Gastroenterología
⭳ Abrir artículo (PDF)1.1 MBEste artículo fue revisado respecto a la Edición 2/2000. Ver esa versión →
Contenido del artículo
IX CIRROSIS HEPATICA
- Manifestaciones clínicas
- Evaluación diagnóstica
- Formas específicas de cirrosis
- CIRROSIS ALCOHOLICA
- CIRROSIS POSNECROTICA
- CIRROSIS BILIAR PRIMARIA
- HEMOCROMATOSIS
- ENFERMEDAD DE WILSON
- DEFICIENCIA DE GLOBULINA a1-ANTITRIPSINA
- ESQUISTOSOMIASIS
- CIRROSIS RELACIONADA CON CORTOCIRCUITOS YEYUNO-ILEALES
- HIGADO GRASO Y CIRROSIS
- CIRROSIS CARDIACA
- CIRROSIS DIVERSAS
- Complicaciones de la cirrosis
- VARICES
- ASCITIS
- PERITONITIS BACTERIANA ESPONTANEA
- SINDROME HEPATORRENAL
- ENCEFALOPATIA HEPATICA
- LITIASIS VESICULAR
- ULCERA PEPTICA
- HIPOXIA
- DESNUTRICION
- CANCER PRIMARIO DEL HIGADO
- Trasplante
IX CIRROSIS HEPATICA
DR. PETER B. GREGORY
La cirrosis es la secuela de un gran número de enfermedades hepáticas crónicas y progresivas. Se desarrolla cuando estas enfermedades llegan a producir tal cicatrización del hígado que la arquitectura normal desaparece, formándose nódulos de regeneración parenquimatosa. El patrón de cicatrización difícilmente permite distinguir la etiología específica del proceso, pero otros cambios histológicos relacionados sí pueden aportar pistas. En la mayoría de las formas de hepatopatía, para establecer un diagnóstico específico generalmente se requiere de la combinación de la historia, exploración física, pruebas de laboratorio y la identificación de los rasgos histológicos característicos.
En los Estados Unidos, la ingesta excesiva de alcohol es con mucho la causa más frecuente de cirrosis hepática. En otros países es la hepatitis viral crónica el primer agente etiológico.
Manifestaciones clínicas
La fatiga, el malestar general y la astenia son comunes en todas las formas de cirrosis; sin embargo, estos síntomas no específicos también son parte del cuadro clínico de prácticamente todos los padecimientos hepáticos, tanto agudos como crónicos. Aunado a ello, no podemos hablar de alguna alteración física que indique sin lugar a dudas que el hígado es cirrótico. Entre los hallazgos característicos, pero no diagnósticos, se encuentran el eritema palmar (una lesión púrpura-rojiza sobre la región hipotenar de la mano) y las telangiectasias. Otros hallazgos típicos son ginecomastia, atrofia testicular, evidencia de hipertensión porta (esplenomegalia, ascitis, hemorroides, várices esofágicas y red venosa colateral en las venas del abdomen). Existen algunas manifestaciones que son frecuentes sólo en algunas formas específicas de cirrosis hepática: contractura de Dupuytren, xantelasma, xantomas, anillo de Kayser-Flescher, coloración bronceada de la piel e hiperpigmentación.
El hígado cirrótico es generalmente grande, y a menudo el lóbulo izquierdo es palpable debajo del apófisis xifoides. Sólo los pacientes con fases avanzadas e inactivas de la enfermedad tienen hígados pequeños y contraídos. Otra característica es la firmeza del órgano a la palpación, que incluso puede tener consistencia pétrea cuando el padecimiento está avanzado. En ocasiones pueden detectarse por palpación los nódulos de regeneración grandes en la superficie del hígado; pero las nodulaciones finas de la cirrosis micronodular son imposibles de detectar.
Evaluación diagnóstica
Es frecuente que la cirrosis solamente pueda ser detectada por medio de biopsia hepática.1 Este procedimiento puede realizarse con seguridad cuando no hay antecedentes de hemorragia anormal después de cirugías, procedimientos dentales o toma de biopsias, y cuando las pruebas de coagulación se encuentran en límites normales o sólo ligeramente anormales. Las recomendaciones más razonables son las siguientes: tiempo de protrombina con una relación internacional normalizada (INR) no mayor de 1.5, tiempo parcial de tromboplastina no mayor de 10 segundos en relación con el control, recuento plaquetario de por lo menos 50,000/mm3 y tiempo de sangrado normal. Bajo circunstancias excepcionales la biopsia hepática puede ser realizada con alteraciones mayores en los factores de la coagulación; sin embargo, antes del procedimiento debe hacerse una corrección vigorosa de cualquier alteración en la coagulación. Otras contraindicaciones relativas para la biopsia hepática son la falta de cooperación del paciente, la ascitis y la neumonía del lóbulo inferior derecho.
El hallazgo de una arquitectura hepática distorcionada, con nódulos regenerativos rodeados por tejido cicatricial, es concluyente sobre la existencia de cirrosis. Sin embargo, la biopsia hepática puede aportar diagnósticos falsos negativos, es decir, puede haber cirrosis aunque la biopsia no lo confirme.
La biopsia hepática ayuda también a identificar la causa. Particularmente la invación de los conductos biliares por tejido granulomatoso y su subsecuente destrucción, permite establecer el diagnóstico de cirrosis biliar primaria; el exceso de hierro en las células de los conductos biliares y en los hepatocitos identifica la hemocromatosis; una reacción hialina alcohólica relacionada con infiltrado polimorfornuclear indica hepatopatía alcohólica.
La disminución de la albúmina sérica y la prolongación del tiempo de protrombina son datos característicos de cirrosis, otras pruebas de función hepática pueden tambier ser anormales. La tomografía computada, la imagen por resonancia magnética (IRM) o el ultrasonido de hígado con estudio de flujo Doppler pueden demostrar datos compatibles con cirrosis. Entre estos destacan esplenomegalia, ascitis no sospechada, una superficie hepática irregular e inversión de la circulación porta ( en el ultrasonido ) o incluso várices intrabdominales. Además, la esofagoscopía demuestra con frecuencia la presencia de várices esofagogástricas.
La medición de la presión en cuña de la vena hepática puede ser de utilidad en el diagnóstico de cirrosis.2 Se desplaza un catéter a través de la vena cava hacia las suprahepáticas y se le enclava en una vena hepática distal pequeña; la presión así registrada corresponde a la intrahepática y es reflejo de la presión portal. En condiciones normales la presión en cuña no debe ser mayor de 4 mm Hg que en la vena hepática libre. Cuando la presión en cuña es normal a pesar de un cuadro clínico evidente de hipertensión porta, deberá pensarse en la posibilidad de obstrucción venosa prehepática. Cuando la presión en cuña está elevada, la posibilidad de cirrosis es muy alta.
La arteriografía puede ser útil cuando es difícil dilucidar la causa de un cuadro de hipertensión portal.3,4 En la fase arterial puede observarse distorsión típica de la arteria hepática en forma de sacacorchos; en la fase venosa se determinará la permeabilidad de las venas porta y esplénica. Pueden realizarse además tomas arteriográficas selectivas de las venas renal y mesentérica cuando se esté considerando la posibilidad de una cirugía con derivación.
En algunas instituciones se acostumbra realizar esplenoportografía con el fin de visualizar las raíces de las venas porta y esplénica. Este procedimiento, que implica puncionar el bazo e inyectar medio de contraste, permite obtener una medición directa de la presión venosa portal; sin embargo, tiene complicaciones en un cinco porciento de los casos y no permite la visualización de las venas renal y mesentérica.
La peritoneoscopía permite observar la superficie del hígado y realizar una biopsia dirigida;5 este es un procedimiento que tiene gran utilidad para la investigación de un posible proceso neoplásico.
Formas específicas de cirrosis
CIRROSIS ALCOHOLICA
Entre los sintomás de la cirrosis alcohólica están: cirrosis portal, de Laennec, nutricional y micronodular. Como el alcohol produce un efecto tóxico sobre el hígado, tal y como se ha demostrado en animales de experimentación, el término cirrosis alcohólica parece ser el más adecuado.6,7 La exploración física comúnmente demuestra que hay hepatomegalia, a veces muy grande; no es raro que en la autopsia se encuentren pesos mayores a 2,000 g. La hepatomegalia refleja inflamación, infiltración grasa y formación cicatricial extensa. El cuadro histológico característico consiste en una cicatriz lineal que separa al perénquima hepático y rodea pequeños nódulos de hepatocitos [ver figura 1]
La cirrosis alcohólica es consecuencia de la ingesta de grandes cantidades de alcohol; para su desarrollo no se requiere que exista desnutrición concomitante, aunque esta situación ocurre casi invariablemente. La desnutrición es sin lugar a dudas reflejo de la sustitución de las calorías normales de la dieta por alcohol.8 Todas las personas que beben consuetudinariamente desarrollan infiltración grasa leve, la cual es totalmente reversible cuando cesa la ingesta alcohólica.9 Aunque para el desarrollo de la cirrosis alcohólica suelen requerirse entre 10 a 15 años de beber intensamente (v.gr., más de 120 ml de whiskey al día), el padecimiento puede aparecer rápidamente. Se han informado casos de hepatopatía alcohólica fatal en adolescentes. Es típico que la cirrosis alcohólica aparezca como consecuencia de cuadros repetitivos de hepatitis alcohólica clínica y subclínica. El término hepatitis alcohólica se refiere al hallazgo histológico de hialinización rodeada por inflamación de células polimorfonucleares [ver figura 3]. Estas lesiones necróticas se acompañan de formación de colágena; en las etapas iniciales las lesiones se encuentran típicamente en una localización pericentral. Cuando el proceso es severo aparece cicatrización de la vena central (necrosis hialina esclerosante).10
Sólo unos cuantos de los casos en los que se demuestra hepatitis
alcohólica histológica se acompañan de un síndrome
clínico agudo, que requiere de hospitalización inmediata, y
consiste en fiebre de 38 C. o mayor, dolor e hipersensibilidad en el cuadrante
superior derecho del abdomen, hepatomegalia significativa, leucositosis e
ictericia notable. No todos estos datos aparecen en cada caso. La mortalidad en
estos pacientes va del 10 al 40 porciento. Aunque la gran mayoría de los
enfermos con hepatitis alcohólica demostrada por biopsia tiene cierto
grado de cirrosis concomitante, en algunos casos no es así.11 La mitad
de los sujetos sin cirrosis mostrarán morfología hepática
normal una vez que suspenda la ingesta alcohólica; el resto
desarrollará cirrosis a pesar de todo. En la hepatopatía
alcohólica es común encontrar telangiectasias, eritema palmar,
esplenomegalia y ascitis.
En los individuos con cirrosis alcohólica descompensada los niveles séricos de bilirrubinas están elevados, frecuentemente en forma importante (20 a 40 mg/dl). La aspartato aminotransferasa (AST, antes TGO) también se eleva, con frecuencia a niveles entre 200 y 300 UI/L. Es característico que la elevación en dicha aminotransferasa sea mayor que la de la alanino aminotransferasa (ALT, antes TGP). La inversión de esta relación o los niveles de aminotransferasas por arriba de 300 UI/L deben hacer pensar en otras posibilidades diagnósticas diferentes a la hepatopatía alcohólica. La fosfatasa alcalina suele elevarse sólo en forma ligera. Comúnmente coexiste disminución de la albúmina sérica por debajo de 3.5 g/dl y prolongación del tiempo de protrombina.
Conforme la enfermedad progresa, por la ingesta alcohólica continua y la desnutrición, el paciente pierde masa muscular en cara, cuello, hombros, antebrazos y manos; aparece pérdida de peso, que puede ser sustituida por el acúmulo de líquido de ascitis. Esta puede detectarse fácilmente por medio de la identificación de matidez cambiante en la exploración física; la onda líquida abdominal es patognomónica de ascitis, pero aparece únicamente cuando la cantidad de líquido acumulado es grande. El enfermo puede tener una red venosa colateral prominente, reflejo de aumento en la circulación a través de las venas periumbilicales. La hepatomegalia y la esplenomegalia son frecuentes. Aparecen eritema palmar y telangiectasias en cara, dorso, brazos y pecho. Hay hemorragias subcutáneas frecuentes como resultado de deficiencias vitamínicas y de la reducción en los factores de coagulación. También son comunes la ginecomastia (que puede ser dolorosa) y la atrofia testicular. Puede desarrollarse encefalopatía hepática. Cuando se les busca con intención, generalmente se encuentran várices.
El único tratamiento definido para los sujetos con hepatopatía alcohólica es la suspensión del hábito alcohólico. Los individuos con cirrosis alcohólica que continúan bebiendo tienden a tener peor pronóstico que los que abandonan dicho hábito. El índice de supervivencia a cinco años para pacientes que continúan bebiendo es menor al 40 porciento, pero puede llegar a ser del 60 al 70 porciento cuando se mantiene la abstinencia.12 Aunque el pesimismo y escepticismo son grandes, hasta un 30 porciento de los casos con hepatopatía alcohólica pueden llegar a mantener una abstinencia total.12 Por lo tanto, debe realizarse un gran énfasis terapéutico en apoyar los esfuerzos que el enfermo hace para dejar de beber. Actualmente existen diferentes grupos de rehabilitación, de apoyo mutuo y diversas técnicas psicoterapéuticas para ello.
Las importantes deficiencias nutricionales observadas en muchos pacientes con cirrosis alcohólica han causado que muchos médicos recomienden el apoyo nutricional durante la enfermedad aguda. Dos estudios extensos cooperativos de la Administración de veteranos en los EUA evaluaron el papel de los suplementos nutricionales enterales así como de la oxandrolona, un esteroide anabólico, en la enfermedad hepática alcohólica descompensada.13,14 Los investigadores demostraron una relación directa entre la ingesta calórica y la supervivencia y encontraron que el apoyo nutricional vigoroso aumentó la supervivencia, en especial en los pacientes con desnutrición importante.
Aunque existen varios estudios sobre el tratamiento con esteroides en la enfermedad hepática alcohólica descompensada, no hay datos convincentes ni constantes que demuestren su eficacia.11,15-20 Además, aunque se han empleado propiltiouracilo y colchicina en pacientes con enfermedad hepática alcohólica, no existen conclusiones que justifiquen el uso de estos medicamentos. Por lo tanto, el tratamiento de la cirrosis alcohólica debe ser de apoyo y dirigido a mejorar la nutrición, alentar la abstinencia y tratar las complicaciones.
CIRROSIS POSNECROTICA
La cirrosis posnecrótica se caracteriza por un hígado reducido que contiene nódulos de regeneración de tamaño variable [ver figura 4]. Sin embargo, el término posnecrótico es poco adecuado, ya que todas las cirrosis son consecuencia de necrosis de los hepatocitos. La causa más frecuente de esta entidad es la hepatitis crónica activa, aunque en muchos casos la causa no puede ser determinada, por lo cual se aplica el término de cirrosis criptogénica. Parecería que en tales casos la cirrosis se desarrolla como consecuencia de una hepatitis crónica activa subclínica de evolución lenta. La cirrosis posnecrótica puede encontrarse también en pacientes alcohólicos, en estos casos se piensa que la imagen patológica revela la remodelación regenerativa del hígado cirrótico micronodular después de que el paciente ha dejado de beber.
