Contenido del artículo
I ORGANOS Y CELULAS DEL SISTEMA INMUNE
DR. JOHN DAVID
DR. COX TERHORST, PH.D.
Características del sistema inmune
El sistema inmune media la relación del individuo con el ambiente. La inmunidad incluye respuestas innatas o naturales, y adquiridas o específicas. La diferencia esencial entre los dos tipos de inmunidad es la manera como se reconocen los microrganismos. En la inmunidad innata, carbohidratos (principalmente) que son únicos de los organismos infecciosos son reconocidos por receptores e superficie celular en los macrófagos y células asesinas naturales (NK) o por el sistema del complemento. En la inmunidad adquirida los linfocitos emplean receptores de antígenos muy específicos para reconocer a los agentes infecciosos. Una vez que un individuo normal ha tenido una infección como varicela o sarampión, el sistema inmunológico la reconoce y evita su recurrencia. Además, el sistema inmune tiene una marcada capacidad para discriminar entre los antígenos, incluso si sus estructuras son muy semejantes.
Existen dos grupos principales de linfocitos, las células T (también denominados linfocitos dependientes del timo o linfocitos T) y las células B (linfocitos derivados de la médula ósea o linfocitos B) [ver figura 1]. Ni las células T ni las B constituyen una población homogénea de células, cada grupo consiste en un número de subgrupos que pueden diferenciarse por sus marcadores de superficie y su función. Además, existe un grupo heterogéneo de linfocitos que no está formado por células T o B. La unión de anticuerpos monoclonales a los marcadores de superficie es la técnica más específica en la actualidad para identificar los principales tipos de estas células.
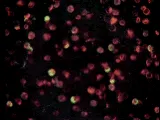
|
| Figura 1 |
| Inmunoglobulinas |
CELULAS T
Todas las células T maduras y los timocitos de la médula expresan CD2 y CD3. CD4 se expresa en solo el 50 a 65% de las células T periféricas y CD8 en el 25 a 35% de las células T periféricas. Aunque CD4 y CD8 se expresan juntos en los timocitos en fase II, solo uno u otro se expresan en los subtipos complementarios de células T maduras (células T CD4+ y CD8+). Las células T CD4+ reconocen antígenos cuando están presentes en asociación con moléculas del CPH de clase II (antígenos HLA-D); las células T CD8+ reconocen antígenos en el contexto de moléculas clase I (antígenos HLA-A, HLA-B y HLA-C).
Las células T CD4+ cooperadoras pueden diferenciarse en Th1 y Th2 según las citocinas que producen. 1,2 Las células TH1 producen IL-2 e interferón gama, que son importantes para la inmunidad mediada por células. Las células Th2 producen IL-4, IL-5, IL-6, IL-10 e IL-13, que son cruciales para la producción de anticuerpos. Las linfocinas producidas por cada uno de estos tipos celulares influyen en el otro tipo celular. El interferón gama producido por las células Th1 puede inhibir la función de las células Th2, mientras que la IL-10, que es producida por las células Th2 (así como por monocitos, macrófagos y células B) puede inhibir la función de las células Th1.
Es importante mencionar que el CD4 en las células T cooperadoras actúa como un correceptor para el VIH, junto con un receptor quimocina. El CCR5, un receptor CC (cisteína-cisteína) o beta-quimocina para RANTES (regulado por la activación y expresado y secretado por las células T normales), o proteína inflamatoria de macrófagos-1a (MIP-1a) y la MIP-1b, es el correceptor para las cepas tempranas del VIH. El uno por ciento de las personas de raza blanca son homocigotas para un defecto en este receptor y son resistentes a la infección por VIH. El CCR5 también se observa en los macrófagos. El CCR4, un receptor alfa-quimocina específico de células T, que es un receptor del factor 1 derivado de células del estroma (SDF-1), participa en la infección de las cepas tardías del VIH. 3-5
Los linfocitos B son las células productoras de inmunoglobulinas del sistema inmune, y pueden ser identificadas por la presencia de inmunoglobulinas en su superficie. Estas células con receptores para inmunoglobulinas positivos en la superficie de su membrana (SmIg+) const ituyen del 5 al 15 por ciento de los linfocitos en sangre periférica [ver figura 1]. La mayor parte de las células B tienen IgM e IgD en su superficie y cerca de la cuarta parte del total de estas células sólo poseen inmunoglobulinas de alguna de estas dos clases. Uno por ciento de las células B muestra IgG o IgA Los linfocitos T y B constituyen del 80 al 95 por ciento de los linfocitos en sangre periférica. El resto de las los linfocitos presentes en sangre periférica constituyen una población heterogénea de células.
Sobre la superficie de las células B se encuentra un receptor del complemento llamado receptor 2 del complemento (CR2), que se une a los factores C3d, C3dg y al factor iC3b.
Las células B también poseen receptores para el virus de Epstein-Barr y para la porción Fc de la IgG. La molécula CD20, una fosfoproteína de 35 kd que se encuentra sobre la superficie de las células B, es un marcador de los linfocitos B utilizado con frecuencia. Algunos aloantígenos están presentes sobre la superficie de los linfocitos B, pero no sobre la superficie de los linfocitos T. Algunos precursores de las células B carecen del receptor SmIg, pero poseen estos aloantígenos.
