Contenido del artículo
V ESCLERODERMIA Y ENFERMEDADES RELACIONADAS
DR. GEROGE MOXLEY
La esclerodermia, o esclerosis sistémica, es una enfermedad reumatológica rara, lentamente progresiva, caracterizada por depósito de tejido conectivo fibroso en la piel y otros tejidos. Suele acompañarse de lesiones vasculares, en especial en la piel, pulmonares y riñones. No se conoce una curación.
La esclerodermia puede ser sistémica o localizada. La enfermedad sistémica puede ser difusa o limitada. La forma limitada de esclerodermia sistémica, o CREST (calcinosis, fenómeno de Raynaud, afección esofágica, esclerodactilia y telangiectasias) afecta los órganos internos con menos frecuencia que la esclerodermia difusa. La esclerodermia sistémica puede ser fatal, excepto cuando existe hipertensión pulmonar, la forma limitada tiene mejor pronóstico que la forma difusa [ver tabla 1].
|
||||||
CREST- calcinosis, Raynaud, disfunción en la motilidad esofágica, esclerodactilia y telangiectasias, AR- artritis reumatoide, LEG- lupus eritematoso generalizado |
La esclerodermia localizada se limita a la piel, tejido subcutáneo y
músculo, y no se asocia con fenómeno de Raynaud, acroesclerosis o
afección visceral. Existen dos formas de esclerodermia localizada:
morfea, que consiste en placas de tamaño variable de induración
en la piel y esclerodermia lineal, que se presenta como bandas de
induración de la piel en la cara o una sola extremidad. La esclerodermia
lineal puede asociarse con atrofia muscular y afección del hueso
subyacente. Suele afectar niños o adultos jóvenes y puede causar
un deterioro significativo en el crecimiento de la parte afectada. En la morfea
las lesiones persisten por meses o años, después de lo cual puede
ocurrir mejoría [ver tabla 1].
Aunque existe posibilidad de desfiguración, la esclerodermia localizada no es una enfermedad severa y suele asociarse con una longevidad normal.
El fenómeno de Raynaud se asocia con esclerodermia [ver adelante, Diagnóstico, Manifestaciones clínicas]. El fenómeno de Raynaud primario (fenómeno de Raynaud sin enfermedad subyacente) es muy común y hasta el 30 por ciento de las mujeres jóvenes tienen estos episodios. 1 Un meta-análisis mostró que la tasa de transición a una enfermedad reumática inflamatoria definida (v.gr., esclerodermia o lupus) es de 3.2 por 100 pacientes-año de observación. El desarrollo eventual de una enfermedad reumatológica inflamatoria ocurrió en el 12.6 por ciento de los individuos. El mejor factor predictor para el desarrollo de enfermedad inflamatoria es un patrón anormal en el lecho capilar ungueal, que tiene un valor predictivo del 47 por ciento. Los anticuerpos antinucleares positivos tienen un valor predictivo del 30 porciento. 2
Los factores raciales, genéticos y ambientales pueden influir en el riesgo de esclerodermia y el patrón de enfermedad. 3 La incidencia global de esclerodermia es de alrededor de 17 por un millón de población por año, con tasas mayores para las mujeres que para los varones. Los afroamericanos típicamente sufren esclerodermia difusa con más frecuencia que otros grupos étnicos y en los blancos se diagnostica más variante de CREST. 4 La prevalencia en los Estados Unidos es de alrededor de 24 por 100,000 habitantes, 5 esta es cuatro a nueve veces la prevalencia en otros países. Los factores de mortalidad incluyen enfermedad difusa, edad avanzada al inicio y afección de órganos internos, en especial pulmón y riñón. Algunos investigadores han sugerido que diversos tipos de exposición ambiental pueden relacionarse con esclerodermia. 6 Estos incluyen polvo de sílice, solventes orgánicos, medicamentos y otras sustancias. La exposición a polvo de sílice tiene una explicación biológica lógica a través de toxicidad a macrófagos. Aunque la exposición al polvo de sílice aumente el riesgo relativo 25 a 74 veces, incluso en estas circunstancias la esclerodermia es una enfermedad poco frecuente. El cloruro de vinilo, el tricloroetileno y otros solventes orgánicos pueden modificar las proteínas nativas de modo que estimulen la autoinmunidad y la activación de las células inmunes. No se ha encontrado que los implantes de mama de silicón y la mamoplastía de aumento tengan relación con la esclerodermia. 7
La etiología de la esclerodermia se desconoce en gran parte, aunque un factor etiológico podría ser un defecto de fibrilina. La fibrilina es una macromolécula que es un componente de las fibras elásticas y se encuentra defectuosa en el síndrome de Marfán. En modelo animal autosómico dominante (el modelo de ratón tsk1 de piel tensa), la esclerodermia es causada por una inserción en el gen de fibrilina que al parecer codifica una región de unión latente para la citocina factor de transformación de crecimiento beta (FTC-b). Una explicación propuesta para la enfermedad murina del tsk1 es que la fibrilina anormal fija un mayor número de factores de crecimiento de fibroblastos, que después se liberan en forma lenta. El conocimiento actual se extiende a pacientes Choctaw de Oklahoma con esclerodermia, que parecen tener un defecto genético en esta región cromosómica dentro o cerca del gen humano de la fibrilina y que quizá heredaron de un ancestro común. Sin embargo, sin duda deben participar otros genes o exposiciones ambientales también.
