, 21 Las células leucémicas agudas de origen linfoide son mieloperoxidasa negativas; se han establecido tres subgrupos, de L1 a L3 [ver tabla 2].
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Nota: ver la referencia 15. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
MARCADORES DE SUPERFICIE CELULAR
La inmunofenotipificación con anticuerpos para marcadores de células T y B pudiera ser útil en el diagnóstico de las leucemias. Los linfocitos B maduros se caracterizan por la presencia de inmunoglobulinas en la superficie de la membrana (SmIg), mientras que sus progenitores, denominadas células pre-B, no tienen SmIg pero contienen cadenas pesadas citoplásmicas (Cµ). Los estudios de maduración diferencial de los linfocitos B, que van desde el más primitivo hasta el más diferenciado, o células plasmáticas, pueden distinguirse en la actualidad por medio de anticuerpos monoclonales que reconocen diferentes antígenos de superficie celular. Del mismo modo, pueden utilizarse anticuerpos monoclonales para identificar a todos los linfocitos T, los subgrupos colaborador/inductor o supresor de linfocitos T, así como los linfocitos T maduros e inmaduros. Se han generado anticuerpos monoclonales que pueden identificar antígenos específicos para la diferenciación de células mieloides, monocitos, plaquetas y formas inmaduras de eritrocitos [ver tabla 3].22
|
||||||||||||||||||
Nota: ver referencia 17 |
OTROS MARCADORES CELULARES
La desoxinucleotidil transferasa terminal (TdT) es una enzima que no existe en los linfocitos periféricos normales de origen T o B ni en las células mieloides. Sin embargo, se encuentran niveles altos en el tejido tímico normal, en los linfoblastos de la mayoría de los pacientes con leucemia linfocítica aguda de la niñez, en las líneas celulares lifoblastoides T humanas, en los blastos malignos en algunos casos de crisis blástica en la LMC y en los linfoblastos de la mayoría de los pacientes con leucemia linfocítica aguda de la infancia.23
Por razones aún no conocidas, la actividad de algunas enzimas intracelulares varía en algunas formas de leucemia linfoblástica aguda. La adenosin diaminasa está elevada en la variante linfocítica T (LLA-T), y la 5'-nucleotidasa y la purina nucléosido fosforilasa están disminuidas en la LLA-T.22
Los niveles altos de la enzima sérica lisozima (muramidasa) indican la presencia de leucemia monocítica o mielomonocítica.
MARCADORES GENETICOS
El análisis citogenético es cada vez más útil en el diagnóstico y evaluación de pronósticos [ver tabla 1]. Se han desarrollado pruebas moleculares para los genes de las inmunoglobulinas y genes de los receptores de las células T que pueden ayudar en la identificación del origen de las células leucémicas. Al madurar las células B, redistribuyen sus genes variables (V), de diversidad (D) y de unión (U) para producir una inmunoglobulina específica. Durante la maduración estos rearreglos ocurren en una secuencia ordenada: primero hay rearreglo de las cadenas pesadas, luego de las cadenas ligeras k y al final de las cadenas ligeras l. Todas las células B que se originan de una sola clona tienen un rearreglo único en los genes de las inmunoglobulinas. Una vez que ha ocurrido la expansión de una línea de linfocitos B monoclonal, o maligna, y que la misma abarca el uno porciento o más de la población total de linfocitos, el rearreglo de los genes que codifican la inmunoglobulina específica puede detectarse por medio de una sonda molecular. Así, dichas sondas permiten determinar si una expansión linfocítica es clonal, es decir, neoplásica, y si se originó en las células B.22, 24 De igual forma, los genes de los receptores de las células T sufren rearreglos durante su diferenciación, y la clonicidad se puede estudiar por sondas para identificar la expansión maligna de una línea de linfocitos T.25-29
Las sondas moleculares ofrecen ventajas sobre los estudios cariotípicos porque pueden utilizarse para analizar la citogenética de las células que no están sometidas a mitosis. Por ejemplo, se ha identificado un área llamada región agrupada de punto y ruptura (bcr) en el sitio del cromosoma 22 donde ocurre la traslocación recíproca con el cromosoma 9 en la LMC. Un nuevo gen de fusión, el bcr-abl, se produce en el sitio del brazo largo del cromosoma 9 en donde el gen c-abl se inserta en forma recíproca en el gen bcr del cromosoma 22. Al emplear sondas para detectar expansión clonal de células leucémicas que expresan el gen bcr-abl, puede identificarse el equivalente a la traslocación 9;22. El uso de RCP para producir amplificación del ADN aumenta mucho la sensibilidad de esta prueba y permite la detección de una célula que porte el marcador leucémico bcr-abl entre 100,000.
Principios de tratamiento
El tratamiento de algunas leucemias ha variado desde los intentos por inducir una remisión completa a los intentos por lograr la curación. En las leucemias agudas y en la leucemia mieloide crónica existe la posibilidad de curación si el conocimiento técnico coincide con las circunstancias apropiadas y la disposición del paciente de someterse a programas agresivos y peligrosos. Estos últimos incluyen programas de intensificación quimioterapéutica o trasplante de médula ósea en las leucemias agudas, y transplante de médula ósea en la leucemia mieloide crónica. Los tratamientos pueden aumentar el riesgo de mortalidad del paciente, por lo que se realizan en centros especializados. Por tanto, al planear el tratamiento debe intentarse llegar al diagnóstico definitivo usando información clínica, morfológica clásica, citoquímica, citogenética, biología molecular y análisis de los marcadores de superficie celular. Posteriormente se debe determinar si es posible tratar de conseguir la curación, dada la naturaleza de la enfermedad, el tratamiento requerido, la edad y condición general del paciente y las actitudes de éste hacia el programa después de que se le ha dado la infomación necesaria. Si se considera imposible obtener curación por la afección general, se trata con agentes citotóxicos para reducir la masa tumoral total y para mejorar la función orgánica y la calidad de vida.
QUIMIOTERAPIA
Muchos de los programas terapéuticos están compuestos por varias fases. Existe una fase de inducción, en la cual la masa tumoral es tratada en forma agresiva para reducir o bloquear la carga de células leucémicas clínicamente detectable, esto es, para inducir la remisión clínica. Sigue una segunda fase, llamada de consolidación o citorreducción, en la que las células leucémicas, que probablemente sobrevivieron a la fase de inducción, de 108 a 109 son reducidas aún más por uno o más ciclos de quimioterapia adicional. A veces se realiza una tercera etapa de quimioterapia; ésta busca destruir las células leucémicas residuales o disminuir su número a un nivel por debajo del cual ya no puedan volver a poblar los tejidos. Esto se logra administrando ciclos intensivos de combinaciones de agentes mientras el paciente está en remisión clínica completa. Los pacientes en remisión pueden tolerar dosis mayores de quimioterapia porque su función medular es esencialmente normal. Los programas de intensificación tempranos y tardíos causan hipoplasia medular y por lo general requieren hospitalización. La evidencia respecto a su eficacia ha sido diversa: las remisiones completas prolongadas observadas por algunos investigadores no han sido reproducidas por algunos investigadores no han sido reproducidas por otros.30 Se esta considerando y evaluando el papel de la terapia de mantenimiento en varias formas de leucemia.
La elaboración de programas de quimioterapia que aprovechan la cinética del ciclo celular en diferentes partes del tratamiento sólo ha demostrado moderada utilidad hasta la fecha.31 Al parecer, en el momento de diagnóstico la mayor parte de los blastos leucémicos están en una fase G1 prolongada o G0 y no pueden ser afectados por los agentes que actúan específicamente en el ciclo celular.
La acción de los programas quimioterapéuticos consiste sólo en la destrucción de una fracción determinada de células en una población; por tanto, mientras mayor sea la carga de células tumorales, más difícil resultará realizarlos. Como la leucemia causa aplasia medular funcional y como los medicamentos que se usan son predominantemente mielosupresores, las infecciones y las hemorragias son problemas clínicos limitantes.
Las combinaciones de medicamentos pueden a veces causar destrucción sinérgica de blastos leucémicos sin destruir un número igual de células normales. Si existen agentes selectivos el trabajo es mucho más fácil. En la LLA, la prednisona, vincristina y L-asparaginasa muestran toxicidad selectiva para los linfoblastos leucémicos. Por el contrario, no hay agentes que actúen selectivamente en la LMA. Es probable que el tratamiento de esta entidad cause la muerte de blastos normales y leucémicos por igual.
Un método alternativo en investigación actual se basa en el uso de agentes que podrían inducir a las células leucémicas inmaduras a sufrir diferenciación terminal de tal forma que no puedan dividirse más y finalmente mueran. El mejor ejemplo es el uso del ácido trans-retinoico total (ATR) para el tratamiento de la leucemia promielocítica aguda (LPA).
Otra posibilidad es la administración de modificadores de la respuesta biológica como el interferón, que se está utilizando para el tratamiento de la leucemia mieloide crónica.
Si la enfermedad produce problemas locales graves puede tratarse con tratamiento local. Por ejemplo, la infiltración del sistema nervioso central puede tratarse con radiación craneoespinal, con methrotexate o citarabina intratecales, o con combinaciones de radiación y quimioterapia intratecal.
La hiperuricemia es un problema común; el aumento del recambio celular produce una carga de ácido úrico, que si no se trata apropiadamente, puede causar nefropatía por uratos y artritis gotosa.32
Es importante mantener o mejorar el estado nutricional de los pacientes que se someten a quimioterapia. Se puede requerir alimentación suplementaria para evitar el desarrollo de hipoalbuminemia y pérdida de la inmunocompetencia, y para apoyar la regeneración óptima de las células tronco. Suele requerirse nutrición parenteral total durante la inducción de remisión en la LMA.
TRASPLANTE DE MEDULA OSEA
El trasplante de médula ósea es una modalidad reconocida de tratamiento de la anemia aplástica, y se ha usado experimentalmente para tratar la talasemia mayor. Sin embargo, en la actualidad se utiliza sobre todo en el tratamiento de enfermedades malignas, en particular las leucemias agudas y la leucemia mieloide crónica.33
El propósito del trasplante de médula ósea es restaurar en la médula ósea las funciones de las células tronco con proliferación normal. En el caso del trasplante alogénico para la leucemia, también se desea el efecto de injerto contra células leucémicas. El donador puede ser el paciente (para transplante autólogo), un gemelo idéntico del paciente (para trasplante isogénico o sinergénico), o un donador histocompatible (para trasplante alogénico), por lo general un hermano. La determinación de la histocompatilidad incluye comparar al donador y al receptor en sus loci para HLA-A-A, -B, -C, y -D. Un paciente determinado tiene del 30 al 40 porciento de probabilidades de tener un donador consanguíneo histocompatible. Sin embargo, un donador histocompatible no es un candidato perfecto para trasplante. Dicho donador es compatible simplemente en los loci de histocompatibilidad mayor; quedan incompatibilidades menores, y los injertos alogénicos suelen ser rechazados, a menos que se modifiquen las respuestas inmunes del receptor.34-36
Trastornos mieloproliferativos
El término mieloproliferativo fue propuesto por Dameshek, quin observó que una proliferación medular exuberante de granulocitos, elementos eritroides, megaloblastos, osteoblastos y fibroblastos, caracterizaba a enfermedades como la leucemia mielocítica crónica, la metaplasia mieloide agnogénica, la policitemia vera, la trombocitopenia esencial, la leucemia mieloblástica aguda y la eritroleucemia (enfermedad de Di Guglielmo). Los transtornos crónicos: leucemia mielocítica crónica, metaplasia mieloide agnogénica, policitemia vera y trombocitemia esencial, tendían a sufrir evolución hacia la crisis blástica, afección parecida a la leucemia mieloblástica aguda. Es más, había interconversiones aparentemente regulares dentro del grupo, de manera que la policitemia vera, concretamente si se trataba con radiofósforo, evolucionaba hacia una enfermedad parecida a la metaplasia mieloide agnogénica.37 Sin embargo, se ha discutido la idea de unificar estas enfermedades como trastornos mieloproliferativos.23,28 Por ejemplo, más del 90 porciento de los pacientes con leucemia mieloide crónica muestran una alteración citogenética, la traslocación 9;22 que produce el cromosoma Filadelfia (Ph1). No obstante, los pacientes con policitemia vera, metaplasia mieloide agnogénica y trombocitemia esencial rara vez, si es que alguna, muestran este patrón citogenético.
La medición de las isoenzimas de la G6PD (Gg,A GdB, a-) en mujeres heterocigotas ha permitido clasificar la situación al demostrar que varios de los tratamientos comparten un patrón de crecimiento clonal similar. Esta técnica ha mostrado un patrón común en las células de sangre periférica de pacientes heterocigotos para la G6PD con policitemia vera, leucemia mielocítica crónica, metaplasia mieloide agnogénica o trombocitemia esencial.39 En un paciente con un trastorno mieloproliferativo, los eritrocitos, granulocitos, plaquetas, al igual que los monocitos y macrófagos circulantes, tienen sólo una isoenzima de G6PD, mientras que la piel y fibroblastos medulares cultivados muestran cantidades aproximadamente iguales de las dos isoenzimas, en general Gg,B GdA. Por lo tanto, es probable que todas las células de la sangre periférica se originen de una misma clona maligna, mientras que los fibroblastos medulares y cutáneos se desarrollen de varias clonas normales. Este dato significa que la fibrosis medular que se ve en pacientes con esta enfermedad tal vez sea reactiva, y no un componente de la proliferación clonal hematopoyética maligna.
Es más, cuando pacientes heterocigotos para G6PD son tratados y entran en remisión clínica, las células de la sangre periférica aún muestran sólo el marcador G6PD de la clona maligna. Por lo tanto, la quimioterapia tradicional para las enfermedades mieloproliferativas crónicas no suprime la clona maligna y permite el crecimiento de las clonas normales. En lugar de ello, el tratamiento induce una mejor respuesta de la clona maligna a los mecanismos reguladores. Los trastornos mieloproliferativos crónicos pueden originarse de una mutación en una sola célula tronco pluripotencial, y los diferentes síndromes clínicos probablemente reflejen diversas respuestas de la clona maligna a los mecanismos reguladores.40
Trastornos mieloproliferativos crónicos
TROMBOCITEMIA ESENCIAL
La trombocitemia esencial es un síndrome clínico caracterizado por episodios tromboembólicos o hemorragias espontáneas repetidas relacionadas con un aumento importante en el número de plaquetas circulantes.
METAPLASIA MIELOIDE AGNOGENICA
Diagnóstico
La metaplasia mieloide agnogénica suele aparecer en personas mayores de edad. El paciente busca atención médica cuando se percata de la presencia de esplenomegalia o tiene síntomas de alteraciones hipermetabólicas y hematológicas, en particular anemia. Los datos clínicos incluyen pérdida de peso como resultado del hipermetabolismo, hiperuricemia acompañada de artritis gotosa o nefropatía por uratos, hepatoesplenomegalia masiva, usualmente causada por hematopoyesis difusa extramedular, linfadenopatías debidas a hematopoyesis extramedular, a veces expansión del volumen plasmático que se manifiesta por edema, derrame pleural y ascitis. El frotis de sangre periférica es leucoeritroblástico y muestra glóbulos rojos distorsionados con cola, glóbulos rojos nucleados, plaquetas gigantes y células mieloides inmaduras, incluyendo mieloblastos. En el examen radiológico se observa osteoesclerosis. La médula ósea no puede ser aspirada, y la biopsia de médula muestra fibrosis,42 aumento en el número de megacariocitos y mielopoyesis con desviación a la izquierda en la cuenta diferencial. Las plaquetas y los glóbulos blancos pueden estar elevados, disminuidos o normales.
La anemia es frecuente y está causada por la combinación de hemodilución secundaria a la expansión de volumen plasmático, la mayor destrucción de eritrocitos en el bazo, la hipoplasia eritroide en la médula ósea fibrótica y la eritropoyesis ineficaz en un bazo aumentado de tamaño o en la médula fibrótica. El tiempo de sangrado puede estar prolongado a pesar de que las cuentas de plaquetas sean normales o estén aumentadas, lo que indica un trastorno en la función plaquetaria. El nivel de fosfatasa alcalina leucocitaria es alto. En raras ocasiones la hematopoyesis extramedular causa hipertensión portal con hemorragia por várices esofágicas, las masas de tejido hematopoyético pueden ocasionar también compresión de la médula espinal.
Ocurre eritromelalgia (una sensación de quemadura en las manos y pies que se alivia con el enfriamiento local) en la metaplasia mieloide agnogénica, lo mismo que en otros padecimientos mieloproliferativos crónicos. Los pacientes afectados, que tienen aumento en las cuentas plaquetarias, responden con prontitud al tratamiento con 0.3 g de aspirina administrados una vez al día.43 Puede desarrollarse hepatomegalia en pacientes con metaplasia mieloide agnogénica, en parte como resultado de la hematopoyesis extramedular, y puede ocurrir fibrosis sinusoidal hepática.44 Los trastornos mieloproliferativos crónicos pueden ocasionar trombosis de la vena hepática y síndrome de Budd-Chiari.45,46
La enfermedad que se debe excluir en el diagnóstico diferencial es el carcinoma diseminado con participación de la médula ósea y que esté causando un frotis leucoeritroblástico.47 La historia clínica y la presencia de células de carcinoma en la muestra de biopsia medular suelen resolver el problema. En ocasiones se ha pensado que la tuberculosis u otras enfermedades granulomatosas pueden producir una entidad similar a la metaplasia mieloide agnogénica, pero no existe evidencia clínica convincente.48 Otras entidades diferentes a la metaplasia mieloide agnogénica que puede causar mielofibrosis llegan a manifestarse clínicamente en menos de cuatro meses. En una gran serie, la leucemia de células peludas (LCP) se diagnosticó erróneamente como metaplasia mieloide agnogénica. La (LCP) puede presentarse con una médula infiltrada y no aspirable que contiene cantidades aumentadas de reticulina, pero a diferencia de la médula ósea de la metaplasia mieloide agnogénica, no muestra evidencia de mieloproliferación.49
Por lo general, la leucemia mielocítica crónica no produce un frotis leucoeritroblástico; otras características distintivas de la LMC son infiltración de la médula con una línea predominantemente granulocítica, la presencia del cromosoma Ph1 en el 80 al 90 porciento de los casos, y un nivel bajo de fosfatasa alcalina de los neutrófilos.
