Contenido del artículo
XI ENFERMEDAD DE ALZHEIMER Y DEMENCIAS
- Definición y clasificación de las demencias
- Etiología
- Epidemiología
- Factores genéticos de riesgo
- Efecto genético en la historia natural y el diagnóstico
- EDAD DE INICIO CONTRA PROGRESION DE LA ENFERMEDAD
- GENOTIPO APOE EN PERSONAS NORMALES
- GENOTIPIFICACION DEL APOE EN PACIENTES CON DEMENCIA SINTOMATICA
- Posibles mecanismos patogénicos
- Manifestaciones clínicas
- Diagnóstico diferencial
- Tratamiento
DR. ALLEN D. ROSES
DRA. ANN M. SAUNDERS, PH.D.
Definición y clasificación de las demencias
Una definición simple y lógica de la demencia es la siguiente el deterioro persistente y adquirido de la función intelectual con compromiso de por lo menos tres de los siguientes aspectos de la actividad mental: lenguaje, memoria, destrezas visuoespaciales, emoción o personalidad y función cognitiva (abstracción, cálculo, juicio, función de ejecución).1 Las presentaciones clínicas de los diferentes síndromes de demencia pueden mostrar áreas características de deterioro. Por ejemplo, la enfermedad de Alzheimer es una demencia cortical típica con un inicio insidioso y progresión lenta. La llamada demencia subcortical, como la de la enfermedad de Huntington o la parálisis supranuclear progresiva también puede comenzar en forma lenta. En muchos casos, pero ciertamente no en todos, pueden existir datos que distingan la demencia cortical de la subcortical. Los síntomas que son característicos de enfermedades demenciales específicas pueden ser sutiles, aunque se vuelven más francos al examinar al paciente durante un tiempo prolongado.
Las demencias corticales, como la enfermedad de Alzheimer, la enfermedad de Pick y la degeneración del lóbulo frontal, muestran problemas de amnesia y aprendizaje, con frecuencia con disfasia progresiva que se agrega a un lenguaje normal. Puede ser difícil distinguir la enfermedad de Alzheimer de la enfermedad de Pick, pero cuando es posible, los datos característicos en las fases tempranas de la enfermedad pueden ser útiles. Los problemas de amnesia parecen frecuentes en la enfermedad de Alzheimer temprana, mientras que la enfermedad de Pick se caracteriza por cambios tempranos de la personalidad y síntomas de comportamiento en presencia de destrezas intelectuales relativamente conservadas. Las dificultades cognitivas de las demencias corticales se observan dentro del contexto de un afecto no alterado a pesar de los problemas de cálculo, abstracción o juicio.1
En las demencias subcorticales, como las asociadas con la enfermedad de Parkinson, la enfermedad de Huntington y la parálisis supranuclear progresiva, el lenguaje disártrico o el deterioro motor pueden interferir con la función normal del lenguaje. La función cognitiva en las demencias subcorticales puede ser lenta y apática, con más problemas del sistema motor que depende de los ganglios basales. Por el contrario, el daño en la memoria y función cognitiva en las demencias corticales se caracteriza por problemas en la recuperación de memoria.
La enfermedad de Alzheimer es la causa más común de demencia progresiva en el grupo de edad mayor de 55 años. Los criterios clínicos para hacer el diagnóstico de enfermedad de Alzheimer posible o probable se establecieron por consenso hace más de una década [ver tabla 1]. Cuando los pacientes con diagnóstico clínico son después sometidos a confirmación por autopsia, se hace el diagnóstico definitivo de enfermedad de Alzheimer con base en criterios neuropatológicos en el 70 a 90 por ciento de los casos. Las definiciones proporcionadas por el National Institute of Neurological and Communicative Disorders and Stroke (NINCDS, Instituto nacional de trastornos neurológicos y de la comunicación, y eventos cerebrovasculares, de los EUA, n. del t.) y de la Alzheimer Disease and Related Disorders Association (ADRDA, Asociación para la enfermedad de Alzheimer y trastornos relacionados de los EUA, n. del t.) han permitido a los investigadores clínicos clasificar a grupos semejantes de pacientes cuando se usa el término de enfermedad de Alzheimer, pero estas definiciones clínicas incluyen otras etiologías.2
|
||
|
La enfermedad de Alzheimer es un trastorno genético complejo en el que mutaciones genéticas múltiples y distintas, genes de susceptibilidad e influencias ambientales participan para producir un fenotipo de enfermedad muy prevalente.3,4 Los factores genéticos etiológicos que causan la enfermedad de Alzheimer se han delimitado mejor últimamente que los de cualquier otra enfermedad médica compleja como la diabetes mellitus, la enfermedad coronaria o la osteoartritis. Existen tres loci genéticos identificados en los que las mutaciones sin sentido causan una forma de inicio temprano de enfermedad de Alzheimer que se hereda como un rasgo autosómico dominante [ver tabla 2]. La mutación más común es la del gen presenilin 1 (PSI), que causa la enfermedad en menos de 200 familias (o casos aislados contados como familias) en el mundo.5 Las mutaciones del gen de la proteína precursora de amiloide (APP, por sus siglas en inglés, n. del t.) ocurre en menos de 20 familias en el mundo, y las mutaciones en el gen presenilin-2 (PS2) se han demostrado en solo tres familias.6,7 Estas mutaciones son muy importantes para la investigación neurobiológica básica porque identifican genes que, cuando mutan, causan el fenotipo de la enfermedad de Alzheimer. Los mecanismos relacionados con estas mutaciones proteicas pueden proporcionar información muy valiosa para las nuevas hipótesis de investigación respecto a los mecanismos de la enfermedad. Sin embargo, por la perspectiva de las formas comunes de enfermedad de Alzheimer que se ven en la práctica clínica, las formas autosómico dominantes representan mucho menos del 1 por ciento de la incidencia o prevalencia. Incluso en el grupo de inicio de 50 a 60 años, la etiología de inicio temprano más común parece relacionarse con la herencia de uno o más alelos de apolipoproteína E e4 como genes de susceptibilidad y no de mutaciones autosómicas dominantes.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Las formas más comunes de enfermedad de Alzheimer suelen observarse como casos esporádicos o en familias grandes en las que pueden agregarse varios casos. Mientras mayor sea la familia, mayor oportunidad de agregación de casos de una enfermedad común.8 Existe un rango muy amplio en la edad de inicio, que varía desde casos poco frecuentes entre los 40 y 50 años a una mayor proporción en cada década hasta el rango de 90 a 100 años. La edad promedio de inicio en los Estados Unidos es de 69 a 71 años, y se calcula que existen tres a cuatro millones de casos en todo el mundo.
Las variaciones polimórficas en el locus de la apolipoproteína E (APOE), que actúan como factores heredados de riesgo, alteran la susceptibilidad genética.8,9 Existen tres variaciones comunes que se heredan en el locus APOE, denominadas e2, e3 y e4. En la mayoría de los grupos blancos la frecuencia del alelo e3 es de 78 por ciento. El siguiente alelo en frecuencia es el e4 (15 por ciento) y el e2 (7 por ciento) es el menos común. Los alelos e2 y e4 difieren cada uno del e3, que tiene una sola cisteína en la posición 112 de 299 aminoácidos. El e4 tiene un residuo arginina en la posición 112 y el e2 tiene una segunda cisteína en la posición 158.10 A pesar de las variaciones aparentemente discretas, los diferentes genotipos pueden tener un efecto importante en el riesgo y edad de inicio de la enfermedad de Alzheimer.
