Hematología
⭳ Abrir artículo (PDF)519.2 KBEste artículo es idéntico en la Edición 2/2000.
Contenido del artículo
V POLICITEMIAS
- Clasificación de las policitemias
- Evaluación inicial
- Policitemia relativa
- Policitemia secundaria
- AUMENTO APROPIADO EN LA PRODUCCION DE ERITROPOYETINA
- Vida a altitud elevada
- Hemoglobina de alta afinidad
- Hipoxia tisular causada por enfermedad cardiopulmonar
- Síndrome de obesidad-hipoventilación
- Apnea obstructiva del sueño
- Aumento en los niveles de carboxihemoglobina
- AUMENTO INAPROPIADO EN LA PRODUCCION DE ERITROPOYETINA
- HIPERSENSIBILIDAD A LA ERITROPOYETINA (POLICITEMIA FAMILIAR CONGENITA)
- USO DE MEDICAMENTOS
- Policitemia vera
V POLICITEMIAS
DRA. VIRGINIA C. BROUDY
Clasificación de las policitemias
La policitemia, también denominada eritrocitosis, es el aumento en el número de eritrocitos circulantes por volumen de sangre, que se refleja por aumento del hematocrito o de la concentración de hemoglobina. Existen tres categorías principales de policitemia: (1) policitemia relativa, (2) policitemia secundaria, y (3) policitemia primaria, o policitemia vera.
En la policitemia relativa la masa eritrocitaria es normal, pero el volumen del plasma está disminuido. La policitemia secundaria está causada por elevación en la concentración sérica de eritropoyetina y puede deberse a diversos trastornos, por ejemplo la presencia de hemoglobina de alta afinidad que no oxigena los tejidos con eficacia, causando hipoxia tisular y mayor producción de eritropoyetina. La policitemia vera es un trastorno neoplásico de los eritrocitos caracterizado por sobreproducción autónoma de eritrocitos, y con frecuencia de leucocitos y plaquetas.
Evaluación inicial
El aumento en la concentración de hemoglobina o del hematocrito justifica una evaluación inmediata para investigar la naturaleza de la policitemia y su causa posible [ver figura 1]. Debe realizarse una historia clínica y examen físico, investigando el antecedente familiar de policitemia y los resultados de los hematocritos durante los años previos. Cuando es posible deben buscarse también los valores de hematocrito durante la adolescencia o el inicio de la edad adulta. Son importantes los datos que sugieran enfermedad cardiaca congénita, enfermedad pulmonar obstructiva crónica (EPOC) severa o síndrome de apnea del sueño, y se investigará la presencia o ausencia de esplenomegalia. Los resultados de una biometría hemática completa son indispensables, incluyendo la cuenta de plaquetas y diferencial de leucocitos. La presencia de leucocitosis, trombocitosis, leucocitos circulantes inmaduros o aumento en los basófilos, son sugestivos de policitemia vera y van en contra de causas secundarias de eritrocitosis.
Un hombre con un hematocrito de 60 porciento o más1 o una mujer con hematocrito de 57 porciento o más, casi siempre tienen un aumento verdadero en la masa eritrocitaria (i.e., policitemia primaria o secundaria). Si el hematocrito del paciente está por debajo de esos valores pero sobre lo normal, debe hacerse un estudio de la masa eritrocitaria para definir si está en realidad aumentada. Para realizar este estudio se marca una muestra de los eritrocitos del paciente con cromo radioactivo (Cr51) ex vivo y se reinyecta al paciente. Se obtiene después una segunda muestra para cuantificar la concentración de eritrocitos marcados entre los eritrocitos no marcados. En forma paralela, se administra al paciente una inyección de albúmina marcada con ioduro radioactivo (I125) para medir el volumen plasmático. Los resultados de la determinación de la masa eritrocitaria marcada con Cr51 identifican si el paciente tiene policitemia relativa por contracción del volumen plasmático o policitemia verdadera (i.e., primaria o secundaria) por aumento en la masa de eritrocitos. Una vez que se demuestra un aumento relativo o absoluto en la masa de eritrocitos debe determinarse el diagnóstico exacto [ver figura 1].
Policitemia relativa
La policitemia relativa también se conoce como síndrome de Gaisböck por la primera descripción clínica. Los pacientes con policitemia relativa suelen ser varones de edad media, obesos, e hipertensos, en ocasiones fumadores intensos.2,3 Con frecuencia inician los síntomas a los 45 a 55 años de edad, una década menos que la típica para policitemia vera [ver adelante, Policitemia vera]. En la mayoría de las series el 80 a 90 porciento de los pacientes con policitemia vera son varones.2,3 Se ha calculado que el 0.5 a 0.7 porciento de la población masculina normal en los Estados Unidos tiene policitemia relativa.4 El uso de diuréticos para el tratamiento de la hipertensión puede exacerbar la deficiencia en el volumen plasmático, y los niveles altos de carboxihemoglobina inducidos por el tabaquismo o la hipoxemia también pueden influir. La policitemia relativa suele ser leve (hematocrito menor de 55 porciento). Es importante tener en cuenta el diagnóstico y medir la masa eritrocitaria y el volumen plasmático en pacientes con hematocrito menor del 60 porciento [ver figura 1] para evitar una investigación extensa y al final frustrante de otras causas de policitemia. Los pacientes con policitemia relativa pertenecen a dos grupos principales: (1) los que tienen masa eritrocitaria normal y disminución clara en el volumen plasmático y (2) los que tienen masa eritrocitaria en el rango superior del normal y volumen plasmático en el límite inferior normal.4 El hematocrito se normaliza después de cierto tiempo en alrededor de la tercera parte de los pacientes.
Policitemia secundaria
AUMENTO APROPIADO EN LA PRODUCCION DE ERITROPOYETINA
Puede ocurrir policitemia cuando la producción de eritropoyetina aumenta como resultado de una compensación adecuada en presencia de hipoxia tisular generalizada. Ocurre hipoxia tisular con la vida a altitud elevada, la presencia de hemoglobina de alta afinidad, la enfermedad cardiopulmonar, el síndrome de obesidad-hipoventilación, la apnea obstructiva del sueño y los niveles elevados de carboxihemoglobina.
Vida a altitud elevada
En 1890 el médico francés Viault visitó un pueblo pequeño en los Andes a 15,000 pies de altura, y realizó la observación histórica de que su cuenta eritrocitaria aumentó de 5 x 106 células/mm3 a 8 x 106 células/mm3 durante un periodo de varias semanas.5 Después se observó que el hematocrito de las personas que viven a mayor altura es un poco mayor que el de los que viven a nivel del mar. La adaptación humana inicial a la altitud incluye aumento en la frecuencia respiratoria, en el gasto cardiaco y en la concentración de 2,3-difosfoglicerato para facilitar la liberación de oxígeno de la hemoglobina en los tejidos [ver figura 2]. Seis a 24 horas después de que la persona asciende aumenta la concentración de eritropoyetina, lo que causa reticulocitosis a las 24 a 48 horas.6 Varios días después la concentración de eritropoyetina se normaliza, pero se mantiene el aumento del hematocrito. Además del aumento en la masa eritrocitaria, en los residentes de grandes alturas puede encontrarse una reducción moderada en el volumen plasmático.7
Hemoglobina de alta afinidad
La hemoglobina de alta afinidad es causada por la sustitución de un solo aminoácido en la cadena alfa o, con más frecuencia, en la cadena b de la globina, lo que impide el cambio de conformación normal durante la unión y liberación del oxígeno.8 Esto provoca una menor capacidad para liberar oxígeno en los tejidos, con hipoxia subsecuente. El estímulo hipóxico es captado por el riñón, que aumenta la producción de eritropoyetina, provocando policitemia. Se han descrito más de 40 mutaciones individuales que causan hemoglobinas de alta afinidad.8 Estas suelen ser familiares y se heredan en forma autosómica dominante, pero en ocasiones son resultado de una mutación espontánea. Las biometrías hemáticas previas de un paciente pueden ser de gran utilidad porque el enfermo con hemoglobina de alta afinidad debe tener evidencia de policitemia de por vida. El hematocrito de los pacientes con hemoglobina de alta afinidad casi siempre es menor de 60 porciento, y las cuentas de leucocitos y plaquetas son normales. Debe medirse la presión parcial de oxígeno a la que la hemoglobina está saturada al 50 porciento (P50), que estará reducida en los pacientes con hemoglobina de alta afinidad [ver figura 2]. Por lo general no es útil obtener una electroforesis de hemoglobina porque muchas de las mutaciones que causan hemoglobinas de alta afinidad son silenciosas desde el punto de vista electroforético. La policitemia en estos pacientes está regulada fisiológicamente para optimizar la oxigenación tisular. Los pacientes con hemoglobina de alta afinidad no suelen tener síntomas de hiperviscosidad, y no requieren tratamiento.
Hipoxia tisular causada por enfermedad cardiopulmonar
A diferencia de la policitemia causada por hemoglobinas de alta afinidad, en la que el defecto primario consiste en un aporte inadecuado de oxígeno a los tejidos, la policitemia causada por defectos cardiopulmonares es causada por deficiente carga de oxígeno en los pulmones. Las cardiopatías congénitas cianógenas (v.gr., complejo de Eisenmenger, corazón univentricular y tetralogía de Fallot) pueden acompañarse de policitemia importante.9 El hematocrito puede variar de 60 a 75 porciento, y existen signos francos de hiperviscosidad, incluyendo cefalea, mareo, alteraciones visuales, fatiga, parestesias y menor agudeza mental.9,10 Un estudio de 124 adultos con cardiopatía congénita cianógena sugirió que pueden identificarse desde el punto de vista clínico dos grupos de pacientes.10 Uno tiene eritrocitosis compensada, y mantiene un hematocrito estable (aunque elevado) sin síntomas de hiperviscosidad muy importantes. Un segundo grupo tiene eritrocitosis descompensada, que se caracteriza por hematocrito que se incrementa e hiperviscosidad sintomática.8,10 Es notable que en este estudio no se observaron eventos cerebrovasculares a pesar de lo elevado de los hematocritos.10 Las investigaciones sobre los efectos hemodinámicos de reducir el hematocrito en pacientes con cardiopatía congénita cianógena han demostrado un aumento en la capacidad de ejercicio máximo, en el gasto cardiaco y en la captación de oxígeno por los tejidos durante el ejercicio.11 Un enfoque práctico de tratamiento consiste en flebotomizar con cuidado a los pacientes con cardiopatía congénita cianógena que tienen un hematocrito mayor del 65 porciento y que causa síntomas de hiperviscosidad.10 La magnitud de la flebotomía dependerá de los síntomas del paciente. Debe excluirse deshidratación aguda que exacerbe la policitemia antes de realizar la flebotomía, y el volumen de sangre que se extrae se repondrá con solución salina isotónica.10
La EPOC severa puede asociarse con policitemia, aunque por lo general predominan los síntomas clínicos de la primera. En pacientes que continúan fumando, tanto la hipoxemia como el aumento en la concentración de carboxihemoglobina pueden contribuir al desarrollo de la policitemia (ver adelante). La reducción del hematocrito en los pacientes con policitemia importante (hematocrito promedio de 59.6 porciento) causada por EPOC mejora la circulación cerebral y alivia los síntomas de cefalea y mareo atribuibles a la hiperviscosidad.12 También se ha reportado mejoría muy importante en el estado de alerta después de la flebotomía en pacientes que tenían un hematocrito previo de 61.3 porciento.13 El análisis de la tolerancia al ejercicio tanto antes como después de la flebotomía mostró un aumento en la duración del ejercicio y el consumo máximo de oxígeno en pacientes con EPOC que tenían un hematocrito inicial mayor de 55 porciento.14 Por estos motivos debe realizarse flebotomía en los pacientes con EPOC y síntomas atribuibles a la policitemia.
Síndrome de obesidad-hipoventilación
El síndrome de obesidad-hipoventilación también se conoce como síndrome de Pickwick en referencia a la astuta descripción de Charles Dickens del chico obeso con somnolencia diurna excesiva.15 Los pacientes con este síndrome suelen ser obesos (con peso corporal mayor del 120 porciento del ideal) y tienen hipoxemia e hipercapnia, en parte por bloqueo de la respuesta ventilatoria a estos estímulos.16 Algunos de estos pacientes tienen también un componente de apnea obstructiva del sueño (ver adelante). La hipoxemia proporciona el estímulo para la mayor producción de eritropoyetina y la policitemia. Otras características clínicas asociadas con el síndrome de obesidad-hiperventilación son la hipersomnolencia nocturna y el cor pulmonale. El tratamiento puede incluir pérdida de peso y tratamiento con progesterona para mejorar el estímulo respiratorio central.
Apnea obstructiva del sueño
Se calcula que el síndrome de apnea obstructiva del sueño se presenta en el cuatro y dos porciento de los varones y mujeres de edad media, respectivamente.17 El síndrome consiste en episodios recurrentes de colapso de las vías respiratorias superiores durante el sueño, que obstruyen el paso del aire, con lo que se presenta hipoxemia nocturna intermitente.18 Puede haber historia de ronquidos que alternan con periodos de silencio que duran de 10 segundos a un minuto, seguido de ruidos de ahogamiento. La fragmentación del sueño causa somnolencia diurna excesiva y menor desempeño laboral. El hematocrito puede estar un poco aumentado,19 y debe pensarse en esta posibilidad en los pacientes con policitemia inexplicable.20 La polisomnografía nocturna puede establecer el diagnóstico. El tratamiento puede incluir pérdida de peso, presión positiva continua a las vías respiratorias por vía nasal y cirugía18,21,22 .