Las características patológicas comunes de los tejidos con esclerodermia son fibrosis progresiva, alteraciones vasculares e inflamación. La fibrosis incluye acúmulo del exceso de colágena y otros constituyentes de la matriz extracelular, como glucosaminoglucanos y fibronectina. Las alteraciones vasculares son hiperplasia de la íntima con depósito de colágena y fibrosis de la adventicia, desaparición, dilatación y tortuosidad de los capilares y ateroesclerosis fibrosa. Los cambios inflamatorios pueden incluir infiltración celular. Se piensa que estas características patológicas son resultado de tres o más componentes que interactúan: autoinmunidad, una alteración endotelial y lesión de los fibroblastos cutáneos. Una explicación alternativa es que estas características son el resultado de un proceso semejante a la enfermedad injerto contra huésped (EICH).
Células inmunes y alteraciones de citocinas
Entre las células que infiltran los tejidos afectados predominan los linfocitos activados derivados del timo, también existen células inflamatorias activadas. Estas células inmunológicas e inflamatorias liberan gran cantidad de citocinas y mediadores solubles. Es importante destacar las citocinas que influyen en la función de los fibroblastos: interleucina-1 (IL-1), IL-4, IL-6, IL-8, factor de necrosis tumoral-a (FNT-a) y FTC-b. La acción del FCT-b causa un patrón de daño tisular semejante al patrón observado en la esclerodermia: el FTC-b estimula los fibroblastos y células musculares lisas de los vasos para que fabriquen colágena, y estimula las células endoteliales para formar endotelina-1 que, a su vez, causa vasoconstricción y producción de colágena. Las células B también se activan en los pacientes con esclerodermia, pero no parecen tener un papel primordial. Aunque existen autoanticuerpos relativamente específicos para la escleroermia, no parecen ocasionar daño tisular.
Como con los linfocitos y los macrófagos que muestran datos de activación en la esclerodermia, los fibroblastos también están metabólicamente activos. 8 Estas células activadas producen colágena y otras moléculas de la matriz extracelular en exceso. Los motivos por los que la expresión del gen de la colágena está aumentada y es sostenida no se saben del todo. Una explicación posible es que la presencia de citocinas como la IL-4 y el FTC-b exacerban la producción de colágena. Aunque los macrófagos activados pueden producir FTC-b al inicio, este factor puede desencadenar su propia producción a partir de fibroblastos blanco. Por lo tanto, la fibrosis progresiva y constante que se observa en la esclerdermia puede deberse a un proceso en el que los macrófagos originan FTC-b, que a su vez activa a los fibroblastos para secretar colágena y otras moléculas de la matriz mientras induce su propia producción a partir de fibroblastos.
Los vasos en los tejidos afectados muestran anatomía y función alterados, con trastorno en la permeabilidad endotelial, adhesión de plaquetas y leucocitos al endotelio y presencia de células inflamatorias. 9 Las células endoteliales expresan un mayor número de moléculas de adhesión, necesarias para la adhesión y extravasación de las células inflamatorias. Los capilares se obliteran, pero no son sustituidos, los tejidos afectados se vuelven isquémicos y después sufren reperfusión. El fenómeno de Raynaud [ver adelante, Diagnóstico, Manifestaciones clínicas] es la lesión vascular característica, y representa una respuesta exagerada de la pared rígida de un vaso a la exposición ambiental típica. El análisis microscópico de los capilares ungueales de los pacientes con esclerodermia y fenómeno de Raynaud revela desaparición y dilatación de los capilares. El flujo vascular normal incluye interacciones entre señales neurales, hormonas circulantes y mediadores liberados por el vaso en sí y por células circulantes. Las señales neurales son principalmente simpáticas y están mediadas por norepinefrina que se une a los receptores vasoconstrictores alfa adrenérgicos o a receptores vasodilatadores beta adrenérgicos. Los receptores alfa adrenérgicos se han subdividido en de tipo 1 y de tipo 2. El vasoespasmo del Raynaud inducido por el frío es bloqueado principalmente por antagonistas selectivos de los receptores a2-adrenérgicos. En la esclerodermia las arterias pequeñas desarrollan fibrosis concéntrica de la íntima y por lo tanto se angostan, esto aumenta en mucho la reactividad vascular a los agentes adrenérgicos a2.