La metaplasia mieloide agnogénica pudiera ser un trastorno de la célula tronco, en la cual la clona anormal se diferencía en granulocitos, glóbulos rojos y plaquetas. Es posible que la fibrosis medular sea un fenómeno secundario, debido a que los fibroblastos no comparten los marcadores cariotípicos ni los marcadores enzimáticos de la clona anormal.50
Tratamiento
El tratamiento se debe dirigir a resolver los problemas clínicos más importantes. Los datos indicadores de mal pronóstico son fiebre inexplicable, pérdida de peso, sudores nocturnos, anemia y trombocitopenia.49 El pronóstico no se afecta por factores como el número de células mieloides inmaduras en sangre periférica, el tamaño hepático o esplénico, o el grado de fibrosis o de celularidad de la médula. Si el paciente está anémico por el defecto en la producción, el tratamiento con andrógenos, en dosis de 150 mg/día de oximetolona o un medicamento equivalente, puede producir expansión de la masa de eritrocitos en dos meses. Los efectos colaterales son hepatitis colestásica en el 15 porciento de los casos, retención de líquidos y virilización en las mujeres.
Si el paciente presenta síntomas relacionados con la esplenomegalia (sensación de plenitud, incapacidad para inclinarse hacia adelante o dolor pleural debido a infarto esplénico), el tratamiento con busulfán, en dosis de 2 a 4 mg/día administrado durante varias semanas o meses, puede reducir el tamaño del bazo (o del hígado). Sin embargo, por lo general dicho tratamiento causa anemia, neutropenia o trombocitopenia intolerables. La hidroxiurea, prescrita por vía oral en dosis de 20 a 30 mg/kg/día, también disminuye el tamaño del hígado y del bazo, pero debe vigilarse estrechamente el recuento sanguíneo con este tratamiento. Las dosis pequeñas de radioterapia local (un total de 200 a 500 cGy (200 a 500 rads) ) pueden en ocasiones reducir el tamaño del bazo, pero en la metaplasia mieloide agnogénica este tratamiento puede causar efectos dañinos sobre tejidos no irradiados y pancitopenia importante, y los efectos benéficos son sólo a corto plazo.51
Se puede realizar esplenectomía, pero es peligrosa en pacientes de edad avanzada y la trombocitopenia posesplenectomía puede causar hemorragia, trombosis, o ambas. Se ha expresado la preocupación de que la esplenectomía elimina la única médula funcional residual en pacientes con metaplasia mieloide agnogénica. Sin embargo, en este trastorno el bazo pudiera ser un lugar inadecuado para la hematopoyesis eficaz. La esplenectomía produce una respuesta satisfactoria en casos de anemia hemolítica; al igual que en la trombocitopenia, se encuentran megacariocitos residuales en la médula. La esplenectomía profiláctica no está justificada; en una serie grande se demostró que al menos la mitad de los pacientes con metaplasia mieloide agnogénica nunca necesitaron este procedimiento.49
En ocasiones los síntomas de hipermetabolismo, como la diaforesis, la fiebre poco intensa y la pérdida de peso, responden a la hidroxiurea, administrada por vía oral en dosis de 20 a 30 mg/kg/día. La hiperuricemia se maneja con 100 mg de alopurinol, administrado tres veces al día con un mínimo de 3 L/día de líquidos. Puede ocurrir trombocitopenia que en ocasiones causa hemorragia, quizá relacionada con el trastorno en la función plaquetaria. La hemorragia aguda puede manejarse transfundiendo seis unidades de plaquetas en forma diaria hasta que cese la hemorragia. La trombosis trombocitémica es más difícil de manejar.
Pueden intentarse los medicamentos antiplaquetarios, como la aspirina, 160 a 320 mg/día. El interferón alfa-2a o alfa-2b, en dosis de 3 millones de unidades/m2/día durante tres a siete días por semana, puede disminuir la cuenta de plaquetas y reducir el tamaño del bazo.51-54 Se ha empleado la plaquetoféresis para disminuir la cuenta plaquetaria en forma temporal, pero no existe una prueba clara de su beneficio en la tromboembolia secundaria a trombocitemia. La quimioterapia con busulfán puede disminuir el grado de mielofibrosis sin alterar el número de megacariocitos. Este efecto parece ir en contra de la teoría de que la mielofibrosis es causada por la liberación de un factor de crecimiento derivado de plaquetas (FCDP) liberado por los megacariocitos que proliferan de modo anormal.55
La metaplasia mieloide agnogénica suele ser, una enfermedad crónica, pero eventualmente entra en una fase agresiva, que ocurre en el 10 al 25 porciento de los pacientes, y que recuerda la crisis blástica en la leucemia mieloide crónica (ver adelante). La fase agresiva se caracteriza por el crecimiento de órganos, aparición de blastos y presencia ocasional de un patrón leucémico en el frotis de sangre periférica, anemia progresiva y trombocitopenia. El tratamiento de esta fase agresiva es muy satisfactorio.56
Existen pacientes con variantes de mieloproliferación que han sido clasificados como metaplasia mieloide agnogénica, policitemia vera o leucemia mieloide subaguda. Estos pacientes tienen diversos grados de hepatoesplenomegalia, y la proliferación mieloide y eritroide en la médula ósea puede ser extensa. Puede presentarse mielofibrosis y seguir un curso que varía de crónico a agresivo. Dichos trastornos se denominaron enfermedades mieloproliferativas no clasificadas y se tratan de acuerdo con los principios delineados para la metaplasia mieloide agnogénica.
LEUCEMIA MIELOIDE CRONICA
La leucemia mieloide crónica, también llamada leucemia mielocítica crónica y leucemia granulocítica crónica, es un trastorno clonal en el que la célula tronco leucémica origina un aumento de eritrocitos, neutrófilos, eosinófilos, basófilos, macrófagos-monocitos, plaquetas,57 linfocitos T y, probablemente, linfocitos B.58 Por lo general, se reconocen tres fases de la enfermedad: una fase crónica estable, una fase acelerada y una fase de crisis blástica.
Casi todos los casos de LMC se caracterizan por la presencia del cromosoma Ph1, producido por una traslocación recíproca entre los cromosomas 9 y 22; el cromosoma 22 acortado, o Ph1, se detecta con facilidad mediante citogenética. El efecto importante de la traslocación se relaciona con la yuxtaposición del oncogén c-abl del cromosoma 9 con el gen bcr del cromosoma 22.59, 60 Los genes yuxtapuestos codifican un ARN bcr/c-abl quimérico, y este ARN anormal es producido incluso en las células de pacientes con LMC que no tienen cromosoma Ph1 detectable por medio de citogenética.61El producto proteico de este ARNm tiene actividad poco usual de cinasa de tirosina.62-64
Aunque en otro tiempo del 5 al 10 porciento de los casos de LMC fueron clasificados como Ph1 negativos, en la actualidad parece que en la LMC el Ph1 negativo es sumamente raro, si es que existe. Como se describió antes, las pruebas moleculares revelan la traslocación 9;22 en algunos pacientes identificados como Ph1 negativos, basándose en estudios citogenéticos. Además, en un estudio se analizaron 25 supuestos casos de LMC Ph1 negativos reclasificándose de nuevo todos los casos exceptuando uno, como otro tipo de trastorno mieloproliferativo, en particular en uno de los síndromes mielodisplásicos [ver adelante Síndromes mielodisplásicos].65 Un dato interesante en este estudio, que también examinó a 50 pacientes con LMC Ph1 positivos, fue que todos los pacientes con cromosoma Filadelfia tenían también basofilia en la sangre periférica (i.e., ³ 250 basófilos/mm3).
Fase crónica estable
Los síntomas iniciales de la LMC se relacionan con hipermetabolismo y esplenomegalia. El paciente tiene pérdida de peso, y rara vez fiebre, y presenta esplenomegalia y hepatomegalia. Generalmente hay anemia moderada con pocos glóbulos rojos nucleados en el frotis de sangre periférica. Las plaquetas pueden ser grandes en el frotis y el recuento plaquetario puede estar aumentado, disminuido o normal. El número de leucocitos suele estar muy aumentado, de 50,000 a 300,000/ml,3 y hay una diferenciación ordenada de mieloblastos hasta neutrófilos. Los elementos granulocíticos no son anormales morfológicamente, y está disminuida la fosfatasa alcalina de los neutrófilos. La médula ósea es hipercelular y es evidente la presencia de granulopoyesis con desviación a la izquierda. El aumento de la masa celular está causado por el retraso en la maduración, de manera que hay retraso en el envejecimiento y muerte celular. Probablemente haya ligero aumento en la producción de granulocitos. Los megacariocitos de la médula ósea pueden estar aumentados, normales o disminuidos en número, y la eritropoyesis suele estar disminuida.
Fase acelerada
Después de un periodo durante el cual la enfermedad parece permanecer estable, el bazo y el hígado comienzan a aumentar de tamaño y se observan anemia y trombocitopenia cada vez mayores. El paciente presenta dolor óseo, fiebre, sudores nocturnos y pérdida de peso. En sangre periférica se observa eosinofilia, basofilia, células mieloides inmaduras o un patrón leucémico. La aparición de afección extramedular en ganglios linfáticos, huesos, piel y tejidos blandos indica enfermedad acelerada.66 El tratamiento que se había utilizado resulta cada vez menos eficaz.57, 59 No existe un límite claro entre esta fase y la fase blástica subsecuente.
Fase blástica de crisis
En la fase blástica la fiebre, el dolor y la órganomegalia llegan a ser más prominentes y la evolución de la anemia y trombocitopenia es más grave. El número de blastos en sangre periférica aumenta y los estudios citogenéticos revelan duplicación del cromosoma Ph1, trisomía 8 y otros cambios cromosómicos.57, 59, 63 Quizá la cuarta o la tercera parte de los pacientes en esta fase de la LMC sufran una crisis blástica linfoide en la que las células blásticas tienen características morfológicas de tipo linfoide, no se tiñen con mieloperoxidasa y son positivas para marcadores linfoides como TdT y el antígeno común de la leucemia linfoblástica aguda.
Tratamiento
Fase crónica estable La hiperuricemia es frecuente y debe controlarse con alopurinol, en dosis de 300 mg/día. El aumento de leucocitos puede originar en ocasiones, trombos de leucocitos intravasculares, y estos agregados se alojan en las vasculatura cerebral, pulmonar y retiniana, produciendo obstrucción, hemorragia e infarto, respectivamente. La leucoforesis reducirá en forma aguda el número de leucocitos, y la daunorrubicina o doxorrubicina intravenosa, en una dosis de 60 mg/m2, causarán en 36 horas una caída rápida de estas células.67 El paciente debe estar hidratado y recibir alopurinol durante este periodo.
Es probable que el trasplante alogénico de la médula osea sea el tratamiento de elección para los pacientes con LMC mejores de 55 años y que tienen un donador compatible (ver adelante), porque ofrece la oportunidad de curación. De acuerdo a los datos del International Bone Marrow Transplant Registry (Registro internacional de trasplantes de médula ósea, n. del t.), la probabilidad de supervivencia sin leucemia después de un trasplante alogénico de un hermano es de 45± 4 porciento si el trasplante se realiza en fase temprana de la etapa crónica estable.68 Parece que el efecto de injerto contra leucemia es importante para la supervivencia libre de enfermedad. Debido a que solo el 30 porciento de los pacientes tienen un hermano donador totalmente compatible, se están realizando estudios con trasplantes alogénicos compatibles de no familiares. Con los donadores no familiares la incidencia de fracaso del injerto es mayor y las consecuencias de la enfermedad injerto contra huésped (EICH) son mucho más severas. Para un receptor de trasplante en la fase crónica estable, la probabilidad de supervivencia sin leucemia a dos años es de solo el 30 porciento. En los pacientes menores de 55 años con LMC en fase crónica estable, el uso de un hermano donador totalmente compatible para el trasplante alogénico se considera como la opción óptima. Cuando se usan donadores no familiares, incluso totalmente compatibles, el tratamiento debe considerarse como experimental.
Los estudios actuales evalúan diversos procedimientos de trasplante autólogo en los que el paciente recibe combinaciones de agentes terapéuticos y factor estimulador de colonias de granulocitos (FEC-G) o factor estimulador de colonias de granulocitos y macrófagos (FEC-GM) para movilizar las células madre. En las técnicas de aféresis, las células madre (por lo general las positivas para el marcador de superficie CD34) son cosechadas de la sangre periférica. Después de remover, congelar y almacenar las células, el paciente recibe tratamiento agresivo con combinaciones de radiación corporal total y quimioterapia. A continuación se realiza el rescate con células madre de sangre periférica. Los resultados de estos estudios de investigación aún están pendientes.69
Las pruebas de RCP han detectado rearreglos bcr-abl en las células de la médula ósea de algunos pacientes que han recibido un trasplante alogénico de médula ósea. Aunque no se conoce con certeza el significado de este hallazgo, no necesariamente predice una recaída. Puede indicar que algunas células malignas no son suficientes para producir enfermedad activa o que debe ocurrir otro evento oncogénico para que exista recaída clínica.70,71 El tratamiento convencional se usa, durante la fase crónica estable de la LMC, para los siguientes pacientes: los que son mayores de 55 años, en los que no puede encontrarse un donador no familiar totalmente compatible y en los que tienen trastornos concomitantes que pueden comprometer la capacidad de sobrevivir ante la complicación de EICH asociada con un trasplante alogénico de un donador no familiar.
El busulfán solía constituir el tratamiento inicial para los pacientes que no eran candidatos a trasplante de médula ósea, pero en un estudio aleatorio que comparó el busulfán con la hidroxiurea, se encontró que esta última era claramente superior para prolongar la supervivencia, y que ocasionaba reacciones adversas (v.gr., fibrosis pulmonar y aplasia persistente de la médula ósea) con menos frecuencia.72
El interferón alfa puede ser el tratamiento de elección en los pacientes que pueden tolerar una dosis de 5 millones de unidades/m2/día por vía subcutánea. Este agente causa disminución en el número de leucocitos, blastos y, en quienes tienen trombocitosis, de plaquetas. También provoca reducción del bazo. Para la evaluación de la respuesta citogenética debe medirse la proporción de células de la médula que presentan el cromosoma Filadelfia en intervalos de tres a seis meses.71 En alrededor del 30 porciento de los pacientes la respuesta citogenética es excelente: las células con metafases anormales se reducen a niveles de 0 a 10 porciento. Estos pacientes tienen una supervivencia libre de enfermedad más larga, a pesar de que las pruebas de RCP detecten en forma constante rearreglos bcr-abl en las células de la médula ósea.
En un estudio aleatorio, se comparó el tratamiento con hidroxiurea (20 a 30 mg/kg/día, con dosis ajustadas para producir una buena respuesta hematológica) con el tratamiento con interferón alfa (con dosis que aumentaron en forma gradual de la siguiente manera: 3 millones de unidades/día durante las semanas 1 y 2, 6 millones de unidades/día durante las semanas 3 y 4, y 9 millones de unidades/día, después). Debido a que el interferón actúa con lentitud, algunos pacientes requieren hidroxiurea en un principio para controlar la leucocitosis. El grupo que recibió interferón alfa tuvo mucho mejores respuestas citogenéticas, un mayor tiempo antes de la fase acelerada, y mayor supervivencia. La mejor evolución se presentó en los pacientes que recibieron interferón alfa que tenían respuestas citogenéticas excelentes; en los pacientes sin respuesta citogenética los resultados fueron comparables a los del grupo de hidroxiurea.
El interferón alfa es caro y tóxico, causa fiebre, malestar, pérdida de peso, depresión y, en ocasiones, trombocitopenia, síndrome nefrótico, hipotiroidismo y alteraciones cardiacas.71 El tratamiento con interferón alfa requiere considerar su costo y efectos adversos, así como realizar estudios citogenéticos cuidadosos en intervalos de tres a seis meses. Si no ocurre respuesta citogenética en seis a 12 meses, no existe motivo para continuar este tratamiento y puede administrarse hidroxiurea o un agente de segunda línea, como busulfán (4 a 6 mg/día por vía oral). No se recomienda la esplenectomía temprana como procedimiento de rutina en la LMC.71,73
Tratamiento de las fases acelerada y blástica La dosis de hidroxiurea puede aumentarse, o puede indicarse el tratamiento con bolos de busulfán cuando la LMC entra en fase acelerada, pero la respuesta suele ser insatisfactoria. El trasplante de médula ósea es mucho menos eficaz en la fase acelerada o en la crisis blástica de la LMC, que en la fase estable.74-76 Sin embargo, con el trasplante alogénico de un hermano, la probabilidad de supervivencia libre de enfermedad, según varios estudios, es de 25 a 35 porciento para los pacientes con fase acelerada de la LMC y de cinco a 15 porciento para los que están en fase de crisis blástica de la LMC.
Por lo general, los pacientes con crisis blástica mieloide responden poco a la quimioterapia.77 Las formas más agresivas de tratamientos, como la citarabina en dosis altas combinada con mitoxantrone, no mejoran el pronóstico en los pacientes con crisis blástica y son muy tóxicas.78
Para los pacientes que tienen una crisis linfoblástica clara, la quimioterapia intensiva con vincristina en dosis altas y prednisona puede producir una respuesta terapéutica. Los linfoblastos pueden identificarse por técnicas morfológicas, por su reacción con anticuerpos monolonales al antígeno común de la leucemia linfoblástica aguda (ACLLA), por ensayos para TdT,79 o por su presencia como una clona de células que tienen rearreglos idénticos en sus genes de inmunoglobulinas.80 El tratamiento, que dura entre 14 y 21 días, consiste en vincristina, 1.4 mg/m2/semana administrados por vía intravenosa (dosis única máxima de 2 mg) y prednisona, 60 mg/m2/día por vía oral.81 Si el tratamiento combinado con vincristina y prednisona reduce la cuenta de blastos y disminuye la organomegalia, el paciente puede cambiarse a un protocolo de LLA [ver adelante, Leucemia linfoblástica aguda].77 La quimioterapia combinada con vincristina, doxorrubicina (Adriamicina) y dexametasona (el esquema VAD), se ha usado también en pacientes con LMC linfoide o indiferenciada en crisis blástica.82 Este enfoque tiene un valor limitado en pacientes con crisis linfoide blástica, pero ninguno en los enfermos con crisis blástica indiferenciada.