Aumento en los niveles de carboxihemoglobina
La exposición crónica a monóxido de carbono causa niveles altos de carboxihemoglobina. El monóxido de carbono se une a la hemoglobina con una afinidad 210 veces mayor que la del oxígeno, disminuyendo así la cantidad de hemoglobina disponible para transportar oxígeno.23 La unión de monóxido de carbono aumenta también la afinidad del resto de los grupos hem por el oxígeno, desplazando la curva de disociación oxígeno-hemoglobina hacia la izquierda [ver figura 2] y disminuyendo la liberación de oxígeno en los tejidos.23 Es por estos mecanismos que la exposición crónica a monóxido de carbono puede causar policitemia. Los fumadores de puro y cigarrillos24 y las personas que tienen exposición ocupacional crónica a gases automotrices en áreas mal ventiladas (v.gr., trabajadores de estacionamientos cubiertos, cargadores de trailers) tienen mayor riesgo.
Los síntomas pueden incluir alteraciones neuropsiquiátricas sutiles y exacerbación de la angina de pecho (probablemente por menor aporte de oxígeno al miocardio). El diagnóstico se establece midiendo el porcentaje de carboxihemoglobina en sangre. Debido a que la vida media de la carboxihemoglobina es de alrededor de cuatro horas, la prueba debe hacerse tarde durante el día, cuando el paciente ha fumado su número habitual de cigarrillos o ha pasado varias horas en el ambiente laboral. En un estudio de 22 fumadores con policitemia, la concentración promedio de carboxihemoglobina fue de 11.6 porciento, cuando el valor normal debe ser menor del uno porciento.24 Los fumadores policitémicos suelen tener tanto aumento en la masa eritrocitaria como disminución del volumen plasmático.24 Estas alteraciones revierten a los tres meses de suspender el tabaquismo.24,25
AUMENTO INAPROPIADO EN LA PRODUCCION DE ERITROPOYETINA
Las policitemias también pueden deberse a aumento en la producción de eritropoyetina como respuesta inadecuada a enfermedades renales y, con menos frecuencia, hepáticas. En la vida adulta, alrededor del 90 porciento de la producción de eritropoyetina ocurre en el riñón y el 10 porciento en el hígado. Los mecanismos moleculares de producción no se conocen con exactitud. Sin embargo, se sabe que una proteína hem que fija oxígeno26 en el riñón detecta la hipoxia tisular, lo que provoca aumento en la producción de eritropoyetina por las células intersticiales peritubulares del riñón.27 La producción aumenta por el reclutamiento de células intersticiales peritubulares adicionales para producir eritropoyetina.28 Debido a la complicada regulación de la producción de eritropoyetina en el riñón, la distorsión de la anatomía renal puede causar policitemia. Existen reportes de casos de policitemia asociada a quistes renales, hidronefrosis, glomerulonefritis focal y síndrome de Bartter.29-32 Alrededor del 10 porciento de los pacientes tienen policitemia temporal o persistente después del trasplante renal. Además, las neoplasias primarias del riñón o del hígado pueden provocar policitemia. Se desarrolla policitemia en alrededor del tres porciento de los pacientes con carcinoma de células renales. Se ha demostrado producción de eritropoyetina por las células de carcinomas renales primarios y de hepatomas.26,33,34 En casos raros los hemangiomas del hígado o cerebelo, los fibroides uterinos, los adenomas suprarrenales y los feocromocitomas pueden causar policitemia.35
HIPERSENSIBILIDAD A LA ERITROPOYETINA (POLICITEMIA FAMILIAR CONGENITA)
Se han descrito y estudiado en forma extensa varias familias con policitemia autosómica dominante. En algunas de estas familias se encontró alteración en el receptor de eritropoyetina36,37 o en la regulación de la producción de la misma.38,39 En otras familias no se ha identificado aún el mecanismo preciso que explica la policitemia.40
La eritropoyetina inicia sus efectos biológicos al unirse a un receptor específico41 que se encuentra en la superficie de las células progenitoras eritroides, las células precursoras y varios otros tipos celulares. La unión con la eritropoyetina desencadena una cascada de eventos, incluyendo la asociación y activación de la proteína-tirosina cinasa JAK2 [ver figura 3].42 La fosforilación tirosina del receptor de eritropoyetina crea sitios de ensamble para otras moléculas de transducción de señales y para la proteína tirosina fosfatasa SH-PTP1, que desfosforila a JAK2 y termina la transducción de señales.43 En una familia finlandesa extensa con policitemia se encontró una mutación puntiforme en el receptor de la eritropoyetina que causó ruptura de 70 aminoácidos en el extremo carboxilo, incluyendo en sitio en el que se une la SH-PTP1 para terminar la señal de transducción.36 Esta mutación vuelve a las células progenitoras eritroides hipersensibles a la eritropoyetina, y pudo contribuir a la obtención de tres medallas de oro en esquí a campo traviesa que obtuvo en los Juegos Olímpicos de 1964 un miembro de esta familia que tenía un hematocrito del 60 porciento.44 En otra familia bien estudiada, una mutación de marco en el receptor de eritropoyetina causó la ruptura del sitio de unión de la SH-PTP1, con hipersensibilidad a la eritropoyetina y policitemia.37
La medición de las concentraciones séricas de eritropoyetina ha sugerido que en algunas familias existe una alteración primaria en el punto umbral del mecanismo censor de oxígeno en el riñón.38,39 En estas familias no pudo encontrarse alteración en la afinidad de la hemoglobina por el oxígeno y los niveles de eritropoyetina eran inapropiadamente altos para el hematocrito.
USO DE MEDICAMENTOS
El uso de andrógenos, como la testosterona, puede asociarse con policitemia.45 Los andrógenos estimulan la producción de eritropoyetina por el riñón. El aumento en el hematocrito suele ser leve, y éste se normaliza dos a tres meses después de suspender los anabólicos. Debido a que en la actualidad se cuenta con eritropoyetina humana recombinante, ha surgido la preocupación de que los atletas de competencia que participan en deportes de resistencia, como ciclismo, esquí a campo traviesa y carrera de fondo, puedan inyectarse esta sustancia para mejorar su rendimiento.46 Se ha demostrado que la reinfusión de sangre autóloga justo antes de una carrera acorta los tiempos en las carreras de 10 km,47 y varios otros estudios han encontrado que la flebotomía seguida de doping con sangre puede mejorar el rendimiento de corredores y esquiadores.48 Puede lograrse un incremento semejante en el hematocrito empleando inyecciones de eritropoyetina. Existe expeculación sobre si la muerte súbita de varios ciclistas de competencia entre 1987 y 1990 se relacionó con el abuso de eritropoyetina.49,50 El incremento no vigilado en la producción de eritrocitos puede causar policitemia importante que, aunada a la deshidratación inducida por el ejercicio, puede causar consecuencias trágicas. Este uso de la eritropoyetina no es ético, es riesgoso y está prohibido por el Comité Olímpico Internacional.
Policitemia vera
La policitemia vera es un trastorno neoplásico que se origina en una célula tronco hematopoyética pluripotencial. Ocurre en alrededor de cinco a 10 personas por millón (0.0005 a 0.0010 porciento),51 es ligeramente más común en hombres que en mujeres (relación hombre: mujer de 1.2:1) y alcanza su incidencia máxima entre los 50 a 75 años de edad.52,53 Sin embargo, alrededor del cinco porciento de los casos se diagnostican en pacientes menores de 40 años.52,53
FISIOPATOLOGIA
En un adulto normal la hematopoyesis es policlonal, de modo que las células que circulan en la sangre periférica derivan de múltiples células tronco. Sin embargo, en los pacientes con policitemia vera muchos o todos los eritrocitos, leucocitos y plaquetas circulantes derivan de una célula tronco neoplásica. Esto se demostró en estudios de mujeres con policitemia vera que eran también heterocigotas para la enzima glucosa-6-fosfato-deshidrogenasa (G/PD), que se codifica en el cromosoma X.54 En estas pacientes, los neutrófilos, eritrocitos y plaquetas circulantes mostraban solo una de las dos isoenzimas de G6PD, lo que indicaba que las células se originaban de una sola célula tronco, mientras que los fibroblastos de la piel contenían proporciones balanceadas de ambas isoenzimas de G6PD A y B.54 Recientemente, el análisis de polimorfismo de ADN ligado al X ha demostrado también la naturaleza clonal de la policitemia vera.55
La policitemia vera es un padecimiento de un grupo de padecimientos relacionados de las células tronco denominados trastornos mieloproliferativos, todos de origen clonal. En éstos se incluyen la leucemia mieloide crónica (LMC), la trombocitemia esencial y la metaplasia mieloide agnogénica. Las características predominantes de la LMC son leucocitosis dramática, presencia de cromosoma Philadelphia y evolución a leucemia mieloide aguda (LMA) o a leucemia linfocítica aguda. La trombocitemia esencial se distingue por la trombocitosis predominante sin eritrocitosis o alteraciones citogenéticas de LMC. La metaplasia mieloide agnogénica se caracteriza por esplenomegalia masiva acompañada de anemia y un frotis de sangre periférica leucoeritroblástico.
La fisiopatología de la policitemia vera aún no se conoce del todo.56 En los adultos normales las células eritroides progenitoras no proliferan en ausencia de eritropoyetina exógena.57 Sin embargo, se ha demostrado que las células progenitoras eritroides de los pacientes con policitemia vera desarrollan colonias in vitro incluso cuando no se agrega eritropoyetina al cultivo.57,58 Estas observaciones forman la base para una de las pruebas diagnósticas de la policitemia vera, el ensayo endógeno de colonias eritroides.59 Los estudios han demostrado que las células eritroides progenitoras de los pacientes con policitemia vera son hipersensibles a los efectos de múltiples citocinas promotoras del crecimiento, incluyendo interleucina-3 y factor de crecimiento semejante a insulina-1.60,61 Debido a estos hallazgos y al hecho de que en la policitemia vera pueden afectarse varias líneas celulares, parece poco probable que la mutación en el receptor de una citocina de acción tardía (v.gr., eritropoyetina) pueda explicar la fisiopatología de esta enfermedad.
MANIFESTACIONES CLINICAS Y DIAGNOSTICO
Durante la década de los 50 y 60, un grupo de pacientes con policitemia vera no tratados tuvieron una supervivencia promedio de alrededor de 18 meses, y la mayoría de las defunciones fueron causadas por eventos cerebrovasculares.62 Sin embargo, en la actualidad la supervivencia promedio de los pacientes tratados es de 10 a 15 años.63,64 Debido a que la policitemia vera se diagnostica con más frecuencia en los pacientes entre la séptima y la octava década de la vida, la expectancia de vida puede no diferir de la del grupo de población de la misma edad.64 Un estudio de pacientes jóvenes con policitemia vera (menores de 40 años) mostró que más del 70 porciento sobrevivió 15 años después del diagnóstico.65
La historia natural de la policitemia vera incluye una fase latente protrombótica relativamente asintomática,53 una fase clínica proliferativa, durante la que se expande la masa de eritrocitos y el paciente puede tener síntomas de hiperviscosidad o hipermetabolismo [ver tabla 1] o trombosis, una fase de deterioro (i.e., metaplasia mieloide pospolicitemia), que se caracteriza por disminución en la producción de eritrocitos y desarrollo de anemia, leucopenia, trombocitopenia, fibrosis medular y hepatoesplenomegalia progresiva. Eventualmente se desarrolla metaplasia mieloide en alrededor del 15 al 20 porciento de los pacientes. La LMA es una complicación natural de la policitemia vera, desarrollándose en alrededor del uno a dos porciento de los pacientes tratados solo con flebotomía,66 y también es una complicación de ciertos tratamientos per se [ver adelante, Complicaciones].
Síntomas
Aunque ciertos síntomas son comunes a todas las policitemias, otros son más específicos de la policitemia vera52 [ver tabla 1]. Muchos pacientes policitémicos tienen cefalea o cambios cognoscitivos vagos, incluyendo cambios en la memoria e incapacidad para realizar cálculos mentales rápidos. Son frecuentes la debilidad y el mareo. El prurito después de un baño o regaderazo caliente es más específico de policitemia vera y puede ser muy molesto. Con menos frecuencia los pacientes con este padecimiento tienen síntomas de hipermetabolismo, incluyendo fiebre, pérdida de peso y sudoración excesiva. La eritromelalgia, el dolor quemante episódico severo y el eritema en dedos de manos y pies se explican por isquemia y sugieren un trastorno mieloproliferativo. Alrededor del 20 porciento de los pacientes con policitemia vera han tenido un evento trombótico arterial o venoso premonitorio.53 En algunos casos los pacientes con policitemia vera están asintomáticos.
Examen físico
El examen físico suele mostrar plétora facial, que es común en todos los pacientes con policitemia real (i.e., primaria o secundaria). Se encuentra esplenomegalia en el 70 porciento de los pacientes con policitemia vera,52 que se debe a hematopoyesis extramedular junto con congestión esplénica por eritrocitos, ya que el bazo es la tumba de estas células. Existe hepatomegalia en el 40 porciento de los casos, que también puede deberse a eritropoyesis extramedular y, en ocasiones, al síndrome de Budd-Chiari. Una tercera parte de los pacientes con policitemia vera tienen hipertensión.