El quimerismo denota un estado en el que una persona tienen células derivadas de dos o más personas. Después de someterse a un transplante alogénico de médula, un paciente tiene sus propias células, así como las de la persona de la que se tomó el trasplante. Muchos pacientes sometidos a trasplantes sufren EICH, un padecimiento semejante a la esclerodermia caracterizado por induración progresiva de la piel con ulceración, posible afección del tubo digestivo, pulmones, músculo esquelético y glándulas salivales y lacrimales. Ocurre EICH cuando las células injertadas reaccionan contra las células nativas del paciente.
El microquimerismo, un nivel bajo de quimerismo, puede ocurrir asociado al embarazo. Las mujeres que dan a luz pueden tener células fetales circulantes por muchos años. Por lo tanto, se ha sugerido que la esclerodermia representa un tipo de EICH en el que las células extrañas que permanecen (fetales o transfundidas) atacan a los constituyentes nativos del organismo. 10,11 Las observaciones que apoyan esta hipótesis incluyen que se encuentra ADN del varón con más frecuencia y en mayor concentración en mujeres con esclerodermia que han tenido un hijo varón que en mujeres sin esclerodermia que también han tenido un hijo varón. Además, se ha encontrado microquimerismo en la piel de madres con esclerodermia y no en la piel de madres sanas. Sin embargo, esta explicación patogénica no explica del todo por qué los pacientes con esclerodermia tienen fenómeno de Raynaud y afección renal y los que sufren EICH crónica no.
Piel Los primeros signos de esclerodermia en la piel son inflamación e induración de los dedos y manos, con o sin afección de la cara, en fases más avanzadas de la enfermedad pueden afectarse otras áreas del cuerpo. La afección del tronco y de los brazos en sentido proximal a los codos se asocia con afección visceral y peor pronóstico. La piel continúa engrosándose durante los primeros 2 a 3 años después del inicio de la enfermedad, después la induración cesa y puede incluso disminuir, dando la impresión de que la piel se está suavizando. En los años subsecuentes ocurre atrofia de la piel, con pérdida concomitante del pelo, glándulas sebáceas y glándulas sudoríparas, así como pérdida de los pliegues. Además la piel queda tensa y fija a las estructuras adyacentes. La afección de la piel suele ser más prominente en las manos y dedos (esclerodactilia), con frecuencia se afecta también la cara. El engrosamiento de la piel de la cara disminuye las líneas de la piel, dando una apariencia lustrosa y disminución en la apertura oral. La fibrosis de la piel puede limitar la movilidad, en especial en los dedos pudiendo desarrollarse contracturas en flexión [ver figura 1]. Otras alteraciones de la piel pueden acompañar estos cambios, con frecuencia ocurren telangiectasias, que pueden ser numerosas [ver figura 2]. Suelen ser más prominentes en la cara, manos y mucosa oral. La calcinosis (el depósito de cristales de hidroxiapatita en las áreas subcutáneas) puede ser limitada o diseminada y se localiza casi siempre alrededor de las cápsulas articulares [ver figura 3]. Las ulceraciones de la piel sobre los depósitos de calcio pueden provocar drenaje de material blanco con una consistencia que recuerda pasta de dientes. El aumento difuso en la pigmentación melanótica oscurece toda la piel, aunque con frecuencia se observan áreas de hipopigmentación.
|
|
|
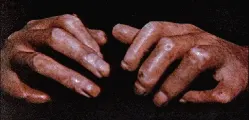

El fenómeno de Raynaud es una manifestación episódica de adormecimiento o dolor aunado a cambio de color en dos o tres fases en los dedos. Estos cambios suelen ser desencadenados por frío o estrés emocional y desaparecen calentando la extremidad. Sin embargo, en los casos severos la relación ambiental puede ser menos obvia. El episodio típicamente comienza con palidez, seguido por cianosis y al final eritema causado por hiperemia reactiva [ver figura 4]. La isquemia prolongada puede causar dolor en los dedos, ulceraciones e incluso gangrena. Casi todos los pacientes (95 por ciento) con esclerodermia difusa o limitada presentan fenómeno de Raynaud. Este se observa en pacientes con otros padecimientos, incluyendo lupus eritematoso generalizado (LEG), polimiositis y varias formas de vasculitis, en pacientes que reciben ciertos medicamentos como bleomicina, derivados del ergot, betabloqueadores y metisergida, y después de la exposición laboral a cloruro de vinilo, temperaturas frías y herramientas que vibran.

|
| Figura 4 |
| Fenómeno de Raynaud |
En la esclerodermia el fenómeno de Raynaud es una vasculopatía que afecta arterias pequeñas y capilares, ocurre no sólo en las extremidades, sino también en algunas vísceras afectadas, como los pulmones y el riñón. Los pacientes con esclerodermia y fenómeno de Raynaud tienen cambios capilares característicos en la microscopía del lecho ungueal. Este procedimiento consiste en la observación de la estructura capilar de los tejidos periungueales con una lupa de magnificación, como la usada en el oftalmoscopio estándar. Los pacientes con esclerodermia presentan pérdida de algunos capilares y dilatación en otras áreas. Estos cambios suelen ocurrir asociados a afección de órganos internos y pueden usarse para predecir el desarrollo de esclerodermia difusa cuando no existe afección visceral clínicamente aparente.
