Neumología
⭳ Abrir artículo (PDF)1.0 MBEste artículo es idéntico en la Edición 2/2000.
Contenido del artículo
V ENFERMEDAD PULMONAR INFILTRATIVA CRONICA DIFUSA
- Generalidades
- Patogenia
- Estudio del paciente con enfermedad pulmonar infiltrativa difusa
- HISTORIA CLINICA
- EXAMEN FISICO
- EXAMENES DE LABORATORIO
- RADIOGRAFIA DE TORAX
- TOMOGRAFIA COMPUTADA DE ALTA RESOLUCION
- PRUEBAS DE FUNCION RESPIRATORIA
- LAVADO BRONCOALVEOLAR
- BIOPSIA PULMONAR
- TRATAMIENTO
- Enfermedad pulmonar infiltrativa difusa de etiología conocida
- ENFERMEDAD INDUCIDA POR MEDICAMENTOS
- NEUMOCONIOSIS
- NEUMONITIS POR HIPERSENSIBILIDAD
- ENFERMEDAD PULMONAR INFILTRATIVA DIFUSA MALIGNA
- Enfermedad infiltrativa difusa de causa desconocida
- SARCOIDOSIS
- Epidemiología
- Patogenia
- Manifestaciones clínicas de la sarcoidosis intratorácica
- Manifestaciones clínicas de la sarcoidosis extratorácica
- Diagnóstico
- Tratamiento
- FIBROSIS PULMONAR IDIOPATICA
- ENFERMEDADES DEL TEJIDO CONJUNTIVO
- Lupus eritematoso generalizado
- Artritis reumatoide
- Esclerosis sistémica progresiva
- Polimiositis y dermatomiositis
- Síndrome de Sjögren
- Enfermedad mixta del tejido conjuntivo
- GRANULOMA EOSINOFILICO DEL PULMON
- NEUMONIA ORGANIZADA CON BRONQUIOLITIS OBLITERANTE
- PROTEINOSIS ALVEOLAR
- NEUMONIA EOSINOFILICA
- Eosinofilia pulmonar simple (síndrome de Löffler)
- Eosinofilia pulmonar prolongada sin asma
- Eosinofilia pulmonar con asma
- Eosinofilia pulmonar tropical
- Vasculitis pulmonar (granulomatosis y angeítis alérgica)
- Síndrome hipereosinofílico
- Neumonía eosinofílica aguda
- HEMORRAGIA ALVEOLAR DIFUSA
- MICROLITIASIS
V ENFERMEDAD PULMONAR INFILTRATIVA CRONICA DIFUSA
DR. GERALD W. STATON. JR.
DR. ROLAND H. INGRAM JR.
Generalidades
El término enfermedad pulmonar infiltrativa crónica difusa se refiere a un grupo heterogéneo de padecimientos que afectan principalmente los alveolos y el intersticio pulmonar, aunque las vías respiratorias o los vasos pulmonares suelen afectarse en forma secundaria al proceso intersticial.
La enfermedad pulmonar infiltrativa crónica difusa afecta a una proporción sorprendentemente grande de la población. Un estudio de Nuevo México sugiere una prevalencia de 81 casos /100,000 en varones y 67 casos /100,000 en mujeres. El diagnóstico más común es fibrosis pulmonar o fibrosis pulmonar idiopática (FPI).1 En los Estados Unidos, algunos de estos trastornos son más comunes en la minorías étnicas, aunque los datos al respecto son incompletos.2
Patogenia
Se cree que ciertos mecanismos patogénicos de lesión pulmonar pueden influir en el desarrollo de la enfermedad pulmonar infiltrativa difusa. Se ha demostrado en estudios en animales que varios agentes causales diferentes (v.gr., bleomicina, paraquat, radiación y ciclofosfamida) producen un patrón característico de lesión que podría ser importante en las variedades idiopáticas más frecuentes de este padecimiento. En estos modelos se presenta daño inicial a las células epiteliales tipo I y a las células del endotelio cepilar. Después de una fase de edema y hemorragia leve, en ocasiones con acúmulo de fibrina, existe acúmulo de neutrófilos. Pocos días después aparecen los linfocitos y los macrófagos, y bajo la influencia de ciertos factores de crecimiento las células tipo II comienzan a replicarse y diseminarse para sustituir al epitelio tipo I dañado. En un periodo de dos semanas existe colágena, elastina y otros componentes extracelulares de la matriz, y al final llega a desarrollarse fibrosis pulmonar extensa. La combinación de exposición ambiental y susceptibilidad genética puede tener un papel importante en la patogenia de la enfermedad.3 Las variaciones en esta vía común causan las entidades patológicas específicas que se analizarán en este capítulo.
Estudio del paciente con enfermedad pulmonar infiltrativa difusa
Aunque se han descrito más de 100 causas de enfermedad pulmonar infiltrativa difusa, un número mucho menor de padecimientos son responsables de la mayoría de los casos. Sin embargo, debido a que los hallazgos clínicos, radiográficos y hasta patológicos pueden ser inespecíficos, es importante analizar el diagnóstico diferencial en una forma organizada. En todos los casos deberá realizarse una historia clínica, examen físico, radiografía de tórax y evaluación de la función pulmonar. Estos estudios simples suelen no dar el diagnóstico definitivo, pero ayudan a orientar al clínico hacia las causas más probables en cada enfermo, sirviendo para limitar las posibilidades en el diagnóstico diferencial.
HISTORIA CLINICA
Es indispensable excluir la infección como causa de la enfermedad pulmonar infiltrativa por la necesidad de tratamiento antimicrobiano específico y porque muchas causas no infecciosas del padecimiento se tratan con esteroides. La historia clínica debe incluir un interrogatorio cuidadoso en relación con la duración de los síntomas, que por lo general consisten en tos y disnea. Con frecuencia los síntomas tienen varios meses o incluso años de duración, lo que casi descarta una causa infecciosa. Por el contrario, una evolución más rápida, de días a semanas, debe sugerir la posibilidad de infección. Puede existir fiebre en varios tipos no infecciosos de enfermedad pulmonar infiltrativa difusa (v.gr., sarcoidosis, neumonitis por hipersensibilidad, enfermedades vasculares del tejido conjuntivo y enfermedad inducida por medicamentos), pero su presencia siempre debe alertar al clínico hacia la posibilidad de infección. La enfermedad pulmonar infiltrativa difusa infecciosa que evoluciona en un periodo de semanas a meses rara vez es causada por virus o bacterias. Sin embargo, las infecciones por micobacterias y hongos patógenos (v.gr., histoplasmosis, coccidioidomicosis y blastomicosis) pueden tener una evolución subaguda y suelen asociarse con fiebre. Quizá la causa infecciosa más frecuente es la neumonía por Pneumocystis carinii en un paciente con síndrome de inmunodeficiencia adquirida (SIDA). Este tipo de neumonía puede presentarse con disnea que progresa en forma lenta durante varias semanas, en ocasiones sin fiebre u otras evidencias de toxicidad sistémica, y este patrón puede recordar al de las causas no infecciosas frecuentes de enfermedad pulmonar infiltrativa difusa. Una historia cuidadosa en relación con los factores de riesgo para la infección por VIH es parte indispensable de la evaluación de todos los pacientes con una enfermedad pulmonar infiltrativa difusa reciente.
Además de establecer la duración de la enfermedad y la presencia o ausencia de fiebre y otros síntomas generales, la historia debe enfocarse hacia la ocupación del paciente y los medicamentos que ha recibido. Muchas diferentes exposiciones de tipo laboral pueden causar enfermedad pulmonar infiltrativa difusa, pero por fortuna la mayoría se encuentran en forma poco frecuente en la clínica. Las variedades de tipo laboral más frecuentes son causadas por la exposición a polvos inorgánicos de asbesto y sílice o a polvos orgánicos que causan neumonitis por hipersensibilidad. La asbestosis suele volverse evidente muchos años después de la exposición, por lo que la historia de las ocupaciones previas es muy importante. La causa más frecuente de neumonitis por hipersensibilidad de tipo laboral es el pulmón del granjero. Deberá interrogarse con gran cuidado a los granjeros respecto a cualquier relación entre los síntomas respiratorios o sistémicos y la exposición a material orgánico (granos o heno enmohecido) que puedan contener actinomicetos termofílicos. La historia debe incluir preguntas sobre la posible exposición a palomas o pájaros y humidificadores, porque este tipo de exposición causa también neumonitis por hipersensibilidad.
Muchos medicamentos han sido reportados como causa de enfermedad pulmonar infiltrativa difusa. La historia debe enfocarse tanto a los medicamentos prescritos como a los que se venden sin receta médica, y debe incluir preguntas sobre los fármacos tomados durante las semanas y meses previos.
Por último, debe investigarse si existen síntomas o signos que puedan indicar la presencia de una enfermedad del tejido conjuntivo o vasculitis. Los datos como fenómeno de Raynaud, fotosensibilidad, erupciones cutáneas o artritis son de gran importancia para el diagnóstico.
EXAMEN FISICO
El examen físico tiene menos utilidad diagnóstica que los antecedentes, pero también es importante. Puede sospecharse un padecimiento del tejido conjuntivo si se encuentra sinovitis, telangiectasias, esclerodactilia o erupción malar. La sarcoidosis afecta también a órganos extrapulmonares y puede causar uveítis, eritema nodoso y lesiones cutáneas en placa que corresponden a sarcoidosis en la piel. La enfermedad pulmonar infiltrativa difusa maligna puede asociarse con hallazgos debidos al tumor primario, como una masa abdominal o en la mama, hepatomegalia o una prueba de guayaco en heces positiva. Por último, el examen cardiovascular es muy importante porque en ocasiones la congestión pulmonar venosa crónica debida a una estenosis mitral oculta o a falla del ventrículo izquierdo puede manifestarse como una enfermedad pulmonar infiltrativa difusa.
EXAMENES DE LABORATORIO
Los exámenes de laboratorio incluyen estudios rutinarios y algunos más especializados. En ocasiones pueden encontrarse datos que indican la causa del padecimiento pulmonar infiltrativo difuso en los exámenes estándar. Puede pensarse en sarcoidosis cuando existen citopenias si está afectada la médula ósea o por la presencia de hipercalcemia o alteración en las pruebas de función hepática. Las enfermedades del tejido conjuntivo y las neoplasias pueden causar también citopenias. La eosinofilia periférica sugiere neumonía eosinofílica crónica. En raras ocasiones el granuloma eosinofílico puede presentarse como un padecimiento pulmonar asociado con evidencia por laboratorio de diabetes insípida.
Otras pruebas de laboratorio pueden ser útiles, dependiendo de los datos clínicos y radiográficos de cada caso. Suelen existir anticuerpos antinucleares o factores reumatoides en el suero de pacientes con padecimientos del tejido conjuntivo, pero también se encuentran en títulos bajos hasta en el 50 porciento de los enfermos con FPI. El aumento en la concentración sérica de la enzima convertidora de angiotensina (ECA) sugiere sarcoidosis, pero esta enzima puede también aumentar en la tuberculosis miliar, la beriliosis, la asbestosis y la silicosis. La neumonitis por hipersensibilidad casi siempre se asocia con anticuerpos séricos contra el antígeno agresor. Sin embargo, la presencia de anticuerpos contra uno de los agentes causales de las neumonitis por hipersensibilidad no demuestra que ésta sea la causa de la enfermedad pulmonar infiltrativa difusa, sino que solo ha existido la suficiente exposición al antígeno para despertar una respuesta inmunológica.
RADIOGRAFIA DE TORAX
Los datos radiográficos suelen ser inespecíficos. En la mayor parte de los casos existe un proceso intersticial simétrico y bilateral. Sin embargo, existen ciertas claves radiográficas que, cuando están presentes, pueden ayudar en el diagnóstico diferencial de la enfermedad pulmonar infiltrativa difusa para disminuir la lista de posibles causas [ver tabla 1].
TOMOGRAFIA COMPUTADA DE ALTA RESOLUCION
El uso de la tomografía computada de alta resolución (TCAR) representa un adelanto importante en la evaluación de la enfermedad pulmonar parenquimatosa difusa. Con el uso de este procedimiento puede determinarse la extensión, localización y patrón de afección pulmonar con gran exactitud [ver figura 1]. Con frecuencia la TCAR puede detectar alteraciones en pacientes que tienen síntomas de enfermedad pulmonar intersticial con radiografía de tórax normal. Cuando se combina con los datos clínicos y la radiografía de tórax, la TCAR de tórax puede identificar el diagnóstico específico en el 60 a 80 porciento de los casos.4 Cuando la TCAR indica un diagnóstico específico (v.gr., granuloma eosinofílico) se elimina la necesidad de una biopsia pulmonar.5
PRUEBAS DE FUNCION RESPIRATORIA
Las pruebas de función respiratoria orientan hacia la presencia de enfermedad pulmonar infiltrativa difusa y son útiles para evaluar la progresión de la enfermedad. Las características en este tipo de trastornos son un patrón ventilatorio restrictivo (volumen pulmonar disminuido), una relación volumen espiratorio forzado en el primer segundo a capacidad vital forzada (VEF1/CVF) normal o aumentada, reducción en la capacidad de difusión para monóxido de carbono (DLCO) y reducción en la tensión arterial de oxígeno (PaO2) asociada con tensión arterial de bióxido de carbono normal o disminuida (PaCO2). Además, suele existir limitación significativa al ejercicio por disminución en la PaO2, alteraciones en la mecánica respiratoria y enfermedad vascular pulmonar asociada.
LAVADO BRONCOALVEOLAR
En la mayoría de los casos la causa no se descubre a pesar de una historia clínica cuidadosa, evaluación fisiológica y estudios radiográficos y de laboratorio. El siguiente paso suele ser realizar una broncoscopía con lavado broncoalveolar (LBA) y biopsia pulmonar transbronquial. Ciertas causas pueden diagnosticarse sólo con el LBA, otras sólo con la biopsia y algunas con cualquiera de ambas técnicas. El LBA es más útil para diagnosticar causas infecciosas, en especial neumonía por P. carinii. La sensibilidad diagnóstica de este procedimiento para neumonía por P. carinii relacionada con SIDA es de alrededor de 90 a 95 porciento. Por lo tanto, éste es el procedimiento de eleccción para diagnosticar el 20 al 50 porciento de los casos de neumonía por P. cariniii asociada a SIDA que no pueden ser diagnosticados por medio del estudio de la expectoración. Otras infecciones oportunistas, como la neumonía por citomegalovirus y la infección micótica o tuberculosa diseminadas, pueden también diagnosticarse por medio del lavado. Causas no infecciosas que pueden diagnosticarse por esta técnica incluyen la proteinosis alveolar, la carcinomatosis linfática y el carcinoma de células alveolares. Además, el LBA puede proporcionar información útil, aunque no totalmente específica, al demostrar algunos de los siguientes cambios: (1) aumento en el número de eosinófilos en la neumonía eosinofílica crónica, (2) cuerpos de asbesto en la asbestosis, (3) los cambios llamados espumosos, con inclusiones laminares en la enfermedad inducida por amiodarona, (4) hiperplasia y atipia de los neumocitos tipo II en la lesión pulmonar inducida por citotóxicos, (5) células de Langerhans en el granuloma eosinófilo, y (6) sangre con hemosiderina abundante en los macrófagos alveolares en la hemorragia alveolar difusa. La cuantificación y distribución de las células inflamatorias (v.gr., macrófagos, linfocitos y neutrófilos) puede sugerir un diagnóstico específico.6 El líquido del lavado alveolar de los no fumadores normales típicamente tiene 84 a 99 porciento de macrófagos, 1 a 14 porciento de linfocitos y 0 a 1 porciento de neutrófilos. Las causas comunes de enfermedad pulmonar infiltrativa difusa no granulomatosa (v.gr., FPI, algunos trastornos del tejido conectivo y asbestosis) suelen caracterizarse por alveolitis neutrofílica, mientras que la sarcoidosis y las neumonitis por hipersensibilidad se asocian con aumento en los linfocitos. Sin embargo, existe sobreposición, y puede existir un paciente con FPI que tenga mayor número de linfocitos, o un enfermo con sarcoidosis y un número normal de linfocitos.
BIOPSIA PULMONAR
En muchos casos el diagnóstico se desconoce hasta que se realiza una biopsia pulmonar. Desde el punto de vista patológico, estos trastornos pueden caracterizarse por varios grados de afección de los tabiques alveolares o los alveolos por células inflamatorias, células mesenquimatosas, fibrosis, granulomas o células neoplásicas. En raros casos la sustancia anormal que se acumula en el parénquima pulmonar es sangre, material proteináceo (proteinosis alveolar), amiloide, músculo liso (linfangiomiomatosis o esclerosis tuberosa) o un material anormal que se deposita como resultado de un trastorno de almacenamiento hereditario (enfermedad de Gaucher, enfermedad de Niemann-Pick o síndrome de Hermansky-Pudlak).
Puede obtenerse tejido pulmonar por biopsia transbronquial durante una broncoscopía [ver tabla 2] o por una de dos técnicas de biopsia abierta: la biopsia abierta tradicional o la toracoscopía asistida por video. La biopsia pulmonar transbronquial es útil sobre todo en el diagnóstico de la enfermedad pulmonar infiltrativa difusa infecciosa y en la sarcoidosis. Pueden demostrarse granulomas no caseosos en muestras de biopsia transbronquial en el 70 a 90 porciento de los pacientes con sarcoidosis. El porcentaje de diagnóstico es alto (alrededor del 90 porciento) en pacientes con enfermedad pulmonar aparente desde el punto de vista radiográfico (estadios II y III) y menor (alrededor del 70 porciento) en enfermos con adenopatía hiliar sola (estadio I). La conveniencia de realizar una biopsia abierta después de una biopsia pulmonar transbronquial inespecífica dependerá de las características clínicas y radiográficas específicas de cada caso, del impacto que tendrá un diagnóstico más preciso en el tratamiento y de la evaluación del riesgo de la biopsia abierta en cada paciente. La frecuencia con la que se realiza una biopsia abierta de pulmón para determinar mejor las características histopatológicas de las formas no infecciosas de la enfermedad varía mucho entre los diferentes centros. El uso de la toracoscopía asistida por video en lugar de la biopsia abierta de pulmón no ha causado la reducción esperada en la morbilidad y el costo.7
TRATAMIENTO
Está indicada la inmunización contra los antígenos neumocócicos y anual contra la influenza. Los pacientes con un defecto obstructivo reversible pueden beneficiarse con el uso de broncodilatadores. La administración de oxígeno suplementario durante el reposo y el ejercicio puede aumentar la tolerancia del paciente al realizar sus actividades cotidianas.8
Enfermedad pulmonar infiltrativa difusa de etiología conocida
ENFERMEDAD INDUCIDA POR MEDICAMENTOS
Se ha reportado que muchos medicamentos diferentes causan enfermedad pulmonar infiltrativa difusa.9,10 Se ha calculado que la enfermedad pulmonar inducida por medicamentos afecta a varios cientos de miles de pacientes cada año. Para minimizar la morbimortalidad de ésta, es indispensable detectarla en etapas tempranas. La suspensión del agente agresor suele ser seguida de mejoría espontánea, mientras que el no detectar la relación causal entre el medicamento y la enfermedad pulmonar puede causar lesión pulmonar irreversible. Por desgracia, ciertos aspectos de la enfermedad inducida por medicamentos pueden dificultar el reconocimiento de esta relación causa-efecto. Primero, el número de diferentes medicamentos que causan enfermedad pulmonar infiltrativa difusa es muy grande como para recordarlos todos, y la enfermedad pulmonar suele ocurrir sólo en un pequeño porcentaje de los enfermos que reciben el medicamento. Segundo, el inicio de la enfermedad pulmonar puede ocurrir semanas a meses después de iniciado el fármaco. En el caso de los citotóxicos, los síntomas respiratorios comienzan muchas semanas después de la última exposición al agente agresor. Por último, los medicamentos que pueden causar enfermedad pulmonar infiltrativa difusa se prescriben con frecuencia para padecimientos que se asocian con un alto riesgo de la enfermedad. Por ejemplo, un paciente que recibe un agente quimiotáctico tiene mayor riesgo de infección y de infiltración tumoral en el pulmón. En forma semejante, los medicamentos antinflamatorios no esteroides que causan enfermedad pulmonar parenquimatosa suelen prescribirse para las enfermedades del tejido conjuntivo que se asocian con enfermedad pulmonar infiltrativa difusa. Por todos estos motivos, el médico debe estar muy alerta respecto a que la enfermedad pulmonar infiltrativa difusa sea causada por medicamentos.
Fármacos citotóxicos
La enfermedad pulmonar infiltrativa difusa es una causa importante de morbimortalidad en pacientes bajo tratamiento por una neoplasia. En algunos casos el medicamento utilizado para tratar la neoplasia es la causa directa de las lesiones pulmonares. Algunos enfermos mueren por toxicidad inducida por medicamentos citotóxicos, y otros experimentan lesión permanente de la función pulmonar a pesar de ser curados de la neoplasia. La mayor parte de los fármacos empleados en el tratamiento de las neoplasias pueden causar enfermedad pulmonar infiltrativa difusa. Los principales son la bleomicina, la ciclosfosfamida, el metotrexate y las nitrosoureas. La ciclofosfamida y el metotrexate se utilizan cada vez más para el manejo de padecimientos no malignos, aunque en dosis mucho menores. La enfermedad pulmonar inducida por medicamentos ocurre con menos frecuencia a estas dosis más bajas, pero también puede existir.
Las características clínicas de la enfermedad pulmonar inducida por fármacos son inespecíficas. La tos y la disnea suelen ser prominentes, y la fiebre no es rara. La radiografía de tórax suele demostrar infiltrados intersticiales bilaterales y simétricos, aunque puede existir asimetría al principio de la enfermedad. La progresión del padecimiento, tanto clínica como radiográfica, es variable. Lo más frecuente es que el principio sea subagudo, con tos y disnea que se presentan en varias semanas. Puede verse también un inicio más explosivo, con datos de síndrome de insuficiencia respiratoria progresiva aguda (SIRPA) y una necesidad urgente de apoyo ventilatorio mecánico. En el otro extremo del espectro, puede existir fibrosis pulmonar por el tratamiento con bleomicina y otros agentes que se desarrolla en forma insidiosa durante muchos meses.
Las características patológicas de la enfermedad inducida por fármacos citotóxicos son típicas, aunque no patognomónicas. Puede observarse infiltrado intersticial de células inflamatorias y fibrosis. Sin embargo, el dato más característico es que los neumocitos tipo II aumentan en número y muestran atipia importante. Esta característica patológica puede ser muy sugestiva de enfermedad inducida por fármacos citotóxicos en los casos pertinentes, pero pueden observarse cambios en las células tipo II con las infecciones virales graves y durante la fase de reparación del SIRPA.
Se observa un cuadro patológico un poco diferente cuando la enfermedad es causada por citarabina o metotrexate. La citarabina se ha asociado con una forma frecuentemente mortal de edema no cardiogénico. La enfermedad inducida por metotrexate se asocia con granulomas, alveolitis linfocítica y un aumento significativo en las células T cooperadoras.
El diagnóstico de enfermedad pulmonar infiltrativa difusa inducida por fármacos citotóxicos se establece si existe la exposición previa al agente agresor, excluyendo una infección como causa del daño pulmonar y demostrando características patológicas que sean compatibles con la lesión inducida por citotóxicos. La relación temporal entre la exposición al fármaco y el inicio de la enfermedad pulmonar es variable. No es raro que exista un intervalo de unas pocas semanas entre la última exposición al medicamento y el inicio de los síntomas; en casos raros este intervalo puede ser hasta de varios meses. Las características clínicas y radiográficas de la enfermedad pulmonar inducida por un medicamento citotóxico son casi indistinguibles de las causadas por una infección oportunista. Por lo tanto, se requiere un enfoque agresivo para establecer el diagnóstico. El no detectar una etiología infecciosa mediante la broncoscopía aumenta la posibilidad de enfermedad inducida por fármacos, en especial si se observan los cambios característicos en las células tipo II en la muestra de biopsia transbronquial. Sin embargo, puede requerirse la biopsia abierta de pulmón para excluir con confianza una infección y establecer mejor las características histopatológicas de la lesión por medicamentos citotóxicos. Incluso con la biopsia a cielo abierto, el diagnóstico de enfermedad inducida por fármacos citotóxicos es inferencial si no se encuentran criterios patognomónicos para establecer el diagnóstico.
La enfermedad pulmonar inducida por medicamentos citotóxicos no se previene con facilidad, ya que estos fármacos casi siempre son indispensables para el tratamiento óptimo de alguna neoplasia potencialmente mortal. Para los pacientes con carcinoma testicular que reciben bleomicina, el riesgo de lesiones pulmonares parece relacionarse con la dosis. La vigilancia de la DLCO y la espirometría durante el tratamiento permite en algunos casos detectar la lesión pulmonar en forma temprana, de modo que el evitar mayor exposición al fármaco disminuya la posibilidad de daño pulmonar permanente y grave. Sin embargo, el tratamiento para el linfoma no-Hodgkin con bleomicina combinada con doxorrubicina, ciclofosfamida, vincristina y prednisona causa enfermedad pulmonar infiltrativa difusa en forma no dependiente de la dosis, lo que anula los esfuerzos para prevenirla. Quizá la estrategia más eficaz para disminuir la morbimortalidad de la enfermedad inducida por citotóxicos no es prevenirla, sino diagnosticarla en una etapa tan temprana que pueda evitarse mayor exposición al fármaco, cambiando a esquemas alternos de quimioterapia siempre que sea posible.
La eliminación de mayor exposición al medicamento es el aspecto crucial del tratamiento de la enfermedad pulmonar inducida por fármacos citotóxicos. Algunos reportes anecdóticos indican que el tratamiento con esteroides se asocia con rápida mejoría del intercambio de gases y normalización de las alteraciones radiográficas en algunos casos. Si la enfermedad inducida por medicamentos citotóxicos es muy severa o parece progresar a pesar de la suspensión del fármaco, es aconsejable administrar un periodo de tratamiento empírico con esteroides.
Fármacos no citotóxicos
La enfermedad pulmonar infiltrativa difusa puede ser causada por la exposición a una gran variedad de fármacos no citotóxicos, incluyendo antibióticos, agentes antinflamatorios, antiarrítmicos y drogas ilícitas.10 Además, este padecimiento puede ser una manifestación del lupus eritematoso generalizado (LEG) inducido por medicamentos.
La nitrofurantoína es un agente antibacteriano utilizado principalmente para el tratamiento de las infecciones de las vías urinarias y constituye una de las causas más frecuentes de enfermedad pulmonar infiltrativa difusa inducida por fármacos.10 Puede presentarse toxicidad aguda o crónica, pero el síndrome agudo es mucho más común. La reacción pleuropulmonar aguda comienza dos a 10 días después de la exposición inicial al fármaco y se manifiesta por disnea, tos y fiebre. Ocurre pleuresía en una tercera parte de los enfermos. La radiografía de tórax muestra un patrón de infiltrado alveolar o intersticial, en ocasiones acompañado de derrame pleural. Puede observarse eosinofilia en sangre periférica. El padecimiento se diagnostica por el antecedente de exposición reciente a nitrofurantoína y resolución espontánea de los datos clínicos y radiográficos después de uno a cuatro días de suspendido el medicamento. La velocidad de resolución es mucho más rápida que en el caso de una infección y sirve para establecer el diagnóstico con un alto grado de certeza. La toxicidad crónica no se asocia con síntomas sistémicos y tiene un cuadro clínico y radiográfico indistinguible de la fibrosis pulmonar idiopática. Si no existe mejoría en dos a tres meses después de suspender el fármaco, está indicado el tratamiento con esteroides.
Amiodarona La amiodarona es un agente antiarrítmico que se utiliza principalmente para el tratamiento de la taquiarritmia ventricular refractaria. Este fármaco se asocia con diversos efectos tóxicos dependientes de la dosis sobre diferentes órganos, pero la principal limitación a su uso es la toxicidad pulmonar, que ocurre en alrededor del 5 a 7 porciento de los casos. La toxicidad pulmonar inducida por amiodarona suele caracterizarse por tos y disnea que en un principio se atribuyen a insuficiencia cardiaca congestiva. Los síntomas sistémicos, incluyendo la fiebre leve, no son raros. La radiografía de tórax puede mostrar un infiltrado intersticial simétrico y bilateral semejante al observado en otros tipos de enfermedad pulmonar infiltrativa difusa. Sin embargo, no son raros otros patrones radiográficos, que pueden incluir alteración unilateral o enfermedad aislada del lóbulo superior, este último hallazgo sugiere tuberculosis. En ocasiones se presenta derrame pleural. El diagnóstico de enfermedad pulmonar inducida por amiodarona se establece principalmente al excluir otras causas probables de enfermedad pulmonar, en especial la infección y la insuficiencia cardiaca. La broncoscopía con LBA y biopsia es útil para excluir la infección y también demuestra la presencia de los llamados macrófagos espumosos con inclusiones laminares (que se observan bajo microscopía electrónica). Estos cambios en los macrófagos indican exposición a la amiodarona, pero no demuestran que este fármaco sea la causa del proceso pulmonar porque se encuentran cambios semejantes en individuos asintomáticos que reciben el medicamento.
La suspensión del medicamento es la base del tratamiento, pero los esteroides parecen haber sido útiles en algunos casos seleccionados. Es interesante que existen varias notificaciones de enfermedad pulmonar aguda inducida por amiodarona en pacientes que fueron sometidos a procedimientos quirúrgicos menores, como la colocación de un desfibrilador automático o la angiografía pulmonar. Con frecuencia la enfermedad tiene un curso fulminante, y la evolución llega a ser mortal. Debido a que la vida media del medicamento es de varias semanas, los pacientes sometidos a estos procedimientos tienen aún riesgo de lesión pulmonar aguda si el medicamento se suspendió solo uno a dos meses antes del procedimiento.
Oro y penicilamina El oro y la penicilamina, utilizadas principalmente para el tratamiento de la artritis reumatoide y otras enfermedades del tejido conjuntivo, pueden causar enfermedad pulmonar infiltrativa difusa. Puede ser difícil determinar si el daño pulmonar es causado por el agente terapéutico o por la enfermedad subyacente, incluso con una biopsia a cielo abierto de pulmón. Se aconseja en estos casos la suspensión del medicamento, junto con el tratamiento empírico con esteroides si los síntomas son graves. La penicilamina también ha sido implicada en casos de hemorragia alveolar difusa con glomerulonefritis, LEG inducido por medicamentos y panbronquiolitis con obstrucción grave de las vías respiratorias.
