Contenido del artículo
II TRASTORNOS DEL EQUILIBRIO ACIDO-BASE Y DEL POTASIO
- Trastornos ácido-base
- FISIOLOGIA ACIDO-BASE NORMAL
- ACIDOSIS METABOLICA
- Brecha aniónica en la acidosis metabólica
- Acidosis metabólica con brecha aniónica aumentada
- Acidosis metabólica con brecha aniónica normal
- Desequilibrio del potasio en la acidosis metabólica
- Diagnóstico
- Tratamiento
- ALCALOSIS METABOLICA
- ACIDOSIS Y ALCALOSIS RESPIRATORIA
- Trastornos del potasio plasmático
DR. ROBERT M. BLACK
Trastornos ácido-base
El pH sanguíneo se mantiene en condiciones normales entre 7.38 y 7.42. Cualquier desviación de este rango indica un cambio en la concentración de iones hidrógeno [H+] porque el pH sanguíneo es el logaritmo negativo de la [H+], como lo expresa la siguiente ecuación:
La [H+] a un pH sanguíneo fisiológico de 7.40 es de 40 nEq/L [ver figura 1]. El aumento en la [H+] (disminución del pH) se llama acidemia. La disminución en la [H+] (aumento en el pH sanguíneo) se denomina alcalemia. Los trastornos que causan estos cambios en el pH sanguíneo son la acidosis y la alcalosis, respectivamente. Debido a que las alteraciones del metabolismo ácido-base se asocian con frecuencia con desequilibrio del potasio, también se analizan en esta sección el estudio clínico de la hipocalemia y la hipercalemia.
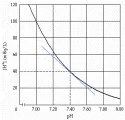
|
| Figura 1 |
| Relación entre [H+] y pH en sangre. |
FISIOLOGIA ACIDO-BASE NORMAL
La dieta de un adulto normal contiene un exceso de 70 a 100 mEq de ácido que deben eliminarse todos los días. Si esto no sucede se produce una caída persistente en la concentración plasmática de bicarbonato (HCO3-), que se expresa en la siguiente ecuación:
|
anhidrasa carbónica |
||||||||
| H+ | + | HCO3- | ¬¾® | H2CO3 | ¬¾¾¾® | CO2 | + | H2O |
La adición de iones hidrógeno dirige la reacción a la derecha, disminuyendo la concentración de bicarbonato en plasma [HCO3-] y aumentando la presión arterial de bióxido de carbono (PaCO2). Como se muestra en la siguiente ecuación, la reducción en la [HCO3-] en plasma aumenta la [H+], disminuyendo el pH sanguíneo:
| [H+] | = | 24 | x | PaCO2 | / | [HCO3-] |
También es aparente en esta ecuación que la pérdida de bicarbonato por el tubo digestivo o los riñones causa que la [H+] aumente.
Reabsorción renal de bicarbonato
La [HCO3-] en plasma es, en condiciones normales, de alrededor de 25 mEq/L. Si la carga filtrada diario de bicarbonato (alrededor de 4,500 mEq) no se reabsorben la [HCO3-] en plasma disminuye, lo mismo que el pH sanguíneo. Por lo tanto, la conservación de una [HCO3-] en plasma normal requiere que virtualmente todo el bicarbonato que se filtra en el glomérulo se reabsorba.
El túbulo contorneado proximal reabsorbe el 85 a 90 por ciento del bicarbonato filtrado [ver figura 2]; por el contrario, en el túbulo contorneado distal se reabsorbe muy poco. Esta diferencia se debe a la mayor cantidad de anhidrasa carbónica luminal en el túbulo proximal.

|
| Figura 2 |
| Reabsorción tubular proximal de bicarbonato |
Excreción renal de ácido
Además de reabsorber todo el bicarbonato filtrado, los riñones excretan la carga diaria de ácido, derivado principalmente de los aminoácidos que contienen azufre. Los iones hidrógeno que se excretan en la orina final son secretados principalmente en los túbulos colectores [ver figura 3]. Este proceso secretorio es facilitado por la aldosterona.

|
| Figura 3 |
| Secreción de H+ en el túbulo colector cortical y reabsorción de Na+. |
Debido a que se requiere un pH en la orina mínimo de alrededor de 4.0 para que las bombas H+-ATPasa de los túbulos colectores secreten iones hidrógeno hacia la luz tubular, los iones se combinan con amortiguadores antes de su eliminación. Existen varios amortiguadores urinarios, de los que el más importante es el amoníaco porque es el único que puede aumentar en forma sustancial en presencia de una carga de ácido.1 La limitación en la capacidad para generar una cantidad adecuada de amoníaco en la orina, como ocurre en la insuficiencia renal, suele provocar acidosis.
El principal sitio de producción de amoníaco en el riñón es el túbulo proximal1 [ver figura 4], el amoníaco se mueve de este sitio al túbulo colector, en donde es eliminado [ver figura 5]. La cantidad de amoníaco producido depende tanto de la acidemia como de la hipocalemia. Por el contrario, la alcalemia y la hipercalemia inhiben la producción de amoníaco.
|
|

Se presenta acidosis metabólica siempre que exista una disminución primaria en la [HCO3-] del plasma. Esta reducción puede deberse a varios factores: administración exógena de ácido, producción endógena, menor secreción renal de hidrógeno y pérdida de bicarbonato por el aparato digestivo o el riñón. El cálculo de la brecha aniónica (anión gap) en el plasma es de gran utilidad para identificar la causa específica de la acidosis metabólica y disminuir el diagnóstico diferencial.
Brecha aniónica en la acidosis metabólica
La brecha aniónica (medida en mEq/L) se refiere a la diferencia que existe entre la concentración plasmática del principal catión mesurable (sodio) y los principales aniones mesurables (cloruro y bicarbonato), según lo muestra la siguiente ecuación:
| Brecha aniónica | = | [Na+] | - | ( [HCO3-] + [Cl-] ) |
El valor normal de la brecha aniónica varía entre 3 y 13 mEq/L, con un promedio de 7 a 10 mEq/L.2 Está compuesta principalmente de proteínas del plasma (en especial albúmina) que portan una carga negativa. Por lo tanto, en los casos de hipoalbuminemia la brecha aniónica basal puede ser menor de 3 mEq/L.
El uso más importante de la brecha aniónica es para identificar la causa de la acidosis metabólica. Los trastornos que causan acidosis metabólica pertenecen a dos categorías: (1) los que producen reducción en la [HCO3-] en el plasma con aumento concomitante de la brecha aniónica y (2) los que causan acidemia con brecha aniónica normal. En este último caso la concentración de cloruro en plasma aumenta para sustituir al bicarbonato depletado.
Acidosis metabólica con brecha aniónica aumentada
Varios padecimientos, así como la ingesta de toxinas, pueden causar acidosis metabólica con aumento de la brecha aniónica [ver tabla 1].
|
||
*Puede también asociarse con acidosis con brecha aniónica normal. |
Insuficiencia renal La insuficiencia renal grave es la causa más frecuente de acidosis metabólica con brecha aniónica aumentada. En este caso, la retención de iones hidrógeno disminuye la [HCO3-] en plasma. Debido a que el sulfato y el fosfato (que son los aniones acompañantes) se excretan en la orina mientras que el cloruro se conserva para mantener la electroneutralidad, la brecha aniónica es normal durante las primeras etapas del padecimiento. Al progresar la falla renal (en forma típica cuando la creatinina plasmática llega a ser de 3.0 mg/dl), estos aniones ingeridos no pueden ya ser excretados en forma normal. En este momento aumenta la brecha aniónica. Por lo tanto, no existe una correlación lineal entre el grado de acidemia (o hiperbicarbonatemia) y el nivel de la brecha aniónica. Sin embargo, en la insuficiencia renal no complicada, la [HCO3-] en plasma pocas veces disminuye a menos de 12 mEq/L y es característico que la brecha aniónica sea menor de 23 mEq/L.
Acidosis láctica La acidosis láctica es la causa más frecuente de acidosis con brecha aniónica aumentada en los pacientes hospitalizados. La producción de ácido láctico suele aumentar como resultado de hipotensión o sepsis, y ambas causan isquemia tisular verdadera o relativa. Las vías oxidativas del metabolismo del piruvato se alteran en forma importante en estados de disfunción mitocondrial, como las inducidas por hipoxemia tisular. Esto aumenta la conversión de piruvato a lactato.
El hígado, y en menor grado el riñón, son los principales órganos que eliminan lactato de la circulación. En ambos órganos el lactato es convertido de nuevo a piruvato y después a bióxido de carbono y agua a través del ciclo del ácido tricarboxílico. El metabolismo del lactato (CH3CHOHCOO-) en estos órganos regenera con rapidez el bicarbonato que se usa al principio para neutralizar la carga de ácido, un proceso que es independiente de la excreción renal de ácido y que se representa en la siguiente ecuación:
| CH3CHOHCOO- | + | 302 | ¾® | 2CO2 | + | 2H2O | + | HCO3- |
La velocidad normal del metabolismo del lactato puede llegar hasta 320 mEq/hr (v.gr., durante el ejercicio), y ésta suele ser mayor que la velocidad de producción de lactato en la acidosis láctica. Estos hallazgos sugieren que para que se acumule ácido láctico debe alterarse algún componente del metabolismo del lactato. Por ejemplo en el estado de choque, la reducción tan importante en la perfusión hepática podría disminuir el metabolismo del lactato.
La brecha aniónica y la concentración de lactato casi siempre son mayores que lo normal en la acidosis láctica. Debido a que el umbral de excreción de lactato es de 6 a 8 mEq/L y la concentración normal de este ácido es de 1 mEq/L, se acumula lactato en exceso en la sangre antes de que se elimine en la orina.
La acidosis láctica-D es una forma poco común de acidosis láctica que se observa principalmente en pacientes que han sido sometidos a derivación ileal o resección del intestino delgado y que ocurre en raras ocasiones en los que reciben tratamiento antibiótico.3,4 En cada uno de estos casos puede ocurrir síndrome de intestino corto, causando aumento en el metabolismo bacteriano de los carbohidratos a ácido láctico-D por proliferación local. En la acidosis láctica-D la brecha aniónica aumenta al principio pero cae con el tiempo porque la reabsorción renal tubular de lactato-D es ineficaz en comparación con la reabsorción de lactato-L. Las pruebas estándar de lactato-L, que usan deshidrogenasa de lactato-L, no miden el ácido láctico-D y por lo tanto indican una concentración de lactato normal. Se requiere un ensayo enzimático específico para lactato-D para confirmar el diagnóstico [ver adelante, Diagnóstico].
Cetoacidosis La acidosis causada por sobreproducción de los cetoácidos (o cuerpos cetónicos) ácido acetoacético y ácido beta-hidroxibutírico ocurre cuando el ayuno, la deficiencia o la resistencia a la insulina alteran el uso de la glucosa.5 En estas circunstancias se producen cuerpos cetónicos en exceso (una condición denominada cetosis), que sirven como fuente alterna de energía en muchas células.
La cetona que se forma en un principio es el ácido acetoacético, que puede ser reducido a ácido beta-hidroxibutírico o decarboxilado en forma no enzimática a acetona. Aunque la acetona es químicamente neutra, las otras cetonas son ácidos orgánicos y su acúmulo causa acidosis metabólica. La siguiente ecuación resume las reacciones:
| Ácido acetoacético | ¬¾® | ácido beta-hidroxibutírico | ¾® | acetona |
En la cetoacidosis aumenta la brecha aniónica en forma característica. El acetoacetato y el beta-hidroxibutirato son los principales aniones no mesurables que se acumulan, aunque puede observarse acidosis láctica concomitante en algunos pacientes. En la cetoacidosis diabética, la [HCO3-] en plasma puede estar muy disminuida. Por el contrario, la acidemia suele ser leve y el bicarbonato en plasma pocas veces es menor a 18 mEq/L en la cetosis del ayuno.
El remplazo de líquido isotónico en la cetoacidosis causa cetonuria. Aunque al principio aumenta la brecha aniónica en la cetoacidosis, esta comienza a disminuir cuando la excreción urinaria de acetoacetato y beta-hidroxibutirato es mayor a la velocidad de producción. Al final esto normaliza la brecha aniónica y el cloruro sustituye los aniones cetónicos perdidos. Debido a que las cetonas urinarias perdidas no regeneran bicarbonato, la [HCO3-] en plasma permanece disminuida.
La tendencia a una acidosis metabólica con brecha aniónica normal durante la recuperación de la cetoacidosis tiene un efecto importante en la velocidad a la que puede corregirse la acidemia. Mientras las cetonas estén circulando y exista insulina, el acetoacetato y el beta-hidroxibutirato pueden ser metabolizados a bicarbonato en los tejidos no hepáticos, como el músculo. Sin embargo, una vez que estos aniones se eliminan por completo a través de la orina, la regeneración de bicarbonato requerirá de la excreción renal de iones hidrógeno. Este proceso, que depende del aumento en la producción de amoníaco, requiere por lo menos 2 a 3 días para operar al máximo. Por lo tanto, la resolución de la acidemia ocurre con más rapidez en pacientes con brecha aniónica más alta (y mayores niveles circulantes de acetoacetato y beta-hidroxibutirato) cuando se comienza tratamiento con insulina.