Diagnóstico de laboratorio
La citología hemática completa de los pacientes que presentan policitemia vera de novo suele mostrar elevación del hematocrito. Un hematocrito de 60 o mayor en un hombre, o de 57 o mayor en una mujer, es casi diagnóstico de policitemia primaria o secundaria. Sin embargo, debido a la alta prevalencia de pérdida de sangre por la vía gastrointestinal en los pacientes con policitemia vera, algunos tienen hematocrito normal. El volumen corpuscular medio de los eritrocitos puede estar disminuido, lo que refleja deficiencia de hierro. Se encuentra leucocitosis en el 45 porciento de los pacientes en el momento del diagnóstico.52 El frotis de sangre periférica puede mostrar basofilia discreta y en ocasiones leucocitos inmaduros en la circulación, incluyendo mielocitos y metamielocitos. Alrededor del 65 porciento de los pacientes con policitemia vera tienen trombocitosis.52 Los principales diagnósticos diferenciales en el paciente con un hematocrito normal, trombocitosis y volumen corpuscular promedio bajo es la policitemia vera con deficiencia de hierro, la trombocitemia esencial o la trombocitosis causada por la deficiencia de hierro en sí. Si el paciente tiene buenas reservas de hierro el diagnóstico más probable es trombocitopenia esencial. La trombocitosis causada por deficiencia de hierro suele responder a la repleción del mismo.
Otros datos de laboratorio incluyen elevación de la concentración de ácido úrico en el 30 a 50 porciento de los pacientes con policitemia vera, causada por producción aumentada en forma crónica y destrucción de los eritrocitos.67 La concentración de vitamina B12 y de proteína fijadora de vitamina B12 suele estar elevada. El nivel de fosfatasa alcalina leucocitaria (FAL) se encuentra aumentado en el 70 porciento de los pacientes con policitemia vera, a diferencia de los pacientes con LMC, en quienes la concentración de FAL está disminuida.
El nivel de eritropoyetina está disminuido como resultado de mecanismos de retroalimentación68 [ver figura 4]. Sin embargo, en algunos pacientes pueden ser normales.68 La eritropoyetina sérica no debe medirse después de una flebotomía vigorosa porque al reducir el hematocrito aumentará la producción de eritropoyetina.
En la policitemia vera no se requiere evaluación de la médula ósea, ni tiene especial utilidad. Los datos de la médula suelen incluir aumento en la celularidad (promedio de 82 porciento), en el número de megacariocitos y bajas reservas de hierro.69 También puede existir aumento en las fibras de reticulina.69
Se encuentran alteraciones citogenéticas en el momento del diagnóstico en alrededor del 10 a 15 porciento de los pacientes con policitemia vera, que comunmente incluyen trisomía 8, 9 y deleción del brazo largo del cromosoma 20.70,71 La presencia de alteraciones citogenéticas en el momento del diagnóstico no identifica a pacientes con mayor riesgo de LMA o que puedan progresar a metaplasia mieloide.70,71 No se realizan estudios citogenéticos de rutina en los pacientes con probable diagnóstico de policitemia vera a menos que exista dificultad para distinguir esta enfermedad de una LMC. En ese caso pueden evaluarse los leucocitos de sangre perif por medio de Southern blot en busca del rearreglo genético bcr-abl, que es característico de LMC.
En 1967, el Polycythemia Vera Study Group (PVSG, Grupo de estudio de la policitemia vera de los EUA, n. del t.) estableció un grupo de criterios diagnósticos para incluir a los pacientes en estudios clínicos multicéntricos.52 Estos criterios se desarrollaron antes de contar con las determinaciones de eritropoyetina en suero y de ensayos de colonias eritroides endógenos. Además, los criterios intentaban ser estrictos, por lo que con frecuencia excluyen a pacientes con policitemia vera temprana que no han desarrollado aún esplenomegalia, leucocitosis o trombocitosis significativas. En un paciente que no satisface los criterios estrictos del PVSG, el encontrar una concentración baja de eritropoyetina puede proporcionar evidencia adicional para apoyar el diagnóstico [ver tabla 2]. La presencia de colonias eritroides endógenas en sangre periférica también es un dato a favor del diagnóstico de policitemia vera [ver tabla 2].
TRATAMIENTO
Existen muchas opciones terapéuticas para la policitemia vera, pero no un consenso sobre el mejor esquema de tratamiento. Este debe individualizarse, tomando en consideración la edad del paciente y su perfil de riesgo de trombosis, además de los riesgos inherentes a cada tipo de manejo.
Para la mayoría de los pacientes el tratamiento inicial debe incluir la flebotomía. En general, se eliminan 500 ml de sangre una o dos veces por semana hasta que se alcanza un valor de menos de 46 porciento. Después se realizan flebotomías periódicas para mantener el hematocrito entre 40 y 45 porciento. En muchos pacientes sometidos a flebotomías se desarrolla deficiencia de hierro, lo que limita la capacidad para producir eritrocitos y reduce la frecuencia de la flebotomía. El paciente debe evitar los multivitamínicos que contienen hierro. Existe controversia sobre si los eritrocitos hipocrómicos, microcíticos, deficientes en hierro tienen un comportamiento hemorreológico que aumenta la viscosidad de la sangre y quizá un poco el riesgo de trombosis.72,73 Por ello, es probable que sea adecuado evitar la inducción de microcitosis severa. En ocasiones los pacientes pueden desarrollar síntomas atribuibles a la deficiencia de hierro, incluyendo pica,74 glositis y fatiga muscular inducida por ejercicio, que responden a la repleción de este mineral. Sin embargo, en la mayoría de los casos es conveniente mantener una deficiencia moderada de hierro causada por las flebotomías porque esto ayuda a mantener el hematocrito en los niveles adecuados.
Los pacientes que tienen riesgo alto de trombosis (v.gr., mayores de 70 años, que han tenido un evento trombótico previo y que requieren flebotomías con gran frecuencia) deben manejarse mejor con un agente mielosupresor, como la hidroxiurea,75 aunado a la flebotomía. Se ha demostrado que la hidroxiurea (en dosis de 500 mg a 1.5 g vía oral diario) y la flebotomía pueden controlar en forma más eficaz el hematocrito en la mayoría de los pacientes con policitemia vera, comparado con la flebotomía sola. La hidroxiurea suele ser bien tolerada; sin embargo, su uso requiere vigilancia frecuente de la citología hemática para evitar una mielosupresión excesiva. También es motivo de controversia si el uso de hidroxiurea aumenta el riesgo de desarrollar leucemia mieloide aguda. En 100 pacientes con policitemia vera que recibieron hidroxiurea durante un promedio de 5.4 años se desarrollo leucemia mieloide aguda con una incidencia baja (uno porciento).77 Sin embargo, una publicación reciente encontró una incidencia de 7.8 porciento de leucemia mieloide aguda en los pacientes tratados con hidroxiurea, lo que es significativamente más alto que la incidencia de uno a dos porciento que se encuentra solo con las flebotomías.78
El busulfán, un agente alquilante, puede ser un mielosupresor alternativo también de administración por vía oral. Un estudio aleatorio extenso sugirió que el riesgo de desarrollar LMA en pacientes tratados con busulfán es semejante al de pacientes tratados con fósforo radioactivo (P32).79 Los riesgos a largo plazo del busulfán incluyen fibrosis pulmonar y daño a las células tronco hematopoyéticas, lo que puede causar supresión medular por tiempo prolongado. Debido a su toxicidad, el busulfán suele reservarse para pacientes que no mejoran con hidroxiurea.
El tratamiento intermitente con P32 (2.3 mCi/m2 I.V. cada 12 semanas según se requiera para controlar el hematocrito, con dosis máxima de 5 mCi) suele permitir un control paulatino del hematocrino, no requiere de vigilancia frecuente y es muy cómodo para los pacientes. Sin embargo, aumenta el riesgo de desarrollar LMA (ver adelante). Este tipo de tratamiento puede ser adecuado para pacientes con policitemia vera de edad avanzada.
El clorambucil se asocia con mayor riesgo de LMA de inicio temprano,80 por lo que no se recomienda para el tratamiento de la policitemia vera.
Comparado con los pacientes tratados con un agente mielosupresor, los que se manejan solo con flebotomías tienen un mayor riesgo de trombosis durante los primeros tres años después del inicio del tratamiento.63 Por este motivo se estudió si la administración de aspirina, 300 mg tres veces al día más dipiridamol (75 mg tres veces al día) aunado a la flebotomía disminuía el riesgo de trombosis, comparando con el tratamiento con P32. Se demostró que la adición de aspirina en esta dosis relativamente alta no evitó las trombosis y aumentó el riesgo de hemorragia gastrointestinal importante.81 Por lo tanto, no puede recomendarse el uso de aspirina a esas dosis como tratamiento inicial de la policitemia vera.
COMPLICACIONES
El periodo posoperatorio es especialmente peligroso para los pacientes con policitemia vera no controlada. En un estudio, el 79 porciento de los pacientes con policitemia vera no controlada (hematocrito mayor de 52 porciento en el momento de la cirugía) tuvieron complicaciones posoperatorias, principalmente hemorragia o trombosis.82 La tasa de complicaciones puede minimizarse logrando un buen control del hematocrito durante por lo menos cuatro meses antes de la cirugía electiva.82
Trombosis
La trombosis es la complicación más frecuente de la policitemia vera. Varias características de este padecimiento parecen contribuir a la mayor incidencia. El valor del hematocrito se relaciona en forma directa con el riesgo de trombosis: al aumentar éste, disminuye el flujo sanguíneo cerebral,83 y aumenta la incidencia de complicaciones trombóticas.84 Esta asociación entre el hematocrito y el riesgo de trombosis forma las bases para una recomendación clave en el tratamiento de la policitemia vera: el hematocrito debe mantenerse en menos de 46 porciento para disminuir el riesgo de trombosis.62 Las plaquetas derivan de una clona neoplásica y pueden no funcionar de modo normal,54 lo que facilita la ocurrencia de trombosis o hemorragia clínica.85 Son comunes las equímosis, la epistaxis y la hemorragia gastrointestinal. Los estudios de agregometría plaquetaria ex vivo demuestran defectos en la agregación, sobre todo en respuesta a la epinefrina.86 Aunque suele haber exceso de plaquetas en la policitemia vera, no existe una relación clara entre la cuenta absoluta de plaquetas y el riesgo de trombosis.85
En el estudio clínico inicial realizado por el PVSG, 431 pacientes recibieron tratamiento con flebotomía sola, P32 o clorambucil para mantener el hematocrito por debajo del 45 porciento.63 En la actualización más reciente de este estudio, el 35 porciento de los pacientes sufrió trombosis.75 Los pacientes con policitemia vera tienen mayor riesgo de trombosis tanto arterial como venosa53,65 [ver tabla 3]. Son problemas clínicos comunes el infarto cerebral, el infarto al miocardio, la trombosis de venas profundas en miembros inferiores, los episodios de isquemia cerebral transitoria y la oclusión arterial periférica. Los pacientes suelen notar exacerbación de su angina conforme aumenta el hematocrito.
Puede ocurrir trombosis de la vena hepática, causando síndrome de Budd-Chiari. En un estudio de 20 pacientes con síndrome de Budd-Chiari, el 80 porciento tuvo colonias eritroides endógenas, y es posible que su trombosis de venas hepáticas haya sido causada por un trastorno mieloproliferativo subyacente.87 Solo dos de estos pacientes tuvieron un trastorno mieloproliferativo evidente. Sin embargo, la baja incidencia del síndrome de Budd-Chiari durante los muchos años de seguimiento del primer estudio del PVSG va en contra de que éste sea una complicación frecuente de la policitemia vera.
Tratamiento Los pacientes con policitemia vera que tienen un evento trombótico agudo y trombocitosis deben someterse a aféresis de plaquetas para reducir con rapidez la cuenta plaquetaria hasta el rango normal. Después debe iniciarse tratamiento inmunosupresor (v.gr., hidroxiurea) para ayudar a mantener una cuenta de plaquetas normal, a pesar de que no exista una relación clara entre la cuenta alta de plaquetas y el riesgo de trombosis. Si el paciente no tiene historia de hemorragia excesiva, debe considerarse también la adición de un agente que inhiba la función plaquetaria (v.gr., aspirina 81 mg/día). Los pacientes con hemorragias de mucosas y trombocitosis deben recibir un agente mielosupresor para disminuir la cuenta de plaquetas al rango normal.
Leucemia mieloide aguda
Una de las complicaciones más temidas de la policitemia vera es la evolución a LMA. La incidencia de LMA en los pacientes manejados con flebotomía sola es de uno a dos porciento (100 veces más que la incidencia en la población adulta de edad semejante). El estudio inicial del PVSG encontró que el tratamiento con clorambucil o P32 se asocia con mayor riesgo de desarrollar LMA.63,80 El inicio de la LMA en los pacientes tratados con clorambucil fue temprano, y la mayoría de los casos ocurrieron menos de cinco años después del inicio del tratamiento. Eventualmente, el 14 porciento de los pacientes desarrollaron LMA.75 Además, el riesgo de desarrollar linfoma no Hodgkin aumentó en los pacientes tratados con clorambucil. En los enfermos que recibieron P32 el inicio de la LMA fue relativamente tardío, y la mayoría de los casos ocurrieron seis a 10 años después del inicio del tratamiento, con una incidencia acumulada de 9.6 porciento.75 Puede ocurrir evolución a mielodisplasia en algunos casos de policitemia vera, y una serie de reportes de casos sugiere que el uso de tratamiento mielosupresor puede aumentar también el riesgo.88 El tratamiento con clorambucil o P32 se asoció también con mayor riesgo de desarrollar una neoplasia no hematológica.75
Tratamiento Si la policitemia vera evoluciona a LMA la posibilidad de lograr una remisión completa y duradera con quimioterapia de inducción estándar es muy baja.75 En estos pacientes se justifica discutir en forma franca las opciones terapéuticas, incluyendo los riesgos y beneficios de la quimioterapia de inducción contra el tratamiento de sostén, en especial porque los pacientes son ancianos y pueden tener otros padecimientos concomitantes.
Gota y nefrolitiasis
Debido a la hiperuricemia los pacientes con policitemia vera tienen mayor riesgo de desarrollar gota o nefrolitiasis67.
