Endocrinología
⭳ Abrir artículo (PDF)477.6 KBEste artículo fue revisado respecto a la Edición 2/2000. Ver esa versión →
Contenido del artículo
VI PARATIROIDES
- Fisiología de la regulación del calcio
- Hiperparatiroidismo
- HIPERPARATIROIDISMO PRIMARIO
- Etiología y patogenia
- Manifestaciones clínicas
- Diagnóstico de laboratorio
- Tratamiento
- Cáncer de las paratioides
- HIPERPARATIROIDISMO SECUNDARIO
- Hipercalcemia
- GUIAS DIAGNOSTICAS
- Aumento en la absorción gastrointestinal de calcio
- Aumento en la resorción ósea
- Aumento en la reabsorción renal de calcio
- Diagnóstico diferencial
- TRATAMIENTO
- Hipocalcemia
- Calcitonina y sus trastornos
VI PARATIROIDES
DR. DANIEL D. FEDERMAN
Los trastornos de las paratiroides solían considerarse raros, difíciles de diagnosticar y frustrantes en cuanto a tratamiento. Sin embargo, las técnicas diagnósticas y quirúrgicas modernas han demostrado que son comunes y susceptibles de tratarse.
Fisiología de la regulación del calcio
Los niveles de calcio en el líquido extracelular y los reservorios óseos son controlados principalmente por la hormona paratiroidea (PTH) y el calcitrol (la forma activa de la vitamina D) [ver tabla 1]. Las hormonas calcitonina y proteína relacionada a la PTH son activas en niños y en casos patológicos, pero tienen escaso papel fisiológico en los adultos.
HORMONA PARATIROIDEA
La PTH es sintetizada inicialmente como parte de una molécula más grande, pre-pro-PTH, que es inmediatamente acortada a pro-PTH. Aunque la pro-PTH tiene una vida más prolongada que su precursora, ninguna de las moléculas es liberada fuera de la glándula y ninguna es biológicamente activa.1 Sin embargo, la ruptura enzimática de pro-PTH deja en libertad a la PTH para su liberación a la circulación, donde es desdoblada por lo menos en dos porciones por los tejidos periféricos, principalmente el hígado y el riñón [ver figura 1]. Además, la paratiroides libera algunos fragmentos de la hormona. De esta forma, la PTH circulante incluye tres moléculas: la molécula completa, el fragmento amino-terminal con una vida media corta y el fragmento carboxi-terminal con una vida media más larga. Sólo la molécula completa y el fragmento amino-terminal son biológicamente activos.2
La síntesis de PTH y su liberación están controlados principalmente por los niveles séricos de calcio: un valor bajo estimula, y uno alto suprime la síntesis y liberación hormonal2 [ver figura 2]. Una cifra sérica elevada de fósforo por la disminución del calcio también estimula la liberación de PTH y es particularmente importante en la insuficiencia renal. La hipomagnesemia puede impedir la liberación de PTH y alterar la respuesta de los tejidos hacia la hormona, aunque estos efectos ocurren únicamente en situaciones patológicas. Algunos otros factores que influyen en la secreción de PTH, incluyendo hormonas alfa y beta adrenérgicas, neurotransmisores, prostaglandinas y otras hormonas, tienen efecto farmacológico, pero no son claros reguladores fisiológicos de la liberación de PTH.
La regulación intracelular de la secreción de PTH está controlada por un receptor de superficie celular recién detectado que fija iones calcio (Ca2+) en una estructura extracelular que recuerda un matamoscas. El receptor está acoplado a una porción intracelular de la célula a través de un dominio de serpentina de siete segmentos que atraviesan la membrana, característicos de la superfamilia de receptores unidos a la proteína G.3,4 Este receptor, el primero que se identificó para un ión extracelular, convierte al calcio en una hormona, o primer mensajero, así como un segundo mensajero intracelular. Esta molécula sensible al calcio también se encuentra en las células C del tiroides, en donde controla la liberación de calcitonina, en el riñón, en donde influye en el intercambio de fósforo y calcio, y en el sistema nervioso central y otros tejidos.5
La PTH mantiene los niveles séricos de calcio por promoción directa e indirecta de la entrada de calcio a la sangre a partir de tres sitios de intercambio: intestino, hueso y riñón. La PTH contribuye a la absorción neta de calcio por el intestino al favorecer la síntesis de calcitrol por el riñón. También promueve la entrada de calcio a la sangre desde el hueso por la inhibición de la función de síntesis de los osteoblastos e indirectamente estimulando la diferenciación hacia células de resorción ósea u osteoclastos. Finalmente, tiene tres efectos en el riñón: estimula la reabsorción tubular de calcio, incrementa la depuración de fósforo y promueve un aumento en la enzima que completa la síntesis de la forma activa de la vitamina D. Como la mayoría de las hormonas peptídicas, la PTH ejerce sus efectos a través de la activación de las adenilato-ciclasas unidas a membranas que generan monofosfato cíclico de adenosina (AMPc).
Los niveles circulantes de PTH (hormona paratiroidea inmunorreactiva o PTHi) se determina por radioinmunoanálisis. Los anticuerpos que detectan la PTH intacta, en especial la prueba inmunorradiométrica con doble anticuerpo son lo bastante sensibles y específicos para uso clínico. Esta prueba ha sustituido en mucho a otras usadas para evaluar eventos mediados por PTH, como la producción de AMPc nefrogénico, en la evaluación de la función paratiroidea [ver adelante, Hiperparatiroidismo, Hiperparatiroidismo primario].
VITAMINA D
La vitamina D es uno de los componentes importantes del sistema endócrino que regula el metabolismo de los minerales óseos. La síntesis de la vitamina D activa se inicia con la radiación ultravioleta sobre el 7-dehidrocolesterol en la piel, produciendo previtamina D3, que es convertida a vitamina D3 (colecalciferol). Esta vitamina D3 y parte de la que es absorbida de la dieta, es hidroxilada en la posición 25 en el hígado para formar 25-(OH)D3 o calcifediol, que es la forma principal de vitamina D circulante; los niveles plasmáticos de calcifediol promedian 30 ng/ml. El calcifediol es 1alfa-hidroxilado para formar calcitriol [1,25-(OH)D3, por una enzima renal cuya actividad es estimulada por la PTH, la hipocalcemia (a través de la estimulación de PTH) y la hipofosfatemia. El calcitriol parece ser la forma más activa de vitamina D; aunque sus niveles séricos de 25 a 40 pg/ml son mucho menores que los de calcifediol, es 100 veces más potente en base a su peso. Las enfermedades hepáticas pueden impedir la 25-hidroxilación y las enfermedades renales pueden suprimir la 1alfa-hidroxilación. La producción de calcitriol es autorregulada hasta cierto grado, y otras hormonas como prolactina, hormona del crecimiento y esteroides sexuales pueden también modular su producción.
El calcitriol ejerce su acción en los mismos tejidos que la PTH [ver figura 2], pero sus sitios subcelulares y su mecanismo de acción son distintos. A diferencia de la PTH, que interactúa con un receptor en la membrana celular, el calcitriol se une a un receptor intracelular y el complejo se fija a sitios específicos en la cromatina. A este respecto, el calcitriol actúa como una hormona esteroidea. El receptor de la vitamina D es un miembro de la superfamilia c-erb A de receptores, son miembros de esta misma familia los receptores que fijan tiroxina, esteroides y ácido retinoico.
En el intestino el calcitriol estimula la síntesis de proteína fijadora de calcio, y también estimula la absorción de calcio y fósforo.6 Al aumentar las concentraciones séricas de calcio y fósforo el calcitriol promueve el depósito de hidroxiapatita en el hueso. En forma paradójica, también moviliza calcio del hueso ya formado. El calcitriol inhibe además la síntesis de pre-pro-PTH y quizá sea un regulador fisiológico de la secreción de PTH. Existen receptores de calcitriol en muchos tejidos además del hueso, riñón e intestino, y entre los órganos y tipos celulares que los tienen se incluyen las glándulas paratiroides, los islotes pancreáticos, la glándula mamaria, los fibroblastos, y otros tejidos que no se conocen como órganos blanco del calcitriol.10 El papel del calcifediol y de la 24,25-(OH)2D3, un producto del metabolismo renal del calcifediol, no está bien definido; es probable que este último compuesto promueva la movilización de mineral óseo.
Los ensayos para calcifediol pueden ser útiles para ayudar en las decisiones clínicas. Esta forma de vitamina D tiene una vida media larga y niveles estables en el plasma, con un rango normal de 15 a 40 ng/ml. La principal aplicación clínica de la prueba de calcifediol es la valoración del metabolismo de la vitamina D: los valores bajos reflejan deficiencia de vitamina D o enfermedad hepática severa y ocurren en el embarazo, en el hipertiroidismo y durante el tratamiento con anticonvulsivos. Las cifras elevadas confirman la intoxicación con vitamina D. Hay menos experiencia con los ensayos de calcitriol, pero sus niveles están regulados en una manera distinta a los de calcifediol; los límites normales de 25 a 40 pg/ml varían con las estaciones (son más bajos en el invierno), con la edad (menores en personas de edad avanzada), los niveles de calcio sérico y otros factores menos claros. Sus valores se elevan por la PTH cuando la función renal está intacta; de esta forma, la medición del calcitriol es útil en el diagnóstico diferencial de la hipercalcemia.
CALCITONINA
El tercer regulador hormonal del balance del calcio es la calcitonina, polipéptido secretado por las células parafoliculares del tiroides. Su principal acción es reducir la reabsorción ósea, realizada por acción directa de los osteoclastos, al antagonizar la PTH y otros estímulos osteoclásticos. El efecto de la calcitonina es directamente proporcional a la tasa de formación y remodelación ósea, y al parecer su función consiste en minimizar la pérdida de calcio del esqueleto durante el crecimiento, el embarazo y la lactancia. Es posible que tenga una función limitada, si acaso tiene alguna, en el adulto. Las cifras de calcitonina disminuyen después de la menopausia y esta deficiencia relativa puede contribuir a la osteoporosis. Sin embargo, debe recordarse que ni la ausencia total ni el exceso extremo de la hormona producen manifestaciones clínicas claras. Este dato es sorprendente porque la calcitonina y sus receptores se encuentran diseminados en el cerebro, riñón y otros sitios.8 El procesamiento alternativo del gen de la calcitonina produce un péptido relacionado que tiene poderosos efectos hemodinámicos y que quizá sea importante en el sistema nervioso central. Este péptido no tiene función en la homeostasis del calcio.
PROTEINA RELACIONADA A LA PTH
Aunque se conoce otra hormona reguladora del calcio, su función precisa no ha sido bien determinada. La proteína relacionada con la hormona paratiroidea (PTHrP por sus siglas en inglés, n. del t.), fue detectada al principio como la causa de la hipercalcemia humoral en las neoplasias malignas (HHM), pero ha sido identificada (o su ARNm) en muchos tejidos, incluyendo la placenta y las glándulas paratiroides del feto, además de en queratinocitos, neuronas, islotes pancreáticos, riñón y hueso en el adulto. La molécula se parece mucho a la de la PTH en su región N-terminal, pero es más larga y más compleja que la PTH en otras regiones: por separación alternativa, sus ocho exones pueden, por medio de separación alterna, producir tres proteínas diferentes. Existen dos carcterísticas únicas de esta hormona. Primero, comparte un receptor único con la PTH y, segundo, parece tener un segundo locus biológicamente activo además de la región similar a la PTH. Este segundo sitio activo participa en el transporte de calcio en la placenta, convirtiendo así a la PTHrP en la PTH del feto. Al parecer,la PTHrP tienen una función principalmente autócrina local, la única alteración clínica significativa que se ha asociado con esta proteína es su secreción excesiva en diversos tumores, provocando la HHM.11
EFECTOS DE LA EDAD
Con la edad ocurren cambios importantes en el metabolismo del calcio y de las hormonas relacionadas: (1) el nivel sérico medio de PTHi es de 20 a 40 porciento más alto en personas mayores de 70 años, (2) los valores de calcitriol parecen disminuir en las personas mayores,12,13 y (3) los niveles de calcitonina disminuyen en las mujeres de edad avanzada. Estos cambios pueden relacionarse con ingesta inadecuada de calcio, porque los niveles de este ión no cambian con la edad. Cuando mujeres ancianas recibieron una dieta con 2,400 mg/día de clacio, sus niveles de PTH fueron semejantes a los de pacientes jóvenes.14,15 La principal expresión clínica de estos cambios es la reducción de la masa ósea relacionada con la edad. En algunos pacientes, esta disminución causa osteoporosis y mayor riesgo de fracturas.
Hiperparatiroidismo
El término hiperparatiroidismo se aplica a cualquier trastorno en el que el organismo está expuesto a niveles elevados de hormona paratiroidea efectiva. El calcio sérico puede estar aumentado, normal o disminuido, dependiendo de otros factores. El término hiperparatiroidismo primario denota un exceso en la producción de PTH que se origina en una lesión (adenoma, hiperplasia o carcinoma) de las paratiroides. El calcio sérico suele estar elevado. El hiperparatiroidismo secundario indica un exceso de adaptación en la producción de PTH estimulada por uno de los reguladores fisiológicos de la función paratiroidea: hipocalcemia, hiperfosfatemia o hipomagnesemia. El calcio sérico se encuentra bajo o, si la compensación es completa, normal.
HIPERPARATIROIDISMO PRIMARIO
Etiología y patogenia
El hiperparatiroidismo primario es causado por un adenoma único en el 85 a 90 porciento de los casos. Las mujeres padecen hiperparatiroidismo con una frecuencia mayor del doble que los hombres, y la incidencia aumenta con la edad: después de los 50 años, ésta es de alrededor de uno por 1,000 por año en varones y de dos a tres por 1,000 por año en mujeres.16 La hiperplasia de células principales es la siguiente causa más frecuente del hiperparatiroidismo primario. Las cuatro glándulas suelen encontrarse afectadas, aunque es común que el crecimiento sea asimétrico. Este trastorno, en el que la hipercalcemia moderada es la regla, puede reflejar el defecto de alguna sustancia paratirotrópica o un reajuste en las concentraciones que dan lugar a las alteraciones en el calcio sérico que modifican la liberación de PTH.
La diferencia entre adenoma e hiperplasia parece tener una base genética. Los adenomas son sobre todo de origen monoclonal. La primera alteración genética molecular identificada en pacientes con adenomas paratiroideos se debe a dos rupturas y una inversión en el cromosoma 11, que mueve la región regulador 5' del gen de la PTH lejos de la secuencia codificadora hacia un gen diferente conocido como PRAD1 [ver figura 3]. La proteína codificada por el PRAD1 es la ciclina que, en combinación con otros activadores del ciclo celular, estimula la proliferación celular sin cambios malignos. El cromosoma no mutado, con sus regiones reguladora 5' y codificadora adyacentes y sin cambio, puede ser el origen del exceso en la secreción de PTH. Este defecto ocurre en alrededor del 5 porciento de los adenomas paratiroideos. Quizá hasta el 25 porciento de los casos muestran pérdida de grandes segmentos de un cromosoma 11, que quizá en asociación con deleciones no visibles o con mutaciones del otro cromosoma 11, causa la formación del adenoma.17,18
Por el contrario, la hiperplasia partiroidea, en especial la observada en la neoplasia endócrina múltiple de tipo I (NEM I) típicamente es policlonal. Esto puede deberse a una mutación heredada en un locus supresor de tumores en el cromosoma 11. Debido a que se hereda por línea germinal, el defecto incluye a todas las células. Cuando aparece un defecto somático en el otro cromosoma 11, la pérdida resultante de la heterocigosidad causa expansión clonal y neoplásica de un adenoma agregado a la hiperplasia. Estos tumores típicamente son grandes y se asocian con hipercalcemia moderadamente severa. Estas diferencias son clínicamente importantes porque el hiperparatiroidismo es el signo más común y con frecuencia el inicial en la NEM I.
La causa menos frecuente de hiperparatiroidismo primario es el carcinoma de las paratiroides, que corresponde a alrededor del 1 porciento de los casos. Estos tumores tienden a crecer lentamente y producen invasión local, pero pueden alcanzar el hígado, el pulmón o el hueso. Por lo menos un defecto molecular, la deleción del gen supresor de tumores del retinoblastoma, favorece el desarrollo del carcinoma paratiroideo.19
Los efectos clínicos del exceso de PTH, diferentes de la hipercalcemia, se comprenden poco. La hipercalcemia puede contribuir a los datos neurológicos, así como a la fatiga general, y ciertamente tiene un papel importante en el daño renal. Sin embargo, no es claro si la hipercalcemia o el aumento en la PTH son los responsables de la mayoría de los otros síntomas. La PTH es directamente responsable de los trastornos esqueléticos por estimular la resorción osteoclástica, principalmente en el hueso cortical.
Manifestaciones clínicas
El síndrome clínico del hiperparatiroidismo primario tiene manifestaciones diversas [ver tabla 2]. Inicialmente el trastorno fue identificado como una combinación de enfermedad ósea severa (osteítis fibrosa quística) con pérdida de estatura, debilidad y emaciación, anemia y disfunción renal. Después se reconoció un síndrome diferente caracterizado por litiasis renal y denominado nefrocalcinosis. Aún después, se demostró sintomatología digestiva asociada, como constipación, dolor abdominal, dispepsia o úlcera péptica y pancreatitis. Ultimamente se han identificado trastornos musculoesqueléticos, incluyendo miopatía, neuropatía, seudogota y debilidad inespecífica. Sin embargo, en la actualidad se sabe que estos síndromes constituyen la minoría de los casos de hiperparatiroidismo primario. Con más frecuencia los nuevos casos se descubren al detectar hipercalcemia en forma incidental durante los escrutinios generales en pacientes adultos asintomáticos o con molestias inespecíficas.16,20
Los signos físicos del hiperparatiroidismo, además de la debilidad generalizada moderada, se encuentran solamente en sujetos muy enfermos e incluyen pérdida de peso, palidez y queratopatía en banda. Ocasionalmente los individuos presentan pérdida de estatura y xifosis secundaria a fracturas vertebrales, y algunos tienen dedos en palillo de tambor por osteítis severa de las falanges terminales, con fracturas y colapso óseo. Pueden también observarse diversos signos neurológicos inespecíficos (v.gr., somnolencia, fatiga y cambios en la personalidad).21
Diagnóstico de laboratorio
La confirmación del diagnóstico de hiperparatiroidismo sigue basándose principalmente en las características distintivas de los resultados de laboratorio. La prueba más útil es la medición del calcio sérico, que se encuentra elevado en el 96 porciento o más de los casos con hiperparatiroidismo primario. Es frecuente la elevación intermitente en la enfermedad leve, por lo que deben repetirse las mediciones cuando la sospecha diagnóstica es fuerte pero la medición inicial de calcio en suero es normal.