Cualquier dosis de insulina que reduzca la glucemia disminuirá el metabolismo de las cetonas porque la concentración de insulina que inhibe la lipólisis por el tejido adiposo es mucho menor que la concentración requerida para la utilización de la glucosa. Aunque el metabolismo de los cetoácidos circulantes restantes ocurre con rapidez, la eliminación de la acetona, que ocurre en parte por los pulmones, es un proceso más lento. Por lo tanto, pueden persistir niveles positivos de cetonas en sangre y orina por más de 24 horas, que no necesariamente indican cetogénesis persistente.
Rabdomiolisis La destrucción masiva de músculo es una causa importante de acidosis metabólica.6 La retención de ácidos metabólicos y de aniones inorgánicos como el fosfato parecen contribuir al aumento de la brecha aniónica. Es más probable que se desarrolle acidosis metabólica cuando existe falla renal asociada.
Agentes y toxinas ingeridos Dentro de la categoría de los agentes y toxinas ingeridos, los salicilatos y los alcoholes etilenglicol (componente de los anticongelantes y solventes) y metanol, o el alcohol de madera (componente de lacas, barnices, soluciones descongelantes y otras preparaciones comerciales), son las causas más frecuentes de acidosis metabólica. La ingestión de alcohol etílico no se asocia con acidosis con brecha aniónica aumentada a menos que se añadan acidosis láctica o cetoacidosis.
La alteración ácido-base más frecuente que se observa con la ingestión de salicilatos en los adultos es la alcalosis respiratoria causada por estimulación directa del centro respiratorio en la médula [ver adelante, Acidosis y alcalosis respiratoria]. En los adultos, la presencia de alcalosis respiratoria parece ser un prerrequisito para el desarrollo de acidosis metabólica porque es rara la acidosis metabólica aislada. Por otro lado, la intoxicación moderada a severa con salicilatos causa un trastorno mixto, alcalosis respiratoria y acidosis metabólica con brecha aniónica aumentada. Otros datos clínicos que pueden observarse con la intoxicación severa por salicilatos incluyen tinnitus, hiperpirexia, vasodilatación que provoca choque y edema periférico o pulmonar.
Los alcoholes metanol y etilenglicol pueden producir intoxicación fatal. Ambos aumentan la brecha osmolar plasmática, que se requiere a la diferencia entre la osmolaridad plasmática (Posm) medida por el laboratorio y la calculada por la siguiente fórmula:
| Posm calculada | = | (2 x [Na+] en plasma) | + | (concentración de glucosa/18) | + | (nitrógeno de urea en sangre/2.8) |
Una brecha osmolar alta (con nivel medido por lo menos 10 mOsm/kg mayor que el valor calculado) puede detectarse solo cuando la Posm se mide por depresión del punto de congelamiento. Por el contrario, la contribución osmótica de los alcoholes volátiles no se incluye cuando se usa un osmómetro de presión de vapor, que supone que solo el agua está en fase de vapor.7 Aún más, la presencia de una brecha osmolar no es específica de esta intoxicación y la confirmación del diagnóstico requerirá de pruebas en plasma para las sustancias individuales [ver adelante, Diagnóstico].
Acidosis metabólica con brecha aniónica normal
Varios trastornos pueden causar acidosis metabólica con brecha aniónica normal, incluyendo la administración de ácidos inorgánicos como ácido hidroclórico, la pérdida de bicarbonato y la menor excreción renal de iones hidrógeno [ver tabla 2]. En estos casos, el bicarbonato plasmático disminuye y es reemplazado por cloruro para mantener la electroneutralidad. En consecuencia, en ocasiones se denomina a estos padecimientos como acidosis hiperclorémicas.
|
|
*Causas raras de acitosis metabólica. |
Administración de ácido y cloruro La infusión de soluciones de aminoácidos durante la nutrición parenteral contiene gran cantidad de ácido clorhídrico. En ciertos casos la administración de una solución de cloruro de sodio puede disminuir también la concentración de bicarbonato en el plasma por dilución (una condición denominada acidosis de expansión), causando reducción en el pH de la sangre.8
Pérdida de bicarbonato El bicarbonato puede perderse del organismo por el tubo digestivo o por el riñón. Comparado con la sangre, el contenido intestinal es alcalino porque las secreciones pancreáticas y biliares agregan bicarbonato, que después es intercambiado por cloruro en el íleon y colon para mantener un equilibrio ácido-base. La pérdida de bicarbonato por el tubo digestivo (o de sus precursores como lactato y acetato) se observa principalmente en pacientes con diarrea severa. Con menor frecuencia la acidosis metabólica es causada por pérdida de bases por fístulas pancreáticas, drenaje biliar o derivaciones urinarias al colon o intestino delgado.9 También ocurre después del trasplante de páncreas en pacientes que pierden bicarbonato a través de anastomosis páncreas-vejiga.10
El diagnóstico de que la diarrea es la causa de la acidosis con brecha aniónica normal suele ser evidente por la historia clínica [ver adelante, Diagnóstico]. La presencia de hipocalemia por las pérdidas de potasio en las evacuaciones apoya el diagnóstico.
Las pérdidas de bicarbonato por el riñón causan la acidemia que se observa en la acidosis tubular renal (ATR) proximal (o tipo 2) y en los pacientes poshipocápnicos. En la ATR proximal el umbral normal para la reabsorción de bicarbonato está disminuido. Por lo tanto, el bicarbonato no puede reabsorberse a la velocidad adecuada para mantener su concentración plasmática de alrededor de 25 mEq/L. Durante esta fase ocurre bicarbonaturia, causando un pH urinario mayor de 5.3. Sin embargo, la pérdida de bicarbonato cesa una vez que la [HCO3-] en plasma se estabiliza a una concentración menor. En este momento el pH urinario puede ser menor de 5.
La hipocapnia disminuye la reabsorción de bicarbonato en el túbulo contorneado proximal. Después de 1 a 3 días la concentración plasmática de bicarbonato disminuye. Debido a que este proceso de adaptación renal requiere del mismo tiempo para dejar de funcionar, el aumento súbito en la PaCO2 no modifica de inmediato la reabsorción renal de bicarbonato. Esta acidosis metabólica poshipocápnica se resuelve en forma espontánea en 24 a 72 horas.
Reducción en la excreción renal de iones hidrógeno Se observa menor excreción renal de iones hidrógeno en tres padecimientos: insuficiencia renal, ATR distal (tipo 1) y ATR tipo 4 (hipoaldosteronismo) [ver tablas 3 y 4]. La acidosis de la insuficiencia renal es causada principalmente por reducción del número de nefronas. Por el contrario, la ATR tipo 1 se caracteriza por reducción en la excreción renal de ácido por cada nefrona. Debido a que la cantidad total de amoníaco que puede sintetizarse también disminuye en la insuficiencia renal, el pH urinario es menor de 5.3 en la mayoría de los pacientes.
|
||||
* Puede causar también ATR tipo [ver tabla 5]
|
|
|||||||
* Puede ocurrir adrenalitis que cause ATR tipo 4 en personas seropositivas para VIH.
|
La ATR tipo 1 ocurre principalmente cuando los iones hidrógeno no pueden ser bombeados hacia el exterior de las células intercaladas alfa y hacia la luz del túbulo contorneado distal [ver figura 3]. Como resultado, la orina no se acidifica al máximo y el pH urinario casi siempre es mayor de 5.3. Aún más, es característico que se asocie hipocalemia clásica por un mayor intercambio Na+-K+ en la nefrona distal, proceso que es necesario para mantener el equilibrio del sodio debido a que no pueden secretarse iones hidrógeno en respuesta a la reabsorción de sodio. También se ha descrito una forma hipercalémica deATR tipo 1, que ocurre principalmente en pacientes que tienen obstrucción de las vías urinarias. A diferencia de la ATR tipo 4 (ver adelante), este trastorno causa incapacidad para acidificar la orina al máximo (pH urinario > 5.3).
La complicación clínica más importante de la ATR tipo 1 hipocalémica es la formación y depósito de sales de fosfato de calcio, que pueden formar cálculos en todo el riñón (nefrocalcinosis). El principal factor que contribuye a la formación de los cristales es la hipocitraturia, que es resultado de aumento en la reabsorción de citrato en el túbulo proximal causado por la acidosis metabólica. Además, la hipocalemia promueve la hipocitraturia.11 Debido a que el citrato de calcio es mucho más soluble que el fosfato de calcio, la disminución en el citrato urinario facilita la precipitación de cristales de fosfato de calcio en la luz del túbulo colector.
La ATR tipo 4 puede ser causada por diferentes medicamentos, incluyendo antinflamatorios no esteroides (AINE), inhibidores de la enzima convertidora de angiotensina (ECA), bloqueadores de la angiotensina II, ciclosporina y heparina. Sin embargo, se observa con más frecuencia en pacientes con diabetes mellitus [ver adelante, Hipercalemia].
La alteración electrolítica más frecuente en la ATR tipo 4 es la hipercalemia ([K+] en plasma > 5.0 mEq/L), que es causada por la menor reabsorción luminal de iones sodio, con reducción en la secreción secundaria de iones de potasio por las células principales del túbulo colector [ver figura 3]. A diferencia de la hipocalemia, la hipercalemia altera la producción de amoniaco.12 La reducción del amortiguador urinario amoniaco limita la secreción de iones hidrógeno en el túbulo colector. A pesar de esta menor capacidad para excretar iones hidrógeno, la orina formada al final está acidificada al máximo (pH < 5.3). Este hallazgo, aparentemente paradójico, ocurre porque la cantidad limitada de amortiguador permite que la [H+] libres exceda la capacidad secretora de las células intercaladas -alfa, limitando un mayor transporte de iones hidrógeno hacia el exterior y hacia el túbulo colector. A diferencia de los pacientes con ATR tipo 1 (ver antes) los enfermos con ATR tipo 4 pueden excretar cierta cantidad de ácido, aunque en cantidades inadecuadas. En consecuencia, el pH urinario suele ser menor de 5.3 en los pacientes con esta enfermedad.
Desequilibrio del potasio en la acidosis metabólica
Es frecuente observar hipercalemia en los pacientes con acidosis metabólica, y también se presenta en enfermos con acidosis respiratoria, aunque en este caso el desplazamiento de potasio es menor. La reducción de la [HCO3-] en plasma provoca un intercambio de iones hidrógeno extracelulares por iones potasio intracelulares. Este mecanismo de defensa permite que los iones hidrógeno sean neutralizados dentro de las células, mientras que el intercambio H+-K+ mantiene la electroneutralidad. Por lo tanto, el paciente que tiene diarrea con acidosis metabólica con brecha aniónica normal y una [K+] en plasma baja tiene mucho mayor depleción de potasio que el paciente con la misma [K+] en plasma pero con un pH sanguíneo normal.
A diferencia de los pacientes con acidosis metabólica y brecha aniónica normal, los movimientos de potasio causados por la acidemia son menos importantes en los enfermos con acidosis orgánica endógena (v.gr., acidosis láctica y cetoacidosis).13 Esto no debe implicar que la hipercalemia sea poco frecuente en los pacientes con estos padecimientos. Por ejemplo, en la acidosis láctica el catabolismo celular permite la liberación de potasio de las células, y en la cetoacidosis la deficiencia de insulina limita la entrada de este ión al intracelular y la hiperglucemia promueve la salida de potasio de las células. En ambos trastornos la [K+] en plasma puede estar aumentada desde la llegada del paciente, aunque la hipercalemia no indica el grado de la acidemia.
Diagnóstico
Manifestaciones clínicas La respiración de Kussmaul sugiere la presencia de acidosis metabólica. Un aumento en el volumen corriente, más que en la frecuencia respiratoria, causa este cambio en la ventilación, que es resultado de la estimulación del sistema respiratorio en el tallo cerebral por el descenso en el pH sanguíneo. Al aumentar la acidemia puede existir náusea y vómito o cambios en el nivel de conciencia, incluyendo estado de coma.
También puede observarse hipotensión secundaria en los individuos con acidemia grave. En estos casos el descenso en la presión arterial se debe a depresión de la contractilidad miocárdica y a vasodilatación arterial, que son inducidos por la reducción del pH en sangre. Al inicio, el aumento en las concentraciones de catecolaminas circulantes contrarrestan los efectos cardiovasculares de la acidemia; sin embargo, con un pH en sangre menor de 7.15 a 7.20, pueden predominar los efectos de la acidemia.14 Pueden ocurrir arritmias por reentrada y reducción en el umbral para la fibrilación ventricular, mientras que el umbral de desfibrilación no se altera.