Metaplasia mieloide
Alrededor del 15 a 20 porciento de los pacientes con policitemia vera desarrollan metaplasia mieloide pospolicitémica,75,89,90 o la llamada fase involutiva o de deterioro de los trastornos mieloproliferativos. La metaplasia mieloide se caracteriza por remplazo insidioso del tejido medular por reticulina fibrosa, y por movimiento gradual de la hematopoyesis de la médula al bazo, el hígado y en ocasiones otros órganos. El bazo e hígado crecen en forma progresiva y pueden adquirir un tamaño enorme. Los pacientes sufren pancitopenia, en parte porque estos órganos son menos eficaces para la hematopoyesis y al parecer también por alteraciones intrínsecas de las células tronco hematopoyéticas. Se encuentran en la circulación eritrocitos en gota y una plétora de células mieloides, lo que causa el frotis leucoeritroblástico de sangre periférica característico de la metaplasia mieloide. Los pacientes con metaplasia mieloide pueden tener dolor esplénico e infartos por la esplenomegalia masiva, y presentar saciedad temprana, con pérdida subsecuente de peso. La fibrosis en la médula de los pacientes con metaplasia mieloide no es parte de la clona neoplásica. En lugar de ello, la fibrosis es reactiva y puede ser inducida por citocinas como el factor de crecimiento derivado de plaquetas, liberado por los megacariocitos anormales. La supervivencia de los pacientes con metaplasia mieloide es corta, alrededor de dos a tres años.
Tratamiento El tratamiento de la metaplasia mieloide incluye manejo de sostén con transfusiones, según se requiera, y uso juicioso de hidroxiurea o busulfán para disminuir el tamaño del bazo. Puede considerarse realizar esplenectomía en los pacientes con esplenomegalia masiva y pérdida de peso por saciedad temprana, dolor por infarto esplénico o trombocitopenia exacerbada por el secuestro de plaquetas en un bazo con crecimiento masivo. La esplenectomía es un gran reto en estos pacientes, y por lo general es seguida de hepatomegalia progresiva. En la actualidad no existen evidencias convincentes de que pueda lograrse regresión de la fibrosis medular con hidroxiurea, P32 o interferón alfa.
Hipermetabolismo y prurito
Los síntomas de hipermetabolismo (v.gr., calosfríos, diaforesis y fiebre no debida a infección) y el prurito suelen responder a la hidroxiurea y a otros agentes mielosupresores. En un paciente con prurito puede ser útil evitar los eventos exacerbantes (v.gr., baños calientes) y administrar bloqueadores de los receptores H1 y H2.
TRATAMIENTOS EXPERIMENTALES
El anagrelide es un vasodilatador activo que inhibe la maduración de los megacariocitos, disminuyendo así la cuenta plaquetaria. En un estudio de más de 400 pacientes con trombocitosis causada por un trastorno mieloproliferativo, se encontró que el anagrelide redujo la cuenta plaquetaria en el 93 porciento, por lo general en dos semanas.91 Los efectos adversos más frecuentes de este medicamento fueron retención hídrica, palpitaciones, cefalea, náusea y diarrea. El anagrelide puede ser útil especialmente en los pacientes con policitemia vera que requieren un agente suplementario para el control óptimo de la trombocitosis. Este medicamento es revisado en la actualidad por la Food and Drug Administration para usarse en la trombocitosis asociada a trastornos mieloproliferativos.
El interferón alfa es un agente de administración parenteral que puede inducir remisión citogenética en un porcentaje de pacientes con LMC. Debido a que la policitemia vera, al igual que la LMC, es un trastorno mieloproliferativo de tipo clonal, se ha postulado que el interferón alfa puede causar también regresión de la clona neoplásica en este padecimiento. Los estudios de pacientes tratados con interferón alfa hasta por seis años han demostrado que el interferón alfa, administrado tres veces por semana, puede controlar en forma eficaz el hematocrito y la cuenta plaquetaria, además de disminuir el tamaño del bazo.92 No se diagnosticaron eventos trombóticos en los pacientes tratados con interferón alfa, a diferencia de la alta incidencia de trombosis en los pacientes con policitemia vera tratados solo con flebotomía.63 Debido a que solo una pequeña fracción de estos pacientes tenía una alteración citogenética, es menos claro cuál fue el efecto del interferón alfa sobre la clona neoplásica. Los efectos adversos de este agente incluyen síntomas de tipo gripal severo que pueden disminuir después de uno a dos meses de tratamiento. Otros problemas incluyen un inicio de acción lento (en meses) y el costo tan alto. Sin embargo, si puede demostrarse que el interferón alfa induce regresión de la clona neoplásica93 o impide la progresión a metaplasia mieloide, sus beneficios superarán sus desventajas. Se está planeando un estudio que comparará el uso de flebotomía, hidroxiurea e interferón alfa, y que ayudará a aclarar el papel de este último en el tratamiento de la policitemia vera.
También se ha investigado la utilidad del trasplante alogénico de médula ósea (que en la actualidad es el tratamiento de elección en los pacientes con LMC) para el tratamiento de la policitemia vera, a pesar de que esta última tiene una evolución más indolente. Solo algunos pacientes con policitemia vera han sido sometidos a tratamiento mieloablativo y trasplante alogénico. Este enfoque puede ser conveniente para pacientes jóvenes con evidencia temprana de fibrosis medular.
Bibliografía
DRA. VIRGINIA C. BROUDY
Clasificación de las policitemias
La policitemia, también denominada eritrocitosis, es el aumento en el número de eritrocitos circulantes por volumen de sangre, que se refleja por aumento del hematocrito o de la concentración de hemoglobina. Existen tres categorías principales de policitemia: (1) policitemia relativa, (2) policitemia secundaria, y (3) policitemia primaria, o policitemia vera.
En la policitemia relativa la masa eritrocitaria es normal, pero el volumen del plasma está disminuido. La policitemia secundaria está causada por elevación en la concentración sérica de eritropoyetina y puede deberse a diversos trastornos, por ejemplo la presencia de hemoglobina de alta afinidad que no oxigena los tejidos con eficacia, causando hipoxia tisular y mayor producción de eritropoyetina. La policitemia vera es un trastorno neoplásico de los eritrocitos caracterizado por sobreproducción autónoma de eritrocitos, y con frecuencia de leucocitos y plaquetas.
Evaluación inicial
El aumento en la concentración de hemoglobina o del hematocrito justifica una evaluación inmediata para investigar la naturaleza de la policitemia y su causa posible [ver figura 1]. Debe realizarse una historia clínica y examen físico, investigando el antecedente familiar de policitemia y los resultados de los hematocritos durante los años previos. Cuando es posible deben buscarse también los valores de hematocrito durante la adolescencia o el inicio de la edad adulta. Son importantes los datos que sugieran enfermedad cardiaca congénita, enfermedad pulmonar obstructiva crónica (EPOC) severa o síndrome de apnea del sueño, y se investigará la presencia o ausencia de esplenomegalia. Los resultados de una biometría hemática completa son indispensables, incluyendo la cuenta de plaquetas y diferencial de leucocitos. La presencia de leucocitosis, trombocitosis, leucocitos circulantes inmaduros o aumento en los basófilos, son sugestivos de policitemia vera y van en contra de causas secundarias de eritrocitosis.

|
| Figura 1 |
| Evaluación de las policitemias |
Un hombre con un hematocrito de 60 porciento o más1 o una mujer con hematocrito de 57 porciento o más, casi siempre tienen un aumento verdadero en la masa eritrocitaria (i.e., policitemia primaria o secundaria). Si el hematocrito del paciente está por debajo de esos valores pero sobre lo normal, debe hacerse un estudio de la masa eritrocitaria para definir si está en realidad aumentada. Para realizar este estudio se marca una muestra de los eritrocitos del paciente con cromo radioactivo (Cr51) ex vivo y se reinyecta al paciente. Se obtiene después una segunda muestra para cuantificar la concentración de eritrocitos marcados entre los eritrocitos no marcados. En forma paralela, se administra al paciente una inyección de albúmina marcada con ioduro radioactivo (I125) para medir el volumen plasmático. Los resultados de la determinación de la masa eritrocitaria marcada con Cr51 identifican si el paciente tiene policitemia relativa por contracción del volumen plasmático o policitemia verdadera (i.e., primaria o secundaria) por aumento en la masa de eritrocitos. Una vez que se demuestra un aumento relativo o absoluto en la masa de eritrocitos debe determinarse el diagnóstico exacto [ver figura 1].
Policitemia relativa
La policitemia relativa también se conoce como síndrome de Gaisböck por la primera descripción clínica. Los pacientes con policitemia relativa suelen ser varones de edad media, obesos, e hipertensos, en ocasiones fumadores intensos.2,3 Con frecuencia inician los síntomas a los 45 a 55 años de edad, una década menos que la típica para policitemia vera [ver adelante, Policitemia vera]. En la mayoría de las series el 80 a 90 porciento de los pacientes con policitemia vera son varones.2,3 Se ha calculado que el 0.5 a 0.7 porciento de la población masculina normal en los Estados Unidos tiene policitemia relativa.4 El uso de diuréticos para el tratamiento de la hipertensión puede exacerbar la deficiencia en el volumen plasmático, y los niveles altos de carboxihemoglobina inducidos por el tabaquismo o la hipoxemia también pueden influir. La policitemia relativa suele ser leve (hematocrito menor de 55 porciento). Es importante tener en cuenta el diagnóstico y medir la masa eritrocitaria y el volumen plasmático en pacientes con hematocrito menor del 60 porciento [ver figura 1] para evitar una investigación extensa y al final frustrante de otras causas de policitemia. Los pacientes con policitemia relativa pertenecen a dos grupos principales: (1) los que tienen masa eritrocitaria normal y disminución clara en el volumen plasmático y (2) los que tienen masa eritrocitaria en el rango superior del normal y volumen plasmático en el límite inferior normal.4 El hematocrito se normaliza después de cierto tiempo en alrededor de la tercera parte de los pacientes.
Policitemia secundaria
AUMENTO APROPIADO EN LA PRODUCCION DE ERITROPOYETINA
Puede ocurrir policitemia cuando la producción de eritropoyetina aumenta como resultado de una compensación adecuada en presencia de hipoxia tisular generalizada. Ocurre hipoxia tisular con la vida a altitud elevada, la presencia de hemoglobina de alta afinidad, la enfermedad cardiopulmonar, el síndrome de obesidad-hipoventilación, la apnea obstructiva del sueño y los niveles elevados de carboxihemoglobina.
Vida a altitud elevada
En 1890 el médico francés Viault visitó un pueblo pequeño en los Andes a 15,000 pies de altura, y realizó la observación histórica de que su cuenta eritrocitaria aumentó de 5 x 106 células/mm3 a 8 x 106 células/mm3 durante un periodo de varias semanas.5 Después se observó que el hematocrito de las personas que viven a mayor altura es un poco mayor que el de los que viven a nivel del mar. La adaptación humana inicial a la altitud incluye aumento en la frecuencia respiratoria, en el gasto cardiaco y en la concentración de 2,3-difosfoglicerato para facilitar la liberación de oxígeno de la hemoglobina en los tejidos [ver figura 2]. Seis a 24 horas después de que la persona asciende aumenta la concentración de eritropoyetina, lo que causa reticulocitosis a las 24 a 48 horas.6 Varios días después la concentración de eritropoyetina se normaliza, pero se mantiene el aumento del hematocrito. Además del aumento en la masa eritrocitaria, en los residentes de grandes alturas puede encontrarse una reducción moderada en el volumen plasmático.7

|
| Figura 2 |
| Curva de disociación oxígeno-hemoglobina |
Hemoglobina de alta afinidad
La hemoglobina de alta afinidad es causada por la sustitución de un solo aminoácido en la cadena alfa o, con más frecuencia, en la cadena b de la globina, lo que impide el cambio de conformación normal durante la unión y liberación del oxígeno.8 Esto provoca una menor capacidad para liberar oxígeno en los tejidos, con hipoxia subsecuente. El estímulo hipóxico es captado por el riñón, que aumenta la producción de eritropoyetina, provocando policitemia. Se han descrito más de 40 mutaciones individuales que causan hemoglobinas de alta afinidad.8 Estas suelen ser familiares y se heredan en forma autosómica dominante, pero en ocasiones son resultado de una mutación espontánea. Las biometrías hemáticas previas de un paciente pueden ser de gran utilidad porque el enfermo con hemoglobina de alta afinidad debe tener evidencia de policitemia de por vida. El hematocrito de los pacientes con hemoglobina de alta afinidad casi siempre es menor de 60 porciento, y las cuentas de leucocitos y plaquetas son normales. Debe medirse la presión parcial de oxígeno a la que la hemoglobina está saturada al 50 porciento (P50), que estará reducida en los pacientes con hemoglobina de alta afinidad [ver figura 2]. Por lo general no es útil obtener una electroforesis de hemoglobina porque muchas de las mutaciones que causan hemoglobinas de alta afinidad son silenciosas desde el punto de vista electroforético. La policitemia en estos pacientes está regulada fisiológicamente para optimizar la oxigenación tisular. Los pacientes con hemoglobina de alta afinidad no suelen tener síntomas de hiperviscosidad, y no requieren tratamiento.