Con mayor frecuencia que en otras formas de cirrosis, en la posnecrótica la biopsia puede ser engañosamente normal.21 La explicación más posible es que la aguja de biopsia atraviesa una banda fibrosa y remueve el centro de un nódulo de regeneración, el cual puede tener una apariencia casi normal.
La cirrosis posnecrótica parece evolucionar en forma insidiosa, incluso cuando se encuentra aparentemente inactiva o cuando el tratamiento de la hepatitis crónica activa se considera exitoso.22 La causa habitual de muerte es la hemorragia gastrointestinal o la insuficiencia hepática. Cerca del 10 o 15 porciento de los casos con cirrosis posnecrótica desarrollan carcinoma primario de hígado.
Cuando la hepatitis asociada está activa, puede estar indicado utilizar esteroides. Sin embargo, si la cirrosis es avanzada y la evidencia de hepatitis es mínima, entonces no hay medidas terapéuticas que puedan utilizarse de manera específica; el tratamiento médico debe ser de apoyo y enfocado a las complicaciones.
CIRROSIS BILIAR PRIMARIA
La cirrosis biliar primaria es una hepatopatía poco común, que se presenta en mujeres entre los 30 y 50 años de edad.23 Se considera que los mecanismos inmunológicos juegan un papel preponderante en la patogenia de este padecimiento, ello en base a la presencia de autoanticuerpos séricos, la relación con otras enfermedades autoinmunes y la semejanza de las lesiones de los conductos biliares con aquellas que se observan en enfermedades crónicas por rechazo a injertos.24 Estudios recientes han demostrado que la cirrosis biliar primaria se asocia con el haplotipo HLA-DR8, lo que sugiere una predisposición genética para la enfermedad.25 Las manifestaciones iniciales son astenia y prurito generalizado; éste puede haberse iniciado meses o años antes de que el paciente busque atención médica por la presencia de piel pigmentada con tono parduzco, que se debe en parte al rascado crónico. La ictericia puede tardar en aparecer hasta cinco a 10 años a partir del inicio del prurito y de los síntomas constitucionales. Algunos enfermos desarrollan dolor óseo, fracturas múltiples y colapso vertebral. La causa habitual es la osteoporosis, que ocurre en 20 a 30 porciento de los pacientes y al parecer es secundaria a una notable reducción en la función de los osteoblastos.26 Con menor frecuencia puede existir osteomalacia concomitante. Entre los factores que condicionan las anomalías óseas están la malabsorción de calcio y fosfato, alteraciones en el metabolismo de la vitamina D, tratamiento con colestiramina y desnutrición. La exploración física revela con frecuencia xantelasma, xantomas, hepatoesplenomegalia, hiper-pigmen-tación y escoriación de la piel [ver figura 5]. Cuando la enfermedad está avanzada también se encontrará ictericia de conjuntivas, ascitis y edema.
Los hallazgos típicos de laboratorio incluyen la elevación de la fosfatasa alcalina (por lo general > a 300 UI/L y con frecuencia > 700 UI/L), colesterol sérico mayor de 300 mg/dl y anticuerpos antimitocondriales positivos (en 90 a 95 porciento de los casos).
Un número moderado de enfermos con cirrosis biliar primaria tiene enfermedades concomitantes como acidosis tubular renal, esclerodermia, síndrome de CREST o síndrome de Sjögren. Se han publicado algunos casos de cirrosis biliar primaria familiar.
La cirrosis biliar primaria puede tener características clínicas y de laboratorio semejantes a las de la cirrosis secundaria a enfermedad crónica de las vías biliares. De hecho, el cáncer de páncreas o de las vías biliares, la pericolangitis crónica secundaria a enfermedad intestinal inflamatoria y la colangitis esclerosante pueden tener datos de laboratorio e histológicos muy semejantes a la cirrosis biliar primaria. En algunos pocos casos es necesario observar en forma directa las vías biliares en los pacientes que tienen datos clínicos, de laboratorio e histológicos típicos de cirrosis biliar primaria.
La biopsia hepática puede mostrar destrucción de los conductos biliares con infiltración linfoplasmacitaria de espacios porta, formación periportal de granulomas y cicatrización con fusión de los espacios porta [ver figura 6]. La proliferación de los conductos es común. Cuando la cicatrización es extensa puede encontrarse formación de nódulos, frecuentemente con retención de las venas centrales. La estasis biliar es periportal en la mayoría de los casos, e indica que el padecimiento está avanzado.
Por desgracia, solo en pocos casos se encuentran los cambios histológicos propios de la cirrosis biliar primaria; en el resto los hallazgos son muy semejantes a los de la hepatitis crónica activa, por lo cual la distinción entre ambos padecimientos debe hacerse en base al cuadro clínico y a los datos de laboratorio.
El pronóstico es variable, pero la evolución suele ser insidiosa. La insuficiencia hepática severa no es frecuente sino hasta que la enfermedad se encuentra muy avanzada. El tiempo promedio de supervivencia para pacientes sintomáticos es de aproximadamente 10 años. Los pacientes asintomáticos pueden vivir más, aunque un estudio reportó un periodo promedio de supervivencia también de 10 años para estos pacientes.27-29
En el tratamiento de la cirrosis biliar primaria se han indicado los esteroides, la azatioprina, la penicilamina, la colchicina y el ursodiol. En un estudio aleatorio, un año de tratamiento con prednisolona mejoró los síntomas, la función hepática y los hallazgos histológicos, pero aceleró la pérdida de hueso.30 Debido a que la enfermedad ósea es frecuente y muchas veces incapacitante en los pacientes con cirrosis biliar primaria, no se recomienda el uso de esteroides a pesar de su aparente efecto benéfico sobre el daño hepático en sí. Se han realizado dos estudios aleatorios para valorar el tratamiento con azatioprina; en el primero, la administración de azatioprina en dosis de 2 mg/kg/día, no tuvo beneficios y se presentaron efectos colaterales en el 27 porciento de los pacientes.31 En el segundo, la azatioprina se administró en dosis más bajas (50 a 100 mg/día), observándose una tendencia al aumento en la supervivencia, pero se presentaron efectos adversos en el 11 porciento de los pacientes tratados.32 La penicilamina, en dosis de 600 a 1,000 mg/día, se evaluó en cinco estudios aleatorios prospectivos.33-37 En cuatro de ellos el fármaco no aumentó la supervivencia, y en el quinto el beneficio fue moderado.
Las complicaciones de la penicilinamina son comunes e incluyen: náusea, vómito, alteraciones en el sentido del gusto, estomatitis, fiebre, mialgias, artralgias, liquen plano, proteinuria y aplasia de médula ósea; incluso se han publicado casos de complicaciones mortales por el uso de este fármaco. Debido a la eficacia dudosa de la azatioprina y de la penicilamina en el tratamiento de la cirrosis biliar primaria, sus efectos colaterales tan frecuentes son factores en contra de su uso. Dos estudios clínicos aleatorios evaluaron la eficacia del tratamiento con colchicina, en dosis de 0.6 mg dos veces al día. Un grupo informó una mejoría leve en las pruebas de funcionamiento hepático y aumento en la supervivencia;38 el otro reportó que la supervivencia tiende a ser mayor.39 La colchicina produjo efectos colaterales leves, que se controlaron con facilidad en estos estudios al reducir la dosis. Aunque el uso de ciclosporina, un polipéptido cíclico de origen micótico con importantes propiedades inmunosupresoras, en pacientes con cirrosis biliar primaria causó mejoría clínica,40,41 se presentaron efectos adversos renales y de hipertensión, y el impacto sobre la supervivencia a largo plazo o la necesidad de trasplante hepático fue solo modesto. Se ha administrado también ursodiol, un ácido biliar hidrofílico, con la idea de que al alterar la composición del reservorio endógeno de ácidos biliares podría lograrse un efecto benéfico consistente en reducir la concentración de ácidos biliares hidrofóbicos endógenos potencialmente tóxicos. En un estudio extenso controlado con placebo, el tratamiento durante dos años con ursodiol en dosis de 13 a 15 mg/kg causó mejoría clínica, bioquímica e histológica con efectos colaterales leves.42 La mayoría de los pacientes reciben en la actualidad ursodiol porque parece ser tan eficaz como otros medicamentos y más seguro, aunque por desgracia este fármaco es costoso.
El tratamiento no específico debe dirigirse a aliviar los síntomas durante la evolución, lenta pero inexorable, de la enfermedad. La colestiramina, resina de intercambio aniónico, puede ayudar a disminuir el prurito, la dosis habitual es de 4 g VO tres veces al día; algunos enfermos pueden mejorar con dosis menores, mientras que otros requieren hasta 16 a 20 g/día. Es frecuente que la colestiramina sea poco tolerada, pero se acepta mejor si se administra con los alimentos o se mezcla con jugos o puré de fruta. La rifampicina (300 a 450 mg/día en dosis divididas), el ursodiol (10 a 15 mg/kg/día) e incluso el tratamiento con luz ultravioleta, han sido eficaces para aliviar el prurito severo.43 Los antihistamínicos rara vez son eficaces, pero su efecto sedante puede permitir que el paciente duerma a pesar del prurito continuo. La terfenadina no se utiliza ya para tratar el prurito en este grupo de pacientes porque ha causado arritmias graves en enfermos con daño hepático.
Se recomienda el uso de complementos vitamínicos. Debe administrarse diariamente una dosis de 5,000 unidades de vitamina A en un preparado hidrosoluble. Si el tiempo de protrombina se alarga, deberá administrarse vitamina K, también en una forma hidrosoluble, a razón de 5 mg/día. Los individuos que desarrollan osteomalacia deben recibir un metabolito activo de la vitamina D, ya sea 25-(OH)D3 (calcifediol) o 1,25-(OH)2D3 (calcitrol). Estos medicamentos han producido mejoría en algunos casos de osteomalacia,44 pero el beneficio que puedan aportar en los casos de osteoporosis aún está por demostrarse.45 Las dosis recomendadas para alteraciones óseas sintomáticas son de 0.5 a 1.0 µg de 1,25-(OH)2D3 o de 40 a 120 µg de 25-(OH)D3, al día. Debe administrarse calcio adicional a dosis de 1 o 2 g/día. Además, el autor recomienda que en pacientes asintomáticos se utilicen esquemas profilácticos con 0.25 µg de 1,25-(OH)2D3 o 20 µg de 25-(OH)D3 al día. Los niveles séricos de calcio deben ser vigilados durante los primeros meses de tratamiento con vitamina D y calcio.
HEMOCROMATOSIS
La hemocromatosis se desarrolla por el depósito de grandes cantidades de hierro en las células parenquimatosas hepáticas [ver figura 7].46 Este acúmulo causa la destrucción de células periportales y cicatrización hepática,lo cual produce cirrosis. El padecimiento ocurre predominantemente en varones (relación de 10:1). Los síntomas aparecen generalmente entre los 40 y 60 años en hombres o después de la menopausia en mujeres; ocasionalmente la enfermedad se hace aparente a una edad más temprana.
La hemocromatosis se hereda con caracter autosómico
recesivo. El gen afectado se localiza entre el locus HLA-A y el locus HLA-B del brazo corto del
cromosoma 6.47 Se calcula que la frecuencia del gen es de 0.056 y
hasta tres de cada 1,000 personas pueden ser homocigotas. Los heterocigotos
pueden tener alteraciones en el depósito de hierro, pero en
circunstancias normales no desarrollan un síndrome clínico. Si se
confirma el diagnóstico de hemocromatosis hereditaria, puede utilizarse
la tipificación del HLA para identificar a los familiares de primer
grado que son normales, heterocigotos y homocigotos.48
Los depósitos de hierro en páncreas y corazón producen disfunción de esos órganos. Al momento de aparición de los síntomas de hepatopatía, cerca de la mitad de los casos con hemocromatosis tiene ya diabetes mellitus, 15 porciento tienen insuficiencia cardiaca congestiva o arritmias y una minoría significativa se queja de rigidez y artralgias. También la impotencia es común, aparentemente debida a disfunción hipofisiaria.49
En las etapas avanzadas el examen físico mostrará coloración bronceada de la piel, producto del depósito de hierro y melanina. El hígado tiende a estar moderadamente crecido; cerca de la mitad de los pacientes tendrá esplenomegalia. En etapas menos avanzadas el color de la piel puede ser normal y el hígado ser apenas palpable. La mayoría de los casos cursan tarde o temprano con hipertensión porta. Entre 15 y 20 porciento de los individuos desarrolla carcinoma primario de hígado.
Los estudios de laboratorio muestran aumento del hierro sérico relacionado con una saturación de transferrina sérica del 80 a 90 porciento (el valor normal de saturación es de 15 a 47 porciento). También la ferritina sérica tiende a elevarse.50 La elevación del coeficiente de atenuación lineal promedio (número de la TC) en la tomografía computada de hígado puede indicar la presencia de depósitos excesivos de hierro en el hígado.51 La RM puede demostrar también el exceso de hierro y la hemocromatosis primaria.52 No son raros los aumentos discretos en la aminotransferasa y fosfatasa alcalina séricas, pero la ictericia es poco frecuente. La concentración sérica de albumina y el tiempo de protrombina tienden a permanecer normales hasta etapas avanzadas de la enfermedad.
La concentración sérica de hierro y la capacidad de fijación total de hierro pueden emplearse para realizar escrutinio de hemocromatosis en la población.53 Una saturación de transferrina en ayuno de 62 porciento o mayor en varones (y quizá de 50 porciento en mujeres) identifica a un alto porcentaje de pacientes que son homocigotos para la hemocromatosis. La concentración de ferritina sérica no ha demostrado ser un estudio útil para escrutinio porque muy pocos individuos homocigotos tienen niveles elevados antes de desarrollar la enfermedad clínica.
Al identificar a un individuo homocigoto se justifica el estudio de sus hermanos porque se espera que el 25 porciento sea homocigoto también. Los hermanos del mismo sexo suelen tener una cantidad comparable de hiero hepático a menos de que tengan antecedente de hemorragias o donaciones de sangre de importancia.54
El nivel elevado de hierro o de ferritina sérica no es diagnóstico, ya que dicho valor puede aumentar en todas las enfermedades hepáticas que se caracterizan por necrosis significativa de las células hepáticas. No es raro que las concentraciones séricas de hierro o ferritina se eleven en la hepatopatía alcohólica descompensada, en la hepatitis viral aguda o en la hepatitis crónica activa. Por lo tanto, la única manera en que puede establecerse el diagnóstico de hemocromatosis es mediante la biopsia hepática.55 El hallazgo característico es el importante depósito de gránulos de hemosiderina en los hepatocitos y las células de los conductos biliares. La fibrosis puede ser desde mínima hasta una cirrosis franca. En ocasiones es difícil distinguir entre una hemocromatosis primaria y una cirrosis con saturación secundaria de hierro. La hemocromatosis primaria se caracteriza por predominio del hierro parenquimatoso en relación con la cantidad de tejido cicatricial, así como por la presencia de hierro en los conductos biliares. La sobrecarga de hierro secundaria a una cirrosis subyacente generalmente se relaciona con una cirrosis avanzada, en la cual predomina el tejido cicatricial sobre el hierro y los conductos biliares están libres de este metal. Cuando el exceso de hierro hepático proviene de una fuente exógena, como transfusiones repetidas para manejo de hemólisis crónica, los depósitos de hierro predominarán en las células de Kupffer. Cuando las características morfológicas de la biopsia hepática no permiten distinguir en forma clara entre una hemocromatosis primaria y la sobrecarga secundaria de hierro, el análisis cuantitativo del contenido hepático de hierro puede ser útil.56 En los pacientes con hemocromatosis las muestras de tejido obtenidas de corazón, páncreas o piel mostrarán también grandes depósitos de hierro.