Los síntomas y signos de la acidosis láctica son característicamente los del padecimiento subyacente que causó el trastorno [ver antes, Acidosis láctica]. Con frecuencia los pacientes tienen evidencia de hipoperfusión, como hipotensión arterial y extremidades frías o marmóreas. Con menos frecuencia la causa es un medicamento (como el metformin).15
Los síntomas de la acidosis causados por la ingestión de metanol o etilenglicol [ver antes, Agentes ingeridos y toxinas] pueden desarrollarse 12 a 36 horas después de la ingestión. Además de los cambios ácido-base, los síntomas iniciales que ocurren después de la ingestión del metanol incluyen debilidad, nausea, cefalea y disminución en la visión, que pueden progresar a ceguera, coma y muerte. El examen de fondo de ojo puede revelar edema retiniano. Después de que se ingiere etilenglicol los primeros datos consisten en alteraciones neurológicas que varían desde ebriedad hasta estado de coma. Si el paciente no es tratado estos cambios pueden ser seguidos por síntomas cardiopulmonares (taquipnea y edema pulmonar) y después por dolor en el flanco e insuficiencia renal debida a depósito de cristales de oxalato de calcio, que pueden observarse en el sedimento urinario.
Varios trastornos se han asociado con la ATR tipo 2 [ver tabla 5]. La mayoría de los adultos con este padecimiento presentan también otras alteraciones en la reabsorción tubular proximal, como glucosuria, aminoaciduria y fosfaturia, conocidas en conjunto como síndrome de Fanconi. Por el contrario, la administración de un inhibidor de la anihidrasa carbónica, como la acetazolamida, se asocia solo con bicarbonaturia.
|
||||||||||||||
*Suele asociarse con ATR tipo 1 clásica o hipercalémica [ver tabla 3] |
Pruebas de laboratorio El diagnóstico de la acidosis metabólica se realiza con relativa facilidad ante la presencia de disminución del pH en sangre y de la [HCO3-] en plasma. La medición de la brecha aniónica puede utilizarse para identificar la causa específica del trastorno (ver antes).
La defensa respiratoria contra la acidemia comienza de inmediato aunque puede no llegar a su máxima intensidad hasta 12 a 24 horas después. Por una fórmula matemática, la respuesta respiratoria esperada (nivel al que puede disminuir la PaCO2) en la acidosis metabólica es igual a los dos últimos dígitos del pH sanguíneo (v.gr., 24 si el pH en sangre es de 7.24). La respuesta respiratoria es inadecuada cuando la PaCO2 es mayor que el valor esperado y excesiva cuando es menor. Cuando ocurren estas respuestas inadecuadas o excesivas existe una acidosis o alcalosis respiratoria, respectivamente, agregada [ver adelante, Acidosis y alcalosis respiratoria].
El diagnóstico de diarrea como causa de acidosis metabólica con brecha aniónica normal suele ser aparente por la historia del paciente y la presencia de hipocalemia (ver antes). Puede observarse un perfil de electrolitos semejante en los pacientes con ATR tipo 1. Estos trastornos suelen poder distinguirse por el pH en la orina, la orina tiende a ser ácida (pH < 5.3) en pacientes con diarrea y alcalina (pH> 5.3) en pacientes con ATR tipo 1. Sin embargo, en algunas personas con diarrea la orina puede ser alcalina, quizá porque la producción de amoníaco (inducida por la hipocalemia) aumenta a tal grado que el amortiguador urinario se produce en mayor cantidad que los iones hidrógeno secretados. El cálculo de la brecha aniónica urinaria, como se muestra en la siguiente ecuación, puede ser muy útil en estas circunstancias:
| Brecha aniónica urinaria | = | ( [Na+) urinario + [K+] urinario ) | - | [Cl-] urinario |
Siempre que los iones hidrógeno secretados se excreten como cloruro de amoníaco ocurre excreción urinaria de cloruro. El aumento en la excreción urinaria de cloruro disminuye la brecha aniónica urinaria, causando una brecha negativa en la mayoría de los pacientes con diarrea. Por comparación, en la ATR tipo 1 la brecha aniónica urinaria es positiva.
Tratamiento
El tratamiento de la acidosis metabólica debe dirigirse a corregir tanto la acidemia como el padecimiento subyacente. La posibilidad de que se requiera la administración de alcalinos y de que ésta sea eficaz depende del pH sanguíneo, de los mecanismos compensadores y del padecimiento subyacente.
Hasta que el pH en sangre arterial cae por debajo de 7.15 a 7.20, los efectos adversos de la acidemia suelen ser compensados por un aumento en la concentración de catecolaminas en plasma [ver antes, Diagnóstico]. Para mantener las reservas adecuadas de amortiguadores, debe utilizarse el tratamiento con alcalinos para conservar una [HCO3-] mayor de 10 a 12 mEq/L. Sin embargo, por lo general no se necesita administrar alcalinos cuando es probable que la acidosis se resuelva en forma espontánea (v.gr., acidosis láctica después de una crisis convulsiva tipo gran mal). Más adelante se analizan aspectos importantes que tratan de las causas específicas de acidosis metabólica.
Insuficiencia renal crónica Mientras la acidosis metabólica sea leve, la mayor parte de los adultos con insuficiencia renal reciben tratamiento con álcalis, en parte por la preocupación de que el bicarbonato de sodio aumente la expansión de volumen y la hipertensión que suelen coexistir.
Sin embargo, estudios recientes han sugerido varias razones por las que este tratamiento podría ser conveniente.16 Entre ellas están la posibilidad de que la acidemia pueda aumentar la destrucción del músculo esquelético,17 disminuir la síntesis de albúmina,18 y (por activación del sistema de complemento) contribuir a la lesión tubulointersticial, además de que el amortiguamiento de los iones hidrógeno por parte del hueso aumente la resorción ósea. Estos datos han hecho que algunos médicos sugieran el uso temprano de alcalinos para mantener la [HCO3-] por arriba de 22 mEq/L.19 Sin embargo, aún se requieren estudios definitivos para comprobar los beneficios de este tratamiento.
Acidosis láctica La corrección del padecimiento subyacente constituye el tratamiento primario de la acidosis láctica. El alivio de la falla circulatoria, de la hipoxemia o de la sepsis reduce la velocidad de producción de lactato y aumenta su eliminación.
Como en el caso de la insuficiencia renal crónica (ver antes), el uso de alcalinos en este padecimiento es motivo de controversia. El beneficio potencial de la administración de bicarbonato es principalmente mantener la homeostasis cardiovascular. Esta posible ventaja debe ser comparada con los efectos deletéreos, como sobrecarga de volumen, hipernatremia y alcalosis (cuando se administra bicarbonato en cantidad excesiva).
Los estudios clínicos sugieren también que el tratamiento con bicarbonato de sodio no mejora ni el pH sanguíneo ni la supervivencia de los pacientes con acidosis láctica. Se ha observado que la administración de bicarbonato de sodio disminuye la función cardiaca en los pacientes con cardiopatía, insuficiencia cardiaca congestiva o infarto agudo del miocardio. Estudios experimentales sugieren que esta falta de eficacia es consecuencia de un aumento asociado en la producción neta de ácido láctico, aunque la hiperosmolaridad de la solución alcalina podría también ser importante.20 El hallazgo de que el dicloroacetato puede disminuir los niveles de lactato y aumentar el pH sanguíneo en pacientes con acidosis láctica sin mejorar la supervivencia apoya el concepto de que tratar la causa subyacente de la acidosis láctica es más importante que tratar la acidemia.21
Debido a estas observaciones, una conducta razonable sería administrar bicarbonato sólo para mantener el pH en sangre arterial por arriba de 7.15 y el bicarbonato en plasma por arriba de 10 mEq/L. Si aparece alguna de las complicaciones del tratamiento con bicarbonato habrá de revalorarse el beneficio de continuar el tratamiento con alcalinos.
Cetoacidosis El tratamiento inicial de la cetoacidosis diabética con volúmenes muy grandes de líquidos ha sido puesto en duda. Después de que se ha restablecido el volumen intravascular, las ventajas de la administración intensiva de líquidos pueden ser limitadas porque la expansión de volúmenes causa la excreción de acetoacetato y beta-hidroxibutirato en la orina. Al parecer es efectivo lograr una tasa máxima de 500 ml de líquido por hora.
Las velocidades mayores de administración de líquidos pueden causar reducción de la brecha aniónica sin aumentar la [HCO3-] en plasma. Cuando esto sucede, la corrección de la acidosis requerirá de la regeneración de bicarbonato por el riñón, proceso que puede tardar 3 o más días. Esto contrasta con el rápido aumento en la [HCO3-] en plasma que puede lograrse cuando las cetonas se metabolizan a bicarbonato en los tejidos extrahepáticos.22 Como resultado, la administración vigorosa de líquido puede retrasar la recuperación de la acidosis, y la velocidad de administración debe disminuirse después de que se ha corregido la deficiencia intravascular (que se manifiesta por hipotensión arterial o aumento en la concentración de creatinina o nitrógeno ureico en sangre).
Aunque ocurren excepciones, la administración de bicarbonato de sodio casi nunca es necesaria en la cetoacidosis, y parece que no existen diferencias en la mortalidad en los pacientes tratados con bicarbonato de sodio y los sujetos controles.23 Sin embargo, cuando existe hipercalemia severa la administración de bicarbonato de sodio puede ser benéfica porque el bicarbonato lleva potasio a las células, disminuyendo así la concentración del mismo en plasma. Debe administrarse una solución salina hipotónica (0.45%) sin bicarbonato a los pacientes menos enfermos.24
ATR tipo 1 La acidemia en la ATR tipo 1 puede corregirse con bicarbonato o con un precursor de éste, como el citrato. Los requerimientos habituales son de 1 a 3 mEq/kg/día. La corrección de la acidemia disminuye la reabsorción tubular de citrato, lo que aumenta su excreción urinaria y reduce la tendencia hacia la nefrolitiasis y la nefrocalcinosis. Por lo general se administra en forma de una sal de potasio, como citrato de potasio, porque esta también reemplaza la deficiencia de potasio. El tratamiento de la acidemia permite el crecimiento en los niños y promueve la curación del raquitismo y osteomalacia, cuando existen.25
ALCALOSIS METABOLICA
La alcalosis metabólica primaria se caracteriza por aumento en la [HCO3-] en plasma y un pH arterial mayor de 7.40. Sin embargo, cuando existe acidosis metabólica concomitante, el pH en sangre puede estar aumentado, disminuido o ser normal. Aún más, la hipercarbonatemia por sí sola no es diagnóstica de alcalosis metabólica primaria, porque ésta puede representar también una respuesta fisiológica adecuada ante una acidosis respiratoria crónica (ver adelante). Por lo general, estas condiciones pueden distinguirse con facilidad al medir el pH en sangre arterial, que está disminuido en la acidosis respiratoria.
Etiología
La alcalosis metabólica es un problema clínico relativamente común que suele ser inducido por tratamiento diurético o por pérdida de las secreciones gástricas como resultado de vómito o succión nasogátrica. La elevación en la [HCO3-] en plasma puede ser resultado de pérdida de hidrógeno, movimiento de hidrógeno al interior de las células, administración y retención de alcalinos o contracción del volumen plasmático alrededor de una cantidad relativamente constante de bicarbonato extracelular (alcalosis por contracción [ver tabla 6].
|
||||||||||
* Estas son las causas mas comunes
|
Pérdida gastrointestinal de hidrógeno La pérdida de iones hidrógeno puede ocurrir a través de las secreciones gastrointestinales o en la orina. Cada 1 mEq de hidrógeno perdido genera 1 mEq de bicarbonato.
El jugo gástrico contiene una alta concentración de ácido clorhídrico y menor cantidad de cloruro de potasio. En las personas normales la secreción de hidrógeno en el estómago no causa alcalosis metabólica porque está equilibrada con la secreción pancreática de bicarbonato, que es estimulada al entrar el ácido al duodeno. Sin embargo, no existe un estímulo para la secreción de bicarbonato cuando el vómito o una sonda nasogástrica evitan que los iones hidrógeno lleguen al duodeno. El vómito puede no ser evidente en algunos casos, como con los pacientes con trastornos de la alimentación.
Pérdida renal de hidrógeno Puede ocurrir un aumento inapropiado en la pérdida renal de ácido cuando existe aumento en la secreción de iones hidrógeno en las nefronas distales. La aldosterona actúa aquí estimulando en forma directa la bomba secretora H+-ATPasa y haciendo la luz tubular más electronegativa por medio de la estimulación de la reabsorción del sodio [ver figura 6]. La secreción distal de potasio también aumenta en estos casos y provoca hipocalemia concomitante.

|
| Figura 6 |
| Función de las células intercaladas en la alcalosis metabólica |
La hipersecreción primaria de aldosterona puede causar alcalosis metabólica, generalmente acompañada por hipertensión e hipocalemia. Por el contrario, los pacientes no tratados con aldosteronismo secundario causado por insuficiencia cardiaca congestiva o cirrosis suelen no presentar alcalosis metabólica o hipocalemia. En estos casos, el efecto de la aldosterona es contrarrestado por el menor aporte distal de sodio (a menos que se administren diuréticos) y el menor volumen de orina. Estos factores limitan la cantidad de ácido y potasio secretados y excretados en la orina final. Sin embargo, si se administra un anión no reabsorbible (como la ticarcilina en dosis altas), se requiere pérdida tanto de ión potasio como de ión hidrógeno para mantener la electroneutralidad. Esto causa alcalosis metabólica hipocalémica.