Otras pruebas bioquímicas son útiles pero no diagnósticas. Con frecuencia los valores de fósforo sérico están disminuidos. Los niveles de cloro sérico suelen ser mayores de 102 mEq/L; esta elevación refleja la influencia del exceso de PTH sobre la excreción renal de bicarbonato, produciendo una acidosis metabólica moderada con niveles de cloro altos y disminución leve en las cifras de bióxido de carbono.
La evaluación definitiva de la función paratiroidea consiste en medir la concentración de la hormona paratiroidea en la circulación. Como se mencionó, las mediciones de anticuerpos aislados no son sensibles ni específicas. Los ensayos de doble anticuerpo medido por inmunorradiometría e inmunoquimoluminiscencia indican que la PTHi está elevada en más del 90 porciento de los casos de hiperparatiroidismo primario, incluso en pacientes con hipercalcemia leve. Además, la PTHi es baja en las hipercalcemias no paratiroideas, en especial en la HHM. Por lo tanto, debe medirse la concentración de PTHi en todos los pacientes con hipercalcemia [ver figura 4]. Los pacientes con niveles de PTHi elevados o inapropiadamente normales deben someterse a una absorciometría de la muñeca, ya que el hueso cortical es más sensible al exceso de PTH. El hueso trabecular, como el de la columna, se afecta mucho menos, y puede incluso estar aumentado en el hiperparatiroidismo. Se encuentra menor densidad del hueso vertebral en el 15 porciento de los pacientes.22 Para los pacientes ocasionales con osteítis fibrosa las placas simples de manos y cráneo son útiles; las últimas deben examinarse con luz brillante para identificar la resorción subperióstica de las falanges medias.
En ausencia de deficiencia de vitamina D existe una correlación razonable entre el tamaño del adenoma paratiroideo y la hipercalcemia, y el aumento en la PTHi que produce. En los pacientes con hiperparatiroidismo leve los niveles de PTHi pueden caer dentro del rango normal superior, pero ser altos en relación con el nivel de calcio sérico; se usa un nomograma para interpretar estos valores. Si la PTHi está muy elevada comparado con el nivel de calcio en suero, es posible que exista un factor agregado, como deficiencia de vitamina D. Debido a que la PTH ejerce parte de su efecto calcémico a través del calcitriol, los pacientes hiperparatiroideos con deficiencia o resistencia simultánea de vitamina D pueden tener una concentración de calcio normal alta o muy poco elevada. Esta combinación de hiperparatiroidismo severo e hipercalcemia mínima puede ocurrir cuando existe deficiencia dietética de calcio, malabsorción, deficiencia de magnesio, enfermedad hepática severa o, más importante, insuficiencia renal crónica. En la última de estas condiciones la normocalcemia es causada más por hiperfosfatemia que por deficiencia de calcitriol.
El uso diseminado del escrutinio multifásico ha revelado una nueva población de pacientes hiperparatiroideos. El término hiperparatiroidismo asintomático se refiere a pacientes que tienen una concentración de calcio en suero elevada pero que no excede 11 mg/dl, un nivel no suprimido de PTHi, no enfermedad ósea, no enfermedad renal por cálculos activa o insuficiencia renal y ausencia de síntomas (incluso al interrogatorio sutil) potencialmente atribuibles al hipertiroidismo. La calidad y sensibilidad del interrogatorio es importante porque la cirugía puede aliviar los síntomas de los pacientes con hipercalcemia leve y porque los síntomas potenciales, incluyendo los psiquiátricos, son muy diversos [ver tabla 2].23 Irónicamente, la incidencia de hiperparatiroidismo parece estar disminuyendo, y el advenimiento de la atención dirigida ha introducido una nueva fase en la evolución de la epidemiología y manejo de este padecimiento. Si las limitaciones sobre el escrutinio progresan, el descubrimiento incidental del hipertiroidismo será menos común y aumentarán los casos sintomáticos.22,24
Tratamiento
El único tratamiento curativo para el hiperparatiroidismo es la resección quirúrgica del tejido paratiroideo. Por lo tanto, en los pacientes sintomáticos deberá elegirse el procedimiento quirúrgico adecuado. Para obtener mejores resultados se necesita de un equipo médico-quirúrgico experimentado. El siguiente programa parece ser razonable para tomar esta decisión:
1. Para el enfermo común con síntomas leves, calcio elevado y fósforo bajo, es razonable un manejo conservador. El cirujano debe explorar un lado del cuello; si encuentra un adenoma paratiroideo grande y una glándula normal, confirmado por el patólogo, el lado contralateral no deberá explorarse y se cierra el cuello después de la extirpación del adenoma. Este método se emplea en el Hospital General de Massachusetts, donde se han realizado más de 700 operaciones para hiperparatiroidismo y en los Institutos Nacionales de Salud de los Estados Unidos (NIH).
2. Para el sujeto con afección hiperplásica de las cuatro glándulas, por lo general es necesaria una paratiroidectomía resecando 7 octavas partes del tejido paratiroideo, para el control de la enfermedad y prevenir la recurrencia. Este procedimiento limita por una parte la aparición de hipoparatiroidismo y por otra evita el hiperparatiroidismo persistente. Algunos cirujanos extirpan las cuatro glándulas y reimplantan una porción de ellas en los músculos del antebrazo. Pueden implantarse rebanadas muy delgadas de tejido paratiroideo durante el procedimiento original, o pueden congelarse y emplearse hasta 18 meses después. Pasan 6 a 8 semanas para que el tejido trasplantado desarrolle vasculatura y comience a funcionar.
3. Cuando el cirujano no está seguro del diagnóstico patológico, conviene exponer al menos tres glándulas; si más de una está agrandada y macroscópicamente afectada, es mejor considerar la enfermedad como hiperplasia. Aunque existen adenomas dobles, estos son raros y uno no debe basar el tratamiento en esta esperanza. Los cirujanos experimentados que usan estos lineamientos para encontrar la lesión corrigen el hiperparatiroidismo en el 90 a 95 porciento de los casos. En general los estudios de imagen preoperatorios no mejoran estos resultados ni se realizan de rutina. Sin embargo, varios centros que han probado con las nuevas modalidades han encontrado que los estudios de imagen preoperatorios positivos pueden ser útiles en pacientes con riesgo alto.26
4. La reintervención quirúrgica en los enfermos que no se curaron con la primera exploración es una decisión difícil de tomar. Los factores cruciales son la experiencia y la habilidad del cirujano. Las consideraciones adicionales son una meticulosa revisión del informe quirúrgico y patológico original, la disección cuidadosa con exposición adecuada y, en algunos casos, contar con estudios de imagen preoperatorios.30 La selección de las técnicas de imagen varía en diferentes instituciones. El ultrasonido y la gammagrafía con isótopos doble da resultado más satisfactorios que la TC, pero ninguna de estas técnicas posee la sensibilidad o especificidad de la habilidad quirúrgica.27 El gamagrama con sestamibi marcado con tecnecio 99m parece ser el método más útil para la localización.28
5. La gran prevalencia de hiperparatiroidismo asintomático, combinado con la negativa de estos pacientes a someterse a cirugía ha aumentado la duda respecto a si la cirugía es realmente necesaria. Una conferencia de consenso de los NIH admitió que la vigilancia no quirúrgica es adecuada para los pacientes sin síntomas, que tienen concentraciones de calcio menores de 12 mg/dl, sin calcificación renal, no enfermedad ósea evidente por radiología, concentraciones de calcio en orina menores de 400 mg/24 horas y depuración de creatinina normal.29
La vigilancia no quirúrgica puede compararse con el informe del Hospital General de Massachusetts acerca del tratamiento quirúrgico de 242 enfermos que padecían hiperparatiroidismo leve. Estos sujetos se consideraban asintomáticos, pero mostraron datos de evolución de la enfermedad por uno o más de los siguientes criterios: niveles de calcio sérico mayores de 11 mg/dl, calcio urinario mayor de 150 mg/24 horas o incremento en los valores séricos de PTHi. La hipercalcemia se corrigió en el 98 porciento de los casos y sólo persistió despues de la cirugía en 4 individuos; no hubo muertes o complicaciones de importancia. Esta experiencia debe tomarse como marco de referencia ideal, más que como un logro quirúrgico común, pero la seguridad quirúrgica actual, incluso en personas de edad avanzada, desafía el tratamiento médico conservador. Por ejemplo, la serie de la Universidad de Michigan describe a 81 pacientes mayores de 65 años que sobrevivieron a la cirugía.30
La vigilancia de los pacientes con hiperparatiroidismo asintomático es de particular importancia en mujeres posmenopáusicas, el mayor grupo de pacientes asintomáticos (y el que se descubre más a menudo de manera incidental). En este grupo, los estrógenos y la noretindrona pueden disminuir el calcio sérico y mejorar el balance de calcio, lo que al parecer protege contra la osteoporosis.31 Si existe osteopenia significativa de la columna es adecuado realizar una densitometría de seguimiento. De igual manera, debe hacerse una medición densitométrica anual de la muñeca (el hueso cortical más afectado por la PTH) para detectar la progresión de la enfermedad paratiroidea. Los pacientes deben evitar el litio y los diuréticos tiacídicos, que pueden aumentar la secreción de PTH y aumentar la concentración sérica de calcio. Deben solicitar atención en caso de náusea o vómito porque la deshidratación puede inducir un aumento rápido y potencialmente peligroso en la concentración sérica de calcio. Por último, el clínico debe considerar la aparición o progresión de síntomas leves, incluyendo depresión, fatiga y dolores inespecíficos, como una justificación para la cirugía, porque el hiperparatiroidismo puede evolucionar de manera sutil; hasta el 65 porciento de los pacientes supuestamente asintomáticos mejoran después de la cirugía.
Manejo pre y posoperatorio La reacción que debe vigilarse en el posoperatorio inmediato es el desarrollo de tetania hipoparatiroidea [ver adelante, Hipocalcemia]; a largo plazo, es necesario vigilar la aparición de datos de hipoparatiroidismo, la recurrencia del hiperparatiroidismo o la afección de otras glándulas endócrinas. Ninguno de estos problemas ocurren ordinariamente en sujetos que tienen un adenoma único, una segunda glándula normal, normalización rápida de los niveles de calcio y ausencia de antecedentes familiares compatibles con neoplasia endócrina múltiple. En tales individuos, lo más adecuado es una revisión anual con determinación única de calcio sérico y glucemia en ayunas. Cuando se encuentra afectada más de una glándula deberá realizarse una investigación cuidadosa en busca de las características clínicas del síndrome de neoplasia endócrina múltiple en el paciente y su familia.
Los enfermos con osteítis fibrosa quística severa ocasionalmente tienen un curso posoperatorio difícil y prolongado, caracterizado por hipocalcemia, hipofosfatemia y aun tetania clínica. Este racimo de complicaciones es atribuible al llamado fenómeno del "hueso hambriento", en el que la formación excesiva de hueso afecta el depósito de calcio y fósfato de una manera que supera la homeostasis del calcio. El tratamiento con vitamina D mejora este problema y debe iniciarse en forma temprana si la hipocalcemia persiste en el posoperatorio, pero debe ser suspendida si se ha dejado tejido paratiroideo funcionante, confirmado por la presencia de niveles elevados de PTHi.
Cáncer de las paratioides
El carcinoma de las paratiroides es causa de hiperparatiroidismo en menos del 1 porciento de los casos. Esta rara enfermedad es típicamente grave: produce síntomas notables, niveles de calcio promedio en pacientes nuevos superiores a 15 mg/dl, enfermedad renal y ósea frecuentes y, en la mitad de los casos, una masa palpable en el cuello.32-34 Estos datos contrastan con la presentación conocida en la actualidad como típica para los casos de hiperparatiroidismo primario.35 En el momento de la cirugía, una clave para el diagnóstico es la adherencia de la glándula paratiroides, aumentada de volumen, a las estructuras vecinas; si el cirujano encuentra estos datos, deberá realizarse en forma muy cuidadosa una resección en bloque, ya que el control satisfactorio de la enfermedad solo se ha logrado con una resección amplia inicial.
Cuando el diagnóstico de cáncer no se realiza inicialmente, puede sospecharse en forma retrospectiva por la reaparición de la hipercalcemia 6 meses o más después de la cirugía; en cambio, cuando se deja una glándula hiperplásica remanente, la hipercalcemia se presenta en forma más temprana. La hipercalcemia persistente o recurrente obliga a revisar los datos histopatológicos originales. Aunque el diagnóstico definitivo de cáncer puede ser difícil, la invasión capsular por el tumor es un dato específico. Además, los estudios moleculares son medios poderosos para distinguir entre las lesiones benignas y malignas.19 La mayoría de los pacientes mueren por hipercalcemia; sin embargo, la reoperación local, la extirpación de las metástasis pulmonares guiada por TC, el tratamiento con bifosfonatos y el control de la hipercalcemia severa son todas medidas útiles.35
HIPERPARATIROIDISMO SECUNDARIO
El hiperparatiroidismo secundario es el resultado de la adaptación de las glándulas paratiroides a un estado de hiperfunción; es decir, la hipersecreción de PTH ocurre como una consecuencia fisiológica de otra enfermedad. Por ejemplo en la insuficiencia renal crónica, la disminución en la formación de calcitriol produce absorción baja de calcio,36 la pérdida del parénquima renal causa hiperfosfatemia y tanto la hipocalcemia como la hiperfosfatemia estimulan la función de las paratiroides. Los niveles bajos de calcitriol pueden permitir que la masa celular paratiroidea aumente, y la supresión de la actividad secretora de las paratiroides parece requerir un nivel más alto de calcio ionizado que el que se encuentra en estados normourémicos. La supresión habitual de la producción de PTH por el calcitriol está bloqueada y el contenido en las células paratiroides de receptores de calcio también está disminuido.37 En la insuficiencia renal avanzada la mayor masa de tejido paratiroideo, aunado a una secreción no suprimible basal por célula, se agrega a la sobreproducción de PTH. Además, puede haber resistencia renal y esquelética a la PTH. En los trastornos gastrointestinales la malabsorción de vitamina D y calcio produce hipocalcemia e hiperparatiroidismo secundario.
Ya que el exceso secundario de PTH es adaptativo, más que autónomo, raras veces produce hipercalcemia. Más bien, se deduce que existe compensación paratiroidea cuando el calcio sérico se encuentra disminuido o dentro de límites normales en circunstancias en las que se espera que se encuentre bajo. Los niveles de PTHi pueden estar muy aumentados. Los signos radiológicos de osteítis fibrosa ofrecen información adicional a este respecto. En los casos severos puede ocurrir hipercalcemia en el hiperparatiroidismo secundario, y puede ser difícil o imposible distinguir los casos primarios de los secundarios.
El mejor recurso para el hiperparatiroidismo secundario es el control de la enfermedad de fondo; cuando esto no es posible, la manipulación farmacológica puede ser de ayuda. En los pacientes con insuficiencia renal crónica los niveles de fosfato pueden ser controlados con la administración de fijadores de fosfato por vía oral (sin embargo, los fármacos que contienen aluminio empeoran la osteomalacia).37 Si este método es insuficiente se puede intentar el tratamiento cuidadoso con dosis pequeñas de vitamina D. Se ha obtenido más experiencia con el calcitriol, que parece aliviar el dolor óseo y la debilidad muscular en enfermos con falla renal crónica e hiperparatiroidismo secundario. La osteítis fibrosa leve y la osteomalacia, que frecuentemente la acompañan, mejoran y los niveles de fosfatasa alcalina y de PTHi descienden gradualmente. Los sujetos con osteítis fibrosa severa y aquellos en quienes la cifras de calcio se encuentran en límites superiores normales antes del tratamiento con calcitriol, presentan episodios de hipercalcemia notable al iniciar el tratamiento. Por lo tanto, requieren de vigilancia estrecha; afortunadamente, la acción del calcitriol es de corta duración y permite una recuperación pronta de sus efectos al suspenderlo.
Un síndrome de osteomalacia relacionado con la diálisis parece ser causado por la exposición al aluminio que se encuentra presente en los dializadores. Este metal parece bloquear la mineralización del hueso; cuando se administra vitamina D, los individuos afectados tienden a presentar hipercalcemia notable, que responde bien a la suspensión de este medicamento, aunque la mejoría en la enfermedad ósea requiere de la quelación del aluminio.
Anteriormente se sugería que en algunos pacientes con hiperparatiroidismo grave secundario de evolución prolongada, una de las glándulas paratiroides podría empezar a funcionar autónomamente y causar hiperparatiroidismo terciario. Los estudios moleculares han demostrado que tanto en la hiperplasia de células principales policlonal primaria como secundaria, ocurren tumores monoclonales. Sin embargo, esto es más frecuente que el síndrome de hiperparatiroidismo terciario, y el significado exacto de esta evolución molecular no es clara aún.38 Se ha obtenido experiencia relevante sobre el hiperparatiroidismo secundario desde la introducción del trasplante renal para la insuficiencia renal crónica. Al principio, algunos pacientes desarrollaban hipercalcemia grave poco después de la cirugía. En ocasiones se realizó paratiroidectomía y los hallazgos indicaron un hiperparatiroidismo secundario severo e irreversible, y aún hiperparatiroidismo terciario. Sin embargo, en muchos de estos casos se corrigió la hipercalcemia después de 2 años o más sin paratiroidectomía, lo que se atribuyó en forma retrospectiva a la regresión de la hiperplasia secundaria. Aunque la hipercalcemia grave que requiere cirugía es menos común en la actualidad, es más frecuente la hipercalcemia moderada pero persistente. Estos casos pueden indicar ya sea un cambio en el punto fijo en el que los niveles de calcio estimulan la secreción de PTH o más probablemente, una masa poco frecuente de tejido paratiroideo hiperplásico que requiere de un tiempo más prolongado para su regresión.