El alelo e4 se asocia con una edad de inicio más temprano de la forma común de enfermedad de Alzheimer.11 El alelo e2 confiere un menor riesgo y se asocia con mayor edad al inicio.l2 Debido a que cada individuo hereda dos alelos, la asociación del riesgo y la distribución del inicio de la enfermedad de Alzheimer varía con los diferentes genotipos [ver figura 1].13 Los homocigotos e4/e4, que constituyen alrededor del 2 por ciento de la población general, tienen el mayor riesgo, y la enfermedad de Alzheimer comienza en el 50 por ciento de los casos antes de los 70 años de edad. En las personas e2/e3, que representan alrededor del 12 a 14 por ciento de la población, la edad promedio de inicio es de más de 90 años. La distribución de la población según la edad de inicio con el genotipo más común, e3/e3, queda en medio. Por lo tanto, con base sólo en el genotipo APOE existen más de dos décadas de diferencia en la edad promedio de inicio. Sin embargo, existen diferencias en la prevalencia de cada genotipo en los diferentes grupos étnicos y raciales.14 En los japoneses y chinos, en los que la frecuencia del alelo e4 es menor (<10 por ciento), aún ocurre asociación con el Alzheimer, pero la enfermedad es menos frecuente. La menor frecuencia de los genotipos e4/e4 (menos de 1 por ciento contra 2 a 3 por ciento) y la mayor proporción relativa de individuos e3/e4 en la población japonesa causa una mayor edad de al inicio que la observada en poblaciones blancas. La asociación de e4 y enfermedad de Alzheimer en afroamericanos e hispánicos sigue siendo tema de controversia y puede no existir en una muestra de casos de este padecimiento en población de Nigeria y sujetos controles.14

|
| Figura 1 |
| Edad de inicio y genotipo APOE |
Efecto genético en la historia natural y el diagnóstico
EDAD DE INICIO CONTRA PROGRESION DE LA ENFERMEDAD
En ocasiones existe confusión respecto, a la edad de inicio contra la velocidad de progresión de la enfermedad de Alzheimer. Como podría esperarse, los pacientes de mayor edad viven con la enfermedad menos tiempo. Debido a que el genotipo e4/e4 se asocia con edad de inicio más temprano, estos pacientes suelen tener una enfermedad más prolongada. Sin embargo, si uno compara los pacientes con inicio dentro del mismo rango de edad (v.gr., e4/e4 y e3/e3 con inicio entre los 65 y 69 años de edad), la duración de la enfermedad es semejante, independientemente de los factores de riesgo APOE.I7 Los datos de tomografías computadas anuales seriadas de la porción medial del lóbulo temporal en pacientes con enfermedad de Alzheimer que fueron seguidos cada año16 muestran que la velocidad de progresión de la enfermedad después del inicio clínico es rápida y autocatalítica [ver figura 2]. Una manera de interpretar estos datos es sugerir que cada persona progresa hacia la enfermedad de Alzheimer a una velocidad heredada, que depende mucho del genotipo APOE. Sin embargo, una vez que se desarrollan los síntomas de la demencia, la progresión es rápida. La duración de la enfermedad parece depender más de la edad de inicio que del genotipo APOE.15

|
| Figura 2 |
| Velocidad de atrofia del lóbulo temporal |
Es probable que aparezcan alteraciones metabólicas en el cerebro muchos años antes del inicio de la enfermedad clínica. En hijos adultos de pacientes con enfermedad de Alzheimer se observó alteración en el metabolismo de la glucosa por tomografía de emisión de positrones 2 décadas antes de la edad esperada de inicio en las personas con genotipo heterocigoto e3/e4.17 Un estudio subsecuente identificó a voluntarios homocigotos e4/e4 de la población general. Cada sujeto con el genotipo e4/e4 fue equiparado con dos sujetos controles con genotipos e3/e3, e2/e3 o ambos. Los voluntarios e4/e4 no tuvieron evidencia de alteración en la función cognitiva o demencia según pruebas neuropsicológicas formales, pero sí alteración en el metabolismo regional de la glucosa. La edad promedio de los voluntarios e4/e4 fue alrededor de 20 años menor que la edad promedio esperada de inicio para el genotipo e4/e4.18 Estos datos apoyan el punto de vista de que la enfermedad de Alzheimer es una enfermedad metabólica activa e insidiosa durante muchos años antes de que comiencen los síntomas de deterioro cognitivo y demencia [ver figura 3].
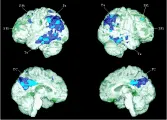
|
| Figura 3 |
| Metabolismo de la glucosa cerebral: TEP |
GENOTIPO APOE EN PERSONAS NORMALES
Aunque el riesgo relativo de enfermedad de Alzheimer aumenta con la presencia de uno o dos alelos e4, no es posible predecir si la enfermedad se presentará y cuando en una persona específica [ver figura 1].19 Hasta que se disponga de estudios epidemiológicos apropiados, los cálculos que cuantifican el riesgo relativo de enfermedad de Alzheimer para cualquier genotipo APOE no pueden ser exactos. Debido a que es posible que alguien con el genotipo e3/e4 viva más de 100 años sin enfermedad de Alzheimer y que una proporción importante de personas con el genotipo e2/e3 inicien antes de los 90 años de edad, el genotipo APOE no puede determinar si una persona con función cognitiva intacta desarrollará enfermedad de Alzheimer ni cuando. Por lo tanto, se acepta que el genotipo APOE no debe usarse como factor predictor en la población general.20-22 Sin embargo, al confirmarse y publicarse estudios epidemiológicos sobre otros genes de susceptibilidad, es posible que la predicción del inicio de enfermedad temprana o tardía dentro de cada genotipo APOE se vuelva más exacta. El papel poco satisfactorio de las pruebas de predicción, con todas sus consideraciones éticas y legales deberá cambiar una vez que se disponga de tratamientos seguros y eficaces para prevenir la enfermedad de Alzheimer.
GENOTIPIFICACION DEL APOE EN PACIENTES CON DEMENCIA SINTOMATICA
Existe importante discusión en la literatura clínica respecto al uso de la genotipificación del APOE en el diagnóstico diferencial de los pacientes con síntomas y signos leves de posible o probable enfermedad de Alzheimer.19-22 Dependiendo de la fase de la evaluación clínica, los resultados pueden ser variables. En general, cuando los síntomas y signos son suficientes para clasificarse como deterioro cognitivo leve pero no satisfacen los criterios para diagnosticar una posible enfermedad de Alzheimer, el alelo e4 será un buen predictor de qué pacientes presentarán demencia franca algunos años después.23 En estudios en los que el deterioro cognitivo está menos definido, la predicción de la demencia inminente usando la presencia de un alelo e4 puede ser menos confiable.