La detección y el tratamiento temprano en los pacientes con hemocromatosis primaria es indispensable. El tratamiento habitual consiste en la eliminación del exceso de hierro por medio de flebotomías semanales.57 Debido a que cada 50 ml de sangre contiene 250 mg de hierro, la extracción de esa cantidad por semana permitirá agotar las reservas en uno a dos años. El objetivo del tratamiento es mantener niveles persistentemente bajos de hierro en suero y ausencia de éste en la biopsia. A partir de entonces pueden hacerse flebotomías subsecuentes cada dos o tres meses para evitar reacúmulo de hierro. Si los pacientes se detectan antes de que se desarrolle la cirrosis y la remoción del hierro corporal total se lleva a cabo en forma satisfactoria, la esperanza de vida se aproxima a lo normal.58 Los pacientes con cirrosis establecida que reciben tratamiento evolucionan mejor que los pacientes no tratados, pero siguen teniendo riesgo de desarrollar cáncer primario de hígado años después de la remoción satisfactoria del hierro. La incapacidad para reducir los depósitos de hierro después de 18 meses de tratamiento es un signo de mal pronóstico. Los signos de hepatopatía ceden en el 70 porciento de los pacientes tratados, pero las alteraciones endócrinas y la artropatía mejoran sólo en un 20 porciento de los casos tratados.
Las flebotomías repetidas resultan obviamente imprácticas en el manejo de la sobrecarga de hierro resultante del tratamiento de las anemias hemolíticas crónicas. En tales casos, la deferoxamina utilizada por vía subcutánea en dosis de 1 a 3 g cada 12 horas produce una pérdida urinaria promedio de hierro de 50 mg por día.59 La adición de ácido ascórbico, 500 mg al día por vía oral, puede duplicar la cifra de excreción urinaria de hierro.
ENFERMEDAD DE WILSON
La enfermedad de Wilson, denominada también degeneración hepatolenticular, es un padecimiento autosómico recesivo que se desarrolla con una frecuencia aproximada de 1 por millón.60 El defecto genético parece encontrarse en el cromosoma 13, banda 14.3, en donde se han identificado cuatro mutaciones específicas de la enfermedad en un gen que codifica para la proteína trifosfatasa de adenosina (ATPasa) tipo P fijadora de cobre.61 La enfermedad parece deberse a una excreción de cobre inadecuada a través de la bilis, lo cual provoca acúmulo de cantidades excesivas de cobre en la mayoría de los tejidos corporales. También está afectada la incorporación de cobre a la ceruloplasmina. La mayoría de los sujetos afectados ya han sufrido algunos síntomas de la enfermedad a los 15 años de edad, producto de disfunción neurológica o hepática. Aunque ha ocurrido que la enfermedad de Wilson tenga sus manifestaciones iniciales hasta los 30 años de edad, estos casos son excepcionales (El autor conoce un paciente que presentó hepatomegalia asintomática a los 62 años de edad). Las manifestaciones iniciales son atribuibles a disfunción hepática en el 40 porciento de los casos. La afección hepática se manifiesta habitualmente como un padecimiento crónico caracterizado por fatiga, ictericia, telangiectasias, ascitis, edema, esplenomegalia y hemorragia por várices esofágicas. La asociación de anemia hemolítica es una de las claves para el diagnóstico. En ocasiones la afección hepática puede semejarse a una hepatitis aguda severa, causando la muerte en días o semanas. Las principales manifestaciones neurológicas incluyen temblores, rigidez, alteraciones en la marcha e incoordinación de movimientos, lenguaje incomprensible y cambios en la personalidad. También se han detectado casos de hipoparatiroidismo.63
El signo patognomónico es el anillo de Kayser-Fleischer, que consiste en un anillo delgado, parduzco, que se encuentra en la periferia de la córnea. Aunque la pigmentación abarca habitualmente toda la circunferencia, puede estar localizada solamente en la parte superior o inferior. En etapas tempranas de la enfermedad puede requerirse de una lámpara de hendidura para detectar dicho anillo, que puede ser particularmente difícil de identificar en la exploración física rutinaria en pacientes con ojos de color café.
Al momento de la detección del padecimiento, por lo menos 50 porciento de los enfermos tienen hepatoespleno-megalia y alteraciones moderadas en las pruebas de funcionamiento hepático. Dos hallazgos de laboratorio con carácter distintivo son la disminución o ausencia de ceruloplasmina sérica y el aumento en la excreción urinaria de cobre, desde el valor normal de menos de 50 µg/día hasta niveles tan elevados como 1,000 µg/día. Hay un pequeño porcentaje de casos con enfermedad de Wilson que pueden tener niveles séricos de ceruloplasmina normales o excreción normal de cobre en orina y ausencia de anillo de Kayser-Fleischer. Es por ello recomendable que se evalúen siempre los tres factores, ya que por lo menos uno de ellos será anormal. En los casos problemáticos, el encontrar un exceso de cobre urinario después de la administración de 1,000 mg de penicilamina ayudará a establecer el diagnóstico.64 Si persiste la duda, el estándar final puede ser un aumento en la concentración de cobre en el tejido hepático, aunque este resultado es concluyente solo si el paciente no tiene colestasis de larga evolución, que también puede aumentar la concentración hepática de cobre.
El tratamiento de la enfermedad de Wilson requiere de la administración de D-penicilamina, agente quelante que se une al cobre y promueve la excreción urinaria de 1,000 a 3,000 µg/día.65 La dosis habitual es de 1 g/día. La mejoría clínica se acompaña de disminución de las reservas de cobre en los tejidos. El tratamiento con D-penicilamina se asocia con efectos colaterales importantes, sobre todo náusea y molestia abdominal que ocurren después de la ingesta del medicamento; entre los más serios están la leucopenia y la anemia, que pueden implicar en algunos casos la existencia de anemia aplásica. Un porcentaje pequeño de enfermos desarrolla síndrome nefrótico. Todos los individuos con enfermedad de Wilson tratados con el medicamento en cuestión deben ser vigilados estrechamente con análisis de orina y biometrías hemáticas, sobre todo durante los primeros meses del tratamiento. Debido a que la D-penicilamina es un antagonista de la piridoxina, deberán administrarse 50 mg de piridoxina por semana. Cuando la D-penicilamina no sea tolerada deberá considerarse el tratamiento con zinc oral. El zinc elemental puede administrarse en forma de acetato en una dosis total de 150 mg al día, en dosis divididas administradas cuando el estómago esté vacío. El tratamiento con zinc aumenta las pérdidas fecales de cobre e induce un balance negativo del metal en los pacientes con enfermedad de Wilson.66 Sin embargo, el inicio de su acción es bastante tardío y se desconocen los beneficios que el tratamiento a largo plazo pueda tener.
DEFICIENCIA DE GLOBULINA a1-ANTITRIPSINA
La deficiencia homocigota de globulina a1-antitripsina se relaciona con un síndrome raro en el cual existe cirrosis progresiva.67 Aunque originalmente se describió en niños con cirrosis juvenil, también en adultos se ha reportado la combinación de deficiencia de esta proteína y cirrosis portal. Los adultos desarrollan además enfisema concomitante.68 El diagnóstico de deficiencia de globulina a1-antitripsina debe sospecharse en todos los casos en los cuales sea difícil dilucidar la causa de una cirrosis hepática.
En las fases iniciales la mayoría de los enfermos tienen hepatomegalia moderada y alteraciones ligeras de la función hepática. La ausencia de globulina a1-antitripsina en la electroforesis de proteínas hace que el diagnóstico sea muy probable. La medición específica de sus niveles en sangre confirma el diagnóstico. Se han encontrado variantes genéticas que reflejan la existencia de por lo menos 25 diferentes alelos para el gen que controla la producción de la proteína.69 El genotipo que habitualmente se relaciona con cirrosis y enfisema ha sido denominado inhibidor de proteasa tipo ZZ (PiZZ). Una sustitución de aminoácido en la variante Z de la proteína permite que las moléculas de a1-antitripsina polimericen dentro del hepatocito, alterando así la excreción de la proteína del hígado. En la biopsia pueden identificarse los cuerpos de inclusión PAS positivos (diastasa-resistentes) anormales y característicos que contienen globulina a1-antitripsina [ver figura 8]. Una proporción significativa de los pacientes homocigotos para PiZZ con enfermedad hepática crónica tienen también evidencia de infección por virus de la hepatitis B o C.70
ESQUISTOSOMIASIS
La cirrosis secundaria e infestación por esquistosoma, rara en los Estados Unidos, es relativamente frecuente en el lejano oriente, Egipto y algunas zonas de Sudamérica.71 En la esquistosomiasis, los huevecillos depositados en las raíces portales desencadenan una reacción granulomatosa fibrótica que ocasionalmente causa fibrosis difusa periportal o a la llamada fibrosis en pipa. La manifestación inicial es generalmente la hemorragia de várices esofágicas; la exploración física mostrará hepatoesplenomegalia prominente. Rara vez se observan estigmas de hepatopatía crónica o de insuficiencia hepática. El tratamiento de la esquistosomiasis no siempre da buenos resultados, sobre todo en etapas avanzadas.72
CIRROSIS RELACIONADA CON CORTOCIRCUITOS YEYUNO-ILEALES
En todos los pacientes sometidos a cortocircuitos yeyuno-ileales hay acumúlo de grasa en el hígado durante el periodo de pérdida rápida de peso que sigue a la cirugía.73 Un número reducido de enfermos desarrollará una hepatopatía progresiva que es indistinguible de la cirrosis alcohólica; generalmente hay hepatomegalia y la función hepática se va deteriorando progresivamente.74,75 Se desconoce la causa de esta grave complicación. Se ha informado sobre un síndrome similar secundario a gastroplastía.76 Aunque algunos comunicados indican que hay mejoría de la función hepática al someter a los sujetos a hiperalimenta-ción parenteral, el único tratamiento efectivo es la reanastomosis del intestino o la corrección de la gastroplastía. Cuando este procedimiento no se efectúa rápidamente, el deterioro de la función hepática puede ser mortal.
HIGADO GRASO Y CIRROSIS
Aunque existen pocas evidencias que indican que el acúmulo de grasa en el hígado puede producir cirrosis, hay informes de un tipo idiopático de cirrosis relacionada con aumento en la grasa hepática.77,78 Esta cirrosis parece ser secundaria a una esteatohepatitis no alcohólica, padecimiento raro que es más frecuente en mujeres y se asocia con obesidad, hiperlipidemia y diabetes mellitus.79 Con frecuencia estos pacientes tienen hepatomegalia considerable, pero las alteraciones funcionales son solamente discretas. Los estigmas de hpatopatías son comunes cuando se ha establecido la cirrosis, pero no en etapas más tempranas. Se desconocen los mecanismos de daño hepático, aunque la enfermedad parece comenzar por un mal control de la diabetes o por la reducción de peso demasiado rápida. La evolución es gradual y no existe un tratamiento específico disponible.
CIRROSIS CARDIACA
La cirrosis cardiaca se ha vuelto cada vez menos frecuente a partir de que se han podido corregir las valvulopatías mediante el remplazo con prótesis. El padecimiento se desarrolla después de años o décadas de insuficiencia cardiaca derecha severa o de insuficiencia tricuspídea. El cuadro histológico consiste en la cicatrización central con conexión entre venas centrales por tejido fibroso, así como la eventual formación de nódulos que frecuentemente contienen una triada portal. El hígado suele estar crecido y puede demostrarse reflujo hepatoyugular. El tratamiento de la cirrosis cardiaca implica la corrección de las alteraciones valvulares que le han dado origen.
Algunos enfermos con síndrome de Budd-Chiari (trombosis de las venas suprahepáticas) pueden sobrevivir al episodio agudo, desarrollando una cirrosis similar a la cardiaca.80
CIRROSIS DIVERSAS
Puede presentarse cirrosis secundaria a exposición crónica a arsénico, methotrexate o a cantidades excesivas de vitamina A.81-83 También puede ocurrir cirrosis después de una enfermedad de vías biliares prolongada, como la colangitis esclerosante, y como una complicación rara de la sarcoidosis hepática. Además, varios padecimientos exclusivos de la niñez pueden desarrollar cirrosis, los más comunes de éstos son la fibrosis quística, las enfermedades por depósitos de glucógeno, la atresia biliar y la fibrosis hepática congénita.84-87
Complicaciones de la cirrosis
VARICES
La hemorragia por várices es una de las complicaciones más graves de la cirrosis. La mortalidad durante el episodio agudo puede ser hasta de 60 a 70 porciento.88 Hay muchos factores relacionados con una cirrosis descompensada que incrementan este riesgo de por sí alto, incluyendo la debilidad generalizada, los defectos de la coagulación y la encefalopatía hepática. El tamaño de las várices correlaciona también con el riesgo de hemorragia.89 La hemorragia recurrente, común en las dos semanas que siguen al episodio inicial, también contribuye a la gran mortalidad. Cuando el paciente sobrevive más allá de seis semanas, el riesgo de hemorragia recurrente disminuye sustancialmente, acercándose al de los cirróticos que nunca han sangrado.90 El sangrado por várices esofágicas se identifica mejor por medio de una endoscopía de la porción superior del tubo digestivo.
La escleroterapia, inyección de un agente esclerosante en las várices esofágicas, es el tratamiento de elección para el control inmediato de la hemorragia por várices. La escleroterapia también es eficaz para el control a largo plazo de la hemorragia recurrente, aunque el efecto de este tratamiento en la supervivencia aún es incierto.91-93 El agente esclerosante causa inflamación necrosante severa de la pared esofágica, seguida de importante reacción fibrótica.94 Las complicaciones son frecuentes, e incluyen dolor retroesternal, ulceración esofágica, hemorragia, derrame pleural y estenosis y perforación esofágicas. La ligadura esofágica es una variante de la escleroterapia semejante a la ligadura de las hemorroides y que se realiza con un endoscopio modificado. En tres estudios aleatorios, la ligadura de várices las obliteró con más rapidez que la escleroterapia, y fue tan eficaz como esta última para controlar la hemorragia. Este procedimeinto también es eficaz en las várices esofágicas y puede usarse como una alternativa a la escleroterapia o cuando ésta no ha sido eficaz.95-97 Sin embargo, el uso del tubo exterior, que se emplea para facilitar la ligadura repetida rápida, se ha asociado con una incidencia excesiva de perforación esofágica, por lo que ha sido retirado del mercado por el fabricante. Esto impide el uso eficaz del tratamiento de ligadura para las várices esofágicas con sangrado activo.