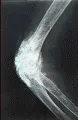
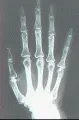
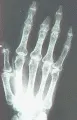
Sistema musculoesquelético Puede ocurrir artritis leve, por lo general simétrica e inflamatoria en la esclerodermia. Son frecuentes las erosiones yuxtarticulares, en especial en las articulaciones interfalángicas distales, pero el grado de destrucción suele ser menor al observado en la artritis reumatoide. Con frecuencia se desarrollan contracturas en flexión de los dedos, que principalmente se explican por fibrosis de los tendones y las cápsulas articulares. La crepitación y los frotes de fricción detectados por palpación o auscultación sobre tendones y bursas son característicos y se relacionan a los cambios fibróticos de los tejidos subyacentes. Otra complicación esquelética es la osteolisis acral, que consiste en la resorción de las falanges terminales y el tejido blando circundante con acortamiento subsecuente de los dedos [ver figura 5]. Puede ocurrir con infección o ulceración.
|
|
Los pacientes con esclerodermia pueden manifestar uno o más de diversos trastornos musculares. 12 Algunos pacientes con esclerodermia sufren fatiga sin evidencia objetiva de daño muscular (i.e., tienen creatina cinasa [CK] normal, así como datos normales en los estudios de electromiografía [EMG] o resonancia magnética). Otros pacientes tienen una miopatía simple con elevación mínima de CK, datos miopáticos en la EMG y engrosamiento o fibrosis de las fascias evidenciada por la IRM. Aún otros presentan una miositis inflamatoria con debilidad sustancial, elevación significativa de la CK, cambios miopáticos en la EMG, infiltrados mononucleares evidenciados en la biopsia muscular y brillantez del músculo en la imagen T2 de la IRM.
Tubo digestivo En la esclerodermia puede afectarse casi cualquier parte del tubo digestivo. 13 Ocurre síndrome de Sjögren, que causa xeroftalmia y xerostomía, en alrededor de la tercera parte de los pacientes con esclerodermia. En más del 90 por ciento de los pacientes, muchos asintomáticos, se observa hipomotilidad esofágica, que se demuestra por examen cinefluoroscópico y estudios de manometría [ver figura 6]. La ausencia de ondas peristálticas normales en los dos tercios inferiores del esófago puede ocasionar disfagia, y la incompetencia del esfínter gastroesofágico provoca esofagitis por reflujo y en ocasiones estenosis esofágica. Puede ocurrir esófago de Barrett, carcinoma esofágico y esofagitis por Candida. En forma semejante, existe hipomotilidad del estómago, intestino delgado, colon y área anorrectal, lo que causa gastroparesia, seudobstrucción, impacto colónico o deterioro de la función anorrectal. Pueden existir telangiectasias en la mucosa gástrica, la del intestino delgado y del colon. El gas puede disecar la pared intestinal (neumatosis intestinal) y fugarse a la cavidad peritoneal, simulando una víscera perforada. Se desarrollan divertículo de boca ancha en el colon, que son patognomónicos de la esclerodermia. Las lesiones tempranas del tubo digestivo pueden ser causadas por disfunción de los nervios autonómicos, y esta disfunción con el tiempo provoca atrofia del músculo liso y fibrosis irreversible del intestino. También se asocian cirrosis biliar primaria y hepatitis inducida por fármacos.
|
| Figura 6 |
| Afección esofágica en la esclerodermia |
Pulmones La afección pulmonar representa una causa importante de morbimortalidad relacionada a la esclerodermia, 14 y el daño pulmonar es la primer causa de muerte en esta enfermedad. Aunque los hallazgos pueden variar desde neoplasias y silicosis a calcinosis y hemorragia, los datos más comunes consisten en enfermedad vascular pulmonar, inflamación intersticial y fibrosis. Típicamente se encuentra hipertensión pulmonar aislada en el síndrome de CREST (la prevalencia de enfermedad severa es de alrededor del 10 por ciento en el tipo cutáneo limitado), y se observa fibrosis pulmonar intersticial en la forma tanto limitada como difusa (en el estudio posmortem la frecuencia es de alrededor del 75 por ciento). La hipertensión pulmonar aislada suele provocar tos, disnea y síncope, con mal pronóstico (supervivencia a 10 años < a 5 por ciento). La hipertensión pulmonar se define por una presión arterial pulmonar media en reposo mayor de 25 mm Hg o una presión arterial pulmonar media durante el ejercicio mayor de 30 mm Hg. Además, la hipertensión pulmonar suele asociarse con capacidad de difusión anormal para el monóxido de carbono. La fibrosis intersticial típicamente se acompaña de disnea y tos, la supervivencia a 5 años es del 45 por ciento. Aunque las radiografías de tórax pueden mostrar alteraciones lineales y reticulares, la TC de alta resolución es el mejor método para detectar afección temprana. En la TC la alveolitis aparece como áreas en parche con apariencia en vidrio despulido. La fibrosis intersticial parece deberse a alveolitis inflamatoria y la liberación de varias citocinas y quimocinas durante el curso del proceso inflamatorio causa activación de los fibroblastos y remodelación de la matriz extracelular.