Hipercalcemia
La hipercalcemia es un problema clínico común que puede constituir una urgencia que ponga en peligro la vida del paciente, o ser el heraldo silencioso de una gran variedad de enfermedades curables.39,40
Más del 90 porciento de los casos se atribuyen a hiperparatiroidismo o se asocian con cáncer; por lo tanto, debe pensarse primero en estas opciones. Por lo demás, el primer paso debe consistir en revisar los mecanismos fisiológicos, más que intentar abarcar todas las posibilidades diagnósticas. Fisiológicamente los niveles de calcio se mantienen por tres procesos: absorción gastrointestinal, resorción osteoclástica de minerales de las reservas óseas y reabsorción tubular de calcio del filtrado glomerular. El control predominante en la absorción gastrointestinal es la vitamina D y el principal control en la esfera renal y ósea es la PTH. Las causas más comunes de hipercalcemia son la absorción gastrointestinal excesiva, la resorción ósea excesiva y un mecanismo incierto que probablemente entraña un aumento en la reabsorción renal de calcio [ver figura 5].
GUIAS DIAGNOSTICAS
Una guía general basada en los tres principales mecanismos productores de hipercalcemia puede ser útil en la interpretación de la sintomatología del paciente.
Aumento en la absorción gastrointestinal de calcio
En la sarcoidosis, algunas otras enfermedades granulomatosas, la enfermedad de Hodgkin y los linfomas no Hodgkin ocurre aumento en la absorción GI de calcio debido a síntesis excesiva de calcitriol. El calcitriol y el calcio inhiben la expresión del gen de PTH, así como su secreción; por lo tanto, la PTHi en plasma es baja.41,42
El síndrome leche-álcali (asociado con ingesta escesiva de leche o alcalinos absorbibles) produce una insuficiencia renal moderada, alcalosis y elevación en los niveles de calcio y fósforo. Puede ser muy difícil distinguir este síndrome del hiperparatiroidismo primario con insuficiencia renal crónica y úlcera péptica secundarias, por lo que la determinación de PTHi debe ser incluida en el estudio de estos pacientes.
La hipervitaminosis D ocurre en enfermos que están recibiendo tratamiento con esta vitamina o entre las personas con manías alimentarias que consumen grandes cantidades de vitamina D. El síndrome se caracteriza por insuficiencia renal, elevación de los niveles de fósforo (en dos tercios de los casos) y disminución del filtrado glomerular. En sujetos que han sido tratados con vitamina D, generalmente por hipoparatiroidismo, la hipercalcemia casi siempre es el resultado de los esfuerzos por corregir la hipocalcemia de una manera precipitada o por un seguimiento inadecuado. Ocasionalmente también ocurre en individuos bien tratados, pero se desconoce la causa en etos casos; en otros puede ser difícil recabar el antecedente de ingestión de vitamina D, aunque los niveles de calcifediol estarán elevados en estos pacientes. Este último dato fue la clave en los casos reportados de pacientes que bebieron leche fortificada en forma errónea con un exceso de vitamina D.42,44
Aumento en la resorción ósea
El hiperparatiroidismo primario ha sido analizado en forma extensa [ver antes, Hiperparatiroidismo] porque es la causa más común de hipercalcemia asintomática, particularmente en los pacientes ambulatorios, y el diagnóstico se hace más probable a medida que aumenta la duración de la hipercalcemia y ninguna otra alternativa se hace aparente.
En los enfermos hospitalizados la hipercalcemia se relaciona a los padecimientos oncológicos con más frecuencia que a ningún otro. Ocurren lesiones osteolíticas secundarias y metástasis en cánceres de mama, pulmón y próstata, así como otras muchas enfermedades malignas. El mieloma múltiple también puede afectar al hueso. Este fenómeno, conocido como hipercalcemia osteolítica local, es causado por estimulación osteoclástica directa por una o más citocinas producidas en los tumores, incluyendo varias interleucinas, factor de necrosis tumoral alfa, factor transformador de crecimiento y linfotoxina. Los tumores metastásicos que causan hipercalcemia generalmente son visibles en las radiografías simples de los huesos, pero los gamagramas óseos pueden ser compatibles con metástasis aun cuando las radiografías no muestren alteraciones.
La causa más frecuente de hipercalcemia severa en las neoplasias es el síndrome conocido como hipercalcemia humoral maligna (HHM). Los cánceres que se asocian con más frecuencia con este síndrome son de células epidermoides y de origen genitourinario o ginecológico. Los pacientes con HHM tienen manifestaciones semejantes a las de los pacientes hiperparatiroideos, incluyendo hipofosfatemia leve y aumento en la concentración urinaria de AMPc. Estos enfermos tienen más posibilidad de tener anemia y menos de tener hipercloremia, pero el mayor contraste consiste en que tanto la concentración de PTHi como de calcitriol están bajas o en límites normales bajos. El compuesto responsable de este cuadro clínico es la PTHrP (ver antes).45-47 Aunque el papel fisiológico endócrino de la PTHrP no se conoce, esta proteína promueve la resorción ósea y la reabsorción renal de calcio cuando se secreta en exceso por las células neoplásicas, produciendo hipercalcemia.45,48,49 La PTHrP también se ha identificado en leucemias causadas por virus linfotrópico a células T humanas tipo I y algunos cánceres de mama. En pocos casos se ha reportado la producción de prostaglandina E2 o PTH.
La enfermedad de Paget del hueso puede producir hipercalcemia, particularmente en enfermos que tienen afección ósea diseminada. Este fenómeno es raro en pacientes ambulatorios, pero es más frecuente en los hospitalizados. El hipertiroidismo se relaciona con hipercalcemia moderada en aproximadamente 10 a 20 porciento de los casos. El mecanismo exacto se desconoce, pero el recambio óseo acelerado parece ser la lesión principal.
La inmovilización, como la que ocurre durante la hospitalización o después de traumatismos graves, puede producir una hipercalcemia marcada; aunque ésta puede ser atribuida a la inactividad per se, es mucho más común cuando hay una causa independiente de recambio óseo acelerado, como la enfermedad de Paget o el crecimiento rápido en adolescentes.
El tratamiento con litio y los diureticos tiacídicos pueden inducir hipercalcemia leve a moderada, generalmente con un aumento correspondiente en los niveles de PTHi; el síndrome es reversible al suspender el medicamento. Los pacientes deben ser vigilados durante 3 meses después de suspender cualquier medicamento. Si la hipercalcemia y el nivel aumentado de PTHi persisten, casi siempre existirá un adenoma paratiroideo o hiperplasia.
Aumento en la reabsorción renal de calcio
Tanto la PTH como la PTHrP estimulan la adenilato ciclasa renal. Por lo tanto, la hipercalcemia que producen, principalmente por resorción ósea, es exacerbada por una mayor reabsorción renal del calcio filtrado. La reabsorción de calcio contribuye también a la hipercalcemia en otras circunstancias. Los diuréticos tiacídicos inducen hipercalcemia de manera impredescible, y las publicaciones difieren en la frecuencia con que la hipercalcemia inducida por las tiacidas constituye una clave diagnóstica para la presencia de hiperparatiroidismo.
La hipercalcemia hipocalciúrica familiar (HHF) es un trastorno autosómico dominante caracterizado por la aparición de hipercalcemia en la niñez, ausencia de hipercalciuria, fatiga, cefalea, artralgias y polidipsia. Los niveles de PTHi y de calcitriol no están aumentados y aunque la enfermedad se acompaña con frecuencia de hiperplasia paratiroidea, responde poco a la paratiroidectomía subtotal. El diagnóstico es especialmente díficil a menos de que la hipercalcemia inicie antes de los 10 años de edad (lo cual es muy raro en el hiperparatiroidismo primario o familiar) o que varios miembros de la familia estén afectados. La HHF es causada por un defecto autosómico dominante en los receptores sensibles a calcio tanto en las paratiroides como en los riñones: la secreción de PTH no es inhibida por el nivel habitual de calcio sérico y su excreción renal es desproporcionadamente baja para el nivel de hipercalcemia. El síntome letal de hiperparatiroidismo neonatal severo es causado por la expresión homocigota del mismo defecto en el receptor.50,51
Diagnóstico diferencial
La hipercalcemia que se descubre de manera incidental en el paciente ambulatorio es causada con mayor frecuencia por hiperparatiroidismo; en el enfermo hospitalizado, suele representar una complicación del cáncer. El hiperparatiroidismo suele acompañarse de acidosis hiperclorémica leve, con un nivel de cloro superior a 102 mEq/L y una cifra de fosfato inorgánico menor de 3 mg/dl. En la hipercalcemia no relacionada con hiperparatiroidismo el nivel de bicarbonato sérico se encuentra a menudo ligeramente elevado. Sin embargo, en la hipercalcemia aguda la presencia de deshidratación, insuficiencia renal y vómito complican las alteraciones electrolíticas.
La determinación de PTHi y de calcitriol constituyen el enfoque más directo para esclarecer la etiología de la hipercalcemia. El nivel de PTHi está elevado en el hiperparatiroidismo y suprimido en el resto de los estados hipercalcémicos. El nivel de calcitriol está elevado en el hiperparatiroidismo, la sarcoidosis, en algunos trastornos malignos y en la intoxicación por vitamina D, y reducido en la HHM. El aumento en la concentración de AMPc urinario puede orientar en casos de hipercalcemia que son difíciles de clasificar.
TRATAMIENTO
Una elevación estable de los niveles de calcio sérico refleja un equilibrio entre el aporte de calcio del intestino y hueso y su pérdida a través del riñón. En algunos pacientes el ritmo de reabsorción ósea supera este balance y el desequilibrio produce hipercalcemia. Cuando los niveles de calcio comienzan a elevarse, la reabsorción de agua y sal en el túbulo contorneado distal se altera. La deficiencia de volumen, frecuentemente agravada por el vómito, ocasiona aumento en la reabsorción de sodio y agua en el túbulo contorneado proximal. Finalmente, al disminuir el filtrado glomerular (y la filtración de calcio), además de la disminución de la entrada de calcio al hueso por la inactividad, se puede producir una hipercalcemia que ponga en peligro la vida del enfermo.
En el equilibrio o en la hipercalcemia estable, cuando los niveles séricos de calcio son menores de 14 mg/dl, el tratamiento de la hipercalcemia dependerá de la enfermedad subyacente. La mayoría de las causas de hipercalcemia pueden ser tratadas específicamente y casi siempre es mejor proceder de esta menra que considerar el tratamiento farmacológico crónico para disminuir los niveles séricos de calcio. Sin embargo, es urgente disminuir los niveles de calcio cuando éstos exceden los 14 mg/dl o cuando están entre 12 y 14 mg/dl y el sujeto está sintomático. Cuando los niveles de calcio se encuentran por abajo de 12 mg/dl es raro que la sintomatología severa pueda atribuirse a la hipercalcemia.
Existen varios métodos disponibles para el manejo de urgencia de la hipercalcemia.39 La medida inicial consiste en restablecer el volumen extracelular y la filtración glomerular, habitualmente con 3 a 4 L de solución salina por día. Una vez que la diuresis es adecuada, si la concentración sérica de calcio está por arriba de 14.5 mg/dl, se agrega furosemide, 40 a 80 mg/día IV. Si el paciente no está totalmente alerta y no es capaz de beber, cada cuarta o quinta botella de líquido debe ser hipotónica para prevenir la hipernatremia. En los pacientes con función ventricular izquierda limítrofe debe vigilarse la presión venosa con una línea central para evitar sobrecargar el corazón. Debe tenerse cuidado de evitar la depleción de potasio y magnesio.
Un nuevo tipo de fármacos sintéticos, los bifosfonatos, inhiben la resorción osteoclástica del hueso; algunos promueven también la formación ósea. El medicamento de elección para tratar la hipercalcemia es el pamidronato, y la dosis eficaz es de 45 a 60 mg en una sola infusión de 24 horas que se inicia en cuando se establece buena diuresis. Se logra la normalización del calcio sérico con esta dosis en el 70 a 100 porciento de los pacientes, y en los que tienen hipercalcemia severa pueden usarse 90 mg en 24 horas.52 Una vez que el calcio es normal, el tratamiento antitumoral puede ser eficaz para prevenir recurrencias de la hipercalcemia. Si no es posible administrar un tratamiento definitivo para el problema primario, puede tratarse con pamidronato IV en forma intermitente. El tratamiento con etidronato por vía oral, en dosis de 1,200 a 1,600 mg/día, suele prevenir las recurrencias de la hipercalcemia, pero no se ha autorizado su uso para este fin.
La calcitonina es otra alternativa para el tratamiento de la hipercalcemia. Actúa pronto, pero sus efectos son parciales y temporales, y su principal utilidad es ganar tiempo cuando no se dispone de otros agentes más potentes. La dosis es de 4 U MRC (Medical Research Council)/kg por vía subcutánea o intramuscular cada 12 horas. Con este tratamiento la concentración de calcio sérico disminuye 1 a 2 mg/dl en algunas horas, pero el efecto desaparece con igual rapidez y casi siempre se requieren otras medidas.
La prednisona, en dosis de 60 mg/día es un excelente suplemento en la intoxicación por vitamina D, mieloma, linfoma o cáncer de mama cuando la hipercalcemia ha sido precipitada por el tratamiento con esteroides sexuales.
La plicamicina es un potente agente antihipercalcémico que puede usarse para el tratamiento urgente de la hipercalcemia severa mientras se realizan los estudios dianósticos. Por lo general se administra una dosis inicial de 15 a 25 µg/kg IV durante 4 a 6 horas; es mejor esperar antes de repetir la dosis hasta que los niveles de calcio comiencen a aumentar de nuevo.
El fosfato inorgánico, 1.0 a 1.5 g/24 horas IV logrará el control temporal de la mayoría de los episodios de hipercalcemia; este tratamiento debe usarse solo si los agentes más potentes no están disponibles o fueron ineficaces. Su empleo está limitado a los pacientes con nivel sérico de fosfato bajo; si la concentración de fosfato se encuentra en límites superiores normales o elevados existe riesgo de calcificación extraesquelética.
La hipercalcemia producida por hiperparatiroidismo agudo suele responder a las medidas antes descritas. Sin embargo en algunos casos el tratamiento conservador no reduce el nivel de calcio en forma eficaz cuando la hipercalcemia es grave y en estos casos la exploración paratiroidea de urgencia puede salvar la vida y debe realizarse en forma pronta.
Hipocalcemia
La hipocalcemia (concentración de calcio en suero por debajo de 8.5 mg/dl) es más frecuente cuando existe deficiencia o resistencia a calcitriol, el hipoparatiroidismo (deficiencia de hormona paratiroidea activa) es una causa mucho menos común.
ETIOLOGIA Y FISIOPATOLOGIA
Hipoparatiroidismo
El hipoparatiroidismo puede deberse a falla en las glándulas para secretar PTH o falla de los tejidos para reaccionar a ella (seudohipoparatiroidismo). Dentro de la amplia gama de trastornos en la secreción y resistencia de los tejidos, hay muchas causas de hipoparatiroidismo [ver tabla 3].
La causa más común de hipoparatoridismo es la cirugía de tiroides. Con frecuencia no se identifican las cuatro glándulas durante la resección del tejido tiroideo, por lo que se considera que la deficiencia sea secundaria a infarto transoperatorio de las mismas.53
La deficiencia paratiroidea idiopática ocurre en varias formas. En una, la insuficiencia se acompaña de deficiencia variable de otras glándulas, sobre todo de suprarrenales y tiroides, y de candidiasis mucocutánea persistente; los anticuerpos contra tejido endócrino son la característica común de esta enfermedad, que tiende a presentarse en familias y que en muchos casos se ha relacionado con un defecto en el cromosoma 21q22.3.54 En una segunda forma, la deficiencia paratiroidea ocurre como parte del síndrome de Di George, que se caracteriza por linfopenia y aplasia de estructuras derivadas del tercer arco branquial, incluyendo el timo. El defecto se localiza en el cromosoma 22q11.56 Por último, el hipoparatiroidismo idiopático puede ocurrir como un defecto aislado, de manera esporádica o hereditaria. En algunos casos se han detectado anticuerpos contra el receptor sensible a calcio.56 Una forma autosómida dominante es causada por una mutación sin sentido en el gen que codifica para el receptor sensible a calcio. Al parecer confiere una mayor afinidad al calcio, causando inadecuada liberación de PTH e hipocalcemia subsecuente.57 Este aumento de función por mutación y su síndrome clínico son lo opuesto al defecto de pérdida de función que produce la hipercalcemia hipocalciúrica familiar. Un motivo importante para identificar este trastorno es que el tratamiento con vitamina D, que parece razonable, suele empeorar la hipercalciuria, el daño renal y causar nefrocalcinosis, y debe evitarse en pacientes sintomáticos.58,59
En la variante ligada al cromosoma X, el defecto se ha localizado en el brazo largo de este cromosoma.60 Es probable que el defecto se encuentre en la regulación de la secreción de PTH, más que en el gen en sí.
Otro mecanismo importante que produce hipoparatiroidismo es la resistencia a la acción de la PTH. Albright y colaboradores fueron los primeros en definir un síndrome en el que la hipocalcemia, la hiperfosfatemia y la hiperplasia tiroidea estaban relacionadas con talla corta, obesidad, cara redonda, huesos metacarpianos cortos y calcificación de los ganglios basales. Estos autores demostraron que la inyección de PTH no elevaba los niveles de calcio sérico ni los de fósforo en orina y acuñaron el término de seudohipoparatiroidismo para esta enfermedad. La PTH no eleva los niveles de AMPc nefrogénico, lo que indica que hay un defecto en el receptor hormonal en el riñón. En algunas familias el defecto se ha localizado en la unidad reguladora de nucleótidos (Gs) que acopla el receptor de membrana a la adenilciclasa. Este defecto también podría ser responsable de la resistencia ocasional a otras hormonas proteicas, como tirotropina, que se observa en estos pacientes.61 Sin embargo, existen algunos aspectos que son inexplicables, incluyendo una asociación inconstante entre la unidad reguladora defectuosa y la capacidad de la PTH para inducir el incremento en el AMPc. De hecho, los pacientes seudohipoparatiroideos pueden presentar alguna deficiencia en la actividad de GS, al igual que los pacientes que aparentan tenerla pero no son hipocalcémicos. Estos individuos normocalcémicos se clasificaban al principio como portadores de seudohipoparatiroidismo. En la actualidad ambos síndromes se conocen como osteodistrofia hereditaria de Albright.62 Estos síndromes de resistencia a la PTH se han subclasificado por sus mecanismos molculares, patrones de herencia, defectos genéticos y resistencia asociada a otras hormonas.63
Los trastornos adquiridos en la secreción de PTH, en los efectos de la PTH o en la activación de la vitamina D, son mucho más comunes que los síndromes descritos previamente.64 La hipomagnesemia altera la liberación de PTH y bloquea sus efectos hipercalcémicos, los niveles de calcio y fósforo séricos están bajos. Estos efectos dependen, al menos en parte, de la gravedad de la deficiencia de magnesio; la corrección de esta deficiencia restaura rápidamente la secreción de PTH, aunque la respuesta a la PTH se recupera en forma más lenta. Ya que los alcohólicos y otros individuos propensos a la desnutrición pueden tener deficiencia de magnesio, y como la tetania puede ocurrir por deficiencia de magnesio o calcio, para diagnosticar este tipo de hiperparatiroidismo se necesitan capacidad clínica y pruebas de laboratorio discriminantes. En forma alterna, la intoxicación alcohólica aguda puede acompañarse de disminución en la concentración sérica de calcio ionizado y de aumento en la excreción urinaria de calcio y magnesio. Estos cambios son consecuencia de una menor secreción de PTH, y suelen ser reversibles.65 Es curioso que la hipermagnesemia también puede suprimir la secreción de PTH y provocar hipocalcemia.66
La insuficiencia renal crónica es probablemente la causa más común de hipocalcemia, y ya se describieron los mecanismos que la producen [ver antes, Hiperparatiroidismo secundario]. Los individuos afectados generalmente tienen niveles elevados de fósforo, creatinina y nitrógeno de urea en sangre.