Hipoxia tisular causada por enfermedad cardiopulmonar
A diferencia de la policitemia causada por hemoglobinas de alta afinidad, en la que el defecto primario consiste en un aporte inadecuado de oxígeno a los tejidos, la policitemia causada por defectos cardiopulmonares es causada por deficiente carga de oxígeno en los pulmones. Las cardiopatías congénitas cianógenas (v.gr., complejo de Eisenmenger, corazón univentricular y tetralogía de Fallot) pueden acompañarse de policitemia importante.9 El hematocrito puede variar de 60 a 75 porciento, y existen signos francos de hiperviscosidad, incluyendo cefalea, mareo, alteraciones visuales, fatiga, parestesias y menor agudeza mental.9,10 Un estudio de 124 adultos con cardiopatía congénita cianógena sugirió que pueden identificarse desde el punto de vista clínico dos grupos de pacientes.10 Uno tiene eritrocitosis compensada, y mantiene un hematocrito estable (aunque elevado) sin síntomas de hiperviscosidad muy importantes. Un segundo grupo tiene eritrocitosis descompensada, que se caracteriza por hematocrito que se incrementa e hiperviscosidad sintomática.8,10 Es notable que en este estudio no se observaron eventos cerebrovasculares a pesar de lo elevado de los hematocritos.10 Las investigaciones sobre los efectos hemodinámicos de reducir el hematocrito en pacientes con cardiopatía congénita cianógena han demostrado un aumento en la capacidad de ejercicio máximo, en el gasto cardiaco y en la captación de oxígeno por los tejidos durante el ejercicio.11 Un enfoque práctico de tratamiento consiste en flebotomizar con cuidado a los pacientes con cardiopatía congénita cianógena que tienen un hematocrito mayor del 65 porciento y que causa síntomas de hiperviscosidad.10 La magnitud de la flebotomía dependerá de los síntomas del paciente. Debe excluirse deshidratación aguda que exacerbe la policitemia antes de realizar la flebotomía, y el volumen de sangre que se extrae se repondrá con solución salina isotónica.10
La EPOC severa puede asociarse con policitemia, aunque por lo general predominan los síntomas clínicos de la primera. En pacientes que continúan fumando, tanto la hipoxemia como el aumento en la concentración de carboxihemoglobina pueden contribuir al desarrollo de la policitemia (ver adelante). La reducción del hematocrito en los pacientes con policitemia importante (hematocrito promedio de 59.6 porciento) causada por EPOC mejora la circulación cerebral y alivia los síntomas de cefalea y mareo atribuibles a la hiperviscosidad.12 También se ha reportado mejoría muy importante en el estado de alerta después de la flebotomía en pacientes que tenían un hematocrito previo de 61.3 porciento.13 El análisis de la tolerancia al ejercicio tanto antes como después de la flebotomía mostró un aumento en la duración del ejercicio y el consumo máximo de oxígeno en pacientes con EPOC que tenían un hematocrito inicial mayor de 55 porciento.14 Por estos motivos debe realizarse flebotomía en los pacientes con EPOC y síntomas atribuibles a la policitemia.
Síndrome de obesidad-hipoventilación
El síndrome de obesidad-hipoventilación también se conoce como síndrome de Pickwick en referencia a la astuta descripción de Charles Dickens del chico obeso con somnolencia diurna excesiva.15 Los pacientes con este síndrome suelen ser obesos (con peso corporal mayor del 120 porciento del ideal) y tienen hipoxemia e hipercapnia, en parte por bloqueo de la respuesta ventilatoria a estos estímulos.16 Algunos de estos pacientes tienen también un componente de apnea obstructiva del sueño (ver adelante). La hipoxemia proporciona el estímulo para la mayor producción de eritropoyetina y la policitemia. Otras características clínicas asociadas con el síndrome de obesidad-hiperventilación son la hipersomnolencia nocturna y el cor pulmonale. El tratamiento puede incluir pérdida de peso y tratamiento con progesterona para mejorar el estímulo respiratorio central.
Apnea obstructiva del sueño
Se calcula que el síndrome de apnea obstructiva del sueño se presenta en el cuatro y dos porciento de los varones y mujeres de edad media, respectivamente.17 El síndrome consiste en episodios recurrentes de colapso de las vías respiratorias superiores durante el sueño, que obstruyen el paso del aire, con lo que se presenta hipoxemia nocturna intermitente.18 Puede haber historia de ronquidos que alternan con periodos de silencio que duran de 10 segundos a un minuto, seguido de ruidos de ahogamiento. La fragmentación del sueño causa somnolencia diurna excesiva y menor desempeño laboral. El hematocrito puede estar un poco aumentado,19 y debe pensarse en esta posibilidad en los pacientes con policitemia inexplicable.20 La polisomnografía nocturna puede establecer el diagnóstico. El tratamiento puede incluir pérdida de peso, presión positiva continua a las vías respiratorias por vía nasal y cirugía18,21,22 .
Aumento en los niveles de carboxihemoglobina
La exposición crónica a monóxido de carbono causa niveles altos de carboxihemoglobina. El monóxido de carbono se une a la hemoglobina con una afinidad 210 veces mayor que la del oxígeno, disminuyendo así la cantidad de hemoglobina disponible para transportar oxígeno.23 La unión de monóxido de carbono aumenta también la afinidad del resto de los grupos hem por el oxígeno, desplazando la curva de disociación oxígeno-hemoglobina hacia la izquierda [ver figura 2] y disminuyendo la liberación de oxígeno en los tejidos.23 Es por estos mecanismos que la exposición crónica a monóxido de carbono puede causar policitemia. Los fumadores de puro y cigarrillos24 y las personas que tienen exposición ocupacional crónica a gases automotrices en áreas mal ventiladas (v.gr., trabajadores de estacionamientos cubiertos, cargadores de trailers) tienen mayor riesgo.
Los síntomas pueden incluir alteraciones neuropsiquiátricas sutiles y exacerbación de la angina de pecho (probablemente por menor aporte de oxígeno al miocardio). El diagnóstico se establece midiendo el porcentaje de carboxihemoglobina en sangre. Debido a que la vida media de la carboxihemoglobina es de alrededor de cuatro horas, la prueba debe hacerse tarde durante el día, cuando el paciente ha fumado su número habitual de cigarrillos o ha pasado varias horas en el ambiente laboral. En un estudio de 22 fumadores con policitemia, la concentración promedio de carboxihemoglobina fue de 11.6 porciento, cuando el valor normal debe ser menor del uno porciento.24 Los fumadores policitémicos suelen tener tanto aumento en la masa eritrocitaria como disminución del volumen plasmático.24 Estas alteraciones revierten a los tres meses de suspender el tabaquismo.24,25
AUMENTO INAPROPIADO EN LA PRODUCCION DE ERITROPOYETINA
Las policitemias también pueden deberse a aumento en la producción de eritropoyetina como respuesta inadecuada a enfermedades renales y, con menos frecuencia, hepáticas. En la vida adulta, alrededor del 90 porciento de la producción de eritropoyetina ocurre en el riñón y el 10 porciento en el hígado. Los mecanismos moleculares de producción no se conocen con exactitud. Sin embargo, se sabe que una proteína hem que fija oxígeno26 en el riñón detecta la hipoxia tisular, lo que provoca aumento en la producción de eritropoyetina por las células intersticiales peritubulares del riñón.27 La producción aumenta por el reclutamiento de células intersticiales peritubulares adicionales para producir eritropoyetina.28 Debido a la complicada regulación de la producción de eritropoyetina en el riñón, la distorsión de la anatomía renal puede causar policitemia. Existen reportes de casos de policitemia asociada a quistes renales, hidronefrosis, glomerulonefritis focal y síndrome de Bartter.29-32 Alrededor del 10 porciento de los pacientes tienen policitemia temporal o persistente después del trasplante renal. Además, las neoplasias primarias del riñón o del hígado pueden provocar policitemia. Se desarrolla policitemia en alrededor del tres porciento de los pacientes con carcinoma de células renales. Se ha demostrado producción de eritropoyetina por las células de carcinomas renales primarios y de hepatomas.26,33,34 En casos raros los hemangiomas del hígado o cerebelo, los fibroides uterinos, los adenomas suprarrenales y los feocromocitomas pueden causar policitemia.35
HIPERSENSIBILIDAD A LA ERITROPOYETINA (POLICITEMIA FAMILIAR CONGENITA)
Se han descrito y estudiado en forma extensa varias familias con policitemia autosómica dominante. En algunas de estas familias se encontró alteración en el receptor de eritropoyetina36,37 o en la regulación de la producción de la misma.38,39 En otras familias no se ha identificado aún el mecanismo preciso que explica la policitemia.40
La eritropoyetina inicia sus efectos biológicos al unirse a un receptor específico41 que se encuentra en la superficie de las células progenitoras eritroides, las células precursoras y varios otros tipos celulares. La unión con la eritropoyetina desencadena una cascada de eventos, incluyendo la asociación y activación de la proteína-tirosina cinasa JAK2 [ver figura 3].42 La fosforilación tirosina del receptor de eritropoyetina crea sitios de ensamble para otras moléculas de transducción de señales y para la proteína tirosina fosfatasa SH-PTP1, que desfosforila a JAK2 y termina la transducción de señales.43 En una familia finlandesa extensa con policitemia se encontró una mutación puntiforme en el receptor de la eritropoyetina que causó ruptura de 70 aminoácidos en el extremo carboxilo, incluyendo en sitio en el que se une la SH-PTP1 para terminar la señal de transducción.36 Esta mutación vuelve a las células progenitoras eritroides hipersensibles a la eritropoyetina, y pudo contribuir a la obtención de tres medallas de oro en esquí a campo traviesa que obtuvo en los Juegos Olímpicos de 1964 un miembro de esta familia que tenía un hematocrito del 60 porciento.44 En otra familia bien estudiada, una mutación de marco en el receptor de eritropoyetina causó la ruptura del sitio de unión de la SH-PTP1, con hipersensibilidad a la eritropoyetina y policitemia.37
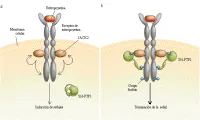
|
| Figura 3 |
| Acción de la eritropoyetina |
La medición de las concentraciones séricas de eritropoyetina ha sugerido que en algunas familias existe una alteración primaria en el punto umbral del mecanismo censor de oxígeno en el riñón.38,39 En estas familias no pudo encontrarse alteración en la afinidad de la hemoglobina por el oxígeno y los niveles de eritropoyetina eran inapropiadamente altos para el hematocrito.
USO DE MEDICAMENTOS
El uso de andrógenos, como la testosterona, puede asociarse con policitemia.45 Los andrógenos estimulan la producción de eritropoyetina por el riñón. El aumento en el hematocrito suele ser leve, y éste se normaliza dos a tres meses después de suspender los anabólicos. Debido a que en la actualidad se cuenta con eritropoyetina humana recombinante, ha surgido la preocupación de que los atletas de competencia que participan en deportes de resistencia, como ciclismo, esquí a campo traviesa y carrera de fondo, puedan inyectarse esta sustancia para mejorar su rendimiento.46 Se ha demostrado que la reinfusión de sangre autóloga justo antes de una carrera acorta los tiempos en las carreras de 10 km,47 y varios otros estudios han encontrado que la flebotomía seguida de doping con sangre puede mejorar el rendimiento de corredores y esquiadores.48 Puede lograrse un incremento semejante en el hematocrito empleando inyecciones de eritropoyetina. Existe expeculación sobre si la muerte súbita de varios ciclistas de competencia entre 1987 y 1990 se relacionó con el abuso de eritropoyetina.49,50 El incremento no vigilado en la producción de eritrocitos puede causar policitemia importante que, aunada a la deshidratación inducida por el ejercicio, puede causar consecuencias trágicas. Este uso de la eritropoyetina no es ético, es riesgoso y está prohibido por el Comité Olímpico Internacional.
Policitemia vera
La policitemia vera es un trastorno neoplásico que se origina en una célula tronco hematopoyética pluripotencial. Ocurre en alrededor de cinco a 10 personas por millón (0.0005 a 0.0010 porciento),51 es ligeramente más común en hombres que en mujeres (relación hombre: mujer de 1.2:1) y alcanza su incidencia máxima entre los 50 a 75 años de edad.52,53 Sin embargo, alrededor del cinco porciento de los casos se diagnostican en pacientes menores de 40 años.52,53
FISIOPATOLOGIA
En un adulto normal la hematopoyesis es policlonal, de modo que las células que circulan en la sangre periférica derivan de múltiples células tronco. Sin embargo, en los pacientes con policitemia vera muchos o todos los eritrocitos, leucocitos y plaquetas circulantes derivan de una célula tronco neoplásica. Esto se demostró en estudios de mujeres con policitemia vera que eran también heterocigotas para la enzima glucosa-6-fosfato-deshidrogenasa (G/PD), que se codifica en el cromosoma X.54 En estas pacientes, los neutrófilos, eritrocitos y plaquetas circulantes mostraban solo una de las dos isoenzimas de G6PD, lo que indicaba que las células se originaban de una sola célula tronco, mientras que los fibroblastos de la piel contenían proporciones balanceadas de ambas isoenzimas de G6PD A y B.54 Recientemente, el análisis de polimorfismo de ADN ligado al X ha demostrado también la naturaleza clonal de la policitemia vera.55
La policitemia vera es un padecimiento de un grupo de padecimientos relacionados de las células tronco denominados trastornos mieloproliferativos, todos de origen clonal. En éstos se incluyen la leucemia mieloide crónica (LMC), la trombocitemia esencial y la metaplasia mieloide agnogénica. Las características predominantes de la LMC son leucocitosis dramática, presencia de cromosoma Philadelphia y evolución a leucemia mieloide aguda (LMA) o a leucemia linfocítica aguda. La trombocitemia esencial se distingue por la trombocitosis predominante sin eritrocitosis o alteraciones citogenéticas de LMC. La metaplasia mieloide agnogénica se caracteriza por esplenomegalia masiva acompañada de anemia y un frotis de sangre periférica leucoeritroblástico.