Corazón La cardiopatía relacionada con la esclerodermia y clínicamente evidente se asocia con mal pronóstico. 15 El miocardio se afecta en alrededor del 20 a 25 por ciento de los casos clínicos de esclerodermia sistémica. La afección miocárdica asociada con esclerodermia se caracteriza en forma típica por áreas en parche de fibrosis miocárdica que sustituyen al músculo normal, esto puede causar hipertrofia y disminución en el gasto cardiaco, en especial con el ejercicio. La fibrosis miocárdica relacionada a la esclerodermia difiere de la encontrada en la enfermedad coronaria ateroesclerótica. No tiene depósitos de hemosiderina y, a diferencia de la fibrosis consecuente con un infarto del miocardio por una lesión coronaria, no se limita a la distribución de una arteria coronaria. Pueden desarrollarse infartos al miocardio en los pacientes con esclerodermia que tienen datos normales en el cateterismo coronario. También puede ocurrir miocarditis en pacientes con miositis inflamatoria asociada a esclerodermia. La fibrosis miocárdica en parches y la afección del sistema de conducción pueden provocar arritmias en parches, así como muerte súbita. La enfermedad pericárdica es común en la autopsia, pero ocurren manifestaciones clínicas en solo el 5 a 16 por ciento de los casos. Esta afección pericárdica puede consistir en pericarditis aguda, arritmias, derrame pericárdico o muerte súbita. En la esclerodermia cutánea limitada, además de una frecuencia similar de arritmias cardiacas y defectos en la conducción, la afección cardiaca suele ser menos frecuente y severa que en la esclerodermia difusa.
Riñón La proteinuria crónica leve y la hipertensión leve son efectos comunes de la esclerodermia, que típicamente no causan disfunción renal significativa. El proceso renal más significativo asociado con la esclerodermia es la crisis renal, 16 que consiste en desarrollo rápido de hipertensión maligna, hiperreninemia, anemia hemolítica microangiopática e insuficiencia renal oligúrica. La crisis renal ocurre en forma típica en la esclerodermia que se caracteriza por afección cutánea difusa y rápidamente progresiva. La crisis renal se consideraba antes la causa más común de muerte en la esclerodermia, pero el tratamiento agresivo con inhibidores de la enzima convertidora de angiotensina (ECA) ha mejorado en mucho la evolución. Incluso en los pacientes que al inicio requieren diálisis, los inhibidores de la ECA pueden restablecer la función renal lo suficiente para que la diálisis no sea ya necesaria.
Afección a otros órganos Aunque la esclerodermia rara vez afecta al sistema nervioso central, puede ocurrir neuropatía periférica. La forma más frecuente es la neuropatía uni o bilateral del trigémino en una o más de sus ramas. La neuropatía se manifiesta por adormecimiento y dolor progresivos. La neuralgia del trigémino suele asociarse con miositis, títulos altos de anticuerpos contra ribonucleoproteína y otras características de una enfermedad mixta del tejido conectivo. Se ha postulado que ocurre atrapamiento nervioso por tejido fibroso y lesiones vasculares, aunque el mecanismo se desconoce. La disfunción diseminada del sistema nervioso autonómico explica la propensión a fenómeno de Raynaud y la afección intestinal. Las alteraciones hematopoyéticas son raras en la esclerodermia.
Se encuentran resultados positivos de anticuerpos antinucleares en más del 85 por ciento de los pacientes con esclerodermia 17 [ver tabla 2]. Los anticuerpos contra ciertos antígenos son específicos para la esclerodermia y cada tipo de anticuerpos se asocia con un patrón clínico particular de la enfermedad. Los anticuerpos específicos se dirigen contra la topoisomerasa I (Scl-70), las proteínas del centrómero, las polimerasas de ARN, la fibrilarina (ribonucleoproteína nucleolar pequeña U3) y las proteínas de las regiones de organización del nucleolo de 90 a 100 kd [ver tabla 2]. Los anticuerpos contra Scl-70 y ARN polimerasas (por lo general ARN polimerasa III) se observan en los pacientes con esclerodermia sistémica. En un meta-análisis, los anticuerpos anti-Scl-70 tuvieron un valor predictivo positivo del 70 por ciento para la esclerodermia cutánea difusa. 18 Los anticuerpos anticentrómero se asocian con esclerodermia limitada (síndrome de CREST), y el meta-análisis indicó un valor predictivo positivo del 88 por ciento para la esclerodermia cutánea limitada. 18 Los anticuerpos contra ribonucleoproteína nuclear (RNPn) se asocian con esclerodermia difusa y limitada. Los anticuerpos contra PM-1 y KU son poco frecuentes. La presencia de estos anticuerpos suele asociarse con manifestaciones clínicas traslapadas de polimiositis y esclerodermia. Alrededor del 50 por ciento de los pacientes con esclerodermia localizada tienen anticuerpos contra histonas [ver tabla 2].