LEUCEMIA EOSINOFILICA CRONICA
La leucemia eosinofílica es un trastorno mieloproliferativo que por lo general puede distinguirse de los síndromes hipereosinofílicos. En algunos casos, los patrones de crecimiento de colonias in vitro y los cariotipos indican la naturaleza clonal y neoplásica de la leucemia eosinofílica.83, 84
Leucemias mieloides agudas
Las leucemias mieloides agudas están entre las enfermedades malignas más agresivas de los humanos; si no se tratan, pueden causar la muerte en un periodo de 40 a 100 días desde el momento del diagnóstico.85 En la siguiente discusión se incluyen varios tipos: leucemia monoblástica aguda, leucemia mielomonocítica aguda, leucemia granulocítica agua, eritroleucemia aguda (M6), leucemia megacariocítica aguda (M7) y leucemia mieloblástica aguda.87
Existe un aumento de la incidencia de leucemia mieloide aguda que parece ser el resultado del tratamiento de enfermedades malignas. La enfermedad de Hodgkin, en particular cuando se trata con radioterapia y quimioterapia según el protocolo MOPP (mecloretamina, vincristina, procarbazina y prednisona), es seguida, varios años después, por la aparicipde formas subagudas y agudas de leucemia mielocítica y mielomonocítica en cerca del 5 porciento de los casos (ver adelante).13 El tratamiento con agentes alquilantes del mieloma múltiple, macroglobulinemia de Waldenström y cáncer de ovario87 también aumenta la incidencia de leucemia mieloide aguda, que suele ser precedida por una fase sideroblástica o eritroleucémica.
LEUCEMIA MIELOBLASTICA AGUDA
Diagnóstico
Aunque la leucemia mieloblástica aguda (LMA), también llamada leucemia no linfoblástica aguda (LNLA), puede progresar en forma relativamente gradual, la mayoría de las veces los pródromos de astenia, hemorragias y fiebre acompañada de infecciones tiene una duración de varios días a varias semanas. El examen físico después del comienzo muestra petequias y dolor a la palpación en la región esternal; algunas veces, adenopatías, esplenomegalia y hepatomegalia, al igual que participación testicular, cutánea y meníngea. En los pacientes con morfología monoblástica, las células leucémicas a menudo invaden la piel, encías, área perineal y sistema nervioso central y meninges. Aunque se ha dicho que la fiebre acompaña a la leucemia aguda; ésta suele deberse a infección.88
El diagnóstico se confirma por medio del estudio de la médula ósea, que revela infiltrado mieloblástico que remplaza por completo a todos los elementos normales. Debido a dicho remplazo medular total, los pacientes están propensos a presentar anemia, hemorragias trombocitopénicas e infecciones neurotropénicas. En esta situación puede aparecer una dermatosis neutrofílica febril denominada síndrome de Sweet que se caracteriza por placas cutáneas rojas, dolorosas, y la biopsia de piel muestra infiltrado de neutrófilos maduros (no blastos). La lesión responde bien al tratamiento con esteroides sistemáticos.89
El análisis citogenético y la inmunotipificación [ver tablas 1,2 y 3] complementan el estudio habitual de morfología del aspirado de la médula ósea y de las muestras de biopsia, y pueden proporcionar información clínica y pronóstica adicional. El uso diseminado del análisis inmunofenotípico ha demostrado que las células de algunos pacientes con LMA tienen características más propias de células linfoides.90 Al hacer el diagnóstico, el hematólogo debe considerar la preponderancia de los marcadores.
El enfoque combinado de análisis citogenético e inmunotipificación ha revelado la existencia de varios síndromes de leucemia mieloblástica aguda,91 y se analizarán las variantes más comunes (ver adelante). Una variante de la leucemia mielomonocítica M4 [ver tabla 2] se caracteriza por eosinófilos anormales en la médula ósea y se asocia con inversión, deleción o traslocación del cromosoma 16. El diagnóstico correcto de esta variante, que se ha denominado M4Eo/inv(16),17 es importante porque la posibilidad de remisión completa sostenida es alta a pesar de la gran frecuencia de afección al sistema nervioso central. Otra variante de LMA que puede tener un fenotipo M4 se asocia con inversión del cromosoma 3 y megacariocitos anormales; por lo general, la cuenta de plaquetas aún es alta en el momento de las manifestaciones clínicas. Una variante M2 de LMA que se asocia con la traslocación 6;9 se caracteriza por basofilia importante de la médula. Con frecuencia se observan bacilos de Auer en los mieloblastos de los pacientes con una variante M2 asociada con la traslocación 8;21. Alrededor del 20 porciento de estos pacientes tienen cloromas y esplenomegalia.
Tratamiento
Debe explicarse la naturaleza del trastorno al paciente, quien debe decidir si se somete o no al tratamiento de inducción [ver adelante Principios psicosociales generales para el manejo de pacientes con leucemia]. Esta no es una decisión trivial: el tratamiento de inducción en la LMA requiere cerca de un mes de hospitalización, lo que genera costos de hasta 200,000 dólares. Durante un período de 10 a 30 días, el paciente estará aplástico y sufrirá de fiebre causada por infecciones repetidas y a veces dolorosas, debidas en un principio a bacterias y después a hongos. El paciente deberá estar aislado en un ambiente protector; sin duda necesitará transfusiones regulares de plaquetas y también transfusiones de glóbulos rojos.92
Si se lleva a cabo un programa convencional de inducción, existe un 65 a 85 porciento de probabilidades de lograr la remisión completa.93 Los pacientes mayores de 60 años y tal vez incluso los mayores de 50 años presentan cifras de remisión completa un poco menores, y duración más corta de la remisión completa. La respuesta menos satisfactoria en los individuos de mayor edad es importantes porque cerca de la mitad de todos los casos nuevos de LMA aparecen en pacientes mayores de 60 años.94 De 1950 a 1990 la supervivencia promedio de los pacientes menores de 60 años mejoró en forma importante, pero esto no fue igual para los mayores de esta edad.95 No debe tomarse la decisión de aplicar tratamiento de inducción para la LMA en los pacientes mayores de 60 años sin considerar estos datos. En algunas series, la duración completa varía desde 16 hasta 24 meses, y alrededor del 20 al 25 porciento de los pacientes en este grupo se encuentran estables en la primera remisión completa después de tres años. Después de cinco a siete años, tal vez el 10 al 20 porciento de los individuos todavía permanecerán vivos y sin evidencia de recaída leucémica. La curva de recurrencia no parece alcanzar una meseta, indicando que no existen nuevas recaídas (la llamada curación) hasta el octavo año de remisión.96, 97 Al evaluar los reportes de supervivencia en la LMA, el médico debe distinguir entre la supervivencia real y actuarial, y debe estar conciente de que se requieren siete o más años de seguimiento para los pacientes que reciben quimioterapia estándar.98
Desde el punto de vista clínico los pacientes con remisión completa están en buen estado. La exploración física es normal al igual que el recuento hemático y la médula ósea. Estas y otras observaciones conducen a la hipótesis de que durante la remisión completa la clona leucémica es incapaz de diferenciación, lo cual permite a las clonas normales multiplicarse, diferenciarse y repoblar la médula. Varios estudios han demostrado la persistencia de una clona anormal durante la remisión, a lo que se denomina remisión clonal.99,100 La incidencia de remisión clonal es quizá hasta del 25 porciento en la LMA, pero no necesariamente presagia una recaída. Aún no se ha determinado el significado de este fenómeno.
Es probable que el tratamiento de inducción para la leucemia mieloblástica aguda deba realizarse en instituciones que posean experiencia con este tratamiento. Los factores que deben considerarse son los requerimientos de plaquetas y tal vez de granulocitos, las dificultades relacionadas con el mantenimiento de una vía vascular no infectada y el problema constante de las infecciones repetidas.101, 102 Debido a que sólo del 10 al 20 porciento de los pacientes logran una supervivencia libre de enfermedad de cinco a siete años con los programas de quimioterapia disponibles en la actualidad, se han evaluado extensos programas basados en el tratamiento alogénico y autólogo de médula ósea.96, 103, 104 El riesgo de recaída leucémica es mucho menor en pacientes tratados con trasplante alogénico de médula ósea que en aquellos que reciben quimioterapia convencional. Sin embargo, la comparación de los datos de supervivencia en los dos grupos produce resultados menos evidentes porque aproximadamente del 20 al 30 porciento de los pacientes que recibieron trasplante alogénico de médula ósea murieron por complicaciones durante el primer año.104-106
El trasplante autólogo obvia la necesidad de un donador histocompatible y evita las complicaciones de la EICH, pero el procedimiento no tiene el beneficio del efecto injerto contra leucemia.107 No es claro si la médula cosechada para trasplante autólogo debe ser purgada con 4-hidroperoxiciclofosfamida u otros agentes. Dos estudios extensos están comparando la quimioterapia con trasplante autólogo y alogénico de médula ósea. En el estudio de la European Organization for Research and Treatment of Cancer (Organización europea para la investigación y el tratamiento del cáncer, n. del t.) (estudio EORTC-AML-8A), todos los pacientes con leucemia mielógena aguda que lograron remisión completa después de la inducción y un curso de consolidación intensiva, fueron sometidos a trasplante alogénico de médula ósea si contaban con un hermano donador con HLA idéntico. El resto de los pacientes fue asignado en forma aleatoria para recibir trasplante autólogo no purgado de médula ósea o un segundo curso de consolidación intensiva quimioterapéutica. En el estudio americano, el Southwest Oncology Group, el Eastern Cooperative Oncology Group, y el Cancer and Leukemia Group B (Grupo de oncología del suroeste, Grupo cooperativo de oncología del este y Grupo B de cáncer y leucemia, de los EUA, n. del t.) están evaluando las mismas alternativas, pero la médula se purga con 4-hidroperoxiciclofosfamida en los trasplantes autólogos de médula ósea y se usa idarrubicina porque parece ser superior a la daunorrubicina [ver figura 1 y tabla 4].108,109

|
| Figura 1 |
| Tratamiento de la leucemia mieloblástica aguda |
|
||
|
Casi cualquier procedimiento de inducción se complica con fiebre, se supone que los episodios iniciales son causados por sepsis bacteriana y se tratan con antibióticos intravenosos mientras se obtienen los cultivos. Después pueden desarollarse otro tipo de infecciones, incluyendo infección por Staphylococcus epidemidis en el sitio del catéter de Hickman, infecciones herpéticas de la boca o el esófago, infección por Candida del esófago, hígado, bazo o sangre, o aspergilosis pulmonar.
Al parecer, la función gonadal en el varón regresa a lo normal después del tratamiento de inducción estándar para la LMA.110
La infiltración leucémica de las meninges en adultos es menos común en la leucemia mieloblástica aguda (siete porciento de casos) que en la leucemia linfocítica aguda (40 porciento de los casos). Debido a que la infiltración leucémica de las meninges sí se presenta en unos cuantos pacientes, es importante darse cuenta de las complicaciones neurológicas.111 El tratamiento con methrotexate intratecal, en dosis de 12 mg/m2 (máximo 12 mg) dos veces a la semana, hasta que el LCR se normalice, ha probado ser eficaz en estos casos. La administración de soluciones diluidas en grandes volúmenes parece potenciar el efecto antileucémico y reducir la toxicidad del medicamento.112, 113 Las soluciones deben diluirse en solución salina de manera que contengan alrededor de 1 mg de methrotexate por ml de solución. En tales circunstancias pudiera ser útil instalar un reservorio de Ommaya. Cuando es necesario tratar la infiltración leucémica de las meninges con methotrexate intratecal, puede evitarse la neurotoxicidad inducida por el medicamento midiendo los niveles de methotrexate en el LCR.114
Puede haber masas cloromatosas en la leucemia mieloblástica aguda en el momento de presentación o como primera manifestación de la enfermedad. No está claro si éste es un signo de mal pronóstico.115
La leucostasis puede presentarse cuando el recuento de blastos se eleva sustancialmente por encima de 50,000/mm3, y es frecuente cuando el recuento de blastos excede los 200,000/mm3. Los trombos y agregados de blastos causan obstrucción vascular acompañada de hemorragia e infarto en vasos cerebrales y pulmonares.117 Los pacientes con recuentos de blastos muy aumentados no deben recibir transfusiones de eritrocitos que elevan la hemoglobina a niveles mayores de 10 g/dl porque la viscosidad mayor puede precipitar la leucostasis cerebral y la muerte.117
La cuenta de blastos puede reducirse en forma aguda por medio de leucoféresis seguida de la administración oral de hidroxiurea (750 mg/m2 cada seis horas hasta que la cuenta de leucocitos sea menor de 50,000/mm3). El aumento del ácido úrico puede manejarse con alopurinol. Para evitar la leucoestasis cerebral en los pacientes con cuentas elevadas de blastos, algunos investigadores recomiendan radioterapia craneal urgente, en dosis única de 400 cGy (400 rads).
El fracaso del tratamiento, definido como la persistencia de blastos leucémicos en la médula, ocurre en el 10 a 20 porciento de los pacientes. Existen varias explicaciones para este fracaso. Los blastos leucémicos que contienen el antígeno CD34, que es un marcador primitivo de células madre, son muy resistentes al tratamiento. Las células leucémicas pueden expresar la proteína bcl-2 en gran cantidad, que bloquea la apoptosis [ver antes, Biología de las leucemias].118 La expresión del gen de resistencia a múltiples medicamentos (mdr-1) puede permitir a las células leucémicas resistir a los agentes terapéuticos como la vincristina, el etopósido, la idarrubicina y otras antraciclinas.119
Después del fracaso del tratamiento pueden intentarse programas de salvamento, siempre que el paciente comprenda que el pronóstico no es muy bueno y que la posibilidad de remisión completa es de 30 porciento o menor. Estas remisiones duran solo cuatro o cinco meses. Los programas suelen combinar mitoxantrone, citosina arabinósido en dosis altas y etopósido. En un estudio se administraron los siguientes agentes en forma diaria durante seis días: mitoxantrona, 6 mg/m2/día como bolo I.V., citarabina, 1 mg/m2/día I.V. en seis horas y etopósido, 80 mg/m2/día I.V. durante una hora.120 Se están realizando estudios que emplean dosis grandes de ciclosporina o de agentes semejantes para bloquear el transportador de resistencia a múltiples fármacos, lo que parece incrementar el efecto de los medicamentos. Debe considerarse la realización de un trasplante alogénico de médula ósea de un donador no familiar compatible si el paciente es jóven, tiene buena salud y ha tenido la suficiente suerte como para encontrar un donador en el Programa nacional de donadores de médula ósea de los EUA.121 Los trasplantes de donadores no familiares se asocian con complicaciones como fracaso del injerto y EICH, así como con recaída de la leucemia. La probabilidad de supervivencia libre de leucemia a los dos años es menor del 20 porciento.
VARIANTES DE LA LEUCEMIA MIELOBLASTICA AGUDA
Leucemia mieloblástica aguda relacionada con el tratamiento
Un trastorno similar a la leucemia mieloblástica aguda puede desarrollarse varios años después de la administración de tratamiento para otro tipo de cáncer. Este trastorno, que se ha denominado leucemia mieloblástica aguda relacionada con el tratamiento (LMA-t) o leucemia no linfocítica aguda relacionada con el tratamiento (NLA-t), es precedida por lo general de un síndrome mielodisplásico caracterizado por citopenias o anemia refractaria con sideroblastos en anillo (ver adelante). En una serie de pacientes con enfermedad de Hodgkin que fueron tratados con medicamentos alquilantes, la incidencia de LMA-t alcanzó el 13 porciento en diez años, después de los cuales no se observaron más casos.122 En otra serie, no aparecieron casos nuevos de LMA-t después de once años.123 Esta variante se asocia con deleción de parte o todo el cromosoma 5 o 7 [ver tabla 1].17, 91, 124
La leucemia mieloblástica aguda relacionada con el tratamiento no responde bien a la quimioterapia: sólo el 20 al 40 porciento de los pacientes alcanzan la remisión completa,125 e incluso en estos casos, la remisión es corta (duración media, cinco meses).126, 127 Si el paciente es menor de 50 años de edad y cuenta con un hermano donante compatible, debe considerarse el trasplante alogénico de médula ósea porque han producido supervivencias satisfactorias libres de enfermedad en algunos casos.128 Las decisiones terapéuticas con respecto a este trastorno requieren el análisis cuidadoso de la relación riesgo-beneficio, evaluación de la enfermedad inicial que requirió el tratamiento previo y la consideración de la edad del paciente y los deseos de someterse a tratamientos nuevos.