Es importante recordar que las inmunoglobulinas producidas por las células B pueden fijarse de modo pasivo a las membranas de superficie de las células no B. Por ejemplo, las células con receptores Fc fijan complejos de inmunoglobulina, además, los anticuerpos fluorescentes contra las inmunoglobulinas, que se usan con frecuencia para identificar células SmIg+, pueden fijarse también a los receptores Fc. Los anticuerpos contra células T aumentarán el número de células SmIg+ que se confunden con células B. Se han encontrado estos anticuerpos antilinfocito en diversas enfermedades, incluyendo lupus eritematoso generalizado, mononucleosis infecciosa, neumonía atípica, artritis reumatoide y varios trastornos linfoproliferativos.
Un subgrupo de células B, los llamados linfocitos B1, se desarrollan en fases tempranas y tienen vida media muy prolongada.6 Se encuentran progenitores de células B1 en el hígado fetal y en el epiplón embrionario, pero no en la médula ósea adulta. Las células B1 que expresan CD5 en su superficie se conocen como B1a, y las que no, se denominan células B1b. Las células B1 se asocian con frecuencia con producción de autoanticuerpos. También elaboran grandes cantidades de IL-10.
Por la influencia de los antígenos, los linfocitos T y células accesorias, las células B se diferencian en células plasmáticas, que son células maduras productoras de anticuerpos. Estas células son de mayor tamaño que los linfocitos y se caracterizan por un núcleo redondo y excéntrico con heterocromatina gruesa distribuida en un patrón en rueda de carreta. Las células plasmáticas tienen un citoplasma altamente basófilo, y un retículo endoplásmico bien desarrollado, con frecuencia organizado en capas concéntricas. Las células plasmáticas pueden estar repletas de material granular, que corresponde al anticuerpo que está produciendo [ver figura 2]. Algunas veces, una o más de las cisternas del retículo endoplásmico se encuentran distendidas por grandes inclusiones llamadas cuerpos de Russell; éstos son agregados de moléculas de inmunoglobulinas formadas de manera incompleta. Las células plasmáticas no tienen ya inmunoglobulinas en su superficie. Estas células son terminales, lo que significa que no se dividen. Los precursores inmaduros de las células plasmáticas, los plasmoblastos, son difíciles de distinguir de los linfoblastos y de los linfocitos grandes. En condiciones normales no se encuentran células plasmáticas en sangre periférica.
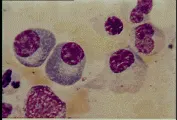
|
| Figura 2 |
| Células plasmáticas |
CELULAS ASESINAS NATURALES
Las células asesinas naturales (NK, por sus siglas en inglés, n. del t.) son linfocitos granulares de gran tamaño que no poseen el complejo RCT-CD3 característico de las células T ni el SmIg de las células B. In vitro, estas células pueden destruir diversas células tumorales o infectadas por virus de modo no específico. Esto es, no requieren de una sensibilización previa o de la presencia de anticuerpos para ser citotóxicas. Sus gránulos contienen proteínas formadoras de poros que pueden mediar la lisis celular. Las células NK no tienen memoria ni muestran restricción de CPH. No se conoce con precisión al precursor de las células NK, pero se piensa que es diferente a la célula tronco tímica (precursora de los linfocitos T) y a la célula pro-B (precursora de los linfocitos B). La IL-12 estimula las células NK para proliferar y producir interferón gama, que es importante en muchas reacciones inmunológicas. 7,8
Las células NK poseen varios marcadores de superficie, incluyendo el CD56, una glucoproteína de 140 kd también denominada NKH1; el CD2, que también está presente en las células T y el CD16, un receptor Fc. El CD16 se asocia con la cadena z del complejo RCT-CD3. Esta cadena participa en la transducción de señales desencadenada por los anticuerpos que se fijan al CD16, lo que activa la célula.
Un gran adelanto ha sido la identificación de receptores NK para las moléculas de clase I del CPH por medio de clonas de células NK-CD3-. Estos receptores difieren en sus pesos moleculares y muestran especificidad alélica para los tres tipos principales de moléculas humanas de clase I codificadas por el HLA-A, HLA-B y HLA-C. Los receptores difieren de los RCT en que no sufren rearreglo de genes y por lo tanto tienen menos diversidad. 9 Al igual que los RCT, los 13 receptores que han sido clonados son proteínas transmembrana de una sola cadena que pertenecen a la superfamilia de los genes de las inmunoglobulinas. Se unen a las moléculas propias de clase I del CPH e inhiben la respuesta citotóxica NK, por lo que se denominan receptores inhibidores de células asesinas. Por otro lado, existen otros receptores en las células NK que pertenecen a la misma superfamilia de genes y que activan a la célula NK cuando se liga a las moléculas de clase I del CPH, denominándose receptores activadores. Se piensa que las alteraciones en las moléculas propias de clase I, sea por virus o por transformación maligna, pueden evitar la detección del automarcador deficiente por los receptores inhibidores de las células NK y permitir que la célula destruya su blanco. 9
Una población de células T citotóxicas no restringida por el CPH puede lisar algunas de las mismas células blanco que destruyen las células NK. Estas células T citotóxicas tienen un marcador de superficie que puede detectarse por medio del anti-CD56, pero no por anti-CD16. Además, expresan el complejo RCT-CD3. 10
Una pequeña proporción de las células nulas de la sangre son precursores mieloides y precursores de los linfocitos T y B inmaduros.
