Contenido del artículo
VI TRASTORNOS DE LA HEMOSTASIA Y COAGULACION
- Hemostasia normal
- PROCOAGULANTES DEL PLASMA
- INHIBIDORES DE LA COAGULACION
- Sistema AT-III-heparán (o AT-III-heparina)
- Sistema proteína C-proteína S- trombomodulina
- Inhibidor de la vía del factor tisular
- SISTEMA FIBRINOLITICO
- Producción y cinética de las plaquetas
- Pruebas de coagulación y su uso
- PRUEBAS DE LOS PROCOAGULANTES
- PRUEBAS DE PLAQUETAS Y DE FUNCION PLAQUETARIA
- PRUEBAS DE INHIBIDORES DE LA HEMOSTASIA
- Estudio del paciente con trastornos hemorrágicos o trombóticos
- Disminución de la cuenta plaquetaria, trombocitopenia
- DEFECTOS EN LA PRODUCCION DE PLAQUETAS
- ELIMINACION PLAQUETARIA ACELERADA POR DESTRUCCION INMUNOLOGICA
- Púrpura trombocitopénica idiopática/púrpura trombocitopénica autoinmune (PTI/PTA)
- Púrpura trombocitopénica secundaria
- Púrpura postransfusional
- Destrucción plaquetaria inmune inducida, por medicamentos
- REMOCION ACELERADA DE LAS PLAQUETAS POR MECANISMOS NO INMUNOLOGICOS
- Púrpura trombocitopénica trombótica y síndrome urémico-hemolítico del adulto
- Coagulación intravascular diseminada
- Trombocitopenia inducida por infección
- Trombocitopenia en el embarazo y perinatal
- Trombocitopenia en la hipotermia
- Lavado de plaquetas y alteraciones del lecho vascular
- SECUESTRO DE PLAQUETAS
- Trastornos de la función plaquetaria
- ALTERACIONES DE TIPO HEREDITARIO
- Trastornos de la membrana plaquetaria
- Trastornos de los gránulos plaquetarios
- Enfermedad de von Willebrand
- TRASTORNOS ADQUIRIDOS
- Púrpuras vasculares
- TELEANGIECTASIA HEMORRAGICA HEREDITARIA
- ESCORBUTO
- EXCESO DE ESTEROIDES
- VASCULITIS LEUCOCITOCLASTICA
- PURPURA SENIL
- PURPURA AUTERITROCITICA
- LESIONES DE LA MICROCIRCULACION POR EMBOLIAS
- Trastornos hereditarios de la coagulación
- ENFERMEDAD DE VON WILLEBRAND
- HEMOFILIA A
- Principios generales de tratamiento en la hemofilia
- Tratamiento de la hemorragia aguda
- Principios del tratamiento sustitutivo
- Cirugía electiva y extracción dental
- Tratamiento cuando existen inhibidores
- OTROS TRASTORNOS HEMORRAGICOS HEREDITARIOS
- Trastornos hemorrágicos adquiridos
- DEFICIENCIA DE VITAMINA K
- HEMORRAGIA INDUCIDA POR MEDICAMENTOS
- DISPROTEINEMIAS
- COAGULACION INTRAVASCULAR DISEMINADA
- FIBRINOLISIS PRIMARIA
- ENFERMEDADES HEPATICAS GRAVES
- INHIBIDORES CIRCULANTES, (ANTICOAGULANTES)
- HEMATOMAS CRONICOS EXPANSIVOS
- HEMORRAGIA DESPUES DE LA DERIVACION CARDIOPULMONAR
- Estados de hipercoagulabilidad
- SINDROME ANTIFOSFOLIPIDO
- DEFICIENCIA DE ANTITROMBINA III
- DEFICIENCIA DE COFACTOR II DE HEPARINA
- DEFICIENCIA DE PROTEINA C Y PROTEINA S
- ALTERACIONES DE LA FIBRINOLISIS
- DISFIBRINOGENEMIA
- HOMOCISTINURIA
- ADMINISTRACION DE FARMACOS
- SINDROME DE TROUSSEAU
- DEFICIENCIA DE FACTOR XII
- Errores quirúrgicos contra enfermedades sistémicas
DR. STANLEY L. SCHRIER
Hemostasia normal
Cuando se rompe una pared vascular, la respuesta hemostática debe ser rápida, localizada y estar regulada en forma cuidadosa. El daño al endotelio activa a las células endoteliales de modo muy específico, y expone la sangre circulante a la laminina, fibronectina y colágena subendoteliales y a la membrana basal. Se adhieren plaquetas en el sitio de la lesión por medio de una serie ordenada de interacciones receptor-ligando, que son influidas por la velocidad de¡ desgarro de la pared. Las interacciones son diferentes en las arterias y en las venas. Para formar el tapón hemostático ocurren interacciones receptor-ligando, muy específicas y dependientes de este daño, entre plaquetas y células endoteliales activadas, y quizá también entre macrófagos.1-3
La respuesta hemostática se inicia cuando el seudópodo de una plaqueta se pone en contacto con el subendotelio. La plaqueta cambia de forma y se extiende sobre el área dañada de modo que expone una gran cantidad de su membrana plasmática al subendotelio.4 La adhesión ocasiona activación de la plaqueta. En este proceso la membrana plasmática de la plaqueta expresa nuevos receptores, algunos de los que existían en forma previa sufren cambios morfológicos que alteran su función, y la plaqueta libera materiales contenidos en sus gránulos. Algunas de las sustancias almacenadas, como el factor de von Willebrand (FvW), el fibrinógeno, la trombospondina (que también existe en el plasma) y el difosfato de adenosina (ADP), inducen a las plaquetas que pasan a adherirse, agregarse y activarse, con lo que liberan también su contenido. Ciertas sustancias secretadas por las plaquetas, como el tromboxano A2, producen vasoconstricción intensa, enlentecimiento del flujo sanguíneo y contribuyen a la respuesta hemostática.
Las células endoteliales tienen una función clave en el control de la hemostasia y del tono vascular. Al activarse, expresan receptores de superficie que son semejantes a los de las plaquetas activadas; también expresan receptores como el GMP- 140 (también conocido como proteína extema de la membrana de los gránulos dependiente de la activación de las plaquetas, o PADGEM, y P-selectina), que median la adherencia de los neutrófilos y monocitos activados a las células endoteliales y a las plaquetas.5 Además, las células endoteliales activadas secretan endotelina,6,7 un péptido que causa vasoconstricción intensa, así como una sustancia denominada factor de relajación derivado del endotelio (FRDE). Se sabe que el FRDE es óxido nítrico, un potente vasodilatador e inhibidor de la agregación plaquetaria. Las células endoteliales arteriales generan óxido nítrico, no así las células endoteliales venosas. 3.8-11
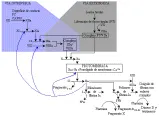
Es probable que el tapón plaquetario constituya la primera línea de defensa contra la hemorragia; mientras tanto, los fibroblastos y los pericitos localizados bajo el sitio del daño vascular liberan factor tisular (FT). El factor tisular y el sistema extrínseco de la coagulación son de vital importancia para formar el coágulo loalizado. El factor tisular unido al factor VII es convertido con rapidez por cantidades diminutas y locales de factor Xa en el complejo muy activo factor fisular/factor VIIa. Este complejo activa de inmediato el factor IX, transformándolo en factor IXa y el factor X a factor Xa.12 A continuación, el factor Xa, unido a factor Va sobre la superficie de las células endoteliales o de las plaquetas, convierte con rapidez la protrombina (factor II) en trombina (factor IIa).13 Comenzando al mismo tiempo, pero en forma un poco más lenta, el factor IXa se une a factor VIII, a y el complejo factor IXa/factor VIIIa también convierte el factor X en factor Xa. La trombina tiene múltiples e importantes funciones, activa los factores VIII, V, XI, las plaquetas, e inicia el ataque proteolítico sobre el fibrinógeno [ver figura 1].
|
| Figura 1 |
| Vías de la coagulación |
Desde el punto de vista fisiológico, parece ser que el sistema intrínseco de la coagulación, iniciado por la activación por contacto del factor XII (factor de Hageman), tiene menos importancia fisiológica. El factor XI puede activarse a factor XI a por trombina, 14 y el factor XIa a su vez puede activar el factor IX a factor IXa, lo mismo que el complejo factor tisular/factor VIIa de la vía extrínseca. La generación de trombina causa también que las plaquetas se adhieran, agreguen y expresen receptores apropiados, que junto con los receptores de las células endoteliales dirigen el proceso. Esta secuencia de eventos induce la formación localizada de fibrina, que se introduce en el tapón plaquetario. Este trombo es compactado por el sistema contráctil de actomiosina de las plaquetas en un proceso conocido como de retracción del coágulo. Las propiedades vasoconstrictoras del tromboxano A2 plaquetario y de la endotelina contribuyen a este proceso al disminuir la luz del vaso y enlentecer el flujo sanguíneo.15, 16
Si no se controlaran las propiedades de autoamplificación de la coagulación, cada hemorragia importante produciría coagulación generalizada y oclusión permanente de los vasos. Sin embargo, la coagulación está modulada por varios mecanismos: la dilución de los procoagulantes en el flujo sanguíneo, la remoción de partículas y factores activados a través del sistema reticuloendotelial, especialmente en el hígado, y el antagonismo de los procoagulantes activados por inhibidores circulantes.
Existen cuatro sistemas de control diferentes y separados que regulan y enfatizan cada fase de la hemostasia. Primero, los efectos vasconstrictor y de agregación plaquetaria de la endotelina y del tromboxano A2 se oponen a las acciones vasodilatadoras y de inhibición de la agregación plaquetaria del óxido nítrico y de la prostaciclina (PGI2). Segundo, la acción de los procoagulantes de proteasa de serina factor Xa y factor Ha, y quizá también de los factores IXa y XIa, es contrarrestada en forma específica por el complejo de heparina (o heparán unido a la superficie) con antitrombina III (AT-111). Tercero, el sistema de proteína C trombomodulina-S divide en forma proteolítica y destruye a los factores VIIIa y Va, los cofactores proteicos no enzimáticos de la hemostasia. Por último, la sustancia antes llamada inhibidor de la vía extrínseca (IVE) o inhibidor de la coagulación asociado a lipoproteína (ICAL), y ahora denominado inhibidor de la vía del factor tisular (IVFT) inhibe el complejo factor tisular/factor VIIa de la vía extrínseca.12
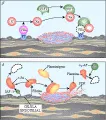
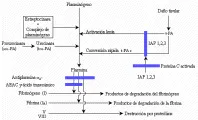
Después de que se ha logrado la hemostasia, debe ocurrir cicatrización de la herida y restablecerse la permeabilidad del vaso trombosado [ver figura 2] . Este último paso depende del sistema fibrinolítico. La fibrinolisis comienza por la liberación de activadores del plasminógeno, como el activador del plasminógeno tisular (t-PA por sus siglas en inglés, n. del t.), a partir de las paredes del vaso lesionado. Estos activadores convierten al plasminógeno circulante en la proteasa activa plasmina [ver figura 3]. La plasmina tiene varias acciones de proteasa y puede lisar coágulos de fibrina, además de fibrinógeno y otros procoagulantes. Los moduladores de la acción fibrinolítica son las antiplasminas circulantes, como la a2-antiplasmina (que antes se denominaba inhibidor de la a2-plasmina) y varias clases de inhibidores de activadores del plasminógeno (IAP).17, 18
|
|
|
|
|
| Figura 3 |
| Sistema de fibrinolisis |
FUNCIONES DE LAS PLAQUETAS Y DE LAS CELULAS ENDOTELIALES EN LA HEMOSTASIA
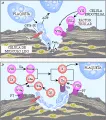
Las células endoteliales tienen un papel complejo de control sobre la hemostasia, Generan óxido nítrico y endotelina, y proporcionan también el sitio en el que el factor Va puede unirse al factor Xa, formando así una protrombinasa (ver adelante). Es probable que los proteoglicanos unidos ala membrana de la célula endotelial, como el heparán sulfato, interactúen con la AT-III para producir el complejo anticoagulante AT-III-heparán. La trombomodulina de la célula endotelial es el sitio de interacción de la proteína C-proteína S. Las células endoteliales pueden también elaborar un tipo de IAP.19
Después de que ocurre el daño a la célula endotelial, quedan expuestas colágena, laminina, fibronectina subendoteliales y otras sustancias relacionadas de la membrana basal. El FvW interactúa con algunas de estas sustancias de una manera que depende del daño vascular y causa que las plaquetas se adhieren al subendotelio por medio de la unión a la glucoproteína plaquetaria GPIb-IX y quizá también a la GPIlb-IIIa. La GPIb-IX, que está ausente en la enfermedad de Bemard-Soulier, y la GPIIb-IIIa son receptores plaquetarios bien definidos. Es probable que las plaquetas también se adhieren por la interación entre otro receptor plaquetario, el GPIa-IIa, y la colágena subendotelial.20 Estas interacciones receptor-ligando activan a las plaquetas, causando que expresen más sitios GPllb-llla, que a su vez forman la base de las interacciones interplaquetarias (agregación) con el fibrinógeno como ligando. El FvW puede también actuar como ligando cuando la velocidad de ruptura de la pared vascular es grande.21 La agregación plaquetaria mediada por fibrinógeno se estabiliza por acción de la trombospondina22 liberada de los gránulos a de la plaqueta, esta sustancia se fija tanto al receptor de la plaqueta como al de la célula endotelial.
En las plaquetas activadas, el factor Va se moviliza de los gránulos a hacia la superficie de la plaqueta, en donde actúa como receptor del factor Xa, proporcionando así una protrombinasa justo en el sitio de la lesión. El contacto de trombina o de la plaqueta con la colágena activa también a la fosfolipasa A2 de superficie de la plaqueta [ver figura 4], catalizando la formación de ácido araquidónico, que es un precursor de los endoperóxidos cíclicos. El tromboxano A2 es un vasoconstrictor potente, y favorece también la agregación plaquetaria. La retracción del coágulo está mediada por la actomiosina plaquetaria que, al activarse la plaqueta, se une a la extensión citosólica de la GPIIb-IIIa. Por lo tanto, la GPIIb-llIa integra interacciones receptorligando que ocurren en la porción exterior de la membrana con eventos citosólicos. La célula endotelial dañada y la plaqueta activada actúan juntas para generar el trombo hemostático en el sitio exacto de daño vascular.
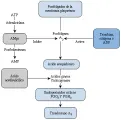
|
| Figura 4 |
| Liberación y agregación de plaquetas |
PROCOAGULANTES DEL PLASMA
Todos los procoagulantes [ver tabla 1], excepto el factor de von Willebrand se sintetizan en el hígado. El FvW se sintetiza en los megacariocitos y en las células endoteliales. Los procoagulantes dependientes de vitamina K son los factores II, VII, IX y X, y los anticoagulantes dependientes de vitamina K son la proteína C y la proteína S. Para cada uno de estos factores, la carboxilación de los residuos del ácido glutámico para formar residuos de ácido y-carboxilación actúa como una señal de reconocimiento que guía la modificación postraducción de la proteína que se requiere para la actividad biológica.23 Esta carboxilación es un proceso que depende de vitamina K.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
* n indica el número de subunidades.
|
Dentro del proceso general de coagulación, la acción de los procoagulantes depende de una proteasa que se une aun cofactor sobre la membrana para producir activación y amplificación del siguiente paso en la secuencia.13 Por ejemplo, la protrombinasa, que consiste en la proteasa de serina factor Xa se une a su cofactor y receptor, el factor Va, sobre las plaquetas y células endoteliales, convirtiendo a la protrombina (factor II) en trombina (factor IIa).1,2 El factor VIII circula como parte de un complejo factor VIII:C (el procoagulante activo del factor VIII)-FvW. El FvW sirve como un acarreador, y puede proteger al factor VIII:C de la degradación proteolítica.
En la actualidad se considera que la vía extrínseca tiene gran importancia para mantener la hemostasia. El complejo factor tisular/factor VIIa tiene un papel crucial dentro de la vía extrínseca al unirse y activar sus dos sustratos, el factor X y el factor IX. La importancia de la activación del factor IX a factor IXa se demuestra por las hemorragias tan graves que ocurren en los pacientes con deficiencia de este factor (hemofilia B o deficiencia de factor Christmas) y de su cofactor proteico, el factor VIII (hemofilia A). En forma semejante, debido a que los pacientes con deficiencia de factor XI no sufren hemorragias graves, sino que estas son variables o mínimas, parece ser que la activación por contacto (la vía intrínseca) tiene menor importancia fisiológica [ver figura 1]2,24
INHIBIDORES DE LA COAGULACION
Sistema AT-III-heparán (o AT-III-heparina)
Los inhibidores de la coagulación más importantes son la antitrombina III-heparán (o AT-III-heparina) y la proteína C. La AT-III antagoniza la acción de los procoagulantes proteasa de serina, o sea los factores XIIa, XIa, IXa, Xa y Ila, al formar con ellos complejos estoiquiométricos. El heparán (o heparina) se une a los residuos lisil de la AT-III, produciendo un cambio morfológico que hace que el sitio activo de la AT-III sea más accesible a los procoagulantes proteasa de serina.25 Esta acción del heparán (o heparina) aumenta la inactivación de la trombina 750 veces, y la inactivación del factor Xa en un grado aún mayor. Sin embargo, tanto la trombina como el factor Xa son protegidos de la inactivación por AT-III-heparán cuando se unen a las plaquetas o a las células endoteliales. Al parecer, los proteoglucanos heparán Sulfato de la superficie luminal de las células endoteliales activan a la AT-III de modo idéntico al de la heparina.25 Estos proteoglucanos se intercalan dentro de la membrana plasmática de las células endoteliales, la porción proteica de la molécula se inserta en el centro de la membrana y la porción polisacárida, que es análoga a la heparina, protruye de la superficie celular hacia el torrente sanguíneo.25
Sistema proteína C-proteína S- trombomodulina
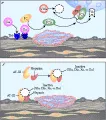
La proteína C y la proteína S son anticoagulantes dependientes de vitamina K que se sintetizan en el hígado, y la trombomodulina es un receptor que fija trombina sobre las células endoteliales y otras células.26 Cuando la trombina (factor Ha) se une a la trombomodulina, es modificada de tal manera que no puede ya convertir al fibrinógeno en fibrina. El complejo trombina-trombomodulina activa a la proteína C, una proteasa de serina que degrada en forma proteolítica al factor Va y al factor VIIIa [ver figura 2e y figura 5]. La proteína C activada actúa en la superficie de la membrana, quizá en asociación con la proteína S y unida al receptor de la célula endotelial. La proteína C activada no sólo degrada a los factores VIIIa y Va, sino que también inhibe uno de los inhibidores de los activadores M plasminógeno, permitiendo así que el t-PA lise los coágulos sin oposición.27-29 Debido a los poderosos efectos de la proteína C, su activación está regulada en forma estrecha [ver figura 5], y su acción es bloqueada por el inhibidor de proteína C. La deficiencia de proteína C o de proteína S ocasiona estados de hipercoagulabilidad clínicamente importantes.30

|
| Figura 5 |
| Proteínas C y S |
El sistema proteína C- proteína S también parece constituir un enlace entre la hemostasia y la inflamación. Por ejemplo, la exposición in vitro de las células endoteliales a interleucina-1 (IL-1) disminuye la concentración de trombomodulina a alrededor del 33 porciento del nivel normal, y la función de la proteína C-proteína S amenos del 10 porciento de los valores controles.27,28
La proteína S circula en dos formas: forma libre, en la que es activa como anticoagulante, y una forma combinada, unida a la proteína C4b del sistema del complemento, que es inactiva. Debido a que el C4b actúa como un reactante de fase aguda, su aumento en los estados de inflamación o infección tiende a disminuir la actividad de la proteína S y, por lo tanto, aumenta la posibilidad de trombosis.
Inhibidor de la vía del factor tisular
Con el descubrimiento del sistema anticoagulante ATIII-heparán (o heparina) (que neutraliza los factores Xa, Ila, IXa y Xla) y del sistema proteína C-proteína Strombomodulina (que degrada a los factores Va y VIIIa), se han identificado anticoagulantes que controlan cada paso de la hemostasia, excepto el complejo factor tisular/ factor VIIa. Como se mencionó antes, en la actualidad se ha identificado un inhibidor específico de este complejo, que se denomina inhibidor de la vía del factor tisular (lVFT).31,32
El IVFT es una lipoproteína que es activada por el factor Xa. El complejo factor Xa/IVFT actúa en forma específica sobre el factor tisular/factor VIIa para formar un complejo cuaternario que provoca inactivación del factor tisular/ factor VHa.12,31,32 El sistema está diseñado en forma admirable para detectar la producción excesiva de factor Xa y usar este acúmulo para detener su sitio de origen, el complejo factor fisular/factor VIIa [ver figura 6]. Debido a que no se ha identificado un estado de deficiencia de lVFT en los humanos, su función biológica se deduce de experimentos realizados in vivo en animales.33
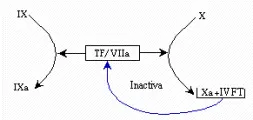
|
| Figura 6 |
| Complejo factor tisular/factor VIIa |
SISTEMA FIBRINOLITICO
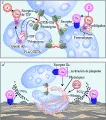
Se supone que el daño a las células endoteliales, lo mismo que la activación de contacto del sistema intrínseco, desencadenan la fibrinolisis. La cicatrización de la herida comienza en cuanto se ha formado el trombo hemostático y establecido un coágulo con enlaces cruzados firmes en el sido del daño vascular. Entonces el coágulo debe ser lisado para restablecer la circulación sanguínea en el área.
El sistema fibrinolítico consiste principalmente del precursor enzimático plasminógeno que circula en el plasma como glu-plasminógeno (el aminoácido N-terminal es glutamato).34 El plaminógeno contiene cinco estructuras de triple asa que son características de los componentes del sistema fibrinolítico. Un proceso de proteólisis limitada convierte al glu-plasminógeno en lis-plasminógeno (ahora el aminoácido N-terminal es lisina) y es el lis-plasminógeno el que se convierte, en forma preferente, en la enzima con actividad fibrinolítica, la plasmina. El plasminógeno interactúa con la fibrina por medio de sitios de unión de lisina, que pueden ser bloqueados por agentes parecidos a este aminoácido (v.gr., ácido e-aminocaproico o AEAC). La conversión crucial de plasminógeno a plasmina está mediada por dos activadores del plasminógeno: el t-PA y la urocinasa (activador del plasminógeno que se encuentra en la orina), y ambos son empleados en la actualidad en la clínica como agentes fibrinolíticos. La molécula de t-PA producida en las células endoteliales y en otros tejidos también tiene dominios de asa, y convierte el plasminógeno en plasmina en forma lenta. Cuando el t-PA se une a la fibrina su acción aumenta cien veces. Esta especificidad por la fibrina es ideal desde el punto de vista fisiológico: un complejo ternario que consiste en fibrina, t-PA y plasminógeno, en el que el t-PA unido a fibrina convierte al plasminógeno asociado a la fibrina en plasmina justo en el sitio en que se encuentra el trombo de fibrina. La plasmina asociada a la fibrina también es protegida de la acción inhibitoria específica de la a2-antiplasmina. La prourocinasa, o activador urinario del plasminógeno de cadena única (scu-PA), es convertida por medio de proteólisis en urocinasa, o activador urinario del plasminógeno de doble cadena (tcu-PA), que tiene un dominio de asa (al igual que el plasminógeno y el t-PA). La urocinasa (tenPA) no tiene especificidad por la fibrina para convertir el plasminógeno en plasmina, mientras que el scu-PA sí la tiene.
Los inhibidores naturales de la fibrinolisis bloquean a la plasmina o a los dos activadores del plasminógeno, el t-PA y la urocinasa. La plasmina es inhibida con rapidez por interacción con su inhibidor, la a2-antiplasminasa, a nivel de su sitio lisina. Sin embargo, cuando la plasmina está unida a fibrina, el sitio lisina está bloqueado y esto la protege en forma parcial contra el ataque de la a2 -antiplasmina. Los activadores del plasminógeno son inhibidos por varios IAP, algunos de los cuales ya se han descrito.35 El IAP-1 se encuentra en el plasma y es el principal inhibidor del t-PA y de la urocinasa. Su expresión y producción dependen de un control complejo, y el aumento en su concentración en plasma se asocia con trombosis. Las deficiencias de IAP- 1 pueden causar fibrinolisis sin oposición y hemorragia.36
Además del t-PA y de la urocinasa, existen por lo menos otras dos preparaciones fibrinolíticas que se usan en el ámbito clínico: la estreptocinasa y el complejo activador de plasminógeno anisolado y estreptocinasa (APSAC, por sus siglas en inglés, n. del t.). La estreptocinasa es una enzima bacteriana que primero se fija al plasminógeno del plasma, exponiendo su sitio activo y causando que parte del mismo se convierta en plasmina. 17, 34 Después, el complejo plasminógeno-estreptocinasa se convierte en estreptocinasa-plasmina, que puede activar con facilidad al plasminógeno porque no es bloqueado por la a2-antiplasmina.17 La acción de la estreptocinasa-plasmina no es específica de la fibrina: este complejo degrada fibrinógeno y otros procoagulantes en plasma, lo mismo que coágulos de fibrina. El APSAC tiene mayor especificidad por la fibrina que la estreptocinasa y depende de la unión del componente lis-plasminógeno a la fibrina para localizar el complejo en el sitio del trombo. Después de que las enzimas tisulares locales desprenden el grupo acilo que cubre el sitio catalítico activo del plasminógeno, el complejo estreptocinasa-plasminógeno puede convertir el plasminógeno a plasmina de una forma relativamente específica por la fibrina.
Lipoproteína (a)
La lipoproteína (a) o Lp (4), es una lipoproteína de baja densidad que tiene una estructura semejante al plasminógeno y que puede ser el enlace entre la hemostasia y la ateroesclerosis.37 La concentración plasmática de Lp (a) es un predictor independiente e importante de enfermedad cardiovascular ateroesclerótica. Se encuentran partículas intactas de Lp (a) en la íntima de las arterias, casi siempre en asociación con placas ateroescleróticas. Es sorprendente que la Lp (a) tiene estructuras de asa semejantes a las del plasminógeno. La Lp(a) puede competir, por medio de estas asas, con el glu-plasminógeno por el sitio de unión en las células endoteliales, desplazando al plasminógeno y disminuyendo su formación en la superficie de la célula endotelial. Además, la Lp (a) aumenta la expresión y síntesis del IAP-1, disminuyendo así la acción del t-PA.38 Como consecuencia de estas dos acciones se presenta una reducción importante en la fibrinolisis, lo que quizá facilite que se produzca el proceso trombótico al no tener oposición.
Producción y cinética de las plaquetas
Las plaquetas derivan de los megacariocitos, que se originan de células tronco mieloides pluripotenciales. Al parecer, la producción de plaquetas está controlada por una trombopoyetina que participa en la maduración final de los megacariocitos. Se han descrito otros factores que pueden tener cierta función en el control del crecimiento y diferenciación de las células formadoras de colonias de megacariocitos. Es posible que exista un solo factor estimulador de colonias de magacariocitos (FEC-Meg), así como un factor potencializador de los mismos (también denominado Pot-Meg), pero estos no han sido aún identificados ni purificados. Puede ser que el factor de células tronco, la interleucina-3 (IL-3), el factor estimulador de colonias de granulocitos y macrófagos (FEC-GM) y la interleucina-6 (IL-6) todos tengan algún papel en el control de la megacariopoyesis.40-43
Los megacariocitos sufren endomitosis, en la que ocurren divisiones nucleares sin que exista división celular y después se presenta fusión nuclear, para formar una célula con un contenido cromosómico de 8n, l6n o 32n. En respuesta a una señal desconocida aún, el citoplasma megacariocítico se transforma en una serie de bandas, delgadas y cilíndricas que después toman una apariencia de gotas. Las constricciones a lo largo de las protrusiones tubulares se vuelven más pronunciados y , al final, los fragmentos se separan para convertirse en porciones de megacariocitos que tienen el tamaño aproximado de las plaquetas.44 El volumen de los megacariocitos correlaciona con la ploidia y madurez citoplásmica, mientras más grandes, producen mayor número de plaquetas.
En los estados trombocilopénicos se observan en sangre periférica plaquetas de gran tamaño llamadas megatrombocitos, sobre todo en la púrpura trombocitopénica idiopática (PTI). Existe gran discusión respecto a si los megatrombocitos son o no plaquetas jóvenes. Algunas plaquetas muy grandes que se observan en pacientes que se recuperan de una trombocitopenia inmune pueden estar separadas en forma incompleta, y al parecer derivan de megacariocitos que maduraron con una rapidez excesiva.45 La presencia de estas llamadas proplaquetas puede ser la responsable del aumento en el volumen plaquetario promedio que ocurre durante la respuesta a, o la recuperación de una trombocitopenia aguda.
Las plaquetas que entran a la circulación sobreviven alrededor de 8.5 a 10 días, y tienen una vida media de alrededor de cuatro días. Alrededor del 30 a 40 porciento de las plaquetas existen en un reservorio esplénico que puede intercambiarse en forma libre con las plaquetas circulantes .46 Cuando aumenta la necesidad de plaquetas la producción puede aumentar siete a ocho veces. Debido a que no existe un reservorio de plaquetas en la médula ósea, esperando ser liberadas, los aumentos en el requerimiento de plaquetas dependen de un aumento en la síntesis. El tiempo de maduración de los megacariocitos no parece acortarse, pero el volumen de los mismos puede aumentar en circunstancias en que se requieren más plaquetas. Al parecer, se obtiene un mayor número de plaquetas, gracias a un mayor número de células activadas provenientes del reservorio de células tronco, y quizá por aumento en el volumen de los megacariocitos.
Pruebas de coagulación y su uso
PRUEBAS DE LOS PROCOAGULANTES
Unas cuantas pruebas de coagulación pueden Ser eficaces para la detección de la función de los procoagulantes. La mayor parte de estas pruebas están basadas en mediciones del tiempo que se requiere pira formar bandas de fibrina, que se pueden detectar por medio de dispositivos ópticos o eléctricos. En cada caso, la prolongación excesiva del tiempo de una de las pruebas puede representar una concentración anormalmente baja de algún factor [ver tabla 1], la presencia de un factor o factores biológicamente inactivos o la presencia de inhibidores.
Tiempo parcial de tromboplastina (TPT) El TPT también llamado tiempo parcial de tromboplastina activada (TPTa), evalúa el sistema intrínseco de la coagulación [ver figura 1]. En esta prueba se añade caolín, que proporciona una superficie extraña, al plasma total 6 minutos antes de la prueba, con el fin de provocar una activación máxima de los factores XII y XI. Enseguida se añade cefalina, un sustituto de los lípidos plaquetarios, al plasma y se registra el tiempo que se requiere para producir fibrina. Los valores normales del TPT varían en diferentes laboratorios, pero en general van de 25 a 39 segundos. Este rango normal tan amplio hace que sea poco práctico expresar el TPT en términos de porcentaje de un valor normal. Como el tiempo normal de protrombina es de 10 segundos y el tiempo que se requiere para formar un coágulo añadiendo cantidades óptimas de trombina al plasma es de 6 segundos, está claro que gran parte del TPT analiza la secuencia que precede a la activación del factor X [ver figura 1]. El TPT es más sensible a las alteraciones y deficiencias de la secuencia de activaciones de los procoagulantes que ocurren antes de la activación del factor X.
Tiempo de protrombina de un paso de Quick (TP) El TP es una prueba del sistema extrínseco que consiste en la agregación de factor tisular al plasma total y en la medición de la fibrina resultante, en condiciones normales, de nueve a 12 segundos. Por lo común, el TP se reporta en segundos, pero también pueden usarse curvas de tiempo y actividad basadas en estándares comerciales para expresar el TP como el porcentaje de la respuesta de una reserva plasmática normal. Un tercer método de información expresa el resultado como una relación de acuerdo con el valor normal de control. Así, se reportaría una relación de 2 si el paciente tuviera un tiempo de protrombina de 20 segundos y el valor control fuera de 10 segundos. El cálculo y notificación de los resultados del TP se complica aún más por el hecho de que las diferentes tromboplastinas comerciales tienen sensibilidad variable respecto a la reducción de los procoagulantes dependientes de vitamina K producida por los antagonistas de la vitamina K que se emplean en la práctica clínica. Por ejemplo, un estudio en Inglaterra puede notificar sobre la eficacia de un esquema de anticoagulación que produce una prolongación de dos a dos veces y media en el TP (usando la tromboplastina británica que es muy sensible). Un esquema de anticoagulación que logrará la misma relación de protrombina en un laboratorio norteamericano (que usa una tromboplastina mucho menos sensible) podría ser peligroso y originar hemorragias. La solución a este problema consiste en emplear la relación normalizada internacional (INR, por sus siglas en inglés, n. del t.) recomendada por la Organización Mundial de la Salud. De esta manera, se calibra la tromboplastina individual empleada en cada laboratorio contra una tromboplastina estándar de referencia, y el resultado se notifica como una relación de protrombina (como si cada laboratorio empleara la tromboplastina estándar de referencia) .41
El TP puede detectar deficiencias de factores 1, 11, V, VII y X. Los antagonistas del sistema extrínseco de la coagulación, incluyendo la antitrombina 111 activada por heparina o los productos de degradación de fibrina, pueden prolongar el tiempo de protrombina.
Tiempo de protrombina y proconvertina (P y P) El P y P no está muy disponible, pero es muy útil para investigar una alteración detectada en el TP. En el P y P, el plasma se diluye 1:10, disminuyendo de esta manera los efectos de la heparina o de los productos de degradación de fibrina. Se agregan fuentes de factores 1 y V a la reacción, por lo que el P y P es sensible a deficiencias de factores II, VII y X. Si el TP y el P y P están prolongados en la misma magnitud, es probable que exista deficiencia del factor 11, VII o X. Si el TP está más prolongado que el P y P, quizá sean anormales los factores 1 y V o existan inhibidores de la conversión de fibrinógeno a fibrina. Dichos inhibidores incluyen productos de degradación de fibrina o antitrombina III activada por heparina. El P y P se reporta tanto en segundos como en porcentaje del normal.
Tiempo de trombina (TT) El TT se usa para investigar alteraciones que afectan la conversión del fibrinógeno en fibrina. Cuando se añaden cantidades óptimas de trombina al plasma, aparece fibrina en un lapso de 5 a 6 segundos. Como la trombina es una enzima muy potente puede bloquear a sus inhibidores ¡n vitro. Sin embargo, si se agregan diluciones de trombina al plasma hasta que el TT normal sea de cerca de 16 segundos, los inhibidores de la conversión de fibrinógeno en fibrina, corno los productos de degradación de fibrina o el cofactor antitrombinaheparina, pueden prolongar claramente el TT. Los pacientes con disfibrinogenemia y paraproteinemias también pueden tener un TT prolongado.
Tiempo de reptilasa (TR) La reptilasa es una enzima parecida a la trombina que convierte M fibrinógeno en fibrina, pero que no resulta afectada por el complejo heparina-antitrombina III. La presencia de productos de degradación de fibrina y fibrinógeno afecta menos al TR que al tiempo de trombina. Sin embargo, el efecto de los estados disfibrinogenémicos es más acentuado en el TR que en el tiempo de trombina. Un tiempo de trombina prolongado y un TR normal indican que existe efecto de heparina48. Algunos laboratorios clínicos utilizan columnas de adsorción de uso comercial que extraen la heparina para establecer si un resultado anormal es consecuencia de este anticoagulante.
Pruebas de la acción de trombina Como la trombina desempeña una función clave en la coagulación al afectar a plaquetas, fibrinógeno, factor V, factor VIII y factor XIII, se han diseñado pruebas para medir la acción de la trombina. La trombina actúa principalmente en el fibrinógeno al separar dos moléculas de los fibrinopéptidos A y B, y dejar una molécula del monómero de fibrina. En general no han tenido éxito los intentos por medir el monómero circulante usando la prueba de gelación de etanol ó la prueba de paracoagulación del plasma con protamina (PPP). Se ha descrito un radioinmunoensayo para el fibrinopéptido A como índice de acción reciente de la trombina. Esta prueba puede usarse para evaluar la actividad de la trombina en una jeringa. Las mediciones del fibrinopéptido A pueden ser un indicador eficaz de la generación de trombina o de la actividad de la trombina.49
Fibrinógeno El fibrinógeno, o factor I, está presente en cantidades que se pueden medir químicamente, y los resultados por lo común se informan como mg/dl.
Productos de degradación de fibrinógeno y fibrina El sistema fibrinolítico es activado por los mismos factores que inician la coagulación [ver figuras 1 y 3]. La plasmina ataca tanto al fibrinógeno como a la fibrina, produciendo productos de degradación de fibrina-fibrinógeno (PDF), que pueden ser detectados por varias técnicas. Un método común utilizado para detectar PDF consiste en medir el fibrinógeno y la fibrina identificable en suero inmunológicamente, Un índice elevado de productos de degradación no indica la existencia de hiperplasminemia generalizada, como ocurre en la coagulación intravascular diseminada (CID), ya que las lesiones extensas o las enfermedades inflamatorias también pueden elevar los PDF,.Existe una prueba más precisa de los productos de degradación que detecta el dímero D de la Fibrina, un fragmento que es liberado por la degradación (mediada por plasmina) de la fibrina completamente polimerizada. Debido a que la coagulación intravascular genera fibrina polimerizada por completo, la detección del dímero D de la fibrina indica que ha ocurrido coagulación intravascular, seguida por la acción de la plasmina. En los casos de fibrinolisis primaria, la degradación del fibrinógeno o de los monómeros de fibrina soluble, por acción de la plasmina, no produce el dímero D [ver figura 1].
Factor XIII El factor XIII se puede detectar midiendo la solubilidad de un coágulo en urca. En vista de que sólo un pequeño porcentaje de la cantidad normal del factor XIII puede dar un resultado normal, esta prueba no debe usarse si el paciente ha sido transfundido dos o tres semanas antes.
Pruebas de plasminógeno, acción de la plasmina y de a-2-antiplasmina La activación del sistema de plasminógeno-plasmina se puede inferir a partir de la demostración de una prolongación del tiempo de trombina, disminución del nivel de fibrinógeno y elevación del nivel de los productos de degradación de la fibrina. Otra prueba utilizada para medir la activación del sistema de plasminógeno-plasmina es el tiempo de lisis de la euglobina. En esta prueba, que es una variante del tiempo de lisis del coágulo, se han eliminado sustancialmente los inhibidores de la fibrinolisis, de manera que se acorte el tiempo necesario para completar el análisis.
En la actualidad es posible determinar la plasmina y el inhibidor específico de plasmina o a-2-antiplasmina en forma directa. Durante la trombosis y fibrinolisis extensa, se consume plasminógeno a medida que se une a la fibrina y que es activado por varios activadores de plasminógeno [ver figura 3]. Si la utilización supera a la producción, los niveles de plasminógeno disminuyen. En la fibrinolisis primaria, los niveles elevados de plasmina circulante son contrarrestados por la a-2-antiplasmina. Este inhibidor inactiva a la plasmina al formar un complejo inerte estable con ella, y cuando el nivel de hiperplasminemia excede al nivel de producción de la a-2-antiplasmina se producirá una disminución en la concentración plasmática del inhibidor. Por consiguiente, las cifras bajas de a-2-antiplasmina suelen indicar hiperplasminemia. Otra prueba de acción de la plasmina, que hasta la fecha sólo existe en laboratorios de investigación, detecta la presencia en plasma del fragmento péptido BBI-42 en plasma [ver figura 1], formado por la acción proteolítica de la plasmina sobre el polímero de fibrina soluble.25
PRUEBAS DE PLAQUETAS Y DE FUNCION PLAQUETARIA
Evaluación del frotis de sangre periférica El examen del frotis de sangre periférica proporciona información rápida y definitiva. En un frotis normal bien teñido habrá de 8 a 12 plaquetas por campo de gran aumento (1,000x), lo que corresponde a una cuenta plaquetaria normal de 150,000 a 300,000/mm3. El frotis de sangre periférica puede examinarse para determinar el grado de granulaciones plaquetarias y la posible existencia de megatrombocitos.