Aunque alrededor del 70 a 90 por ciento de los casos en la mayoría de las series de probable enfermedad de Alzheimer se confirman eventualmente por autopsia, el 10 a 30 por ciento de los casos no corresponden a este diagnóstico. Por lo tanto, los cálculos de la exactitud de la genotipificación APOE en las series clínicas están muy limitados por la exactitud del diagnóstico clínico y la variabilidad entre las series. Parte de la variabilidad parece deberse a diferencias entre grupos étnicos y raciales en relación con la frecuencia de los alelos APOE.
Sin embargo, es posible mejorar la variabilidad de los cálculos realizando mediciones directas. La genotipificación APOE puede examinarse en series longitudinales de pacientes con enfermedad de Alzheimer posible o probable que se confirman posteriormente por autopsia. Los estudios iniciales proporcionaron una medición exacta de la sensibilidad, especificidad y valor predictivo positivo de la genotipificación del APOE al definir en forma directa a los pacientes sin enfermedad de Alzheimer en cada serie.24-28 Los estudios de casos confirmados por autopsia demostraron un valor predictivo positivo de los genotipos e4/e4 y e3/e4 que alcanzaron más del 99 por ciento.24-28 Los pacientes sin enfermedad de Alzheimer en estas series rara vez portaron el alelo e4, y las series iniciales mostraron una especificidad mayor del 99 por ciento para el genotipo e4/e4 (definida como el número de pacientes sin enfermedad de Alzheimer y que no tuvieron la enfermedad) y una especificidad no esperada muy alta para el genotipo e3/e4 (> 98 por ciento). Estos dos genotipos APOE son responsables de alrededor del 65 por ciento de los pacientes con enfermedad de Alzheimer esporádica.9 Un estudio de más de 2,000 pacientes que fueron referidos a uno de 26 centros para estudio de enfermedad de Alzheimer evaluó los diagnósticos clínicos de autopsia y el genotipo APOE.27 El estudio concluyó que la genotipificación APOE, cuando se usa como adyuvante diagnóstico en combinación con los criterios clínicos, mejora la especificidad del diagnóstico. La genotipificación no proporciona suficiente sensibilidad o especificidad para usarse sola como prueba diagnóstica. Debido a que una proporción significativa de pacientes con enfermedad de Alzheimer confirmada por autopsia no portan el alelo e4, la ausencia de éste no descarta la posibilidad de la enfermedad, ni es útil para diferenciar la enfermedad de Alzheimer de otras demencias.24,27,28 Cuando se agrupan los pacientes con e4 y sin este alelo, los pacientes con enfermedad diferente al Alzheimer se incluirán preferentemente en el grupo no e4.
Posibles mecanismos patogénicos
Las placas neuriticas extracelulares y las marañas neurofibrilares (MNF) observadas en la autopsia por Alois Alzheimer en 1907 se han considerado como relacionados en el inicio de la enfermedad.29 Las placas tiñen para amiloide, un término genérico para la apariencia birrefringente a la luz polarizada reflejada por tejidos teñidos con químicos intercalados, como el rojo Congo. Muchas proteínas se agregan en las placas neuríticas; la proteína amiloide beta (bA4 o Ab), un fragmento de la APP, es un constituyente importante de la placa. Las mutaciones sin sentido de la APP son la causa de una de las formas autosómicas dominantes raras de la enfermedad de Alzheimer. Aunque se deposita Ab en las placas en las formas comunes de enfermedad de Alzheimer, las mutaciones sin sentido de la APP no son frecuentes.2 En la actualidad se estudia el metabolismo de la APP en las formas autosómico dominantes de enfermedad de Alzheimer. Sin embargo, la generalización de las formas comunes de enfermedad de Alzheimer aún es muy especulativa.29 Las evidencias clínicas y de autopsia han llevado a la conclusión de que las placas Ab en la enfermedad de Alzheimer son un marcador específico pero no un componente importante en la patogenia. De hecho, en estudios epidemiológicos de ancianos, la presencia de depósitos significativos de amiloide en las placas neuríticas fue común, independientemente de si los individuos tenían deterioro cognitivo.30 Los datos sugieren que tanto la presenilina como la Ab participan en la apoptosis neuronal.29
Se encuentran más de 30 proteínas agregadas en las placas, incluyendo el receptor (RLBD) relacionado a la lipoproteína de baja densidad (LBD) y todas las proteínas diversas que se sabe se unen al RLBD, incluyendo la ApoE.31 La unión e internalización por el RLBD parece ser el principal medio por el que la ApoE entra a las neuronas.32 La Ab, unida a las isoformas de ApoE de modo variable, puede ser el blanco en la unión Ab al RLBD (u otros miembros de la familia de receptores de LBD) en los procesos neurales o gliales encontrados en las placas. Por lo tanto, la expresión de las isoformas del locus ApoE de mayor susceptibilidad puede participar directamente en la formación de las placas. Aún es motivo de debate si las placas son consecuencia de una alteración en el metabolismo que causa la enfermedad de Alzheimer o si son las lesiones causales primarias.l3,28,33,34
Otros autores hacen énfasis en la formación de las MNF35,36 con argumentos apoyados por la identificación de mutaciones tau en la demencia frontotemporal con herencia autosómica dominante.37 Las MNF son agregados de proteína tau hiperfosforilada, una de las proteínas asociadas a los microtúbulos en las neuronas. La tau humana puede unirse a sí misma, así como a a-tubulina, para estabilizar los microtúbulos. Cuando la tau se une a sí misma, forma filamentos pareados helicoidales que ya no son capaces de estabilizar los microtúbulos. Estos filamentos pueden hiperfosforilarse en muchos sitios y agregarse para formar los cambios neurofibrilares que constituyen las MNF. En las series grandes de autopsia, se ha observado que los cambios neurofibrilares se desarrollan por etapas durante décadas.38 El desarrollo inicial de cambios neurofibrilares se observa en personas con un alelo e4 a una edad que es varias décadas menor que la de la presentación de la enfermedad de Alzheimer.39
EFECTO DE APOE EN EL METABOLISMO CEREBRAL
Con el descubrimiento del APOE como un locus importante de susceptibilidad para la forma común de enfermedad de Alzheimer, en la actualidad es posible examinar la manera como las diferencias específicas en la isoforma de la ApoE pueden afectar el metabolismo cerebral.32,40,41
Se desconoce cuáles son las moléculas que se fijan a los sitios de unión de lípidos de la ApoE en condiciones normales. La ApoE deslipidizada in vitro se une más rápidamente a la Ab, lo que es compatible con los datos neuropatológicos obtenidos de pacientes con enfermedad de Alzheimer.41,42 Los pacientes con el padecimientos que son homocigotos para e4/e4 tienen un depósito Ab más denso que los homocigotos e3/e3 con duración semejante de la enfermedad.42 La ApoE se sintetiza en las células gliales y se transfiere al citoplasma de ciertas neuronas humanas, al parecer a través de receptores LRP.32,43 Cuando ratones transgénicos deficientes en ApoE portan isoformas específicas de ApoE del genoma humano (sin agregar promotores para la expresión específica neuronal o glial), aparece ApoE citoplásmica en las neuronas.44,45 Apenas comienza a darse importancia y a estudiarse la biología de las múltiples proteínas que terminan en diferentes localizaciones intracelulares dentro del sistema nervioso central de lo esperado, pero los datos pueden aportar mucho al conocimiento de los mecanismos de las enfermedades.