Los monocitos pertenecen al sistema fagocítico mononuclear, antes llamado sistema reticuloendotelial. Son grandes células mononucleares que constituyen del 3 al 8 por ciento de los leucocitos en sangre periférica. Su citoplasma es mucho más abundante que el de los linfocitos. Su núcleo es a menudo excéntrico y oval o reniforme [ver figura 3]. Los lisosomas llenos de enzimas degradantes aparecen como pequeñas vacuolas en el citoplasma. Los monocitos se originan a partir de los promonocitos, los cuales son precursores con una tasa elevada de división y que se encuentran en la médula ósea. Cuando las células maduras entran a la sangre periférica son llamadas monocitos; cuando abandonan la sangre e infiltran los tejidos, sufren cambios adicionales y entonces son conocidas como macrófagos.
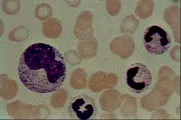
|
| Figura 3 |
| Monocito |
Los macrófagos desempeñan un papel importante en la inducción de la respuesta inmunológica al ser uno de los tipos de células que presenta el antígeno a los linfocitos. También actúan como células efectoras, atacando ciertos microrganismos y células neoplásicas, y eliminando material extraño.
Los macrófagos contienen receptores específicos para anticuerpos y complemento, lo que incrementa su capacidad para fagocitar microrganismos cubiertos con estas moléculas. Los receptores de anticuerpos son para las porciones Fc de la IgG1 y de la IgG3; existe también un receptor Fc para la IgE. Tienen dos receptores del complemento: CR1 y CR3. El CR1 tiene alta afinidad por el componente C3b del complemento y menor afinidad por iC3b y C4b. El CR3, también llamado MAC-1, interactúa con iC3b y con ciertas moléculas de carbohidratos, incluyendo los antígenos que contienen carbohidratos del protozoario Leishmania.
Una pequeña proporción de los antígenos de clase II del CPH está presente en los monocitos; la expresión de las moléculas de clase II del CPH se incrementa considerablemente cuando se activan los macrófagos, lo cual puede ocurrir por varias linfocinas, incluyendo el interferón gama, el factor estimulador de colonias de granulocitos y macrófagos (FEC-GM), el factor activador de los macrófagos (FAM) y el factor inhibidor de la migración (FIM). Citocinas como la interleucina-4 y el factor-ß transformador del crecimiento (FCT-ß) antagonizan esta activación. El antígeno Mo3e, una proteína de 50 kd, es un receptor de urocinasa. Este antígeno ha sido identificado por medio de anticuerpos monoclonales y parece ser específico de los macrófagos activados. El Mo3e no se ha encontrado en otras células hematopoyéticas.11
Los macrófagos producen un gran número de sustancias solubles que son importantes en la respuesta inmunológica y en el proceso de inflamación.12 Estas sustancias incluyen enzimas como el factor activador del plasminógeno y la elastasa, factores de crecimiento como el FEC-GM, citocinas como la interleucina-1 (IL-1), IL-6, IL-10, IL-12 y el factor de necrosis tumoral-a (FNT-a), factores que son críticos para combatir a los microrganismos, como los metabolitos del oxígeno y el óxido nítrico, componentes del complemento de la vía clásica y alterna, proteínas inflamatorias de macrófagos (PIM), y factores que facilitan la reparación tisular, como el factor de crecimiento de fibroblastos (FCF).
Organos linfoides y circulación de linfocitos
El sistema inmune consta de diversos órganos linfoides, incluyendo el timo, los ganglios linfáticos, el bazo y las amígdalas, agregados de tejido linfático en órganos no linfoides, como las placas de Peyer en el intestino, grupos de células linfoides dispersas en todos los tejidos conjuntivos y epiteliales del organismo, así como en la médula ósea y la sangre, y una variedad de células individuales que circulan entre los diversos órganos linfoides y el resto del organismo. Aunque las células inmunológicamente activas de este sistema son los linfocitos, que se dividen en varios tipos, muchas otras células, incluyendo monocitos, macrófagos, granulocitos (i.e., neutrófilos, eosinófilos, basófilos y células cebadas) y plaquetas, desempeñan funciones accesorias importantes en el sistema inmunológico [ver figura 4].
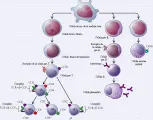
|
| Figura 4 |
| Desarrollo de las células del sistema inmunológico |
TIMO
El timo, que se origina en el embrión a partir de la tercera y cuarta bolsas faríngeas, se localiza en el mediastino anterior y está formado por múltiples lóbulos, cada uno de los cuales tiene una médula y una corteza [ver figura 5].