|
|||||||||||||||||||||||||||||||||
* Requiere una línea de carcinoma epitelial humano (Hep-2). |
La crisis renal es una manifestación grave de la esclerodermia susceptible de tratamiento. El médico debe instruir a las personas con esclerodermia difusa temprana para vigilar en forma diaria su presión arterial e iniciar tratamiento antihipertensivo cuando la presión arterial excede 130/80 mm Hg. El tratamiento de esta complicación grave con inhibidores de la ECA y otros antihipertensivos potentes parece controlar la hipertensión y detener el deterioro de la función renal. Sin embargo, este tratamiento ha demostrado ser más útil cuando se inicia antes de que la creatinina sérica exceda 3 mg/dl. En algunos pacientes la falla renal progresiva a pesar del buen control de la presión arterial, ameritando diálisis y quizá trasplante.
La afección pulmonar puede detectarse midiendo la capacidad vital forzada, la capacidad de difusión de bióxido de carbono y los niveles de gases en sangre arterial durante el ejercicio. Los pacientes con alteraciones deben evaluarse por medio de lavado broncoalveolar, cuando sea posible, y TC de alta resolución para detectar inflamación en las vías aéreas inferiores. En los pacientes con alveolitis fibrosante temprana la progresión a fibrosis pulmonar puede detenerse por medio de tratamiento a largo plazo con ciclofosfamida vía oral y prednisona en dosis bajas (< 10 mg/día). 19 La ciclofosfamida es un inmunosupresor potente, su uso se asocia con eventos adversos como infecciones bacterianas, herpes zoster, infección por virus varicela zoster, cistitis intersticial y neoplasias como el carcinoma de células transicionales de vejiga. La hipertensión pulmonar es una manifestación grave de la esclerodermia que en general no se controla con el tratamiento con vasodilatadores u otros agentes.
La mayoría de los pacientes con fenómeno de Raynaud deben tratarse con medidas como evitar las temperaturas frías y dejar de fumar. Además, los pacientes deben evitar agentes vasoconstrictores (v.gr., descongestivos, cafeína, anfetaminas, betabloqueadores y alcaloides del ergot). Para las personas con episodios de Raynaud tan frecuentes o severos como para interferir con las actividades diarias o con riesgo de necrosis cutánea puede emplearse el tratamiento farmacológico. Los bloqueadores de los canales del calcio promueven la vasodilatación y en general reducen la frecuencia y severidad del fenómeno. 20 Aunque la nifedipina en dosis de 30 a 60 mg al día es eficaz en la mayoría de los pacientes, alrededor de la tercera parte no responden y algunos sufren efectos adversos. En los estudios doble ciego y controlados con placebo otros bloqueadores de los canales del calcio han demostrado ser útiles para el fenómeno de Raynaud, como amlodipina, 5 a 10 mg al día y felodipina, 5 a 10 mg al día. Las ulceras digitales isquémicas pueden ser difíciles de tratar y pueden progresar a gangrena de las puntas de los dedos. El iloprost, un análogo de la prostaglandina I2, ha sido evaluado como agente para el fenómeno de Raynaud en la esclerodermia. Un estudio doble ciego, a corto plazo y controlado con placebo de infusión de iloprost mostró reducción en el número de episodios de fenómeno de Raynaud, así como reducción en la severidad. También se observó mejoría en la cicatrización de úlceras digitales. 21 Sin embargo, estudios semejantes no han encontrado diferencia entre el iloprost oral y el placebo. 22 La simpatectomía debe reservarse para las crisis isquémicas severas.
Debido a que el reflujo gastroesofágico es prácticamente universal en la esclerodermia, los clínicos deben indicar a los pacientes elevar la cabecera de la cama y administrar antagonistas de los receptores H2 o inhibidores de la bomba de protones. La motilidad esofágica puede aumentarse por medio de metoclopramida. En caso de que existan estenosis puede requerirse de dilatación esofágica. Los pacientes con menor vaciamiento gástrico deben tomar alimentos en cantidad pequeña y frecuente y pueden emplear medicamentos procinéticos como la cisaprida y la metoclopramida. Para la proliferación bacteriana con malabsorción se recomienda el tratamiento antibiótico cíclico con ciprofloxacina, metronidazol, doxiciclina o eritromicina.