Drogas ilícitas Las drogas ilícitas pueden también causar enfermedad pulmonar infiltrativa difusa. Puede existir granulomatosis inducida por talco y fibrosis pulmonar por la inyección de cristales y anfetaminas disueltas, o por pastillas de narcóticos que tienen talco como excipiente. La enfermedad puede progresar años después de la última exposición, quizá porque el talco persiste en los pulmones y continúa ocasionando una respuesta inflamatoria. La biopsia abierta o transbronquial demuestra inflamación granulomatosa con partículas abundantes de talco, que pueden ser observadas por microscopía polarizada. El no analizar los granulomas con microscopio polarizado puede ocasionar un diagnóstico erróneo de sarcoidosis. El diagnóstico puede también hacerse al detectar talco en la retina. La heroína puede causar edema pulmonar agudo, pero no causa per se una enfermedad pulmonar infiltrativa crónica difusa. La cocaína también se ha asociado a edema pulmonar agudo, y se ha reportado que causa hemorragia alveolar difusa y un síndrome de infiltrado pulmonar con eosinofilia. Las reacciones pulmonares relacionadas con la cocaína no ocasionan lesiones crónicas.
L-triptofano El síndrome de mialgia-eosinofilia relacionado con L-triptofano, una enfermedad caracterizada por mialgias severas, fatiga y diversas erupciones, puede tener también manifestaciones pulmonares . Muchos pacientes con este síndrome refieren disnea, y se han observado infiltrados intersticiales bilaterales, bronquiolitis folicular, hipertensión pulmonar y derrames pleurales. Las pruebas de función pulmonar revelan restricción con reducción en la capacidad de difusión, hipoxemia y evidencia de debilidad de los músculos respiratorios. Las biopsias pulmonares muestran inflamación intersticial con eosinofilia parenquimatosa y vasculitis prominente de los vasos pequeños. Algunos pacientes con este síndrome han respondido a esteroides. Dos años después del inicio la mayoría de los síntomas han mejorado o desaparecido.11
NEUMOCONIOSIS
La exposición ambiental o laboral a material particulado puede causar varias neumoconiosis que se manifiestan como asma, bronquitis crónica o enfermedad parenquimatosa difusa (sobre todo intersticial). Estas neumoconiosis incluyen asbestosis, silicosis, neumoconiosis del trabajador de carbón (NTC) y beriliosis.
Asbestosis
La exposición a asbesto, un silicato usado para aislamiento en superficies de fricción y para endurecer materiales, se asocia con el desarrollo de cambios pleurales (placas y derrame), mayor incidencia de neoplasias (carcinoma broncogénico y mesotelioma) y fibrosis intersticial difusa. Solo la fibrosis intersticial difusa se denomina asbestosis.
La asbestosis se caracteriza por disnea de inicio gradual 20 a 30 años después de la exposición al asbesto. Suele existir tos, pero ésta es no productiva a menos que el paciente haya sido fumador y tenga una bronquitis crónica asociada. Pueden auscultarse estertores finos al final de la inspiración antes de que existan alteraciones en la radiografía de tórax, y es común la presencia de dedos en palillo de tambor. En la fase tardía de la asbestosis se desarrollan signos de cor pulmonale.
Al principio de la enfermedad la radiografía de tórax puede ser normal, pero gradualmente aparecen sombras lineales, pequeñas e irregulares, en las porciones inferiores de los campos pulmonares. La captación pulmonar de galio-67 puede estar aumentada, pero es inespecífica. Las pruebas de función pulmonar suelen mostrar restricción, reducción en la DLCO e hipoxemia inducida por el ejercicio. El diagnóstico de asbestosis se realiza cuando un paciente tiene historia de exposición a asbestos, placas pleurales (indicador objetivo de la exposición) y, en casos dudosos, exceso de asbesto en las muestras obtenidas (por LBA, biopsia pulmonar transbronquial o biopsia abierta). No existe tratamiento específico para los pacientes con asbestosis.
Silicosis
La silicosis es una enfermedad pulmonar fibrótica crónica causada por la exposición a sílice cristalino libre. Se ha demostrado la exposición laboral significativa a sílice en la industria minera, el corte, pulido y esculpido de piedra, el trabajo de fundición y la limpieza abrasiva (con arena). Se requiere la exposición durante alrededor de 5 años para el desarrollo de la silicosis, a menos que la exposición sea muy intensa. Existen dos formas de silicosis: la nodular simple, en la que muchos pacientes están asintomáticos o sufren solo de bronquitis crónica secundaria al uso de tabaco, y la fibrosis progresiva masiva (FPM), en la que se desarrolla disnea incapacitante. No suelen existir datos físicos. Desde el punto de vista radiográfico la silicosis nodular simple se caracteriza por opacidades difusas, redondas y pequeñas que tienden a ser más prominentes en los lóbulos superiores. Puede observarse crecimiento hiliar, y en ocasiones las llamadas calcificaciones en cascarón de huevo. En los pacientes con FPM las opacidades coalescen para formar masas grandes e irregulares. La función pulmonar en los pacientes con silicosis nodular simple puede ser normal o manifestarse por un patrón mixto de obstrucción y restricción. En los enfermos que tienen FPM se desarrolla restricción e hipoxemia severas, así como hipertensión pulmonar.
La tuberculosis y las infecciones por micobacterias atípicas, que ocurren con mayor frecuencia en los enfermos con silicosis, pueden confundirse con progresión de la silicosis, y el diagnóstico definitivo es difícil. Debido a que la inmunidad mediada por células parece ser normal en los pacientes con silicosis, la prueba cutánea de tuberculina puede ser muy útil para identificar a los pacientes en los que debe investigarse el diagnóstico de tuberculosis.
El diagnóstico de silicosis casi siempre se realiza si un paciente tienen antecedente de exposición y una radiografía de tórax compatible. En los casos atípicos la presencia de sílice en el LBA o en las muestras de biopsia de pulmón pueden establecer el diagnóstico y excluir el de tuberculosis y carcinoma.
Se ha pensado que la silicosis es intratable, aunque un reporte sugiere que los esteroides por vía oral disminuyen la inflamación y mejoran la función pulmonar.12 La prevención de la mayor exposición al sílice y el tratamiento de la tuberculosis activa son aspectos muy importantes en la atención al paciente.
Neumoconiosis del trabajador de carbón
La NTC es una causa poco frecuente de fibrosis pulmonar en los trabajadores que están expuestos a polvo de carbón y grafito. La mayoría de estos pacientes son mineros que con frecuencia han trabajado bajo la superficie. La influencia del tabaquismo y de materiales como el sílice en la producción de los síntomas respiratorios es motivo de controversia. Muchos pacientes con NTC tienen tos crónica que en ocasiones es productiva, con expectoración gris o negra, y que puede ser causada por bronquitis crónica que se relaciona con polvo de tabaco o carbón. En forma semejante a la silicosis, la NTC incluye a la forma simple y a la FPM. La primera no se asocia con una mayor incidencia de disnea, mientras que la fibrosis progresiva masiva se caracteriza por disnea severa. Los cambios radiográficos de la forma simple consisten en opacidades pequeñas y redondeadas. Cuando las opacidades son mayores de 1 cm se clasifican en forma arbitraria como FPM. La NTC no progresa si se elimina la exposición al polvo, mientras que la FPM progresa aún después de suspendida la exposición. No existe tratamiento específico para ninguna forma de NTC.
Beriliosis
El berilio es un metal raro que se usa en las industrias de alta tecnología moderna por su bajo peso, su alto punto de fundición, su resistencia tensil, sus excelentes propiedades aislantes y su capacidad para retardar la fisión nuclear. La exposición de alta intensidad al berilio puede ocasionar una bronquitis aguda y neumonitis químicas, que son muy raras en la actualidad por los controles ambientales tan estrictos. Sin embargo, aún se ve con frecuencia beriliosis crónica. Esta se caracteriza por enfermedad granulomatosa multisistémica que tiene muchas semejanzas con la sarcoidosis y que ocurre meses a años después de la exposición. El número de casos crónicos parece estar disminuyendo, quizá por una mayor atención hacia la enfermedad y por los controles ambientales. Se piensa que el berilio se une a proteínas del huésped que son transportadas por todo el organismo, estos complejos de berilio-proteína estimulan una respuesta de hipersensibilidad tardía, que produce inflamación granulomatosa en los sitios de actividad de la enfermedad.
Los síntomas de la beriliosis son disnea, tos, dolor torácico, fatiga, pérdida de peso y artralgias. Los estertores no son comunes al inicio. Los datos radiográficos, que pueden preceder a los síntomas clínicos, incluyen opacidades nodulares e irregulares mal definidas que en ocasiones se asocian con adenopatía hiliar. Al progresar la enfermedad los pulmones disminuyen de tamaño y adquieren el aspecto de un panal de abejas. Las pruebas de función pulmonar pueden ser normales; sin embargo, con más frecuencia se caracterizan por una baja DLCO con o sin restricción o demuestran obstrucción leve. Otras alteraciones incluyen hiperuricemia, hipercalcemia, hipercalciuria y aumento en la concentración sérica de ECA.
Los criterios diagnósticos incluyen una historia bien demostrada de exposición o la demostración de berilio en exceso en muestras obtenidas del paciente, evidencia radiográfica y en las pruebas de función pulmonar de una enfermedad de vías respiratorias bajas compatible con beriliosis, y la demostración de inflamación granulomatosa en el pulmón, los ganglios linfáticos y otras áreas de actividad de la enfermedad. La diferenciación entre beriliosis y sarcoidosis puede ser difícil. Debe pensarse en el diagnóstico de sarcoidosis por la presencia de uveítis y eritema nodoso, adenopatía hiliar sin infiltrados parenquimatosos o por mejoría después de un periodo breve de administración de esteroides. Es más posible el diagnóstico de beriliosis si existe una prueba de transformación de linfocitos positiva, realizada en la sangre o células obtenidas por LBA, que se positiviza en respuesta a compuestos de berilio.
El paso más importante en el tratamiento de la beriliosis es la prevención de una mayor exposición. Se piensa que los esteroides mejoran la evolución de la enfermedad, pero estos agentes deben administrarse de por vida.
NEUMONITIS POR HIPERSENSIBILIDAD
El término de neumonitis por hipersensibilidad se refiere a una enfermedad pulmonar intersticial que resulta de la inhalación de antígenos orgánicos. Parece ser que la patogenia de la neumonitis por hipersensibilidad tiene una base inmunológica y no es resultado de una lesión tóxica directa al pulmón por la sustancia inhalada. Varios antígenos orgánicos causan este síndrome, pero la forma más frecuente de neumonitis por hipersensibilidad, llamada pulmón del granjero, es causada por la inhalación de Actinomyces termofílico, que está presente en el heno o granos enmohecidos. Otras causas frecuentes de neumonitis por hipersensibilidad son la enfermedad de criadores de palomas y de criadores de pájaros, en la que las proteínas séricas de las palomas o periquitos, al ser inhaladas, inducen el síndrome. La enfermedad pulmonar por humidificadores es un tipo de neumonitis por hipersensibilidad causada por la exposición a sistemas de ventilación contaminados. El antígeno responsable en estos casos es, como en el pulmón del granjero, un organismo Actinomyces termofílico, pero puede incluir también organismos Aureobasidium y varias amibas. Las otras formas de neumonitis de hipersensibilidad además del pulmón del granero, la enfermedad de criadores de palomas, la enfermedad de cuidadores de pájaros y la enfermedad por humidificadores, son muy poco frecuentes y suelen resultar de la exposición a antígenos en ocupaciones arcaicas (v.gr., corte de paprika y amortajamiento de momias).
Patogenia y patología
La patogenia de la neumonitis por hipersensibilidad no se conoce bien. La hipótesis actual consiste en que el material inhalado causa la activación directa de la vía alterna del complemento, ocasionando la formación de factores quimiotácticos para neutrófilos y, en consecuencia, una alveolitis neutrofílica aguda. Los productos de la activación del complemento estimulan también a los macrófagos alveolares para producir monocinas, causando una secuencia de eventos que al final provoca formación de granulomas.
Los cambios patológicos en la neumonitis por hipersensibilidad han sido bien definidos al examinar biopsias de pulmón a cielo abierto de gran número de pacientes con pulmón del granjero. Todos los pacientes tienen una neumonitis intersticial, y dos tercios muestran granulomas aislados. Puede ocurrir fibrosis en las formas más crónicas de la enfermedad, y hasta el 50 porciento de los pacientes tiene bronquiolitis obliterante. El cuadro patológico de la neumonitis intersticial linfocítica con formación de granulomas y bronquiolitis obliterante es característico de la neumonitis por hipersensibilidad [ver figura 2]. Los estudios de LBA han determinado el tipo de población linfocitaria en el pulmón, y han demostrado que los linfocitos predominantes son de la subclase T supresora-citotóxica. Esta inversión en la relación entre células T cooperadoras y supresoras-citotóxicas en la neumonitis por hipersensibilidad contrasta con el predominio de células T cooperadoras en los pacientes con sarcoidosis. El hallazgo de una alveolitis linfocítica intensa con inversión de la relación entre células T cooperadoras-citotóxicas sugiere neumonitis por hipersensibilidad, pero este patrón no puede considerarse específico porque puede observarse en otros padecimientos (v.gr., FPI asociada con alveolitis linfocítica).
Manifestaciones clínicas y diagnóstico
La neumonitis por hipersensibilidad existe básicamente en dos formas clínicas distintas: aguda y crónica.
La neumonitis aguda por hipersensibilidad se caracteriza por fiebre, calosfríos, tos, disnea y malestar, que ocurren típicamente 4 a 8 horas después de la exposición antigénica. La mayoría de los pacientes con neumonitis aguda por hipersensibilidad mejoran en forma espontánea a menos que persista la exposición al antígeno. En ocasiones el paciente presenta un episodio más grave caracterizado por hipoxemia e insuficiencia respiratoria considerable que requiere hospitalización. Debido a la importancia de los síntomas sistémicos durante el episodio agudo, puede pensarse en forma equivocada que el paciente tiene una infección respiratoria. Ante la exposición continua al antígeno inhalado (por ejemplo, en una granja) el paciente puede tener episodios diarios de neumonitis aguda, que se superponen unos con otros. Si ocurre este patrón sintomático, el granjero podrá referir que ha tenido una enfermedad gripal durante varias semanas. Los síntomas respiratorios como tos y sibilancias, que ocurren inmediatamente después de la exposición a material orgánico, no deben atribuirse a neumonitis por hipersensibilidad, sino a una respuesta irritante dentro de las vías respiratorias.
En la neumonitis crónica por hipersensibilidad no suele haber síntomas generales, sino que el cuadro clínico se caracteriza por disnea crónica y tos durante el ejercicio. La presentación clínica es la de cualquier enfermedad pulmonar intersticial de etiología incierta.
No existe una sola prueba que pueda utilizarse para establecer el diagnóstico inequívoco de neumonitis por hipersensibilidad. Por lo tanto, deberá analizarse la combinación de la historia clínica, los estudios serológicos y la exclusión de otras causas posibles. La historia de episodios previos de neumonitis aguda por hipersensibilidad es de gran valor diagnóstico. Al realizar la historia, el clínico debe enfocarse a establecer la presencia de una exposición antigénica relevante y a tratar de determinar si esta exposición ha causado síntomas típicos agudos varias horas después. Este tipo de antecedente es más frecuente cuando existe una exposición intensa (v.gr., heno mohoso o un palomar), pero es menos frecuente en casos de exposición de baja intensidad (v.gr., un humidificador o un solo pájaro). En esta última circunstancia la exposición es más o menos continua y es muy difícil establecer una relación causal entre la exposición al antígeno y los síntomas. Los pacientes deben ser interrogados en forma cuidadosa sobre si los síntomas se resolvieron cuando estuvieron de vacaciones o alejados de la posible fuente de exposición. Por lo menos el 50 porciento de los pacientes con la forma crónica de la enfermedad niega antecedente de episodios agudos. Es posible que éstos sí hayan ocurrido, pero que el paciente haya atribuido los síntomas a una infección de las vías respiratorias. El paciente puede mejorar en forma espontánea cuando se le hospitaliza para evaluarle por el padecimiento respiratorio, sólo para presentar recurrencia de los síntomas en cuanto regresa a su casa. Este tipo de antecedente debe siempre despertar la sospecha de una neumonitis por hipersensibilidad como causa de los síntomas.
La apariencia radiográfica del tórax en un paciente con enfermedad aguda varía desde la ausencia de cambios hasta infiltrados alveolares o lobares. Al desarrollarse la enfermedad crónica puede observarse un infiltrado reticular, nodular o combinado, lo mismo que fibrosis si la exposición al antígeno continúa. La TCAR, que es más sensible que la radiografía de tórax, puede sugerir el diagnóstico y distinguir la neumonitis por hipersensibilidad de otros tipos de enfermedad pulmonar difusa.13
Los estudios serológicos son útiles para determinar si ha existido exposición a un antígeno específico. Casi todos los pacientes con pulmón del granjero y enfermedad de criadores de palomas tienen anticuerpos contra organismos Actinomyces termofílicos y suero de palomas, respectivamente. Sin embargo, los anticuerpos pueden también existir en el suero de un porcentaje significativo de individuos expuestos y asintomáticos. Por lo tanto, la presencia de anticuerpos en el suero no establece el diagnóstico de neumonitis por hipersensibilidad, sino que demuestra que ha existido exposición significativa al antígeno.
Los enfermos que sufren neumonitis por hipersensibilidad subaguda o crónica deben ser sometidos a una broncoscopía diagnóstica con lavado broncoalveolar (LBA) y biopsia transbronquial. El líquido del lavado en la neumonitis por hipersensibilidad suele mostrar linfocitosis intensa, con más de 50 porciento de linfocitos. Como se mencionó antes, estos son principalmente linfocitos T supresores-citotóxicos. Al pasar el tiempo desde la última exposición al antígeno el LAB tiende a normalizarse.14 Se espera que la biopsia pulmonar transbronquial muestre una neumonitis intersticial inespecífica en la mayoría de los enfermos; sin embargo, puede no haber evidencia de granulomas ni de bronquiolitis obliterante. Incluso en las dos terceras partes de los casos en los que existen granulomas las lesiones están más aisladas que los granulomas sarcoides y, por lo tanto, no es tan probable demostrarlas en la biopsia transbronquial. La biopsia de pulmón a cielo abierto suele reservarse para casos especialmente difíciles y confusos, en los que no puede establecerse el diagnóstico con base en la historia clínica, los estudios serológicos y la broncoscopía. Pocos pacientes que tienen neumonitis por hipersensibilidad requerirán de una biopsia a cielo abierto para establecer el diagnóstico.
Tratamiento
El tratamiento de la neumonitis por hipersensibilidad incluye principalmente suspender la exposición al agente causal. Esto es relativamente fácil en el caso de la enfermedad pulmonar por un humidificador o por cuidado de pájaros, pero es más difícil en pacientes con pulmón del granjero. En ocasiones los granjeros pueden contar con un ayudante que realice las labores que incluyan exposición al heno enmohecido. Las mejoras en las técnicas de trabajo en el campo, con mejor ventilación, y la atención cuidadosa a otros factores que puedan disminuir el contenido de humedad del heno, reducirán en forma significativa la carga antigénica. Es adecuado referir al paciente a una institución que trate enfermedad pulmonar por agricultura.
Los esteroides se utilizan principalmente para la neumonitis aguda por hipersensibilidad que se caracteriza por síntomas sistémicos graves y alteraciones en el intercambio de gases. Los pacientes con neumonitis por hipersensibilidad subaguda o crónica pueden mejorar con más rapidez si se les administra tratamiento con esteroides durante varias semanas. Mientras se administran los esteroides debe intentarse eliminar al máximo la exposición antigénica. De otro modo, los síntomas serán enmascarados por el uso de los esteroides y la exposición al antígeno aumentará la lesión pulmonar. Cuando cesa la exposición al antígeno la mayoría de los pacientes mejoran en forma espontánea. Los pacientes que están ya en la fase fibrótica de la enfermedad pueden no mejorar mucho, y existe un pequeño número de pacientes que parece empeorar a pesar de que, aparentemente, se haya eliminado el antígeno. En este último caso puede ser útil vigilar los niveles séricos de anticuerpos porque, si no existe mayor exposición, estos deben disminuir en forma gradual. Un título que no cambia o que aumenta puede sugerir que la exposición al antígeno continúa.
ENFERMEDAD PULMONAR INFILTRATIVA DIFUSA MALIGNA
La afección difusa del pulmón por una neoplasia ocurre en la carcinomatosis linfangítica, el cáncer de células alveolares, el linfoma y la leucemia. La carcinomatosis linfangítica pulmonar es causada con más frecuencia por adenocarcinomas, y menos por cánceres epidermoides. Los adenocarcinomas suelen originarse en la mama, el aparato digestivo o el pulmón. Los síntomas atribuibles al tumor linfangítico, tos y disnea, son los mismos que los de la enfermedad pulmonar infiltrativa difusa. Sin embargo, el padecimiento progresa con mucho más rapidez que otras enfermedades intersticiales pulmonares, y es rara la supervivencia por más de 3 a 6 meses. La radiografía de tórax muestra infiltrados reticulares o reticulonodulares bilaterales, y con frecuencia líneas B de Kerley. Este último dato debe hacer sospechar, en ausencia de insuficiencia cardiaca, que una neoplasia es responsable del padecimiento pulmonar. También puede observarse derrame pleural y adenopatía hiliar. Con menos frecuencia la carcinomatosis linfangítica se presenta con afección unilateral o con una radiografía de tórax totalmente normal a pesar de que la disnea y la hipoxemia sean graves. En pacientes con carcinomatosis linfangítica pero con radiografía de tórax normal, con frecuencia se encuentran células tumorales abundantes en los pequeños vasos pulmonares, que pueden en ocasiones causar hipertensión pulmonar severa (síndrome de embolia tumoral). El diagnóstico suele hacerse por broncoscopía, ya sea por LBA o con biopsia transbronquial. Un método alternativo para establecer el diagnóstico es por examen citológico de la sangre aspirada a través del cáteter arterial pulmonar en cuña. Esta técnica puede considerarse como opción para los pacientes con disnea muy severa en los que el cateterismo cardiaco derecho puede tolerarse mejor que una broncoscopía. En ocasiones se requiere biopsia abierta de pulmón para diagnosticar la carcinomatosis linfangítica.
El carcinoma de células alveolares puede manifestarse como un nódulo o masa único, nódulos múltiples, o como una enfermedad ocupativa del espacio aéreo que puede ser focal, multifocal o difusa. Cuando el tumor es difuso el diagnóstico diferencial incluye causas no malignas de ocupación alveolar difusa, como el edema pulmonar, y la proteinosis, sarcoidosis y hemorragia alveolares. El diagnóstico de carcinoma de células alveolares se establece por broncoscopía con LBA y biopsia transbronquial.
Los linfomas y la leucemia pueden causar enfermedad pulmonar infiltrativa difusa. La afección linfomatosa del parénquima pulmonar suele observarse en asociación con linfadenopatía mediastinal y evidencia de linfoma extratorácico.15 En casos raros la enfermedad pulmonar aislada es la manifestación inicial del linfoma. Cuando la leucemia subyacente está mal controlada puede observarse infiltración leucémica del pulmón, que puede contribuir a la muerte al causar insuficiencia respiratoria. El diagnóstico de infiltración leucémica o linfomatosa del pulmón puede requerir de una biopsia a cielo abierto para establecer en forma adecuada el tipo de proceso infiltrativo y excluir por completo una infección. Sin embargo, en algunos casos la broncoscopía es suficiente para establecer el diagnóstico.
Enfermedad infiltrativa difusa de causa desconocida
SARCOIDOSIS
La sarcoidosis es una enfermedad multisistémica de etiología desconocida que se caracteriza desde el punto de vista patológico por la presencia de granulomas no caseosos en varios tejidos. Aunque casi cualquier órgano puede afectarse, la gran mayoría de los pacientes solicitan atención médica por afección intratorácica que se manifiesta por síntomas respiratorios o por alteraciones asintomáticas en la radiografía de tórax. Menos del 5 porciento de los pacientes con sarcoidosis tienen una radiografía de tórax normal. Además, la morbimortalidad que se asocia con la sarcoidosis se debe principalmente a afección pulmonar. Por lo tanto, es adecuado considerar a esta enfermedad como un padecimiento de las vías respiratorias y que puede tener manifestaciones extrapulmonares.
Muchos padecimientos infecciosos y no infecciosos se asocian con formación de granulomas y un cuadro histopatológico indistinguible de sarcoidosis. Por lo tanto, aunque las características clínico-patológicas en cada paciente sean muy sugestivas de sarcoidosis, el diagnóstico requiere de la exclusión de otras enfermedades que causen granulomas. Ejemplos de padecimientos con características clínico-patológicas idénticas a las de la sarcoidosis son la beriliosis y la histoplasmosis [ver antes, Estudio del paciente con enfermedad pulmonar infiltrativa difusa].
Epidemiología
La sarcoidosis ocurre en todo el mundo y grupos étnicos, pero existen importantes diferencias regionales y étnicas en su incidencia y prevalencia. Por ejemplo, en los Estados Unidos la sarcoidosis es 10 veces más frecuente entre afroamericanos que entre blancos, además de más severa. Sin embargo, esta mayor prevalencia no se observa entre los negros en Europa. Se ha calculado que la prevalencia de la sarcoidosis en los Estados Unidos puede ser entre uno en 10,000 y uno en 2,500. La enfermedad es más frecuente en individuos entre 20 y 40 años de edad, pero puede presentarse en niños y ancianos. La sarcoidosis tiene una frecuencia muy similar en ambos sexos, con una ligera mayor prevalencia entre las mujeres. En algunos casos se ha reportado sarcoidosis familiar; con menos frecuencia se ha notificado de un matrimonio con el padecimiento, lo que podría sugerir un factor ambiental común en ciertos casos.
Patogenia
Existe una secuencia específica de eventos que causan la formación del granuloma en la sarcoidosis16 [ver figura 3]. La expansión de la población de linfocitos T cooperadores en los pulmones parece ocurrir por la activación de linfocitos T por macrófagos a través de interleucina -1 (IL-1), con la consecuente liberación de interleucina-2 (también conocida como factor de crecimiento de linfocitos T) por la célula T cooperadora. La IL-2 causa replicación de la población de células T existente. Esta población de linfocitos T cooperadores, que están muy aumentados en número y nivel de actividad, recluta monocitos de la sangre periférica hacia el pulmón al liberar un factor quimiotáctico de monocitos, participando así en la formación del granuloma. Los monocitos se convierten en macrófagos tisulares y, al final, en células epitelioides y células gigantes multinucleadas, que forman el centro del granuloma. Otro efecto de la población expandida y activada de linfocitos T cooperadores en los sitios de actividad de la enfermedad es que la estimulación local de linfocitos B produce inmunoglobulinas, que causan la hipergamaglobulinemia que con frecuencia se observa en este padecimiento.
Manifestaciones clínicas de la sarcoidosis intratorácica
Las manifestaciones más frecuentes de la sarcoidosis intratorácica son la linfadenopatía y la enfermedad pulmonar infiltrativa difusa. Se ha creado un sistema de estadificación con base en la presencia o ausencia de estas dos manifestaciones en la radiografía: estadio I, solo linfadenopatía hiliar bilateral; estadio II, linfadenopatía hiliar bilateral y enfermedad pulmonar infiltrativa difusa; y estadio III, solo enfermedad pulmonar infiltrativa difusa [ver figura 4]. Los síntomas secundarios a la afección del parénquima pulmonar incluyen disnea y tos. Se encuentran alteraciones en la función respiratoria en casi todos los pacientes sintomáticos y en algunos asintomáticos. Las anomalías fisiológicas en los enfermos con sarcoidosis sintomática consisten en forma típica en reducción de la Dlco y de la capacidad vital sin obstrucción al flujo del aire. La Dlco suele alterarse antes de la capacidad vital, pero ambas medidas se modifican en las personas con síntomas moderados a severos. La obstrucción al flujo del aire es relativamente rara, excepto en pacientes con enfermedad avanzada o con afección endobronquial de vías respiratorias grandes. En casos raros los granulomas endobronquiales difusos en las vías respiratorias pequeñas causan una alteración de predominio obstructivo en pacientes con enfermedad en estadio I.
La mayoría de los enfermos con sarcoidosis en estadio I
están asintomáticos y la enfermedad se detecta en una
radiografía de tórax de rutina. Cuando la enfermedad en estadio I
es sintomática, los síntomas suelen ser extrapulmonares, y
consisten en fiebre, malestar general, artralgias o eritema nodoso. La
presencia de fiebre, adenopatía hiliar bilateral, artralgias o artritis
y eritema nodoso se conoce como síndrome de Lögfren. Alrededor del
10 porciento de los enfermos con enfermedad en estadio I tienen evidencia de
afección extrapulmonar (v.gr., ocular, del sistema nervioso o de las
glándulas lacrimales). Alrededor del 75 porciento de los enfermos con
sarcoidosis en estadio I están en remisión a los 2 años
después del comienzo. El eritema nodoso como forma inicial de
presentación aumenta la posibilidad de remisión
espontánea. Los enfermos en los que la linfadenopatía hiliar
bilateral no remite en 2 años pueden permanecer estables, tener
remisión espontánea más tarde, o desarrollar enfermedad
pulmonar progresiva. No más de 10 a 15 porciento de los enfermos
desarrollan un padecimiento pulmonar progresivo.
La frecuencia de los síntomas es más alta en pacientes con enfermedad en estadio II, pero los casos asintomáticos no son raros. Los síntomas pueden ser principalmente de tipo general, como en el estadio I, o pueden originarse de la afección pulmonar. No es raro que existan alteraciones radiográficas extensas asociadas con síntomas respiratorios mínimos. Alrededor del 50 porciento de los pacientes con sarcoidosis en estadio II tendrán remisión a los 2 años después del inicio. Existe más probabilidad de que la enfermedad en estadio II sea progresiva y siga una evolución sintomática crónica que la enfermedad en estadio I.
En el 5 a 15 porciento de los pacientes se encuentra enfermedad en estadio III en el momento del diagnóstico. Los síntomas respiratorios son frecuentes pero, como en la enfermedad en estadio II, la radiografía de tórax puede verse mucho peor que el paciente. Sólo una tercera parte de los pacientes con sarcoidosis en estadio III tendrán remisión a los 2 años después de su presentación. Es frecuente la evolución crónica que causa fibrosis pulmonar. Ciertas manifestaciones extrapulmonares de la sarcoidosis crónica, como la enfermedad cutánea infiltrativa, son mucho más frecuentes en los pacientes con enfermedad en estadio III que en los enfermos en estadio I o II. La evolución crónica de la enfermedad en estadio III no es sorprendente porque los pacientes que son diagnosticados en esta etapa pueden representar a los que antes tuvieron linfadenopatía hiliar bilateral (estadios I y II) y que no tuvieron una remisión espontánea.