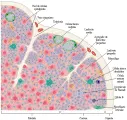
|
| Figura 5 |
| Estructura del timo |
Las células pre-T derivadas de la médula ósea entran al timo. Sus células hijas pueden expresar el complejo RCT-CD3 (RCD es el receptor de la célula T y CD significa grupo de diferenciación) y posteriormente adquieren la capacidad de reaccionar con diferentes péptidos unidos al complejo principal de histocompatibilidad (CPH). Estos timocitos sufren después un proceso de selección negativa o positiva. Solo un pequeño porcentaje de los timocitos con selección positiva, la mayoría de los cuales tienen restricción por el CPH (ver adelante, Propio y no propio), migran a la médula y después se movilizan hacia el sistema linfoide periférico.
Dos a 3 días después de que las células tronco entran al timo los linfocitos migran a través de la pared de las vénulas poscapilares de la médula, después ingresan al torrente sanguíneo y se albergan en los órganos linfoides periféricos. Una vez ahí los linfocitos dejan la circulación, de nuevo a través de las vénulas poscapilares y entran a regiones dependientes del tipo en el sistema linfoide periférico: la corteza interna de los ganglios linfáticos, las vainas periarteriales del bazo y las áreas intranodulares en las placas de Peyer, las amígdalas y el apéndice. Algunas células T en la mucosa intestinal (linfocitos intraepiteliales) pueden no derivar del timo y diferenciarse in situ.
Si no existe timo al nacimiento el paciente tendrá linfocitopenia, con depleción marcada o ausencia de células T. Las áreas dependientes del timo del sistema linfoide periférico tampoco contienen linfocitos. Existe alteración franca en la inmunidad celular y las respuestas de anticuerpo que requieren cooperación de las células T (excepto la respuesta IgM) son deficientes.
El timo involuciona con la edad, lo que puede explicar el desarrollo de deficiencias del sistema inmunológico en los ancianos.
Las evidencias actuales indican que en los humanos las células tronco se diferencian a células B en la médula ósea [ver figura 4] y en los órganos linfoides periféricos.
El principal sitio de activación inicial de la célula T es el ganglio linfático, en donde convergen linfocitos provenientes de la sangre y antígenos de la linfa, mediadores solubles y otras células [ver figura 6]. Los linfáticos aferentes llevan la linfa y células al ganglio y entran en la región subcapsular vaciando su contenido al seno subcapsular. La linfa y las células salen a través de los linfáticos eferentes en el hilio.
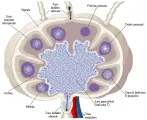
|
| Figura 6 |
| Estructura del ganglio linfático |
La infraestructura del ganglio linfático consiste en una red extensa en donde las células presentadoras de antígeno (CPA) y los linfocitos T se encuentran e interactúan. Por ejemplo, las células dendríticas en la piel captan el antígeno y viajan a través de los linfáticos hacia el ganglio linfático que drena. Las células migran entonces a través del piso del seno subcapsular del ganglio linfático, por las regiones interfoliculares y se establecen en la red de la paracorteza en forma de células dendríticas interdigitantes (CDI), Las células T de la sangre migran a través de vénulas poscapilares especializadas, conocidas como vénulas endoteliales altas (VEA) y migran a lo largo de la misma red, en donde se ponen en contacto con numerosas CDI presentadoras de antígeno. 13
Los centros germinales contienen células B y células dendríticas foliculares (CDF) características, que no derivan de la médula ósea y que pueden retener complejos antígeno-anticuerpo durante mucho tiempo. Los centros germinales activos son rodeados por un manto de células B (células foliculares en manto) que expresan IgD en su superficie y que pueden madurar hacia células plasmáticas que producen solo anticuerpos de IgM. Las células B en el centro germinal (centrocitos) sufren un cambio de clase para producir otros isotipos, como IgG, IgA e IgE. Las células B con anticuerpos de alta afinidad en su superficie parecen ser seleccionadas al unirse a las CDF que retienen antígenos. Los linfocitos B que no son seleccionados sufren apoptosis (muerte celular programada). La célula B más madura, la célula de memoria, se localiza en el centro germinal y puede evolucionar a células plasmáticas productoras de todos los isotipos.
Las células dendríticas derivadas de la médula ósea, que se encuentran en las áreas paracorticales del ganglio linfático, tienen un papel importante para iniciar respuestas inmunológicas dependientes de células T. 14 Estas células se encuentran también como células inmaduras en los órganos no linfoides, en especial en la epidermis (en donde se denominan células de Langherhans). Fagocitan y procesan antígenos y expresan receptores de superficie de clase II del CPH, Fc y de manosa. Las citocinas inflamatorias como la interleucina-1b (Il-1b) y el factor de necrosis tumoral-a (FNT-a), promueven la migración de las células dendríticas por los linfáticos aferentes a los ganglios linfáticos, en donde maduran, pierden su capacidad para fagocitar y expresan moléculas coestimuladoras cruciales como CD80 (B7-1) y CD86 (B7-2), dando a las células dendríticas la capacidad para presentar antígenos a las células T. También se encuentran células dendríticas inmaduras en los ganglios linfáticos, en donde pueden fagocitar antígenos que provienen de los linfáticos aferentes y que después maduran a CDA.
En el bazo los linfocitos se concentran en la pulpa blanca que rodea a las arteriolas centrales [ver figura 7].