Deficiencia de vitamina D
Los precursores de vitamina D se absorben tanto de la piel como por el tubo digestivo. La deficiencia de vitamina D suele deberse a la combinación de exposición inadecuada a la luz solar y una dieta pobre en pescado y productos lácteos.67 Se conocen varios defectos del metabolismo de la vitamina D. En el raquitismo resistente a vitamina D de tipo I existe menor conversión de 25-(OH)D3 a calcitriol; en el raquitismo resistente a vitamina D de tipo II el defecto se encuentra en la unión del calcitriol a su receptor.
Ocurre malabsorción de calcio y vitamina D en las enfermedades del intestino delgado, y ocasionalmente en la deficiencia pancreática. La esteatorrea, no siempre obvia, hace necesaria la cuantificación de la grasa en las heces, y realizar estudios radiográficos del intestino delgado para confirmarla. En pacientes que se encuentran en los límites de deficiencia de vitamina D y con malabsorción de calcio, una diarrea aguda puede precipitar tetania.
El tratamiento con fenitoína y otros medicamentos anticonvulsivos ha sido relacionado como causa de deficiencia de vitamina D. Aunque la hipocalcemia franca no se observa frecuentemente, los enfermos que están tomando estos medicamentos pueden tener niveles séricos de calcio en límites inferiores normales, fósforo sérico bajo, elevación moderada de la fosfatasa alcalina y calcio urinario bajo, todos compatibles con osteomalacia. Las pocas biopsias de hueso disponibles muestran vetas osteoides sin calcificar, que es una característica distintiva de la osteomalacia. La patogenia de este defecto no se ha dilucidado por completo, pero la hipocalcemia puede agravar cualquier trastorno convulsivo y los niveles de calcio en límites inferiores normales, que en otras personas no tienen importancia, pueden ser peligrosos para enfermos con epilepsia.
Otras causas de hipocalcemia
Las causas diversas de hipocalcemia incluyen la pancreatitis aguda, en la que los mecanismos precisos no se han aclarado; la enfermedad metastásica osteoblástica, en la que se supone que hay desplazamiento de calcio y fósforo de líquido extracelular por un rápido depósito en las metástasis osteoblásticas; las transfusiones excesivas con sangre tratada con citrato, en donde la formación de complejos del calcio con el citrato infundido es el factor que produce el daño, y la insuficiencia renal inducida por la rabdomiolisis.68
ASPECTOS CLINICOS DE LA HIPOCALCEMIA
Los rasgos clínicos de la hipocalcemia dependen en parte de la causa de fondo, de la rapidez con que se desarrollan y de la presencia de enfermedades relacionadas. En casos de instalación aguda, como después de una tiroidectomía, la sintomatología común incluye parestesias peribucales, inquietud, depresión, y los signos de Chvostek o Trousseau. En los pacientes que desarrollan hipocalcemia en forma más gradual la fatiga, los calambres musculares y las mialgias son la sintomatología prominente. Sin embargo, estos síntomas son sutiles y es común confundirlos con rasgos inespecíficos de depresión o astenia. La prolongación del intervalo QT en el electrocardiograma o el descubrimiento casual de calcio sérico bajo pueden aclarar el diagnóstico.
El enfermo con hipocalcemia deberá ser evaluado bajo las bases fisiológicas básicas que han sido discutidas y que se resumen en la tabla 3. Como ya se mencionó, la concentración de calcio sérico se eleva por absorción gastrointestinal, resorción ósea, o reabsorción renal, este último proceso es el menos importante. Si los niveles de calcio sérico se encuentran disminuidos, el defecto radica en la mayoría de los casos en el intestino o en el hueso y las causas principales son la deficiecia de vitamina D y la ineficacia de la PTH. A menudo el trastorno de fondo es aparente: tiroidectomía previa, insuficiencia renal crónica o pancreatitis aguda grave. Cuando el mecanismo productor no es obvio, está indicado determinar los valores séricos de vitamina D y de PTHi; si la PTHi se encuentra elevada en presencia de hipocalcemia, es probable que la hormona sea ineficaz por carencia de vitamina D o a la resistencia de los órganos blanco a una o ambas hormonas. La concentración de 25-(OH)D3 refleja la ingesta y reservas de vitmina D.69 La depleción de vitamina D puede compensarse por un hiperparatiroidismo secundario, que puede aumentar la síntesis de calcitriol y por lo tanto, mantener los niveles de calcio sérico normales. Cuando la compensación es inadecuada, el calcio y fósforo sérico disminuyen y se desarrolla osteomalacia [ver tabla 3].
TRATAMIENTO DE LA HIPOCALCEMIA
La PTH no es práctica para uso clínico, pero las acciones muy similares de la vitamina D ofrecen una alternativa satisfactoria. La hipocalcemia generalmente es tratada con una combinación de calcio y vitamina D.
Existe gran cantidad de literatura sobre el uso de análogos sintéticos de la vitamina D.6 Para los pacientes con enfermedad hepática o malabsorción, el calcifediol puede usarse esperando que el riñón añada el grupo 1-alfa-H. Pueden emplearse dosis entre 50 y 300 µg/día. La 1alfa-(OH)D3 puede usarse en el hipoparatiroidismo y también en pacientes con enfermedad renal porque la 25-hidrolasa hepática usará la 1-(OH)D3 como sustrato, la dosis es de 2 a 4 µg/día. También se dispone de calcitriol, y se han empleado dosis de 0.25 a 1.0 µg/día.70 Las nuevas preparaciones parecen ser eficaces para aumentar la concentración de calcio sérico en pacientes hipoparatiroideos, pero son más costosas que la vitamina D estándar y ofrecen pocas ventajas para la mayoría de los pacientes. La concentración sérica de calcio debe vigilarse para usar todas las formas de vitamina D en forma segura. Los análogos 1-OH de la vitamina D son los que más producen hipercalcemia por la pérdida de la regulación habitual de la 1alfa-OH hidroxilasa. Por este motivo se prefiere el calcitriol. La dosis requerida disminuye al normalizarse el calcio sérico y sanar las lesiones óseas. El calcitriol, usado en cantidades fisiológicas, y la 1-(OH)D3 tienen un inicio y cese de acción rápidos, en caso de que la toxicidad obligara a suspenderlos. Es demasiado pronto para afirmar si las ventajas reales y posibles de los análogos sintéticos de la vitamina D justifican su mayor costo, pero su introducción marca una aplicación clínica excitante de adelantos científicos recientes.
Hipocalcemia después de tiroidectomía
Los síntomas agudos de la hipocalcemia después de una tiroidectomía pueden tratarse con gluconato de calcio; una dosis de 10 ml de solución al 10 porciento, administrados lentamente por vía intravenosa, producen alivio casi inmediato. En casos leves se puede utilizar 1 g de cloruro de calcio tres veces al día durante 1 a 3 semanas, que después se reducen en forma gradual. Si se necesitan más de 4 días de tratamiento, por lo general se requiere la restitución permanente y bastarán 25,000 a 50,000 unidades de vitamina D diarios. Los niveles de calcio sérico deberán determinarse cada 2 semanas a partir del inicio del tratamiento y cada 6 meses después de habrer alcanzado la estabilización de los valores, ya que la hipercalcemia episódica es común en estos pacientes. Deben mantenerse las cifras de calcio en 8.5 mg/dl, porque la excreción urinaria de calcio en hipoparatiroideos tratados con vitamina D es muy alta. El calcio urinario debe determinarse de manera periódica; si se encuentra por arriba de 250 mg/día, el individuo deberá mantener una ingestión de líquidos elevada para evitar la formación de cálculos renales. La clortalidona y la restricción moderada de sodio pueden ser útiles como tratamiento complementario en pacientes que desarrollan hipercalciuria problemática o litiasis renal mientras se encuentran en tratamiento con vitamina D.
Hipocalcemia después de la paratiroidectomía
En enfermos hiperapartiroideos que no padecen osteítis severa la hipocalcemia después de la paratiroidectomía generalmente refleja la supresión transitoria de las otras glándulas; la restitución temporal de calcio ayudará al sujeto hasta que la secreción de PTH se restablezca en las otras glándulas. Esta sustitución temporal mantendrá el bienestar del paciente hasta que se reanude la secreción de PTH. En individuos con osteítis severa la evolución posoperatoria suele caracterizarse por osteomalacia de varios meses de evolución con niveles séricos bajos de calcio y fósforo y fosfatasa alcalina elevada. El tratamiento con vitamina D acelerará la curación pero puede suspenderse después de 3 a 6 meses. Los casos que desarrollan tetania después de la resección de siete octavos de las paratiroides generalmente requerirán de tratamiento permanente con vitamina D.
Calcitonina y sus trastornos
La calcitonina es codificada por un gen que se localiza en el cromosoma 11. La secreción de calcitonina aumenta por el aumento súbito del calcio sérico, por dosis farmacológicas de algunos péptidos gastrointestinales y por los agonistas beta adrenérgicos. Su secreción es inhibida por la hipocalcemia aguda y por los antagonistas beta. El papel fisiológico de estas influencias no es aún muy claro.
Ha sido difícil determinar cuál es el papel fisiológico de esta hormona.8 En cantidades farmacológicas suprime la resorción ósea, actuando en forma directa sobre los osteoclastos. Por tanto, modula la concentración sérica de calcio durante el recambio rápido de hueso, disminuyendo la hipercalcemia e, incluso en ocasiones, reduciendo concentraciones normales de calcio sérico. Sin embargo, la ausencia de calcitonina (v.gr., después de la tiroidectomía total) no se asocia con hipercalcemia, y cantidades excesivas de la hormona (v.gr., en el carcinoma medular del tiroides) no causan hipocalcemia.
El gen que codifica la síntesis de calcitonina puede, por trascripción alterna del ARNm, producir también otros ocho péptidos. La mayor parte de estos péptidos parecen tener función de neurotransmisores o parácrina, pero no hormonal. Uno de estos, conocidos como péptido relacionado con el gen de la calcitonia, es un neuropéptido y el más potente vasodilatador natural conocido.71,72
En ciertas enfermedades se presentan cantidades enormes de calcitonina. En el carcinoma medular del tiroides la hipercalcitonemia es un marcador tumoral constante. Se ha notificado producción ectópica de calcitonina en muchos tumores, incluyendo de páncreas, pulmón, próstata, útero, vejiga, mama y melanoma.
La calcitonina es de gran utilidad terapéutica en la enfermedad de Paget del hueso. En este trastorno, cuya lesión básica es la osteolisis franca, la calcitonina alivia el dolor óseo, reduce el calcio urinario y antagoniza la hipercalcemia producida por la inmovilización; reduce también los niveles urinarios de hidroxiprolina y séricos de fosfatasa alcalina, que son indicadores generales de la gravedad de esta enfermedad. Se afirma que en algunos pacientes ha ocurrido mejoría de las lesiones óseas, aunque este beneficio es dudoso en la mayoría de las series publicadas. El tratamiento prolongado con calcitonina parece ser seguro y la hormona sigue siendo el agente más potente contra la enfermedad.
Reconocimientos
Figuras 3 a 5 Talar Agasyan
DR. DANIEL D. FEDERMAN
Los trastornos de las paratiroides solían considerarse raros, difíciles de diagnosticar y frustrantes en cuanto a tratamiento. Sin embargo, las técnicas diagnósticas y quirúrgicas modernas han demostrado que son comunes y susceptibles de tratarse.
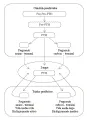
Fisiología de la regulación del calcio
Los niveles de calcio en el líquido extracelular y los reservorios óseos son controlados principalmente por la hormona paratiroidea (PTH) y el calcitrol (la forma activa de la vitamina D) [ver tabla 1]. Las hormonas calcitonina y proteína relacionada a la PTH son activas en niños y en casos patológicos, pero tienen escaso papel fisiológico en los adultos.
|
||||||||
|
HORMONA PARATIROIDEA
La PTH es sintetizada inicialmente como parte de una molécula más grande, pre-pro-PTH, que es inmediatamente acortada a pro-PTH. Aunque la pro-PTH tiene una vida más prolongada que su precursora, ninguna de las moléculas es liberada fuera de la glándula y ninguna es biológicamente activa.1 Sin embargo, la ruptura enzimática de pro-PTH deja en libertad a la PTH para su liberación a la circulación, donde es desdoblada por lo menos en dos porciones por los tejidos periféricos, principalmente el hígado y el riñón [ver figura 1]. Además, la paratiroides libera algunos fragmentos de la hormona. De esta forma, la PTH circulante incluye tres moléculas: la molécula completa, el fragmento amino-terminal con una vida media corta y el fragmento carboxi-terminal con una vida media más larga. Sólo la molécula completa y el fragmento amino-terminal son biológicamente activos.2
|
| Figura 1 |
| Síntesis de la hormona paratiroidea |
La síntesis de PTH y su liberación están controlados principalmente por los niveles séricos de calcio: un valor bajo estimula, y uno alto suprime la síntesis y liberación hormonal2 [ver figura 2]. Una cifra sérica elevada de fósforo por la disminución del calcio también estimula la liberación de PTH y es particularmente importante en la insuficiencia renal. La hipomagnesemia puede impedir la liberación de PTH y alterar la respuesta de los tejidos hacia la hormona, aunque estos efectos ocurren únicamente en situaciones patológicas. Algunos otros factores que influyen en la secreción de PTH, incluyendo hormonas alfa y beta adrenérgicas, neurotransmisores, prostaglandinas y otras hormonas, tienen efecto farmacológico, pero no son claros reguladores fisiológicos de la liberación de PTH.
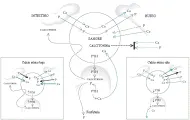
|
| Figura 2 |
| Metabolismo de calcio y fósforo |
La regulación intracelular de la secreción de PTH está controlada por un receptor de superficie celular recién detectado que fija iones calcio (Ca2+) en una estructura extracelular que recuerda un matamoscas. El receptor está acoplado a una porción intracelular de la célula a través de un dominio de serpentina de siete segmentos que atraviesan la membrana, característicos de la superfamilia de receptores unidos a la proteína G.3,4 Este receptor, el primero que se identificó para un ión extracelular, convierte al calcio en una hormona, o primer mensajero, así como un segundo mensajero intracelular. Esta molécula sensible al calcio también se encuentra en las células C del tiroides, en donde controla la liberación de calcitonina, en el riñón, en donde influye en el intercambio de fósforo y calcio, y en el sistema nervioso central y otros tejidos.5
La PTH mantiene los niveles séricos de calcio por promoción directa e indirecta de la entrada de calcio a la sangre a partir de tres sitios de intercambio: intestino, hueso y riñón. La PTH contribuye a la absorción neta de calcio por el intestino al favorecer la síntesis de calcitrol por el riñón. También promueve la entrada de calcio a la sangre desde el hueso por la inhibición de la función de síntesis de los osteoblastos e indirectamente estimulando la diferenciación hacia células de resorción ósea u osteoclastos. Finalmente, tiene tres efectos en el riñón: estimula la reabsorción tubular de calcio, incrementa la depuración de fósforo y promueve un aumento en la enzima que completa la síntesis de la forma activa de la vitamina D. Como la mayoría de las hormonas peptídicas, la PTH ejerce sus efectos a través de la activación de las adenilato-ciclasas unidas a membranas que generan monofosfato cíclico de adenosina (AMPc).
Los niveles circulantes de PTH (hormona paratiroidea inmunorreactiva o PTHi) se determina por radioinmunoanálisis. Los anticuerpos que detectan la PTH intacta, en especial la prueba inmunorradiométrica con doble anticuerpo son lo bastante sensibles y específicos para uso clínico. Esta prueba ha sustituido en mucho a otras usadas para evaluar eventos mediados por PTH, como la producción de AMPc nefrogénico, en la evaluación de la función paratiroidea [ver adelante, Hiperparatiroidismo, Hiperparatiroidismo primario].
VITAMINA D
La vitamina D es uno de los componentes importantes del sistema endócrino que regula el metabolismo de los minerales óseos. La síntesis de la vitamina D activa se inicia con la radiación ultravioleta sobre el 7-dehidrocolesterol en la piel, produciendo previtamina D3, que es convertida a vitamina D3 (colecalciferol). Esta vitamina D3 y parte de la que es absorbida de la dieta, es hidroxilada en la posición 25 en el hígado para formar 25-(OH)D3 o calcifediol, que es la forma principal de vitamina D circulante; los niveles plasmáticos de calcifediol promedian 30 ng/ml. El calcifediol es 1alfa-hidroxilado para formar calcitriol [1,25-(OH)D3, por una enzima renal cuya actividad es estimulada por la PTH, la hipocalcemia (a través de la estimulación de PTH) y la hipofosfatemia. El calcitriol parece ser la forma más activa de vitamina D; aunque sus niveles séricos de 25 a 40 pg/ml son mucho menores que los de calcifediol, es 100 veces más potente en base a su peso. Las enfermedades hepáticas pueden impedir la 25-hidroxilación y las enfermedades renales pueden suprimir la 1alfa-hidroxilación. La producción de calcitriol es autorregulada hasta cierto grado, y otras hormonas como prolactina, hormona del crecimiento y esteroides sexuales pueden también modular su producción.