Leucemia promielocítica aguda
Casi todos los casos de leucemia promielocítica aguda, designada como M3 en la clasificación FAB, muestran la traslocación 15;17 [ver tabla 1]. Aunque la LPA es responsable de solo el 10 porciento de todos los casos de LMA, existe gran interés en esta variante. En la LPA el gen para el receptor-a del ácido retinoico (RAR-a) en el brazo largo del cromosoma 17, se trasloca al cromosoma 15 en el sitio que contiene el gen PML. Esta traslocación provoca la fusión de los genes PML y RAR-a (PMR-RAR-a). Además, el descubrimiento de que el ATR causa diferenciación de los promielocitos leucémicos in vitro e in vivo ha provocado cambios dramáticos en el manejo de la LPA y para evitar las complicaciones hemorrágicas graves.129
El diagnóstico de LPA se basa, desde el punto de vista morfológico, al identificar la invasión de la médula ósea por promielocitos atípicos o al detectar la traslocación 15;17 por medio de citogenética o análisis con sondas. Con frecuencia ocurre coagulación intravascular diseminada (CID). El tratamiento comienza con ATR (disponible en el protocolo que realiza el Instituto Nacional contra el Cáncer de los EUA), que suele administrarse en una sola dosis oral de 45 mg/m2 o en dos dosis divididas, aunque no se ha determinado la dosis correcta.130 En 30 a 40 días el 80 a 90 porciento de los pacientes logran la remisión completa. La mayoría de los efectos adversos del ATR, como prurito, cefalea y fiebre ocasional, no son graves, pero puede ocurrir toxicidad hepática e insuficiencia cardiaca congestiva. Se desarrolla seudotumor cerebri en algunos pacientes, y alrededor del 25 porciento sufren el llamado síndrome del ácido retinoico, que consiste en fiebre, hiperleucocitosis y deterioro respiratorio por infiltración pulmonar. Este síndrome se trata con 10 mg de dexametasona administrados por vía intravenosa cada 12 horas durante por lo menos tres días. Incluso con el uso de medidas diseñadas para disminuir la cuenta de leucocitos, hasta una tercera parte de los pacientes con síndrome de ácido retinoico fallecen.
Debido a que la duración de la remisión completa inducida por el ATR es muy corta, el tratamiento debe ser seguido por programas de inducción de quimioterapia estándar basados en antraciclinas.131,132 Los pacientes en remisión completa pueden tolerar un esquema de inducción como el usado en la LMA [ver tabla 4] con menos lisis del tumor y menos hemorragia secundaria a la CID.132 Incluso con el empleo del ATR, los pacientes con LPM tienen un mayor riesgo de CID y, en consecuencia, de hemorragias. Ocurre hemorragia petequial en el sitio de aspiración de la médula ósea y en los sitios de punción venosa, pueden presentarse hemorragias del sistema nervioso central y de las áreas del cuello y paratraqueales, lo que puede causar muerte súbita. Hay evidencias clínicas y de laboratorio de CID. Se piensa que los promielocitos malignos liberan factor tisular de sus gránulos o de su membrana plasmática cuando son destruídos, y que un bolo de esta sustancia puede iniciar la CID. Debido a que la gravedad de la coagulopatía se relaciona en forma estrecha con la reducción del inhibidor a2-de plasmina (a2-PI), parece ser que las sustancias liberadas por las células de la LPA estimulan también al sistema fibrinolítico en forma directa. En los pacientes que sufren sangrado, la a2-PI es menor al 30 porciento de lo normal, lo que sugiere que este factor se consume al contrarrestar la plasmina libre circulante.132,133
Si existe CID, ésta se trata por medio de heparina intravenosa continua infundida a velocidad de 250 a 500 unidades/hr y con remplazo agresivo de plaquetas, con objeto de mantener la cuenta plaquetaria en más de 50,000/mm3. La dosis de heparina se calcula vigilando el nivel de fibrinógeno y de productos de degradación de fibrina y fibrinógeno.134 Si el paciente continúa sangrando, puede añadirse con precaución el agente antifibrinolítico, ácido e-aminocaproico (AEAC), en dosis de 0.5 g/hr, a la infusión de heparina. Si pueden medirse las concentraciones de plasmina y de a2-PI, ésto permitirá decidir cuando agregar el AEAC. Varios estudios indican que a diferencia de la LMA, es posible lograr la remisión completa en la LPA sin producir aplasia total de la médula ósea.134,135
Leucemia megacarioblástica aguda
La leucemia megaloblástica aguda,20, 21 designada M7 en la clasificación de la FAB, es un trastorno reconocido recientemente que es probable que incluya muchos casos que antes se designaban como mielofibrosis aguda. El diagnóstico de la M7 se basa en el análisis citoquímico de la sangre periférica porque con frecuencia la médula está fibrótica y no es posible aspirarla [ver tabla 2]. Los pacientes tienen pancitopenia, mielofibrosis y un infiltrado en la médula ósea compuesto probablemente de megacarioblastos. No hay esplenomegalia y existe poca evidencia de leucoeritroblastosis en sangre periférica y hematopoyesis extramedular.
Se ha publicado un caso de mielofibrosis aguda después de tratamiento citotóxico y se publicó otro después de una combinación de tratamiento citotóxico y radioterapia.136 Se ha intentado un tratamiento de inducción similar al usado en la leucemia mieloblástica aguda; sin embargo, la respuesta terapéutica ha sido en general mala. El trasplante alogénico ha producido remisión prolongada.
Eritroleucemia aguda
La eritroleucemia aguda, también llamada mielosis eritrocítica aguda, es una variante de la leucemia mieloblástica aguda, que se presenta como un trastorno espontáneo, generalmente en pacientes mayores de edad, o como una supuesta complicación del tratamiento alquilante, como en los casos que se presentan después del tratamiento con melfalán para el mieloma múltiple. Los pacientes tienen anemia por eritropoyesis ineficaz y pancitopenia. La eritroleucemia aguda es la variedad M6 en la clasificación FAB [ver tabla 2]. Los pacientes suelen tratarse con programas de inducción para LMA. En un estudio se obtuvo remisión completa en seis de 14 pacientes.137-138
Leucemias agudas bifenotípicas híbridas
Las células leucémicas de algunos pacientes muestran características linfoides mediante los métodos citoquímicos convencionales y técnicas de inmunofenotipificación. Diferentes términos se han utilizado para describir estas leucemias, incluyendo bifenotípica, híbrida, biclonal y bilineal. La variante en la cual las células malignas individuales muestran marcadores mieloides y linfoides es denominada leucemia bifenotípica. En otra variante, conocida como leucemia aguda híbrida, los blastos leucémicos son heterogéneos, ya que algunos muestran marcadores mieloides y otros marcadores linfoides. A esta variante también se han aplicado los términos biclonal bilineal.139 El análisis citogénico muestra la translocación 4;11 en algunos pacientes con leucemia bifenotípica.91 El pronóstico de los pacientes con leucemia aguda híbrida o bifenotípica es malo. Aun cuando dichos pacientes alcanzan remisión completa, a menudo como respuesta a la inducción utilizada para la leucemia linfoblástica aguda, la duración de la remisión es corta.
Síndromes mielodisplásicos
Pueden considerarse juntas varias designaciones clínicas: leucemia mieloide subaguda, leucemia mielomonocítica crónica, mielosis eritrémica crónica, síndromes mielodisplásicos, preleucemia (displasia hemopoyética) y leucemia mieloide latente.140 Los pacientes con estos trastornos tienen leucemia con un cuadro clínico que cae en algún lugar entre las entidades mejor definidas de leucemia mieloide crónica y la leucemia mieloide aguda. En general, los pacientes son hombres en su séptima u octava década de la vida, y sus problemas clínicos se relacionan con la presencia, esencialmente uniforme, de citopenias aisladas o múltiples y la disfunción celular asociada. Puede haber un periodo prodrómico relativamente largo.
El frotis de sangre periférica contiene neutrófilos hipogranulados que en ocasiones muestran la seudoanomalía de Pelger-Huet (neutrófilos bilobulados que parecen neutrófilos juveniles). Además, puede haber blastos en el frotis de sangre periférica, al igual que un patrón leucémico. La morfología del eritrocito puede ser normal, o puede haber una hipocromía impresionante y macrocitos gigantes. El número de plaquetas está a menudo disminuido, y éstas generalmente son grandes y con disminución de las granulaciones. La médula puede ser hipercelular, pero suele ser normocelular, e incluso en ocasiones es hipocelular; en cualquier caso, el mecanismo usual de desplazamiento físico no explica la pancitopenia. Las anormalidades en el desarrollo de una o las tres líneas celulares son: pobre crecimiento granulocítico, presencia de megacariocitos enanos bilobulados [ver figura 2], y ocasionalmente sideroblastos en forma de anillo junto con normoblastos gigantes multinucleados. Puede estar aumentado el número de mastocitos en la médula.141 Una búsqueda cuidadosa puede revelar grupos de mieloblastos atípicos y promielocitos.142
|
| Figura 2 |
| Megacariocitos displásicos |
El grupo de la FAB ha propuesto una clasificación para los síndromes mielodisplásicos [ver tabla 5],143 pero su utilidad todavía no se ha determinado. Existen varios problemas en esta clasificación. Por ejemplo, la diferencia entre anemia refractaria con exceso de blastos en transformación (AREB-T) y la eritroleucemia, o M6 depende en parte de la capacidad para distinguir entre un nivel del 20 al 30 porciento de blastos y un nivel mayor al 30 porciento de blastos en el material aspirado de la médula. Los rasgos morfológicos utilizados para distinguir las cinco clases de mielodisplasia en la médula ósea se observan sólo en el material aspirado pero no tienen su contrapartida en las biopsias de médula ósea.144 Por otra parte, las biopsias, pueden revelar si existen grupos de mieloblastos, dato que puede tener importancia diagnóstica y pronóstica.145, 146 Otra deficiencia propuesta es el hecho de que los cinco subtipos sólo presentan dos comportamientos biológicos en general: (1) los pacientes con anemia refractaria (AR) y los que tienen anemia refractaria con sideroblastos en anillo (ARSA) tienen supervivencia promedio de 64 y 71 meses, respectivamente, y (2) los pacientes con anemia refractaria con exceso de blastos (AREB), los pacientes con anemia refractaria y exceso de blastos en transformación y los enfermos con leucemia mielomonocítica crónica (LMMC) tienen supervivencias promedio de siete, cinco y ocho meses, respectivamente.147 Un estudio que incluyó a 336 pacientes encontró que no era posible distinguir con claridad entre la leucemia aguda y el síndrome mielodisplásico con base en una proporción específica de blastos en la médula ósea. Sin embargo, no fue sorprendente que, a mayor porcentaje de blastos en la médula, más breve la supervivencia de los pacientes.148
|
||||||||||||||||||
|
Los estudios citogenéticos demuestran que las alteraciones cromosómicas ocurren con más frecuencia en la AREB, AREB-T y LMMC, que representan los síndromes más evidentemente leucémicos. Se observan tanto similitudes como diferencias entre las alteraciones citogenéticas en los síndromes mielodisplásicos y los de la LMA, variantes de LMA y LMC. La trisomía 8 es común en los síndromes mielodisplásicos, así como la pérdida de los cromosomas 5, 7 y Y; sin embargo, ninguno de los pacientes con estos síndromes tuvieron los patrones de la LMA, o sea t(8;21) o t(15;17) [ver tabla 1].149
El síndrome 5q-, trastorno mielodisplásico relativamente raro, caracterizado por la pérdida de una pieza del brazo largo del cromosoma 5, se asocia con un cuadro clínico muy característico consistente en aumento del número de megacariocitos anormales y enanos en la médula [ver figura 2], anemia macroovalocítica con eritrocitos de forma anormal, y recuento plaquetario normal o elevado. La médula ósea muestra características similares a la de pacientes con AR, ARSA o AREB [ver tabla 5]. Debido a que la porción q del cromosoma 5 contiene los genes para el FEC-GM, el factor estimulante de colonias de los macrófagos (FEC-M), la interleucina-3 (IL-3), el FCDP y otros factores de crecimiento y sus receptores,150 se ha considerado que la pérdida de uno o más de estos genes controlados conduce de alguna manera a esta variante mielodisplásica.151
La frecuencia de los síndromes mielodisplásicos es cada vez mayor debido a que las leucemias relacionadas con quimioterapia11-14 van en aumento y dichos trastornos primero evolucionan hacia un cuadro clínico mielodisplásico, que a menudo se asocia con deleción de los cromosomas 5 y 7.152, 153 El cuadro clínico combinado se conoce como síndrome SMD-t/LNLA-t debido a la regularidad del patrón evolutivo. Aunque el tratamiento con medicamentos alquilantes es el denominador común en las leucemias relacionadas con la quimioterapia, un estudio indica que es probable que el melfalán sea un leucemógeno más potente que la ciclofosfamida.154 Otro estudio indica que el tratamiento con procarbazina o nitrosoureas puede ser más leucemogénico que la ciclofosfamida.155
El curso clínico está marcado por hemorragia e infecciones. Si los pacientes presentan un recuento leucocitario aumentado o plaquetario disminuido, la supervivencia media es de sólo un mes y medio.156
La mayoría de los pacientes mueren en esta etapa de la enfermedad, antes de que se transforme en una entidad más obvia de leucemia mieloblástica aguda, hecho que ocurre en el 10 al 40 porciento de los pacientes. Los estudios de clonalidad indican que la mayoría de los síndromes mielodisplásicos derivan de una célula madre que puede diferenciarse hacia la línea linfoide o hacia la línea mieloide.157
TRATAMIENTO
La anemia hemolítica, manifestación poco frecuente de los síndromes mielodisplásicos, en ocasiones responde a la prednisona en dosis de 30 a 50 mg/día, pero este tratamiento se asocia con riesgo de infección. Los antimetabolitos, como la mercaptopurina y la hidroxiurea, se han empleado para controlar los síntomas sistémicos, las organomegalias y las cuentas elevadas de leucocitos, pero no alteran la historia natural del padecimiento.
La mayoría de los pacientes con síndromes mielodisplásicos son de edad avanzada, pero para aquellos pocos que son menores de 50 años de edad y cuentan con un donante histocompatible, el trasplante alogénico de médula ósea ofrece la mejor oportunidad para una vida normal.152, 158
Un grupo de investigadores ha tratado de manera agresiva a pacientes con síndrome mielodisplásico grave utilizando protocolos de LMA. En individuos mayores de 50 años de edad la respuesta es mala, y sólo el 25 porciento entró en remisión completa. En pacientes menores de 50 años, en el 86 porciento se logró la remisión completa, pero la duración de la remisión parece ser corta.159 Cuando llega a emplearse la quimioterapia intensiva, debe reservarse para los pacientes más jóvenes con enfermedad agresiva que comprenden que, incluso si se logra la remisión completa, ésta será de breve duración.160
Debido a que la mayoría de los pacientes con SMD tienen citopenia, la atención se ha enfocado a corrregir las citopenias individuales y sus consecuencias. En los pacientes neutropénicos con infección, la cuenta absoluta de neutrófilos puede aumentarse, quizá con beneficio clínico, por medio del tratamiento con FEC-G, 1-3 µg/kg/día; en ocasiones se requiere una dosis de 5 a 10 µg/kg/día.161 Existe preocupación de que la administración de FEC-G pueda aumentar la conversión del SMD a leucemia aguda, pero estas conversiones parecen tener la misma frecuencia que las espontáneas. En un estudio, la anemia asociada con el SMD respondió a la combinación de FEC-G y eritropoyetina en el 40 porciento de los pacientes. El FEC-G se inició en dosis de 1 µg/kg/día, administrado por vía subcutánea, y después se ajustó la dosis para normalizar o duplicar la cuenta absoluta de neutrófilos. Cuando la cuenta de neutrófilos alcanzó cualquiera de estos niveles, se añadió eritropoyetina, al inicio en dosis de 100 U/kg/día por vía subcutánea, que se aumentó a 300 U/kg/día en caso necesario.162 Este uso del FEC-G no ha sido aún autorizado por la Food and Drug Administration de los EUA, y los seguros pueden no cubrir su costo. En la actualidad no existe tratamiento útil con factores de crecimiento para los pacientes trombocitopénicos con SMD.
Para inducir diferenciación, que puede alterar la evolución de las formas severas de SMD, se estudiaron pacientes con AREB y AREB-T en un estudio de fase III. No se demostraron beneficios en el grupo tratado con FEC-G.
Leucemias crónicas y subagudas originadas de células B
Las leucemias linfocíticas incluyen una forma aguda, leucemia linfoblástica aguda [ver adelante, Leucemia linfoblástica aguda], así como varias formas subagudas y crónicas. Existen evidencias inequívocas indicando que las leucemias subagudas y crónicas deben tener una subdivisión adicional de acuerdo a su origen en las células T o B porque a menudo existe una correlación entre la clase de leucemia y su comportamiento biológico.
LEUCEMIA LINFOCITICA CRONICA
Fisiopatología
La leucemia linfocítica crónica (LLC) es un trastorno clonal maligno de linfocitos relativamente maduros, con predominio de células B; sin embargo, un trastorno maligno de células T (LLC-T) causa alrededor del cinco porciento de los casos. En la LLC la clona anormal remplaza a las células B o inhibe su crecimiento y maduración, reduciendo los niveles de inmunoglobulinas y alterando la inmunidad humoral. Al detectar el marcador clonal G6PD, un grupo de investigación parece haber demostrado el origen clonal a partir de linfocitos B en dos pacientes con LLC. Sin embargo, eritrocitos, plaquetas, granulocitos y linfocitos T de sangre periférica mostraron dos tipos de G6PD, lo que indica que estas células surgieron de precursores heterogéneos.163 Se ha identificado una anomalía citogenética, la trisomía 12, en las células B malignas en cerca del 50 porciento de los pacientes con LLC;164 también ocurren anomalías del cromosoma 14 pero con menos frecuencia que la trisomía 12. Típicamente, las células de la LLC tienen un nivel bajo de SmIg y por lo tanto exhiben sólo una ligera inmunofluorescencia de superficie.
Los linfocitos malignos parecen tener una supervivencia prolongada y la capacidad de recircular desde la sangre a la médula ósea y ganglios linfáticos y de nuevo de regreso a la sangre. En algunos casos los linfocitos anormales responden mal a la estimulación con fitohemaglutinina. Alrededor del 25 porciento de los pacientes con LLC presentan hipogamaglobulinemia, y hasta el 10 porciento hipergamaglobulinemia. Ambos estados reflejan al parecer una función anormal de las células B. Cerca del 5 porciento de los pacientes cursan con gamopatía monoclonal,165 fundamentalmente gamopatía IgM, parecida a la producida en la macroglobulinemia de Waldenström. Sin embargo, por lo general la producción de IgM no alcanza un nivel que pueda causar problemas de viscosidad.