Entre las manifestaciones poco usuales de la sarcoidosis intratorácica se incluyen el derrame pleural, los infiltrados alveolares, las opacidades nodulares grandes, la cavitación, las atelectasias y la calcificación. La afección pleural ocurre en 1 a 4 porciento de los pacientes y consiste en derrame pleural o engrosamiento de la pleura visceral y parietal. Los derrames pocas veces son importantes y suelen contener un alto porcentaje de linfocitos. La biopsia pleural demuestra granulomas, y el principal diagnóstico diferencial a tener en cuenta en pacientes con sarcoidosis pleural es la tuberculosis. La sarcoidosis alveolar tiene varias presentaciones que van desde infiltrados en parches en pacientes asintomáticos hasta áreas extensas de consolidación del espacio aéreo en enfermos con insuficiencia respiratoria. Pueden observarse nódulos de 2 a 10 cm de diámetro, que en algunas ocasiones se cavitan. El diagnóstico diferencial de la sarcoidosis nodular incluye a las infecciones micóticas, la tuberculosis y, en especial, los tumores metastásicos. Las atelectasias son causadas por sarcoidosis endobronquial. La calcificación de los ganglios linfáticos es una manifestación tardía de la enfermedad, que se observa en el 5 porciento de los pacientes.
Es probable que la granulomatosis sarcoide necrosante, caracterizada por masas de granulomas confluentes con cierto grado de vasculitis, sea una variante de la sarcoidosis. Ambos trastornos parecen tener muchas características en común, incluyendo linfadenopatía hiliar e inflamación granulomatosa extrapulmonar.
Manifestaciones clínicas de la sarcoidosis extratorácica
Casi cualquier órgano puede afectarse en la sarcoidosis. Aunque la sarcoidosis extratorácica es mucho menos frecuente como causa de morbilidad que la intratorácica, puede dominar el cuadro clínico en algunos pacientes.
El eritema nodoso, una paniculitis no granulomatosa, es la manifestación cutánea más frecuente de la sarcoidosis. Más del 90 porciento de los pacientes con sarcoidosis que desarrollan eritema nodoso tienen una radiografía en estadio I; el otro 10 porciento está en estadio II. Ocurren lesiones cutáneas granulomatosas en el 10 a 30 porciento de los pacientes, en especial en los afroamericanos en los Estados Unidos. El lupus pernio es una lesión amoratada y edematosa sobre la nariz, mejillas, pabellones auriculares, dedos de manos y pies, labios o rodillas, que puede desfigurar a la persona. Las placas cutáneas tienden a ser violáceas, angulares y planas, con borde elevado. También pueden observarse placas semejantes a psoriasis. Los nódulos pueden ser elevados o encontrarse dentro del tejido subcutáneo. Como regla, estas manifestaciones cutáneas granulomatosas de la sarcoidosis se observan en la enfermedad pulmonar en estadio III que es crónica y persistente.
Ocurre afección ocular en alrededor del 25 porciento de los pacientes con sarcoidosis. El tracto uveal es el más afectado, y la sarcoidosis es responsable del dos a cuatro porciento de todos los casos de uveítis. La enfermedad del tracto uveal anterior con uveociclitis suele manifestarse por epífora, fotofobia y poco o ningún dolor. El síndrome de Heertford, o fiebre uveoparotídea, es una manifestación poco frecuente de la sarcoidosis que consiste en uveítis, crecimiento parotídeo, parálisis de nervios craneales, meningitis subaguda y síntomas sistémicos. La uveítis posterior (coriorretinitis) causa visión borrosa, o puede ser asintomática. La coriorretinitis puede ser difícil de detectar cuando existe uveítis anterior.
El sistema nervioso se afecta en menos del 5 porciento de los pacientes con sarcoidosis. Las manifestaciones del sistema nervioso central incluyen meningitis crónica, encefalopatía, lesiones hipotalámicas, afección de nervios craneales y crisis convulsivas. Puede observarse neuropatía periférica sensorial y motora. La meningitis crónica en la sarcoidosis puede ser difícil de distinguir de la tuberculosa o la micótica y suele asociarse con linfocitosis en el líquido cefalorraquídeo (LCR), aumento en las proteínas del LCR y, en ocasiones, hipoglucorraquia. La sarcoidosis puede también causar masas que ocupan espacio en diversas localizaciones, provocando diferentes alteraciones neurológicas.
Se encuentra evidencia de sarcoidosis cardiaca en la necropsia con mucho más frecuencia de lo que se sospecha por las manifestaciones clínicas. Las alteraciones del ritmo son la manifestación más frecuente de la sarcoidosis cardiaca. Pueden ocurrir taquiarritmias ventriculares y bloqueo cardiaco completo, que pueden ocasionar muerte súbita. Otros datos menos frecuentes de sarcoidosis cardiaca incluyen insuficiencia cardiaca, enfermedad pericárdica, disfunción del músculo papilar y aneurismas ventriculares en ausencia de enfermedad coronaria. El ecocardiograma, la vigilancia con Holter y el gamagrama con talio-201 puede mostrar alteraciones, pero no necesariamente indican que existen granulomas en el miocardio. Deben tenerse en cuenta factores como la edad y otros padecimientos concomitantes al evaluar la posibilidad de sarcoidosis miocárdica.
Se presenta afección ósea o articular en 1 a 10 porciento de los pacientes. La enfermedad ósea se caracteriza por lesiones quísticas, y tiende a restringirse a los dedos de manos y pies. Puede observarse edema digital. La sarcoidosis sinovial puede presentarse como una artritis crónica mono o poliarticular.
La afección de las vías respiratorias superiores suele manifestarse por síntomas nasales o faríngeos. La epistaxis y la congestión nasal, que pueden confundirse con una rinitis alérgica o vasomotora, son síntomas frecuentes. Debe pensarse en sarcoidosis cuando la obstrucción nasal sea refractaria al tratamiento convencional. La sarcoidosis laríngea afecta a las estructuras supraglóticas, y en pocas ocasiones a las cuerdas vocales. La disfonía es el síntoma más común, pero puede ocurrir también obstrucción de las vías respiratorias superiores.
Las alteraciones endócrinas en la sarcoidosis incluyen hipercalcemia y disfunción hipofisiaria, que puede presentarse como diabetes insípida. Se observa hipercalcemia con mucho menos frecuencia que hipercalciuria. La hipercalcemia e hipercalciuria en la sarcoidosis parecen ser causadas por una mayor síntesis de 1,25-dihidroxivitamina D [1,25-(OH)2D] por macrófagos activados en los granulomas, que ocasiona un aumento secundario en la absorción de calcio en el intestino.
Diagnóstico
El diagnóstico de sarcoidosis se establece por la demostración de granulomas no caseosos en el tejido [ver figura 5] y la exclusión de otras causas de granulomas. La sarcoidosis intratorácica se diagnostica con más frecuencia por medio de una broncoscopía con biopsia transbronquial. La utilidad de la biopsia transbronquial depende del estadio radiográfico. En los pacientes con infiltrados pulmonares (estadios II y III) la biopsia pulmonar transbronquial demostrará granulomas no caseosos en alrededor del 90 porciento de los casos. En la enfermedad en estadio I se obtiene positividad en el 60 a 70 porciento de los casos. Si no logra establecerse el diagnóstico por este método, está indicado realizar una mediastinoscopía, que dará el diagnóstico en el 95 al 100 porciento de los casos. La biopsia de tejidos extrapulmonares puede utilizarse para el diagnóstico cuando tiene indicación clínica (v.gr., en pacientes con crecimiento de ganglios linfáticos periféricos o lesiones cutáneas).
Cuando se encuentran granulomas no caseosos en el tejido deberán siempre tenerse en cuenta otras posibles causas de la formación de granulomas. Las causas infecciosas de granulomas en los pulmones y ganglios linfáticos mediastinales son de las más importantes, en especial la tuberculosis y la histoplasmosis. Está indicado realizar una prueba cutánea de tuberculina, estudios serológicos para hongos y tinciones especiales para el estudio del material de biopsia. Por ejemplo, los hallazgos como adenopatía hiliar, infiltrados pulmonares y granulomas no caseosos en los pulmones pueden orientar a un diagnóstico precipitado de sarcoidosis antes de que estén los resultados de los estudios especiales. Si se detecta un resultado positivo en la prueba de fijación del complemento para histoplasmosis, el diagnóstico deberá modificarse a histoplasmosis.
En el momento de la broncoscopía suele realizarse un LBA además de la biopsia. En la sarcoidosis activa el líquido del lavado suele mostrar mayor número de linfocitos, y la relación entre linfocitos T cooperadores y supresores-citotóxicos está aumentada. Otros padecimientos pulmonares, tanto infecciosos como no infecciosos, pueden asociarse con linfocitosis, por lo que este dato no tiene especificidad diagnóstica. La neumonitis por hipersensibilidad puede confundirse con sarcoidosis porque ambos padecimientos se asocian con linfocitosis en el líquido del lavado y granulomas no caseosos en el tejido. Sin embargo, es típico observar inversión en la relación entre los linfocitos T cooperadores y supresores-citotóxicos en la neumonitis por hipersensibilidad.
Por lo general, la concentración de ECA aumenta en la sarcoidosis activa. Se ha sugerido que la medición de la concentración de la ECA es de utilidad en el diagnóstico de sarcoidosis. Sin embargo, no es claro si esta prueba tiene un valor diagnóstico extra sobre los criterios patológicos descritos antes. No se recomienda hacer el diagnóstico de sarcoidosis con base sólo en la concentración de la ECA, sin la confirmación del diagnóstico por demostración de granulomas no caseosos en el tejido. También se ha detectado aumento en los niveles de ECA en la tuberculosis miliar, la lepra, la cirrosis biliar primaria, la silicosis y la asbestosis. Con excepción de la tuberculosis miliar, no es probable que estas enfermedades se confundan clínicamente con la sarcoidosis. La medición periódica de la concentración de la ECA puede ser útil porque correlaciona con la actividad de la enfermedad.
El diagnóstico diferencial de la sarcoidosis en estadio I incluye principalmente al linfoma y a las infecciones granulomatosas como la tuberculosis y la histoplasmosis. Son causas menos frecuentes de linfadenopatía hiliar bilateral los cánceres metastásicos (en especial el de células renales) y la amiloidosis. La gran mayoría de los pacientes que tienen linfoma que se manifiesta como linfadenopatía hiliar bilateral tendrá linfadenopatía periférica, esplenomegalia o síntomas sistémicos, como fiebre, pérdida de peso o diaforesis nocturna. Los adultos jóvenes que presentan adenopatía hiliar bilateral asintomática, sin linfadenopatía periférica ni esplenomegalia, es muy poco probable que tengan un linfoma. Por lo tanto, en estas circunstancias es razonable hacer el diagnóstico presuntivo de sarcoidosis en estadio I sin confirmarlo examinando una muestra de tejido. Sin embargo, deberá realizarse una prueba cutánea de tuberculina y estudios serológicos para hongos, lo mismo que vigilancia clínica y radiográfica para asegurarse de que no existe progresión de la enfermedad. Si se observa progresión, es indispensable realizar una biopsia de tejido para confirmar el diagnóstico.
Tratamiento
La administración de esteroides es el tratamiento estándar para la sarcoidosis sintomática, aunque no existen prueba definitivas de que el tratamiento influya sobre la evolución a largo plazo. La mayoría de los pacientes responden muy bien al tratamiento, y con frecuencia las dosis bajas de prednisona (v.gr., 15 mg cada tercer día) son eficaces para suprimir la actividad de la enfermedad. Suele administrarse una dosis más alta (v.gr., 40 a 60 mg/día) durante algunas semanas para inducir remisión de la actividad de la enfermedad. La principal indicación para los esteroides es la afección pulmonar sintomática. Otras indicaciones incluyen síntomas generales importantes (v.gr., fiebre o pérdida de peso), hipercalcemia y afección de tejidos extrapulmonares que causen deterioro funcional o riesgo de éste. No se ha establecido qué tiempo debe durar el tratamiento. En la práctica se trata a la sarcoidosis pulmonar con prednisona durante un año, y después se disminuye progresivamente el medicamento, observando si la enfermedad ha remitido. Las recurrencias se tratan de modo semejante, con dosis más altas al principio, que después se reducen hasta una dosis de mantenimiento. Algunos pacientes presentarán recurrencias frecuentes al suspender la prednisona, por lo que requerirán un tratamiento supresor con dosis bajas en forma indefinida.
Si está absolutamiente contraindicado el uso de esteroides o si el paciente no mejora con el tratamiento esteroideo, puede intentarse la administración de metotrexate17 o de clorambucil. Con frecuencia la cloroquina es útil en los pacientes con enfermedad cutánea.
FIBROSIS PULMONAR IDIOPATICA
La fibrosis pulmonar idiopática (alveolitis fibrosante) es una de las causas más frecuentes de enfermedad pulmonar infiltrativa difusa crónica.18 Aunque los datos clínicos, radiográficos, fisiológicos e histopatológicos de este padecimiento son característicos, ninguno es patognomónico. Por lo tanto, el diagnóstico de FPI requiere no sólo de un cuadro clinicopatológico compatible, sino también de la exclusión de todas las otras causas de enfermedad difusa. Por ejemplo, las manifestaciones pulmonares de la FPI pueden ser indistinguibles de la enfermedad difusa asociada a los padecimientos del tejido conjuntivo, la asbestosis o la fibrosis pulmonar inducida por quimioterapia.
Patogenia
La patogenia de la fibrosis pulmonar idiopática no está bien establecida. Se ha sospechado una causa viral en algunos casos porque una minoría de los pacientes relacionan el inicio de sus síntomas respiratorios con una enfermedad gripal. También puede ser importante la susceptibilidad genética. Es posible que la FPI se presente cuando un individuo genéticamente susceptible se exponga a un agente viral o ambiental que desencadene una cascada de eventos inflamatorios, inmunológicos y de fibrosis en el pulmón.
La alveolitis de la FPI temprana se caracteriza principalmente por aumento en el número de neutrófilos activados y la presencia de macrófagos en el líquido del LBA. Los neutrófilos pueden ser atraídos hacia el pulmón por un desequilibrio entre las sustancias quimiotácticas derivadas de macrófagos (v.gr., factor de necrosis tumoral alfa [FNT-alfa], IL-8) y sustancias antinflamatorias (v.gr., interferón gama, IL-10).19 En algunos casos las sustancias quimiotácticas puedn ser liberadas en respuesta a la presencia de complejos inmunes locales. Se han detectado complejos inmunes en el tejido pulmonar, en el líquido de lavado y en sangre periférica en pacientes que tienen una alveolitis neutrofílica intensa. Una vez en el pulmón, los neutrófilos pueden causar lesión tisular local al liberar varias sustancias potencialmente tóxicas, como radicales de oxígeno y enzimas proteolíticas. La inflamación pulmonar puede ir seguida eventualmente por fibrosis extensa. Es posible que la inflamación neutrofílica crónica de la FPI tenga un papel en el desarrollo de la fibrosis pulmonar. Sin embargo, el proceso fibrogénico puede ser secundario en gran parte a la actividad de los macrófagos pulmonares. Los macrófagos de los pacientes con fibrosis pulmonar idiopática liberan en forma espontánea factores de crecimiento para fibroblastos, como el factor transformador de crecimiento-beta, causando un aumento en la población de fibroblastos en el pulmón. Además, en este padecimiento el macrófago libera fibronectina, una sustancia que promueve la fijación y migración de los fibroblastos. Estos macrófagos liberan en forma espontánea factores estimuladores de fibroblastos, como IL-1ß y factor de necrosis tumoral-alfa, que causan que los fibroblastos secreten mayores cantidades de constituyentes de la matriz extracelular. Por lo tanto, los macrófagos alveolares parecen tener un papel crucial en el desarrollo de la FPI.
Manifestaciones clínicas
Como en otros tipos de enfermedad pulmonar infiltrativa difusa, la disnea con el ejercicio y la tos son los síntomas más importantes. El examen físico suele demostrar estertores inspiratorios finos (estertores de Velcro). Puede observarse cianosis si la hipoxemia es severa, y las características del cor pulmonale (v.gr., aumento en la presión venosa yugular, edema y un segundo ruido cardiaco pulmonar prominente) indican enfermedad avanzada. Se observan dedos en palillo de tambor en el 40 al 75 porciento de los casos, aunque casi nunca en las etapas tempranas del padecimiento. No existen otras manifestaciones extratorácicas de la enfermedad. La evidencia clínica de enfermedad extrapulmonar (v.gr., artritis, enfermedad cutánea o serositis) sugiere un padecimiento sistémico, como sarcoidosis o alguna enfermedad del tejido conjuntivo, más que FPI.
Las características radiográficas son inespecíficas y suelen consistir en un infiltrado reticular o reticulonodular bilateral que tiene predilección por la porción inferior de los campos pulmonares. La llamada opacidad en vidrio despulido, que indica un proceso de ocupación acinar, puede observarse en la neumonitis intersticial descamativa (ver adelante). La radiografía de tórax es normal hasta en el 10 porciento de los pacientes con FPI sintomática. Es probable que la TCAR demuestre alteraciones en los casos en que la radiografía de tórax es normal. En la enfermedad avanzada se observa el llamado pulmón en panal de abeja, que indica fibrosis pulmonar en estadio terminal. Los hallazgos radiográficos en la FPI se limitan a los campos pulmonares.
Las alteraciones fisiológicas de la fibrosis pulmonar idiopática son básicamente las mismas que las descritas para la enfermedad pulmonar infiltrativa difusa en general. El cuadro fisiológico típico consiste en reducción de la DLCO, restricción de los volúmenes pulmonares, desaturación de oxígeno inducida por ejercicio y ausencia de obstrucción a la circulación del aire. Algunas alteraciones correlacionan con los hallazgos patológicos.20 Por ejemplo, la reducción en la DLCO se relaciona con cambios descamativos, y la disminución en la capacidad pulmonar total con la celularidad global. Aunque muy significativas desde el punto de vista estadístico, la dispersión es demasiado grande como para que estas correlaciones sean útiles en la clínica.
Las características histopatológicas son inespecíficas: se observan datos histopatológicos semejantes en la enfermedad pulmonar infiltrativa difusa asociada con enfermedades del tejido conjuntivo, diversos tipos de enfermedad difusa asociada a fármacos y otros padecimientos. El infiltrado de células inflamatorias y la fibrosis crónica, que son los datos patológicos claves en la FPI, representan reacciones comunes a diversos agentes.
Se han observado dos patrones histopatológicos básicos en la fibrosis pulmonar idiopática: la neumonitis intersticial habitual (NIH) y la neumonitis intersticial descamativa (NID). La NIH, el patrón más frecuente, se caracteriza por grados diversos de un infiltrado celular inflamatorio, intersticial y redondeado, engrosamiento de los septos alveolares y fibrosis [ver figura 6a]. La distribución de la lesión es irregular: áreas de inflamación intensa y fibrosis que pueden coexistir con áreas de pulmón casi normal en la misma muestra tomada de una biopsia a cielo abierto. La NID se caracteriza por un patrón más o menos homogéneo en el que los espacios alveolares se llenan por macrófagos alveolares [ver figura 6b].
Aún es motivo de controversia si las formas descamativa y habitual
de la neumonitis intersticial son entidades distintas o representan diferentes
patrones histopatológicos de un mismo proceso. Se piensa que esta
última es la opción más probable, y se ha observado la
coexistencia de patrón habitual y descamativo de una neumonitis
intersticial en un mismo paciente. El proceso descamativo puede representar una
lesión más temprana, y por lo general tiene mejor respuesta a los
esteroides que la neumonitis intersticial habitual.
Diagnóstico
El diagnóstico de FPI se establece por la correlación entre los datos histopatológicos y clínicos. Desde el punto de vista clínico se sospecha FPI cuando ocurre una enfermedad pulmonar infiltrativa difusa sin afección de otros órganos y sin relación aparente a infecciones, exposición ambiental o medicamentos. La radiografía de tórax no muestra evidencia de adenopatía hiliar o derrame pleural. Las pruebas de laboratorio en la fibrosis pulmonar idiopática suelen ser irrelevantes, excepto por la presencia de anticuerpos antinucleares positivos o factor reumatoide positivo en hasta el 50 porciento de los casos. Por lo tanto, los anticuerpos antinucleares y el factor reumatoide séricos no son útiles para diferenciar entre la fibrosis pulmonar idiopática y la enfermedad infiltrativa difusa de los padecimientos del tejido conjuntivo.
El enfoque inicial del diagnóstico en la FPI consiste en excluir la mayoría de las causas posibles de enfermedad pulmonar infiltrativa difusa usando la historia clínica y el examen físico. Sin embargo, existen ciertos padecimientos que no pueden descartarse en forma certera sin una biopsia pulmonar. Entre estos se incluyen la sarcoidosis en estadio III, la carcinomatosis linfangítica, la linfangiomatosis, la histiocitosis X y las formas más difusas de bronquiolitis obliterante con neumonía organizada (BONO). La TCAR puede ser útil en esta fase de la evaluación porque el patrón de las alteraciones observadas en la FPI es específico, y muchas de las otras causas tienen patrones característicos y muy diferentes.21 Además, la TCAR en ocasiones sugiere si el patrón patológico es de NIH o de NID.
La biopsia pulmonar transbronquial es una técnica muy sensible para el diagnóstico de sarcoidosis y cáncer linfangítico, y puede demostrar datos patológicos sugestivos de fibrosis pulmonar idiopática (i.e., fibrosis y ensanchamiento de los septos alveolares con un infiltrado de células inflamatorias). Se requiere de la biopsia pulmonar a cielo abierto para excluir otras causas de enfermedad difusa. Será decisión del clínico realizar una biopsia a cielo abierto en los casos en que la evolución clínica y la biopsia transbronquial sean muy sugestivas de FPI. Se recomienda realizar la biopsia cuando existen dudas respecto a la presencia de una causa infecciosa. También debe practicarse este tipo de biopsia en pacientes jóvenes para establecer el diagnóstico del padecimiento subyacente con un grado de certeza razonable. Por otro lado, en los pacientes mayores que tienen datos clínicos típicos y una biopsia transbronquial compatible puede evitarse la biopsia abierta de pulmón por la morbilidad que causa. En ciertos casos puede estar justificado diagnosticar una FPI sin una biopsia tisular. Por ejemplo, no se requerirá de la biopsia para el diagnóstico en un paciente de edad avanzada que tiene disnea progresiva al ejercicio de meses a años de duración, estertores en Velcro y dedos en palillo de tambor a la exploración física, infiltrados intersticiales gruesos bilaterales, un título bajo de anticuerpos antinucleares y ninguna evidencia de un padecimiento de tipo ambiental o relacionado a medicamentos.
Pronóstico y tratamiento
La mortalidad a 5 años para los pacientes con fibrosis pulmonar idiopática es de alrededor del 50 porciento. La mayor parte de las defunciones son causadas por insuficiencia respiratoria progresiva. Se emplea la prednisona para tratar la enfermedad progresiva y alrededor del 20 porciento de los pacientes que la reciben muestran mejoría sintomática, radiográfica y funcional. Sin embargo, incluso los que responden no tienen una mejoría muy significativa, a diferencia de los pacientes con sarcoidosis sintomática que son tratados con esteroides. La respuesta a la prednisona es mejor si la fibrosis es menos evidente que la inflamación en la biopsia a cielo abierto, pero la respuesta suele ser mala si predomina la fibrosis. Sin embargo, debido a que la afección pulmonar es heterogénea, los datos histopatológicos pueden variar en diferentes regiones del organismo. Por lo tanto, no está indicado realizar una biopsia a cielo abierto para decidir si tratar o no a un paciente sintomático con esteroides. Los estudios sobre la relación entre los resultados de la TCAR y la mejoría en la función pulmonar sugieren que la extensión del patrón de atenuación en vidrio despulido identifica al cuadro patológico de la NID, que correlaciona con respuesta a los esteroides. Uno de los mejores factores de predicción de la evolución a largo plazo es la respuesta a corto plazo (6 a 8 semanas) a la prednisona, quienes responden tienen mejor pronóstico. Se ha usado la ciclofosfamida y la azatioprina aunadas a la prednisona, por lo general cuando la prednisona por sí sola ha sido ineficaz. Los factores de predicción de una respuesta benéfica al tratamiento citotóxico incluyen breve duración de los síntomas y una buena respuesta temporal al tratamiento esteroideo.22 En ocasiones los pacientes que no mejoran con el esteroide sí lo hacen con el tratamiento inmunosupresor combinado o con colchicina.23 También se han realizado trasplantes de un solo pulmón con éxito para la fibrosis en estadio terminal.
ENFERMEDADES DEL TEJIDO CONJUNTIVO
Con frecuencia las enfermedades del tejido conjuntivo afectan el pulmón. Hasta dos terceras partes de los pacientes tienen evidencia clínica de afección pleuropulmonar, y casi todos tienen alteraciones en la autopsia. El mecanismo inmunológico parece ser el depósito de complejos inmunes, pero la evidencia de esto es más convincente en algunos padecimientos que en otros. Las pruebas de función pulmonar en los pacientes con infiltrados intersticiales y fibrosis muestran restricción, obstrucción de las vías respiratorias de pequeño calibre y reducción de la DLCO con hipoxemia al reposo que empeora durante el ejercicio. La TCAR del tórax suele detectar enfermedad intersticial sutil que no se observa en la radiografía de tórax de rutina. Aunque la enfermedad intersticial crónica que se observa en estos padecimientos suele ser indistinguible desde el punto de vista patológico de la fibrosis pulmonar idiopática, los pacientes con una enfermedad del tejido conjuntivo suelen tener mejor pronóstico que los que padecen FPI,24 esto es cierto incluso en los pacientes en los que se desarrolla una enfermedad de tejido conjuntivo después de la FPI.25
Lupus eritematoso generalizado
El lupus eritematoso generalizado es uno de los padecimientos del tejido conjuntivo más prevalentes, en especial entre mujeres afroamericanas (en Estados Unidos) en edad reproductiva. Sin embargo, hasta el 18 porciento de los casos ocurren después de la quinta década de la vida. El LEG inducido por medicamentos es un problema clínico común, sobre todo entre ancianos con cardiopatía. Los fármacos que con más frecuencia causan LEG incluyen la hidralazina, la procainamida, la isoniacida, la fenitoína, la quinidina, la metildopa y varios de los agentes bloqueadores beta. Se han implicado otros medicamentos, pero las evidencias son menos claras.
Con frecuencia ocurren complicaciones pleuropulmonares en el LEG (en 38 a 89 porciento de los pacientes), que deben diferenciarse de infecciones. La enfermedad pleural, las atelectasias, las neumonitis lúpicas agudas y la hemorragia masiva son los problemas clínicos más comunes. Además, la enfermedad pleuropulmonar puede ser la manifestación inicial en el LEG inducido por medicamentos.
La neumonitis lúpica aguda se manifiesta por inicio súbito de tos con disnea, y en la radiografía aparecen opacidades basales en parches. En ocasiones la radiografía muestra solo sombras lineales horizontales que se han atribuido a pequeñas áreas de atelectasia. Los pacientes con esta imagen suelen responder bien al uso de esteroides.
Los pacientes con LEG pueden sufrir hemorragia alveolar masiva o hemorragia oculta durante la evolución de su enfermedad. El LEG severo se asocia con una mortalidad del 70 porciento. Los síntomas consisten en tos y disnea con o sin hemoptisis y la radiografía de tórax muestra infiltrados alveolares esponjosos que pueden ser en parches o confluentes. El tratamiento es semejante al del síndrome de Goodpasture e incluye plasmaféresis e inmunosupresores.
Artritis reumatoide
Aunque la incidencia de artritis reumatoide es más alta en mujeres que en varones, las complicaciones pleuropulmonares son más frecuentes en éstos últimos. Este grupo de complicaciones son clínicamente aparentes en el 14 porciento de los enfermos en los primeros 2 años de evolución26 y en alrededor del 50 porciento durante el curso de la enfermedad; un porcentaje mucho mayor tiene afección desde el punto de vista patológico. En casos ocasionales la afección torácica precede a las lesiones articulares, lo que dificulta el diagnóstico.
Los problemas pleuropulmonares más frecuentes son enfermedad pulmonar intersticial, enfermedad pleural y nódulos reumatoides parenquimatosos. Estos problemas suelen asociarse con la presencia de artritis activa, títulos altos de factor reumatoide, complejos inmunes circulantes y crioglobulinemia.
La neumonitis intersticial difusa con fibrosis es el problema pulmonar más frecuente y serio en los pacientes con artritis reumatoide. La disnea, en ocasiones con tos, es el síntoma más frecuente. Pueden existir dedos en palillo de tambor. La radiografía de tórax evoluciona de nodularidad fina a reticulación gruesa y, al final, a un patrón en panal de abeja. Los resultados del LBA pueden ser anormales, demostrando aumento en el número de linfocitos, neutrófilos y eosinófilos, o los tres, pero este hallazgo no parece ayudar en el tratamiento. En los pacientes con enfermedad intersticial que se caracteriza por biopsia con intensa celularidad o la presencia de síntomas progresivos deben administrarse esteroides.
Pueden encontrarse nódulos reumatoides (necrobióticos) en muchos órganos, incluyendo el parénquima pulmonar, la mucosa endobronquial y la pleura, y en cualquiera de estos sitios pueden cavitarse. Las lesiones tienen una apariencia histopatológica característica y, a menos que se realice biopsia por punción percutánea transtorácica o biopsia abierta de pulmón, pueden ser difíciles de distinguir de neoplasias o tuberculosis. La mayoría de los nódulos reumatoides no causan síntomas, y rara vez se requiere tratamiento si ya se ha establecido el diagnóstico.
Esclerosis sistémica progresiva
La esclerosis sistémica progresiva (ESP), o esclerodermia, es un trastorno que predomina en las mujeres en la cuarta a sexta décadas de la vida. El pronóstico es desfavorable, en especial entre las personas de origen afroamericano, los varones y los enfermos con daño pulmonar. Con frecuencia se desarrollan complicaciones pulmonares, sobre todo enfermedad pulmonar intersticial, que ocurren hasta en el 90 porciento de los pacientes en quienes se realizan pruebas de función pulmonar o estudios histológicos. La enfermedad pulmonar intersticial en la ESP se caracteriza por infiltrados reticulares basales o reticulonodulares que se vuelven más prominentes conforme progresa la enfermedad. Además, la radiografía puede mostrar reducción del volumen con el tiempo, y puede ocurrir neumotórax. La evidencia de hipertensión pulmonar puede ser mayor de la esperada por el grado de alteración radiológica o de alteración en las pruebas de función respiratoria, en especial si el fenómeno de Raynaud es parte del síndrome clínico.