Cuando se trata a los pacientes con diuréticos tiacida o de asa, ocurre tanto llegada adecuada distal de cloruro de sodio como mayor secreción de aldosterona. El aumento en la secreción distal de iones hidrógeno y la contracción de volumen, si la diuresis ha sido abundante, contribuyen al desarrollo de la alcalosis metabólica.
La acidosis respiratoria crónica causa un aumento apropiado en la secreción de hidrógeno, lo mismo que el aumento en la [HCO3-] en plasma aumenta el pH hacia lo normal. La reducción rápida en la PaCO2, por lo general por ventilación mecánica, causa alcalosis metabólica, ya que el paciente queda con una [HCO3-] en plasma elevada. Esta alteración se denomina alcalosis metabólica poshipercápnica.
La hipercalemia aumenta la reabsorción tubular renal de bicarbonato. Sin embargo, se observa alcalosis metabólica significativa en los pacientes hipercalémicos con síndrome de leche-álcali. En este síndrome la mayor carga de alcalinos (causada por la ingestión de bicarbonato de calcio) y la insuficiencia renal inducida por la hipercalemia aumentan la producción de bicarbonato y disminuyen su excreción.26,27
Desplazamiento de iones hidrógeno La hipocalemia es un dato frecuente en pacientes con alcalosis metabólica. Tanto el vómito como el tratamiento diurético inducen pérdida directa tanto de potasio como de hidrógeno. La hipocalemia produce un desplazamiento transcelular en el que el potasio deja las células para repletar las reservas extracelulares. Para mantener la electroneutralidad entra hidrógeno a las células. Este desplazamiento no solo aumenta el pH extracelular sino que también disminuye el pH intracelular, lo que promueve reabsorción tubular proximal de bicarbonato y secreción distal de iones hidrógeno.
Administración de alcalinos La administración de bicarbonato de sodio en dosis tan altas como 1,000 mEq/día no induce alcalosis metabólica en las personas normales porque el exceso de bicarbonato es excretado con rapidez en la orina. Sin embargo, si se altera la capacidad para excretar bicarbonato, ocurre alcalosis metabólica cuando se administra en forma aguda una gran cantidad de bicarbonato o de sus precursores (v.gr., lactato, citrato o acetato), como ocurre con el citrato en las transfusiones de sangre en gran cantidad.
Alcalosis por contracción La alcalosis por contracción se desarrolla cuando ocurre pérdida de volúmenes relativamente grandes de líquido sin bicarbonato. En este caso aumenta la [HCO3-] en plasma porque el volumen extracelular se contrae alrededor de una cantidad relativamente constante de bicarbonato.
La causa más común de alcalosis por contracción es la administración de un diurético de asa para inducir eliminación rápida de líquido en un paciente con edema importante. En forma similar, la alcalosis por contracción ocurre en otras condiciones en las que se pierde líquido con alta concentración de cloruro y baja de [HCO3-]. Entre estos casos se encuentra el uso de diuréticos tiacídicos, la pérdida de secreciones gástricas (incluso en pacientes con aclorhidria), la pérdida por sudor en pacientes con fibrosis quística y la diarrea en algunos pacientes con adenomas vellosos o cloridorrea congénita, un padecimiento raro caracterizado por un defecto específico en la reabsorción intestinal de cloruro y la secreción de bicarbonato.28,29
Patogenia
Deben existir dos alteraciones para que se desarrolle y mantenga la alcalosis metabólica. Primero, debe ocurrir un aumento inicial en la [HCO3-] en plasma causada por pérdida de hidrógeno en las secreciones gastrointestinales o la orina, movimiento de hidrógeno hacia las células, administración de alcalinos o alcalosis por contracción. Segundo, debe existir uno de tres factores (en ausencia de insuficiencia renal avanzada) para mantener la [HCO3-] alta una vez que ha terminado el evento inicial: depleción real del volumen circulante, depleción de cloruro e hipocloremia o hipocalemia.
Depleción real del volumen Tanto la reducción en la velocidad de filtración glomerular (VFG) como la avidez por sodio asociada con la hipovolemia limitan la excreción de bicarbonato de sodio. La mayoría del bicarbonato se reabsorbe en el túbulo proximal [ver figura 2]. Un estímulo importante para la mayor reabsorción en este segmento de la nefrona es la mayor actividad de la compuerta Na+-H+ en las membranas de las células tubulares. La contracción del volumen promueve el intercambio de Na+-H+ en este segmento de la nefrona, en parte por liberación de angiotensina II. Los iones hidrógeno secretados hacia la luz se combinan con el bicarbonato filtrado, causando una mayor tasa de trasnporte inverso hacia las células tubulares. Una cantidad aún mayor de bicarbonato regresa a la sangre venosa a nivel de los túbulos colectores, en parte por la influencia del aldosteronismo secundario.
Depleción de cloruro El vómito y el tratamiento con diuréticos se asocian con pérdida tanto de hidrógeno como de cloruro. La depleción de cloruro puede promover la regeneración de bicarbonato y disminuir su secreción distal.30 La generación de bicarbonato en las células intercaladas alfa en el túbulo colector cortical está mediada por secreción de iones hidrógeno a través de bombas H+-ATPasa en la membrana luminal [ver figura 6]. Se requiere de cosecreción pasiva de cloruro para mantener la electroneutralidad cuando el bicarbonato intracelular regresa a la circulación sistémica.
Por el contrario, las células intercaladas beta en el túbulo colector cortical (que aumentan en número cuando se desarrolla alcalosis metabólica) son capaces de secretar bicarbonato en forma directa al revertir la localización de los transportadores [ver figura 6]. Por lo tanto los sistemas de intercambio Cl- -HCO3- se localizan en la membrana luminal, causando secreción de bicarbonato hacia la luz tubular. Las bombas de H+-ATPasa se localizan en la membrana basolateral. Aunque la actividad de estas células aumenta en forma apropiada por la alcalemia en un intento por excretar el exceso de bicarbonato, la caída asociada en la concentración tubular de cloruro disminuye el gradiente favorable para el ingreso de cloruro, disminuyendo así la secreción de bicarbonato.
Hipocalemia La hipocalemia aumenta en forma directa la reabsorción de bicarbonato por lo menos por dos mecanismos diferentes. Primero, la caída en la [K+] en plasma desplaza potasio de las células e hidrógeno hacia su interior. La acidosis intracelular resultante estimula la secreción de hidrógeno y la reabsorción de bicarbonato en los túbulos proximales y colectores.
Segundo, la secreción distal de hidrógeno y potasio está mediada por un intercambio por sodio en la luz [ver figura 6]. En estado de depleción de potasio la velocidad de secreción de hidrógeno que se intercambia por sodio aumenta. Como resultado, la hipocalemia y el hiperaldosteronismo, que estimulan la secreción de iones hidrógeno, pueden tener un efecto potenciador en el desarrollo y mantenimiento de la alcalosis metabólica.31
Diagnóstico
El diagnóstico de la alcalosis metabólica suele ser evidente por la historia de vómito del paciente o administración de diuréticos. Sin embargo, en algunos casos la causa no es aparente. En estas circunstancias el diagnóstico más probable es vómito subrepticio causado por un trastorno de la alimentación, uso de diuréticos o una de las causas del exceso de mineralocorticoides (como aldosteronismo primario). Los primeros dos factores inducen depleción eficaz de volumen, mientras que el aldosteronismo suele asociarse con expansión leve de volumen como resultado del efecto estimulador de la aldosterona sobre la reabsorción renal de sodio.
Concentraciones urinarias de sodio y cloruro La medición de la concentración de sodio en una muestra de orina aleatoria (UNa) se usa en muchas condiciones para distinguir entre la depleción de volumen (UNa < 20 mEq/L) y la euvolemia (UNa > 40 mEq/L). Sin embargo, la alcalosis metabólica es una de las condiciones en las que la depleción de volumen puede no causar una UNa baja. La capacidad para retener sodio en estos casos puede ser antagonizada por la necesidad de excretar bicarbonato (como sal de sodio) en un intento por corregir la alcalosis.
Es más probable que ocurra pérdida de sodio durante los primeros días del vómito, cuando la [HCO3-] en plasma y, por lo tanto, el bicarbonato filtrado están aumentados. En las fases iniciales la capacidad para aumentar la reabsorción de bicarbonato no se ha establecido. Los efectos netos son una UNa alta, alta concentración de potasio en orina y un pH urinario mayor de 7.0 causado por la bicarbonaturia.
Como resultado, la UNa no es necesariamente un reflejo exacto del estado volumétrico del paciente en la alcalosis metabólica. La presencia de hipovolemia subyacente puede detectarse en forma más exacta al encontrar una concentración urinaria de cloruro de menos de 25 mEq/L. La conservación adecuada de cloruro es causada tanto por depleción de volumen como por hipocloremia inducida por pérdidas de cloruro en las secreciones gástricas. Sin embargo, la concentración de cloruro en la orina puede estar inapropiadamente elevada si existe un defecto en la reabsorción de cloruro. Este tipo de defectos ocurre con más frecuencia en pacientes que reciben tratamiento diurético y es temporal, ya que el cloruro se conserva en forma apropiada una vez que cesa el efecto del fármaco. Por lo tanto, en los pacientes con alcalosis metabólica causada por vómito la concentración de cloruro en la orina es típicamente baja y es mayor cuando se administran diuréticos. En ambas circunstancias la UNa puede estar elevada.
Síndrome de Bartter y síndrome de Gitelman El síndrome de Bartter es un trastorno raro que causa alcalosis metabólica hipocalémica. Debido a que la UNa y la concentración de cloruro en orina suelen ser mayores de 25 mEq/L, el uso subrepticio de diuréticos es el principal trastorno a considerar en el diagnóstico diferencial. El síndrome de Bartter clásico suele presentarse a inicios de la vida y puede asociarse con retraso mental y del crecimiento. El espectro de datos, incluyendo hipercalciuria, es compatible con un defecto primario en la reabsorción de cloruro de sodio en la porción gruesa medular de la rama ascendente del asa de Henle.32
El síndrome de Gitelman es una condición más benigna que puede heredarse pero puede no diagnosticarse hasta finales de la niñez o incluso la edad adulta. A diferencia de los pacientes con síndrome de Bartter, que tienen un defecto en la capacidad de concentración urinaria (se requiere de la función normal del asa de Henle para generar un gradiente intersticial osmótico alto), los pacientes con síndrome de Gitelman pueden tener capacidad de concentración urinaria normal e hipocalciuria. Este dato sugiere que el defecto puede residir en el túbulo distal.33 El síndrome de Bartter y el síndrome de Gitelman se diagnostican solo después de haber excluido una causa mucho más común, el uso de diuréticos.
Tratamiento
En los pacientes con depleción real de volumen causada por vómito, succión nasogástrica, adenomas vellosos o tratamiento diurético, la alcalosis metabólica puede corregirse por la administración de cloruro de sodio.34 El aumento resultante en la excreción de bicarbonato de sodio es causado por dos eventos principales que ocurren en los túbulos colectores35 (menor generación de bicarbonato y aumento en su secreción), así como por una reducción en la reabsorción de bicarbonato en el túbulo proximal. La administración de cloruro de potasio a los pacientes con hipocalemia concomitante contribuye también a la corrección de la alcalemia.
Estados edematosos El tratamiento es diferente para los pacientes que tienen edema causado por insuficiencia cardiaca, cor pulmonale o enfermedad hepática avanzada. En estos trastornos el cloruro de sodio está contraindicado porque aumenta el grado de edema. La administración del inhibidor de la anhidrasa carbónica acetazolamida, puede ser especialmente eficaz. Este fármaco inhibe en forma preferencial la reabsorción tubular proximal de bicarbonato de sodio, corrigiendo así tanto la alcalosis como la sobrecarga de líquido.
Un efecto adverso potencial del tratamiento con un inhibidor de la anhidrasa carbónica es el desarrollo o empeoramiento de la hipocalemia. Aunque la hipocalemia puede tratarse con suplementos de potasio, un enfoque alternativo consiste en administrar un diurético ahorrador de potasio (v.gr., espironolactona) en lugar de un inhibidor de la anhidrasa carbónica. Los diuréticos ahorradores de potasio alteran la reabsorción de sodio en los túbulos colectores y, como resultado, limitan aún más la secreción de potasio e hidrógeno [ver figura 6]. En los pacientes con hepatopatía avanzada la espironolactona puede ser el diurético más eficaz. En raras ocasiones la alcalosis metabólica puede ser tan severa que se requiere de la administración de ácido clorhídrico por una vena central o de diálisis36 para corregir el problema.
Síndrome de Bartter y síndrome de Gitelman El defecto tubular en los pacientes con síndrome de Bartter o síndrome de Gitelman no puede corregirse. Como resultado, el tratamiento está dirigido a minimizar las alteraciones de electrolitos y metabólicas. La combinación de un AINE (porque los niveles de prostaglandinas están aumentados en forma secundaria) y un diurético ahorrador de potasio puede mejorar la concentración plasmática de potasio y revertir en gran parte la alcalosis metabólica.37 Sin embargo, la mayoría de los pacientes requieren de suplementos continuos de potasio y magnesio por vía oral porque el tratamiento farmacológico rara vez es totalmente eficaz.