El calcitriol ejerce su acción en los mismos tejidos que la PTH [ver figura 2], pero sus sitios subcelulares y su mecanismo de acción son distintos. A diferencia de la PTH, que interactúa con un receptor en la membrana celular, el calcitriol se une a un receptor intracelular y el complejo se fija a sitios específicos en la cromatina. A este respecto, el calcitriol actúa como una hormona esteroidea. El receptor de la vitamina D es un miembro de la superfamilia c-erb A de receptores, son miembros de esta misma familia los receptores que fijan tiroxina, esteroides y ácido retinoico.
En el intestino el calcitriol estimula la síntesis de proteína fijadora de calcio, y también estimula la absorción de calcio y fósforo.6 Al aumentar las concentraciones séricas de calcio y fósforo el calcitriol promueve el depósito de hidroxiapatita en el hueso. En forma paradójica, también moviliza calcio del hueso ya formado. El calcitriol inhibe además la síntesis de pre-pro-PTH y quizá sea un regulador fisiológico de la secreción de PTH. Existen receptores de calcitriol en muchos tejidos además del hueso, riñón e intestino, y entre los órganos y tipos celulares que los tienen se incluyen las glándulas paratiroides, los islotes pancreáticos, la glándula mamaria, los fibroblastos, y otros tejidos que no se conocen como órganos blanco del calcitriol.10 El papel del calcifediol y de la 24,25-(OH)2D3, un producto del metabolismo renal del calcifediol, no está bien definido; es probable que este último compuesto promueva la movilización de mineral óseo.
Los ensayos para calcifediol pueden ser útiles para ayudar en las decisiones clínicas. Esta forma de vitamina D tiene una vida media larga y niveles estables en el plasma, con un rango normal de 15 a 40 ng/ml. La principal aplicación clínica de la prueba de calcifediol es la valoración del metabolismo de la vitamina D: los valores bajos reflejan deficiencia de vitamina D o enfermedad hepática severa y ocurren en el embarazo, en el hipertiroidismo y durante el tratamiento con anticonvulsivos. Las cifras elevadas confirman la intoxicación con vitamina D. Hay menos experiencia con los ensayos de calcitriol, pero sus niveles están regulados en una manera distinta a los de calcifediol; los límites normales de 25 a 40 pg/ml varían con las estaciones (son más bajos en el invierno), con la edad (menores en personas de edad avanzada), los niveles de calcio sérico y otros factores menos claros. Sus valores se elevan por la PTH cuando la función renal está intacta; de esta forma, la medición del calcitriol es útil en el diagnóstico diferencial de la hipercalcemia.
CALCITONINA
El tercer regulador hormonal del balance del calcio es la calcitonina, polipéptido secretado por las células parafoliculares del tiroides. Su principal acción es reducir la reabsorción ósea, realizada por acción directa de los osteoclastos, al antagonizar la PTH y otros estímulos osteoclásticos. El efecto de la calcitonina es directamente proporcional a la tasa de formación y remodelación ósea, y al parecer su función consiste en minimizar la pérdida de calcio del esqueleto durante el crecimiento, el embarazo y la lactancia. Es posible que tenga una función limitada, si acaso tiene alguna, en el adulto. Las cifras de calcitonina disminuyen después de la menopausia y esta deficiencia relativa puede contribuir a la osteoporosis. Sin embargo, debe recordarse que ni la ausencia total ni el exceso extremo de la hormona producen manifestaciones clínicas claras. Este dato es sorprendente porque la calcitonina y sus receptores se encuentran diseminados en el cerebro, riñón y otros sitios.8 El procesamiento alternativo del gen de la calcitonina produce un péptido relacionado que tiene poderosos efectos hemodinámicos y que quizá sea importante en el sistema nervioso central. Este péptido no tiene función en la homeostasis del calcio.
PROTEINA RELACIONADA A LA PTH
Aunque se conoce otra hormona reguladora del calcio, su función precisa no ha sido bien determinada. La proteína relacionada con la hormona paratiroidea (PTHrP por sus siglas en inglés, n. del t.), fue detectada al principio como la causa de la hipercalcemia humoral en las neoplasias malignas (HHM), pero ha sido identificada (o su ARNm) en muchos tejidos, incluyendo la placenta y las glándulas paratiroides del feto, además de en queratinocitos, neuronas, islotes pancreáticos, riñón y hueso en el adulto. La molécula se parece mucho a la de la PTH en su región N-terminal, pero es más larga y más compleja que la PTH en otras regiones: por separación alternativa, sus ocho exones pueden, por medio de separación alterna, producir tres proteínas diferentes. Existen dos carcterísticas únicas de esta hormona. Primero, comparte un receptor único con la PTH y, segundo, parece tener un segundo locus biológicamente activo además de la región similar a la PTH. Este segundo sitio activo participa en el transporte de calcio en la placenta, convirtiendo así a la PTHrP en la PTH del feto. Al parecer,la PTHrP tienen una función principalmente autócrina local, la única alteración clínica significativa que se ha asociado con esta proteína es su secreción excesiva en diversos tumores, provocando la HHM.11
EFECTOS DE LA EDAD
Con la edad ocurren cambios importantes en el metabolismo del calcio y de las hormonas relacionadas: (1) el nivel sérico medio de PTHi es de 20 a 40 porciento más alto en personas mayores de 70 años, (2) los valores de calcitriol parecen disminuir en las personas mayores,12,13 y (3) los niveles de calcitonina disminuyen en las mujeres de edad avanzada. Estos cambios pueden relacionarse con ingesta inadecuada de calcio, porque los niveles de este ión no cambian con la edad. Cuando mujeres ancianas recibieron una dieta con 2,400 mg/día de clacio, sus niveles de PTH fueron semejantes a los de pacientes jóvenes.14,15 La principal expresión clínica de estos cambios es la reducción de la masa ósea relacionada con la edad. En algunos pacientes, esta disminución causa osteoporosis y mayor riesgo de fracturas.
Hiperparatiroidismo
El término hiperparatiroidismo se aplica a cualquier trastorno en el que el organismo está expuesto a niveles elevados de hormona paratiroidea efectiva. El calcio sérico puede estar aumentado, normal o disminuido, dependiendo de otros factores. El término hiperparatiroidismo primario denota un exceso en la producción de PTH que se origina en una lesión (adenoma, hiperplasia o carcinoma) de las paratiroides. El calcio sérico suele estar elevado. El hiperparatiroidismo secundario indica un exceso de adaptación en la producción de PTH estimulada por uno de los reguladores fisiológicos de la función paratiroidea: hipocalcemia, hiperfosfatemia o hipomagnesemia. El calcio sérico se encuentra bajo o, si la compensación es completa, normal.
HIPERPARATIROIDISMO PRIMARIO
Etiología y patogenia
El hiperparatiroidismo primario es causado por un adenoma único en el 85 a 90 porciento de los casos. Las mujeres padecen hiperparatiroidismo con una frecuencia mayor del doble que los hombres, y la incidencia aumenta con la edad: después de los 50 años, ésta es de alrededor de uno por 1,000 por año en varones y de dos a tres por 1,000 por año en mujeres.16 La hiperplasia de células principales es la siguiente causa más frecuente del hiperparatiroidismo primario. Las cuatro glándulas suelen encontrarse afectadas, aunque es común que el crecimiento sea asimétrico. Este trastorno, en el que la hipercalcemia moderada es la regla, puede reflejar el defecto de alguna sustancia paratirotrópica o un reajuste en las concentraciones que dan lugar a las alteraciones en el calcio sérico que modifican la liberación de PTH.
La diferencia entre adenoma e hiperplasia parece tener una base genética. Los adenomas son sobre todo de origen monoclonal. La primera alteración genética molecular identificada en pacientes con adenomas paratiroideos se debe a dos rupturas y una inversión en el cromosoma 11, que mueve la región regulador 5' del gen de la PTH lejos de la secuencia codificadora hacia un gen diferente conocido como PRAD1 [ver figura 3]. La proteína codificada por el PRAD1 es la ciclina que, en combinación con otros activadores del ciclo celular, estimula la proliferación celular sin cambios malignos. El cromosoma no mutado, con sus regiones reguladora 5' y codificadora adyacentes y sin cambio, puede ser el origen del exceso en la secreción de PTH. Este defecto ocurre en alrededor del 5 porciento de los adenomas paratiroideos. Quizá hasta el 25 porciento de los casos muestran pérdida de grandes segmentos de un cromosoma 11, que quizá en asociación con deleciones no visibles o con mutaciones del otro cromosoma 11, causa la formación del adenoma.17,18
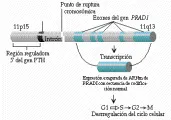
|
| Figura 3 |
| Estructura molecular del gen de PTH/PRAD1 |
Por el contrario, la hiperplasia partiroidea, en especial la observada en la neoplasia endócrina múltiple de tipo I (NEM I) típicamente es policlonal. Esto puede deberse a una mutación heredada en un locus supresor de tumores en el cromosoma 11. Debido a que se hereda por línea germinal, el defecto incluye a todas las células. Cuando aparece un defecto somático en el otro cromosoma 11, la pérdida resultante de la heterocigosidad causa expansión clonal y neoplásica de un adenoma agregado a la hiperplasia. Estos tumores típicamente son grandes y se asocian con hipercalcemia moderadamente severa. Estas diferencias son clínicamente importantes porque el hiperparatiroidismo es el signo más común y con frecuencia el inicial en la NEM I.
La causa menos frecuente de hiperparatiroidismo primario es el carcinoma de las paratiroides, que corresponde a alrededor del 1 porciento de los casos. Estos tumores tienden a crecer lentamente y producen invasión local, pero pueden alcanzar el hígado, el pulmón o el hueso. Por lo menos un defecto molecular, la deleción del gen supresor de tumores del retinoblastoma, favorece el desarrollo del carcinoma paratiroideo.19
Los efectos clínicos del exceso de PTH, diferentes de la hipercalcemia, se comprenden poco. La hipercalcemia puede contribuir a los datos neurológicos, así como a la fatiga general, y ciertamente tiene un papel importante en el daño renal. Sin embargo, no es claro si la hipercalcemia o el aumento en la PTH son los responsables de la mayoría de los otros síntomas. La PTH es directamente responsable de los trastornos esqueléticos por estimular la resorción osteoclástica, principalmente en el hueso cortical.
Manifestaciones clínicas
El síndrome clínico del hiperparatiroidismo primario tiene manifestaciones diversas [ver tabla 2]. Inicialmente el trastorno fue identificado como una combinación de enfermedad ósea severa (osteítis fibrosa quística) con pérdida de estatura, debilidad y emaciación, anemia y disfunción renal. Después se reconoció un síndrome diferente caracterizado por litiasis renal y denominado nefrocalcinosis. Aún después, se demostró sintomatología digestiva asociada, como constipación, dolor abdominal, dispepsia o úlcera péptica y pancreatitis. Ultimamente se han identificado trastornos musculoesqueléticos, incluyendo miopatía, neuropatía, seudogota y debilidad inespecífica. Sin embargo, en la actualidad se sabe que estos síndromes constituyen la minoría de los casos de hiperparatiroidismo primario. Con más frecuencia los nuevos casos se descubren al detectar hipercalcemia en forma incidental durante los escrutinios generales en pacientes adultos asintomáticos o con molestias inespecíficas.16,20
|
||||||||||||||||||||
* Nota :La presentación más común del hiperparatoidismo es la detección incidental de hipercalcemia leve durante el escrutinio multifásico |
Los signos físicos del hiperparatiroidismo, además de la debilidad generalizada moderada, se encuentran solamente en sujetos muy enfermos e incluyen pérdida de peso, palidez y queratopatía en banda. Ocasionalmente los individuos presentan pérdida de estatura y xifosis secundaria a fracturas vertebrales, y algunos tienen dedos en palillo de tambor por osteítis severa de las falanges terminales, con fracturas y colapso óseo. Pueden también observarse diversos signos neurológicos inespecíficos (v.gr., somnolencia, fatiga y cambios en la personalidad).21
Diagnóstico de laboratorio
La confirmación del diagnóstico de hiperparatiroidismo sigue basándose principalmente en las características distintivas de los resultados de laboratorio. La prueba más útil es la medición del calcio sérico, que se encuentra elevado en el 96 porciento o más de los casos con hiperparatiroidismo primario. Es frecuente la elevación intermitente en la enfermedad leve, por lo que deben repetirse las mediciones cuando la sospecha diagnóstica es fuerte pero la medición inicial de calcio en suero es normal.
Otras pruebas bioquímicas son útiles pero no diagnósticas. Con frecuencia los valores de fósforo sérico están disminuidos. Los niveles de cloro sérico suelen ser mayores de 102 mEq/L; esta elevación refleja la influencia del exceso de PTH sobre la excreción renal de bicarbonato, produciendo una acidosis metabólica moderada con niveles de cloro altos y disminución leve en las cifras de bióxido de carbono.
La evaluación definitiva de la función paratiroidea consiste en medir la concentración de la hormona paratiroidea en la circulación. Como se mencionó, las mediciones de anticuerpos aislados no son sensibles ni específicas. Los ensayos de doble anticuerpo medido por inmunorradiometría e inmunoquimoluminiscencia indican que la PTHi está elevada en más del 90 porciento de los casos de hiperparatiroidismo primario, incluso en pacientes con hipercalcemia leve. Además, la PTHi es baja en las hipercalcemias no paratiroideas, en especial en la HHM. Por lo tanto, debe medirse la concentración de PTHi en todos los pacientes con hipercalcemia [ver figura 4]. Los pacientes con niveles de PTHi elevados o inapropiadamente normales deben someterse a una absorciometría de la muñeca, ya que el hueso cortical es más sensible al exceso de PTH. El hueso trabecular, como el de la columna, se afecta mucho menos, y puede incluso estar aumentado en el hiperparatiroidismo. Se encuentra menor densidad del hueso vertebral en el 15 porciento de los pacientes.22 Para los pacientes ocasionales con osteítis fibrosa las placas simples de manos y cráneo son útiles; las últimas deben examinarse con luz brillante para identificar la resorción subperióstica de las falanges medias.

|
| Figura 4 |
| PTH y calcio en diferentes padecimientos |
En ausencia de deficiencia de vitamina D existe una correlación razonable entre el tamaño del adenoma paratiroideo y la hipercalcemia, y el aumento en la PTHi que produce. En los pacientes con hiperparatiroidismo leve los niveles de PTHi pueden caer dentro del rango normal superior, pero ser altos en relación con el nivel de calcio sérico; se usa un nomograma para interpretar estos valores. Si la PTHi está muy elevada comparado con el nivel de calcio en suero, es posible que exista un factor agregado, como deficiencia de vitamina D. Debido a que la PTH ejerce parte de su efecto calcémico a través del calcitriol, los pacientes hiperparatiroideos con deficiencia o resistencia simultánea de vitamina D pueden tener una concentración de calcio normal alta o muy poco elevada. Esta combinación de hiperparatiroidismo severo e hipercalcemia mínima puede ocurrir cuando existe deficiencia dietética de calcio, malabsorción, deficiencia de magnesio, enfermedad hepática severa o, más importante, insuficiencia renal crónica. En la última de estas condiciones la normocalcemia es causada más por hiperfosfatemia que por deficiencia de calcitriol.
El uso diseminado del escrutinio multifásico ha revelado una nueva población de pacientes hiperparatiroideos. El término hiperparatiroidismo asintomático se refiere a pacientes que tienen una concentración de calcio en suero elevada pero que no excede 11 mg/dl, un nivel no suprimido de PTHi, no enfermedad ósea, no enfermedad renal por cálculos activa o insuficiencia renal y ausencia de síntomas (incluso al interrogatorio sutil) potencialmente atribuibles al hipertiroidismo. La calidad y sensibilidad del interrogatorio es importante porque la cirugía puede aliviar los síntomas de los pacientes con hipercalcemia leve y porque los síntomas potenciales, incluyendo los psiquiátricos, son muy diversos [ver tabla 2].23 Irónicamente, la incidencia de hiperparatiroidismo parece estar disminuyendo, y el advenimiento de la atención dirigida ha introducido una nueva fase en la evolución de la epidemiología y manejo de este padecimiento. Si las limitaciones sobre el escrutinio progresan, el descubrimiento incidental del hipertiroidismo será menos común y aumentarán los casos sintomáticos.22,24
Tratamiento
El único tratamiento curativo para el hiperparatiroidismo es la resección quirúrgica del tejido paratiroideo. Por lo tanto, en los pacientes sintomáticos deberá elegirse el procedimiento quirúrgico adecuado. Para obtener mejores resultados se necesita de un equipo médico-quirúrgico experimentado. El siguiente programa parece ser razonable para tomar esta decisión:
1. Para el enfermo común con síntomas leves, calcio elevado y fósforo bajo, es razonable un manejo conservador. El cirujano debe explorar un lado del cuello; si encuentra un adenoma paratiroideo grande y una glándula normal, confirmado por el patólogo, el lado contralateral no deberá explorarse y se cierra el cuello después de la extirpación del adenoma. Este método se emplea en el Hospital General de Massachusetts, donde se han realizado más de 700 operaciones para hiperparatiroidismo y en los Institutos Nacionales de Salud de los Estados Unidos (NIH).
2. Para el sujeto con afección hiperplásica de las cuatro glándulas, por lo general es necesaria una paratiroidectomía resecando 7 octavas partes del tejido paratiroideo, para el control de la enfermedad y prevenir la recurrencia. Este procedimiento limita por una parte la aparición de hipoparatiroidismo y por otra evita el hiperparatiroidismo persistente. Algunos cirujanos extirpan las cuatro glándulas y reimplantan una porción de ellas en los músculos del antebrazo. Pueden implantarse rebanadas muy delgadas de tejido paratiroideo durante el procedimiento original, o pueden congelarse y emplearse hasta 18 meses después. Pasan 6 a 8 semanas para que el tejido trasplantado desarrolle vasculatura y comience a funcionar.