Se piensa que los esteroides no son útiles como tratamiento de la enfermedad pulmonar intersticial en la ESP; la ciclofosfamida puede ser eficaz en los pacientes que tienen alveolitis activa por LBA.27 Como resultado de los trastornos en la motilidad del esófago puede ocurrir aspiración de contenido gástrico y esofágico, sobre todo cuando los pacientes están en posición de decúbito. La aspiración puede evitarse elevando la porción superior del tronco hasta una posición casi vertical.
El síndrome de CREST (calcinosis, fenómeno de Raynaud, trastornos esofágicos, esclerodactilia y telangiectasias) es una variante de la ESP. Los pacientes con síndrome de CREST tienen enfermedad vascular pulmonar prominente. La hipertensión pulmonar puede asociarse con fibrosis intersticial, como en la ESP, o puede ocurrir como un evento aislado.
Polimiositis y dermatomiositis
La polimiositis y la dermatomiositis son padecimientos autoinmunes que se caracterizan por debilidad y en ocasiones por dolor en los músculos proximales y del cuello, erupciones dermatológicas (dermatomiositis) y neoplasias. La complicación pulmonar más frecuente de la poli-dermatomiosisits es la insuficiencia respiratoria causada por debilidad de los músculos respiratorios y la aspiración secundaria a debilidad faríngea posterior y esofágica proximal.
Se observa enfermedad pulmonar intersticial en una minoría de los pacientes, que puede asociarse con anticuerpos anti-Jo-1, un autoanticuerpo que es específico contra la enzima celular histidil-ARNt sintetasa. Estos pacientes pueden tener NOBO, fibrosis pulmonar o daño alveolar difuso.28 A diferencia de la enfermedad intersticial asociada con muchas de las otras enfermedades del tejido conjuntivo, la asociada con poli-dermatomiositis responde bien al tratamiento esteroideo.
Síndrome de Sjögren
El síndrome de Sjögren consiste en la triada de queratoconjuntivitis sicca, xerostomía y edema recurrente de las glándulas parótidas, y con frecuencia (en el 60 porciento de los casos) se asocia con otra enfermedad del tejido conjuntivo. Este síndrome incluye con frecuencia problemas pleuropulmonares, que en la mayoría de los casos son secundarios a la enfermedad del tejido conjuntivo subyacente. Las complicaciones torácicas que pueden relacionarse en forma directa con el síndrome de Sjögren incluyen infiltrados intersticiales, con frecuencia de tipo linfocítico, pleuresía con o sin derrame, bronquiolitis folicular y sequedad de las vías traqueobronquiales, produciendo bronquiectasias y bronquitis o neumonitis recurrente.
La disnea es el síntoma principal de la enfermedad intersticial asociada con el síndrome de Sjögren, mientras que la disfonía y la tos productiva con expectoración espesa son datos de afección laríngea y traqueobronquial. La radiografía de tórax puede mostrar infiltrados intersticiales con un componente nodular prominente. Estos infiltrados tienen un patrón linfoplasmacitario que puede ser difícil de distinguir del observado en pacientes con linfoma. Los resultados de las pruebas de función respiratoria demuestran patrones restrictivos, obstructivos o mixtos. La neumonitis intersticial linfocítica y la bronquiolitis asociadas con síndrome de Sjögren responden al tratamiento esteroideo o al inmunosupresor.29
Enfermedad mixta del tejido conjuntivo
Varios reportes han descrito pacientes que tienen características de LEG, ESP y polimiositis. Una característica de esta enfermedad mixta del tejido conjuntivo es la presencia de títulos altos de anticuerpos contra ribonucleoproteínas nucleares extraíbles (anti-nRNP). Este síndrome suele ser una enfermedad relativamente benigna que responde bien a los esteroides, aunque algunos casos tienen una evolución menos benigna, algunos de los cuales se han asociado con enfermedad pulmonar intersticial difusa o hipertensión pulmonar.
GRANULOMA EOSINOFILICO DEL PULMON
El granuloma eosinofílico del pulmón, o histiocitosis X del pulmón, es una causa poco frecuente de enfermedad pulmonar infiltrativa difusa, y afecta principalmente a individuos entre 20 y 50 años de edad. La enfermedad es muy poco frecuente en afroamericanos y extraordinariamente rara en asiáticos. La etiología del granuloma eosinófilo es deconocida, pero existe una intensa asociación con el uso de cigarrillos. En los niños y los adultos jóvenes el granuloma eosinófilo es un padecimiento sistémico con apariencia histológica semejante, y se puede observar diabetes insípida por afección hipofisiaria o granuloma eosinofílico del hueso en el 15 al 20 porciento de los pacientes.
La mayoría de los enfermos con granuloma eosinófilo pulmonar tienen tos o disnea, pero hasta una cuarta parte de los individuos afectados tienen sólo alteraciones radiográficas asintomáticas. Entre el 10 y 20 porciento de los enfermos desarrollan neumotórax, y ésta puede ser la manifestación inicial del padecimiento. La asociación de neumotórax espontáneo con evidencia radiográfica de infiltrados intersticiales difusos sugiere el diagnóstico de granuloma eosinófilo o de neumonía por P. carinii asociada a SIDA.
El patrón radiográfico o por TCAR del granuloma eosinófilo es variable. En la etapa temprana de la enfermedad son típicas las opacidades nodulares y en vidrio despulido con quistes de paredes gruesas. Después se observan quistes de paredes delgadas, opacidades lineales y lesiones enfisematosas.30
El granuloma eosinófilo causa con frecuencia reducción en la DLCO y en la capacidad vital. Sin embargo, a diferencia de la mayoría de las principales causas de enfermedad pulmonar infiltrativa difusa, la obstrucción al flujo del aire es un dato característico, sobre todo en las etapas avanzadas de la enfermedad. La alta incidencia de obstrucción al flujo del aire puede ser secundaria a afección bronquiolar en las primeras fases de la enfermedad y a bulas en las etapas tardías. El tabaquismo también puede ser un factor que contribuye a la frecuencia de obstrucción aérea en este padecimiento. Es frecuente el deterioro significativo en la capacidad para realizar ejercicio, que es mayor al esperado por el grado de restricción de volumen. Debido a que existe un incremento mínimo en la DLCO y un aumento en la relación de ventilación desperdiciada (VD-VT) con el ejercicio, se ha deducido que las alteraciones vasculares pulmonares son la causa de la poca tolerancia al ejercicio.31
Las características histológicas del granuloma eosinófilo son típicas, e incluyen la presencia de células que recuerdan a las células de Langerhans en la piel. Estas células se tiñen en forma positiva con anticuerpos monoclonales contra CD1a, y se ha demostrado con microscopia electrónica que contienen las inclusiones características denominadas cuerpos X o gránulos de Birbeck.32 Estas células pueden observarse en otros tipos de enfermedad pulmonar infiltrativa difusa, pero en mucho menor número que en el granuloma eosinófilo. Los estudios sugieren que existen células T CD4+ que se encuentran en aposición a las células de Langherhans y un mayor número de células neuroendócrinas en los pacientes con granuloma eosinófilo.33,34 Se han encontrado factor estimulador de colonias de granulocitos y macrófagos y factor transformador de crecimiento -ß(FTC-ß) en las lesiones, lo que sugiere que estos factores son mediadores potenciales de la estimulación de las células de Langerhans y de los fibroblastos, respectivamente. Estos datos sugieren que la patogenia incluye interacciones entre las células que causan estimulación de los fibroblastos y fibrosis.
El diagnóstico de granuloma eosinófilo y su diferenciación de otros padecimientos que causan enfermedad pulmonar difusa puede hacerse con mayor certeza si se realiza una biopsia de pulmón a cielo abierto. Sin embargo, se ha sugerido que el encontrar más de 5 porciento de células CD1a+ en el líquido del LBA, aunado a un cuadro clínico y radiográfico compatible, puede ser suficiente para el diagnóstico.32 Como se mencionó antes, la TCAR puede ser una técnica no cruenta y útil para el diagnóstico del granuloma eosinófilo.
Varios estudios de casos han reportado pacientes con granuloma eosinófilo que mejoraron después de suspender el tabaquismo, y se ha informado que el tratamiento con esteroides ha sido eficaz. Sin embargo, ningún tratamiento se ha estudiado aún lo suficiente o ha producido evidencias convincentes de mejoría como para justificar su recomendación definitiva.
Los pacientes con granuloma eosinófilo tienen una sobrevida promedio de 6 años después del diagnóstico. La mayor edad, la obstrucción al flujo del aire y la hiperinflación son predictores de un mal pronóstico.35
NEUMONIA ORGANIZADA CON BRONQUIOLITIS OBLITERANTE
La neumonía organizada con bronquiolitis obliterante, también llamada neumonía organizada criptogénica,36 es un padecimiento que ocurre parte en el intersticio, parte en el alveolo y parte en las vías aéreas pequeñas. En algunos pacientes puede diagnosticarse de modo incorrecto como FPI y enfermedad pulmonar obstructiva crónica. La enfermedad tiene tres patrones clínicos, idiopática sintomática, secundaria y focal asintomática.37 La NOBO puede ser causada por diversos trastornos, incluyendo inhalación de humos tóxicos (i.e., óxidos de nitrógeno), infecciones virales, padecimientos del tejido conjuntivo y otras enfermedades pulmonares, como la FPI y la neumonitis por hipersensibilidad. Aunque una historia clínica cuidadosa puede identificar estas causas, existe un grupo de pacientes con NOBO idiopática en quienes se requiere una biopsia de pulmón a cielo abierto para distinguir la enfermedad de otros procesos parenquimatosos difusos.
Los pacientes con NOBO suelen tener entre 50 y 60 años de edad. La enfermedad comienza en forma súbita y se caracteriza por tos seca y disnea, fiebre, malestar y fatiga, que son más frecuentes en los pacientes con NOBO que en los enfermos con FPI. En ocasiones el examen físico demuestra estertores asociados a sibilancias. No existen dedos en palillo de tambor, que son comunes en la FPI. La radiografía de tórax muestra, en forma típica, infiltrados alveolares bilaterales en parche o en vidrio despulido, a diferencia de los infiltrados intersticiales que ocurren en la FPI. La TCAR demuestra áreas bilaterales de consolidación que afectan principalmente la región subpleural, peribroncovascular o ambas, que son muy sugestivas del diagnóstico.38 Las pruebas de función pulmonar indican obstrucción, restricción o un patrón mixto con reducción de la capacidad de difusión e hipoxemia.
El tratamiento con esteroides ha sido útil, y muchos pacientes responden en forma dramática en unos cuantos días. Sin embargo, cuando se reduce la dosis muchos pacientes recaen y vuelven a tener síntomas generales, infiltrados e hipoxemia. Los pacientes con NOBO asintomática focal no requieren tratamiento.
PROTEINOSIS ALVEOLAR
La proteinosis alveolar es una enfermedad de etiología desconocida que se caracteriza por el acúmulo intralveolar de un material celular lipoproteináceo que recuerda al surfactante.39 El aumento de este material puede ser ocasionado por mayor producción o menor degradación del surfactante por los macrófagos. Se ha propuesto que la menor utilización del surfactante, con menor regreso desde el alveolo a las células tipo II, puede superar la capacidad de los macrófagos normales para eliminar esta sustancia. Además, el material lipoproteináceo puede inhibir la defensa de los macrófagos contra la infección, favoreciendo la mayor incidencia de infecciones por patógenos intracelulares que se observa en los enfermos con proteinosis alveolar. Ocurre un trastorno clínicamente semejante a la proteinosis alveolar idiopática en individuos con exposición aguda al sílice (por lo general en la limpieza con arena) y en pacientes con trastornos hematológicos graves.
Los cambios patológicos en la proteinosis alveolar incluyen la presencia de material granular eosinofílico dentro de los espacios alveolares, que se tiñe en forma positiva con el agente ácido-Schiff (PAS) [ver figura 7]. Se ha detectado inflamación y fibrosis en algunos casos, pero éstas no suelen ser características importantes. La microscopía electrónica demuestra que la sustancia intralveolar contiene numerosos cuerpos laminares, semejantes a los encontrados en las células epiteliales tipo II.
La proteinosis alveolar se presenta en una de tres formas: como una radiografía de tórax anormal asintomática, como el inicio súbito de tos, fiebre y dolor torácico causados por el padecimiento y complicados por infecciones oportunistas (con especiesNocardia, hongos o especiesMycobacterium), o como un padecimiento gradual con disnea y tos (a veces productiva). Los datos físicos suelen ser mínimos. La radiografía de tórax o la TCAR muestra un proceso bilateral de ocupación alveolar que semeja al edema pulmonar pero sin líneas B de Kerley, derrame pleural o crecimiento de la vasculatura central. En las etapas tardías pueden observarse marcas intersticiales gruesas causadas por fibrosis agregada. El aumento en la deshidrogenasa láctica del suero sin elevación de otras enzimas séricas es un dato que puede ayudar al médico a realizar el diagnóstico.
El diagnóstico de la proteinosis alveolar pulmonar se hace mejor por medio de LBA. El líquido se observa turbio, y su tinción demuestra gran cantidad de material PAS-positivo [ver figura 7]. La neumonía por P. carinii causa acúmulo de una sustancia intralveolar semejante, y debe descartarse esta posibilidad por medio de tinciones especiales. El líquido del lavado debe cultivarse para especiesNocardia, hongos y especiesMycobacterium.
El tratamiento de la proteinosis alveolar está indicado si existe disnea significativa en el momento del diagnóstico o si los síntomas leves no remiten en forma espontánea después de un periodo de observación de varios meses. Debido a que algunos enfermos remiten en forma espontánea, siempre que sea posible será mejor esperar varios meses antes de instituir el tratamiento. El tratamiento para la proteinosis alveolar pulmonar consiste en el lavado pulmonar total con solución salina estéril. El pulmón no lavado se protege por medio de una sonda endotraqueal de doble luz. La mayoría de los pacientes mejoran en forma importante después de eliminar el material alveolar por medio del lavado, y el pulmón contralateral puede lavarse después.
NEUMONIA EOSINOFILICA
Los síndromes de infiltrados pulmonares con eosinofilia en sangre periférica (IPE) se caracterizan por eosinofilia en el parénquima pulmonar, eosinofilia en sangre periférica, o ambas, y suelen asociarse con infiltrados en la radiografía.40 Se piensa que los mecanismos de hipersensibilidad aguda participan en la patogenia por la presencia de eosinófilos y la ocurrrencia ocasional de broncoespasmo. En muchos de estos padecimientos las sustancias secretadas por los eosinófilos pueden ayudar a causar la lesión al parénquima pulmonar, a las vías aéreas o a ambas. Como propuso Crofton en 1952, los IPE incluyen cinco síndromes: eosinofilia pulmonar simple (síndrome de Löffler), eosinofilia pulmonar prolongada sin asma, eosinofilia pulmonar con asma, eosinofilia pulmonar tropical y vasculitis pulmonar (granulomatosis y angeítis alérgica). Aunque no se incluye en esta clasificación, el síndrome hipereosinofílico se asocia en ocasiones con infiltrados pulmonares. Recientemente se ha descrito un trastorno nuevo, la neumonía eosinofílica aguda. Otros trastornos que puecen causar infiltrados pulmonares con eosinofilia periférica incluyen infecciones (v.gr., tuberculosis, brucelosis y micosis), neoplasias (v.gr., carcinoma broncogénico, enfermedad de Hodgkin y linfadenopatía inmunoblástica) y procesos inmunológicos (v.gr., enfermedad pulmonar reumatoidea y sarcoidosis).
Eosinofilia pulmonar simple (síndrome de Löffler)
El síndrome de Löffler se caracteriza por infiltrados transitorios y en ocasiones migratorios y por síntomas leves que duran menos de un mes. Los infiltrados suelen ser de densidad homogénea, únicos o múltiples. En algunos casos no existen síntomas, aunque pueden desarrollarse disnea y tos seca. El examen físico revela el acúmulo intersticial e intralveolar de eosinófilos, macrófagos y líquido. El síndrome de Löffler puede ser idiopático o puede ser causado por una infestación parasitaria (v.gr., con Ascaris o Strongyloides) o por medicamentos (v.gr., nitrofurantoína o penicilina). El tratamiento consiste en suspender el fármaco agresor o tratar la infestación parasitaria; en la mayoría de los casos no se requiere más. En ocasiones los pacientes muy sintomáticos con enfermedad idiopática pueden requerir tratamiento esteroideo.
Eosinofilia pulmonar prolongada sin asma
La eosinofilia pulmonar prolongada (neumonía eosinofílica crónica) es un padecimiento idiopático que afecta de modo predominante a mujeres de edad media y que se caracteriza por tos productiva, disnea, malestar, pérdida de peso, diaforesis nocturna y fiebre asociada con infiltrados pulmonares periféricos progresivos. Pueden existir hemoptisis y sibilancias, que hacen que el diagnóstico se confunda con otras causas de IPE. Las muestras de la biopsia abierta de pulmón muestran infiltrados inflamatorios mixtos masivos que tienen alto contenido de eosinófilos. Algunos vasos pulmonares pueden tener escasas células inflamatorias, pero no existe una vasculitis real.
El diagnóstico se basa en el síndrome clínico, la radiografía de tórax y si existe o no eosinofilia en sangre. Algunos investigadores han sugerido que si los resultados de la radiografía muestran los infiltrados periféricos densos típicos sin afección central, el diagnóstico puede hacerse sin necesidad de biopsia. Por lo general es posible confirmar el diagnóstico con facilidad por medio de un LBA y biopsia pulmonar transbronquial.
Ocurre remisión espontánea en hasta el 10 porciento de los casos, pero puede también ocurrir insuficiencia respiratoria. El tratamiento con esteroides es muy eficaz. Debido a que son frecuentes las recaídas, es común que se administre tratamiento hasta por 5 años.
Eosinofilia pulmonar con asma
La aspergilosis broncopulmonar alérgica puede causar infiltrados pulmonares en un paciente con asma.
Eosinofilia pulmonar tropical
Debe pensarse en el diagnóstico de eosinofilia pulmonar tropical cuando existe asma de inicio reciente, fiebre, eosinofilia importante en sangre y la presencia de un infiltrado basal mixto reticulonodular y alveolar en una persona que ha viajado recientemente al Lejano Oriente. Es posible que esta enfermedad represente una forma de filariasis (causada por Wuchereria bancrofti u otros organismos). A pesar del tratamiento con dietilcarbamacina, muchos pacientes con este padecimiento tienen inflamación persistente y desarrollan enfermedad intersticial crónica.
Vasculitis pulmonar (granulomatosis y angeítis alérgica)
La granulomatosis y angeítis alérgica es un padecimiento multisistémico que se caracteriza por vasculitis e inflamación granulomatosa necrosante que afecta a los pulmones, el sistema nervioso central y la piel. Aunque los riñones, el corazón, el bazo y el tubo digestivo se afecten con mucho menos frecuencia, casi cualquier órgano o tejido puede estar afectado. La eosinofilia prominente y el incremento de los niveles de IgE sugieren que mecanismos de hipersensibilidad inmediata deben participar de modo importante en la patogenia.
Síndrome hipereosinofílico
El síndrome hipereosinofílico se caracteriza por un amplio rango de manifestaciones clínicas que ocurren cuando los eosinófilos maduros infiltran diversos órganos.
Neumonía eosinofílica aguda
Recientemente se ha descrito una enfermedad pulmonar eosinofílica no detectada antes. Se denomina neumonía eosinofílica aguda y se caracteriza por el inicio súbito de tos, disnea, fiebre, taquipnea y estertores; los pacientes con frecuencia requieren de ventilación mecánica.42 No existe asociación clara con el tabaquismo, la exposición ambiental o la ingesta de medicamentos, y los pacientes tienden a ser jóvenes. Se observa eosinofilia en sangre en la mayoría de los casos, pero este dato no es útil para el diagnóstico. La radiografía de tórax y la TC muestran combinaciones inespecíficas de infiltrados alveolares (en vidrio despulido) e infiltrados alveolares e intersticiales mixtos que en ocasiones se asocian con derrames pleurales. El diagnóstico puede hacerse por lavado broncoalveolar, que muestra un aumento dramático en el porcentaje de eosinófilos, con frecuencia mayor del 20 porciento (la cuenta normal es menor del 1 porciento). La administración de esteroides causa mejoría rápida y son raras las recaídas después de la suspensión de los esteroides.
HEMORRAGIA ALVEOLAR DIFUSA
Los síndromes de hemorragia alveolar se caracterizan por hemorragia difusa parenquimatosa en ausencia de aspiración de sangre, coagulopatía, aumento de la presión venosa pulmonar o alguna causa local identificable (i.e., embolia pulmonar, cáncer o bronquitis). Con frecuencia la hemorragia está mediada por una alteración inmunológica y casi siempre se asocia con glomerulonefritis.
Las enfermedades que causan este síndrome son la enfermedad por anticuerpos contra la membrana basal glomerular (anti-MBG) o síndrome de Goodpasture, la glomerulonefritis idiopática rápidamente progresiva con o sin complejos inmunes, la vasculitis (v.gr., granulomatosis de Wegener), el LEG y la hemosiderosis pulmonar idiopática.43 Aunque cada uno de estos padecimientos tienen características clínicas sugestivas, en muchos casos no puede hacerse el diagnóstico por clínica. Además, muchos pacientes con hemorragia alveolar difusa están graves, lo que hace urgente una evaluación inmediata. El enfoque diagnóstico que se sugiere en estos pacientes consiste en:
1. Obtener suero para buscar anticuerpos anti-MBG, anticuerpos antinucleares y anti-ADN, anticuerpos citoplasmáticos antineutrófilo, niveles de complemento y complejos inmunes.
2. Realizar lavado broncoalveolar con tinción de hierro del tejido obtenido. Si existen macrófagos con hemosiderina se confirma el diagnóstico de hemorragia alveolar.
3. Si el paso 1 no confirma el diagnóstico o los resultados no están disponibles, realizar biopsia de riñón, pulmón u otro sitio afectado. Debe incluirse inmunofluorescencia y microscopía electrónica además del estudio histopatológico de rutina.
Casi todos los casos de hemorragia alveolar pueden diagnosticarse con rapidez si se siguen estos pasos. Deberá iniciarse tratamiento con dosis altas de esteroides por vía intravenosa mientras la evaluación está en progreso porque la hemorragia alveolar puede ser de extrema gravedad.
Enfermedad por anticuerpos anti-MBG
La enfermedad por anticuerpos anti-MBG ocurre en varones jóvenes que fuman y se caracteriza por glomerulonefritis y hemorragia alveolar difusa.44 Es causada por anticuerpos citotóxicos contra la cadena alfa3 de la colágena tipo IV en las membranas basales glomerular y alveolar.
Muchos pacientes presentan hemoptisis, que puede ser masiva; sin embargo, algunos episodios de hemorragia no se asocian con ella. La mayoría de los enfermos refieren disnea y hematuria macroscópica. Existe anemia por deficiencia de hierro en casi todos los pacientes, e insuficiencia renal desde la etapa inicial en el 50 porciento de los casos.
La radiografía de tórax muestra al principio sombras acinares perihiliares esponjosas, y después cambios intersticiales. Las pruebas de función respiratoria indican un patrón restrictivo que mejora con la resolución clínica. La DLCO puede aumentar por la captación de monóxido de carbono por la hemoglobina extravascular. Al volverse crónica la enfermedad pueden persistir los cambios radiográficos intersticiales y las alteraciones restrictivas de la función pulmonar.
El diagnóstico se confirma cuando se detectan anticuerpos circulantes anti-MBG (el 95 porciento de los pacientes tienen anticuerpos positivos) o depósitos lineales de IgG (más raro de IgA) en la membrana basal glomerular o alveolar. También pueden obtenerse anti-MBG del tejido afectado.
Las recomendaciones actuales de tratamiento incluyen agentes inmunosupresores, esteroides y plasmaféresis. A pesar de este tratamiento agresivo, el 50 porciento de los pacientes fallece o requiere diálisis a largo plazo después de 2 años. La muerte suele ser causada por un episodio de hemorragia alveolar masiva que con frecuencia es precipitado por infección.
Hemosiderosis pulmonar idiopática
La hemosiderosis pulmonar idiopática es un padecimiento caracterizado por hemorragia alveolar recurrente que ocurre sobre todo en niños (casi siempre menores de 10 años) y en adultos jóvenes. Cuando se presentan adultos, los hombres se afectan más que las mujeres. La presentación de la enfermedad es semejante a la del síndrome de Goodpasture, excepto por la ausencia de enfermedad renal. Con el tiempo ocurren episodios recurrentes de hemorragia clínica y subclínica, que causan daño pulmonar que puede ser obstructivo o restrictivo. El tejido pulmonar muestra cambios de hemorragia aguda y crónica, pero no vasculitis, complejos inmunes o depósitos con tinción lineal.
Los pacientes con hemosiderosis pulmonar idiopática se tratan con esteroides. Evidencias anecdóticas apoyan el uso de agentes inmunosupresores y plasmaféresis. La supervivencia promedio es de 3 a 5 años, pero ocurren remisiones espontáneas con supervivencia prolongada.
Linfangiomiomatosis
El inicio de la linfangiomiomatosis, que afecta en forma casi exclusiva a mujeres en edad reproductiva, suele caracterizarse por hemoptisis, neumotórax y quilotórax.45 Al principio de los síntomas puede no existir alteración radiográfica difusa, aunque en ocasiones se observa un infiltrado reticulonodular fino que predomina en las bases pulmonares. Puede existir un patrón intersticial difuso asociado a hiperinflación. A diferencia de la mayoría de las enfermedades pulmonares infiltrativas difusas, este padecimiento se caracteriza por fisiología obstructiva con aumento en los volúmenes pulmonares que se ha atribuido a hipertrofia del músculo liso peribronquial. Además, la capacidad de difusión suele estar disminuida.
El diagnóstico puede sospecharse en una mujer no fumadora con quilotórax, obstrucción fija de las vías respiratorias y un infiltrado intersticial difuso en la radiografía. La TCAR muestra quistes difusos de pared delgada, y es diagnóstica. Estos datos son idénticos a los de la esclerosis tuberosa.46 La biopsia de pulmón a cielo abierto demuestra proliferación de células de músculo liso en el pulmón y pleura visceral, con formación de quistes, quizá por obstrucción bronquial. Alrededor del 50 porciento de los pacientes tienen también angiomiolipomas como dato asociado, que suelen detectarse por TC.48 Más del 75 porciento de los pacientes sobreviven por más de 8 años.47 Existe alguna evidencia de que este proceso depende de hormonas, y se ha reportado una evolución más lenta después del tratamiento con ooforectomía, progesterona o la combinación de ambas. Debe considerarse la posibilidad de trasplante pulmonar en los casos en etapa terminal. Aunque el trasplante puede salvar la vida, se ha reportado recurrencia en el pulmón trasplantado, lo que destaca la naturaleza sistémica del padecimiento.
MICROLITIASIS
La microlitiasis es un padecimiento excesivamente raro, en ocasiones familiar, que suele diagnosticarse en un paciente asintomático por una radiografía de tórax de rutina. Esta demuestra nódulos pequeños calcificados difusos que, al examen histopatológico, son esferas calcificadas que ocupan el espacio alveolar.48 Esta enfermedad puede progresar hasta la insuficiencia respiratoria. En un solo paciente se reportó una posible respuesta al tratamiento con etidronato de sodio, un difosfonato.49
Reconocimientos
Figura 1 Cortesía del Dr. Sam Aguayo, Veterans Affairs Medical Center, Decatur, Georgia.
Figura 3 Andy Christie.
DR. GERALD W. STATON. JR.
DR. ROLAND H. INGRAM JR.
Generalidades
El término enfermedad pulmonar infiltrativa crónica difusa se refiere a un grupo heterogéneo de padecimientos que afectan principalmente los alveolos y el intersticio pulmonar, aunque las vías respiratorias o los vasos pulmonares suelen afectarse en forma secundaria al proceso intersticial.
La enfermedad pulmonar infiltrativa crónica difusa afecta a una proporción sorprendentemente grande de la población. Un estudio de Nuevo México sugiere una prevalencia de 81 casos /100,000 en varones y 67 casos /100,000 en mujeres. El diagnóstico más común es fibrosis pulmonar o fibrosis pulmonar idiopática (FPI).1 En los Estados Unidos, algunos de estos trastornos son más comunes en la minorías étnicas, aunque los datos al respecto son incompletos.2
Patogenia
Se cree que ciertos mecanismos patogénicos de lesión pulmonar pueden influir en el desarrollo de la enfermedad pulmonar infiltrativa difusa. Se ha demostrado en estudios en animales que varios agentes causales diferentes (v.gr., bleomicina, paraquat, radiación y ciclofosfamida) producen un patrón característico de lesión que podría ser importante en las variedades idiopáticas más frecuentes de este padecimiento. En estos modelos se presenta daño inicial a las células epiteliales tipo I y a las células del endotelio cepilar. Después de una fase de edema y hemorragia leve, en ocasiones con acúmulo de fibrina, existe acúmulo de neutrófilos. Pocos días después aparecen los linfocitos y los macrófagos, y bajo la influencia de ciertos factores de crecimiento las células tipo II comienzan a replicarse y diseminarse para sustituir al epitelio tipo I dañado. En un periodo de dos semanas existe colágena, elastina y otros componentes extracelulares de la matriz, y al final llega a desarrollarse fibrosis pulmonar extensa. La combinación de exposición ambiental y susceptibilidad genética puede tener un papel importante en la patogenia de la enfermedad.3 Las variaciones en esta vía común causan las entidades patológicas específicas que se analizarán en este capítulo.
Estudio del paciente con enfermedad pulmonar infiltrativa difusa
Aunque se han descrito más de 100 causas de enfermedad pulmonar infiltrativa difusa, un número mucho menor de padecimientos son responsables de la mayoría de los casos. Sin embargo, debido a que los hallazgos clínicos, radiográficos y hasta patológicos pueden ser inespecíficos, es importante analizar el diagnóstico diferencial en una forma organizada. En todos los casos deberá realizarse una historia clínica, examen físico, radiografía de tórax y evaluación de la función pulmonar. Estos estudios simples suelen no dar el diagnóstico definitivo, pero ayudan a orientar al clínico hacia las causas más probables en cada enfermo, sirviendo para limitar las posibilidades en el diagnóstico diferencial.