Tiempo de sangrado El tiempo de sangrado, realizado en forma cuidadosa y estandarizado, proporciona información útil acerca de la cuenta plaquetaria y su función. Existe un dispositivo, activado por un resorte, con el que puede efectuarse una incisión uniforme a una presión estándar sobre los capilares. El tiempo de sangrado suele ser normal cuando la cuenta plaquetaria es mayor de 100,000/mm3 y se prolonga de manera lineal inversa cuando se relaciona con cuentas inferiores. Un tiempo de sangrado prolongado y una cuenta plaquetaria mayor de 100,000 indican alteración en la función plaquetaria. Aunque el tiempo de sangrado es una herramienta tradicional en la evaluación de los pacientes, parece ser que un tiempo de sangrado normal no predice la seguridad al realizar varios procedimientos quirúrgico o invasores, ni el tiempo de sangrado anormal predice con exactitud la propensión a hemorragias.50 El tiempo de sangrado puede ser útil para estudiar familias con enfermedad de von Willebrand (EvW) o con ciertas alteraciones funcionales de las plaquetas, pero hasta que existan datos de estudios prospectivos mejor controlados, no debe emplearse para predecir la posibilidad de hemorragia. Una excepción puede ser el estudio de pacientes con formas graves de EvW que van a someterse a cirugía, pero los datos respecto al uso de esta prueba no son aún claros.
Retracción del coágulo La retracción del coágulo es un método burdo que se puede usar para evaluar la función del mecanismo contráctil de las plaquetas.
Factor 3 plaquetario Hay métodos complicados para detectar el factor 3 plaquetario, el lípido de las plaquetas disponible para la coagulación en el punto de interacción de los factores IXa, VIII y en el punto de interacción de los factores Xa, V y II [ver figura 1], la prueba pocas veces tiene utilidad clínica.
Agregometría plaquetaria Los agregómetros plaquetarios son dispositivos simples que sirven para registrar la trasmisión de luz a través de una suspensión de plaquetas. Cuando las plaquetas se agregan, la luz pasa con mayor facilidad a través de la suspensión. Para probar la agregación se añaden concentraciones diluidas de ADP a plasma citrado rico en plaquetas.
Después de que se agrega ADP se presenta una onda de agregación, seguida por una segunda onda. La segunda onda de agregación es estimulada por las sustancias liberadas de las plaquetas que participan en la agregación inicial inducida por ADP [ver figura 7].51 La segunda onda refleja la inducción de la reacción de liberación e indica el estado de las reservas de nucleótidos en las plaquetas. La ristocetina agrega plaquetas en presencia de FvW, y causa que las plaquetas no estimuladas expresen GPIb, que a su vez sirve como receptor del FvW.52 Después de que el FvW se fija a la GPIb, las plaquetas liberan ADP y serotonina, y ambos son agentes agregantes plaquetarios muy eficaces.

|
| Figura 7 |
| Prueba de agregación plaquetaria |
PRUEBAS DE INHIBIDORES DE LA HEMOSTASIA
Antitrombina III La actividad de la AT-III puede evaluarse por bioensayo e inmunoensayo.53 Es importante contar con los dos tipos de estudio porque algunos pacientes que parecen tener valores normales de AT-III en las pruebas de inmunoensayo presentan niveles anormales cuando el factor se determina por pruebas funcionales.
Proteína C y proteína S En la actualidad existen métodos inmunológicos y funcionales de uso comercial para medir estos moduladores de la coagulación dependientes de la vitamina K. Ambos tipos de pruebas son útiles, sobre todo para la evaluación de la deficiencia de la proteína S.54 Los niveles de proteína C y S pueden determinarse incluso en pacientes con insuficiencia hepática o que reciben warfarina. Si se le notifica con anticipación, el laboratorio puede informar el nivel de proteína C y proteína S en relación con el nivel de protrombina o de factor X, determinados con precisión mediante la prueba inmunoabsorbente ligada a enzimas (ELISA). Si el valor de proteína C o de proteína S está disminuido como resultado de insuficiencia hepática o de la ingestión de antagonistas de la vitamina K como la warfarina, la relación de cualquiera de estos anticoagulantes con la protrombina o el factor X será de 1 aproximadamente; sin embargo, cuando el paciente tiene una deficiencia oculta de proteína C o S, la relación será considerablemente menor de 1.
Estudio del paciente con trastornos hemorrágicos o trombóticos
Los datos clínicos guían el diagnóstico. Es de primordial importancia distinguir los trastornos hemorrágicos hereditarios de los trastornos adquiridos. Por lo tanto, son útiles las preguntas acerca de hemorragia cuando se practicó circuncisión, después de cirugía y, en especial, hemorragias durante extracciones dentales. La ausencia de hemorragia importante en el momenla extracciónto de la extracción de muelas del juicio impactadas es fuerte evidencia contra el diagnóstico de un trastorno hemorrágico congénito. El sangrado petequial sugiere trombocitopenia o trastornos vasculares y puede ocurrir en los miembros inferiores o en las membranas bucales. Puede haber hemorragias en mucosas como resultado de trombocitopenia o por sobredosis de warfarina. En los hemofílicos se ven hemorragias de tejidos profundos, típicamente en la forma de hemartrosis. La aspirina puede alterar de manera significativa la hemostasia al inhibir la cicloxigenasa plaquetaria [ver figura 4]. Es un problema identificar a la aspirina como causa de un trastorno hemorrágico porque varios cientos de fórmulas contienen aspirina y a menudo el nombre M preparado no informa en relación con su contenido de salicilatos.
En pacientes con trastornos trombóticos, la historia clínica debe enfocarse a investigar tabaquismo, hipertensión arterial, trombosis puerperal, cirugía y procedimientos ortopédicos. Otros datos clínicos importantes para el diagnóstico incluyen el antecedente de uso de anticonceptivos orales que contengan estrógenos, antecedentes familiares de tromboembolias venosas recurrentes o tromboembolias de aparición en la juventud. La presencia de trombosis en un sitio atípico indica la existencia de un estado de hipercoagulabilidad de fondo. La trombosis de la vena hepática, de la vena mesentérica o la necrosis cutánea posterior a la administración de warfarina debe orientar hacia la búsqueda de un estado hipercoagulable; la trombosis espontánea de la vena axilar también indica la presencia de este problema (ver adelante Estados de hipercoagulabilidad).
Disminución de la cuenta plaquetaria, trombocitopenia
La trombocitopenia puede ser causada por producción anormal de plaquetas, aumento en su eliminación debido a mecanismos inmunológicos o no imunológicos, secuestro de plaquetas en el bazo, o a una combinación de todos estos mecanismos.
La presentación clínica de la trombocitopenia varía dependiendo de factores tales como la presencia o ausencia de pancitopenia y la etiología del trastorno. La característica clínica de la trombocitopenia son las petequias, que reflejan hemorragias probablemente a nivel del capilar o de la vénu la poscapilar. Las petequias por lo general ocurren en sitios de mayor presión intravascular. Por ejemplo, se desarrollan en las extremidades inferiores supuestamente debido a la mayor presión hidrostática en las venas de las piernas; aparecen en la mucosa oral de los carrillos porque los maseteros generan una fuerza enorme sobre la mucosa durante la masticación, y se encuentran en sitios donde las prendas de vestir ajustadas, como el sostén, producen aumento en la presión intravascular.
Si la cuenta de plaquetas es muy baja, también puede observarse púrpura, hemorragias mucosas y aún hemorragias en los tejidos profundos. No existe un nivel umbral de plaquetas bien determinado por encima del cual los pacientes puedan considerarse a salvo de hemorragias secundarias a trombocitopenia. En general, los pacientes con púrpura trombocitopénica idiopática (PTI) sangran menos con el mismo nivel de plaquetas que los pacientes con anemia aplásica. La medición de los volúmenes plaquetarios promedio usando contadores celulares automatizados han aclarado este hecho. Los pacientes con trombocitopenia importante (i.e., cuenta plaquetaria < 20,000/mm3) y un volumen plaquetario bajo ( menor 6.4 femtolitros por plaqueta) sangran más que las enfermos con volúmenes plaquetarios más grandes. Cuando las, plaquetas se regeneran con rapidez, como ocurre en la PTI o durante la recuperación después de supresión de la médula ósea, se liberan plaquetas muy grandes a la circulación. Es posible que estas plaquetas grandes tengan un papel más eficaz en la hemostasia porque sean más activas desde el punto de vista metabólico.35 Los pacientes de edad avanzada o que tienen otras enfermedades, sangran más que los pacientes jóvenes o que sólo sufren trombocitopenia. Los trastornos asociados, como la insuficiencia hepática o las enfermedades del tejido conjuntivo, aumentan el riesgo de presentar una hemorragia grave. Las petequias o la purpura sólas pocas veces son causa de preocupación clínica importante. En ocasiones puede presentarse una hemorragia transvaginal severa, pero el principal peligro lo constituyen las hemorragias intracraneanas, y esta preocupación es la que dirige el tratamiento.
El análisis de las causas clínicas comunes de la trorribocitopenia [ver figura 2] provee un marco de referencia para el diagnóstico diferencial. Debe preguntarse al paciente sobre los antecedentes de ingesta de medicamentos (incluyendo el abuso de sustancias por vía intravenosa), patrones de actividad sexual y antecedente de transfusiones; y se le explorará en busca de evidencia de anemia, infección neutropénica, enfermedad del tejido conjuntivo, linfoma o estado de inmunodeficiencia. La evaluación del tamaño del bazo merece especial atención. El frotis de sangre periférica establece con rapidez si existe trombocitopenia, revela las, alteraciones morfológicas de las plaquetas e indica si existe policromatofilia, neutropenia, linfopenia, blastos, esferocitosis o eritrocitos fragmentados microangiopáticos. La cuenta plaquetaria, la citología hemática completa y la cuenta de reticulocitos confirman el grado de trombocitopenia e indican si existe regeneración de critrocitos por anemia o cualquier alteración de leucocitos. El volumen plaquetario promedio, determinado por contadores automatizados de células sanguíneas, puede proporcionar un dalo adicional respecto a la causa de la trombocitopenia. Los volúmenes plaquetarios disminuidos (< de 6.4 fentolitros) indican defectos de producción, mientras que los volúmenes elevados sugieren regeneración plaquetaria rápida o producción displásica de plaquetas:55 La aspiración de la médula ósea, con o sin biopsia de Jamshidi, es decisiva para el diagnóstico diferencial.
DEFECTOS EN LA PRODUCCION DE PLAQUETAS
.El hallazgo de una médula hipoplásica en la cual la celularidad total está reducida, con disminución concomitante en los megacariocitos, implica anemia aplásica. Se debe investigar toxicidad por medicamentos. La médula que está fibrosada o infiltrada con células leucémicas u otras células malignas representa el síndrome de pancitopenia por médula infiltrada.
Una biopsia medular que muestra celularidad y maduración normales de los precursores eritroides y mieloides, con disminución de] número de megacariocitos que aparentemente son normales, sugiere que el paciente ha ingerido un medicamento que afecta específicamente a las células progenitoras megacariocíticas o a las células precursoras de esta línea.56-60 El etanol produce también megacariopoyesis ineficaz.56 Aún no está claro si el oro tiene efectos pancitopénicos generales o si deprime en forma selectiva las líneas megacariocíticas. La ingestión de medicamentos también puede causar trombocitopenia al aumentar ha eliminación de plaquetas, y en estas condiciones el número de megacariocitos puede estar aumentado.
En la deficiencia de vitamina B12 y de folatos están afectadas las tres líneas celulares de la médula ósea. En consecuencia, además de la eritropoyesis ineficaz megaloblástica, los pacientes afectados también tienen megacariopoyesis ineficaz, y sus frotis de médula ósea muestran muchos megacariocitos grandes hiperlobulados. Algunos trastornos mieloproliferativos se caracterizan por megacariopoyesis ineficaz con megacariocitos binucleados. atípicos.
Tratamiento
Si se identifica un agente agresor, se debe suspender su administración. Se ha usado dimercaprol (13 AL en aceite) para tratar la trombocitopenia inducida por oro, pero con resultados benéficos objetivos solo ocasionales. Para las deficiencias de vitamina B12 y de folatos se requieren tratamientos sustitutivos específicos.
Cuando la trombocitopenia ocasiona hemorragias importantes se requerirá la administración de plaquetas hasta que el problema pueda complementarse con otras formas de tratamiento. Cada unidad de concentrado plaquetario transfundido de donadores al azar, eleva la cuenta plaquetaria aproximadamente 10,000/mm3/m2 de superficie corporal. Por lo tanto, la administración de 5 o 6 paquetes plaquetarios debe aumentar la cuenta plaquetaria aproximadamente de 40,000 a 50,000/mm3. La mitad de las plaquetas transfundidas estarán circulando cerca de 24 horas. Se debe continuar la transfusión de plaquetas, 2 a 3 veces por semana, para evitar hemorragias hasta lograr una mejoría o hasta que el paciente se haya aloinmunizado a las plaquetas. Se ha preconizado que la administración continua de prednisona en dosis de 20 a 30 mg/día mantiene la integridad vascular, pero es cuestionable si los corticoesteroides deben usarse en vista de su eficacia no probada y de sus efectos colaterales indeseables.
ELIMINACION PLAQUETARIA ACELERADA POR DESTRUCCION INMUNOLOGICA
Cuando un paciente tiene trombocitopenia a pesar de gran cantidad de megacariocitos normales en la médula, es probable que el mecanismo del trastorno sea la eliminación acelerada de plaquetas, aunque en ocasiones también puede deberse a una escasa producción de plaquetas.61 Las pruebas que miden la supervivencia de plaquetas marcadas con cromio-51 o indio-111-oxina, cuando están disponibles, pueden demostrar la velocidad de la pérdida de las plaquetas. En condiciones normales las plaquetas sobreviven 10 días y tienen una vida media de alrededor de 4 días. En los estados de eliminación acelerada, como en la PTI, la vida media plaquetaria puede ser tan corta como de 30 a 60 minutos. La cuenta de plaquetas refleja entonces el equilibrio logrado entre la eliminación plaquetaria acelerada y la megacariopoyesis compensatoria, que puede aumentar aproximadamente siete veces sobre su valor normal.
Los estudios de supervivencia plaquetaria no están disponibles en todos los laboratorios y en general no se necesitan para determinar si existe remoción acelerada de plaquetas. En casos en los que el mecanismo de la trombocitopenia esté en duda, la infusión de plaquetas de donadores al azar puede usarse como procedimiento diagnóstico y terapéutico. Cuando la remoción plaquetaria acelerada es responsable de la trombocitopenia, la transfusión de 6 unidades de concentrado plaquetario sólo aumenta un poco la cuenta plaquetaria, que enseguida vuelve a valores basales en menos de 24 horas. Sin embargo, esta prueba terapéutica se vuelve poco confiable si el paciente ha sido previamente aloinmunizado a las plaquetas por transfusiones de sangre o de plaquetas, o por embarazos múltiples.
Cuando la trombocitopenia del paciente parezca ser causada por remoción acelerada, debe hacerse un diagnóstico diferencial rápido [ver tabla 2]. Es fundamental investigar el antecedente de ingestión de medicamentos, como lo es una historia de lupus eritematosodiseminado. linfoma o anemia hemolítica adquirida Coombs-positiva. En forma similar, infecciones tales como septicemia, varicela, mononucleosis infecciosa o por el virus de inmunodeficiencia humana (VIH), pueden ser importantes para la etiología. Las alteraciones concomitantes de las pruebas de los procoagulantes indican coagulación intravascular diseminada, mientras que la evidencia dehemólisis intravascular con glóbulos rojos mieroangiopáticos sugiere lesión vascular, como puede ocurrir en la púrpura trombocitopénica trombótica o en el síndrome urémico hemolítico.
|
||||||||||||||||||||||
|
Púrpura trombocitopénica idiopática/púrpura trombocitopénica autoinmune (PTI/PTA)
Fisiopatología En 1951, en un intento por determinar las bases de la púrpura trombocitopénica idiopática, el hematólogo norteamericano William Harrington se transfundió a sí mismo plasma de un paciente con este trastorno. El hecho de que después haya desarrollado trombocitopenia severa fue muy sugestivo de la participación de un factor antiplaquetario destructivo circulante. La observación de que las mujeres con PTI a menudo tenían hijos trombocitopénicos también apoyaba esta hipótesis. Estudios subsecuentes62 han demostrado que cerca del 90 porciento de los pacientes diagnosticados como portadores de PTI tienen cantidades aumentadas de inmunoglobulinas unidas a las plaquetas, la llamada IgG relacionada con las plaquetas (IgG-RP).63 El nivel de IgG plaquetaria se relaciona directamente con la severidad de la trombocitopenia.63 Aunque la destrucción acelerada de las plaquetas es el fenómeno fisiopatológico clave en la PTI, la disminución en la producción de plaquetas contribuye a la gravedad de la trombocitopenia en muchos pacientes.64
Con más frecuencia la inmunoglobulina que se localiza sobre la membrana de la plaqueta es de tipo IgG, y dirigida contra la glucoproteína plaquetaria GPIIb-llIa o GPlb.61 Cuando la densidad del anticuerpo es lo suficientemente grande, también puede fijarse complemento. En ocasiones también se encuentran anticuerpos de IgA e IgM. La IgG1 es la subclase de IgG que se encuentra con más frecuencia sobre las plaquetas en la PTI/PTA, pero también pueden encontrarse IgG2, IgG3 e IgG4; la subclase IgG3 produce una enfermedad más grave. Estos anticuerpos no se absorben como complejos inmunes en la superficie plaquetaria debido a que no se unen a los sitios receptores de la porción Fc de la plaqueta, sino que están unidos a la membrana plaquetaria por sus porciones Fab y parecen dirigirse contra antígenos de la membrana plaquetaria.
En la púrpura trombocitopénica idiopática aparecen confrecuencia complejos inmunes circulantes que pueden reflejar la unión de anticuerpos antiplaquetarios a antígenos solubilizados (le las plaquetas. Tales complejos inmunes se unen en seguida a los receptores Fc plaquetarios. La uniónde los complejos inmunes a las plaquetas puede no ejercer efectos patológicos, puede reforzar el ataque de los macrófagos o puede llegar a ser el mecanismo principal que induce el ataque de macrófagos y la destrucción de las plaquetas cubiertas. Los complejos inmunes circulantes componentes del complemento unidos a las plaquetas. Los complejos inmunes séricos que pueden fijarse y destruir a las plaquetas en estos pacientes parecen consistir en fragmentos F (ab')2 de anticuerpo el anticuerpo antiplaquetario se forma principalmente en las células linfoides esplénicas, se sintetiza en los linfocitos medulares, lo que explica en parte los complejos efectos de la esplenectomía. La IG-RP causa que la plaqueta afectada sea atrapada y destruida en forma extravascular por el sistema monocito-macrófago y puede interferir con la producción de plaquetas por los megacariocitos.,61 Este sistema reconoce o la porción Fc del anticuerpo unido o el C3b, cuando también se fija complemento. Los macrófagos esplénicos son los más activos para eliminar y destruir a las plaquetas porque en condiciones normales más de una tercera parte de éstas se localizan en el bazo, la circulación esplénica es lenta y el anticuerpo antiplaquetario se elabora en forma local.65,68 Los macrófagos hepáticos pueden también eliminar a las plaquetas cubiertas por anticuerpos, pero sólo cuando el padecimiento es grave.69
Por consiguiente, la púrpura trombocitopénica idiopática se caracteriza sobre todo por la destrucción rápida de plaquetas. La médula ósea responde a la trombocitopenia, sobre todo cuando es grave, aumentando la producción plaquetaria. Sin embargo, las mediciones indican que la médula ósea puede aumentar la producción de plaquetas sólo de 2.3 a 5.0 veces, en lugar de llegar al máximo teórico de siete veces.65 Parece ser que el mismo anticuerpo antiplaquetario reacciona también con los antígenos de los megacariocitos supuestamente parecidos al antígeno plaquetario. Esta reacción antígenoanticuerpo inhibe la megacariopoyesis y de esta manera se explica en forma parcial la respuesta medular subóptima. De hecho, existen informes de pacientes con PTI que aparentemente no tienen megacariocitos identificables en la médula ósea.70 En tales casos, el anticuerpo antiplaquetario inhibió el crecimiento de las unidades formadoras de colonias de megacariocitos.71
Es probable que exista predisposición genética para el desarrollo de púrpura trombocitopénica idiopática, pero los estudios del HLA aún no han resuelto esta situación. Sin duda el hallazgo de PTI en dos gemelos monocigotos indica la existencia de una base genética para la enfermedad.72 Por lo general, las plaquetas producidas en la púrpura trombocitopénica idiopática son grandes y con gran actividad metabólica; también son eficaces para controlar el tiempo de sangrado.68,73 En ocasiones, sin embargo, se ha informado que las plaquetas de pacientes con PTI funcionan en forma anormal. Es probable que un paciente con PTI, recuento plaquetario de 50,000 a 100,000/mm3 y hemorragias, presente una prolongación del tiempo desangrado que indique la presencia de una alteración en el funcionamiento de las plaquetas. En un estudio se demostró que las plaquetas de tres pacientes con estos datos clínicos mostraron agregación anormal y trastornos en el metabolismo del ácido araquidónico en su conversión atromboxano A2 [ver figura 4].68, 74 Estudios realizados en otros enfermos con PTI y plaquetas anormales revelan la presencia de autoanticuerpos dirigidos contra los compo nentes de la membrana plaquetaria GPIb o GPIIb/IIIa, que inhiben la adhesión o agregación plaquetarias.63
En ocasiones ocurren remisiones clínicas aparentes en pacientes con púrpura trombocitopénica idiopática. En tales casos, el recuento plaquetario vuelve a la normalidad aunque continúa la remoción acelerada de plaquetas.73 Esta situación, llamada trombolisis compensada, puede compararse conceptualmente a la hemólisis compensada. Es probable que su existencia explique las remisiones y exacerbaciones espontáneas, que caracterizan la evolución clínica del trastorno crónico. Cuando las plaquetas están siendo destruidas a una velocidad acelerada, o incluso con pequeñas variaciones en la producción de las plaquetas, debidas a medicamentos o a ciertas infecciones virales, puede precipitarse una recaída clínica. Asimismo, la función reforzada de los monocitos durante las infecciones virales puede acelerar aún más la remoción de plaquetas, siendo responsable de tales recaídas.65 En forma similar, las infecciones pueden aumentar la producción de anticuerpos plaquetarios al causar activación policlonal de linfocitos B.63
Diagnóstico La púrpura trombocitopénica idiopática y el púrpura trombocitopénica autoinmune aparecen de manera típica en mujeres jóvenes, pero otros grupos también pueden resultar afectados. La epidemia de SIDA ha tenido un efecto importante en la prevalencia de esta enfermedad. En algunas comunidades la prevalencia de PTI en mujeres jóvenes ha sido superada por su aparición en varones seropositivos para la infección por VIII. Tales pacientes incluyen hemofílicos dependientes de transfusiones, homosexuales y adictos a drogas por vía intravenosa.75,76 No existe antecedente relevante de ingestión de medicamentos. La mononucleosis infecciosa puede contribuir a la destrucción plaquetaria autoinmune, y es importante buscar datos clínicos de este trastorno.68 La enfermedad de Graves y la tiroiditis de Hashimoto también se asocian con púrpura trombocitopénica idiopática y con trombocitopénica autoinmune.77,78 Algunos invetigadores recomiendan incluso realizar pruebas de función tiroidea en todos los pacientes con PTI para evitar llevar a cabo 1 esplenectomía en un paciente con hipertiroidismo no diagnosticado.
En las mujeres afectadas puede haber sangrado uterino importante. La presencia de, vesículas hemáticas en la boca es un dato indicador de trombocitopenia grave. Las hemorragias retinianas son raras. En todos los pacientes debe realizarse una exploración neurológica completa y cuida dosa ante la posibilidad de hemorragia M sistema nervioso central.
En los pacientes con púrpura trombocitopénica idiopática típica no complicada el bazo no es palpable. La presencia de un bazo palpable indica la posibilidad de que entre las causas de trombocitopenia se encuentren el lupus eritematoso generalizado, el linfoma, el síndrome de Evans, la mononucleosis infecciosa o el hiperesplenismo.
El frotis de sangre periférica no muestra alteraciones de los leucocitos a menos que la hemorragia a través de las mucosas haya sido tan grave para producir deficiencia de hierro; las pocas plaquetas que se encuentran son grandes y muy granulares. La médula ósea ¡nuestra abundantes megacariocitos, muchos de los cuales son jóvenes; los precursores eritroides y mieloides permanecen normales. Los resultados de las pruebas para investigar lupus eritematoso generalizado son negativos.
Los niveles de inmunoglobulina G relacionada con las plaquetas están elevados, pero también se incrementan en otras formas de púrpura trombocitopénica. La utilidad de las pruebas para determinar la IG-RP en el diagnóstico de M es motivo de discusión. Aunque la sensibilidad de las diversas pruebas se aproxima al 90 porciento, su especificidades tan solo del 25 porciento.63 Por lo tanto, algunos expertosen la materia no consideran que la prueba de IgG-RP tengautilidad clínica.63,79 Sin embargo, otros investigadores sostienen que la determinación cuidadosa de la IgG-RP puededistinguir entre los casos de trombocitopenia inmune y los que no tienen una causa inmunológica.80 La IgG relacionada con la plaquetas existe en dos reservorios: (1) en granulos alfa, como resultado de endocitosis, y (2) sobre la super Oficie plaquetaria. Los anticuerpos de superficie son responsables del 0.5 porciento de las inmunoglobulinasplaquetarias totales y quizá estén compuestas principalmente de anticuerpos plaquetarios más otras inmunoglobulinas de superficie asociadas. Incluso las mediciones cuidadosas de las inmunoglobulinas en la superficie plaquetaria no distinguen en forma invariable la PTI de otras formas de trombocitopenia que se caracterizanpor eliminación acelerada de plaquetas y por la presenciade plaquetas grandes recién formadas, en la circulación.81 De cualquier modo, las pruebas de IgG-RP no son necesarias, ya que a menudo el diagnóstico de PTI se realiza concerteza con base en los siguientes datos: presencia de trombocitopenia con manifestaciones clínicas, a pesar de encontrar una cifra normal o aumentada de megacariocitos de apariencia normal en la médula ósea y en ausencia de datos clínicos característicos de otras enfermedades (ver antes).79 En los casos en que el diagnóstico de PTI está en duda, la prueba de IgG-RP es aún menos específica.
La púrpura trombocitopénica idiopática aguda es más común en niños y adultos jóvenes y con frecuencia la precede una enfermedad viral. En menos de tres meses se observa remisión espontánea permanente. La púrpura trombocitopénica idiopática crónica de tipo adulto es una afección que persiste por más de tres meses. Aunque la forma crónica presenta remisiones y recaídas espontáneas, son raras las remisiones espontáneas a largo plazo.
Tratamiento La púrpura trombocitopénica idiopática es un trastorno relativamente benigno que tiene una tasa de mortalidad de alrededor de uno a cinco porciento; la mayoría de las muertes son causadas por hemorragia intracraneana.65,68 El tratamiento varía dependiendo de la presentación clínica de las hemorragias, el paciente puede presentar solo petequias, hemorragias mucosas moderadas, hemorragias mucosas graves o hemorragias del sistema nervioso central. El tratamiento de los pacientes con PTI refractaria o durante el embarazo requiere atención especial. Los pacientes de edad avanzada pueden necesitar un tratamiento más, cuidadoso que los enfermos más jóvenes. Por ejemplo, los pacientes mayores de 60 años tienen un riesgo 20 veces mayor de sufrir hemorragias graves que los individuos menores de 40 años de edad que tienen la misma cuenta plaquetaria.82
Presentación con petequias solamente ("púrpura seca") Los pacientes que sólo presentan petequias no necesitan tratamiento activo. Se les puede vigilar y simplemente indicarles que estén alertas ante cualquier hemorragia en mucosas o la aparición de nuevas petequias, de manera que de inmediato pueda instituirse el tratamiento en caso de que se deteriore su estado. Las infecciones virales o bacterianas pueden provocar una recaída causando activación de los macrófagos, supresión megacariocítrica o incremento en la actividad de las células B.63 Es aconsejable evitar la aspirina.
Presentación con hemorragia moderada en las mucosas ("púrpura húmeda") El tratamiento para los pacientes con hemorragia moderada de las mucosas se inicia con esteroides, por ejemplo, prednisona en una dosis de 60 a 100 mg/día en porciones divididas. A menos que la hemorragia sea severa, el paciente no necesita ser hospitalizado y puede guardar reposo en casa. Se debe evitar la actividad física intensa, en particular cualquier actividad relacionada con la maniobra de Valsalva, para no aumentar la presión intracraneana. Se debe hacer hincapié sobre el riesgo de tomar aspirina y otros agentes antiplaquetarios. Si es necesario, se pueden transfundir glóbulos rojos, pero rara vez es necesario transfundir plaquetas en tales casos.
Los esteroides interfieren con al ataque de los macrófagos a las plaquetas y eventualmente reducen la cantidad de anticuerpo antiplaquetario que producen las células linfoides esplénicas y la médula. Por lo general la cuenta plaquetaria aumenta en un lapso de varios días a 2 o 3 semanas después del tratamiento. Cuando la cuenta plaquetaria ha alcanzado niveles normales, se pueden ir disminuyendo las dosis de esteroides en un periodo de 3 a 4 semanas. Aunque algunos investigadores han publicado remisiones completas a largo plazo con el tratamiento a base sólo de esteroides hasta en un 20 porciento de casos,65,68 los autores pocas veces han observado tales respuestas en el Hospital de la Univesidad de Stanford.
La esplenectomía está indicada cuando la cuenta plaquetaria empieza a disminuir de nuevo. El procedimiento produce remisiones prolongadas en 65 a 80 porciento de los pacientes con PTI.65,68,83 Lo mejor es reiniciar los esteroides antes de la esplenectomía, de manera que el paciente tenga una cuenta plaquetaria de por lo menos 30,000 a 50,000/mm3 en el momento de la cirugía. En general, la cuenta plaquetaria empieza a aumentar al primer día de posoperatorio, frecuentemente sobrepasando los valores normales para la segunda semana. Aunque hay publicaciones contradictorias en la literatura, el autor no cree que las mediciones del secuestro esplénico de plaquetas marcadas con radioisótopos pueda predecir en forma precisa el éxito o fracaso de la esplenectomía. El efecto de la esplenectomía depende no sólo de la eliminación de la función de atrapamiento y filtrado del bazo, sino también de la remoción de la reserva de células linfoides esplénicas, que son una fuente importante de anticuerpos antiplaquetarios.84
En pacientes de edad avanzada o en mal estado general, y que por lo tanto no telerarían la esplenectomía, se puede tratar de controlar la enfermedad administrando la cantidad mínima de esteroides necesaria para aumentar el recuento plaquetario de 30,000 a50,OOO/mm3, un nivel por arriba del cual rara vez ocurren hemorragias graves. Como alternativa, se pueden administrar dosis de 1 a 2 rng de vincristina intravenosa en intervalos de cinco a siete días. Es de esperarse que la respuesta ocurra entre tres a seis semanas; por lo tanto, no se recomienda prolongar el tratamiento con vincristina más alla de este periodo. Aún cuando ocurra una respuesta satisfactoria a la vincristina, a menudo ésta es transitoria. Es preferible administrar la vincristina o la vinblastina por infusión lenta, en lugar de la inyección en bolo, de tal manera que cuando las pocas plaquetas restantes cubiertas de inmunoglobulinas capten el medicamento y sean fagocitadas por los macrófagos, éstos sean destruidos. En este método, la vincristina en dosis de 0.03 mg/kg, 0 la vinblastina en dosis de 0. 1 mg/kg, se disuelven en 500 a 1,000 ml de solución salina isotónica y se administran por bomba de infusión continua por un periodo de seis a ocho horas. El envase y el equipo de venoclisis se cubren con aluminio para evitar la degradación de los medicamentos por la luz ultravioleta. El tratamiento debe repetirse cada cinco a 10 días, y un fármaco puede sustituirse por otro, dependiendo del desarrollo de toxicidad: neurotoxicidad en el caso de la vincristina, y mielosupresión con leucopenia en el caso de la vinblastina. Aunque la infusión prolongada es atractiva en principio, puede constituir un problema cuando se adapta a un programa de cuidados ambulatorios.85 La utilidad de fármacos potencialmente peligrosos como ciclofosfamida o azatioprina en el tratamiento de los casos clasificados como fracasos terapéuticos, sigue siendo dudoso, en vista de que estos enfermos evolucionan en forma satisfactoria.
El danazol se ha utilizado en la PTI crónica. Puede administrarse solo o en combinación con esteroides para permitir la reducción en la dosis de los últimos. La dosis habitual de danazol es de 200 mg tres a cuatro veces al día (10 a 15 mg/kg/día v.o.). Cuando hay respuesta satisfactoria, suele presentarse hasta después de dos o tres meses del tratamiento. El efecto benéfico del danazol podría deberse a la reducción en el número de receptores Fe situados sobre los monocitos. Es probable que la vinblastina tenga una acción similar, y que altere la capacidad de los monocitos y quiza de los macrófagos que fagocitan las plaquetas cubiertas de IgG.63,86
La administración de vacuna neumocócica a los pacientes esplenectomizados es una práctica clínica común. Sin embargo, esta vacuna ha provocado recaídas de la PTI cuando se administra a pacientes que estaban en remisión.87 Un método para resolver este problema es no administrar la vacuna y vigilar cuidadosamente al paciente en busca de signos de infección neumocócica. Sin embargo, quizá sea preferible administrar la vacuna antes de la cirugía.68
Presentación con hemorragia severa de las mucosas o del sistema nervioso central La hemorragia severa de las mucosas o del sistema nervioso central es una auténtica urgencia médica, que requiere hospitalización. Al paciente se le debe colocar en una unidad capaz de proporcionar las atenciones adecuadas. Se transfunden glóbulos rojos según se requieran, y se administra prednisona inmediatamente, iniciando con una dosis de 100 rng y continuando con 25 rng cada 6 horas. Se transfunden plaquetas con la esperanza de que tengan algún efecto durante su breve circulación, aun cuando sean destruidas junto con las plaquetas del propio paciente y no produzcan un aumento mesurable en la cuenta plaquetaria. Se desconoce la cantidad apropiada de plaquetas que deben transfundirse, pero parece ser razonable la administración de 6 a 20 unidades de plaquetas de donadores al azar, cada 6 a 12 horas.88 El autor acostumbra administrar 2 rng de vincristina por vía intravenosa en infusión continua durante 6 horas,85 y repetir la dosis en 5 días sí la cuenta plaquetaria no ha empezado a aumentar. No hay datos para apoyar el uso de vincristina en tales circunstancias, pero los riesgos de este tratamiento son bastante menores en relación con los beneficios potenciales. Cuando existe hemorragia uterina severa puede administrarse una sola dosis intravenosa de 25 mg de estrógenos conjugados para controlar la hemorragia, después de lo cual se puede realizar la esplenectomía.
Puede realizarse esplenectomía de urgencia en casos de hemorragia severa e incontrolable del tubo digestivo o vaginal. Al iniciar la cirugía se administran al paciente 100 mg de metilprednisolona por vía intravenosa, seguidos de 6 unidades de plaquetas de donadores al azar. Se transfunden otras 6 unidades de plaquetas después de que el cirujano divide el pedículo esplénico. Se administran también esteroides, en dosis de 60 a 100 rng de prednisona o prednisolona, hasta que la cuenta plaquetaria comience a normalizarse. En el estado agudo, algunos investigadores recomiendan el tratamiento con megadosis de esteroides, esto es, la infusión de 1 g de metilprednisolona durante un periodo de 30 minutos cada día durante tres días consecutivos.89 La transfusión de plaquetas después del bolo de metilprednisolona puede ser benéfico, cuando la transfusión de plaquetas no fue eficaz sin este esteroide. También se pueden considerar otras alternativas de tratamiento, además de la esplenectomía de urgencia (ver adelante).
Si se sospecha hemorragia en el sistema nervioso central, puede llevarse a cabo una punción lumbar usando una aguja del No. 20; el procedimiento debe hacerse con cuidado a causa de la trombocitopenia severa. Si los datos clínicos sugieren la posibilidad o probabilidad de una hemorragia del sistema nervioso central en evolución, se puede realizar una tomografía computada para confirmar el diagnóstico. La decisión de llevar a cabo una tomografía computada es motivo de controversia, pero el autor recomienda no transferir al paciente fuera de la unidad para dicho procedimiento, ya que podría ser desastroso retirarlo de la vigilancia minuciosa y del tratamiento en ese momento.
Cuando se sospecha la existencia de una hemorragia inminente o establecida en el SNC o una hemorragia no controlada en otros sitios, el médico tiene varias opciones terapéuticas: esplenectomía de urgencia (ver antes), plasmaféresis o administración intravenosa de gamaglobulina humana en dosis altas. Aunque no hay estudios controlados comparando estas tres modalidades, el autor prefiere la administración de IgG humana por vía intravenosa, lo que ha producido una mejoría transitoria de la trombocitopenia. La IgG por lo general se aplica en dosis de 18 a 25 g diarios en un lapso de 30 a 60 minutos; la dosis total recomendada en un periodo de cuatro a cinco días es de 1.0a 1.5 g/kg.90,91 Otro grupo de investigadores administra IgG intravenosa en dosis hasta de 0.8 g/kg/día, durante cinco días. Una dosis tan alta deI IgG, que representa l0 veces la cantidad de IgG sintetizada en un día, produce una respuesta temporal en alrededor de cinco a siete días.95 En caso de urgencia puede administrarse IgG por vía intravenosa en dosis de 0.4 g/kg así como ocho a 10 unidades de plaquetas de donadores al azar en forma de bolo una vez completa la infusión.93 La transfusión de- plaquetas después de administrar IgG por vía intravenosa produce un aumento mayor y más prolongado en el recuento plaquetario. La administración de IgG por vía endovenosa puede producir bloqueo reticuloendotelial al cubrir los receptores de IgG-Fc situados sobre los monocitos-macrófagos.63
En ocasiones la administración de IgG en dosis altas produce una remisión prolongada, pero no está clara la explicación de este hecho. Aunque muy costosa, la infusión de IgG en dosis altas parece controlar la hemorragia en algunos casos, en particular en pacientes jóvenes con PTI. A continuación, puede llevarse a cabo una esplenectomía, en condiciones más estables.
Ha habido publicaciones anecdóticas de resultados favorables con la plasmaféresis terapéutica, un método alternativo o suplementario si no es eficaz la IgG en dosis altas. La plasmaféresis puede ser más útil para manejar las hemorragias graves en la púrpura trombocitopénica idiopática aguda,94 que para tratarla hemorragia recurrente en la PTI crónica. Como la plasmaféresis es más eficaz para retirar complejos inmunes grandes, esta técnica es más apropiada para el tratamiento de la trombocitopenia causada por enfermedad por complejos inmunes que para la causada por ataque por autoanticuerpos a las plaquetas.68
PTI refractaria Se considera que un paciente tiene púrpura trombocitopénica idiopática refractaria si después de la esplenectomía y del tratamiento con esteroides persiste con trombocitopenia grave, o si entra en remisión pero posteriormente recae y ya no responde a dosis altas de prednisona. Como pocos pacientes con enfermedad trombocitopénica crónica padecen hemorragias graves cuando sus cuentas plaquetarias son mayores de 30,0001 mm3, frecuentemente es prudente aceptar una respuesta incompleta y no administrar formas más tóxicas de tratamiento.95 Sin embargo, si la cuenta plaquetaria es muy baja u ocurre hemorragia importante, es necesario realizar otros procedimientos. En estos casos, se administran agentes inmunosupresores, por lo común en la siguiente secuencia:
1. Vincristina intravenosa, 1 a 2 mg/semana, administrada durante tres a seis semanas. La vincristina también puede estimular la producción de megacariocitos. En forma alterna puede administrarse vinblastina en dosis de 0.1 mg/kg. Ambos medicamentos pueden administrarse como un bolo o en una infusión intravenosa continua durante seis a ocho horas.85
2. Si la combinación de vincristina y prednisona no mantiene la cuenta plaquetaria del paciente esplenectomizado en límites razonables (de 50,000 a 100,000/mm3), se debe iniciar azatioprina en una dosis de 100 a 150 mg/día. Se debe vigilar la citología hemática y la cuenta plaquetaria durante tres a seis semanas. Este régimen ocasionalmente produce un efecto ahorrador de prednisona, después del cual se puede disminuir la dosis de prednisona. También puede disminuirse la dosis de azatioprina.