ACIDOSIS Y ALCALOSIS RESPIRATORIA
La ventilación alveolar proporciona el oxígeno necesario para el metabolismo oxidativo y elimina el bióxido de carbono producido por estos procesos metabólicos. Por lo tanto es adecuado que el principal estímulo fisiológico de la respiración sea la reducción en la PaO2 (denominada hipoxemia) y el aumento en la PaCO2. El bióxido de carbono estimula la ventilación principalmente en las áreas quimiosensibles del centro respiratorio localizado en la médula que responden a los cambios en el pH del intersticio cerebral causados por el bióxido de carbono. Por el contrario, el aumento de la ventilación mediado por la hipoxia depende principalmente de los quimiorreceptores de los cuerpos carotídeos, que están localizados cerca de la bifurcación de las arterias carótidas. Diversos padecimientos pueden ser responsables de la acidosis y alcalosis respiratorias aguda y crónica [ver tablas 7 y 8].
|
||||||||||||
|
|
||||||||
* La alcalosis respiratoria es el trastorno inicial, aunque después se desarrolla acidosis metabólica si la intoxicación es grave. |
Manifestaciones clínicas
La acidosis respiratoria grave puede causar diversas alteraciones neurológicas. Los síntomas iniciales incluyen cefalea, visión borrosa, inquietud y ansiedad, que pueden progresar a temblor, asterixis, delirio y un estado de somnolencia, llamado narcosis por bióxido de carbono. Algunas de estas manifestaciones, incluyendo el papiledema, son causadas por el aumento en la circulación sanguínea cerebral inducido por la acidemia. En general, estos signos parecen resultar de la reducción en el pH del líquido cefalorraquídeo y no de cambios en el pH arterial o de la PaCO2.
Los síntomas producidos por la alcalosis respiratoria se relacionan con mayor irritabilidad de los sistemas nerviosos central y periférico e incluyen sensación de cabeza ligera, alteración de la conciencia, parestesias de las extremidades y del área peribucal, calambres, espasmo carpopedal (indistinguible del causado por hipocalcemia) y síncope. En los pacientes graves pueden ocurrir diversas arritmias. Se piensa que estas alteraciones se relacionan con la capacidad de la alcalosis para reducir la circulación sanguínea cerebral y aumentar la excitabilidad de la membrana. La disminución en el calcio o magnesio ionizados también contribuye a la excitabilidad de la membrana.
Tratamiento
Por lo general, la acidosis respiratoria aguda es consecuencia de una falla respiratoria, por lo que está indicado el tratamiento, pudiendo requerir el paciente ventilación mecánica. Por el contrario, el tratamiento de la alcalemia respiratoria suele no ser necesario, y la evaluación del paciente debe dirigirse a establecer el diagnóstico y corregir la causa subyacente.
Trastornos del potasio plasmático
El potasio es el principal catión intracelular. Solo alrededor del 2 por ciento del potasio corporal se localiza en el espacio extracelular, en donde su concentración (3.5 a 5.0 mEq/L) es mucho menor que en el interior de las células (123 a 140 mEq/L). Esta diferencia de concentración es mantenida por la bomba ATPasa de Na+-K+, que transporta en forma activa iones sodio hacia el exterior y iones potasio hacia el interior en la mayor parte de las células. A pesar de la relativamente pequeña cantidad de potasio extracelular, los cambios discretos en su concentración pueden tener efectos importantes sobre la contracción muscular y la conducción nerviosa porque la diferencia en la concentración de potasio intra y extracelular es el principal determinante de la excitabilidad de la membrana.
HOMEOSTASIS DEL POTASIO
La ingesta dietética de potasio varía entre 40 y 120 mEq/día en los Estados Unidos. En condiciones normales, alrededor del 90 por ciento del potasio ingerido se excreta por la orina y el resto se elimina en las heces. Aunque las pérdidas de potasio gastrointestinales aumentan en los pacientes con insuficiencia renal, el significado de esta adaptación no es clara aún.38
Después de la administración de una carga de potasio oral o intravenosa, sólo alrededor del 50 por ciento del potasio aparece en la orina durante las primeras 4 horas. Podría ocurrir hipercalemia potencialmente grave si el resto del potasio se localiza en el líquido extracelular, porque este volumen es sólo de 14 L en el varón de 70 kg. El transporte de la mayor parte de este potasio hacia el interior de las células antes de su excreción en la orina disminuye el riesgo de aumento en la concentración plasmática de este ión.
Los dos factores principales que intervienen en el transporte de potasio hacia el interior de las células son la insulina y la estimulación adrenérgica beta2. La insulina estimula a la bomba ATPasa de Na+-K+, aumentando la velocidad de entrada de potasio. En forma semejante, la activación de los receptores adrenérgicos beta2 promueve el movimiento de potasio del plasma hacia las células. La aldosterona es la hormona que influye más en la secreción de potasio por las superficies epiteliales, incluyendo la secreción por las células epiteliales del túbulo renal. Al parecer la aldosterona es menos importante en el transporte de potasio en otro tipo de células.
Regulación renal de la secreción de potasio
El potasio se filtra en forma libre por el glomérulo, por lo que la concentración de iones que entran al túbulo proximal es de alrededor de 4 mEq/L, idéntica a la [K+] en plasma. Para el momento en que el filtrado glomerular alcanza el túbulo distal, el 90 por ciento del potasio filtrado se ha reabsorbido. Por lo tanto, la excreción renal de potasio ocurre casi en forma exclusiva por secreción en el túbulo colector.39
La secreción de potasio en el túbulo colector se lleva a cabo en las células principales [ver figura 3]. El movimiento de los iones potasio de la célula tubular hacia la luz es controlada por (1) la ingesta de potasio en la dieta, (2) la reabsorción de sodio inducida por aldosterona, que genera un gradiente eléctrico negativo, permitiendo que el potasio pase hacia la luz, y (3) el flujo urinario distal, que mantiene un mayor gradiente de concentración entre la célula tubular y la luz al eliminar el potasio secretado.
La aldosterona producida en las glándulas suprarrenales entra a la célula principal a través de la superficie antiluminal o capilar. Una vez dentro, la hormona se fija a receptores que aumentan el número de canales abiertos de sodio en la membrana luminal de la célula. También aumenta el número y actividad de la bomba ATPasa de Na+-K+ en la membrana celular. El aumento subsecuente en el potasio celular ocasiona su secreción hacia la luz del túbulo colector a favor de un gradiente de concentración y electroquímico.
En casos de disminución de potasio, la secreción por las células principales se reduce (siempre y cuando el riñón no sea el sitio de pérdida del ión), y se estimula su reabsorción. Este último proceso ocurre en las células intercaladas adyacentes [ver figura 3].
HIPOCALEMIA
Etiología
La hipocalemia, que puede definirse como una [K+] en plasma menor de 3.5 mEq/L, puede ser causada por baja ingesta, desplazamiento de potasio hacia las células o pérdida de potasio del organismo. En la mayor parte de los individuos las pérdidas de potasio a través del tubo digestivo, piel o riñones son las responsables de la reducción en la [K+] en plasma.
Baja ingesta de potasio La depleción de potasio causada por ingesta inadecuada es rara porque el potasio es abundante en la mayoría de los alimentos. Aún más, si la ingesta disminuye, las pérdidas urinarias e intestinales pueden reducirse a menos de 15 mEq/día.
Sin embargo, en algunas zonas rurales dos factores dietéticos pueden combinarse para producir depleción de potasio. Primero, la ingesta de potasio puede ser sólo de 25 mEq/día en esta población, en parte porque los alimentos que contienen potasio son relativamente caros. Segundo, la ingesta crónica de arcilla, que no es rara en zonas rurales del sureste de los Estados Unidos, fija el potasio en el intestino, limitando su absorción.
Distribución alterada de potasio Incluso en presencia de reservas normales de potasio en el organismo, varios padecimientos pueden causar transporte de potasio hacia el interior de las células y así disminuir la [K+] en plasma.
|
||||||||
* Las pérdidas gástricas también contribuyen, pero son menores |
La alcalosis metabólica, y en mucho menor grado la alcalosis respiratoria aguda, pueden asociarse con hipocalemia. El principal mecanismo en este caso es la transferencia de iones hidrógeno hacia el exterior de las células como parte de una respuesta de neutralización que minimiza el aumento en el pH extracelular, y la neutralidad eléctrica se mantiene en parte por la entrada de potasio a las células. La relación entre el grado de hipocalemia y el aumento en el pH sanguíneo varía mucho. La pérdida de potasio del organismo participa también en la hipocalemia observada en la alcalosis metabólica causada por vómito o uso de diuréticos.
Un exceso de catecolaminas, como el que ocurre durante un infarto al miocardio o el delirium tremens, puede causar desplazamiento agudo de potasio hacia el intracelular, que es causado por el estímulo de los receptores adrenérgicos beta2. Este fenómeno, por el que la epinefrina puede convertir a la hipocalemia leve en grave, puede contribuir a la aparente mayor mortalidad coronaria observada en estudios de pacientes con hipertrofia del ventrículo izquierdo e hipertensión leve que han sido tratados con diuréticos tiacídicos. La estimulación beta adrenérgica con terbutalina usada en la labor pretérmino también puede reducir la [K+] en plasma a menos de lo normal.40
Es típico que la [K+] disminuye durante la administración de insulina en pacientes con cetoacidosis diabética, a pesar de que su concentración sea normal o aumentada cuando se ve al paciente por vez primera. Este cambio es causado en parte por la transferencia (inducida por insulina) de potasio al interior de la célula. Puede ocurrir un problema parecido cuando se administra dextrosa por vía intravenosa. Por ejemplo, en un paciente hipocalémico, la administración de 20 mEq de potasio en 1 L de dextrosa y agua puede empeorar la hipocalemia en forma temporal y quizá causar arritmias cardiacas.
Entre las causas poco frecuentes de hipocalemia inducida por redistribución se incluyen la administración de ácido fólico o vitamina B12 en el tratamiento de las anemias megaloblásticas (en las que la rápida producción de células nuevas causa captación del potasio del líquido extracelular) y el envenenamiento con sales de bario (que bloquean los canales de la membrana celular que en condiciones normales permiten la salida de potasio de las células). Los pacientes sometidos a procedimientos radiográficos no tienen riesgo de esta última complicación porque el sulfato de bario que se utiliza en los estudios gastrointestinales no se absorbe hacia la circulación sistémica. La sobredosis grave de teofilina puede también causar hipocalemia por desplazamiento de potasio inducido por catecolaminas o por liberación de insulina inducida por glucosa. Se ha reportado un dato similar después de la intoxicación con cloroquina.41
La parálisis periódica hipocalémica es un padecimiento raro de causa desconocida que se caracteriza por episodios potencialmente fatales de debilidad o parálisis muscular causados por el movimiento súbito de potasio hacia el interior de las células.42 Los episodios agudos suelen ser precipitados por reposo después del ejercicio, estrés o una comida abundante en carbohidratos, eventos que suelen asociarse con mayor liberación de epinefrina o insulina.
El trastorno puede ser familiar y el gen anormal en la mayoría de los pacientes parece codificar la parte del canal del calcio en el músculo esquelético que es bloqueada por bloqueadores de los canales del calcio dihidropiridina (v.gr., nifedipina).43 Por el contrario, algunos pacientes con hipertiroidismo, en especial varones asiáticos, adquieren la enfermedad.44 En estos pacientes el exceso de hormona tiroidea puede predisponer a episodios paralíticos al aumentar la actividad de la Na+-K+-ATPasa en las células [ver adelante, Tratamiento].
Pérdida de potasio del organismo Lo más frecuente es que la hipocalemia sea causada por pérdida de potasio a través del tubo digestivo, piel o riñones.
El tubo digestivo puede ser un sitio importante de pérdida de potasio, sobre todo si existe vómito o diarrea. Sin embargo, la [K+] en las secreciones gástricas (5 a 10 mEq/L) es mucho menor que la de las secreciones intestinales, que puede ser hasta de 75 mEq/L. Como resultado, se requiere la pérdida de grandes volúmenes de secreciones gástricas para producir disminución importante en el potasio. La menor [K+] que se observa con el vómito es resultado principalmente de la mayor pérdida urinaria, más que de la pérdida gástrica (ver adelante).
En dos circunstancias puede observarse pérdida cutánea de potasio que cause hipocalemia: el ejercicio en un ambiente cálido y húmedo, y las quemaduras graves. Los individuos que realizan ejercicio físico intenso pueden perder hasta 10 L de sudor por día. Esto puede significar una pérdida importante de potasio a pesar de que la [K+] en el sudor es de sólo 5 mEq/L. Por el contrario, la [K+] del líquido que se pierde a través de la piel que sufre una quemadura extensa puede exceder con mucho la concentración plasmática, porque se destruyen los tejidos locales, lo que libera gran cantidad de potasio de las células.