Quizá una de las piezas de evidencia más importante de que la ApoE tiene un papel en el depósito de amiloide es la observación de que la falta de ApoE reduce en forma dramática el depósito Ab en un modelo transgénico de enfermedad de Alzheimer.46 En ratones de 6 meses de edad homocigotos para la un transgén mutado APP y el gen APOE nativo del ratón, la corteza cerebral y el hipocampo tuvo gran cantidad de placas de amiloide. Por el contrario, en ratones de 6 meses de edad homocigotos para la mutación APP pero que no tenían el gen APOE nativo del ratón no hubo depósitos de amiloide.
Los cambios neurofibrilares correlacionan con la demencia en la enfermedad de Alzheimer, en especial en los pacientes que portan e4.39 Los cambios neurofibrilares parecen ser causados por la formación relativamente temprana de filamentos helicoidales pareados y MNF en los portadores e4. Las proteínas ApoE2 y ApoE3 se unen a las proteínas asociadas a microtúbulos tau y PAM-2 mejor que la ApoE4.39,47 La ApoE3 se une a los dominios de la tau que fijan PAM, y se postula que cuando solo existe (y se hereda) ApoE4, los dominios de unión de la PAM pueden unirse con más facilidad entre sí para formar filamentos helicoidales pareados que no pueden ya estabilizar la a-tubulina en los microtúbulos.48 Con el tiempo, esto parecer crear mayor inestabilidad de los microtúbulos y una plasticidad menos eficaz de los mismos en respuesta al estrés. Esta hipótesis es apoyada por una serie de estudios de cultivo de tejido neuronal que demostraron menor crecimiento neurítico con ApoE4 en el medio que con ApoE3.49,50 La menor densidad sináptica es una característica de la enfermedad de Alzheimer,51 y estas alteraciones en la densidad de la sinapsis pueden ser más importantes para la enfermedad clínica que la presencia de placas y marañas. Además de los cambios en el crecimiento neuríticos con la ApoE4, pudo demostrarse agregación de la a-tubulina.49 Por lo tanto, es posible que exista una tendencia hacia la desestabilización de los microtúbulos y la formación de marañas en individuos que heredan los alelos e4. La interacción de ApoE2 y ApoE3 con tau puede regular la función y metabolismo de tau. Esta regulación podría estar ausente en individuos que codifican solo para la proteína ApoE4 y disminuiría en individuos heterocigotos, causando una menor vida funcional de las neuronas. Esto sugiere una estrategia terapéutica que podría incluir el desarrollo de una compuerta que pueda entrar al citoplasma de las neuronas en concentraciones muy bajas y simular la función de la ApoE2 y ApoE3, aumentando la eficacia de las respuestas de los microtúbulos y quizá retrasando el inicio de la enfermedad de Alzheimer.
Los síntomas de la enfermedad de Alzheimer se desarrollan en forma lenta e insidiosa. Es difícil definir el momento de inicio. Durante un periodo variable de tiempo se presentan alteraciones cognitivas y de memoria que pueden progresar hasta el punto de interferir con actividades específicas que eran normales para el paciente, presentándose un problema por el que se solicita atención médica. Durante muchos años ha existido un enorme debate entre los científicos respecto a si la pérdida de memoria es una consecuencia normal del envejecimiento, lo que dificulta distinguir entre los cambios cognitivos dependientes de la edad y las llamadas dificultades patológicas características de la enfermedad. De hecho, el conocimiento de que la susceptibilidad causa un deterioro cognitivo más rápido y funcionalmente importante es más claro con el descubrimiento de las distribuciones de edad de inicio en función del genotipo APOE específico. Por lo tanto, la pérdida de la función cognitiva y de memoria puede progresar en función de la edad en todas las personas con todos los genotipos, y algunas personas empeoran con más rapidez y a una edad más temprana que otras [ver figura 1 y 2].
Muchas personas tienen molestias cognitivas y de memoria, pero no pueden diagnosticarse porque no satisfacen los criterios establecidos para el diagnóstico de posible enfermedad de Alzheimer. Datos recientes han demostrado que en los pacientes con dificultades cognitivas tempranas la presencia de un alelo e4 es el mejor predictor de qué pacientes progresarán hasta satisfacer las definiciones de posible enfermedad de Alzheimer. En forma semejante, cuando los pacientes con síntomas y signos de demencia temprana portan un alelo e4, puede suponerse que el diagnóstico patológico final será enfermedad de Alzheimer en el 95 por ciento de los casos. Varios ejemplos pueden ilustrar la dificultad para establecer la edad precisa de inicio en los pacientes con deterioro de la función cognitiva o alteraciones progresivas de la memoria. Una de las quejas iniciales de algunos pacientes es la desorientación, reportada con frecuencia al médico después de que el paciente se perdió mientras viajaba por una ruta familiar. Un relato cuidadoso del esposo o del acompañante puede descubrir episodios previos de dificultades, y las pruebas neuropsicológicas formales confirmarán los problemas de orientación. Por lo tanto, es posible que existan síntomas que precedan por meses a la queja por la que se solicitó atención médica.
En forma semejante, las dificultades en el cálculo pueden desencadenar un problema financiero que desencadena la solicitud de evaluación médica. No es común que los pacientes que se han hecho cargo de las financias familiares durante años cometan errores serios por los que se solicite una evaluación. En estos casos pueden existir datos cronológicos anotados en los registros financieros o herramientas semejantes que demuestren que las dificultades en los cálculos datan de muchos meses o años. Estos datos son útiles no sólo para demostrar con más exactitud el inicio del problema, sino para establecer la progresión gradual de la enfermedad.
Las quejas de falta de memoria pueden ser más frecuentes en adultos de edad media. Suelen hacerse bromas respecto al posible desarrollo de enfermedad de Alzheimer cuando se pierden las llaves, se olvidan nombres familiares o existen otros lapsos de memoria. La progresión de la alteración en la memoria, que se vuelve tan frecuente que comienza a interferir con la conversación o empleo de una persona, suele orillar a solicitar una evaluación médica.
Por lo tanto, la interfase entre los problemas del envejecimiento normal y una progresión más rápida de las dificultades cognitivas y de memoria puede constituir un área clínica difícil. Los polimorfismos de susceptibilidad, como el APOE y otros aún no descubiertos, proporcionarán cierta orientación sobre la velocidad de progresión de los síntomas en función de la edad. Al desarrollarse cada vez más tratamientos para los síntomas de la enfermedad de Alzheimer, se vuelve importante el diagnóstico exacto. Otros factores no genéticos que pueden usarse para predecir el desarrollo de enfermedad de Alzheimer, como las pruebas de sangre o líquido cefalorraquídeo que pueden relacionarse con la velocidad de progresión de la enfermedad, se convertirán en adyuvantes importantes para la evaluación y tratamiento de la demencia clínica. Una vez que se ha demostrado progresión de la demencia y se satisfacen los criterios clínicos de enfermedad de Alzheimer, el curso se vuelve autocatalítico, con acortamiento en los intervalos de tiempo que demuestran empeoramiento clínico. La progresión de problemas emocionales, cognitivos y de memoria caracterizan a la evolución de la enfermedad de Alzheimer [ver tabla 1].