3. Como alternativa, o si la azatioprina no funciona, se agrega ciclofosfamida en una dosis de 100 a 150 mg/día y se continúa diariamente durante tres a seis semanas. La respuesta a este agente puede ser similar a la que sigue a la azatioprina. La ciclofosfamida y la azatioprina se deben usar con precaución. Ambos medicamentos son mielosupresores, y se han asociado con el desarrollo subsecuente de linfoma histiocítico y de leucemia mieloide aguda. Si se debe continuar con prednisona, se puede instituir un programa de vías, alternas con objeto de reducir los efectos del hipercorticismo.
4. Se ha informado que el danazol, en dosis diaria de 400 a 800 rng durante un periodo de dos a tres meses, es benéfico en algunos casos.86,96 Se han notificado algunas. respuestas en pacientes que reciben dosis mucho más bajas de este medicamento (50 mg/día).
5. Algunos reportes anecdóticos indican que la ciclosporina, en dosis de 4 a 12 mg/kg/día, en dos dosis divididas, puede ayudar a aumentar la cuenta de plaquetas al actuar como un agente inmunomodulador.98
6. Se ha informado que la administración de interferón alfa durante 12 días en dosis de 3 millones U/día es benéfica en alrededor de la cuarta parte de los pacientes refractarios,89, 99 pero el autor no ha observado, en su práctica clínica, buenos resultados con este medicamento.
7. Con base en la experiencia con la inmunoglobulina G humana intravenosa, se ha administrado anticuerpos antiD a pacientes Rh+ (D+) con PTI con la teoría de que los eritrocitos cubiertos con anticuerpos podrían fijarse a los receptores Fc sobre los macrófagos y evitar la eliminación acelerada de las plaquetas. 100 El tratamiento con un anti-D (WinRho) intravenoso en dosis de 25 g/kg el día 1, con dosis repetidas según se requiera los días 3 y 4, produjo un aumento moderado y breve de las plaquetas, con toxicidad mínima y disminución de la hemoglobina de solo 1 a 2 g/ dl (por hemólisis inmunológica). Se han logrado resultados semejantes al administrar otra preparación anti-D (RhoGAM) por vía subcutánea en dosis de 1,800 ug (alrededor de 25 ug/kg )en tres ocasiones separadas (con dos o tres días de diferencia). 101 Esta forma de tratamiento puede administrarse en casa por medio de ¡inyecciones intramusculares semanales.102 Debido a que este método de tratamiento tiene una toxicidad muy baja y evita los efectos inmunosupresores asociados con otros agentes, puede ser útil para pacientes con M asociada a infección por VIII.
Un reporte no confirmado sugiere que la radiación esplénica con una dosis total de 75 a 1,370 cGy (75 a 1,370 rads) administrada en una a seis semanas, puede estabilizar la cuenta de plaquetas a un nivel más seguro en los pacientes ancianos,103 pero el autor no tiene experiencia con este tipo de tratamiento.
Es importante examinar la sangre en el paciente esplenectomizado refractario para buscar la presencia continua de cuerpos de Howell-Jolly, que aparecen en los glóbulos rojos después de la esplenectomía. La desaparición de los cuerpos de Howell-Jolly sugiere la presencia de un bazo accesorio remanente o de un bazo regenerado. De hecho, se han encontrado tales bazos accesorios en pacientes cuyos glóbulos rojos contenían cuerpos de Howell-Jolly en forma persistente.104 Tres de cinco de dichos pacientes tuvieron una respuesta plaquetaria favorable después de someterse a una segunda esplenectomía para extirpar los bazos accesorios. Por lo tanto, si la situación clínica lo requiere, es probable que deba llevarse a cabo la búsqueda de un bazo accesorio en pacientes esplenectomizados refractarios que siguen teniendo cuerpos de Howell-Jolly en los glóbulos rojos de la sangre periférica. El rastreo esplénico se realiza usando glóbulos rojos marcados ya sea con Tc99mm o con Cr 51, 104
Rara vez, los pacientes con PTI no tienen megacariocitos detectables en la médula ósea, debido a que los anticuerpos antiplaquetarios presentan reacción cruzada con antígenos de los megacariocitos, causando supresión en el crecimiento y desarrollo de los megacariocitos. En tales casos el diagnóstico es difícil y casi siempre es necesario descartar un síndrome mielodisplásico. Un paciente con este trastorno respondió después de 30 días de tratamiento con ciclofosfamida, administrada en dosis de 50 a 100 rng V.O.70 En otro estudio, el paciente respondió primero a la plasmaféresis extensa y después a la administración de vinblastina intravenosa.71
El tratamiento de la PTI en el complejo relacionado al SIDA (CRS) se enfrenta a problemas especiales. Debido a que el VIII ataca y destruye a las células encargadas de la inmunocompetencia, el uso de agentes inmunosupresores en estos pacientes puede ser peligroso. Con frecuencia los pacientes con CRS y PTI están concientes de esto y es difícil que acepten el tratamiento con medicamentos inmunosupresores. En los casos en que la disminución en la cuenta plaquetaria es moderada no se requiere tratamiento. Cuando la trombocitopenia es grave puede administrarse un tratamiento breve con prednisona, seguido de esplenectomía. Este sistema ha causado buenas respuesta,, en varios casos sin inducir complicaciones infecciosas graves.75,76,105
La hemorragia trombocitopénica aguda en la PTI asociada con infección por VIII puede tratarse administrando IgG intravenosa en una dosis única de 1 g/kg.106 La PTI crónica asociada a VIII puede responder con la administración de zidovudina (también conocida como AZT) en dosis de 250 rng cada seis horas por vía oral o, si se toleran, 500 mg cada ocho horas durante 12 semanas. La respuesta a la zidovudina sugiere que el VIH tiene un papel importante en el desarrollo del de PTI.107 El anti-D puede también ser útil en el tratamiento de la PTI crónica asociada con VIII.100-102 La dapsona, administrada en dosis de 50 a 125 mg/día
durante dos a 43 meses, parece ser eficaz para aumentar la cuenta de plaquetas en pacientes con PTI asociada a infección por VIH.108
PTI en el embarazo Si aparece trombocitopenia por primera vez durante el embarazo, el diagnóstico diferencia] inicial debe incluir preeclampsia al igual que las otras causas usuales de este trastorno [ver tabla 2]. Una vez que se hace el diagnóstico de PTI, las opciones terapéuticas son limitadas debido a que la esplenectomía puede causar aborto espontáneo y los agentes inmunosupresores pueden dañar los órganos fetales en desarrollo. De esta manera, los esteroides siguen siendo la modalidad terapéutica principal . Sin embargo, en casos de hemorragia trombociopénica severa, el autor recomendaría todos los tratamientos disponibles para poder proteger la vida y bienestar de la madre.
Como el anticuerpo antiplaquetario en la PTI tiene amplia especificidad y caso siempre es una IgG, puede cruzar la placenta y producir trombocitopenia en el feto. Durante un parto vaginal, la presión aplicada a la cabeza de un feto trombocitopénico puede inducir una hemorragia intracraneana. La preocupación acerca de esto ha llevado a muchos expertos a recomendar la operación cesárea temprana en mujeres con historia de PTI o enfermedad activa. Es difícil estimar la frecuencia de muertes perinatales que ocurren en las mujeres con PTI; se ha publicado que la mortalidad en los recién nacidos de estas mujeres es M siete porcientol.109
Aunque ayudan a minimizar la mortalidad fetal, las operaciones cesáreas pueden causar hemorragia importante en la madre trombocitopénica.109 En un esfuerzo para limitar la frecuencia del procedimiento en dichos casos, se han hecho intentos para correlacionar la trombocitopenia de¡ recién nacido con las cuentas plaquetarias y con los niveles de IgG-AP materna; no obstante, estos factores no han probado ser indicadores confiables.110 Sin embargo, el nivel de anticuerpos antiplaquetarios en el plasma materno sí se correlacionó en forma notable con el recuento plaquetario del recién nacido110 y podría usarse como marcador para seleccionar a las pacientes para operación cesárea.
Algunos investigadores109,111 han encontrado que la administración de prednisona en una dosis de 20mg/día de 10 a 14 días antes del parto aumenta el recuento plaquetario del recién nacido y produce un pronóstico neonatal razonablemente bueno; se supone que esto sucede porque los esteroides cruzan la placenta y ejercen un efecto terapéutico en el feto. Sin embargo, dicha administración materna de prednisona no siempre beneficia al recién nacido.110
Mientras se completan los estudios para una mejor resolución de estos problemas, lo siguiente parece representar un tratamiento razonable. Si se dispone de ensayos para IgG antiplaquetaria sérica o plasmática, pueden utilizarse éstos como indicadores para seleccionara las pacientes que deberán someterse a una operación cesárea. En caso de no contar con éstos, el autor recomienda tratara todas las pacientes con historia de PTI o PTI activa con prednisona o prednisolona en dosis de 20 a 30 mg/día, iniciando dos semanas antes de la fecha probable del parto. Durante el parto deben hacerse intentos para obtener tan pronto como sea posible muestras de la sangre venosa del cuero cabelludo fetal con el fin de realizar cuentas plaquetarias.112 Si la cuenta plaquetaria del recién nacido es de 50,000/mm3 o menor, debe interrumpirse el parto vagina¡ y proceder a una operación cesárca. Ciertas unidades obstétricas equipadas en forma especial pueden obtener muestras de sangre fetal por medio de una técnica percutánea hacia la vena umbilical justo antes del nacimiento. Si la cuenta de plaquetas del feto es normal, puede inducirse el trabajo de parto y permitirse un parto vaginal, pero si la cuenta de plaquetas es baja (v.gr., < 50,000/mm3) se realizará una cesárca o se transfundirán plaquetas al feto. Si el niño tiene trombocito penia después del parto, debe recibir prednisona en una dosis diaria de alrededor de 2 mg/kg.
Aunque los pasos descritos antes constituyen las normas generales de tratamiento de la trombocitopenia durante el embarazo, en una reunión internacional reciente se aportaron datos que sugieren un cambio en el manejo. Investigadores canadienses obtuvieron las cuentas plaquetarias de más de 10,000 mujeres embarazadas y sanas y encontraron que del cinco al siete porciento de éstas tenían cuentas disminuidas. Se consideró que el 80 porciento de estas pacientes tenían una entidad denominada trombocitopenia incidental del embarazo, caracterizada por plaquetas entre 70,000 y 150,000/mm3. De 500 de estas pacientes, ninguna tuvo problemas hemorrágicos, y casi todas tuvieron partos vaginales; sólo el cuatro porciento de los recién nacidos sufrió trombocitopenia moderada, y ninguno de estos infantes tuvocuentas plaquetarias menores a 50,000/mm3. El grupo de investigación de Canadá recomienda intervenir sólo en los casos en que existan complicaciones graves como CID o preclampsia. Además, recomiendan que se administre IgG intravenosa a la madre si la cuenta de plaquetas disminuye más de 50,000/mm3, lo que sugiere que pudiera tener PTI, más que trombocitopenia incidental del embarazo. Se consideró que, en ocasiones, las cuentas de plaquetas obtenidas de muestras venosas del cuero cabelludo fetal fueron más bajas de lo real por errores en la muestra, y se concluyó que el peligro asociado con la toma percutánea de muestras de la vena umbilical es equivalente al beneficio que pueda causar. 113-115
Púrpura trombocitopénica secundaria
Los pacientes con lupus eritematoso diseminado, enfermedad de Hodkin o linfoma no Hodgkin pueden presentar un cuadro clínico idéntico al visto en la púrpura trombocitopénica idiopática. El estudio diagnóstico y tratamiento son los mismos que en la PTI.
El llamado síndrome de PTI en estas condiciones se puede complicar con otras manifestaciones del trastomo primario. El hígado puede estar agrandado y es posible que aumente el secuestro esplénico. En los pacientes con linfoma, la médula puede estar infiltrada con la neoplasia maligna y el tratamiento antineoplásico o inmunosupresor puede reducir más la actividad megacariocítica compensatoria. Varios pacientes con el síndrome de PTI tienen niveles de IgG-AP elevados en forma similar a los vistos en pacientes con PTI.116 Los pacientes con lupus eritematoso generalizado o linfoma pueden tener síndrome de Evans, o una asociación de PTI con anemia hemolítica autoinmune. El tratamiento del síndrome de Evans es el mismo que para la PTI y anemia hemolítica autoinmune.
El síndrome de PTI que se asocia con la enfermedad de Hodgkin puede ocurrir en pacientes que no muestran datos de actividad de la enfermedad neoplásica, previamente diagnosticada y tratada. Este patrón contrasta con el que se observa en casos de anemia hemolítica autoinmune asociada con enfermedad de 1 Hodgkin. En la última, el inicio y exacerbación de la hemólisis parece coincidir con la exacerbación del trastorno principal.
Se ha publicado una causa mediada por autoinmunidad de destrucción plaquetaria y trombocitopenia en pacientes con tumores sólidos en los cuales se ha descartado coagulación intravascular diseminada.117 La trombocitopenia en estos pacientes asemeja a la PTI en cuanto a a que los megacariocitos medulares y la IgG-AP están aumentados, la supervivencia plaquetaria es corta y el número de plaquetas se eleva después del tratamiento con esteroides. No está claro que el tumor sea un factor causal; estos casos pueden simplemente representar alteraciones simultáneas de dos enfermedades.118 El diagnóstico y tratamiento son similares a los de la PTI.
Púrpura postransfusional
La púrpura postransfusional (ver adelante Accidentes quirúrgicos contra enfermedades sistémicas, Causas frecuentes de hemorragia posoperatoria), puede ocurrir dos a10 días después de la transfusion de sangre total o de componentes que contengan plaquetas. De manera típica, la cuenta plaquetaria es muy baja, menor de 10,000/mm3, y hay abundantes megacariocitos en la médula ósea. En el diagnóstico diferencial deben considerarse la trombocitopenia séptica, la CID y la PTI inducida por heparina. La hemorragia puede ser grave y la trombocitopenia suele durar cerca de cuatro semanas, aunque se han informado casos en los que la trombocitopenia y la hemorragia persistieron hasta 120 días.
La fisiopatología del trastorno no es clara. En la mayoría de los casos el paciente ha estado expuesto con anterioridad a aloantígenos plaquetarios durante el embarazo o el parto o como consecuencia de una transfusión. Los estudios revelan que la mayoría de los pacientes tienen anticuerpos contra el antígeno plaquetario Zwa, el cual se encuentra presente sobre las plaquetas del 90 porciento de los individuos normales. Los pacientes que desarrollan púrpura postransfusional casi siempre son negativos para el antígeno Zwa. Por lo tanto, resulta difícil comprender cómo es que un aloanticuerpo dirigido contra un antígeno presente en las plaquetas de donadores al azar (o en los fragmentos plaquetarios de la suspensión de glóbulos rojos) causa la destrucción de las plaquetas autólogas, del paciente, que no expresan al antígeno. No han tenido éxito los intentos para tratar este problema mediante la transfusión de plaquetas negativas para el Zwa.
No existen estudios clínicos controlados que evalúen el tratamiento de la púrpura postransfusional, pero en estos casos parece razonable administrar IgG por vía endovenosa en dosis de 0.4 a 0.8 g/kg durante 30 a 60 minutos, seguida por la transfusión de ocho a 12 unidades de plaquetas de donadores al azar. Este enfoque se basa en la esperanza de que el bloqueo del sistema reticuloendotelial inducido, por la administración intravenosa de IgG permitirá que las plaquetas transfundidas sobrevivan el tiempo suficiente para producir un efecto hemostático.119 Otro procedimiento terapéutico, quizás menos eficaz que el descrito, consiste en la administración de prednisona en dosis inicial diaria de 2 mg/kg y aumentándola hasta dosis masivas para obtener respuesta. Si los resultados no son satisfactorios y la situación del enfermo pone en peligro su vida, puede ser útil la plasmaféresis terapéutica.120
Destrucción plaquetaria inmune inducida, por medicamentos
El cuadro clínico de la destrucción plaquetaria inmune inducida por medicamentos es indistinguible del de la púrpura trombocitopénica idiopática, aunque con más frecuencia se ven en la boca vesículas llenas de sangre. Puede haber síntomas generales. Se debe buscar con insistencia el antecedente de ingestión de fármacos.
La médula ósea muestra abundantes megacariocitos. Se ha propuesto como prueba la inhibición por medicamentos de la retracción del coágulo, pero es difícil de llevar a cabo y con frecuencia da resultados falsos negativos. En laboratorios especiales se puede detectar la presencia de anticuerpos contra fármacos.
Púrpura por quinidina y quinina Los anticuerpos patógenos en casos de púrpura por quinidina o quinina se desarrollan en tan poco tiempo como 12 días después de la exposición al medicamento responsable.121 Los anticuerpos se unen a las plaquetas a través de sus receptores Fab (el locus del reconocimiento del antígeno).122 Existe una hipótesis que afirma que las plaquetas cubiertas con el fármaco expresan un neoantígeno inestable que enseguida es estabilizado por la unión al anticuerpo, En vista de que el medicamento es necesario para la expresión contínua del neoantígeno hipotético, el anticuerpo parece ser dependiente del fármaco.123 Los anticuerpos contra las plaquetas dependientes del medicamento inducidos por quinina o quinidina parecen interactuar en forma preferente con la glucoproteína GPIb-IX y, en menor grado, con la GPII-IIIa.124
Resulta obvio que el fármaco responsable (quinidina o quinina) debe suspenderse. En la púrpura por quinidina o quinina, ni el tratamiento con esteroides ni la esplenectomía de urgencia han demostrado beneficios. La plasmaféresis para extraer estos fármacos y los anticuerpos parecería ser el tratamiento lógico; pero no se cuenta con estudios sistemáticos sobre su eficacia. Las plaquetas transfundidas son eliminadas con la misma rapidez que las plaquetas del propio receptor. No obstante, puede intentarse la transfusión plaquetaria para controlar las hemorragias que pongan en peligro la vida.
Aunque no se cuenta con estudios controlados, la práctica de los autores consiste en tratar la trombocitopenia intensa inducida por quinidina o quinina asociada con hemorragias con la administración de prednisona en dosis diarias de 100 rng y la infusión de IgG por vía intravenosa en dosis de 0.4 a 0.8 g/kg durante una hora. Al terminar la infusión, se administran de 10 a 12 unidades de plaquetas de donadores al azar. El grupo de los autores ha obtenido buenos resultados con este enfoque en tres casos.
Trombocitopenia por heparina Es probable que existan dos tipos de trombocitopenia inducida por heparina. Una forma leve ocurre en los primeros cuatro días de la administración de heparina en cerca del cinco porciento de los casos.126 Incluso los catéteres bañados con heparina contienen una cantidad suficiente para causar este síndrome clínico. 21 Existe una disminución leve, no progresiva, en el recuento plaquetario, que no requiere intervención.126 La forma más grave de trombocitopenia inducida por heparina produce disminuciones del recuento plaquetario importantes desde el punto de vista clínico, a menudo menores de 50,000/mm3, y se relaciona con tromboembolias, sobre todo arteriales. 125,126 Parece ser que un anticuerpo antiplaquetario de IgG dependiente de heparina es el responsable de esta forma grave de trombocitopenia. Estudios realizados in vitro han demostrado que estos anticuerpos pueden producir agregación extensa de las plaquetas normales en presencia de heparina. 126 Un estudio sugiere que la heparina se fija a la superficie de la plaqueta, exponiendo un neoantígeno que en algunos pacientes despierta una respuesta de autoanticuerpos.128 Se supone que la formación de estos agregados in vivo es responsable de la disminución en el recuento plaquetario y del desarrollo de trombosis arterial. En algunos pacientes con trombocitopenia por heparina se desarrollan anticuerpos que reaccionan con la heparina unida a las células endoteliales o con el sulfato de heparán sintetizado en este sitio. Por lo tanto, estos pacientes presentan una lesión doble: daño a las células endoteliales mediado por una reacción inmunológica, inducida por heparina, combinado con agregación plaquetaria inducida también por la heparina.129
El tratamiento de la trombocitopenia por heparina puede ser muy difícil, sobre todo cuando ha ocurrido trombosis arterial. Puede hacerse el intento de cambiar de tipo de heparina, en especial si se ha empleado la bovina, pero si este cambio no es benéfico, será necesario suspender el medicamento. Sin embargo, el paciente casi siempre requiere de continuar la anticoagulación, o incluso de un esquema más intenso. El ácido acetilsalicílico puede ser útil para prevenir la agregación plaquetaria, y algunos investigadores recomiendan la administración intravenosa de 500 rng de solución de dextrán al seis porciento. Existen reportes anecdóticos acerca de que la administración de IgG por vía intravenosa produce un aumento en la cuenta plaquetaria.130 Algunos especialistas recomiendan usar al anticoagulante ancrod, un derivado de veneno de víbora que causa desfibrinación rápida y que no tiene relación química con la heparina. En los pacientes con trombocitopenia por heparina y que requieren de anticoagulación, se inicia ancrod en dosis de 1 a 2 U/kg por vía subcutánea o por infusión intravenosa durante seis a 24 horas, teniendo como meta lograr una concentración de fibrinógeno de 50 a 100 mg/dl. La dosis se ajusta para mantener esta concentración de Fibrinógeno; por lo general es necesario administrar alrededor de 1 a 2 U/kg cada periodo de 24 horas. El tratamiento con ancrod se mantiene hasta que se logren niveles terapéuticos de warfarina.131 Los heparinoides de bajo peso molecular han demostrado ser útiles en el ámbito clínico, pero no han sido autorizados aún por la IDA de los EE.UU.132, 133El problema de la trombosis arterial inducida por heparina puede evitarse iniciando el tratamiento con warfarina al mismo tiempo que la heparinización. La trombocitopenia por heparina que causa trombosis arterial suele presentarse hasta después de cuatro a seis días de tratamiento, y en ese momento la warfarina ya tiene efecto terapéutico y la heparina puede suspenderse con seguridad.
Trombocitopenia por oro Es probable que la trombocitopenia por oro deba considerarse como otro ejemplo de destrucción plaquetaria inmune inducida por fármacos, aunque no hay datos compatibles con una reacción por anticuerpos tipo fármaco-antifármaco, como sucede en la trombocitopenia por quinidina y quinina. Sin embargo, el tratamiento con sales de oro para la artritis reumatoide produce trombocitopenia en uno a tres porciento de los pacientes. Esta complicación se caracteriza por aumento de los megacariocitos en la médula ósea, disminución de la supervivencia de las plaquetas y presencia ocasional de anticuerpos plaquetarios. La mayoría de los sujetos responden al tratamiento con 60 rng de prednisona al día. No se ha establecido la utilidad del dimercaprol como agente quelante del oro. Los pacientes que no responden a los esteroides parecen beneficiarse con la esplenctomía.134
Trombocitopenia asociada con cocaína Se ha notificado un síndrome semejante a la PTI en los adictos a cocaína intravenosa. Estos individuos responden a un manejo parecido al empleado en la PTI, incluyendo esteroides, IgG intravenosa y esplenectomía. No se ha identificado el mecanismo causa] de la trombocitopenia en estos casos.135
REMOCION ACELERADA DE LAS PLAQUETAS POR MECANISMOS NO INMUNOLOGICOS
Hay varios mecanismos posibles no inmunológicos que pueden ser responsables de la remoción de cantidades suficientes de plaquetas como para causar trombocitopenia importante. Las alteraciones de los vasos sanguíneos o daño a las células endoteliales pudieran inducir intensa activación plaquetaria; la aparición en el plasma de factores activadores de plaquetas o de agentes aglutinantes podría ocasionar que se formaran cantidades iinportantes de conglomerados de plaquetas; y el aumento de los niveles de trombina en el plasma, en forma local o sistémica, podría producir coagulación intravascular con consumo de plaquetas, al igual que activación y agregación plaquetaria intensas.
La diferencia entre las formas inmunológica y no inmunológica de destrucción plaquetaria es arbitraria, y existe superposición considerable. Por ejemplo, el nivel de IgG-APestá aumentado en varias de estas entidades. Dicho dato puede reflejar la unión de complejos inmunes a los receptores Fc plaquetarios, lo cual podría ocurrir bajo circunstancias muy diferentes a las del mecanismo inmunológico primario.
Varias de las entidades situadas en esta categoría son raras, pero se describen con algún detalle debido a que pueden ser rápidamente mortales, a menos que se inicie un tratamiento eficaz inmediato.
Púrpura trombocitopénica trombótica y síndrome urémico-hemolítico del adulto
Cuadro clínico y diagnóstico Es probable que el término púrpura trombocitopénica trombótica (PTI) no denote una entidad clínica única y coherente. En lugar de ello, parece comprender un grupo de síndromes clínicos en los cuales algún evento primario lesiona las paredes de los vasos sanguíneos pequeños, causando la aprición de las cinco manifestaciones principales de la TTP: (1) anemia hemolítica grave asociada con una concentración muy aumentada de deshidrogenasa láctica (DHL) y un frotis de sangre periférica que muestra los esquistocitos y células en casco característicos de la hemólisis microangiopática, (2) trombocitopenia grave, con aumento de los megacariocitos en médula ósea, disminución de la supervivencia plaquetaria y aumento de la B-tromboglobulina sérica, que indican destrucción o activación plaquetaria intravascular, (3) fiebre, en ocasiones muy elevada, (4) síntomas y signos neurológicos notables que suelen iniciarse con agitación pasajera, cefalea y desorientación, pero que a menudo evolucionan en forma rápida hacia hemisparesia, afasia, crisis convulsivas, datos de focalización y muerte, y (5) enfermedad renal, casi siempre leve y que produce elevación moderada de la creatinina sérica y de las proteínas urinarias. Otras características clínicas menos frecuentes incluyen insuficiencia hepática grave, infartos isquémicos ocasionales de la piel, del intestino o de los huesos e insuficiencia cardiaca congestiva con arritmias cardiacas. La enfermedad suele aparecer entre los 10 y los 40 años de edad, con una incidencia máxima a los 25 años y muy rara vez ocurre en personas mayores de 50 años de edad; hay ligero predominio en mujeres.136,137 Una variante de este trastorno aparece en el posparto y las manifestaciones neurológicas pueden confundirse inicialmente con depresión posparto, lo cual puede ser de consecuencias trágicas.
La forma adulta del síndrome urémico-hemolítico (SUII) pudiera ser parte del mismo espectro clínico de la PTT, o puede representar un trastorno diferente. Las características comunes de la púrpura trombocitopénica trombótica y del síndrome urémico-hemolítico incluyen hemólisis microangiopática, trombocitopenia y presencia de agregados de plaquetas y trombos de plaquetas, y fibrina en los pequeños vasos.138 Sin embargo, el síndrome urémicohemolítico tiende a presentarse en pacientes más jóvenes y muestra variaciones tan sorprendentes en severidad e incidencia local que su aparición sugiere en ocasiones la presencia de una epidemia. La participación renal es uniformemente grave en el SUH, mientras que la enfermedad del SNC es menos prominente que en la PTT. Como el tratamiento de la PTT y del SUH del adulto es similar, los dos trastornos se considerarán juntos.
Tanto la PTTcomo el SUH necesitan ser diferenciados del lupus eritematoso diseminado y del síndrome de Evans. El dato de hemólisis microangiopática combinada con leucocitos neutrofílica importante y una prueba de Coombs directa negativa (prueba directa de antiglobulina) es muy compatible con el diagnóstico de PTT o SUH. Las pruebas de coagulación no revelan en general alteraciones importantes, y los niveles séricos de DHL y B-tromboglobulina están aumentados. La biopsia de médula ósea realizada para evaluar el número y la morfología de los megacariocitos, puede mostrar los trombos hialinos de plaquetas característicos, pero no patognomónicos, rodeados por fibrina, que se encuentra principalmente en las arterias pequeñas y arteriolas.139 La biopsia gingival no parece ayudar en forma importante en el diagnóstico.139, 140
La complejidad de la púrpura trombocitopénica trombótica y el síndrome urémico-hemolítico está enfatizada por el hecho de que el tratamiento agresivo con intercambio de plasma puede producir una remisión tan prolongada en algunos pacientes que parecen estar curados de esta grave enfermedad. Otros pacientes, que clínicamente no parecen diferentes, recaen meses o años después de dicho tratamiento. Esta observación sugiere que puede haber formas crónicas y agudas de PTT/SUH.
Varias circunstancias pueden predisponer al desarrollo de púrpura trombocitopénica trombótica o síndrome urémico hemolítico. En forma anecdótica se ha publicado desarrollo del SUH del adulto después de partos normales,141´ desprendimiento prematuro de placenta normoinserta o preeclampsia, en pacientes con retención de placenta e incluso en asociación con el uso de anticonceptivos orales.142 Un caso de PTT pareció desarrollarse como complicación de la enfermedad de los Legionarios.´ Se ha informado PTT/SUH en pacientes con cáncer, algunos de estos pacientes han sido tratados con mitomicina C, que quizá tenga alguna relación etiológica.144, 145 También la ciclosporina ha sido implicada como causa de este síndrome,146 y se ha notificado SUH en pacientes sometidos a trasplante autólogo o alogénico de médula ósea por neoplasias hematológicas, incluso en algunos que no recibieron ciclosporina.147
Parece ser que el agente antiplaquetario ticlopidina también puede causar un síndrome semejante a la M. La incidencia del mismo Parece ser baja. pero la evolución ha sido mala en los pacientes afectados. 148
Fisiopatología Dos mecanismos, que no son mutuamente excluyentes, se han propuesto para explicar la PTT/SUII.138 Una hipótesis postula que el mecanismo primario es la liberación de un factor que aglutina las plaquetas o las estimula para que se agreguen. Los acúmulos de plaquetas entonces ocluirían y dañarían las arteriolas y podrían producir efectos isquémicos distales. La adherencia subsecuente de mayor número de plaquetas contribuiría a aumentar el daño vascular, trombocitopenia y hemólisis mieroangiopática. La segunda hipótesis sugiere que el defecto primario es un daño endotelial arteriolar extenso. La activación y adherencia plaquetarias, oclusión y formación de bandas de fibrina subsecuentes causarían entonces trombocitopenia y hemólisis microangiopática.
Algunos autores han informado que el suero de pacientes con púrpura trombocitopénica trombótica contiene un anticuerpo lgG que es citotóxico para las, células endoteliales humanas en cultivo.149 Otros informes han documentado la presencia de un factor aglutinante de plaquetas en el plasma de pacientes con PTT.149, 150
Un estudio indica que en la forma crónica recurrente de la M el complejo factor VIII-factor de von Willebrand existen en forma de multímeros más grandes de lo normal.151 El complejo circulante factor VIII-FvW expresa por lo común varias funciones, incluyendo las propiedades anticoagulantes del factor VIII y la actividad agregante del cofactor ristocetina. Los grandes multímeros encontrados en los pacientes con PTT pudieran hacer que las plaquetas se agregaran en presencia de sustancias naturales con propiedades parecidas a las de la ristocetina. En contraste con su mecanismo de acción normal, los multímeros del FvW encontrados en el suero (le los pacientes con PTT no producen agregación al unirse a la GPIb de la membrana plaquetaria;152 sin embargo, pueden provocar agregación plaquetaria al unirse a la GPIIb-IIIa.146 En los pacientes estudiados, las concentraciones de estos multímeros excesivamente grandes aumentan durante la remisión y disminuyen en las recaídas; estos hallazgos sugieren que los multímeros se consumen en el proceso de agregación plaquetaria.151 Es posible que los pacientes con PTT crónica recurrente sean capaces de producir multímeros de tamaño normal. Algunas situaciones como el embarazo o las infecciones pueden precipitar la liberación de sustancias parecidas a la ristocetina, que podrían inducir la agregación intravascular de plaquetas. Es posible que la presencia de multímeros grandes, que pueden ser extraídos en forma eficaz por medio del intercambio del pluma, sea la responsable de los beneficios (antes inexplicables) de esta técnica en la PTT/SUH. Los beneficios ocasionales descritos después de la infusión del plasma normal pueden deberse a enzimas proteolíticas que degradan los grandes multímeros a un tamaño normal.150 Se ha identificado un complejo inmune agregador de plaquetas en casos de PTT/SUH relacionados con cáncer.145
Se ha sugerido que tanto el daño a las células endoteliales como la agregación plaquetaria contribuyen al desarrollo de PTT/SUH. Está hipótesis propone que el daño o estimulación de la célula endotelial provoca que ésta libere los multímeros de factor von Willebrand de mayor tamaño que, como resultado de un procesamiento defectuoso en el plasma, causan gran agregación plaquetaria.146
Algunos investigadores encontraron que los pacientes con PTT carecen por completo del activador del plasminógeno que normalmente está presente en la íntima vascular. De esta manera, la lisis del coágulo que por lo común ocurriría, está ausente en la PTF, lo que pudiera explicar la acumulación y aún propagación de trombos en este trastorno.153
Dos productos del metabolismo de los endoperóxidos, el tromboxano A. y la prostaciclina, desempeñan funciones importantes en la función de las plaquetas: el tromboxano A2 es un agente agregante muy potente [ver figura 4], y la prostaciclina es un agente antiagregante muy eficaz [ver figura 2]. Por tanto, se han hecho intentos para identificar alteraciones del metabolismo de los endoperóxidos en pacientes con M, pero no ha habido evidencia concluyente de dichas anormalidades.154, 155
La dificultad para distinguir entre los mecanismos inmunológicos y no inmunológicos de la trombocitopenia se ilustra en la PTT. Aunque la destrucción plaquetaria intravascular parece resultar de mecanismos no inmunológicos, tales como daño endotelial o agregación plaquetaria, algunos pacientes con PTT también tienen niveles elevados de IgG-AP.68 Por lo tanto, este ensayo de anticuerpos no pueden usarse para diferenciar entre PTT y PTI.
La coagulación intravascular diseminada ya no se considera un mecanismo de producción de la PTT/SUH.
Tratamiento Se han usado gran cantidad de agentes en el tratamiento de la PTT. La variedad de métodos refleja la evolución tan grave de la PTT (la mayor parte de los pacientes mueren en los 10 días siguientes al diagnóstico) y su alta tasa de mortalidad (de 60 a 80 porciento). No se han realizado aún ensayos clínicos clásicos. Dentro del tratamiento se administran esteroides para controlar la inflamación vascular y dipiridamol, aspirina, sulfinpirazona y dextranos para bloquear la agregación plaquetaria (antes se empleaba heparina, cuando se pensaba que la coagulación intravascular diseminada podía estar relacionada con la FM. En la actualidad se han introducido técnicas de infusión e intercambio de plasma, que causan mejoría sustancial en la respuesta, produciendo remisiones clínicas duraderas y disminuyendo en forma franca la mortalidad. El intercambio de plasma permite extraer sustancias que son dañinas para las células endoteliales o que causan agregación plaquetaria. La infusión de plasma puede proporcionar un inhibidor deficiente o una sustancia que degrade los multímeros grandes de factor VIII-FvW a su tamaño normal.156 Un estudio aleatorio encontró que el intercambio de plasma era superior a la infusión del mismo corno tratamiento para la PTT.157
Los aspectos terapéuticos que se describen a continuación reflejan la práctica actual en el tratamiento de la PTT/SUH. El estado del SNC del paciente es crucial para determinar la intensidad el tratamiento. Datos adicionales útiles incluyen la evaluación seriada de la DHL, del frotis de sangre periférica y de la cuenta de reticulocitos. El tratamiento se inicia con 60 rng de prednisona al día en dosis hasta de 200 mg/día,158 0.3 g de aspirina cada 12 horas y 100 rng de dipiridamol cada 6 horas. Algunos especialistas administran vincristina en dosis de 2 rng intravenosos en cuatro horas y repiten esta dosis en intervalos de cuatro a siete días si no ha habido mejoría.159,160 Como la toxicidad de la vincristina no es alta, el autor la añadiría al programa de tratamiento dependiendo de la evolución del paciente.
Si la trombocitopenia es moderada y el paciente no presenta deterioro neurológico, puede intentarse la infusión simple de plasma. La dosis indicada equivale a un volumen plasmático simple (2,500 a 3,000 mi, cerca de 45 ml/kg, o 10 a 12 unidades de plasma fresco congelado) en las primeras 24 horas, y después tres unidades de plasma fresco congelado por día.161 En el estudio aleatorio que comparó la infusión con el intercambio de plasma,157 la dosis de plasma utilizada fue de 30 ml/kg I.V. durante las primeras 24 horas y después 15 ml/kg/día. Si el paciente presenta datos mínimos de confusión mental o deterioro neurológico, o si las plaquetas disminuyen o se eleva la DHL, debe inciarse plasmaféresis intensiva, extrayendo el equivalente de un volumen plasmático y sustituyéndolo por plasma fresco congelado compatible. En el estudio aleatorio,157 se eliminó 1.5 veces el volumen plasmático calculado, y se remplazó con plasma fresco congelado durante cada uno de los tres primeros días de tratamiento, posteriormente se intercambió un volumen plasmático por día durante un mínimo de siete días. Es necesario vigilar con gran cuidado los efectos del intercambio plasmático para garantizar que los electrolitos, proteínas plasmáticas y procoagulantes permanezcan en sus niveles normales. Aunque existen informes aislados de pacientes que respondieron a la infusión sóla del plasma, en los pacientes con afección grave debe utilizarse plasmaféresis intensiva combinada con infusión de plasma.
Como la plasmaféresis tiende a disminuir más la cuenta plaquetaria en un paciente que ya está trombocitopénico, surge el problema de la transfusión de plaquetas. Algunos autores han observado que la infusión de plaquetas en la PTT puede causar exacerbación de la enfermedad.162 Otros usan transfusiones de plaquetas según se necesite,163 medida que también utiliza el autor. No se ha establecido el volumen óptimo ni la frecuencia de la plasmaféresis y del reemplazo de plasma. Si la condición del paciente empieza a mostrar mejoría, los autores continuan con el programa. Si la condición del paciente sigue deteriorándose, se intensifica el programa de plasmaféresis extrayendo dos veces el volumen plasmático (de 5,000 a 6,000 mildía, o aproximadamente 80 ml/kg/día). Una vez que se ha logrado beneficio terapéutico, valorado por la restauración de la función normal del SNC, aumento de las cuentas plaquetarias y disminución de los niveles de DHL, se puede reducirla intensidad y frecuencia del intercambio de plasma, optando por intercambios de un solo volumen de plasma, primero 3 y luego 2 veces a la semana. A continuación se pueden disminuirlos esteroides, la dosis de salicilatos se puede reducir a 0.3 g al día y posteriormente 0.3 g cada tercer día, y finalmente se puede disminuir y suspender el dipiridamol.
La mieroangiopatía puede persistir semanas o meses después de que las manifestaciones clínica,,, de la enfermedad han desaparecido. Algunos pacientes han permanecido en remisión clínica completa por años, lo que indica que sufrieron un ataque agudo y autolimitado precipitado por algún evento particular. En comparación, otros pacientes que en apariencia tenían manifestaciones idénticas, han recaido en meses o años, y parecen tener una forma crónica y recurrente de PTT. Alrededor del 10 a 30 porciento de los pacientes que sufren un episodio de PTT siguen teniendo alguna evidencia de actividad de la enfermedad146,157,158 y requieren tratamiento de mantenimiento con esteroides, infusión o intercambio de plasma, o alguna combinación de estas modalidades.
Varios médicos experimentados han recomendado ampliamente la esplenectomía 164,165, y el autor ha tenido un éxito notable con este tratamiento. Sin embargo, también ha habido varios fracasos que estuvieron complicados por los efectos de la esplenectomía en una situación clínica dificil de antemano, por lo que los autores ya no practican la esplenectomía sistemática en pacientes CM PTT. 146,158
Se ha preconizado la infusión del agente antiplaquetario dextrán 164. Queda por determinar la utilidad de la prostaciclina. 164 Las mismas modalidades de tratamiento usadas en la M se han usado también en el tratamiento del SUH, junto con hemodiálisis para la insuficiencia renal"' y tratamiento médico de la hipertensión.