Los pacientes con leucemia linfocítica crónica tienen una incidencia baja, pero clara, de anemia hemolítica autoinmune y una incidencia similar de púrpura trombocitopénica inmune, pero los autoanticuerpos que causan estos trastornos no se derivan de la clona de células B malignas.164 En el pasado se creía que los pacientes con LLC tenían mayor riesgo de neoplasias no hematológicas, pero esta opinión se ha puesto en duda.166
Manifestaciones clínicas
Los pacientes con LLC presentan un gran espectro de signos y síntomas en relación a la amplia variación del curso de la enfermedad. En un examen de rutina se puede descubrir que los pacientes tienen una linfocitosis absoluta con linfocitos maduros de 10,000/mm3 o hasta diez veces más. El examen físico de rutina puede mostrar adenopatías no dolorosas generalizadas y una esplenomegalia moderada. Por el contrario, los pacientes pueden tener pancitopenia profunda, crecimientos ganglionares impresionantes, organomegalia e infecciones repetidas.
La evolución clínica está determinada, sobre todo, por el grado de infiltración de la médula y de desplazamiento de elementos normales, que produce aplasia funcional y pancitopenia periférica. La neutropenia y el deterioro de la inmunidad humoral causan susceptibilidad e infecciones repetidas. La anemia hemolítica autoinmune y entidades equivalentes, al igual que el hiperesplenismo, pueden contribuir a la pancitopenia. La anemia y trombocitopenia pueden ser signos pronósticos malos. Las adenomegalias rara vez causan problemas, pero pueden obstruir los uréteros. Suelen no presentarse síntomas generales. En comparación con la leucemia mielocítica crónica, en muy raras ocasiones hay crisis blásticas en la leucemia linfocítica crónica.167
En ocasiones, en las etapas tardías de la LLC, los pacientes desarrollan fiebre, sudoración nocturna, pérdida de peso y crecimiento rápido de masas ganglionares y extraganglionares. En la sangre periférica se observan linfocitos inmaduros con nucleolos. Esta transformación, que semeja al linfoma difuso de células grandes, se denomina síndrome de Richter.164 Este síndrome responde poco al tratamiento, incluso a los programas agresivos que se utilizan para el tratamiento del linfoma difuso de células grandes. Algunos investigadores distinguen entre la transformación de Richter, que se caracteriza por una inmadurez linfoide mínima en sangre periférica, y una notable transformación prolinfocítica (ver adelante), que se caracteriza por una población distinta de células inmaduras en sangre periférica.168
Diagnóstico
El diagnóstico se confirma al encontrar linfocitosis absoluta en sangre periférica, infiltrado linfocitario en la médula ósea y biopsia de ganglios linfáticos que muestra el patrón de un linfoma difuso bien diferenciado. La electroforesis de proteínas séricas y la determinación cuantitativa de inmunoglobulinas muestran a menudo panhipogamaglobulinemia y rara vez un pequeño pico de proteínas M de IgM. Debe tenerse cuidado al interpretar los aspirados de médula ósea porque en condiciones normales hay nódulos linfoides. Sin embargo, el infiltrado por leucemia linfocítica revelará la presencia de desplazamiento de la médula en varias espículas. La biopsia con aguja de Jamshidi también demostrará el grado de alteración de la arquitectura por los linfocitos invasores. No está claro que existan diferencias clínicas entre la LLC y el linfoma difuso bien diferenciado.169 Por lo general, el análisis inmunofenotípico de la sangre periférica o de la médula ósea muestra proliferación de las células B con fluorescencia de bajo grado cuando se tiñe con inmunoglobulinas de membrana de superficie. Se demuestra que la proliferación es clonal por el hecho de que las células se tiñen sólo con los reactivos de cadenas ligeras k o l. Las células expresan con frecuencia IgM o tanto IgM como IgD; en este último caso, los idiotipos son idénticos. La mayoría de las células B en la LLC expresan también CD5, que se consideraba como un marcador de células T.166 Es probable que el CD5 refleje el origen primitivo de la célula B en la LLC. Los estudios de rearreglo de genes de las inmunoglobulinas muestran también evidencia de expansión clonal.
Estadificación
Se han propuesto varios sistemas de estadificación para la LLC basándose en el concepto de que los principales factores pronósticos son el número de áreas ganglionares afectadas, el grado de hepatoesplenomegalia, el patrón de desplazamiento medular y el grado de anemia y trombocitopenia.170, 171 En la clasificación de Rai,172 el estadio 0 representa una linfocitosis absoluta de 15,000/mm3 o mayor en la sangre periférica y linfocitosis del 40 porciento en la médula; el estadio I indica linfocitosis con linfadenomegalia; en el estadio II se presenta linfocitosis con aumento de tamaño del hígado o el bazo; en el estadio III existe linfocitosis con concentración de hemoglobina menor de 11 g/dl, y en el IV, linfocitosis con plaquetas por debajo de 100,000/mm3. Se considera que el estadio 0 predice una supervivencia a largo plazo de unos diez años, mientras que la mayoría de los pacientes en estadios III/IV mueren antes de los cuatro años.
La clasificación de Binet173 reduce el número de estadios de la LLC de cinco a tres y considera el aumento de cinco áreas específicas, bazo, hígado y ganglios linfáticos (ya sea unilateral o bilateral) axilares, cervicales e inguinales, además del grado de anemia y trombocitopenia. Los pacientes del grupo A no tienen anemia ni trombocitopenia y menos de tres áreas de aumento de tejido linfoide. Los pacientes del grupo B también tienen niveles normales de hemoglobina y plaquetas pero tienen tres o más áreas de linfadenomgalia. Los pacientes clasificados en el estadio C tienen un nivel de hemoglobina inferior de 10 g/dl o un recuento plaquetario menor de 100,000/mm3, sin considerar el número de áreas con crecimiento linfoide. Los pacientes en etapa C tienen el peor pronóstico.
Se ha propuesto una clasificación que se basa en parte en la histología de la médula.174 Esta distingue dos tipos de patrones de infiltrado medular, difuso y nodular; aquellos pacientes con el peor pronóstico, correspondiente al estadio III/IV de Rai, o etapa C de Binet, presentan una médula infiltrada en forma densa y difusa.
El análisis de series relativamente grandes de pacientes con LLC indican que otros factores también afectan el pronóstico de manera importante. El nivel de inmunoglobulinas séricas parece ser importante debido a que la supervivencia se reduce en pacientes con valores de gamaglobulina menores de 700 mg/ml. Al parecer los niveles de IgA, y en menor grado de IgG, se correlacionan más estrechamente con el pronóstico.175 Otro estudio encontró que el nivel de linfocitos en sangre periférica superior a 60,000/mm3 indica mala evolución.176 Es probable que estos factores constituyan importantes componentes de los sistemas de clasificación futuros.
Tratamiento
Los síndromes de anemia hemolítica autoinmune y púrpura trombocitopénica se tratan de la misma manera que en pacientes sin leucemia linfocítia crónica. Las infecciones repetidas se tratan con antibióticos, en particular si el paciente presenta infecciones crónicas del aparato urinario o enfermedades como bronquiectasias. Se ha demostrado en estudios controlados que pacientes con infecciones recurrentes relacionadas con hipogamaglobulinemia (niveles de IgG menores de 400 a 640 mg/dl)177 presentan menos infecciones cuando reciben tratamiento de IgG intravenosa a la dosis de 0.4 g/kg cada tres semanas.177, 178 Sin embargo, estos estudios no han demostrado ventajas en la supervivencia. Debido a que la IgG I.V. es cara, su uso en la LLC necesita mejores justificaciones.166 La leucostasis es muy rara, aún en presencia de cifras muy elevadas de leucocitos. La enfermedad local puede tratarse con irradiación local paliativa.
Es difícil decidir si se trata o no a un paciente que al parecer tiene crecimiento ganglionar y orgánico progresivo, y cuya pancitopenia se está haciendo más profunda como consecuencia aparente de infiltrado medular progresivo. En comparación con la leucemia mieloide crónica, el número de leucocitos en la leucemia linfocítica crónica no es un indicador preciso del grado de la enfermedad. Los estudios del tratamiento de la leucemia linfocítica crónica han sido difíciles de evaluar debido a que casos aparentemente similares parecen haber tenido evoluciones notablemente diferentes, variando de meses hasta más de 15 años.
Debido a que los tratamientos disponibles son mielosupresores, y de esta manera complican la pancitopenia preexistente y deterioran la inmunidad humoral,la estrategia preferida es no administrar tratamiento general hasta que existan síntomas y signos de pancitopenia progresiva. Los sistemas de estadificación son los que indican si tratar o no a los pacientes de mayor edad. Los enfermos con estadio A de Binet o estadio 0 o 1 de Rai no deben ser tratados, mientras que los que tienen estadios C de Binet o III o IV de Rai sí. Las decisiones respecto al tratamiento de algunos pacientes con estadio II de Rai o B de Binet deben hacerse en forma individual, aunque un grupo experimentado trata a todos los pacientes en estdio B de Binet.166
Se han obtenido algunos resultados favorables con radiación corporal total, pero un ensayo de radiación mediastínica indicó que dicho tratamiento tiene efectos colaterales de pancitopenia muy grave.179 Los esteroides no destruyen los linfocitos de la leucemia linfocítica crónica, sino que simplemente los redistribuyen desde los ganglios hasta la sangre periférica. Por esto, los agentes alquilantes son la piedra angular del tratamiento en los programas de un solo medicamento. El tratamiento consiste en dosis de 0.1 mg/kg al día de clorambucil, o dosis de 2 mg/kg al día de ciclofosfamida. El medicamento se administra con precaución, y durante el tratamiento se lleva un control de citologías hemáticas, ganglios linfáticos y tamaños de órganos. También se puede administrar clorambucil cada dos semanas, iniciando con una sola dosis de 0.4 mg/kg y aumentando 0.1 mg/kg hasta que exista mielotoxicidad. Algunos especialistas combinan el clorambucil con un curso de cinco días de prednisona (80 mg/día por vía oral). No reducen la dosis de prednisona en forma gradual.
Un grupo de investigadores en Francia llevó a cabo un estudio clínico único, al azar, en pacientes con LLC.180 Pacientes en etapa A de Binet recibieron tratamiento diario con clorambucil por vía oral, en dosis de 0.1 mg/kg durante un periodo indefinido, o no recibieron tratamiento. El estudio encontró que el tratamiento con clorambucil era dañino para los pacientes en etapa A.181 Los pacientes en etapa B recibieron al azar clorambucil por vía oral diariamente o CVP (i.e., ciclofosfamida, 300 mg/m2/día por vía oral en los días 1 a 5; vincristina, 1 mg/m2 por vía I.V. en el día 1; y prednisona, 40 mg/m2/día por vía oral dividido en cuatro dosis en los días 1 a 5, el ciclo se repetía mensualmente); la supervivencia a los dos años para los dos grupos fue la misma (83 porciento). Desgraciadamente, debido a que no hubo un grupo en estadio B que no recibiera tratamiento, no se estudió la posibilidad de que los pacientes en estadio B no necesiten tratamiento sistémico. Los pacientes en estadio C recibieron al azar terapia con CVP o con CHOP, que es igual al protocolo CVP pero se le agrega dexorrubicina, en dosis de 25 mg/m2 administrados por vía intravenosa el día 1 de cada ciclo mensual. Para los pacientes en etapa C, los dos programas produjeron diferencias significativas en la supervivencia: a los dos años, el 77 porciento de los individuos que recibían CHOP estaban vivos, en comparación con sólo un 44 porciento del grupo de CVP. No se ha confirmado esta aparente ventaja de la doxorrubicina.166
Se han usado varios análogos del nucleósido purina en pacientes con LLC. La fludarabina, un agente muy eficaz en la LLC avanzada, se administra en dosis de 25 mg/m2/día I.V. durante 30 minutos por cinco días, y el ciclo se repite cada mes. En estudios en fase III se está evaluando si el tratamiento debe comenzar con programas basados en fludarabina o clorambucil. La fludarabina suele tardar dos a tres ciclos para producir cualquier efecto, y se asocia con mielotoxicidad considerable. Además, la fludarabina interfiere con la deaminasa de adenosina, produciendo supresión profunda de las células T, en especial de las CD4 (T cooperadoras). La adminstración de fludarabina en combinación con prednisona se ha asociado con un mayor riesgo de infección por Listeria monocytogenes.182
La pentostatina, análogo de un nucleósido que inhibe la deaminasa de adenosina, se ha probado en pacientes en estadio B y C, aproximadamente el 25 al 30 porciento de los pacientes han presentado buena respuesta. A pesar del efecto supresor del medicamento sobre las células T, no se han presentado complicaciones infecciosas. El papel de la pentostatina en el tratamiento de la LLC no está claro.183 La 2-clorodesoxiadenosina, otro agente que interfiere con la acción de la deaminasa de adenosina, ha sido autorizada por la FDA para su empleo en leucemia de células peludas (ver adelante) pero no de LLC. Varios reportes indican que la 2-clorodesoxiadenosina es útil en pacientes con LLC, sobre todo en los que son refractarios a la fludarabina. La dosis de este medicamento en la LLC no se ha establecido aún. Un esqumea para la LLC es idéntido al usado para la leucemia de células peludas, otro consiste en 0.12 mg/kg/día administrado por vía intravenosa durante dos horas por cinco días consecutivos, y el ciclo se repite cada mes.184,185
En ocasiones la esplenectomía puede ser importante en el tratamiento de la LLC. Las indicaciones para ésta son: esplenomegalia dolorosa, anemia intensa que requiere cada vez más transfusiones, y trombocitopenia importante. En determinadas circunstancias, la esplenectomía puede producir mejoría clínica; sin embargo, ésta se ha relacionado con una morbilidad aproximada del 20 porciento, mortalidad de cerca del 5 porciento, y no prolonga la supervivencia.186
LEUCEMIA CRONICA DE CELULAS DE LINFOSARCOMA
Las células en la leucemia crónica de células de linfosarcoma son más grandes que las de la LLC, y sus nucleolos se distinguen por lo general con facilidad. Los frotis de la sangre periférica de los pacientes con esta enfermedad muestran en ocasiones muchos linfocitos pequeños hendidos. Esta es la fase leucémica de los linfomas foliculares o difusos de células hendidas pequeñas. La biopsia de ganglio linfático o de médula ósea con análisis de marcadores inmunofenotípicos suele revelar el diagnóstico primario, que determina el tratamiento de elección.
LEUCEMIA PROLINFOCITICA
La leucemia prolinfocítica es una variante rara caracterizada por síntomas generales, esplenomegalia masiva y linfocitos grandes [ver figura 3]. El número de leucocitos está muy aumentado, por lo general por encima de 100,000/mm3,164 y las células expresan mucho más SmIg que las células de la LLC. La reactividad a CD5 es baja.
|
|
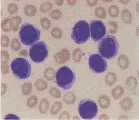
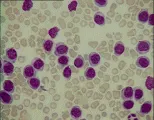
Puede usarse la esplenectomía combinada con quimioterapia usando el esquema CHOP (ver antes) o un programa semejante. En algunos pacientes con LLC ocurre progresión prolinfocítica (LPL). Este síndrome, llamado LLC/LPL se manifiesta por fiebre, esplenomegalia, citopenias y aumento en el número de prolinfocitos en la sangre periférica. No es claro si la LPL es diferente al síndrome de Richter, una transformación maligna muy agresiva que incluye los mismos marcadores clonales expresados en la línea original de la LLC.188 Algunos pacientes con LLC/LPL responden a la fludarabina, pero la respuesta es corta y la evolución mala.
LEUCEMIA DE CELULAS PELUDAS (RETICULOENDOTELIOSIS LEUCEMICA)
La célula maligna en la leucemia de células peludas [ver figura 4] es un linfocito B que infiltra y remplaza la médula ósea o infiltra el bazo, causando esplenomegalia.189 La combinación del desplazamiento medular y esplenomegalia ocasiona pancitopenia (incluyendo monocitopenia). Es muy característica la morfología en el frotis de sangre periférica, en la biopsia de médula ósea y en la biopsia esplénica. La prueba positiva de la actividad de la fosfatasa ácida resistente es útil pero no patognomónica.
|
| Figura 4 |
| Leucemia de células peludas |
Las complicaciones más importantes son las infecciones, al parecer como consecuencia de la neutropenia; pueden aparecer infecciones por micobacterias atípicas con frecuencia superior a la normal.190 La vasculitis en la leucemia de células peludas, suele relacionarse con alguna infección, y suele responder al tratamiento con interferón alfa.191, 192
La esplenectomía para corregir la pancitopenia ha sido la base del tratamiento de la leucemia de células peludas. Esta modalidad terapéutica ha sido sustituida, y casi abandonada, por el uso del interferón alfa, que tiene un efecto benéfico en casi el 75 porciento de los pacientes, pero con importante toxicidad [ver antes, Leucemia mieloide crónica]. La introducción de la 2-clorodesoxiadenosina tiene un impacto importante en el tratamiento porque una sola administración, que consiste en infusión I.V. continua durante siete días en dosis de 0.1 mg/kg/día, produce remisión completa duradera en alrededor del 80 porciento de los pacientes. La toxicidad, que es relativamente leve, consiste en fiebre, pero también leucopenia y trombocitopenia en algunos pacientes. La 2-clorodesoxiadenosina es inmunosupresora, causando caída en el número de células T CD4+. La respuesta terapéutica ocurre durante un periodo de varias semanas y en ocasiones se asocia con el síndrome de lisis tumoral. Algunos pacientes pueden requerir un segundo curso de 2-clorodesoxiadenosina.193
Leucemias crónicas y subagudas originadas de células T
Algunos autores proponen que existen cuatro categorías de trastornos de células T maduras malignas: el síndrome de Sézary (la fase leucémica de las micosis fungoides), la leucemia/linfoma de células T del adulto, la leucemia prolinfocítica de células T y la variante de linfocitos granulares grandes (LGG), que incluye la LLC de células T.