La hemorragia por várices gástricas es menos frecuente en pacientes con cirrosis, pero más difícil de tratar en forma eficaz, excepto por cirugía.98
Si la hemorragia persiste o recurre, la colocación de una sonda de Sengstaken-Blakemore podrá interrumpirla, cuando menos temporalmente, en más del 90 porciento de los casos.99 Muchas de las dificultades relacionadas con este recurso pueden ser evitadas si el paciente es vigilado en una unidad de cuidados intensivos. El procedimiento adecuado implica colocar la sonda a través de la nariz o la boca, inflar el balón gástrico con 250 o 300 ml. de aire y colocarlos ejerciendo presión contra la unión cardioesofágica. En muchos pacientes este medio por sí solo sera suficiente para detener la hemorragia; cuando no sea así, debe inflarse el balón esofágico a una presión de 30 a 40 mm. Hg. Es conveniente colocar una sonda de succión nasogástrica por arriba del balón esofágico para prevenir el acúmulo de sangre y moco. Dado que la posibilidad de complicaciones por el uso de esta sonda aumenta en relación al tiempo en que el balón esofágico esté inflado, se deben hacer intentos por reducir su presión tras un periodo de 24 hrs.; si la hemorragia se ha detenido, la sonda podrá retirarse en otras 24 horas.
La vasopresina intrarterial puede reducir temporalmente e incluso detener la hemorragia; sin embargo, no mejora el índice de supervivencia y su administración requiere de procedimientos angiográficos especializados que rara vez están disponibles. La administración de vasopresina por infusión intravenosa continua a pacientes con hemorragia activa no parece ser efectiva.100
La obliteración transhepática percutánea de las várices puede controlar una hemorragia varicosa activa en 70 porciento de los casos; sin embargo, las recurrencias son muy frecuentes una vez que se ha terminado con el procedimiento.101
Las hemorragias recurrentes o continuas pueden ser indicación para realizar una derivación portosistémica. Esta cirugía mayor se relaciona con una mortalidad del 40 porciento cuando se practica en forma urgente.102 Cuando el sangrado se detiene y la cirugía se puede realizar en forma electiva, la mortalidad se reduce sustancialmente. Aunque las derivaciones portosistémicas no mejoran la supervivencia, sí reducen los episodios de hemorragia en forma satisfactoria. La complicación mayor de este tipo de cirugía es la insuficiencia hepática con encefalopatía intratable.103 El procedimiento quirúrgico de elección será aquel con el cual el cirujano esté más familiarizado. Aunque las derivaciones grandes son más efectivas para prevenir nuevas hemorragias, las pequeñas se relacionan con una menor incidencia de insuficiencia y encefalopatía hepática; sin embargo, estas derivaciones tienen una mayor tendencia a trombosarse, lo cual causará nuevos episodios de hemorragia. Una derivación esplenorrenal distal con devasculariza-ción selectiva gastroesofágica concomitante, provee una descompresión selectiva de las várices esofágicas al mismo tiempo que mantiene el flujo sanguíneo mesentérico al hígado. La mayoría de los estudios demostraron que la incidencia de encefalopatía severa se reducía significativa-mente en los dos o tres años posteriores a la cirugía cuando se utilizaba la derivación esplenorrenal distal; ello comparándola con los métodos convencionales de derivación.104-106. Sin embargo, el procedimiento tiene una gran dificultad técnica, y sólo el tiempo mostrará si tiene ventajas a largo plazo.
Se han hecho trabajos comparando las derivaciones portocava de urgencia con la escleroterapia en pacientes con hepatopatía alcohólica muy descompensada y hemorragia activa de várices esofágicas.107 Aunque los requerimientos transfusionales fueron inicialmente mayores en los enfermos sometidos a derivación, la recurrencia subsecuente de hemorragias fue mucho menor en esos pacientes que en los sometidos a escleroterapia. La incidencia de encefalopatía fue un poco mayor en los casos con derivación, pero los índices de supervivencia fueron semejantes para ambos grupos.
El propranolol produce reducciones sustanciales de la presión portal en pacientes con cirrosis, por lo que es de esperarse que disminuya el riesgo de hemorragia por várices esofágicas.108 En un estudio se encontró una reducción notable en los episodios de resangrado y mejoría en la supervivencia a dos años cuando se administraba propranolol en una dosis suficiente como para reducir la frecuencia cardiaca en reposo en un 25 porciento.109 Sin embargo, otro trabajo no corroboró dicha disminución en los episodios de hemorragia utilizando dosis similares y se encontró además que el bloqueo beta producido por el propranolol hacía sumamente díficil la reanimación de los enfermos con hemorragia.110 En sujetos susceptibles, el medicamento puede además inducir el desarrollo de encefalopatía hepática.111 Este medicamento parece ser menos eficaz que la escleroterapia para prevenir el sangrado recurrente por várices esofágicas.112 Tanto la escleroterapia como el propranolol tienen efectos adversos significativos, y la elección de uno u otro tratamiento dependerá de la tolerancia y cumplimiento por parte del paciente.
La colocación de una derivación portosistémica intrahepática a través de la vía transyugular (conocida como procedimiento TIPS por sus siglas en inglés, n. del t.) se ha convertido en una técnica muy aceptada para el tratamiento de las várices esofágicas sangrantes.113 Este procedimiento crea una derivación quirúrgica, pero evita las complicaciones de la cirugía mayor. Sus beneficios y complicaciones a largo plazo se desconocen todavía. Parece razonable dejar esta forma de tratamiento a las instituciones que puedan reunir una experiencia de magnitud considerable y para los pacientes que sean malos candidatos para cirugía y refractarios a la escleroterapia.
Debido a que el primer episodio de hemorragia por várices puede tener una gran morbimortalidad, ha existido gran interés en relación con el tratamiento profiláctico de las várices esofágicas. Las derivaciones portosistémicas profilácticas disminuyen el sangrado recurrente, pero no aumentan la supervivencia.114 En varias instituciones se han realizado estudios sobre escleroterapia profiláctica con resultados contradictorios.115-118 En el estudio más extenso, que se limitó a pacientes alcohólicos, este procedimiento fue incluso dañino.119 La experiencia con los antagonistas beta adrenérgicos propranolol y nadolol ha sido alentadora porque ambos medicamentos parecen prevenir el primer episodio de hemorragia y reducir la mortalidad asociada con hemorragia en los pacientes con várices esofágicas moderadas o graves.120 Si un paciente que se sabe tienen várices de gran tamaño está bien motivado y tolera el medicamento, debe considerarse administrarle antagonistas beta adrenérgicos. El mononitrato-5 de isosorbide, un nitrato de acción prolongada, puede también ayudar a prevenir la primera hemorragia por várices.121 Sin embargo, este medicamentos debe emplearse con precaución porque daña la función renal, en especial en pacientes con ascitis.122
El tratamiento de las várices esofágicas incluye también el manejo de sostén habitual, con infusión de líquidos intravenosos para restablecer el volumen plasmático y el balance de electrolitos y administración de sangre. Cuando las alteraciones de la coagulación son importantes deberá administrarse también vitamina K por vía subcutánea, plasma fresco congelado y plaquetas.
ASCITIS
La ascitis es una secuela habitual en muchas formas de cirrosis, y puede ser detectada cuando se encuentra matidez cambiante o una onda líquida en la exploración abdominal. En ocasiones la ascitis debe sospecharse en pacientes con derrame pleural derecho.123 Los factores que contribuyen a la formación de ascitis son la hipertensión porta, la disminución en la albúmina sérica con la consecuente pérdida de presión oncótica vascular e intersticial y la retención renal de sodio y agua.
Aunque la ascitis de origen infeccioso, pancreático o neoplásico no es frecuente, no deben olvidarse estas posibilidades porque el tratamiento y pronóstico son diferentes para cada situación.124 Para excluir esas posibilidades debe obtenerse una muestra de líquido de ascitis por medio de una aguja delgada. La apariencia macroscópica del líquido puede sugerir una etiología poco habitual: por ejemplo, el líquido turbio sugiere una etiología infecciosa, el hemorrágico, un tumor, y el líquido lechoso, obstrucción linfática. Los estudios de laboratorio del líquido deben incluir citología diferencial, determinaciones de proteínas y amilasa, citología y cultivo. En la ascitis trasudativa secundaria a cirrosis, la cuenta total de proteínas es menor a 2.5 g/dl, la cuenta leucocitaria es menor a 300/mm3, la proporción de granulocitos es menor al 30 porciento, la amilasa está en un nivel menor al sérico y tanto la citología como el cultivo son negativos. Alrededor del cinco porciento de los pacientes con ascitis atribuible a cirrosis tienen una concentración total de proteínas mayor a 2.5 g/dl.
El tratamiento de la ascitis no complicada secundaria a cirrosis debe ser directo.125 En primer lugar, deben suspenderse todos los medicamentos que inhiben la síntesis de prostaglandinas, ya que reducen la velocidad de filtración glomerular, la excreción urinaria basal de sodio y bloquean la respuesta natriurética a los diuréticos.126,127 La indometacina y la aspirina son los inhibidores de prostaglandinas más utilizados. Una vez que esos fármacos han sido suspendidos, debe instituirse la restricción de agua y sodio en la dieta. Aunque en medios hospitalarios puede mantenerse una restricción extrema de agua y sodio, tal medida no es generalmente necesaria, ya que no podrá ser mantenida en el hogar del enfermo. Una dieta en la que se limita el sodio a 2 g por día y el agua a 2,000 ml suele ser bien tolerada.128 Cuando la restricción de sodio y el reposo no inducen diuresis, el diurético de elección será la espironolactona.129 Setenta y cinco porciento de los sujetos hospitalizados por ascitis mejoran sustancialmente con el simple uso de espironolactona. La eficacia de este medicamento en cirróticos con ascitis contrasta con su débil efecto en quienes tienen retención hídrica de otro origen. Dado que la espironolactona inhibe la acción de la aldosterona, tiende a prevenir la excreción renal de potasio (efecto farmacológico deseable en la cirrosis). Sin embargo, por esta razón el medicamento no es recomendable para enfermos con insuficiencia renal. El uso a largo plazo de dosis altas produce ginecomastia en 20 a 30 porciento de los pacientes.
La espironolactona debe administrarse a una dosis inicial de 50 mg dos veces al día y puede incrementarse a 100 mg dos veces al día a los dos a tres días si no ha producido diuresis. Aunque la espironolactona puede administrarse hasta a 400 mg/día o más, el medicamento se tolera poco en estas dosis. Por lo tanto, si con 100 mg dos veces al día no se obtiene una diuresis adecuada, es mejor añadir furosemide, 40 mg por la mañana. La dosis de furosemide puede aumentarse cada día 40 mg (en una dosis por la mañana) hasta lograr la diuresis. La mayoría de los pacientes comienza a responder antes de que se alcancen dosis de 120 a 160 mg de furosemide y 200 mg de espironolactona. El objetivo es lograr una diuresis adecuada con las menores dosis posibles.
La diuresis máxima del líquido de ascitis no debe exceder los 1,000 ml/día.130 En base a ello, la pérdida ponderal en pacientes cirróticos no debe ser mayor a 0.5 a 1 kg/día. Una diuresis mayor conduce a la disminución peligrosa de los volúmenes intravascular e intracelular, especialmente en los individuos con edema periférico mínimo o ausente.
Entre las complicaciones del tratamiento diurético están las alteraciones electrolíticas severas, la encefalopatía, la uremia y la deshidratación. Estas son más comunes cuando la diuresis ha sido exagerada o cuando se ha continuado el tratamiento y el paciente ya estaba libre del exceso de líquido corporal.
En cerca del 5 porciento de los casos no se logrará obtener una respuesta diurética a pesar de utilizar dosis máximas o dicha respuesta se logrará sacrificando la función renal. En tales enfermos deberá considerarse la posibilidad de colocar una derivación peritoneovenosa de LeVeen.131 Dicha derivación hace pasar el líquido de ascitis del peritoneo a la vena yugular interna por medio de un tubo subcutáneo que tiene una válvula de una sola vía. Comparado con el tratamiento médico aislado, la derivación peritoneovenosa permite una resolución más rápida de la ascitis, estancias hospitalarias más breves y recurrencia más tardía de la ascitis. Además, la ascitis refractaria puede resolverse con la aplicación de dicha derivación, aunque el procedimiento no parece alterar la supervivencia. Debido a que la mayoría de los pacientes continúan requiriendo de diuréticos, aunque a dosis menores, lo más probable es que la derivación ejerza su efecto benéfico aumentando el flujo renal.
En los pacientes con derivación pueden desarrollarse complicaciones graves, e incluso mortales, incluyendo infección bacteriana del peritoneo, coagulación intravascular diseminada y ruptura de várices esofágicas. Además, la derivación puede trombosarse y requerir un remplazo. La derivación no debe ser colocada en los enfermos que puedan ser controlados con restricción dietética y medicamentos; tampoco debe utilizarse cuando no se espera que la ascitis tenga una duración prolongada, como es el caso en episodios de hepatitis alcohólica o después de una cirugía de derivación portocava. En general, no se debe considerar que un individuo tiene ascitis refractaria hasta que ésta persista por varios meses. Finalmente, no hay motivo para colocar una derivación peritoneovenosa a menos que el sujeto esté incapacitado seriamente por la ascitis crónica.
La paracentesis de grandes volúmenes se ha vuelto un procedimiento popular para el tratamiento de pacientes con ascitis. En un estudio se realizaron con seguridad paracentesis de 4 a 6 L/día, con estancias hospitalarias más cortas y menos complicaciones que con el tratamiento diurético convencional.132 Estudios subsecuentes han demostrado que la administración de 40 g de albúmina por vía intravenosa después de cada procedimiento evita la insuficiencia renal y la hiponatremia inducidas por la paracentesis.133 Por lo general los pacientes aceptan las paracentesis porque alivian sus molestias en forma importante. También proporcionan al médico líquido de ascitis para fines diagnósticos.
Se ha comparado la derivación peritoneovenosa con las paracentesis repetidas de grandes volúmenes para el tratamiento de la ascitis refractaria.134 Ambos procedimientos fueron eficaces para aliviar la ascitis. La derivación permitió un control a más largo plazo, pero con frecuencia tuvo que remplazarse y no proporcionó ventajas de supervivencia.
Se han realizado anastomosis portocava para el tratamiento de la ascitis refractaria, pero este procedimiento no puede ser recomendado mas que para casos extremos porque se asocia con una mortalidad y morbilidad muy elevadas. Por lo general es preferible recurrir a la derivación peritoneovenosa, que es más simple.