Coagulación intravascular diseminada
La activación intensa del mecanismo de coagulación causa hipertrombinemia con activación plaquetaria, coagulación intravascular y remoción de plaquetas. La coagulación intravascular diseminada (CID), que representa otro mecanismo no inmunológico de trombocitopenia, se discute con mayor detalle después (ver adelante Trastornos hemorrágicos adquiridos, Coagulación intravascular diseminada).
Trombocitopenia inducida por infección
Infecciones virales, bacterianas, micóticas o parasitarias graves pueden producir CID y, en consecuencia trombocitopenia. Sin embargo, mecanismos diferentes ala CID pueden también causar trombocitopenia asociada a infecciones.
Infecciones virales Algunas infecciones virales como el dengue y la rubéola congenita pueden dañar directarnente los megacariocitos. 167 La varicela puede causar una forma de trombocitopenia que tiene las características de una reacción inmune: aumento en la cantidad de megacariocitos, ausencia de datos de CID y presencia de IgG-AP o IgM-AP.168 Por lo general no se requiere tratamiento.
La trombocitopenia aguda de la mononucleosis infecciosa probablemente está mediada por mecanismos inmunológicos, como lo muestra el aumento en los megacariocitos de la médula, el aumento en la IgG-AP y la respuesta favorable a los esteroides. En ocasiones la trombocitopenia es grave, pero, como se ha hecho notar, responde a los esteroides.167
Septicemia bacteriana Los pacientes con septicemia grave por gram negativos y cuentas plaquetarias por debajo de 50,000/mm3 tienen evidencia de CID .117 Sin embargo, muchos pacientes con septicemia por gram negativos y gram positivos, y cuentas plaquetarias entre 50,000 y 150,000/mm3 no tienen evidencia de CID. En estos casos puede estar participando un mecanismo inmunológico, puesto que los niveles de IgG-AP de los pacientes a menudo están elevados, y el grado de aumento se correlaciona con la gravedad de la trombocitopenia.
La clave para controlar la trombocitopenia es establecer el tratamiento adecuado para la infección. Si hay CID, debe tratarse como se ha descrito (ver adelante Trastornos hemorrágicos adquiridos, Coagulación intravascular diseminada) con control cuidadoso de la hipotensión y del volumen sanguíneo. Si hay trombocitopenia clínicamente significativa no causada por CID, se deben administrar transfusiones de plaquetas, según las necesidades, para evitar hemorragias.
Infección por protozoarios En el paludismo es común la trombocitopenia, aunque la CID es rara. La supervivencia plaquetaria es corta, y se ha encontrado que la IgG-AP está elevada. Los anticuerpos de IgG al parecer se unen a través de su porción Fab a un antígeno de la membrana plaquetaria.169 Las plaquetas poseen receptores para antígenos palúdicos, y durante la parasitemia, estos antígenos se unen a la superficie plaquetaria. A continuación, los anticuerpos de IgG antipalúdicos producidos durante la respuesta inmune a la infección atacan a los antígenos que están unidos a las plaquetas. Si el paciente presenta trombocitopenia severa y no hay evidencia de CID deben transfundirse plaquetas.
Trombocitopenia en el embarazo y perinatal
Además de sufrir hipertensión, proteinuria y evidencia de cambios patológicos en los riñones, hígado, sistema nervioso central y placenta, aproximadamente el 15 porciento de las pacientes con preeclampsia tienen trombocitopenia moderada. Sólo algunas de las pacientes con preeclampsia y trombocitopenia tienen datos de laboratorio de CID. El número de megacariocitos está aumentado y la supervivencia de las plaquetas está poco acortada. Algunas pacientes con preeclampsia y trombocitopenia también tienen hemólisis microangiopática, lo que sugiere que los vasos sanguíneos dañados que contienen bandas de fibrina están destruyendo glóbulos rojos y plaquetas. El aumento en los niveles de IgG-AP en algunas pacientes indica la posibilidad de un mecanismo inmunológico o de un mecanismo mediado por complejos inmunes. También puede participar el vasoespasmo intenso que causa daño endotelial y activación, adherencia y destrucción plaquetaria.
El tratamiento consiste en el cuidado prenatal de la preeclampsia y los esfuerzos para detectar trombocitopenia tan pronto como sea posible. La trombocitopenia que se identifica por primera vez en el momento del parto puede causar problemas especiales. El diagnóstico diferencial en dichos casos incluye preeclampsia contra PTI aguda; si la paciente tiene el último trastorno, también debe considerarse la posibilidad de trombocitopenia en el recién nacido. En estas circunstancias puede ser imposible hacer un diagnóstico diferencial preciso. Si la trombocitopenia es grave, puede ser necesario iniciar la administración de esteroides y transfundir plaquetas, y después atender el parto de manera que se minimicen los riesgos y la trombocitopenia del recién nacido (ver antes).170
Alrededor del cinco porciento de las mujeres tienen cuentas plaquetarias menores de 136,000/mm3 durante el periodo prenatal y posparto, por lo que se considera que tienen trombocitopenia [ver antes, PTI en el embarazo]. El recuento plaquetario suele volver a cifras normales en término de una semana después del parto, y no tiene consecuencias clínicas graves. Algunas de estas pacientes trombocitopénicas cursan con niveles elevados de IgGRP, pero lo más frecuente es que aumente la concentración de C3 relacionado con las plaquetas. No se conoce la causa de la trombocitopenia en estas pacientes.171
Síndrome de HELLP El síndrome de HELLP (por sus siglas en inglés, n. del t.) se refiere a un trastorno que ocurre durante el embarazo y que se caracteriza por hemólisis, aumento en las concentraciones de enzimas hepáticas y cuenta de plaquetas disminuida. No es claro si este trastorno grave es una forma de preeclampsia, de CID o es otro tipo de fenómeno. Las pacientes afectadas sufren trombocitopenia en algún momento entre la 23 y la 39 semanas de gestación, con cuentas plaquetarias menores de 100,000/mm3, hemólisis microangiopática, pruebas anormales de función hepática y, en ocasiones, hipertensión. 172,173 Los resultados de las pruebas estándar para CID [ver adelante, Trastornos hemorrágicos adquiridos, Coagulación intravascular diseminada] son normales, aunque exista cierta elevación en la concentración de productos de degradación de la fibrina y disminución de los niveles de AT-III.173 Con frecuencia las pacientes están muy graves y algunas sufren pulmón de choque, hematomas subcapsulares de hígado con ruptura, insuficiencia circulatoria, paro respiratorio, insuficiencia renal aguda, hemorragias posparto, crisis convulsivas y ceguera. El síndrome se trata suspendiendo el embarazo, casi siempre por parto, y proporcionando tratamiento cuidadoso de sostén. La etiología del síndrome se desconoce.
Trombocitopenia en la hipotermia
La trombocitopenia puede ocurrir en ocasiones en la hipotermia inducida durante la cirugía cardiaca. La hipotermia en el anciano al parecer también puede causar trombocitopenia; en estos casos se han informado niveles de plaquetas tan bajos como 30,000/mm3, 174 Los mecanismos propuestos para explicar la trombocitopenia incluyen CID y secuestro hepático y esplénico. Una vez que la temperatura corporal vuelve a niveles normales, el recuento plaquetario retorna a valores normales en forma espontánea, en el lapso de una dos semanas.
Lavado de plaquetas y alteraciones del lecho vascular
El lavado transoperatorio era antes una causa frecuente de trombocitopenia no inmunológica. Los pacientes que presentaban hemorragia súbita durante la cirugía y a los que se les transfundía con sangre total almacenada, desarrollaban trombocitopenia después de que había recibido cerca de 10 unidades de sangre almacenada. Esto se debía a que se sometía al paciente a una exsanguineotransfusión con sangre que contenía plaquetas no viables. El trastorno se desarrolla con rapidez porque no existen reservas de plaquetas en la médula. La cuenta de plaquetas disminuye y las pruebas de TP, TPT y TT son normales. Por lo tanto, es necesario vigilar la cuenta de plaquetas en los pacientes que reciben transfusiones masivas (v.gr., 10 unidades de eritrocitos o sangre total). Si la concentración de éstas cae por debajo de 100,000/mm3 y el paciente es sometido a cirugía u otro reto hemostático, deberán administrarse plaquetas
Las plaquetas también pueden ser extravasadas a través de un lecho vascular anormal. En los hemangiomas gigantes existe un flujo sanguíneo lento a través de conductos con un endotelio inadecuado. Estas superficies pueden producir una coagulación intravascular diseminada leve. La septicemia por gram negativos con endotoxemia puede causar trombocitopenia aguda transitoria, supuestamente haciendo que las plaquetas se adhieran al endotelio dañado.
SECUESTRO DE PLAQUETAS
El tercer mecanismo importante de trombocitopenia es el secuestro de plaquetas. Se ve trombocitopenia relativamente moderada, con cuentas plaquetarias del orden de 40,000 a 80,000/mm3, en pacientes con esplenomegalia significativa. Rara vez ocurre hemorragia clínicamente importante, a menos que exista un trastorno hemorrágico coexistente, como pudiera ocurrir en la hepatopatía grave con esplenomegalia congestiva. Puede haber anemia y leucopenia asociadas.
El secuestro de plaquetas parece ser consecuencia del aumento en el lecho vascular esplénico. En condiciones normales, del 20 al 30 porciento de las plaquetas circulantes están reversiblemente secuestradas en el lecho vascular esplénico, y se supone que estas plaquetas se intercambian con las plaquetas de la circulación. En el hiperesplenismo, del 50 al 80 porciento del volumen plaquetario total puede estar secuestrado en el bazo.46
El tiempo de supervivencia de las plaquetas está ligeramente acortado, hasta cerca de dos o uno y medio días, pero la producción medular de plaquetas sólo aumenta de uno y uno y medio hasta dos veces lo normal.73 El hecho de que la médula ósea no lleve a cabo una respuesta máxima orienta en cuanto al mecanismo que determina la capacidad de la producción plaquetaria. El mecanismo aparentemente no es un contador que mide la concentración de plaquetas por unidad de volumen, sino es más bien un receptor que responde a la masa plaquetaria total o a los productos metabólicos de la masa plaquetaria. Ciertas enfermedades causan hiperesplenismo que puede producir o contribuir al desarrollo de trombocitopenia [ver tabla 3].
|
||||||||
|
El tratamiento se dirige hacia la enfermedad primaria. En pocas ocasiones se justifica la esplenectomía con base en la hemorragia trombocitopénica aislada.
Trastornos de la función plaquetaria
El dato clave en cuanto a la existencia de un defecto de la función de las plaquetas es el dato de hemorragia clínica en presencia de tiempo de sangrado prolongado y una cuenta plaquetaria mayor de 100,000/mm3. Son raras las petequias. La morfología de las plaquetas y, en ocasiones, las pruebas sencillas de función plaquetaria como la retracción del coágulo, pueden ser anormales [ver tabla 4].
|
|||||||||||||||
|
ALTERACIONES DE TIPO HEREDITARIO
Trastornos de la membrana plaquetaria
Las alteraciones hereditarias de la función de la membrana plaquetaria son raras. En la enfermedad de Bernard Soulier se observan plaquetas gigantes en el frotis de sangre periférica. El tiempo de sangrado está prolongado y hay trombocitopenia moderada. Puede haber hemorragia mortal, generalmente a partir de superficies mucosas. El defecto consiste en la ausencia de la glucoproteína plaquetaria (GPIb-IX), que es el sitio de unión del factor de von Willebrand en la plaqueta activada, causando deterioro en la agregación de la plaqueta a las superficies de las heridas. Además, como la aglutinación plaquetaria inducida por ristocetina depende de la unión del FvW con GPIb, dicha aglutinación también es anormal y no se corrige al agregar plasma normal que contenga factor de von Willebrand.175 Las hemorragias agudas pueden tratarse con transfusiones de plaquetas normales de donadores al azar. Si se requieren transfusiones frecuentes, se deben obtener las plaquetas de un hermano o de otro donador que tenga HLA compatible con el paciente.
También se han observado hemorragias mortales de las superficies mucosas en la trombastenia de Glanzmann, un trastorno autosómico recesivo raro. La morfología y el recuento plaquetario son normales, pero el tiempo de sangrado está prolongado. Como está ausente la glucoproteína GP Ilb/IIla fundamental que forma el sitio de unión plaquetario para el fibrinógeno, las plaquetas no se agregan cuando son estimuladas con ADP o colágena. Sin embargo, la aglutinación inducida por ristocetina es normal. 171 La unión de la fibrinonectina y del FvW a las plaquetas trombasténicas también es defectuosa, tal vez porque estos factores también tienen un sitio de unión en algún lugar del complejo GPlIb/IIIa.176 La hemorragia aguda ve trata como en la enfermedad de Bernard-Soulier.
Trastornos de los gránulos plaquetarios
Síndrome de la plaqueta gris y de deficiencia de gránulos densos Los pacientes con el síndrome de la plaqueta gris, un trastorno raro, presentan hemorragias mucosas, equimosis y petequias. Hay trombocitopenia moderada y el tiempo de sangrado está prolongado. Las plaquetas son más grandes de lo normal, y agranulares por la ausencia de gránulos alfa. Como el contenido de los gránulos alfa está reducido en forma importante, son deficientes la adhesión plaquetaria y la coagulación apoyada por las plaquetas. La agregación plaquetaria con colágena es anormal. Los episodios hemorrágicos deben ser tratados con transfusión de plaquetas normales.175
Otro tratorno raro, el síndrome de deficiencia de gránulos densos, se caracteriza por hemorragias mucosas asociadas con un recuento plaquetario normal, morfología plaquetaria normal y prolongación variable del tiempo de sangrado. La agregación plaquetaria con ADP y colágena es anormal. La disminución del contenido de ADP en los gránulos densos deteriora los eventos mediados por ADP. La hemorragia se trata con transfusiones de plaquetas.175
Un tratamiento alternativo para los pacientes con trastornos primarios de las plaquetas, como los síndromes de la plaqueta gris y deficiencia de gránulos densos que requieren cirugía, consiste en el tratamiento con 1-desamino-8-D-arginina vasopresina (DDAVP), en dosis de 0.3 ug/kg en solución salina, administrados justo antes de la cirugía y 12 a 24 horas después si el tiempo de sangrado vuelve a estar prolongado. Es probable que la DDAVP actúe al liberar multímeros del FvW desde las células endoteliales.177
Enfermedad de von Willebrand
Como la enfermedad de von Willebrand es causada por una deficiencia del factor plasmático, el factor de von Willebrand, será discutida junto con los trastornos hereditarios de la coagulación (ver adelante Trastornos hereditarios de la coagulación). Sin embargo, el vWF interviene en la adhesión plaquetaria a las superficies subendoteliales, de manera que la enfermedad de von Willebrand también puede ser considerada como un trastorno hereditario de la función plaquetaria [ver tabla 4].
TRASTORNOS ADQUIRIDOS
Enfermedades mieloproliferativas
Pueden ocurrir alteraciones de la función plaquetaria en enfermedades mieloproliferativas: leucemia mieloide crónica, policitemia vera, trombocitemia esencial y leucemia aguda. La cuenta plaquetaria en los trastornos mieloproliferativos crónicos está con frecuencia muy aumentada, pero el tiempo de sangrado puede estar prolongado y pueden ocurrir hemorragias manifestadas por hemorragias mucosas y hematomas. Las alteraciones se parecen a las de los defectos adquiridos por almacenamiento. Los megacariocitos a menudo son anormales con núcleos separados; las plaquetas de la sangre periférica son grandes y ocasionalmente no granuladas.
El tratamiento de la hemorragia aguda consiste en la transfusión de plaquetas normales para aumentar el nivel normal de plaquetas hasta 50,000/mm3 Las plaquetas defectuosas del paciente son incapaces de sufrir la reacción de liberación, aunque pueden agregarse razonablemente bien. las plaquetas normales transfundidas llevan acabo la reacción de liberación, proporcionando el ADP, que puede inducir la agregación de las plaquetas anormales del paciente. Se requiere la transfusión de cerca de seis unidades de concentrado plaquetario si la enfermedad no se acompaña de trombocitopenia.
Uremia
Se ha demostrado que en la uremia se prolonga el tiempo de sangrado y pueden existir hemorragias clínicas a pesar de que la cuenta de plaquetas sea normal. La causa de este trastorno no es clara, pero parece relacionarse con un defecto en las plaquetas urémicas que evita que éstas se distribuyan en forma normal sobre las superficies endoteliales expuestas, un proceso que es mediado por la interacción entre el FvW y la GPIIb-llIa de las plaquetas.178 Este defecto en la adhesión de las plaquetas urémicas puede estar mediado en parte por óxido nítrico, el factor de relajación derivado del endotelio.179
Antes se usaba hemodiálisis para retirar supuestos productos inhibitorios, o transfusiones de plaquetas para el tratamiento de la hemorragia urémica espontánea o para preparar a un paciente urémico para cirugía. Sin embargo, la observación de que la infusión de 10 unidadesde crioprecipitado puede controlar el sangrado urémico grave y acortar el tiempo de sangrado ha motivado la introducción de nuevos procedimientos.180 En la actualidad se ha demostrado que un análogo no vasoconstrictor de la vasopresina, DDAVP, también es eficaz para controlar la hemorragia urémica.181 Se ha usado la DDAVP para tratar la enfermedad de FvW y la hemofilia leves debido a que hace que el FvW almacenado sea liberado de las células endoteliales hacia el plasma. La infusión intravenosa de DDAVP, en dosis de 0.3 11g/kg en 50 mi de solución salina en un periodo de 30 minutos, acorta el tiempo de sangrado de los pacientes urémicos en un lapso de una hora después de su administración. La infusión de DDAVP produce aumento en la actividad plasmática de FvW, y particularmente en los multímeros mayores de FvW. El aumento en el nivel de multímeros tan grandes puede aumentar la adhesión plaquetaria en pacientes urémicos, aun cuando tales pacientes tienen por lo común niveles endógenos normales o elevados de actividad de FvW.
El hematócrito debe mantenerse arriba de 30 porciento en pacientes urémicos con hemorragias debido a que el tiempo de sangrado es más prolongado cuando el hematocrito es menor de 26.184 Si el paciente con uremia presenta hemorragias de importancia clínica con tiempo de sangrado prolongado o requiere cirugía, la hemorragia debe controlarse administrando estrógenos conjugados. En un estudio, la administración de estrógenos conjugados por vía oral o intravenosa en dosis diaria de 10 a 15 mg durante aproximadamente 10 días acortó el tiempo de sangrado después dedos a seis días y detuvo la hemorragia. El tiempo de sangrado permanece más acortado que el valor basal durante tres a 10 días después de la suspensión de los estrógenos.185 En un estudio confirmatorio, otro estrógeno conjugado (Emopremarin) administrado en dosis diarias de 0.6 mg/kg I.V. durante cinco días consecutivos, también acortó el tiempo de sangrado, por lo general, a las seis horas después de la infusión inicial.
El mecanismo de acción de los estrógenos se desconoce, pero al parecer el tratamiento estrogénico puede ayudar al control de la hemorragia urémica en circunstancias definidas, y evita el uso de productos sanguíneos, que tienen el riesgo de infección. Debido a que el comienzo del efecto benéfico de los estrógenos ocurre después de seis horas, debe utilizarse DDAVP o crioprecipitado para obtener hemostasia inmediata en casos de hemorragia urémica aguda y severa. La infusión de estrógenos conjugados debe comenzarse al mismo tiempo para alcanzar un efecto más prolongado.186
Enfermedad hepática
Además de hiperesplenismo y síntesis defectuosa de procoagulantes, hay evidencia de que ocurre CID leve y persistente en casos de enfermedad hepática severa. El deterioro de la depuración de activadores de plasminógeno puede contribuir a elevar los niveles de productos de degradación de fibrinógeno y fibrina. Estos productos pueden interferir con la función plaquetaria, y su nivel se correlaciona con las hemorragias que aparecen en la cirrosis hepática grave. El tratamiento debe dirigirse a la atención de la causa primaria.
Macroglobulinemia y otras disproteinemias
La presencia de concentraciones altas de proteínas viscosas produce efectos complicados en todo el mecanismo hemostático. Las proteínas parecen cubrir las plaquetas e interfieren con la adhesión y quizá con la agregación. Se han informado casos en los que la proteína sérica anormal interactuaba con la colágena y la cubría, interfiriendo de esta manera con la interacción plaqueta-colágena. El tratamiento debe dirigirse a la enfermedad primaria, pero si la hiperviscosidad y la hemorragia son importantes, se puede requerir plasmaféresis inmediata para disminuir la concentración de la proteína anormal y corregir al trastorno hemorrágico.
Trastornos inducidos por medicamentos
Aspirina y otros agentes antiinflamatorios no esteroides La ingestión de 0.6 g de aspirina prolonga el tiempo de sangrado de 2 a 3 minutos en los sujetos normales. Las plaquetas son afectadas en forma irreversible. Los endoperóxidos cíclicos y el tromboxano A2 son inductores potentes de las reacciones de liberación y agregación plaquetaria [ver figura 4]. La aspirina acetila e inhibe la cicloxigenasa que cataliza la síntesis de los endoperóxidos. Algunos sujetos aparentemente normales muestran notable sensibilidad a la acción de la asprina, de manera que sus tiempos de sangrado se prolongan mucho y tienen hemorragias clínicas graves, en particular durante o después de una cirugía o traumatismo. Estos pacientes pueden tener una forma leve de enfermedad por almacenamiento.
Los pacientes urémicos son en especial sensibles al sangrado inducido por ácido acetilsalicílico. Las dosis pequeñas de ácido acetilsalicílico (100 mg/m2 v.o.) no prolongan el tiempo de sangrado en individuos normales pero producen prolongación importante, a menudo hasta de 15 minutos, en pacientes urémicos.187 Se desconoce el mecanismo por el cual el ácido acetilsacilico prolonga el tiempo de sangrado en la uremia, pero parece depender de una que vía que no incluye la inhibición de la cicloxigenasa.188 La combinación de alcohol y ácido acetilsalicílico también es peligrosa por su capacidad para prolongar el tiempo de sangrado.
La hemorragia inducida por aspirina se diagnostica determinando que existe un defecto de la función plaquetaria de tipo adquirido (una cuenta plaquetaria de más de 100,000/mm3 prolongación de¡ tiempo de sangrado, sin historia previa de sangrados), y luego buscando evidencia de ingestión de aspirina. Debido a que cerca de 300 compuestos en el mercado contienen aspirina, el antecedente negativo debe ser complementado con las siguientes pruebas: niveles séricos de salicilatos, agregación de cloruro férrico a la orina, que se vuelve de color azul-negro si hay salicilato presente, y, el mejor método, detección de un patrón anormal de agregación por colágena que vuelve a lo normal en 7 días, lo cual es característico de la ingestión de salicilatos.
Las plaquetas modificadas por ácido acetilsalicílico pueden agregarse cuando se exponen a ADP, pero no llevan a cabo la reacción de liberación. Por lo tanto, el tratamiento de la hemorragia aguda inducida por ácido acetisalicílico incluye la transfusión de suficientes plaquetas normales para aumentar el recuento plaquetario a 50,000/mm3 Las plaquetas transfundidas realizan la reacción de liberación, después de la cual se pueden agregar las plaquetas modificadas por ácido acetilsalicílico. En términos generales, se necesitan seis unidades de concentrado plaquetario, y la administración se puede repetir diariamente o en días alternos hasta que cese la hemorragia o hasta que ya no se modifique la mayor parte de las plaquetas del paciente. En vista de que el-ácido acetilsalicílico acetila las plaquetas en forma irreversible, el daño hemostático podría durar cuatro a cinco días después de haber suspendido su administración.
Si el paciente necesita analgesia, se pueden usar acetaminofén o codeína, porque ninguno de ellos prolonga el tiempo de sangrado. Si el paciente aún requiere dosis elevadas de salicilatos, como en el tratamiento de la artritis reumatoide, se prefiere el salicilato de sodio, ya que sólo modifica ligeramente a las plaquetas.
Alcohol Además de producir trombocitopenia al suprimir la producción plaquetaria, el consumo de alcohol también puede causar defectos funcionales.189 El tiempo de sangrado se prolonga en los alcohólicos crónicos incluso cuando la cuenta plaquetaria es normal. Estudios realizados in vitro han demostrado que el alcohol impide la agregación plaquetaria y la liberación de tromboxano A 2.
La función plaquetaria vuelve a ser normal después de dos o tres semanas de abstinencia.
Dextranos U forma de dextrán de 40,000 P se excreta con rapidez, pero el dextrán de 70,000 PM puede persistir en la circulación durante tres días e interferir con la acción de la superficie plaquetaria. Además de prolongar el tiempo de sangrado, los pacientes que reciben dextrán de 70,000 PM tienen evidencias de agregación de eritrocitos. El tratamiento de estos enfermos incluye apoyo hasta que se excrete el dextrán. El dextrán en el plasma afecta a las plaquetas transfundidas.
Antibióticos La carbenicilina y la ticarcilina pueden inhibir la agregación plaquetaria y contribuir a un trastorno hemorrágico, al igual que las dosis masivas de penicilina. Las dosis masivas de penicilina prolongan el tiempo de sangrado y deterioran la agregación plaquetaria inducida por colágena y ristocetina. La situación clínica es particularmente desconcertante cuando un paciente pancitopénico tratado por septicemia desarrolla un defecto de la función plaquetaria que se agrega a la hemorragia. Una maniobra razonable consiste en suspender los antibióticos o reducir la dosis.190
El moxalactam, un antibiótico B-lactámico con estructura similar a la carbenicilina, puede causar hemorragia por varios mecanismos aparentemente diferentes.191 Esta cefalosporina de tercera generación es capaz de producir un trastorno funcional plaquetario después de tres a cinco días de tratamiento en una dosis diaria de 4 g o mayor. Este trastorno se caracteriza por un tiempo de sangrado prolongado y una cuenta plaquetaria normal. Se supone que el moxalactan se une a un sitio en la membrana plaquetaria que interfiere con el desarrollo de la lectina inducida por ADP que se requiere para la agregación plaquetaria. Si se diagnostica un trastorno hemorrágico relacionado como moxalactam, debe suspenderse el medicamento. Debe evitarse el uso combinado de moxalactam con agentes antinflamatorios no esteroides que inhiben la cicloxigenasa plaquetaria porque estas dos clases de inhibidores pueden tener efectos aditivos y potencialmente desastrosos en la función plaquetaria.
Muchas de las otras cefalosporinas de tercera generación prolongan en forma mínima el tiempo de sangrado; las acilureidopenicilinas parecen ser más seguras que la carbenicilina o el moxalactam. Sin embargo, parece haber cierta variación en la respuesta de individuo a individuo. Por lo tanto, si se desarrollo sangrado en el paciente que está siendo tratado con estos agentes antimicrobianos, debe realizarse tiempo desangrado y recuento plaquetario; si el tiempo de sangrado está prolongado y no hay otras causas posibles, debe suspenderse el antibiótico hasta la resolución del problema.192 Algunos de estos antimicrobianos también producen hemorragia al interferir con la función dependiente de la vitamina K.
También se ha descrito un raro síndrome, parecido a la PTI, después de la administración de moxalactam. Después de establecido el diagnóstico y de haber suspendido el moxalactam, se debe iniciar el tratamiento con esteroides si existe evidencia clínica de hemorragia.
Agentes diversos Una gran variedad de otros agentes pueden modificar la función plaquetaria [ver tabla 5].193
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
* "Agregación plaquetaria anormal in vitro"indica una alteración cuando se añade el agente a plasma rico en
plaquetas.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Trombocitosis y trombocitemia
Una cuenta plaquetaria elevada, es decir, mayor de 500,000/mm3, puede ser el resultado de diversas alteraciones clínicas y se denomina trombocitosis reactiva [ver tabla 6]. En la trombocitosis reactiva las pruebas de función plaquetaria, incluyendo el tiempo de sangrado y las mediciones de las adhesión y agregación plaquetaria, son en general normales, y los pacientes no sufren de hemorragias clínicas o tromboembolia, incluso con cuentas plaquetarias mayores de 1,000,000/mm3. 194
|
|
|
|
Por comparación, el recuento plaquetario puede estar aumentado en forma autónoma en las enfermedades mieloproliferativos como la leucemia mieloide crónica, la metaplasia mieloide agnogénica con mielofibrosis, la policitemia vera y la trombocitemia esencial.195 En este último padecimiento, la cantidad de unidades formadoras de colonias de la serie de los megacariocitos (UFC-Meg) está aumentada en la médula ósea. Al igual que en los enfermos con policitemia vera, en los que las unidades formadoras de colonias de la serie eritroide (UFCe) pueden desarrollarse sin agregar eritropoyetina, alguna de las CFU-Meg en los enfermos con trombocitemia esencial pueden desarrollarse sin necesidad de agregar un medio de cultivo específico.196
La cuenta plaquetaria puede variar desde 1,000,000 hasta 3,000,000/mm3 o más, y las pruebas de función plaquetaria, incluyendo el tiempo de sangrado, con frecuencia son anormales [ver antes. Trastornos de la función plaquetaria]. Algunos de estos pacientes parecen mostrar un aumento de la predisposición para desarrollar hemorragias y tromboembolias. No hay un nivel definido de trombocitos que se correlacione bien con las manifestaciones clínicas. En un paciente, la cuenta plaquetaria permaneció en 14,000,000/mm3 por tres años, pero no hubo otros signos o síntomas anormales. En la policitemia vera, las complicaciones tromboembólicas pueden ser explicadas, por el aumento en la masa de glóbulos rojos, pero en las otras enfermedades mieloproliferativas es evidente que las plaquetas con trastornos en su función predisponen a la hemorragia y, paradójicamente a trombosis. Ni el número de plaqueta,,; ni las mediciones de la función plaquetaria predicen el grado de trombosis o hemorragia. La actividad de la lipoxigenasa plaquetaria siempre está normal en pacientes con trombocitosis reactiva, pero es deficiente en algunos pacientes con la trombocitemia de los trastornos mieloproliferativos. Curiosamente, esto.,, pacientes con trombocitemia tienen más complicaciones de tipo hemorrágico (67 porciento de los pacientes) que complicaciones de tipo trombótico (13 porciento). 197, 198
Desde el punto de vista clínico, los signos hemorrágicos incluyen hemorragias mucosas (en particular gastrointestinales), hematomas y equímosis. Puede haber trombosis de la vena esplénica, trombosis de la vena porta o mesentérica y trombosis recurrente de venas profundas con o sin embolia pulmonar. Es menos común la trombosis arterial. En la trombocitemia el problema clave es una función plaquetaria defectuosa, si bien el aumento absoluto en el número de plaquetas puede desempeñar un papel adicional, aunque mal definido. Algunos investigadores creen que la quimioterapia podría suprimir las clonas megacariocíticas más displásicas, permitiendo de esta manera que las clonas más diferenciadas y normales produzcan plaquetas. Los pacientes con trombocitenia esencial y policitemia vera pueden manifestar eritromelalgia debilitante (ardor y prurito en los dedos de las manos y de los pies) que puede evolucionar a la acrocianosis isquémica. 199 este grupo de síntomas parece ser una consecuencia de oclusión e inflamación de las, arteriolas por agregados plaquetarios. La administración de ácido acetilsalicílico o indometacina produce alivio en término de horas. La administración de ácido acetilsalicílico en dosis de 0.3 a 0.6 g cada tercer día puede producir beneficio prolongado.
El Grupo de Estudio de la Policitemia Vera en los Estados Unidos ha propuesto criterios para el diagnóstico de la trombocitopemia esencial. 200 Tales criterios consisten en un recuento plaquetario mayor de 600,000/mm3 y la exclusión de otras causas posibles, como otros trastornos mieloproliferativos o trombocitosis reactiva. Los hallazgos que indicarían la presencia de otra enfermedad de fondo incluyen un nivel elevado de hemoglobina y de la masa eritrocitaria, datos de deficiencia de hierro, la presencia del cromosoma Filadelfia en el examen citogenético (leucemia mieloide crónica) y fibrosis extensa de la médula ósea (metaplasia mieloide agnogénica).
La clave en el tratamiento de la trombocitopenia consiste en reconocer que no existe un nivel plaquetario específico que pueda predecir hemorragia o trombosis y que cualquier complicación es poco común, incluyendo a pacientes con cuentas plaquetarias de 1,000,000/mm3. 194 El estudio de una serie de enfermos reveló que aunque la hemorragia no ocurrió a menudo, se desarrolló en pacientes que al mismo tiempo recibían ácido acetilsalicílico o esteroides. En un paciente con trobocitemia esencial que tiene hemorragias o trombosis clínicamente significativas, puede lograrse un buen control de la cuenta plaquetaria administrando hidroxiurea por vía oral, 15 mg/kg/día con los ajustes necesarios en la dosis para disminuir la cuenta plaquetaria. A deferencia del tratamiento con alquilantes, la hidroxiurea no parece aumentar el riesgo de un segundo padecimiento neoplásico200El tratamiento con hidroxiurea obliga a vigilar en forma cuidadosa la citología hemática, y las consecuencias a largo plazo del mismo se desconocen. Entre los nuevos tratamientos para la trombocitopenia se incluyen el uso de anegrelide, un agente muy potente para disminuir el número de plaquetas que aún no ha sido autorizado por la FDA de los EE.UU.201 Parece ser que el interferón alfa, en dosis de 3 a 5 millones Uldía por vía subcutánea es eficaz para disminuir la cuenta plaquetaria cuando se administra durante un periodo de uno a tres meses. Cuando se logra el número deseado de plaquetas, se administra una dosis de mantenimiento de 1 a 3 millones U/ día, que incluso puede normalizar el número de megacariocitos en la médula ósea durante un periodo de varios meses.202
Un problema muy común y difícil es la selección del tratamiento apropiado en aquellos pacientes con cuentas plaquetarias muy elevadas (mayores de 1,000,000/mm 3 ) causadas por un trastorno mieloproliferativo crónico, pero que no presentan signos de hemorragia o trombosis, están asintomáticos o manifiestan síntomas inespecíficos como debilidad, malestar general o cefalea leve. En tales casos el autor prefiere, apoyado en parte por otro estudio, 203 no utilizar agentes citotóxicos a menos de que existan evidencias claras de hemorragia o trombosis y recomienda que los pacientes eviten el uso de ácido acetisalicílico y de otros agentes antinflamatorios que pueden inducir hemorragia. Si después desarrollan tromboembolias recurrentes, puede hacerse el intento de disminuir la cuenta plaquetaria con hidroxiurea. También deben administrarse dosis bajas de ácido acetilsalicílico (300 mg cada 6 horas). Las dosis altas de este medicamento causan hemorragias en pacientes con policitemia vera y trombocitemia.200 En casos de hemorragia trombocitopénica recurrente debe intentarse el tratamiento con hidroxiurea, interferón alfa o anagrelide, si existe tiempo suficiente (se requiere entre uno y tres meses de tratamiento para producir un descenso importante en el número de plaquetas). Sin embargo, cuando la hemorragia es grave puede requerirse la transfusión de seis a 10 unidades de plaquetas normales para corregir la función plaquetaria defectuosa. Si el paciente sufre tromboembolias o hemorragias incontrolables, el número de plaquetas puede disminuirse con rapidez (hasta cerca de 400,000/mm3) mediante plaquetoféresis. Algunos especialistas indican que en estas situaciones pueden utilizarse bolos de mostaza nitrogenada. Sin embargo, debido a que la vida media de las plaquetas es de cerca de 10 días, no se logrará una reducción sustancial en el recuento plaquetario durante varios días, incluso si se abate por completo la megacariopoyesis. Por ejemplo, en los pacientes con cuentas plaquetarias de 3,000,000/mm 3 , esta seguirá siendo elevada, aproximándose a 2,500,000/mm3 , al día siguiente de la administración nitrogenada.
Púrpuras vasculares
Las púrpuras vasculares Son un grupo muy heterogéneo de trastornos [ver tabla 7] que por lo general se caracterizan por hemorragia cutáneas, en ocasiones asociadas con hemorragia en mucosa. La filtración ocurre a partir de las arteriolas terminales, capilares y vénulas poscapilares.204 Los resultados de las pruebas plaquetarias, cuantitativas y cualitativas, son normales, así como las pruebas funcionales de los procoagulantes.
|
||||||||||||
|
TELEANGIECTASIA HEMORRAGICA HEREDITARIA
La teleangiectasia hemorrágica hereditaria se trasmite como un rasgo autosómico dominante y se manifiesta como epistaxis 205 o por hemorragias gastrointestinales. El examen físico revela teleangiectasias en los pulpejos de los dedos, mucosa oral, lengua y bordes de los labios. Puede haber cortocircuitos arteriovenosos en el hígado y pulmones. La pruebas de coagulación son normales, aunque pueden ser anormales algunas pruebas de función plaquetaria. El manejo es difícil y con frecuencia incluye el taponamiento nasal recordando que la epistaxis vuelve a ocurrir cuando se retira un taponamiento nasal que ha estado colocado durante más, de 24 horas. La hemorragia gastrointestinal se trata, cuando es posible, con preparaciones de hierro; esta circunstancia es única en el sentido de que puede requerir la administración de hierro intravenoso. Se ha propuesto el tratamiento con estrógenos, pero no se ha demostrado que produzca un beneficio real.
Las malformaciones pulmonares arteriovenosas que ocurren en algunos pacientes con telangiectasias hemorrágicas hereditarias pueden producir disnea, hemoptisis, disminución de la tensión arteria¡ de oxígeno (Pa02) y eritrema secundaria. El diagnóstico se confirma por angiografía pulmonar. Si los cortocircuitos son grandes y significativos desde el punto de vista clínico, pueden tratarse por medio de emboloterapia con balón.206 Pueden ocurrir émbolos paradójicos con daño vascular cerebral en los pacientes con telangiectasia hemorrágica hereditaria que tienen cortocircuitos arteriovenosos y malformaciones pulmonares.206,207
ESCORBUTO
La vitamina C es necesaria para el metabolismo normal de la colágena, de los folatos y quizá de] hierro. El paciente con escorbuto sufre sobre todo de trastornos en la síntesis de la colágena. La carencia de un tejido de sostén de colágena adecuado para la microcirculación producirá hemorragias perifoliculares, gingivorragias e incluso hematomas en tejidos profundos. Al parecer defectos similares de la colágena en otras partes M organismo producen el llamado cabello en sacacorcho y las queratosis relacionadas con este trastorno.208,209 El cuadro clínico característico en un individuo desnutrido orienta al diagnóstico. Los niveles de ácido ascórbico en plasma o en la capa de leucocitos centrifugados se encuentran disminuidos y también suele haber otras deficiencias vitamínicas. El tratamiento eficaz consiste en la administración de 1 g diario de ácido ascórbico en dosis divididas.
EXCESO DE ESTEROIDES
El exceso de esteroides, de causa endógena o exógena, produce hemorragias cutáneas, probablemente como consecuencia de] catabolismo de proteínas en los tejidos de apoyo vascular, inducido por los esteroides.
En la amiloidosis pueden observarse equimosis cutáneas con predilección por el cuello y la región superior del tórax. La biopsia del sitio afectado muestra la sustancia amiloide, que por infiltración puede debilitar las paredes de los vasos o interferir con la superficie de activación de las plaquetas, de los procoagulantes o de ambos.