El deterioro cognitivo es un síntoma de varios padecimientos tratables que deben descartarse antes de establecer el diagnóstico de enfermedad de Alzheimer. Una causa común de alteración cognitiva susceptible de tratamiento es una forma de delirio crónico que ocurre cuando los pacientes ancianos toman demasiados medicamentos combinados (polifarmacia); esto puede causar confusión, desorientación y en ocasiones lo que parece ser una demencia progresiva. Pueden ocurrir problemas de memoria reciente que fácilmente son confundidos con demencia temprana. En todos los pacientes que se evalúan por problemas cognitivos progresivos debe realizarse una historia cuidadosa de medicamentos, incluyendo los que no requieren receta médica.
Las dificultades cognitivas pueden ser también resultado de exposición a toxinas en el ambiente. Cuando miembros múltiples de la misma familia, en especial jóvenes, tienen problemas en forma simultánea, debe considerarse la posible exposición a bióxido o monóxido de carbono en el hogar o medio de transporte. Es común el mal funcionamiento de los calentadores domésticos, en especial en áreas rurales, y las concentraciones bajas de humo son un problema importante. Las personas ancianas pueden sufrir dificultades cognitivas insidiosas, mientras que los miembros más jóvenes de la familia pueden quejarse de cefalea, náusea y otros síntomas. El inicio de las molestias puede ser gradual y pasar desapercibido, ocurriendo con más frecuencia cuando el clima es frío. Un adyuvante diagnóstico útil y no caro es la compra de un detector de monóxido de carbono para el hogar.
Otros ejemplos de toxinas ambientales comunes incluyen los pesticidas y los materiales de jardinería. La exposición tóxica a órganofosforados o metales pesados puede causar confusión y problemas de memoria, en especial en los ancianos. Aunque estas toxinas ambientales se identifican poco en la práctica clínica, debe realizarse un esfuerzo por obtener historias confiables de pasatiempos u otras actividades que permitan la detección temprana de las causas reversibles de deterioro cognitivo. Los síntomas vagos en otras personas en la misma casa pueden usarse para identificar a los pacientes con riesgo alto. Para que exista mejoría deben evitase las actividades asociadas con riesgo de contacto con la toxina.
Varios problemas metabólicos bien conocidos aunque poco comunes pueden simular una demencia, incluyendo el hipotiroidismo, la deficiencia de vitamina B12, la deficiencia de tiamina (asociada con frecuencia con alcoholismo) y el deterioro del estado mental causado por otros trastornos metabólicos como la insuficiencia hepática o renal. Las pruebas para estos problemas son componentes de rutina de la mayoría de las evaluaciones por demencia. Durante las últimas décadas los estudios sobre demencia se han concentrado en descartar condiciones reversibles. Solo recientemente se cuenta con pruebas genéticas que ayudan a establecer el diagnóstico de enfermedad de Alzheimer, Huntington o Creutzfeldt-Jakob con una buena capacidad predictiva.3,52
Siempre deben tenerse en cuenta las lesiones ocupativas dentro del estudio del deterioro cognitivo. La demencia como único síntoma es muy rara. Por lo general los tumores se asocian con cefalea, crisis convulsivas y síntomas de focalización. De nuevo, es importante revisar la historia clínica cuando se sospechen tumores, hematomas subdurales u otras lesiones ocupativas. El examen neurológico cuidadoso suele descubrir signos focales o patológicos, y debe ser un componente indispensable en el paciente con evaluación por deterioro cognitivo. Por desgracia, en los Estados Unidos, en donde la tomografía computada y la resonancia magnética son estudios fácilmente disponibles, la historia clínica y el examen neurológico de rutina suelen ser sustituidos por costosos estudios de imagen. En el Reino Unidos los médicos generales no tienen este acceso a los estudios de imagen y suelen tener más experiencia en el examen neurológico. Sin embargo, no existen datos que sugieran que es frecuente que en el Reino Unido las lesiones ocupativas pasen desapercibidas con facilidad. Cuando la demencia es el único síntoma inicial de un tumor, como en los llamados gliomas en mariposa que infiltran el cuerpo calloso y los lóbulos frontales en forma bilateral, el tumor puede no detectarse en la TC, aunque puede causar signos neurológicos patológicos. El costo económico de los estudios de imagen de rutina es cada vez más importante en los Estados Unidos, debido a que una mayor proporción de la atención médica es prestada a través de sistemas de atención u organizaciones de mantenimiento de la salud. Las evaluaciones diagnósticas deben considerar las causas específicas más importantes, en lugar de realizar un escrutinio de todas las causas posibles. Las pruebas de susceptibilidad genética y los exámenes neurológicos de seguimiento tendrán un papel cada vez más importante en la atención médica de los pacientes con daño cognitivo.
EVALUACION DE LOS PACIENTES CON DETERIORO COGNITIVO
Las pruebas más útiles para evaluar el deterioro cognitivo de los pacientes son una buena historia clínica, examen físico y examen neurológico. Una vez que se han eliminado otras causas, como la polifarmacia, se realizan pruebas estándar para causas poco frecuentes de demencia. Estas incluyen panel tiroideo, vitamina B12, serología, virus de inmunodeficiencia humana, química sanguínea y otras pruebas comunes a la evaluación médica de rutina. En ocasiones, cuando el deterioro cognitivo se acompaña de signos neurológicos sugestivos, deben realizarse las pruebas genéticas diagnósticas para otras enfermedades demenciales, como la enfermedad de Huntington y la atrofia dentatorrubropalidolusiana (DRPLA).
La genotipificación APOE es un adyuvante útil para diagnosticar posible o probable enfermedad de Alzheimer en alrededor del 65 al 75 por ciento de los pacientes. Sin embargo, sin un alelo e4, la genotipificación APOE no proporciona otra información útil. En otras palabras, el valor predictivo positivo de la genotipificación del APOE es muy alto, pero el valor predictivo negativo es prácticamente nulo. El análisis de mutaciones en los genes APP y PS1 es adecuado en individuos con enfermedad de Alzheimer de inicio temprano e historia familiar positiva.22 El encontrar una mutación en el gen APP o el PS1 tiene un alto valor predictivo para el desarrollo eventual de la enfermedad. Debido a que la edad de inicio en las familias PS1 y APP positivas no varía mucho, puede hacerse una predicción exacta del riesgo y de la edad aproximada de inicio. La enfermedad de Alzheimer de inicio temprano debida a mutaciones PS2 es extraordinariamente rara y debe considerarse realizar este análisis solo si el paciente es familiar de una de las tres familias Volga-germanas. Las pruebas genéticas siempre deben realizarse aunadas a un consejo genético pre y posprueba.
