Nefrología
⭳ Abrir artículo (PDF)791.9 KBEste artículo es idéntico en la Edición 2/2000.
Contenido del artículo
VII ENFERMEDADES VASCULARES DEL RIÑON
- Complicaciones renales de la vasculitis sistémica
- Anticuerpos anticitoplasma de neutrófilos
- GRANULOMATOSIS DE WEGENER
- POLIARTERITIS NODOSA Y SINDROME DE CHURG-STRAUSS
- VASCULITIS POR HIPERSENSIBILIDAD
- CAUSAS DIVERSAS DE VASCULITIS RENAL
- Complicaciones renales de la esclerodermia
- Síndromes urémico-hemolíticos
- Complicaciones renales en el embarazo: preeclampsia e hipertensión inducida por el embarazo
- CAMBIOS FISIOLOGICOS DURANTE EL EMBARAZO
- PATOGENIA Y PATOLOGIA
- MANIFESTACIONES CLINICAS Y DIAGNOSTICO
- Diferencias entre la preeclampsia y otras causas de hipertensión arterial del embarazo
- Diferencia entre la preeclampsia y la nefropatía primaria
- TRATAMIENTO
- PRONOSTICO
- Enfermedad tromboembólica de las arterias renales
- Necrosis de la corteza renal
- Nefropatía por células falciformes
- HEMATURIA
- INFARTO RENAL Y NECROSIS PAPILAR
- CAPACIDAD DE CONCENTRACION DISMINUIDA
- SECRECION RENAL BAJA DE HIDROGENO Y POTASIO
- INSUFICIENCIA RENAL AGUDA
- Nefritis por radiación
VII ENFERMEDADES VASCULARES DEL RIÑON
DR. ROBERT M. BLACK
Las enfermedades vasculares del riñón producen oclusión parcial o completa de los vasos de pequeño, mediano y gran calibre. Algunas de estas enfermedades, como la vasculitis, tienen una base inmunológica; en otros padecimientos, como la tromboembolia, participan mecanismos no inmunológicos. Las manifestaciones clínicas de estos trastornos son muy variables. Sin embargo, por lo general la función renal se afecta como resultado de la disminución del flujo sanguíneo renal. El diagnóstico se realiza correlacionando los antecedentes, los hallazgos de la exploración física y los datos de laboratorio con las alteraciones renales.
Complicaciones renales de la vasculitis sistémica
Un buen método para la clasificación de la vasculitis sistémica se basa en la extensión y el sitio de la afección vascular [ver tabla 1].1 Este método es sobre todo útil porque se conoce poco sobre la etiología y patogenia de los diversos trastornos. Es importante distinguir entre estas enfermedades porque tienen una evolución y una respuesta al tratamiento diferentes.2
Anticuerpos anticitoplasma de neutrófilos
En los últimos años se han observado un grupo nuevo de autoanticuerpos dirigidos contra antígenos citoplásmicos de los neutrófilos (ANCA, por sus siglas en inglés, n. del t.) en pacientes con granulomatosis de Wegener y otras vasculitis relacionadas.3,4 Los anticuerpos anticitoplasma de neutrófilo citoplasmáticos (c-ANCA) están dirigidos contra una enzima llamada proteinasa 3. Los anticuerpos anticitoplasma de neutrófilo perinucleares (p-ANCA) suelen estar dirigidos contra mieloperoxidasa o, con menos frecuencia, elastasa. En consecuencia, se observan dos diferentes patrones de inmunofluorescenia, la tinción citoplásmica y la tinción perinuclear, al incubar el suero del paciente con neutrófilos.
Es posible detectar ANCA en casi el 90 porciento de los pacientes con granulomatosis de Wegener.4 En este caso los c-ANCA son los más comunes. También se encuentran ANCA en dos padecimientos relacionados con datos histológicos renales idénticos a los de la granulomatosis de Wegener: la poliarteritis microscópica y la glomerulonefritis necrosante (con medias lunas) rápidamente progresiva. Sin embargo, en estos dos padecimientos son más comunes los p-ANCA.
Muchos estudios sugieren que los títulos de ANCA en granulomatosis de Wegener y enfermedades relacionadas tienden a ser paralelos a la evolución clínica de la vasculitis, aumentando con la actividad de la enfermedad y disminuyendo con la remisión.4 Además, se ha observado un aumento en los títulos que puede preceder al inicio de una recidiva clínica en muchos pacientes.5 Sin embargo, a pesar de estas interesantes observaciones, no se ha demostrado el papel patogénico de los ANCA.
Si los ANCA favorecen el desarrollo de vasculitis, el mecanismo es incierto. Se ha observado que los neutrófilos normales sufren degranulación y producen radicales libres de oxígeno después de incubarse con ANCA;6 estos neutrófilos estimulados pueden dañar a las células endoteliales vasculares.7 Como alternativa, es posible que los ANCA inhiban la neutralización de enzimas polimorfonucleares después de su liberación de los neutrófilos, ocasionando así daño tisular por actividad enzimática no controlada.8
GRANULOMATOSIS DE WEGENER
La granulomatosis de Wegener es una vasculitis rara pero bien definida que afecta de manera característica el aparato respiratorio superior e inferior y los riñones, aunque pueden afectarse casi todos los órganos. La lesión renal se caracteriza por granulonefritis necrosante, que suele acompañarse de la formación de medias lunas [ver figura 1]. Por lo general la tinción inmunofluorescente de los glomérulos y los vasos renales no muestra depósitos de complejos inmunes. Sin embargo, su ausencia no excluye la participación del sistema inmune, porque es posible que los ANCA dañen en forma directa a los vasos (ver antes).6 Los granulomas necrosantes, que a menudo se encuentran en el tejido del aparato respiratorio superior e inferior de individuos afectados, pueden obaservarse en pocas ocasiones en las muestras de biopsia renal.
La granulomatosis de Wegener se desarrolla con mayor frecuencia en los adultos de mediana edad, pero también puede observarse en pacientes más jóvenes.1 En el momento del diagnóstico suelen existir manifestaciones clínicas de afección renal. El sedimento urinario característico muestra hematuria, cilindros eritrocitarios y proteinuria. Las concentraciones de creatinina plasmática y nitrógeno ureico de la sangre (BUN) suelen elevarse, lo que indica lesión renal considerable. Aunque se ha descrito una forma limitada de la granulomatosis de Wegener, caracterizada por afección respiratoria, pero no renal, en la mayoría de estos casos al final se desarrolla la enfermedad renal típica.
Los hallazgos de laborotorio pueden incluir anemia, leucocitosis y velocidad de sedimentación globular elevada, aunque no suelen ser diagnósticos. Por lo general, las concentraciones del complemento son normales y se detectan anticuerpos antinucleares, lo mismo que (como ya se mencionó) ANCA.
Por lo general se requiere de la biopsia del tejido para confirmar el diagnóstico. Es indispensable realizar una exploración cuidadosa de los senos del cráneo y la nasofaringe, en vista de que es posible encontrar lesiones vasculíticas en estas zonas, incluso en pacientes asintomáticos, y porque la toma de biopsia de las lesiones se efectúa con facilidad. Los hallazgos de la biopsia renal pueden ser muy sugestivos de granulomatosis de Wegener, aunque por lo general no son patognomónicos por dos razones. En primer lugar, en forma característica las muestras de tejido renal no presentan granulomas y en segundo lugar, también se observa glomerulonefritis necrosante focal con tinción inmunofluorescente negativa en pacientes con poliarteritis nodosa y como un trastorno idiopático en el que no puede identificarse vasculitis renal o extrarrenal.9 Sin embargo, algunos de los pacientes con glomerulonefritis con el tiempo desarrollan signos de granulomatosis de Wegener por lo que tal vez requieran modificaciones en el tratamiento.
Sin tratamiento, el pronóstico de los pacientes con granulomatosis de Wegener es muy malo, en vista de que hasta el 90 porciento mueren en los primeros dos años después del inicio del padecimiento.1,10 Aunque en algunos pacientes la administración de esteroides produce mejoría clínica y en los resultados de laboratorio, esta respuesta suele ser temporal. Por el contrario, con el uso de fármacos citotóxicos pueden producirse remisiones prolongadas en la mayoría de los pacientes, sobre todo con la ciclofosfamida.1,11 Es muy importante el inicio temprano del tratamiento en vista de que la necrosis tisular no puede revertirse una vez que ocurra. Sin embargo, la aparición de insuficiencia renal lo bastante grave para requerir diálisis durante la fase aguda de la enfermedad no necesariamente impide el tratamiento agresivo, ya que la función renal puede mejorar lo suficiente para permitir la suspensión de la diálisis. El tratamiento debe continuarse durante uno a dos años después de la remisión en vista de que las recaídas son comunes y pueden ocasionar suficiente necrosis tisular para impedir la recuperación de la función renal.
En algunos pacientes se han usado tratamientos alternativos. La administración de trimetoprim-sulfametoxazol (una tableta de dosis doble dos veces al día) ha sido benéfica para un número pequeño de pacientes.12,13 Al parecer es más eficaz en pacientes con síntomas sistémicos leves y enfermedad que se limita a las vías respiratorias superiores. El papel de este agente combinado en la granulomatosis de Wegener debe definirse aún en un número mayor de pacientes.
Existen también publicaciones no controladas y preliminares que sugieren que la inmunoglobulina intravenosa puede ser útil en la granulomatosis de Wegener y en otras formas de vasculitis sistémica.14 No se conoce aún el mecanismo de acción.14,15
Un estudio abierto indica que el methotrexate en dosis bajas puede ser útil en pacientes seleccionados,16 alcanzándose la remisión en casi el 70 porciento de los pacientes, aunque se requieren más estudios antes de que pueda recomendarse el uso habitual de este medicamento.
No es rara la progresión a insuficiencia renal, que puede ser causada por recidivas de la vasculitis o por glomeruloesclerosis progresiva por cicatrización de las lesiones glomerular y vascular iniciales. Con menos frecuencia se ha observado glomerulonefritis de novo, como nefropatía por IgA.17
La experiencia con el trasplante renal en la granulomatosis de Wegener es limitada. Reportes aislados indican que tanto las manifestaciones renales como extrarrenales pueden recurrir después del trasplante,18-20 asociándose con títulos altos de ANCA en plasma.21 El riesgo de recaídas puede ser mayor en pacientes que solo reciben prednisona y azatioprina, comparando con pacientes tratados con ciclosporina.20 El aumentar la dosis de prednisona no suele ser útil. Por otro lado, la sustitución de la azatioprina por ciclofosfamida o el cambio a ciclosporina suele causar la remisión de las alteraciones clínicas, en forma semejante a lo que se observa en la enfermedad primaria.18-20
POLIARTERITIS NODOSA Y SINDROME DE CHURG-STRAUSS
Se han descrito cuatro formas de poliarteritis nodosa, también conocida como periarteritis nodosa. Estos trastornos pueden estar relacionados entre sí y con la granulomatosis de Wegener; por lo menos en algunos pacientes con estos padecimientos se han detectado ANCA.4
1.Poliarteritis nodosa clásica, que afecta las arterias musculares de pequeño y mediano calibre y ocasiona la formación de aneurismas.
2.Poliarteritis nodosa microscópica, que afecta vasos más pequeños que la poliarteritis nodosa clásica.
3.Síndrome de Churg-Strauss, que se caracteriza por la formación de granulomas extravasculares e infiltración eosinofílica en arterias y vénulas.
4.Síndrome de sobreposición, compuesto por signos y síntomas característicos de más de una forma de poliarteritis nodosa.
Las manifestaciones clínicas para diferenciar estos trastornos se revisan en otro capítulo. La mayoría de los pacientes tienen síntomas sistémicos inespecíficos, que pueden incluir fiebre, pérdida de peso, artralgias, polineuropatía asimétrica (mononeuritis múltiple) o anorexia. La afección del aparato respiratorio es menos común en la poliarteritis nodosa clásica y microscópica que en la granulomatosis de Wegener. Sin embargo, en el síndrome de Churg-Strauss la enfermedad pulmonar es común, sobre todo el broncoespasmo, aunque la afección renal y pulmonar no siempre se desarrollan al mismo tiempo. La cardiopatía también es más común en el síndrome de Churg-Strauss que en las otras formas de poliarteritis nodosa.
La hipertensión arterial resultante de la vasculitis renal es común en la poliarteritis nodosa clásica y en ocasiones se manifiesta como hipertensión maligna. Por el contrario, la presión arterial en pacientes con poliarteritis nodosa microscópica es normal a menos que la función renal esté muy afectada. La causa de esta diferencia no se ha determinado, aunque puede estar relacionada con el tamaño de los vasos afectados. En la forma clásica, la inflamación de las arterias de mayor calibre puede causar isquemia distal, lo que ocasiona la activación del sistema renina-angiotensina. La poliarteritis nodosa microscópica se acompaña de inflamación de los capilares glomerulares, por lo que es menos probable que active este sistema.22
Las manifestaciones de la afección renal varían en las diferentes formas de poliarteritis nodosa. La forma microscópica a menudo se caracteriza por un sedimento urinario activo con eritrocitos, cilindros eritrocitarios y proteinuria. Estos hallazgos son resultado de la glomerulonefritis necrosante segmentaria focal. Es posible que las lesiones observadas en la biopsia renal no puedan distinguirse de las lesiones de la granulomatosis de Wegener [ver figura 1]. La poliarteritis nosoda clásica puede mostrar un patrón diferente a la forma microscópica de la enfermedad. En vista de que la forma clásica afecta las arterias renales de mayor calibre más que los vasos pequeños que irrigan los glomérulos, es posible no observar necrosis del ovillo glomerular; como consecuencia, en algunas ocasiones el sedimento urinario es normal, ya que la reducción de la velocidad de filtración glomerular se debe sobre todo a la disminución de la perfusión glomerular y no a inflamación. La lesión renal suele ser menos grave en el síndrome de Churg-Strauss que en otras variantes de la poliarteritis nodosa.23
El diagnóstico de poliarteritis nodosa se basa en los antecedentes clínicos, los hallazgos de la exploración física y en el tipo de afección de los órganos. En casos raros pueden palparse aneurismas en los vasos superficiales. Las observaciones preliminares sugieren que existe más posibilidad de encontrar ANCA positivos en la forma microscópica que en la poliarteritis nodosa clásica. La frecuencia con la que se observan estos anticuerpos en el síndrome de Churg-Strauss se desconoce, pero tambien se han detectado.
Al igual que en la granulomatosis de Wegener, por lo general es necesario el diagnóstico histológico.24 Si el examen clínico y los estudios de conducción sugieren afección nerviosa, está indicada la biopsia del nervio sural. Por otra parte, la angiografía celiaca y renal es casi diagnóstica de poliarteritis nodosa clásica si demuestra la presencia de microaneurismas y estenosis segmentaria e irregular en los vasos de mayor calibre, además de disminución progresiva y oclusión de las arterias intrarrenales de menor calibre.
El pronóstico global de la poliarteritis nodosa sin tratamiento es malo. La supervivencia a un año en pacientes con la forma clásica de la enfermedad es alrededor del 50 porciento, y la supervivencia a cinco años es del 13 porciento.25 El pronóstico de la variante microscópica puede ser un poco mejor, aunque la supervivencia a largo plazo en pacientes con este trastorno también es limitada sin un tratamiento eficaz.
El tratamiento con esteroides puede ser eficaz en algunos individuos. Algunos pacientes con poliarteritis nodosa, incluyendo los que no responden a los esteroides, han tenido remisiones prolongadas con el uso de ciclofosfamida.1,26 Es importante resaltar que el tratamiento puede mejorar la función renal, incluso en pacientes que presentan insuficiencia renal avanzada y que requieren diálisis. Por consiguiente, la gravedad inicial del padecimiento no impide proporcionar un tratamiento eficaz, aunque la respuesta inicial puede ser seguida por el desarrollo tardío de aterosclerosis si la resolución de las lesiones ocasiona estenosis progresiva de la luz de los vasos. En los pacientes que responden debe continuarse el tratamiento de mantenimiento durante uno a dos años después de la remisión para disminuir el riesgo de recaídas.
VASCULITIS POR HIPERSENSIBILIDAD
La vasculitis por hipersensibilidad se caracteriza por inflamación de los vasos sanguíneos pequeños como las arteriolas, los capilares y, sobre todo, las vénulas poscapilares.27 Los cambios inflamatorios son muy notables en la piel, donde existe infiltración considerable de neutrófilos alrededor de los vasos sanguíneos de la dermis, hemorragia local y edema. Este cuadro histológico recibe el nombre de vasculitis leucocitoclástica que se manifiesta como exantema purpúrico palpable [ver figura 2]. El exantema casi siempre es el principal signo de esta enfermedad.
Las tres principales variantes de la vasculitis por hipersensibilidad que afectan al riñón son la púrpura de Henoch-Schönlein, la crioglobulinemia mixta esencial y la enfermedad del suero. Con menos frecuencia, se observa vasculitis por hipersensibilidad en pacientes con lupus eritematoso sistémico, síndrome de sobreposición de poliarteritis nodosa u otros padecimientos más raros.
Púrpura de Henoch-Schönlein
La púrpura de Henoch-Schönlein se caracteriza por la tétrada clásica de dolor abdominal, exantema cutáneo purpúrico, artritis o artralgias y afección renal. Es más común en niños, aunque también ocurre en adultos.28 Las lesiones cutáneas, que son más marcadas en las superficies extensoras y en declive, se observan en prácticamente todos los pacientes, anque pueden no existir durante el examen inicial.29
La afección renal es común y suele ser evidente en los primeros días o meses después del inicio de los síntomas. El examen general de orina muestra de manera característica hematuria microscópica o macroscópica, además de cilindros eritrocitarios y proteinuria. En la mayoría de los pacientes la concentración de creatinina plasmática es normal o sólo ligeramente elevada al inicio de la enfermedad. Sin embargo, puede existir afección renal más importante, caracterizada por proteinuria en rango nefrótico, hipertensión arterial e insuficiencia renal.
Los cambios característicos en el riñón son idénticos a los que se observan en la nefropatía por IgA. Existe expansión del mesangio glomerular por depósitos de IgA, que puede detectarse por medio de tinción inmunofluorescente. En los casos de afección renal grave puede haber formación de medias lunas.
El diagnóstico de púrpura de Henoch-Schönlein por lo general es evidente por las manifestaciones clínicas clásicas. La biopsia renal no suele ser necesaria. Cuando el diagnóstico es dudoso, la biopsia tanto de la piel afectada como de la sana pueden mostrar depósitos de IgA en las paredes de los vasos sanguíneos, y de esta forma se evita la necesidad de la biopsia renal, que implica un riesgo mayor.27,30
En general, las manifestaciones clínicas de los pacientes con púrpura de Henoch-Schönlein desaparecen de manera espontánea, aunque pueden recurrir los episodios de púrpura o glomerulonefritis. No existe una relación constante entre la recurrencia de la enfermedad y el pronóstico, o entre la gravedad de la glomerulonefritis y la de las manifestaciones extrarrenales. Más bien, el grado de afección renal es el factor más importante del pronóstico a largo plazo. Los pacientes con síndrome nefrótico e insuficiencia renal tienen el mayor riesgo de desarrollar insuficiencia renal progresiva.
Existen pocas evidencias de que el tratamiento convencional con esteroides o ciclofosfamida tenga efectos favorables sobre la enfermedad cutánea o la nefropatía.31 Estos medicamentos no parecen reducir la frecuencia de las recaídas ni tampoco modificar la duración de la actividad de la enfermedad. A pesar de ello, suele administrarse un esquema que consiste en pulsos intravenosos de metilprednisolona, seguido de prednisona oral y ciclofosfamida, y en ocasiones de plasmaféresis, en los pacientes con riesgo alto, insuficiencia renal aguda y glomerulonefritis con medias lunas.1,31 Este esquema tiene por objeto revertir el proceso inflamatorio, como el infiltrado por macrófagos, más que el depósito de IgA en sí.
Estudios no controlados sugieren que la dapsona puede ser útil para disminuir la severidad y duración de las lesiones cutáneas en pacientes con vasculitis leucocitoclásticas.32 Sin embargo, su eficacia en pacientes con púrpura de Henoch-Schönlein no se ha estudiado.
El trasplante renal puede tener éxito en los pocos pacientes que progresan hasta la enfermedad renal terminal. Es común el depósito recurrente de IgA en el mesangio del riñón trasplantado, aunque la enfermedad es subclínica en la mayoría de los pacientes.18 En menos del 15 porciento de estos enfermos se observa hematuria, proteinuria y púrpura.33,34
Crioglobulinemia mixta
Las crioglobulinas son anticuerpos que se precipitan con la temperatura fría y se disuelven al exponerlos de nuevo al calor. En la crioglobulinemia mixta esencial, prototipo de este trastorno, las crioglobulinas contienen tanto una IgG policlonal como un factor reumatoide IgM monoclonal dirigido contra la IgG.
Aunque se ha implicado al virus de Epstein-Barr y al virus de la hepatitis B como estímulos antigénicos en algunos casos, estudios recientes sugieren que la infección por virus de la hepatitis C (VHC) es la causa subyacente más común.35,36 A pesar de este dato, existen pocas evidencias de hepatitis activa en el momento de la presentación del paciente.
Los síntomas de la crioglobulinemia mixta suelen ser inespecíficos. La fatiga y las artralgias son muy comunes. Los hallazgos que más sugieren el diagnóstico incluyen exantema purpúrico palpable, linfadenopatía, hepatoesplenomegalia, mononeuritis y fenómeno de Raynaud. Por lo general existe hipocomplementemia, a diferencia de las concentraciones normales del complemento que se observan en los pacientes con púrpura de Henoch-Schönlein.
Ocurre afección renal en el 30 a 60 porciento de los pacientes.37 En estos casos la biopsia renal muestra en forma característica proliferación difusa del endotelio y de las células mesangiales, un patrón indicativo de glomerulonefritis membranoproliferativa. Resulta de particular importancia el hallazgo frecuente de trombos intraluminales compuestos de crioglobulinas precipitadas [ver figura 3].
Debe pensarse en el diagnóstico de crioglobulinemia mixta en cualquier persona con exantema purpúrico palpable, sobre todo si el exantema se presenta en la superficies extensoras o los glúteos y se acompaña de hipocomplementemia y afección renal.37,38 Las pruebas de funcionamiento hepático por lo común son normales, sobre todo si la infección viral incitante ha sido crónica. El diagnóstico se confirma por la demostración de crioglobulinas circulantes. En la crioglobulinemia mixta, la inmunoelectroforesis sérica revela un título alto de crioglobulinas mixtas IgM-IgG (o, en algunos casos, un título alto de IgM-IgA) con un componente monoclonal. En muchos individuos existen anticuerpos contra el VHC. La biopsia renal debe reservarse para los pacientes con enfermedad progresiva o en los que el diagnóstico está en duda.
El pronóstico de la enfermedad renal en la crioglobulinemia mixta es variable. Alrededor de la tercera parte de los pacientes presenta remisión parcial o completa, mientras que el resto tiene un curso lentamente progresivo que puede complicarse por exacerbaciones periódicas.37-39
En el pasado la quimioterapia agresiva se reservaba para los pacientes con crioglobulinemia mixta y enfermedad aguda grave manifestada por un nivel de crioglobulinas en aumento, insuficiencia renal progresiva, necrosis distal o neuropatía avanzada. En estos casos se ha usado plasmaféresis para eliminar las crioglobulinas circulantes junto con esteroides y ciclofosfamida para evitar la formación de más anticuerpos.1,37,38,40 El plasma reinfundido durante la plasmaféresis debe calentarse para evitar la precipitación de las crioglobulinas circulantes. Por lo general este esquema permite estabilizar la función renal, aunque la muerte por vasculitis activa sigue siendo un gran problema.38
En contraste, con el uso de quimioterapia y plasmaféresis, existen evidencias en un número cada vez mayor de pacientes con crioglobulinemia mixta sobre que el interferón alfa puede reducir las concentraciones circulantes de crioglobulinas e inducir remisión clínica.36,41,42 Sin embargo, en la mayoría de estos pacientes ocurren recaídas al suspender el tratamiento.36,43 A pesar de estas observaciones, el interferón alfa puede ser el tratamiento inicial de elección en pacientes con crioglobulinemia mixta, en especial porque los títulos de VHC pueden aumentar después del tratamiento inmunosupresor con ciclofosfamida.
Enfermedad del suero
La enfermedad del suero a menudo es ocasionada por la administración de medicamentos, incluyendo penicilinas, derivados de sulfonamidas y difenihidantoína, que parecen actuar como haptenos y estimular una respuesta inmune. También puede observarse en presencia de algunas infecciones, como la hepatitis B.44 Las manifestaciones iniciales típicas incluyen fiebre, exantema, artralgias y linfadenopatía. En la mayoría de los casos el exantema es urticariano. En algunas ocasiones existe edema. Los síntomas aparecen en forma característica en un periodo de siete a 10 días después del contacto con el antígeno. En algunos pacientes puede haber hipocomplementemia.
La afección renal se manifiesta por alteraciones en el examen general de orina caracterizadas por proteinuria, eritrocitos y cilindros celulares. La insuficiencia renal aguda es rara, pero puede ocurrir, sobre todo con la exposición prolongada al antígeno. En este caso la biopsia renal muestra la típica glomerulonefritis por complejos inmunes con proliferación celular difusa y depósitos de IgG y del componente C3 del complemento en las paredes de los capilares glomerulares.