PERITONITIS BACTERIANA ESPONTANEA
Se desarrolla peritonitis bacteriana espontánea en el 10 a 25 porciento de los pacientes cirróticos que se vigilan en forma prospectiva por lo menos durante un año.135,136 Habitualmente la cirrosis se encuentra avanzada y activa, manifestada por encefalopatía hepática, várices esofágicas e ictericia marcada. La incidencia de peritonitis bacteriana espontánea es especialmente alta en pacientes con concentraciones de proteínas en el líquido de ascitis menores a 1.0 g/dl y de bilirrubinas séricas por arriba de 2.5 mg/dl. Estos hallazgos podrían explicar el mayor riesgo de infección del líquido de ascitis porque la actividad antibacteriana del líquido de ascitis, medida por actividad de opsoninas, es proporcional a la concentración de proteínas en el mismo. Se desconoce la patogenia exacta; se supone que existe siembra hematógena del líquido de ascitis, el cual funciona como un medio ideal de cultivo. La cirrosis indudablemente facilita el proceso permitiendo que los organismos entéricos penetren a la circulación venosa portal a través de las colaterales portosistémicas, evitando así el sistema reticuloendotelial del hígado.
El cuadro típico de peritonitis bacteriana se manifiesta por la presencia de fiebre, leucocitosis periférica, dolor abdominal, peristalsis disminuida o ausente y rebote. No todos los enfermos tienen estos datos, y algunos no tendrán ni siquiera uno de ellos. Por ello, se debe realizar un estudio de líquido de ascitis siempre que las condiciones de un cirrótico empeoren súbitamente.
El líquido de ascitis se verá turbio debido a la leucocitosis y a la proliferación bacteriana. Son comunes las cifras de leucocitos superiores a 1,000 células/mm3 con más del 85 porciento de granulocitos.137 Prácticamente todos los casos tienen más de 300 leucocitos/mm3 y más de la mitad de estas células corresponderán a polimorfonucleares. Sin embargo, no todos los pacientes con leucocitosis en líquido de ascitis tienen peritonitis bacteriana. En la práctica es recomendable dar antibióticos a todos los individuos con un cuadro clínico sugerente de peritonitis y con una cifra de leucocitos en líquido de ascitis superior a 500 células/mm3.138 El criterio diagnóstico definitivo es la demostración de bacterias, ya sea por medio de la tinción de Gram (una cuarta parte de los casos) o por medio de cultivo. Para maximizar la detección de los organismos infecciosos responsables deben inyectarse 5 ml de líquido de ascitis en botellas de hemocultivo aeróbico y anaeróbico al lado de la cama del paciente. Dos terceras partes de los agentes etiológicos son enterales, siendo E. Coli y Klebsiella los más comunes. Los neumococos y estreptococos son responsables de hasta el 20 porciento de los casos. En cerca de la mitad de los pacientes el hemocultivo es positivo para el mismo microrganismo encontrado en el líquido de ascitis.
La cefotaxima, administrada por vía intravenosa en dosis de 2 g cada seis horas, es el tratamiento inicial apropiado para los episodios de peritonitis bacteriana espontánea.140 En los pacientes con insuficiencia renal la dosis se ajusta según sea necesario. Con este esquema se resuelve el 75 a 80 porciento de los episodios. Algunos requieren un cambio de esquema. Sin embargo, el 20 a 25 porciento de los pacientes fallecen antes de que la infección se resuelva. El tratamiento tiene la misma eficacia si se administra por cinco o por 10 días.141 Los pacientes deben ser vigilados en forma estrecha, reanalizando el líquido de ascitis por lo menos en una ocasión para segurarse que se está tratando la infección en forma adecuada. A pesar de un tratamiento óptimo, 40 a 60 porciento de estos enfermos fallece; la mitad como consecuencia directa de la peritonitis, mientras que el resto por otras complicaciones de la hepatopatía. El tratamiento a largo plazo con norfloxacina (400 mg/día) reduce la incidencia de peritonitis bacteriana espontánea recurrente atribuíble a bacterias aeróbicas gram negativas y debe considerarse en los pacientes con alto riesgo de recurrencia.142
SINDROME HEPATORRENAL
El síndrome hepatorrenal se define como una insuficiencia renal funcional relacionada con cirrosis bien establecida y habitualmente descompensada. Cuando aparece, el síndrome hepatorrenal generalmente tiene consecuencias mortales.143 El paciente típico suele tener ictericia profunda, está prácticamente moribundo y tendrá además ascitis a tensión, hipoalbuminemia e hipoprotrombinemia, y encefalopatía. Conforme la hepatopatía progresa, los volúmenes urinarios y la excreción de sodio disminuyen y la creatinina y nitrógeno de urea (BUN) séricos aumentan antes de la muerte. Bajo estas circunstancias, podemos decir que la insuficiencia renal es uno más de los incidentes relacionados con una hepatopatía severa. Sin embargo, en 10 a 20 porciento de los casos la hepatopatía puede estar razonablemente estable, por lo cual la insuficiencia renal aguda será la principal amenaza seria contra la vida del enfermo.
La patogenia del síndrome hepatorrenal no ha sido bien aclarada; tanto la reducción en el flujo sanguíneo renal como en el índice de filtración glomerular pueden preceder a la insuficiencia renal manifiesta incluso por varios meses. Paradójicamente, estas alteraciones ocurren combinadas con un aumento en el volumen plasmático.144 También ocurre un fenómeno llamado cortocircuito intrarrenal que se caracteriza por un incremento en el flujo a la médula renal a expensas de la corteza. Debido a que muchos pacientes presentan además hipotensión y no responden a agentes presores, se han implicado a falsos neuro-transmisores en la patogenia del síndrome hepatorrenal.
Aunque hay muchos padecimientos que pueden afectar al mismo tiempo al hígado y el riñón, la historia del enfermo con oliguria, retención importante de sal y hepatopatía severa generalmente limita las posibilidades diagnósticas a dos: síndrome hepatorrenal o azoemia prerrenal. Dado que estas dos alteraciones son indistinguibles desde el punto de vista clínico y de laboratorio, es indispensable que todos los enfermos sean tratados inicialmente como si tuvieran azoemia prerrenal.145 Debe suspenderse la medicación antidurética, cualquier pérdida sanguínea debe ser remplazada y el plasma se expandirá por medio de soluciones salinas o glucosadas. Estas medidas deben ser vigiladas estrechamente con la medición de la presión venosa central, y deben ser suspendidas si no se obtiene diuresis a pesar de que la presión venosa central haya ascendido sastisfactoriamente. Una vez que se ha descartado la presencia de azoemia prerrenal con dichas acciones, el tratamiento del síndrome hepatorrenal debe basarse en medidas de apoyo básicamente conservadoras. Aunque es poco frecuente, puede haber una corrección del síndrome cuando la hepatopatía mejora.
Se han informado casos de corrección del síndrome hepatorrenal después de la colocación de una derivación peritoneovenosa. Tal procedimiento debe considerarse en el pequeño grupo de pacientes en el que las manifestaciones de insuficiencia renal predominan sobre las de falla hepática; sin embargo no es raro que estos enfermos desarrollen complicaciones posiblemente mortales. Entre otras medidas que se han intentado sin algún beneficio están la derivación portocava de urgencia, los esteroides, la fenoxibenzamina, el metaraminol y la metildopa.
ENCEFALOPATIA HEPATICA
El diagnóstico de encefalopatía hepática se basa en la demostración de alteraciones mentales, asterixis y hedor hepático.146 Este último consiste en un olor desagradable, como de frutas fermentadas, del aliento. La asterixis se refiere a la flexión irregular de las extremidades, fácilmente demostrable pidiéndole al paciente que sostenga su brazo horizontalmente con las manos extendidas a la altura de la cintura. El movimiento de aleteo producido por la pérdida intermitente del tono extensor es un rasgo diferencial de la encefalopatía hepática, aunque de hecho el signo puede encontrarse también en la uremia y en las neumopatías severas. Un electroencefalograma con ondas lentas y aplanadas confirma el diagnóstico.
La encefalopatía hepática puede ser clasificada a grandes rasgos en cuatro etapas: la primera consiste en agitación sin hallazgos físicos acompañantes; los sujetos en esta etapa suelen recibir por error sedantes que rápidamente profundizan la encefalopatía. En la segunda el paciente estará moderadamente confuso pero aún reactivo, pudiéndose detectar asterixis. En la tercera etapa, el enfermo estará estuporoso y prácticamente sin respuesta. En la cuarta, el individuo se encontrará en coma profundo. En la tercera y cuarta etapas puede no haber asterixis.
Es indudable que la causa de la encefalopatía es multifactorial. En el origen de este síndrome se ha implicado a las concentraciones elevadas de amonio en suero, a los ácidos grasos de cadena corta, a falsos neurotransmisores y a ciertos aminoácidos.147 Además puede existir en la insuficiencia hepática una sustancia circulante que tiene propiedades semejantes a los agonistas benzodiacepínicos y que pueden potencializar la acción del ácido g-aminobutírico, contribuyendo a la encefalopatía hepática. Algunos pacientes con encefalopatía parecen responder a la administración de un antagonista del receptor de benzodiacepinas,148 lo que apoya la hipótesis de que la sustancia semejante a las benzodiacepinas participa en este transtorno. A la potogenia contribuyen la derivación de sangre alrededor del hígado que ocurre como consecuencia de la hipertensión porta y el deterioro en la función hepática. También es cierto que el sistema nervioso central de los pacientes con cirrosis parece ser más sensible a los efectos sedantes de productos endógenos y de medicamentos exógenos.
En la mayoría de los casos es posible identificar algún factor presipitante de la encefalopatía hepática.149 Entre los eventos causales están la hemorragia gastrointestinal, las alteraciones electrolíticas, los transtornos ácido-base, la hipoxia, la retención de CO2, las infecciones y el uso inadecuado de diuréticos, sedantes y otros fármacos.
El aspecto más importante es la eliminación o corrección de la causa presipitante. Debe investigarse con cuidado sobre los antecedentes de ingesta de sedantes, que deberán suspenderse. Una vez que se han tomado dichas medidas, el tratamiento estándar de la encefalopatía hepática incluye restricción de las proteínas de la dieta y la administración de lactulosa y neomicina. El criterio habitual es limitar las proteínas a 40-60 g/día producirá desnutrición proteica.
El medicamento de elección es la lactulosa, disacárido que llega sin haber sido digerido al colon, en donde sufre degradación bacteriana a ácidos de dos o tres carbonos que reducen el pH intraluminal y producen diarrea; la dosis habitual es de 30 ml. tres o cuatro veces al día. No se conoce con certeza el mecanismo de acción de la lactulosa; aparentemente la reducción en el pH de las heces hace que el amonio sea reducido a su forma iónica ( NH4 ), que se absorbe poco y es excretada con la materia fecal.
Se ha demostrado que la lactulosa es tan eficaz como la neomicina en el tratamiento de la encefalopatía hepática crónica o recurrente. Cerca del 80 porciento de los casos responde a uno de estos medicamentos.150 En algunos pacientes la neomicina puede ser eficaz aun cuando la lactulosa no lo haya sido. La dosis habitual de neomicina es de 1 g. VO c/6 hrs., que puede ser aumentada hasta 12 g. por día. Aunque la absorción intestinal de neomicina es muy pobre, se han publicado casos de otoxicidad y daño renal; por ello, dado que la lactulosa es menos tóxica, debe ser intentada en primer lugar. Aunque desde el punto de vista teórico podría parecer que la neomicina interfiera con la lactulosa, la evidencia existente indica que ambos medicamentos pueden actuar en forma sinérgica.
Tanto la lactulosa como la neomicina son eficaces en la encefalopatía crónica o recurrente, sin embargo, sus efectos benéficos pueden no funcionar en la enfalopatía que acompaña a hepatopatías severas agudas. Algunos enfermos pueden desarrollar una encefalopatía crónica refractaria, que generalmente se debe a cortocircuitos portosistémicos.
LITIASIS VESICULAR
En la cirrosis se incrementa la incidencia de litiasis biliar, probablemente como resultado del aumento en la producción de bilirrubina secundaria a la anemia hemolítica crónica y al hiperesplenismo.151 Es por ello que siempre debe pensarse en la posibilidad de coledocolitiasis cuando encontremos a un paciente cirrótico con ictericia. Afortunadamente esta complicación es rara, porque el diagnóstico de dicha entidad puede ser sumamente difícil en un cirrótico descompensado. La ultrasonografía es un procedimiento simple que detecta con certeza la dilatación de los conductos biliares en un 90 porciento de los casos.152 Este recurso se debe usar con premura siempre que se sospeche litiasis. La colangiografía intravenosa no tiene ninguna utilidad, ya que no se puede visualizar el colédoco en un sujeto con cirrosis e ictericia. Si es necesario descartar la litiasis biliar en forma concluyente deberá pensarse en la necesidad de una colangiografía percutánea o en una colangiografía endoscópica retrógrada.153 A menos que la sospecha diagnóstica sea muy alta, siempre será recomendable evitar estos procedimientos.
ULCERA PEPTICA
Los pacientes cirróticos tienen una incidencia mayor de úlcera péptica que la población general. El diagnóstico deberá considerarse en todos los casos en los que exista dolor abdominal o hemorragia del aparato digestivo. Debido a la importancia de definir si el origen de una hemorragia son las várices esofágicas o una úlcera péptica, habitualmente es necesario realizar endoscopía en todo cirrótico que desarrolle hemorragia gastrointestinal de importancia.
HIPOXIA
La hipoxia es una complicación frecuente en enfermos con cirrosis avanzada, en los que no son raras las cifras de tensión de oxígeno (pO2) de 60 a 70 mm Hg. La ascitis dificulta la ventilación y puede haber angiomas pulmonares responsables de cortocircuitos sanguíneos de derecha a izquierda.154 Dado que muchos cirróticos fuman al mismo tiempo que beben, es común que haya cierto grado de neumopatía obstructiva complicando el cuadro.
DESNUTRICION
Casi todos los pacientes con enfermedad hepática avanzada tienen algún grado de desnutrición proteico-calórica. La desnutrición severa contribuye a la retención de sal y agua, a una respuesta inmunológica defectuosa y a recuperación más tardía de la función hepática. Debe realizarse una evaluación nutricional en todos los pacientes con hepatopatía avanzada, y administrarse suplementos nutricionales cuando estén indicados. Los suplementos a la nutrición pueden proporcionarse añadiendo fórmulas estándar enterales a la dieta, y en algunos pacientes graves pueden indicarse suplementos parenterales. Existen pocas evidencias de que las fórmulas enriquecidas con aminoácidos de cadena ramificada tengan alguna ventaja sobre las fórmulas con composición estándar de aminoácidos, y las primeras son bastante más costosas. Por lo general los suplementos son bien tolerados, por vía oral o parenteral. Se espera con ellos un retorno más rápido a un balance positivo de nitrógeno y mejoría en la función hepática. No se ha demostrado mejoría convincente en la supervivencia.155
CANCER PRIMARIO DEL HIGADO
El cáncer primario del hígado se desarrolla en cinco a 20 porciento de los pacientes con cirrosis. Si no es tratada, la enfermedad evoluciona hacia el deterioro con rapidez.
Trasplante
La gran disponibilidad del trasplante de hígado ha mejorado en forma importante el diagnóstico de casi todas las formas de hepatopatía terminal.156,157 Debido a que los porcentajes de supervivencia a uno y cinco años llegan a ser de 90 y 70 porciento, respectivamente, en varias instituciones, esta forma de tratamiento es muy aceptada. (Algunos pacientes han vivido más de 20 años después de un trasplante de hígado). Las únicas excepciones para ser candidato a este procedimiento son la presencia de otras enfermedades debilitantes y la mayoría de los padecimientos neoplásicos del hígado. La principal limitante de este procedimiento es la disponibilidad de órganos.