VASCULITIS LEUCOCITOCLASTICA
Las lesiones purpúricas en pacientes con vasculitis leucocitoclástica pueden ser elevadas (púrpura palpable), En la biopsia, estas lesiones pueden mostrar degranulación de células cebadas y, cuando se tiñen en forma apropiada, depósitos de complejos inmunes. Se supone que los complejos proveen la estimulación quimiotática que conduce a la congregación de neutrófilos. El daño a la microcirculación es causado por el ataque del complemento y por la liberación del contenido de los gránulos de los neutrófilos. El componente inflamatorio produce la púrpura palpable.210
El ejemplo clásico de vasculitis es la púrpura de Henoch Schönlen (púrpura alérgica o anafilactoide), que es más común en niños que en adultos. La enfermedad consiste en lesiones purpúricas elevadas pruriginosas, edema subcutáneo, poliartritis, síntomas gastrointestinales que incluyen hemorragia y glomerulonefritis aguda. También existen hipergamaglobulinemia policlonal y crioglobulinemia.211 El examen de la lesión purpúrica por biopsia muestra vasculitis aguda con glóbulos rojos que pasan a través de las paredes vasculares. Las tinciones inmunofluorescentes muestran depósitos de IgA y componentes del complemento; también se han demostrado IgG e IgM, aunque con menos frecuencia. La búsqueda de medicamentos u otros alergenos, como agentes etiológicos, no ha tenido en general buenos resultados. El tratamiento se basa en el reposo en cama y evitar aspirina y fenacetina. Estudios no controlados con esteroides, azatioprina y ciclofosfamida para el manejo de la nefritis aguda no han demostrado su beneficio. 212
Las reacciones medicamentosas pueden producir una púrpura vasculítica con aparición de lesiones purpúricas pruríticas elevadas. Se han usado antihistamínicos y esteroides para tratar la erupción, sin resultados favorables.
PURPURA SENIL
Los pacientes con púrpura senil tienen hemorragias cutáneas en el dorso de la mano, la muñeca y los brazos, y ocasionalmente, en las pantorrillas. No ocurren hemorragias graves y no se requiere tratamiento. Se supone que esta entidad representa deterioro del tejido de sostén vascular, que depende de la edad.
PURPURA AUTERITROCITICA
La púrpura auteritrocítica es un trastorno poco frecuente y extraño que ocurre casi exclusivamente en mujeres de edad media. Las pacientes tienen hematomas subcutáneos dolorosos, y en ocasiones enormes, que algunas veces se asocian con síntomas generales. El trastorno deteriora en forma clara la calidad de vida de la paciente. La evaluación psiquiátrica revela por lo común patrones de agresividad reprimida. Aunque algunos estudios han indicado que el trastorno es facticio, en muchos casos no se puede demostrar automutilación. El nombre se deriva de la observación de que las equimosis dolorosas se pueden reproducir si los glóbulos rojos de la misma paciente se inyectan subcutáneamente. El tratamiento con esteroides, antihistamínicos, cloroquina, dietas bajas en alérgenos y psicoterapia ha sido ineficaz.
LESIONES DE LA MICROCIRCULACION POR EMBOLIAS
La CID y M pueden causar oclusiones vasculares localizadas que producen lesiones de la microcirculación y filtración de glóbulos rojos. Lesiones similares pueden producirse por embolias originadas en las válvulas cardiacas infectadas. La embolia grasa es una complicación de las fracturas de la pelvis y los huesos largos. El síndrome consiste en fiebre, confusión mental y aparición de petequias, lesiones purpúricas o ambas, en cuello, tórax, cara y axilas. La embolia por colesterol también puede causar petequias, por lo general en las extremidades inferiores. De manera típica se presenta en pacientes con aterosclerosis severa sometidos recientemente a algún procedimiento invasivo que afecta la aorta abdominal o las arterias renales. La biopsia de las lesiones purpúricas muestra cristales de colesterol cuando se aplica la tinción apropiada.
Trastornos hereditarios de la coagulación
Los trastornos de la coagulación se manifiestan clínicamente como hemorragias espontáneas o sangrado profuso después de sufrir traumatismos o de una cirugía. Los antecedentes indican por lo común si el trastorno es congénito o adquirido. Los padecimientos hereditarios se caracterizan por su aparición al principio de la vida y por la presencia de una sola alteración que puede ser responsable de todo el cuadro clínico. 213
ENFERMEDAD DE VON WILLEBRAND
La enfermedad de von Willebrand (EvW), que puede ser el trastorno de procoagulantes hereditario más común, se produce por la presencia de factor de von Willebrand deficiente o defectuoso en el plasma. Un estudio epidemiológico realizado en Italia demostró que el 0.5 a 1.0 porciento de los niños estudiados tuvieron criterios para el diagnóstico de EvW tipo I heterocigota clásica. 214 El gen que codifica el FvW se localiza en el cromosoma 12. Después de un prolongado proceso y de remodelación, el pre-pro-FvW sintetizado por este gen surge como una subunidad de 270 kd que tiene dominios específicos para fijarse al factor VIII:C, a la heparina (dos sitios), a la colágena (dos sitios), y a la GPlb y GPllb-Illa plaquetarias. Estos dominios se relacionan en forma directa con las siguientes funciones de] FvW: (1) su acción como una molécula acarreadora del factor VIII:C, protegiendo al procoagulante de la proteólisis y otros insultos, (2) su promoción de la adhesión primaria de las plaquetas cuando la lesión de la pared es rápida, al unir plaquetas (por medio de su receptor de GPlb) a los tejidos subendoteliales en el sitio de la lesión, y (3) cuando la velocidad de lesión de la pared es baja, apoya la agregación plaquetaria al unir plaquetas por medio de sus receptores de GPIIb-llIa.215,216 El FvW circula como multímeros que varían en tamaño de 0.5 millones de daltons (el dímero) a 20 millones de daltons. Pueden existir multímeros más grandes no circulantes dentro de las células endoteliales, almacenados dentro de los cuerpos de Weibel-Palade. El FvW puede ser procesado mientras es liberado, quizá en respuesta a la trombina o la fibrina. Este factor es liberado hacia la circulación o hacia la pared, en donde se une a la colágena subendotelial y facilita la unión plaquetaria. Los gránulos alfa de las plaquetas también contienen FvW, que se libera cuando estas células se activan. Es probable que los multímeros de FvW mayores de 12 millones de daltons sean más eficaces para aumentar la adhesión plaquetaria.
Existen numerosas variantes de la enfermedad de von Willebrand que difieren en sus manifestaciones clínicas, alteraciones de laboratorio y tratamiento. Por lo tanto, se requieren pruebas específicas para identificar el tipo de enfermedad y su gravedad. Las pruebas comienzan con TPTa, cuenta de plaquetas y tiempo de sangrado. Debido a que el FvW es un acarreador proteico del factor VIII:C, la concentración de este último será baja y el TPTa estará prolongado cuando la concentración de FvW sea baja. La cuenta de plaquetas casi siempre es normal y con frecuencia el tiempo de sangrado está prolongado. Existen dos aspectos importantes respectos a las prueba¿ para la EvW: (1) los resultados de laboratorio son muy variables, sobre todo en los caso,, leves, y los valores del TPTa y del tiempo de sangrado pueden variar desde el límite normal bajo hasta el anormal, y (2) el grupo sanguíneo del paciente afecta la concentración de FvW; los pacientes con grupo sanguíneo 0 tienen concentraciones más bajas que los enfermos con grupos A o B.215,217 Después de las pruebas iniciales, se mide la concentración de FvW por medio de varios métodos inmunológicos. El resultado casi siempre se notifica como antígeno del FvW y se expresa como porcentaje de un control normal que equivale al 100 porciento. Debido a que el FvW circula en muchas formas multiméricas, de las cuales los multímeros más grande: son los que tienen mayor importancia fisiológica, es necesario determinar la composición multimérica del FvW en el plasma del paciente. En la actualidad ya existen en los laboratorios comerciales procedimientos para hacer esta determinación.
También se investiga la capacidad funciona¡ del FvW, en parte con el tiempo de sangrado, que no es muy confiable, y por la prueba de aglutinación de plaquetas con ristocetina. En esta última prueba se agrega ristocetina a plasma rico en plaquetas del paciente, lo que provoca que las plaquetas expresen el receptor de GPlb de modo que puedan unirse al FvW para causar aglutinación plaquetaria. Por lo tanto, la prueba de aglutinación con ristocetina evalúalas plaquetas del paciente y quizá el tipo y cantidad de FvW en los gránulos alfa de las plaquetas. Una variante de esta prueba es la de cofactor de ristocetina, que mide la cantidad y tipo de FvW en el plasma del paciente al añadir a éste cantidades crecientes de una mezcla estándar de ristocetina y plaquetas normales.217
La enfermedad de von Willebran de tipo 1 se hereda como un rasgo autosómico dominante y casi siempre aparece en la forma heterocigota. Los pacientes con EvW clásica tipo I tienen antecedentes de toda la vida de hemorragias leves o moderadas, sobre todo de mucosas. Sin embargo, los pacientes pueden no detectar el trastorno hemorrágico sino hasta que se someten a una cirugía o sufren un traumatismo y presentan una hemorragia más grave de lo esperado. En la EvW tipo I existen multímeros de todos los tamaños.218 La forma homocigota o heterocigota doble se denomina EvW tipo III, es poco frecuente y se caracteriza por hemorragias graves, TPT prolongado y concentración de factor VIII:C menor del cinco porciento. En la enfermedad tipo 111 el FvW no es detectable.
La EvW tipo 11 se caracteriza por alteraciones cualitativas del FvW, con disminución en los multímeros de gran tamaño, más iinportantes fisiológicamente o con alteraciones funcionales del FvW. Se han descrito varios subtipos de ésta. En el tipo TIA se altera la formación de los multímeros de gran tamaño. En el tipo IIB existen multímeros de tamaño pequeño e intermedio, pero los multímeros de gran tamaño son defectuosos y quiza están disminuidos en número porque se unen en forma excesiva alas plaquetas. La aglutinación con ristocetina está aumentada.
Se ha descrito una forma plaquetaria de enfermedad de von Willebran, o seudoenfermedad de von Willebrand, en la que la unión excesiva de FvW plasmático normal a plaquetas anormales no estimuladas induce la agregación plaquetaria. Esta interacción puede explicar la trombocitopenia intermitente y la disminución de los multímeros de gran tamaño del FvW que se observa en los pacientes con este trastorno.219,220 Los diversos tipos de enfermedad de von Willebrand tienen ciertas características distintivas de laboratorio [ver tabla 8].
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
De modo tradicional, la enfermedad de von Willebrand ha sido tratada con infusiones de crioprecipitado porque esto proporciona los multímeros de FvW de gran tamaño, los cuales faltan en los concentrados purificados de factor VIII. Después de que se han infundido alrededor de 10 bolsas de criprecipitado (1,000 a 1,250 unidades de factor VIII:C), los beneficios potenciales pueden vigilarse midiendo el tiempo de sangrado o el nivel de actividad del cofactor de ristocetina. Algunos investigadores consideran que la meta terapéutica debe ser obtener una concentración de cofactor de ristocetina mayor del 50 porciento.221 Este tratamiento puede repetirse cada 12 horas hasta que se controla la hemorragia grave. Debe evitarse el uso de aspirina. Muchos concentra dos de factor VIII tienen muy poco FvW, o no contienen suficientes multímeros de gran tamaño para que el tratamiento con estos concentrados sea eficaz, aunque algunos, como el Humate-P, si son útiles.221 Para controlar las hemorragias graves que no responden al tratamiento con crioprecipitado o concentrados de factor VIII, puede intentarse una transfusión con plaquetas normales con la esperanza de que el FvW plaquetario sea hemostáticamente eficaz.222 Se han probado varios preparados comerciales de FvW concentrado, pero ninguno ha sido autorizado aún por la FDA de los EE.UU. Estos preparados elevan en forma eficaz la concentración de FvW, pero la mejoría en el tiempo de sangrado es incompleta.223
La diamino-8-D-arginina vasopresina (DDAVP) también ha demostrado ser eficaz en el tratamiento de la enfermedad de von Willebrand. La DDAVP administrada por vía intravenosa en dosis de 0.3 ug/kg en un lapso de 15 a 30 minutos causa la liberación de VIII:C y FvW de las reservas de las células endoteliales.224 Mientras el nivel de FvW del paciente sea mayor de cinco porciento, la infusión de DDAVP elevará en forma importante los niveles de FvW y en ocasiones acortará el tipo de sangrado. La inyección puede repetirse varias horas después. Los sujetos con las formas más leves de enfermedad de von Willebrand también pueden ser preparados para cirugía abdominal general usando infusiones de DDAVp.225 El incremento en el FvW logrado por la DDAVP tiene vida media de cerca de 8 horas y en muchos pacientes las inyecciones repetidas producen nuevas elevaciones de VII:C y FvW.160 Debido a que existe considerable variación individual en la respuesta a la DDAVP, la infusión de este agente puede no producir beneficio uniforme. Es probable que una buena práctica consista en determinar el patrón de respuesta de nuevos pacientes con enfermedad de von Willebrand leve a moderada, mediante la infusión de DDAVP y seguimiento del cambio en VIII:C, FvW y tiempo de sangrado. La DDAVP es eficaz en la enfermedad de von Willebrand tipo 1 y tipo II,162 pero no debe emplearse en la afección tipo IIb, debido a que causa trombocitopenia en esta variante.
Ya que la DDAVP parece aumentar la fibrinolisis al producir la liberación de activadores del e-aminocaproico plasminógeno [ver figura 3], puede ser aconsejable la administración simultánea de ácido e-aminocaproico oral, 4 g cada cuatro horas (24 g/día), en particular en circunstancias en que puede anticiparse la fibrinolisis local, como en caso de extracción dental.226
Existe un aerosol nasal de DDAVP que parece promisorio para el tratamiento ambulatorio de los pacientes con EvW, tanto para el manejo de los episodios hemorrágicos como para preparación para cirugías menores. Se administra una dosis de 150 ug de DDAVP en cada narina.227
Los efectos colaterales de la DDAVP en dosis de o.3 ug/ kg durante un periodo de 15 a 30 minutos son leves y consisten en retención hídrica, enrojecimiento, taquicardia y cefalea. La actividad del FvW tiende a elevarse durante el embarazo e incluso puede alcanzar niveles normales bajos, ayudando así en el tratamiento.
HEMOFILIA A
La hemofilia A, trastorno raro que afecta a uno de cada 10,000 varones, es el ejemplo de alteración hemorrágica hereditaria más estudiada. Se caracteriza por deficiencia o por defecto funcional del factor VIII:C. Dado que el gen de la actividad coagulante del factor VIII:C se encuentra en el cromosoma X, la enfermedad se manifiesta en hombres hemocigotos. Todas las hijas del varón hemofílico serán portadoras, mientras que la mitad de los hijos de la madre que lleva el rasgo de la hemofilia serán hemofílicos y la mitad de sus hijas serán portadoras.
El gen del factor VIII, que se localiza en el cromosoma X en Xq28, es enorme y contiene 26 exones. Codifica una proteína de peso molecular variable, por lo general de alrededor de 333,000 daltons, que circula unida y protegida por el FvW.228,229 En la actualidad se han estudiado más de 600 genes de factor VIII anormal y las alteraciones encontradas, que incluyen mutaciones puntiformes, inserciones y deleciones genéticas, causan deficiente producción del factor VIII:C o generación de un factor VIII:C cuya función es anormal.
Todos los pacientes con hemofilia A tienen niveles normales de FvW. Las mujeres portadoras230 pueden ser detectadas con facilidad porque su nivel de VIII:C es de cerca de la mitad de lo normal, mientras que su concentración de FvW es normal. Por lo tanto, la relación VIII:C a FvW en los portadores es de 0.5.
Las diferentes familias se afectan en distintos grados, dependiendo de la naturaleza específica del defecto genético,228,229 la gravedad clínica de la hemofilia A correlaciona con las concentraciones medidas de actividad coagulante del factor VIII [ver tabla 9].
|
||||||||||
|
El diagnóstico se establece por el cuadro clínico, los antecedentes familiares (positivos en el 75 porciento de los casos) y el nivel de actividad coagulante de factor VIII. Una historia familiar típica y un tiempo de sangrado normal descartan el diagnóstico de enfermedad de von Willebrand que, a diferencia de la hemofilia A, se trasmite en forma autosómica.
Principios generales de tratamiento en la hemofilia
Los aspectos psicosociales de la hemofilia son complejos debido a la preocupación y sentimiento de culpa de los padres. Los niños afectados por la enfermedad se ausentan con frecuencia de la escuela, están propensos a sufrir deformidades incapacitantes y corren el riesgo de drogadicción debido al dolor intenso que en ocasiones presentan.
Los concentrados de factor VIII han demostrado ser eficaces para controlar la hemorragia espontánea y traumática en la hemofilia severa. A principios de 1980 muchos hemofílicos con enfermedad severa fueron tratados con programas profilácticos basados en la autoadministración en el hogar de concentrado de factor VIII tres veces por semana. Tales programas parecerían permitir al hemofílico severo llevar una vida más normal, aun cuando el uso de los concentrados se relacionaran con mayor probabilidad de infección por hepatitis B o hepatitis C. Incluso en los últimos años, en los que los concentrados de factor VIII son tratados con calor para inactivar al virus de la hepatitis C, muchos hemofílicos siguen teniendo infección persistente por este virus.231
Las desventajas de este método aumentaron cuando se descubrió que los concentrados liofilizados de factor VIII, formados a partir de ¡nuestras de plasma de hasta 2,000 individuos donadores,232 se encontraban muy contaminados con VIH. Hacia la mitad de la década de los SO, cuando menos el 75 porciento de los receptores de concentrados de factor VIII residentes en Estados Unidos y Europa occidental habían desarrollado anticuerpos contra el VIH.233-236 Algunos de estos pacientes presentaron también PTI asociada a la infección por VIII, una complicación especialmente grave para un hemofílico.88 Los hemofílicos con vida sexual activa pueden trasmitir la infección por VIII a sus compañeros sexuales.237,239 A pesar del tratamiento con zidovudina y de la profilaxis con pentamidina, que disminuyen la progresión de la enfermedad, se están presentando cuadros clínicos evidentes de SIDA en forma inexorable en los hemofílicos infectados por el VIH.240 Esto ha anulado la mejoría en la expectancia de vida de los hemofílicos, que llegó a ser de 66 años de edad durante los años 70. En la década de los 80 la expectancia de vida disminuyó a 49 años de edad.241
La contaminación de los concentrados de factor VIII con VIII ha tenido un efecto notable sobre los pacientes con hemofilia severa, que ahora deben sopesar en forma muy cuidadosa las opciones terapéuticas. La contaminación de los concentrados de factor VIII:C con VIH y con virus de la hepatitis B y C ha estimulado los esfuerzos por desarrollar preparaciones concentradas libres de virus. En la actualidad ya existen varios de estos preparados, y el problema consiste en equilibrar su costo con el beneficio probable. Se están elaborando preparaciones muy purificadas de factor VIII:C por medio de anticuerpos monoclonales y comatografía de afinidad, como el Monoclate-P (Armour) y el Hemofil M (Baxter healthcare).242
El factor VIH ya ha sido clonado y se está probando en la clínica el factor recombinante, observándose que tiene una actividad biológica y vida media en plasma semejante al producto original.243 En diciembre de 1992, la FDA autorizó el uso del factor VIII recombinante para la prevención y control de las hemorragias excesivas, y para su administración en personas con homofilia A que requieren cirugía.
La profilaxis dental es de gran importancia porque la hemofilia casi siempre causa complicaciones si se requiere de cirugía dental. Deben evitarse los medicamentos que interfieren con la función plaquetaria como la aspirina, porque causan hemorragias graves. Debe considerarse la revacunación contra el virus de la hepatitis B en los hemofílicos recién diagnosticados.242
El consejo genético debe formar parte del programa de tratamiento. Dadas las dificultades que sufren los individuos con hemofilia severa, muchas mujeres optarían por terminar el embarazo si tuvieran la certeza de su estado de portador y conocieran que su feto está afectado.244 Se cuenta con varías estrategias para detectar a las portadoras. Un caso resulta obvio, la hija de un padre hemofílico es portadora obligatoria. Un método para detectar portadoras en otros casos consiste en la medición de la relación factor VIII:C-antígeno de FvW: esta relación se aproxima al 0.5 en las portadoras,234,245 pero la incidencia de error para esta prueba es de 10 a 17 porciento.246
Se ha informado que el método que utiliza polimorfismos de longitud por fragmento de restricción (PLFR) dentro del gen del factor VIII:C para detectar a las portadoras en casos esporádicos,244,246 tiene una precisión tan alta como 96 porciento.247 Estas sondas moleculares para PLFR están ahora siendo utilizada% para determinar el estado del feto. La medición en el plasma fetal del factor VIII:C del antígeno del FvW y del antígeno del factor VIII, sólo puede obtenerse a través de fetoscopía en la semana 18 a 21, lo que resulta relativamente tardío en la gestación. Además, la fetoscopía en este estadío se relaciona con una incidencia de aborto espontáneo del seis porciento.247, 248 El tejido fetal para análisis de ADN se puede obtener con más, seguridad mediante amniocentesis hacia la semana 12 de gestación248 y por el muestreo de vellosidades coriónicas, que suele realizarse entre la semana 8 y 12 del embarazo.247,248 La amplificación especifica de las secuencias del genoma por un método enzimático mejora en forma notable la sensibilidad de estas técnicas .249,250
Tratamiento de la hemorragia aguda
Las hemorragias de tejidos profundos, las hemartrosis, y la hematuria son las formas más comunes de hemorragia clínica en la hemofilia A. Las hemorragias retroperitoneales, las hemorragias por la boca, lengua, o cuello que impiden la permeabilidad de la vía aérea, y las hemorragias intracraneanas, amenazan en forma aguda la vida. Se puede utilizar ultrasonido251 o tomografía computada para identificar hematomas retroperitoneales e intramusculares. El tratamiento se basa en la localización y extensión de la hemorragia.
Principios del tratamiento sustitutivo
Un nivel de procoagulante plasmático de 100 porciento indica que hay una unidad de procoagulante por mililitro de plasma. La mayoría de las personas tienen 40 mi de plasma por kilogramo de peso corporal. De esta manera, determinado el volumen plasmático y el nivel de procoagulante de un paciente, se puede calcular la cantidad de factor VIII requerida para remplazo. Por ejemplo, en el caso de un muchacho de 60 kg que tiene una hemartrosis no complicada en la rodilla y un nivel basal de factor VIII menor de 1 porciento, bastará con aumentar el nivel de factor VIII aproximadamente 25 porciento (0.25 Ulmi) por dos o tres días. Este paciente tiene un volumen plasmático de 60 kg X 40 mi, o 600 unidades de factor VIII como bolo inicial. Otra forma de hacer este cálculo se basa en el hecho de que la infusión de una unidad de factor VIII:C por kilogramo aumenta el nivel de factor VIII:C en dos porciento o 0.02 U/ml. En el ejemplo citado, un aumento del 25 porciento (0.25 U/ml) requerirá (25 x 60) - 2, o 750 unidades, de factor VIII:C.252
La cantidad deseada se puede administrar en seis a ocho bolsas de crioprecipitado (hay alrededor de 100 unidades por bolsa) o como concentrado purificado, en el que se indica el número de unidades en la etiqueta. La vida media inicial del factor VIII:C durante la fase de equilibrio es de sólo unas cuatro horas,252 pero la vida media biológica después del equilibrio es de 12 horas. Por consiguiente, cerca de la mitad de la dosis de carga inicial se administra después de las primeras seis horas y luego cada 12 horas durante el tiempo que sea necesario para controlar la hemorragia. Para el tratamiento de la hemartrosis, el nivel del factor VIII debe mantenerse durante dos a tres días. Un estudio controlado determinó que la dosis correcta para la mayoría de las hemartrosis agudas de rodilla, tobillo y codo fue de 14 unidades de factor VIII:C por kilogramo o cerca de 1,000 unidades de factor VIII en un adulto con 70 kg de peso. Esta infusión debe elevar el nivel de factor VIII:C en 28 a 36 porciento. En caso de hemorragias más graves en tejidos profundos, como hemorragia retroperitoneal o sangrado del paladar blando, el nivel del factor VIII:C debe elevarse a 0.5 U/ml (50 porciento).
Cirugía electiva y extracción dental
Los trabajos dentales deben ser realizados por dentistas con experiencia en el tratamiento de hemofílicos. Antes de una extracción dental, se administra factor VIII para elevar el nivel al .50 porciento y el inhibidor fibrinolítico ácido e-aminocaproico (AEAC), en dosis de 0.1 g/kg por aplicación intravenosa. Después del trabajo dental, se continúa con el AEAC por vía intravenosa en una dosis de 0. 1 g/kg cada 6 horas por 10 días. En general no se necesita mayor administración de factor VIII.
Antes de llevar a cabo cualquier cirugía electiva, el valor del factor VIII:C debe elevarse 50 a 100 porciento (0.5 a 1.0 U/ml) y después mantenerlo arriba del 50 porciento durante los próximos 10 a 14 días.252,253 La cantidad de concentrado de factor VIII:C puede disminuirse si se administra en infusión continua en lugar de bolos, en parte porque la infusión continua mantiene el factor arriba del nivel plasmático al cual pueden ocurrir hemorragias.253
Debido al riesgo persistente de trasmitir infecciones virales con las preparaciones biológica% diferentes al factor VIII:C recombinante, se han buscado medios alternativos para tratar a los pacientes hemofílicos. Un estudio notificó que el esteroide anabólico atenuado danazol, administrado en dosis de 200 rng tres veces al día, aumentó la concentración plasmática del factor VIII:C,254 sobre todo en individuos con hemofilia moderada, hasta un nivel suficiente para producir una disminución de cinco veces en la frecuencia de los episodios hemorrágicos.255 Sin embargo, otros tres estudios han encontrado que el danazol no aumentó, o lo hizo en forma mínima, la concentración de factor VIII:C, y causó aumento en el sangrado, quizá por su capacidad para favorecer la fibrinolisis.256-258 Por lo tanto, el autor no recomienda tratar a los hemofílicos con danazol sino hasta contar con más experiencia al respecto.
La DDAVP se ha empleado para tratar las hemorragias traumáticas agudas en hemofílicos leves o moderados, e incluso para preparar a estos pacientes para cirugía menor.224,225 La DDAVP, que parece producir la liberación de factor VIII:C a partir de las reservas de las células endoteliales,224-226,252 no puede emplearse en forma repetida durante muchos días porque estas reservas se agotan. Por lo tanto, en la preparación preoperatoria de los individuos con hemofilia leve o moderada, la DDAVP debe alternarse con concentrados de factor VIII:C.252 La DDAVP se infunde en dosis de 0.3 ug/kg en 50 mi de solución salina durante 15 a 30 minutos, y produce aumento inmediato del factor VIII:C. La vida media biológica del factor VIII:C liberado es de 11 a 12 horas.224,252 Debido a que la DDAVP libera también activador del plasminógeno tisular [ver figura 3], es recomendable la administración concomitante de AEAC, en dosis de 4 g por vía oral cada cuatro horas, o de ácido tranexámico, 1.5 g por vía oral tres veces al día.
Tratamiento cuando existen inhibidores
Es difícil determinar qué porcentaje de hemofílicos desarrollan anticuerpos inhibidores contra el factor VIII:C. Los inhibidores tienden a ocurrir en los pacientes más afectados y en los que reciben mayor número de concentrados de factor VIII. En un estudio prospectivo se determinó que el 33 porciento de los pacientes con hemofilia moderada o grave (i.e., aquellos con concentraciones de factor VIII:C menores del cinco porciento) desarrollaron inhibidores alrededor de los seis años de edad, o después de seis años de comenzar su tratamiento con concentrados. El 50 porciento de los hemofílicos con enfermedad grave desarrollaron inhibidores. No todos los inhibidores tuvieron una concentración suficiente como para producir problemas clínicos. Se ha sugerido que el tratamiento con factor VIII:C recombinante puede inducir mayor producción de inhibidores que las preparaciones actuales, pero esto no se ha comprobado aún.243,259 Es necesario investigar en forma periófica la presencia de inhibidores en todos los pacientes con hemofilia grave.
En ocasiones la hemorragia puede poner en peligro la vida y debe ser tratada con diversos procedimientos. En aquellos pacientes con hemorragias graves pero que no pongan en peligro la vida (i.e., no intracraneanas), con títulos de inhibidor menores de cinco unidades Bethesda y que no presentan respuestas adecuadas de anticuerpos, se debe administrar una cantidad elevada de concentrado de factor VIII en un intento por exceder el anticuerpo. La cantidad de concentrado de factor VIII administrada es de cerca de 40 Ulkg más 20 Ulkg adicionales por cada unidad Bethesda del inhibidor.252 De esta manera, el paciente que presenta un título de inhibidor de cinco unidades Bethesda recibiría 40 Ulkg + 5 x 20 Uikg, o 140 Ulkg. Debe medirse el nivel postinfusión de factor VIII:C para determinar sise ha alcanzado el punto final anticipado, de 0.3 U/ml de plasma.260
En aquellos pacientes con hemorragias más graves o con hemorragias de menor gravedad pero con niveles elevados de inhibidor contra el factor VIII humano, los concentrados de factor VIII:C porcino (Hyate:C) pueden ser el tratamiento de elección. La dosis recomendada es de 20 a 50 U/ kg de factor VIII:C porcino en caso de hemorragia moderada o cuando el sangrado ocurre en pacientes con título de inhibidor de menos de cinco unidades Bethesda; 50 a 100 Ulkg en caso de hemorragia severa o cuando el sangrado ocurre en pacientes con título de inhibidor de cinco a 50 unidades Bethesda; y 100 Ulkg en individuos con títulos de inhibidor mayores de 50 unidades Bethesda. La medición del título de inhibidor contra el factor VIII:C porcino brinda información útil: si el título es mayor de 13 unidades Oíd Oxford, la administración del factor VIII:C porcino no producirá beneficios terapéuticos.261,262
Ocasionalmente puede usarse plasmaféresis agresiva para reducir los niveles del inhibidor,190 seguida de la administración de concentrados de factor VIII:C. Sin embargo, este procedimiento es laborioso cono para usarse en un hemofílico que tiene una hemorragia potencialmente mortal o incapacitante. Se han usado concentrados de complejo de protrombina (CCP), tales como Proplex o Konyne, en el tratamiento de pacientes con títulos aumentados de inhibidores con base en que los factores presentes en dichos concentrados pudieran corregir la deficiencia de factor VIII:C. Un ensayo controlado demostró la eficacia de Konyne o Proplex, administrados como una dosis única' de 75 Ulkg, en el tratamiento de hemartrosis.264
Se ha introducido el uso de concentrados de protrombina activados (CCPA), tales como el complejo coagulante antiinhibidor (Autoplex), con la esperanza de que los factores activados IXa o VIIa presentes en estas preparaciones pudieran sereficaces para establecer la hemostasia;265 el CCPA se administró en una dosis de alrededor de 75 U/ kg. En algunas pruebas clínicas controladas, los CCPA lograron resultados ligeramente mejores que los CCP,266,267 pero también se han informado fracasos.268
Se han usado dosis altas de IgG intravenosa para tratar a pacientes no hemofílicos con inhibidores del factor VIII:C, y este tratamiento también ha sido benéfico en algunas ocasiones en pacientes con hemofilia.269
OTROS TRASTORNOS HEMORRAGICOS HEREDITARIOS
Deficiencia de factor IX
La deficiencia de factor IX (enfermedad de Christmas, hemofilia B) es un padecimiento ligado al cromosoma X que es indistinguible, desde el punto de vista clínico, de la hemofilia A. El gen del factor IX se localiza en el cromosoma X y produce un péptido de 56,000 daltos que, al igual que otros factores dependientes de vitamina K, tiene una región rica en ácidos glutámicos y-carboxilados. Al parecer el Ca+2 une esta región a las membranas de las células endoteliales, en donde el factor IX interactúa con el factor VIIIa para formar un complejo asociado a la membrana que puede convertir el factor X en factor Xa [ver figura 2]. Se han detectado gran número de inserciones, rearreglos y deleciones en el gen del factor IX, y el síndrome de hemofilia B es muy heterogéneo.228,229
Para hacer el diagnóstico se requiere una prueba de determinación de factor IX. Los principios de tratamiento son los mismos que para la hemofilia A. El factor IX es remplazado con plasma fresco congelado o con concentrados de complejo de protrombina. Estos concentrados estaban antes contaminados, en varios grados, con VIH y virus de la hepatitis. Además, pueden también contener factores Xa y VIIa, que vuelven al material trombogénico. Se ha esterilizado uno de los nuevos concentrados de factor IX (Armour Mononine) y tiene una excelente actividad específica y una vida media biológica muy conveniente, de 18 a 34 horas.269
La concentración de factor IX que se requiere para el control de la hemostasia en los pacientes con hemofilia B es un poco más baja que la concentración de factor VIII:C requerida para el tratamiento de la hemofilia A, alrededor de 0. 15 a 0.20 Ulmi, a diferencia de 0.3 a 0.5 U/ml de factor VIII:C. El factor IX es una molécula más pequeña que el factor VIII:C, y se distribuye en el espacio de la albúmina. Al hacer los cálculos de remplazo, debe suponerse que una unidad de factor IX por kilogramo aumentará la concentración plasmática en uno porciento, o en 0.01 U/ml. Este factor tiene una vida media bifásica, y su concentración plasmática puede mantenerse, en los pacientes con hemofilia B, infundiendo el concentrado cada 24 horas durante el episodio de hemorragia aguda. En la actualidad, las técnicas de clonación genética pueden detectar el estado de portador de deficiencia de factor IX, lo que permite proporcionar consejo genético adecuado.270,272
Deficiencia leve de factor VII
En ocasiones las pruebas de escrutinio preoperatorias revelan que el paciente presenta prolongación leve de¡ tiempo de protrombina en ausencia de hepatopatía, ingesta de alimentos inadecuada o administración de antibióticos. Se llega a demostrar que algunos de estos pacientes son heterocigotos para estados de deficiencia de factor VII, como lo confirman los estudios en familias y la determinación M nivel de] antígeno de factor VII en el plasma.273 No es necesario el tratamiento a menos que se contemple alguna cirugía mayor, en cuyo caso el factor VII puede suministrarse en forma de concentrados de complejos. de protrombina o plasma fresco congelado.
Alteraciones fibrinolíticas
Dos trastornos hemorrágicos congénitos se han relacionado con alteraciones de la fibrinolisis [ver figura 3].274 La deficiencia de a2-antiplasmina, el principal inhibidor de la plasmina, provoca una actividad no controlada de plasmina con la consecuente hemorragia.274 Se ha descrito otro trastorno hemorrágico que ocurre después de traumatismos que causan aumento de la actividad fibrinolítica. En este trastorno está implicado un exceso de activador circulante de plasminógeno, lo que lleva a la degradación del fibrinógeno y la fibrina. El tratamiento para ambos tipos de alteraciones fibrinolíticas consiste en el agente antifibrinolítico ácido tranexámico, en dosis de 1.5 g administrados por vía oral tres veces al día [ver figura 3].275 Como profilaxis para trabajos dentales, se administra una infusión intravenosa de 50 mg/kg de ácido tranexámico. Los agentes antifibrinolíticos, tales como el AEAC y el ácido tranexámico actúan bloqueando la unión e interacción del plasminógeno y plasmina con la fibrina.274
Los demás trastornos hereditarios de la coagulación ocurren en muy rara,,; ocasiones [ver tabla 10].
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
Trastornos hemorrágicos adquiridos
Además de los trastornos hereditarios de la coagulación, se han identificado varias alteraciones que pueden producir hemorragia generalizada [ver tabla 11].
|
|
|
DEFICIENCIA DE VITAMINA K
La carboxilasa dependiente de vitamina K sintetiza en el hígado ácido y-carboxiglutámico, necesario para la función biológica de los factores II, VII, IX y X.276,277 En ausencia de vitamina K se sintetiza el factor II, o protrombina anormal, que carece de residuos y-carboxiglutámicos.278 Los inmunoensayos específicos llevados a cabo en pacientes con deficiencia de vitamina K revelan una disminución importante en la protrombina normal y aumento concomitante en la des-y-carboxiprotrombina anormal. La misma alteración molecular ocurre con los factores X, IX y VII.
La deficiencia de vitamina K ocurre en la desnutrición grave, la malabsorción intestinal y la ictericia obstructiva [ver tabla 11]. En la ictericia obstructiva, las sales biliares, necesarias para la emulsificación y absorción de las vitaminas liposolubles (vitaminas A, D, E, y K), no pueden entrar al intestino. La ingestión crónica de antibióticos por vía oral parece suprimir la producción de vitamina K por los organismos intestinales. El antimicrobiano moxalactam es en particular eficaz para suprimir la flora bacteriana intestina] que provee de vitamina K. El efecto de este agente es de especial importancia en pacientes que, debido a su enfermedad, son incapaces de consumir una dicta completa y nutritiva [ver antes Trastornos de la función plaquetaria, Trastornos inducidos por medicamentos). La deficiencia de vitamina K produce disminución de la actividad biológica de los factores II, VII, IX y X. Los pacientes con cáncer que reciben combinaciones de cefoperazona y mezlocilina parecen estar propensos en particular a la hipoprotrombinemia.279 La combinación de cefoxitina y azlocilina produce hipotrombinemia similar.280 Si los niveles de procoagulante disminuyen por debajo de 10 a 15 porciento de los normales, se presentan hemorragias de mucosas y equímosis.
El diagnóstico puede confirmarse si se prolonga el tiempo de protrombina y proconvertina, así como el tiempo de protrombina; el tiempo parcial de tromboplastina es normal o está un poco prolongado y el nivel de fibrinógeno es normal. El tratamiento con análogos de la vitamina K, administrando de 10 a 25 rng de fitonadiona diarios por vía oral durante dos a tres días o bien a base de fitonadiona por vía parenteral, en la ictericia obstructiva, corregirá las alteraciones en cerca de seis a 24 horas. Sin embargo, cuando el agente antimicrobiano tiene una cadena lateral similar a la de la warfarina, pueden pasar hasta 36 horas antes de que el tratamiento restablezca los valores de protrombina a lo normal.280 Si el paciente presenta hemorragias severas, el retraso puede ser peligroso, por lo que el paciente debe ser tratado con plasma fresco congelado para restaurar los niveles de procoagulantes al menos a 30 porciento de lo normal [ver antes, Principios de tratamiento sustitutivo]. En tales casos se requieren aproximadamente tres unidades (tres bolsas) de plasma fresco congelado.
HEMORRAGIA INDUCIDA POR MEDICAMENTOS
En la sobredosis de warfarina, el evento precipitante puede ser una dosis mal administrada de warfarina o la ingestión simultánea de agentes que potencían la acción anticoagulante de este medicamento. Los efectos de administrar un antibiótico fluoroquinolona a un paciente en tratamiento con warfarina pueden ser devastadores. El autor ha observado varias hemorragias graves dependientes de vitamina K dos días después de la administración de dosis habituales de ciprofloxacina en un paciente que recibe una dosis continua y estable de warfarina.281 Lo habitual es que ocurran hemorragias en mucosas, equímosis o hemorragias subserosas en la pared intestinal de estos pacientes. El TP y P y P están prolongados. Si la hemorragia es importante, debe iniciarse el tratamiento descrito calculado para restaurar los niveles de procoagulante a 30 porciento de lo normal con plasma fresco congelado. Si no existe urgencia puede administrarse fitonadiona como se describió (ver antes). Como es de esperarse, el uso de warfarina se relaciona con disminución en la protrombina nativa y aumento concomitante en el nivel de des-y-carboxiprotrombina anormal, que no coagula.278 La ingestión subrepticia de warfarina puede identificarse con el análisis sérico de la misma, disponible en laboratorios especializados.
La sobredosis de heparina puede no ser obvia. Causa hemorragias subcutáneas y hematomas en los tejidos profundos y se asocia con TPT, TP y TT muy prolongados, aunque el tiempo de reptilasa es normal. La administración de protamina intravenosa en dosis de 1 rng por 100 unidades de heparina administrada corregirá el trastorno. Como la vida media de desaparición de la protamina es más rápida que la de la heparina, puede presentarse un "rebote de heparina" que requiere una segunda administración de protamina. Las preparaciones de heparina de bajo peso molecular se asocian con el mismo riesgo de hemorragia que la heparina habitual no fraccionada.282
En la actualidad se emplea el tratamiento trombolítico en el infarto agudo del miocardio y en algunos casos de embolia pulmonar. Las complicaciones de este tipo de tratamiento son de tipo hemorrágico. Por lo general la hemorragia se limita a rezumo de Sangre alrededor de los sitios de invasión vascular, 113 pero pueden ocurrir hematomas subdurales, infarto cerebral y hemorragia intracraneana. Los agentes trombolíticos, incluso los diseñados para ser relativamente específicos a la fibrina, causan en ocasiones grados peligrosos de lisis del fibrinógeno. Además, la generación de productos de degradación del fibrinógeno interfiere con la formación de un coágulo firme y con la función plaquetaria. Los agentes trombolíticos pueden también destruir al factor VIII y al factor V.