La fisiopatología de la policitemia vera aún no se conoce del todo.56 En los adultos normales las células eritroides progenitoras no proliferan en ausencia de eritropoyetina exógena.57 Sin embargo, se ha demostrado que las células progenitoras eritroides de los pacientes con policitemia vera desarrollan colonias in vitro incluso cuando no se agrega eritropoyetina al cultivo.57,58 Estas observaciones forman la base para una de las pruebas diagnósticas de la policitemia vera, el ensayo endógeno de colonias eritroides.59 Los estudios han demostrado que las células eritroides progenitoras de los pacientes con policitemia vera son hipersensibles a los efectos de múltiples citocinas promotoras del crecimiento, incluyendo interleucina-3 y factor de crecimiento semejante a insulina-1.60,61 Debido a estos hallazgos y al hecho de que en la policitemia vera pueden afectarse varias líneas celulares, parece poco probable que la mutación en el receptor de una citocina de acción tardía (v.gr., eritropoyetina) pueda explicar la fisiopatología de esta enfermedad.
MANIFESTACIONES CLINICAS Y DIAGNOSTICO
Durante la década de los 50 y 60, un grupo de pacientes con policitemia vera no tratados tuvieron una supervivencia promedio de alrededor de 18 meses, y la mayoría de las defunciones fueron causadas por eventos cerebrovasculares.62 Sin embargo, en la actualidad la supervivencia promedio de los pacientes tratados es de 10 a 15 años.63,64 Debido a que la policitemia vera se diagnostica con más frecuencia en los pacientes entre la séptima y la octava década de la vida, la expectancia de vida puede no diferir de la del grupo de población de la misma edad.64 Un estudio de pacientes jóvenes con policitemia vera (menores de 40 años) mostró que más del 70 porciento sobrevivió 15 años después del diagnóstico.65
La historia natural de la policitemia vera incluye una fase latente protrombótica relativamente asintomática,53 una fase clínica proliferativa, durante la que se expande la masa de eritrocitos y el paciente puede tener síntomas de hiperviscosidad o hipermetabolismo [ver tabla 1] o trombosis, una fase de deterioro (i.e., metaplasia mieloide pospolicitemia), que se caracteriza por disminución en la producción de eritrocitos y desarrollo de anemia, leucopenia, trombocitopenia, fibrosis medular y hepatoesplenomegalia progresiva. Eventualmente se desarrolla metaplasia mieloide en alrededor del 15 al 20 porciento de los pacientes. La LMA es una complicación natural de la policitemia vera, desarrollándose en alrededor del uno a dos porciento de los pacientes tratados solo con flebotomía,66 y también es una complicación de ciertos tratamientos per se [ver adelante, Complicaciones].
|
||||
|
Síntomas
Aunque ciertos síntomas son comunes a todas las policitemias, otros son más específicos de la policitemia vera52 [ver tabla 1]. Muchos pacientes policitémicos tienen cefalea o cambios cognoscitivos vagos, incluyendo cambios en la memoria e incapacidad para realizar cálculos mentales rápidos. Son frecuentes la debilidad y el mareo. El prurito después de un baño o regaderazo caliente es más específico de policitemia vera y puede ser muy molesto. Con menos frecuencia los pacientes con este padecimiento tienen síntomas de hipermetabolismo, incluyendo fiebre, pérdida de peso y sudoración excesiva. La eritromelalgia, el dolor quemante episódico severo y el eritema en dedos de manos y pies se explican por isquemia y sugieren un trastorno mieloproliferativo. Alrededor del 20 porciento de los pacientes con policitemia vera han tenido un evento trombótico arterial o venoso premonitorio.53 En algunos casos los pacientes con policitemia vera están asintomáticos.
Examen físico
El examen físico suele mostrar plétora facial, que es común en todos los pacientes con policitemia real (i.e., primaria o secundaria). Se encuentra esplenomegalia en el 70 porciento de los pacientes con policitemia vera,52 que se debe a hematopoyesis extramedular junto con congestión esplénica por eritrocitos, ya que el bazo es la tumba de estas células. Existe hepatomegalia en el 40 porciento de los casos, que también puede deberse a eritropoyesis extramedular y, en ocasiones, al síndrome de Budd-Chiari. Una tercera parte de los pacientes con policitemia vera tienen hipertensión.
Diagnóstico de laboratorio
La citología hemática completa de los pacientes que presentan policitemia vera de novo suele mostrar elevación del hematocrito. Un hematocrito de 60 o mayor en un hombre, o de 57 o mayor en una mujer, es casi diagnóstico de policitemia primaria o secundaria. Sin embargo, debido a la alta prevalencia de pérdida de sangre por la vía gastrointestinal en los pacientes con policitemia vera, algunos tienen hematocrito normal. El volumen corpuscular medio de los eritrocitos puede estar disminuido, lo que refleja deficiencia de hierro. Se encuentra leucocitosis en el 45 porciento de los pacientes en el momento del diagnóstico.52 El frotis de sangre periférica puede mostrar basofilia discreta y en ocasiones leucocitos inmaduros en la circulación, incluyendo mielocitos y metamielocitos. Alrededor del 65 porciento de los pacientes con policitemia vera tienen trombocitosis.52 Los principales diagnósticos diferenciales en el paciente con un hematocrito normal, trombocitosis y volumen corpuscular promedio bajo es la policitemia vera con deficiencia de hierro, la trombocitemia esencial o la trombocitosis causada por la deficiencia de hierro en sí. Si el paciente tiene buenas reservas de hierro el diagnóstico más probable es trombocitopenia esencial. La trombocitosis causada por deficiencia de hierro suele responder a la repleción del mismo.
Otros datos de laboratorio incluyen elevación de la concentración de ácido úrico en el 30 a 50 porciento de los pacientes con policitemia vera, causada por producción aumentada en forma crónica y destrucción de los eritrocitos.67 La concentración de vitamina B12 y de proteína fijadora de vitamina B12 suele estar elevada. El nivel de fosfatasa alcalina leucocitaria (FAL) se encuentra aumentado en el 70 porciento de los pacientes con policitemia vera, a diferencia de los pacientes con LMC, en quienes la concentración de FAL está disminuida.
El nivel de eritropoyetina está disminuido como resultado de mecanismos de retroalimentación68 [ver figura 4]. Sin embargo, en algunos pacientes pueden ser normales.68 La eritropoyetina sérica no debe medirse después de una flebotomía vigorosa porque al reducir el hematocrito aumentará la producción de eritropoyetina.
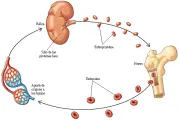
|
| Figura 4 |
| Aporte de oxígeno y producción de eritropoyetina |
En la policitemia vera no se requiere evaluación de la médula ósea, ni tiene especial utilidad. Los datos de la médula suelen incluir aumento en la celularidad (promedio de 82 porciento), en el número de megacariocitos y bajas reservas de hierro.69 También puede existir aumento en las fibras de reticulina.69
Se encuentran alteraciones citogenéticas en el momento del diagnóstico en alrededor del 10 a 15 porciento de los pacientes con policitemia vera, que comunmente incluyen trisomía 8, 9 y deleción del brazo largo del cromosoma 20.70,71 La presencia de alteraciones citogenéticas en el momento del diagnóstico no identifica a pacientes con mayor riesgo de LMA o que puedan progresar a metaplasia mieloide.70,71 No se realizan estudios citogenéticos de rutina en los pacientes con probable diagnóstico de policitemia vera a menos que exista dificultad para distinguir esta enfermedad de una LMC. En ese caso pueden evaluarse los leucocitos de sangre perif por medio de Southern blot en busca del rearreglo genético bcr-abl, que es característico de LMC.
En 1967, el Polycythemia Vera Study Group (PVSG, Grupo de estudio de la policitemia vera de los EUA, n. del t.) estableció un grupo de criterios diagnósticos para incluir a los pacientes en estudios clínicos multicéntricos.52 Estos criterios se desarrollaron antes de contar con las determinaciones de eritropoyetina en suero y de ensayos de colonias eritroides endógenos. Además, los criterios intentaban ser estrictos, por lo que con frecuencia excluyen a pacientes con policitemia vera temprana que no han desarrollado aún esplenomegalia, leucocitosis o trombocitosis significativas. En un paciente que no satisface los criterios estrictos del PVSG, el encontrar una concentración baja de eritropoyetina puede proporcionar evidencia adicional para apoyar el diagnóstico [ver tabla 2]. La presencia de colonias eritroides endógenas en sangre periférica también es un dato a favor del diagnóstico de policitemia vera [ver tabla 2].
|
|
* Se diagostica policitemia vera si se satisfacen los criterios 1,2 y 3 o solo criterios 1 y 2 más de los dos criterios 4 a 8. |
TRATAMIENTO
Existen muchas opciones terapéuticas para la policitemia vera, pero no un consenso sobre el mejor esquema de tratamiento. Este debe individualizarse, tomando en consideración la edad del paciente y su perfil de riesgo de trombosis, además de los riesgos inherentes a cada tipo de manejo.
Para la mayoría de los pacientes el tratamiento inicial debe incluir la flebotomía. En general, se eliminan 500 ml de sangre una o dos veces por semana hasta que se alcanza un valor de menos de 46 porciento. Después se realizan flebotomías periódicas para mantener el hematocrito entre 40 y 45 porciento. En muchos pacientes sometidos a flebotomías se desarrolla deficiencia de hierro, lo que limita la capacidad para producir eritrocitos y reduce la frecuencia de la flebotomía. El paciente debe evitar los multivitamínicos que contienen hierro. Existe controversia sobre si los eritrocitos hipocrómicos, microcíticos, deficientes en hierro tienen un comportamiento hemorreológico que aumenta la viscosidad de la sangre y quizá un poco el riesgo de trombosis.72,73 Por ello, es probable que sea adecuado evitar la inducción de microcitosis severa. En ocasiones los pacientes pueden desarrollar síntomas atribuibles a la deficiencia de hierro, incluyendo pica,74 glositis y fatiga muscular inducida por ejercicio, que responden a la repleción de este mineral. Sin embargo, en la mayoría de los casos es conveniente mantener una deficiencia moderada de hierro causada por las flebotomías porque esto ayuda a mantener el hematocrito en los niveles adecuados.
Los pacientes que tienen riesgo alto de trombosis (v.gr., mayores de 70 años, que han tenido un evento trombótico previo y que requieren flebotomías con gran frecuencia) deben manejarse mejor con un agente mielosupresor, como la hidroxiurea,75 aunado a la flebotomía. Se ha demostrado que la hidroxiurea (en dosis de 500 mg a 1.5 g vía oral diario) y la flebotomía pueden controlar en forma más eficaz el hematocrito en la mayoría de los pacientes con policitemia vera, comparado con la flebotomía sola. La hidroxiurea suele ser bien tolerada; sin embargo, su uso requiere vigilancia frecuente de la citología hemática para evitar una mielosupresión excesiva. También es motivo de controversia si el uso de hidroxiurea aumenta el riesgo de desarrollar leucemia mieloide aguda. En 100 pacientes con policitemia vera que recibieron hidroxiurea durante un promedio de 5.4 años se desarrollo leucemia mieloide aguda con una incidencia baja (uno porciento).77 Sin embargo, una publicación reciente encontró una incidencia de 7.8 porciento de leucemia mieloide aguda en los pacientes tratados con hidroxiurea, lo que es significativamente más alto que la incidencia de uno a dos porciento que se encuentra solo con las flebotomías.78
El busulfán, un agente alquilante, puede ser un mielosupresor alternativo también de administración por vía oral. Un estudio aleatorio extenso sugirió que el riesgo de desarrollar LMA en pacientes tratados con busulfán es semejante al de pacientes tratados con fósforo radioactivo (P32).79 Los riesgos a largo plazo del busulfán incluyen fibrosis pulmonar y daño a las células tronco hematopoyéticas, lo que puede causar supresión medular por tiempo prolongado. Debido a su toxicidad, el busulfán suele reservarse para pacientes que no mejoran con hidroxiurea.
El tratamiento intermitente con P32 (2.3 mCi/m2 I.V. cada 12 semanas según se requiera para controlar el hematocrito, con dosis máxima de 5 mCi) suele permitir un control paulatino del hematocrino, no requiere de vigilancia frecuente y es muy cómodo para los pacientes. Sin embargo, aumenta el riesgo de desarrollar LMA (ver adelante). Este tipo de tratamiento puede ser adecuado para pacientes con policitemia vera de edad avanzada.
El clorambucil se asocia con mayor riesgo de LMA de inicio temprano,80 por lo que no se recomienda para el tratamiento de la policitemia vera.
Comparado con los pacientes tratados con un agente mielosupresor, los que se manejan solo con flebotomías tienen un mayor riesgo de trombosis durante los primeros tres años después del inicio del tratamiento.63 Por este motivo se estudió si la administración de aspirina, 300 mg tres veces al día más dipiridamol (75 mg tres veces al día) aunado a la flebotomía disminuía el riesgo de trombosis, comparando con el tratamiento con P32. Se demostró que la adición de aspirina en esta dosis relativamente alta no evitó las trombosis y aumentó el riesgo de hemorragia gastrointestinal importante.81 Por lo tanto, no puede recomendarse el uso de aspirina a esas dosis como tratamiento inicial de la policitemia vera.