Bibliografía
DR. PETER B. GREGORY
La cirrosis es la secuela de un gran número de enfermedades hepáticas crónicas y progresivas. Se desarrolla cuando estas enfermedades llegan a producir tal cicatrización del hígado que la arquitectura normal desaparece, formándose nódulos de regeneración parenquimatosa. El patrón de cicatrización difícilmente permite distinguir la etiología específica del proceso, pero otros cambios histológicos relacionados sí pueden aportar pistas. En la mayoría de las formas de hepatopatía, para establecer un diagnóstico específico generalmente se requiere de la combinación de la historia, exploración física, pruebas de laboratorio y la identificación de los rasgos histológicos característicos.
En los Estados Unidos, la ingesta excesiva de alcohol es con mucho la causa más frecuente de cirrosis hepática. En otros países es la hepatitis viral crónica el primer agente etiológico.
Manifestaciones clínicas
La fatiga, el malestar general y la astenia son comunes en todas las formas de cirrosis; sin embargo, estos síntomas no específicos también son parte del cuadro clínico de prácticamente todos los padecimientos hepáticos, tanto agudos como crónicos. Aunado a ello, no podemos hablar de alguna alteración física que indique sin lugar a dudas que el hígado es cirrótico. Entre los hallazgos característicos, pero no diagnósticos, se encuentran el eritema palmar (una lesión púrpura-rojiza sobre la región hipotenar de la mano) y las telangiectasias. Otros hallazgos típicos son ginecomastia, atrofia testicular, evidencia de hipertensión porta (esplenomegalia, ascitis, hemorroides, várices esofágicas y red venosa colateral en las venas del abdomen). Existen algunas manifestaciones que son frecuentes sólo en algunas formas específicas de cirrosis hepática: contractura de Dupuytren, xantelasma, xantomas, anillo de Kayser-Flescher, coloración bronceada de la piel e hiperpigmentación.
El hígado cirrótico es generalmente grande, y a menudo el lóbulo izquierdo es palpable debajo del apófisis xifoides. Sólo los pacientes con fases avanzadas e inactivas de la enfermedad tienen hígados pequeños y contraídos. Otra característica es la firmeza del órgano a la palpación, que incluso puede tener consistencia pétrea cuando el padecimiento está avanzado. En ocasiones pueden detectarse por palpación los nódulos de regeneración grandes en la superficie del hígado; pero las nodulaciones finas de la cirrosis micronodular son imposibles de detectar.
Evaluación diagnóstica
Es frecuente que la cirrosis solamente pueda ser detectada por medio de biopsia hepática.1 Este procedimiento puede realizarse con seguridad cuando no hay antecedentes de hemorragia anormal después de cirugías, procedimientos dentales o toma de biopsias, y cuando las pruebas de coagulación se encuentran en límites normales o sólo ligeramente anormales. Las recomendaciones más razonables son las siguientes: tiempo de protrombina con una relación internacional normalizada (INR) no mayor de 1.5, tiempo parcial de tromboplastina no mayor de 10 segundos en relación con el control, recuento plaquetario de por lo menos 50,000/mm3 y tiempo de sangrado normal. Bajo circunstancias excepcionales la biopsia hepática puede ser realizada con alteraciones mayores en los factores de la coagulación; sin embargo, antes del procedimiento debe hacerse una corrección vigorosa de cualquier alteración en la coagulación. Otras contraindicaciones relativas para la biopsia hepática son la falta de cooperación del paciente, la ascitis y la neumonía del lóbulo inferior derecho.
El hallazgo de una arquitectura hepática distorcionada, con nódulos regenerativos rodeados por tejido cicatricial, es concluyente sobre la existencia de cirrosis. Sin embargo, la biopsia hepática puede aportar diagnósticos falsos negativos, es decir, puede haber cirrosis aunque la biopsia no lo confirme.
La biopsia hepática ayuda también a identificar la causa. Particularmente la invación de los conductos biliares por tejido granulomatoso y su subsecuente destrucción, permite establecer el diagnóstico de cirrosis biliar primaria; el exceso de hierro en las células de los conductos biliares y en los hepatocitos identifica la hemocromatosis; una reacción hialina alcohólica relacionada con infiltrado polimorfornuclear indica hepatopatía alcohólica.
La disminución de la albúmina sérica y la prolongación del tiempo de protrombina son datos característicos de cirrosis, otras pruebas de función hepática pueden tambier ser anormales. La tomografía computada, la imagen por resonancia magnética (IRM) o el ultrasonido de hígado con estudio de flujo Doppler pueden demostrar datos compatibles con cirrosis. Entre estos destacan esplenomegalia, ascitis no sospechada, una superficie hepática irregular e inversión de la circulación porta ( en el ultrasonido ) o incluso várices intrabdominales. Además, la esofagoscopía demuestra con frecuencia la presencia de várices esofagogástricas.
La medición de la presión en cuña de la vena hepática puede ser de utilidad en el diagnóstico de cirrosis.2 Se desplaza un catéter a través de la vena cava hacia las suprahepáticas y se le enclava en una vena hepática distal pequeña; la presión así registrada corresponde a la intrahepática y es reflejo de la presión portal. En condiciones normales la presión en cuña no debe ser mayor de 4 mm Hg que en la vena hepática libre. Cuando la presión en cuña es normal a pesar de un cuadro clínico evidente de hipertensión porta, deberá pensarse en la posibilidad de obstrucción venosa prehepática. Cuando la presión en cuña está elevada, la posibilidad de cirrosis es muy alta.
La arteriografía puede ser útil cuando es difícil dilucidar la causa de un cuadro de hipertensión portal.3,4 En la fase arterial puede observarse distorsión típica de la arteria hepática en forma de sacacorchos; en la fase venosa se determinará la permeabilidad de las venas porta y esplénica. Pueden realizarse además tomas arteriográficas selectivas de las venas renal y mesentérica cuando se esté considerando la posibilidad de una cirugía con derivación.
En algunas instituciones se acostumbra realizar esplenoportografía con el fin de visualizar las raíces de las venas porta y esplénica. Este procedimiento, que implica puncionar el bazo e inyectar medio de contraste, permite obtener una medición directa de la presión venosa portal; sin embargo, tiene complicaciones en un cinco porciento de los casos y no permite la visualización de las venas renal y mesentérica.
La peritoneoscopía permite observar la superficie del hígado y realizar una biopsia dirigida;5 este es un procedimiento que tiene gran utilidad para la investigación de un posible proceso neoplásico.
Formas específicas de cirrosis
CIRROSIS ALCOHOLICA
Entre los sintomás de la cirrosis alcohólica están: cirrosis portal, de Laennec, nutricional y micronodular. Como el alcohol produce un efecto tóxico sobre el hígado, tal y como se ha demostrado en animales de experimentación, el término cirrosis alcohólica parece ser el más adecuado.6,7 La exploración física comúnmente demuestra que hay hepatomegalia, a veces muy grande; no es raro que en la autopsia se encuentren pesos mayores a 2,000 g. La hepatomegalia refleja inflamación, infiltración grasa y formación cicatricial extensa. El cuadro histológico característico consiste en una cicatriz lineal que separa al perénquima hepático y rodea pequeños nódulos de hepatocitos [ver figura 1]

|
| Figura 1 |
| Cirrosis alcohólica: biopsia |
La cirrosis alcohólica es consecuencia de la ingesta de grandes cantidades de alcohol; para su desarrollo no se requiere que exista desnutrición concomitante, aunque esta situación ocurre casi invariablemente. La desnutrición es sin lugar a dudas reflejo de la sustitución de las calorías normales de la dieta por alcohol.8 Todas las personas que beben consuetudinariamente desarrollan infiltración grasa leve, la cual es totalmente reversible cuando cesa la ingesta alcohólica.9 Aunque para el desarrollo de la cirrosis alcohólica suelen requerirse entre 10 a 15 años de beber intensamente (v.gr., más de 120 ml de whiskey al día), el padecimiento puede aparecer rápidamente. Se han informado casos de hepatopatía alcohólica fatal en adolescentes. Es típico que la cirrosis alcohólica aparezca como consecuencia de cuadros repetitivos de hepatitis alcohólica clínica y subclínica. El término hepatitis alcohólica se refiere al hallazgo histológico de hialinización rodeada por inflamación de células polimorfonucleares [ver figura 3]. Estas lesiones necróticas se acompañan de formación de colágena; en las etapas iniciales las lesiones se encuentran típicamente en una localización pericentral. Cuando el proceso es severo aparece cicatrización de la vena central (necrosis hialina esclerosante).10
|
|


En los individuos con cirrosis alcohólica descompensada los niveles séricos de bilirrubinas están elevados, frecuentemente en forma importante (20 a 40 mg/dl). La aspartato aminotransferasa (AST, antes TGO) también se eleva, con frecuencia a niveles entre 200 y 300 UI/L. Es característico que la elevación en dicha aminotransferasa sea mayor que la de la alanino aminotransferasa (ALT, antes TGP). La inversión de esta relación o los niveles de aminotransferasas por arriba de 300 UI/L deben hacer pensar en otras posibilidades diagnósticas diferentes a la hepatopatía alcohólica. La fosfatasa alcalina suele elevarse sólo en forma ligera. Comúnmente coexiste disminución de la albúmina sérica por debajo de 3.5 g/dl y prolongación del tiempo de protrombina.
Conforme la enfermedad progresa, por la ingesta alcohólica continua y la desnutrición, el paciente pierde masa muscular en cara, cuello, hombros, antebrazos y manos; aparece pérdida de peso, que puede ser sustituida por el acúmulo de líquido de ascitis. Esta puede detectarse fácilmente por medio de la identificación de matidez cambiante en la exploración física; la onda líquida abdominal es patognomónica de ascitis, pero aparece únicamente cuando la cantidad de líquido acumulado es grande. El enfermo puede tener una red venosa colateral prominente, reflejo de aumento en la circulación a través de las venas periumbilicales. La hepatomegalia y la esplenomegalia son frecuentes. Aparecen eritema palmar y telangiectasias en cara, dorso, brazos y pecho. Hay hemorragias subcutáneas frecuentes como resultado de deficiencias vitamínicas y de la reducción en los factores de coagulación. También son comunes la ginecomastia (que puede ser dolorosa) y la atrofia testicular. Puede desarrollarse encefalopatía hepática. Cuando se les busca con intención, generalmente se encuentran várices.
El único tratamiento definido para los sujetos con hepatopatía alcohólica es la suspensión del hábito alcohólico. Los individuos con cirrosis alcohólica que continúan bebiendo tienden a tener peor pronóstico que los que abandonan dicho hábito. El índice de supervivencia a cinco años para pacientes que continúan bebiendo es menor al 40 porciento, pero puede llegar a ser del 60 al 70 porciento cuando se mantiene la abstinencia.12 Aunque el pesimismo y escepticismo son grandes, hasta un 30 porciento de los casos con hepatopatía alcohólica pueden llegar a mantener una abstinencia total.12 Por lo tanto, debe realizarse un gran énfasis terapéutico en apoyar los esfuerzos que el enfermo hace para dejar de beber. Actualmente existen diferentes grupos de rehabilitación, de apoyo mutuo y diversas técnicas psicoterapéuticas para ello.
Las importantes deficiencias nutricionales observadas en muchos pacientes con cirrosis alcohólica han causado que muchos médicos recomienden el apoyo nutricional durante la enfermedad aguda. Dos estudios extensos cooperativos de la Administración de veteranos en los EUA evaluaron el papel de los suplementos nutricionales enterales así como de la oxandrolona, un esteroide anabólico, en la enfermedad hepática alcohólica descompensada.13,14 Los investigadores demostraron una relación directa entre la ingesta calórica y la supervivencia y encontraron que el apoyo nutricional vigoroso aumentó la supervivencia, en especial en los pacientes con desnutrición importante.
Aunque existen varios estudios sobre el tratamiento con esteroides en la enfermedad hepática alcohólica descompensada, no hay datos convincentes ni constantes que demuestren su eficacia.11,15-20 Además, aunque se han empleado propiltiouracilo y colchicina en pacientes con enfermedad hepática alcohólica, no existen conclusiones que justifiquen el uso de estos medicamentos. Por lo tanto, el tratamiento de la cirrosis alcohólica debe ser de apoyo y dirigido a mejorar la nutrición, alentar la abstinencia y tratar las complicaciones.
CIRROSIS POSNECROTICA
La cirrosis posnecrótica se caracteriza por un hígado reducido que contiene nódulos de regeneración de tamaño variable [ver figura 4]. Sin embargo, el término posnecrótico es poco adecuado, ya que todas las cirrosis son consecuencia de necrosis de los hepatocitos. La causa más frecuente de esta entidad es la hepatitis crónica activa, aunque en muchos casos la causa no puede ser determinada, por lo cual se aplica el término de cirrosis criptogénica. Parecería que en tales casos la cirrosis se desarrolla como consecuencia de una hepatitis crónica activa subclínica de evolución lenta. La cirrosis posnecrótica puede encontrarse también en pacientes alcohólicos, en estos casos se piensa que la imagen patológica revela la remodelación regenerativa del hígado cirrótico micronodular después de que el paciente ha dejado de beber.
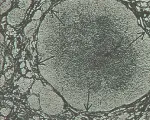
|
| Figura 4 |
| Cirrosis posnecrótica: biopsia |
Con mayor frecuencia que en otras formas de cirrosis, en la posnecrótica la biopsia puede ser engañosamente normal.21 La explicación más posible es que la aguja de biopsia atraviesa una banda fibrosa y remueve el centro de un nódulo de regeneración, el cual puede tener una apariencia casi normal.
La cirrosis posnecrótica parece evolucionar en forma insidiosa, incluso cuando se encuentra aparentemente inactiva o cuando el tratamiento de la hepatitis crónica activa se considera exitoso.22 La causa habitual de muerte es la hemorragia gastrointestinal o la insuficiencia hepática. Cerca del 10 o 15 porciento de los casos con cirrosis posnecrótica desarrollan carcinoma primario de hígado.
Cuando la hepatitis asociada está activa, puede estar indicado utilizar esteroides. Sin embargo, si la cirrosis es avanzada y la evidencia de hepatitis es mínima, entonces no hay medidas terapéuticas que puedan utilizarse de manera específica; el tratamiento médico debe ser de apoyo y enfocado a las complicaciones.