El uso de la TC o de la RM como parte del escrutinio diagnóstico es común en los Estados Unidos, pero puede no estar justificado. Muchos médicos que atienden pacientes con deterioro cognitivo pueden sentirse incómodos con la interpretación y confiabilidad solo del estudio neurológico. Cualquier paciente con demencia y un signo neurológico focal u otros datos patológicos es candidato para estudios de imagen. Además, cualquier paciente con historia de hemorragia subaracnoidea, traumatismo craneal, meningitis u otras enfermedades inflamatorias que puedan afectar el líquido cefalorraquídeo debe investigarse para buscar hidrocefalia comunicante (normotensa). Sin embargo, las imágenes cerebrales que se consideran compatibles con el diagnóstico de enfermedad de Alzheimer no suelen ser seguidas de pruebas que apoyen la presencia de hidrocefalia normotensa. La sensibilidad o especificidad para establecer este diagnóstico con las imágenes de rutina son bajas. A pesar de ello, la hidrocefalia normotensa es muy poco común si no existe una historia predisponente y solo el 2 por ciento de los pacientes con demencia se espera que tengan el genotipo e4/e4 (21 por ciento tendrán el e3/e4) con base en las frecuencias de población.
La literatura describe varias pruebas en el líquido cefalorraquídeo que ayudan a establecer el diagnóstico de enfermedad de Alzheimer. Los niveles en líquido cefalorraquídeo de Ab1-42 (bajos) y tau (altos) se han correlacionado con el diagnóstico de enfermedad de Alzheimer, aunque aún no se reportan series confirmadas por autopsia. Una concentración alta de tau y baja de Ab1-42 en LCR puede ser útil para distinguir a los pacientes que tienen enfermedad de Alzheimer de los que no, en especial en los no portadores de e4.53,54 Es posible considerar realizar estas pruebas de laboratorio para enfermedad de Alzheimer en los pacientes que no portan un alelo e4. En forma semejante, las decisiones respecto a las pruebas de imágenes múltiples pueden tener su mayor beneficio al evaluar a los pacientes no e4. Es importante enfatizar que los estudios clínicos de seguimiento constituyen el principal indicador para el uso subsecuente de recursos diagnósticos costosos. Por ejemplo, un paciente con demencia leve y genotipo e3/e4 que presenta una alteración neurológica focal durante el estudio de seguimiento que no existía al inicio debe, por supuesto, estudiarse con estudios de imagen apropiados en busca de otra patología. Los proyectos actuales de investigación en servicios de salud están evaluando el costo-beneficio de las pruebas diagnósticas de uso frecuente, incluyendo los estudios de imagen únicas y múltiples, y podrán determinar normas para la evaluación futura de la demencia. Es claro que el examen físico y neurológico cuidadoso es un procedimiento con muy buena relación costo-eficacia, y con considerable especificidad para diversas etiologías.
En la actualidad no existen tratamientos preventivos aceptados para la enfermedad de Alzheimer, ni agentes que se consideren eficaces en forma constante para prevenir la progresión de la enfermedad. El tratamiento sintomático puede ser muy útil, en especial en las fases tempranas de la demencia. La depresión puede ser un síntoma psiquiátrico importante en los pacientes con enfermedad de Alzheimer porque puede interferir con la función ya en si deteriorada del paciente. La depresión y la ansiedad pueden constituir un problema para los familiares y los cuidadores del paciente.
Deben evitarse los medicamentos con efectos anticolinérgicos. Por lo general el tratamiento se inicia con agentes heterocíclicos como nortriptilina, doxepina o desipramina. Después pueden desarrollarse alteraciones del comportamiento o psicosis, y el tratamiento con agentes neurolépticos poco potentes, como haloperidol, flufenacina o risperidone55,56 puede ser útil.
El tacrine es un inhibidor de la anticolinesterasa y fue el primer compuesto autorizado para el tratamiento de la enfermedad de Alzheimer en los Estados Unidos. Parece tener efectos benéficos en algunos pacientes en las etapas tempranas de la enfermedad, pero también tiene efectos adversos importantes.55,57 El tacrine requiere de dosificación frecuente, por lo general cuatro veces al día, lo mismo que vigilancia por su toxicidad hepática.
El donepezil fue el segundo compuesto autorizado para el tratamiento del Alzheimer. También es un inhibidor de la anticolinesterasa pero tiene la ventaja de administrarse una vez al día. La toxicidad hepática no parece constituir un problema, de modo que no se requieren pruebas sanguíneas periódicas. Otros agentes anticolinesterasa se están probando en la clínica, al igual que otros compuestos. Por lo tanto, es posible que en los próximos años se observe un cambio significativo en la terapéutica de esta enfermedad, en especial en las fases tempranas. Estos cambios enfatizarán las pruebas diagnósticas tempranas y el desarrollo de indicadores de actividad de la enfermedad o efectos terapéuticos sobre la misma.
Las hipótesis sobre el papel de las placas y marañas, así como la etiología genética, sugieren también diversos caminos de intervención terapéutica. Estudios epidemiológicos recientes han sugerido que los medicamentos antinflamatorios, el tratamiento sustitutivo con estrógenos y los depuradores de radicales libres, como la vitamina E, pueden tener un efecto protector. En la actualidad se realizan numerosos estudios clínicos para evaluar si estos tratamientos retrasan la progresión de la enfermedad de Alzheimer.55,58
TRATAMIENTO EN EL AMBITO FAMILIAR
Los pacientes y sus familias pueden ser ayudados en forma considerable si se pone atención a las dificultades sociales y psicológicas que hacen que la adaptación a esta enfermedad sea difícil. El personal de atención para la salud ha desarrollado estrategias de adaptación, y es un área de especial importancia continuar la investigación psicológica para manejar la pérdida progresiva de capacidad y los problemas a largo plazo, en especial debido a que la frecuencia de la enfermedad de Alzheimer aumenta al envejecer la población.
Suele ser muy importante para el paciente y sus familiares realizar un diagnóstico temprano. Al inicio del padecimiento el paciente es capaz de tomar decisiones sobre su atención y disposiciones para el futuro. Con frecuencia la participación del paciente es útil no sólo para él mismo, sino para los familiares y cuidadores, que de otra manera tienen que tomar decisiones dolorosas en etapas avanzadas del padecimiento. Al desarrollarse tratamientos sintomáticos más eficaces, puede anticiparse que su utilidad será más evidente en los síntomas tempranos. Por lo tanto, cada vez se emplean más en los pacientes sintomáticos las pruebas con valor predictivo positivo, como la genotipificación del APOE. Durante el curso de la atrofia cerebral los estudios de imagen seriados pueden confirmar un proceso autocatalítico. Sin embargo, estos datos requieren de varios años más de seguimiento para establecer un valor predictivo positivo en comparación con la identificación del alelo e4 en pacientes sintomáticos.
Cuando las pruebas genéticas se convierten en un adyuvante común para el diagnóstico, la familia se enfrenta al dilema adicional de la información colateral. Las posibilidades de que un hermano o hijo de un paciente con enfermedad de Alzheimer que porta el e4 haya heredado también este factor de riesgo son mayores. En las condiciones autosómico dominantes, una mutación específica puede predecir con exactitud el desarrollo de la enfermedad. Debido a que es posible portar un alelo e4 y no adquirir enfermedad de Alzheimer hasta los 100 años de edad o después, el peso colateral de la información sobre el gen de susceptibilidad es muy diferente de la predicción en el caso de las mutaciones. No se puede decir a un individuo normal con genotipo e3/e4 si desarrollará enfermedad de Alzheimer a los 55 años o nunca. Los médicos y familiares deben aprender sobre este nuevo paradigma de susceptibilidad genética. Debe hacerse un gran esfuerzo para demostrar el valor de las pruebas de polimorfismo genético para el diagnóstico de los pacientes con deterioro cognitivo a pesar de la falta de información para la predicción individual en personas asintomáticas. Debido a que la genotipificación APOE representa el primer polimorfismo de susceptibilidad que se usa para una enfermedad común, el contexto epidemiológico de esta información, más que el paradigma estándar de las mutaciones genéticas dominantes, es poco conocido en la comunidad médica. Debe aumentarse la educación para los médicos y las familias, y esto debe constituir parte importante del manejo.