COMPLICACIONES
El periodo posoperatorio es especialmente peligroso para los pacientes con policitemia vera no controlada. En un estudio, el 79 porciento de los pacientes con policitemia vera no controlada (hematocrito mayor de 52 porciento en el momento de la cirugía) tuvieron complicaciones posoperatorias, principalmente hemorragia o trombosis.82 La tasa de complicaciones puede minimizarse logrando un buen control del hematocrito durante por lo menos cuatro meses antes de la cirugía electiva.82
Trombosis
La trombosis es la complicación más frecuente de la policitemia vera. Varias características de este padecimiento parecen contribuir a la mayor incidencia. El valor del hematocrito se relaciona en forma directa con el riesgo de trombosis: al aumentar éste, disminuye el flujo sanguíneo cerebral,83 y aumenta la incidencia de complicaciones trombóticas.84 Esta asociación entre el hematocrito y el riesgo de trombosis forma las bases para una recomendación clave en el tratamiento de la policitemia vera: el hematocrito debe mantenerse en menos de 46 porciento para disminuir el riesgo de trombosis.62 Las plaquetas derivan de una clona neoplásica y pueden no funcionar de modo normal,54 lo que facilita la ocurrencia de trombosis o hemorragia clínica.85 Son comunes las equímosis, la epistaxis y la hemorragia gastrointestinal. Los estudios de agregometría plaquetaria ex vivo demuestran defectos en la agregación, sobre todo en respuesta a la epinefrina.86 Aunque suele haber exceso de plaquetas en la policitemia vera, no existe una relación clara entre la cuenta absoluta de plaquetas y el riesgo de trombosis.85
En el estudio clínico inicial realizado por el PVSG, 431 pacientes recibieron tratamiento con flebotomía sola, P32 o clorambucil para mantener el hematocrito por debajo del 45 porciento.63 En la actualización más reciente de este estudio, el 35 porciento de los pacientes sufrió trombosis.75 Los pacientes con policitemia vera tienen mayor riesgo de trombosis tanto arterial como venosa53,65 [ver tabla 3]. Son problemas clínicos comunes el infarto cerebral, el infarto al miocardio, la trombosis de venas profundas en miembros inferiores, los episodios de isquemia cerebral transitoria y la oclusión arterial periférica. Los pacientes suelen notar exacerbación de su angina conforme aumenta el hematocrito.
|
||||||||||
|
Puede ocurrir trombosis de la vena hepática, causando síndrome de Budd-Chiari. En un estudio de 20 pacientes con síndrome de Budd-Chiari, el 80 porciento tuvo colonias eritroides endógenas, y es posible que su trombosis de venas hepáticas haya sido causada por un trastorno mieloproliferativo subyacente.87 Solo dos de estos pacientes tuvieron un trastorno mieloproliferativo evidente. Sin embargo, la baja incidencia del síndrome de Budd-Chiari durante los muchos años de seguimiento del primer estudio del PVSG va en contra de que éste sea una complicación frecuente de la policitemia vera.
Tratamiento Los pacientes con policitemia vera que tienen un evento trombótico agudo y trombocitosis deben someterse a aféresis de plaquetas para reducir con rapidez la cuenta plaquetaria hasta el rango normal. Después debe iniciarse tratamiento inmunosupresor (v.gr., hidroxiurea) para ayudar a mantener una cuenta de plaquetas normal, a pesar de que no exista una relación clara entre la cuenta alta de plaquetas y el riesgo de trombosis. Si el paciente no tiene historia de hemorragia excesiva, debe considerarse también la adición de un agente que inhiba la función plaquetaria (v.gr., aspirina 81 mg/día). Los pacientes con hemorragias de mucosas y trombocitosis deben recibir un agente mielosupresor para disminuir la cuenta de plaquetas al rango normal.
Leucemia mieloide aguda
Una de las complicaciones más temidas de la policitemia vera es la evolución a LMA. La incidencia de LMA en los pacientes manejados con flebotomía sola es de uno a dos porciento (100 veces más que la incidencia en la población adulta de edad semejante). El estudio inicial del PVSG encontró que el tratamiento con clorambucil o P32 se asocia con mayor riesgo de desarrollar LMA.63,80 El inicio de la LMA en los pacientes tratados con clorambucil fue temprano, y la mayoría de los casos ocurrieron menos de cinco años después del inicio del tratamiento. Eventualmente, el 14 porciento de los pacientes desarrollaron LMA.75 Además, el riesgo de desarrollar linfoma no Hodgkin aumentó en los pacientes tratados con clorambucil. En los enfermos que recibieron P32 el inicio de la LMA fue relativamente tardío, y la mayoría de los casos ocurrieron seis a 10 años después del inicio del tratamiento, con una incidencia acumulada de 9.6 porciento.75 Puede ocurrir evolución a mielodisplasia en algunos casos de policitemia vera, y una serie de reportes de casos sugiere que el uso de tratamiento mielosupresor puede aumentar también el riesgo.88 El tratamiento con clorambucil o P32 se asoció también con mayor riesgo de desarrollar una neoplasia no hematológica.75
Tratamiento Si la policitemia vera evoluciona a LMA la posibilidad de lograr una remisión completa y duradera con quimioterapia de inducción estándar es muy baja.75 En estos pacientes se justifica discutir en forma franca las opciones terapéuticas, incluyendo los riesgos y beneficios de la quimioterapia de inducción contra el tratamiento de sostén, en especial porque los pacientes son ancianos y pueden tener otros padecimientos concomitantes.
Gota y nefrolitiasis
Debido a la hiperuricemia los pacientes con policitemia vera tienen mayor riesgo de desarrollar gota o nefrolitiasis67.
Metaplasia mieloide
Alrededor del 15 a 20 porciento de los pacientes con policitemia vera desarrollan metaplasia mieloide pospolicitémica,75,89,90 o la llamada fase involutiva o de deterioro de los trastornos mieloproliferativos. La metaplasia mieloide se caracteriza por remplazo insidioso del tejido medular por reticulina fibrosa, y por movimiento gradual de la hematopoyesis de la médula al bazo, el hígado y en ocasiones otros órganos. El bazo e hígado crecen en forma progresiva y pueden adquirir un tamaño enorme. Los pacientes sufren pancitopenia, en parte porque estos órganos son menos eficaces para la hematopoyesis y al parecer también por alteraciones intrínsecas de las células tronco hematopoyéticas. Se encuentran en la circulación eritrocitos en gota y una plétora de células mieloides, lo que causa el frotis leucoeritroblástico de sangre periférica característico de la metaplasia mieloide. Los pacientes con metaplasia mieloide pueden tener dolor esplénico e infartos por la esplenomegalia masiva, y presentar saciedad temprana, con pérdida subsecuente de peso. La fibrosis en la médula de los pacientes con metaplasia mieloide no es parte de la clona neoplásica. En lugar de ello, la fibrosis es reactiva y puede ser inducida por citocinas como el factor de crecimiento derivado de plaquetas, liberado por los megacariocitos anormales. La supervivencia de los pacientes con metaplasia mieloide es corta, alrededor de dos a tres años.
Tratamiento El tratamiento de la metaplasia mieloide incluye manejo de sostén con transfusiones, según se requiera, y uso juicioso de hidroxiurea o busulfán para disminuir el tamaño del bazo. Puede considerarse realizar esplenectomía en los pacientes con esplenomegalia masiva y pérdida de peso por saciedad temprana, dolor por infarto esplénico o trombocitopenia exacerbada por el secuestro de plaquetas en un bazo con crecimiento masivo. La esplenectomía es un gran reto en estos pacientes, y por lo general es seguida de hepatomegalia progresiva. En la actualidad no existen evidencias convincentes de que pueda lograrse regresión de la fibrosis medular con hidroxiurea, P32 o interferón alfa.
Hipermetabolismo y prurito
Los síntomas de hipermetabolismo (v.gr., calosfríos, diaforesis y fiebre no debida a infección) y el prurito suelen responder a la hidroxiurea y a otros agentes mielosupresores. En un paciente con prurito puede ser útil evitar los eventos exacerbantes (v.gr., baños calientes) y administrar bloqueadores de los receptores H1 y H2.
TRATAMIENTOS EXPERIMENTALES
El anagrelide es un vasodilatador activo que inhibe la maduración de los megacariocitos, disminuyendo así la cuenta plaquetaria. En un estudio de más de 400 pacientes con trombocitosis causada por un trastorno mieloproliferativo, se encontró que el anagrelide redujo la cuenta plaquetaria en el 93 porciento, por lo general en dos semanas.91 Los efectos adversos más frecuentes de este medicamento fueron retención hídrica, palpitaciones, cefalea, náusea y diarrea. El anagrelide puede ser útil especialmente en los pacientes con policitemia vera que requieren un agente suplementario para el control óptimo de la trombocitosis. Este medicamento es revisado en la actualidad por la Food and Drug Administration para usarse en la trombocitosis asociada a trastornos mieloproliferativos.
El interferón alfa es un agente de administración parenteral que puede inducir remisión citogenética en un porcentaje de pacientes con LMC. Debido a que la policitemia vera, al igual que la LMC, es un trastorno mieloproliferativo de tipo clonal, se ha postulado que el interferón alfa puede causar también regresión de la clona neoplásica en este padecimiento. Los estudios de pacientes tratados con interferón alfa hasta por seis años han demostrado que el interferón alfa, administrado tres veces por semana, puede controlar en forma eficaz el hematocrito y la cuenta plaquetaria, además de disminuir el tamaño del bazo.92 No se diagnosticaron eventos trombóticos en los pacientes tratados con interferón alfa, a diferencia de la alta incidencia de trombosis en los pacientes con policitemia vera tratados solo con flebotomía.63 Debido a que solo una pequeña fracción de estos pacientes tenía una alteración citogenética, es menos claro cuál fue el efecto del interferón alfa sobre la clona neoplásica. Los efectos adversos de este agente incluyen síntomas de tipo gripal severo que pueden disminuir después de uno a dos meses de tratamiento. Otros problemas incluyen un inicio de acción lento (en meses) y el costo tan alto. Sin embargo, si puede demostrarse que el interferón alfa induce regresión de la clona neoplásica93 o impide la progresión a metaplasia mieloide, sus beneficios superarán sus desventajas. Se está planeando un estudio que comparará el uso de flebotomía, hidroxiurea e interferón alfa, y que ayudará a aclarar el papel de este último en el tratamiento de la policitemia vera.
También se ha investigado la utilidad del trasplante alogénico de médula ósea (que en la actualidad es el tratamiento de elección en los pacientes con LMC) para el tratamiento de la policitemia vera, a pesar de que esta última tiene una evolución más indolente. Solo algunos pacientes con policitemia vera han sido sometidos a tratamiento mieloablativo y trasplante alogénico. Este enfoque puede ser conveniente para pacientes jóvenes con evidencia temprana de fibrosis medular.
Bibliografía
-
Pearson TC, Botterill CA, Glass UH, et al: Interpretation
of measured red cell mass and plasma volume in males with elevated venous
PCV values. Scand J Haematol 33:68, 1984 [PMID
6463587]
-
Messinezy M, Pearson TC: A retrospective study of apparent
and relative polycythaemia: associated factors and early outcome. Clin
Lab Haematol 12:121, 1990
-
Watts EJ, Lewis SM: Spurious polycythaemia: a study
of 35 patients. Scand J Haematol 31:241, 1983 [PMID
6879107]
-
Copelan EA, Balcerzak SP: Secondary polycythemia. Polycythemia
Vera and the Myeloproliferative Disorders. Wasserman LR, Berk PD, Berlin
NI, Eds. WB Saunders Co, Philadelphia, 1995, p 195
-
Erslev AJ: Blood and mountains. Blood, Pure and Eloquent.
Wintrobe MM, Ed. McGraw-Hill Book Co, New York, 1980, p 257
-
Faura J, Ramos J, Reynafarje C, et al: Effect of altitude
on erythropoiesis. Blood 33:668, 1969 [PMID
4976192]
-
Sanchez C, Merino C, Figallo M: Simultaneous measurement
of plasma volume and cell mass in polycythemia of high altitude. J Appl
Physiol 28:775, 1970
-
Hemoglobinopathy due to abnormal oxygen binding. Hemoglobin:
Molecular, Genetic and Clinical Aspects. Bunn HF, Forget BG. WB Saunders
Co, Philadelphia, 1986, p 595
-
Rosove MH, Hocking WG, Canobbio MM, et al: Chronic
hypoxaemia and decompensated erythrocytosis in cyanotic congenital heart
disease. Lancet 2:313, 1986
-
Perloff JK, Rosove MH, Child JS, et al: Adults with
cyanotic congenital heart disease: hematologic management. Ann Intern Med
109:406, 1988 [PMID
3044212]
-
Oldershaw PJ, St John Sutton MG: Haemodynamic effects
of haematocrit reduction in patients with polycythaemia secondary to cyanotic
congenital heart disease. Br Heart J 44:584, 1980 [PMID
7437201]
-
York EL, Jones RL, Menon D, et al: Effects of secondary
polycythemia on cerebral blood flow in chronic obstructive pulmonary disease.
Am Rev Respir Dis 121:813, 1980
-
Wade JPH, Pearson TC, Ross Russell RW, et al: Cerebral
blood flow and blood viscosity in patients with polycythaemia secondary
to hypoxic lung disease. Br Med J 283:689, 1981
-
Chetty KG, Brown SE, Light RW: Improved exercise tolerance
of the polycythemic lung patient following phlebotomy. Am J Med 74:415,
1983 [PMID
6402930]
-
Burwell CS, Robin ED, Whaley RD, et al: Extreme obesity
associated with alveolar hypoventilation: a Pickwickian syndrome. Am J
Med 21:811, 1956
-
Zwillich CW, Sutton FD, Pierson DJ, et al: Decreased
hypoxic ventilatory drive in the obesity-hypoventilation syndrome. Am J
Med 59:343, 1975 [PMID
1163544]
-
Young T, Palta M, Dempsey J, et al: The occurrence
of sleep-disordered breathing among middle-aged adults. N Engl J Med 328:1230,
1993 [PMID
8464434]
-
Strollo PJ Jr, Rogers RM: Obstructive sleep apnea.