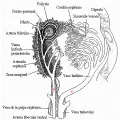
|
| Figura 7 |
| Estructura del bazo |
OTROS TEJIDOS LINFOIDES
Los linfocitos se encuentran también en otros lugares. El tejido linfoide relacionado con el intestino incluye las placas de Peyer y el apéndice. Estos tejidos contienen regiones con predominio de linfocitos T o B semejantes a los encontrados en los centros germinales. Las células epiteliales especializadas denominadas células M parecen tener una capacidad única para captar y presentar antígenos a los linfocitos adyacentes. 15 Las células M se encuentran cercanas a las placas de Peyer. Otros linfocitos en el intestino son los linfocitos de la lámina propia (LLP), los encontrados en las vellosidades y los linfocitos intraepiteliales (LIE), que se encuentran entre las células epiteliales. La migración y adherencia de los LLP depende en parte de las integrinas y las selectinas. Las células linfoides asociadas a las mucosas se localizan en las vías respiratorias y genitourinarias, pero no en estructuras inductivas reconocidas.
Existen tres tipos principales de circulación de los linfocitos: (1) la siembra de las células tronco provenientes del hígado fetal o de la médula ósea en los órganos linfoides primarios y la subsecuente diferenciación y distribución de estas células en el sistema linfoide periférico, (2) la recirculación sangre-linfa-sangre de linfocitos y (3) la distribución de células efectoras a regiones particulares del organismo. Los linfocitos circulan en forma continua de la sangre a los tejidos y de regreso a la sangre. Sin embargo, el tráfico de los linfocitos vírgenes (CD45RA) es diferente al de las células efectoras activadas o de memoria (CD45RO). Los linfocitos nativos recirculan a través de los tejidos linfoides secundarios, como los ganglios linfáticos, el bazo, las amígdalas y las placas de Peyer, hacia microambientes especiales en los que se encuentran antígenos, citocinas y otras células que ocasionan su activación. Por el contrario, las células efectoras activadas o de memoria pueden tener también tráfico a sitios extralinfoides en diversos tejidos, como la piel y la lámina propia del intestino. 16
La disposición de los linfocitos se controla por la expresión de diversos receptores en la superficie celular y contrarreceptores en el endotelio vascular [ver figura 8]. Para detener el flujo de células en los vasos, ocurre una adhesión inicial entre los receptores de los linfocitos y los de las microvellosidades de las células, como la L-selectina y su contrarreceptor en el endotelio, la adresina de ganglios linfáticos periféricos (PNAd). Si la interacción inicial no es seguida de interacciones subsecuentes, una proteasa desnaturalizará la L-selectina, liberando a la célula. 17 Otros receptores celulares permitirán la fijación a la E-selectina y P-selectina endoteliales. Posteriormente las células pueden fijarse y rodar usando sus receptores de superficie de integrina-Ig, como a4b7 y a4b1, que se fijan a la molécula de adhesión celular adresina-1 de la mucosa endotelial (MAdCAM-1) y la molécula-1 de adhesión vascular (VCAM-1), respectivamente. Estas interacciones pueden causar una detención estable que incluye un receptor que desencadena la adhesión a través de señales intracelulares que involucran una proteína fijadora de guanosina trifosfato (GTP). La cooperación entre las interacciones de los receptores es indispensable porque la interacción inicial con la L-selectina puede ser demasiado débil para inducir una interacción estable LFA-1/ICAM-1 (antígeno asociado a la función leucocitaria-1/molécula de adhesión intercelular-1) y por lo tanto se requiere la interacción a4b7/MadCAM-1. Por el contrario, cuando los tejidos expresan niveles altos del receptor L-selectina, el contacto y rodamiento mediado por la L-selectina puede ser suficiente para lograr una detención estable mediada por LFA-1. Las citocinas producidas durante la inflamación pueden aumentar la expresión de receptores de adhesión y así incrementar la infiltración de células al sitio.
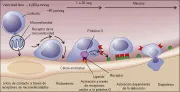
|
| Figura 8 |
| Modelo de reconocimiento y reclutamiento de linfocitos |
Al parecer las variaciones en estos mecanismos se deben a los diferentes subtipos de linfocitos que responden en diversos microambientes en el ganglio linfático, como los centros germinales, las áreas paracorticales y la zona T, en donde las células, los antígenos y los factores solubles provocan una respuesta inmunológica particular. Estos miroambientes son regulados además por diversas citocinas, como el factor de transformación de crecimiento b1 (FTC-b1), que puede aumentar la expresión de integrinas y mediar la unión de las células B a las CPA. Las citocinas como la IL-2 y la IL-10, y en especial las muchas quimocinas generadas por el proceso inflamatorio, también regulan a los receptores de adhesión linfocitaria como el CD44, y otras citocinas influyen en la actividad de los linfocitos al atravesar los tejidos.
Memoria y especificidad inmunológica
La capacidad de los linfocitos T y de los anticuerpos producidos por los linfocitos B para discriminar entre los antígenos está determinada por los genes de la región variable conocidos como V (variable), D (diversidad) y J (de unión). Durante la maduración de los linfocitos T y B ocurre rearreglo de las secuencias de ADN en estos genes, además de mutaciones somáticas adicionales en las células B. Los varios cientos de genes de la regiones V, D y J codifican las inmunoglobulinas (anticuerpos) y los RCT, que pueden distinguir alrededor de un billón de diferentes especificidades antigénicas. Cada linfocito tiene un receptor de superficie que reconoce un solo determinante antigénico o epítope. Los receptores de las células B reconocen antígenos nativos. Después de que las células B interactúan con un antígeno, proliferan y se diferencian en células de memoria y en células plasmáticas para la producción de anticuerpos.