3. Cuando el cirujano no está seguro del diagnóstico patológico, conviene exponer al menos tres glándulas; si más de una está agrandada y macroscópicamente afectada, es mejor considerar la enfermedad como hiperplasia. Aunque existen adenomas dobles, estos son raros y uno no debe basar el tratamiento en esta esperanza. Los cirujanos experimentados que usan estos lineamientos para encontrar la lesión corrigen el hiperparatiroidismo en el 90 a 95 porciento de los casos. En general los estudios de imagen preoperatorios no mejoran estos resultados ni se realizan de rutina. Sin embargo, varios centros que han probado con las nuevas modalidades han encontrado que los estudios de imagen preoperatorios positivos pueden ser útiles en pacientes con riesgo alto.26
4. La reintervención quirúrgica en los enfermos que no se curaron con la primera exploración es una decisión difícil de tomar. Los factores cruciales son la experiencia y la habilidad del cirujano. Las consideraciones adicionales son una meticulosa revisión del informe quirúrgico y patológico original, la disección cuidadosa con exposición adecuada y, en algunos casos, contar con estudios de imagen preoperatorios.30 La selección de las técnicas de imagen varía en diferentes instituciones. El ultrasonido y la gammagrafía con isótopos doble da resultado más satisfactorios que la TC, pero ninguna de estas técnicas posee la sensibilidad o especificidad de la habilidad quirúrgica.27 El gamagrama con sestamibi marcado con tecnecio 99m parece ser el método más útil para la localización.28
5. La gran prevalencia de hiperparatiroidismo asintomático, combinado con la negativa de estos pacientes a someterse a cirugía ha aumentado la duda respecto a si la cirugía es realmente necesaria. Una conferencia de consenso de los NIH admitió que la vigilancia no quirúrgica es adecuada para los pacientes sin síntomas, que tienen concentraciones de calcio menores de 12 mg/dl, sin calcificación renal, no enfermedad ósea evidente por radiología, concentraciones de calcio en orina menores de 400 mg/24 horas y depuración de creatinina normal.29
La vigilancia no quirúrgica puede compararse con el informe del Hospital General de Massachusetts acerca del tratamiento quirúrgico de 242 enfermos que padecían hiperparatiroidismo leve. Estos sujetos se consideraban asintomáticos, pero mostraron datos de evolución de la enfermedad por uno o más de los siguientes criterios: niveles de calcio sérico mayores de 11 mg/dl, calcio urinario mayor de 150 mg/24 horas o incremento en los valores séricos de PTHi. La hipercalcemia se corrigió en el 98 porciento de los casos y sólo persistió despues de la cirugía en 4 individuos; no hubo muertes o complicaciones de importancia. Esta experiencia debe tomarse como marco de referencia ideal, más que como un logro quirúrgico común, pero la seguridad quirúrgica actual, incluso en personas de edad avanzada, desafía el tratamiento médico conservador. Por ejemplo, la serie de la Universidad de Michigan describe a 81 pacientes mayores de 65 años que sobrevivieron a la cirugía.30
La vigilancia de los pacientes con hiperparatiroidismo asintomático es de particular importancia en mujeres posmenopáusicas, el mayor grupo de pacientes asintomáticos (y el que se descubre más a menudo de manera incidental). En este grupo, los estrógenos y la noretindrona pueden disminuir el calcio sérico y mejorar el balance de calcio, lo que al parecer protege contra la osteoporosis.31 Si existe osteopenia significativa de la columna es adecuado realizar una densitometría de seguimiento. De igual manera, debe hacerse una medición densitométrica anual de la muñeca (el hueso cortical más afectado por la PTH) para detectar la progresión de la enfermedad paratiroidea. Los pacientes deben evitar el litio y los diuréticos tiacídicos, que pueden aumentar la secreción de PTH y aumentar la concentración sérica de calcio. Deben solicitar atención en caso de náusea o vómito porque la deshidratación puede inducir un aumento rápido y potencialmente peligroso en la concentración sérica de calcio. Por último, el clínico debe considerar la aparición o progresión de síntomas leves, incluyendo depresión, fatiga y dolores inespecíficos, como una justificación para la cirugía, porque el hiperparatiroidismo puede evolucionar de manera sutil; hasta el 65 porciento de los pacientes supuestamente asintomáticos mejoran después de la cirugía.
Manejo pre y posoperatorio La reacción que debe vigilarse en el posoperatorio inmediato es el desarrollo de tetania hipoparatiroidea [ver adelante, Hipocalcemia]; a largo plazo, es necesario vigilar la aparición de datos de hipoparatiroidismo, la recurrencia del hiperparatiroidismo o la afección de otras glándulas endócrinas. Ninguno de estos problemas ocurren ordinariamente en sujetos que tienen un adenoma único, una segunda glándula normal, normalización rápida de los niveles de calcio y ausencia de antecedentes familiares compatibles con neoplasia endócrina múltiple. En tales individuos, lo más adecuado es una revisión anual con determinación única de calcio sérico y glucemia en ayunas. Cuando se encuentra afectada más de una glándula deberá realizarse una investigación cuidadosa en busca de las características clínicas del síndrome de neoplasia endócrina múltiple en el paciente y su familia.
Los enfermos con osteítis fibrosa quística severa ocasionalmente tienen un curso posoperatorio difícil y prolongado, caracterizado por hipocalcemia, hipofosfatemia y aun tetania clínica. Este racimo de complicaciones es atribuible al llamado fenómeno del "hueso hambriento", en el que la formación excesiva de hueso afecta el depósito de calcio y fósfato de una manera que supera la homeostasis del calcio. El tratamiento con vitamina D mejora este problema y debe iniciarse en forma temprana si la hipocalcemia persiste en el posoperatorio, pero debe ser suspendida si se ha dejado tejido paratiroideo funcionante, confirmado por la presencia de niveles elevados de PTHi.
Cáncer de las paratioides
El carcinoma de las paratiroides es causa de hiperparatiroidismo en menos del 1 porciento de los casos. Esta rara enfermedad es típicamente grave: produce síntomas notables, niveles de calcio promedio en pacientes nuevos superiores a 15 mg/dl, enfermedad renal y ósea frecuentes y, en la mitad de los casos, una masa palpable en el cuello.32-34 Estos datos contrastan con la presentación conocida en la actualidad como típica para los casos de hiperparatiroidismo primario.35 En el momento de la cirugía, una clave para el diagnóstico es la adherencia de la glándula paratiroides, aumentada de volumen, a las estructuras vecinas; si el cirujano encuentra estos datos, deberá realizarse en forma muy cuidadosa una resección en bloque, ya que el control satisfactorio de la enfermedad solo se ha logrado con una resección amplia inicial.
Cuando el diagnóstico de cáncer no se realiza inicialmente, puede sospecharse en forma retrospectiva por la reaparición de la hipercalcemia 6 meses o más después de la cirugía; en cambio, cuando se deja una glándula hiperplásica remanente, la hipercalcemia se presenta en forma más temprana. La hipercalcemia persistente o recurrente obliga a revisar los datos histopatológicos originales. Aunque el diagnóstico definitivo de cáncer puede ser difícil, la invasión capsular por el tumor es un dato específico. Además, los estudios moleculares son medios poderosos para distinguir entre las lesiones benignas y malignas.19 La mayoría de los pacientes mueren por hipercalcemia; sin embargo, la reoperación local, la extirpación de las metástasis pulmonares guiada por TC, el tratamiento con bifosfonatos y el control de la hipercalcemia severa son todas medidas útiles.35
HIPERPARATIROIDISMO SECUNDARIO
El hiperparatiroidismo secundario es el resultado de la adaptación de las glándulas paratiroides a un estado de hiperfunción; es decir, la hipersecreción de PTH ocurre como una consecuencia fisiológica de otra enfermedad. Por ejemplo en la insuficiencia renal crónica, la disminución en la formación de calcitriol produce absorción baja de calcio,36 la pérdida del parénquima renal causa hiperfosfatemia y tanto la hipocalcemia como la hiperfosfatemia estimulan la función de las paratiroides. Los niveles bajos de calcitriol pueden permitir que la masa celular paratiroidea aumente, y la supresión de la actividad secretora de las paratiroides parece requerir un nivel más alto de calcio ionizado que el que se encuentra en estados normourémicos. La supresión habitual de la producción de PTH por el calcitriol está bloqueada y el contenido en las células paratiroides de receptores de calcio también está disminuido.37 En la insuficiencia renal avanzada la mayor masa de tejido paratiroideo, aunado a una secreción no suprimible basal por célula, se agrega a la sobreproducción de PTH. Además, puede haber resistencia renal y esquelética a la PTH. En los trastornos gastrointestinales la malabsorción de vitamina D y calcio produce hipocalcemia e hiperparatiroidismo secundario.
Ya que el exceso secundario de PTH es adaptativo, más que autónomo, raras veces produce hipercalcemia. Más bien, se deduce que existe compensación paratiroidea cuando el calcio sérico se encuentra disminuido o dentro de límites normales en circunstancias en las que se espera que se encuentre bajo. Los niveles de PTHi pueden estar muy aumentados. Los signos radiológicos de osteítis fibrosa ofrecen información adicional a este respecto. En los casos severos puede ocurrir hipercalcemia en el hiperparatiroidismo secundario, y puede ser difícil o imposible distinguir los casos primarios de los secundarios.
El mejor recurso para el hiperparatiroidismo secundario es el control de la enfermedad de fondo; cuando esto no es posible, la manipulación farmacológica puede ser de ayuda. En los pacientes con insuficiencia renal crónica los niveles de fosfato pueden ser controlados con la administración de fijadores de fosfato por vía oral (sin embargo, los fármacos que contienen aluminio empeoran la osteomalacia).37 Si este método es insuficiente se puede intentar el tratamiento cuidadoso con dosis pequeñas de vitamina D. Se ha obtenido más experiencia con el calcitriol, que parece aliviar el dolor óseo y la debilidad muscular en enfermos con falla renal crónica e hiperparatiroidismo secundario. La osteítis fibrosa leve y la osteomalacia, que frecuentemente la acompañan, mejoran y los niveles de fosfatasa alcalina y de PTHi descienden gradualmente. Los sujetos con osteítis fibrosa severa y aquellos en quienes la cifras de calcio se encuentran en límites superiores normales antes del tratamiento con calcitriol, presentan episodios de hipercalcemia notable al iniciar el tratamiento. Por lo tanto, requieren de vigilancia estrecha; afortunadamente, la acción del calcitriol es de corta duración y permite una recuperación pronta de sus efectos al suspenderlo.
Un síndrome de osteomalacia relacionado con la diálisis parece ser causado por la exposición al aluminio que se encuentra presente en los dializadores. Este metal parece bloquear la mineralización del hueso; cuando se administra vitamina D, los individuos afectados tienden a presentar hipercalcemia notable, que responde bien a la suspensión de este medicamento, aunque la mejoría en la enfermedad ósea requiere de la quelación del aluminio.
Anteriormente se sugería que en algunos pacientes con hiperparatiroidismo grave secundario de evolución prolongada, una de las glándulas paratiroides podría empezar a funcionar autónomamente y causar hiperparatiroidismo terciario. Los estudios moleculares han demostrado que tanto en la hiperplasia de células principales policlonal primaria como secundaria, ocurren tumores monoclonales. Sin embargo, esto es más frecuente que el síndrome de hiperparatiroidismo terciario, y el significado exacto de esta evolución molecular no es clara aún.38 Se ha obtenido experiencia relevante sobre el hiperparatiroidismo secundario desde la introducción del trasplante renal para la insuficiencia renal crónica. Al principio, algunos pacientes desarrollaban hipercalcemia grave poco después de la cirugía. En ocasiones se realizó paratiroidectomía y los hallazgos indicaron un hiperparatiroidismo secundario severo e irreversible, y aún hiperparatiroidismo terciario. Sin embargo, en muchos de estos casos se corrigió la hipercalcemia después de 2 años o más sin paratiroidectomía, lo que se atribuyó en forma retrospectiva a la regresión de la hiperplasia secundaria. Aunque la hipercalcemia grave que requiere cirugía es menos común en la actualidad, es más frecuente la hipercalcemia moderada pero persistente. Estos casos pueden indicar ya sea un cambio en el punto fijo en el que los niveles de calcio estimulan la secreción de PTH o más probablemente, una masa poco frecuente de tejido paratiroideo hiperplásico que requiere de un tiempo más prolongado para su regresión.
Hipercalcemia
La hipercalcemia es un problema clínico común que puede constituir una urgencia que ponga en peligro la vida del paciente, o ser el heraldo silencioso de una gran variedad de enfermedades curables.39,40
Más del 90 porciento de los casos se atribuyen a hiperparatiroidismo o se asocian con cáncer; por lo tanto, debe pensarse primero en estas opciones. Por lo demás, el primer paso debe consistir en revisar los mecanismos fisiológicos, más que intentar abarcar todas las posibilidades diagnósticas. Fisiológicamente los niveles de calcio se mantienen por tres procesos: absorción gastrointestinal, resorción osteoclástica de minerales de las reservas óseas y reabsorción tubular de calcio del filtrado glomerular. El control predominante en la absorción gastrointestinal es la vitamina D y el principal control en la esfera renal y ósea es la PTH. Las causas más comunes de hipercalcemia son la absorción gastrointestinal excesiva, la resorción ósea excesiva y un mecanismo incierto que probablemente entraña un aumento en la reabsorción renal de calcio [ver figura 5].
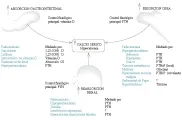
|
| Figura 5 |
| Patogenia de la hipercalcemia |
GUIAS DIAGNOSTICAS
Una guía general basada en los tres principales mecanismos productores de hipercalcemia puede ser útil en la interpretación de la sintomatología del paciente.
Aumento en la absorción gastrointestinal de calcio
En la sarcoidosis, algunas otras enfermedades granulomatosas, la enfermedad de Hodgkin y los linfomas no Hodgkin ocurre aumento en la absorción GI de calcio debido a síntesis excesiva de calcitriol. El calcitriol y el calcio inhiben la expresión del gen de PTH, así como su secreción; por lo tanto, la PTHi en plasma es baja.41,42
El síndrome leche-álcali (asociado con ingesta escesiva de leche o alcalinos absorbibles) produce una insuficiencia renal moderada, alcalosis y elevación en los niveles de calcio y fósforo. Puede ser muy difícil distinguir este síndrome del hiperparatiroidismo primario con insuficiencia renal crónica y úlcera péptica secundarias, por lo que la determinación de PTHi debe ser incluida en el estudio de estos pacientes.
La hipervitaminosis D ocurre en enfermos que están recibiendo tratamiento con esta vitamina o entre las personas con manías alimentarias que consumen grandes cantidades de vitamina D. El síndrome se caracteriza por insuficiencia renal, elevación de los niveles de fósforo (en dos tercios de los casos) y disminución del filtrado glomerular. En sujetos que han sido tratados con vitamina D, generalmente por hipoparatiroidismo, la hipercalcemia casi siempre es el resultado de los esfuerzos por corregir la hipocalcemia de una manera precipitada o por un seguimiento inadecuado. Ocasionalmente también ocurre en individuos bien tratados, pero se desconoce la causa en etos casos; en otros puede ser difícil recabar el antecedente de ingestión de vitamina D, aunque los niveles de calcifediol estarán elevados en estos pacientes. Este último dato fue la clave en los casos reportados de pacientes que bebieron leche fortificada en forma errónea con un exceso de vitamina D.42,44
Aumento en la resorción ósea
El hiperparatiroidismo primario ha sido analizado en forma extensa [ver antes, Hiperparatiroidismo] porque es la causa más común de hipercalcemia asintomática, particularmente en los pacientes ambulatorios, y el diagnóstico se hace más probable a medida que aumenta la duración de la hipercalcemia y ninguna otra alternativa se hace aparente.
En los enfermos hospitalizados la hipercalcemia se relaciona a los padecimientos oncológicos con más frecuencia que a ningún otro. Ocurren lesiones osteolíticas secundarias y metástasis en cánceres de mama, pulmón y próstata, así como otras muchas enfermedades malignas. El mieloma múltiple también puede afectar al hueso. Este fenómeno, conocido como hipercalcemia osteolítica local, es causado por estimulación osteoclástica directa por una o más citocinas producidas en los tumores, incluyendo varias interleucinas, factor de necrosis tumoral alfa, factor transformador de crecimiento y linfotoxina. Los tumores metastásicos que causan hipercalcemia generalmente son visibles en las radiografías simples de los huesos, pero los gamagramas óseos pueden ser compatibles con metástasis aun cuando las radiografías no muestren alteraciones.
La causa más frecuente de hipercalcemia severa en las neoplasias es el síndrome conocido como hipercalcemia humoral maligna (HHM). Los cánceres que se asocian con más frecuencia con este síndrome son de células epidermoides y de origen genitourinario o ginecológico. Los pacientes con HHM tienen manifestaciones semejantes a las de los pacientes hiperparatiroideos, incluyendo hipofosfatemia leve y aumento en la concentración urinaria de AMPc. Estos enfermos tienen más posibilidad de tener anemia y menos de tener hipercloremia, pero el mayor contraste consiste en que tanto la concentración de PTHi como de calcitriol están bajas o en límites normales bajos. El compuesto responsable de este cuadro clínico es la PTHrP (ver antes).45-47 Aunque el papel fisiológico endócrino de la PTHrP no se conoce, esta proteína promueve la resorción ósea y la reabsorción renal de calcio cuando se secreta en exceso por las células neoplásicas, produciendo hipercalcemia.45,48,49 La PTHrP también se ha identificado en leucemias causadas por virus linfotrópico a células T humanas tipo I y algunos cánceres de mama. En pocos casos se ha reportado la producción de prostaglandina E2 o PTH.
La enfermedad de Paget del hueso puede producir hipercalcemia, particularmente en enfermos que tienen afección ósea diseminada. Este fenómeno es raro en pacientes ambulatorios, pero es más frecuente en los hospitalizados. El hipertiroidismo se relaciona con hipercalcemia moderada en aproximadamente 10 a 20 porciento de los casos. El mecanismo exacto se desconoce, pero el recambio óseo acelerado parece ser la lesión principal.
La inmovilización, como la que ocurre durante la hospitalización o después de traumatismos graves, puede producir una hipercalcemia marcada; aunque ésta puede ser atribuida a la inactividad per se, es mucho más común cuando hay una causa independiente de recambio óseo acelerado, como la enfermedad de Paget o el crecimiento rápido en adolescentes.
El tratamiento con litio y los diureticos tiacídicos pueden inducir hipercalcemia leve a moderada, generalmente con un aumento correspondiente en los niveles de PTHi; el síndrome es reversible al suspender el medicamento. Los pacientes deben ser vigilados durante 3 meses después de suspender cualquier medicamento. Si la hipercalcemia y el nivel aumentado de PTHi persisten, casi siempre existirá un adenoma paratiroideo o hiperplasia.