Si se sospecha que la causa de la hemorragia es el tratamiento trombolítico, debe obtenerse de inmediato sangre para medir TPTa, tiempo de trombina, tiempo de reptilasa, concentración de fibrinógeno y a2-antiplasmina. Si el tratamiento trombolítico es la causa, el TPTa estará prolongado, la concentración de fibrinógeno será menor de 50 mg/dl, el tiempo de trombina y de reptilasa estarán prolongados (por la presencia de productos de degradación de la fibrina) y la a2-antiplamina estará disminuida.213 Esta complicación se trata con crioprecipitado para aumentar la concentración de fibrinógeno a alrededor de 100 mg/dl, con aproximadamente dos unidades de plasma fresco congelado, que pueden aumentarse hasta seis unidades según se requiera (para remplazar factor V y otros procoagulantes), y con alrededor de seis paquetes de plaquetas. Si estas medidas no detienen la hemorragia debe considerarse el uso de un agente antifibrinolítico específico, como el AEAC. El AEAC se administra en dosis de 5 g en bolo I.V. durante 30 a 60 minutos, y después en dosis de 1 g/hr por medio de infusión I.V. continua.283
El tratamiento de los trastornos de lípidos con preparaciones de niacina de liberación sostenida puede en ocasiones causar hepatotoxicidad, que se manifiesta por prolongación del tiempo de protrombina.285
DISPROTEINEMIAS
Las proteínas anormales asociadas con el mieloma y la macroglobulinemia pueden interferir con la función plaquetaria y causar hemorragias clínicas. Estas proteínas o la enfermedad principal pueden causar de igual manera alteraciones en las pruebas de coagulación. Las proteínas de mieloma IgG e IgA pueden prolongar el tiempo de trombina sin hemorragia clínica. En el mieloma de IgA ocasionalmente ocurre deficiencia de factor V. Las disproteinemias de IgA e IgM pueden causar hemorraagias al reducir la actividad del factor VIII.
El manejo está orientado a la enfermedad primaria. La plasmaféresis corrige rápidamente los defectos, pues disminuye notablemente el nivel de proteína anormal.286
COAGULACION INTRAVASCULAR DISEMINADA
El sistema de coagulación completo, iniciado por la vía extrínseca o por la vía intrínseca, está perfectamente coordinado para culminar en un brote de actividad de trombina (factor IIA) en el sitio de la lesión vascular. Por sus acciones catalíticas sobre las plaquetas y los factores I, V, VIII y XIII, la trombina causa actividad hemostática en el sitio de la lesión, induciendo cambios plaquetarios y causando el depósito de fibrina con uniones cruzadas para formar el tapón hemostático. En condiciones normales los efectos nocivos de la coagulación intravascular descontrolada (sobre todo por una acción no controlada de la trombina) están modulados por los efectos dilucionales del flujo sanguíneo, por antitrombinas circulantes, por la remoción de partículas y de factores activados por las células reticuloendoteliales en el hígado, por la antiplasmina, y por factores que regulan la hemostasia: AT-III los sistema de proteína C y proteína S, y el lVFT.
Estos mecanismos, cuidadosamente controlados, pueden ser superados bajo las siguientes circunstancias:
1. Daño tisular masivo, que libera cantidades enormes de materiales tromboplásticos tisulares, causando activación extensa del sistema extrínseco [ver figura 1].
2. Alteración extensa del endotelio vascular, que expone cantidades iinportantes de los procoagulantes intrínsecos a los iniciadores subendoteliales.
3. Estado de choque, asociado con disminución del flujo Sanguíneo y pérdida de los efectos benéficos de la hemodilución.
4. Deterioro de la perfusión o función hepática, que causa una inadecuada remoción hepática de las partículas circulantes y procoagulantes activados.
Existen muchas circunstancias diferentes que pueden causar CID [ver tabla 12] En cada caso, la activación masiva de mecanismos hermostáticos supera a los mecanismos inhibidores [ver figura 9]. Esta fuga de la hemostasia y las respuestas fisiológicas a la misma conducen a la propagación cíclica de esta catástrofe en evolución. La coagulación masiva consume procoagulantes y plaquetas, causando hemorragias. La generación no controlada de trombina ocasiona trombosis en los lechos arteriales y venosos, dando lugar a infartos isquémicos y necrosis que intensifican el daño y activan aún más el sistema hemostático. El daño tisular y los depósitos de fibrina [ver figura 3] producen la liberación y activación de activadores de plasminógeno y la generación de plasmina en cantidades que superan a su inhibidor, la a2- antiplasmina. La plasmina degrada los factores VIII, V y I y produce productos de degradación de fibrinógeno y fibrina; estas sustancias, así como los productos de fibrina polimerizada en forma incompleta, impiden la función plaquetaria y la polimerización normal de la fibrina [ver figura 1].
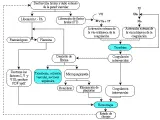
|
| Figura 9 |
| Coagulación intravascular diseminada |
|
||||
|
La liberación de endotoxinas durante la septicemia por gram negativos aumenta la expresión del factor tisular, acelerando con ello la activación procoagulante [ver figura 1] mientras que suprimen la expresión de trombomodulina [ver figura 5]. Estas acciones deterioran la regulación del sistema de proteína C-proteína S, promoviendo aún más la tendencia a la CID.287
También se ha observado CID en pacientes con hemangiomas múltiples o solitarios asociados con trombocitopenia (síndrome de Kassabach-Merrit). En estos tumores se consumen plaquetas y fibrinógeno, parece ser que la fibrinolisis aumenta,288 y esto puede causar hemorragias y muerte. La CID relacionada con estos tumores parece iniciarse por el contacto prolongado de las células endoteliales neoplásicas anormales con la sangre en regiones de estasis vascular.
Ciertas mordeduras de víbora también pueden producir CID; se han identificado varios mecanismos. Por ejemplo, el veneno de la víbora Russel contiene un potente procoagulante que produce desfibrinación casi instantánea. El veneno también contiene una nefrotoxina altamente mortal al igual que hemorraginas que dañan el endotelio vascular y causan alteraciones plaquetarias.289
Las consecuencias de la coagulación intravascular diseminada dependen de su causa y de la rapidez con la que se propague el suceso inicial. Si la activación ocurre lentamente, se produce un exceso de productos activados, predisponiendo a trombosis. La situación clínica consiste en infartos vasculares o en trombosis venosas. A veces se conoce esta entidad como CID compensada o crónica. Si la reacción es brusca y explosiva, el cuadro clínico está dominado por la coagulación intravascular, 1, 11, V, VIII y XIII (y quizás de factor VII), y la producción por acción de la plasmina de productos de degradación de fibrina, que interfieren aún más con la hemostasia. Las consecuencias clínicas son hemorragias alrededor de los sitios de heridas, líneas intravenosas y catéteres, al igual que hemorragias en tejidos profundos. Las bandas intravasculares de fibrina producen anemia hemolítica microangiopática.
Diagnóstico
El frotis de sangre periférica con eritrocitos mieroangiopáticos y una cuenta plaquetaria baja, y que disminuye cada vez más a pesar de existir una cantidad adecuada de megacariocitos en la médula ósea, orientan al diagnóstico. Debido a la pérdida de procoagulantes, las pruebas de TPT, TP y P y P son anormales. Como los productos de degradación de la fibrina interfieren con la polimerización de la misma y producen complejos no coagulantes, también se prolonga el tiempo de trombina y de reptilasa. La concentración de productos de degradación de la fibrina está elevada. Las pruebas para valorar el sistema fibrinolítico, como el tiempo de lisis de la euglobulina, muestran aumento de la actividad fibrinolítica y disminución de los niveles de plasminógeno. La medición del plasminógeno, y en menor grado de los niveles de la proteína C, permiten confirmar el diagnóstico por laboratorio de CID.290 También la a2-antiplasmina se depleta en los pacientes en quienes existe hiperplasminemia.291 La CID puede distinguirse de la fibrinolisis primaria, una enfermedad muy rara, con la prueba del dímero D de la fibrina. Esta prueba detecta los fragmentos de fibrina polimerizados que ocurren en la CID, pero no en la fibrinolisis primaria. Existe otro método experimental que permite diferenciar entre el exceso de polimerización normal de la fibrina, como ocurre en la CID, y la destrucción de fibrina soluble o parcialmente polimerizada mediada por la plasmina [ver figura 1]. La medición de la relación del fibrinopéptido B (FPB), derivado de la polimerización de la fibrina, con el fragmento péptido B B1-42, producto del ataque de la plasmina, ayuda a determinar si el evento principal es CID o fibrinolisis.25
Es sumamente importante la repetición a intervalos regulares de las pruebas de coagulación antes enumeradas, ya que las pruebas repetidas constituyen un indicador cinético que ayuda mucho en el diagnóstico. Por ejemplo, un paciente con vasculitis agresiva o con rechazo agudo de injerto puede tener un nivel alto de fibrinógeno y una cuenta plaquetaria alta debido al componente inflamatorio de la enfermedad principal. Sin embargo, una sola determinación puede mostrar un nivel de fibrinógeno normal y una cuenta plaquetaria normal, así que puede desorientar en este paciente, ya que los valores pudieran ser mucho mayores varias horas antes. La disminución en los niveles de procoagulantes y plaquetas mayor de lo que esperaría con base en su vida media biológica, indica su remoción rápida (y probablemente su consumo acelerado). De manera similar, si los intentos de reponer dichos factores producen resultados substancialmente inferiores a los calculados, es probable que esté ocurriendo un consumo acelerado.
Tratamiento
El tratamiento debe estar dirigido a la enfermedad primaria para de esta forma detener el evento inicial. Este enfoque puede relacionarse con el uso de quimioterapia en enfermos con cáncer, tratamiento de infecciones o vaciamiento uterino cuando las complicaciones del embarazo han iniciado el problema. El apoyo hemodinámico es indispensable. Se debe evitar el uso de inhibidores de plasminógeno y plasmina, como el AEAC o la aprotinina; estos agentes bloquean el sistema fibrinolítico y por ello pueden causar el acumulo continuo de múltiples trombos y provocar infartos vasculares extensos. La infusión de fibrinógeno significa sólo echar leña al fuego, ya que la plasmina convertirá el fibrinógeno recién añadido en productos de degradación del fibrinógeno, que interferirán aún más con la coagulación. Por lo general son suficientes el apoyo cardiopulmonar y la atención al trastorno primario.
La pregunta decisiva consiste en cuándo usar heparina, que antagoniza en forma directa la trombina por su interacción específica con la antitrombina III.292 El tratamiento con heparina no se justifica con base sólo en pruebas de coagulación anormales. Sin embargo, puede considerarse la heparinización en caso de hemorragia que amenace la vida, hemorragias en heridas, en tejidos profundos o hacia cavidades corporales, y también si se puede realizar una maniobra clínica específica hacia la enfermedad básica con una probabilidad razonable de controlarla en dos a tres días. Debe enfatizarse que con la posible excepción de la CID relacionada con la leucemia promielocítica aguda y otras formas de LMA, no hay evidencias claras para apoyar el uso de la heparina. La práctica del autor consiste en evitar el uso de la heparina. La práctica del autor consiste en evitar el uso de la heparina con excepción de las situaciones específicas señaladas.
No existen estudios clínicos aleatorios en los que pueda basarse la dosis. Se puede iniciar el tratamiento con 500 unidades/hr de heparina, administrada por vía intravenosa por medio de una bomba de infusión continua, con el bolo intravenoso inicial de 500 a 1,000 unidades o sin éste. Después de haber administrado heparina por dos horas, se puede iniciar la reposición de procoagulantes con la protección de este fármaco. Se pueden administrar de 2 a 3 unidades de plasma fresco congelado si el sistema cardiovascular del paciente tolera la carga de volumen. Esta dosis proveerá de 550 a 750 unidades de procoagulantes y cofactor antitrombina III-heparina. También se puede añadir de cuatro a ocho unidades de plaquetas. Hay que evitar el uso de Proplex, un concentrado de factores II, VII, IX y X pues contiene factores activados que aumentan la coagulación intravascular.
Una vez que se inicia el tratamiento con heparina, el tiempo de protrombina, el tiempo parcial de tromboplastina y el tiempo de trombina no se pueden interpretar. Es útil realizar pruebas periódicas de productos de degradación de la fibrina (en especial de dímero D), tiempo de reptilasa, medición cuantitativa de fibrinógeno y cuenta de plaquetas con intervalos de seis a 12 horas. Los productos de degradación de fibrina son depurados con más rapidez que la producción de plaquetas o la síntesis de fibrinógeno. Por lo tanto, un resultado favorable suele ir precedido por disminución de los productos de degradación de la fibrina y por un nivel estable de plaquetas y fibrinógeno. El remplazo subsecuente de factores se calcula con base en estos resultados. Si el nivel de fibrinógeno es muy bajo se puede usar crioprecipitado como fuente de reposición de fibrinógeno. Cada bolsa de crioprecipitado proporciona alrededor de 400 mg de fibrinógeno y de 80 a 100 unidades de factor VIII. Si continúan las hemorragias clínicas y los resultados de los exámenes no muestran mejoría, entonces puede aumentarse la infusión continua de heparina a 750-1,000 U/hr. Después de que cesa la hemorragia clínica y se estabiliza el estado del paciente, se reduce poco a poco la dosis de heparina, y se vigilan los resultados de las pruebas hematológicas hasta que pueda suspenderse la heparina.293
El tratamiento de la CID asociada con hemangiomas solitarios o múltiples plantea problemas específicos. Cuando los hemangiomas están localizados, pueden extirparse; en ocasiones muestran buena respuesta a la radiación local. Los intentos por controlar la CID utilizando heparina, esteroides, aspirina, sulfinpirazona, estrógenos y dipiridamol no han sido satisfactorios.288 Sin embargo, la infusión de crioprecipitado, 10 bolsas diarias durante tres días, junto con 36 g de AEAC al día (el doble de la dosis usual recomendada), produjo respuesta excelente en un caso.288 Se supone que el crioprecipitado restituye la deficiencia de fibrinógeno lo suficiente para permitir que ocurra trombosis local en los hemangiomas; la administración simultánea de AEAC evita entonces la lisis de estos coágulos. De manera subsecuente, los niveles de procoagulantes y plaquetas mejoran en forma sustancial.288
La clave para el tratamiento satisfactorio de la CID relacionada con la mordedura de ciertas víboras consiste en la indentificación de la víbora y la administración inmediata del antídoto apropiado.
FIBRINOLISIS PRIMARIA
Los casos de fibrinolisis primaria generalizada son raros, y muchas de las publicaciones iniciales de fibrinolisis primaria es probable que representaran fibrinolisis secundaria relacionada con la CID [ver figura 3 y 9]. Como se señaló, la fibrinolisis primaria puede distinguirse de la CID utilizando la prueba del dímero D de la fibrina, que detecta los productos de degradación de la fibrina polimerizada que ocurren en la CID. La medición del fragmento B B1-42, un producto específico del ataque de la plasmina sobre los complejos solubles de fibrina,25 puede ofrecer un nuevo método para detectar hiperplasminemia.
El veneno de serpiente puede producir afibrinogenemia por dos mecanismos diferentes. En uno, la destrucción extensa de tejido causada por lipasas y proteasas del veneno desencadena CID, disminuyendo los niveles de fibrinógeno. Otros venenos, v.gr., el de la víbora de cascabel occidental, Crotalus atrox, liberan activador de plasminógeno endógeno, induciendo de esta manera fibrinolisis primaria y afibrinogenemia [ver figura 3].294
La hematuria posprostatectomía puede constituir un ejemplo verdadero de hemorragia debida a fibrinolisis localizada. En esta entidad la concentración elevada de urocinasa en la orina hace que el plasminógeno sea convertido en plasmina [ver figura 3], con la consecuente lisis del coágulo. Si se pueden descartar otras causas de hematuria posoperatoria persistente, la entidad puede tratarse con una dosis de 0.1 g/kg de ácido e-aminocaproico administrado por vía oral o intravenosa. La dosis se repite cada seis horas hasta que se controla la hemorragia.295 También resulta eficaz la instilación local de AEAC por la sonda uretral.
ENFERMEDADES HEPATICAS GRAVES
Los pacientes con enfermedades hepáticas graves pueden presentar hemorragias que pongan en peligro su vida por deterioro extenso de sus mecanismos hemostáticos [ver tabla 13]. Además de várices sangrantes, gastritis y úlcera péptica, las hemorragias clínicas incluyen hemorragias en las mucosas y en los tejidos.
|
|
|
Los pacientes con cirrosis tienen disminución de la cantidad de protrombina y aumento en los niveles de fibrinógeno anormal.278 En forma característica, la supervivencia plaquetaria está disminuida, y el secuestro esplénico de las mismas está aumentado. La mayoría de los pacientes cirróticos tienen función plaquetaria normal, como lo indican el tiempo de sangrado y estudios de agregación plaquetaria normales Se acorta el tiempo de recambio de fibrinógeno, pero el nivel de fibrinógeno se mantiene por lo general en el límite normal. No aumentan la fibrina ni los productos de degradación de fibrinógeno. En la cirrosis se acorta la vida media del plasminógeno y sus niveles disminuyen.296 Las pruebas más fidedignas de detección para esta enfermedad incluyen el tiempo de protrombina, el tiempo de protrombina y proconvertina (P y P), la cuenta plaquetaria y el tiempo de Sangrado.
La sustitución se logra administrando 1,000 m] de plasma fresco congelado y plaquetas según se requiera. No deben usarse concentrados de complejos de protrombina porque no contienen factor V y tienen productos activados que el hígado dañado no puede retirar en forma eficaz, lo que aumenta el riesgo de CID. Si la hemorragia no es grave puede administrarse vitamina K en dosis de 10 mg/día durante tres días.297
INHIBIDORES CIRCULANTES, (ANTICOAGULANTES)
Además de los anticuerpos circulantes inhibidores que se observan en las hemofilias graves A y B, las hemorragias clínicas ocasionalmente están causadas por inhibidores circulantes de la coagulación que al parecer aparecen de manera espontánea. Un tipo de inhibidor se dirige contra factores específicos tales como el factor VIII.
Debido a que los autoanticuerpos contra el factor VIII:C son los que se han estudiado más, la descripción que sigue a continuación se refiere a este inhibidor inespecífico, pero muchos de estos mismos principios se aplican también a otros inhibidores. Los autoanticuerpos contra el factor VIII:C suelen ser de tipo IgG, sobre todo IgG4, por lo que no fijan complemento. Por lo general están dirigidos contra los sitios epítopes activos del factor VIII:C, y aunque se unan a otros sitios, alteran la función del factor VIII:C al disminuir en forma importante su vida media biológica y disminuir su concentración.298 Alrededor de la mitad de los pacientes con inhibidor de factor VIII:C clínicamente significativo no tienen trastornos asociados detectables, pero se han identificado muchas alteraciones en el resto. Entre ellas se encuentran trastornos autoinmunes como lupus eritematoso generalizado, padecimientos linfoproliferativos, neoplasias de células malignas, reacciones a medicamentos (v.gr., a penicilina), estados posparto y trastornos de la piel.299
El cuadro clínico de hemorragias mucosas, hematomas y equimosis requiere pruebas de coagulación. Si hay un inhibidor, el tiempo parcial de tromboplastina, tiempo de protrombina o tiempo de trombina estarán selectivamente prolongados dependiendo del sitio de acción del inhibidor. La incapacidad de corregir el resultado anormal de la prueba mezclando partes iguales del plasma del paciente y plasma normal es muy sugestiva de la presencia de un inhibidor. Los laboratorios especializados en pruebas de coagulación están equipados para detectar inhibidores, identificar su sustrato y cuantificar su actividad.
Clínicamente la hemorragia puede poner en peligro la vida. Los intentos por sustituir los factores suelen no tener éxito porque el inhibidor inactiva el procoagulante remplazado. Sin embargo, las cantidades masivas de procoagulantes pueden en ocasiones sobrepasar la acción del inhibidor. Puede ser posible extraer el inhibidor por plasmaféresis. Algunos informes sugieren que la administración de azatioprina, en dosis de 100 a 150 mg/día o ciclofosfamida, en dosis de 100 a 150 mg/día, combinados con 60 mg/día de prednisona o sin ésta, puede reducir el nivel del inhibidor.' La combinación de quimioterapia con ciclofosfamida y prednisona ha sido probada en un estudio controlado en el que se administró cada medicamento en una dosis de 2 mg/kg/día durante tres a seis semanas.´ El autor empleó con éxito, en un paciente con 900 unidades Bethesda de inhibidor, un programa de tratamiento de 50 a 100 Ulkg de concentrado de factor VIII:C seguidos de 500 rng de ciclofosfamida el día 1 y de 200 mg los días 2 a 5, además de vincristina, en dosis de 2 mg/kg I.V. el día 1 y 100 rng de prednisona los días 1 y 5. El esquema se repitió cada tres a cuatro semanas.302
En el caso de hemorragia aguda que pone en peligro la vida en el que no existe tiempo suficiente para tratar de disminuir la concentración de inhibidor, puede administrarse factor VIII:C porcino (Hyate:C, Porton Products, Ltd.) porque por lo general los autoanticuerpos tienen una reactividad cruzada muy baja con el factor VIII:C porcino. La dosis inicial es de alrededor de 100 Ulkg de factor VIII:C porcino.303 La otra alternativa de tratamiento en caso de hemorragia aguda es la administración de CCP, que parece saltarse el bloqueo del inhibidora] proporcionar grandes cantidades de factor X, factor VII y quizá factor VIIIa, aumentando así la hemostasia a través del sistema extrínseco [ver figuras 1 y 2] y disminuyendo la dependencia al complejo del factor VIIIa-IXa. Estos CCP, como el Proplex T (Baxter Healthcare), o los CCP activados, como el Autoplex T (Baxter Healthcare) o Feiba VII Immuno (Immuno-U.S.), administrados en dosis de 75 Ulkg, pueden controlar las hemorragias en algunos pacientes; sin embargo, es probable que estas preparaciones contengan virus de hepatitis B y C, y puedan ser trombogénicas.304
De manera sorprendente, las dosis altas de IgG pueden producir beneficio de larga duración en pacientes no hemofílicos con autoanticuerpos contra el factor VIII:C.305, Los pacientes han sido tratados con IgG I.V. en dosis diarias de 0.4 g/kg durante cinco días consecutivos,305 o en dosis diaria de 0.5 g/kg durante ocho días consecutivos.306 La infusión produjo disminución en el nivel de inhibidor y aumento en el factor VIII:C, y esta respuesta parece haberse mantenido. Algunos pacientes con hemofilia que tuvieron el aloanticuerpo inhibidor del factor VIII:C también
respondieron a este tratamiento.307 Tales preparaciones almacenadas de IgG I.V. contienen anticuerpos antiidiotípicos que pueden reaccionar e inactivar los autoanticuerpos contra el factor VIII:C encontrado en el plasma de los pacientes.308
Anticoagulante lúpico
Otro tipo de inhibidor se denomina inhibidor tipo LE G, o anticuagulante lúpico, porque se identificó inicialmente en pacientes con esta enfermedad y su aparición, en dichos pacientes, se correlaciona con un resultado falso positivo de la reacción biológica de Wassermann para la sífilis. El clínico debe estar alerta de la posible presencia de este inhibidor en pacientes sin trastornos hemorrágicos importantes que presentan TPT muy prolongado. En un estudio, el 33 porciento de los sujetos con este trastorno también tuvieron tiempo de protrombina prolongado y el 25 porciento tuvieron tiempo de trombina prolongado.309 Al llevar a cabo el estudio combinado utilizando el plasma del paciente y plasma normal se demostrará de inmediato la prolongación de TPT.
Al parecer el anticoagulante lúpico está dirigido contra la cefalina añadida como fuente accesoria del componente fosfolípido del complejo protrombinasa en el tiempo parcial de tromboplastina [ver figura 1]. Si las plaquetas lavadas son sustituidas por el fosfolípido agregado, el TPT se normaliza en los pacientes con anticoagulante lúpico.309 Por consiguiente, el anticoagulante 1 úpico que se encuentra en pacientes con muchos tipos de trastornos además del LEG, es en realidad un fenómeno in vitro.
El anticoagulante lúpico es un anticuerpo que muestra heterogeneidad importante, ocurriendo como IgM o IgG policlonal310 y en ocasiones como IgM monoclonal.309 A menos que las personas con este inhibidor tengan también otra alteración que afecte el mecanismo hemostático, no tendrán trastorno hemorrágico, y no hay necesidad de limitar el uso de estudios invasivos en tales casos.311 El tratamiento crónico con clorpromacina puede causar la aparición del anticoagulante lúpico; su ocurrencia se correlaciona con la dosis total y la duración del tratamiento con clorpromacina.312 Muchos pacientes con SIDA tienen también anticoagulante lúpico,310 que suele aparecer durante las infecciones oportunistas y luego desaparecer con la resolución clínica. Dada la heterogeneidad del anticuerpo anticoagulante lúpico, no es sorprendente que la presencia de este anticuerpo se relacione con diversos cuadros clínicos.309,310
El anticuerpo anticoagulante lúpico y el anticuerpo que se asocia con una prueba serológica falsa positiva para sífilis, la anticardiolipina, están dirigidos contra fosfolípidos. Los anticuerpos denominados anticardiolipina, que en la actualidad se detectan por prueba de ELISA, están dirigidos contra un complejo compuesto por la cardiolipina agregada y una proteína plasmática denominada glucoproteína 1 o apolipoproteina 11; es posible que la glucoproteína 1 tenga un efecto anticoagulante.313 La presencia de anticoagulante lúpico y de anticuerpo anticardiolipina define el síndrome antifosfolípido, que se caracteriza por trombosis recurrentes en las venas profundas de las extremidades inferiores, trombosis en las venas renales y hepáticas, hipertensión pulmonar, oclusión arteria] cerebral asociada a infartos y episodios de isquemia cerebral transitoria, datos neurológicos que recuerdan la demencia por infartos múltiples, epilepsia y trombocitopenia moderada. Otras manifestaciones del síndrome antifosfolípido incluyen alteraciones dermatológicas como livedo reticularis, úlceras en los miembros inferiores y púrpura necrosante y, en mujeres embarazadas, abortos espontáneos y muerte fetal.314 Los pacientes con este síndrome que sufren tromboembolias venosas o arteriales requieren de tratamiento anticoagulante.315,316 Sin embargo, no se ha establecido cuál es el tratamiento adecuado para las mujeres que presentan abortos espontáneos y muerte fetal. Se ha notificado que el tratamiento combinado con prednisona, 50 mg/día y ácido acetilsalicílico, 80 mg/día, es eficaz.317 Sin embargo, la prednisona indujo una incidencia relativamente alta de preeclampsia y causó también problemas fetales. En la actualidad no existe un esquema terapéutico bien establecido, y se usan por lo menos cuatro regímenes diferentes: (1) ácido acetilsalicílico en dosis de 80 mg/día, (2) heparina subcutánea durante todo el embarazo en una dosis suficiente para prolongar el TPTa uno o una y media veces el valor control, (3) combinación de ácido acetilsalicílico y heparina y (4) adición de dosis bajas de prednisona, por ejemplo, 10 a 20 mg/día, o la combinación de ácido acetilsalicílico y heparina.
HEMATOMAS CRONICOS EXPANSIVOS
En algunos casos se desarrollan hematomas después de cirugía o traumatismos, en ausencia de una anormalidad hemostática. Dichas lesiones pueden seguir expandiéndose debido a su tamaño y propiedades inflamatorias. Es importante reconocer la posibilidad de un hematoma expansivo crónico porque la lesión puede ser extirpada mediante cirugía.318
HEMORRAGIA DESPUES DE LA DERIVACION CARDIOPULMONAR
Los pacientes sometidos a cirugía cardiaca con derivación cardiopulmonar presentan con frecuencia episodios trans y posperatorios de hemorragias graves, en ausencia de consumo de procoagulantes o de sobredosis de heparina.319 Parece existir en estos casos un trastorno adquirido de la función plaquetaria, quizá causado por el contacto entre las plaquetas y el aparato oxigenador. Además de la liberación del contenido de los gránulos alfa de las plaquetas, puede ocurrir activación de la fibrinolisis y depleción moderada de los factores procoagulante.297
En estos casos la hemorragia se asocia con tiempo de sangrado prolongado y a menudo responde alas transfusiones de plaquetas. Sin embargo, dichas transfusiones son costosas, consumen tiempo y se asocian con riesgo de trasmisión de hepatitis, SIDA y otras infecciones virales.
En un estudio,320 pacientes sometidos a colocación de injerto de derivación en las arterias coronarias fueron tratados con 0.3 ug/kg de DDAVP en 50 mi, administrados por vía intravenosa durante 15 minutos después de concluir la derivación cardiopulmonar y de que el efecto de la heparina había sido revertido con protamina. La disminución de alrededor del 40 porciento en la pérdida sanguínea posoperatoria se relacionó con elevación en los niveles del factor de von Willebrand. Es posible que este incremento en la concentración del FvW haya compensado la función plaquetaria defectuosa. Se alcanzó una disminución semejante en la pérdida sanguínea con la administración de DDAVP a pacientes sometidos a cirugía de fusión de la columna vertebral con barras de Harrington.321 Sin embargo, no todos los estudios confirman los beneficios de la DDAVP en estas situaciones clínicas.297 En especial, dos estudios no confirmaron los efectos útiles de la DDAVP en la cirugía cardiovascular.322,323
Estados de hipercoagulabilidad
Muchos casos de trombosis son causados por un evento local, como el daño a la pared vascular (debido a ateroesclerosis o vasculitis) o estasis, pero un problema reológico también puede predisponer a trombosis. Por ejemplo, el hematocrito elevado en la policitemia vera puede retardar el flujo sanguíneo, y los eritrocitos rígidos indeformables presentes en hemoglobinopatías, tales como la enfermedad de células falciformes o la enfermedad por hemoglobina C, pueden inducir trombosis en los pulmones o en la mierociruculación. Puede haber trombosis en enfermedades mieloproliferativas crónicas relacionadas con una cuenta de plaquetas muy elevada, y aun así es difícil demostrar una correlación entre los episodios trombóticos y la cuenta plaquetaria, las alteraciones funcionales plaquetarias o el nivel espontáneo de agregación plaquetaria.195 Entre los problemas que indican la existencia de una alteración hemostática de fondo se encuentran los antecedentes familiares de trombosis o la aparición de trombosis recurrente o en sitios atípicos en personas jóvenes por lo demás sanas [ver Tabla 14].
|
|
|
SINDROME ANTIFOSFOLIPIDO
No se ha determinado cuáles el mecanismo por el que el anticoagulante lúpico, el anticuerpo anticardiolipina u otros anticuerpos aún no identificados, causan un estado de hipercoagulabilidad en los individuos con el síndrome de anticuerpos antifosfolípido (ver antes).214,324,325
Los pacientes con anticoagulante lúpico [ver antes, Trastornos hemorrágicos adquiridos, Inhibidores circulantes (anticoagulantes)] tienen un aumento paradójico en la incidencia de trombosis, y la causa de esto no es clara.326 En un caso, el inhibidor lúpico inhibió la activación de la proteína C a proteína C activada (PCA) mediada por trombina-trombomodulina, estableciendo así un estado de hipercoagulabilidad.327
Los pacientes con anticoagulante lúpico y que requieren cirugía deben ser manejados con minidosis profilácticas de heparina o programas antitrombóticos parecidos. Los enfermos con el anticoagulante lúpico y que sufren trombosis de las venas profunda: son difíciles de tratar porque el TPT ya está muy prolongado, y en ocasiones el TP también lo está. El anticoagulante lúpico tiene una ligera asociación con disminución en la concentración de protrombina y una asociación más frecuente con trombocitopenia [ver antes, Trastornos adquiridos de la hemostasia, Anticoagulante lúpico]. Pueden usarse esquemas de dosis fijas de heparina en un intento por controlarla trombosis; la warfarina oral se inicia después, y el tiempo de P y P puede ser muy útil para ajustar la dosis.
DEFICIENCIA DE ANTITROMBINA III
La deficiencia de antitrombina III [ver figura 5], que puede ser hereditaria o adquirida, deteriora la inactivación de los procoagulantes activados, factores Xa y Ila, causando trombosis.53. La ATIII al parecer inactiva a los factores XIIa, XIa, IXa y Xa al formar un complejo estoiquiométrico estable con cada uno de estos factores, siendo el mismo mecanismo por el cual la AT-III inactiva a la trombina. La AT-III está presente en el plasma en cantidades suficientes para inactivar toda la trombina en un volumen de plasma determinado, pero lo hace en forma lenta a menos de que sea activada por el heparán de las membranas celulares o la administración de heparina.328 El nivel normal de AT-III activada por el heparán presente en forma natural [ver figura 2] ejerce un efecto modulador estable en la hemostasia al neutralizar el exceso de factor Xa. Los pacientes con deficiencia hereditaria de AT-III tienen mayor incidencia de trombosis venosa; 328 rara vez, llegan a ocurrir trombosis arteriales.329 Un hallazgo típico es la trombosis de la vena mesentérica.
Por lo general hay antecedentes familiares de tromboembolias recurrentes que suelen comenzar en la juventud y a menudo se relacionan con cirugía o traumatismos. El embarazo y el uso de anticonceptivos orales aumentan el riesgo de trombosis en pacientes con deficiencia de AT-III.328 La tendencia a la trombosis aumenta con la edad, de manera que a los 50 años sólo el 10 porciento de los pacientes afectados están asintomáticos.53 Los individuos con esta deficiencia tienen, de manera sorprendente, reducción leve de la AT-III. Los valores medidos por bioanálisis y por inmunoanálisis varían entre 25 y 60 porciento de lo normal. En general, el trastorno se diagnostica cuando los médicos, con alto índice de sospecha, solicitan un bioensayo o un inmunoensayo para AT-III.
El estudio de una familia extensa con deficiencia de ATIII indica que la profilaxis con anticoagulantes no está justificada para los portadores asintomáticos de esta deficiencia. Estos portadores deben recibir anticoagulación profiláctica sólo cuando se exponen a situaciones que aumenten el riesgo de trombosis, como la cirugía general abdominal.330 Sin embargo, una vez que ocurre un evento trombótico, es probable que los pacientes requieran tratamiento de por vida con anticoagulantes cumarínicos por vía oral, que en ocasiones pueden normalizar los niveles de AT-III. Los episodios agudos de trombosis deben tratarse con heparina. Debido a que la deficiencia de AT-III puede volver a la heparina relativamente ineficaz, el médico debe estar alerta ante la posibilidad de resistencia a la heparina, que se manifiesta por prolongación mínima M TPT después de la administración de dosis terapéuticas de este medicamento, es decir, 15 U/kg/hr I.V. después de un bolo inicial de 5,000 U I.V.
Si ocurre resistencia a la heparina deberá administrarse AT-III junto con la heparina, ya sea en forma de concentrados de AT-III o como plasma fresco congelado. En la actualidad existe un concentrado purificado de AT-III. La administración de 1 Ulkg de peso corporal de AT-III suele aumentar la actividad en plasma de esta sustancia en alrededor de dos porciento, y la vida media de la AT-III es de 60 horas. Esta preparación puede usarse para tratar al paciente con deficiencia de AT-III durante una cirugía o un parto. Un bolo de 20 a 50 Ulkg, dependiendo de la concentración basal, será suficiente para elevar la concentración de AT-III hasta alrededor M 100 porciento. Después se mide la concentración de AT-III y se repite la infusión cada 24 horas para mantener una concentración de AT-III normal durante cinco a seis días después de un parto, o durante una semana después de cirugía. El embarazo es difícil de manejar en las pacientes con deficiencia de AT-III porque la warfarina, el anticoagulante clave en el tratamiento, atraviesa la placenta y es teratogénico. Parece ser que la combinación de heparina subcutánea, en dosis de 15,000 U cada 12 horas y de AT-III, al comenzar el trabajo de parto y durante todo éste, es eficaz.331 La warfarina sigue siendo el medicamento de elección en el tratamiento crónico de pacientes con deficiencia de AT-III y tromboembolias recurrentes. El danazol, en dosis de 200 mg dos a tres veces al día, aumenta la concentración de AT-III, pero no se ha demostrado que este cambio proteja a los pacientes contra las tromboembolias.
Puede ocurrir deficiencia adquirida de AT-III en pacientes que reciben heparina intravenosa durante más de tres días, en la CID, en la hepatopatía severa, en el síndrome nefrótico y después del tratamiento con asparaginasa.53
DEFICIENCIA DE COFACTOR II DE HEPARINA
La AT-III es responsable de la mayor parte de la actividad de antitrombina del plasma, pero otro inhibidor de la coagulación, llamado cofactor 11 de heparina, interactúa con la heparina para inactivar la trombina. Este cofactor no afecta a otras proteasas de serina (factores IXa, XIa, Xa). Para su activación, el cofactor II de heparina requiere cerca de 10 veces más de heparina que la AT-III, y puede ser activado también por el sulfato de dermatán. Una vez activado forma un complejo estable con la trombina inactivándola.332,333
La deficiencia hereditaria de cofactor 11 causa un estado de hipercoagulabilidad. Los miembros de las familias afectadas tienen aproximadamente la mitad del valor normal de este cofactor y sufren de trombosis venosas y arteriales.332,333 La deficiencia adquirida del cofactor II de heparina ocurre en la CID y en la hepatopatía severa, pero no se ha determinado el papel fisiopatológico de la deficiencia en estas situaciones.334
DEFICIENCIA DE PROTEINA C Y PROTEINA S
La deficiencia o defecto en la proteína C o proteína S [ver figura 5] causa pérdida de la capacidad para inactivar el exceso de factor VIIIa y factor Va y para mantener una respuesta fibrinolítica apropiada.25,54 La proteína C, junto con la proteína S, inactiva al inhibidor del activador de plasminógeno.335 Los niveles de proteína C están bajos en pacientes con CID y hepatopatía, quizá porque la activación de la hemostasia consume este factor.336
La deficiencia hemocigota de la proteína C causa trombosis mortal en la infancia.337 Es probable que la deficiencia heterocigota de la proteína C ocurra con una prevalencia de uno en 200 a uno en 300. Los niveles sanguíneos de la proteína C en pacientes con la deficiencia heterocigota se superponen con el límite inferior de los valores normales. Muchos heterocigotos e individuos con valores normales inferiores de proteína C no tienen antecedente de trombosis.338Sin embargo, otros heterocigotos muestran tendencia definida a la trombosis venosa aún cuando sus niveles de proteína C se encuentren en límites de 40 a 50 porciento.339 La trombosis venosa cerebral parece ser la responsable de los casos de infarto hemorrágico cerebral que ocurren en adultos jóvenes con esta deficiencia.341 La necrosis cutánea inducida por warfarina se relaciona con la deficiencia de esta proteína.342
La warfarina oral es el tratamiento de elección para evitar la trombosis a pesar de que disminuye aún más los niveles de proteína C. Debido a que la vida media de la proteína C es sólo de seis a siete horas,54 mucho menor que la de la protrombina, factor X o factor IX [ver tabla 1], un periodo de mayor hipercoagulabilidad sigue al inicio del tratamiento con warfarina en pacientes con deficiencia de proteína C. La necrosis cutánea durante el tratamiento con warfarina sugiere la presencia de deficiencia de proteína C. Debe administrase heparina junto con la warfarina durante el inicio de la anticoagulación en estos pacientes, y después la heparina puede suspenderse.
La deficiencia de proteína S [ver figura 5] también induce la formación de trombosis venosa, incluyendo trombosis de la vena mesentérica.342,355 El embarazo y el uso de anticonceptivos orales disminuyen el nivel de proteína S, lo que puede explicar algunos de los casos de tromboembolias que ocurren en tales circunstancias.346 La deficiencia de proteína S también ocurre en pacientes con síndrome nefrótico y es parcialmente causada por la pérdida selectiva de la proteína S en la orina. Esta deficiencia puede contribuir a los episodios tromboembólicos observados en estos pacientes.347
En los laboratorios de investigación existen pruebas para medir la proteína C y la proteína S.