Los familiares deben conocer los recursos disponibles en la comunidad. En los Estados Unidos existen más de 250 capítulos de Asociaciones para Alzheimer locales, con acceso directo a gran variedad de servicios. En muchas comunidades existen grupos de apoyo para padres, esposos, hijos y cuidadores. Pueden existir unidades de atención diurna y otros tipos de asistencia. También existen servicios de enfermería, en especial para los pacientes con enfermedad más avanzada, dentro de las unidades para atención del Alzheimer y otros programas especiales, de modo que el costo de un servicio de enfermería o asilo final sea menos oneroso. Al crecer el número de pacientes con Alzheimer, la importancia de los recursos financieros y emocionales para la familia también aumenta.
En los servicios de la comunidad se desarrollan estrategias para una mejor adaptación a las necesidades de cada paciente y su familia. El comportamiento agresivo de los pacientes hacia sus cuidadores puede mejorar en un ambiente calmado y si la atención se enfoca a otros aspectos. La molestia de la familia respecto a las actitudes negativas del paciente se vuelve menos grave cuando los cuidadores saben que no están solos y que otras personas se enfrentan a problemas semejantes.
Las actitudes potencialmente peligrosas deben modificarse en forma adecuada. Por ejemplo, el cocinar puede aumentar el riesgo de un incendio si el paciente se olvida de apagar los quemadores. Esta actividad puede continuar si el paciente es acompañado y vigilado durante la misma. El vagabundeo es muy común, y puede evitarse cerrando las puertas o proporcionando un ambiente confortable con límites bien establecidos. La Asociación de Alzheimer de los EUA, con el apoyo de donadores voluntarios, ha iniciado un programa de seguridad en el que los pacientes deben usar un brazalete con información que les identifica en caso que se pierdan. Existen materiales educativos sobre las actitudes negativas, de modo que las familias puedan aprender sobre los problemas potenciales antes de que los sufran.
También existen muchos folletos y publicaciones periódicas sobre los problemas relacionados con la atención de pacientes con demencia, incluyendo una guía de 1999 elaborada por el American Medical Association's Program on Aging and Community Health.59 La Asociación de Alzheimer es la más grande de su tipo dedicada a la investigación y a aspectos de atención al paciente. Proporciona una serie de folletos que hablan sobre los problemas legales, médicos, sociales y de otro tipo. La Asociación de Alzheimer cuenta con una dirección en Internet (http://www.alz.org/) para difundir información sobre la enfermedad de Alzheimer, los capítulos locales y la disponibilidad de recursos. Las familias y el personal médico encontrarán aquí información actualizada. Puede obtenerse más información en la oficina nacional de la asociación (919 North Michigan Avenue, Suite 1000, Chicago, IL 60611-1676; el número sin costo para llamar dentro de los Estados Unidos es 800-272-3900).
La investigación sobre la enfermedad de Alzheimer se ha diversificado, permitiendo adelantos en el análisis genético que han producido múltiples hipótesis nuevas respecto a los mecanismos de patogenia, con investigadores que defienden en forma apasionada cada una de ellas. Con el descubrimiento de otros genes de susceptibilidad y autosómico dominantes, se generarán nuevas hipótesis que brindarán esperanza para descubrir o diseñar compuestos preventivos y terapéuticos.
Figuras 1 y 2 Marcia Kammerer.
Bibliografía
- Cummings JL, Benson DF: Dementia: a Clinical Approach, 2nd ed. Butterworth-Heinemann, Stoneham, Massachussets, 1992
- McKhann G, Drachman D, Folstein M, et al: Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 34:939, 1984 [PMID 6610841 ]
- Roses AD: Apolipoprotein E alleles as risk factors in Alzheimer's disease. Annu Rev Med 47:387, 1996
- Cruts M, Van Broeckhoven C: Molecular genetics of Alzheimer's Disease. Ann Med_30:560, 1998
- Sherrington R, Rogaev EI, Liang Y, et al: Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 375:754, 1995 [PMID 7596406 ]
- Rogaev E, Sherrington R, Rogaeva EA, et al: Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 376:775, 1995 [PMID 7651536 ]
- Levy-Lahad E, Wijsman EM, Nemens E, et al: A familial Alzheimer's disease locus on chromosome 1. Science 269:970, 1995 [PMID 7638621 ]
- Pericak-Vance MA, Bebout JL, Gaskell PC Jr, et al: Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet 48:1034, 1991 [PMID 2035524 ]
- Saunders AM, Strittmatter WJ, Schmechel D, et al: Association of apolipoprotein E allele e4 with late-onset familial and sporadic Alzheimer's disease. Neurology 43:1467, 1993 [PMID 8350998 ]
- Weisgraber KH: Apolipoprotein E: structure-function relationships. Adv Protein Chem 45:249, 1994
- Corder EH, Saunders AM, Strittmatter WJ, et al: Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261:921, 1993 [PMID 8346443 ]
- Corder EH, Saunders AM, Risch NJ, et al: Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer's disease. Nat Genet 7:180, 1994 [PMID 7920638 ]
- Roses AD: Apolipoprotein E affects the rate of Alzheimer disease expression: b-amyloid burden is a secondary consequence dependent on APOE genotype and duration of disease. J Neuropathol Exp Neurol 53:429, 1994
- Farrer LA, Cupples LA, Haines JL, et al: Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278:1349, 1997
- Corder EH, Saunders AM, Strittmatter WJ, et al: Apolipoprotein E, survival in Alzheimer's disease patients, and the competing risks of death and Alzheimer's disease. Neurology 45:1323, 1995 [PMID 7617191 ]
- Jobst KA, Smith AD, Szatmari M, et al: Rapidly progressing atrophy of medial temporal lobe in Alzheimer's disease. Lancet 343:829, 1994 [PMID 7908080 ]
- Small GW, Mazziotta JC, Collins MT, et al: Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer's disease. JAMA 273:942, 1995 [PMID 7884953 ]
- Reiman EM, Caselli RJ, Yun LS, et al: Preclinical evidence of Alzheimer's disease in persons homozygous for the e4 allele for apolipoprotein E. N Engl J Med 334:752, 1996 [PMID 8592548 ]
- Roses AD: Apolipoprotein E genotyping in differential diagnosis, not prediction, of Alzheimer's disease. Ann Neurol 38:6, 1995
- Consensus statement: apolipoprotein E genotyping in Alzheimer's disease. National Institute on Aging/Alzheimer's Association Working Group. Lancet 347:1091, 1996
- Statement on use of apolipoprotein E testing for Alzheimer disease. American College of Medical Genetics/American Society of Human Genetics Working Group on ApoE and Alzheimer disease. JAMA 274:1627, 1995