Aumento en la reabsorción renal de calcio
Tanto la PTH como la PTHrP estimulan la adenilato ciclasa renal. Por lo tanto, la hipercalcemia que producen, principalmente por resorción ósea, es exacerbada por una mayor reabsorción renal del calcio filtrado. La reabsorción de calcio contribuye también a la hipercalcemia en otras circunstancias. Los diuréticos tiacídicos inducen hipercalcemia de manera impredescible, y las publicaciones difieren en la frecuencia con que la hipercalcemia inducida por las tiacidas constituye una clave diagnóstica para la presencia de hiperparatiroidismo.
La hipercalcemia hipocalciúrica familiar (HHF) es un trastorno autosómico dominante caracterizado por la aparición de hipercalcemia en la niñez, ausencia de hipercalciuria, fatiga, cefalea, artralgias y polidipsia. Los niveles de PTHi y de calcitriol no están aumentados y aunque la enfermedad se acompaña con frecuencia de hiperplasia paratiroidea, responde poco a la paratiroidectomía subtotal. El diagnóstico es especialmente díficil a menos de que la hipercalcemia inicie antes de los 10 años de edad (lo cual es muy raro en el hiperparatiroidismo primario o familiar) o que varios miembros de la familia estén afectados. La HHF es causada por un defecto autosómico dominante en los receptores sensibles a calcio tanto en las paratiroides como en los riñones: la secreción de PTH no es inhibida por el nivel habitual de calcio sérico y su excreción renal es desproporcionadamente baja para el nivel de hipercalcemia. El síntome letal de hiperparatiroidismo neonatal severo es causado por la expresión homocigota del mismo defecto en el receptor.50,51
Diagnóstico diferencial
La hipercalcemia que se descubre de manera incidental en el paciente ambulatorio es causada con mayor frecuencia por hiperparatiroidismo; en el enfermo hospitalizado, suele representar una complicación del cáncer. El hiperparatiroidismo suele acompañarse de acidosis hiperclorémica leve, con un nivel de cloro superior a 102 mEq/L y una cifra de fosfato inorgánico menor de 3 mg/dl. En la hipercalcemia no relacionada con hiperparatiroidismo el nivel de bicarbonato sérico se encuentra a menudo ligeramente elevado. Sin embargo, en la hipercalcemia aguda la presencia de deshidratación, insuficiencia renal y vómito complican las alteraciones electrolíticas.
La determinación de PTHi y de calcitriol constituyen el enfoque más directo para esclarecer la etiología de la hipercalcemia. El nivel de PTHi está elevado en el hiperparatiroidismo y suprimido en el resto de los estados hipercalcémicos. El nivel de calcitriol está elevado en el hiperparatiroidismo, la sarcoidosis, en algunos trastornos malignos y en la intoxicación por vitamina D, y reducido en la HHM. El aumento en la concentración de AMPc urinario puede orientar en casos de hipercalcemia que son difíciles de clasificar.
TRATAMIENTO
Una elevación estable de los niveles de calcio sérico refleja un equilibrio entre el aporte de calcio del intestino y hueso y su pérdida a través del riñón. En algunos pacientes el ritmo de reabsorción ósea supera este balance y el desequilibrio produce hipercalcemia. Cuando los niveles de calcio comienzan a elevarse, la reabsorción de agua y sal en el túbulo contorneado distal se altera. La deficiencia de volumen, frecuentemente agravada por el vómito, ocasiona aumento en la reabsorción de sodio y agua en el túbulo contorneado proximal. Finalmente, al disminuir el filtrado glomerular (y la filtración de calcio), además de la disminución de la entrada de calcio al hueso por la inactividad, se puede producir una hipercalcemia que ponga en peligro la vida del enfermo.
En el equilibrio o en la hipercalcemia estable, cuando los niveles séricos de calcio son menores de 14 mg/dl, el tratamiento de la hipercalcemia dependerá de la enfermedad subyacente. La mayoría de las causas de hipercalcemia pueden ser tratadas específicamente y casi siempre es mejor proceder de esta menra que considerar el tratamiento farmacológico crónico para disminuir los niveles séricos de calcio. Sin embargo, es urgente disminuir los niveles de calcio cuando éstos exceden los 14 mg/dl o cuando están entre 12 y 14 mg/dl y el sujeto está sintomático. Cuando los niveles de calcio se encuentran por abajo de 12 mg/dl es raro que la sintomatología severa pueda atribuirse a la hipercalcemia.
Existen varios métodos disponibles para el manejo de urgencia de la hipercalcemia.39 La medida inicial consiste en restablecer el volumen extracelular y la filtración glomerular, habitualmente con 3 a 4 L de solución salina por día. Una vez que la diuresis es adecuada, si la concentración sérica de calcio está por arriba de 14.5 mg/dl, se agrega furosemide, 40 a 80 mg/día IV. Si el paciente no está totalmente alerta y no es capaz de beber, cada cuarta o quinta botella de líquido debe ser hipotónica para prevenir la hipernatremia. En los pacientes con función ventricular izquierda limítrofe debe vigilarse la presión venosa con una línea central para evitar sobrecargar el corazón. Debe tenerse cuidado de evitar la depleción de potasio y magnesio.
Un nuevo tipo de fármacos sintéticos, los bifosfonatos, inhiben la resorción osteoclástica del hueso; algunos promueven también la formación ósea. El medicamento de elección para tratar la hipercalcemia es el pamidronato, y la dosis eficaz es de 45 a 60 mg en una sola infusión de 24 horas que se inicia en cuando se establece buena diuresis. Se logra la normalización del calcio sérico con esta dosis en el 70 a 100 porciento de los pacientes, y en los que tienen hipercalcemia severa pueden usarse 90 mg en 24 horas.52 Una vez que el calcio es normal, el tratamiento antitumoral puede ser eficaz para prevenir recurrencias de la hipercalcemia. Si no es posible administrar un tratamiento definitivo para el problema primario, puede tratarse con pamidronato IV en forma intermitente. El tratamiento con etidronato por vía oral, en dosis de 1,200 a 1,600 mg/día, suele prevenir las recurrencias de la hipercalcemia, pero no se ha autorizado su uso para este fin.
La calcitonina es otra alternativa para el tratamiento de la hipercalcemia. Actúa pronto, pero sus efectos son parciales y temporales, y su principal utilidad es ganar tiempo cuando no se dispone de otros agentes más potentes. La dosis es de 4 U MRC (Medical Research Council)/kg por vía subcutánea o intramuscular cada 12 horas. Con este tratamiento la concentración de calcio sérico disminuye 1 a 2 mg/dl en algunas horas, pero el efecto desaparece con igual rapidez y casi siempre se requieren otras medidas.
La prednisona, en dosis de 60 mg/día es un excelente suplemento en la intoxicación por vitamina D, mieloma, linfoma o cáncer de mama cuando la hipercalcemia ha sido precipitada por el tratamiento con esteroides sexuales.
La plicamicina es un potente agente antihipercalcémico que puede usarse para el tratamiento urgente de la hipercalcemia severa mientras se realizan los estudios dianósticos. Por lo general se administra una dosis inicial de 15 a 25 µg/kg IV durante 4 a 6 horas; es mejor esperar antes de repetir la dosis hasta que los niveles de calcio comiencen a aumentar de nuevo.
El fosfato inorgánico, 1.0 a 1.5 g/24 horas IV logrará el control temporal de la mayoría de los episodios de hipercalcemia; este tratamiento debe usarse solo si los agentes más potentes no están disponibles o fueron ineficaces. Su empleo está limitado a los pacientes con nivel sérico de fosfato bajo; si la concentración de fosfato se encuentra en límites superiores normales o elevados existe riesgo de calcificación extraesquelética.
La hipercalcemia producida por hiperparatiroidismo agudo suele responder a las medidas antes descritas. Sin embargo en algunos casos el tratamiento conservador no reduce el nivel de calcio en forma eficaz cuando la hipercalcemia es grave y en estos casos la exploración paratiroidea de urgencia puede salvar la vida y debe realizarse en forma pronta.
Hipocalcemia
La hipocalcemia (concentración de calcio en suero por debajo de 8.5 mg/dl) es más frecuente cuando existe deficiencia o resistencia a calcitriol, el hipoparatiroidismo (deficiencia de hormona paratiroidea activa) es una causa mucho menos común.
ETIOLOGIA Y FISIOPATOLOGIA
Hipoparatiroidismo
El hipoparatiroidismo puede deberse a falla en las glándulas para secretar PTH o falla de los tejidos para reaccionar a ella (seudohipoparatiroidismo). Dentro de la amplia gama de trastornos en la secreción y resistencia de los tejidos, hay muchas causas de hipoparatiroidismo [ver tabla 3].
|
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
La causa más común de hipoparatoridismo es la cirugía de tiroides. Con frecuencia no se identifican las cuatro glándulas durante la resección del tejido tiroideo, por lo que se considera que la deficiencia sea secundaria a infarto transoperatorio de las mismas.53
La deficiencia paratiroidea idiopática ocurre en varias formas. En una, la insuficiencia se acompaña de deficiencia variable de otras glándulas, sobre todo de suprarrenales y tiroides, y de candidiasis mucocutánea persistente; los anticuerpos contra tejido endócrino son la característica común de esta enfermedad, que tiende a presentarse en familias y que en muchos casos se ha relacionado con un defecto en el cromosoma 21q22.3.54 En una segunda forma, la deficiencia paratiroidea ocurre como parte del síndrome de Di George, que se caracteriza por linfopenia y aplasia de estructuras derivadas del tercer arco branquial, incluyendo el timo. El defecto se localiza en el cromosoma 22q11.56 Por último, el hipoparatiroidismo idiopático puede ocurrir como un defecto aislado, de manera esporádica o hereditaria. En algunos casos se han detectado anticuerpos contra el receptor sensible a calcio.56 Una forma autosómida dominante es causada por una mutación sin sentido en el gen que codifica para el receptor sensible a calcio. Al parecer confiere una mayor afinidad al calcio, causando inadecuada liberación de PTH e hipocalcemia subsecuente.57 Este aumento de función por mutación y su síndrome clínico son lo opuesto al defecto de pérdida de función que produce la hipercalcemia hipocalciúrica familiar. Un motivo importante para identificar este trastorno es que el tratamiento con vitamina D, que parece razonable, suele empeorar la hipercalciuria, el daño renal y causar nefrocalcinosis, y debe evitarse en pacientes sintomáticos.58,59
En la variante ligada al cromosoma X, el defecto se ha localizado en el brazo largo de este cromosoma.60 Es probable que el defecto se encuentre en la regulación de la secreción de PTH, más que en el gen en sí.
Otro mecanismo importante que produce hipoparatiroidismo es la resistencia a la acción de la PTH. Albright y colaboradores fueron los primeros en definir un síndrome en el que la hipocalcemia, la hiperfosfatemia y la hiperplasia tiroidea estaban relacionadas con talla corta, obesidad, cara redonda, huesos metacarpianos cortos y calcificación de los ganglios basales. Estos autores demostraron que la inyección de PTH no elevaba los niveles de calcio sérico ni los de fósforo en orina y acuñaron el término de seudohipoparatiroidismo para esta enfermedad. La PTH no eleva los niveles de AMPc nefrogénico, lo que indica que hay un defecto en el receptor hormonal en el riñón. En algunas familias el defecto se ha localizado en la unidad reguladora de nucleótidos (Gs) que acopla el receptor de membrana a la adenilciclasa. Este defecto también podría ser responsable de la resistencia ocasional a otras hormonas proteicas, como tirotropina, que se observa en estos pacientes.61 Sin embargo, existen algunos aspectos que son inexplicables, incluyendo una asociación inconstante entre la unidad reguladora defectuosa y la capacidad de la PTH para inducir el incremento en el AMPc. De hecho, los pacientes seudohipoparatiroideos pueden presentar alguna deficiencia en la actividad de GS, al igual que los pacientes que aparentan tenerla pero no son hipocalcémicos. Estos individuos normocalcémicos se clasificaban al principio como portadores de seudohipoparatiroidismo. En la actualidad ambos síndromes se conocen como osteodistrofia hereditaria de Albright.62 Estos síndromes de resistencia a la PTH se han subclasificado por sus mecanismos molculares, patrones de herencia, defectos genéticos y resistencia asociada a otras hormonas.63
Los trastornos adquiridos en la secreción de PTH, en los efectos de la PTH o en la activación de la vitamina D, son mucho más comunes que los síndromes descritos previamente.64 La hipomagnesemia altera la liberación de PTH y bloquea sus efectos hipercalcémicos, los niveles de calcio y fósforo séricos están bajos. Estos efectos dependen, al menos en parte, de la gravedad de la deficiencia de magnesio; la corrección de esta deficiencia restaura rápidamente la secreción de PTH, aunque la respuesta a la PTH se recupera en forma más lenta. Ya que los alcohólicos y otros individuos propensos a la desnutrición pueden tener deficiencia de magnesio, y como la tetania puede ocurrir por deficiencia de magnesio o calcio, para diagnosticar este tipo de hiperparatiroidismo se necesitan capacidad clínica y pruebas de laboratorio discriminantes. En forma alterna, la intoxicación alcohólica aguda puede acompañarse de disminución en la concentración sérica de calcio ionizado y de aumento en la excreción urinaria de calcio y magnesio. Estos cambios son consecuencia de una menor secreción de PTH, y suelen ser reversibles.65 Es curioso que la hipermagnesemia también puede suprimir la secreción de PTH y provocar hipocalcemia.66
La insuficiencia renal crónica es probablemente la causa más común de hipocalcemia, y ya se describieron los mecanismos que la producen [ver antes, Hiperparatiroidismo secundario]. Los individuos afectados generalmente tienen niveles elevados de fósforo, creatinina y nitrógeno de urea en sangre.
Deficiencia de vitamina D
Los precursores de vitamina D se absorben tanto de la piel como por el tubo digestivo. La deficiencia de vitamina D suele deberse a la combinación de exposición inadecuada a la luz solar y una dieta pobre en pescado y productos lácteos.67 Se conocen varios defectos del metabolismo de la vitamina D. En el raquitismo resistente a vitamina D de tipo I existe menor conversión de 25-(OH)D3 a calcitriol; en el raquitismo resistente a vitamina D de tipo II el defecto se encuentra en la unión del calcitriol a su receptor.
Ocurre malabsorción de calcio y vitamina D en las enfermedades del intestino delgado, y ocasionalmente en la deficiencia pancreática. La esteatorrea, no siempre obvia, hace necesaria la cuantificación de la grasa en las heces, y realizar estudios radiográficos del intestino delgado para confirmarla. En pacientes que se encuentran en los límites de deficiencia de vitamina D y con malabsorción de calcio, una diarrea aguda puede precipitar tetania.
El tratamiento con fenitoína y otros medicamentos anticonvulsivos ha sido relacionado como causa de deficiencia de vitamina D. Aunque la hipocalcemia franca no se observa frecuentemente, los enfermos que están tomando estos medicamentos pueden tener niveles séricos de calcio en límites inferiores normales, fósforo sérico bajo, elevación moderada de la fosfatasa alcalina y calcio urinario bajo, todos compatibles con osteomalacia. Las pocas biopsias de hueso disponibles muestran vetas osteoides sin calcificar, que es una característica distintiva de la osteomalacia. La patogenia de este defecto no se ha dilucidado por completo, pero la hipocalcemia puede agravar cualquier trastorno convulsivo y los niveles de calcio en límites inferiores normales, que en otras personas no tienen importancia, pueden ser peligrosos para enfermos con epilepsia.
Otras causas de hipocalcemia
Las causas diversas de hipocalcemia incluyen la pancreatitis aguda, en la que los mecanismos precisos no se han aclarado; la enfermedad metastásica osteoblástica, en la que se supone que hay desplazamiento de calcio y fósforo de líquido extracelular por un rápido depósito en las metástasis osteoblásticas; las transfusiones excesivas con sangre tratada con citrato, en donde la formación de complejos del calcio con el citrato infundido es el factor que produce el daño, y la insuficiencia renal inducida por la rabdomiolisis.68
ASPECTOS CLINICOS DE LA HIPOCALCEMIA
Los rasgos clínicos de la hipocalcemia dependen en parte de la causa de fondo, de la rapidez con que se desarrollan y de la presencia de enfermedades relacionadas. En casos de instalación aguda, como después de una tiroidectomía, la sintomatología común incluye parestesias peribucales, inquietud, depresión, y los signos de Chvostek o Trousseau. En los pacientes que desarrollan hipocalcemia en forma más gradual la fatiga, los calambres musculares y las mialgias son la sintomatología prominente. Sin embargo, estos síntomas son sutiles y es común confundirlos con rasgos inespecíficos de depresión o astenia. La prolongación del intervalo QT en el electrocardiograma o el descubrimiento casual de calcio sérico bajo pueden aclarar el diagnóstico.
El enfermo con hipocalcemia deberá ser evaluado bajo las bases fisiológicas básicas que han sido discutidas y que se resumen en la tabla 3. Como ya se mencionó, la concentración de calcio sérico se eleva por absorción gastrointestinal, resorción ósea, o reabsorción renal, este último proceso es el menos importante. Si los niveles de calcio sérico se encuentran disminuidos, el defecto radica en la mayoría de los casos en el intestino o en el hueso y las causas principales son la deficiecia de vitamina D y la ineficacia de la PTH. A menudo el trastorno de fondo es aparente: tiroidectomía previa, insuficiencia renal crónica o pancreatitis aguda grave. Cuando el mecanismo productor no es obvio, está indicado determinar los valores séricos de vitamina D y de PTHi; si la PTHi se encuentra elevada en presencia de hipocalcemia, es probable que la hormona sea ineficaz por carencia de vitamina D o a la resistencia de los órganos blanco a una o ambas hormonas. La concentración de 25-(OH)D3 refleja la ingesta y reservas de vitmina D.69 La depleción de vitamina D puede compensarse por un hiperparatiroidismo secundario, que puede aumentar la síntesis de calcitriol y por lo tanto, mantener los niveles de calcio sérico normales. Cuando la compensación es inadecuada, el calcio y fósforo sérico disminuyen y se desarrolla osteomalacia [ver tabla 3].
TRATAMIENTO DE LA HIPOCALCEMIA
La PTH no es práctica para uso clínico, pero las acciones muy similares de la vitamina D ofrecen una alternativa satisfactoria. La hipocalcemia generalmente es tratada con una combinación de calcio y vitamina D.