ALTERACIONES DE LA FIBRINOLISIS
Se han descrito varios casos de plasminógenos anormales desde el punto de vista estructural en sujetos con tromboembolias recurrentes. Los plasminógenos no fueron funcionales biológicamente [ver figura 3], causando al parecer actividad fibrinolítica inadecuada e incremento en la trombosis. También se han descrito pacientes con hipoplasminogenia.342 El diagnóstico de displasminogenemia e hipoplasminogenemia puede establecerse por el hallazgo de actividad de Plasminógeno baja (menos de 50 porciento de lo normal). Los individuos con displasminogenemia tienen niveles normales de plasminógeno identificable en forma inmunológica.342 Estos pacientes responden al tratamiento con warfarina.274
Una causa adicional de estado hipercoagulable es la deficiencia o acción defectuosa de los activadores de plasminógeno. En tales casos la hemostasia local no es contrarrestada por la fibrinolisis adecuada. La fibrinólisis defectuosa puede ser un factor importante en algunos casos de tromboembolias recurrentes.348 Un estudio que examinó 100 pacientes consecutivos con trombosis venosas profundas y embolia pulmonar, encontró que 11 liberaban niveles muy bajos de t-PA durante la oclusión venosa convencional. En otros22 sujetos la fibrinolisis fue defectuosa porque el t-PA que era liberado en cantidades normales, era inactivado por un exceso de inhibidor de t-PA.348 En la actualidad las pruebas para determinar t-PA y el inhibidor de t-PA están disponibles sólo en laboratorios de investigación, pero los hallazgos de este estudio indican la necesidad de programas destinados a aumentarla liberación local de t-PA o bloquear el exceso local de inhibidores de t-PA.
La alteración adquirida de la actividad fibrinolítica puede ser un factor predictivo de trombosis posoperatorias.349
DISFIBRINOGENEMIA
Un caso de disfrinogenemia congénita se ha relacionado con enfermedad trombótica. Aunque los coágulos formados en los pacientes con este trastorno fueron defectuosos, no pudieron ser removidos porque estaban muy rígidos y resistentes a la lisis mediada por plasmina. Las pruebas de laboratorio convencionales como el tiempo de trombina y el tiempo de reptilasa deben estar prolongados, pero se requieren pruebas llevadas a cabo en laboratorios de investigación para hacer el diagnóstico. La polimerización incompleta de la fibrina debe causar una prueba positiva para los productos de degradación de la fibrina.350
HOMOCISTINURIA
La homocistinuria es un trastorno hereditario caracterizado por incapacidad para metabolizar en forma normal la homocisteína por una deficiencia en la enzima cistationina b-sintetasa. Debido a este defecto, el nivel plasmático de homocisteína aumenta hasta el punto en el cual interfiere con los enlaces cruzados de la colágena, lo que finalmente causa daño endotelial y subendotelial. La homocisteína inhibe también la expresión de la trombomodulina y la activación de la proteína C [ver figura 5], y ambos efectos provocan hipercoagulabilidad.351 El daño vascular causado por las altas concentraciones de homocisteína provocan trombosis venosa: y arteriales, y quiza ateroesclerosis acelerada. El diagnóstico debe sospecharse en caso de tromboembolias recurrentes en personas jóvenes, sobre todo cuando el problema se asocia con retraso mental y luxación de] cristalino. La homocistinuria hormocigota es poco frecuente, pero el estado heterocigoto que produce hiperhomocistinemia leve puede ocurrir en el uno a dos porciento de la población general. Se ha demostrado que la hiperhomocistinemia se asocia en forma estrecha con enfermedad cerebrovascular, coronaria y vascular periférica en personas menores de 55 años. Las personas heterocigotas pueden detectarse por medio de una prueba de carga de metionina, en la que se administra L-metionina (110 mg/kg) después de un ayuno de una noche. Se obtienen muestras de sangre cuatro, seis y ocho horas después de la administración de la metionina para medir las concentraciones séricas de la homocisteína no unida a proteínas. Los pacientes con deficiencia parcial de cistationina b-sintetasa tienen concentraciones séricas mucho más altas de homocisteína que los sujetos controles.352 Cuando se identifica a estos pacientes debe tratárseles con piridoxina o betaína en un esfuerzo para evitar mayor daño vascular.
ADMINISTRACION DE FARMACOS
Algunos fármacos tienden a causa trombosis. Los anticonceptivos orales a base de estrógenos parecen aumentar el riesgo de enfermedad tromboembolica.353 La explicación para esta relación es desconocida, pero las mujeres que toman anticonceptivos orales tienen niveles disminuidos de AT-III y de proteína S, factor de Hageman elevado (factor XII), y de manera paradójica, aumento de la actividad fibrinolítica.
La trombosis relacionada con la heparina puede ser un evento devastador que ocurre en pacientes que requieren tratamiento anticoagulante y que suele relacionarse con trombocitopenia inducida por heparina (ver antes, Cuenta plaquetaria disminuida; Trombocitopenia, Trombocitopenia por heparina).256,257
Los pacientes que están recibiendo ciclosporina después M trasplante renal de donador de cadáver muestran mayor incidencia de tromboembolias venosas.356 La ciclosporina también puede causar una variante del síndrome urémico hemolítico, pero no está claro que las dos alteraciones trombóticas asociadas con el uso de la ciclosporina estén relacionadas.
SINDROME DE TROUSSEAU
Algunos pacientes con cáncer muestran mayor tendencia a desarrollar tromboembolias migratorias recurrentes, a esta asociación se le denomina síndrome de Trousseau. Este síndrome ocurre en alrededor M 20 porciento de los pacientes con mesoteliomas peritoneales.357 Se considera que el evento fisiopatológico de fondo consiste en una forma subclínica de CID y que los procoagulantes activados en la CID producen aumento en las trombosis.358 Puede ser que el factor tisular liberado por el tumor participe también.359, 360 La CID franca en estos, pacientes es poco frecuente,361 pero cuando ocurre se necesita tratamiento continuo con heparina para prevenir los episodios recurrentes de trombosis arterial y venosa. Es probable que la warfarina sea ineficaz en esta situación.362 Los exámenes de laboratorio pueden mostrar niveles bajos de fibrinógeno y plaquetas, tiempo de protrombina prolongado y aumento en los niveles de productos de degradación del fibrinógeno y la fibrina. La clave para el tratamiento consiste en el diagnóstico y tratamiento del tumor oculto, lo que debe permitir el control de la CID. Sin embargo, como la CID subclínica se asocia con trombosis, es razonable instituir tratamiento con heparina mientras se realiza el tratamiento definitivo del tumor.
El síndrome de Trousseau representa una situación clínica muy difícil y frustrante porque con frecuencia la neoplasia está oculta y no se identifica a pesar de la búsqueda extensa y agresiva. Existe recurrencia de las tromboembolias venosas a pesar del uso de dosis totales y controladas de warfarina, combinadas con aspirina o sulfinpirazona. Puede ser útil la administración intravenosa de heparina o por vía subcutánea cada 12 horas.362 Al final, la neoplasia a menudo se manifiesta en forma explosiva y no llega a responder a los tratamientos habituales.
DEFICIENCIA DE FACTOR XII
La deficiencia de factor XII no produce un trastorno hemorrágico, a pesar de que el TPTa se prolonga mucho. Sin embargo, y en forma paradójica, los pacientes homocigotos a la deficiencia pueden tener mayor frecuencia de tromboembolias. Es, posible que el factor XII actúe en la activación de contacto de la fibrinolisis, y que este proceso sea defectuoso en los pacientes con deficiencia de factor XII. El tratamiento de estos pacientes es el de los enfermos con padecimientos tromboembólicos.363-365
Errores quirúrgicos contra enfermedades sistémicas
Una hemorragia grave durante o después de la cirugía representa un problema clínico complicado que requiere diagnóstico rápido e intervención inmediata.
Si el paciente sólo presenta hemorragia en la herida quirúrgica, puede ser difícil decidir si hay un trastorno hemorrágico general o no. Un dato revelador en cuanto a un trastorno general es la existencia de hemorragias en múltiples sitios, particularmente en áreas diferentes a la herida quirúrgica. Las hemorragias que ocurren alrededor de un catéter, en sitios de venopunción y por venodisecciones son muy sugestivas de trastornos hemorrágicos.
Resulta obligado hacer una evaluación rápida de todo el cuadro clínico: ¿padece el paciente alguna enfermedad renal, hepática o maligna?, ¿la cirugía ha requerido de bomba extracorpórea, de la inducción de hipotermia o el paciente ha estado en estado de choque o hipertérmico?, ¿cuántas unidades de sangre y de productos de la sangre se han administrado y durante cuánto tiempo?, ¿se practicaron exámenes iniciales de procoagulantes como referencia antes de la cirugía?, ¿se dispone M plasma congelado del paciente?
La resolución rápida requiere inevitablemente de pruebas de coagulación realizadas en forma cuidadosa, que incluyen tiempo parcial de tromboplastina, tiempo de protrombina por el método de un paso de Quick, tiempo de trombina, ensayo para fibrinógeno, medición de productos de degradación de fibrinógeno y fibrina, prueba de dímero de fibrina D, cuenta de plaquetas, un frotis de sangre bien teñido para evaluar la morfología plaquetaria y tiempo de sangrado (de preferencia por el método de Ivy modificado). Esta batería de exámenes debe realizarse de inmediato. Se pueden obtener estudios más especializados si existe evidencia de un trastorno específico.
CAUSAS FRECUENTES DE HEMORRAGIA POSOPERATORIA
Causas relacionadas con las plaquetas
Lavado de plaquetas Siempre que un paciente pequeño o de tamaño promedio ha sido transfundido rápidamente con 8 o más unidades de sangre existe el riesgo de que ocurra hemorragia trombocitopénica por lavado de plaquetas, [ver antes, Lavado de plaquetas y alteraciones del lecho vascular].
Formas inmunológicas de trombocitopenia aguda (púrpura postransfusional) En algunos casos ocurre trombocitopenia aguda devastadora después de transfusiones de sangre total o de componentes sanguíneos que contienen plaquetas. En ciertos casos se ha mostrado que el receptor desarrolla una respuesta con anticuerpos contra un antígeno denominado antígeno Zwa, (antes llamado PIaI) en las plaquetas del donador.366 Las plaquetas del propio paciente también resultan afectadas, aunque carezcan del antígeno Zwa, lo que ocasiona púrpura trombocitopénica inmunológica aguda. El mecanismo de esta reacción no se ha esclarecido.
La administración de esteroides en las dosis usuales, la esplenectomía y la transfusión plaquetaria no suelen ser eficaces. Se ha propuesto el tratamiento con esteroides, desde dosis convencionales, hasta 2 g de metilprednisolona al día durante tres días.368 También se ha utilizado la plasmaféresis en un intento por extraer los anticuerpos.368 Sin embargo, el tratamiento de elección en la actualidad consiste en la infusión de IgG por vía intravenosa, en dosis de 0.4 a 0.8 g/kg durante un periodo de 60 minutos, seguido de la administración de cerca de 10 a 12 unidades de plaquetas de donadores al azar. Sin tratamiento médico, el recuento plaquetario vuelve a los valores normales en un periodo aproximado de dos semanas.
Trastornos de la función plaquetaria Si en la cirugía se ha utilizado algún procedimiento de derivación extracorpórea prolongada o sí la sangre del paciente ha estado en contacto estrecho con cualquier tipo de prótesis, pueden ocurrir hemorragias en el posoperatorio como resultado de un trastorno cualitativo de las plaquetas.
Las plaquetas que han tenido contacto con superficies extrañas pueden haber sufrido una liberación parcial y por tanto, haber liberado cantidades importantes de su material granular. Aunque aún pueden agregarse, la capacidad de éstas para desarrollar la reacción de liberación estará alterada. Esta disfunción plaquetaria se caracteriza por una prolongación del tiempo de sangrado, el hallazgo de plaquetas degranuladas en un frotis bien teñido y una cuenta plaquetaria relativamente normal. Algunas enfermedades como las nefropatías agudas o crónicas y en algunos casos, los trastornos hepáticos agudos, también pueden causar trastornos funcionales en las plaquetas.
Las plaquetas disfuncionales no desarrollarán la reacción de liberación; sin embargo, si hay materiales agregantes apropiados, las plaquetas dañadas pueden agregarse y contribuir a la formación M tapón hemostático. Por lo tanto, es necesario añadir una cantidad mínima de plaquetas normales, que se puedan adherir y producir la reacción de liberación. La transfusión de alrededor de 4 o 6 unidades de plaquetas debe elevar la cuenta plaquetaria de 30,000 a 40,000/mm3 y debe controlar la hemorragia. Si el paciente tiene un trastorno de función plaquetaria relacionado con uremia aguda, puede ser necesaria una hemodiálisis rápida para controlar la hemorragia (ver antes, Trastornos de la función plaquetaria, Uremia).
Causas de hemorragia transoperatoria relacionadas con defectos de los procoagulantes
Heparina Con frecuencia se utiliza heparina durante los procedimientos quirúrgicos de derivación; también se emplea para mantener permeables las líneas endovenosas. En general la heparina se neutraliza después de un procedimiento quirúrgico por medio de la infusión de protamina. Como la heparina es inactivada por las enzimas hepática% y excretada por los riñones, el daño a cualquiera de estos órganos aumentará la duración de la actividad de la heparina. La prolongación del TPT, TP y TT junto con un tiempo de sangrado y de reptilasa normal y una cuenta plaquetaria normal sugieren el diagnóstico de hemorragia por anticoagulación inadvertida con heparina.
La administración intravenosa de protamina en una dosis de 50 mg cada 1 a 2 horas debe corregir el TT y TPT a lo normal y detener la hemorragia.
Coagulación intravascular diseminada La CID es una secuencia catastrófica de la coagulación, por lo general secundaria a traumatismos extensos, estado de choque o intervenciones quirúrgicas. [ver antes, Trastornos hemorrágicos adquiridos, Coagulación intravascular diseminada].
Deterioro del funcionamiento hepático El hígado es el sitio de síntesis de todos los procoagulantes con excepción del vFW. Las células reticuloendoteliales del hígado retiran las partículas y otros materiales tromboplásticos que pueden circular en pacientes que sufran choque o traumatismos. Las enfermedades hepáticas graves suelen relacionarse con deficiencia de procoagulantes y cierto grado de CID.
Como existe deficiencia de múltiples factores, el principal diagnóstico diferencial es entre deficiencia de procoagulantes por enfermedad hepática y CID. La situación se complica más por el hecho de que la enfermedad hepática severa produce por lo común cierto grado de CID, que puede tener una importancia clínica variable. El hallazgo de un TT normal es útil porque excluye tanto el efecto de heparina como la acción de los productos de degradación de fibrina-fibrinógeno que se producen en la CID. Una determinación normal de fibrinógeno es otro criterio en contra del diagnóstico de CID, y una cuenta plaquetaria normal es rara en la coagulación intravascular diseminada. Se debe usar plasma fresco congelado para sustituir los procoagulantes.
Hemofilia y enfermedad de von Willebrand no diagnosticadas Durante la cirugía, la hemofilia y la enfermedad de von Willebrand pueden causar hemorragias. La hemorragia generalmente es severa, pero se limita al sitio de la herida.
Si el paciente tiene hemofilia leve o moderada y ha recibido múltiples transfusiones de sangre y productos sanguíneos, puede no sangrar en el posoperatorio inmediato, pero sí dos o tres días después del procedimiento quirúrgico, cuando los niveles de los procoagulantes, que se habían elevado en forma transitoria con las transfusiones hayan disminuido [ver antes, Trastornos hereditarios de la coagulación, Hemofilia A.].
Puede ser difícil diagnosticar la enfermedad de von Willebrand durante el periodo posoperatorio. Si el paciente recibió plasma, las pruebas de TPT y factor VIII pueden ser normales, y el tiempo desangrado puede o no estar prolongado. En estos casos la prueba de ristocetina es útil. En la deficiencia de factor VII clásica, el paciente debe recibir infusiones de crioprecipitado o concentrado de factor VIII [ver antes, Trastornos hereditarios de la coagulación, principios del tratamiento sustitutivo]. El tratamiento de la enfermedad de von Willebrand puede ser más difícil que el de la hemofilia.
Fibrinolisis primaria
En algunos casos se debe considerar la fibrinolisis primaria en el diagnóstico diferencial de la coagulación intravascular diseminada, en particular en el paciente que se ha sometido a cirugía por cáncer prostático y que ahora muestra evidencia de lisis del coágulo. [ver figura 3]. La fibrinolisis suele ocurrir como respuesta al estímulo de la CID. Sin embargo, la fibrinolisis primaria puede ocurrir en la leucemia promielocítica aguda y en otras circunstancias en las que la acción del t-PA está aumentada o sin oposición. La prueba del dímero de la fibrina es útil para distinguir la CID de la fibrinolisis primaria. Debido a que el fragmento dímero D es producido sólo a partir de fibrina polimerizada por completo, su presencia indica CID. El AFAC debe emplearse con cuidado en el tratamiento de la fibrinolisis en caso de cirugía urológica y genital [ver antes, Trastornos hemorrágicos adquiridos, Fibrinolisis primaria]. También existen informes anecdóticos de efectos benéficos con el ácido e-aminocaproico en el tratamiento de la hemorragia posoperatoria en niños con cardipatías congénitas y datos de fibrinolisis.
Alteraciones falsas en los resultados de las pruebas de procoagulantes
Algunas veces los resultados de las pruebas de coagulación, particularmente el TPT y el TT, están muy prolongados en el posoperatorio. El examen del paciente puede mostrar o no hemorragias importantes. En este momento, debe investigarse el método usado para recolectar la muestra de sangre.267 Los pacientes con hemorragia posoperatoria significativa pueden estar en choque y a menudo tienen catéteres intravenosos. La muestra para el examen puede haber sido obtenida de un catéter intravenoso que se mantiene permeable con concentraciones bajas de heparina. Es más, la sangre puede haber sido diluida con soluciones salinas, soluciones de glucosa, o ambas. La muestra de sangre puede haberse coagulado durante la extracción. Un tiempo de trombina prolongado que se puede corregir con la adición de protrombina, es evidencia de contaminación con heparina. Las soluciones glucosadas mezcladas con sangre causan deformaciones de los eritrocitos en los frotis de sangre. Estos problemas pueden evitarse obteniendo muestras sanguíneas directamente de una vena.369
Las pruebas son costosas y pueden fallar, y casi siempre surge la duda de utilizar o no pruebas de coagulación de escrutinio antes de la cirugía.370 La importancia de los antecedentes se pone de manifiesto por estudios que demuestran que el TPT no puede utilizarse como prueba general de escrutinio para predecir cuales pacientes sufrirán hemorragias; sin embargo, el TPT sí tiene valor predictivo cuando se utiliza en pacientes que son seleccionados por historia clínica como sujetos con alto riesgo de sangrado.371 En estos casos, se recomienda el siguiente procedimiento preoperatorio:
1. Preguntar específicamente si hay antecedente de hemorragia en la familia o si el paciente alguna vez ha sangrado en forma excesiva durante cirugías o extracciones dentales.
2. Solicitar una serie de pruebas que. incluyan tiempo de sangrado, cuenta plaquetaria, examen de plaquetas en un frotis de sangre periférica, TPT, TP y el cálculo de la concentración de fibrinógeno.
3. Congelar un tubo de plasma y guardarlo durante 24 horas después de la cirugía. De esta manera, si el paciente sangra, se pueden examinar muestras preoperatorias al igual que posoperatorias. Este método facilita en gran medida el descubrimiento de hemofilia y enfermedad de von Willebrand previamente no diagnosticadas.
Figura I Janet Betries. Adaptada de "The Pathophysiology of the Prethrombotic State in Humans: Insights Gained from Studies Using Markers of Hemostatic System Activation," por K.A. Bauer y R.D. Rosenberg, en Blood 70:343, 1987. Utilizado con autorización.
Figuras 2, 4 y 7 Dana Bums.
Figuras 3, 5, 6 y 9 Janet Betries.
Tabla I Adaptada de "The Molecular Basis of Blood Coagulation," por B. Furie y B.C. Furie, en Cell 53:505. 1998. Utilizado con autorización. Tabla 5 Adaptada de "Medical Progress: The Clinical Importance of Acquired Abnormalities of Platelet Function," por J.N. George y S. Shattil, en The New England Journal of Medicine 324: 31, 199 1.
Utilizado con autorización.
Tabla 8 Adaptada de "Von Willebrand Factor: Clinical Feature of Inherited and Acquired Disorders," por A.L Bloom, en Mayo Clinic Proceeding 66: 746, 199 1. Utilizado con autorización.
- Roth GJ: Developing relationships: arterial platelet adhesion, glycoprotein lb, and leucine-rich glycoproteins. Blood 77:5,1991
- Furie B, Furie BC: The molecular basis of blood coagulation. Cell 53:505,1988
- Yang Z, stulz P, von Segesser L, et al: Different interactions of platelets with arterial and venous coronary bypass vessels. Lancet 337:939,1991
- Turitto VT, Weiss HJ, Baumgartner HR: Decreased platelet adhesion on vessel segments in von Willebrand's disease: a defect in initial platelet attachment: J Lab Clin Med 102:551,1983
- Larsen E, Alessandro C, Gilbert GE, et al: PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell 59:305, 1989
- Endothelins (editorial). Lancet 337:79,1991
- Lerman A, Hildebrand FL Jr, Margulies KB, et al: Endothelin: a new cardiovascular regulatory peptide. Mayo Clin Proc 65:1441,1990
- The endothelium-modulator of vascular smooth-muscle tone. N Engl J Med 319:512,1988
- Ignarro LI, Ross G, Tillisch J: Pharmacology of endothelium-derived nitric oxide and nitrovasodilators. West J Med 154:51,1991
- Bennett Js: Mechanisms of platelet adhesion and aggregation: an update. Hosp Pract 27(4):124,1992
- Wu KK: Endothelial cells in hemostasis, thrombosis, and inflammation. Hosp Pract 27(4):145,1992
- Rapaport SI: Regulation of the tissue factor pathway. Progress in Vascular Biology, Hemostasis, and Thrombosis. Ruggeri ZM, Fulcher CA, Ware J, Eds.New York Academy of Sciences,NewYork, 1991, p 51
- Mann KG, Bovill EG, Krishnaswamy S, et al: surface-dependent reactions in the propagation phase of blood coagulation. Progress in Vascular Biology, Hemostasis, and Thrombosis. Ruggeri ZM, Fulcher CA, Ware J, Eds. New York Academy of Sciences, New York, 1991, p 63
- Gailani D, Broze GJ Jr: Factor XI activation in a revised model of blood coagulation. science 253:909,1991
- Roberts HR, Lozier JN: New perspectives on the coagulation cascade. Hosp Pract 27(1):97,1992
- Flier Js, Underhill LH: Molecular and cellular biology of blood coagulation. N Engl J Med 326:800, 1992
- Collen D, Lijnen HR: Basic and clinical aspects of fibrinolysis and thrombolysis. Blood 78:3114,1991
- Mosesson MW: Fibrin polymerization and its regulatory role in hemostasis. J Lab Clin Med 116:8,1990
- Nachman RL, Hajjar KA: Endothelial cell fibrinolytic assembly. Progress in Vascular Biology , Hemostasis, and Thrombosis. Ruggeri ZM, Fulcher CA, Ware J, Eds. New York Academy of sciences, New York, 1991, p 240
- Weiss HJ, Hawiger J, Ruggeri ZM, et al: Fibrinogen-independent platelet adhesion and thrombus formation on subendothelium mediated by glycoprotein IlB-IIIa complex at high shear rate. J Clin Invest 83:288,1989
- Ikeda Y, Handa M, Kawano K, et al: The role of von Willebrand factor and fibrinogen in platelet aggregation under varying shear stress. J Clin Invest 87:1234,1991
- Leung LLK, Nachman RL: Complex formation of platelet thrombospondin with fibrinogen. J Clin Invest 70:542,1982
- Furie B, Furie BC: Molecular basis of gamma-carboxylation: role of propeptide in the vitamin K-dependent proteins. Ann NY Acad Sci 614:1,1991
- Repke D, Gemmell CH, Gaha A, et al: Hemophilia as a defect of the tissue factor pathway of blood coagulation: effect of factors VIII and IX on factor X activation in a continuous-flow reactor. Proc Natl Acad Sci USA 87:7623,1990
- Rosenberg RD, Bauer KA: New insights into hypercoagulable states. Hosp Pract 21(3):131,1986
- Adams GA: Building bridges in molecular rheoIogy: the role of the microenvironment in platelet thrombogenesis. J Lab Clin Med 7:613, 1990
- Esmon CT: The regulation of natural anticoagulant pathways. Science 235:1348,1987
- Fulcher CA, Gardiner JE, Griffin JH, et al: Proteolytic inactivation of human factor VIII procoagulant protein by activated human protein C and its analogy with factor V. Blood 63:486,1984
- Esmon CT , Johnson AE, Esmon NL, et al: Initiation of the protein C pathway. Ann NY Acad Sci USA 614:30,1991
- Walker FJ,Fay PJ: Regulation of blood coagulation by theprotein C system. FASEB J 6:2561, 1991
- Rapaport SI: Inhibition of factor VIla/tissue factor-induced blood coagulation: with particular emphasis upon a factor Xa-dependent inhibitory mechanism. Blood 73:359,1989
- NovotnyWF, Palmier M, Wun TC, etal: Purification and properties of heparin-releasable lipoprotein-associated coagulation inhibitor. Blood 78:394,1991
- Sandset PM, Wam-Cramer BJ, Rao LVM,et al: Depletion of extrinsic pathway inhibitor (EPI) sensitizes rabbits to disseminated intravascular coagulation induced with tissue factor: evidence supporting a physiologic role for EPI as a natural anticoagulant. Proc Natl Acad Sci USA 88:708,1991
- Becker RL: Seminars in thrombosis, thrombolysis, and vascular biology: IV. Fibrinolysis. Cardiology 79:188,1991
- Vassalli J-D, Sappino A-P, Belin D: The plasminogen activator /plasmin system. J Clin Invest 88:1067,1991
- Sawdey MS, Loskutoff DJ: Regulation of murine type 1 plasminogen activator inhibitor gene expression in vivo: tissue specificity and induction by lipopolysaccharide, tumor necrosis factor-lX, and transforming growth factor-J3. J Clin Invest 88:1346,1991
- Scanu AM, Lawn RM, Berg K: Lipoprotein(a) and atherosclerosis. Ann Intern Med 115:209,1991
- Etingin OR, Hajjar DP, Hajjar KA, et al: Lipoprotein (a) regulates plasminogen activator inhibitor-1 expression in endothelial ceils: a potential mechanism in thrombogenesis. J Biol Chem 266:2459,1991
- Nachman RL: Thrombosis and atherogenesis: molecular connections. Blood 79:1897,1992
- Levin J: An overview of megakaryocytopoiesis. Molecular Biology and Differentiation of Megakaryócytes. Wiley-Liss, Inc, New York, 1990,p1
- Levin J, Vainchenker W: Regulation of megakaryocytopoiesis. Molecular Biology and Differentiation of Megakaryocytes. Wiley-Liss, Inc, New York, 1990, p 217
- Hoffman R, Straneva J, Hudson N, et al: Regulation of human megakaryocytopoiesis by interacting cytokines. Molecular Biology and Differentiation of Megakaryocytes. Wiley-Liss, Inc, New York, 1990, p 199
- Han ZC, Bellucci S, Wan HY, et al: New insights into the regulation of megakaryocytopoiesis by haematopoietic and fibroblastic growth factors and transforming growth factors and transforming growth factor beta-1. Br J HaematoI 81:1, 1992
- Haller CJ, Radley JM: Time-lapse cinernicrography and scanning electron microscopy of platelet formation by megakaryocytes. Blood Cells 9:407,1983
- Tong M, Seth P, Penington DG: Proplatelets and stress platelets. Blood 69:522,1987
- Hill-Zobel RL, McCandless B,Kang SA, et al: Organ distribution and fate of human platelets: studies of asplenic and splenomegalic patients. Am J HematoI 23:231, 1986
- HirshJ: Oral anticoagulant drugs. N Engl J Med 324:1865,1991
- FunkC,GmürJ, Herold R,et al: Reptilase-R-anew reagentin blood coagulation. Br J HaematoI 21:43, 1971
- Nossel HL: Radioimmunoassay of fibrinopeptides in relation to intravascular coagulation and thrombosis. N Engl J Med 295:428,1976
- The bleeding time (editorial). Lancet 337:1447,1991
- Weiss H: Platelet physiology and abnorrnalities of platelet function (pts 1 and 2). N Engl J Med 293:531, 580, 1975
- Kroll MH, Harris TS, Moake JL, et al: von Willebrand factor binding to platelet Gp1b initiates signals for platelet activation. J Clin lnvest 88:1568,1991
- Familial antithrombin III deficiency (editorial). Lancet 1:1021,1983
- Clouse LH, Comp PC: The regulation of hemostasis: the protein C system. N Engl J Med 314:1298,1986
- Eldor A, Avitzour M, Or R, et al: Prediction of haemorrhagic diathesis in thrombocytopenia by mean platelet volume. Br Med J 285:397,1982
- Gewirtz AM, Hoffman R: Transitory hypomegakaryocytic thrombocytopenia: etiological association with ethanol abuse and complications regarding regulation of human megakaryocytopoiesis. Br J HaematoI 62:333, 1986
- Straus DJ: Hematologic aspects of alcoholism. Semin Hematol 10:183,1973
- Miescher PA: Drug-induced thrombocytopenia. Semin Hematol 10:311,1973
- De Gruchy GC: Drug-1nduced Blood Disorders. Blackwell Scientific Publications, Oxford, 1975
- Levine RF, Spivak JL, Meagher RC, et al: Effect of ethanol on thrombopoiesis. Br J HaematoI 62:345, 1986
- Tomer A, Hanson S, Harker LA, et al: Autologous platelet kinetics in patients with severe thrombocytopenia: discrimination between disorders of production and destruction. J Lab Clin Med 118:546,1991
- Dixon R, Rosse W, Ebbert L: Quantitative deterrnination of antibody in idiopathic thrombocytopenic purpura: correlation of serum and platelet-bound antibody with clinical response. N Engl J Med 292:230,1975
- Kelton JG: Advances in the diagnosis and management of ITP.Hosp Pract 20(6):95,1985
- Ballem PJ, Segal GM, Stratton JR, et al: Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura: evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 80:33,1987
- McMillan R: Chronic idiopathic thrombocytopenic purpura. N Engl J Med 304:1135,1981
- Yu J-R, Lennette ET, Karpatkin S: Anti-F(ab')2 antibodies in thrombocytopenic patients at risk for acquired immunodeficiency syndrome. J Clin Invest 77:1756,1986
- Karpatkin S, Nardi M: Autoimmune anti-HIV-1gp120 antibody with antiidiotype-like activity in sera and immune complexes of HIV-1-related immunologic thrombocytopenia. J Clin Invest 89:356, 1992
- Kelton JG, Bibbons S: Autoimmune platelet destruction: idiopathic thrombocytopenic purpura. Semin Thromb Hemost 8:83,1982
- Kernoff LM, Blake KCH, Shackleton D: Influence of the amount of platelet-bound IgG on platelet survival and site of sequestration in autoimmune thrombocytopenia. Blood 55:730,1980
- El Saghir NS, Geltman RL: Treatment of acquired amegakaryocytic thrombocytopenic purpura with cyclophosphamide. Am J Med 81:139,1986
- Hoffman R, Zaknoen S, Yang HH, et al: An antibody cytotoxic to megakaryocyte progenitor cells in a patient with immune thrombocytopenic purpura. N Engl J Med 312:1170,1985
- Laster AJ, Conley CL, Keckler TS, et al: Chronic immune thrombocytopenic purpura in monozygotic twins: genetic factors predisposing to ITP. N Engl J Med 307:1495,1982
- Harker LA: Hemostasis Manual, 2nd ed. FA Davis Co, Philadelphia, 1974
- Stuart MJ, Kelton JG, Allen JB: Abnormal platelet function and arachidonate metabolism in chronic idiopathic thrombocytopenic purpura. Blood 5:326,1981
- Walsh C, Krigel R, Lennette E, et al: Thrombocytopenia in homosexual patients. Ann Intern Med 103:542, 1985
- Savona S, Nardi MA, Lennette ET, et al: Thrombocytopenic purpura in narcotics addicts. Ann Intern Med 102:737,1985
- Adrouny A, Sandler RM, Carmel R: Variable presentation of thrombocytopenia in Graves' disease. Arch Intem Med 142:1460,1982
- Hymes K, Blaum M, Lackner H, et al: Easy bruising, thrombocytopenia, and elevated platelet immunoglobulin G in Graves' disease and Hashimoto' s thyroiditis. Ann Intern Med 94:27,1981
- Harrington WJ: Are platelet-antibody tests worthwhile? (editorial). N Engl J Med 316:211,1987
- Court Ws, Bozeman JM, soong S, et al: Platelet surface-bound IgG in patients with immune and nonimmune thrombocytopenia. Blood 69:278, 1987
- George JN: Platelet immunoglobulin G: its significance for the evaluation of thrombocytopenia and for understanding the origin of a-granule proteins. Blood 76:859,1990
- Cortelazzo S, Finazzi G, Buelli M, et al: High risk of severe bleeding in aged patients with chronic idiopathic thrombocytopenic purpura. Blood 77:31,1991
- Coon WW: Splenectomy for idiopathic thrombocytopenic purpura. surg Gynecol Obstet 164:225,1987
- Chronic idiopathic thrombocytopenic purpura (editorial). Br Med J 282:1497,1981
- Ahn YS, Harrington WJ, Mylvaganam R, et al: slow infusion of vinca alkaloids in the treatment of idiopathic thrombocytopenic purpura. Ann Intern Med 100:192,1984
- schreiber AD, Chien P, T omaski A, et al: Effect of danazol in immune thrombocytopenic purpura. N Engl J Med 316:503,1987
- Kelton JG: Vaccination-associated relapse of immune thrombocytopenia. JAMA 245:369, 1981
- Carr JM, Kruskall MS, Kaye JA, et al: Efficacy of platelet transfusions in immune thrombocytopenia. Am J Med 80:1051,1986
- Berchtold P, McMillan R: Therapy of chronic idiopathic thrombocytopenic purpura in adults. Blood 74:2309,1989
- Fehr J, Hofmann V, Kappeler U: Transient reversal of thrombocytopenia in idiopathic thrombocytopenic purpura by high-dose intravenous gamma globulin. N Engl J Med 306:1254,1982
- Newland AC, Treleaven JG, Minchinton RM, et al: High-dose intravenous IgG in adults with autoimmune thrombocytopenia. Lancet 1:84,1983
- Schaison G, Clauvel JP, Tobelem G, et al: High dose intra veinous immunoglobulin for thrombocytopenic purpura (abstr). Blood 60(suppl1):190a,1982
- Baumann MA, Menitove JE, Aster RH, et al: Urgent treatment of idiopathic thrombocytopenic purpura with single-dose gammaglobulin infusion followed by platelet transfusion. Ann Intern Med 104:808,1988
- Marder VJ, Nusbacher J, Anderson FW: One-year follow-up of plasma exchange therapy in 14 patients with idiopathic thrombocytopenic purpura. Transfusion 21:291,1981
- Picozzi VJ, Roeske WR, Creger WP: Fate of therapy failures in adult idiopathic thrombocytopenic purpura. Am J Med 69:690,1980
- Ahn YS, Harrington WJ, Simon SR, et al: Danazol for the treatment of idiopathic thrombocytopenic purpura. N Engl J Med 308:1396,1983
- Ahn YS, Mylvaganam R, Garcia RO, et al: Low-dose danazol therapy in idiopathic thrombocytopenic purpura. Ann Intern Med 107:177,1987
- Kelsey PR, schofield KP, Geary CG: Refractory idiopathic thrombocytopenic purpura (ITP) treated with cyclosporine. Br J Haematol 60:197,1985
- Treatment of refractory idiopathic thrombocytopenic purpura in adults. Br J HaematoI79:143, 1991
- Bussel JB, Graziano JN, Kimberly RP, et al: Intravenous anti-D treatment of immune thrombocytopenic purpura: analysis of efficacy, toxicity, and mechanism of effect. Blood 77:1884,1992
- Anti-D for the treatment of splenectomized patients with immune thrombocytopenic purpura (letter). Blood 78:2786,1991
- Gringeri A, Cattaneo M, Santagostino E, et al: Intramuscular anti-D immunoglobulins for home treatment of chronic immune thrombocytopenic purpura. Br J Haematol 80:337 , 1992
- Calverley DC, Jones GW, Kelton JG: splenic radiation for corticoste-roid-resistant immune thrombocytopenia. Ann Intern Med 116:977,1992
- DiFino SM, Lachant NA, Kirshner JJ, et al: Adult idiopathic thrombocytopenic purpura: clinical findings and response to therapy. Am J Med 69:430,1980
- Oksenhendler E, Bierling P, Farcet J-P, et al: Response to therapy in 37 patients with HIV-related thrombocytopenic purpura. Br J Haematol 66:491,1987
- Perkocha LA, Rodgers GM: Hematologic aspects of human immunodeficiency virus infection: laboratory and clinical considerations. Am J HematoI 29:94, 1988
- Oksenhendler E, Bierling P, Ferchal F, et al: Zidovudine for thrombocytopenic purpura related to human immunodeficiency virus (HIV) infection. Ann Intern Med 110:365,1989
- Durand JM, Lefévre P, Hovette P, et al: Dapsone for thrombocytopenic purpura related to human immunodeficiency virus infection. Am J Med 90:675,1991
- O'Reilly RA, Taber B-Z: Immunologic thrombocytopenic purpura and pregnancy: six new cases. Obstet GynecoI 51:590, 1978
- Cines DB, Dusak B, Tomaski A, et al: Immune thrombocytopenic purpura and pregnancy .N Engl J Med 306:836, 1982
- Karpatkin M, Porges RF, Karpatkin S: Platelet counts in infants of women with autoimmune thrombocytopenia: effect of steroid administration on the mother. N Engl J Med 305:936, 1981
- Scott JR, Cruikshank DP, Kochenour NK, et al: Fetal platelet counts in the obstetric management of immunologic thrombocytopenic purpura. Am J Obstet GynecoI136:495, 1980
- Burrows RF, Kelton JG: Thrombocytopenia at delivery: a prospective survey of 6715 deliveries. Am J Obstet GynecoI 162:731, 1990
- Burrows RF, Kelton JG: Incidentally detected thrombocytopenia in healthy mothers and their infants. N Engl J Med 305:936, 1981
- Burrows R.F, Kelton JG: Low fetal risks in pregnancies associated with idiopathic thrombocytopenic purpura. Am J Obstet Gynecol 163:1147,1990
- Kaden BR, Rosse WF, Hauch TW: Immune thrombocytopenia in lymhoproliferative diseases. Blood 53:545, 1979
- Gamick MB, Griffin JD: Idiopathic thrombocytopenia in association with extragonadal germ cell cancer. Ann Intern Med 98:926,1983
- Schwartz KA, Slichter SJ, Harker LA: Immune-mediated platelet destruction and thrombocytopenia in patients with solid tumours.Br J HaematoI 51:17, 1982
- Mueller-Eckhardt C: Post-transfusion purpura. Br J Haematol 64:419,1986
- Slichter SJ: Post-transfusion purpura: response to steroids and association with red blood cell and lymphocytotoxic antibodies. Br J HaematoI 50:599,1982
- Garty M,Ilfeld D, Kelton JG: Correlation of a quinidine-induced platelet-specific antibody with development of thrombocytopenia. Am J Med 79:253,1985
- Smith ME, Reid DM, Jones CE, et al: Binding of quinine- and quinidine-dependent drug antibodies to platelets is mediated by the Fab domain of the immunoglobulin G and is not Fc dependent. J Clin Invest 79:912, 1987
- Christie DJ, Mullen PC, Aster RH: Fab-mediated binding of drugdependent antibodies to platelets in quinidine- and quinine-in-duced thrombocytopenia. J Clin Invest 75:310,1985
- Visentin GP, Newman PJ, Aster RH: Characteristics of quinine- and quinidine-induced antibodies specific for platelet glycoproteins Iib and IIIa. Blood 77:2668, 1991
- King DJ, Kelton JG: Heparin-associated thrombocytopenia. Ann Intern Med 100:535, 1984
- Chong BH, Castaldi PA: Heparin-induced thrombocytopenia: further studies of the effects of heparin-dependent antibodies on platelets. Br J Haematol 64:347, 1986
- Laster JL, Nichols WK, silver D: Thrombocytopenia associated with heparin-coated catheters in patients with heparin-associated antiplatelet antibodies. Arch Intern Med 149:2285,1989
- Anderson GP: Insights into heparin-induced thrombocytopenia. Br J HaematoI 80:504, 1992
- Cines DB, Tomaski A, Tannenbaum S: Immune endothelial-cell injury in heparin-associated thrombocytopenia. N Engl J Med 316:581,1987
- Frame JN, Mulvey KP, Phares JC, et al: Correction of severe heparin-associated thrombocytopenia with intravenous immunoglobulin. Ann Intern Med 111:946,1989
- Demers C,Ginsberg Js, Brill-EdwardsP ,et al: Rapid anticoagulation using ancrod for heparin-induced thrombocytopenia. Blood 78:2194,1991
- Chong BH, Ismail F, Cade J, et al: Heparin-induced thrombocytopenia: studies with a new low molecular weight heparinoid, Org 10172. Blood 73:1592, 1989
- Hirsh J, Levine MN: Low molecular weight heparin. Blood 79:1,1992
- Coblyn JS, Weinblatt M, Holdsworth D, et al: Gold-induced thrombocytopenia. Ann Intern Med 95:178,1981
- Burday MJ, Martin SE: Cocaine-associated thrombocytopenia. Am J Med 91:656,1992
- Ridolfi RL, Bell WR: Thrombotic thrombocytopenic purpura: report of 25 casesand review of the 1iterature. Medicine (Baltimore) 60:413,1981
- Crain SM, Choudhury AM: Thrombotic thrombocytopenic purpura: a reappraisal. JAMA 246:1243,1981
- Brain MC, Neame PB: Thrombotic thrombocytopenic purpura and the hemolytic uremic syndrome. Semin Thromb Hemost 8:186,1982
- Berkowitz LR, Dalldorf FG, Blatt PM: Thrombotic thrombocytope- nic purpura: pathology review. JAMA 241:1709,1979
- Goodman A, Ramos R, Petrelli. M, et al: Gingival.biopsy in thrombotic thrombocytopenic purpura. Ann Intern Med 89:501, 1978
- Nissenson AR, Krumlovsky FA, del Greco F: Postpartum hemolytic uremic syndrome: late recovery after prolonged maintenance dialysis. JAMA 242:173,1979
- Ponticelli C, Rivolta E, Imbasciati E, et al: Hemolytic uremic syndrome in adults. Arch Intern Med 140:353,1980
- Riggs SA, Wray NP, Waddell CC, et al: Thrombotic thrombocytopenic purpura complicating Legionnaires' disease. Arch Intern Med 142:2275,1982
- Cantrell JE Jr, Phillips TM, Smith FP,et al: Immune complex analysis and plasmapheresis in cancer related thrombotic thrombocytopenic purpura (TTP)/hemolytic uremic syndrome (HUS) syndrome (abstr). Blood 60(suppI 1):185a, 1982
- Lyman NW , Michaelson R, Viscuso RL, et al: Mitomycin-induced hemolytic-uremic syndrome: successful treatment with corticosteroids and intense plasma exchange. Arch Intern Med 143:1617;1983
- TTP-desperation, empiricism, progress. N Engl J Med 325:426.1991
- Rabinowe SN, Soiffer RJ, Tarbell NJ, et al: Hemolytic-uremic syndrome following bone marrow transplantation in adults for hematologic malignancies. Blood :1837,1991
- Page Y, Tardy B, Zeni F, et al: Thrombotic thrombocytopenic purpura related to ticlopidine. Lancet 337:774,1991
- Bums ER, Zucker-Franklin D: Pathologic effects of plasma from patients with thrombotic thrombocytopenic purpura on platelets and cultured vascular endothelial cells. Blood 60:1030, 1982
- Marcus AJ: Moschcowitz revisited (editorial). N Engl J Med 307:1447,1982!