HISTORIA CLINICA
Es indispensable excluir la infección como causa de la enfermedad pulmonar infiltrativa por la necesidad de tratamiento antimicrobiano específico y porque muchas causas no infecciosas del padecimiento se tratan con esteroides. La historia clínica debe incluir un interrogatorio cuidadoso en relación con la duración de los síntomas, que por lo general consisten en tos y disnea. Con frecuencia los síntomas tienen varios meses o incluso años de duración, lo que casi descarta una causa infecciosa. Por el contrario, una evolución más rápida, de días a semanas, debe sugerir la posibilidad de infección. Puede existir fiebre en varios tipos no infecciosos de enfermedad pulmonar infiltrativa difusa (v.gr., sarcoidosis, neumonitis por hipersensibilidad, enfermedades vasculares del tejido conjuntivo y enfermedad inducida por medicamentos), pero su presencia siempre debe alertar al clínico hacia la posibilidad de infección. La enfermedad pulmonar infiltrativa difusa infecciosa que evoluciona en un periodo de semanas a meses rara vez es causada por virus o bacterias. Sin embargo, las infecciones por micobacterias y hongos patógenos (v.gr., histoplasmosis, coccidioidomicosis y blastomicosis) pueden tener una evolución subaguda y suelen asociarse con fiebre. Quizá la causa infecciosa más frecuente es la neumonía por Pneumocystis carinii en un paciente con síndrome de inmunodeficiencia adquirida (SIDA). Este tipo de neumonía puede presentarse con disnea que progresa en forma lenta durante varias semanas, en ocasiones sin fiebre u otras evidencias de toxicidad sistémica, y este patrón puede recordar al de las causas no infecciosas frecuentes de enfermedad pulmonar infiltrativa difusa. Una historia cuidadosa en relación con los factores de riesgo para la infección por VIH es parte indispensable de la evaluación de todos los pacientes con una enfermedad pulmonar infiltrativa difusa reciente.
Además de establecer la duración de la enfermedad y la presencia o ausencia de fiebre y otros síntomas generales, la historia debe enfocarse hacia la ocupación del paciente y los medicamentos que ha recibido. Muchas diferentes exposiciones de tipo laboral pueden causar enfermedad pulmonar infiltrativa difusa, pero por fortuna la mayoría se encuentran en forma poco frecuente en la clínica. Las variedades de tipo laboral más frecuentes son causadas por la exposición a polvos inorgánicos de asbesto y sílice o a polvos orgánicos que causan neumonitis por hipersensibilidad. La asbestosis suele volverse evidente muchos años después de la exposición, por lo que la historia de las ocupaciones previas es muy importante. La causa más frecuente de neumonitis por hipersensibilidad de tipo laboral es el pulmón del granjero. Deberá interrogarse con gran cuidado a los granjeros respecto a cualquier relación entre los síntomas respiratorios o sistémicos y la exposición a material orgánico (granos o heno enmohecido) que puedan contener actinomicetos termofílicos. La historia debe incluir preguntas sobre la posible exposición a palomas o pájaros y humidificadores, porque este tipo de exposición causa también neumonitis por hipersensibilidad.
Muchos medicamentos han sido reportados como causa de enfermedad pulmonar infiltrativa difusa. La historia debe enfocarse tanto a los medicamentos prescritos como a los que se venden sin receta médica, y debe incluir preguntas sobre los fármacos tomados durante las semanas y meses previos.
Por último, debe investigarse si existen síntomas o signos que puedan indicar la presencia de una enfermedad del tejido conjuntivo o vasculitis. Los datos como fenómeno de Raynaud, fotosensibilidad, erupciones cutáneas o artritis son de gran importancia para el diagnóstico.
EXAMEN FISICO
El examen físico tiene menos utilidad diagnóstica que los antecedentes, pero también es importante. Puede sospecharse un padecimiento del tejido conjuntivo si se encuentra sinovitis, telangiectasias, esclerodactilia o erupción malar. La sarcoidosis afecta también a órganos extrapulmonares y puede causar uveítis, eritema nodoso y lesiones cutáneas en placa que corresponden a sarcoidosis en la piel. La enfermedad pulmonar infiltrativa difusa maligna puede asociarse con hallazgos debidos al tumor primario, como una masa abdominal o en la mama, hepatomegalia o una prueba de guayaco en heces positiva. Por último, el examen cardiovascular es muy importante porque en ocasiones la congestión pulmonar venosa crónica debida a una estenosis mitral oculta o a falla del ventrículo izquierdo puede manifestarse como una enfermedad pulmonar infiltrativa difusa.
EXAMENES DE LABORATORIO
Los exámenes de laboratorio incluyen estudios rutinarios y algunos más especializados. En ocasiones pueden encontrarse datos que indican la causa del padecimiento pulmonar infiltrativo difuso en los exámenes estándar. Puede pensarse en sarcoidosis cuando existen citopenias si está afectada la médula ósea o por la presencia de hipercalcemia o alteración en las pruebas de función hepática. Las enfermedades del tejido conjuntivo y las neoplasias pueden causar también citopenias. La eosinofilia periférica sugiere neumonía eosinofílica crónica. En raras ocasiones el granuloma eosinofílico puede presentarse como un padecimiento pulmonar asociado con evidencia por laboratorio de diabetes insípida.
Otras pruebas de laboratorio pueden ser útiles, dependiendo de los datos clínicos y radiográficos de cada caso. Suelen existir anticuerpos antinucleares o factores reumatoides en el suero de pacientes con padecimientos del tejido conjuntivo, pero también se encuentran en títulos bajos hasta en el 50 porciento de los enfermos con FPI. El aumento en la concentración sérica de la enzima convertidora de angiotensina (ECA) sugiere sarcoidosis, pero esta enzima puede también aumentar en la tuberculosis miliar, la beriliosis, la asbestosis y la silicosis. La neumonitis por hipersensibilidad casi siempre se asocia con anticuerpos séricos contra el antígeno agresor. Sin embargo, la presencia de anticuerpos contra uno de los agentes causales de las neumonitis por hipersensibilidad no demuestra que ésta sea la causa de la enfermedad pulmonar infiltrativa difusa, sino que solo ha existido la suficiente exposición al antígeno para despertar una respuesta inmunológica.
RADIOGRAFIA DE TORAX
Los datos radiográficos suelen ser inespecíficos. En la mayor parte de los casos existe un proceso intersticial simétrico y bilateral. Sin embargo, existen ciertas claves radiográficas que, cuando están presentes, pueden ayudar en el diagnóstico diferencial de la enfermedad pulmonar infiltrativa difusa para disminuir la lista de posibles causas [ver tabla 1].
|
||||||||||||
|
TOMOGRAFIA COMPUTADA DE ALTA RESOLUCION
El uso de la tomografía computada de alta resolución (TCAR) representa un adelanto importante en la evaluación de la enfermedad pulmonar parenquimatosa difusa. Con el uso de este procedimiento puede determinarse la extensión, localización y patrón de afección pulmonar con gran exactitud [ver figura 1]. Con frecuencia la TCAR puede detectar alteraciones en pacientes que tienen síntomas de enfermedad pulmonar intersticial con radiografía de tórax normal. Cuando se combina con los datos clínicos y la radiografía de tórax, la TCAR de tórax puede identificar el diagnóstico específico en el 60 a 80 porciento de los casos.4 Cuando la TCAR indica un diagnóstico específico (v.gr., granuloma eosinofílico) se elimina la necesidad de una biopsia pulmonar.5
|
|
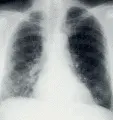
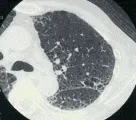
Las pruebas de función respiratoria orientan hacia la presencia de enfermedad pulmonar infiltrativa difusa y son útiles para evaluar la progresión de la enfermedad. Las características en este tipo de trastornos son un patrón ventilatorio restrictivo (volumen pulmonar disminuido), una relación volumen espiratorio forzado en el primer segundo a capacidad vital forzada (VEF1/CVF) normal o aumentada, reducción en la capacidad de difusión para monóxido de carbono (DLCO) y reducción en la tensión arterial de oxígeno (PaO2) asociada con tensión arterial de bióxido de carbono normal o disminuida (PaCO2). Además, suele existir limitación significativa al ejercicio por disminución en la PaO2, alteraciones en la mecánica respiratoria y enfermedad vascular pulmonar asociada.
LAVADO BRONCOALVEOLAR
En la mayoría de los casos la causa no se descubre a pesar de una historia clínica cuidadosa, evaluación fisiológica y estudios radiográficos y de laboratorio. El siguiente paso suele ser realizar una broncoscopía con lavado broncoalveolar (LBA) y biopsia pulmonar transbronquial. Ciertas causas pueden diagnosticarse sólo con el LBA, otras sólo con la biopsia y algunas con cualquiera de ambas técnicas. El LBA es más útil para diagnosticar causas infecciosas, en especial neumonía por P. carinii. La sensibilidad diagnóstica de este procedimiento para neumonía por P. carinii relacionada con SIDA es de alrededor de 90 a 95 porciento. Por lo tanto, éste es el procedimiento de eleccción para diagnosticar el 20 al 50 porciento de los casos de neumonía por P. cariniii asociada a SIDA que no pueden ser diagnosticados por medio del estudio de la expectoración. Otras infecciones oportunistas, como la neumonía por citomegalovirus y la infección micótica o tuberculosa diseminadas, pueden también diagnosticarse por medio del lavado. Causas no infecciosas que pueden diagnosticarse por esta técnica incluyen la proteinosis alveolar, la carcinomatosis linfática y el carcinoma de células alveolares. Además, el LBA puede proporcionar información útil, aunque no totalmente específica, al demostrar algunos de los siguientes cambios: (1) aumento en el número de eosinófilos en la neumonía eosinofílica crónica, (2) cuerpos de asbesto en la asbestosis, (3) los cambios llamados espumosos, con inclusiones laminares en la enfermedad inducida por amiodarona, (4) hiperplasia y atipia de los neumocitos tipo II en la lesión pulmonar inducida por citotóxicos, (5) células de Langerhans en el granuloma eosinófilo, y (6) sangre con hemosiderina abundante en los macrófagos alveolares en la hemorragia alveolar difusa. La cuantificación y distribución de las células inflamatorias (v.gr., macrófagos, linfocitos y neutrófilos) puede sugerir un diagnóstico específico.6 El líquido del lavado alveolar de los no fumadores normales típicamente tiene 84 a 99 porciento de macrófagos, 1 a 14 porciento de linfocitos y 0 a 1 porciento de neutrófilos. Las causas comunes de enfermedad pulmonar infiltrativa difusa no granulomatosa (v.gr., FPI, algunos trastornos del tejido conectivo y asbestosis) suelen caracterizarse por alveolitis neutrofílica, mientras que la sarcoidosis y las neumonitis por hipersensibilidad se asocian con aumento en los linfocitos. Sin embargo, existe sobreposición, y puede existir un paciente con FPI que tenga mayor número de linfocitos, o un enfermo con sarcoidosis y un número normal de linfocitos.
BIOPSIA PULMONAR
En muchos casos el diagnóstico se desconoce hasta que se realiza una biopsia pulmonar. Desde el punto de vista patológico, estos trastornos pueden caracterizarse por varios grados de afección de los tabiques alveolares o los alveolos por células inflamatorias, células mesenquimatosas, fibrosis, granulomas o células neoplásicas. En raros casos la sustancia anormal que se acumula en el parénquima pulmonar es sangre, material proteináceo (proteinosis alveolar), amiloide, músculo liso (linfangiomiomatosis o esclerosis tuberosa) o un material anormal que se deposita como resultado de un trastorno de almacenamiento hereditario (enfermedad de Gaucher, enfermedad de Niemann-Pick o síndrome de Hermansky-Pudlak).
Puede obtenerse tejido pulmonar por biopsia transbronquial durante una broncoscopía [ver tabla 2] o por una de dos técnicas de biopsia abierta: la biopsia abierta tradicional o la toracoscopía asistida por video. La biopsia pulmonar transbronquial es útil sobre todo en el diagnóstico de la enfermedad pulmonar infiltrativa difusa infecciosa y en la sarcoidosis. Pueden demostrarse granulomas no caseosos en muestras de biopsia transbronquial en el 70 a 90 porciento de los pacientes con sarcoidosis. El porcentaje de diagnóstico es alto (alrededor del 90 porciento) en pacientes con enfermedad pulmonar aparente desde el punto de vista radiográfico (estadios II y III) y menor (alrededor del 70 porciento) en enfermos con adenopatía hiliar sola (estadio I). La conveniencia de realizar una biopsia abierta después de una biopsia pulmonar transbronquial inespecífica dependerá de las características clínicas y radiográficas específicas de cada caso, del impacto que tendrá un diagnóstico más preciso en el tratamiento y de la evaluación del riesgo de la biopsia abierta en cada paciente. La frecuencia con la que se realiza una biopsia abierta de pulmón para determinar mejor las características histopatológicas de las formas no infecciosas de la enfermedad varía mucho entre los diferentes centros. El uso de la toracoscopía asistida por video en lugar de la biopsia abierta de pulmón no ha causado la reducción esperada en la morbilidad y el costo.7
|
|
BPT - biopsia pulmonar transbronquial |
TRATAMIENTO
Está indicada la inmunización contra los antígenos neumocócicos y anual contra la influenza. Los pacientes con un defecto obstructivo reversible pueden beneficiarse con el uso de broncodilatadores. La administración de oxígeno suplementario durante el reposo y el ejercicio puede aumentar la tolerancia del paciente al realizar sus actividades cotidianas.8
Enfermedad pulmonar infiltrativa difusa de etiología conocida
ENFERMEDAD INDUCIDA POR MEDICAMENTOS
Se ha reportado que muchos medicamentos diferentes causan enfermedad pulmonar infiltrativa difusa.9,10 Se ha calculado que la enfermedad pulmonar inducida por medicamentos afecta a varios cientos de miles de pacientes cada año. Para minimizar la morbimortalidad de ésta, es indispensable detectarla en etapas tempranas. La suspensión del agente agresor suele ser seguida de mejoría espontánea, mientras que el no detectar la relación causal entre el medicamento y la enfermedad pulmonar puede causar lesión pulmonar irreversible. Por desgracia, ciertos aspectos de la enfermedad inducida por medicamentos pueden dificultar el reconocimiento de esta relación causa-efecto. Primero, el número de diferentes medicamentos que causan enfermedad pulmonar infiltrativa difusa es muy grande como para recordarlos todos, y la enfermedad pulmonar suele ocurrir sólo en un pequeño porcentaje de los enfermos que reciben el medicamento. Segundo, el inicio de la enfermedad pulmonar puede ocurrir semanas a meses después de iniciado el fármaco. En el caso de los citotóxicos, los síntomas respiratorios comienzan muchas semanas después de la última exposición al agente agresor. Por último, los medicamentos que pueden causar enfermedad pulmonar infiltrativa difusa se prescriben con frecuencia para padecimientos que se asocian con un alto riesgo de la enfermedad. Por ejemplo, un paciente que recibe un agente quimiotáctico tiene mayor riesgo de infección y de infiltración tumoral en el pulmón. En forma semejante, los medicamentos antinflamatorios no esteroides que causan enfermedad pulmonar parenquimatosa suelen prescribirse para las enfermedades del tejido conjuntivo que se asocian con enfermedad pulmonar infiltrativa difusa. Por todos estos motivos, el médico debe estar muy alerta respecto a que la enfermedad pulmonar infiltrativa difusa sea causada por medicamentos.
Fármacos citotóxicos
La enfermedad pulmonar infiltrativa difusa es una causa importante de morbimortalidad en pacientes bajo tratamiento por una neoplasia. En algunos casos el medicamento utilizado para tratar la neoplasia es la causa directa de las lesiones pulmonares. Algunos enfermos mueren por toxicidad inducida por medicamentos citotóxicos, y otros experimentan lesión permanente de la función pulmonar a pesar de ser curados de la neoplasia. La mayor parte de los fármacos empleados en el tratamiento de las neoplasias pueden causar enfermedad pulmonar infiltrativa difusa. Los principales son la bleomicina, la ciclosfosfamida, el metotrexate y las nitrosoureas. La ciclofosfamida y el metotrexate se utilizan cada vez más para el manejo de padecimientos no malignos, aunque en dosis mucho menores. La enfermedad pulmonar inducida por medicamentos ocurre con menos frecuencia a estas dosis más bajas, pero también puede existir.
Las características clínicas de la enfermedad pulmonar inducida por fármacos son inespecíficas. La tos y la disnea suelen ser prominentes, y la fiebre no es rara. La radiografía de tórax suele demostrar infiltrados intersticiales bilaterales y simétricos, aunque puede existir asimetría al principio de la enfermedad. La progresión del padecimiento, tanto clínica como radiográfica, es variable. Lo más frecuente es que el principio sea subagudo, con tos y disnea que se presentan en varias semanas. Puede verse también un inicio más explosivo, con datos de síndrome de insuficiencia respiratoria progresiva aguda (SIRPA) y una necesidad urgente de apoyo ventilatorio mecánico. En el otro extremo del espectro, puede existir fibrosis pulmonar por el tratamiento con bleomicina y otros agentes que se desarrolla en forma insidiosa durante muchos meses.
Las características patológicas de la enfermedad inducida por fármacos citotóxicos son típicas, aunque no patognomónicas. Puede observarse infiltrado intersticial de células inflamatorias y fibrosis. Sin embargo, el dato más característico es que los neumocitos tipo II aumentan en número y muestran atipia importante. Esta característica patológica puede ser muy sugestiva de enfermedad inducida por fármacos citotóxicos en los casos pertinentes, pero pueden observarse cambios en las células tipo II con las infecciones virales graves y durante la fase de reparación del SIRPA.
Se observa un cuadro patológico un poco diferente cuando la enfermedad es causada por citarabina o metotrexate. La citarabina se ha asociado con una forma frecuentemente mortal de edema no cardiogénico. La enfermedad inducida por metotrexate se asocia con granulomas, alveolitis linfocítica y un aumento significativo en las células T cooperadoras.
El diagnóstico de enfermedad pulmonar infiltrativa difusa inducida por fármacos citotóxicos se establece si existe la exposición previa al agente agresor, excluyendo una infección como causa del daño pulmonar y demostrando características patológicas que sean compatibles con la lesión inducida por citotóxicos. La relación temporal entre la exposición al fármaco y el inicio de la enfermedad pulmonar es variable. No es raro que exista un intervalo de unas pocas semanas entre la última exposición al medicamento y el inicio de los síntomas; en casos raros este intervalo puede ser hasta de varios meses. Las características clínicas y radiográficas de la enfermedad pulmonar inducida por un medicamento citotóxico son casi indistinguibles de las causadas por una infección oportunista. Por lo tanto, se requiere un enfoque agresivo para establecer el diagnóstico. El no detectar una etiología infecciosa mediante la broncoscopía aumenta la posibilidad de enfermedad inducida por fármacos, en especial si se observan los cambios característicos en las células tipo II en la muestra de biopsia transbronquial. Sin embargo, puede requerirse la biopsia abierta de pulmón para excluir con confianza una infección y establecer mejor las características histopatológicas de la lesión por medicamentos citotóxicos. Incluso con la biopsia a cielo abierto, el diagnóstico de enfermedad inducida por fármacos citotóxicos es inferencial si no se encuentran criterios patognomónicos para establecer el diagnóstico.
La enfermedad pulmonar inducida por medicamentos citotóxicos no se previene con facilidad, ya que estos fármacos casi siempre son indispensables para el tratamiento óptimo de alguna neoplasia potencialmente mortal. Para los pacientes con carcinoma testicular que reciben bleomicina, el riesgo de lesiones pulmonares parece relacionarse con la dosis. La vigilancia de la DLCO y la espirometría durante el tratamiento permite en algunos casos detectar la lesión pulmonar en forma temprana, de modo que el evitar mayor exposición al fármaco disminuya la posibilidad de daño pulmonar permanente y grave. Sin embargo, el tratamiento para el linfoma no-Hodgkin con bleomicina combinada con doxorrubicina, ciclofosfamida, vincristina y prednisona causa enfermedad pulmonar infiltrativa difusa en forma no dependiente de la dosis, lo que anula los esfuerzos para prevenirla. Quizá la estrategia más eficaz para disminuir la morbimortalidad de la enfermedad inducida por citotóxicos no es prevenirla, sino diagnosticarla en una etapa tan temprana que pueda evitarse mayor exposición al fármaco, cambiando a esquemas alternos de quimioterapia siempre que sea posible.
La eliminación de mayor exposición al medicamento es el aspecto crucial del tratamiento de la enfermedad pulmonar inducida por fármacos citotóxicos. Algunos reportes anecdóticos indican que el tratamiento con esteroides se asocia con rápida mejoría del intercambio de gases y normalización de las alteraciones radiográficas en algunos casos. Si la enfermedad inducida por medicamentos citotóxicos es muy severa o parece progresar a pesar de la suspensión del fármaco, es aconsejable administrar un periodo de tratamiento empírico con esteroides.
Fármacos no citotóxicos
La enfermedad pulmonar infiltrativa difusa puede ser causada por la exposición a una gran variedad de fármacos no citotóxicos, incluyendo antibióticos, agentes antinflamatorios, antiarrítmicos y drogas ilícitas.10 Además, este padecimiento puede ser una manifestación del lupus eritematoso generalizado (LEG) inducido por medicamentos.
La nitrofurantoína es un agente antibacteriano utilizado principalmente para el tratamiento de las infecciones de las vías urinarias y constituye una de las causas más frecuentes de enfermedad pulmonar infiltrativa difusa inducida por fármacos.10 Puede presentarse toxicidad aguda o crónica, pero el síndrome agudo es mucho más común. La reacción pleuropulmonar aguda comienza dos a 10 días después de la exposición inicial al fármaco y se manifiesta por disnea, tos y fiebre. Ocurre pleuresía en una tercera parte de los enfermos. La radiografía de tórax muestra un patrón de infiltrado alveolar o intersticial, en ocasiones acompañado de derrame pleural. Puede observarse eosinofilia en sangre periférica. El padecimiento se diagnostica por el antecedente de exposición reciente a nitrofurantoína y resolución espontánea de los datos clínicos y radiográficos después de uno a cuatro días de suspendido el medicamento. La velocidad de resolución es mucho más rápida que en el caso de una infección y sirve para establecer el diagnóstico con un alto grado de certeza. La toxicidad crónica no se asocia con síntomas sistémicos y tiene un cuadro clínico y radiográfico indistinguible de la fibrosis pulmonar idiopática. Si no existe mejoría en dos a tres meses después de suspender el fármaco, está indicado el tratamiento con esteroides.
Amiodarona La amiodarona es un agente antiarrítmico que se utiliza principalmente para el tratamiento de la taquiarritmia ventricular refractaria. Este fármaco se asocia con diversos efectos tóxicos dependientes de la dosis sobre diferentes órganos, pero la principal limitación a su uso es la toxicidad pulmonar, que ocurre en alrededor del 5 a 7 porciento de los casos. La toxicidad pulmonar inducida por amiodarona suele caracterizarse por tos y disnea que en un principio se atribuyen a insuficiencia cardiaca congestiva. Los síntomas sistémicos, incluyendo la fiebre leve, no son raros. La radiografía de tórax puede mostrar un infiltrado intersticial simétrico y bilateral semejante al observado en otros tipos de enfermedad pulmonar infiltrativa difusa. Sin embargo, no son raros otros patrones radiográficos, que pueden incluir alteración unilateral o enfermedad aislada del lóbulo superior, este último hallazgo sugiere tuberculosis. En ocasiones se presenta derrame pleural. El diagnóstico de enfermedad pulmonar inducida por amiodarona se establece principalmente al excluir otras causas probables de enfermedad pulmonar, en especial la infección y la insuficiencia cardiaca. La broncoscopía con LBA y biopsia es útil para excluir la infección y también demuestra la presencia de los llamados macrófagos espumosos con inclusiones laminares (que se observan bajo microscopía electrónica). Estos cambios en los macrófagos indican exposición a la amiodarona, pero no demuestran que este fármaco sea la causa del proceso pulmonar porque se encuentran cambios semejantes en individuos asintomáticos que reciben el medicamento.
La suspensión del medicamento es la base del tratamiento, pero los esteroides parecen haber sido útiles en algunos casos seleccionados. Es interesante que existen varias notificaciones de enfermedad pulmonar aguda inducida por amiodarona en pacientes que fueron sometidos a procedimientos quirúrgicos menores, como la colocación de un desfibrilador automático o la angiografía pulmonar. Con frecuencia la enfermedad tiene un curso fulminante, y la evolución llega a ser mortal. Debido a que la vida media del medicamento es de varias semanas, los pacientes sometidos a estos procedimientos tienen aún riesgo de lesión pulmonar aguda si el medicamento se suspendió solo uno a dos meses antes del procedimiento.
Oro y penicilamina El oro y la penicilamina, utilizadas principalmente para el tratamiento de la artritis reumatoide y otras enfermedades del tejido conjuntivo, pueden causar enfermedad pulmonar infiltrativa difusa. Puede ser difícil determinar si el daño pulmonar es causado por el agente terapéutico o por la enfermedad subyacente, incluso con una biopsia a cielo abierto de pulmón. Se aconseja en estos casos la suspensión del medicamento, junto con el tratamiento empírico con esteroides si los síntomas son graves. La penicilamina también ha sido implicada en casos de hemorragia alveolar difusa con glomerulonefritis, LEG inducido por medicamentos y panbronquiolitis con obstrucción grave de las vías respiratorias.
Drogas ilícitas Las drogas ilícitas pueden también causar enfermedad pulmonar infiltrativa difusa. Puede existir granulomatosis inducida por talco y fibrosis pulmonar por la inyección de cristales y anfetaminas disueltas, o por pastillas de narcóticos que tienen talco como excipiente. La enfermedad puede progresar años después de la última exposición, quizá porque el talco persiste en los pulmones y continúa ocasionando una respuesta inflamatoria. La biopsia abierta o transbronquial demuestra inflamación granulomatosa con partículas abundantes de talco, que pueden ser observadas por microscopía polarizada. El no analizar los granulomas con microscopio polarizado puede ocasionar un diagnóstico erróneo de sarcoidosis. El diagnóstico puede también hacerse al detectar talco en la retina. La heroína puede causar edema pulmonar agudo, pero no causa per se una enfermedad pulmonar infiltrativa crónica difusa. La cocaína también se ha asociado a edema pulmonar agudo, y se ha reportado que causa hemorragia alveolar difusa y un síndrome de infiltrado pulmonar con eosinofilia. Las reacciones pulmonares relacionadas con la cocaína no ocasionan lesiones crónicas.
L-triptofano El síndrome de mialgia-eosinofilia relacionado con L-triptofano, una enfermedad caracterizada por mialgias severas, fatiga y diversas erupciones, puede tener también manifestaciones pulmonares . Muchos pacientes con este síndrome refieren disnea, y se han observado infiltrados intersticiales bilaterales, bronquiolitis folicular, hipertensión pulmonar y derrames pleurales. Las pruebas de función pulmonar revelan restricción con reducción en la capacidad de difusión, hipoxemia y evidencia de debilidad de los músculos respiratorios. Las biopsias pulmonares muestran inflamación intersticial con eosinofilia parenquimatosa y vasculitis prominente de los vasos pequeños. Algunos pacientes con este síndrome han respondido a esteroides. Dos años después del inicio la mayoría de los síntomas han mejorado o desaparecido.11
NEUMOCONIOSIS
La exposición ambiental o laboral a material particulado puede causar varias neumoconiosis que se manifiestan como asma, bronquitis crónica o enfermedad parenquimatosa difusa (sobre todo intersticial). Estas neumoconiosis incluyen asbestosis, silicosis, neumoconiosis del trabajador de carbón (NTC) y beriliosis.
Asbestosis
La exposición a asbesto, un silicato usado para aislamiento en superficies de fricción y para endurecer materiales, se asocia con el desarrollo de cambios pleurales (placas y derrame), mayor incidencia de neoplasias (carcinoma broncogénico y mesotelioma) y fibrosis intersticial difusa. Solo la fibrosis intersticial difusa se denomina asbestosis.
La asbestosis se caracteriza por disnea de inicio gradual 20 a 30 años después de la exposición al asbesto. Suele existir tos, pero ésta es no productiva a menos que el paciente haya sido fumador y tenga una bronquitis crónica asociada. Pueden auscultarse estertores finos al final de la inspiración antes de que existan alteraciones en la radiografía de tórax, y es común la presencia de dedos en palillo de tambor. En la fase tardía de la asbestosis se desarrollan signos de cor pulmonale.
Al principio de la enfermedad la radiografía de tórax puede ser normal, pero gradualmente aparecen sombras lineales, pequeñas e irregulares, en las porciones inferiores de los campos pulmonares. La captación pulmonar de galio-67 puede estar aumentada, pero es inespecífica. Las pruebas de función pulmonar suelen mostrar restricción, reducción en la DLCO e hipoxemia inducida por el ejercicio. El diagnóstico de asbestosis se realiza cuando un paciente tiene historia de exposición a asbestos, placas pleurales (indicador objetivo de la exposición) y, en casos dudosos, exceso de asbesto en las muestras obtenidas (por LBA, biopsia pulmonar transbronquial o biopsia abierta). No existe tratamiento específico para los pacientes con asbestosis.
Silicosis
La silicosis es una enfermedad pulmonar fibrótica crónica causada por la exposición a sílice cristalino libre. Se ha demostrado la exposición laboral significativa a sílice en la industria minera, el corte, pulido y esculpido de piedra, el trabajo de fundición y la limpieza abrasiva (con arena). Se requiere la exposición durante alrededor de 5 años para el desarrollo de la silicosis, a menos que la exposición sea muy intensa. Existen dos formas de silicosis: la nodular simple, en la que muchos pacientes están asintomáticos o sufren solo de bronquitis crónica secundaria al uso de tabaco, y la fibrosis progresiva masiva (FPM), en la que se desarrolla disnea incapacitante. No suelen existir datos físicos. Desde el punto de vista radiográfico la silicosis nodular simple se caracteriza por opacidades difusas, redondas y pequeñas que tienden a ser más prominentes en los lóbulos superiores. Puede observarse crecimiento hiliar, y en ocasiones las llamadas calcificaciones en cascarón de huevo. En los pacientes con FPM las opacidades coalescen para formar masas grandes e irregulares. La función pulmonar en los pacientes con silicosis nodular simple puede ser normal o manifestarse por un patrón mixto de obstrucción y restricción. En los enfermos que tienen FPM se desarrolla restricción e hipoxemia severas, así como hipertensión pulmonar.
La tuberculosis y las infecciones por micobacterias atípicas, que ocurren con mayor frecuencia en los enfermos con silicosis, pueden confundirse con progresión de la silicosis, y el diagnóstico definitivo es difícil. Debido a que la inmunidad mediada por células parece ser normal en los pacientes con silicosis, la prueba cutánea de tuberculina puede ser muy útil para identificar a los pacientes en los que debe investigarse el diagnóstico de tuberculosis.
El diagnóstico de silicosis casi siempre se realiza si un paciente tienen antecedente de exposición y una radiografía de tórax compatible. En los casos atípicos la presencia de sílice en el LBA o en las muestras de biopsia de pulmón pueden establecer el diagnóstico y excluir el de tuberculosis y carcinoma.