La pérdida de potasio en la orina es más frecuente en los pacientes que o están vomitando o reciben tratamiento con diuréticos. La depleción de volumen extracelular aumenta la liberación de aldosterona, causando un mayor intercambio de Na+-K+ en el túbulo colector [ver figura 3]. El hiperaldosteronismo por sí solo es insuficiente para aumentar la excreción de potasio, y debe conservarse la diuresis distal para que el potasio secretado se elimine. Se conserva el flujo urinario distal cuando existe vómito o acción de un diurético porque parte del sodio y agua escapan a la reabsorción en el túbulo proximal; los diuréticos inhiben la reabsorción de agua y sodio en el asa de Henle o el túbulo distal, mientras que la mayor carga de bicarbonato que existe durante el vómito persistente permite que el sodio y agua escapen a la reabsorción en el túbulo proximal. Este último mecanismo explica también la hipocalemia que se observa con las dosis altas de penicilina porque ésta, al igual que el bicarbonato, es un anión no reabsorbible. La inhibición directa de la reabsorción de potasio contribuye también a la hipocalemia que se observa con los diuréticos de asa.
Hasta el 40 por ciento de los pacientes hipocalémicos tienen hipomagnesemia. En ocasiones la alteración que causó la pérdida de potasio, como el tratamiento diurético o con cisplatino, es responsable de la menor reabsorción de magnesio. Sin embargo, los estudios han demostrado que la depleción de potasio casi nunca puede corregirse sin restablecer el equilibrio del magnesio.45 Esto parece ser porque el magnesio puede actuar como un bloqueador de los canales de potasio; por ejemplo, en el asa de Henle el potasio puede escapar a través de las membranas celulares hacia la luz tubular cuando los niveles de magnesio están bajos, contribuyendo así a las pérdidas urinarias.
Varias causas menos frecuentes de disfunción tubular pueden también causar pérdidas urinarias de potasio. Entre estas se incluyen la ATR tipo 1 (ver antes), medicamentos (v.gr., cisplatino, anfotericina B y aminoglucósidos), leucemia aguda y con menos frecuencia crónica, y la administración de levodopa. La menor reabsorción de sodio, que causa depleción de volumen, estimula la secreción de aldosterona y participa en muchos de estos padecimientos. Sin embargo, debe también pensarse en hiperaldosteronismo primario cuando se detecte pérdida no provocada de potasio en un paciente hipertenso.
Diagnóstico
Manifestaciones clínicas En la mayor parte de los casos, la hipocalemia leve (con [K+] en plasma entre 3.0 a 3.5 mEq/L) no causa síntomas. Las principales alteraciones que acompañan a la deficiencia de potasio importante son consecuencia de cambios en las funciones cardiovascular, neuromuscular y renal. La toxicidad cardiaca se manifiesta por arritmias graves, que ocurren porque la hiperpolarización de la membrana de la célula miocárdica prolonga el periodo refractario y aumenta la propensión a arritmias por reentrada. Otros cambios electrocardiográficos de la hipocalemia incluyen depresión de la onda T y ondas U prominentes [ver figura 7].

|
| Figura 7 |
| Cambios en el ECG por hipo o hipercalemia |
La hiperpolarización también retrasa la conducción nerviosa y la contracción muscular, lo que puede contribuir a la debilidad muscular, calambres y parestesias, aunque estos síntomas suelen no presentarse hasta que la [K+] en plasma es menor de 2.5 mEq/L. Cuando es grave, la hipocalemia puede deteriorar la función de los músculos respiratorios, provocando hipoventilación. Debido a que el potasio ocasiona, en condiciones normales, vasodilatación en respuesta a la contracción muscular, la hipocalemia grave puede también causar rabdomiolisis.
Las principales manifestaciones renales de la hipocalemia son la poliuria, resultado del aumento en la sed, y la resistencia a la acción de la hormona antidurética, a la que se le denomina diabetes insípida nefrogénica. El aumento en la sed es una respuesta apropiada a la poliuria, pero también es causada por estímulo directo del centro hipotalámico de la sed. La resistencia a la hormona antidiurética es causada por una reducción inducida por la hipocalemia en el número de canales de agua en el túbulo colector.46 La hipocalemia crónica puede ocasionar nefritis intersticial crónica y deterioro de la filtración glomerular.
Cuando la hipocalemia es grave ([K+] en plasma < 2 mEq/L), la capacidad de los túbulos renales de reabsorber sodio y cloruro puede alterarse. Esto puede causar depleción de volumen y pérdida de sodio y cloruro en la orina incluso en estados de menor perfusión renal. El mecanismo puede consistir en menor actividad de los transportadores tubulares de cloruro, muchos de los cuáles facilitan también la reabsorción de sodio.
Examen físico y pruebas de laboratorio La historia clínica y el examen físico suelen casi siempre sugerir el diagnóstico en el paciente con hipocalemia. La excreción de más de 30 mEq de potasio al día indica cierta pérdida renal de potasio. Los enfermos con pérdidas extrarrenales excretan menos potasio al día, lo mismo que los pacientes que han suspendido el tratamiento con diuréticos.
Una vez que se ha medico la excreción de potasio en orina, deben considerarse las siguientes probabilidades diagnósticas en el paciente con hipocalemia de origen incierto:
1. En un paciente asintomático, la acidosis metabólica con una baja excreción urinaria de potasio sugieren pérdidas gastrointestinales bajas por diarrea, abuso de laxantes o adenomas vellosos.
2. La acidosis metabólica con pérdida renal de potasio suele ser causada por cetoacidosis diabética o ATR tipo 1 (ver antes).
3. La alcalosis metabólica con baja excreción urinaria de potasio se debe a vómito subrepticio (fase tardía) o uso de diuréticos (en el que la muestra de orina se obtuvo después de que se eliminó el efecto del diurético).
4. La alcalosis metabólica con pérdida de potasio y una presión arterial normal suele ser causada por vómito subrepticio (fase inicial), uso de diuréticos o síndrome de Bartter. En estos casos la medición de la concentración de cloruro en la orina es útil porque en los pacientes que han vomitado es baja, mientras que la UNa y la excreción urinaria de potasio es relativamente alta. Este diagnóstico puede determinarse al lado de la cama del paciente a partir del pH de la orina, que debe ser de 7.0 o mayor si existe bicarbonaturia significativa.
5. La alcalosis metabólica con pérdida de potasio e hipertensión sugiere uso subrepticio de diuréticos en un paciente con hipertensión subyacente, hipertensión renovascular, hipertensión maligna o aldosteronismo primario.
Tratamiento
En la mayoría de los pacientes está indicado normalizar la [K+] en plasma, aunque los métodos y la cantidad y vía de la administración del potasio varían mucho. En ausencia de factores que causen desplazamientos transcelulares de potasio, existe una relación más o menos predecible entre el grado de hipocalemia y la magnitud de depleción de potasio en el organismo. Por cada 1 mEq/L de disminución en la [K+] en plasma, las reservas de potasio disminuyen en 200 a 400 mEq, hasta que la [K+] plasmática cae por debajo de 2.0 mEq/L. En este momento el déficit corporal total puede ser mayor de 1,000 mEq.
Con el reemplazo oral y parenteral, el potasio entra al plasma antes de ser transferido a las células. En consecuencia, los suplementos de potasio, sobre todo cuando se administran por vía intravenosa, se asocian con riesgo de hipercalemia si la dosis es demasiado grande o la velocidad de administración excesiva. Por lo tanto, si es posible, es mejor administrar el potasio por vía oral.
El reemplazo oral de potasio por medio de alimentos ricos en este elemento puede ser eficaz cuando existen acidosis metabólica o insuficiencia renal. Por el contrario, el potasio de la dieta no corrige la deficiencia que ocurre en la alcalosis metabólica porque en la mayor parte de los alimentos el potasio está unido en forma débil a aniones poco reabsorbibles, como el fosfato.47 Por ejemplo, en el caso de la alcalosis metabólica causada por diuréticos, se requieren sales de cloruro, como cloruro de potasio, para restablecer el déficit.
El potasio puede también administrarse por vía intravenosa a través de una vena periférica en concentraciones hasta de 40 mEq/L. Las concentraciones más altas pueden causar flebitis, y deben infundirse sólo en venas de gran calibre, como la subclavia o la femoral. Excepto en algunos casos, como en el de hipocalemia grave con arritmias intratables, la velocidad de administración no debe ser mayor de 20 a 40 mEq/hr, aunque se han usado dosis tan altas como 100 mEq/h en pacientes seleccionados con parálisis o arritmias graves.48 Deben evitarse las soluciones que contienen glucosa porque la estimulación de la insulina puede introducir potasio a las células, aumentando la hipocalemia.
Para pacientes que reciben diuréticos por hipertensión arterial existen varias alternativas para corregir la hipocalemia sin utilizar suplementos de cloruro de potasio. Debido a que el cumplimiento del paciente disminuye cuando se aumentan los medicamentos prescritos (incluyendo los suplementos de potasio), debe intentarse primero la disminución de la dosis de diurético (a 12.5 mg de hidroclorotiacida, por ejemplo) o la sustitución con un agente alterno.
Los individuos edematosos o con hiperaldosteronismo primario que también desarrollan hipocalemia pueden ser tratados con diuréticos ahorradores de potasio (amiloride o espironolactona) hasta que pueda establecerse un tratamiento definitivo (v.gr., la extirpación quirúrgica del adenoma suprarrenal en el hiperaldosteronismo).
En los pacientes con parálisis periódica hipocalémica [ver antes, Alteración en la distribución del potasio], la administración de cloruro de potasio puede abortar los ataques agudos en minutos. Cuando existe, debe tratarse el hipertiroidismo, y la administración de un bloqueador beta adrenérgico no selectivo (como propranolol) puede prevenir los episodios en pacientes con la forma familiar del padecimiento.
HIPERCALEMIA
La hipercalemia es un trastorno electrolítico frecuente. Debido a que gran cantidad de medicamentos de uso común pueden interferir con la homeostasis del potasio, es frecuente que la hipercalemia sea iatrogénica y, por tanto, previsible. La hipercalemia severa se asocia con alteración en la función neuromuscular o cardiaca.
Etiología
La hipercalemia puede ser causada por ingesta excesiva, mayor liberación de las células o disminución de la excreción renal [ver tabla 10].
|
||||||||||
* La hipercalemia persistente que se asocia con aumento de la indigesta casi siempre se acompaña de una menor excreción renal. |
Aumento en la ingesta El aumento en la [K+] en plasma que ocurre después de una carga oral o intravenosa de potasio depende de cuatro factores: la cantidad de potasio administrado, la posibilidad de que parte del exceso de potasio entre a las células, la excreción urinaria y la concentración previa de potasio ingerido (un proceso denominado adaptación). El aumento en la [K+] es mínima si la ingesta es lenta, porque en este caso la excreción renal, y quizá la entrada a las células, es más eficaz. El aumento en la aldosterona y en la actividad de la ATPasa de Na+-K+ en el túbulo colector cortical tienen una función importante en este proceso. Incluso si no ha existido adaptación, el riñón excreta al final el exceso de potasio, aunque en forma más lenta. Por lo tanto, la hipercalemia causada por una carga súbita es transitoria, a menos que la excreción urinaria de potasio esté reducida en forma concomitante.
Mayor liberación de las células Tanto la deficiencia de insulina como el bloqueo adrenérgico beta2 pueden aumentar la [K+] en plasma. Por ejemplo, en pacientes con nefropatía terminal que estan en hemodiálisis, el propranolol o el labetalol, pero no el bloqueador adrenérgico selectivo beta1 atenolol, pueden aumentar el potasio plasmático alrededor de 1 mEq/L.49 Sin embargo, en dosis más altas el atenolol puede perder su efecto selectivo.
La elevación en la osmolaridad plasmáctica causa un movimiento osmótico de agua de las células hacia el líquido extracelular. Este evento se acompaña de movimiento de potasio desde las células, que ocurre por dos mecanismos. Primero, la pérdida de agua eleva la concentración celular de potasio, creando un gradiente favorable para la salida pasiva de potasio a través de los canales de potasio en la membrana celular. Segundo, las fuerzas de fricción entre el solvente (agua) y el soluto pueden provocar que el potasio sea transportado junto con el agua a través de los poros de agua de la membrana celular.
También se sabe que la acidemia aguda aumenta la [K+] en plasma al desplazar potasio de las células hacia el plasma. Es posible que este movimiento ocurra cuando se pierde bicarbonato en casos de diarrea y durante la infusión de ácidos. Por ejemplo, cuando se infunde HCl arginina, se intercambia potasio por arginina catiónica que penetra a las células. Sin embargo, este desplazamiento parece ser importante sólo en el caso de ácidos inorgánicos (ver antes). En comparación, la hipercalemia en la acidosis láctica es causada principalmente por destrucción celular, mientras que la hipercalemia en la cetoacidosis es ocasionada por deficiencia de insulina e hiperglucemia.