- Moake JL, Rudy CK, Troll JH, et al: Unusually large plasma factor VIII: von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N EnglJ Med 307:1432,1982
- Kelton JG, Moore JC, Murphy WG: studies investigating platelet aggregation and release initiated by sera from patients with thrombotic thrombocytopenic purpura. Blood 69:924, 1987
- Kwaan HC: Thrombotic thrombocytopenic purpura (editorial). JAMA 247:3119,1982
- Cocchetto DM, Cook L, Cato AE, et al: Rationale and proposal for use of prostacyclin in thrombotic thrombocytopenic purpura therapy. Semin Thromb Hemost 7:43,1981
- Lee SH, Wainscoat JS, Zeitlin H, et al: Prostacyclin and thromboxane A2 in thrombotic thrombocytopenic purpura. Br Med J 283:1351,1981
- Bukowski RM, Hewlett JS, Reimer RR, et al: Therapy of thrombotic thrombocytopenic purpura: an overview. Semin Thromb Hemost 7:1,1981 .
- Rock GA, Shumack KH, Buskard NA, et al: Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 325:393,1991
- Bell WR, Braine HG, Ness PM, et al: Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome:clinical experience in 108 patients. N Engl J Med 325:398,1991
- Gutterman LA, Stevenson TD: Treatment of thrombotic thrombocytopenic purpura with vincristine. JAMA 247:1433,1982
- Gutterman LA, Meek LE, Stevenson m: Treatment of thrombotic thrombocytopenic purpura (TfP) and hemolytic-uremic syndrome (HUS) with vinca alkaloids (abstr). Blood 60(suppl1):186a, 1982
- Byrnes JJ: Plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Semin Thromb Hemost 7:9, 1981
- Harkness DR, Byrnes JJ, Lian EC-Y, et al: Hazard of platelet transfusion in thrombotic thrombocytopenic purpura. JAMA 246:1931,1981
- Taft EG, Baldwin ST: Plasma exchange transfusion. Semin Thromb Hemost 7:15,1981
- Cuttner J: Thrombotic thrombocytopenic purpura: a ten-year experience. Blood 56:302, 1980
- Onundarson PT, Rowe JM, Heal JM, et al: Response to plasma exchange and splenectomy in thrombotic thrombocytopenic purpura: a 10-year experience at a single institution. Arch Intern Med 152:791,1992
- Misiani R, Appiani AC, Edefonti A, et al: Haemolytic uraemic syndrome: therapeutic effect of plasma infusion. Br Med J 285:1304,1982
- Wilson JJ, Neame PB, Kelton JG: Infection-induced thrombocytopenia. Semin Thromb Hemost 8:217, 1982
- Feusner JH, Slichter SJ, Harker LA: Mechanisms of thrombocytopenia in varicella. Am J HematoI 7:255, 1979
- Kelton JG, Keystone J, Moore J, et al: Immune-mediated thrombocytopenia of malaria. J Clin Invest 71 :832, 1983
- Gibson B, Hunter D, Neame PB, et al: Thrombocytopenia in preeclampsia and eclampsia. Semin Thromb Hemost 8:234,1982
- Freedman J, Musclow E, Garvey B, et al: Unexplained periparturient thrombocytopenia. Am J HematoI 21:397, 1986
- The HELLP syndrome: a serious complication of pregnancy with hemolysis, elevated levels of liver enzymes, and low platelet count. Am J Med 85:590, 1988
- Van Dam PA, Renier M, Baekelandt M, et al: Disseminated intravascular coagulation and the syndrome of hemolysis, elevated liver enzymes, and low platelets in severe preeclampsia. Obstet Gynecol 73:97,1989
- Easterbrook PJ, Davis HP: Thrombocytopenia in hypothermia: a common but poorly recognised complication. Br Med J 291 :23, 1985
- George JN, Nurden AT, Phillips DR: Molecular defects in interactions of platelets with the vessel wall. N Engl J Med 311:1084,1984
- Leung L; Nachman R: Molecular mechanisms of platelet aggregation. Annu Rev Med 37:179,1986
- Kentro TB, Lottenberg R, Kitchens CS: Clinical efficacy of desmopressin acetate for hemostatic control in patients with primary platelet disorders undergoing surgery .Am J Hematol 24:215,1987
- Escolar G, Cases A, Bastida E, et al: Uremic platelets have a functional defect affecting the interaction of von Willebrand factor with glycoprotein IIb-IIIa. Blood 76:1336,1990
- Remuzzi G, Perico N, Zoja C, et al: Role of endothelium-derived nitric oxidein the bleeding tendency of uremia. J Clin Invest 86:1768,1990
- Janson PA, Jubelirer SJ, Weinstein MJ, et al: Treatment of the bleeding tendency in uremia with cryoprecipitate. N Engl J Med 303:1318,1980
- Mannucci PM, Remuzzi G, Pusineri F, et al: Deamino-8-D-arginine vasopressin shortens the bleeding time in uremia. N Engl J Med 308:8,1983
- Mannucci PM: Desmopressin: a nontransfusional form of treatment for congenital and acquired bleeding disorders. Blood 72:1449, 1988
- Cattaneo M, Moia M, Della Valle P, et al: DDAVP shortens the prolonged bleeding times of patients with severe von Willebrand disease treated with cryoprecipitate: evidence for a mechanism of action independent of released von Willebrand factor. Blood 74:1972,1989
- Fernandez F, Goudable C, Sie P, et al: Low haematocrit and prolonged bleeding time in uraemic patients: effect of red cell transfusions. Br J HaematoI 59:139, 1985
- Liu YK, Kosfeld RE, Marcum SG: Treatment of uraemic bleeding with conjugated oestrogen. Lancet 2:887,1984
- Livio M, Mannucci PM, Vigano G, et al: Conjugated estrogens for the management of bleeding associated with renal failure. N Engl J Med 315:731,1986
- Livio M, Benigni A, Vigano G, et al: Moderate doses of aspirin and risk of bleeding in renal failure. Lancet 2:414,1986
- Gaspari F, Vigano G, Orisio S, et al: Aspirin prolongs bleeding time in uremia by a mechanism distinct from platelet cyclooxygenase inhibition. J Clin lnvest 79:1788,1987
- Mikhailidis DP, Jenkins WJ, Barradas MA, et al: Platelet function defects in chronic alcoholism. Br Med J 293:715,1986
- Antimicrobials and haemostasis (editorial). Lancet 1:510,1983
- Weitekamp MR, Aber RC: Prolonged bleeding times and bleeding diathesis associated with moxalactam administration. JAMA 249:69, 1983
- Sattler FR, Weitekamp MR, Ballard JO: Potential for bleeding with the new beta-Iactam antibiotics. Ann Intern Med 105:924,1986
- George JN, Shattil SJ: The clinical importance of acquired abnormalities of platelet function. N Engl J Med 324:27,1991
- Buss DH, Stuart JJ, Lipscomb GE: The incidence of thrombotic and hemorrhagic disorders in association with extreme thrombocytosis: an analysis of 129 cases. Am J HematoI20:365, 1985
- Silverstein MN: Primary or hemorrhagic thrombocythemia. Arch Intern Med 122:18,1968
- Kimura H, Ishibashi T, Sato T, et al: Megakaryocytic colony formation (CFV-Meg) in essential thrombocythemia: quantitative and qualitative abnormalities of bone marrow CFV-Meg. Am J Hematol 24:23,1987
- Schafer Al: Deficiency of platelet lipoxygenase activity in myeloproliferative disorders. N Engl J Med 306:381, 1982
- Hirsh J: Hypercoagulability. Semin HematoI 14:409, 1977
- Michiels JJ, Abels J, Steketee J, et al: Erythromelalgia caused by platelet-mediated arteriolar inflammation and thrombosis in thrombocythemia. Ann Intern Med 102:466,1985
- Murphy S, Iland H, Rosenthal D, et al: Essential thrombocythemia: an interim report from the Polycythemia Vera Study Group. Semin HematoI 23:177,1986
- Anagrelide Study Group: Anagrelide, a therapy for thrombocythemic states: experience in 577 patients. Am J Med 92:69,1992
- Copenhagen M: Therapeutic use of interferon alpha-2b. Eur J HaematoI 45(suppl):15,1990
- Kessler CM, Klein HG, Havlik RJ: Uncontroned thrombocytosis in chronic myeloproliferative disorders. Br J Haematol 50:157 , 1982
- Kitchens CS: The purpuric disorders. Semin Thromb Hemost 10:173, 1984
- A Assar OS, Friedman CM, White RI Jr: The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 101:977,1991
- White RI Jr, Lynch-Nyhan A, Terry P, et al: Pulmonary arteriovenous malformation: techniques and long-term outcome of embolotherapy. Radiology 169:663,1988
- Hewes RC, Auster M, White RI Jr: Cerebral embolism-first mani-festation of pulmonary arteriovenous malformation in patients with hereditary hemorrhagic telangiectasia. Cardiovasc Intervent Radiol 8:151,1985
- Wanerstein RO Wanerstein RO Jr: Scurvy. Semin HematoI 13:211,1976
- Vilter RW: Nutritional aspects of ascorbic acid: uses and abuses. West J Med 133:485,1980
- Pierson KK: Leukocytoclastic vasculitis viewed as a phase of immune-mediated vasculopathy. Semin Thromb Hemost 10:196,1984
- Quismorio FP Jr, Blau RH,AkmaI M,et al: Palpable leg purpuraand renal failure in a 23-year-old man. West J Med 140:60,1984
- Henoch-Schonlein purpura (editorial). Br Med J 1:190,1977
- Rizza CR: The clinical features of clotting factor deficiencies. Human Blood Coagulation, Haemostasis and Thrombosis. Biggs R, Ed. Blackwen Scientific Publications, Oxford, 1972, p 210
- Rodeghiero F, Castaman G, Dini E: Epidemiological investigation of the prevalence of von Winebrand's disease. Blood 69:454,1987
- Ruggeri ZM: Structure and function of von Willebrand factor: relationship to von Winebrand's disease. Mayo Clin Proc 66:847,1991
- Bloom AL: Von Willebrand factor: clinical features of inherited and acquired disorders. Mayo Clin Proc 66:743,1991
- Triplett DA: Laboratory diagnosis of von Willebrand's disease. Mayo Clin Proc 66:832,1991
- Ruggeri ZM, Zimmerman TS: Von Winebrand factor and von Winebrand disease. Blood 70:895,1987
- Miller JL,Kupinski JM,Castella A,et al: Von Willebrand factor binds to platelets and induces aggregation in platelet-type but not type B von Willebrand disease. J Clin Invest 72:1532,1983
- Miller JL, Boselli BD, Kupinski JM: In vivo interaction of von Willebrand factor with platelets following cryoprecipitate transfusion in platelet-type von Willebrand's disease. Blood 63:226,1984
- Aledort LM: Treatment of von Willebrand's disease. MayoClin Proc 66:841,1991
- Castillo R, Monteagudo J, Escolar G, et al: Hemostatic effect of normal platelet transfusion in severe von Willebrand disease patients. Blood 77:1901,1991
- Goudemand J, Mazurier C, Marey A, et al: Clinical and biological eyaluation in von Willebrand's disease of a von Willebrand factor concentrate with low factor VIII activity. Br J Haematol 80:214, 1992
- Mannucci PM, Canciani MT, Rota L, et al: Response of factor VIII/von Willebrand factor to DDA VP in healthy subjects and patients with haemophilia A and von Willebrand's disease. Br J HaematoI 47:283,1981
- DDAVP in haemophilia and von Willebrand's disease (editorial). Lancet 2:774,1983
- De la Fuente B, Kasper CK, Rickles FR, et al: Response of patients with mild and moderate hemophilia A and von Willebrand´s disease to treatment with desmopressin. Ann Intern Med 103:6,1985
- Rose EH, Aledort LM: Nasal spray desmopressin (DDA VP) for mild hemophilia A and von Willebrand disease. Ann Intern Med 114:563, 1991
- Furie B, Furie BC: Molecular basis of hemophilia. Semin Hematol 27:270, 1990
- Kurachi K, Yao S-N, Furukawa M, et al: Deficiencies in factors IX and VIII: what is now known. Hosp Pract 27(2):41,1992
- Hoyer LW: The Factor VIII complex: structure and function. Blood 58:1,1981
- Allain J-P, Dailey SH, Laurian Y, et al: Evidence for persistent hepatitis C virus (HCV) infection in hemophiliacs. J Clin Invest 88:1672,1991
- Ratnoff OD: Some complications of the therapy of classic hemophilia. J Lab Clin Med 103:653, 1984
- Pneumocystis carinii pneumonia among persons with hemophilia A. MMWR 31:365,1982
- Human T-Iymphotropic retrovirus type 1II/Iymphadenopathy-associated virus antibody: association with hemophiliacs' immune status and blood component usage. JAMA 253:3409,1985
- Rouzioux C, Brun-Vezinet F, Couroucé AM, et al: Immunoglobulin G antibodies to lymphadenopathy-associated virus in differently treated French and Belgian hemophiliacs. Ann Intern Med 102:476,1985
- Surveillance of hemophilia-associated acquired immunodeficiency syndrome. National Hemophilia Foundation and Associated Hemophilia Treatment Centers; Division of Host Factors, Center for Infectious Diseases, CDC. JAMA 256:3205, 1986
- Kreiss JK, Kitchen LW, Prince HE, et al: Antibody to human Tlymphotropic virus type III in wives of hemophiliacs: evidence of heterosexual transmission. Ann Intern Med 102:623,1985
- Goldsmith MF: Hemophilia, beaten on one front, is beset on others. JAMA 256:3200,1986
- Ragni MV, Spero JA, Bontempo FA, et al: Recurrent infections and lymphadenopathy in the child of a hemophiliac :a survey of children of hemophiliacs positive for human immunodeficiency virus antibody. Ann Intern Med 105:886,1986
- Lee CA, Phillips AN, Elford J, et al. Progression of HIV disease in a haemophilic cohort follqwed for 11 years and the effect of treatment. Br Med J [Clin Res] 303:1093,1991
- Jones PK, Ratnoff OD: The changing prognosis of classic hemophilia (factor VIII HdeficiencyH). Ann Intern Med 114:641, 1991
- Brettler DB, Levine PH: Factor concentrates of hemophilia: which one to choose? Blood 73:2067,1989
- Schwartz RS, Abildgaard CF, Aledorf LM, et al: Human recombinant DNA-derived antihemophilic factor (factor VIII) in the treatment of hemophilia A. N Engl J Med 323:1800,1990
- Robertson M: The heredity of haemophilia. Nature 314:674,1985
- Green PP, Mannucci PM, Briet E, et al: Carrier detection in hemophilia A: a cooperative international study: II. The efficacy of a universal discriminant. Blood 67:1560,1986
- Janco RL, Phillips JA III, Orlando P, et al: Carrier testing strategy in haemophilia A. Lancet 1:148,1986
- Oberle I, Camerino G, Heilig R, et al: Genetic screening for hemophilia A (classic hemophilia) with a polymorphic DNA probe. N Engl J Med 312:682,1985
- Antonarakis SE, Carpenter RJ, Hoyer LW, et al: Prenatal diagnosis of haemophilia A by factor VIII gene analysis. Lancet 1:1407,1985
- Kogan SC, Doherty M, Gitschier J: An improved method for prenatal diagnosis of genetic diseases by analysis of amplified DNA sequences: application in hemophilia A. N Engl J Med 317:985,1987
- White GC II, shoemaker CB: Factor VIII gene and hemophilia A. Blood 78:1,1989
- McVerry BA, Voke J, Vicary FR, et al: Ultrasonography in the management of haemophilia. Lancet 1:872,1977
- Kasper CK, Dietrich SL: Comprehensive management of haemophilia. Clin HaematoI 14:489, 1985
- Kasper CK, Boylen AL, Ewing NP , et al: Hematologic management of hemophilia A for surgery. JAMA 253:1279,1985
- Gralnick HR, Rick ME: Danazol increases factor VIII and factor IX in classic hemophilia and Christmas disease. N Engl J Med 308:1393, 1983
- Gralnick HR, Maisonneuve P, sultan Y, et al: Benefits of danazol treatment in patients with hemophilia A (classic hemophilia). JAMA 253:1151,1985
- Garewal HS, Corrigan JJ Jr, Durie BGM, et al: Effect of danazol on coagulation parameters and bleeding in hemophilia. JAMA 253:1154,1985 .
- Nugent DJ, Bray GL, Counts RB, et al: Danazol fails to increase factor VIII or IX levels in a double-blind crossover study of patients with haemophila A and B. Br J Haematol 64:493, 1986
- Saidi P, Lega BZ, Kim HC, et al: Effect of danazol on clotting factor levels, bleeding incidence, factor infusion requirements, and immune parameters in hemophilia. Blood 68:673,1986
- Ehrenforth S, Kreuz W, Scharrer L et al: lncidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 339:594,1992
- Kasper CK: Prothrombin complex concentrates and inhibitors: a reappraisal (editorial). JAMA 251:68,1984
- Kemoff PBA, Thomas ND, Liley PA, et al: Clinical experience with polyelectrolyte-fractionated porcine factor VIII concentrate in the treatment of hemophiliacs with antibodies to factor VIII. Blood 63:31,1984
- Ciavarella N, Antoncecchi S, Ranieri P: Efficacy of porcine factor VIII in the management of haemophiliacs with inhibitors. Br J Haematol 58:641,1984
- Wensley RT, stevens RF, Bum AM, et al: Plasma exchange and human factor VIII concentrate in managing haemophilia A with factor VIII inhibitors. Br Med J 281:1388, 1980
- Lusher JM, shapiro SS, Palascak JE, et al: Efficacy of prothrombincomplex concentrates in hemophiliacs with antibodies to factor VIII. N Engl J Med 303:421,1980
- Abildgaard CF, Penner JA, Watson-Williams EJ: Anti-inhibitor Coagulant Complex (Autoplex) for treatment of factor VIII inhibitors in hemophilia. Blood 56:978,1980
- Sjamsoedin LJM, Heijnen L, Mauser-Bunschoten EP, et al: The effect of activated prothrombin-complex concentrate (FEIBA) on joint and muscle bleeding in patients with hemophilia A and antibodies to factor VIII: a double-blind clinical trial. N Engl J Med 305:717, 1981
- Lusher JM, Blatt PM, Penner JA, et al: Autoplex versus Proplex: a controlled, double-blind study of effectiveness in acute hemarthroses in hemophiliacs with inhibitors to factor VIII. Blood 62:1135,1983
- Blatt PM, White GC II, McMillan CW, et al: Failure of activated prothrombin complex concentrates in a hemophiliac with an antifactor VIII antibody. JAMA 251:67,1984
- Bloom AL: Progress in the clinical management of haemophilia. Thromb Haemost 66:166,1991
- Peake IR, Furlong BL, Bloom AL: Carrier detection by direct gene analysis in a family with haemophilia B (factor IX deficiency). Lancet 1:242,1984
- Gianelli F, Anson DS, Choo KH, et al: Characterisation and use of an intragenic polymorphic marker for detection of carriers of haemophilia B (factor IX deficiency). Lancet 1:239,1984
- Hay CW, Robertson KA, Yong S-L, et al: Use of a BamH1 polymorphism in the factor IX gene for the determination of hemophilia B carrier status. Blood 67:1508,1986
- Mazzucconi MG, Mariani G, Chistolini A, et al: Evaluation of the nature of mildly prolonged prothrombin times. Am J Hematol24:37 , 1987
- Aoki N: Genetic abnormalities of the fibrinolytic system. Semin Thromb Hemost 10:42,1984
- Booth NA, Bennett B, Wijngaards G, et al: A new life-long hemorrhagic disorder due to excess plasminogen activator. Blood 61:267,1983
- Stenflo J: Vitamin K, prothrombin and [gamma]-carboxyglutamic acid. N Engl J Med 296:624, 1977
- Friedman PA: Vitamin K-dependent proteins (editorial). N Engl J Med 310:1458,1984
- Blanchard RA, Furie BC, Jorgensen M, et al: Acquired vitamin K-dependent carboxylation deficiency in liver disease. N Engl J Med 305:242,1981
- Jones PG, Strother SV, Rolston KVI, et al: Hypoprothrombinemia in patients with cancer receiving cefoperazone and mezlocillin. Arch Intern Med 146:1397,1986
- Sattler FR, Weitekamp MR, Ballard JO: Potential for bleeding with the new beta-Iactam antibiotics. Ann Intern Med 105:924,1986
- Jolson HM, Tanner LA, Green L, et al: Adverse reaction reporting of interaction between warfarin and fluoroquinolones. Arch Intern Med 151:1003, 1991
- Thomas DP: Bleeding after low-molecular-weight heparin. Lancet 339:1119,1992
- Sane DC, Califf RM, Topol EJ, et al: Bleeding during thrombolytic therapy for acute myocardial infarction: mechanisms and management. Ann Intern Med III:I0I0, 1989
- Gore JM, Sloan M, Price TR, et al: Intracerebral hemorrhage, cerebral infarction, and subdural hematoma after acute myocardial infarction and thrombolytic therapy in the thrombolysis in myocardial infarction study: Thrombolysis in Myocardial Infarction, Phase II, pilot and clinical trial. Circulation 83:448,1991
- Dearing BD, Lavie CJ, Lohmann TP, et al: Niacin-induced clotting factor synthesis deficiency with coagulopathy. Arch Intern Med 152:861,1992
- Perkins HA, MacKenzie MR, Fudenberg HH: Hemostatic defects in dysproteinemias. Blood 35:695,1970
- Moore KL, AndreoIl SP, Esmon NL, et al: Endotoxin enhances tissue factor and suppresses thrombomodulin expression of human vascular endothelium in vivo. J Clin Invest 79:l24, 1987
- WarreIl RP, Kempin SJ: Treatment of severe coagulopathy in the Kasabach-Merritt syndrome with aminocaproic acid and cryoprecipitate. N Engl J Med 313:309,1985
- Win MI, Warrell DA, PhiIlips RE, et al: Bites by RusseIl's viper (Vipera russelli siamensis) in Burma: haemostatic, vascular; and renal disturbances and response to treatment. Lancet 2:l259, 1985
- Cembrowski GS, Griffin JH, Mosher DF: Diagnostic efficacy of six plasma proteins in evaluating consumptive coagulopathies: use of receiver operating characteristic curves to compare antithrombin III, plasminogen, [alpha]2-plasmin inhibitor, fibronectin, prothrombin, and protein C. Arch Intern Med 146:1997,1986
- Williams EC: Plasma [alpha]2-antiplasrnin activity: role in the evaluation and management of fibrinolytic states and other bleeding disorders. Arch Intern Med 149:1769.1989
- Rosenberg R: Actions and interactions of antithrombin and heparin. N Engl J Med 292:146,1975
- Deykin D: The clinical challenge of disseminated intravascular coagulation. N Engl J Med 283:686, 1970
- Budzynski AZ, Pandya BV, Rubin RN, et al: Fibrinogenolytlc afibrinogenemia after envenomation by western diamondback rartlesnake (Crotalus atrox). Blood 63:1,1984
- Vinnecombe J, shuttleworth KED: Aminocaproic acid in the control of hemorrhage after prostatectomy: safety of aminocaproic acid-a controlled trial. Lancet 1:232,1966
- Stein SF, Harker LA: Kinetic and functional studies of platelets, fibrinogen, and plasminogen in patients with hepatic cirrhosis. J Lab Clin Med 99:217,1982
- Bolan CD, Alving BM: Pharmacologic agents in the management of bleeding disorders. Transfusion 30:541,1990
- Kessler CM: An introduction to factor VIII inhibitors: the detection and quantitation. Am J Med 91(suppI 5A):5A-1s, 1991
- Hultin MB: Acquired inhibitors malignant and nonmalignant disease states. Am J Med 91 (suppI5A):5A-9S, 1991
- Feinstein DI, Rapaport SI: Acquired inhibitors of blood coagulation. Progress in Hemostasis and Thrombosis. Spaet T, Ed. Grune & Stratton, Inc, New York, 1972, p 75
- Green D: Cytotoxic suppression of acquired factor VIlI:C inhibitorS. Am J Med 91(suppI5A):5A-14S, 1991
- LianC-Y, Larcada AF, Chiu AY-Z: Combination immunosuppressive therapy after factor VIII infusion for acquired factor VIII inhibitor. Ann Intem Med 110:774,1989
- Kemoff PBA: Rationale and evolution of therapy with porcine factor VlII:C. Am J Med 91 (suppI5A):5A-2OS, 1991
- Lusher JM: Perspectives on the use of factor IX complex concentrates in the treatment of bleeding in persons with acquired factor VIII inhibition. Am J Med 91 (suppI5A):5A-3OS, 1991
- Sultan Y, Kazatchkine MD, Maisonneuve P, et al: Anti-idiotypic suppression of autoantibodies to factor VIlI (antihaemophilic factor) by high-dose intravenous gammaglobulin. Lancet 2:765,1984
- Zimmermann R, Kommerell B, Harenberg J, et al: Intravenous IgG for patients with Spontaneous inhibitor to factor VIII. Lancet 1:273, 1985
- Sultan Y, Kazatchkine MD, Nydegger U, et al: Intravenous immunoglobulin in the treatment of spontaneously acquired factor VlII:C inhibitorS. Am J Med 91(suppI5A):5A-35S, 1991
- Sultan Y, Rossi F, Kazatchkine MD: Recovery from anti-VIII:C (antihemophilic factor) autoimmune disease is dependent on generation of antiidiotypeS against antiVIII:C autoantibodies. ProC Natl Acad Sci USA 84:828, 1987
- Gastineau DA, Kazmier FJ, Nichols WL, et al: Lupus anticoagulant: an analysis of the clinical and laboratory features of 219 cases. Am J Hematol 19:265, 1985
- Cohen AJ, Philips TM, Kessler CM: Circulating coagulation inhibitors in the acquired immunodeficiency syndrome. Ann Intern Med 104:175,1986
- Bloom EJ, Abrams DI, Rodgers G: Lupus anticoagulant in the acquired immunodeficiency syndrome. JAMA 256:491,1986
- Zarrabi MH, Zucker S, Miller F, et al: Immunologic and coagulation disorders in chlorpromazine-treated patients. Ann Intern Med 91:194,1979
- Roubey RAS, Pratt CW, Buyon JP,etal: Lupus anticoagulant activity of autoimmune antiphospholipid antibodies is dependent upon [beta]2-glycoprotein I. J Clm Invest 90:1100, 1992
- Stephens CJM: The antiphospholipid syndrome: clinical correlations, cutaneous features, mechanism of thrombosis and treatment of patients with the lupus anticoagulant and anticardiolipin antibodies. Br J Dermatol 125:199, 1991
- Eisenberg GM: Antiphospholipid syndrome: the reality and implications. Hosp Pract 27(6):119,1992
- Rosove MH, Brewer MC: Antiphospholipid thrombosis: clinical course after the first thrombotic event in 70 patients. Ann Intern Med 117:303,1992
- Branch DW, Scott JR, KoChenour NK, et al: Obstetric complications associated with the lupus anticoagulant. N Engl J Med 313:1322, 1985
- Reid JD, Kommareddi S, Lankerani M, et al: Chronic expanding hematomas: a clinicopathologic entity. JAMA 244:2441,1980
- Harker LA: Bleedmg after cardiopulmonary bypass. N Engl J Med 314:1446,1986
- Salzman EW, Weinstein MJ, Weintraub RM, et al: Treatment with desmopressin acetate to reduce blood loss after cardiac surgery. A doubleblind randomized trial. N Engl J Med 314:1402,1986
- Kobrinsky NL, Letts RM, Patel LR, et al: 1-Desammo-8-D-arginine vasopressin (desmopressin) decreases operative blood loss in patients having Harrington rod spinal fusion surgery: a randomized, double-blinded, controlled trial. Ann Intern Med 107:446,1987
- Rocha E, Llorens R, Paramo JA, et al: Does desmopressin acetate reduce blood loss after surgery in patients on cardiopulmonary bypass? Circulation 77:1319,1988
- Hackmann T, Gascoyne R, Naiman SC, et al: A trial of desmopressin to reduce blood loss in uncomplicated cardiac surgery. N EnglJ Med 321:1437,1989
- Harris EN: Antiphospholipid antibodies. Br J Haematol 74:251, 1990
- McCrae KR, DeMichele A, Samuels P, et al: Detection of endothelial cell-reactive immunoglobulin in patients with anti-phospholipid antibodies. Br J Haernatol 79:595, 1991
- Boey ML, Colaco CB, Gharavi AE, et al: Thrombosis in systemic lupus erythematosus: striking association with the presence of cir. culating lupus anticoagulant. Br Med J 287:1021,1983
- Comp PC, DeBault LE, Esmon NL, et al: Human thrombomodulin is inhibited by IgG from two patients with non-specific anticoagulants. Blood 62(suppI 1):299a, 1983
- Cosgriff TM, Bishop DT, Hershgold EJ, et al: Familial antithrombin III deficiency: its natural history , genetics, diagnosis and treatment. Medicine (Baltimore) 62:209,1983
- Shapiro ME, Rodvien R, Bauer KA, et al: Acute aortic thrombosis in antithrombin III deficiency. JAMA 245:1759,1981
- Demers C, Ginsberg Js, Hirsh J, et al: Thrombosis in antithrombin III-deficient persons: report of a large kindred and literature review. Ann Intern Med 116:754,1992
- Menache D, O'Malley JP, Schorr JB, et al: Evaluation of the safety, recovery , half-life, and clinical efficacy of antithrombin III (human) in patients with hereditary antithrombin III deficiency .Blood 75:33,1990
- Sie P, Dupouy D, Pichon J, et al: Constitutional heparin co-factor II deficiency associated with recurrent thrombosis. Lancet 2:414, 1985
- Tran TH, Marbet GA, Duckert F: Association of hereditary heparin co-factor II deficiency with thrombosis. Lancet 2:413,1985
- Tollefsen DM, Pestka CA: Heparin cofactor II activity in patients with disseminated intravascular coagulation and hepatic failure. Blood 66:769,1985
- D' Angelo A, Lockhart MS, D' Angelo SV, et al: Protein S is a cofactor for activated protein C neutralization of an inhibitor of plasminogen activation released from platelets. Blood 69:231,1987
- Griffin JH, Mosher DF, Zimmerman TS, et al: Protein C, an anti-thrombotic protein, is reduced in hospitalized patients with intravascular coagulation. Blood 60:261,1982
- Seligsohn U, Berger A, Abend M, et al: Homozygous protein C deficiency manifested by massive venous thrombosis in the newborn. N EnglJ Med 310:559,1984
- Miletich J, Sherman L, Broze G Jr: Absence of thrombosis in subjects with heterozygous protein C deficiency. N Engl J Med317:991, 1987
- Esmon CT: Protein-C: biochemistry, physiology, and clinical implications. Blood 62:1155,1983
- Marder VJ: Molecular bad actors and thrombosis (editorial). N Engl J Med 310:588,1984
- Wintzen AR, Broekmans AW, Bertina RM, et al: Cerebral haemorrhagic infarction in young patients with hereditary protein C deficiency: evidence for "spontaneous" cerebral venous thrombosis. Br Med J 290:350,1985
- Rodgers GM, Shuman MA: Congenital thrombotic disorders. Am J HematoI 21:419,1986
- Comp PC, Esmon CT: Recurrent venous thromboembolism in patients with a partial deficiency of protein S. N Engl J Med 311:1525, 1984
- Broekmans AW, van Rooyen W, Westerveld BD, et al: Mesenteric vein thrombosis as presenting manifestation of hereditary protein S deficiency. Gastroenterology 92:240,1987
- Engesser L, Broekmans AW, Briet E, et al: Hereditary protein S deficiency: clinical manifestations. Ann Intern Med 106:677,1987
- Boerger LM, Morris PC, Thurnau GR, et al: Oral contraceptives and gender affect protein S status. Blood 69:692,1987
- Vigano-D' Angelo S, D' Angelo A, Kaufman CE Jr, et al: Protein S deficiency occurs in the nephrotic syndrome. Ann Intern Med 107:42,1987
- Nilsson IM, Ljungner H, Tengborn L: Two different mechanisms in patients with venous thrombosis and defective fibrinolysis: low concentration of plasminogen activator or increased concentration of plasminogen activator inhibitor. Br Med J 290:1453,1985
- Prins MH, Hirsh J: A critical review of the evidence supporting a relationship between impaired fibrinolytic activity and venous thromboembolism. Arch Intern Med 151:1721,1991
- Carrell N, Gabriel DA, Blatt PM, et al: Hereditary dysfibrinogenemia in a patient with thrombotic disease. Blood 62:439,1983
- Lentz SR, Sadler JE: Inhibition of thrombomodulin surface expression and protein C activation by the thrombogenic agent homocysteine. J Clin Invest 88:1906,1991
- Clarke R, Daly L, Robinson K, et al: Hyperhomocysteinemia: an independent risk factor for vascular disease. N Engl J Med 324:1149,1991
- Gordon EM, Ratnoff OD, Saito H, et al: Rapid fibrinolysis, aug~mented Hageman factor (factor XII) titers, and decreased C1 esterase inhibitor titers in women taking oral contraceptives. J Lab Clin Med 96:762,1980
- Phelan BK: Heparin-associated thrombosis without thrombocytopenia. Ann Intem Med 99:637, 1983
- King DJ, Kelton JG: Heparin-assodated thrombocytopenia. Ann Intem Med 100:535,1984
- Vanrenterghem Y, Roels L, Lerut T, et al: Thromboembolic complications and haemostatic changes in cyclosporin-treated cadaveric. kidney allograft recipients. Lancet 1:999,1985
- Antman KH, Pomfret EA, Aisner J, et al: Peritoneal mesotheliorna: natural history and response to chernotherapy. J Clin Oncol 1:386,1983
- Sack GH Jr, Levin J, Bell WR: Trousseau's syndrorne and other rnanifestations of chronic disserninated coagulopathy in patients with neoplasrns: clinical, thophysiologic, and therapeutic features. Medicine (Baltirnore) 56:1,1977
- silverstein RK, Nachrnan RL: Cancer and clotting-Trousseau's warning. N Engl J Med 327:1163,1992
- Callander Ns, Varki N, Rao VM: Imrnunohistochernical identification of tissue factor in solid tumors. Cancer 70:1194,1992
- Rickles FR, Edwards RL: Activation of blood coagulation in cancer: Trousseau's syndrome revisited. Blood 62:14,1983
- Bell WR, starksen NF, Tong S, et al: Trousseau's syndrorne: devastating coagulopathy in the absence of heparin. Am J Med 79:423,1985
- Levi M, Hack CE, de Boer JP, et al: Reduction of contact activation related fibrinolytic activity in factor XII deficient patients: further evidence for the role of the contact systern in fibrinolysis in vivo. J Clin Invest88:1155, 1991
- Rodeghiero F, Castarnan G, Ruggeri M, et al: Thrornbosis in subjects with hornozygous and heterozygous factor XII deficiency .Thrornb Haernos 67:590,1992
- Liirnmle B, Furlan M: Rebuttal to the letter of F. Rodeghiero et al.-thrornbosis in subjects with hornozygous and heterozygous factor XII deficiency. Thrornb Haernos 67:591,1992
- Howard JE, Glassberg AB, Perkins HA: Post-transfusion thrornbocytopenic purpura: a case report. Am J Hematol1:339, 1976
- Vaughan-Neil EF, Ardeman S, Bevan G, et aJ: Post-transfusion purpura associated with unusual platelet antibody (anti-PIBIB1. Br Med J 1:436,1975
- Abrarnson N, Eisenberg PD, Aster RH: Post-transfusion purpura: immunologic aspects and therapy. N EnglJ Med 291:1163,1974
- Barton JC, Poon M-C: Coagulation testing of Hickrnan catheter blood in patients with acute leukemia. Arch Intern Med 146:2165,1986
- Rapaport SI: Preoperative hemostatic evaluation: which tests, if any? Blood 61:229,1983
- Suchman AL, Mushlin Al: How well does the activated partial thromboplastin time predict postoperative hemorrhage? JAMA 256:750,1986