Se ha pensado que la silicosis es intratable, aunque un reporte sugiere que los esteroides por vía oral disminuyen la inflamación y mejoran la función pulmonar.12 La prevención de la mayor exposición al sílice y el tratamiento de la tuberculosis activa son aspectos muy importantes en la atención al paciente.
Neumoconiosis del trabajador de carbón
La NTC es una causa poco frecuente de fibrosis pulmonar en los trabajadores que están expuestos a polvo de carbón y grafito. La mayoría de estos pacientes son mineros que con frecuencia han trabajado bajo la superficie. La influencia del tabaquismo y de materiales como el sílice en la producción de los síntomas respiratorios es motivo de controversia. Muchos pacientes con NTC tienen tos crónica que en ocasiones es productiva, con expectoración gris o negra, y que puede ser causada por bronquitis crónica que se relaciona con polvo de tabaco o carbón. En forma semejante a la silicosis, la NTC incluye a la forma simple y a la FPM. La primera no se asocia con una mayor incidencia de disnea, mientras que la fibrosis progresiva masiva se caracteriza por disnea severa. Los cambios radiográficos de la forma simple consisten en opacidades pequeñas y redondeadas. Cuando las opacidades son mayores de 1 cm se clasifican en forma arbitraria como FPM. La NTC no progresa si se elimina la exposición al polvo, mientras que la FPM progresa aún después de suspendida la exposición. No existe tratamiento específico para ninguna forma de NTC.
Beriliosis
El berilio es un metal raro que se usa en las industrias de alta tecnología moderna por su bajo peso, su alto punto de fundición, su resistencia tensil, sus excelentes propiedades aislantes y su capacidad para retardar la fisión nuclear. La exposición de alta intensidad al berilio puede ocasionar una bronquitis aguda y neumonitis químicas, que son muy raras en la actualidad por los controles ambientales tan estrictos. Sin embargo, aún se ve con frecuencia beriliosis crónica. Esta se caracteriza por enfermedad granulomatosa multisistémica que tiene muchas semejanzas con la sarcoidosis y que ocurre meses a años después de la exposición. El número de casos crónicos parece estar disminuyendo, quizá por una mayor atención hacia la enfermedad y por los controles ambientales. Se piensa que el berilio se une a proteínas del huésped que son transportadas por todo el organismo, estos complejos de berilio-proteína estimulan una respuesta de hipersensibilidad tardía, que produce inflamación granulomatosa en los sitios de actividad de la enfermedad.
Los síntomas de la beriliosis son disnea, tos, dolor torácico, fatiga, pérdida de peso y artralgias. Los estertores no son comunes al inicio. Los datos radiográficos, que pueden preceder a los síntomas clínicos, incluyen opacidades nodulares e irregulares mal definidas que en ocasiones se asocian con adenopatía hiliar. Al progresar la enfermedad los pulmones disminuyen de tamaño y adquieren el aspecto de un panal de abejas. Las pruebas de función pulmonar pueden ser normales; sin embargo, con más frecuencia se caracterizan por una baja DLCO con o sin restricción o demuestran obstrucción leve. Otras alteraciones incluyen hiperuricemia, hipercalcemia, hipercalciuria y aumento en la concentración sérica de ECA.
Los criterios diagnósticos incluyen una historia bien demostrada de exposición o la demostración de berilio en exceso en muestras obtenidas del paciente, evidencia radiográfica y en las pruebas de función pulmonar de una enfermedad de vías respiratorias bajas compatible con beriliosis, y la demostración de inflamación granulomatosa en el pulmón, los ganglios linfáticos y otras áreas de actividad de la enfermedad. La diferenciación entre beriliosis y sarcoidosis puede ser difícil. Debe pensarse en el diagnóstico de sarcoidosis por la presencia de uveítis y eritema nodoso, adenopatía hiliar sin infiltrados parenquimatosos o por mejoría después de un periodo breve de administración de esteroides. Es más posible el diagnóstico de beriliosis si existe una prueba de transformación de linfocitos positiva, realizada en la sangre o células obtenidas por LBA, que se positiviza en respuesta a compuestos de berilio.
El paso más importante en el tratamiento de la beriliosis es la prevención de una mayor exposición. Se piensa que los esteroides mejoran la evolución de la enfermedad, pero estos agentes deben administrarse de por vida.
NEUMONITIS POR HIPERSENSIBILIDAD
El término de neumonitis por hipersensibilidad se refiere a una enfermedad pulmonar intersticial que resulta de la inhalación de antígenos orgánicos. Parece ser que la patogenia de la neumonitis por hipersensibilidad tiene una base inmunológica y no es resultado de una lesión tóxica directa al pulmón por la sustancia inhalada. Varios antígenos orgánicos causan este síndrome, pero la forma más frecuente de neumonitis por hipersensibilidad, llamada pulmón del granjero, es causada por la inhalación de Actinomyces termofílico, que está presente en el heno o granos enmohecidos. Otras causas frecuentes de neumonitis por hipersensibilidad son la enfermedad de criadores de palomas y de criadores de pájaros, en la que las proteínas séricas de las palomas o periquitos, al ser inhaladas, inducen el síndrome. La enfermedad pulmonar por humidificadores es un tipo de neumonitis por hipersensibilidad causada por la exposición a sistemas de ventilación contaminados. El antígeno responsable en estos casos es, como en el pulmón del granjero, un organismo Actinomyces termofílico, pero puede incluir también organismos Aureobasidium y varias amibas. Las otras formas de neumonitis de hipersensibilidad además del pulmón del granero, la enfermedad de criadores de palomas, la enfermedad de cuidadores de pájaros y la enfermedad por humidificadores, son muy poco frecuentes y suelen resultar de la exposición a antígenos en ocupaciones arcaicas (v.gr., corte de paprika y amortajamiento de momias).
Patogenia y patología
La patogenia de la neumonitis por hipersensibilidad no se conoce bien. La hipótesis actual consiste en que el material inhalado causa la activación directa de la vía alterna del complemento, ocasionando la formación de factores quimiotácticos para neutrófilos y, en consecuencia, una alveolitis neutrofílica aguda. Los productos de la activación del complemento estimulan también a los macrófagos alveolares para producir monocinas, causando una secuencia de eventos que al final provoca formación de granulomas.
Los cambios patológicos en la neumonitis por hipersensibilidad han sido bien definidos al examinar biopsias de pulmón a cielo abierto de gran número de pacientes con pulmón del granjero. Todos los pacientes tienen una neumonitis intersticial, y dos tercios muestran granulomas aislados. Puede ocurrir fibrosis en las formas más crónicas de la enfermedad, y hasta el 50 porciento de los pacientes tiene bronquiolitis obliterante. El cuadro patológico de la neumonitis intersticial linfocítica con formación de granulomas y bronquiolitis obliterante es característico de la neumonitis por hipersensibilidad [ver figura 2]. Los estudios de LBA han determinado el tipo de población linfocitaria en el pulmón, y han demostrado que los linfocitos predominantes son de la subclase T supresora-citotóxica. Esta inversión en la relación entre células T cooperadoras y supresoras-citotóxicas en la neumonitis por hipersensibilidad contrasta con el predominio de células T cooperadoras en los pacientes con sarcoidosis. El hallazgo de una alveolitis linfocítica intensa con inversión de la relación entre células T cooperadoras-citotóxicas sugiere neumonitis por hipersensibilidad, pero este patrón no puede considerarse específico porque puede observarse en otros padecimientos (v.gr., FPI asociada con alveolitis linfocítica).
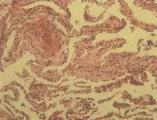
|
| Figura 2 |
| Neumonitis por hipersensibilidad |
Manifestaciones clínicas y diagnóstico
La neumonitis por hipersensibilidad existe básicamente en dos formas clínicas distintas: aguda y crónica.
La neumonitis aguda por hipersensibilidad se caracteriza por fiebre, calosfríos, tos, disnea y malestar, que ocurren típicamente 4 a 8 horas después de la exposición antigénica. La mayoría de los pacientes con neumonitis aguda por hipersensibilidad mejoran en forma espontánea a menos que persista la exposición al antígeno. En ocasiones el paciente presenta un episodio más grave caracterizado por hipoxemia e insuficiencia respiratoria considerable que requiere hospitalización. Debido a la importancia de los síntomas sistémicos durante el episodio agudo, puede pensarse en forma equivocada que el paciente tiene una infección respiratoria. Ante la exposición continua al antígeno inhalado (por ejemplo, en una granja) el paciente puede tener episodios diarios de neumonitis aguda, que se superponen unos con otros. Si ocurre este patrón sintomático, el granjero podrá referir que ha tenido una enfermedad gripal durante varias semanas. Los síntomas respiratorios como tos y sibilancias, que ocurren inmediatamente después de la exposición a material orgánico, no deben atribuirse a neumonitis por hipersensibilidad, sino a una respuesta irritante dentro de las vías respiratorias.
En la neumonitis crónica por hipersensibilidad no suele haber síntomas generales, sino que el cuadro clínico se caracteriza por disnea crónica y tos durante el ejercicio. La presentación clínica es la de cualquier enfermedad pulmonar intersticial de etiología incierta.
No existe una sola prueba que pueda utilizarse para establecer el diagnóstico inequívoco de neumonitis por hipersensibilidad. Por lo tanto, deberá analizarse la combinación de la historia clínica, los estudios serológicos y la exclusión de otras causas posibles. La historia de episodios previos de neumonitis aguda por hipersensibilidad es de gran valor diagnóstico. Al realizar la historia, el clínico debe enfocarse a establecer la presencia de una exposición antigénica relevante y a tratar de determinar si esta exposición ha causado síntomas típicos agudos varias horas después. Este tipo de antecedente es más frecuente cuando existe una exposición intensa (v.gr., heno mohoso o un palomar), pero es menos frecuente en casos de exposición de baja intensidad (v.gr., un humidificador o un solo pájaro). En esta última circunstancia la exposición es más o menos continua y es muy difícil establecer una relación causal entre la exposición al antígeno y los síntomas. Los pacientes deben ser interrogados en forma cuidadosa sobre si los síntomas se resolvieron cuando estuvieron de vacaciones o alejados de la posible fuente de exposición. Por lo menos el 50 porciento de los pacientes con la forma crónica de la enfermedad niega antecedente de episodios agudos. Es posible que éstos sí hayan ocurrido, pero que el paciente haya atribuido los síntomas a una infección de las vías respiratorias. El paciente puede mejorar en forma espontánea cuando se le hospitaliza para evaluarle por el padecimiento respiratorio, sólo para presentar recurrencia de los síntomas en cuanto regresa a su casa. Este tipo de antecedente debe siempre despertar la sospecha de una neumonitis por hipersensibilidad como causa de los síntomas.
La apariencia radiográfica del tórax en un paciente con enfermedad aguda varía desde la ausencia de cambios hasta infiltrados alveolares o lobares. Al desarrollarse la enfermedad crónica puede observarse un infiltrado reticular, nodular o combinado, lo mismo que fibrosis si la exposición al antígeno continúa. La TCAR, que es más sensible que la radiografía de tórax, puede sugerir el diagnóstico y distinguir la neumonitis por hipersensibilidad de otros tipos de enfermedad pulmonar difusa.13
Los estudios serológicos son útiles para determinar si ha existido exposición a un antígeno específico. Casi todos los pacientes con pulmón del granjero y enfermedad de criadores de palomas tienen anticuerpos contra organismos Actinomyces termofílicos y suero de palomas, respectivamente. Sin embargo, los anticuerpos pueden también existir en el suero de un porcentaje significativo de individuos expuestos y asintomáticos. Por lo tanto, la presencia de anticuerpos en el suero no establece el diagnóstico de neumonitis por hipersensibilidad, sino que demuestra que ha existido exposición significativa al antígeno.
Los enfermos que sufren neumonitis por hipersensibilidad subaguda o crónica deben ser sometidos a una broncoscopía diagnóstica con lavado broncoalveolar (LBA) y biopsia transbronquial. El líquido del lavado en la neumonitis por hipersensibilidad suele mostrar linfocitosis intensa, con más de 50 porciento de linfocitos. Como se mencionó antes, estos son principalmente linfocitos T supresores-citotóxicos. Al pasar el tiempo desde la última exposición al antígeno el LAB tiende a normalizarse.14 Se espera que la biopsia pulmonar transbronquial muestre una neumonitis intersticial inespecífica en la mayoría de los enfermos; sin embargo, puede no haber evidencia de granulomas ni de bronquiolitis obliterante. Incluso en las dos terceras partes de los casos en los que existen granulomas las lesiones están más aisladas que los granulomas sarcoides y, por lo tanto, no es tan probable demostrarlas en la biopsia transbronquial. La biopsia de pulmón a cielo abierto suele reservarse para casos especialmente difíciles y confusos, en los que no puede establecerse el diagnóstico con base en la historia clínica, los estudios serológicos y la broncoscopía. Pocos pacientes que tienen neumonitis por hipersensibilidad requerirán de una biopsia a cielo abierto para establecer el diagnóstico.
Tratamiento
El tratamiento de la neumonitis por hipersensibilidad incluye principalmente suspender la exposición al agente causal. Esto es relativamente fácil en el caso de la enfermedad pulmonar por un humidificador o por cuidado de pájaros, pero es más difícil en pacientes con pulmón del granjero. En ocasiones los granjeros pueden contar con un ayudante que realice las labores que incluyan exposición al heno enmohecido. Las mejoras en las técnicas de trabajo en el campo, con mejor ventilación, y la atención cuidadosa a otros factores que puedan disminuir el contenido de humedad del heno, reducirán en forma significativa la carga antigénica. Es adecuado referir al paciente a una institución que trate enfermedad pulmonar por agricultura.
Los esteroides se utilizan principalmente para la neumonitis aguda por hipersensibilidad que se caracteriza por síntomas sistémicos graves y alteraciones en el intercambio de gases. Los pacientes con neumonitis por hipersensibilidad subaguda o crónica pueden mejorar con más rapidez si se les administra tratamiento con esteroides durante varias semanas. Mientras se administran los esteroides debe intentarse eliminar al máximo la exposición antigénica. De otro modo, los síntomas serán enmascarados por el uso de los esteroides y la exposición al antígeno aumentará la lesión pulmonar. Cuando cesa la exposición al antígeno la mayoría de los pacientes mejoran en forma espontánea. Los pacientes que están ya en la fase fibrótica de la enfermedad pueden no mejorar mucho, y existe un pequeño número de pacientes que parece empeorar a pesar de que, aparentemente, se haya eliminado el antígeno. En este último caso puede ser útil vigilar los niveles séricos de anticuerpos porque, si no existe mayor exposición, estos deben disminuir en forma gradual. Un título que no cambia o que aumenta puede sugerir que la exposición al antígeno continúa.
ENFERMEDAD PULMONAR INFILTRATIVA DIFUSA MALIGNA
La afección difusa del pulmón por una neoplasia ocurre en la carcinomatosis linfangítica, el cáncer de células alveolares, el linfoma y la leucemia. La carcinomatosis linfangítica pulmonar es causada con más frecuencia por adenocarcinomas, y menos por cánceres epidermoides. Los adenocarcinomas suelen originarse en la mama, el aparato digestivo o el pulmón. Los síntomas atribuibles al tumor linfangítico, tos y disnea, son los mismos que los de la enfermedad pulmonar infiltrativa difusa. Sin embargo, el padecimiento progresa con mucho más rapidez que otras enfermedades intersticiales pulmonares, y es rara la supervivencia por más de 3 a 6 meses. La radiografía de tórax muestra infiltrados reticulares o reticulonodulares bilaterales, y con frecuencia líneas B de Kerley. Este último dato debe hacer sospechar, en ausencia de insuficiencia cardiaca, que una neoplasia es responsable del padecimiento pulmonar. También puede observarse derrame pleural y adenopatía hiliar. Con menos frecuencia la carcinomatosis linfangítica se presenta con afección unilateral o con una radiografía de tórax totalmente normal a pesar de que la disnea y la hipoxemia sean graves. En pacientes con carcinomatosis linfangítica pero con radiografía de tórax normal, con frecuencia se encuentran células tumorales abundantes en los pequeños vasos pulmonares, que pueden en ocasiones causar hipertensión pulmonar severa (síndrome de embolia tumoral). El diagnóstico suele hacerse por broncoscopía, ya sea por LBA o con biopsia transbronquial. Un método alternativo para establecer el diagnóstico es por examen citológico de la sangre aspirada a través del cáteter arterial pulmonar en cuña. Esta técnica puede considerarse como opción para los pacientes con disnea muy severa en los que el cateterismo cardiaco derecho puede tolerarse mejor que una broncoscopía. En ocasiones se requiere biopsia abierta de pulmón para diagnosticar la carcinomatosis linfangítica.
El carcinoma de células alveolares puede manifestarse como un nódulo o masa único, nódulos múltiples, o como una enfermedad ocupativa del espacio aéreo que puede ser focal, multifocal o difusa. Cuando el tumor es difuso el diagnóstico diferencial incluye causas no malignas de ocupación alveolar difusa, como el edema pulmonar, y la proteinosis, sarcoidosis y hemorragia alveolares. El diagnóstico de carcinoma de células alveolares se establece por broncoscopía con LBA y biopsia transbronquial.
Los linfomas y la leucemia pueden causar enfermedad pulmonar infiltrativa difusa. La afección linfomatosa del parénquima pulmonar suele observarse en asociación con linfadenopatía mediastinal y evidencia de linfoma extratorácico.15 En casos raros la enfermedad pulmonar aislada es la manifestación inicial del linfoma. Cuando la leucemia subyacente está mal controlada puede observarse infiltración leucémica del pulmón, que puede contribuir a la muerte al causar insuficiencia respiratoria. El diagnóstico de infiltración leucémica o linfomatosa del pulmón puede requerir de una biopsia a cielo abierto para establecer en forma adecuada el tipo de proceso infiltrativo y excluir por completo una infección. Sin embargo, en algunos casos la broncoscopía es suficiente para establecer el diagnóstico.
Enfermedad infiltrativa difusa de causa desconocida
SARCOIDOSIS
La sarcoidosis es una enfermedad multisistémica de etiología desconocida que se caracteriza desde el punto de vista patológico por la presencia de granulomas no caseosos en varios tejidos. Aunque casi cualquier órgano puede afectarse, la gran mayoría de los pacientes solicitan atención médica por afección intratorácica que se manifiesta por síntomas respiratorios o por alteraciones asintomáticas en la radiografía de tórax. Menos del 5 porciento de los pacientes con sarcoidosis tienen una radiografía de tórax normal. Además, la morbimortalidad que se asocia con la sarcoidosis se debe principalmente a afección pulmonar. Por lo tanto, es adecuado considerar a esta enfermedad como un padecimiento de las vías respiratorias y que puede tener manifestaciones extrapulmonares.
Muchos padecimientos infecciosos y no infecciosos se asocian con formación de granulomas y un cuadro histopatológico indistinguible de sarcoidosis. Por lo tanto, aunque las características clínico-patológicas en cada paciente sean muy sugestivas de sarcoidosis, el diagnóstico requiere de la exclusión de otras enfermedades que causen granulomas. Ejemplos de padecimientos con características clínico-patológicas idénticas a las de la sarcoidosis son la beriliosis y la histoplasmosis [ver antes, Estudio del paciente con enfermedad pulmonar infiltrativa difusa].
Epidemiología
La sarcoidosis ocurre en todo el mundo y grupos étnicos, pero existen importantes diferencias regionales y étnicas en su incidencia y prevalencia. Por ejemplo, en los Estados Unidos la sarcoidosis es 10 veces más frecuente entre afroamericanos que entre blancos, además de más severa. Sin embargo, esta mayor prevalencia no se observa entre los negros en Europa. Se ha calculado que la prevalencia de la sarcoidosis en los Estados Unidos puede ser entre uno en 10,000 y uno en 2,500. La enfermedad es más frecuente en individuos entre 20 y 40 años de edad, pero puede presentarse en niños y ancianos. La sarcoidosis tiene una frecuencia muy similar en ambos sexos, con una ligera mayor prevalencia entre las mujeres. En algunos casos se ha reportado sarcoidosis familiar; con menos frecuencia se ha notificado de un matrimonio con el padecimiento, lo que podría sugerir un factor ambiental común en ciertos casos.
Patogenia
Existe una secuencia específica de eventos que causan la formación del granuloma en la sarcoidosis16 [ver figura 3]. La expansión de la población de linfocitos T cooperadores en los pulmones parece ocurrir por la activación de linfocitos T por macrófagos a través de interleucina -1 (IL-1), con la consecuente liberación de interleucina-2 (también conocida como factor de crecimiento de linfocitos T) por la célula T cooperadora. La IL-2 causa replicación de la población de células T existente. Esta población de linfocitos T cooperadores, que están muy aumentados en número y nivel de actividad, recluta monocitos de la sangre periférica hacia el pulmón al liberar un factor quimiotáctico de monocitos, participando así en la formación del granuloma. Los monocitos se convierten en macrófagos tisulares y, al final, en células epitelioides y células gigantes multinucleadas, que forman el centro del granuloma. Otro efecto de la población expandida y activada de linfocitos T cooperadores en los sitios de actividad de la enfermedad es que la estimulación local de linfocitos B produce inmunoglobulinas, que causan la hipergamaglobulinemia que con frecuencia se observa en este padecimiento.
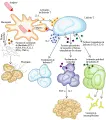
|
| Figura 3 |
| Patogenia actual de la sarcoidosis |
Manifestaciones clínicas de la sarcoidosis intratorácica
Las manifestaciones más frecuentes de la sarcoidosis intratorácica son la linfadenopatía y la enfermedad pulmonar infiltrativa difusa. Se ha creado un sistema de estadificación con base en la presencia o ausencia de estas dos manifestaciones en la radiografía: estadio I, solo linfadenopatía hiliar bilateral; estadio II, linfadenopatía hiliar bilateral y enfermedad pulmonar infiltrativa difusa; y estadio III, solo enfermedad pulmonar infiltrativa difusa [ver figura 4]. Los síntomas secundarios a la afección del parénquima pulmonar incluyen disnea y tos. Se encuentran alteraciones en la función respiratoria en casi todos los pacientes sintomáticos y en algunos asintomáticos. Las anomalías fisiológicas en los enfermos con sarcoidosis sintomática consisten en forma típica en reducción de la Dlco y de la capacidad vital sin obstrucción al flujo del aire. La Dlco suele alterarse antes de la capacidad vital, pero ambas medidas se modifican en las personas con síntomas moderados a severos. La obstrucción al flujo del aire es relativamente rara, excepto en pacientes con enfermedad avanzada o con afección endobronquial de vías respiratorias grandes. En casos raros los granulomas endobronquiales difusos en las vías respiratorias pequeñas causan una alteración de predominio obstructivo en pacientes con enfermedad en estadio I.
|
|
|
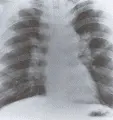
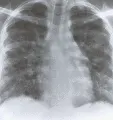
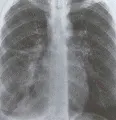
La frecuencia de los síntomas es más alta en pacientes con enfermedad en estadio II, pero los casos asintomáticos no son raros. Los síntomas pueden ser principalmente de tipo general, como en el estadio I, o pueden originarse de la afección pulmonar. No es raro que existan alteraciones radiográficas extensas asociadas con síntomas respiratorios mínimos. Alrededor del 50 porciento de los pacientes con sarcoidosis en estadio II tendrán remisión a los 2 años después del inicio. Existe más probabilidad de que la enfermedad en estadio II sea progresiva y siga una evolución sintomática crónica que la enfermedad en estadio I.
En el 5 a 15 porciento de los pacientes se encuentra enfermedad en estadio III en el momento del diagnóstico. Los síntomas respiratorios son frecuentes pero, como en la enfermedad en estadio II, la radiografía de tórax puede verse mucho peor que el paciente. Sólo una tercera parte de los pacientes con sarcoidosis en estadio III tendrán remisión a los 2 años después de su presentación. Es frecuente la evolución crónica que causa fibrosis pulmonar. Ciertas manifestaciones extrapulmonares de la sarcoidosis crónica, como la enfermedad cutánea infiltrativa, son mucho más frecuentes en los pacientes con enfermedad en estadio III que en los enfermos en estadio I o II. La evolución crónica de la enfermedad en estadio III no es sorprendente porque los pacientes que son diagnosticados en esta etapa pueden representar a los que antes tuvieron linfadenopatía hiliar bilateral (estadios I y II) y que no tuvieron una remisión espontánea.
Entre las manifestaciones poco usuales de la sarcoidosis intratorácica se incluyen el derrame pleural, los infiltrados alveolares, las opacidades nodulares grandes, la cavitación, las atelectasias y la calcificación. La afección pleural ocurre en 1 a 4 porciento de los pacientes y consiste en derrame pleural o engrosamiento de la pleura visceral y parietal. Los derrames pocas veces son importantes y suelen contener un alto porcentaje de linfocitos. La biopsia pleural demuestra granulomas, y el principal diagnóstico diferencial a tener en cuenta en pacientes con sarcoidosis pleural es la tuberculosis. La sarcoidosis alveolar tiene varias presentaciones que van desde infiltrados en parches en pacientes asintomáticos hasta áreas extensas de consolidación del espacio aéreo en enfermos con insuficiencia respiratoria. Pueden observarse nódulos de 2 a 10 cm de diámetro, que en algunas ocasiones se cavitan. El diagnóstico diferencial de la sarcoidosis nodular incluye a las infecciones micóticas, la tuberculosis y, en especial, los tumores metastásicos. Las atelectasias son causadas por sarcoidosis endobronquial. La calcificación de los ganglios linfáticos es una manifestación tardía de la enfermedad, que se observa en el 5 porciento de los pacientes.
Es probable que la granulomatosis sarcoide necrosante, caracterizada por masas de granulomas confluentes con cierto grado de vasculitis, sea una variante de la sarcoidosis. Ambos trastornos parecen tener muchas características en común, incluyendo linfadenopatía hiliar e inflamación granulomatosa extrapulmonar.
Manifestaciones clínicas de la sarcoidosis extratorácica
Casi cualquier órgano puede afectarse en la sarcoidosis. Aunque la sarcoidosis extratorácica es mucho menos frecuente como causa de morbilidad que la intratorácica, puede dominar el cuadro clínico en algunos pacientes.
El eritema nodoso, una paniculitis no granulomatosa, es la manifestación cutánea más frecuente de la sarcoidosis. Más del 90 porciento de los pacientes con sarcoidosis que desarrollan eritema nodoso tienen una radiografía en estadio I; el otro 10 porciento está en estadio II. Ocurren lesiones cutáneas granulomatosas en el 10 a 30 porciento de los pacientes, en especial en los afroamericanos en los Estados Unidos. El lupus pernio es una lesión amoratada y edematosa sobre la nariz, mejillas, pabellones auriculares, dedos de manos y pies, labios o rodillas, que puede desfigurar a la persona. Las placas cutáneas tienden a ser violáceas, angulares y planas, con borde elevado. También pueden observarse placas semejantes a psoriasis. Los nódulos pueden ser elevados o encontrarse dentro del tejido subcutáneo. Como regla, estas manifestaciones cutáneas granulomatosas de la sarcoidosis se observan en la enfermedad pulmonar en estadio III que es crónica y persistente.
Ocurre afección ocular en alrededor del 25 porciento de los pacientes con sarcoidosis. El tracto uveal es el más afectado, y la sarcoidosis es responsable del dos a cuatro porciento de todos los casos de uveítis. La enfermedad del tracto uveal anterior con uveociclitis suele manifestarse por epífora, fotofobia y poco o ningún dolor. El síndrome de Heertford, o fiebre uveoparotídea, es una manifestación poco frecuente de la sarcoidosis que consiste en uveítis, crecimiento parotídeo, parálisis de nervios craneales, meningitis subaguda y síntomas sistémicos. La uveítis posterior (coriorretinitis) causa visión borrosa, o puede ser asintomática. La coriorretinitis puede ser difícil de detectar cuando existe uveítis anterior.
El sistema nervioso se afecta en menos del 5 porciento de los pacientes con sarcoidosis. Las manifestaciones del sistema nervioso central incluyen meningitis crónica, encefalopatía, lesiones hipotalámicas, afección de nervios craneales y crisis convulsivas. Puede observarse neuropatía periférica sensorial y motora. La meningitis crónica en la sarcoidosis puede ser difícil de distinguir de la tuberculosa o la micótica y suele asociarse con linfocitosis en el líquido cefalorraquídeo (LCR), aumento en las proteínas del LCR y, en ocasiones, hipoglucorraquia. La sarcoidosis puede también causar masas que ocupan espacio en diversas localizaciones, provocando diferentes alteraciones neurológicas.
Se encuentra evidencia de sarcoidosis cardiaca en la necropsia con mucho más frecuencia de lo que se sospecha por las manifestaciones clínicas. Las alteraciones del ritmo son la manifestación más frecuente de la sarcoidosis cardiaca. Pueden ocurrir taquiarritmias ventriculares y bloqueo cardiaco completo, que pueden ocasionar muerte súbita. Otros datos menos frecuentes de sarcoidosis cardiaca incluyen insuficiencia cardiaca, enfermedad pericárdica, disfunción del músculo papilar y aneurismas ventriculares en ausencia de enfermedad coronaria. El ecocardiograma, la vigilancia con Holter y el gamagrama con talio-201 puede mostrar alteraciones, pero no necesariamente indican que existen granulomas en el miocardio. Deben tenerse en cuenta factores como la edad y otros padecimientos concomitantes al evaluar la posibilidad de sarcoidosis miocárdica.
Se presenta afección ósea o articular en 1 a 10 porciento de los pacientes. La enfermedad ósea se caracteriza por lesiones quísticas, y tiende a restringirse a los dedos de manos y pies. Puede observarse edema digital. La sarcoidosis sinovial puede presentarse como una artritis crónica mono o poliarticular.
La afección de las vías respiratorias superiores suele manifestarse por síntomas nasales o faríngeos. La epistaxis y la congestión nasal, que pueden confundirse con una rinitis alérgica o vasomotora, son síntomas frecuentes. Debe pensarse en sarcoidosis cuando la obstrucción nasal sea refractaria al tratamiento convencional. La sarcoidosis laríngea afecta a las estructuras supraglóticas, y en pocas ocasiones a las cuerdas vocales. La disfonía es el síntoma más común, pero puede ocurrir también obstrucción de las vías respiratorias superiores.
Las alteraciones endócrinas en la sarcoidosis incluyen hipercalcemia y disfunción hipofisiaria, que puede presentarse como diabetes insípida. Se observa hipercalcemia con mucho menos frecuencia que hipercalciuria. La hipercalcemia e hipercalciuria en la sarcoidosis parecen ser causadas por una mayor síntesis de 1,25-dihidroxivitamina D [1,25-(OH)2D] por macrófagos activados en los granulomas, que ocasiona un aumento secundario en la absorción de calcio en el intestino.