SINDROME DE SEZARY (FASE LEUCEMICA DE LAS MICOSIS FUNGOIDES)
El linfoma/leucemia cutáneo de células T (LCCT) es un padecimiento crónico de las células T cooperadoras-inductoras. Cuando la sangre periférica contiene muchas células linfoides con nucleos en forma de nalgas o cerebriformes, el trastorno se denomina síndrome de Sézary [ver figura 5]. Existe enfermedad cutánea en casi todos los casos. Cuando el padecimiento se confina a la piel, se maneja en forma local y se han logrado resultados excelentes con tratamiento de la piel con haz de electrones.194 Sin embargo, más del 20 porciento de los linfocitos que existen en los frotis de sangre periférica son del tipo anormal de Sézary [ver figura 5], y estos pacientes tienen peor pronóstico.195 Aunque los estudios de rearreglo de receptores de células T muestran que la célula circulatoria de Sézary suele ser una célula T de una clona idéntica a la encontrada en las biopsias de piel, el análisis de genes de receptores muestra que las células de Sézary CD3+ que circulan en algunos pacientes son policlonales.196 No es claro cuál es el significado de estas variantes policlonales del síndrome de Sézary.
|
|
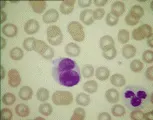
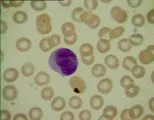
Se ha descrito en Japón, las Indias Occidentales y el sureste de Estados Unidos un síndrome rápidamente mortal conocido como linfoma/leucemia de células T del adulto (LLTA), que produce leucocitosis, hepatoesplenomegalia, linfadenopatía, lesiones cutáneas e hipercalcemia. Se considera que las células malignas son linfocitos T maduros colaboradores/inductores. Un retrovirus humano, denominado virus del linfoma/leucemia de células T humano (HTLV-I) se ha identificado en varios cultivos celulares de pacientes, uno de los cuales tenía linfoma cutáneo de células T. Los pacientes con LLTA tienen anticuerpos contra el HTLV-I.197
En los Estados Unidos los pacientes se presentan generalmente con varias lesiones cutáneas o con hipercalcemia manifestada por malestar general, náusea, confusión, sed y poliuria. Los niveles elevados de fosfatasa alcalina sérica, los rastreos óseos anormales y las lesiones líticas visibles a los rayos X indican que la causa probable de la hipercalcemia es un recambio óseo acelerado. Sorprendentemente, existe poca participación tumoral en los lugares de destrucción de hueso, lo que sugiere que la destrucción ósea representa una forma de activación paraneoplásica de los osteoblastos. Es frecuente encontrar células malignas circulantes que tienen núcleos lobulados y que son similares a las del síndrome de Sézary. Aunque son frecuentes las linfadenopatías periféricas, no hay adenopatía mediastínica, y de esta manera puede distinguirse de la LLA de los linfocitos T o del linfoma linfoblástico. Son frecuentes las infecciones oportunistas debidas a agentes como Pneumocystis carinii y citomegalovirus, lo mismo que la afección del sistema nervioso central.
El diagnóstico puede realizarse por medio de biopsia cutánea, de algún ganglio linfático que muestre linfoma difuso o mediante examen de la sangre periférica. La quimioterapia combinada intensiva puede producir mejoría transitoria.5 Los principales indicadores de mal pronóstico son un estado funcional poco satisfactorio, los niveles elevados de deshidrogenasa láctica (DHL) y un cuadro leucémico.198
LEUCEMIA PROLINFOCITICA DE CELULAS T
La LPL de células T (LPL-T) es una variante distinta de la LPL que se asocia con más adenopatías, lesiones cutáneas y afección del sistema nervioso central, en comparación con la LPL de células B (LPL-B).199 Este es un padecimiento agresivo que responde poco a los agentes alquilantes o a la radioterapia del mediastino o bazo. En una serie de pacientes con alteraciones citogenéticas relativamente constantes que incluyeron el cromosoma 14, alrededor de la mitad de los pacientes tuvieron respuesta parcial a la 2-desoxicoformicina (pentostatin).200 La dosis estándar de pentostatin suele ser de 4 mg/m2 administrados por vía intravenosa una vez a la semana durante tres semanas. Después se reevalúa la necesidad de tratamiento adicional.201
LINFOCITOSIS DE LINFOCITOS GRANULARES GRANDES
Los términos empleados para describir a la linfocitosis de LGG incluyen LLC-T, LLC de células asesinas naturales (NK) y trastorno linfoproliferativo de LGG. La característica de este padecimiento es la proliferación de linfocitos de gran tamaño que contienen gránulos azurófilos, y que pueden observarse en sangre periférica [ver figura 6].202 Parece que existen dos variantes de clonas: las células CD3+, que son células T y las células CD3-, que son células NK. La primera, que es la forma más común, debe denominarse leucemia de LGG-T (CD3+), y la segunda debe llamarse leucemia de LGG-NK (CD3-).
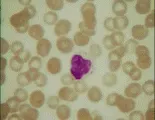
|
| Figura 6 |
| Linfocito granular grande |
El cuadro clínico es variable, pero de forma característica los pacientes presentan infecciones frecuentes o hemorragias. La sangre periférica muestra linfocitosis de LGG superior a 3,000/mm3 y neutropenia, trombocitopenia o evidencia de hemólisis.203, 204 El bazo puede ser palpable y la médula normal, o mostrar colecciones focales de linfocitos. El desplazamiento de la médula nunca es tan extenso como para explicar la presencia de citopenia. Los anticuerpos antiplaquetarios y antineutrófilos son en apariencia los responsables de la neutropenia y la trombocitopenia; también se ha descrito la presencia de anemia hemolítica Coombs positiva. Algunos pacientes presentan títulos séricos elevados de factor reumatoide. Es probable que algunos casos que se han clasificado como síndrome de Felty atípico en realidad son debidos a linfocitosis de LGG. En un caso, al parecer la infección con el retrovirus HTLV-I produjo la expansión clonal de los linfocitos granulares grandes asociados con un cuadro clínico similar al de la enfermedad linfoproliferativa-g T crónica. Las células infectadas al parecer liberaron linfocinas que a su vez produjeron el síndrome de aplasia pura de glóbulos rojos.205
Leucemia de linfocitos granulares grandes-T
Los pacientes con LGG-T puedn tener también artritis reumatoide. El diagnóstico de leucemia de LGG-T se basa en la morfología característica en los frotis de sangre periférica y en los análisis de inmunotipificación y de médula ósea que muestran el patrón CD3+, CD8+, CD16+ y CD57+. La clonalidad puede establecerse por rearreglo del gen del receptor de la célula T. El tratamiento para la leucemia de LGG-T suele dirigirse a la neutropenia y a las infecciones asociadas. Puede ser útil el FEC-G.202
Leucemia de linfocitos granulares grandes-asesinos naturales
La variante de LGG-NK (CD3-) ocurre en pacientes más jóvenes que no tienen artritis reumatoide pero que sufren una evolución tórpida caracterizada por fiebre, cuentas altas de LGG, hepatoesplenomegalia, ascitis y, en ocasiones, alteraciones de la coagulación. El cuadro clínico, la morfología característica y el patrón inmunofenotípico que muestra CD3-, CD4-, CD8-, CD56+ y CD57- permite hacer el diagnóstico. No existen reportes de tratamiento eficaz.
En algunos pacientes existe un incremento absoluto en los LGG de fenotipo CD3+ o CD3-, pero el análisis de los genes de receptores de las células T no muestra signos de clonalidad. Este patrón es especialmente válido en la variante CD3-, porque no existen marcadores clonales específicos de las células NK además de las alteraciones citogenéticas. Se desconoce el significado de estas variantes.
POSIBLES TRASTORNOS CLONALES DE CELULAS T
Reticulosis medular histicítica
El raro trastorno denominado reticulosis medular histiocítica se parece a la leucemia, ya que produce fiebre elevada, organomegalia y, con frecuencia, pancitopenia. La médula está infiltrada por grandes células macrofágicas que ingieren eritrocitos y plaquetas. Al menos en algunos casos de reticulosis medular histiocítica el paciente presenta un linfoma de células T subyacente cuyo origen central se ha confirmado por el rearreglo de los genes que codifican la cadena ß del receptor de células T.206, 207 Parece que el linfoma puede secretar linfocinas que activan y reclutan monocitos y macrófagos normales en el lugar de la afección, generando con ello la apariencia morfológica característica de la reticulosis medular histiocítica. Algunos pacientes responden rápidamente al tratamiento con el programa CHOP,208 pero en general los resultados son malos. No se cuenta con estudios controlados al respecto.
Linfadenopatía angioinmunoblástica
La linfadenopatía angioinmunoblástica consiste en la proliferación de células linfoides que pueden ser benignas o malignas. Los pacientes se presentan con fiebre, erupciones cutáneas, linfadenopatía, hiperglobulinemia, esplenomegalia, anemia hemolítica, anemia hemolítica autoinmune y trombocitopenia. El cuadro clínico es coherente con una proliferación de células B y al principio se consideró que era un trastorno clonal de células B. En algunos pacientes se han detectado rearreglos de genes de inmunoglobulinas, y esta observación apoya la idea de que la linfadenopatía angioinmunoblástica es un trastorno clonal de células B. Sin embargo, en otros pacientes se ha observado evidencia de rearreglo del gen de la cadena ß del receptor de células T, lo cual indica la presencia de trastorno clonal de células T.207, 209 La inmunotipificación de los ganglios afectados en pacientes con linfadenopatía angioinmunoblástica muestra con frecuencia un fenotipo de células T. Es benéfico el tratamiento con prednisona (2mg/kg/día por vía oral durante cuatro a ocho semanas). El paciente debe recibir profilaxis para neumonía por P. carinii. Si fracasa el tratamiento con prednisona, pueden considerarse programas agresivos de poliquimioterapia, tomando siempre en cuenta los riesgos y la morbilidad.
Leucemia linfoblástica aguda
En los adultos, la leucemia linfoblástica aguda (LLA) representa del 10 al 15 porciento de los casos de leucemia aguda en adultos. La LLA se ha subdividido basándose en marcadores inmunológicos y enzimáticos en varios tipos [ver tabla 3]: la variante común (LLAc), una forma de células pre-B (LLA pre-B), una variante de células maduras (LLA-B) y una variante de células T (LLA-T) [ver figura 7]. Estas subclases pueden tener implicaciones terapéuticas, pero el protocolo terapéutico unificado que se menciona más adelante se basa en la suposición de que la LLA del adulto siempre es una enfermedad de alto riesgo.211
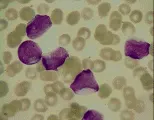
|
| Figura 7 |
| Linfoblastos |
La fisiopatología de la enfermedad y sus manifestaciones clínicas no son muy diferentes a las de la LMA, excepto que la incidencia de infiltración al SNC es mayor en la LLA.112, 212, 213 Es crucial hacer la distinción morfológica porque no hay fármacos tumor-específicos para el tratamiento de la LMA, mientras que la L-asparginasa, la vincristina y la prednisona atacan específicamente los linfoblastos leucémicos. Son signos de mal pronóstico el recuento de blastos en sangre periférica mayor de 20,000/mm3, la edad relativamente avanzada (i.e., mayores de 30 años), la leucemia de Burkitt (L3),214 el inmunofenotipo ALLAC, la traslocación 9;22 o la evidencia de la variante LLA del rearreglo bcr-abl, y la traslocación 4;11.213, 215
Están surgiendo resultados que han originado esperanzas de que las formas agresivas del tratamiento pueden producir remisiones prolongadas sin enfermedad, y aún la curación. El tratamiento se realiza con un protocolo de inducción con cuatro medicamentos seguidos de profilaxis del SNC y ciclos secuenciales de tratamiento citorreductor intenso.
Los programas de inducción han causado remisión completa en el 85 a 90 porciento de los pacientes.216,217 La adición de ciclofosfamida y citosina arabinósido a los programas de consolidación parece bloquear la resistencia previa a la LLA-T. No son claras la necesidad y duración del tratamiento de mantenimiento, pero es probable que sea útil el tratamiento durante dos años [ver tabla 6]. El uso del protocolo estándar [ver tabla 6] causa remisión completa y estable en alrededor del 35 a 40 porciento de los pacientes. Aunque puede inducirse remisión en pacientes con traslocaciones 9;22 y 4;11, ésta no dura ni un año. Por lo tanto, los pacientes con estas traslocaciones y los enfermos que no logren la remisión completa durante la inducción deben ser candidatos, cuando es posible, a trasplante alogénico de médula ósea de un hermano. Se están evaluando los trasplantes autólogos, al igual que el uso de un donador no relacionado para trasplante alogénico. También se ha propuesto el trasplante autólogo con médula purgada por medio de anticuerpos monoclonales, pero aún no se ha establecido por completo la utilidad de este procedimiento en la LLA.218,219
|
||
|
La quimioterapia de inducción para la LLA deprime en forma severa la función gonadal reproductiva, pero no la gonadal endócrina, en los pacientes varones. La quimioterapia para la LLA no causó deterioro en la función reproductiva o gonadal en mujeres.220
Principios psicosociales generales para el tratamiento de pacientes con leucemia
El médico que asume la atención primaria de los pacientes con leucemia sabe que hay dificultades reales para responder a las necesidades emocionales del paciente y su familia.
Las siguientes sugerencias pueden ser útiles. Son provechosas las discusiones abiertas con el paciente y su familia, pero nadie más que el paciente debe ser el receptor de la información médica directa. Básicamente, usted, el médico, es el indicado para educar al paciente acerca de la historia natural de la enfermedad y sus consecuencias.
Después de que el paciente conoce y comprende el pronóstico, debe ofrecérsele la seguridad de que usted seguirá siendo el médico tratante, aún si el paciente decide suspender la quimioterapia agresiva. Antes del primer tratamiento inductivo, usted sabe más acerca del proceso de lo que sabe el paciente. Sin embargo, en el momento de la reinducción, el paciente ha tenido la experiencia de pasar por el tratamiento.
Es bueno permitir que el paciente sepa que el control del dolor es importante para usted y que usted desea usar medicamentos potentes para mantener al paciente sin dolor. Un corolario de esta proposición es que le puede decir al paciente que usted tiene los medios para controlar el dolor si el paciente lo desea, pero los medicamentos pueden producir aturdimiento.
Incluso después de horas de discusión y explicación con el paciente y su familia, es probable que las enfermeras o los amigos del paciente le hagan saber que el paciente opina que usted no habla lo suficiente con él o ella. No hay necesidad de sentirse ofendido. Los pacientes moribundos y sus familiares a menudo no pueden referir o evaluar información, reconocer que han recibido información, o saber apreciar el tiempo invertido. Uno puede suponer que se necesitarán múltiples conversaciones y nuevas explicaciones. Se debe pedir al paciente y a su familia que hagan listas por escrito de sus preguntas, y usted debe responder a las preguntas de forma tan simple y directa como sea posible.
Después de tomar una decisión firme acerca del tratamiento, el paciente puede de repente cambiar su posición. Es la vida del paciente y tiene derecho a hacer estos cambios, aunque él o ella no haya tenido experiencia previa tomando este tipo de decisiones.
Usted puede oír de amigos, psiquiátras, sacerdotes, o enfermeras que el paciente quiere que usted suspenda el tratamiento y deje que llegue la muerte. Con frecuencia esta información no refleja con precisión los deseos del paciente. El paciente puede estar dejando que estas ideas floten como globos de prueba, es mejor hablar directamente con él y ver si es que estos deseos surgen espontáneamente.
Los pacientes no mueren programados. Es mejor no suponer o dejar que la familia suponga que debido a que usted y el paciente han decidido suspender la quimioterapia agresiva, los antibióticos o transfusiones, el paciente morirá en breve tiempo. Todos deben estar preparados para la capacidad del cuerpo de sobrevivir a pesar de la suspensión del apoyo terapéutico. Es inteligente no dar programas o fechas al paciente ni a la familia.
El autor ha encontrado varios casos en los cuales después de que todas las opciones terapéuticas razonables han fracasado, los pacientes no aceptan la recomendación de que el tratamiento debe retirarse y que los esfuerzos futuros se centrarán en el mantenimiento de la comodidad y la calidad de vida con la familia y los amigos. En estos casos los pacientes solicitan un tratamiento más agresivo, incluso cuando se le ha señalado que ya no existe ninguno disponible y no existen protocolos experimentales en el que pudiera ingresar. Esta situación es la inversa del punto de vista más difundido donde el médico a menudo insiste en la necesidad de que el paciente continúe con el tratamiento, mientras que el paciente desea que sea suspendido. Se considera que el consejo y concordancia con el paciente puede evitar esta situación, pero en la experiencia del autor, no siempre es posible.
El paciente tiene derechos que deben reconocerse. Aún si se compromete el cuidado, es la enfermedad del paciente y él o ella tiene derecho a tomar ciertas decisiones.