- Consensus report of the Working Group on: "Molecular and Biochemical Markers of Alzheimer's Disease." The Ronald and Nancy Reagan Institute of the Alzheimer's Association and the National Institute on Aging Working Group (review). Neurobiol Aging 19:109, 1998
- Petersen RC, Smith GE, Ivnik RJ, et al: Apolipoprotein E status as a predictor of the development of Alzheimer disease in memory-impaired individuals. JAMA 273:1274, 1995 [PMID 7646655 ]
- Saunders AM, Hulette C, Welsh-Bohmer KA, et al: Specificity, sensitivity, and predictive value of apolipoprotein-E genotyping for sporadic Alzheimer's disease. Lancet 348:90, 1996 [PMID 8676723 ]
- Kakulas BA, Wilton SD, Fabian VA, et al: Apolipoprotein-E genotyping in the diagnosis of Alzheimer's disease in an autopsy confirmed series. Lancet 348:483, 1996 [PMID 8709818 ]
- Smith AD, Jobst KA, Johnston C, et al: Apolipoprotein-E genotyping in diagnosis of Alzheimer's disease. Lancet 348:483, 1996 [PMID 8709819 ]
- Mayeux R, Saunders AM, Shea S, et al: The utility of the apolipoprotein E genotype in the diagnosis of Alzheimer's disease. Alzheimer's Disease Centers Consortium on Apolipoprotein E and Alzheimer's Disease. N Engl J Med 338:506, 1998
- Welsh-Bohmer KA, Gearing M, Saunders AM, et al: Apolipoprotein E genotypes in a neuropathological series from the Consortium to Establish a Registry for Alzheimer's disease (CERAD). Ann Neurol 42:319, 1996
- Selkoe DJ: The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol 8:447, 1998
- Polvikoski T, Sulkava R, Haltia M, et al: Apolipoprotein E, dementia, and cortical deposition of b-amyloid protein. N Engl J Med 333:1242, 1995 [PMID 7566000 ]
- Rebeck GW, Harr SD, Strickland DK, et al: Multiple, diverse senile plaque-associated proteins are ligands of an apolipoprotein E receptor, the a<->2-macroglobulin receptor/low-density-lipoprotein receptor-related protein. Ann Neurol 37:211, 1995 [PMID 7531418 ]
- Roses AD: Perspective: on the metabolism of apolipoprotein E and the Alzheimer diseases. Exp Neurol 132:149, 1995
- Goedert M, Strittmatter WJ, Roses AD: Alzheimer's disease: risky apolipoprotein in brain. Nature 372:45, 1994 [PMID 7969418 ]
- Yankner BA: Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron 16:921, 1996
- Goedert M, Spillantini MG, Cairns NJ, et al: Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron 8:159, 1992 [PMID 1530909 ]
- Mandelkow EM, Biernat J, Drewes G, et al: Microtubule-associated protein tau, paired helical filaments, and phosphorylation. Ann NY Acad Sci 695:209, 1993 [PMID 7694533 ]
- Hutton M, Lendon CL, Rizzu P, et al: Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702, 1998 [PMID 9641683 ]
- Braak H, Braak E: The human entorhinal cortex: normal morphology and lamina-specific pathology in various diseases. Neurosci Res 15:6, 1992 [PMID 1336586 ]
- Ohm TG, Kirca M, Bohl J, et al: Apolipoprotein E polymorphism influences not only cerebral senile plaque load but also Alzheimer-type neurofibrillary tangle formation. Neuroscience 66:583, 1995 [PMID 7644022 ]
- Strittmatter WJ, Burke JR, DeSerrano VS, et al: Protein:protein interactions in Alzheimer disease and CAG triplet repeat diseases. Cold Spring Harb Symp Quant Biol 61:597, 1996 [PMID 9246486 ]
- Strittmatter WJ, Weisgraber KH, Huang DY, et al: Binding of human apolipoprotein E to synthetic amyloid b peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA 90:8098, 1993 [PMID 8367470 ]
- Schmechel DE, Saunders AM, Strittmatter WJ, et al. Increased amyloid b-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA 90:9649, 1993
- Rebeck GW, Reiter JS, Strickland DK, et al: Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions. Neuron 11:575, 1993 [PMID 8398148 ]
- Xu P-T, Schmechel D, Rothrock-Christian T, et al: Human apolipoprotein E2, E3 and E4 isoform specific transgenic mice: human-like pattern of neuronal immunoreactivity in central nervous system not observed in wild type mice. Neurobiol Dis 3:229, 1996
- Xu PT, Gilbert JR, Qui HL, et al: Specific regional transcription of apolipoprotein E in human brain neurons. Am J Pathol 154:601, 1999 [PMID 10027417 ]
- Bales KR, Verina T, Dodel RC, et al: Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet 17:263, 1997 [PMID 9354781 ]
- Strittmatter WJ, Weisgraber KH, Goedert M, et al: Hypothesis: microtubule instability and paired helical filament formation in the Alzheimer disease brain are related to apolipoprotein E genotype. Exp Neurol 125:163, 1994 [PMID 8313935 ]
- Huang DY, Weisgraber KH, Goedert M, et al: ApoE3 binding to tau tandem repeat I is abolished by tau serine262 phosphorylation. Neurosci Lett 192:209, 1995 [PMID 7566652 ]
- Nathan BP, Chang K-C, Bellosta S, et al: The inhibitory effect of apolipoprotein E4 on neurite outgrowth is associated with microtubule depolymerization. J Biol Chem 270:19791, 1995 [PMID 7649988 ]
- Holtzman DM, Pitas RE, Kilbridge J, et al: Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc Natl Acad Sci USA 92:9480, 1995 [PMID 7568158 ]
- Terry RD, Masliah E, Salmon DP, et al: Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572, 1991 [PMID 1789684 ]
- Martin JB: Molecular genetics in neurology. Ann Neurol 34:757, 1993
- Vigo-Pelfrey C, Seubert P, Barbour R, et al: Elevation of microtubule-associated protein tau in the cerebrospinal fluid of patients with Alzheimer's disease. Neurology 45:788, 1995 [PMID 7723971 ]
- Nitsch RM, Rebeck GW, Deng M, et al: Cerebrospinal fluid levels of amyloid b-protein in Alzheimer's disease: inverse correlation with severity of dementia and effect of apolipoprotein E genotype. Ann Neurol 37:512, 1995 [PMID 7717688 ]
- Mayeux R, Sano M: Treatment of Alzheimer's disease. N Engl J Med 341:1671, 1999
- De Deyn PP, Rabheru K, Rasmussen A, et al: A randomized trial of risperidone, placebo, and haloperidol for behavioral symptoms of dementia. Neurology 53:946, 1999. [PMID 10496251 ]
- Poirier J, Delisle M-C, Quirion R, et al: Apolipoprotein E4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer disease. Proc Natl Acad Sci USA 92:12260, 1995 [PMID 8618881 ]
- Whitehouse PJ, Kittner B, Roessner M, et al: Clinical trial designs for demonstrating disease-course-altering effects in dementia. Alz Dis Assoc Disord 12:281, 1998
- Diagnosis, Management and Treatment of Dementia: A Practical Guide for Primary Care Physicians. Guttman R, Seleski M, Eds. American Medical Association, Chicago, 1999