Diagnóstico
El diagnóstico de sarcoidosis se establece por la demostración de granulomas no caseosos en el tejido [ver figura 5] y la exclusión de otras causas de granulomas. La sarcoidosis intratorácica se diagnostica con más frecuencia por medio de una broncoscopía con biopsia transbronquial. La utilidad de la biopsia transbronquial depende del estadio radiográfico. En los pacientes con infiltrados pulmonares (estadios II y III) la biopsia pulmonar transbronquial demostrará granulomas no caseosos en alrededor del 90 porciento de los casos. En la enfermedad en estadio I se obtiene positividad en el 60 a 70 porciento de los casos. Si no logra establecerse el diagnóstico por este método, está indicado realizar una mediastinoscopía, que dará el diagnóstico en el 95 al 100 porciento de los casos. La biopsia de tejidos extrapulmonares puede utilizarse para el diagnóstico cuando tiene indicación clínica (v.gr., en pacientes con crecimiento de ganglios linfáticos periféricos o lesiones cutáneas).
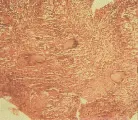
|
| Figura 5 |
| Sarcoidosis: granulomas no caseosos |
Cuando se encuentran granulomas no caseosos en el tejido deberán siempre tenerse en cuenta otras posibles causas de la formación de granulomas. Las causas infecciosas de granulomas en los pulmones y ganglios linfáticos mediastinales son de las más importantes, en especial la tuberculosis y la histoplasmosis. Está indicado realizar una prueba cutánea de tuberculina, estudios serológicos para hongos y tinciones especiales para el estudio del material de biopsia. Por ejemplo, los hallazgos como adenopatía hiliar, infiltrados pulmonares y granulomas no caseosos en los pulmones pueden orientar a un diagnóstico precipitado de sarcoidosis antes de que estén los resultados de los estudios especiales. Si se detecta un resultado positivo en la prueba de fijación del complemento para histoplasmosis, el diagnóstico deberá modificarse a histoplasmosis.
En el momento de la broncoscopía suele realizarse un LBA además de la biopsia. En la sarcoidosis activa el líquido del lavado suele mostrar mayor número de linfocitos, y la relación entre linfocitos T cooperadores y supresores-citotóxicos está aumentada. Otros padecimientos pulmonares, tanto infecciosos como no infecciosos, pueden asociarse con linfocitosis, por lo que este dato no tiene especificidad diagnóstica. La neumonitis por hipersensibilidad puede confundirse con sarcoidosis porque ambos padecimientos se asocian con linfocitosis en el líquido del lavado y granulomas no caseosos en el tejido. Sin embargo, es típico observar inversión en la relación entre los linfocitos T cooperadores y supresores-citotóxicos en la neumonitis por hipersensibilidad.
Por lo general, la concentración de ECA aumenta en la sarcoidosis activa. Se ha sugerido que la medición de la concentración de la ECA es de utilidad en el diagnóstico de sarcoidosis. Sin embargo, no es claro si esta prueba tiene un valor diagnóstico extra sobre los criterios patológicos descritos antes. No se recomienda hacer el diagnóstico de sarcoidosis con base sólo en la concentración de la ECA, sin la confirmación del diagnóstico por demostración de granulomas no caseosos en el tejido. También se ha detectado aumento en los niveles de ECA en la tuberculosis miliar, la lepra, la cirrosis biliar primaria, la silicosis y la asbestosis. Con excepción de la tuberculosis miliar, no es probable que estas enfermedades se confundan clínicamente con la sarcoidosis. La medición periódica de la concentración de la ECA puede ser útil porque correlaciona con la actividad de la enfermedad.
El diagnóstico diferencial de la sarcoidosis en estadio I incluye principalmente al linfoma y a las infecciones granulomatosas como la tuberculosis y la histoplasmosis. Son causas menos frecuentes de linfadenopatía hiliar bilateral los cánceres metastásicos (en especial el de células renales) y la amiloidosis. La gran mayoría de los pacientes que tienen linfoma que se manifiesta como linfadenopatía hiliar bilateral tendrá linfadenopatía periférica, esplenomegalia o síntomas sistémicos, como fiebre, pérdida de peso o diaforesis nocturna. Los adultos jóvenes que presentan adenopatía hiliar bilateral asintomática, sin linfadenopatía periférica ni esplenomegalia, es muy poco probable que tengan un linfoma. Por lo tanto, en estas circunstancias es razonable hacer el diagnóstico presuntivo de sarcoidosis en estadio I sin confirmarlo examinando una muestra de tejido. Sin embargo, deberá realizarse una prueba cutánea de tuberculina y estudios serológicos para hongos, lo mismo que vigilancia clínica y radiográfica para asegurarse de que no existe progresión de la enfermedad. Si se observa progresión, es indispensable realizar una biopsia de tejido para confirmar el diagnóstico.
Tratamiento
La administración de esteroides es el tratamiento estándar para la sarcoidosis sintomática, aunque no existen prueba definitivas de que el tratamiento influya sobre la evolución a largo plazo. La mayoría de los pacientes responden muy bien al tratamiento, y con frecuencia las dosis bajas de prednisona (v.gr., 15 mg cada tercer día) son eficaces para suprimir la actividad de la enfermedad. Suele administrarse una dosis más alta (v.gr., 40 a 60 mg/día) durante algunas semanas para inducir remisión de la actividad de la enfermedad. La principal indicación para los esteroides es la afección pulmonar sintomática. Otras indicaciones incluyen síntomas generales importantes (v.gr., fiebre o pérdida de peso), hipercalcemia y afección de tejidos extrapulmonares que causen deterioro funcional o riesgo de éste. No se ha establecido qué tiempo debe durar el tratamiento. En la práctica se trata a la sarcoidosis pulmonar con prednisona durante un año, y después se disminuye progresivamente el medicamento, observando si la enfermedad ha remitido. Las recurrencias se tratan de modo semejante, con dosis más altas al principio, que después se reducen hasta una dosis de mantenimiento. Algunos pacientes presentarán recurrencias frecuentes al suspender la prednisona, por lo que requerirán un tratamiento supresor con dosis bajas en forma indefinida.
Si está absolutamiente contraindicado el uso de esteroides o si el paciente no mejora con el tratamiento esteroideo, puede intentarse la administración de metotrexate17 o de clorambucil. Con frecuencia la cloroquina es útil en los pacientes con enfermedad cutánea.
FIBROSIS PULMONAR IDIOPATICA
La fibrosis pulmonar idiopática (alveolitis fibrosante) es una de las causas más frecuentes de enfermedad pulmonar infiltrativa difusa crónica.18 Aunque los datos clínicos, radiográficos, fisiológicos e histopatológicos de este padecimiento son característicos, ninguno es patognomónico. Por lo tanto, el diagnóstico de FPI requiere no sólo de un cuadro clinicopatológico compatible, sino también de la exclusión de todas las otras causas de enfermedad difusa. Por ejemplo, las manifestaciones pulmonares de la FPI pueden ser indistinguibles de la enfermedad difusa asociada a los padecimientos del tejido conjuntivo, la asbestosis o la fibrosis pulmonar inducida por quimioterapia.
Patogenia
La patogenia de la fibrosis pulmonar idiopática no está bien establecida. Se ha sospechado una causa viral en algunos casos porque una minoría de los pacientes relacionan el inicio de sus síntomas respiratorios con una enfermedad gripal. También puede ser importante la susceptibilidad genética. Es posible que la FPI se presente cuando un individuo genéticamente susceptible se exponga a un agente viral o ambiental que desencadene una cascada de eventos inflamatorios, inmunológicos y de fibrosis en el pulmón.
La alveolitis de la FPI temprana se caracteriza principalmente por aumento en el número de neutrófilos activados y la presencia de macrófagos en el líquido del LBA. Los neutrófilos pueden ser atraídos hacia el pulmón por un desequilibrio entre las sustancias quimiotácticas derivadas de macrófagos (v.gr., factor de necrosis tumoral alfa [FNT-alfa], IL-8) y sustancias antinflamatorias (v.gr., interferón gama, IL-10).19 En algunos casos las sustancias quimiotácticas puedn ser liberadas en respuesta a la presencia de complejos inmunes locales. Se han detectado complejos inmunes en el tejido pulmonar, en el líquido de lavado y en sangre periférica en pacientes que tienen una alveolitis neutrofílica intensa. Una vez en el pulmón, los neutrófilos pueden causar lesión tisular local al liberar varias sustancias potencialmente tóxicas, como radicales de oxígeno y enzimas proteolíticas. La inflamación pulmonar puede ir seguida eventualmente por fibrosis extensa. Es posible que la inflamación neutrofílica crónica de la FPI tenga un papel en el desarrollo de la fibrosis pulmonar. Sin embargo, el proceso fibrogénico puede ser secundario en gran parte a la actividad de los macrófagos pulmonares. Los macrófagos de los pacientes con fibrosis pulmonar idiopática liberan en forma espontánea factores de crecimiento para fibroblastos, como el factor transformador de crecimiento-beta, causando un aumento en la población de fibroblastos en el pulmón. Además, en este padecimiento el macrófago libera fibronectina, una sustancia que promueve la fijación y migración de los fibroblastos. Estos macrófagos liberan en forma espontánea factores estimuladores de fibroblastos, como IL-1ß y factor de necrosis tumoral-alfa, que causan que los fibroblastos secreten mayores cantidades de constituyentes de la matriz extracelular. Por lo tanto, los macrófagos alveolares parecen tener un papel crucial en el desarrollo de la FPI.
Manifestaciones clínicas
Como en otros tipos de enfermedad pulmonar infiltrativa difusa, la disnea con el ejercicio y la tos son los síntomas más importantes. El examen físico suele demostrar estertores inspiratorios finos (estertores de Velcro). Puede observarse cianosis si la hipoxemia es severa, y las características del cor pulmonale (v.gr., aumento en la presión venosa yugular, edema y un segundo ruido cardiaco pulmonar prominente) indican enfermedad avanzada. Se observan dedos en palillo de tambor en el 40 al 75 porciento de los casos, aunque casi nunca en las etapas tempranas del padecimiento. No existen otras manifestaciones extratorácicas de la enfermedad. La evidencia clínica de enfermedad extrapulmonar (v.gr., artritis, enfermedad cutánea o serositis) sugiere un padecimiento sistémico, como sarcoidosis o alguna enfermedad del tejido conjuntivo, más que FPI.
Las características radiográficas son inespecíficas y suelen consistir en un infiltrado reticular o reticulonodular bilateral que tiene predilección por la porción inferior de los campos pulmonares. La llamada opacidad en vidrio despulido, que indica un proceso de ocupación acinar, puede observarse en la neumonitis intersticial descamativa (ver adelante). La radiografía de tórax es normal hasta en el 10 porciento de los pacientes con FPI sintomática. Es probable que la TCAR demuestre alteraciones en los casos en que la radiografía de tórax es normal. En la enfermedad avanzada se observa el llamado pulmón en panal de abeja, que indica fibrosis pulmonar en estadio terminal. Los hallazgos radiográficos en la FPI se limitan a los campos pulmonares.
Las alteraciones fisiológicas de la fibrosis pulmonar idiopática son básicamente las mismas que las descritas para la enfermedad pulmonar infiltrativa difusa en general. El cuadro fisiológico típico consiste en reducción de la DLCO, restricción de los volúmenes pulmonares, desaturación de oxígeno inducida por ejercicio y ausencia de obstrucción a la circulación del aire. Algunas alteraciones correlacionan con los hallazgos patológicos.20 Por ejemplo, la reducción en la DLCO se relaciona con cambios descamativos, y la disminución en la capacidad pulmonar total con la celularidad global. Aunque muy significativas desde el punto de vista estadístico, la dispersión es demasiado grande como para que estas correlaciones sean útiles en la clínica.
Las características histopatológicas son inespecíficas: se observan datos histopatológicos semejantes en la enfermedad pulmonar infiltrativa difusa asociada con enfermedades del tejido conjuntivo, diversos tipos de enfermedad difusa asociada a fármacos y otros padecimientos. El infiltrado de células inflamatorias y la fibrosis crónica, que son los datos patológicos claves en la FPI, representan reacciones comunes a diversos agentes.
Se han observado dos patrones histopatológicos básicos en la fibrosis pulmonar idiopática: la neumonitis intersticial habitual (NIH) y la neumonitis intersticial descamativa (NID). La NIH, el patrón más frecuente, se caracteriza por grados diversos de un infiltrado celular inflamatorio, intersticial y redondeado, engrosamiento de los septos alveolares y fibrosis [ver figura 6a]. La distribución de la lesión es irregular: áreas de inflamación intensa y fibrosis que pueden coexistir con áreas de pulmón casi normal en la misma muestra tomada de una biopsia a cielo abierto. La NID se caracteriza por un patrón más o menos homogéneo en el que los espacios alveolares se llenan por macrófagos alveolares [ver figura 6b].
|
|
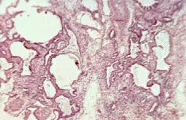
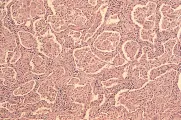
Diagnóstico
El diagnóstico de FPI se establece por la correlación entre los datos histopatológicos y clínicos. Desde el punto de vista clínico se sospecha FPI cuando ocurre una enfermedad pulmonar infiltrativa difusa sin afección de otros órganos y sin relación aparente a infecciones, exposición ambiental o medicamentos. La radiografía de tórax no muestra evidencia de adenopatía hiliar o derrame pleural. Las pruebas de laboratorio en la fibrosis pulmonar idiopática suelen ser irrelevantes, excepto por la presencia de anticuerpos antinucleares positivos o factor reumatoide positivo en hasta el 50 porciento de los casos. Por lo tanto, los anticuerpos antinucleares y el factor reumatoide séricos no son útiles para diferenciar entre la fibrosis pulmonar idiopática y la enfermedad infiltrativa difusa de los padecimientos del tejido conjuntivo.
El enfoque inicial del diagnóstico en la FPI consiste en excluir la mayoría de las causas posibles de enfermedad pulmonar infiltrativa difusa usando la historia clínica y el examen físico. Sin embargo, existen ciertos padecimientos que no pueden descartarse en forma certera sin una biopsia pulmonar. Entre estos se incluyen la sarcoidosis en estadio III, la carcinomatosis linfangítica, la linfangiomatosis, la histiocitosis X y las formas más difusas de bronquiolitis obliterante con neumonía organizada (BONO). La TCAR puede ser útil en esta fase de la evaluación porque el patrón de las alteraciones observadas en la FPI es específico, y muchas de las otras causas tienen patrones característicos y muy diferentes.21 Además, la TCAR en ocasiones sugiere si el patrón patológico es de NIH o de NID.
La biopsia pulmonar transbronquial es una técnica muy sensible para el diagnóstico de sarcoidosis y cáncer linfangítico, y puede demostrar datos patológicos sugestivos de fibrosis pulmonar idiopática (i.e., fibrosis y ensanchamiento de los septos alveolares con un infiltrado de células inflamatorias). Se requiere de la biopsia pulmonar a cielo abierto para excluir otras causas de enfermedad difusa. Será decisión del clínico realizar una biopsia a cielo abierto en los casos en que la evolución clínica y la biopsia transbronquial sean muy sugestivas de FPI. Se recomienda realizar la biopsia cuando existen dudas respecto a la presencia de una causa infecciosa. También debe practicarse este tipo de biopsia en pacientes jóvenes para establecer el diagnóstico del padecimiento subyacente con un grado de certeza razonable. Por otro lado, en los pacientes mayores que tienen datos clínicos típicos y una biopsia transbronquial compatible puede evitarse la biopsia abierta de pulmón por la morbilidad que causa. En ciertos casos puede estar justificado diagnosticar una FPI sin una biopsia tisular. Por ejemplo, no se requerirá de la biopsia para el diagnóstico en un paciente de edad avanzada que tiene disnea progresiva al ejercicio de meses a años de duración, estertores en Velcro y dedos en palillo de tambor a la exploración física, infiltrados intersticiales gruesos bilaterales, un título bajo de anticuerpos antinucleares y ninguna evidencia de un padecimiento de tipo ambiental o relacionado a medicamentos.
Pronóstico y tratamiento
La mortalidad a 5 años para los pacientes con fibrosis pulmonar idiopática es de alrededor del 50 porciento. La mayor parte de las defunciones son causadas por insuficiencia respiratoria progresiva. Se emplea la prednisona para tratar la enfermedad progresiva y alrededor del 20 porciento de los pacientes que la reciben muestran mejoría sintomática, radiográfica y funcional. Sin embargo, incluso los que responden no tienen una mejoría muy significativa, a diferencia de los pacientes con sarcoidosis sintomática que son tratados con esteroides. La respuesta a la prednisona es mejor si la fibrosis es menos evidente que la inflamación en la biopsia a cielo abierto, pero la respuesta suele ser mala si predomina la fibrosis. Sin embargo, debido a que la afección pulmonar es heterogénea, los datos histopatológicos pueden variar en diferentes regiones del organismo. Por lo tanto, no está indicado realizar una biopsia a cielo abierto para decidir si tratar o no a un paciente sintomático con esteroides. Los estudios sobre la relación entre los resultados de la TCAR y la mejoría en la función pulmonar sugieren que la extensión del patrón de atenuación en vidrio despulido identifica al cuadro patológico de la NID, que correlaciona con respuesta a los esteroides. Uno de los mejores factores de predicción de la evolución a largo plazo es la respuesta a corto plazo (6 a 8 semanas) a la prednisona, quienes responden tienen mejor pronóstico. Se ha usado la ciclofosfamida y la azatioprina aunadas a la prednisona, por lo general cuando la prednisona por sí sola ha sido ineficaz. Los factores de predicción de una respuesta benéfica al tratamiento citotóxico incluyen breve duración de los síntomas y una buena respuesta temporal al tratamiento esteroideo.22 En ocasiones los pacientes que no mejoran con el esteroide sí lo hacen con el tratamiento inmunosupresor combinado o con colchicina.23 También se han realizado trasplantes de un solo pulmón con éxito para la fibrosis en estadio terminal.
ENFERMEDADES DEL TEJIDO CONJUNTIVO
Con frecuencia las enfermedades del tejido conjuntivo afectan el pulmón. Hasta dos terceras partes de los pacientes tienen evidencia clínica de afección pleuropulmonar, y casi todos tienen alteraciones en la autopsia. El mecanismo inmunológico parece ser el depósito de complejos inmunes, pero la evidencia de esto es más convincente en algunos padecimientos que en otros. Las pruebas de función pulmonar en los pacientes con infiltrados intersticiales y fibrosis muestran restricción, obstrucción de las vías respiratorias de pequeño calibre y reducción de la DLCO con hipoxemia al reposo que empeora durante el ejercicio. La TCAR del tórax suele detectar enfermedad intersticial sutil que no se observa en la radiografía de tórax de rutina. Aunque la enfermedad intersticial crónica que se observa en estos padecimientos suele ser indistinguible desde el punto de vista patológico de la fibrosis pulmonar idiopática, los pacientes con una enfermedad del tejido conjuntivo suelen tener mejor pronóstico que los que padecen FPI,24 esto es cierto incluso en los pacientes en los que se desarrolla una enfermedad de tejido conjuntivo después de la FPI.25
Lupus eritematoso generalizado
El lupus eritematoso generalizado es uno de los padecimientos del tejido conjuntivo más prevalentes, en especial entre mujeres afroamericanas (en Estados Unidos) en edad reproductiva. Sin embargo, hasta el 18 porciento de los casos ocurren después de la quinta década de la vida. El LEG inducido por medicamentos es un problema clínico común, sobre todo entre ancianos con cardiopatía. Los fármacos que con más frecuencia causan LEG incluyen la hidralazina, la procainamida, la isoniacida, la fenitoína, la quinidina, la metildopa y varios de los agentes bloqueadores beta. Se han implicado otros medicamentos, pero las evidencias son menos claras.
Con frecuencia ocurren complicaciones pleuropulmonares en el LEG (en 38 a 89 porciento de los pacientes), que deben diferenciarse de infecciones. La enfermedad pleural, las atelectasias, las neumonitis lúpicas agudas y la hemorragia masiva son los problemas clínicos más comunes. Además, la enfermedad pleuropulmonar puede ser la manifestación inicial en el LEG inducido por medicamentos.
La neumonitis lúpica aguda se manifiesta por inicio súbito de tos con disnea, y en la radiografía aparecen opacidades basales en parches. En ocasiones la radiografía muestra solo sombras lineales horizontales que se han atribuido a pequeñas áreas de atelectasia. Los pacientes con esta imagen suelen responder bien al uso de esteroides.
Los pacientes con LEG pueden sufrir hemorragia alveolar masiva o hemorragia oculta durante la evolución de su enfermedad. El LEG severo se asocia con una mortalidad del 70 porciento. Los síntomas consisten en tos y disnea con o sin hemoptisis y la radiografía de tórax muestra infiltrados alveolares esponjosos que pueden ser en parches o confluentes. El tratamiento es semejante al del síndrome de Goodpasture e incluye plasmaféresis e inmunosupresores.
Artritis reumatoide
Aunque la incidencia de artritis reumatoide es más alta en mujeres que en varones, las complicaciones pleuropulmonares son más frecuentes en éstos últimos. Este grupo de complicaciones son clínicamente aparentes en el 14 porciento de los enfermos en los primeros 2 años de evolución26 y en alrededor del 50 porciento durante el curso de la enfermedad; un porcentaje mucho mayor tiene afección desde el punto de vista patológico. En casos ocasionales la afección torácica precede a las lesiones articulares, lo que dificulta el diagnóstico.
Los problemas pleuropulmonares más frecuentes son enfermedad pulmonar intersticial, enfermedad pleural y nódulos reumatoides parenquimatosos. Estos problemas suelen asociarse con la presencia de artritis activa, títulos altos de factor reumatoide, complejos inmunes circulantes y crioglobulinemia.
La neumonitis intersticial difusa con fibrosis es el problema pulmonar más frecuente y serio en los pacientes con artritis reumatoide. La disnea, en ocasiones con tos, es el síntoma más frecuente. Pueden existir dedos en palillo de tambor. La radiografía de tórax evoluciona de nodularidad fina a reticulación gruesa y, al final, a un patrón en panal de abeja. Los resultados del LBA pueden ser anormales, demostrando aumento en el número de linfocitos, neutrófilos y eosinófilos, o los tres, pero este hallazgo no parece ayudar en el tratamiento. En los pacientes con enfermedad intersticial que se caracteriza por biopsia con intensa celularidad o la presencia de síntomas progresivos deben administrarse esteroides.
Pueden encontrarse nódulos reumatoides (necrobióticos) en muchos órganos, incluyendo el parénquima pulmonar, la mucosa endobronquial y la pleura, y en cualquiera de estos sitios pueden cavitarse. Las lesiones tienen una apariencia histopatológica característica y, a menos que se realice biopsia por punción percutánea transtorácica o biopsia abierta de pulmón, pueden ser difíciles de distinguir de neoplasias o tuberculosis. La mayoría de los nódulos reumatoides no causan síntomas, y rara vez se requiere tratamiento si ya se ha establecido el diagnóstico.
Esclerosis sistémica progresiva
La esclerosis sistémica progresiva (ESP), o esclerodermia, es un trastorno que predomina en las mujeres en la cuarta a sexta décadas de la vida. El pronóstico es desfavorable, en especial entre las personas de origen afroamericano, los varones y los enfermos con daño pulmonar. Con frecuencia se desarrollan complicaciones pulmonares, sobre todo enfermedad pulmonar intersticial, que ocurren hasta en el 90 porciento de los pacientes en quienes se realizan pruebas de función pulmonar o estudios histológicos. La enfermedad pulmonar intersticial en la ESP se caracteriza por infiltrados reticulares basales o reticulonodulares que se vuelven más prominentes conforme progresa la enfermedad. Además, la radiografía puede mostrar reducción del volumen con el tiempo, y puede ocurrir neumotórax. La evidencia de hipertensión pulmonar puede ser mayor de la esperada por el grado de alteración radiológica o de alteración en las pruebas de función respiratoria, en especial si el fenómeno de Raynaud es parte del síndrome clínico.
Se piensa que los esteroides no son útiles como tratamiento de la enfermedad pulmonar intersticial en la ESP; la ciclofosfamida puede ser eficaz en los pacientes que tienen alveolitis activa por LBA.27 Como resultado de los trastornos en la motilidad del esófago puede ocurrir aspiración de contenido gástrico y esofágico, sobre todo cuando los pacientes están en posición de decúbito. La aspiración puede evitarse elevando la porción superior del tronco hasta una posición casi vertical.
El síndrome de CREST (calcinosis, fenómeno de Raynaud, trastornos esofágicos, esclerodactilia y telangiectasias) es una variante de la ESP. Los pacientes con síndrome de CREST tienen enfermedad vascular pulmonar prominente. La hipertensión pulmonar puede asociarse con fibrosis intersticial, como en la ESP, o puede ocurrir como un evento aislado.
Polimiositis y dermatomiositis
La polimiositis y la dermatomiositis son padecimientos autoinmunes que se caracterizan por debilidad y en ocasiones por dolor en los músculos proximales y del cuello, erupciones dermatológicas (dermatomiositis) y neoplasias. La complicación pulmonar más frecuente de la poli-dermatomiosisits es la insuficiencia respiratoria causada por debilidad de los músculos respiratorios y la aspiración secundaria a debilidad faríngea posterior y esofágica proximal.
Se observa enfermedad pulmonar intersticial en una minoría de los pacientes, que puede asociarse con anticuerpos anti-Jo-1, un autoanticuerpo que es específico contra la enzima celular histidil-ARNt sintetasa. Estos pacientes pueden tener NOBO, fibrosis pulmonar o daño alveolar difuso.28 A diferencia de la enfermedad intersticial asociada con muchas de las otras enfermedades del tejido conjuntivo, la asociada con poli-dermatomiositis responde bien al tratamiento esteroideo.
Síndrome de Sjögren
El síndrome de Sjögren consiste en la triada de queratoconjuntivitis sicca, xerostomía y edema recurrente de las glándulas parótidas, y con frecuencia (en el 60 porciento de los casos) se asocia con otra enfermedad del tejido conjuntivo. Este síndrome incluye con frecuencia problemas pleuropulmonares, que en la mayoría de los casos son secundarios a la enfermedad del tejido conjuntivo subyacente. Las complicaciones torácicas que pueden relacionarse en forma directa con el síndrome de Sjögren incluyen infiltrados intersticiales, con frecuencia de tipo linfocítico, pleuresía con o sin derrame, bronquiolitis folicular y sequedad de las vías traqueobronquiales, produciendo bronquiectasias y bronquitis o neumonitis recurrente.
La disnea es el síntoma principal de la enfermedad intersticial asociada con el síndrome de Sjögren, mientras que la disfonía y la tos productiva con expectoración espesa son datos de afección laríngea y traqueobronquial. La radiografía de tórax puede mostrar infiltrados intersticiales con un componente nodular prominente. Estos infiltrados tienen un patrón linfoplasmacitario que puede ser difícil de distinguir del observado en pacientes con linfoma. Los resultados de las pruebas de función respiratoria demuestran patrones restrictivos, obstructivos o mixtos. La neumonitis intersticial linfocítica y la bronquiolitis asociadas con síndrome de Sjögren responden al tratamiento esteroideo o al inmunosupresor.29
Enfermedad mixta del tejido conjuntivo
Varios reportes han descrito pacientes que tienen características de LEG, ESP y polimiositis. Una característica de esta enfermedad mixta del tejido conjuntivo es la presencia de títulos altos de anticuerpos contra ribonucleoproteínas nucleares extraíbles (anti-nRNP). Este síndrome suele ser una enfermedad relativamente benigna que responde bien a los esteroides, aunque algunos casos tienen una evolución menos benigna, algunos de los cuales se han asociado con enfermedad pulmonar intersticial difusa o hipertensión pulmonar.
GRANULOMA EOSINOFILICO DEL PULMON
El granuloma eosinofílico del pulmón, o histiocitosis X del pulmón, es una causa poco frecuente de enfermedad pulmonar infiltrativa difusa, y afecta principalmente a individuos entre 20 y 50 años de edad. La enfermedad es muy poco frecuente en afroamericanos y extraordinariamente rara en asiáticos. La etiología del granuloma eosinófilo es deconocida, pero existe una intensa asociación con el uso de cigarrillos. En los niños y los adultos jóvenes el granuloma eosinófilo es un padecimiento sistémico con apariencia histológica semejante, y se puede observar diabetes insípida por afección hipofisiaria o granuloma eosinofílico del hueso en el 15 al 20 porciento de los pacientes.
La mayoría de los enfermos con granuloma eosinófilo pulmonar tienen tos o disnea, pero hasta una cuarta parte de los individuos afectados tienen sólo alteraciones radiográficas asintomáticas. Entre el 10 y 20 porciento de los enfermos desarrollan neumotórax, y ésta puede ser la manifestación inicial del padecimiento. La asociación de neumotórax espontáneo con evidencia radiográfica de infiltrados intersticiales difusos sugiere el diagnóstico de granuloma eosinófilo o de neumonía por P. carinii asociada a SIDA.
El patrón radiográfico o por TCAR del granuloma eosinófilo es variable. En la etapa temprana de la enfermedad son típicas las opacidades nodulares y en vidrio despulido con quistes de paredes gruesas. Después se observan quistes de paredes delgadas, opacidades lineales y lesiones enfisematosas.30
El granuloma eosinófilo causa con frecuencia reducción en la DLCO y en la capacidad vital. Sin embargo, a diferencia de la mayoría de las principales causas de enfermedad pulmonar infiltrativa difusa, la obstrucción al flujo del aire es un dato característico, sobre todo en las etapas avanzadas de la enfermedad. La alta incidencia de obstrucción al flujo del aire puede ser secundaria a afección bronquiolar en las primeras fases de la enfermedad y a bulas en las etapas tardías. El tabaquismo también puede ser un factor que contribuye a la frecuencia de obstrucción aérea en este padecimiento. Es frecuente el deterioro significativo en la capacidad para realizar ejercicio, que es mayor al esperado por el grado de restricción de volumen. Debido a que existe un incremento mínimo en la DLCO y un aumento en la relación de ventilación desperdiciada (VD-VT) con el ejercicio, se ha deducido que las alteraciones vasculares pulmonares son la causa de la poca tolerancia al ejercicio.31
Las características histológicas del granuloma eosinófilo son típicas, e incluyen la presencia de células que recuerdan a las células de Langerhans en la piel. Estas células se tiñen en forma positiva con anticuerpos monoclonales contra CD1a, y se ha demostrado con microscopia electrónica que contienen las inclusiones características denominadas cuerpos X o gránulos de Birbeck.32 Estas células pueden observarse en otros tipos de enfermedad pulmonar infiltrativa difusa, pero en mucho menor número que en el granuloma eosinófilo. Los estudios sugieren que existen células T CD4+ que se encuentran en aposición a las células de Langherhans y un mayor número de células neuroendócrinas en los pacientes con granuloma eosinófilo.33,34 Se han encontrado factor estimulador de colonias de granulocitos y macrófagos y factor transformador de crecimiento -ß(FTC-ß) en las lesiones, lo que sugiere que estos factores son mediadores potenciales de la estimulación de las células de Langerhans y de los fibroblastos, respectivamente. Estos datos sugieren que la patogenia incluye interacciones entre las células que causan estimulación de los fibroblastos y fibrosis.