Antes de ser reconocido por las células T, el antígeno es captado por una célula presentadora de antígeno (CPA) (v.gr., una célula dendrítica o un macrófago), que lo fragmenta en péptidos pequeños. En la CPA ciertos fragmentos o epítopes son tomados por las moléculas clase II del CPH y transportados hacia la superficie de la CPA. La necesidad de que el antígeno sea presentado en asociación con una molécula del CPH se denomina restricción del CPH. Por lo tanto, los RCT no reconocen antígenos nativos, sino solo partes procesadas del mismo. Después de que las células T interactúan con un antígeno, proliferan y se diferencian a células de memoria. También se vuelven reguladoras en la producción de anticuerpos por las células B y en las respuestas efectoras mediadas por células de otras células T.
Una característica de la respuesta inmunológica es su capacidad para incrementar el número de linfocitos antígeno-específicos después de un estímulo antigénico; por lo tanto, una exposición posterior al mismo antígeno proporciona una respuesta más rápida y de mayor intensidad (i.e., respuesta anamnésica). La base de esta respuesta aumentada es la proliferación de linfocitos antígeno-específicos y la producción de células de memoria después de la interacción con el antígeno. Estas respuestas están mediadas por la producción de citocinas por los linfocitos y otras células. Los mecanismos de respuesta inmunológica también se amplifican por la liberación de sustancias mediadoras de células cebadas y basófilos cubiertos de anticuerpos, por la activación de proteínas del complemento y por la expresión de moléculas de integrina en las células. La alteración en la permeabilidad vascular, la expresión de receptores para leucocitos en las células endoteliales y la liberación de factores quimiotácticos por estos mecanismos secundarios atrae otros tipos de célula a la reacción. Estas células contribuyen en mucho al proceso inflamatorio al incrementar el proceso fagocítico y la eliminación de antígenos extraños.
Un grupo de células B, las células de memoria, aumentan la respuesta inmunológica a los antígenos ya conocidos, lo que se conoce como respuesta inmune secundaria, respuesta anamnésica o respuesta de refuerzo. Estas células B de memoria sufren mutación somática en las regiones variables de sus genes de inmunoglobulina. Cuando ocurre esta mutación somática en los centros germinales de los ganglios linfáticos que contienen antígenos unidos a células dendríticas, origina la selección de células B de memoria que tienen receptores de alta afinidad para los antígenos.
Es importante que la respuesta inmunológica sea capaz de distinguir entre lo propio y lo no propio. Cuando no, las células T y los anticuerpos atacarán en forma constante a células y componentes tisulares autólogos. En los años 50, Sir Frank Macfarlane Burnet propuso por primera vez que en el estado prenatal la interacción de los autoantígenos con los linfocitos específicos para estos antígenos causaba la eliminación de los linfocitos autorreactivos18 y tolerancia inmunológica. Cuando la tolerancia inmunológica se pierde los anticuerpos y células sensibilizados (reactivos contra antígenos) que están dirigidos contra autoantígenos causan las enfermedades autoinmunes. 19
Las respuesas inmunológicas están controladas por tres grandes familias de genes: (1) los genes que codifican para los elementos variables de las inmunoglobulinas, (2) los genes que codifican los RCT y (3) los genes que codifican los antígenos del CPH. En cada persona existe un número enorme de genes que codifican para los elementos variables de las inmunoglobulinas y de los RCT, permitiendo el reconocimiento específico de millones de antígenos. Sin embargo, la extrema variabilidad del CPH se aplica a las poblaciones como un todo y existen muy pocas variaciones en un individuo determinado.
Los genes que controlan la producción de los antígenos del CPH (también conocidos como antígenos de histocompatibilidad o antígenos HLA, en los humanos) se localizan muy cerca entre sí en el cromosoma 6. Los antígenos del HLA identifican a las células autólogas como propias y las distinguen de las de otros individuos. El gen Ir en el ratón y en gen HLA en los humanos constituyen parte del CPH, que tiene un papel crucial en la respuesta inmunológica. Las características del CPH explica por qué algunos individuos pueden no responder a ciertos antígenos. Por ejemplo, aunque los RCT reconocen epítopes que están unidos a las moléculas del CPH, algunos péptidos antigénicos pueden no acomodarse en el surco de la molécula CPH específica de un individuo. Por lo tanto, el tipo apropiado de célula T no reaccionará al epítope y el individuo no será capaz de montar una respuesta inmunológica en su contra.