Se han evaluado diversos tratamientos potencialmente modificadores de la enfermedad en la esclerodermia, pero pocos han demostrado ser eficaces. 23 La penicilamina parece ser útil para tratar el endurecimiento de la piel y la fibrosis pulmonar, pero no se ha evaluado en estudios bien controlados. Se ha sugerido que el interferón gama reduce la proliferación de fibroblastos y la proliferación de colágena. En un estudio aleatorio, doble ciego, controlado con placebo, en pacientes con esclerodermia establecida, 24 el interferón alfa tuvo beneficios modestos en los pacientes con afección cutánea y síntomas subjetivos. Por otro lado, fue dañino en la esclerodermia difusa temprana. 25 Un estudio de 24 semanas de duración de tratamiento con metotrexate, doble ciego y controlado con placebo, mostró una respuesta favorable modesta, principalmente en las manifestaciones cutáneas. 26 Un estudio aleatorio y controlado con placebo de infusiones de dexametasona mensual (10 mg) causó mejoría en la afección cutánea en los casos de esclerodermia difusa, pero otras manifestaciones de la enfermedad no cambiaron. 27 En la actualidad aún no existe un tratamiento realmente eficaz.
En el síndrome de CREST la afección cutánea es relativamente limitada, afectando por lo general solo las manos y cara, el pronóstico de los pacientes con este síndrome suele ser favorable a menos que se afecten órganos internos. Sin embargo, incluso la variante limitada de CREST tiende a ser irreversible y lentamente progresiva. Muchos pacientes sufren una evolución indolente, con pocos cambios durante muchos años, aunque la progresión puede ser más rápida. La esclerodermia difusa es muy variable en su evolución y manifestaciones, por lo que su velocidad de progresión es muy difícil de predecir. Con excepción de algunos de los cambios esclerodermatosos en la enfermedad mixta del tejido conectivo, la esclerodermia difusa rara vez remite por completo. La afección de órganos como el corazón, pulmón o especialmente el riñón indica mal pronóstico. Otras dos características de la esclerodermia indican mal pronóstico: (1) inflamación activa, manifestada por aumento en la velocidad de sedimentación globular, y (2) evidencia de afección cardiopulmonar, renal o ambas en el primer año de diagnóstico. 28 Las complicaciones graves de la esclerodermia, afección cutánea severa, fibrosis pulmonar y crisis renal, suelen ocurrir en los primeros 2 a 5 años después del inicio de la enfermedad. Después de este intervalo la enfermedad tiende a seguir un curso más indolente, 29 la supervivencia a 5 años para los pacientes con esclerodermia difusa es de alrededor del 50 por ciento.
La fascitis eosinofílica puede recordar a la esclerodermia. Se caracteriza por dolor y edema en las extremidades, con induración posterior de la piel y tejidos subcutáneos. La movilidad articular puede estar disminuida, pero no ocurren fenómeno de Raynaud, esclerodactilia ni otras manifestaciones de esclerodermia. Las alteraciones de laboratorio incluyen eosinofilia en sangre periférica, que puede ser importante, elevación de la velocidad de sedimentación globular e hiperglobulinemia. Los resultados de anticuerpos antinucleares y factor reumatoide son negativos. Las muestras de biopsia de las áreas afectadas han demostrado inflamación y engrosamiento de la fascia profunda a los tejidos subcutáneos. La piel se observa normal, pero la fascia profunda subyacente está infiltrada con linfocitos, células plasmáticas, histiocitos y en ocasiones eosinófilos. La fascitis eosinofílica parece ser autolimitada o responder a dosis bajas de esteroides. Su etiología se desconoce, aunque se han reportado algunos casos después de un ejercicio muscular extenuante.
Síndrome de mialgia-eosinofilia
En 1989 apareció un síndrome antes desconocido asociado con la ingestión de L-triptofano contaminado. 30 El síndrome de mialgia-eosinofilia se caracteriza por eosinofilia periférica, mialgias severas e incapacitantes y fatiga de varias semanas de duración. También pueden existir disnea y tos. La afección cutánea consiste en erupciones variables, edema y cambios esclerodermatosos, por lo general sin manifestaciones viscerales. Pueden ocurrir infiltrados pulmonares intersticiales, hipoxia, hipertensión pulmonar y neumonitis por hipersensibilidad. Se ha descrito polineuropatía con patrón de mononeuritis múltiple. Se han reportado trastornos neurocognitivos, como alteraciones de la memoria y dificultad en la concentración. La mayoría de los pacientes sigue manifestando síntomas 2 a 4 años después del inicio, pero sin nuevos signos de inflamación.
Figura 1 Reimpresa de la Colección de Diapositivas sobre Enfermedades
Reumáticas, c 1991, 1995, 1997. Utilizada con autorización del
American College of Rheumatology.