La destrucción tisular masiva que ocurre durante la rabdomiolisis, la formación de un absceso, la necrosis tisular (v.gr., infarto intestinal) o la quimioterapia en casos de leucemias o linfomas, libera potasio hacia el plasma. Causas menos frecuentes de aumento en la [K+] en plasma incluyen la intoxicación por glucósidos cardiacos (como resultado de la inhibición parcial de la ATPasa de Na+-K+), la succinilcolina (que despolariza la membrana celular y permite que el potasio abandone las células) y una forma rara de parálisis periódica hipercalémica.50
Deterioro de la excreción renal El riñón, el principal sitio excretor del potasio, tiene una gran capacidad para aumentar la velocidad de excreción cuando aumenta la [K+] en plasma. Por lo tanto, la hipercalemia persistente casi siempre se asocia con cierto deterioro en la eliminación renal de este ión. Es frecuente que también esté alterada la secreción distal de iones hidrógeno, lo que causa una acidosis metabólica con brecha aniónica normal, debido a que el túbulo colector es el sitio en que se secretan tanto el potasio como los iones hidrógeno. La excreción renal de potasio puede alterarse en la insuficiencia renal oligoanúrica, la reducción de volumen arterial efectivo, el hipoaldosteronismo y la ATR tipo 1 hipercalémica.
La falla renal oligoanúrica disminuye, pero no suspende del todo, la excreción de potasio. Aunque la insuficiencia renal predispone a la retención de potasio, la excreción del potasio de la dieta suele conservarse en la insuficiencia renal crónica hasta que la VFG es menor de 5 a 10 ml/min. Aunque menos eficaz en la insuficiencia renal oligoanúrica, la excreción renal se mantiene porque la mayoría del potasio urinario deriva de secreción tubular y no de filtración glomerular.
Cuando disminuye el volumen arterial efectivo también se reduce el aporte de sodio y agua hacia el túbulo colector. La conservación de un aporte distal adecuado de sodio y agua es indispensable para que la excreción de potasio sea normal. Por ejemplo, el aporte distal está disminuido en pacientes con insuficiencia cardiaca congestiva grave o con cirrosis. Tanto la disminución en la filtración glomerular como el aumento en la reabsorción proximal de sodio y agua, mediada en parte por angiotensina II, pueden contribuir en estas circunstancias. Aunque la aldosterona aumenta la reabsorción de sodio y la secreción de potasio en estas condiciones, el aumento es limitado porque existe menos sodio disponible para su reabsorción. Aún más, el potasio secretado no se eliminará en forma total si la diuresis está muy disminuida.
La deficiencia o resistencia de aldosterona (i.e., ATR tipo 4) es responsable de la hipercalemia en más del 75 a 85 por ciento de los casos en los que los pacientes tienen hipercalemia persistente en ausencia de cualquier causa aparente, como insuficiencia renal avanzada, un medicamento agresor o administración de potasio.
El hipoaldosteronismo también se asocia con menor secreción de iones hidrógeno, que es causada en parte por alteración de la amoniogénesis en el túbulo proximal (causada a su vez por alcalosis intracelular inducida por la hipercalemia). El resulado es menor aporte de amoníaco en los segmentos más distales de la nefrona.51 En consecuencia, la falta de amortiguadores suficientes en la orina influye en forma importante en la menor secreción de iones hidrógeno y explica porqué el pH urinario es característicamente ácido (< 5.3) en este padecimiento.
Varios trastornos causan ATR tipo 4 [ver tabla 4]. La ATR tipo 4 hiporreninémica es la forma más frecuente de esta alteración y ocurre principalmente en pacientes con insuficiencia renal leve. Con frecuencia se asocia a nefropatía diabética o nefritis intersticial crónica. Los medicamentos como los AINE pueden también contribuir a este padecimiento.
La fisiopatología de la ATR tipo 4 en los pacientes hiporreninémicos no se conoce del todo. Es obvio que la menor producción de angiotensina II es importante porque el potasio y la angiotensina II son los principales estímulos fisiológicos para la liberación de aldosterona. Sin embargo, las evidencias sugieren que por lo menos algunos pacientes deben tener un defecto suprarrenal concomitante porque hay observaciones que indican que la glándula suprarrenal normal puede liberar angiotensina II.52 Se ha sugerido también que en algunos pacientes la reducción en la producción de aldosterona está causada por disminución en la prostaciclina, que en condiciones normales estimula la liberación de renina, o por aumento en el factor natriurético auricular, un péptido que inhibe en forma intensa tanto la liberación de renina como de aldosterona.
En otros casos de ATR tipo 4 el defecto en la síntesis y liberación de aldosterona parece confinarse a las glándulas suprarrenales. La heparina (o quizá su preservativo, el clorbutol) es un inhibidor potente de la producción de aldosterona, incluso cuando se administra por vía subcutánea en dosis bajas.53 Por lo general, el uso de heparina no se asocia con hipercalemia porque la deficiencia moderada de aldosterona no es suficiente para causar retención importante de potasio en la mayoría de los pacientes que tienen una función renal normal. Sin embargo, puede ocurrir hipercalemia si existe nefropatía subyacente o si se administran al mismo tiempo otros agentes que alteren el sistema renina-angiotensina-aldosterona, como AINE o inhibidores de la ECA.
Debe analizarse la posibilidad de insuficiencia suprarrenal crónica (i.e., deficiencia tanto de aldosterona como de cortisol, también llamada enfermedad de Addison) antes de realizar el diagnóstico de deficiencia selectiva de aldosterona. Cuando existe insuficiencia suprarrenal crónica, el hipocorticismo altera la excreción de agua, causando hiponatremia además de la hipercalemia causada por la deficiencia de aldosterona.
Aunque la ATR tipo 1 se asocia con hipocalemia (ver antes), algunos pacientes con deterioro de la secreción de iones hidrógeno por el túbulo colector no pueden secretar el potasio en forma normal, y sufren la llamada ATR tipo 1 hipercalémica. Este padecimiento se observa con más frecuencia asociado a obstrucción de las vías urinarias, pero puede ser causado también por daño túbulointersticial como el de la nefropatía de células falciformes, la nefritis lúpica o el rechazo de trasplante. Estos datos son causados por daño tanto a las células principales que secretan potasio como a las células intercaladas que secretan ácido. La ATR tipo 1 hipercalémica puede distinguirse de la tipo 4 por la orina persistentemente alcalina (pH >5.3) y por un nivel de aldosterona en plasma normal o aumentado [ver tabla 11].
|
||||||||
UNa - concentración urinaria de sodio
|
Por último, es importante recordar que los efectos de los medicamentos pueden limitar la secreción de potasio en forma directa. Es obvia la acción de los diuréticos ahorradores de potasio como el amiloride, triamtireno y espironolactona. Otro agente menos común es el trimetoprim-sulfametoxazol. El trimetoprim, combinado o solo, puede disminuir la secreción de potasio al bloquear el transporte de sodio de la luz tubular hacia la células del túbulo colector, en un mecanismo semejante al del amiloride.
Manifestaciones clínicas
Los síntomas causados por la hipercalemia se relacionan con alteración en la trasmisión neuromuscular. La facilitad de generar un potencial de acción (llamada excitabilidad de membrana) se relaciona tanto con la magnitud del potencial de reposo de membrana como con el estado de activación de los canales de sodio de la membrana; la apertura de estos canales causa difusión pasiva del sodio extracelular hacia las células, y es el primer paso en este proceso.
De acuerdo con la ecuación de Nernst, el potencial de reposo de membrana se relaciona con la concentración extracelular de potasio. La elevación en la [K+] en plasma (extracelular) disminuye esta relación y, por lo tanto, despolariza la membrana celular (i.e., hace al potencial de reposo de membrana menos electronegativo). Este cambio al inicio aumenta la excitabilidad de la membrana porque se requiere un estímulo despolarizante menor para generar un potencial de acción. Sin embargo, la despolarización persistente inactiva los canales de sodio en la membrana, causando una reducción neta en la excitabilidad que puede manifestarse clínicamente por menor conducción cardiaca, debilidad muscular o parálisis.
En general, los síntomas severos de hipercalemia no ocurren hasta que la [K+] en plasma es mayor de 7.5 mEq/L. Sin embargo, existe gran variabilidad individual porque factores como la hipocalcemia y la acidosis metabólica pueden aumentar la toxicidad del exceso de potasio. Por lo tanto, está indicada la vigilancia cuidadosa del ECG y de la fuerza muscular para evaluar las consecuencias funcionales de la hipercalemia. Con una [K+] en plasma mayor de 7.5 a 8.0 mEq/L ocurre debilidad muscular severa o cambios en el ECG potencialmente fatales, que requieren tratamiento inmediato con prácticamente todas las modalidades disponibles [ver adelante, Tratamiento].
El aumento simétrico en el tamaño de la onda T es el primer cambio electrocardiográfico causado por la hipercalemia. Esta alteración es seguida por disminución del voltaje de la onda P y ensanchamiento de los complejos QRS [ver figura 7]. Si no se trata, la hipercalemia grave puede causar un patrón ECG sinusoidal, con una oscilación representada por un complejo QRS ancho y la onda complementaria, una onda T anormal. Los cambios ECG no suelen aparecer sino hasta que la [K+] en plasma es mayor a 6.5 mEq/L, y es más posible que se desarrollen cuando el potasio aumenta con rapidez. Sin embargo, no existe una relación predictiva entre la severidad de la alteración electrolítica y el patrón del ECG. En raros casos se han observado ECG sin cambio en pacientes con [K+] en plasma mayor a 9 mEq/L.
Las manifestaciones neuromusculares de la hipercalemia son inespecíficas. Los signos más tempranos consisten en parestesias y debilidad, que pueden progresar a parálisis que afecte los músculos respiratorios. Estos síntomas son semejantes a los observados en la hipocalemia. Sin embargo, es típico que la función de los nervios craneanos permanezca normal.
Cuando la hipercalemia es constante la menor eliminación renal suele ser la causa [ver tabla 10]. Cuando se demuestra ATR tipo 4 debe medirse la concentración plasmática de cortisol para determinar si existe insuficiencia suprarrenal completa.
Seudohipercalemia
Debe pensarse en seudohipercalemia cuando exista evidencia de hemólisis en la muestra de sangre o cuando la cuenta de plaquetas o leucocitos esté muy aumentada. A diferencia de la hipercalemia verdadera, que se asocia con alteración en la función neuromuscular o cardiaca (ver antes), la seudohipercalemia no constituye un riesgo para el paciente.
En este trastorno la medición de [K+] en suero en el laboratorio clínico está aumentada por liberación de K durante la coagulación que ocurre después de obtenida la muestra de sangre. Se debe principalmente a hemólisis por obtención traumática, pero también ocurre cuando existe trombocitosis importante (cuenta de plaquetas > 1,000 000/µl) o leucocitosis (cuenta > 100,000 /µl).
El diagnóstico de seudohipercalemia se realiza demostrando que la [K+] es normal es una muestra de plasma no hemolizada; esta muestra se obtiene colocando la sangre en un tubo heparinizado, lo que evita que se coagule. No está indicado tratar la hipercalemia porque la [K+] in vivo (i.e., del plasma) es normal.
Tratamiento
En la hipercalemia verdadera, en especial cuando es severa ([K+] en plasma > 6.0 mEq/L) es obligatorio suspender todos los medicamentos que afecten en forma adversa el equilibrio del potasio. Estos incluyen los betabloqueadores no selectivos, los inhibidores de la ECA, los diuréticos ahorradores de potasio, los AINE y el trimetoprim. También deben evitarse los sustitutos de la sal, que contienen cloruro de potasio. Las personas con hipercalemia leve (potasio < 6.0 mEq/L) casi siempre pueden ser tratados en forma conservadora disminuyendo la ingesta de potasio a menos de 2 g/día y, si está indicado, añadiendo un diurético de asa.
El tratamiento activo para disminuir la [K+] en plasma o antagonizar los efectos del ión sobre la membrana celular debe comenzar cuando la [K+] en plasma aumente en forma súbita a 6.0 mEq/L o más, sobre todo si existen cambios ECG de hipercalemia. Existen varias opciones terapéuticas [ver tabla 12].
|
||||||||
|
La infusión de calcio normaliza el ECG con gran rapidez. El aumento en la [Ca2+] eleva el umbral del potencial, normalizando así la excitabilidad de la membrana. Sin embargo, sólo debe administrarse calcio si existen manifestaciones en el ECG que puedan preceder a la fibrilación ventricular. Debido a que el calcio puede exacerbar o precipitar las arritmias inducidas por glucósidos cardiacos, sólo se empleará cuando sea realmente necesario y con gran cuidado en los pacientes que reciban digoxina u otros derivados de digital.
El calcio se administra por vía intravenosa como gluconato o sal cloruro (10 ml de gluconato de calcio al 10 por ciento durante 2 o 3 minutos, bajo vigilancia ECG). La normalización del ECG puede durar menos de 60 minutos, y este tratamiento no altera a la [K+] o las reservas corporales del mismo. En consecuencia, en forma simultánea deberán iniciarse medidas para disminuir la concentración plasmática y eliminar potasio del organismo.