Existen pocas pruebas de que el tratamiento inmunosupresor con esteroides o agentes citotóxicos como la ciclofosfamida eviten el desarrollo o modifiquen la evolución de este trastorno. La eliminación del agente estimulante suele dar como resultado la resolución inmediata de los signos y síntomas en algunos días o semanas.
CAUSAS DIVERSAS DE VASCULITIS RENAL
Existen varias vasculitis que en algunas ocasiones se acompañan de afección renal importante incluyendo la vasculitis urticariana con hipocomplementemia, la vasculitis reumatoide y la policondritis recidivante. La vasculitis de vasos grandes, como la arteritis de la temporal o la arteritis de Takayasu, pueden afectar las arterias renales principales, pero no suelen producir lesiones en los vasos intrarrenales pequeños.45,46
Complicaciones renales de la esclerodermia
La esclerodermia es una enfermedad poco frecuente caracterizada por cambios notables en la piel y evidencias de afección multisistémica, por lo general de los pulmones, el aparato digestivo, el corazón y los riñones. Se observa con mayor frecuencia en mujeres, con inicio entre los 20 y 50 años de edad. Las manifestaciones de la enfermedad son principalmente causadas por la isquemia secundaria a las lesiones vasculares o por el aumento local en el acúmulo de colágena.
La afección renal en la esclerodermia se caracteriza por lesiones arteriales obstructivas que afectan sobre todo a las arterias interlobulares. También es común el engrosamiento concéntrico (llamado en capas de cebolla) de la pared vascular, que es resultado de la proliferación de las células de músculo liso en la capa media, que después se deplazan hacia la íntima [ver figura 4]. Estas alteraciones histológicas son similares a las lesiones observadas en la hipertensión maligna y en los síndromes urémico-homolíticos [ver adelante, Síndromes urémico-hemolíticos].
En cerca del 20 al 25 porciento de los pacientes con esclerodermia se observa afección renal importante. La complicación renal más grave es la crisis renal esclerodérmica, que se caracteriza por la aparición súbita de insuficiencia renal aguda. Por lo general el sedimiento urinario sólo muestra proteinuria leve y algunas células o cilindros. Estos hallazgos reflejan el origen no inflamatorio de la enfermedad vascular. En casos raros puede haber hematuria ocasionada por necrosis glomerular isquémica o por hemorragia de las hemangiomas localizados en el aparato urinario. Si no se trata, la crisis renal esderodérmica progresa hasta la insuficiencia renal avanzada en un periodo de uno a dos meses.
La crisis renal esclerodérmica casi siempre se desarrolla en los primeros cuatro años después del inicio de las manifestaciones extrarrenales, sin embargo, en pocas ocasiones es la manifestación inicial de la escleroderma.47,48 En la mayoría de los pacientes se observan elevaciones importantes de la presión arterial como resultado de la activación del sistema renina-angiotensina por la isquemia. Sin embargo, la presencia de hipertensión leve o el aumento en la actividad de la renina plasmática no indican necesariamente que los pacientes con esclerodermia con el tiempo desarrollarán esta complicación. Por otra parte, el desarrollo simultáneo de hipertensión mediada por angiotensina parece ser un factor secundario en la patogenia de la esclerodermia renal, en vista de que en ocasiones la insuficiencia renal aguda no se acompaña de hipertensión arterial.49
El diagnóstico de la crisis renal esclerodérmica suele ser evidente por los antecedentes y la exploración física. En algunas ocasiones la insuficiencia renal aguda es la manifestación inicial. En presencia de insuficiencia renal aguda, deben excluirse otros padecimientos con características similares, incluyendo la hipertensión maligna, los síndromes urémicos-hemolíticos y la vasculitis sistémica (v.gr., poliarteritis nodosa). El antecedente de fenómeno de Raynaud y alteraciones cutáneas, como telangiectasias y capilares periungueales tortuosos y muy dilatados, indica la presencia de esclerodermia.
Aparecen anticuerpos antinucleares hasta en el 90 porciento de los pacientes con esclerodermia. En más del 85 porciento de los casos es posible identificar el antígeno contra el que se dirigen estos anticuerpos.50
La insuficiencia renal representa cerca del 40 porciento de las muertes por esclerodermia. El resto de la mortalidad se debe a insuficiencia respiratoria, insuficiencia cardiaca o complicaciones gastrointestinales isquémicas, como la perforación visceral. Aunque el tratamiento para las complicaciones extrarrenales de la esclerodermia es limitado, el control de la hipertensión en pacientes con crisis renal esclerodérmica puede alterar en forma notable la evolución. El tratamiento antihipertensivo eficaz puede disminuir de manera considerable la incidencia de insuficiencia renal si se inicia antes que aparezcan lesiones vasculares irreversibles.
Los inhibidores de la enzima convertidora de angiotensina (ECA) son los fármacos de elección para el tratamiento de la hipertensión en los pacientes con esclerodermia.51 Los inhibidores de la ECA disminuyen la presión arterial en hasta el 90 porciento de los pacientes al revertir la vasoconstricción inducida por la angiotensina II. Estudios preliminares sugieren también que estos agentes pueden mejorar la evolución de los pacientes en crisis renales normotensas.52
No se conoce del todo porqué los inhibidores de la ECA son muchos más eficaces que otros antihipertensivos para mejorar la evolución renal en la esclerodermia.53 Sin embargo, debe recordarse que casi la mitad de los pacientes con crisis renal esclerodérmica progresan hasta la insuficiencia renal terminal.51
La enfermedad renal por esclerodermia consiste esencialmente en una estenosis de la arteria renal bilateral. Por lo tanto, es posible que la concentración plasmática de creatinina aumente en algunos pacientes por una reducción en la resistencia arteriolar eferente inducida por el inhibidor de la ECA, con menor presión intraglomerular consecuente.
Los enfermos que progresan hasta insuficiencia renal terminal pueden dializarse en forma satisfactoria, aunque a veces se presentan algunos problemas. Por ejemplo, la hemodiálisis puede resultar difícil si existen pocas vías de acceso vascular como resultado de la enfermedad vascular periférica. Este problema puede evitarse con la diálisis peritoneal, aunque la depuración peritoneal a menudo es baja en la esclerodermia como consecuencia de la alteración en el flujo sanguíneo peritoneal. Además, la depuración peritoneal puede disminuir con el descenso de la temperatura ambiente; por consiguiente, suele ser menor durante los meses de invierno.54
Existe poca experiencia con el trasplante renal en la esclerodermia, y éste suele estar limitado por la gravedad de las manifestaciones extrarrenales.55,56 Se ha calculado que la tasa de recurrencia es de alrededor del 20 porciento.56,57 Los pacientes trasplantados tienden a seguir una evolución agresiva, semejante a la de la enfermedad renal primaria aguda. La frecuencia exacta de recurrencia es difícil de valorar porque los cambios histológicos primarios inducidos por la esclerodermia (engrosamiento mucoide de la íntima de las arterias interlobulares y necrosis fibrinoide de las arteriolas glomerulares) pueden ser difíciles de distinguir del rechazo vascular agudo o crónico.
Síndromes urémico-hemolíticos
Los síndromes urémico-hemolíticos son un grupo de padecimientos poco frecuentes con manifestaciones clínicas que se traslapan y que se caracterizan por anemia hemolítica microangiopática, trombocitopenia y grados variables de insuficiencia renal. La oclusión de arterias y arteriolas por trombos de plaquetas y fibrina desempeña una función importante en las manifestaciones clínicas de estos trastornos. Existen tres tipos de síndrome urémico-hemolítico: la púrpura trombocitopénica trombótica, el síndrome urémico-hemolítico infantil y el síndrome urémico-hemolítico del adulto. Todos varían un poco en el cuadro clínico; sin embargo, tienen muchas características en común.58 Por ejemplo, los cambios histológicos en el riñón son similares en los tres síndromes. Los pacientes en la fase aguda presentan oclusión de arterias pequeñas, arteriolas y capilares glomerulares por trombos de plaquetas y fibrina [ver figura 5]. Los pacientes con una evolución más subaguda y crónica desarrollan lesiones obstructivas prominentes en las arterias interlobulares; las lesiones se caracterizan por engrosamiento concéntrico en capas de cebolla, similares a los cambios observados en la esclerodermia o la hipertensión maligna [ver figura 4].
Los síndromes urémicos-hemolíticos parecen ser trastornos aislados del consumo de plaquetas. No se conoce bien porqué ocurre consumo de plaquetas en niños y adultos con síndrome urémico-hemolítico o con púrpura trombocitopénica trombótica, pero se han sugerido tres hipótesis principales, cada una de las cuales puede explicar el trastorno en pacientes seleccionados. Una hipótesis sugiere que en algunos casos existe aumento en el nivel sanguíneo de un factor agregador de plaquetas.59,60 Se han postulado diferentes sustancias como factor agregador, y éstos pueden variar en los distintos pacientes.60-64 De acuerdo con la segunda hipótesis, es posible que exista menor nivel circulante de un inhibidor normal de la agregación plaquetaria, lo que explica tanto la formación de trombos plaquetarios como la remisión ocasional de la enfermedad después de la infusión de plasma.64 No es claro si esta alteración es causada por niveles deficientes de IgG en algunos pacientes.65 La tercera hipótesis sugiere que la activación plaquetaria puede ser una respuesta secundaria al daño endotelial. Por ejemplo, este podría explicar el desarrollo de síndrome urémico-hemolítico en algunos pacientes que reciben ciclosporina.66
Un factor pronóstico importante es el sitio principal de afección renal, en vista de que pueden predominar las lesiones glomerulares o las arteriales.67 En los niños pequeños con síndrome urémico-hemolítico, el glomérulo es el principal sitio afectado, y es común la recuperación espontánea. Por el contrario, los adultos tienen lesiones sobre todo en las arterias, aunque también puede ocurrir daño glomerular. Este tipo de síndrome se acompaña de hipertensión más grave y menor incidencia de recuperación espontánea.
El cuadro clínico de los tres tipos de síndrome urémico-hemolítico varía. La púrpura trombocitopénica trombótica afecta sobre todo a mujeres entre los 10 y 50 años de edad. Se caracteriza por cinco manifestaciones: fiebre, trombocitopenia (a menudo acompañada de púrpura), alteraciones neurológicas focales, anemia hemolítica microangiopática e insuficiencia renal.68 Suele comenzar como una enfermedad gripal, pero finalmente progresa hasta la púrpura trombocitopénica trombótica que produce lesiones purpúricas en la piel y síntomas neurológicos.
El síndrome urémico-hemolítico infantil comienza de manera característica después de un episodio de gastroenteritis viral o bacteriana. La diarrea suele ser sanguinolenta. La presencia de una verocitotoxina, similar a la toxina Shiga, secretada por Escherichia coli (sobre todo el serotipo O157:H7) o por otras bacterias, parece relacionarse con el desarrollo del síndrome urémico-hemofílico en muchos de estos pacientes.66,69,70 Por lo general la insuficiencia renal es más grave que en la púrpura trombocitopénica trombótica, y ocurre anuria hasta en el 50 porciento de los casos. Asimismo, la hipertensión arterial es común.
El síndrome urémico-hemolítico del adulto a menudo es idiopático, aunque en algunas ocasiones pueden identificarse trastornos primarios. Por ejemplo, este síndrome se ha relacionado con el embarazo, sobre todo durante el periodo posparto, y en este caso el anticoagulante lúpico puede desempeñar una función patogénica importante. También se ha asociado con el uso de anticonceptivos orales que contienen estrógenos, con los adenocarcinomas mucinosos del aparato gastrointestinal, páncreas y próstata, con la quimioterapia con mitomicina C o la combinación de bleomicina y cisplatino, y con la administración de ciclosporina. Puede ocurrir un cuadro previo de gastroenteritis similar al observado en la forma infantil del síndrome, aunque no es común.
Las características de los tres tipos de síndrome urémico-hemolítico pueden confundirse, en vista de que es posible que tengan una causa común. Por ejemplo, en un estudio realizado en cinco pacientes tratados con bleomicina y cisplatino, tres presentaron manifestaciones comunes del síndrome urémico-hemolítico (esto es, insuficiencia renal grave sin disfunción neurológica), mientras los otros dos pacientes tuvieron síntomas típicos de púrpura trombocitopénica trombótica.71
El examen del sedimento urinario en pacientes con púrpura trombocitopénica trombótica en ocasiones muestra eritrocitos y cilindros eritrocitarios. Por el contrario, el examen general de orina en niños y adultos con síndrome urémico-hemolítico suele ser normal, tal vez porque este trastorno se caracteriza por disfunción renal más grave, y porque la oliguria o la anuria pueden disminuir la excreción urinaria de eritrocitos. Sin embargo, es importante resaltar que el examen general de orina no es específico para diferenciar un tipo de síndrome urémico-hemolítico de otro.
El diagnóstico del síndrome urémico-hemolítico se sugiere con base en los hallazgos clínicos y por las condiciones generales. En la mayoría de los pacientes, no existen datos de coagulación intravascular diseminada en vista de que este trastorno se acompaña de consumo aislado de plaquetas; el tiempo de protrombina y el tiempo de tromboplastina parcial suelen ser normales. Por lo general, las concentraciones de complemento son normales, aunque se ha informado hipocomplementemia en algunos niños con síndrome urémico-hemolítico; por lo tanto, este hallazgo no excluye el diagnóstico.72
El objetivo del tratamiento en los síndromes urémico-hemolíticos consiste en revertir el consumo aislado de plaquetas que es responsable tanto de la necrosis isquémica (por la formación de trombos) como de la hemorragia (por la trombocitopenia). Puede intentarse el uso de infusiones de plasma, intercambio plasmático y otras modalidades.73-78
La eficacia del tratamiento se ha evaluado mejor en la púrpura trombocitopénica trombótica. Los resultados de estudios recientes extensos75,77 sugieren que la plasmaféresis más el intercambio plasmático con plasma fresco congelado son más eficaces que la infusión aislada de plasma. Sin embargo, durante estos estudios los pacientes tratados con intercambio plasmático recibieron alrededor de tres veces la cantidad de plasma que los pacientes tratados solo con infusión de plasma. Por lo tanto, el beneficio aparente del intercambio plasmático puede ser resultado de la infusión de más plasma fresco congelado y no de la remoción de alguna sustancia tóxica.
El intercambio de plasma se realiza diario hasta que se normaliza la cuenta plaquetaria y cesa la hemoólisis, en promedio este proceso requiere de siete a ocho intercambios.75,77 Desde el punto de vista clínico, los síntomas neurológicos tienden a abatirse primero (a los dos a tres días), después se reduce la concentración de deshidrogenasa láctica y la cuenta de plaquetas comienza a aumentar para el quinto día. Se observa estabilización o mejoría de la función renal alrededor del séptimo día.
Los pacientes con enfermedad muy leve pueden responder a dosis altas de prednisona sola (200 mg/día),77 aunque debe considerarse realizar intercambio plasmático si no existe aumento en la cuenta plaquetaria 48 horas después. Los agentes antiplaquetarios como la aspirina y el dipiridamol no son eficaces cuando se administran en forma aislada.79 Pueden tener algún beneficio cuando se agregan a la plasmaféresis.74
Son comunes las recaídas, sobre todo durante los primeros 30 a 60 días, pero en ocasiones hasta años después.77,78,80 Por lo general es eficaz repetir el recambio plasmático.80
Es necesario hacer énfasis en dos posibles complicaciones de la transfusión de plaquetas. Los agentes antiplaquetarios pueden inducir hemorragia en algunos pacientes,79 y las transfusiones de plaquetas pueden causar empeoramiento de los síntomas neurológicos o síntomas nuevos e insuficiencia renal aguda, al parecer por formación de trombos nuevos o expansión de los mismos cuando se consumen las plaquetas infundidas.82,83
Aunque la mayoría de los niños con síndrome urémico-hemolítico posdiarreico tienen una recuperación clínica completa,84,85 el seguimiento a largo plazo suele revelar evidencia de daño renal irreversible, manifestado por proteinuria, hipertensión o insuficiencia renal. Estos datos ocurren en hasta una tercera parte de los niños con enfermedad inicial prolongada, definida por anuria durante más de ocho días u oliguria por más de 15 días.84,85
También existen niños que no se recuperan en forma espontánea del episodio agudo. Los factores pronósticos malos incluyen ausencia del antecedente de gastroenteritis, no mejoría en dos semanas y enfermedad recurrente o familiar.86,87 Existen reportes anecdóticos de tratamiento exitoso en niños con enfermedad fulminante por medio de infusiones e intercambio de plasma.74,86
Complicaciones renales en el embarazo: preeclampsia e hipertensión inducida por el embarazo
CAMBIOS FISIOLOGICOS DURANTE EL EMBARAZO
De manera característica la presión arterial disminuye al inicio de la gestación, y durante el segundo trimestre del embarazo por lo general se encuentra 10 mmHg por abajo del valor basal, con cifras promedio de 105/60 mm Hg.88,89 Los factores responsable sde la caída en la resistencia vascular sistémica no se conocen del todo, pero parece que la menor respuesta a la acción presora de la angiotensina II y de la epinefrina es un factor importante.90 Al final de la gestación, estos cambios disminuyen en forma gradual y la presión arterial se normaliza.
También ocurre reducción del tono del músculo liso en los sistemas colectores renales y en los ureteros, lo que causa un cuadro radiológico que puede confundirse con obstrucción de las vías urinarias.88 La importancia de este cambio durante el embarazo normal es que es más probable que la bacteriuria asintomática provoque infección de las vías urinarias bajas o pielonefritis.88
El flujo plasmático renal y la velocidad de filtración glomerular (VFG) aumentan al principio del embarazo. El aumento en la VFG alcanza alrededor de un 40 a 50 porciento sobre los niveles basales hacia el final del primer trimestre.91 Se observa una reducción concomitante en la concentración plasmática de creatinina, por lo que una concentración de 0.8 mg/dl (que se considera normal fuera del embarazo) puede representar nefropatía subyacente. La VFG regresa a los niveles previos al embarazo durante los primeros tres meses después del alumbramiento.91
Ocurre hiponatremia leve durante el embarazo normal, y la concentración plasmática de sodio disminuye alrededor de 5 mEq/L. Este cambio en la osmolaridad plasmática es resultado de un ajuste en el osmostato hipotalámico, quizá inducido por la mayor liberación de gonadotrofina coriónica humana durante el embarazo.92,93 Los intentos para corregir la hiponatremia son tanto innecesarios (porque el cambio es leve y no causa síntomas) como ineficaces. La concentración plasmática de sodio aumenta en forma espontánea a los niveles previos al embarazo durante el primer o segundo mes posparto.94,95
Aunque existe cierta actividad excesiva de la hormona antidiurética (HAD) durante el embarazo normal, la liberación de vasopresinas por parte de la placenta causa un mayor metabolismo de la HAD.95,96 Este cambio no es importante desde el punto de vista clínico en la mayoría de las mujeres, aunque algunas desarrollan poliuria sintomática.96-98 La mayor diuresis en estos casos suele ser resistente a la administración de HAD nativa (vasopresina) por su rápido catabolismo. Sin embargo, la mayoría de las pacientes con diabetes insípida inducida por el embarazo responden a la 1-desamino-8-D-arginina vasopresina, un análogo de HAD que es menos sensible a la vasopresinasa.96 En pocos pacientes el desarrollo de poliuria se retrasa hasta el momento del parto o incluso uno a dos días después, quizá por actividad prolongada de la vasopresinasa derivada de la placenta.
También es frecuente observar durante el embarazo hiperventilación persistente que causa una reducción modesta en la presión de bióxido de carbono (PCO2). Esta alcalosis respiratoria crónica parece deberse a estimulación directa del centro respiratorio por la progesterona.99
PATOGENIA Y PATOLOGIA
Ocurre preeclampsia en el cinco al 10 porciento de todos los embarazos, aunque es más común durante el último trimestre del primer embarazo. En general, ésta se caracteriza por la aparición gradual de hipertensión, proteinuria y edema después de la vigésima semana de gestación. Puede progresar hasta la fase convulsiva llamada eclampsia. Es importante hacer énfasis en que ninguno de estos hallazgos por sí sólo es diagnótico de preeclampsia. Por ejemplo, el edema es común en el embarazo normal, en parte como resultado de la compresión de la vena cava inferior por el crecimiento uterino. Además, en pacientes con embarazo molar la preeclampsia puede observarse antes de la vigésima semana de gestación, a menudo durante el primer trimestre.
La patogenia de la preeclampsia no se conoce con claridad. La primera alteración patológica ocurre en la circulación uteroplacentaria y se caracteriza por menor dilatación de las arterias espirales.100,101 La isquemia uterina resultante, que también puede ser consecuencia de enfermedades vasculares primarias, es seguida de alteraciones en el metabolismo de las prostaglandinas, activación intravascular del sistema de la coagulación y mayor respuesta a los agentes presores [ver figura 6]. Es posible que el daño a la célula endotelial, inducido por un factor liberado por la placenta isquémica, tenga un papel importante en la génesis de estos cambios.100
Los factores inmunológicos también parecen ser importantes en la patogenia de la preeclampsia. Se ha propuesto que el desarrollo de preeclampsia puede reflejar un estado de inmunoprotección inadecuado o alterado del tejido fetoplacentario. En mujeres que utilizaban métodos anticonceptivos que evitaban el contacto con el esperma y el líquido seminal (v.gr., preservativo y diafragma) se observó un aumento en la incidencia de preeclampsia 2.4 veces mayor que en las mujeres que usaban otro tipo de anticonceptivos (anticonceptivos orales).102 Este hallazgo sugiere que el contacto con los antígenos paternos en el semen antes del embarazo puede hacer posible el desarrollo de anticuerpos que permitan bloquear una respuesta inmune subsecuente dirigida contra el tejido del feto o la placenta. En apoyo de esta hipótesis está el hecho de que la preeclampsia es más común en los primeros embarazos y en mujeres que tienen un embarazo posterior, pero con diferente pareja.103
Los principales cambios patológicos en el riñón ocurren en los glomérulos. Al examen con microscopio de luz se observa edema de las células endoteliales con el consencuente estrechamiento u obliteración de la luz de los capilares glomerulares. En la mayoría de los pacientes con preeclampsia las alteraciones renales patológicas comienzan a desaparecer poco después del parto, y de manera característica la histología renal se normaliza al cabo de dos a tres semanas.104 Sin embargo, puede no haber mejoría rápida si existen lesiones renales más graves y depósitos extensos de plaquetas y fibrina.
MANIFESTACIONES CLINICAS Y DIAGNOSTICO
El cuadro clínico de la preeclampsia es variable. La mayoría de las veces la presión arterial comienza a aumentar en forma gradual después de la vigésima semana de gestación. Clasicamente la presión arterial es superior a 140/90 mm Hg en el tercer trimestre, sobre todo durante el último mes del embarazo. En la mayoría de las pacientes con preeclampsia, además de la hipertensión arterial, existe edema y proteinuria. La excreción urinaria de proteínas aumenta en forma progresiva, a menudo alcanzando un nivel en rango nefrótico de más de 3.5 g/día, aunque el sedimento urinario suele ser casi normal, ya que contiene algunos eritrocitos o leucocitos. A pesar de todo, es raro que exista reducción importante de la función renal; la concentración de creatinina plasmática aumenta solo en 0.2 a 0.3 mg/dl, a menos que ocurra coagulación intravenosa diseminada con necrosis cortical bilateral u otro trastorno (como el síndrome de HELLP, ver adelante).
En pacientes con una enfermedad más grave pueden observarse otras manifestaciones clínicas. Los signos y síntomas comunes incluyen cefalea, alteraciones visuales e hiperactividad de los reflejos osteotendinosos. El dolor epigástrico o subcostal derecho también es común, y es ocasionado por distensión de la cápsula hepática por edema o hemorragia del hígado. En algunas pacientes puede observarse el desarrollo del síndrome de HELLP (hemólisis con un frotis microangiocítico, enzimas hepáticas elevadas y una cuenta de plaquetas bajas). Estos hallazgos pueden indicar isquemia hepática producida por obstrucción vascular. La aparición de convulsiones se considera criterio diagnóstico de eclampsia.
El diagnóstico de preeclampsia se sospecha por el desarrollo de hipertensión y proteinuria en mujeres primigrávidas, sobre todo en el tercer trimestre del embarazo. La presencia de estos signos al inicio de la gestación o el desarrollo de uno u otro en mujeres multigrávidas sugiere un trastorno primario como hipertensión arterial esencial, nefropatía primaria, diabetes mellitus o mola hidatiforme.105 En un grupo numeroso de pacientes, el diagnóstico clínico de preeclampsia pudo confirmarse por biopsia renal posparto en el 85 porciento de las mujeres primigrávidas, pero solo en el 38 porciento de las multigrávidas, la mayoría de las cuales tenían nefrosclerosis benigna u otra nefropatía primaria.106
Diferencias entre la preeclampsia y otras causas de hipertensión arterial del embarazo
Se considera que existe hipertensión arterial durante el embarazo si la presión arterial es mayor de 140/90 mm Hg. Es importante reconocer que una presión arterial de 120/80 mm Hg, que sería considerada normal en mujeres no embarazadas, puede ser alta durante el embarazo. Por lo tanto, todas las pacientes deben evaluarse en forma individual y los valores de la presión arterial compararse con las mediciones registradas antes y durante las primeras fases del embarazo.