Bibliografía
- Wigley FM, Flavahan NA: Raynaud's phenomenon. Rheum Dis Clin North Am 22:765, 1996 [PMID 8923595 ]
- Spencer-Green G: Outcomes in primary Raynaud phenomenon: a meta-analysis of the frequency, rates, and predictors of transition to secondary diseases. Arch Intern Med 158:595, 1998
- Mayes MD: Epidemiology of systemic sclerosis and related diseases. Curr Opin Rheumatol 9:557, 1997
- Michet C: Update in the epidemiology of the rheumatic diseases. Curr Opin Rheumatol 10:129, 1998
- Tan FK, Stivers DN, Foster MW, et al: Association of microsatellite markers near the fibrillin 1 gene on human chromosome 15q with scleroderma in a native American population. Arthritis Rheum 41:1729, 1998 [PMID 9778214 ]
- Silman AJ, Hochberg MC: Occupational and environmental influences on scleroderma. Rheum Dis Clin North Am 22:737, 1996 [PMID 8923593 ]
- Hochberg MC, Perlmutter DL: The association of augmentation mammoplasty with connective tissue disease, including systematic sclerosis (scleroderma): a meta-analysis. Curr Top Microbiol Immunol 210:411, 1996 [PMID 8565585 ]
- Jimenez SA, Hitraya E, Varga J: Pathogenesis of scleroderma: collagen. Rheum Dis Clin North Am 22:647, 1996 [PMID 8923589 ]
- LeRoy EC: Systemic sclerosis: a vascular perspective. Rheum Dis Clin North Am 22:675, 1996
- Nelson JL: Microchimerism and the pathogenesis of systemic sclerosis. Curr Opin Rheumatol 10:564, 1998
- Evans PC, Lambert N, Maloney S, et al: Long-term fetal microchimerism in peripheral blood mononuclear cell subsets in healthy women and women with scleroderma. Blood 93:2033, 1999 [PMID 10068676 ]
- Olsen NJ, King LEJ, Park JH: Muscle abnormalities in scleroderma. Rheum Dis Clin North Am 22:783, 1996 [PMID 8923596 ]
- Sjögren RW: Gastrointestinal features of scleroderma. Curr Opin Rheumatol 8:569, 1996
- Silver RM: Scleroderma: clinical problems-the lungs. Rheum Dis Clin North Am 22:825, 1996
- Deswal A, Follansbee WP: Cardiac involvement in scleroderma. Rheum Dis Clin North Am 22:841, 1996 [PMID 8923599 ]
- Steen VD: Scleroderma renal crisis. Rheum Dis Clin North Am 22:861, 1996
- Okano Y: Antinuclear antibody in systemic sclerosis (scleroderma). Rheum Dis Clin North Am 22:709, 1996
- Spencer-Green G, Alter D, Welch HG: Test performance in systemic sclerosis: anti-centromere and anti-Scl-70 antibodies. Am J Med 103:242, 1997 [PMID 9316557 ]
- Akesson A: Cyclophosphamide therapy for scleroderma. Curr Opin Rheumatol 10:579, 1998
- Sturgill MG, Seibold JR: Rational use of calcium-channel antagonists in Raynaud's phenomenon. Curr Opin Rheumatol 10:584, 1998 [çPMID 9812220 ]
- Wigley FM, Wise RA, Seibold JR, et al: Intravenous iloprost infusion in patients with Raynaud phenomenon secondary to systemic sclerosis: a multicenter, placebo-controlled, double-blind study. Ann Intern Med 120:199, 1994 [PMID 7506013 ]
- Wigley FM, Korn JH, Csuka ME, et al: Oral iloprost treatment in patients with Raynaud's phenomenon secondary to systemic sclerosis: a multicenter, placebo-controlled, double-blind study. Arthritis Rheum 41:670, 1998 [PMID 9550476 ]
- Pope JE: Treatment of systemic sclerosis. Rheum Dis Clin North Am 22:893, 1996
- Grassegger A, Schuler G, Hessenberger G, et al: Interferon-gamma in the treatment of systemic sclerosis: a randomized controlled multicentre trial. Br J Dermatol 139:639, 1998 [PMID 9892907 ]
- Black CM, Silman AJ, Herrick AI, et al: Interferon-alpha does not improve outcome at one year in patients with diffuse cutaneous scleroderma: results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 42:299, 1999 [PMID 10025924 ]
- van den Hoogen FH, Boerbooms AM, Swaak AJ, et al: Comparison of methotrexate with placebo in the treatment of systemic sclerosis: a 24 week randomized double-blind trial, followed by a 24 week observational trial. Br J Rheumatol 35:364, 1996 [PMID 8624641]
- Sharada B, Kumar A, Kakker R, et al: Intravenous dexamethasone pulse therapy in diffuse systemic sclerosis: a randomized placebo-controlled study. Rheumatol Int 14:91, 1994 [PMID 7839076 ]
- Bulpitt KJ, Clements PJ, Lachenbruch PA, et al: Early undifferentiated connective tissue disease: III. Outcome and prognostic indicators in early scleroderma (systemic sclerosis). Ann Intern Med 118:602, 1993
- Clements PJ: Measuring disease activity and severity in scleroderma (editorial). Curr Opin Rheumatol 7:517, 1995
- Varga J, Kahari VM: Eosinophilia-myalgia syndrome, eosinophilic fasciitis, and related fibrosing disorders. Curr Opin Rheumatol 9:562, 1997 [PMID 9375286 ]