El bicarbonato de sodio, la glucosa (con o sin insulina) y la estimulación de los receptores adrenérgicos beta2, disminuyen la [K+] en plasma al favorecer la entrada de potasio del líquido extracelular a las células.54 Al parecer el bicarbonato y los agonistas adrenérgicos beta2 son menos eficaces que la glucosa e insulina, sobre todo en pacientes con insuficiencia renal avanzada.55 Aún más, el bocarbonato no parece potencializar la acción hipocalémica de los agonistas adrenérgicos beta2 o de la glucosa e insulina, que son eficaces en este caso.56
El efecto hipocalémico de la insulina se observa en 30 a 60 minutos y puede lograrse por la administración simple de 25 g de glucosa por vía intravenosa, porque la infusión de glucosa estimula una rápida liberación de insulina. En forma alterna, pueden administrarse 6 a 8 unidades de insulina con esta carga de glucosa a los pacientes con diabetes, aunque este esquema puede causar hipoglucemia en los individuos normales. Independientemente del método que se emplee, debe tenerse cuidado para no producir una hiperglucemia grave con la infusión de glucosa, porque las glucemias altas pueden exacerbar la hipercalemia al desplazar agua, que contiene potasio, fuera del compartimiento intracelular y hacia el plasma.
Al igual que la administración de calcio, la administración de glucosa e insulina, bicarbonato y agonistas adrenérgicos beta2, son medidas temporales porque las reservas corporales totales de potasio no disminuyen. Por lo tanto, se requieren medidas adicionales para asegurarse de que la [K+] en plasma no regresa a los niveles previos al tratamiento. En algunos pacientes, la reducción de la concentración de potasio en plasma con estos agentes poco antes de la diálisis puede causar menor eliminación dialítica de potasio, lo que podría asociarse con hipercalemia de rebote después de terminado el tratamiento dialítico.57 El aumento de la diuresis con diuréticos puede ser útil, pero la insuficiencia renal suele limitar su eficacia. Puede administrarse poliestireno sulfonato sódico (Kayexalate), una resina de intercambio catiónico de Na+-K+, en sorbitol (para causar diarrea) por vía oral o como un enema de retención, y este medicamento casi siempre es eficaz. En los casos de hipercalemia asintomática el poliestireno sulfonato sódico puede utilizarse como único tratamiento, aunque requiere de por lo menos 2 a 4 horas antes de que la [K+] en plasma comience a disminuir. En casos raros la resina se ha asociado con necrosis intestinal. Esta complicación seria tiende a ocurrir en el periodo posoperatorio temprano en pacientes quirúrgicos, aunque el mecanismo causal no se conoce del todo. Es posible que la naturaleza hipertónica de la solución dañe la mucosa intestinal.58
Debe pensarse en realizar diálisis para los casos graves o rebeldes a tratamiento, sobre todo si existe insuficiencia renal terminal. Cuando está disponible, la hemodiálisis es preferible a la diálisis peritoneal en los casos agudos porque elimina el potasio con mucha mayor rapidez que la diálisis peritoneal.
Cuando la hipercalemia es crónica y no se controla en forma adecuada por dieta o diuréticos, puede ser útil el remplazo de aldosterona si se ha demostrado deficiencia. Sin embargo, debido a que su efecto puede tardar varios días en comenzar, no se emplea como único medicamento en el tratamiento de la hipercalemia aguda grave.
Figura 1 Janet Betries
Figuras 2 a 6 Tom Moore.
Figuras 7 Los electrocardiogramas fueron proporcionados por el Dr. David Spodick, Profesor de Medicina en la University of Massachusetts Medical School, Worcester.
Bibliografía
- Schoolwerth AC: Regulation of renal ammoniagenesis in metabolic acidosis. Kidney Int 40:961, 1991
- Winter SD, Pearson R, Gabow PA, et al: The fall of the serum anion gap. Arch Intern Med 150:311, 1990 [PMID 2302006]
- Vella A, Farrugia G: D-lactic acidosis: pathologic consequence of saprophytism. Mayo Clin Proc 73:451, 1998 [PMID 9581587]
- Coronado BE, Opal SM, Yoburn DC: Antibiotic-induced lactic acidosis. Ann Intern Med 122:839, 1995
- Kamel SK, Lin SH, Cheema-Dhadli S, et al: Prolonged total fasting: a feast for the integrative physiologist. Kidney Int 53:531, 1998 [PMID 9507196]
- Venuto RC: Pigment-associated acute renal failure: is the water clearer 50 years later? J Lab Clin Med 119:452, 1992
- Sweeney T, Beuchat C: Limitation of methods of osmometry: measuring the osmolality of biological fluids (editorial). Am J Physiol 264:R469, 1993 [PMID 8456999]
- Jaber BL, Madias NE: Marked dilutional acidosis complicating management of right ventricular myocardial infarction. Am J Kidney Dis 30:561, 1997 [PMID 9328373]
- Cruz DN, Huot SJ: Metabolic complications of urinary diversions: an overview. Am J Med 102:477, 1997 [PMID 9217645]
- Newell KA, Bruce DS, Cronin DC, et al: Comparison of pancreas transplantation with portal venous and enteric exocrine drainage to the standard technique utilizing bladder drainage of exocrine secretions. Transplantation 62:1353, 1996 [PMID 8932284]
- Levi M, McDonald LA, Preisig PA, et al: Chronic K depletion stimulates rat renal brush-border membrane Na-citrate cotransporter. Am J Physiol 261:F767, 1991 [PMID 1683169]
- Dubose TD Jr, Good DW: Effects of chronic hyperkalemia on renal production and proximal tubular transport of ammonium in rats. Am J Physiol 260:F680, 1991 [PMID 2035655]
- Graber M: A model of hyperkalemia produced by metabolic acidosis. Am J Kidney Dis 22:436, 1993
- Orchard CH, Cingolani HE: Acidosis and arrhythmias in cardiac muscle. Cardiovasc Res 28:1312, 1994 [PMID 7954638]
- Cohen RD: Lactic acidosis: new perspectives on origins and treatment. Diabetes Rev 2:86, 1994
- Alpern RJ, Sakhaee K: The clinical spectrum of chronic metabolic acidosis: homeostatic mechanisms produce significant morbidity. Am J Kidney Dis 29:291, 1997 [PMID 9016905]
- Garibotto G, Russo R, Sofia A, et al: Skeletal muscle protein synthesis and degradation in patients with chronic renal failure. Kidney Int 45:1432, 1994 [PMID 8072256]
- Ballmer PE, McNurlan MA, Hulter HN, et al: Chronic metabolic acidosis decreases albumin synthesis and induces negative nitrogen balance in humans. J Clin Invest 95:39, 1995
- Graham KA, Reaich D, Channon SM, et al: Correction of acidosis in CAPD decreases whole-body protein degradation. Kidney Int 49:1396, 1996; erratum 51:1662, 1997
- Kette F, Weil MH, Gazmuri RJ: Buffer solutions may compromise cardiac resuscitation by reducing coronary perfusion pressure. JAMA 266:2121, 1991 [PMID 1920701]
- Stacpoole PW, Wright EC, Baumgartner TG, et al: A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. N Engl J Med 327:1564, 1992 [PMID 1435883]
- Lebovitz HE: Diabetic ketoacidosis. Lancet 345:767, 1995
- Okuda Y, Adrogue HJ, Field JB, et al: Counterproductive effects of sodium bicarbonate in ketoacidosis. J Clin Endocrinol Metab 81:314, 1996 [PMID 8550770]
- Rother KI, Schwenk WF: Effect of rehydration fluid with 75 mmol/L of sodium on serum sodium concentration and serum osmolality in young patients with diabetic ketoacidosis. Mayo Clin Proc 69:1149, 1994 [PMID 7967775]
- Ori Y, Lee SG, Krieger NS, et al: Osteoblastic intracellular pH and calcium in metabolic and respiratory acidosis. Kidney Int 47:1790, 1995 [PMID 7643550]
- Abreo K, Adlakha A, Kilpatrick S, et al: The milk-alkali syndrome: a reversible form of acute renal failure. Arch Intern Med 153:1005, 1993 [PMID 8481062]
- Beall DP, Scofield RH: Milk-alkali syndrome associated with calcium carbonate consumption. Medicine (Baltimore) 74:89, 1995 [PMID 7891547]
- Hoglund P, Haila S, Socha J, et al: Mutations of the down-regulated adenoma (DRA) gene cause congenital chloride diarrhoea. Nat Genet 14:316, 1996 [PMID 8896562]
- Aichbichler BW, Zerr CH, Santa Ana CA, et al: Proton-pump inhibition of gastric chloride secretion in congenital chloridorrhea. N Engl J Med 336:106, 1997 [PMID 8988888]
- Galla JH, Gifford JD, Luke RG, et al: Adaptations to chloride-depletion alkalosis. Am J Physiol 261:R771, 1991 [PMID 1928424]
- Elam-Ong S, Kurtzman N, Sabatini S: Regulation of collecting tubule adenosine triphosphatases by aldosterone and potassium. J Clin Invest 91:2385, 1993
- Simon DB, Karet FE, Hamdan JM, et al: Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 13:183, 1996 [PMID 8640224]
- Simon DB, Nelson-Williams C, Bia MJ, et al: Gitelman's variant of Bartter's syndrome, inherited hypokalemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12:24, 1996 [PMID 8528245]
- Adrogue HJ, Madias NE: Management of life-threatening acid-base disorders. N Engl J Med 338:107, 1998
- Wesson D, Dolson G: Enhanced HCO3 secretion by distal tubule contributes to Na Cl-induced correction of chronic alkalosis. Am J Physiol 264:F899, 1993 [PMID 8498543]
- Gerhardt RE, Keothe JD, Glickman JD, et al: Acid dialysate correction of metabolic alkalosis in renal failure. Am J Kidney Dis 25:343, 1995 [PMID 7847364]
- Colussi G, Rombola G, De Ferrari M, et al: Correction of hypokalemia with antialdosterone therapy in Gitelman's syndrome. Am J Nephrol 14:127, 1994 [PMID 8080005]
- Agarwal R, Afzalpurkar R, Fordtran JS: Pathophysiology of potassium absorption and secretion by the human intestine. Gastroenterology 107:548, 1994 [PMID 8039632]
- Halperin ML, Cheema-Dhadli S, Phillip LE: Potassium excretion: a story that is easy to digest. J Am Soc Nephrol 5:523, 1994
- Braden GL, von Oeyen PT, Germain MJ, et al: Ritodrine- and terbutaline-induced hypokalemia in preterm labor: mechanisms and consequences. Kidney Int 51:1867, 1997
- Clemessy JL, Favier C, Borron SW, et al: Hypokalaemia related to acute chloroquine ingestion. Lancet 346:877, 1995 [PMID 7564673]
- Fontaine B, Lapie P, Plassart E, et al: Periodic paralysis and voltage-gated ion channels. Kidney Int 49:9, 1996 [PMID 8770943]
- Ptacek LJ, Tawil R, Griggs RC, et al: Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell 77:863, 1994 [PMID 8004673]
- Ko GT, Chow CC, Yeung VT, et al: Thyrotoxic periodic paralysis in a Chinese population. QJM 89:463, 1996 [PMID 8758050]
- Whang R, Whang D, Ryan M: Refractory potassium depletion: a consequence of magnesium deficiency. Arch Intern Med 152:40, 1992 [PMID 1728927]
- Marples D, Frokiaer J, Dorup J, et al: Hypokalemia-induced downregulation of aquaporin-2 water channel expression in rat kidney medulla and cortex. J Clin Invest 97:1960, 1996
- 47. Gennari FJ: Hypokalemia. N Engl J Med 339:451, 1998
- Kruse J, Carlson R: Rapid correction of hypokalemia using concentrated intravenous potassium chloride infusions. Arch Intern Med 150:613, 1990 [PMID 2310280]
- Allon M: Hyperkalemia in end-stage renal disease: Mechanisms and management. J Am Soc Nephrol 6:1134, 1995
- Griggs R, Ptacek L: The periodic paralysis. Hosp Pract (Off Ed) 27:123, 1992 [PMID 1331137]
- Dubose T Jr, Good D: Chronic hyperkalemia impairs ammonium transport and accumulation in the inner medulla of the rat. J Clin Invest 90:1443, 1992
- Gupta P, Franco-Saenz R, Mulrow PJ: Locally generated angiotensin II in the adrenal gland regulates basal, corticotropin-, and potassium-stimulated aldosterone secretion. Hypertension 25:443, 1995 [PMID 7875770]
- Oster JR, Singer I, Fishman LM: Heparin-induced aldosterone suppression and hyperkalemia. Am J Med 98:575, 1995 [PMID 7778574]
- Liou HH, Chiang SS, Wu SC: Hypokalemic effects of intravenous infusion or nebulization of salbutamol in patients with chronic renal failure: comparative study. Am J Kidney Dis 23:266, 1994
- Blumberg A, Weidmann P, Ferrari P: Effect of prolonged bicarbonate administration on plasma potassium in terminal renal failure. Kidney Int 41:369, 1992 [PMID 1552710]
- Allon MA, Shanklin N: Effect of bicarbonate administration on plasma potassium in dialysis patients: interactions with insulin and albuterol. Am J Kidney Dis 28:508, 1996
- Allon M, Shanklin N: Effect of albuterol treatment on subsequent dialytic potassium removal. Am J Kidney Dis 26:607, 1995 [PMID 7573015]
- Emmett D, Emmett M, Borejdo J, et al: Na polystyrene sulfonate resin/hypertonic sorbitol enema toxicity (abstr). J Am Soc Nephrol 6:439, 1995