- Clarkson BD: Acute myelocytic leukemia in adults. Cancer 30:1572, 1972
- Cohen JJ: Apoptosis. Immunol Today 14:126, 1993
- Sachs L, Lotem J: Control of programmed cell death in normal and leukemic cells: new implications for therapy. Blood 82:15, 1993
- MoxleyJH, Perrys, WeissGH, et al: Return of leucocytes to the bone marrow in chronic myelogenous leukaemia. Nature 208:128l. 1965
- Bunn PA Jr, Schechter GP, Jaffe E, et al: Clinical course of retrovirusassociated adult T -cell lyrnphorna in the United States. N Engl J Med 309:257,1983
- Modan B, Lubin E: Radiation induced leukernia in man. Series Haematologica 7:2:192,1974
- Boice JD Jr: The danger of x-rays-real or apparent? (editorial). N Engl J Med 315:828,1986
- Forni A, Vigliani EC: Chemical leukernogenesis in man. Series Haematologica 7:2:21l. 1974
- Aksoy M, DincoJ K, Erdern S, et al: Acute leukernia due to chronic exposure to benzene. Am J Med 52:160,1972
- Shu XO, Gao YT, Linet MS, et al: Chlorarnphenicol use and childhood leukaemia in Shanghai. Lancet 2:934,1987
- Potolsky A, Creger WP: Radiation and drug therapies, and leukernia. Annu Rev Med 24:75,1973
- Greene MH, Boice JD Jr, Greer BE, et al: Acute non-Iyrnphocytic leukernia after therapy with aIkylating agents for ovarian cancer: a study of five randornized clinical trials. N Engl J Med 307:1416,1982
- Secondary rnalignancy after treatrnent of Hodgkin's disease: an evolving picture (editorial). J Clin OncoI 4:82l. 1986
- Valagussa P, Santoro A, Fossati-Bellani F, et al: Second acute leukernia and other malignancies following treatment for Hodgkin's disease. J Clin OncoI4:830, 1986
- Gill Super HJ, McCabe NR, Thirman MJ, et al: Rearrangements of the MLL gene in therapy-related acute myeloid leukernia in patients previously treated with agents targeting DNA-topoisomerase II. Blood 82:3705,1993
- Drabkin HA: The molecular biology of chromosorne alterations in myelogenous leukemia. West J Med 143:825,1985
- Morphologic, immunologic and cytogenetic (MIC) working classification of the acute myeloid leukaernias. Second MIC Cooperative Study Group. Br J Haematol 68:487, 1988
- Gralnick HR, Galton DAG, Catovsky D, et al: Classification of acute leukernia. Ann Intern Med 87:740,1977
- Bennett JM, Catovsky D, Daniel MT , et al: Proposals for the classification of the acute leukaemias. Br J HaematoI 33:451, 1976
- Bennett JM, Catovsky D, Daniel MT, et al: Criteria for the diagnosis of acute leukemia of megakaryocyte lineage (M7): a report of the French-American-British (FAB) Cooperative Group. Ann Intem Med 103:460, 1985
- FAB M7: acute megakaryoblastic leukernia-beyond morphology (editorial). Ann Intern Med 103:450, 1985
- Foon KA, Todd RF IIl: Immunologic classification of leukemia and lymphoma. Blood 68:1,1986
- Janossy G, Robert M, Greaves MF: Target cell in chronic myeloid leukaemia and its relationship to acute lymphoid leukaemia. Lancet 2:1058,1976
- Arnold A, Cossman J, Bakhshi A, et al: Immunoglobulin gene rearrangements as unique clonal markers in human lymphoid neoplasms. N Engl J Med 309:1593,1983
- Aisenberg AC, Krontiris TG, Mak TW , et al: Rearrangement of the gene for the beta chain of the T-ce1l receptor in T-cell chronic lymphocytic leukemia and related disorders. N Engl J Med 313:529,1985
- Waldmann TA, Davis MM, Bongiovanni KF, et al: Rearrangements of genes for the antigen receptor on T cells as markers of lineage and clonality in human lymphoid neoplasms. N Engl J Med 313:776,1985
- Bertness V, Kirsch I, Hollis G, et al: T -cell receptor gene rearrangementsas clinical markers ofhuman T-cell lymphomas. N Engl J Med 313:534,1985
- Davey MP, Waldmann TA: Clonality and lymphoproliferative lesions (editorial). N Engl J Med 315:509,1986
- Griesser H, Tkachuk D, Reis MD, et al: Gene rearrangements and translocations in lymphoproliferative diseases. Blood 73:1402, 1989
- Crowther D: Blood and neoplastic diseases: a rational approach to the chemotherapy of human malignant disease-I. Br Med J 4:156,1974
- Hiddemann W, Buchner T, Andreeff M, et al: Cell kinetics in acute leukemia: a critical reevaluation based on new data. Cancer 50:250,1982
- Crowther D: Blood and neoplastic diseases: a rational approach to the chemotherapy of human malignant disease-lI. Br Med J 4:216,1974
- O'Reilly RJ: Allogeneic bone marrow transplantation: current status and future directions. Blood 62:941, 1983
- Thomas ED: Current status of marrow transplantation for aplastic anemia and acute leukemia: the Philip Levine Award lecture. Am J Clin Pathol72:887, 1979
- Thomas ED: Marrow transplantation for marrow failure or leukemia. Blood Disorders 6:69,1980
- Thomas ED: The role of marrow transplantation in the eradication of malignant disease. Cancer 49: 1963, 1982
- Gilbert H, Dameshek W: The myeloproliferative disorders. DM (October):52,1970
- Ward HP, Vautrin E, Kumick J: Presence of a myeloproliferative factor in patients with polycythemia vera and agnogenic myeloid metaplasia: I. Expansion of the erythropoietin-responsive stem cell compartment. Proc Soc Exp Biol Med 147:305,1974
- Fialkow PJ, Faguet GB, Jacobson RJ, et al: Evidence that essential thrombocythemia is a clonal disorder with origin in a multipotent stem cell. Blood 58:916,1981
- Adamson JW, Fialkow PJ: The pathogenesis of myeloproliferative syndromes. Br J HaernatoI 38:299, 1978
- Glew RH, Haese WH, McIntyre PA: Myeloid metaplasia with myelofibrosis: the clinical spectrum of extramedullary hematopoiesis and tumor formation. Johns Hopkins Med J 132:253,1973
- Roberts BE, Miles DW, Woods CG: Polycythaemia vera and myelosclerosis: a bone marrow study. Br J HaematoI16:75, 1969
- Kurzrock R, Cohen PR: Erythromelalgia: review of clinical characteristics and pathophysiology. Am J Med 91:416,1991
- Tsao M-S: Hepatic sinusoidal fibrosis in agnogenic myeloid metaplasia. Am J Clin PathoI 91:302, 1989
- Valla D, Casadevall N, LacombeC, et al: Primary myeloproliferative disorder and hepatic vein thrombosis: a prospective study of erythroid colony formation in vitro in 20 patients with Budd-Chiari syndrome. Ann Intern Med 103:329,1985
- Boughton BJ: Hepatic and portal vein thrombosis: closely associated with myeloproliferative disorders. Br Med J 302:192,1991
- O'Keane JC, Wolf BC, Neiman RS: The pathogenesis of splenic extramedullary hematopoiesis in metastatic carcinoma. Cancer 63:1539,1989
- André J, Schwartz R, Dameshek W: Tuberculosis and myelosclerosis with myeloid metaplasia: report of three cases.JAMA 178:1169, 1961
- Varki A, Lottenberg R, Griffith R, et al: The syndrome of idiopathic myelofibrosis: a clinicopathologic review with emphasis on the prognostic variables predicting survival. Medicine (Baltimore) 62:353,1983
- Jacobson RJ, Fialkow PJ: Idiopathic myelofibrosis: stem cell abnormality and probable neoplastic origin (abstr). ClinRes 24:439A, 1976
- Manoharan A: Myelofibrosis: prognostic factors and treatment. Br J HaematoI 69:295,1988
- Gilbert HS: Persistence of remission of myeloid metaplasia after treatment with recombinant interferon alpha-2b. Blood 72 (suppl):200a,1988
- Gisslinger H, Ludwig H, Linkesch W: Long-term interferon therapy for thrombocytosis in myeloproliferative diseases. Lancet 1:634,1989
- Talpaz M, Kurzrock R, Kantarjian H, et al: Recombinant interferonalpha therapy of Philadelphia chromosome-negative myeloproliferative disorders with thrombocytosis. Am J Med 86:554, 1989
- Reversible myelofibrosis? (editorial). Lancet 1:497,1985
- Ward HP, Block MH: The natural history of agnogenic myeloid metaplasia (AMM) and a critical evaluation of its relationship with the myeloproliferative syndrome. Medicine (Baltimore) 50:357,1971
- Koeffler HP, Golde DW: Chronic myelogenous leukemia-new concepts (pt 1). N Engl J Med 304:l201, 1981
- Fauser AA, Kanz L, Bross KJ, et al: T cells and probably B cells arise from the malignant clone in chronic myelogenous leukernia. J Clin Invest 75:1080,1985
- Deisseroth AB, Purohit S, Lopez AR: Chronic myelogenous leukemia-recent advances in treatment and pathogenesis. West J Med 144:338,1986
- Stam K Jr, Heisterkamp N, Grosveld G, et al: Evidence of a new chimeric bcr/c-abl mRNA in patients with chronic myelocytic leukemia and the Philadelphia chromosome. N Engl J Med 313:1429, 1985
- The molecular biology of chronic myelogenous leukaemia. Br J Haematol 60:395,1985
- Chronic leukemias: oncogenes, chromosomes, and advances in therapy. Champlin R, moderator. Ann Intern Med 104:671,1986
- Molecular biology and chronic granulocytic leukaemia (editorial). Lancet 2:666,1986
- Kurzrock R, Gutterman JU, Talpaz M: The molecular genetics of Philadelphia chromosome-positive leukemias. N Engl J Med 319:990,1988
- Pugh WC, Pearson M, Vardiman JW, et al: Philadelphia chromo-some-negative chronic myelogenous leukaemia: a morphological reassessment. Br J Haematol 60:457, 1985
- Terjanian T, Kantarjian H, Keating M, et al: Clinical and prognostic features of patients with Philadelphia chromosome-positive chronic myelogenous leukelriia and extramedullary disease. Cancer 59:297,1987
- Mehta AB, Goldham JM, Kohner E: Hyperleucocytic retinopathy in chronic granulocytic leukaemia: the role of intensive leucapheresis. Br J HaematoI 56:661, 1984
- Clift RA, Appelbaum FR, Thomas ED: Treatment of chronic myeloid leukemia by marrow transplantation (editorial). Blood 82:1954, 1993
- Dunbar CE, Stewart FM: Separating the wheat from the chaff: selection of benign hematopoietic cells in chronic myeloid leukemia. Blood 79:1107,1992
- Lion T, Henn T, Gaiger A, et al: Early detection of relapse after bone marrow transplantation in patients with chronic myelogenous leukaemia. Lancet 341:275,1993
- Kantarjian HM, Deisseroth A, Kurzrock R, et al: Chronic myelogenous leukemia: a concise update. Blood 82:691,1993
- Hehlmann R, Heimpel H, Hasford J, et al: Randomized comparison of busulfan and hydroxyurea in chronic myelogenous leukemia: prolongation of survival by hydroxyurea. Blood 82:398,1993
- Results of a prospective randomized trial of early splenectomy in chronic myeloid leukemia: the Italian Cooperative Study Group on Chronic Myeloid Leukemia. Cancer 54:333,1984
- Thomas ED, C1ift RA: Indications for marrow transplantation in chronic myelogenous leukemia. Blood 73:861,1989
- Goldman JM, Gale RP, Horowitz MM, et al: Bone marrow transplantation for chronic myelogenous leukemia in chronic phase. Ann Intern Med 108:806,1988
- Thomas ED, Clift RA, Fefer A, et al: Marrow transplantation for the treatment of chronic myelogenous leukemia. Ann Intern Med 104:155,1986
- Griffin JD: Management of chronic myelogenous leukemia. Semin Hematol 23(suppl 1):20,1986
- Kantarjian HM, Walters RS, Keating MJ, et al: Treatment of the blastic phase of chronic myelogenous leukemia with mitoxantrone and high-dose cytosine arabinoside. Cancer 62:672,1988
- Marks SM, Baltimore D, McCaffrey R: Terminal transferase as a predictor of initial responsiveness to vincristine and prednisone in blastic chronic myelogenous leukemia: a co-operative study. N Engl J Med 298:812, 1978
- Bakhshi A, Minowada J, Arnold A, et al: Lymphoid blast crises of chronic myelogenous leukemia represent stages in the development of B-cell precursors. N Engl J Med 309:1118,1983
- Spiers ASD: Blood and neoplastic diseases: chronic granulocytic leukaemia and chronic Iymphocytic leukaemia. Br Med J 4:460,1974
- Walters Rs, Kantarjian HM, Keating MJ, et al: Therapy of Iymphoid and undifferentiated chronic myelogenous leukemia in blast crisis with continuous vincristine and adriamycin infusions plus highdose decadron. Cancer 60:1708,1987
- Parreira L, Tavares de Castro J, Hibbin JA, et al: Chromosome and ceIl culture studies in eosinophilic leukaemia. Br J HaematoI62:659, 1986
- Goh KO, Ho FSC, Tso SC, et al: Is hypereosinophilic syndrome a malignant disease? Cancer 55:2395,1985
- Freireich EJ: Grounds for optimism in treating acute granulocytic leukemia (adult acute leukemia): a look at the data. Arch Intern Med 136:1375,1976
- Moore MAS, spitzer G, Williams N, et al: Agar culture studies in 127 cases of untreated acute leukemia: the prognostic value of reclassification of leukemia according to in vitro growth characteristics. Blood 44:1, 1974
- Einhom N: Acute leukemia after chemotherapy (melphalan). Cancer 41:444,1978
- lnfectious complications in haematological diseases. Bodey GP, Ed. Clin HaematoI 5:2:227, 1976
- Cohen PR, Talpaz M, Kurzrock R: Malignancy-associated sweet.s syndrome: review ofthe world literature. J Clin OncoI 6:1887, 1988
- Paietta E, Van Ness B, Bennett J, et al: Lymphoid lineage-associated features in acute myeloid leukaemia: phenotypic and genotypic correlations. Br J Haematol 82:324,1992
- Koeffler HP: syndromes of acute nonlymphocytic leukemia. Ann lntern Med 107:748,1987
- Pizzo P A: Combating infections in neutropenic patients. Hosp Pract 24(7):93, 1989
- Foon KA, Gale RP: Controversies in the therapy of acute myelogenous leukemia. Am J Med 72:963,1982
- Lazarus HM, Vogler WR, Burns CP, et al: High-dose cytosine arabinoside and daunorubicin as primary therapy in elderly patients with acute myelogenous leukemia:a phase I-II study of the southeastem Cancer study Group. Cancer 63:1055,1989
- Sorenson JT, Gerald K, Bodensteiner D, et al: Effect of age on survival in acute leukemia, 1950-1990. Cancer 72:1602,1993
- TaIlman Ms, Kopecky KJ, Amos D, et al; Analysis of prognostic factors for the outcome of marrow transplantation or further chemotherapy for patients with acute nonlymphocytic leukemia in first remission. J Clin Oncol 7:326,1989
- Preisler HD, Anderson K, Rai AK, et al: The frequency of long-term remission in patientswith acute myelogenous leukaemia treated with conventional maintenance chemotherapy: a study of 760 patients with a minimal foIlow-up time of 6 years. Br J Haematol 71:189,1989
- Bandini G, Zuffa E, Rosti G, et al: Long-term outcome of adults with acute myelogenous leukaemia: results of a prospective, randomized study of chemotherapy with minimal foIlow-up of 7 years. Br J Haematol 77:486,1991
- Fialkow PJ, Janssen JWG, Bartram CR: Clonal remissions in acute nonlymphocytic leukemia: evidence for a multistep pathogenesis of he malignancy. Blood 77:1415,1991
- Busque L, Gilliland DG: Clonal evolution in acute myeloid leukemia. Blood 82:337,1993
- Schwartz RS, Mackintosh FR, Schrier SL, et al: Multivariate analysis of factors associated with nvasive fungal disease during remission induction therapy for acute myelogenous leukemia. Cancer 53:411, 1984
- Zander AR, Keating M, Dicke K, et al: Acomparison of marrow transplantation with chemotherapy for adults with acute leukemia of poor prognosis in first complete remission. J Clin Oncol 6:1548, 1988
- Allogeneic transplantation versus intensive chemotherapy in firstremission acute leukemia: is there a "best choice?" (editorial). J Clin OncoI6:1532,1988
- Reiffers J, Gaspard MH, Maraninchi D, et al: Comparison of allogeneic or autologous bone marrow transplantation and chemotherapy in patients with acute myeloid leukaemia in first remission: a prospective controlled trial. Br J Haematol 72:57, 1989
- Chemotherapy versus transplantation in acute leukaemia (annotation). Br J HaematoI 72:1, 1989
- Intemational Bone Marrow Transplant Registry: Transplant or chemotherapy in acute myelogenous leukaemia. Lancet 1:1119, 1989
- Petersen FB, Lynch MHE, Clift RA, et al: Autologous marrow transplantation for patients with acute myeloid leukemia in untreated first relapse or in second complete remission. J Clin Oncol 11:1353, 1993
- Berman E, Heller G, Santorsa J, et al: Results of a randomized trial comparing idarubicin and cytosine arabinoside with daunorubicin and cytosine arabinoside in adult patients with newly diagnosed acute myelogenous leukemia. Blood 77:1666, 1991
- Vogler WR, Velez-Garcia E, Weiner RS, et al: A phase III trial comparing idarubicin and daunorubicin in combination with cytarabine in acute myelogenous leukemia: a Southeastem Cancer study Group study. J Clin OncoI10:ll03, 1992
- Waxman J, Terry Y, Rees LH,et al: Gonadalfunction in men treated for acute leukaemia. Br Med J 287:1093,1983
- Wolk RW, Masse SR, Conklin R, et al: The incidence of central nervous system leukemia in adults with acute leukemia. Cancer 33:863,1974
- Armentrout SA, Ulrich RF: Neurological manifestations of acute granulocytic leukemia. West J Med 121:1, 1974