REARREGLO GENETICO DEL RECEPTOR DE LA CELULA T
Los genes que codifican para el rearreglo de los RCT y los genes de los segmentos V, D, J y C (constante) interaccionan para formar dos tipos de RCT, cada uno consistente en dos cadenas. Cada tipo de RCT se asocia con una especificidad particular. Los genes que codifican las cadenas g y d se rearreglan para formar RCT-gd y los que codifican para las cadenas a y b constituyen RCT-ab. Después de que ocurre el rearreglo genético el RCT se expresa en la superficie de la célula T, junto con las proteínas CD3. Al mismo tiempo que ocurre el rearreglo genético se expresan varias moléculas accesorias importantes, incluyendo CD4, CD8, CD2 y CD3. 20,21 Relativamente pocas células T evolucionan a CD4-CD8+ RCT-gd, cuya función no se conoce del todo. La mayoría de las células T se vuelven RCT-ab que expresan primero tanto CD4 como CD8( y después de la selección positiva se vuelven CD4+CD8-RCT-ab (cooperadoras) o CD4-CD8+RCT-ab (con función citotóxica, supresora o ambas). Los timocitos maduros CD4+, CD8+ expresan niveles altos del complejo RCT-CD3.
La selección positiva está controlada por células epiteliales de la corteza tímica y CPA dirigidas, como macrófagos, células dendríticas y células interdigitantes. Muchas de estas células del estroma se localizan en la unión corticomedular. 22 Debido a que las células T pueden reaccionar con antígenos solo en asociación con el CPH propio, solo las células T que tengan un RCT que pueda unirse con el CPH propio serán seleccionadas. Cuando estas células reaccionan con el CPH propio en las células tímicas del estroma, las células CD4+CD8+ RCT-ab que se unen a moléculas de clase II del CPH se transforman en células CD4+CD8-, desapareciendo la expresión de CD8 y aumentando la del complejo RCT-CD3. Por el contrario, las células CD4+CD8+ que se unen a moléculas de clase I del CPH disminuyen su expresión de CD4 y se transforman en células CD4-CD8+ RCT-ab. De esta manera se seleccionan las células CD4+RCT-ab y las células CD8+RCT-ab restringidas por el CPH. Se supone que las células T que se unen incluso en forma débil a las moléculas del CPH por medio del ligando CD4 o CD( serán capaces de fijarse en forma más intensa a las CPA, por lo que no se necesita la presentación de los antígenos extraños para la selección positiva. La mayoría de las células T no interactúan con el CPH propio y sufren muerte programada, o apoptosis.
El proceso de selección negativa elimina las células T que tienen un RCT con afinidad intensa por los autoantígenos. Si no se eliminan, estas células pueden causar enfermedad autoinmune seria. Existen muchos autoantígenos en las células del epitelio tímico y los estudios muestran que ciertos autoantígenos pueden ser presentados por diversas CPA en el timo, como los macrófagos y las células dendríticas. Es posible que la selección negativa de ciertos autoantígenos ocurra cuando las células T se trasladan al sistema linfoide periférico dejando el timo. Esta interacción de alta afinidad entre el autoantígeno presentado por el CPH y el RCT sobre la célula T inmadura al parecer desencadena la muerte celular. Por el contrario, la interacción entre el antígeno presentado en la molécula del CPH y el RCT en la célula T madura causa activación. Se desconoce la manera como se seleccionan los RCT gd en forma positiva o negativa.
La apoptosis tiene un papel crucial en el desarrollo y función del sistema linfoide. Las señales apoptóticas que actúan sobre la membrana celular ocasionan formación de ampollas, encogimiento de la célula y condensación y destrucción de la cromatina nuclear. En minutos a horas la célula es destruida y eliminada por macrófagos.
Los principales receptores sobre las células linfoides para la apoptosis son Fas (CD95/APO-1), activado por el ligando de Fas, 23 y el receptor del factor de necrosis tumoral-1 (RFNT-1), activado por el FNT-a y la linfotoxina-a. También puede participar el receptor de la linfotoxina b. Es importante mencionar que el RFNT-1 puede iniciar vías que ocasionan la activación del factor nuclear kB (FN-kB), que protege contra la apoptosis, 24,25 y de la cinasa terminal Jun N (JNK), que activa genes y no participa en la apoptosis. Estas respuestas están mediadas por vías independientes. 26
Las señales de apoptosis eventualmente actúan sobre una familia de proteasas de cisteína semejantes a la enzima convertidora de IL-1b (ECI), el prototipo que actúa sobre el precursor de la citocina IL-1b y que lo convierte en la citocina activa. Las proteasas relacionadas a la ECI activan otras proteasas, que después actúan sobre diversos sustratos y a su vez causan apoptosis.
Las células T citotóxicas también usan las vías apoptósicas de Fas y RFNT-1 para destruir células blanco. Además de esta vía no secretora, las células T citotóxicas pueden mediar la apoptosis en sus células blanco por una vía secretora a través de perforinas, que facilitan la entrada de proteasas citotóxicas como granzima A y B a la célula blanco [ver figura 9]. La granzima B ha demostrado activar el CPP32, el precursor de la proteasa responsable de escindir la poli (ADP-ribosa) polimerasa. 27 La apoptosis también puede ser inducida en las células B y T por esteroides. Uno de los mecanismos por los que los esteroides inducen apoptosis es por aumentar la expresión del receptor tipo 3 de inositol 1,4,5-trifosfato (IP3R3) en la membrana plasmática. 28 El IP3R3 permite el aumento en el calcio intracelular que facilita la apoptosis de las células T y B.