El diagnóstico de granuloma eosinófilo y su diferenciación de otros padecimientos que causan enfermedad pulmonar difusa puede hacerse con mayor certeza si se realiza una biopsia de pulmón a cielo abierto. Sin embargo, se ha sugerido que el encontrar más de 5 porciento de células CD1a+ en el líquido del LBA, aunado a un cuadro clínico y radiográfico compatible, puede ser suficiente para el diagnóstico.32 Como se mencionó antes, la TCAR puede ser una técnica no cruenta y útil para el diagnóstico del granuloma eosinófilo.
Varios estudios de casos han reportado pacientes con granuloma eosinófilo que mejoraron después de suspender el tabaquismo, y se ha informado que el tratamiento con esteroides ha sido eficaz. Sin embargo, ningún tratamiento se ha estudiado aún lo suficiente o ha producido evidencias convincentes de mejoría como para justificar su recomendación definitiva.
Los pacientes con granuloma eosinófilo tienen una sobrevida promedio de 6 años después del diagnóstico. La mayor edad, la obstrucción al flujo del aire y la hiperinflación son predictores de un mal pronóstico.35
NEUMONIA ORGANIZADA CON BRONQUIOLITIS OBLITERANTE
La neumonía organizada con bronquiolitis obliterante, también llamada neumonía organizada criptogénica,36 es un padecimiento que ocurre parte en el intersticio, parte en el alveolo y parte en las vías aéreas pequeñas. En algunos pacientes puede diagnosticarse de modo incorrecto como FPI y enfermedad pulmonar obstructiva crónica. La enfermedad tiene tres patrones clínicos, idiopática sintomática, secundaria y focal asintomática.37 La NOBO puede ser causada por diversos trastornos, incluyendo inhalación de humos tóxicos (i.e., óxidos de nitrógeno), infecciones virales, padecimientos del tejido conjuntivo y otras enfermedades pulmonares, como la FPI y la neumonitis por hipersensibilidad. Aunque una historia clínica cuidadosa puede identificar estas causas, existe un grupo de pacientes con NOBO idiopática en quienes se requiere una biopsia de pulmón a cielo abierto para distinguir la enfermedad de otros procesos parenquimatosos difusos.
Los pacientes con NOBO suelen tener entre 50 y 60 años de edad. La enfermedad comienza en forma súbita y se caracteriza por tos seca y disnea, fiebre, malestar y fatiga, que son más frecuentes en los pacientes con NOBO que en los enfermos con FPI. En ocasiones el examen físico demuestra estertores asociados a sibilancias. No existen dedos en palillo de tambor, que son comunes en la FPI. La radiografía de tórax muestra, en forma típica, infiltrados alveolares bilaterales en parche o en vidrio despulido, a diferencia de los infiltrados intersticiales que ocurren en la FPI. La TCAR demuestra áreas bilaterales de consolidación que afectan principalmente la región subpleural, peribroncovascular o ambas, que son muy sugestivas del diagnóstico.38 Las pruebas de función pulmonar indican obstrucción, restricción o un patrón mixto con reducción de la capacidad de difusión e hipoxemia.
El tratamiento con esteroides ha sido útil, y muchos pacientes responden en forma dramática en unos cuantos días. Sin embargo, cuando se reduce la dosis muchos pacientes recaen y vuelven a tener síntomas generales, infiltrados e hipoxemia. Los pacientes con NOBO asintomática focal no requieren tratamiento.
PROTEINOSIS ALVEOLAR
La proteinosis alveolar es una enfermedad de etiología desconocida que se caracteriza por el acúmulo intralveolar de un material celular lipoproteináceo que recuerda al surfactante.39 El aumento de este material puede ser ocasionado por mayor producción o menor degradación del surfactante por los macrófagos. Se ha propuesto que la menor utilización del surfactante, con menor regreso desde el alveolo a las células tipo II, puede superar la capacidad de los macrófagos normales para eliminar esta sustancia. Además, el material lipoproteináceo puede inhibir la defensa de los macrófagos contra la infección, favoreciendo la mayor incidencia de infecciones por patógenos intracelulares que se observa en los enfermos con proteinosis alveolar. Ocurre un trastorno clínicamente semejante a la proteinosis alveolar idiopática en individuos con exposición aguda al sílice (por lo general en la limpieza con arena) y en pacientes con trastornos hematológicos graves.
Los cambios patológicos en la proteinosis alveolar incluyen la presencia de material granular eosinofílico dentro de los espacios alveolares, que se tiñe en forma positiva con el agente ácido-Schiff (PAS) [ver figura 7]. Se ha detectado inflamación y fibrosis en algunos casos, pero éstas no suelen ser características importantes. La microscopía electrónica demuestra que la sustancia intralveolar contiene numerosos cuerpos laminares, semejantes a los encontrados en las células epiteliales tipo II.
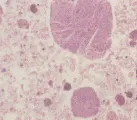
|
| Figura 7 |
| Proteinosis alveolar |
La proteinosis alveolar se presenta en una de tres formas: como una radiografía de tórax anormal asintomática, como el inicio súbito de tos, fiebre y dolor torácico causados por el padecimiento y complicados por infecciones oportunistas (con especiesNocardia, hongos o especiesMycobacterium), o como un padecimiento gradual con disnea y tos (a veces productiva). Los datos físicos suelen ser mínimos. La radiografía de tórax o la TCAR muestra un proceso bilateral de ocupación alveolar que semeja al edema pulmonar pero sin líneas B de Kerley, derrame pleural o crecimiento de la vasculatura central. En las etapas tardías pueden observarse marcas intersticiales gruesas causadas por fibrosis agregada. El aumento en la deshidrogenasa láctica del suero sin elevación de otras enzimas séricas es un dato que puede ayudar al médico a realizar el diagnóstico.
El diagnóstico de la proteinosis alveolar pulmonar se hace mejor por medio de LBA. El líquido se observa turbio, y su tinción demuestra gran cantidad de material PAS-positivo [ver figura 7]. La neumonía por P. carinii causa acúmulo de una sustancia intralveolar semejante, y debe descartarse esta posibilidad por medio de tinciones especiales. El líquido del lavado debe cultivarse para especiesNocardia, hongos y especiesMycobacterium.
El tratamiento de la proteinosis alveolar está indicado si existe disnea significativa en el momento del diagnóstico o si los síntomas leves no remiten en forma espontánea después de un periodo de observación de varios meses. Debido a que algunos enfermos remiten en forma espontánea, siempre que sea posible será mejor esperar varios meses antes de instituir el tratamiento. El tratamiento para la proteinosis alveolar pulmonar consiste en el lavado pulmonar total con solución salina estéril. El pulmón no lavado se protege por medio de una sonda endotraqueal de doble luz. La mayoría de los pacientes mejoran en forma importante después de eliminar el material alveolar por medio del lavado, y el pulmón contralateral puede lavarse después.
NEUMONIA EOSINOFILICA
Los síndromes de infiltrados pulmonares con eosinofilia en sangre periférica (IPE) se caracterizan por eosinofilia en el parénquima pulmonar, eosinofilia en sangre periférica, o ambas, y suelen asociarse con infiltrados en la radiografía.40 Se piensa que los mecanismos de hipersensibilidad aguda participan en la patogenia por la presencia de eosinófilos y la ocurrrencia ocasional de broncoespasmo. En muchos de estos padecimientos las sustancias secretadas por los eosinófilos pueden ayudar a causar la lesión al parénquima pulmonar, a las vías aéreas o a ambas. Como propuso Crofton en 1952, los IPE incluyen cinco síndromes: eosinofilia pulmonar simple (síndrome de Löffler), eosinofilia pulmonar prolongada sin asma, eosinofilia pulmonar con asma, eosinofilia pulmonar tropical y vasculitis pulmonar (granulomatosis y angeítis alérgica). Aunque no se incluye en esta clasificación, el síndrome hipereosinofílico se asocia en ocasiones con infiltrados pulmonares. Recientemente se ha descrito un trastorno nuevo, la neumonía eosinofílica aguda. Otros trastornos que puecen causar infiltrados pulmonares con eosinofilia periférica incluyen infecciones (v.gr., tuberculosis, brucelosis y micosis), neoplasias (v.gr., carcinoma broncogénico, enfermedad de Hodgkin y linfadenopatía inmunoblástica) y procesos inmunológicos (v.gr., enfermedad pulmonar reumatoidea y sarcoidosis).
Eosinofilia pulmonar simple (síndrome de Löffler)
El síndrome de Löffler se caracteriza por infiltrados transitorios y en ocasiones migratorios y por síntomas leves que duran menos de un mes. Los infiltrados suelen ser de densidad homogénea, únicos o múltiples. En algunos casos no existen síntomas, aunque pueden desarrollarse disnea y tos seca. El examen físico revela el acúmulo intersticial e intralveolar de eosinófilos, macrófagos y líquido. El síndrome de Löffler puede ser idiopático o puede ser causado por una infestación parasitaria (v.gr., con Ascaris o Strongyloides) o por medicamentos (v.gr., nitrofurantoína o penicilina). El tratamiento consiste en suspender el fármaco agresor o tratar la infestación parasitaria; en la mayoría de los casos no se requiere más. En ocasiones los pacientes muy sintomáticos con enfermedad idiopática pueden requerir tratamiento esteroideo.
Eosinofilia pulmonar prolongada sin asma
La eosinofilia pulmonar prolongada (neumonía eosinofílica crónica) es un padecimiento idiopático que afecta de modo predominante a mujeres de edad media y que se caracteriza por tos productiva, disnea, malestar, pérdida de peso, diaforesis nocturna y fiebre asociada con infiltrados pulmonares periféricos progresivos. Pueden existir hemoptisis y sibilancias, que hacen que el diagnóstico se confunda con otras causas de IPE. Las muestras de la biopsia abierta de pulmón muestran infiltrados inflamatorios mixtos masivos que tienen alto contenido de eosinófilos. Algunos vasos pulmonares pueden tener escasas células inflamatorias, pero no existe una vasculitis real.
El diagnóstico se basa en el síndrome clínico, la radiografía de tórax y si existe o no eosinofilia en sangre. Algunos investigadores han sugerido que si los resultados de la radiografía muestran los infiltrados periféricos densos típicos sin afección central, el diagnóstico puede hacerse sin necesidad de biopsia. Por lo general es posible confirmar el diagnóstico con facilidad por medio de un LBA y biopsia pulmonar transbronquial.
Ocurre remisión espontánea en hasta el 10 porciento de los casos, pero puede también ocurrir insuficiencia respiratoria. El tratamiento con esteroides es muy eficaz. Debido a que son frecuentes las recaídas, es común que se administre tratamiento hasta por 5 años.
Eosinofilia pulmonar con asma
La aspergilosis broncopulmonar alérgica puede causar infiltrados pulmonares en un paciente con asma.
Eosinofilia pulmonar tropical
Debe pensarse en el diagnóstico de eosinofilia pulmonar tropical cuando existe asma de inicio reciente, fiebre, eosinofilia importante en sangre y la presencia de un infiltrado basal mixto reticulonodular y alveolar en una persona que ha viajado recientemente al Lejano Oriente. Es posible que esta enfermedad represente una forma de filariasis (causada por Wuchereria bancrofti u otros organismos). A pesar del tratamiento con dietilcarbamacina, muchos pacientes con este padecimiento tienen inflamación persistente y desarrollan enfermedad intersticial crónica.
Vasculitis pulmonar (granulomatosis y angeítis alérgica)
La granulomatosis y angeítis alérgica es un padecimiento multisistémico que se caracteriza por vasculitis e inflamación granulomatosa necrosante que afecta a los pulmones, el sistema nervioso central y la piel. Aunque los riñones, el corazón, el bazo y el tubo digestivo se afecten con mucho menos frecuencia, casi cualquier órgano o tejido puede estar afectado. La eosinofilia prominente y el incremento de los niveles de IgE sugieren que mecanismos de hipersensibilidad inmediata deben participar de modo importante en la patogenia.
Síndrome hipereosinofílico
El síndrome hipereosinofílico se caracteriza por un amplio rango de manifestaciones clínicas que ocurren cuando los eosinófilos maduros infiltran diversos órganos.
Neumonía eosinofílica aguda
Recientemente se ha descrito una enfermedad pulmonar eosinofílica no detectada antes. Se denomina neumonía eosinofílica aguda y se caracteriza por el inicio súbito de tos, disnea, fiebre, taquipnea y estertores; los pacientes con frecuencia requieren de ventilación mecánica.42 No existe asociación clara con el tabaquismo, la exposición ambiental o la ingesta de medicamentos, y los pacientes tienden a ser jóvenes. Se observa eosinofilia en sangre en la mayoría de los casos, pero este dato no es útil para el diagnóstico. La radiografía de tórax y la TC muestran combinaciones inespecíficas de infiltrados alveolares (en vidrio despulido) e infiltrados alveolares e intersticiales mixtos que en ocasiones se asocian con derrames pleurales. El diagnóstico puede hacerse por lavado broncoalveolar, que muestra un aumento dramático en el porcentaje de eosinófilos, con frecuencia mayor del 20 porciento (la cuenta normal es menor del 1 porciento). La administración de esteroides causa mejoría rápida y son raras las recaídas después de la suspensión de los esteroides.
HEMORRAGIA ALVEOLAR DIFUSA
Los síndromes de hemorragia alveolar se caracterizan por hemorragia difusa parenquimatosa en ausencia de aspiración de sangre, coagulopatía, aumento de la presión venosa pulmonar o alguna causa local identificable (i.e., embolia pulmonar, cáncer o bronquitis). Con frecuencia la hemorragia está mediada por una alteración inmunológica y casi siempre se asocia con glomerulonefritis.
Las enfermedades que causan este síndrome son la enfermedad por anticuerpos contra la membrana basal glomerular (anti-MBG) o síndrome de Goodpasture, la glomerulonefritis idiopática rápidamente progresiva con o sin complejos inmunes, la vasculitis (v.gr., granulomatosis de Wegener), el LEG y la hemosiderosis pulmonar idiopática.43 Aunque cada uno de estos padecimientos tienen características clínicas sugestivas, en muchos casos no puede hacerse el diagnóstico por clínica. Además, muchos pacientes con hemorragia alveolar difusa están graves, lo que hace urgente una evaluación inmediata. El enfoque diagnóstico que se sugiere en estos pacientes consiste en:
1. Obtener suero para buscar anticuerpos anti-MBG, anticuerpos antinucleares y anti-ADN, anticuerpos citoplasmáticos antineutrófilo, niveles de complemento y complejos inmunes.
2. Realizar lavado broncoalveolar con tinción de hierro del tejido obtenido. Si existen macrófagos con hemosiderina se confirma el diagnóstico de hemorragia alveolar.
3. Si el paso 1 no confirma el diagnóstico o los resultados no están disponibles, realizar biopsia de riñón, pulmón u otro sitio afectado. Debe incluirse inmunofluorescencia y microscopía electrónica además del estudio histopatológico de rutina.
Casi todos los casos de hemorragia alveolar pueden diagnosticarse con rapidez si se siguen estos pasos. Deberá iniciarse tratamiento con dosis altas de esteroides por vía intravenosa mientras la evaluación está en progreso porque la hemorragia alveolar puede ser de extrema gravedad.
Enfermedad por anticuerpos anti-MBG
La enfermedad por anticuerpos anti-MBG ocurre en varones jóvenes que fuman y se caracteriza por glomerulonefritis y hemorragia alveolar difusa.44 Es causada por anticuerpos citotóxicos contra la cadena alfa3 de la colágena tipo IV en las membranas basales glomerular y alveolar.
Muchos pacientes presentan hemoptisis, que puede ser masiva; sin embargo, algunos episodios de hemorragia no se asocian con ella. La mayoría de los enfermos refieren disnea y hematuria macroscópica. Existe anemia por deficiencia de hierro en casi todos los pacientes, e insuficiencia renal desde la etapa inicial en el 50 porciento de los casos.
La radiografía de tórax muestra al principio sombras acinares perihiliares esponjosas, y después cambios intersticiales. Las pruebas de función respiratoria indican un patrón restrictivo que mejora con la resolución clínica. La DLCO puede aumentar por la captación de monóxido de carbono por la hemoglobina extravascular. Al volverse crónica la enfermedad pueden persistir los cambios radiográficos intersticiales y las alteraciones restrictivas de la función pulmonar.
El diagnóstico se confirma cuando se detectan anticuerpos circulantes anti-MBG (el 95 porciento de los pacientes tienen anticuerpos positivos) o depósitos lineales de IgG (más raro de IgA) en la membrana basal glomerular o alveolar. También pueden obtenerse anti-MBG del tejido afectado.
Las recomendaciones actuales de tratamiento incluyen agentes inmunosupresores, esteroides y plasmaféresis. A pesar de este tratamiento agresivo, el 50 porciento de los pacientes fallece o requiere diálisis a largo plazo después de 2 años. La muerte suele ser causada por un episodio de hemorragia alveolar masiva que con frecuencia es precipitado por infección.
Hemosiderosis pulmonar idiopática
La hemosiderosis pulmonar idiopática es un padecimiento caracterizado por hemorragia alveolar recurrente que ocurre sobre todo en niños (casi siempre menores de 10 años) y en adultos jóvenes. Cuando se presentan adultos, los hombres se afectan más que las mujeres. La presentación de la enfermedad es semejante a la del síndrome de Goodpasture, excepto por la ausencia de enfermedad renal. Con el tiempo ocurren episodios recurrentes de hemorragia clínica y subclínica, que causan daño pulmonar que puede ser obstructivo o restrictivo. El tejido pulmonar muestra cambios de hemorragia aguda y crónica, pero no vasculitis, complejos inmunes o depósitos con tinción lineal.
Los pacientes con hemosiderosis pulmonar idiopática se tratan con esteroides. Evidencias anecdóticas apoyan el uso de agentes inmunosupresores y plasmaféresis. La supervivencia promedio es de 3 a 5 años, pero ocurren remisiones espontáneas con supervivencia prolongada.
Linfangiomiomatosis
El inicio de la linfangiomiomatosis, que afecta en forma casi exclusiva a mujeres en edad reproductiva, suele caracterizarse por hemoptisis, neumotórax y quilotórax.45 Al principio de los síntomas puede no existir alteración radiográfica difusa, aunque en ocasiones se observa un infiltrado reticulonodular fino que predomina en las bases pulmonares. Puede existir un patrón intersticial difuso asociado a hiperinflación. A diferencia de la mayoría de las enfermedades pulmonares infiltrativas difusas, este padecimiento se caracteriza por fisiología obstructiva con aumento en los volúmenes pulmonares que se ha atribuido a hipertrofia del músculo liso peribronquial. Además, la capacidad de difusión suele estar disminuida.
El diagnóstico puede sospecharse en una mujer no fumadora con quilotórax, obstrucción fija de las vías respiratorias y un infiltrado intersticial difuso en la radiografía. La TCAR muestra quistes difusos de pared delgada, y es diagnóstica. Estos datos son idénticos a los de la esclerosis tuberosa.46 La biopsia de pulmón a cielo abierto demuestra proliferación de células de músculo liso en el pulmón y pleura visceral, con formación de quistes, quizá por obstrucción bronquial. Alrededor del 50 porciento de los pacientes tienen también angiomiolipomas como dato asociado, que suelen detectarse por TC.48 Más del 75 porciento de los pacientes sobreviven por más de 8 años.47 Existe alguna evidencia de que este proceso depende de hormonas, y se ha reportado una evolución más lenta después del tratamiento con ooforectomía, progesterona o la combinación de ambas. Debe considerarse la posibilidad de trasplante pulmonar en los casos en etapa terminal. Aunque el trasplante puede salvar la vida, se ha reportado recurrencia en el pulmón trasplantado, lo que destaca la naturaleza sistémica del padecimiento.
MICROLITIASIS
La microlitiasis es un padecimiento excesivamente raro, en ocasiones familiar, que suele diagnosticarse en un paciente asintomático por una radiografía de tórax de rutina. Esta demuestra nódulos pequeños calcificados difusos que, al examen histopatológico, son esferas calcificadas que ocupan el espacio alveolar.48 Esta enfermedad puede progresar hasta la insuficiencia respiratoria. En un solo paciente se reportó una posible respuesta al tratamiento con etidronato de sodio, un difosfonato.49
Figura 1 Cortesía del Dr. Sam Aguayo, Veterans Affairs Medical Center, Decatur, Georgia.
Figura 3 Andy Christie.
Bibliografía
- Coultas DB, Zumwalt RE, Black WC, et al: The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med 150:967, 1994 [PMID 7921471]
- Respiratory diseases disproportionately affecting minorities. The NHLBI Working Group. Chest 108:1380, 1995
- Lympany PA, du Bois RM: Diffuse lung disease: product of genetic susceptibility and environmental encounters. Thorax 52:92, 1997 [PMID 9039244]
- Grenier P, Chevret S, Beigelman C, et al: Chronic diffuse infiltrative lung disease: determination of the diagnostic value of clinical data, chest radiography, and CT and bayesian analysis. Radiology 191:383, 1994 [PMID 8153310]
- Raghu G: Interstitial lung disease: a diagnostic approach: are CT scan and lung biopsy indicated in every patient? Am J Respir Crit Care Med 151:909, 1995
- Drent M, van Nierop MAMF, Gerritsen FA, et al: A computer program using BALF-analysis results as a diagnostic tool in interstitial lung diseases. Am J Respir Crit Care Med 153:736, 1996
- Molin LJ, Steinberg JB, Lanza LA: VATS increases costs in patients undergoing lung biopsy for interstitial lung disease. Ann Thorac Surg 58:1595, 1994 [PMID 7979720]
- Harris-Eze AO, Sridhar G, Clemens RE, et al: Oxygen improves maximal exercise performance in interstitial lung disease. Am J Respir Crit Care Med 150:1616, 1994 [PMID 7952624]
- Cooper JA Jr, White DA, Matthay RA: Drug-induced pulmonary disease: I. Cytotoxic drugs. Am Rev Respir Dis 133:321, 1986
- Cooper JA Jr, White DA, Matthay RA: Drug-induced pulmonary disease: II. Noncytotoxic drugs. Am Rev Respir Dis 133:488, 1986
- Hertzman PA, Clauw DJ, Kaufman LD, et al: The eosinophilia-myalgia syndrome: status of 205 patients and results of treatment 2 years after onset. Ann Intern Med 122:851, 1995
- Sharma SK, Pande JN, Verma K: Effect of prednisolone treatment in chronic silicosis. Am Rev Respir Dis 143:814, 1991 [PMID 2008993]
- Lynch DA, Newell JD, Logan PM, et al: Can CT distinguish hypersensitivity pneumonitis from idiopathic pulmonary fibrosis? AJR Am J Roentgenol 165:807, 1995
- Drent M, van Velzen-Blad H, Diamant M, et al: Bronchoalveolar lavage in extrinsic allergic alveolitis: effect of time elapsed since antigen exposure. Eur Respir J 6:1276, 1993
- Bragg DG, Chor PJ, Murray KA, et al: Lymphoproliferative disorders of the lung: histopathology, clinical manifestations, and imaging features. AJR Am J Roentgenol 163:273, 1994
- Newman LS, Rose CS, Maier LA: Sarcoidosis. N Engl J Med 336:1224, 1997 [PMID 9110911]
- Lower EE, Baughman RP: Prolonged use of methotrexate for sarcoidosis. Arch Intern Med 155:846, 1995
- Chan-Yeung M, Muller NL: Cryptogenic fibrosing alveolitis. Lancet 350:651, 1997 [PMID 9288060]
- Coker RK, Laurent GJ: Anticytokine approaches in pulmonary fibrosis: bringing factors into focus. Thorax 52:294, 1997 [PMID 9093352]
- Cherniack RM, Colby TV, Flint A, et al: Correlation of structure and function in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 151:1180, 1995 [PMID 7697250]
- Hansell DM, Wells AU: CT evaluation of fibrosing alveolitis: applications and insights. J Thorac Imaging 11:231, 1996 [PMID 8892193]
- Van Oortegem K, Wallaert B, Marquette CH, et al: Determinants of response to immunosuppressive therapy in idiopathic pulmonary fibrosis. Eur Respir J 7:1950, 1994 [PMID 7875264]
- Douglas WW, Ryu JH, Bjoraker JA, et al: Colchicine versus prednisone as treatment of usual interstitial pneumonia. Mayo Clin Proc 72:201, 1997 [PMID 9070193]
- Agusta C, Xaubet A, Roca J, et al: Interstitial pulmonary fibrosis with and without associated collagen vascular disease: results of a two year follow up. Thorax 47:1035, 1992
- Homma Y, Ohtsuka Y, Tanimura K, et al: Can interstitial pneumonia as the sole presentation of collagen vascular diseases be differentiated from idiopathic interstitial pneumonia? Respiration 62:248, 1995
- Gabbay E, Tarala R, Will R, et al: Interstitial lung disease in recent onset rheumatoid arthritis. Am J Respir Crit Care Med 156:528, 1997 [PMID 9279235]
- Behr J, Vogelmeier C, Beinert T, et al: Bronchoalveolar lavage for evaluation and management of scleroderma disease of the lung. Am J Respir Crit Care Med 154:400, 1996 [PMID 8756813]
- Tazelaar HD, Viggiano RW, Pickersgill J, et al: Interstitial lung disease in polymyositis and dermatomyositis: clinical features and prognosis as correlated with histologic findings. Am Rev Respir Dis 141:727, 1990 [PMID 2310101]
- Deheinzelin D, Capelozzi VL, Kairalla RA, et al: Interstitial lung disease in primary Sjogren's syndrome: clinical-pathological evaluation and response to treatment. Am J Respir Crit Care Med 154:794, 1996 [PMID 8810621]
- Brauner MW, Grenier P, Tijani K, et al: Pulmonary Langerhans cell histiocytosis: evolution of lesions on CT scans. Radiology 204:497, 1997 [PMID 9240543]
- Crausman RS, Jennings CA, Tuder RM, et al: Pulmonary histiocytosis X: pulmonary function and exercise pathophysiology. Am J Respir Crit Care Med 153:426, 1996 [PMID 8542154]
- Kambouchner M, Valeyre D, Soler P, et al: Pulmonary Langerhans' cell granuloma tosis (histiocytosis X). Annu Rev Med 43:105, 1992
- Aguayo SM, King TE Jr, Waldron JA Jr, et al: Increased pulmonary neuroendocrine cells with bombesin-like immunoreactivity in adult patients with eosinophilic granuloma. J Clin Invest 86:838, 1990 [PMID 2394833]
-
Tazi A, Bonay M, Grandsaigne M, et al: Surface phenotype
of Langerhans cells and lymphocytes in granulomatous lesions from patients
with pulmonary histiocytosis X. Am Rev Respir Dis 147:1531, 1993 [PMID
8503566]
-
Delobbe A, Durieu J, Duhamel A, et al: Determinants
of survival in pulmonary Langerhans' cell granulomatosis (histiocytosis
X). Groupe d'Etude en Pathologie Interstitielle de la Societe de Pathologie
Thoracique du Nord. Eur Respir J 9:2002, 1996
-
Alasaly K, Muller N, Ostrow DN, et al: Cryptogenic
organizing pneumonia: a report of 25 cases and a review of the literature.
Medicine (Baltimore) 74:201, 1995 [PMID
7623655]
-
Lohr RH, Boland BJ, Douglas WW, et al: Organizing pneumonia:
features and prognosis of cryptogenic, secondary, and focal variants. Arch
Intern Med 157:1323, 1997 [PMID
9201006]
-
Lee KS, Kullnig P, Hartman TE, et al: Cryptogenic organizing
pneumonia: CT findings in 43 patients. AJR Am J Roentgenol 162:543, 1994
[PMID
8109493]
-
Wang BM, Stern EJ, Schmidt RA, et al: Diagnosing pulmonary
alveolar proteinosis: a review and an update. Chest 111:460, 1997 [PMID
9041997]
-
Allen JN, Davis WB: Eosinophilic lung diseases. Am
J Respir Crit Care Med 150:1423, 1994
-
Durieu J, Wallaert B, Tonnel AB: Long-term follow-up of pulmonary
function in chronic eosinophilic pneumonia. Groupe d'Etude en Pathologie
Interstitielle de la Societe de Pathologie Thoracique du Nord. Eur Respir
J 10:286, 1997
-
Pope-Harman A, Davis W, Allen E, et al: Acute eosinophilic
pneumonia: a summary of 15 cases and review of the literature. Medicine
(Baltimore) 75:334, 1996 [PMID
8982150]
-
Green RJ, Ruoss SJ, Kraft SA, et al: Pulmonary capillaritis
and alveolar hemorrhage: update on diagnosis and management. Chest 110:1305,
1996 [PMID
8915239]
-
Kelly PT, Haponik EF: Goodpasture syndrome: molecular and
clinical advances. Medicine (Baltimore) 73:171, 1994 [PMID
8041241]
-
Kalassian KG, Doyle R, Kao P, et al: Lymphangioleiomyomatosis:
new insights. Am J Respir Crit Care Med 155:1183, 1997
-
Lenoir S, Grenier P, Brauner MW, et al: Pulmonary lymphangiomyomatosis
and tuberous sclerosis: comparison of radiographic and thin-section CT
findings. Radiology 175:329, 1990
-
Kitaichi M, Nishimura K, Itoh H, et al: Pulmonary lymphangioleiomyomatosis:
a report of 46 patients including a clinicopathologic study of prognostic
factors. Am J Respir Crit Care Med 151:527, 1995
-
Prakash UB, Barham SS, Rosenow EC 3rd, et al: Pulmonary
alveolar microlithiasis: a review including ultrastructural and pulmonary
function studies. Mayo Clin Proc 58:290, 1983 [PMID
6341721]
-
Gocmen A, Toppare MF, Kiper N, et al: Treatment of
pulmonary alveolar microlithiasis with a diphosphonate-preliminary results
of a case. Respiration 59:250, 1992 [PMID
1485012]