N Engl J Med 334:99, 1996
-
Hoffstein V, Herridge M, Mateika S, et al: Hematocrit
levels in sleep apnea. Chest 106:787, 1994 [PMID
8082360]
-
Moore-Gillon JC, Treacher DF, Gaminara EJ, et al: Intermittent
hypoxia in patients with unexplained polycythaemia. Br Med J 293:588, 1986
-
Smith PL, Gold AR, Meyers DA, et al: Weight loss in
mildly to moderately obese patients with obstructive sleep apnea. Ann Intern
Med 103:850, 1985 [PMID
3933396]
-
Riley RW, Powell NB, Guilleminault C, et al: Obstructive
sleep apnea: trends in therapy. West J Med 162:143, 1995 [PMID
7725686]
-
Carboxyhemoglobin and carboxyhemoglobinemia. Hemoglobin:
Molecular, Genetic and Clinical Aspects. Bunn HF, Forget BG. WB Saunders
Co, Philadelphia, 1986, p 663
-
Smith JR, Landaw SA: Smokers' polycythemia. N Engl
J Med 298:6, 1978
-
Sagone AL Jr, Balcerzak SP: Smoking as a cause of erythrocytosis.
Ann Intern Med 82:512, 1975 [PMID
235231]
-
Goldberg MA, Dunning SP, Bunn HF: Regulation of the
erythropoietin gene: evidence that the oxygen sensor is a heme protein.
Science 242:1412, 1988 [PMID
2849206]
-
Krantz SB: Erythropoietin. Blood 77:419, 1991
-
Koury ST, Koury MJ, Bondurant MC, et al: Quantitation
of erythropoietin-producing cells in kidneys of mice by in situ hybridization:
correlation with hematocrit, renal erythropoietin mRNA, and serum erythropoietin
concentration. Blood 74:645, 1989 [PMID
2752138]
-
The Kidney: Physiology and Pathophysiology, 2nd ed.
Seldin DW, Giebisch G, Eds. Raven Press, New York, 1992
-
Rosse WF, Waldmann TA, Cohen P: Renal cysts, erythropoietin
and polycythemia. Am J Med 34:76, 1963
-
Hirsch I, Leiter E: Hydronephrosis and polycythemia. Urology
21:345, 1983 [PMID
6836818]
-
Erkelens DW, Statius Van Eps LW: Bartter's syndrome
and erythrocytosis. Am J Med 55:711, 1973 [PMID
4356102]
-
Da Silva J-L, Lacombe C, Bruneval P, et al: Tumor cells
are the site of erythropoietin synthesis in human renal cancers associated
with polycythemia. Blood 75:577, 1990
-
Okabe T, Urabe A, Kato T, et al: Production of erythropoietin-like
activity by human renal and hepatic carcinomas in cell culture. Cancer
55:1918, 1985 [PMID
4038914]
-
Doll DC, Weiss RB: Neoplasia and the erythron. J Clin
onCol 3:429, 1985 [PMID
3919163]
-
de la Chappelle A, Treskelin A-L, Juvonen E: Truncated
erythropoietin receptor causes dominantly inherited benign human erythrocytosis.
Proc Natl Acad Sci USA 90:4495, 1993
-
Sokol L, Luhovy M, Guan Y, et al: Primary familial
polycythemia: a frameshift mutation in the erythropoietin receptor gene
and increased sensitivity of erythroid progenitors to erythropoietin. Blood
86:15, 1995 [PMID
7795221]
-
Whitcomb WH, Peschle C, Moore M, et al: Congenital
erythrocytosis: a new form associated with an erythropoietin-dependent
mechanism. Br J Haematol 44:17, 1980
-
Distelhorst CW, Wagner DS, Goldwasser E, et al: Autosomal
dominant familial erythrocytosis due to autonomous erythropoietin production.
Blood 58:1155, 1981 [PMID
7306703]
-
Emanuel PD, Eaves CJ, Broudy VC, et al: Familial and
congenital polycythemia in three unrelated families. Blood 79:3019, 1992
[PMID
1316790]
-
Youssoufian H, Longmore G, Neumann D, et al: Structure,
function, and activation of the erythropoietin receptor. Blood 81:2223,
1993 [PMID
8481505]
-
Witthuhn BA, Quelle FW, Silvennoinen O, et al: JAK2
associates with the erythropoietin receptor and is tyrosine phosphorylated
and activ ated following stimulation with erythropoietin. Cell 74:227,
1993 [PMID
8343951]
-
Klingmuller U, Lorenz U, Cantley LC, et al: Specific
recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation
of JAK2 and termination of proliferative signals. Cell 80:729, 1995 [PMID
7889566]
-
Juvonen E, Ikkala E, Fyhrquist F, et al: Autosomal
dominant erythrocytosis caused by increased sensitivity to erythropoietin.
Blood 78:3066, 1991 [PMID
1954391]
-
Besa EC: Hematologic effects of androgens revisited:
an alternative therapy in various hematologic conditions. Semin Hematol
31:134, 1994
-
Scott WC: The abuse of erythropoietin to enhance athletic
performance (letter). JAMA 264:1660, 1990
-
Brien J, Simon TL: The effects of red blood cell transfusion
on 10-km race time. JAMA 257:2761, 1987
-
Simon TL: Induced erythrocythemia and athletic performance.
Semin Hematol 31:128, 1994
-
Eichner ER: Sports anemia, iron supplements, and blood
doping. Med Sci Sports Exerc 24:S315, 1992
-
Gareau R, Audran M, Barnes R, et al: Erythropoietin
abuse in athletes (letter). Nature 380:113, 1996 [PMID
8600382]
-
Prochazka AV, Markowe HLJ: The epidemiology of polycythaemia
rubra vera in England and Wales 1968-1982. Br J Cancer 53:59, 1986 [PMID
3947516]
-
Berlin NI: Diagnosis and classification of the polycythemias.
Semin Hematol 12:339, 1975
-
Polycythemia vera: the natural history of 1213 patients
followed for 20 years. Gruppo Italiano Studio Policitemia. Ann Intern Med
123:656, 1995
-
Adamson JW, Fialkow PJ, Murphy S, et al: Polycythemia
vera: stem-cell and probable clonal origin of the disease. N Engl J Med
295:913, 1976 [PMID
967201]
-
Lucas GS, Padua RA, Masters GS, et al: The application
of X-chromosome gene probes to the diagnosis of myeloproliferative disease.
Br J Haematol 72:530, 1989
-
Prchal JT, Prchal JF: Evolving understanding of the
cellular defect in polycythemia vera: implications for its clinical diagnosis
and molecular pathophysiology. Blood 83:1, 1994 [PMID
8274728]
-
Lacombe C, Casadevall N, Varet B: Polycythemia vera:
in vitro studies of circulating erythroid progenitors. Br J Haematol
44:189, 1980 [PMID
7378297]
-
Prchal JF, Adamson JW, Murphy S, et al: Polycythemia
vera: the in vitro response of normal and abnormal stem cell lines to erythropoietin.
J Clin Invest 61:1044, 1978 [PMID
659576]
-
Shih L-Y, Lee C-T: Identification of masked polycythemia
vera from patients with idiopathic marked thrombocytosis by endogenous
erythroid colony assay. Blood 83:744, 1994
-
Dai CH, Krantz SB, Means RT Jr, et al: Polycythemia
vera blood burst-forming units-erythroid are hypersensitive to interleukin-3.
J Clin Invest 87:391, 1991
-
Correa PN, Eskinazi D, Axelrad AA: Circulating erythroid
progenitors in polycythemia vera are hypersensitive to insulin-like growth
actor-1 in vitro: studies in an improved serum-free medium. Blood 83:99, 1994 [PMID 8274756]
-
Chievitz E, Thiede T: Complications and causes of death
in polycythaemia vera. Acta Med Scand 172:513, 1962
-
Berk PD, Goldberg JD, Donovan PB, et al: Therapeutic
recommendations in polycythemia vera based on Polycythemia Vera Study Group
protocols. Semin Hematol 23:132, 1986 [PMID
3704665]
-
Rozman C, Giralt M, Feliu E, et al: Life expectancy
of patients with chronic nonleukemic myeloproliferative disorders. Cancer
67:2658, 1991 [PMID
2015567]
-
Najean Y, Mugnier P, Dresch C, et al: Polycythaemia
vera in young people: an analysis of 58 cases diagnosed before 40 years.
Br J Haematol 67:285, 1987 [PMID
3689694]
-
Landaw SA: Acute leukemia in polycythemia vera. Semin
Hematol 23:156, 1986
-
Yu T: Urate metabolism in the polycythemias. Polycythemia
Vera and the Myeloproliferative Disorders. Wasserman LR, Berk PD, Berlin
NI, Eds. WB Saunders Co, Philadelphia, 1995, p 130
-
Cotes PM, Doro CJ, Yin JAL, et al: Determination of
serum immunoreactive erythropoietin in the investigation of erythrocytosis.
N Engl J Med 315:283, 1986
-
Peterson P, Ellis JT: The bone marrow in polycythemia
vera. Polycythemia Vera and the Myeloproliferative Disorders. Wasserman
LR, Berk PD, Berlin NI, Eds. WB Saunders Co, Philadelphia, 1995, p 31
-
Pierre RV, Whang-Peng J: Cytogenetics. Polycythemia
Vera and the Myeloproliferative Disorders. Wasserman LR, Berk PD, Berlin
NI, Eds. WB Saunders Co, Philadelphia, 1995, p 91
-
Swolin B, Weinfeld A, Westin J: A prospective long-term
cytogenetic study in polycythemia vera in relation to treatment and clinical
course. Blood 72:386, 1988 [PMID
3401588]
-
Milligan DW, MacNamee R, Roberts BE, et al: The influence
of iron-deficient indices on whole blood viscosity in polycythaemia. Br
J Haematol 50:467, 1982
-
Van de Pette JEW, Guthrie DL, Pearson TC: Whole blood
viscosity in polycythaemia: the effect of iron deficiency at a range of
haemoglobin and packed cell volumes. Br J Haematol 63:369, 1986
-
Rector WG Jr, Fortuin NJ, Conley CL: Non-hematologic
effects of chronic iron deficiency: a study of patients with polycythemia
vera treated solely with venesections. Medicine (Baltimore) 61:382, 1982
[PMID
7144531]
-
Berk PD, Wasserman LR, Fruchtman SM, et al: Treatment
of polycythemia vera: a summary of clinical trials conducted by the Polycythemia
Vera Study Group. Polycythemia Vera and the Myeloproliferative Disorders.
Wasserman LR, Berk PD, Berlin NI, Eds. WB Saunders Co, Philadelphia, 1995,
p 166
-
Kaplan ME, Mack K, Goldberg JD, et al: Long-term management
of polycythemia vera with hydroxyurea: a progress report. Semin Hematol
23:167, 1986
-
West WO: Hydroxyurea in the treatment of polycythemia
vera: a prospective study of 100 patients over a 20-year period. South
Med J 80:323, 1987
-
Landaw SA: Acute leukemia in polycythemia vera. Polycythemia
Vera and the Myeloproliferative Disorders. Wasserman LR, Berk PD, Berlin
NI, Eds. WB Saunders Co, Philadelphia, 1995, p 154
-
Treatment of polycythaemia vera by radiophosphorus
or busulphan: a randomized trial. "Leukemia and Hepatosarcoma" Cooperative
Group, European Organization for Research on Treatment of Cancer (E.O.R.T.C.).
Br J Cancer 44:75, 1981
-
Berk PD, Goldberg JD, Silverstein MN, et al: Increased
incidence of acute leukemia in polycythemia vera associated with chlorambucil
therapy. N Engl J Med 304:441, 1981
-
Tartaglia AP, Goldberg JD, Berk PD, et al: Adverse
effects of antiaggregating platelet therapy in the treatment of polycythemia
vera. Semin Hematol 23:172, 1986
-
Wasserman LR, Gilbert HS: Surgical bleeding in polycythemia
vera. Ann N Y Acad Sci 115:122, 1964
-
Thomas DJ, du Boulay GH, Marshall J: Effect of haematocrit
on cerebral blood-flow in man. Lancet 2:941, 1977 [PMID
72286]
-
Pearson TC, Wetherley-Mein G: Vascular occlusive episodes
and venous haematocrit in primary proliferative polycythaemia. Lancet 2:1219,
1978 [PMID
82733]
-
Schafer AI: Bleeding and thrombosis in the myeloproliferative
disorders. Blood 64:1, 1984
-
Holme S, Murphy S: Platelet abnormalities in myeloproliferative
disorders. Clin Lab Med 10:873, 1990 [PMID
2272179]
-
Valla D, Casadevall N, Lacombe C, et al: Primary myeloproliferative
disorder and hepatic vein thrombosis: a prospective study of erythroid
colony formation in vitro in 20 patients with Budd-Chiari syndrome. Ann
Intern Med 103:329, 1985
-
Shamdas GJ, Spier CM, List AF: Myelodysplastic transformation
of polycythemia vera: case report and review of the literature. Am J Hematol
37:45, 1991
-
Silverstein MN: The evolution into and the treatment
of late stage polycythemia vera. Semin Hematol 13:79, 1976
-
Najean Y, Deschamps A, Dresch C, et al: Acute leukemia
and myelodysplasia in polycythemia vera. Cancer 61:89, 1988 [PMID
3334954]
-
Anagrelide, a therapy for thrombocythemic states: experience
in 577 patients. Anagrelide Study Group. Am J Med 92:69, 1992
-
Silver RT: Interferon-a2b:
a new treatment for polycythemia vera. Ann Intern Med 119:1091, 1993
-
Sacchi S, Leoni P, Liberati M, et al: A prospective
comparison between treatment with phlebotomy alone and with interferon-alpha
in
patients with polycythemia vera. Ann Hematol 68:247, 1994 [PMID 8018766]