CIRROSIS BILIAR PRIMARIA
La cirrosis biliar primaria es una hepatopatía poco común, que se presenta en mujeres entre los 30 y 50 años de edad.23 Se considera que los mecanismos inmunológicos juegan un papel preponderante en la patogenia de este padecimiento, ello en base a la presencia de autoanticuerpos séricos, la relación con otras enfermedades autoinmunes y la semejanza de las lesiones de los conductos biliares con aquellas que se observan en enfermedades crónicas por rechazo a injertos.24 Estudios recientes han demostrado que la cirrosis biliar primaria se asocia con el haplotipo HLA-DR8, lo que sugiere una predisposición genética para la enfermedad.25 Las manifestaciones iniciales son astenia y prurito generalizado; éste puede haberse iniciado meses o años antes de que el paciente busque atención médica por la presencia de piel pigmentada con tono parduzco, que se debe en parte al rascado crónico. La ictericia puede tardar en aparecer hasta cinco a 10 años a partir del inicio del prurito y de los síntomas constitucionales. Algunos enfermos desarrollan dolor óseo, fracturas múltiples y colapso vertebral. La causa habitual es la osteoporosis, que ocurre en 20 a 30 porciento de los pacientes y al parecer es secundaria a una notable reducción en la función de los osteoblastos.26 Con menor frecuencia puede existir osteomalacia concomitante. Entre los factores que condicionan las anomalías óseas están la malabsorción de calcio y fosfato, alteraciones en el metabolismo de la vitamina D, tratamiento con colestiramina y desnutrición. La exploración física revela con frecuencia xantelasma, xantomas, hepatoesplenomegalia, hiper-pigmen-tación y escoriación de la piel [ver figura 5]. Cuando la enfermedad está avanzada también se encontrará ictericia de conjuntivas, ascitis y edema.
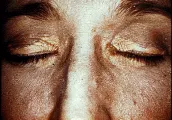
|
| Figura 5 |
| Cirrosis biliar primaria |
Los hallazgos típicos de laboratorio incluyen la elevación de la fosfatasa alcalina (por lo general > a 300 UI/L y con frecuencia > 700 UI/L), colesterol sérico mayor de 300 mg/dl y anticuerpos antimitocondriales positivos (en 90 a 95 porciento de los casos).
Un número moderado de enfermos con cirrosis biliar primaria tiene enfermedades concomitantes como acidosis tubular renal, esclerodermia, síndrome de CREST o síndrome de Sjögren. Se han publicado algunos casos de cirrosis biliar primaria familiar.
La cirrosis biliar primaria puede tener características clínicas y de laboratorio semejantes a las de la cirrosis secundaria a enfermedad crónica de las vías biliares. De hecho, el cáncer de páncreas o de las vías biliares, la pericolangitis crónica secundaria a enfermedad intestinal inflamatoria y la colangitis esclerosante pueden tener datos de laboratorio e histológicos muy semejantes a la cirrosis biliar primaria. En algunos pocos casos es necesario observar en forma directa las vías biliares en los pacientes que tienen datos clínicos, de laboratorio e histológicos típicos de cirrosis biliar primaria.
La biopsia hepática puede mostrar destrucción de los conductos biliares con infiltración linfoplasmacitaria de espacios porta, formación periportal de granulomas y cicatrización con fusión de los espacios porta [ver figura 6]. La proliferación de los conductos es común. Cuando la cicatrización es extensa puede encontrarse formación de nódulos, frecuentemente con retención de las venas centrales. La estasis biliar es periportal en la mayoría de los casos, e indica que el padecimiento está avanzado.

|
| Figura 6 |
| Cirrosis biliar primaria: biopsia |
Por desgracia, solo en pocos casos se encuentran los cambios histológicos propios de la cirrosis biliar primaria; en el resto los hallazgos son muy semejantes a los de la hepatitis crónica activa, por lo cual la distinción entre ambos padecimientos debe hacerse en base al cuadro clínico y a los datos de laboratorio.
El pronóstico es variable, pero la evolución suele ser insidiosa. La insuficiencia hepática severa no es frecuente sino hasta que la enfermedad se encuentra muy avanzada. El tiempo promedio de supervivencia para pacientes sintomáticos es de aproximadamente 10 años. Los pacientes asintomáticos pueden vivir más, aunque un estudio reportó un periodo promedio de supervivencia también de 10 años para estos pacientes.27-29
En el tratamiento de la cirrosis biliar primaria se han indicado los esteroides, la azatioprina, la penicilamina, la colchicina y el ursodiol. En un estudio aleatorio, un año de tratamiento con prednisolona mejoró los síntomas, la función hepática y los hallazgos histológicos, pero aceleró la pérdida de hueso.30 Debido a que la enfermedad ósea es frecuente y muchas veces incapacitante en los pacientes con cirrosis biliar primaria, no se recomienda el uso de esteroides a pesar de su aparente efecto benéfico sobre el daño hepático en sí. Se han realizado dos estudios aleatorios para valorar el tratamiento con azatioprina; en el primero, la administración de azatioprina en dosis de 2 mg/kg/día, no tuvo beneficios y se presentaron efectos colaterales en el 27 porciento de los pacientes.31 En el segundo, la azatioprina se administró en dosis más bajas (50 a 100 mg/día), observándose una tendencia al aumento en la supervivencia, pero se presentaron efectos adversos en el 11 porciento de los pacientes tratados.32 La penicilamina, en dosis de 600 a 1,000 mg/día, se evaluó en cinco estudios aleatorios prospectivos.33-37 En cuatro de ellos el fármaco no aumentó la supervivencia, y en el quinto el beneficio fue moderado.
Las complicaciones de la penicilinamina son comunes e incluyen: náusea, vómito, alteraciones en el sentido del gusto, estomatitis, fiebre, mialgias, artralgias, liquen plano, proteinuria y aplasia de médula ósea; incluso se han publicado casos de complicaciones mortales por el uso de este fármaco. Debido a la eficacia dudosa de la azatioprina y de la penicilamina en el tratamiento de la cirrosis biliar primaria, sus efectos colaterales tan frecuentes son factores en contra de su uso. Dos estudios clínicos aleatorios evaluaron la eficacia del tratamiento con colchicina, en dosis de 0.6 mg dos veces al día. Un grupo informó una mejoría leve en las pruebas de funcionamiento hepático y aumento en la supervivencia;38 el otro reportó que la supervivencia tiende a ser mayor.39 La colchicina produjo efectos colaterales leves, que se controlaron con facilidad en estos estudios al reducir la dosis. Aunque el uso de ciclosporina, un polipéptido cíclico de origen micótico con importantes propiedades inmunosupresoras, en pacientes con cirrosis biliar primaria causó mejoría clínica,40,41 se presentaron efectos adversos renales y de hipertensión, y el impacto sobre la supervivencia a largo plazo o la necesidad de trasplante hepático fue solo modesto. Se ha administrado también ursodiol, un ácido biliar hidrofílico, con la idea de que al alterar la composición del reservorio endógeno de ácidos biliares podría lograrse un efecto benéfico consistente en reducir la concentración de ácidos biliares hidrofóbicos endógenos potencialmente tóxicos. En un estudio extenso controlado con placebo, el tratamiento durante dos años con ursodiol en dosis de 13 a 15 mg/kg causó mejoría clínica, bioquímica e histológica con efectos colaterales leves.42 La mayoría de los pacientes reciben en la actualidad ursodiol porque parece ser tan eficaz como otros medicamentos y más seguro, aunque por desgracia este fármaco es costoso.
El tratamiento no específico debe dirigirse a aliviar los síntomas durante la evolución, lenta pero inexorable, de la enfermedad. La colestiramina, resina de intercambio aniónico, puede ayudar a disminuir el prurito, la dosis habitual es de 4 g VO tres veces al día; algunos enfermos pueden mejorar con dosis menores, mientras que otros requieren hasta 16 a 20 g/día. Es frecuente que la colestiramina sea poco tolerada, pero se acepta mejor si se administra con los alimentos o se mezcla con jugos o puré de fruta. La rifampicina (300 a 450 mg/día en dosis divididas), el ursodiol (10 a 15 mg/kg/día) e incluso el tratamiento con luz ultravioleta, han sido eficaces para aliviar el prurito severo.43 Los antihistamínicos rara vez son eficaces, pero su efecto sedante puede permitir que el paciente duerma a pesar del prurito continuo. La terfenadina no se utiliza ya para tratar el prurito en este grupo de pacientes porque ha causado arritmias graves en enfermos con daño hepático.
Se recomienda el uso de complementos vitamínicos. Debe administrarse diariamente una dosis de 5,000 unidades de vitamina A en un preparado hidrosoluble. Si el tiempo de protrombina se alarga, deberá administrarse vitamina K, también en una forma hidrosoluble, a razón de 5 mg/día. Los individuos que desarrollan osteomalacia deben recibir un metabolito activo de la vitamina D, ya sea 25-(OH)D3 (calcifediol) o 1,25-(OH)2D3 (calcitrol). Estos medicamentos han producido mejoría en algunos casos de osteomalacia,44 pero el beneficio que puedan aportar en los casos de osteoporosis aún está por demostrarse.45 Las dosis recomendadas para alteraciones óseas sintomáticas son de 0.5 a 1.0 µg de 1,25-(OH)2D3 o de 40 a 120 µg de 25-(OH)D3, al día. Debe administrarse calcio adicional a dosis de 1 o 2 g/día. Además, el autor recomienda que en pacientes asintomáticos se utilicen esquemas profilácticos con 0.25 µg de 1,25-(OH)2D3 o 20 µg de 25-(OH)D3 al día. Los niveles séricos de calcio deben ser vigilados durante los primeros meses de tratamiento con vitamina D y calcio.
HEMOCROMATOSIS
La hemocromatosis se desarrolla por el depósito de grandes cantidades de hierro en las células parenquimatosas hepáticas [ver figura 7].46 Este acúmulo causa la destrucción de células periportales y cicatrización hepática,lo cual produce cirrosis. El padecimiento ocurre predominantemente en varones (relación de 10:1). Los síntomas aparecen generalmente entre los 40 y 60 años en hombres o después de la menopausia en mujeres; ocasionalmente la enfermedad se hace aparente a una edad más temprana.
|
|

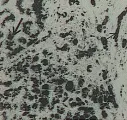
Los depósitos de hierro en páncreas y corazón producen disfunción de esos órganos. Al momento de aparición de los síntomas de hepatopatía, cerca de la mitad de los casos con hemocromatosis tiene ya diabetes mellitus, 15 porciento tienen insuficiencia cardiaca congestiva o arritmias y una minoría significativa se queja de rigidez y artralgias. También la impotencia es común, aparentemente debida a disfunción hipofisiaria.49
En las etapas avanzadas el examen físico mostrará coloración bronceada de la piel, producto del depósito de hierro y melanina. El hígado tiende a estar moderadamente crecido; cerca de la mitad de los pacientes tendrá esplenomegalia. En etapas menos avanzadas el color de la piel puede ser normal y el hígado ser apenas palpable. La mayoría de los casos cursan tarde o temprano con hipertensión porta. Entre 15 y 20 porciento de los individuos desarrolla carcinoma primario de hígado.
Los estudios de laboratorio muestran aumento del hierro sérico relacionado con una saturación de transferrina sérica del 80 a 90 porciento (el valor normal de saturación es de 15 a 47 porciento). También la ferritina sérica tiende a elevarse.50 La elevación del coeficiente de atenuación lineal promedio (número de la TC) en la tomografía computada de hígado puede indicar la presencia de depósitos excesivos de hierro en el hígado.51 La RM puede demostrar también el exceso de hierro y la hemocromatosis primaria.52 No son raros los aumentos discretos en la aminotransferasa y fosfatasa alcalina séricas, pero la ictericia es poco frecuente. La concentración sérica de albumina y el tiempo de protrombina tienden a permanecer normales hasta etapas avanzadas de la enfermedad.
La concentración sérica de hierro y la capacidad de fijación total de hierro pueden emplearse para realizar escrutinio de hemocromatosis en la población.53 Una saturación de transferrina en ayuno de 62 porciento o mayor en varones (y quizá de 50 porciento en mujeres) identifica a un alto porcentaje de pacientes que son homocigotos para la hemocromatosis. La concentración de ferritina sérica no ha demostrado ser un estudio útil para escrutinio porque muy pocos individuos homocigotos tienen niveles elevados antes de desarrollar la enfermedad clínica.
Al identificar a un individuo homocigoto se justifica el estudio de sus hermanos porque se espera que el 25 porciento sea homocigoto también. Los hermanos del mismo sexo suelen tener una cantidad comparable de hiero hepático a menos de que tengan antecedente de hemorragias o donaciones de sangre de importancia.54
El nivel elevado de hierro o de ferritina sérica no es diagnóstico, ya que dicho valor puede aumentar en todas las enfermedades hepáticas que se caracterizan por necrosis significativa de las células hepáticas. No es raro que las concentraciones séricas de hierro o ferritina se eleven en la hepatopatía alcohólica descompensada, en la hepatitis viral aguda o en la hepatitis crónica activa. Por lo tanto, la única manera en que puede establecerse el diagnóstico de hemocromatosis es mediante la biopsia hepática.55 El hallazgo característico es el importante depósito de gránulos de hemosiderina en los hepatocitos y las células de los conductos biliares. La fibrosis puede ser desde mínima hasta una cirrosis franca. En ocasiones es difícil distinguir entre una hemocromatosis primaria y una cirrosis con saturación secundaria de hierro. La hemocromatosis primaria se caracteriza por predominio del hierro parenquimatoso en relación con la cantidad de tejido cicatricial, así como por la presencia de hierro en los conductos biliares. La sobrecarga de hierro secundaria a una cirrosis subyacente generalmente se relaciona con una cirrosis avanzada, en la cual predomina el tejido cicatricial sobre el hierro y los conductos biliares están libres de este metal. Cuando el exceso de hierro hepático proviene de una fuente exógena, como transfusiones repetidas para manejo de hemólisis crónica, los depósitos de hierro predominarán en las células de Kupffer. Cuando las características morfológicas de la biopsia hepática no permiten distinguir en forma clara entre una hemocromatosis primaria y la sobrecarga secundaria de hierro, el análisis cuantitativo del contenido hepático de hierro puede ser útil.56 En los pacientes con hemocromatosis las muestras de tejido obtenidas de corazón, páncreas o piel mostrarán también grandes depósitos de hierro.
La detección y el tratamiento temprano en los pacientes con hemocromatosis primaria es indispensable. El tratamiento habitual consiste en la eliminación del exceso de hierro por medio de flebotomías semanales.57 Debido a que cada 50 ml de sangre contiene 250 mg de hierro, la extracción de esa cantidad por semana permitirá agotar las reservas en uno a dos años. El objetivo del tratamiento es mantener niveles persistentemente bajos de hierro en suero y ausencia de éste en la biopsia. A partir de entonces pueden hacerse flebotomías subsecuentes cada dos o tres meses para evitar reacúmulo de hierro. Si los pacientes se detectan antes de que se desarrolle la cirrosis y la remoción del hierro corporal total se lleva a cabo en forma satisfactoria, la esperanza de vida se aproxima a lo normal.58 Los pacientes con cirrosis establecida que reciben tratamiento evolucionan mejor que los pacientes no tratados, pero siguen teniendo riesgo de desarrollar cáncer primario de hígado años después de la remoción satisfactoria del hierro. La incapacidad para reducir los depósitos de hierro después de 18 meses de tratamiento es un signo de mal pronóstico. Los signos de hepatopatía ceden en el 70 porciento de los pacientes tratados, pero las alteraciones endócrinas y la artropatía mejoran sólo en un 20 porciento de los casos tratados.