En las mujeres embarazadas deben considerarse tres causas importantes de hipertensión arterial. En primer lugar, la hipertensión del embarazo puede ser ocasionada por preeclampsia. En segundo lugar, el aumento de la presión arterial durante la gestación puede indicar una tendencia hipertensiva subyacente. Por último, en algunas mujeres, la hipertensión arterial gestacional tardía o transitoria es una complicación aislada, que desaparece después del parto.
Cuando no existe el antecedente de hipertensión arterial, puede resultar difícil diferenciar la preeclampsia de la hipertensión esencial crónica por la disminución característica de la presión arterial en el segundo trimestre de la gestación. Por consiguiente, las pacientes con hipertensión crónica previa pero no diagnosticada pueden tener una presión arterial que se encuentre en el rango normal cuando son atendidas por primera vez por el médico. A medida que la presión aumenta durante el tercer trimestre, pueden ser útiles los siguientes datos para determinar el diagnóstico: edad de la paciente, número de embarazo, grado de excreción urinaria de proteínas y calcio, y concentración de ácido úrico en plasma [ver tabla 3].
En ocasiones se observa aparición de hipertensión leve aislada durante el tercer trimestre del embarazo sin la presencia de proteinuria coexistente. Este hallazgo parece tener pocos efectos adversos en la madre y el feto, aunque en algunas pacientes puede ser un indicador del desarrollo futuro de hipertensión esencial.
Diferencia entre la preeclampsia y la nefropatía primaria
La preeclampsia es el tipo más común de nefropatía que ocurre en las últimas etapas del embarazo. Por lo general, puede diferenciarse con facilidad de otras causas de nefropatía durante el embarazo porque estos trastornos a menudo se acompañan de una reducción moderada o grave en la velocidad de filtración glomerular y de proteinuria leve o ausente.
La insuficiencia renal aguda puede presentarse en cualquier etapa del embarazo como resultado de diversos trastornos [ver tabla 4], pero rara vez es secundaria a preeclampsia. Las principales causas de insuficiencia renal aguda en la primera mitad del embarazo incluyen la necrosis tubular aguda ocasionada por un aborto séptico y el daño prerrenal por hiperemesis gravídica. Al final del embarazo o durante el periodo posparto, la insuficiencia renal aguda puede ser producida por preeclampsia o por algunos de los síndromes urémico-hemofílicos [ver antes, Síndromes urémico-hemolíticos]; en estos casos varias características distinguen la preeclampsia del síndrome urémico-hemofílico posparto [ver tabla 5]. La insuficiencia renal aguda también puede ser causada por necrosis tubular aguda o necrosis cortical renal en pacientes con desprendimiento de placenta o placenta previa. El hígado graso agudo del embarazo es una complicación poco común, pero se acompaña de insuficiencia renal aguda hasta en el 60 porciento de las pacientes y disminuye de manera característica la función renal por el desarrollo de necrosis tubular aguda o por la reducción de la perfusión renal.107
La obstrucción del aparato urinario es otra causa posible de insuficiencia renal durante las últimas etapas del embarazo. La relajación del músculo liso ureteral, mediada en parte por los defectos de las prostaglandinas y por la presión ejercida sobre los ureteros por el útero, produce cierta dilatación del sistema caliceal durante la gestación normal. Esta hidronefrosis considerada funcional puede detectarse por ultrasonografía renal, y no es diagnóstica de obstrucción de las vías urinarias como causa de la insuficiencia renal en este caso. Sin embargo, rara vez el útero puede ocasionar suficiente obstrucción como para producir disminución de la velocidad de filtración glomerular.108 La normalización de la función renal con el decúbito lateral y su alteración con el decúbito supino debe sugerir la posibilidad de este diagnóstico.
TRATAMIENTO
La preeclampsia no tratada se asocia con una incidencia alta de muertes fetales y neonatales. En pacientes que progresan hasta la preeclampsia grave o la eclampsia, la mortalidad materna se incrementa, sobre todo por la hemorragia cerebral. El tratamiento definitivo requiere la extracción del feto y la placenta. La única razón para retrasar el nacimiento en pacientes con diagnóstico de preeclampsia es la evidencia de inmadurez fetal. En este caso, puede utilizarse reposo en cama en decúbito lateral y administración de fármacos antihipertensivos (si la presión arterial diastólica es mayor de 95 a 100 mm Hg) hasta que la interrumpción del embarazo se lleve a cabo con seguridad, siempre que no existan otras alteraciones que pongan en peligro la vida de la madre, como hipertensión de difícil control, trastornos visuales, convulsiones, síndrome de HELLP o coagulación intravascular deseminada.
La hipertensión leve aislada que aparece durante el tercer trimestre de la gestación no suele requerir la interrupción inmediata del embarazo. Aunque se desconoce el grado mínimo de elevación de la presión arterial que puede lesionar al feto, es raro que ocurran efectos adversos por la hipertensión leve en pacientes que no desarrollan preeclampsia.109
Los datos sobre el tratamiento de la hipertensión crónica durante el embarazo son incompletos. La disminución notable de la presión arterial materna producida por el tratamiento puede afectar el flujo sanguíneo uteroplacentario, comprometiendo de este modo la viabilidad del feto. Por el contrario, cuando la presión arterial diastólica de la paciente es mayor de 84 mm Hg, la mortalidad perinatal aumenta.110 A pesar de estos problemas, es conveniente comenzar el tratamiento en mujeres con hipertensión crónica si la presión arterial diastólica es mayor de 90 a 95 mm Hg.111 Asimismo, la presión arterial diastólica por arriba de 100 a 110 mm Hg, ya sea aguda o crónica, debe tratarse para reducir el riesgo de hemorragia intracerebral materna. Es importante hacer énfasis en que estas recomendaciones son una guía y que el grado de presión arterial en el que se debe iniciar la administración de medicamentos no se conoce con claridad. Por otra parte, las pacientes que reciben tratamiento antihipertensivo antes del embarazo deben continuarlo durante toda la gestación. Como regla general, independientemente del esquema terapéutico utilizado antes del embarazo, este deberá continuarse, con excepción de los fármacos en que se ha demostrado un efecto adverso en la viabilidad del feto (ver adelante).
Una vez que se ha tomado la decisión de iniciar el tratamiento farmacológico, deben administrarse sólo los medicamentos antihipertensivos que se consideran seguros en el embarazo, como metildopa, betabloqueadores e hidralacina. Los diuréticos por lo general deben envitarse, en vista de que la preeclampsia tiende a acompañarse de un volumen plasmático bajo, por lo que una mayor disminución del volumen plasmático pudiera exacerbar la isquemia placentaria. Se han usado con éxito bloqueadores de los canales del calcio en hipertensión inducida por el embarazo, pero su toxicidad potencial para el feto se ha estudiado durante un tiempo más breve que la de otros agentes.112,113
Por el contrario, datos obtenidos en animales y humanos sugieren que la administración de inhibidores de la ECA o nitroprusiato se relaciona con mayor riesgo para el feto. Al parecer aumenta la incidencia de parto prematuro, bajo peso al nacer y muerte fetal en las pacientes tratadas con estos medicamentos durante el embarazo.114,115 Se ha descrito una fetopatía caracterizada por hipotensión, hipoplasia pulmonar, displasia tubular renal e hipocalvaria asociada al uso de inhibidores de la ECA.116 En algunos fetos ha ocurrido reducción importante en la VFG, causando oliguria, oligohidramnios y, por la menor cantidad de líquido amniótico, alteraciones traumáticas del desarrollo.117,118 Como resultado de estos hallazgos, está contraindicada la administración de inhibidores de la ECA durante el embarazo.114 Aunque el riesgo fetal por los inhibidores de la ECA parece ser mayor durante el segundo y tercer trimestres, estos agentes deben suspenderse en cuanto se sospeche o confirme un embarazo.
El nitroprusiato se ha asociado con toxicidad por cianuro tanto en la paciente como en el producto. No debe usarse durante el embarazo a menos que las otras alternativas sean ineficaces.
El tratamiento ideal de la preeclampsia consiste en prevenirla. Las alteraciones frecuentes en el metabolismo de los prostanoides y en la función plaquetaria han provocado la realización de estudios aleatorios que evalúan los efectos de la aspirina en dosis bajas (60 a 150 mg/día) en mujeres con mayor riesgo de preeclampsia.119-124 En los estudios iniciales, el riesgo relativo de hipertensión inducida por el embarazo se redujo en las pacientes tratadas con aspirina, lo mismo que la incidencia de productos de muy bajo peso al nacer y la necesidad de cesáreas. En contraste con estos estudios, el estudio reciente cooperativo sobre aspirina en dosis bajas durante el embarazo (CLASP, por sus siglas en inglés, n. del t.), que estudió a más de 9,000 mujeres con riesgo de preeclampsia, no observó este beneficio.125 Es posible que la diferencia sea resultado del menor riesgo de preeclampsia en las pacientes del CLASP (seis a ocho porciento) en comparación con las de otros estudios (17 a 52 porciento). Aunque las discrepancias entre estos estudios no permiten hacer recomendaciones absolutas, al parecer la aspirina en dosis bajas es relativamente segura tanto para la paciente como para el producto.
Además de la aspirina en dosis bajas, se han usado los suplementos de calcio (1.5 a 2.0 g de CaCO3/día) en un intento por prevenir las complicaciones hipertensivas durante el embarazo. La justificación de este tratamiento se basa en parte en la acción hipotensora potencial del calcio. Un estudio controlado demostró reducción del riesgo tanto de hipertensión inducida por el embarazo como de preeclampsia en el grupo tratado.126 Como en el caso de la aspirina, es necesario definir el papel general de los suplementos de calcio.
PRONOSTICO
La preeclampsia en primigrávidas suele ser una enfermedad autolimitada. La incidencia de hipertensión crónica o nefropatía en estas pacientes no es mayor que en las mujeres embarazadas de edad similar del grupo control.127 Además, el riesgo de desarrollar preeclampsia en los siguientes embarazos es bajo. Sin embargo, es posible que estas observaciones generales no se apliquen a primigrávidas con preeclampsia grave o eclampsia o a multigrávidas preeclámpticas; estas mujeres parecen tener un riesgo elevado de hipertensión y preeclampsia recurrente en embarazos posteriores. La hipertensión crónica también es más probable que ocurra en multigrávidas con preeclampsia, que a menudo tienen hipertensión esencial o nefropatía.
La hipertensión leve aislada del embarazo, en la que la presión arterial diastólica es menor de 90 a 95 mm Hg y no existe proteinuria o insuficiencia renal, no tiene efectos adversos sobre el feto o la madre. Sin embargo, es necesaria la vigilancia periódica en vista de que el riesgo de hipertensión de inicio tardío parece estar aumentado, sobre todo en pacientes en las que la presión arterial no se ha normalizado al décimo día del posparto.111
Enfermedad tromboembólica de las arterias renales
La formación de trombos o la embolización pueden afectar la circulación venosa o arterial del riñón. Los principales trastornos arteriales incluyen la trombosis, los émbolos de coágulos sanguíneos y la ateroembolia. La principal afección venosa es la trombosis de la vena renal, que ocurre con mayor frecuencia en pacientes con síndrome nefrótico.
La tromboembolia arterial o el aneurisma disecante de la aorta pueden causar infarto o atrofia isquémica de los riñones, dependiendo del grado de oclusión vascular, la rapidez con la que se desarrolla la oclusión y si las lesiones vasculares son resultado de coágulos o aterombolia. La circulación renal es muy susceptible a las enfermedades embólicas en vista de que los riñones en condiciones normales reciben el 20 porciento del gasto cardiaco.
OCLUSION AGUDA NO ATEROMATOSA DE LAS ARTERIAS RENALES
Diversos padecimientos se relacionan con el desarrollo de émbolos o trombosis, que pueden producir oclusión no ateromatosa de las arterias renales. Las principales fuentes de émbolos renales son los trombos murales, que son comunes sobre todo en pacientes con arritmias auriculares o infarto del miocardio previo, y con vegetaciones relacionadas con la endocarditis bacteriana. Los émbolos tumorales o los émbolos de grasa se observan con menos frecuencia. La trombosis de la arteria renal suele agregarse a lesiones ateromatosas estenóticas primarias o aparecer después de desgarros traumáticos de la íntima. En raras ocasiones la trombosis se presenta en forma espontánea.
El infarto en forma de cuña que se extiende hacia afuera del vaso afectado es el hallazgo patológico clásico que se observa en el riñón. La magnitud de la lesión renal depende del grado y la duración de la oclusión vascular. Por lo general se presenta necrosis irreversible si la oclusión total de la arteria renal dura dos horas o más.128 Sin embargo, la oclusión total de una duración menor de dos horas puede ocasionar necrosis tubular aguda, más que infarto.
Las manifestaciones clínicas de la oclusión de la arteria renal son variables. Los pacientes pueden estar asintomáticos si la oclusión es incompleta; sin embargo, cuando existen síntomas los más comunes son náusea, vómito, dolor en el flanco y fiebre. A la exploración física puede haber dolor en el flanco o abdominal a la palpación; durante la exploración debe llevarse a cabo una búsqueda cuidadosa de signos de embolización extrarrenal, como lesiones cutáneas o deficiencias neurológicas focales. Además, a menudo pueden detectarse factores subyacentes que predisponen a la embolización renal, como la fibrilación auricular o los infartos del miocardio recientes. En muchos pacientes la presión arterial aumenta poco tiempo después del infarto agudo como resultado de la estimulación del sistema renina-angiotensina por la isquemia renal. La presión arterial suele regresar al nivel basal después de dos a tres semanas.129
La mayor parte de las pruebas de laboratorio de rutina no son diagnósticas. La hematuria macroscópica o microscópica se observa solo en cerca de un tercio de los pacientes; lo frecuente de su ausencia puede reflejar la disminución notable en la irrigación sanguínea de la zona infartada, que ocasiona la interrupción de la filtración glomerular y el flujo de orina.130 Sin embargo, en condiciones clínicas apropiadas existe un hallazgo de laboratorio muy sugestivo de infarto renal: la elevación notable (a menudo mayor de cinco veces el límite superior normal) de la concentración de deshidrogenasa láctica (DHL) y, en ocasiones, un aumento leve de la concentración de aminotransferasa sérica.131 El renograma con radioisótopos es el procedimiento de elección para demostrar la disminución segmentaria o generalizada de la perfusión renal. Además, es un método diagnóstico no cruento que puede evitar la realización de la arteriografía renal o la tomografía computada con material de contraste. Por lo general, la pielografía intravenosa es menos segura y puede ser normal en pacientes con lesiones segmentarias.
Hasta ahora no se ha determinado el tratamiento óptimo para el infarto renal causado por un émbolo, pero por lo general se prefiere el tratamiento médico. Aunque la cirugía puede restaurar la permeabilidad vascular, se relaciona con una mayor mortalidad y no mejor recuperación de la función de la que se observa sólo con el tratamiento anticoagulante.130,132 Este último tratamiento puede causar mejoría importante incluso en pacientes con embolia bilateral e insuficiencia renal avanzada. La cirugía temprana sigue siendo el tratamiento de elección en pacientes con trombosis traumática de la arteria renal, en especial si la cirugía se realiza durante las primeras 24 horas.133
Es conveniente que el tratamiento sea inmediato para conservar el máximo de parénquima renal funcional. Por desgracia, el diagnóstico suele ser tardío por que no se reconocen los datos característicos descritos antes. Por ejemplo, en un estudio de 17 pacientes, el tiempo de diagnóstico fue de tres a seis días, con solo cinco casos diagnosticados el primer día.130
El tratamiento anticoagulante más común consiste en la administración de heparina intravenosa seguida de warfarina. Una alternativa tal vez más eficaz es iniciar el tratamiento con un fármaco trombolítico, como la estreptocinasa, con objeto de disolver el coágulo oclusivo.134,135 La infusión local intrarterial, en lugar de la administración intravenosa, puede disminuir la dosis total necesaria y, por lo tanto, reducir el riesgo de hemorragia sistémica.135
La cirugía, que no suele indicarse como tratamiento inicial para este trastorno, puede considerarse en pacientes con insuficiencia renal grave que no muestran mejoría en la función renal después de cuatro a seis semanas de tratamiento anticoagulante. La embolectomía tardía ha mejorado la función renal en algunos pacientes, indicando que es posible revertir en parte la atrofia isquémica.136
A pesar de la eficacia del tratamiento médico, la mortalidad temprana y tardía por los coágulos embólicos sigue siendo alta tanto por la embolización extrarrenal (en especial hacia el cerebro y el intestino) como por la enfermedad subyacente (v.gr., ateroesclerosis).130
TROMBOEMBOLIA RENAL
La enfermedad ateroembólica renal ocurre en forma característica en pacientes con ateroesclerosis ulcerada y generalizada. La mayoría de las veces la ateroembolia clínicamente importante es resultado de la manipulación de la aorta o de otras arterias de gran calibre durante la arteriografía, la angioplastía o la cirugía, incluyendo el trasplante de órganos.137-139 La ateroembolia también puede ser producida por el tratamiento crónico con warfarina, en vista de que la anticoagulación puede interferir con la cicatrización de las placas ateromatosas ulceradas, o por el tratamiento trombolítico intravenoso o intrarterial, ya que la superficie de la placa puede denudarse por la disolución de la fibrina.140 La ateroembolia espontánea, que en estudios de autopsia se demuestra hasta en el 12 porciento de los pacientes mayores de 80 años, suele ser asintomática y no se relaciona con una disminución considerable de la función renal.
El examen del tejido renal por microscopía de luz a menudo es patognomónico de ateroembolia. Las muestras características contienen fragmentos de material acelular y hendiduras bicóncavas en las arterias renales de pequeño y mediano calibre [ver figura 7]. Las hendiduras bicóncavas representan cristales de colesterol que han sido disueltos durante la fijación con parafina. Los émbolos tienen una forma irregular y no se distienden; como resultado, a menudo producen oclusión incompleta y atrofia isquémica secundaria en lugar de infarto. Con el tiempo suele desarrollarse una reacción a cuerpo extraño, que ocasiona proliferación de la íntima y mayor estrechamiento de la luz del vaso. Es frecuente que existan émbolos similares en otros órganos.
Es importante resaltar que la localización de los émbolos dentro del riñón es regional. Por consiguiente, debido a errores en la selección del sitio adecuado para la biopsia renal percutánea, es posible que no se tomen muestras de lesiones renales características, incluso en pacientes en los que se comprueba en el estudio de autopsia la presencia de ateroémbolos en otros órganos o en el riñón.
La enfermedad vascular erosiva grave que ocasiona la formación de ateroémbolos se observa con mayor frecuencia en pacientes mayores de 50 años de edad. En vista de que la oclusión vascular es incompleta, los ateroémbolos no suelen producir infarto renal ni sus manifestaciones clínicas o hallazgos de laboratorio característicos, incluyendo dolor en flanco y elevación de la concentración de DHL sérica. Por lo tanto, la mayoría de los pacientes no tienen síntomas relacionados con los riñones.
Los antecedentes clínicos y la exploración física pueden mostrar signos y síntomas característicos de ateroembolia extrarrenal. Puede haber deficiencias visuales, dolor abdominal causado por pancreatitis aguda o infarto esplénico, y mialgias. Otras alteraciones características de embolia periférica que pueden observarse incluyen placas color naranja en las arteriolas de la retina, lívido reticular en las extremidades inferiores y zonas de gangrena en los dedos de los pies en presencia de pulsos pedios normales.141 La hipertensión mediada por los efectos de la angiotensina II también es frecuente y puede ser grave.
Los hallazgos de laboratorio son inespecíficos e incluyen el aumento súbito en la concentración de creatinina plasmática poco tiempo después de la realización de procedimientos quirúrgicos o radiológicos. El examen general de orina suele ser normal o casi normal, observándose algunas células o cilindros, que son compatibles con la presencia de atrofia isquémica. Sin embargo, algunos pacientes tienen sedimento urinario activo caracterizado por hematuria y cilindros celulares, incluyendo cilindros eritrocitarios. Es frecuente la presencia de eosinofilia en sangre periférica y en algunos casos puede haber hipocomplementemia; es probable que estos dos hallazgos reflejen activación del sistema inmune por la exposición de la superficie ateroembólica. Es importante distinguir la enfermedad ateroembólica de otros padecimientos que se acompañan de eosinofilia o hipocomplementemia, como glomerulonefritis, vasculitis sistémica y nefritis intersticial aguda.142,143
El desarrollo de insuficiencia después de la realización de cirugía vascular, arteriografía o angioplastía puede sugerir el diagnóstico de ateroembolia renal, aunque en este caso es más probable el diagnóstico de necrosis tubular aguda. Sin embargo, la presencia de embolia extrarrenal favorece el diagnóstico de enfermedad ateroembólica. La ateroembolia renal y la necrosis tubular aguda pueden diferenciarse con mayor precisión por su evolución típica. Los pacientes con necrosis tubular aguda presentan de manera característica un aumento progresivo en la concentración de creatinina plasmática antes de que la función renal se estabilice, pero al final regresa en forma espontánea al nivel basal en un periodo variable de cuatro a 21 días. Por el contrario, los pacientes con ateroembolia a menudo tienen una disminución aguda en la función renal que, dependiendo del grado de embolización, es autolimitada; sin embargo, en vista de que no es posible la disolución de los ateroémbolos, la insuficiencia renal por lo general es irreversible. En comparación con la insuficiencia renal aguda inducida por medio de contraste, el deterioro en la función renal que ocurre con la ateroembolia suele comenzar varios días después del evento desencadenante (v.gr., angiografía coronaria).138 En algunas ocasiones los pacientes pueden tener cierta mejoría en la función renal con el tiempo, tal vez por la resolución de la necrosis tubular aguda coexistente o por el desarrollo de flujo colateral hacia las zonas isquémicas.141,144
El diagnóstico puede ser difícil, sobre todo cuando no es posible identificar factores precipitantes, como ocurre en la ateroembolia espontánea, o cuando la evolución de la enfermedad es atípica. Por ejemplo, puede presentarse insuficiencia renal progresiva subaguda si los pacientes tienen cicatrización fibrótica en respuesta a la ateroembolia o si ocurren episodios nuevos de embolización. En este caso puede ser necesaria la biopsia de un órgano afectado como la piel o el riñón, sobre todo si las manifestaciones extrarrenales, un sedimento urinario activo, hipocomplementemia o eosinofilia, indican un trastorno sistémico. Sin embargo, el resultado negativo de la biopsia renal no excluye necesariamente el diagnóstico de ateroembolia, en vista de que es posible que en la muestra no existan vasos afectados.
Hasta ahora no existe tratamiento específico para la ateroembolia renal. En pacientes con alto riesgo deben evitarse procedimientos como la arteriografía, si existen técnicas menos cruentas que puedan proporcionar la misma información. Además, el tratamiento anticoagulante no está indicado en vista de que puede agravar el trastorno al favorecer la ateroembolia recurrente.142
Necrosis de la corteza renal
La necrosis de la corteza renal es un estado clinicopatológico que se caracteriza por insuficiencia renal aguda, por lo general anúrica, y grados variables de necrosis o infarto de los componentes corticales, túbulos y glomérulos. Este trastorno se relaciona con mayor frecuencia con el desprendimiento de placenta o alguna otra complicación al final del embarazo, como la placenta previa. También puede ocurrir en presencia de choque séptico u otros padecimientos que ocasionan hipovolemia, como las quemaduras o la pancreatitis hemorrágica aguda.145
La principal alteración patológica en esta enfermedad es la necrosis de todos los componentes de la corteza renal. También puede observarse necrosis de los vasos sanguíneos y trombos dispersos en las arterias interlobulares, arteriolas y glomérulos. En casos avanzados la corteza puede estar destruida por completo y las arterias presentar necrosis y trombosis extensas. Sin embargo, incluso en estos casos suele mantenerse intacto el tejido renal en las zonas justo por abajo de la cápsula, en la región yuxtamedular y en la médula.146 Las nefronas por abajo de la cápsula no son funcionales, ya que sus segmentos intratorácicos están necrosados. En experimentos realizados en animales, las nefronas corticales profundas se mantienen intactas, tal vez porque las regiones yuxtamedulares son perfundidas por las arterias arcuatas, más que por los vasos interlobulares trombosados. Se desconoce si existen hallazgos similares en humanos que puedan explicar la recuparación parcial de la función renal en algunos pacientes.147
La patogenia de la necrosis cortical renal no se conoce por completo. Sin embargo, la coagulación intravascular diseminada y la isquemia renal grave se consideran importantes en ella. En los casos de necrosis cortical se encuentran trombos de fibrina en los vasos renales. Este hallazgo es similar al cuadro histológico de la reacción generalizada de Shwartzman, en la que se administran endotoxinas en forma periódica a animales, ocasionando trombosis generalizada y hemorragia que a su vez produce necrosis de la corteza renal. Esta reación ocurre con más facilidad en animales preñados en comparación con grupos control de animales no preñados.148 Aunque la mayor parte de las evidencias indican que la isquemia renal grave es el principal mecanismo patogénico en la mayoría de los pacientes que desarrollan necrosis cortical, en algunos casos la coagulopatía producida por la liberación de tromboplastina a la circulación produce depósito de fibrina intravascular y una mayor reducción de la perfusión renal.