- Weiss HW, Walker MD, Wiernik PH: Neurotoxicity of commonly used antineoplastic agents (pts 1 and 2). N Engl J Med 291:75, 127, 1974
- Stewart DJ, Smith TL, Keating MJ, et al: Remission from central nervous system involvement in adults with acute leukemia: effect of intensive therapy and prognostic factors. Cancer 56:632,1985
- Reardon G, Moloney WC: Chloroma and related myeloblastic neoplasms. Arch Intern Med 108:864,1961
- McKee LC Jr, Collins RD: Intravascular leukocyte thrombi and aggregates as a cause of morbidity and mortality in leukemia. Medicine (Baltimore) 53:463,1974
- Harris AL: Leukostasis associated with blood transfusion in acute myeloid leukaemia. Br Med J 1:1169,1978
- Campos L, Roualt J-P, Sabido O, et al: High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 81:3091,1993
- Marie J-P, Zittoun R, Sikic BI: Multidrug resistance (mdr 1) gene expression in adult acute leukemias: correlations with treatment outcome and in vitro drug sensitivity. Blood 78:586, 1991
- Amadori S, Arcese W, Isacchi G, et al: Mitoxantrone, etoposide, and intermediate-dose cytarabine: an effective and tolerable regimen for the treatment of refractory acute myeloid leukemia. J Clin Oncol 9:l210,1991
- Keman NA, Bartsch G, Ash RC, et al: Analysis of 462 transplantations from unrelated donors facilitated by the National Marrow Donor Program. N Engl J Med 328:593,1993
- Pedersen-Bjergaard J, Specht L, Larsen SO, et al: Risk of therapy-re-lated leukaemia and preleukaemia after Hodgkin's disease: relation to age, cumulative dose of alkylating agents, and time from chemotherapy. Lancet 2:83,1987
- Blayney DW, Longo DL, Young RC, et al: Decreasing risk of leukemia with prolonged follow-up after hemotherapy and radiotherapy for Hodgkin's disease. N Engl J Med 316:710,1987
- Schiffer CA, Lee EJ, Tomiyasu T, et al: Drastic impact of cytogenetic abnormalities in patients with de novo acute nonlymphocytic leukemia. Blood 73:263, 1989
- Champlin R, Gale RP: Acute myelogenous leukemia: recent advances in theTapy. Blood 69:1551,1987
- Larson RA, Wemli M, Le Beau MM, et al: short remission durations in therapy-related leukemia despite cytogenetic complete responses to high-dose cytarabine. Blood 72:1333,1988
- Hoyle CF, De Bastos M, Wheatley K, et al: AML associated with previous cytotoxic therapy, MDS or myeloproliferative disorders: results from the MRC's 9th AML trial. Br J HaematoI 72:45, 1989
- Geller RB, Vogelsang GB, Wingard JR, et al: successful marrow transplantation for acute myelocytic leukemia following therapy for Hodgkin's disease. J Clin OncoI 6:1558, 1988
- Warrell RP Jr, de Thé H, Wang Z-Y, et al: Acute promyelocytic leukemia. N Engl J Med 329:177,1993
- Castaigne S, Lefebvre P, Chomienne C, et al: Effectiveness and pharmacokinetics of low-dose all-trans retinoic acid (25 mg/m2 )in acute promyelocytic leukemia. Blood 82:3560, 1993
- Fenaux P, Le Deley MC, Castaigne S, et al: Effect of all transretinoic acid in newly diagnosed acute promyelocytic leukemia: results of a multicenter randomized trial. Blood 82:324l, 1993
- Acute promyelocytic leukemia (annotation). Br J Haematol 81:315, 1992
- Williams EC, Schwartz Bs, Conlan MG, et al: Treatment of coagulopathy in acute promyelocytic leukemia with epsilon amino caproic add and heparin (abstr). Blood 66(suppl1):211a, 1985
- Kantarjian HM, Keating MJ, Walters RS, et al: Acute promyelocytic leukemia: M.D. Anderson Hospital experience. Am J Med 80:789,1986
- Stone RM, Maguire ME, Goldberg MA, et al: Acute promyelocytic leukemia (APL): complete remission (CR) can occur without bone marrow aplasia (abstr). Blood 66(suppl 1):209a, 1985
- Alio NO, Janes WO: Malignant myelosclerosis (acute myelofibrosis): report of two cases following cytotoxic chemotherapy. Cancer 43:121l, 1979
- Tamura K, Preisler HD: Treatment of erythroleukemia with anthracycline antibiotics and cytosine arabinoside. Cancer 51:1795, 1983
- Olopade OI, Thangavelu M, Larson RA, et al: ClinicaI, morphologic, and cytogenetic characteristics of 26 patients with acute erythroblastic leukemia. Blood 80:2873,1992
- Gale RP, Ben-Bassat I: Hybrid acute leukaemia. Br J Haematol 65:26l, 1987
- Greenberg PL: The smoldering myeloid leukemic states: clinical and biologic features. Blood 61:1035,1983
- Prokocimer M, Polliack A: Increased bone marrow mast cells in preleukemic syndromes, acute leukemia, and lymphoproliferative disorders. Am J Clin Pathol 75:34, 1981
- Doll DC, List AF: Myelodysplastic syndromes: introduction. Semin OncoI 19:l, 1992
- Bennett JM, Catovsky D, Daniel MT, et al: Proposals for the classifcation of the myelodysplastic syndromes. Br J Haematol 51:189 , 1982
- Tricot G, de Wolf-Peeters C, Hendrickx B, et al: Bone marrow histology in myelodysplastic syndromes: I. Histological findings in myelodysplastic syndromes and comparison with bone marrow smears. Br J HaematoI57:423, 1984
- Tricot G, Boogaerts MA, de Wolf-Peeters C, et al: The myelodysplastic syndromes: different evolution patterns based on sequential morphological and cytogenetic investigations. Br J HaematoI 59:659,1985
- Goasguen JE, Bennett JM: Classificat:ion and morphologic features of the myelodysplastic syndromes. Semin OncoI 19:4, 1992
- Foucar K, Langdon RM II, Armitage JO, et al: Myelodysplastic syndromes: a clinical and pathologic analysis of 109 cases. Cancer 56:553,1985
- Coiffier B, Adeleine P, Gentilhomme O, et al: Myelodysplastic syndromes: a multiparametric study of prognostic factors in 336 patients. Cancer 60:3029,1987
- Knapp RH, Dewald GW, Pierre RV: Cytogenetic studies in 174 consecutive patients with preleukemic or myelodysplastic syndromes. Mayo Clin Proc 60:507, 1985
- Nimer SD, Golde DW: The 5q- abnormality. Blood 70:1705,1987
- Mathew P, Tefferi A, Dewald GW, et al: The 5q- syndrome: a single-institution study of 43 consecutive patients. Blood 81:1040, 1993
- Larson RA: Management of myelodysplastic syndromes. Ann Intern Med 103:136,1985
- Le Beau MM, Albain Ks, Larson RA, et al: Clinical and cytogenetic correlations in 63 patients with therapy-related myelodysplastic syndromes and acute nonlymphocytic leukemia: further evidence for characteristic abnormalities of chromosomes no.5 and 7. J Clin OncoI 4:325,1986
- Greene MH, Harris EL, Gershenson DM, et al: Melphalan may be a more potent leukemogen than cyclophosphamide. Ann Intern Med 105:360,1986
- Kantarjian HM, Keating MJ, Walters RS, et al: Therapy-related leukemia and myelodysplastic syndrome: clinical, cytogenetic, and prognostic features. J Clin OncoI4:1748, 1986
- Cohen JR, Creger WP , Greenberg PL, et al: Subacute myeloid leukemia. Am J Med 66:959, 1979
- Tsukamoto N, Morita K, Maehara T, et al: Clonality in myelodysplastic syndromes: demonstration of pluripotent stem cell origin using X-linked restriction fragment length polymorphisms. Br J Haematol 83:589, 1993
- Bone marrow transplantation for myelodysplastic syndrome and secondary leukaemias (annotation). Br J Haematol 84:36l, 1993
- Tricot G, Boogaerts MA: The role of aggressive chemotherapy in the treatment of the myelodysplastic syndromes. Br J HaematoI 63:477 ,1986
- Management of myelodysplastic syndromes (clinical annotation). Br J Haematol 84:19l, 1993
- Negrin RS, Haeuber DH, Nagler A, et al: Maintenance treatment of patients with myelodysplastic syndromes using recombinant human granulocyte colony-stimulating factor. Blood 76:36, 1990
- Negrin RS, Stein R, Vardiman J, et al: Treatment of the anemia of myelodysplastic syndromes using recombinant human granulocyte colony-stimulating factor in combination with erythropoietin. Blood 82:737,1993
- Fialkow PJ, Najfeld V, Reddy AL, et al: Chronic lymphocytic leukaemia: clonal origin in a committed B-lymphocyte progenitor. Lancet 2:444,1978
- Gale RP, Foon KA: Chronic lymphocytic leukemia. Ann Intem Med 103:10l, 1985
- Miller RH, Linet MS, Van Natta ML, et al: Serum protein electrophoresis pattems in chronic lymphocytic leukemia: clinical and epidemiologic correlations. Arch Intern Med 147:1614, 1987
- Dighiero G, Travade P, Chevret S, et al: B-cell chronic lymphocyticleukemia: present status and future directions. Blood 78:190l, 1991
- Brouet J-C, Preud'homme J-L, Seligmann M, et al: Blast cells with monoclonal surface immunoglobulin in two cases of acute blast crisis supervening on chronic lymphocytic leukaemia. Br Med J 4:23,1973
- Ghani AM, Krause JR, Brody JP: Prolymphocytic transformation of chronic lymphocytic leukemia: a report of three cases and review of the literature. Cancer 57:75,1986
- Sweet DL, Golomb HM, Ultmann JE: Chronic lymphocytic leukaemia and its relationship to other lymphoproliferative disorders. Clin HaematoI 6:14l, 1977
- Intemational Workshop on Chronic Lymphocytic Leukemia: Chronic lymphocytic leukemia: recommendations for diagnosis, staging, and response criteria. Ann Intern Med 110:236,1989
- Orfao A. Gonzalez M, San Miguel JF, et al: B-cell chronic lymphocytic leukaemia: prognostic value of the immunophenotype and the clinicohaematological features. Am J HematoI 31:26, 1989
- Rai KR, Sawitsky A, Cronkite EP, et al: Clinical staging of chronic lymphocytic leukemia. Blood 46:219,1975
- Binet J-L, Catovsky D, Chandra P, et al: Chronic lymphocytic leukaemia: proposals for a revised prognostic staging system. Br J Haematol 48:365,1981
- Bartl R, Frisch B, Burkhardt R, et al: Assessment of marrow trephine in relation to staging in chronic lymphocytic leukaemia. Br J Haematol 51:1, 1982
- Rozman C, Montserrat E, Vinolas N: Serum immunoglobulins in [beta]-chronic lymphocytic leukemia. Cancer 61:279,1988
- Mandelli F, De Rossi G, Mancini P, et al: Prognosis in chronic lymphocytic leukemia: a retrospective multicentric study from the GIMEMA group. J Clin OncoI 5:398, 1987
- Griffiths H, Brennan V, Lea J, et al: Crossover study of immunoglobulin replacement therapy in patients with low-grade B-cell tumors. Blood 73:366, 1989
- Cooperative Group for the Study of Immunoglobulin in Chronic Lymphocytic Leukemia: Intravenous immunoglobulin for the prevention of infection in chronic lymphocytic leukemia: a randomized, controlled clinical trial. N Engl J Med 319:902,1988
- Sawitsky A, Rai KR, Aral I et al: Mediastinal irradiation for chronic lymphocytic leukemia. Am J Med 61:892,1976
- Effectiveness of "CHOP" regimen in advanced untreated chronic lymphocytic leukaemia: French Cooperative Group on Chronic Lymphocytic Leukaemia. Lancet 1:1346,1986
- A randomized comparison of no treatment versus dailychlorambucil in 612 patients with stage A CLL. French Cooperative Group on CLL. Blood 72(suppl):191a, 1988
- Anaissie E, Kontoyiannis DP, Kantarjian H, et al: Listeriosis in patients with chronic lymphocytic leukernia who were treated with fludarabine and prednisone. Ann Intern Med 117:466, 1992
- Dillman RO, Mick R, McIntyre OR: Pentostatin in chronic lymphocytic leukernia: a phase II trial of cancer and leukemia group B. J Clin OncoI7:433,1989
- Juliusson G, Elmhom-Rosenborg A, Liliemark J: Response to 2-chorodeoxyadenisine in patients with B-cell chronic lymphocytic leukemia resistant to fludarabine. N Engl J Med 327:1056,1992
- Juliusson G, Liliemark J: High complete remission rate from 2- chloro-2'-deoxyadenosine in previously treated patients with B-cell chronic lymphocytic leukemia: response predicted by rapid derease of blood lymphocyte count. J Clin Oncoll 1:679, 1993
- Delpero JR, Gastaut JA, Letreut YP, et al: The value of splenectomy in chronic lymphocytic leukemia. Cancer 59:340, 1987
- Melo JV, Catovsky D, Galton DAG: The relationship between chronic lymphocytic leukaernia and prolymphocytic leukaernia: I. Clinical and laboratory features of 300 patients and characterization of an intermediate group. Br J Haematol 63:377, 1986
- Robertson LE, Pugh W, O'Brien S, et al: Richter's syndrome: a report on 39 patients. J Clin Oncoll 1:1985, 1993
- Catovsky D: Hairy-cell leukaernia and prolymphocytic leukaemia. Clin HaematoI 6:245, 1977
- Bennett C, Vardiman J, Golomb H: Disseminated atypical mycobacterial infection in patients with hairy cell leukemia. Am J Med 80:891,1986
- Farcet JP, Weschsler J, Wirquin V, et al: Vasculitis in hairy-cell leukemia. Arch Intern Med 147:660,1987
- Gómez-Almaguer D, Herrera-Garza JL, Garcia-Guajardo BM, et al: Vasculitis in hairy-cell leukernia: rapid response to interferon alpha. Am J HematoI 30:261, 1989
- Saven A, Piro LD: Treatment of hairy cell leukemia. Blood 79:1111, 1992
- Winkler CF, Sausville EA, Ihde DC, et al: Combined modality treatment of cutaneous T cell lymphoma: results of a 6-year follow-up. J Clin OncoI 4:1094, 1986
- Sausville EA, Eddy JL, Makuch RW, et al: Histopathologic staging at initial diagnosis of mycosis fungoides and the Sézary syndrome. Ann Intem Med 109:372, 1988
- Whittaker SJ, smith NP, Russell Jones R, et al: Analysis of [beta], [gamma], and [delta] T -cell receptor genes in mycosis fungoides and Sezary syndrome. Cancer 6B:1572, 1991
- Neely SM: Adult T-cell leukemia-lymphoma. West J Med 150:557, 1989
- Shimoyama M, Ota K, Kikuchi M, et al: Major prognostic factors of adult patients with advanced T-cell lymphoma/leukemia. J Clin OncoI 6:1088,1988
- Brunning RD: T-prolymphocytic leukemia. Blood 78:3111, 1991
- Matutes E, Brito-Babapulle V, swansbury J, et al: Clinical and laboratory features of 78 cases of T -prolymphocytic leukemia. Blood 78:3269,1991 .
- Dillman RO, Mick R, McIntyre OR: Pentostatin in chronic lymphocytic leukemia: a phase II trial of cancer and leukemia group B. J Clin OncoI 7:433,1989
- Loughran TP Jr: Clonal diseases of large granular lymphocytes. Blood 82:1, 1993
- Chan WC, Winton EF, Waldmann T A: Lymphocytosis of large granular lymphocytes. Arch Intern Med 146:1201,1986
- Loughran TP Jr, Kadin ME, Starkebaum G, et al: Leukemia of large granular lymphocytes: association with clonal chromosomal abnormalities and autoimmune neutropenia, thrombocytopenia, and hemolytic anemia. Ann Intern Med 102:169,1985
- Levitt LJ, Reyes GR, Moonka DK, et al: Human T cell leukemia virus-I-associated T -suppressor cell inhíbition of erythropoiesis in a patient with pure red cell aplasia and chronic T[gamma]-Iymphoproliferative disease. J Clin Invest 81:538,1988
- Jaffe ES, Costa J, Fauci AS, et al: Malignant lymphoma and erythrophagocytosis simulating malignant histiocytosis. Am J Med 75:741,1983
- Trends in lymphoma diagnosis. Lancet 1:249,1989
- Alexander M, Daniels JR: Chemotherapy of malignant histiocytosis in adults. Cancer 39:1011, 1977
- Angioimmunoblastic lymphadenopathy with dysproteinemia. NIH Conference. Steinberg ED,moderator. Ann Intern Med 108:575,1988
- Siegert W, Agthe A, Griesser H, et al: Treatment of angioimmunoblastic lymphadenopathy (AILD)-type T -cell lymphoma using prednisone with or without the COPBLAM/IMVP-16 regi- men. Ann Intern Med 117:364,1992
- Champlin R, Gale RP: Acute lymphoblastic leukemia: recent advances in biology and therapy. Blood 73:265,1989
- Kantarjian HM, Walters RS, Smith TL, et al: Identification of risk groups for development of central nervous system leukemia in adults with acute lymphocytic leukemia. Blood 72:1784,1988
- Hoelzer D, Thiel E, Löffler H, et al: Prognostic factors in a multicenter study for treatment of acute lymphoblastic leukemia in adults. Blood 71:123, 1988
- Fenaux P, Lai JL, Miaux O, et al: Burkitt cell acute leukaemia (L3 ALL) in adults: a report of 18 cases. Br J HaematoI 71:371, 1989
- Gaynor J, Chapman D, Uttle C, et al: A cause-specific hazard rate analysis of prognostic factors among 199 adults with acute lymphoblastic leukemia: the Memorial Hospital experience since 1969. J Clin OncoI 6:1014, 1988
- Acute lymphoblastic leukemia-progress in children, less in adults (editorial). N Engl J Med 329:1343,1993
- The challenge of acute lymphoblastic leukaemia in adults (annotation). Br J Haematol 85:641, 1993
- Proctor SJ, Hamilton PJ, Taylor P, et al: A comparative study of combination chemotherapy versus marrow transplant in first remission in adult acute lymphoblastic leukaemia. Br J Haematol 69:35,1988
- Kersey JH, Weisdorf D, Nesbit ME, et al: Comparison of autologous and .allogeneic bone marrow transplantation for treatment of highrisk refractory acute lymphoblastic leukemia. N Engl J Med 317:461,1987
- Kreuser ED, Hetzel WD, Heit W, et al: Reproductive and endocrine gonadal functions in adults following multidrug chemotherapy for acute lymphoblastic or ndifferentiated leukemia. J Clin Oncol 6:588, 1988