Existe gran cantidad de literatura sobre el uso de análogos sintéticos de la vitamina D.6 Para los pacientes con enfermedad hepática o malabsorción, el calcifediol puede usarse esperando que el riñón añada el grupo 1-alfa-H. Pueden emplearse dosis entre 50 y 300 µg/día. La 1alfa-(OH)D3 puede usarse en el hipoparatiroidismo y también en pacientes con enfermedad renal porque la 25-hidrolasa hepática usará la 1-(OH)D3 como sustrato, la dosis es de 2 a 4 µg/día. También se dispone de calcitriol, y se han empleado dosis de 0.25 a 1.0 µg/día.70 Las nuevas preparaciones parecen ser eficaces para aumentar la concentración de calcio sérico en pacientes hipoparatiroideos, pero son más costosas que la vitamina D estándar y ofrecen pocas ventajas para la mayoría de los pacientes. La concentración sérica de calcio debe vigilarse para usar todas las formas de vitamina D en forma segura. Los análogos 1-OH de la vitamina D son los que más producen hipercalcemia por la pérdida de la regulación habitual de la 1alfa-OH hidroxilasa. Por este motivo se prefiere el calcitriol. La dosis requerida disminuye al normalizarse el calcio sérico y sanar las lesiones óseas. El calcitriol, usado en cantidades fisiológicas, y la 1-(OH)D3 tienen un inicio y cese de acción rápidos, en caso de que la toxicidad obligara a suspenderlos. Es demasiado pronto para afirmar si las ventajas reales y posibles de los análogos sintéticos de la vitamina D justifican su mayor costo, pero su introducción marca una aplicación clínica excitante de adelantos científicos recientes.
Hipocalcemia después de tiroidectomía
Los síntomas agudos de la hipocalcemia después de una tiroidectomía pueden tratarse con gluconato de calcio; una dosis de 10 ml de solución al 10 porciento, administrados lentamente por vía intravenosa, producen alivio casi inmediato. En casos leves se puede utilizar 1 g de cloruro de calcio tres veces al día durante 1 a 3 semanas, que después se reducen en forma gradual. Si se necesitan más de 4 días de tratamiento, por lo general se requiere la restitución permanente y bastarán 25,000 a 50,000 unidades de vitamina D diarios. Los niveles de calcio sérico deberán determinarse cada 2 semanas a partir del inicio del tratamiento y cada 6 meses después de habrer alcanzado la estabilización de los valores, ya que la hipercalcemia episódica es común en estos pacientes. Deben mantenerse las cifras de calcio en 8.5 mg/dl, porque la excreción urinaria de calcio en hipoparatiroideos tratados con vitamina D es muy alta. El calcio urinario debe determinarse de manera periódica; si se encuentra por arriba de 250 mg/día, el individuo deberá mantener una ingestión de líquidos elevada para evitar la formación de cálculos renales. La clortalidona y la restricción moderada de sodio pueden ser útiles como tratamiento complementario en pacientes que desarrollan hipercalciuria problemática o litiasis renal mientras se encuentran en tratamiento con vitamina D.
Hipocalcemia después de la paratiroidectomía
En enfermos hiperapartiroideos que no padecen osteítis severa la hipocalcemia después de la paratiroidectomía generalmente refleja la supresión transitoria de las otras glándulas; la restitución temporal de calcio ayudará al sujeto hasta que la secreción de PTH se restablezca en las otras glándulas. Esta sustitución temporal mantendrá el bienestar del paciente hasta que se reanude la secreción de PTH. En individuos con osteítis severa la evolución posoperatoria suele caracterizarse por osteomalacia de varios meses de evolución con niveles séricos bajos de calcio y fósforo y fosfatasa alcalina elevada. El tratamiento con vitamina D acelerará la curación pero puede suspenderse después de 3 a 6 meses. Los casos que desarrollan tetania después de la resección de siete octavos de las paratiroides generalmente requerirán de tratamiento permanente con vitamina D.
Calcitonina y sus trastornos
La calcitonina es codificada por un gen que se localiza en el cromosoma 11. La secreción de calcitonina aumenta por el aumento súbito del calcio sérico, por dosis farmacológicas de algunos péptidos gastrointestinales y por los agonistas beta adrenérgicos. Su secreción es inhibida por la hipocalcemia aguda y por los antagonistas beta. El papel fisiológico de estas influencias no es aún muy claro.
Ha sido difícil determinar cuál es el papel fisiológico de esta hormona.8 En cantidades farmacológicas suprime la resorción ósea, actuando en forma directa sobre los osteoclastos. Por tanto, modula la concentración sérica de calcio durante el recambio rápido de hueso, disminuyendo la hipercalcemia e, incluso en ocasiones, reduciendo concentraciones normales de calcio sérico. Sin embargo, la ausencia de calcitonina (v.gr., después de la tiroidectomía total) no se asocia con hipercalcemia, y cantidades excesivas de la hormona (v.gr., en el carcinoma medular del tiroides) no causan hipocalcemia.
El gen que codifica la síntesis de calcitonina puede, por trascripción alterna del ARNm, producir también otros ocho péptidos. La mayor parte de estos péptidos parecen tener función de neurotransmisores o parácrina, pero no hormonal. Uno de estos, conocidos como péptido relacionado con el gen de la calcitonia, es un neuropéptido y el más potente vasodilatador natural conocido.71,72
En ciertas enfermedades se presentan cantidades enormes de calcitonina. En el carcinoma medular del tiroides la hipercalcitonemia es un marcador tumoral constante. Se ha notificado producción ectópica de calcitonina en muchos tumores, incluyendo de páncreas, pulmón, próstata, útero, vejiga, mama y melanoma.
La calcitonina es de gran utilidad terapéutica en la enfermedad de Paget del hueso. En este trastorno, cuya lesión básica es la osteolisis franca, la calcitonina alivia el dolor óseo, reduce el calcio urinario y antagoniza la hipercalcemia producida por la inmovilización; reduce también los niveles urinarios de hidroxiprolina y séricos de fosfatasa alcalina, que son indicadores generales de la gravedad de esta enfermedad. Se afirma que en algunos pacientes ha ocurrido mejoría de las lesiones óseas, aunque este beneficio es dudoso en la mayoría de las series publicadas. El tratamiento prolongado con calcitonina parece ser seguro y la hormona sigue siendo el agente más potente contra la enfermedad.
Figuras 3 a 5 Talar Agasyan
Bibliografía
- Pocotte SL, Ehrenstein G, Fitzpatrick LA: Regulation of parathyroid hormone secretion. Endocr Rev 12:291, 1991 [PMID 1935823]
- Brown EM: Extracellular Ca2+ sensing, regulation of parathyroid cell function, and role of Ca2+ and other ions as extracellular (first) messengers. Physiol Rev 71:371, 1991
- Brown EM, Gamba G, Riccardi D, et al: Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 366:575, 1993 [PMID 8255296]
- Conklin BR, Bourne HR: Marriage of the flytrap and the serpent. Nature 367: 22, 1994 [PMID 8107768]
- Chattopadhyay N, Mithal A, Brown EM: The calcium-sensing receptor: a window into the physiology and pathophysiology of mineral ion metabolism. Endocr Rev 17:289, 1996 [PMID 8854047]
- Walters MR: Newly identified actions of the vitamin D endocrine system. Endocr Rev 13:719, 1992
- McDermott MT: Calcitonin and its clinical applications. The Endocrinologist 2:366, 1992
- Horne WC, Shyu J-F, Chakraborty M, et al: Signal transduction by calcitonin: multiple ligands, receptors, and signaling pathways. TEM 5:395, 1994
- de Papp AE, Stewart AF: Parathyroid hormone-related protein: a peptide of diverse physiologic functions. TEM 4:181, 1993
- Dunbar ME, Wysolmerski JJ, Broadus AE: Parathyroid hormone-related protein: from hypercalcemia of malignancy to developmental regulatory molecule. Am J Med Sci 312: 287, 1996 [PMID 8969618]
- Mallette LE: The parathyroid polyhormones: new concepts in the spectrum of peptide hormone action. Endocr Rev 12:110, 1991
- . Ebeling PR, Sandgren ME, DiMagno EP, et al: Evidence of an age-related decrease in intestinal responsiveness to vitamin D: relationship between serum 1, 25-dihydroxyvitamin D3 and intestinal vitamin D receptor concentrations in normal women. J Clin Endocrinol Metab 75:176, 1992 [PMID 1320048]
- Khaw K-T, Sneyd M-J, Compston J: Bone density parathyroid hormone and 25-hydroxyvitamin D concentrations in middle aged women. BMJ 305:273, 1992
- McKane WR, Khosla S, Egan KS, et al: Role of calcium intake in modulating age-related increases in parathyroid function and bone resorption. J Clin Endocrinol Metab 81:1699, 1996
- Heaney RP: Calcium, parathyroid function, bone, and aging (editorial). J Clin Endocrinol Metab 81:1697, 1996
- Potts JT: Hyperparathyroidism and other hypercalcemic disorders. Adv Intern Med 41:165, 1996
- Arnold A: Genetic basis of endocrine disease 5: molecular genetics of parathyroid gland neoplasia. J Clin Endocrinol Metab 77:1108, 1993
- Arnold A, Staunton CE, Kim HG, et al: Monoclonality and abnormal parathyroid hormone genes in parathyroid adenomas. N Engl J Med 318:658, 1988 [PMID 3344017]
- Cryns VL, Thor A, Xu H-J, et al: Loss of the retinoblastoma tumor-suppressor gene in parathyroid carcinoma. N Engl J Med 330:757, 1994 [PMID 7906387]
- Fischer JA: "Asymptomatic" and symptomatic primary hyperparathyroidism. Clin Investig 71:505, 1993
- Tonner DR, Schlechte JA: Neurologic complications of thyroid and parathyroid disease. Med Clin North Am 77:251, 1993 [PMID 8419721]
- Silverberg SJ, Bilezikian JP: Primary hyperparathyroidism: still evolving? J Bone Miner Res 12:856, 1997
- Kleerekoper M, Bilezikian JP: A cure in search of a disease: parathyroidectomy for nontraditional features of primary hyperparathyroidism. Am J Med 96:99, 1994
- Wermers RA, Khosla S, Atkinson EJ, et al: The rise and fall of primary hyperparathyroidism: a population-based study in Rochester, Minnesota, 1965-1992. Ann Intern Med 126:433, 1997
- Lairmore TC, Wells SA Jr: Parathyroid surgery and autotransplantation. The Endocrinologist 4:99, 1994
- Pearl AJ, Chapnik JS, Freeman JL, et al: Pre-operative localization of 25 consecutive parathyroid adenomas: a prospective imaging/surgical correlative study. J Otolaryngol 22:301, 1993
- Stevens SK, Chang J-M, Clark OH, et al: Detection of abnormal parathyroid glands in postoperative patients with recurrent hyperparathyroidism: sensitivity of MR imaging. AJR Am J Roentgenol 160:607, 1993 [PMID 8430565]
- Yousem DM: Parathyroid and thyroid imaging. Neuroimaging Clin North Am 6:435, 1996
- Diagnosis and management of asymptomatic primary hyperparathyroidism: consensus development conference statement. NIH Consensus Development Conference Panel. Ann Intern Med 114:593, 1991
- Brothers TE, Thompson NW: Surgical treatment of primary hyperparathyroidism in elderly patients. Acta Chir Scand 153:175, 1987 [PMID 3604520]
- Avioli LV: Hyperparathyroidism, estrogens, and osteoporosis. Hosp Pract (Off Ed) 26(1):115, 1991
- Shane E, Bilezikian JP: Parathyroid carcinoma: a review of 62 patients. Endocr Rev 3:218, 1982
- Wynne AG, van Heerden J, Carney JA, et al: Parathyroid carcinoma: clinical and pathologic features in 43 patients. Medicine (Baltimore) 71:197, 1992 [PMID 1518393]
- Obara T, Okamoto T, Kanbe M, et al: Functioning parathyroid carcinoma: clinicopathologic features and rational treatment. Semin Surg Oncol 13:134, 1997 [PMID 9088069]
- Vetto JT, Brennan MF, Woodruf J, et al: Parathyroid carcinoma: diagnosis and clinical history. Surgery 114:882, 1993 [PMID 8236009]
- Slatopolsky E, Delmez JA: Pathogenesis of secondary hyperparathyroidism. Nephrol Dial Transplant 11(suppl 3):130, 1996
- Malluche H, Faugere M-C: Renal bone disease 1990: an unmet challenge for the nephrologist. Kidney Int 38:193, 1990 [PMID 2205749]
- Arnold A, Brown MF, Urena P, et al: Monoclonality of parathyroid tumors in chronic renal failure and in primary parathyroid hyperplasia. J Clin Invest 95:2047, 1995 [PMID 7738171]
- Chan FKW, Koberle LMC, Thys-Jacobs S, et al: Differential diagnosis, causes, and management of hypercalcemia. Curr Prob Surg 34:445, 1997
- Nussbaum SR: Pathophysiology and management of severe hypercalcemia. Endocrinol Metab Clin North Am 22:343, 1993
- Seymour JF, Gagel RF: Calcitriol: the major humoral mediator of hypercalcemia in Hodgkin's disease and non-Hodgkin's lymphomas. Blood 82:1383, 1993 [PMID 8364192]
- Cox M, Haddad JG: Lymphoma, hypercalcemia, and the sunshine vitamin (editorial). Ann Intern Med 21:709, 1994
- Jacobus CH, Holick MF, Shao Q, et al: Hypervitaminosis D associated with drinking milk. N Engl J Med 326:1173, 1992 [PMID 1313547]
- Haddad JG: Vitamin D-solar rays, the Milky Way, or both (editorial)? N Engl J Med 326:1213, 1992
-
. Wysolmerski JJ, Broadus AE: Hypercalcemia of malignancy:
the central role of parathyroid hormone-related protein. Annu Rev Med 45:189,
1994 [PMID
8198376]
-
Mundy GR, Guise TA: Hypercalcemia of malignancy. Am
J Med 103:134, 1997 [PMID
9274897]
-
Goltzman D, Henderson JE: Parathyroid hormone-related
peptide and hypercalcemia of malignancy. Endocrine Neoplasms. Arnold A,
Ed. Kluwer Academic Publishers, Boston 1997, p 192
-
Martin TJ, Grill V: Hypercalcemia in cancer. J Steroid
Biochem Mol Biol 43: 123, 1992 [PMID
1525053]
-
Heath DA: Parathyroid hormone related protein. Clin
Endocrinol (Oxf) 38:135, 1993
-
Pearce SHS, Brown EM: Calcium-sensing receptor mutations:
insights into a structurally and functionally novel receptor (editorial).
J Clin Endocrinol Metab 81: 1309, 1996
-
Health H III, Odelberg S, Jackson CE, et al: Clustered
inactivating mutations and benign polymorphisms of the calcium receptor
gene in familial benign hypocalciuric hypercalcemia suggest receptor functional
domains. J Clin Endocrinol Metab 81:1312, 1996
-
Aaltonen J, Bjšrses, Sandkuijl L, et al: An autosomal
locus causing autoimmune disease: autoimmune polyglandular disease type
1 assigned to chromosome 21. Nat Genet 8: 83, 1994 [PMID
7987397]
-
Carey AH, Kelly D, Halford S, et al. Molecular genetic study
of the frequency of monosomy 22q11 in DiGeorge syndrome. Am J Med Genet
51:964, 1992
-
Li Y, Song Y-H, Rais N, et al: Autoantibodies to the
extracellular domain of the calcium sensing receptor in patients with acquired
hypoparathyroidism. J Clin Invest 97: 910, 1996 [PMID
8613543]
-
Pollak MR, Brown EM, Estep HL, et al: Autosomal dominant
hypocalcaemia caused by a Ca2+-sensing receptor gene mutation.
Nat Genet 8:303, 1994 [PMID
7874174]
-
Heath D: Familial hypocalcemia - not hypoparathyroidism
(editorial): N Engl J Med 335:1145, 1996
-
Pearce SHS, Williamson C, Kifor O, et al: A familial
syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing
receptor. N Engl J Med 335:1115, 1996
-
Thakker RV, Davies KE, Whyte MP, et al: Mapping the
gene causing X-linked recessive idiopathic hypoparathyroidism to Xq26-Xq27
by linkage studies. J Clin Invest 86:40, 1990
-
Faull CM, Welbury RR, Paul B, et al: Pseudohypoparathyroidism:
its phenotypic variability and associated disorders in a large family.
Q J Med 287:251, 1991
-
Schwindinger WF, Levine MA: Albright hereditary osteodystrophy.
The Endocrinologist 4:17, 1994
-
Mallette LE: Pseudohypoparathyroidism. Current Therapy
in Endocrinology and Metabolism, 6th ed. Bardin CW, Ed. CV Mosby Co, St.
Louis, 1997, p 577
-
Zaloga GP: Hypocalcemia in critically ill patients. Crit
Care Med 20:251, 1992
-
Laitinen K, Lamberg-Allardt C, Tunninen R, et al: Transient
hypoparathyroidism during acute alcohol intoxication. N Engl J Med 324:721,
1991 [PMID
1997837]
-
Cholst IN, Steinberg SF, Tropper PJ, et al: The influence
of hypermagnesemia on serum calcium and parathyroid hormone levels in human
subjects. N Engl J Med 310:1221, 1984
-
Anderson JJB, Toverud SU: Diet and vitamin D: a review
with an emphasis on human function. J Nutr Biochem 5:58, 1994
-
Llach F, Felsenfeld AJ, Haussler MR: The pathophysiology
of altered calcium metabolism in rhabdomyolysis-induced acute renal failure:
interactions of parathyroid hormone, 25-hydroxycholecalciferol, and 1,25-dihydroxycholecalciferol.
N Engl J Med 305: 117, 1981 [PMID
6894630]
-
Katz BS, Jackson GJ, Hollis BW, et al: Diagnostic criteria
of vitamin D deficiency. The Endocrinologist 3:248, 1993
-
Markowitz ME, Rosen JF, Smith C, et al: 1,25-Dihydroxyvitamin
D3-treated hypoparathyroidism: 35 patient years in 10 children.
J Clin Endocrinol Metab 55:727, 1982
-
Calcitonin gene related peptide (editorial). BMJ 302:739,
1991
-
Shekhar YC, Anand IS, Sarma R, et al: Effects of prolonged
infusion of human alpha calcitonin gene-related peptide on hemodynamics,
renal blood flow and hormone levels in congestive heart failure. Am J Cardiol
67:732, 1991 [PMID
2006623]
-
Kao PC, van Heerden, JA, Grant CS, et al: Clinical
performance of parathyroid hormone immunometric assays. Mayo Clin Proc
67:637, 1992 [PMID
1434896]
-
Lockwood K, Bruun E, Transbl I: Disease of
the parathyroid glands. Adv Surg 9:177, 1975 [PMID
1103593]