La necrosis de la corteza renal se caracteriza por la aparición súbita de oliguria o anuria. También es frecuente que exista hematuria macroscópica, dolor en el flanco e hipertensión arterial. En más del 50 porciento de los casos este tratorno es causado por el desprendimiento de la placenta u otra complicación del tercer trimestre del embarazo, como la placenta previa.149
Los hallazgos de laboratorio en la necrosis cortical son inespecíficos. El BUN y la concentración de creatinina plasmática aumentan diariamente, lo cual es reflejo de la disminución aguda de la velocidad de filtración glomerular. Además de la hematuria y la proteinuria leve, el examen general de orina puede mostrar leucocitos, células del epitelio tubular y cilindros granulares. Estos hallazgos son similares a los que se observan en la necrosis tubular aguda. Pueden existir alteraciones hematológicas relacionadas con la coagulación intravascular deseminada, sobre todo en pacientes con complicaciones obstétricas primarias.
El diagnóstico diferencial de la necrosis incluye la necrosis tubular aguda, otras causas de insuficiencia renal aguda al final del embarazo [ver tabla 4] y algunas otras causas de anuria. Varias manifestaciones clínicas pueden ayudar a diferenciar la necrosis cortical de la necrosis tubular aguda. En pacientes con necrosis de la corteza renal son comunes el dolor en el flanco, la hematuria macroscópica y la anuria, pero son raros en los casos de necrosis tubular aguda. Asimismo, en la necrosis tubular aguda la función renal comienza a mejorar en un periodo de cuatro a 21 días y con el tiempo la recuperación es casi completa en la mayoría de los pacientes. Por el contrario, la anuria o la oliguria prolongada es una manifestación característica de la necrosis de la corteza renal, además de la falta de mejoría en la función renal o su recuperación parcial y tardía.150
Aunque la insuficiencia renal aguda tiene muchas causas, solo algunas se acompañan de anuria. Los padecimientos que deben considerarse en presencia de insuficiencia renal aguda y anuria, además de la necrosis cortical, se incluyen el estado de choque, que puede causar hipoperfusión importante de los riñones y necrosis tubular aguda agreda, la obstrucción completa del aparato urinario, que por lo general puede excluirse mediante la ultrasonografía renal, alguno de los síndromes urémico-hemolíticos [ver antes, Síndromes urémico-hemolíticos], y en raras ocasiones, la glomerulonefritis grave, la vasculitis o la oclusión vascular bilateral completa. Las condiciones típicas en las que ocurre la necrosis de la corteza renal a menudo permiten distinguirla de estos trastornos. Sin embargo, cuando el diagnóstico es dudoso pueden estar indicados los estudios radiográficos o la biopsia renal. La observación de calcificaciones renales en la radiografía simple de abdomen sugiere la presencia de necrosis de la corteza renal, aunque es frecuente que no se visualicen depósitos de calcio durante un lapso de uno a dos meses, limitando de esta forma su utilidad como criterio diagnóstico.151
Hasta ahora no se ha demostrado que algún tratamiento específico afecte la evolución de la necrosis cortical. En cerca de una tercera parte de los pacientes la diuresis aumenta y la función renal mejora en un periodo de dos a cuatro meses, establizándose a un nivel del 15 al 50 porciento de lo normal.150 Esta recuperación parcial, que puede ser suficiente para suspender la diálisis, tal vez ocurre, al menos en parte, gracias a que las nefronas yuxtamedulares no suelen presentar necrosis isquémica irreversible. Sin embargo, la fase de recuperación puede ser seguida por el deterioro gradual de la función renal durante un periodo de algunos años; esta alteración puede reflejar daño hemodinámico que afecta a los glomérulos supervivientes.152
Nefropatía por células falciformes
La afección renal es común en los pacientes cuyos eritrocitos contienen hemoglobina S, o hemoglobina falciforme. Este tipo de hemoglobina es responsable de la anemia de células falciformes, el rasgo falciforme y otras hemoglobinopatías. La principal manifestación renal en individuos con el rasgo falciforme es la hematuria recurrente; sin embargo, en personas con anemia de células falciformes se presentan de manera característica diversas alteraciones patológicas y funcionales [ver tabla 6]. Por consiguiente, la nefropatía por células falciformes siempre debe considerarse en el diagnóstico diferencial en los pacientes de raza negra que presentan hematuria y necrosis papilar.
La médula es la porción del riñón más afectada en la nefropatía por células falciformes. Existen zonas localizadas de hemorragia o necrosis que finalmente producen inflamación intersticial, atrofia tubular y fibrosis. También son comunes las lesiones glomerulares (ver adelante).
El mecanismo patogénico primario en esta nefropatía parece ser la formación de células falciformes a partir de eritrocitos en los capilares medulares. La osmolaridad elevada y la presión arterial de oxígeno baja presentes en esta región contribuyen a la transformación de los eritrocitos en células falciformes. Como resultado de estas alteraciones medulares, puede haber pérdida de la función, lo que ocasiona anomalías tubulares similares a las observadas en los animales después de la extirpación quirúrgica de las papilas. Estas alteraciones tubulares incluyen la disminución en la capacidad de concentración, el deterioro en la acidificación urinaria y la reducción en la secreción de potasio (ver adelante).
Las manifestaciones clínicas de la nefropatía por células falciformes varían desde alteraciones subclínicas en la excreción de agua y potasio hasta hematuria macroscópica o síndrome nefrótico. El diagnóstico de cualquiera de estas alteraciones se basa en la confirmación de la presencia de hemoglobina S mediante estudios apropiados, como la electroforesis de hemoglobina.
HEMATURIA
La hematuria macroscópica indolora es una de las manifestaciones renales más comunes en pacientes con el rasgo falciforme.153 No se conoce con claridad porqué la hematuria es más común en individuos con el rasgo falciforme que en los pacientes con enfermedad de células falciformes, pero tal vez se relaciona con la prevalencia mucho mayor del rasgo falciforme.
La hematuria puede aparecer en forma espontánea o presentarse después de un traumatismo leve. Por lo general es recurrente y a menudo proviene de un solo riñón; puede ser microscópica o macroscópica. En la mayoría de los casos la hematuria es leve y autolimitada, persistiendo varios días o semanas, pero sin requerir tratamiento específico. En algunos individuos, la hemorragia puede ser más profusa o prolongada, por lo que requiere tratamiento.154
INFARTO RENAL Y NECROSIS PAPILAR
La necrosis papilar es resultado de la trombosis o la precipitación de sedimento en los vasos rectos y constituye una de las manifestaciones más comunes de la anemia de células falciformes [ver figura 9]. La mayoría de los pacientes con esta complicación están asintomáticos y su función renal es normal. Sin embargo, la formación de cicatrices puede predisponer al desarrollo de infecciones del aparato urinario.
Los pacientes con enfermedad de de células falciformes o rasgo falciforme pueden presentar infartos renales. En estos casos se ha informado la presencia de grandes hematomas perirrenales. Es posible que los hematomas se desarrollen debido a que las arterias renales principales permanecen permeables y continúan irrigando las zonas infartadas.155
CAPACIDAD DE CONCENTRACION DISMINUIDA
La disminución en la capacidad para concentrar la orina es un hallazgo general y temprano en pacientes con enfermedad de células falciformes. La formación de células falciformes en los capilares de los vasos interfiere con el intercambio de contracorriente, causando atrapamiento ineficaz de solutos en la médula interna, que es el sitio donde se genera la máxima osmolaridad urinaria en presencia de hormona antidiurética. Por consiguiente, la osmolaridad urinaria máxima en niños de 10 años de edad con anemia de células falciformes es solo de 400 a 450 mOsm/kg, que es mucho menor que el valor normal de 900 a 1,400 mOsm/kg.156 Este defecto en la capacidad de concentración es reversible, después de múltiples transfusiones, en pacientes homocigotos para el gen falciforme y menores de 10 ó 15 años de edad, pero es irreversible en individuos de mayor edad. Estas alteraciones son mucho menos graves y aparecen a una edad mayor en individuos con el rasgo falciforme o con la enfermedad de células falciformes-hemoglobina C.
El principal síntoma producido por la alteración en la capacidad para concentrar la orina es la poliuria. Esta manifestación puede ser molesta, sobre todo en la noche por la nicturia intermitente. Sin embargo, puede mantenerse un balance de líquidos normal mientras la sed sea normal y el aporte de agua suficiente.
SECRECION RENAL BAJA DE HIDROGENO Y POTASIO
La lesión del tubo colector cortical, que es el sitio de secreción de potasio y acidificación urinaria máxima, puede producir hipercalemia y acidosis metabólica.157 En vista de que la concentración de aldosterona es normal en la mayoría de los pacientes con anemia de células falciformes, la lesion isquémica de las células es el mecanismo más probable que ocasiona estas alteraciones. Por el contrario, en pacientes con el rango falciforme el control del potasio no está afectado.158
INSUFICIENCIA RENAL AGUDA
En pacientes con enfermedad de células falciformes o con el rasgo falciforme es poco común la disminución rápida de la función renal, pero puede ocurrir en dos situaciones. En primer lugar, la obstrucción completa del aparato urinario, causada en ocasiones por el desprendimiento bilateral de las papilas, produce insuficiencia renal aguda. En segundo lugar, puede ocurrir insuficiencia renal aguda como consecuencia del desarrollo de rabdomiolisis por el ejercicio vigoroso. En este caso la rabdomiolisis es resultado del aumento en la formación de células falciformes, que puede producir isquemia muscular.
Nefritis por radiación
Al igual que otros órganos, los niños son sensibles a la radiación, sobre todo cuando está expuesto a una dosis acumulativa de más de 2,300 cGy (2,300 rads).159 La nefritis por radiación, que fue un trastorno común en décadas anteriores, en la actualidad se observa en raras ocasiones. La desaparición casi completa de este tipo de nefropatía fue resultado del perfeccionamiento en las técnicas de radioterapia localizada, de la disminución del uso de radiaciones en las enfermedades no malignas y del empleo generalizado de la quimioterapia en lugar de la radiación total del abdomen en los padecimientos malignos intrabdominales. En la actualidad la mayoría de los casos de nefritis por radiación ocurren como resultado de la radioterapia de los tumores malignos retroperitoneales diseminados o después de la preparación para los trasplantes de médula ósea. En este último caso, que implica el uso de quimioterapia y radioterapia, pueden identificarse lesiones vasculares hasta en el 50 porciento de los pacientes [ver antes, Síndromes urémico-hemolíticos].
En general, predominan las lesiones vasculares y glomerulares, que son indistinguibles de las lesiones observadas en el síndrome urémico-hemolítico.160 También suelen existir alteraciones tubulointersticiales de gravedad variable. Con el tiempo, estas alteraciones inflamatorias progresan hasta la fibrosis intersticial, la atrofia tubular y la glomeruloesclerosis. Sin embargo, antes de que sea evidente la disfunción renal hay un intervalo de tiempo variable sin alteraciones. La nefritis aguda por radiación puede aparecer incluso a los seis meses después de la radioterapia, mientras que la nefritis crónica puede presentarse 12 o más años después de la exposición a la radiación.161,162
Además de la insuficiencia renal, también suele observarse hipertensión arterial en pacientes con nefritis por radiación. El aumento de la presión arterial parece ser mediado sobre todo por el sistema renina-angiotensina, por lo que es probable que responda al tratamiento con inhibidores de la enzima conversora de angiotensina.163 La hipertensión arterial maligna puede desarrollarse durante el episodio agudo o después de algunos meses o años.
Bibliografía
DR. ROBERT M. BLACK
Las enfermedades vasculares del riñón producen oclusión parcial o completa de los vasos de pequeño, mediano y gran calibre. Algunas de estas enfermedades, como la vasculitis, tienen una base inmunológica; en otros padecimientos, como la tromboembolia, participan mecanismos no inmunológicos. Las manifestaciones clínicas de estos trastornos son muy variables. Sin embargo, por lo general la función renal se afecta como resultado de la disminución del flujo sanguíneo renal. El diagnóstico se realiza correlacionando los antecedentes, los hallazgos de la exploración física y los datos de laboratorio con las alteraciones renales.
Complicaciones renales de la vasculitis sistémica
Un buen método para la clasificación de la vasculitis sistémica se basa en la extensión y el sitio de la afección vascular [ver tabla 1].1 Este método es sobre todo útil porque se conoce poco sobre la etiología y patogenia de los diversos trastornos. Es importante distinguir entre estas enfermedades porque tienen una evolución y una respuesta al tratamiento diferentes.2
|
||||||||||||||||||||||||||||
c-ANCA anticuerpos contra citoplasma de neutrófilos con patrón citoplasmático p-ANCA- anticuerpos contra citoplasma de neutrófilos con patrón perinuclear |
Anticuerpos anticitoplasma de neutrófilos
En los últimos años se han observado un grupo nuevo de autoanticuerpos dirigidos contra antígenos citoplásmicos de los neutrófilos (ANCA, por sus siglas en inglés, n. del t.) en pacientes con granulomatosis de Wegener y otras vasculitis relacionadas.3,4 Los anticuerpos anticitoplasma de neutrófilo citoplasmáticos (c-ANCA) están dirigidos contra una enzima llamada proteinasa 3. Los anticuerpos anticitoplasma de neutrófilo perinucleares (p-ANCA) suelen estar dirigidos contra mieloperoxidasa o, con menos frecuencia, elastasa. En consecuencia, se observan dos diferentes patrones de inmunofluorescenia, la tinción citoplásmica y la tinción perinuclear, al incubar el suero del paciente con neutrófilos.
Es posible detectar ANCA en casi el 90 porciento de los pacientes con granulomatosis de Wegener.4 En este caso los c-ANCA son los más comunes. También se encuentran ANCA en dos padecimientos relacionados con datos histológicos renales idénticos a los de la granulomatosis de Wegener: la poliarteritis microscópica y la glomerulonefritis necrosante (con medias lunas) rápidamente progresiva. Sin embargo, en estos dos padecimientos son más comunes los p-ANCA.
Muchos estudios sugieren que los títulos de ANCA en granulomatosis de Wegener y enfermedades relacionadas tienden a ser paralelos a la evolución clínica de la vasculitis, aumentando con la actividad de la enfermedad y disminuyendo con la remisión.4 Además, se ha observado un aumento en los títulos que puede preceder al inicio de una recidiva clínica en muchos pacientes.5 Sin embargo, a pesar de estas interesantes observaciones, no se ha demostrado el papel patogénico de los ANCA.
Si los ANCA favorecen el desarrollo de vasculitis, el mecanismo es incierto. Se ha observado que los neutrófilos normales sufren degranulación y producen radicales libres de oxígeno después de incubarse con ANCA;6 estos neutrófilos estimulados pueden dañar a las células endoteliales vasculares.7 Como alternativa, es posible que los ANCA inhiban la neutralización de enzimas polimorfonucleares después de su liberación de los neutrófilos, ocasionando así daño tisular por actividad enzimática no controlada.8
GRANULOMATOSIS DE WEGENER
La granulomatosis de Wegener es una vasculitis rara pero bien definida que afecta de manera característica el aparato respiratorio superior e inferior y los riñones, aunque pueden afectarse casi todos los órganos. La lesión renal se caracteriza por granulonefritis necrosante, que suele acompañarse de la formación de medias lunas [ver figura 1]. Por lo general la tinción inmunofluorescente de los glomérulos y los vasos renales no muestra depósitos de complejos inmunes. Sin embargo, su ausencia no excluye la participación del sistema inmune, porque es posible que los ANCA dañen en forma directa a los vasos (ver antes).6 Los granulomas necrosantes, que a menudo se encuentran en el tejido del aparato respiratorio superior e inferior de individuos afectados, pueden obaservarse en pocas ocasiones en las muestras de biopsia renal.
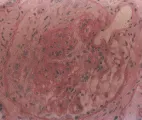
|
| Figura 1 |
| Glomerulonefritis de medias lunas: biopsia |
La granulomatosis de Wegener se desarrolla con mayor frecuencia en los adultos de mediana edad, pero también puede observarse en pacientes más jóvenes.1 En el momento del diagnóstico suelen existir manifestaciones clínicas de afección renal. El sedimento urinario característico muestra hematuria, cilindros eritrocitarios y proteinuria. Las concentraciones de creatinina plasmática y nitrógeno ureico de la sangre (BUN) suelen elevarse, lo que indica lesión renal considerable. Aunque se ha descrito una forma limitada de la granulomatosis de Wegener, caracterizada por afección respiratoria, pero no renal, en la mayoría de estos casos al final se desarrolla la enfermedad renal típica.
Los hallazgos de laborotorio pueden incluir anemia, leucocitosis y velocidad de sedimentación globular elevada, aunque no suelen ser diagnósticos. Por lo general, las concentraciones del complemento son normales y se detectan anticuerpos antinucleares, lo mismo que (como ya se mencionó) ANCA.
Por lo general se requiere de la biopsia del tejido para confirmar el diagnóstico. Es indispensable realizar una exploración cuidadosa de los senos del cráneo y la nasofaringe, en vista de que es posible encontrar lesiones vasculíticas en estas zonas, incluso en pacientes asintomáticos, y porque la toma de biopsia de las lesiones se efectúa con facilidad. Los hallazgos de la biopsia renal pueden ser muy sugestivos de granulomatosis de Wegener, aunque por lo general no son patognomónicos por dos razones. En primer lugar, en forma característica las muestras de tejido renal no presentan granulomas y en segundo lugar, también se observa glomerulonefritis necrosante focal con tinción inmunofluorescente negativa en pacientes con poliarteritis nodosa y como un trastorno idiopático en el que no puede identificarse vasculitis renal o extrarrenal.9 Sin embargo, algunos de los pacientes con glomerulonefritis con el tiempo desarrollan signos de granulomatosis de Wegener por lo que tal vez requieran modificaciones en el tratamiento.
Sin tratamiento, el pronóstico de los pacientes con granulomatosis de Wegener es muy malo, en vista de que hasta el 90 porciento mueren en los primeros dos años después del inicio del padecimiento.1,10 Aunque en algunos pacientes la administración de esteroides produce mejoría clínica y en los resultados de laboratorio, esta respuesta suele ser temporal. Por el contrario, con el uso de fármacos citotóxicos pueden producirse remisiones prolongadas en la mayoría de los pacientes, sobre todo con la ciclofosfamida.1,11 Es muy importante el inicio temprano del tratamiento en vista de que la necrosis tisular no puede revertirse una vez que ocurra. Sin embargo, la aparición de insuficiencia renal lo bastante grave para requerir diálisis durante la fase aguda de la enfermedad no necesariamente impide el tratamiento agresivo, ya que la función renal puede mejorar lo suficiente para permitir la suspensión de la diálisis. El tratamiento debe continuarse durante uno a dos años después de la remisión en vista de que las recaídas son comunes y pueden ocasionar suficiente necrosis tisular para impedir la recuperación de la función renal.
En algunos pacientes se han usado tratamientos alternativos. La administración de trimetoprim-sulfametoxazol (una tableta de dosis doble dos veces al día) ha sido benéfica para un número pequeño de pacientes.12,13 Al parecer es más eficaz en pacientes con síntomas sistémicos leves y enfermedad que se limita a las vías respiratorias superiores. El papel de este agente combinado en la granulomatosis de Wegener debe definirse aún en un número mayor de pacientes.
Existen también publicaciones no controladas y preliminares que sugieren que la inmunoglobulina intravenosa puede ser útil en la granulomatosis de Wegener y en otras formas de vasculitis sistémica.14 No se conoce aún el mecanismo de acción.14,15
Un estudio abierto indica que el methotrexate en dosis bajas puede ser útil en pacientes seleccionados,16 alcanzándose la remisión en casi el 70 porciento de los pacientes, aunque se requieren más estudios antes de que pueda recomendarse el uso habitual de este medicamento.
No es rara la progresión a insuficiencia renal, que puede ser causada por recidivas de la vasculitis o por glomeruloesclerosis progresiva por cicatrización de las lesiones glomerular y vascular iniciales. Con menos frecuencia se ha observado glomerulonefritis de novo, como nefropatía por IgA.17
La experiencia con el trasplante renal en la granulomatosis de Wegener es limitada. Reportes aislados indican que tanto las manifestaciones renales como extrarrenales pueden recurrir después del trasplante,18-20 asociándose con títulos altos de ANCA en plasma.21 El riesgo de recaídas puede ser mayor en pacientes que solo reciben prednisona y azatioprina, comparando con pacientes tratados con ciclosporina.20 El aumentar la dosis de prednisona no suele ser útil. Por otro lado, la sustitución de la azatioprina por ciclofosfamida o el cambio a ciclosporina suele causar la remisión de las alteraciones clínicas, en forma semejante a lo que se observa en la enfermedad primaria.18-20
POLIARTERITIS NODOSA Y SINDROME DE CHURG-STRAUSS
Se han descrito cuatro formas de poliarteritis nodosa, también conocida como periarteritis nodosa. Estos trastornos pueden estar relacionados entre sí y con la granulomatosis de Wegener; por lo menos en algunos pacientes con estos padecimientos se han detectado ANCA.4
1.Poliarteritis nodosa clásica, que afecta las arterias musculares de pequeño y mediano calibre y ocasiona la formación de aneurismas.
2.Poliarteritis nodosa microscópica, que afecta vasos más pequeños que la poliarteritis nodosa clásica.
3.Síndrome de Churg-Strauss, que se caracteriza por la formación de granulomas extravasculares e infiltración eosinofílica en arterias y vénulas.
4.Síndrome de sobreposición, compuesto por signos y síntomas característicos de más de una forma de poliarteritis nodosa.
Las manifestaciones clínicas para diferenciar estos trastornos se revisan en otro capítulo. La mayoría de los pacientes tienen síntomas sistémicos inespecíficos, que pueden incluir fiebre, pérdida de peso, artralgias, polineuropatía asimétrica (mononeuritis múltiple) o anorexia. La afección del aparato respiratorio es menos común en la poliarteritis nodosa clásica y microscópica que en la granulomatosis de Wegener. Sin embargo, en el síndrome de Churg-Strauss la enfermedad pulmonar es común, sobre todo el broncoespasmo, aunque la afección renal y pulmonar no siempre se desarrollan al mismo tiempo. La cardiopatía también es más común en el síndrome de Churg-Strauss que en las otras formas de poliarteritis nodosa.
La hipertensión arterial resultante de la vasculitis renal es común en la poliarteritis nodosa clásica y en ocasiones se manifiesta como hipertensión maligna. Por el contrario, la presión arterial en pacientes con poliarteritis nodosa microscópica es normal a menos que la función renal esté muy afectada. La causa de esta diferencia no se ha determinado, aunque puede estar relacionada con el tamaño de los vasos afectados. En la forma clásica, la inflamación de las arterias de mayor calibre puede causar isquemia distal, lo que ocasiona la activación del sistema renina-angiotensina. La poliarteritis nodosa microscópica se acompaña de inflamación de los capilares glomerulares, por lo que es menos probable que active este sistema.22
Las manifestaciones de la afección renal varían en las diferentes formas de poliarteritis nodosa. La forma microscópica a menudo se caracteriza por un sedimento urinario activo con eritrocitos, cilindros eritrocitarios y proteinuria. Estos hallazgos son resultado de la glomerulonefritis necrosante segmentaria focal. Es posible que las lesiones observadas en la biopsia renal no puedan distinguirse de las lesiones de la granulomatosis de Wegener [ver figura 1]. La poliarteritis nosoda clásica puede mostrar un patrón diferente a la forma microscópica de la enfermedad. En vista de que la forma clásica afecta las arterias renales de mayor calibre más que los vasos pequeños que irrigan los glomérulos, es posible no observar necrosis del ovillo glomerular; como consecuencia, en algunas ocasiones el sedimento urinario es normal, ya que la reducción de la velocidad de filtración glomerular se debe sobre todo a la disminución de la perfusión glomerular y no a inflamación. La lesión renal suele ser menos grave en el síndrome de Churg-Strauss que en otras variantes de la poliarteritis nodosa.23
El diagnóstico de poliarteritis nodosa se basa en los antecedentes clínicos, los hallazgos de la exploración física y en el tipo de afección de los órganos. En casos raros pueden palparse aneurismas en los vasos superficiales. Las observaciones preliminares sugieren que existe más posibilidad de encontrar ANCA positivos en la forma microscópica que en la poliarteritis nodosa clásica. La frecuencia con la que se observan estos anticuerpos en el síndrome de Churg-Strauss se desconoce, pero tambien se han detectado.
Al igual que en la granulomatosis de Wegener, por lo general es necesario el diagnóstico histológico.24 Si el examen clínico y los estudios de conducción sugieren afección nerviosa, está indicada la biopsia del nervio sural. Por otra parte, la angiografía celiaca y renal es casi diagnóstica de poliarteritis nodosa clásica si demuestra la presencia de microaneurismas y estenosis segmentaria e irregular en los vasos de mayor calibre, además de disminución progresiva y oclusión de las arterias intrarrenales de menor calibre.
El pronóstico global de la poliarteritis nodosa sin tratamiento es malo. La supervivencia a un año en pacientes con la forma clásica de la enfermedad es alrededor del 50 porciento, y la supervivencia a cinco años es del 13 porciento.25 El pronóstico de la variante microscópica puede ser un poco mejor, aunque la supervivencia a largo plazo en pacientes con este trastorno también es limitada sin un tratamiento eficaz.