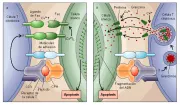
|
| Figura 9 |
| Mecanismo de apoptosis de las células T |
Figura 1 Cortesía del Dr. Curtis B. Wilson, Scripps Clinic and Research Foundation.
Figuras 2 y 3 Cortesía del Dr. Arthur T. Skarin, Harvard Medical School and Dana-Farber Cancer Institute.
Figuras 4, 5, 6, 8 y 9 Seward Hung.
Figura 7 Carol Donner.
Bibliografía
- Mosmann TR, Coffman RL: Heterogeneity of cytokine secretion patterns and functions of helper T cells. Adv Immunol 46:111, 1989
- Romagnani S: Human TH1 and TH2 subsets: doubt no more. Immunol Today 12:256, 1991
- Feng Y, Broder CC, Kennedy PE, et al: HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872, 1996 [PMID 8629022]
- Choe H, Farzan M, Sun Y, et al: The b-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85:1135, 1996 [PMID 8674119]
- Doranz BJ, Rucker J, Yi Y, et al: A dual-tropic primary HIV-1 isolate that uses fusin and the b-chemokine receptors CKR-5, CKR-3, and CKR-2 as fusion cofactors. Cell 85:1149, 1996 [PMID 8674120]
- Qin XF, Schwers S, Yu W, et al: Secondary V(D)J recombination in B-1 cells. Nature 397:355, 1999 [PMID 9950428]
- Wolf SF, Temple PA, Kobayashi M, et al: Cloning of cDNA for natural killer cell stimulatory factor, a heterodimeric cytokine with multiple biologic effects on T and natural killer cells. J Immunol 146:3074, 1991 [PMID 1673147]
- Williams NS, Klem J, Puzanov IJ, et al: Natural killer cell differentiation: insights from knockout and transgenic mouse models and in vitro systems. Immunol Rev 165:47, 1998 [PMID 9850851]
- Rolstad B, Naper C, Vaage JT: Natural killer (NK) cell recognition of MHC class I molecules. The Immunologist 4:165, 1996
- Lanier LL, Le AM, Cwirla S, et al: Antigenic, functional, and molecular genetic studies of human natural killer cells and cytotoxic T lymphocytes not restricted by the major histocompatibility complex. Fed Proc 45:2823, 1986 [PMID 3095151]
- Todd RF III, Liu DY: Mononuclear phagocyte activation: activation-associated antigens. Fed Proc 45:2829, 1986
- Auger MJ, Ross JA: The biology of the macrophage. The Macrophage. Lewis CE, McGee JO'D, Eds. Oxford University Press, Oxford, England, 1992, p 1
- Gretz JE, Kaldjian EP, Anderson AO, et al: Sophisticated strategies for information encounter in the lymph node: the reticular network as a conduit of soluble information and a highway for cell traffic. J Immunol 157:495, 1996 [PMID 8752893]
- Austyn JM: New insights into the mobilization of phagocytic activity of dendritic cells. J Exp Med 183:1287, 1996
- Neutra MR, Frey A, Kraehenbuhl J-P: Epithelial M cells: gateways for mucosal infection and immunization. Cell 86:345, 1996 [PMID 8756716]
- Watson SR, Bradley LM: The recirculation of naive and memory lymphocytes. Cell Adhes Commun 6:105, 1998 [PMID 9823460]
- Steeber DA, Tang ML, Zhang XQ, et al: Efficient lymphocyte migration across high endothelial venules of mouse Peyer's patches requires overlapping expression of L-selectin and beta 7 integrin. J Immunol 161:6638, 1998 [PMID 9862692]
- Burnet M: The Clonal Selection Theory of Acquired Immunity. Cambridge University Press, London, 1959
- Ring GH, Lakkis FG: Breakdown of self-tolerance and pathogenesis of autoimmunity. Semin Nephrol 19:25, 1999 [PMID 9952278]
- Knapp W, Dörken B, Gilks WR, et al, Eds: Leukocyte Typing IV: White Cell Differentiation Antigens. Oxford University Press, Oxford, England, 1989
- Schlossman SF, Boumsell L, Gilks W, et al: Update: CD antigens 1993. J Immunol 152:1, 1994 [PMID 7902854]
- Cosgrove D, Chan SH, Waltzinger C, et al: The thymic compartment responsible for positive selection of CD4+ T cells. Int Immunol 4:707, 1992 [PMID 1352128]
- Fraser A, Evan G: A license to kill. Cell 85:781, 1996 [PMID 8681372]
- Beg AA, Baltimore D: An essential role for NF-kB in preventing TNF-a-induced cell death. Science 274:782, 1996 [PMID 8864118]
- Van Antwerp DJ, Marting SJ, Kafri T, et al: Suppression of TNF-a-induced apoptosis by NF-kB. Science 274:787, 1996 [PMID 8864120]
- Lui Z-g, Hsu H, Goeddel DV, et al: Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kB activation prevents cell death. Cell 87:565, 1996
- Darmon AJ, Nicholson DW, Bleackley RD: Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature 377:446, 1995 [PMID 7566124]
- Khan AA, Soloski MJ, Sharp AH, et al: Lymphocyte apoptosis: mediation by increased type 3 inositol 1, 4, 5-trisphosphate receptor. Science 273:503, 1996 8662540