El tratamiento con esteroides puede ser eficaz en algunos individuos. Algunos pacientes con poliarteritis nodosa, incluyendo los que no responden a los esteroides, han tenido remisiones prolongadas con el uso de ciclofosfamida.1,26 Es importante resaltar que el tratamiento puede mejorar la función renal, incluso en pacientes que presentan insuficiencia renal avanzada y que requieren diálisis. Por consiguiente, la gravedad inicial del padecimiento no impide proporcionar un tratamiento eficaz, aunque la respuesta inicial puede ser seguida por el desarrollo tardío de aterosclerosis si la resolución de las lesiones ocasiona estenosis progresiva de la luz de los vasos. En los pacientes que responden debe continuarse el tratamiento de mantenimiento durante uno a dos años después de la remisión para disminuir el riesgo de recaídas.
VASCULITIS POR HIPERSENSIBILIDAD
La vasculitis por hipersensibilidad se caracteriza por inflamación de los vasos sanguíneos pequeños como las arteriolas, los capilares y, sobre todo, las vénulas poscapilares.27 Los cambios inflamatorios son muy notables en la piel, donde existe infiltración considerable de neutrófilos alrededor de los vasos sanguíneos de la dermis, hemorragia local y edema. Este cuadro histológico recibe el nombre de vasculitis leucocitoclástica que se manifiesta como exantema purpúrico palpable [ver figura 2]. El exantema casi siempre es el principal signo de esta enfermedad.
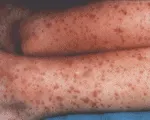
|
| Figura 2 |
| Erupción purpúrica en vasculitis por hipersensibilidad |
Las tres principales variantes de la vasculitis por hipersensibilidad que afectan al riñón son la púrpura de Henoch-Schönlein, la crioglobulinemia mixta esencial y la enfermedad del suero. Con menos frecuencia, se observa vasculitis por hipersensibilidad en pacientes con lupus eritematoso sistémico, síndrome de sobreposición de poliarteritis nodosa u otros padecimientos más raros.
Púrpura de Henoch-Schönlein
La púrpura de Henoch-Schönlein se caracteriza por la tétrada clásica de dolor abdominal, exantema cutáneo purpúrico, artritis o artralgias y afección renal. Es más común en niños, aunque también ocurre en adultos.28 Las lesiones cutáneas, que son más marcadas en las superficies extensoras y en declive, se observan en prácticamente todos los pacientes, anque pueden no existir durante el examen inicial.29
La afección renal es común y suele ser evidente en los primeros días o meses después del inicio de los síntomas. El examen general de orina muestra de manera característica hematuria microscópica o macroscópica, además de cilindros eritrocitarios y proteinuria. En la mayoría de los pacientes la concentración de creatinina plasmática es normal o sólo ligeramente elevada al inicio de la enfermedad. Sin embargo, puede existir afección renal más importante, caracterizada por proteinuria en rango nefrótico, hipertensión arterial e insuficiencia renal.
Los cambios característicos en el riñón son idénticos a los que se observan en la nefropatía por IgA. Existe expansión del mesangio glomerular por depósitos de IgA, que puede detectarse por medio de tinción inmunofluorescente. En los casos de afección renal grave puede haber formación de medias lunas.
El diagnóstico de púrpura de Henoch-Schönlein por lo general es evidente por las manifestaciones clínicas clásicas. La biopsia renal no suele ser necesaria. Cuando el diagnóstico es dudoso, la biopsia tanto de la piel afectada como de la sana pueden mostrar depósitos de IgA en las paredes de los vasos sanguíneos, y de esta forma se evita la necesidad de la biopsia renal, que implica un riesgo mayor.27,30
En general, las manifestaciones clínicas de los pacientes con púrpura de Henoch-Schönlein desaparecen de manera espontánea, aunque pueden recurrir los episodios de púrpura o glomerulonefritis. No existe una relación constante entre la recurrencia de la enfermedad y el pronóstico, o entre la gravedad de la glomerulonefritis y la de las manifestaciones extrarrenales. Más bien, el grado de afección renal es el factor más importante del pronóstico a largo plazo. Los pacientes con síndrome nefrótico e insuficiencia renal tienen el mayor riesgo de desarrollar insuficiencia renal progresiva.
Existen pocas evidencias de que el tratamiento convencional con esteroides o ciclofosfamida tenga efectos favorables sobre la enfermedad cutánea o la nefropatía.31 Estos medicamentos no parecen reducir la frecuencia de las recaídas ni tampoco modificar la duración de la actividad de la enfermedad. A pesar de ello, suele administrarse un esquema que consiste en pulsos intravenosos de metilprednisolona, seguido de prednisona oral y ciclofosfamida, y en ocasiones de plasmaféresis, en los pacientes con riesgo alto, insuficiencia renal aguda y glomerulonefritis con medias lunas.1,31 Este esquema tiene por objeto revertir el proceso inflamatorio, como el infiltrado por macrófagos, más que el depósito de IgA en sí.
Estudios no controlados sugieren que la dapsona puede ser útil para disminuir la severidad y duración de las lesiones cutáneas en pacientes con vasculitis leucocitoclásticas.32 Sin embargo, su eficacia en pacientes con púrpura de Henoch-Schönlein no se ha estudiado.
El trasplante renal puede tener éxito en los pocos pacientes que progresan hasta la enfermedad renal terminal. Es común el depósito recurrente de IgA en el mesangio del riñón trasplantado, aunque la enfermedad es subclínica en la mayoría de los pacientes.18 En menos del 15 porciento de estos enfermos se observa hematuria, proteinuria y púrpura.33,34
Crioglobulinemia mixta
Las crioglobulinas son anticuerpos que se precipitan con la temperatura fría y se disuelven al exponerlos de nuevo al calor. En la crioglobulinemia mixta esencial, prototipo de este trastorno, las crioglobulinas contienen tanto una IgG policlonal como un factor reumatoide IgM monoclonal dirigido contra la IgG.
Aunque se ha implicado al virus de Epstein-Barr y al virus de la hepatitis B como estímulos antigénicos en algunos casos, estudios recientes sugieren que la infección por virus de la hepatitis C (VHC) es la causa subyacente más común.35,36 A pesar de este dato, existen pocas evidencias de hepatitis activa en el momento de la presentación del paciente.
Los síntomas de la crioglobulinemia mixta suelen ser inespecíficos. La fatiga y las artralgias son muy comunes. Los hallazgos que más sugieren el diagnóstico incluyen exantema purpúrico palpable, linfadenopatía, hepatoesplenomegalia, mononeuritis y fenómeno de Raynaud. Por lo general existe hipocomplementemia, a diferencia de las concentraciones normales del complemento que se observan en los pacientes con púrpura de Henoch-Schönlein.
Ocurre afección renal en el 30 a 60 porciento de los pacientes.37 En estos casos la biopsia renal muestra en forma característica proliferación difusa del endotelio y de las células mesangiales, un patrón indicativo de glomerulonefritis membranoproliferativa. Resulta de particular importancia el hallazgo frecuente de trombos intraluminales compuestos de crioglobulinas precipitadas [ver figura 3].
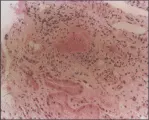
|
| Figura 3 |
| Crioglobulinemia mixta esencial: biopsia |
Debe pensarse en el diagnóstico de crioglobulinemia mixta en cualquier persona con exantema purpúrico palpable, sobre todo si el exantema se presenta en la superficies extensoras o los glúteos y se acompaña de hipocomplementemia y afección renal.37,38 Las pruebas de funcionamiento hepático por lo común son normales, sobre todo si la infección viral incitante ha sido crónica. El diagnóstico se confirma por la demostración de crioglobulinas circulantes. En la crioglobulinemia mixta, la inmunoelectroforesis sérica revela un título alto de crioglobulinas mixtas IgM-IgG (o, en algunos casos, un título alto de IgM-IgA) con un componente monoclonal. En muchos individuos existen anticuerpos contra el VHC. La biopsia renal debe reservarse para los pacientes con enfermedad progresiva o en los que el diagnóstico está en duda.
El pronóstico de la enfermedad renal en la crioglobulinemia mixta es variable. Alrededor de la tercera parte de los pacientes presenta remisión parcial o completa, mientras que el resto tiene un curso lentamente progresivo que puede complicarse por exacerbaciones periódicas.37-39
En el pasado la quimioterapia agresiva se reservaba para los pacientes con crioglobulinemia mixta y enfermedad aguda grave manifestada por un nivel de crioglobulinas en aumento, insuficiencia renal progresiva, necrosis distal o neuropatía avanzada. En estos casos se ha usado plasmaféresis para eliminar las crioglobulinas circulantes junto con esteroides y ciclofosfamida para evitar la formación de más anticuerpos.1,37,38,40 El plasma reinfundido durante la plasmaféresis debe calentarse para evitar la precipitación de las crioglobulinas circulantes. Por lo general este esquema permite estabilizar la función renal, aunque la muerte por vasculitis activa sigue siendo un gran problema.38
En contraste, con el uso de quimioterapia y plasmaféresis, existen evidencias en un número cada vez mayor de pacientes con crioglobulinemia mixta sobre que el interferón alfa puede reducir las concentraciones circulantes de crioglobulinas e inducir remisión clínica.36,41,42 Sin embargo, en la mayoría de estos pacientes ocurren recaídas al suspender el tratamiento.36,43 A pesar de estas observaciones, el interferón alfa puede ser el tratamiento inicial de elección en pacientes con crioglobulinemia mixta, en especial porque los títulos de VHC pueden aumentar después del tratamiento inmunosupresor con ciclofosfamida.
Enfermedad del suero
La enfermedad del suero a menudo es ocasionada por la administración de medicamentos, incluyendo penicilinas, derivados de sulfonamidas y difenihidantoína, que parecen actuar como haptenos y estimular una respuesta inmune. También puede observarse en presencia de algunas infecciones, como la hepatitis B.44 Las manifestaciones iniciales típicas incluyen fiebre, exantema, artralgias y linfadenopatía. En la mayoría de los casos el exantema es urticariano. En algunas ocasiones existe edema. Los síntomas aparecen en forma característica en un periodo de siete a 10 días después del contacto con el antígeno. En algunos pacientes puede haber hipocomplementemia.
La afección renal se manifiesta por alteraciones en el examen general de orina caracterizadas por proteinuria, eritrocitos y cilindros celulares. La insuficiencia renal aguda es rara, pero puede ocurrir, sobre todo con la exposición prolongada al antígeno. En este caso la biopsia renal muestra la típica glomerulonefritis por complejos inmunes con proliferación celular difusa y depósitos de IgG y del componente C3 del complemento en las paredes de los capilares glomerulares.
Existen pocas pruebas de que el tratamiento inmunosupresor con esteroides o agentes citotóxicos como la ciclofosfamida eviten el desarrollo o modifiquen la evolución de este trastorno. La eliminación del agente estimulante suele dar como resultado la resolución inmediata de los signos y síntomas en algunos días o semanas.
CAUSAS DIVERSAS DE VASCULITIS RENAL
Existen varias vasculitis que en algunas ocasiones se acompañan de afección renal importante incluyendo la vasculitis urticariana con hipocomplementemia, la vasculitis reumatoide y la policondritis recidivante. La vasculitis de vasos grandes, como la arteritis de la temporal o la arteritis de Takayasu, pueden afectar las arterias renales principales, pero no suelen producir lesiones en los vasos intrarrenales pequeños.45,46
Complicaciones renales de la esclerodermia
La esclerodermia es una enfermedad poco frecuente caracterizada por cambios notables en la piel y evidencias de afección multisistémica, por lo general de los pulmones, el aparato digestivo, el corazón y los riñones. Se observa con mayor frecuencia en mujeres, con inicio entre los 20 y 50 años de edad. Las manifestaciones de la enfermedad son principalmente causadas por la isquemia secundaria a las lesiones vasculares o por el aumento local en el acúmulo de colágena.
La afección renal en la esclerodermia se caracteriza por lesiones arteriales obstructivas que afectan sobre todo a las arterias interlobulares. También es común el engrosamiento concéntrico (llamado en capas de cebolla) de la pared vascular, que es resultado de la proliferación de las células de músculo liso en la capa media, que después se deplazan hacia la íntima [ver figura 4]. Estas alteraciones histológicas son similares a las lesiones observadas en la hipertensión maligna y en los síndromes urémico-homolíticos [ver adelante, Síndromes urémico-hemolíticos].
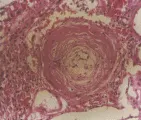
|
| Figura 4 |
| Lesiones en piel de cebolla: esclerodermia |
En cerca del 20 al 25 porciento de los pacientes con esclerodermia se observa afección renal importante. La complicación renal más grave es la crisis renal esclerodérmica, que se caracteriza por la aparición súbita de insuficiencia renal aguda. Por lo general el sedimiento urinario sólo muestra proteinuria leve y algunas células o cilindros. Estos hallazgos reflejan el origen no inflamatorio de la enfermedad vascular. En casos raros puede haber hematuria ocasionada por necrosis glomerular isquémica o por hemorragia de las hemangiomas localizados en el aparato urinario. Si no se trata, la crisis renal esderodérmica progresa hasta la insuficiencia renal avanzada en un periodo de uno a dos meses.
La crisis renal esclerodérmica casi siempre se desarrolla en los primeros cuatro años después del inicio de las manifestaciones extrarrenales, sin embargo, en pocas ocasiones es la manifestación inicial de la escleroderma.47,48 En la mayoría de los pacientes se observan elevaciones importantes de la presión arterial como resultado de la activación del sistema renina-angiotensina por la isquemia. Sin embargo, la presencia de hipertensión leve o el aumento en la actividad de la renina plasmática no indican necesariamente que los pacientes con esclerodermia con el tiempo desarrollarán esta complicación. Por otra parte, el desarrollo simultáneo de hipertensión mediada por angiotensina parece ser un factor secundario en la patogenia de la esclerodermia renal, en vista de que en ocasiones la insuficiencia renal aguda no se acompaña de hipertensión arterial.49
El diagnóstico de la crisis renal esclerodérmica suele ser evidente por los antecedentes y la exploración física. En algunas ocasiones la insuficiencia renal aguda es la manifestación inicial. En presencia de insuficiencia renal aguda, deben excluirse otros padecimientos con características similares, incluyendo la hipertensión maligna, los síndromes urémicos-hemolíticos y la vasculitis sistémica (v.gr., poliarteritis nodosa). El antecedente de fenómeno de Raynaud y alteraciones cutáneas, como telangiectasias y capilares periungueales tortuosos y muy dilatados, indica la presencia de esclerodermia.
Aparecen anticuerpos antinucleares hasta en el 90 porciento de los pacientes con esclerodermia. En más del 85 porciento de los casos es posible identificar el antígeno contra el que se dirigen estos anticuerpos.50
La insuficiencia renal representa cerca del 40 porciento de las muertes por esclerodermia. El resto de la mortalidad se debe a insuficiencia respiratoria, insuficiencia cardiaca o complicaciones gastrointestinales isquémicas, como la perforación visceral. Aunque el tratamiento para las complicaciones extrarrenales de la esclerodermia es limitado, el control de la hipertensión en pacientes con crisis renal esclerodérmica puede alterar en forma notable la evolución. El tratamiento antihipertensivo eficaz puede disminuir de manera considerable la incidencia de insuficiencia renal si se inicia antes que aparezcan lesiones vasculares irreversibles.
Los inhibidores de la enzima convertidora de angiotensina (ECA) son los fármacos de elección para el tratamiento de la hipertensión en los pacientes con esclerodermia.51 Los inhibidores de la ECA disminuyen la presión arterial en hasta el 90 porciento de los pacientes al revertir la vasoconstricción inducida por la angiotensina II. Estudios preliminares sugieren también que estos agentes pueden mejorar la evolución de los pacientes en crisis renales normotensas.52
No se conoce del todo porqué los inhibidores de la ECA son muchos más eficaces que otros antihipertensivos para mejorar la evolución renal en la esclerodermia.53 Sin embargo, debe recordarse que casi la mitad de los pacientes con crisis renal esclerodérmica progresan hasta la insuficiencia renal terminal.51
La enfermedad renal por esclerodermia consiste esencialmente en una estenosis de la arteria renal bilateral. Por lo tanto, es posible que la concentración plasmática de creatinina aumente en algunos pacientes por una reducción en la resistencia arteriolar eferente inducida por el inhibidor de la ECA, con menor presión intraglomerular consecuente.
Los enfermos que progresan hasta insuficiencia renal terminal pueden dializarse en forma satisfactoria, aunque a veces se presentan algunos problemas. Por ejemplo, la hemodiálisis puede resultar difícil si existen pocas vías de acceso vascular como resultado de la enfermedad vascular periférica. Este problema puede evitarse con la diálisis peritoneal, aunque la depuración peritoneal a menudo es baja en la esclerodermia como consecuencia de la alteración en el flujo sanguíneo peritoneal. Además, la depuración peritoneal puede disminuir con el descenso de la temperatura ambiente; por consiguiente, suele ser menor durante los meses de invierno.54
Existe poca experiencia con el trasplante renal en la esclerodermia, y éste suele estar limitado por la gravedad de las manifestaciones extrarrenales.55,56 Se ha calculado que la tasa de recurrencia es de alrededor del 20 porciento.56,57 Los pacientes trasplantados tienden a seguir una evolución agresiva, semejante a la de la enfermedad renal primaria aguda. La frecuencia exacta de recurrencia es difícil de valorar porque los cambios histológicos primarios inducidos por la esclerodermia (engrosamiento mucoide de la íntima de las arterias interlobulares y necrosis fibrinoide de las arteriolas glomerulares) pueden ser difíciles de distinguir del rechazo vascular agudo o crónico.
Síndromes urémico-hemolíticos
Los síndromes urémico-hemolíticos son un grupo de padecimientos poco frecuentes con manifestaciones clínicas que se traslapan y que se caracterizan por anemia hemolítica microangiopática, trombocitopenia y grados variables de insuficiencia renal. La oclusión de arterias y arteriolas por trombos de plaquetas y fibrina desempeña una función importante en las manifestaciones clínicas de estos trastornos. Existen tres tipos de síndrome urémico-hemolítico: la púrpura trombocitopénica trombótica, el síndrome urémico-hemolítico infantil y el síndrome urémico-hemolítico del adulto. Todos varían un poco en el cuadro clínico; sin embargo, tienen muchas características en común.58 Por ejemplo, los cambios histológicos en el riñón son similares en los tres síndromes. Los pacientes en la fase aguda presentan oclusión de arterias pequeñas, arteriolas y capilares glomerulares por trombos de plaquetas y fibrina [ver figura 5]. Los pacientes con una evolución más subaguda y crónica desarrollan lesiones obstructivas prominentes en las arterias interlobulares; las lesiones se caracterizan por engrosamiento concéntrico en capas de cebolla, similares a los cambios observados en la esclerodermia o la hipertensión maligna [ver figura 4].
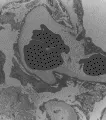
|
| Figura 5 |
| Púrpura trombocitopénica trombótica: daño renal |
Los síndromes urémicos-hemolíticos parecen ser trastornos aislados del consumo de plaquetas. No se conoce bien porqué ocurre consumo de plaquetas en niños y adultos con síndrome urémico-hemolítico o con púrpura trombocitopénica trombótica, pero se han sugerido tres hipótesis principales, cada una de las cuales puede explicar el trastorno en pacientes seleccionados. Una hipótesis sugiere que en algunos casos existe aumento en el nivel sanguíneo de un factor agregador de plaquetas.59,60 Se han postulado diferentes sustancias como factor agregador, y éstos pueden variar en los distintos pacientes.60-64 De acuerdo con la segunda hipótesis, es posible que exista menor nivel circulante de un inhibidor normal de la agregación plaquetaria, lo que explica tanto la formación de trombos plaquetarios como la remisión ocasional de la enfermedad después de la infusión de plasma.64 No es claro si esta alteración es causada por niveles deficientes de IgG en algunos pacientes.65 La tercera hipótesis sugiere que la activación plaquetaria puede ser una respuesta secundaria al daño endotelial. Por ejemplo, este podría explicar el desarrollo de síndrome urémico-hemolítico en algunos pacientes que reciben ciclosporina.66
Un factor pronóstico importante es el sitio principal de afección renal, en vista de que pueden predominar las lesiones glomerulares o las arteriales.67 En los niños pequeños con síndrome urémico-hemolítico, el glomérulo es el principal sitio afectado, y es común la recuperación espontánea. Por el contrario, los adultos tienen lesiones sobre todo en las arterias, aunque también puede ocurrir daño glomerular. Este tipo de síndrome se acompaña de hipertensión más grave y menor incidencia de recuperación espontánea.
El cuadro clínico de los tres tipos de síndrome urémico-hemolítico varía. La púrpura trombocitopénica trombótica afecta sobre todo a mujeres entre los 10 y 50 años de edad. Se caracteriza por cinco manifestaciones: fiebre, trombocitopenia (a menudo acompañada de púrpura), alteraciones neurológicas focales, anemia hemolítica microangiopática e insuficiencia renal.68 Suele comenzar como una enfermedad gripal, pero finalmente progresa hasta la púrpura trombocitopénica trombótica que produce lesiones purpúricas en la piel y síntomas neurológicos.
|
||||||||||||||||||||||
|
El síndrome urémico-hemolítico infantil comienza de manera característica después de un episodio de gastroenteritis viral o bacteriana. La diarrea suele ser sanguinolenta. La presencia de una verocitotoxina, similar a la toxina Shiga, secretada por Escherichia coli (sobre todo el serotipo O157:H7) o por otras bacterias, parece relacionarse con el desarrollo del síndrome urémico-hemofílico en muchos de estos pacientes.66,69,70 Por lo general la insuficiencia renal es más grave que en la púrpura trombocitopénica trombótica, y ocurre anuria hasta en el 50 porciento de los casos. Asimismo, la hipertensión arterial es común.
El síndrome urémico-hemolítico del adulto a menudo es idiopático, aunque en algunas ocasiones pueden identificarse trastornos primarios. Por ejemplo, este síndrome se ha relacionado con el embarazo, sobre todo durante el periodo posparto, y en este caso el anticoagulante lúpico puede desempeñar una función patogénica importante. También se ha asociado con el uso de anticonceptivos orales que contienen estrógenos, con los adenocarcinomas mucinosos del aparato gastrointestinal, páncreas y próstata, con la quimioterapia con mitomicina C o la combinación de bleomicina y cisplatino, y con la administración de ciclosporina. Puede ocurrir un cuadro previo de gastroenteritis similar al observado en la forma infantil del síndrome, aunque no es común.
Las características de los tres tipos de síndrome urémico-hemolítico pueden confundirse, en vista de que es posible que tengan una causa común. Por ejemplo, en un estudio realizado en cinco pacientes tratados con bleomicina y cisplatino, tres presentaron manifestaciones comunes del síndrome urémico-hemolítico (esto es, insuficiencia renal grave sin disfunción neurológica), mientras los otros dos pacientes tuvieron síntomas típicos de púrpura trombocitopénica trombótica.71
El examen del sedimento urinario en pacientes con púrpura trombocitopénica trombótica en ocasiones muestra eritrocitos y cilindros eritrocitarios. Por el contrario, el examen general de orina en niños y adultos con síndrome urémico-hemolítico suele ser normal, tal vez porque este trastorno se caracteriza por disfunción renal más grave, y porque la oliguria o la anuria pueden disminuir la excreción urinaria de eritrocitos. Sin embargo, es importante resaltar que el examen general de orina no es específico para diferenciar un tipo de síndrome urémico-hemolítico de otro.
El diagnóstico del síndrome urémico-hemolítico se sugiere con base en los hallazgos clínicos y por las condiciones generales. En la mayoría de los pacientes, no existen datos de coagulación intravascular diseminada en vista de que este trastorno se acompaña de consumo aislado de plaquetas; el tiempo de protrombina y el tiempo de tromboplastina parcial suelen ser normales. Por lo general, las concentraciones de complemento son normales, aunque se ha informado hipocomplementemia en algunos niños con síndrome urémico-hemolítico; por lo tanto, este hallazgo no excluye el diagnóstico.72
El objetivo del tratamiento en los síndromes urémico-hemolíticos consiste en revertir el consumo aislado de plaquetas que es responsable tanto de la necrosis isquémica (por la formación de trombos) como de la hemorragia (por la trombocitopenia). Puede intentarse el uso de infusiones de plasma, intercambio plasmático y otras modalidades.73-78
La eficacia del tratamiento se ha evaluado mejor en la púrpura trombocitopénica trombótica. Los resultados de estudios recientes extensos75,77 sugieren que la plasmaféresis más el intercambio plasmático con plasma fresco congelado son más eficaces que la infusión aislada de plasma. Sin embargo, durante estos estudios los pacientes tratados con intercambio plasmático recibieron alrededor de tres veces la cantidad de plasma que los pacientes tratados solo con infusión de plasma. Por lo tanto, el beneficio aparente del intercambio plasmático puede ser resultado de la infusión de más plasma fresco congelado y no de la remoción de alguna sustancia tóxica.
El intercambio de plasma se realiza diario hasta que se normaliza la cuenta plaquetaria y cesa la hemoólisis, en promedio este proceso requiere de siete a ocho intercambios.75,77 Desde el punto de vista clínico, los síntomas neurológicos tienden a abatirse primero (a los dos a tres días), después se reduce la concentración de deshidrogenasa láctica y la cuenta de plaquetas comienza a aumentar para el quinto día. Se observa estabilización o mejoría de la función renal alrededor del séptimo día.
Los pacientes con enfermedad muy leve pueden responder a dosis altas de prednisona sola (200 mg/día),77 aunque debe considerarse realizar intercambio plasmático si no existe aumento en la cuenta plaquetaria 48 horas después. Los agentes antiplaquetarios como la aspirina y el dipiridamol no son eficaces cuando se administran en forma aislada.79 Pueden tener algún beneficio cuando se agregan a la plasmaféresis.74
Son comunes las recaídas, sobre todo durante los primeros 30 a 60 días, pero en ocasiones hasta años después.77,78,80 Por lo general es eficaz repetir el recambio plasmático.80
Es necesario hacer énfasis en dos posibles complicaciones de la transfusión de plaquetas. Los agentes antiplaquetarios pueden inducir hemorragia en algunos pacientes,79 y las transfusiones de plaquetas pueden causar empeoramiento de los síntomas neurológicos o síntomas nuevos e insuficiencia renal aguda, al parecer por formación de trombos nuevos o expansión de los mismos cuando se consumen las plaquetas infundidas.82,83
Aunque la mayoría de los niños con síndrome urémico-hemolítico posdiarreico tienen una recuperación clínica completa,84,85 el seguimiento a largo plazo suele revelar evidencia de daño renal irreversible, manifestado por proteinuria, hipertensión o insuficiencia renal. Estos datos ocurren en hasta una tercera parte de los niños con enfermedad inicial prolongada, definida por anuria durante más de ocho días u oliguria por más de 15 días.84,85
También existen niños que no se recuperan en forma espontánea del episodio agudo. Los factores pronósticos malos incluyen ausencia del antecedente de gastroenteritis, no mejoría en dos semanas y enfermedad recurrente o familiar.86,87 Existen reportes anecdóticos de tratamiento exitoso en niños con enfermedad fulminante por medio de infusiones e intercambio de plasma.74,86
Complicaciones renales en el embarazo: preeclampsia e hipertensión inducida por el embarazo
CAMBIOS FISIOLOGICOS DURANTE EL EMBARAZO
De manera característica la presión arterial disminuye al inicio de la gestación, y durante el segundo trimestre del embarazo por lo general se encuentra 10 mmHg por abajo del valor basal, con cifras promedio de 105/60 mm Hg.88,89 Los factores responsable sde la caída en la resistencia vascular sistémica no se conocen del todo, pero parece que la menor respuesta a la acción presora de la angiotensina II y de la epinefrina es un factor importante.90 Al final de la gestación, estos cambios disminuyen en forma gradual y la presión arterial se normaliza.
También ocurre reducción del tono del músculo liso en los sistemas colectores renales y en los ureteros, lo que causa un cuadro radiológico que puede confundirse con obstrucción de las vías urinarias.88 La importancia de este cambio durante el embarazo normal es que es más probable que la bacteriuria asintomática provoque infección de las vías urinarias bajas o pielonefritis.88
El flujo plasmático renal y la velocidad de filtración glomerular (VFG) aumentan al principio del embarazo. El aumento en la VFG alcanza alrededor de un 40 a 50 porciento sobre los niveles basales hacia el final del primer trimestre.91 Se observa una reducción concomitante en la concentración plasmática de creatinina, por lo que una concentración de 0.8 mg/dl (que se considera normal fuera del embarazo) puede representar nefropatía subyacente. La VFG regresa a los niveles previos al embarazo durante los primeros tres meses después del alumbramiento.91
Ocurre hiponatremia leve durante el embarazo normal, y la concentración plasmática de sodio disminuye alrededor de 5 mEq/L. Este cambio en la osmolaridad plasmática es resultado de un ajuste en el osmostato hipotalámico, quizá inducido por la mayor liberación de gonadotrofina coriónica humana durante el embarazo.92,93 Los intentos para corregir la hiponatremia son tanto innecesarios (porque el cambio es leve y no causa síntomas) como ineficaces. La concentración plasmática de sodio aumenta en forma espontánea a los niveles previos al embarazo durante el primer o segundo mes posparto.94,95
Aunque existe cierta actividad excesiva de la hormona antidiurética (HAD) durante el embarazo normal, la liberación de vasopresinas por parte de la placenta causa un mayor metabolismo de la HAD.95,96 Este cambio no es importante desde el punto de vista clínico en la mayoría de las mujeres, aunque algunas desarrollan poliuria sintomática.96-98 La mayor diuresis en estos casos suele ser resistente a la administración de HAD nativa (vasopresina) por su rápido catabolismo. Sin embargo, la mayoría de las pacientes con diabetes insípida inducida por el embarazo responden a la 1-desamino-8-D-arginina vasopresina, un análogo de HAD que es menos sensible a la vasopresinasa.96 En pocos pacientes el desarrollo de poliuria se retrasa hasta el momento del parto o incluso uno a dos días después, quizá por actividad prolongada de la vasopresinasa derivada de la placenta.
También es frecuente observar durante el embarazo hiperventilación persistente que causa una reducción modesta en la presión de bióxido de carbono (PCO2). Esta alcalosis respiratoria crónica parece deberse a estimulación directa del centro respiratorio por la progesterona.99
PATOGENIA Y PATOLOGIA
Ocurre preeclampsia en el cinco al 10 porciento de todos los embarazos, aunque es más común durante el último trimestre del primer embarazo. En general, ésta se caracteriza por la aparición gradual de hipertensión, proteinuria y edema después de la vigésima semana de gestación. Puede progresar hasta la fase convulsiva llamada eclampsia. Es importante hacer énfasis en que ninguno de estos hallazgos por sí sólo es diagnótico de preeclampsia. Por ejemplo, el edema es común en el embarazo normal, en parte como resultado de la compresión de la vena cava inferior por el crecimiento uterino. Además, en pacientes con embarazo molar la preeclampsia puede observarse antes de la vigésima semana de gestación, a menudo durante el primer trimestre.
La patogenia de la preeclampsia no se conoce con claridad. La primera alteración patológica ocurre en la circulación uteroplacentaria y se caracteriza por menor dilatación de las arterias espirales.100,101 La isquemia uterina resultante, que también puede ser consecuencia de enfermedades vasculares primarias, es seguida de alteraciones en el metabolismo de las prostaglandinas, activación intravascular del sistema de la coagulación y mayor respuesta a los agentes presores [ver figura 6]. Es posible que el daño a la célula endotelial, inducido por un factor liberado por la placenta isquémica, tenga un papel importante en la génesis de estos cambios.100

|
| Figura 6 |
| Tensión arterial y embarazo |
Los factores inmunológicos también parecen ser importantes en la patogenia de la preeclampsia. Se ha propuesto que el desarrollo de preeclampsia puede reflejar un estado de inmunoprotección inadecuado o alterado del tejido fetoplacentario. En mujeres que utilizaban métodos anticonceptivos que evitaban el contacto con el esperma y el líquido seminal (v.gr., preservativo y diafragma) se observó un aumento en la incidencia de preeclampsia 2.4 veces mayor que en las mujeres que usaban otro tipo de anticonceptivos (anticonceptivos orales).102 Este hallazgo sugiere que el contacto con los antígenos paternos en el semen antes del embarazo puede hacer posible el desarrollo de anticuerpos que permitan bloquear una respuesta inmune subsecuente dirigida contra el tejido del feto o la placenta. En apoyo de esta hipótesis está el hecho de que la preeclampsia es más común en los primeros embarazos y en mujeres que tienen un embarazo posterior, pero con diferente pareja.103
Los principales cambios patológicos en el riñón ocurren en los glomérulos. Al examen con microscopio de luz se observa edema de las células endoteliales con el consencuente estrechamiento u obliteración de la luz de los capilares glomerulares. En la mayoría de los pacientes con preeclampsia las alteraciones renales patológicas comienzan a desaparecer poco después del parto, y de manera característica la histología renal se normaliza al cabo de dos a tres semanas.104 Sin embargo, puede no haber mejoría rápida si existen lesiones renales más graves y depósitos extensos de plaquetas y fibrina.
MANIFESTACIONES CLINICAS Y DIAGNOSTICO
El cuadro clínico de la preeclampsia es variable. La mayoría de las veces la presión arterial comienza a aumentar en forma gradual después de la vigésima semana de gestación. Clasicamente la presión arterial es superior a 140/90 mm Hg en el tercer trimestre, sobre todo durante el último mes del embarazo. En la mayoría de las pacientes con preeclampsia, además de la hipertensión arterial, existe edema y proteinuria. La excreción urinaria de proteínas aumenta en forma progresiva, a menudo alcanzando un nivel en rango nefrótico de más de 3.5 g/día, aunque el sedimento urinario suele ser casi normal, ya que contiene algunos eritrocitos o leucocitos. A pesar de todo, es raro que exista reducción importante de la función renal; la concentración de creatinina plasmática aumenta solo en 0.2 a 0.3 mg/dl, a menos que ocurra coagulación intravenosa diseminada con necrosis cortical bilateral u otro trastorno (como el síndrome de HELLP, ver adelante).
En pacientes con una enfermedad más grave pueden observarse otras manifestaciones clínicas. Los signos y síntomas comunes incluyen cefalea, alteraciones visuales e hiperactividad de los reflejos osteotendinosos. El dolor epigástrico o subcostal derecho también es común, y es ocasionado por distensión de la cápsula hepática por edema o hemorragia del hígado. En algunas pacientes puede observarse el desarrollo del síndrome de HELLP (hemólisis con un frotis microangiocítico, enzimas hepáticas elevadas y una cuenta de plaquetas bajas). Estos hallazgos pueden indicar isquemia hepática producida por obstrucción vascular. La aparición de convulsiones se considera criterio diagnóstico de eclampsia.
El diagnóstico de preeclampsia se sospecha por el desarrollo de hipertensión y proteinuria en mujeres primigrávidas, sobre todo en el tercer trimestre del embarazo. La presencia de estos signos al inicio de la gestación o el desarrollo de uno u otro en mujeres multigrávidas sugiere un trastorno primario como hipertensión arterial esencial, nefropatía primaria, diabetes mellitus o mola hidatiforme.105 En un grupo numeroso de pacientes, el diagnóstico clínico de preeclampsia pudo confirmarse por biopsia renal posparto en el 85 porciento de las mujeres primigrávidas, pero solo en el 38 porciento de las multigrávidas, la mayoría de las cuales tenían nefrosclerosis benigna u otra nefropatía primaria.106
Diferencias entre la preeclampsia y otras causas de hipertensión arterial del embarazo
Se considera que existe hipertensión arterial durante el embarazo si la presión arterial es mayor de 140/90 mm Hg. Es importante reconocer que una presión arterial de 120/80 mm Hg, que sería considerada normal en mujeres no embarazadas, puede ser alta durante el embarazo. Por lo tanto, todas las pacientes deben evaluarse en forma individual y los valores de la presión arterial compararse con las mediciones registradas antes y durante las primeras fases del embarazo.
En las mujeres embarazadas deben considerarse tres causas importantes de hipertensión arterial. En primer lugar, la hipertensión del embarazo puede ser ocasionada por preeclampsia. En segundo lugar, el aumento de la presión arterial durante la gestación puede indicar una tendencia hipertensiva subyacente. Por último, en algunas mujeres, la hipertensión arterial gestacional tardía o transitoria es una complicación aislada, que desaparece después del parto.
Cuando no existe el antecedente de hipertensión arterial, puede resultar difícil diferenciar la preeclampsia de la hipertensión esencial crónica por la disminución característica de la presión arterial en el segundo trimestre de la gestación. Por consiguiente, las pacientes con hipertensión crónica previa pero no diagnosticada pueden tener una presión arterial que se encuentre en el rango normal cuando son atendidas por primera vez por el médico. A medida que la presión aumenta durante el tercer trimestre, pueden ser útiles los siguientes datos para determinar el diagnóstico: edad de la paciente, número de embarazo, grado de excreción urinaria de proteínas y calcio, y concentración de ácido úrico en plasma [ver tabla 3].
|
|||||||||||||||||||||||||||
|
En ocasiones se observa aparición de hipertensión leve aislada durante el tercer trimestre del embarazo sin la presencia de proteinuria coexistente. Este hallazgo parece tener pocos efectos adversos en la madre y el feto, aunque en algunas pacientes puede ser un indicador del desarrollo futuro de hipertensión esencial.
Diferencia entre la preeclampsia y la nefropatía primaria
La preeclampsia es el tipo más común de nefropatía que ocurre en las últimas etapas del embarazo. Por lo general, puede diferenciarse con facilidad de otras causas de nefropatía durante el embarazo porque estos trastornos a menudo se acompañan de una reducción moderada o grave en la velocidad de filtración glomerular y de proteinuria leve o ausente.
La insuficiencia renal aguda puede presentarse en cualquier etapa del embarazo como resultado de diversos trastornos [ver tabla 4], pero rara vez es secundaria a preeclampsia. Las principales causas de insuficiencia renal aguda en la primera mitad del embarazo incluyen la necrosis tubular aguda ocasionada por un aborto séptico y el daño prerrenal por hiperemesis gravídica. Al final del embarazo o durante el periodo posparto, la insuficiencia renal aguda puede ser producida por preeclampsia o por algunos de los síndromes urémico-hemofílicos [ver antes, Síndromes urémico-hemolíticos]; en estos casos varias características distinguen la preeclampsia del síndrome urémico-hemofílico posparto [ver tabla 5]. La insuficiencia renal aguda también puede ser causada por necrosis tubular aguda o necrosis cortical renal en pacientes con desprendimiento de placenta o placenta previa. El hígado graso agudo del embarazo es una complicación poco común, pero se acompaña de insuficiencia renal aguda hasta en el 60 porciento de las pacientes y disminuye de manera característica la función renal por el desarrollo de necrosis tubular aguda o por la reducción de la perfusión renal.107
|
||||
|
|
|||||||||||||||
|
La obstrucción del aparato urinario es otra causa posible de insuficiencia renal durante las últimas etapas del embarazo. La relajación del músculo liso ureteral, mediada en parte por los defectos de las prostaglandinas y por la presión ejercida sobre los ureteros por el útero, produce cierta dilatación del sistema caliceal durante la gestación normal. Esta hidronefrosis considerada funcional puede detectarse por ultrasonografía renal, y no es diagnóstica de obstrucción de las vías urinarias como causa de la insuficiencia renal en este caso. Sin embargo, rara vez el útero puede ocasionar suficiente obstrucción como para producir disminución de la velocidad de filtración glomerular.108 La normalización de la función renal con el decúbito lateral y su alteración con el decúbito supino debe sugerir la posibilidad de este diagnóstico.
TRATAMIENTO
La preeclampsia no tratada se asocia con una incidencia alta de muertes fetales y neonatales. En pacientes que progresan hasta la preeclampsia grave o la eclampsia, la mortalidad materna se incrementa, sobre todo por la hemorragia cerebral. El tratamiento definitivo requiere la extracción del feto y la placenta. La única razón para retrasar el nacimiento en pacientes con diagnóstico de preeclampsia es la evidencia de inmadurez fetal. En este caso, puede utilizarse reposo en cama en decúbito lateral y administración de fármacos antihipertensivos (si la presión arterial diastólica es mayor de 95 a 100 mm Hg) hasta que la interrumpción del embarazo se lleve a cabo con seguridad, siempre que no existan otras alteraciones que pongan en peligro la vida de la madre, como hipertensión de difícil control, trastornos visuales, convulsiones, síndrome de HELLP o coagulación intravascular deseminada.
La hipertensión leve aislada que aparece durante el tercer trimestre de la gestación no suele requerir la interrupción inmediata del embarazo. Aunque se desconoce el grado mínimo de elevación de la presión arterial que puede lesionar al feto, es raro que ocurran efectos adversos por la hipertensión leve en pacientes que no desarrollan preeclampsia.109
Los datos sobre el tratamiento de la hipertensión crónica durante el embarazo son incompletos. La disminución notable de la presión arterial materna producida por el tratamiento puede afectar el flujo sanguíneo uteroplacentario, comprometiendo de este modo la viabilidad del feto. Por el contrario, cuando la presión arterial diastólica de la paciente es mayor de 84 mm Hg, la mortalidad perinatal aumenta.110 A pesar de estos problemas, es conveniente comenzar el tratamiento en mujeres con hipertensión crónica si la presión arterial diastólica es mayor de 90 a 95 mm Hg.111 Asimismo, la presión arterial diastólica por arriba de 100 a 110 mm Hg, ya sea aguda o crónica, debe tratarse para reducir el riesgo de hemorragia intracerebral materna. Es importante hacer énfasis en que estas recomendaciones son una guía y que el grado de presión arterial en el que se debe iniciar la administración de medicamentos no se conoce con claridad. Por otra parte, las pacientes que reciben tratamiento antihipertensivo antes del embarazo deben continuarlo durante toda la gestación. Como regla general, independientemente del esquema terapéutico utilizado antes del embarazo, este deberá continuarse, con excepción de los fármacos en que se ha demostrado un efecto adverso en la viabilidad del feto (ver adelante).
Una vez que se ha tomado la decisión de iniciar el tratamiento farmacológico, deben administrarse sólo los medicamentos antihipertensivos que se consideran seguros en el embarazo, como metildopa, betabloqueadores e hidralacina. Los diuréticos por lo general deben envitarse, en vista de que la preeclampsia tiende a acompañarse de un volumen plasmático bajo, por lo que una mayor disminución del volumen plasmático pudiera exacerbar la isquemia placentaria. Se han usado con éxito bloqueadores de los canales del calcio en hipertensión inducida por el embarazo, pero su toxicidad potencial para el feto se ha estudiado durante un tiempo más breve que la de otros agentes.112,113
Por el contrario, datos obtenidos en animales y humanos sugieren que la administración de inhibidores de la ECA o nitroprusiato se relaciona con mayor riesgo para el feto. Al parecer aumenta la incidencia de parto prematuro, bajo peso al nacer y muerte fetal en las pacientes tratadas con estos medicamentos durante el embarazo.114,115 Se ha descrito una fetopatía caracterizada por hipotensión, hipoplasia pulmonar, displasia tubular renal e hipocalvaria asociada al uso de inhibidores de la ECA.116 En algunos fetos ha ocurrido reducción importante en la VFG, causando oliguria, oligohidramnios y, por la menor cantidad de líquido amniótico, alteraciones traumáticas del desarrollo.117,118 Como resultado de estos hallazgos, está contraindicada la administración de inhibidores de la ECA durante el embarazo.114 Aunque el riesgo fetal por los inhibidores de la ECA parece ser mayor durante el segundo y tercer trimestres, estos agentes deben suspenderse en cuanto se sospeche o confirme un embarazo.
El nitroprusiato se ha asociado con toxicidad por cianuro tanto en la paciente como en el producto. No debe usarse durante el embarazo a menos que las otras alternativas sean ineficaces.
El tratamiento ideal de la preeclampsia consiste en prevenirla. Las alteraciones frecuentes en el metabolismo de los prostanoides y en la función plaquetaria han provocado la realización de estudios aleatorios que evalúan los efectos de la aspirina en dosis bajas (60 a 150 mg/día) en mujeres con mayor riesgo de preeclampsia.119-124 En los estudios iniciales, el riesgo relativo de hipertensión inducida por el embarazo se redujo en las pacientes tratadas con aspirina, lo mismo que la incidencia de productos de muy bajo peso al nacer y la necesidad de cesáreas. En contraste con estos estudios, el estudio reciente cooperativo sobre aspirina en dosis bajas durante el embarazo (CLASP, por sus siglas en inglés, n. del t.), que estudió a más de 9,000 mujeres con riesgo de preeclampsia, no observó este beneficio.125 Es posible que la diferencia sea resultado del menor riesgo de preeclampsia en las pacientes del CLASP (seis a ocho porciento) en comparación con las de otros estudios (17 a 52 porciento). Aunque las discrepancias entre estos estudios no permiten hacer recomendaciones absolutas, al parecer la aspirina en dosis bajas es relativamente segura tanto para la paciente como para el producto.
Además de la aspirina en dosis bajas, se han usado los suplementos de calcio (1.5 a 2.0 g de CaCO3/día) en un intento por prevenir las complicaciones hipertensivas durante el embarazo. La justificación de este tratamiento se basa en parte en la acción hipotensora potencial del calcio. Un estudio controlado demostró reducción del riesgo tanto de hipertensión inducida por el embarazo como de preeclampsia en el grupo tratado.126 Como en el caso de la aspirina, es necesario definir el papel general de los suplementos de calcio.
PRONOSTICO
La preeclampsia en primigrávidas suele ser una enfermedad autolimitada. La incidencia de hipertensión crónica o nefropatía en estas pacientes no es mayor que en las mujeres embarazadas de edad similar del grupo control.127 Además, el riesgo de desarrollar preeclampsia en los siguientes embarazos es bajo. Sin embargo, es posible que estas observaciones generales no se apliquen a primigrávidas con preeclampsia grave o eclampsia o a multigrávidas preeclámpticas; estas mujeres parecen tener un riesgo elevado de hipertensión y preeclampsia recurrente en embarazos posteriores. La hipertensión crónica también es más probable que ocurra en multigrávidas con preeclampsia, que a menudo tienen hipertensión esencial o nefropatía.
La hipertensión leve aislada del embarazo, en la que la presión arterial diastólica es menor de 90 a 95 mm Hg y no existe proteinuria o insuficiencia renal, no tiene efectos adversos sobre el feto o la madre. Sin embargo, es necesaria la vigilancia periódica en vista de que el riesgo de hipertensión de inicio tardío parece estar aumentado, sobre todo en pacientes en las que la presión arterial no se ha normalizado al décimo día del posparto.111
Enfermedad tromboembólica de las arterias renales
La formación de trombos o la embolización pueden afectar la circulación venosa o arterial del riñón. Los principales trastornos arteriales incluyen la trombosis, los émbolos de coágulos sanguíneos y la ateroembolia. La principal afección venosa es la trombosis de la vena renal, que ocurre con mayor frecuencia en pacientes con síndrome nefrótico.
La tromboembolia arterial o el aneurisma disecante de la aorta pueden causar infarto o atrofia isquémica de los riñones, dependiendo del grado de oclusión vascular, la rapidez con la que se desarrolla la oclusión y si las lesiones vasculares son resultado de coágulos o aterombolia. La circulación renal es muy susceptible a las enfermedades embólicas en vista de que los riñones en condiciones normales reciben el 20 porciento del gasto cardiaco.
OCLUSION AGUDA NO ATEROMATOSA DE LAS ARTERIAS RENALES
Diversos padecimientos se relacionan con el desarrollo de émbolos o trombosis, que pueden producir oclusión no ateromatosa de las arterias renales. Las principales fuentes de émbolos renales son los trombos murales, que son comunes sobre todo en pacientes con arritmias auriculares o infarto del miocardio previo, y con vegetaciones relacionadas con la endocarditis bacteriana. Los émbolos tumorales o los émbolos de grasa se observan con menos frecuencia. La trombosis de la arteria renal suele agregarse a lesiones ateromatosas estenóticas primarias o aparecer después de desgarros traumáticos de la íntima. En raras ocasiones la trombosis se presenta en forma espontánea.
El infarto en forma de cuña que se extiende hacia afuera del vaso afectado es el hallazgo patológico clásico que se observa en el riñón. La magnitud de la lesión renal depende del grado y la duración de la oclusión vascular. Por lo general se presenta necrosis irreversible si la oclusión total de la arteria renal dura dos horas o más.128 Sin embargo, la oclusión total de una duración menor de dos horas puede ocasionar necrosis tubular aguda, más que infarto.
Las manifestaciones clínicas de la oclusión de la arteria renal son variables. Los pacientes pueden estar asintomáticos si la oclusión es incompleta; sin embargo, cuando existen síntomas los más comunes son náusea, vómito, dolor en el flanco y fiebre. A la exploración física puede haber dolor en el flanco o abdominal a la palpación; durante la exploración debe llevarse a cabo una búsqueda cuidadosa de signos de embolización extrarrenal, como lesiones cutáneas o deficiencias neurológicas focales. Además, a menudo pueden detectarse factores subyacentes que predisponen a la embolización renal, como la fibrilación auricular o los infartos del miocardio recientes. En muchos pacientes la presión arterial aumenta poco tiempo después del infarto agudo como resultado de la estimulación del sistema renina-angiotensina por la isquemia renal. La presión arterial suele regresar al nivel basal después de dos a tres semanas.129
La mayor parte de las pruebas de laboratorio de rutina no son diagnósticas. La hematuria macroscópica o microscópica se observa solo en cerca de un tercio de los pacientes; lo frecuente de su ausencia puede reflejar la disminución notable en la irrigación sanguínea de la zona infartada, que ocasiona la interrupción de la filtración glomerular y el flujo de orina.130 Sin embargo, en condiciones clínicas apropiadas existe un hallazgo de laboratorio muy sugestivo de infarto renal: la elevación notable (a menudo mayor de cinco veces el límite superior normal) de la concentración de deshidrogenasa láctica (DHL) y, en ocasiones, un aumento leve de la concentración de aminotransferasa sérica.131 El renograma con radioisótopos es el procedimiento de elección para demostrar la disminución segmentaria o generalizada de la perfusión renal. Además, es un método diagnóstico no cruento que puede evitar la realización de la arteriografía renal o la tomografía computada con material de contraste. Por lo general, la pielografía intravenosa es menos segura y puede ser normal en pacientes con lesiones segmentarias.
Hasta ahora no se ha determinado el tratamiento óptimo para el infarto renal causado por un émbolo, pero por lo general se prefiere el tratamiento médico. Aunque la cirugía puede restaurar la permeabilidad vascular, se relaciona con una mayor mortalidad y no mejor recuperación de la función de la que se observa sólo con el tratamiento anticoagulante.130,132 Este último tratamiento puede causar mejoría importante incluso en pacientes con embolia bilateral e insuficiencia renal avanzada. La cirugía temprana sigue siendo el tratamiento de elección en pacientes con trombosis traumática de la arteria renal, en especial si la cirugía se realiza durante las primeras 24 horas.133
Es conveniente que el tratamiento sea inmediato para conservar el máximo de parénquima renal funcional. Por desgracia, el diagnóstico suele ser tardío por que no se reconocen los datos característicos descritos antes. Por ejemplo, en un estudio de 17 pacientes, el tiempo de diagnóstico fue de tres a seis días, con solo cinco casos diagnosticados el primer día.130
El tratamiento anticoagulante más común consiste en la administración de heparina intravenosa seguida de warfarina. Una alternativa tal vez más eficaz es iniciar el tratamiento con un fármaco trombolítico, como la estreptocinasa, con objeto de disolver el coágulo oclusivo.134,135 La infusión local intrarterial, en lugar de la administración intravenosa, puede disminuir la dosis total necesaria y, por lo tanto, reducir el riesgo de hemorragia sistémica.135
La cirugía, que no suele indicarse como tratamiento inicial para este trastorno, puede considerarse en pacientes con insuficiencia renal grave que no muestran mejoría en la función renal después de cuatro a seis semanas de tratamiento anticoagulante. La embolectomía tardía ha mejorado la función renal en algunos pacientes, indicando que es posible revertir en parte la atrofia isquémica.136
A pesar de la eficacia del tratamiento médico, la mortalidad temprana y tardía por los coágulos embólicos sigue siendo alta tanto por la embolización extrarrenal (en especial hacia el cerebro y el intestino) como por la enfermedad subyacente (v.gr., ateroesclerosis).130
TROMBOEMBOLIA RENAL
La enfermedad ateroembólica renal ocurre en forma característica en pacientes con ateroesclerosis ulcerada y generalizada. La mayoría de las veces la ateroembolia clínicamente importante es resultado de la manipulación de la aorta o de otras arterias de gran calibre durante la arteriografía, la angioplastía o la cirugía, incluyendo el trasplante de órganos.137-139 La ateroembolia también puede ser producida por el tratamiento crónico con warfarina, en vista de que la anticoagulación puede interferir con la cicatrización de las placas ateromatosas ulceradas, o por el tratamiento trombolítico intravenoso o intrarterial, ya que la superficie de la placa puede denudarse por la disolución de la fibrina.140 La ateroembolia espontánea, que en estudios de autopsia se demuestra hasta en el 12 porciento de los pacientes mayores de 80 años, suele ser asintomática y no se relaciona con una disminución considerable de la función renal.
El examen del tejido renal por microscopía de luz a menudo es patognomónico de ateroembolia. Las muestras características contienen fragmentos de material acelular y hendiduras bicóncavas en las arterias renales de pequeño y mediano calibre [ver figura 7]. Las hendiduras bicóncavas representan cristales de colesterol que han sido disueltos durante la fijación con parafina. Los émbolos tienen una forma irregular y no se distienden; como resultado, a menudo producen oclusión incompleta y atrofia isquémica secundaria en lugar de infarto. Con el tiempo suele desarrollarse una reacción a cuerpo extraño, que ocasiona proliferación de la íntima y mayor estrechamiento de la luz del vaso. Es frecuente que existan émbolos similares en otros órganos.
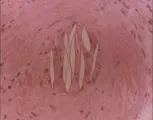
|
| Figura 7 |
| Ateroembolia renal |
Es importante resaltar que la localización de los émbolos dentro del riñón es regional. Por consiguiente, debido a errores en la selección del sitio adecuado para la biopsia renal percutánea, es posible que no se tomen muestras de lesiones renales características, incluso en pacientes en los que se comprueba en el estudio de autopsia la presencia de ateroémbolos en otros órganos o en el riñón.
La enfermedad vascular erosiva grave que ocasiona la formación de ateroémbolos se observa con mayor frecuencia en pacientes mayores de 50 años de edad. En vista de que la oclusión vascular es incompleta, los ateroémbolos no suelen producir infarto renal ni sus manifestaciones clínicas o hallazgos de laboratorio característicos, incluyendo dolor en flanco y elevación de la concentración de DHL sérica. Por lo tanto, la mayoría de los pacientes no tienen síntomas relacionados con los riñones.
Los antecedentes clínicos y la exploración física pueden mostrar signos y síntomas característicos de ateroembolia extrarrenal. Puede haber deficiencias visuales, dolor abdominal causado por pancreatitis aguda o infarto esplénico, y mialgias. Otras alteraciones características de embolia periférica que pueden observarse incluyen placas color naranja en las arteriolas de la retina, lívido reticular en las extremidades inferiores y zonas de gangrena en los dedos de los pies en presencia de pulsos pedios normales.141 La hipertensión mediada por los efectos de la angiotensina II también es frecuente y puede ser grave.
Los hallazgos de laboratorio son inespecíficos e incluyen el aumento súbito en la concentración de creatinina plasmática poco tiempo después de la realización de procedimientos quirúrgicos o radiológicos. El examen general de orina suele ser normal o casi normal, observándose algunas células o cilindros, que son compatibles con la presencia de atrofia isquémica. Sin embargo, algunos pacientes tienen sedimento urinario activo caracterizado por hematuria y cilindros celulares, incluyendo cilindros eritrocitarios. Es frecuente la presencia de eosinofilia en sangre periférica y en algunos casos puede haber hipocomplementemia; es probable que estos dos hallazgos reflejen activación del sistema inmune por la exposición de la superficie ateroembólica. Es importante distinguir la enfermedad ateroembólica de otros padecimientos que se acompañan de eosinofilia o hipocomplementemia, como glomerulonefritis, vasculitis sistémica y nefritis intersticial aguda.142,143
El desarrollo de insuficiencia después de la realización de cirugía vascular, arteriografía o angioplastía puede sugerir el diagnóstico de ateroembolia renal, aunque en este caso es más probable el diagnóstico de necrosis tubular aguda. Sin embargo, la presencia de embolia extrarrenal favorece el diagnóstico de enfermedad ateroembólica. La ateroembolia renal y la necrosis tubular aguda pueden diferenciarse con mayor precisión por su evolución típica. Los pacientes con necrosis tubular aguda presentan de manera característica un aumento progresivo en la concentración de creatinina plasmática antes de que la función renal se estabilice, pero al final regresa en forma espontánea al nivel basal en un periodo variable de cuatro a 21 días. Por el contrario, los pacientes con ateroembolia a menudo tienen una disminución aguda en la función renal que, dependiendo del grado de embolización, es autolimitada; sin embargo, en vista de que no es posible la disolución de los ateroémbolos, la insuficiencia renal por lo general es irreversible. En comparación con la insuficiencia renal aguda inducida por medio de contraste, el deterioro en la función renal que ocurre con la ateroembolia suele comenzar varios días después del evento desencadenante (v.gr., angiografía coronaria).138 En algunas ocasiones los pacientes pueden tener cierta mejoría en la función renal con el tiempo, tal vez por la resolución de la necrosis tubular aguda coexistente o por el desarrollo de flujo colateral hacia las zonas isquémicas.141,144
El diagnóstico puede ser difícil, sobre todo cuando no es posible identificar factores precipitantes, como ocurre en la ateroembolia espontánea, o cuando la evolución de la enfermedad es atípica. Por ejemplo, puede presentarse insuficiencia renal progresiva subaguda si los pacientes tienen cicatrización fibrótica en respuesta a la ateroembolia o si ocurren episodios nuevos de embolización. En este caso puede ser necesaria la biopsia de un órgano afectado como la piel o el riñón, sobre todo si las manifestaciones extrarrenales, un sedimento urinario activo, hipocomplementemia o eosinofilia, indican un trastorno sistémico. Sin embargo, el resultado negativo de la biopsia renal no excluye necesariamente el diagnóstico de ateroembolia, en vista de que es posible que en la muestra no existan vasos afectados.
Hasta ahora no existe tratamiento específico para la ateroembolia renal. En pacientes con alto riesgo deben evitarse procedimientos como la arteriografía, si existen técnicas menos cruentas que puedan proporcionar la misma información. Además, el tratamiento anticoagulante no está indicado en vista de que puede agravar el trastorno al favorecer la ateroembolia recurrente.142
Necrosis de la corteza renal
La necrosis de la corteza renal es un estado clinicopatológico que se caracteriza por insuficiencia renal aguda, por lo general anúrica, y grados variables de necrosis o infarto de los componentes corticales, túbulos y glomérulos. Este trastorno se relaciona con mayor frecuencia con el desprendimiento de placenta o alguna otra complicación al final del embarazo, como la placenta previa. También puede ocurrir en presencia de choque séptico u otros padecimientos que ocasionan hipovolemia, como las quemaduras o la pancreatitis hemorrágica aguda.145
La principal alteración patológica en esta enfermedad es la necrosis de todos los componentes de la corteza renal. También puede observarse necrosis de los vasos sanguíneos y trombos dispersos en las arterias interlobulares, arteriolas y glomérulos. En casos avanzados la corteza puede estar destruida por completo y las arterias presentar necrosis y trombosis extensas. Sin embargo, incluso en estos casos suele mantenerse intacto el tejido renal en las zonas justo por abajo de la cápsula, en la región yuxtamedular y en la médula.146 Las nefronas por abajo de la cápsula no son funcionales, ya que sus segmentos intratorácicos están necrosados. En experimentos realizados en animales, las nefronas corticales profundas se mantienen intactas, tal vez porque las regiones yuxtamedulares son perfundidas por las arterias arcuatas, más que por los vasos interlobulares trombosados. Se desconoce si existen hallazgos similares en humanos que puedan explicar la recuparación parcial de la función renal en algunos pacientes.147
La patogenia de la necrosis cortical renal no se conoce por completo. Sin embargo, la coagulación intravascular diseminada y la isquemia renal grave se consideran importantes en ella. En los casos de necrosis cortical se encuentran trombos de fibrina en los vasos renales. Este hallazgo es similar al cuadro histológico de la reacción generalizada de Shwartzman, en la que se administran endotoxinas en forma periódica a animales, ocasionando trombosis generalizada y hemorragia que a su vez produce necrosis de la corteza renal. Esta reación ocurre con más facilidad en animales preñados en comparación con grupos control de animales no preñados.148 Aunque la mayor parte de las evidencias indican que la isquemia renal grave es el principal mecanismo patogénico en la mayoría de los pacientes que desarrollan necrosis cortical, en algunos casos la coagulopatía producida por la liberación de tromboplastina a la circulación produce depósito de fibrina intravascular y una mayor reducción de la perfusión renal.
La necrosis de la corteza renal se caracteriza por la aparición súbita de oliguria o anuria. También es frecuente que exista hematuria macroscópica, dolor en el flanco e hipertensión arterial. En más del 50 porciento de los casos este tratorno es causado por el desprendimiento de la placenta u otra complicación del tercer trimestre del embarazo, como la placenta previa.149
Los hallazgos de laboratorio en la necrosis cortical son inespecíficos. El BUN y la concentración de creatinina plasmática aumentan diariamente, lo cual es reflejo de la disminución aguda de la velocidad de filtración glomerular. Además de la hematuria y la proteinuria leve, el examen general de orina puede mostrar leucocitos, células del epitelio tubular y cilindros granulares. Estos hallazgos son similares a los que se observan en la necrosis tubular aguda. Pueden existir alteraciones hematológicas relacionadas con la coagulación intravascular deseminada, sobre todo en pacientes con complicaciones obstétricas primarias.
El diagnóstico diferencial de la necrosis incluye la necrosis tubular aguda, otras causas de insuficiencia renal aguda al final del embarazo [ver tabla 4] y algunas otras causas de anuria. Varias manifestaciones clínicas pueden ayudar a diferenciar la necrosis cortical de la necrosis tubular aguda. En pacientes con necrosis de la corteza renal son comunes el dolor en el flanco, la hematuria macroscópica y la anuria, pero son raros en los casos de necrosis tubular aguda. Asimismo, en la necrosis tubular aguda la función renal comienza a mejorar en un periodo de cuatro a 21 días y con el tiempo la recuperación es casi completa en la mayoría de los pacientes. Por el contrario, la anuria o la oliguria prolongada es una manifestación característica de la necrosis de la corteza renal, además de la falta de mejoría en la función renal o su recuperación parcial y tardía.150
Aunque la insuficiencia renal aguda tiene muchas causas, solo algunas se acompañan de anuria. Los padecimientos que deben considerarse en presencia de insuficiencia renal aguda y anuria, además de la necrosis cortical, se incluyen el estado de choque, que puede causar hipoperfusión importante de los riñones y necrosis tubular aguda agreda, la obstrucción completa del aparato urinario, que por lo general puede excluirse mediante la ultrasonografía renal, alguno de los síndromes urémico-hemolíticos [ver antes, Síndromes urémico-hemolíticos], y en raras ocasiones, la glomerulonefritis grave, la vasculitis o la oclusión vascular bilateral completa. Las condiciones típicas en las que ocurre la necrosis de la corteza renal a menudo permiten distinguirla de estos trastornos. Sin embargo, cuando el diagnóstico es dudoso pueden estar indicados los estudios radiográficos o la biopsia renal. La observación de calcificaciones renales en la radiografía simple de abdomen sugiere la presencia de necrosis de la corteza renal, aunque es frecuente que no se visualicen depósitos de calcio durante un lapso de uno a dos meses, limitando de esta forma su utilidad como criterio diagnóstico.151
Hasta ahora no se ha demostrado que algún tratamiento específico afecte la evolución de la necrosis cortical. En cerca de una tercera parte de los pacientes la diuresis aumenta y la función renal mejora en un periodo de dos a cuatro meses, establizándose a un nivel del 15 al 50 porciento de lo normal.150 Esta recuperación parcial, que puede ser suficiente para suspender la diálisis, tal vez ocurre, al menos en parte, gracias a que las nefronas yuxtamedulares no suelen presentar necrosis isquémica irreversible. Sin embargo, la fase de recuperación puede ser seguida por el deterioro gradual de la función renal durante un periodo de algunos años; esta alteración puede reflejar daño hemodinámico que afecta a los glomérulos supervivientes.152
Nefropatía por células falciformes
La afección renal es común en los pacientes cuyos eritrocitos contienen hemoglobina S, o hemoglobina falciforme. Este tipo de hemoglobina es responsable de la anemia de células falciformes, el rasgo falciforme y otras hemoglobinopatías. La principal manifestación renal en individuos con el rasgo falciforme es la hematuria recurrente; sin embargo, en personas con anemia de células falciformes se presentan de manera característica diversas alteraciones patológicas y funcionales [ver tabla 6]. Por consiguiente, la nefropatía por células falciformes siempre debe considerarse en el diagnóstico diferencial en los pacientes de raza negra que presentan hematuria y necrosis papilar.
|
|
|
La médula es la porción del riñón más afectada en la nefropatía por células falciformes. Existen zonas localizadas de hemorragia o necrosis que finalmente producen inflamación intersticial, atrofia tubular y fibrosis. También son comunes las lesiones glomerulares (ver adelante).
El mecanismo patogénico primario en esta nefropatía parece ser la formación de células falciformes a partir de eritrocitos en los capilares medulares. La osmolaridad elevada y la presión arterial de oxígeno baja presentes en esta región contribuyen a la transformación de los eritrocitos en células falciformes. Como resultado de estas alteraciones medulares, puede haber pérdida de la función, lo que ocasiona anomalías tubulares similares a las observadas en los animales después de la extirpación quirúrgica de las papilas. Estas alteraciones tubulares incluyen la disminución en la capacidad de concentración, el deterioro en la acidificación urinaria y la reducción en la secreción de potasio (ver adelante).
Las manifestaciones clínicas de la nefropatía por células falciformes varían desde alteraciones subclínicas en la excreción de agua y potasio hasta hematuria macroscópica o síndrome nefrótico. El diagnóstico de cualquiera de estas alteraciones se basa en la confirmación de la presencia de hemoglobina S mediante estudios apropiados, como la electroforesis de hemoglobina.
HEMATURIA
La hematuria macroscópica indolora es una de las manifestaciones renales más comunes en pacientes con el rasgo falciforme.153 No se conoce con claridad porqué la hematuria es más común en individuos con el rasgo falciforme que en los pacientes con enfermedad de células falciformes, pero tal vez se relaciona con la prevalencia mucho mayor del rasgo falciforme.
La hematuria puede aparecer en forma espontánea o presentarse después de un traumatismo leve. Por lo general es recurrente y a menudo proviene de un solo riñón; puede ser microscópica o macroscópica. En la mayoría de los casos la hematuria es leve y autolimitada, persistiendo varios días o semanas, pero sin requerir tratamiento específico. En algunos individuos, la hemorragia puede ser más profusa o prolongada, por lo que requiere tratamiento.154
INFARTO RENAL Y NECROSIS PAPILAR
La necrosis papilar es resultado de la trombosis o la precipitación de sedimento en los vasos rectos y constituye una de las manifestaciones más comunes de la anemia de células falciformes [ver figura 9]. La mayoría de los pacientes con esta complicación están asintomáticos y su función renal es normal. Sin embargo, la formación de cicatrices puede predisponer al desarrollo de infecciones del aparato urinario.

|
| Figura 9 |
| Riñón normal vs enfermedad de células falciformes |
Los pacientes con enfermedad de de células falciformes o rasgo falciforme pueden presentar infartos renales. En estos casos se ha informado la presencia de grandes hematomas perirrenales. Es posible que los hematomas se desarrollen debido a que las arterias renales principales permanecen permeables y continúan irrigando las zonas infartadas.155
CAPACIDAD DE CONCENTRACION DISMINUIDA
La disminución en la capacidad para concentrar la orina es un hallazgo general y temprano en pacientes con enfermedad de células falciformes. La formación de células falciformes en los capilares de los vasos interfiere con el intercambio de contracorriente, causando atrapamiento ineficaz de solutos en la médula interna, que es el sitio donde se genera la máxima osmolaridad urinaria en presencia de hormona antidiurética. Por consiguiente, la osmolaridad urinaria máxima en niños de 10 años de edad con anemia de células falciformes es solo de 400 a 450 mOsm/kg, que es mucho menor que el valor normal de 900 a 1,400 mOsm/kg.156 Este defecto en la capacidad de concentración es reversible, después de múltiples transfusiones, en pacientes homocigotos para el gen falciforme y menores de 10 ó 15 años de edad, pero es irreversible en individuos de mayor edad. Estas alteraciones son mucho menos graves y aparecen a una edad mayor en individuos con el rasgo falciforme o con la enfermedad de células falciformes-hemoglobina C.
El principal síntoma producido por la alteración en la capacidad para concentrar la orina es la poliuria. Esta manifestación puede ser molesta, sobre todo en la noche por la nicturia intermitente. Sin embargo, puede mantenerse un balance de líquidos normal mientras la sed sea normal y el aporte de agua suficiente.
SECRECION RENAL BAJA DE HIDROGENO Y POTASIO
La lesión del tubo colector cortical, que es el sitio de secreción de potasio y acidificación urinaria máxima, puede producir hipercalemia y acidosis metabólica.157 En vista de que la concentración de aldosterona es normal en la mayoría de los pacientes con anemia de células falciformes, la lesion isquémica de las células es el mecanismo más probable que ocasiona estas alteraciones. Por el contrario, en pacientes con el rango falciforme el control del potasio no está afectado.158
INSUFICIENCIA RENAL AGUDA
En pacientes con enfermedad de células falciformes o con el rasgo falciforme es poco común la disminución rápida de la función renal, pero puede ocurrir en dos situaciones. En primer lugar, la obstrucción completa del aparato urinario, causada en ocasiones por el desprendimiento bilateral de las papilas, produce insuficiencia renal aguda. En segundo lugar, puede ocurrir insuficiencia renal aguda como consecuencia del desarrollo de rabdomiolisis por el ejercicio vigoroso. En este caso la rabdomiolisis es resultado del aumento en la formación de células falciformes, que puede producir isquemia muscular.
Nefritis por radiación
Al igual que otros órganos, los niños son sensibles a la radiación, sobre todo cuando está expuesto a una dosis acumulativa de más de 2,300 cGy (2,300 rads).159 La nefritis por radiación, que fue un trastorno común en décadas anteriores, en la actualidad se observa en raras ocasiones. La desaparición casi completa de este tipo de nefropatía fue resultado del perfeccionamiento en las técnicas de radioterapia localizada, de la disminución del uso de radiaciones en las enfermedades no malignas y del empleo generalizado de la quimioterapia en lugar de la radiación total del abdomen en los padecimientos malignos intrabdominales. En la actualidad la mayoría de los casos de nefritis por radiación ocurren como resultado de la radioterapia de los tumores malignos retroperitoneales diseminados o después de la preparación para los trasplantes de médula ósea. En este último caso, que implica el uso de quimioterapia y radioterapia, pueden identificarse lesiones vasculares hasta en el 50 porciento de los pacientes [ver antes, Síndromes urémico-hemolíticos].
En general, predominan las lesiones vasculares y glomerulares, que son indistinguibles de las lesiones observadas en el síndrome urémico-hemolítico.160 También suelen existir alteraciones tubulointersticiales de gravedad variable. Con el tiempo, estas alteraciones inflamatorias progresan hasta la fibrosis intersticial, la atrofia tubular y la glomeruloesclerosis. Sin embargo, antes de que sea evidente la disfunción renal hay un intervalo de tiempo variable sin alteraciones. La nefritis aguda por radiación puede aparecer incluso a los seis meses después de la radioterapia, mientras que la nefritis crónica puede presentarse 12 o más años después de la exposición a la radiación.161,162
Además de la insuficiencia renal, también suele observarse hipertensión arterial en pacientes con nefritis por radiación. El aumento de la presión arterial parece ser mediado sobre todo por el sistema renina-angiotensina, por lo que es probable que responda al tratamiento con inhibidores de la enzima conversora de angiotensina.163 La hipertensión arterial maligna puede desarrollarse durante el episodio agudo o después de algunos meses o años.
- Balow J: Renal vasculitis. Kidney Int 27:954, 1985
- Conn DL: Update on systemic necrotizing vasculitis. Mayo Clin Proc 64:535, 1989
- Jennette JC, Falk RJ: The coming age of serologic testing for antineutrophil cytoplasmic antibodies. Mayo Clin Proc 69:908, 1994
- Kallenberg C, Brouwer E, Weening J, et al: Anti-neutrophil cytoplasmic antibodies: current diagnostic and pathophysiological potential. Kidney Int 46:1,1994
- Cohen-Tervaert JW, Goldschmeding R, Elema JD, et al: Autoantibodies against myeloid lysosomal enzymes in crescentic glomerulonephritis. Kidney Int 37:799, 1990
- Falk RJ, Terrell RS, Charles LA, et al: Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA 87:4115, 1990
- Ewert B, Jennette J, Falk R: Anti-myeloperoxidase antibodies stimulate neutrophils to damage human endothelial cells. Kidney Int 41:375, 1992
- Lockwood C: Specificity and pathogenicity of antineutrophil cytoplasm antibodies. Exp Nephrol 1:13, 1993
- Weiss MA, Crissman JD: Segmental necrotizing glomerulonephritis: diagnostic, prognostic, and therapeutic significance. Am J Kidney Dis 6:199, 1985
- Fauci AS, Haynes BF, Katz P: The spectrum of vasculitis: clinical, pathologic, immunologic, and therapeutic considerations. Ann Intem Med 89:660, 1978
- Fauci AS, Haynes BF, Katz P, et al: Wegener's granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med 98:76, 1983
- Israel H: sulfamethoxazole-trimethoprim therapy for Wegener's granulomatosis. Arch Intern Med 148:2293, 1988
- Valeriano-Marcet J, Spiera H: Treatment of Wegener's granulomatosis with sulfamethoxazole-trimethoprim. Arch Intern Med 151:1649, 1991
- Jayne DRW, Davies MJ, Fox CJV , et al: Treatment of systemic vasculitis with pooled intravenous immunoglobulin. Lancet 337:1137, 1991
- Buckley RH, Schiff RI: The use of .intravenous immune globulin in immunodeficiency diseases. N Engl J Med 325:l10, 1991
- Hoffman GS, Leavitt RY, Kerr GS, et al: The treatment of Wegener's granulomatosis with glucocorticoids and methotrexate. Arthritis Rheum 35:1322, 1992
- Andrassy K, Waldherr R, Erb A, et al: De novo glomerulonephritis during remission from Wegener's granulomatosis. Clin Nephrol 38:295, 1992
- Mathew T: Recurrence of disease after renal transplantation. Am J Kidney Dis 12:85, 1988
- Steinman T, Jaffe B, Monaco A, et al: Recurrence of Wegener's granulomatosis after kidney transplantation. Am J Med 68:458, 1980
- Clarke A, Bitton A, Eappen R, et al: Treatment of Wegener's granulomatosis after renal transplantation: is cyclosporine the preferred treatrnent? Transplantation 50:1047, 1990
- Aunsholt N, Ahlbom G: Recurrence of Wegener's granulomatosis after kidney transplantation involving the kidney graft. Clin Transplantation 3:159, 1989
- Stockigt JR, Topliss DJ, Hewett MJ: High-renin hypertension in necrotizing vasculitis (Ietter). N Engl J Med 300:1218, 1979
- Lanham JG, Elkon KB, Pusey CD, et al: Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore) 63:65, 1984
- Albert DA, Rimon D, Silverstein MD: The diagnosis of polyarteritis nodosa: I. A literature-based decision analysis approach. Arthritis Rheum 31:1117, 1988
- Frohnert PP, Sheps SG: Long-term follow-up study of periarteritis nodosa. Am J Med 43:8, 1967
- Fauci.AS, Katz P, Haynes BF, et al: Cyclophosphamide therapy of severe systemic necrotizing vasculitis. N Engl J Med 301:235, 1979
- Gibson LE: Cutaneous vasculitis: approach to diagnosis and systemic associations. Mayo Clin Proc 65:221, 1990
- Rieu P, Pruna A, Nochy D, et al: Henoch-Schönlein nephritis (HSN) in adults (abstr). J Am Soc NephroI 5:359, 1994
- Cameron JS: Henoch-Schönlein purpura: clinical presentation. Contrib Nephrol 40:246, 1984
- Hené RJ, Velthuis P, van de Wiel A, et al: The relevance of IgA deposits in vessel walls of clinically normal skin. Arch Intern Med 146:745, 1986
- Austin H III, Balow J: Henoch-Schönlein nephritis: long-term prognostic features and the challenge of therapy. Am J Kidney Dis 2:512, 1983
- Fredenberg M, Malkinson F: Sulfone therapy in the treatment of leukocytoclastic vasculitis. J Am Acad DermatoI 16:772, 1989
- Nast C, Ward H, Koyle M, et al: Fate of renal grafts with recurrent Henoch-Schonlein purpura following renal transplantation. Am J Kidney Dis 9:39,1987
- Hasegawa A, Kawamura T, Ito H, et al: Fate of renal grafts with recurrent Henoch-Schönlein purpura nephritis in children. Transplant Proc 21:2130, 1989
- Mesiani R, Bellavita P, Fenili D, et al: Hepatitis C virus infection in patients with essential mixed cryoglobulinemia. Ann Intern Med 117:573, 1992
- Agnello V, Chung RT, Kaplan LM: A role for hepatitis C virus infection in type II cryoglobulinemia. N Engl J Med 327:1490, 1992
- D'Amico G, Colasanti G, Ferrario F, et al: Renal involvement in essential mixed cryoglobulinemia. Kidney Int 35:1004, 1989
- Frankel AH, Singer DRJ, Winearls CG, et al: Type II essential mixed cryoglobulinernia: presentation, treatment and outcome in 13 patients. Q J Med 82:101, 1992
- Gorevic PD, Kassab HJ, Levo Y, et al: Mixed cryoglobulinemia: clinical aspects and long-term follow-up of 40 patients. Am J Med 69:287, 1980
- Ferri C, Moriconi L, Gremignai G, et al: Treatment of the renal involvement in rnixed cryoglobulinemia with prolonged plasma exchange. Nephron 43:246, 1986
- Bonomo L, Casato M, Afeltra A, et al: Treatment of idiopathic mixed cryoglobulinemia with alpha interferon. Am J Med 83:726, 1987
- Shindo M, Di Bisceglie AM, Cheung L, et al: Decrease in serum hepatitis C viral RNA during alpha-interferon therapy for chronic hepatitis C. Ann Intern Med 115:700, 1991
- Johnson R, Gretch D, Couser W, et al: Hepatitis C virus-associated membranoproliferative glomerulonephritis: effect of (-interferon therapy (abstr). J Am Soc Nephrol 5:352, 1994
- Parker CW: Allergic reactions in man. Pharmacol Rev 34:85, 1982
- Truong L, Kopelman RG, Williams GS, et al: Temporal arteritis and renal disease: case report and review of the literature. Am J Med 78:171, 1985
- Lai KN, Chan KW , Ho CD: Glomerulonephritis associated with Takayasu's arteritis: report of three cases and review of literature. AmJ Kidney Dis 7:197, 1986
- Steen VD, Medsger TA Jr, Osial TA Jr, et al: Factors predicting development of renal involvement in progressive systemic sclerosis. Am J Med 76:779, 1984
- GougeSF, Wilder K, Welch P, et al: Scleroderma renal crisis prior to scleroderma. Am J Kidney Dis 14:236, 1989
- Cannon PJ, Hassar M, Case DB, et al: The relationship of hypertension and renal failure in scleroderma (progressive systemic sclerosis) to structural and functional abnormalities of the renal cortical circulation. Medicine (Baltimore) 53:1, 1974
- Okano Y, Steen VD, Medsger TA Jr: Autoantibody reactive with RNA polymerase III in systemic sclerosis. Ann lntern Med 119:1005, 1993
- Steen VD, Constantino JP, Shapiro AP, et al: Outcome of renal crisis in systemic sclerosis: relationship to availability of angiotensin converting enzyme (ACE) inhibitors. Ann Intern Med 113:352, 1990
- Helfrich DJ, Danner B, Steen VD, et al: Normotensive renal failure in systemic sclerosis. Arthritis Rheum 32:1128, 1989
- Donohoe J: Scleroderma and the kidney. Kidney lnt 41:462, 1992
- Copley JA, Smith BJ: Continuous ambulatory peritoneal dialysis and scleroderma. Nephron 40:353, 1985
- Paul M, Bear R, Sugar L: Renal transplantation in scleroderma. J Rheumatoll1:406,1984
- Merino G, Sutherland D, Kjellstrand C, et al: Renal transplantation for progressive systemic sclerosis with renal failure. Am J Surg 133:745, 1977
- Woodhall P, McCoy R, Gunnels C, et al: Apparent recurrence of progressive sclerosis in a renal allograft. JAMA 236:1032, 1976
- Remuzzi G, Tognoni G: Thrombotic thrombocytopenic purpura (TTP) and haemolytic uraemic syndrome (HUS): a clinical puzzle or a methodological challenge? J NephroI 2:77, 1989
- Moake J, Rudy C, Troll J, et al: Unusually large factor VIII:von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med 307:1432, 1982
- Murphy WG, Moore JC, Kelton JG: Calcium-dependent cysteine protease activity in the sera of patients with thrombotic thrombocytopenic purpura. Blood 70:1683, 1987
- Case records of the Massachusetts General Hospital: Case 30-1991. N Engl J Med 325:265, 1991
- Moore JC, Murphy WG, Kelton JG: Calpain proteolysis of von Willebrand factor enhances its binding to platelet membrane glycoprotein IIb/IIIa: an explanation for platelet aggregation in thrombotic thrombocytopenic purpura. Br J Haematol 74:457, 1990
- Bergstein JM, Riley M, Bang NU: Role of plasminogen-activator inhibitor type 1 in the pathogenesis and outcome of the hemolytic uremic syndrome. N Engl J Med 327:755, 1992
- Case records of the Massachusetts General Hospital: Case 33-1994. N Engl J Med 331:661, 1994
- Siddiqui FA, Lian EC- Y: Platelet-agglutinating protein P37 from a thrombotic thrombocytopenic purpura plasma forms a complex with human immunoglobulin G. Blood 71:299, 1988
- Fitzpatrick MM, Shah V, Trompeter RS, et al: lnterleukin-8 and polymorphophil leucocyte activation in hemolytic uremic syndrome of childhood. Kidney lnt 42:951, 1992
- Morel-Maroger L, Kanfer A, Solez K, et al: Prognostic importance of vascular lesions in acute renal failure with microangiopathic hemolytic anemia (hemolytic-uremic syndrome): clinicopathologic study in 20 adults. Kidney lnt 15:548, 1979
- Amorosi EL, Ultmann JE: Thrombotic thrombocytopenic purpura: report of 16 cases and review of the literature. Medicine (Baltimore) 45:139, 1966
- Pavia AT, Nichols CR, Green DP ,et al: Hemolytic-uremic syndrome during an outbreak of Escherichia coli 0157:H7 infections in institutions for mentally retarded persons: clinical and epidemiologic observations. J Pediatr 116:544, 1990
- Milford DV, Taylor CM, Guttridge B, et al: Haemolytic uraemic syndromes in the British Isles 1985-8: association with verocytotoxin producing Escherichia coli: I. Clinical and epidemiological aspects. Arch Dis Child 65:716, 1990
- Jackson AM, Rose BD, Graff LG, et al: Thrombotic microangiopathy and renal failure associated with antineoplastic chemotherapy. Ann lntern Med 101:41, 1984
- Monnens L, Molenaar J, Lambert PH, et al: The complement system in hemolytic-uremic syndrome in childhood. Clin NephroI 13:168, 1980
- Thompson CE, Damon LE, Reis CA, et al: Thrombotic microangiopathies in the 1980s: clinical features, response to treatment, and the impact of the human immunodeficiency virus epidemic. Blood 80:1890, 1992
- Chauveau D, Dessasis JF, Choukroun G, et al: Hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP) in seven HIV patients (abstr). J Am Soc Nephrol 5:389, 1994
- Rock GA, Shumak KH, Buskard NA, et al: Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 325:393, 1991
- Byrnes J, Moake J, Klug P, et al: Effectiveness of the cryosupernatant fraction of plasma in the treatment of refractory thrombotic thrombocytopenic purpura. Am J Hematol 34:169, 1990
- Bell WR, Braine HG, Ness PM, et al: Improved survival of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: clinical experience in 108 patients. N Engl J Med 325:398, 1991
- Moake J: TTP-desperation, empiricism, progress. N Engl J Med 325:426, 1991
- Resove MH, Ho WG, Goldfinger D: Ineffectiveness of aspirin and dipyridamole in the treatment of thrombotic thrombocytopenic purpura. Ann Intern Med 96:27, 1982
- Onundarson PT, Rowe JM, Heal JM, et al: Response to plasma exchange and splenectomy in thrombotic thrombocytopenic purpura: a 10-year experience in a single institution. Arch Intern Med 152:791, 1992
- Wong P, Itoh K, Yoshida S: Treatment of thrombotic thrombocytopenic purpura with intravenous gamma globulin (letter). N Engl J Med 314:385, 1986
- Lind S: Thrombocytopenic purpura and platelet transfusion (letter). Ann Intern Med 106:478, 1987
- Harkness DR, Byrnes JJ, Lian EC-Y, et al: Hazard of platelet transfusion in thrombotic thrombocytopenic purpura. JAMA 246:1931, 1981
- Siegler RL, Milligan MK, Burningham TH, et al: Long-term outcome and prognostic indicators in the hemolytic-uremic syndrome. J Pediatr 118:195, 1991
- Fitzpatrick M, Shah V, Trompeter R, et al: Long-term renal outcome of childhood haemolytic uraemic syndrome. BMJ 303:498, 1991
- Remuzzi G: HUS and TTP: variable expression of a single entity. Kidney Int 32:292, 1987
- Kaplan B, Drummund K: The hemolytic-uremic syndrome is a syndrome. N Engl J Med 298:964, 1978
- Brown M, Whitworth J: The kidney in hypertensive pregnancies¾victim and villain. Am J Kidney Dis 20:427, 1992
- Lindheimer M, Katz A: Sodium and diuretics in pregnancy. N Engl J Med 288:891, 1973
- Gant NF, Worley RJ, Everett RB, et al: Control of vascular responsiveness during human pregnancy. Kidney Int 18:253, 1980
- Davison JM, Dunlop W: Renal hemodynamics and tubular function in normal human pregnancy. Kidney Int 18:152, 1980
- Davison JM, Shiells EA, Philips PR, et al: Influence of humoral and volume factors on altered osmoregulation of normal human pregnancy. Am J Physiol 258:F900, 1990
- Davison JM, Shiells EA, Philips P, et al: Serial evaluation of vasopressin release and thirst in human pregnancy: role of human chorionic gonadotrophin in the osmoregulatory changes of gestation. J Clin Invest 81:798, 1988
- Lindheimer MD, Barron WM, Davison JM: Osmoregulation of thirst and vasopressin release in pregnancy. Am J Physiol 257:F159, 1989
- Lindheimer M, Barron W, Davison J: Osmotic and volume control of vasopressin release in pregnancy .Am J Kidney Dis 17:105, 1991
- Davison JM, Shiells EA, Barron WM, et al: Changes in the metabolic clearance of vasopressin and plasma vasopressinase throughout human pregnancy. J Clin Invest 83:1313, 1989
- Durr JA, Hoggard JG, Hunt JM, et al: Diabetes insipidus in pregnancy associated with abnormally high circulating vasopressinase activity. N EnglJ Med 316:1070, 1987
- Iwasaki Y, Oiso Y, Kondo K, et al: Aggravation of subclinical diabetes insipidus during pregnancy. N Engl J Med 324:522, 1991
- Lim V, Katz A, Lindheimer M: Acid-base regulation in pregnancy. Am J Physiol 231:1764, 1976
- Roberts J, Taylor R, Goldfen A: Clinical and biochemical evidence of endothelial cell dysfunction in the pregnancy syndrome preeclampsia. Am J Hypertens 4:700, 1991
- Redman C: Platelets and the beginnings of preeclampsia {editorial). N Engl J Med 323:478, 1990
- Klonoff-Cohen HS, Savitz DA, Cefalo RC, et al: An epidemiologic study of contraception and preeclampsia. JAMA 262:3143, 1989
- Need JA: Pre-eclampsia in pregnancies by different fathers: immunological studies. Br Med J 1:548, 1975
- Packham DK, Mathews DC, Fairley KF , et al: Morphometric analysis of pre-eclampsia in women biopsied in pregnancy and post-partum. Kidney Int 34:704, 1988
- lhle BU, Long P, Oats J: Early-onset pre-eclampsia: recognition of underlying renal disease. Br Med J 294:79, 1987
- Fisher KA, Luger A, Spargo BH, et al: Hypertension in pregnancy: clinical-pathological correlations and remote prognosis. Medicine (Baltimore) 60:267, 1981
- Grunfeld J-P, Pertuiset N: Acute renal failure in pregnan'cy: 1987. Am J Kidney Dis 9:359, 1987
- Homans DC, Blake GD, Harrington JT, et al: Acute renal failure caused by ureteral obstruction by a gravid uterus. JAMA 246:1230, 1981
- Sibai BM, Abdella TM, Anderson GD: Pregnancy outcome in 211 patients with mild chronic hypertension. Obstet Gynecol 61:571, 1983
- Ferris TF: Hypertension in pregnancy. The Kidney 23:1, 1990
- Lindheimer MD, Katz AI: Hypertension in pregnancy .N Engl J Med 313:675, 1985
- Fenakel K, Fenakel G, Appelman Z, et al: Nifedipine in the treatment of severe preeclampsia. Obstet Gynecol 77:33l, 1991
- Carbonne B, Jannet D, Touboul C, et al: Nicardipine treatment of hypertension during pregnancy. Obstet Gynecol 81:908, 1993
- Are ACE inhibitors safe in pregnancy? (editorial). Lancet 2:482, 1989
- Millan M, Carvallo P, Izumi -S-I et al: Novel sites of expression of functional angiotensin II receptors in the late gestation fetus. science 244:1340, 1989
- Pryde P, sedman A, Nugent C, et al: Angiotensin-converting enzyme inhibitor fetopathy. J Am Soc NephroI 3:1575, 1993
- Schubiger G, Flurry G, Nussberger J: Enalapril for pregnancy-induced hypertension: acute renal failure in a neonate. Ann Intern Med 108:215, 1988
- Mehta N, Modi N: ACE inhibitors in pregnancy (letter). Lancet 2:96, 1989
- Benigni A, Gregorini G, Frusca T, et al: Effect of low-dose aspirin on fetal and maternal generation of thromboxane by platelets in women at risk for pregnancy-induced hypertension. N Engl J Med 321:357, 1989
- Schiff E, Peleg E, Goldenberg M, et al: The use of aspirin to prevent pregnancy-induced hypertension and cover the ratio of thromboxane A2 to prostacyclin in relatively high risk pregnancies. N Engl J Med 321:35l, 1989
- Wallenburg HCS, Makovitz JW, Dekker GA, et al: Low-doseaspirin prevents pregnancy-induced hypertension and pre-eclampsia in angiotensin-sensitive primigravidae. Lancet 1:1, 1986
- Beaufils M, Uzan S, Donsimoni R, et al: Prevention of pre-eclampsia by early antiplatelet therapy. Lancet 1:840, 1985
- McParland P, Pearce JM, Chamberlain GVP: Doppler ultrasound and aspirin in recognition and prevention of pregnancy-induced hypertension. Lancet 335:1552, 1990
- Imperiale TF, Petrulis AS: A meta-analysis of low-dose aspirin for the prevention of pregnancy-induced hypertensive disease. JAMA 266:237, 1991
- CLASP (Collaborative Low-dose Aspirin study in Pregnancy) CoIlaborative Group: CLASP: a randomised trial of low-dose aspirin for the prevention and treatment of pre-eclampsia among 9364 pregnant women. Lancet 343:619, 1994
- Belzian JM, Villar J, Gonzalez L, et al: Calcium supplementation to prevent hypertensive disorders of pregnancy. N Engl J Med 325:1399, 1991
- Sibai BM, EI-Nazer A, Gonzalez-Ruiz A: Severe preeclampsia-eclampsia in young primigravid women: subsequent pregnancy outcome and remote prognosis. Am J Obstet GynecoI 155:1011, 1986
- Hoffman RM, Stieper KW , Johnson RWG, et al: Renal ischemic tolerance. Arch Surg 109:550, 1974
- Margolin E, Merrill J, Harrison J: Diagnosis of hypertension due to occlusion of the renal artery. N Engl J Med 292:1387, 1975
- Lessman RK, Johnson SF, Coburn JW , et al: Renal artery embolism: clinical features and long-term follow-up of 17 cases. Ann Intern Med 89:477, 1978
- Winzelberg GG, Hull JD, Agar JWM, et al: Elevation of serum lactate dehydrogenase levels in renal infarction. JAMA 242:268, 1979
- Moyer JO, Rao CN, Widrich WC, et al: Conservative management of renal artery embolus. J Urol 109:138, 1973
- Crosby RL, Miller PD, Schrier RW: Traumatic renal artery thrombosis. Am J Med 81:890, 1986
- Steckel A, Johnston J, Fraley DS, et al: The use of streptokinase to treat renal artery thromboembolism. Am J Kidney Dis 4:166, 1984
- Fisher C, Konnak J, Cho K, et al: Renal artery embolism: therapy with intra-arterial streptokinase infusion. J Urol 125:402, 1981
- Perkins R, Jacobsen D, Feder F, et al: Return of renal function after late embolectomy. N Engl J Med 276:1194, 1967
- Aujla ND, Greenberg A, Banner BF, et al: Atheroembolic involvement of renal allografts. Am J Kidney Dis 13:329, 1989
- Smith MC, Ghosé MK, Henry AR: The clinical spectrum of renal cholesterol embolization. Am J Med 71:174, 1981
- Saklayen M, Suryaprasad A, Gupta S, et al: Incidence of atheroembolic renal failure after coronary angiogram (abstr). J Am Soc NephroI 5:403, 1994
- Ridker PM, Michel T: Streptokinase therapy and cholesterol embolization. Am J Med 87:357, 1989
- McGowan JA, Greenberg A: Cholesterol atheroembolic renal disease: report of 3 cases with emphasis on diagnosis by skin biopsy and extended survival. Am J NephroI 6:135, 1986
- Bidani A, Kasinath BS, Corwin MM, et al: Eosinophilia in the diagnosis of atheroembolic renal disease (abstr). Kidney Int 27:134, 1985
- Cosio FG, Zager RA, Sharma HM: Atheroembolic renal disease causes hypocomplementaemia. Lancet 2:118, 1985
- Smith MC, Ghose MK, Henry AR: The clinical spectrum of renal cholesterol embolization. Am J Med 71:174, 1981
- Wells J, Margolin E, Gall E: Renal cortical necrosis: clinical and pathologic features in twenty-one cases. Am J Med 29:257 1960
- Solez K: Cortical necrosis. Pathology of the Kidney, 3rd ed. Heptinstall RH, Ed. Little, Brown & Co, Boston, 1983, p 1121
- Martinez-Maldonado M, López-Novoa JM, Femández-Repollet E, et al: Functional studies in experimental renal cortical necrosis in the rat. Am J PhysioI 257:F925, 1989
- Conger JD, Falk SA, Guggenheim SJ: Glomerular dynamics and morphologic changes in the generalized Shwartzman reaction in postpartum rats. J Clin Invest 67:1334, 1981
- Matlin RA, Gary NE: Acute cortical necrosis: case report and review of the literature. Am J Med 56:110, 1974
- Kleinknecht D, Grunfeld JP, Gomez PC, et al: Diagnostic procedures and long-term prognosis in bilateral renal cortical necrosis. Kidney Int 4:390, 1973
- Smith LE, Adelman RD: Early detection of renal cortical calcification in acute renal cortical necrosis in a child. Nephron 29:155, 1981
- Walser M: Progression of chronic renal failure in man. Kidney Int 37:1195, 1990
- Alleyne G, Statius van Eps L, Addae S, et al: The kidney in sickle cell anemia. Kidney Int 7:371, 1975
- Knochel JP: Hematuria in sickle cell trait: the effect of intravenous administration of distilled water, urinary alkalinization, and diuresis. Arch Intern Med 123:160, 1969
- Sickles EA, Korobkin M: Perirenal hematoma as a complication of renal infarction in sickle-cell trait: a case report. Am J Roentgenol 122:800, 1974
- Statius van Eps LW, Schouten H, Haar Romeny-Wachter CC, et al: The relation between age and renal concentrating capacity in sickle cell disease and hemoglobin C disease. Clin Chim Acta 27:501, 1970
- Batlle D, Itsarayoungyuen K, Arruda JAL, et al: Hyperkalemic hyperchloremic metabolic acidosis in sickle cell hemoglobinopathies. Am J Med 72:188, 1982
- Oster JR, Lanier DC Jr, Vaamonde CA: Renal response to potassium loading in sickle cell trait. Arch Intern Med 140:534, 1980
- Kunkler PB, Farr RF, Luxton RW: The limit of renal tolerance to x-rays: an investigation into renal damage occurring following the treatrnent of tumors of the testis by abdorninal baths. Br J Radiol 25:190, 1952
- Keane WF, Crosson JT, Staley NA, et al: Radiation-induced renal disease: a clinicopathologic study. Am J Med 60:127, 1976
- Loescher LJ, Welch-McCaffrey D, Leigh SA, et al: Surviving adult cancers: I. Physiologic effects. Ann Intern Med 111:411, 1989
- Luxton RW: Radiation nephritis: a long term study of 54 patients. Lancet 2:1221, 1961
- Shapiro AP, Cavallo T, Cooper W, et al: Hypertension in radiation nephritis: report of a patient with unilateral disease, elevated renin activity levels, and reversal after unilateral nephrectomy. Arch Intern Med 137:848, 1977
- Laffay D, Tubbs R, Valenzuela M, et al: Chronic glomerular microangiopathy and metastatic carcinoma. Hum Pathol 10:433, 1979
- Antignac C, Gubler MC, Leverger G, et al: Delayed renal failure with extensive mesangiolysis following bone marrow transplantation. Kidney Int 35:1336, 1989
- Love PE, Santoro SA: Antiphospholipid antibodies: anticardiolipid and the lupus anticoagulant in systemic lupus erythematosus (SLE) and non-SLE disorders: prevalence and clinical significance. Ann Intern Med 112:682, 1990
- McNeil HP, Simpson RJ, Chesterman CN, et al: Antiphospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: (2-glycoprotein (apolipoprotein H). Proc Nat1 Acad Sci USA 87:4120, 1990
- Hayslett JP: Postpartum renal failure. N Engl J Med 312:1556, 1985
- Hauglustaine D, van Damme B, van Renterghem Y,et al: Recurrent hemolytic-uremic syndrome during oral contraception. Clin NephroI 15:148, 1981
- Wolfe J, McCann R, Sanfilippo F: Cyclosporine-associated microangiopathy in renal transplantation: a severe but potentially reversible form of early graft injury. Transplantation 41:541, 1986
- Beaufils H, de Groc F, Gubler M, et al: Hemolytic uremic syndrome in Behçet's disease treated with cyclosporin A: report of 2 cases. Clin Nephrol 34:157, 1990
- Cooper B, Husberg B, Goldstein R, et al: FK506-associated thrombotic thrombocytopenic purpura. Transplantation 55:206, 1993
- Abramowicz D, Pradier O, Marchant A: Induction of thromboses within renal grafts by high-dose prophylactic OKT3. Lancet 339:777, 1992
- Begue R, Dennehy P, Peter G: Hemolytic uremic syndrome assodated with Streptococcus pneumoniae (letter). N Engl J Med 325:133, 1991
- Page Y, Tardy B, Zeni F, et al: Thrombotic thrombocytopenic purpura related to ticlopidine. Lancet 337:774, 1991
- Aster RH: Quinine sensitivity: a new cause of the hemolytic uremic syndrome. Ann Intern Med 119:215, 1993