Las flebotomías repetidas resultan obviamente imprácticas en el manejo de la sobrecarga de hierro resultante del tratamiento de las anemias hemolíticas crónicas. En tales casos, la deferoxamina utilizada por vía subcutánea en dosis de 1 a 3 g cada 12 horas produce una pérdida urinaria promedio de hierro de 50 mg por día.59 La adición de ácido ascórbico, 500 mg al día por vía oral, puede duplicar la cifra de excreción urinaria de hierro.
ENFERMEDAD DE WILSON
La enfermedad de Wilson, denominada también degeneración hepatolenticular, es un padecimiento autosómico recesivo que se desarrolla con una frecuencia aproximada de 1 por millón.60 El defecto genético parece encontrarse en el cromosoma 13, banda 14.3, en donde se han identificado cuatro mutaciones específicas de la enfermedad en un gen que codifica para la proteína trifosfatasa de adenosina (ATPasa) tipo P fijadora de cobre.61 La enfermedad parece deberse a una excreción de cobre inadecuada a través de la bilis, lo cual provoca acúmulo de cantidades excesivas de cobre en la mayoría de los tejidos corporales. También está afectada la incorporación de cobre a la ceruloplasmina. La mayoría de los sujetos afectados ya han sufrido algunos síntomas de la enfermedad a los 15 años de edad, producto de disfunción neurológica o hepática. Aunque ha ocurrido que la enfermedad de Wilson tenga sus manifestaciones iniciales hasta los 30 años de edad, estos casos son excepcionales (El autor conoce un paciente que presentó hepatomegalia asintomática a los 62 años de edad). Las manifestaciones iniciales son atribuibles a disfunción hepática en el 40 porciento de los casos. La afección hepática se manifiesta habitualmente como un padecimiento crónico caracterizado por fatiga, ictericia, telangiectasias, ascitis, edema, esplenomegalia y hemorragia por várices esofágicas. La asociación de anemia hemolítica es una de las claves para el diagnóstico. En ocasiones la afección hepática puede semejarse a una hepatitis aguda severa, causando la muerte en días o semanas. Las principales manifestaciones neurológicas incluyen temblores, rigidez, alteraciones en la marcha e incoordinación de movimientos, lenguaje incomprensible y cambios en la personalidad. También se han detectado casos de hipoparatiroidismo.63
El signo patognomónico es el anillo de Kayser-Fleischer, que consiste en un anillo delgado, parduzco, que se encuentra en la periferia de la córnea. Aunque la pigmentación abarca habitualmente toda la circunferencia, puede estar localizada solamente en la parte superior o inferior. En etapas tempranas de la enfermedad puede requerirse de una lámpara de hendidura para detectar dicho anillo, que puede ser particularmente difícil de identificar en la exploración física rutinaria en pacientes con ojos de color café.
Al momento de la detección del padecimiento, por lo menos 50 porciento de los enfermos tienen hepatoespleno-megalia y alteraciones moderadas en las pruebas de funcionamiento hepático. Dos hallazgos de laboratorio con carácter distintivo son la disminución o ausencia de ceruloplasmina sérica y el aumento en la excreción urinaria de cobre, desde el valor normal de menos de 50 µg/día hasta niveles tan elevados como 1,000 µg/día. Hay un pequeño porcentaje de casos con enfermedad de Wilson que pueden tener niveles séricos de ceruloplasmina normales o excreción normal de cobre en orina y ausencia de anillo de Kayser-Fleischer. Es por ello recomendable que se evalúen siempre los tres factores, ya que por lo menos uno de ellos será anormal. En los casos problemáticos, el encontrar un exceso de cobre urinario después de la administración de 1,000 mg de penicilamina ayudará a establecer el diagnóstico.64 Si persiste la duda, el estándar final puede ser un aumento en la concentración de cobre en el tejido hepático, aunque este resultado es concluyente solo si el paciente no tiene colestasis de larga evolución, que también puede aumentar la concentración hepática de cobre.
El tratamiento de la enfermedad de Wilson requiere de la administración de D-penicilamina, agente quelante que se une al cobre y promueve la excreción urinaria de 1,000 a 3,000 µg/día.65 La dosis habitual es de 1 g/día. La mejoría clínica se acompaña de disminución de las reservas de cobre en los tejidos. El tratamiento con D-penicilamina se asocia con efectos colaterales importantes, sobre todo náusea y molestia abdominal que ocurren después de la ingesta del medicamento; entre los más serios están la leucopenia y la anemia, que pueden implicar en algunos casos la existencia de anemia aplásica. Un porcentaje pequeño de enfermos desarrolla síndrome nefrótico. Todos los individuos con enfermedad de Wilson tratados con el medicamento en cuestión deben ser vigilados estrechamente con análisis de orina y biometrías hemáticas, sobre todo durante los primeros meses del tratamiento. Debido a que la D-penicilamina es un antagonista de la piridoxina, deberán administrarse 50 mg de piridoxina por semana. Cuando la D-penicilamina no sea tolerada deberá considerarse el tratamiento con zinc oral. El zinc elemental puede administrarse en forma de acetato en una dosis total de 150 mg al día, en dosis divididas administradas cuando el estómago esté vacío. El tratamiento con zinc aumenta las pérdidas fecales de cobre e induce un balance negativo del metal en los pacientes con enfermedad de Wilson.66 Sin embargo, el inicio de su acción es bastante tardío y se desconocen los beneficios que el tratamiento a largo plazo pueda tener.
DEFICIENCIA DE GLOBULINA a1-ANTITRIPSINA
La deficiencia homocigota de globulina a1-antitripsina se relaciona con un síndrome raro en el cual existe cirrosis progresiva.67 Aunque originalmente se describió en niños con cirrosis juvenil, también en adultos se ha reportado la combinación de deficiencia de esta proteína y cirrosis portal. Los adultos desarrollan además enfisema concomitante.68 El diagnóstico de deficiencia de globulina a1-antitripsina debe sospecharse en todos los casos en los cuales sea difícil dilucidar la causa de una cirrosis hepática.
En las fases iniciales la mayoría de los enfermos tienen hepatomegalia moderada y alteraciones ligeras de la función hepática. La ausencia de globulina a1-antitripsina en la electroforesis de proteínas hace que el diagnóstico sea muy probable. La medición específica de sus niveles en sangre confirma el diagnóstico. Se han encontrado variantes genéticas que reflejan la existencia de por lo menos 25 diferentes alelos para el gen que controla la producción de la proteína.69 El genotipo que habitualmente se relaciona con cirrosis y enfisema ha sido denominado inhibidor de proteasa tipo ZZ (PiZZ). Una sustitución de aminoácido en la variante Z de la proteína permite que las moléculas de a1-antitripsina polimericen dentro del hepatocito, alterando así la excreción de la proteína del hígado. En la biopsia pueden identificarse los cuerpos de inclusión PAS positivos (diastasa-resistentes) anormales y característicos que contienen globulina a1-antitripsina [ver figura 8]. Una proporción significativa de los pacientes homocigotos para PiZZ con enfermedad hepática crónica tienen también evidencia de infección por virus de la hepatitis B o C.70
|
|


La cirrosis secundaria e infestación por esquistosoma, rara en los Estados Unidos, es relativamente frecuente en el lejano oriente, Egipto y algunas zonas de Sudamérica.71 En la esquistosomiasis, los huevecillos depositados en las raíces portales desencadenan una reacción granulomatosa fibrótica que ocasionalmente causa fibrosis difusa periportal o a la llamada fibrosis en pipa. La manifestación inicial es generalmente la hemorragia de várices esofágicas; la exploración física mostrará hepatoesplenomegalia prominente. Rara vez se observan estigmas de hepatopatía crónica o de insuficiencia hepática. El tratamiento de la esquistosomiasis no siempre da buenos resultados, sobre todo en etapas avanzadas.72
CIRROSIS RELACIONADA CON CORTOCIRCUITOS YEYUNO-ILEALES
En todos los pacientes sometidos a cortocircuitos yeyuno-ileales hay acumúlo de grasa en el hígado durante el periodo de pérdida rápida de peso que sigue a la cirugía.73 Un número reducido de enfermos desarrollará una hepatopatía progresiva que es indistinguible de la cirrosis alcohólica; generalmente hay hepatomegalia y la función hepática se va deteriorando progresivamente.74,75 Se desconoce la causa de esta grave complicación. Se ha informado sobre un síndrome similar secundario a gastroplastía.76 Aunque algunos comunicados indican que hay mejoría de la función hepática al someter a los sujetos a hiperalimenta-ción parenteral, el único tratamiento efectivo es la reanastomosis del intestino o la corrección de la gastroplastía. Cuando este procedimiento no se efectúa rápidamente, el deterioro de la función hepática puede ser mortal.
HIGADO GRASO Y CIRROSIS
Aunque existen pocas evidencias que indican que el acúmulo de grasa en el hígado puede producir cirrosis, hay informes de un tipo idiopático de cirrosis relacionada con aumento en la grasa hepática.77,78 Esta cirrosis parece ser secundaria a una esteatohepatitis no alcohólica, padecimiento raro que es más frecuente en mujeres y se asocia con obesidad, hiperlipidemia y diabetes mellitus.79 Con frecuencia estos pacientes tienen hepatomegalia considerable, pero las alteraciones funcionales son solamente discretas. Los estigmas de hpatopatías son comunes cuando se ha establecido la cirrosis, pero no en etapas más tempranas. Se desconocen los mecanismos de daño hepático, aunque la enfermedad parece comenzar por un mal control de la diabetes o por la reducción de peso demasiado rápida. La evolución es gradual y no existe un tratamiento específico disponible.
CIRROSIS CARDIACA
La cirrosis cardiaca se ha vuelto cada vez menos frecuente a partir de que se han podido corregir las valvulopatías mediante el remplazo con prótesis. El padecimiento se desarrolla después de años o décadas de insuficiencia cardiaca derecha severa o de insuficiencia tricuspídea. El cuadro histológico consiste en la cicatrización central con conexión entre venas centrales por tejido fibroso, así como la eventual formación de nódulos que frecuentemente contienen una triada portal. El hígado suele estar crecido y puede demostrarse reflujo hepatoyugular. El tratamiento de la cirrosis cardiaca implica la corrección de las alteraciones valvulares que le han dado origen.
Algunos enfermos con síndrome de Budd-Chiari (trombosis de las venas suprahepáticas) pueden sobrevivir al episodio agudo, desarrollando una cirrosis similar a la cardiaca.80
CIRROSIS DIVERSAS
Puede presentarse cirrosis secundaria a exposición crónica a arsénico, methotrexate o a cantidades excesivas de vitamina A.81-83 También puede ocurrir cirrosis después de una enfermedad de vías biliares prolongada, como la colangitis esclerosante, y como una complicación rara de la sarcoidosis hepática. Además, varios padecimientos exclusivos de la niñez pueden desarrollar cirrosis, los más comunes de éstos son la fibrosis quística, las enfermedades por depósitos de glucógeno, la atresia biliar y la fibrosis hepática congénita.84-87
Complicaciones de la cirrosis
VARICES
La hemorragia por várices es una de las complicaciones más graves de la cirrosis. La mortalidad durante el episodio agudo puede ser hasta de 60 a 70 porciento.88 Hay muchos factores relacionados con una cirrosis descompensada que incrementan este riesgo de por sí alto, incluyendo la debilidad generalizada, los defectos de la coagulación y la encefalopatía hepática. El tamaño de las várices correlaciona también con el riesgo de hemorragia.89 La hemorragia recurrente, común en las dos semanas que siguen al episodio inicial, también contribuye a la gran mortalidad. Cuando el paciente sobrevive más allá de seis semanas, el riesgo de hemorragia recurrente disminuye sustancialmente, acercándose al de los cirróticos que nunca han sangrado.90 El sangrado por várices esofágicas se identifica mejor por medio de una endoscopía de la porción superior del tubo digestivo.
La escleroterapia, inyección de un agente esclerosante en las várices esofágicas, es el tratamiento de elección para el control inmediato de la hemorragia por várices. La escleroterapia también es eficaz para el control a largo plazo de la hemorragia recurrente, aunque el efecto de este tratamiento en la supervivencia aún es incierto.91-93 El agente esclerosante causa inflamación necrosante severa de la pared esofágica, seguida de importante reacción fibrótica.94 Las complicaciones son frecuentes, e incluyen dolor retroesternal, ulceración esofágica, hemorragia, derrame pleural y estenosis y perforación esofágicas. La ligadura esofágica es una variante de la escleroterapia semejante a la ligadura de las hemorroides y que se realiza con un endoscopio modificado. En tres estudios aleatorios, la ligadura de várices las obliteró con más rapidez que la escleroterapia, y fue tan eficaz como esta última para controlar la hemorragia. Este procedimeinto también es eficaz en las várices esofágicas y puede usarse como una alternativa a la escleroterapia o cuando ésta no ha sido eficaz.95-97 Sin embargo, el uso del tubo exterior, que se emplea para facilitar la ligadura repetida rápida, se ha asociado con una incidencia excesiva de perforación esofágica, por lo que ha sido retirado del mercado por el fabricante. Esto impide el uso eficaz del tratamiento de ligadura para las várices esofágicas con sangrado activo.
La hemorragia por várices gástricas es menos frecuente en pacientes con cirrosis, pero más difícil de tratar en forma eficaz, excepto por cirugía.98
Si la hemorragia persiste o recurre, la colocación de una sonda de Sengstaken-Blakemore podrá interrumpirla, cuando menos temporalmente, en más del 90 porciento de los casos.99 Muchas de las dificultades relacionadas con este recurso pueden ser evitadas si el paciente es vigilado en una unidad de cuidados intensivos. El procedimiento adecuado implica colocar la sonda a través de la nariz o la boca, inflar el balón gástrico con 250 o 300 ml. de aire y colocarlos ejerciendo presión contra la unión cardioesofágica. En muchos pacientes este medio por sí solo sera suficiente para detener la hemorragia; cuando no sea así, debe inflarse el balón esofágico a una presión de 30 a 40 mm. Hg. Es conveniente colocar una sonda de succión nasogástrica por arriba del balón esofágico para prevenir el acúmulo de sangre y moco. Dado que la posibilidad de complicaciones por el uso de esta sonda aumenta en relación al tiempo en que el balón esofágico esté inflado, se deben hacer intentos por reducir su presión tras un periodo de 24 hrs.; si la hemorragia se ha detenido, la sonda podrá retirarse en otras 24 horas.
La vasopresina intrarterial puede reducir temporalmente e incluso detener la hemorragia; sin embargo, no mejora el índice de supervivencia y su administración requiere de procedimientos angiográficos especializados que rara vez están disponibles. La administración de vasopresina por infusión intravenosa continua a pacientes con hemorragia activa no parece ser efectiva.100
La obliteración transhepática percutánea de las várices puede controlar una hemorragia varicosa activa en 70 porciento de los casos; sin embargo, las recurrencias son muy frecuentes una vez que se ha terminado con el procedimiento.101
Las hemorragias recurrentes o continuas pueden ser indicación para realizar una derivación portosistémica. Esta cirugía mayor se relaciona con una mortalidad del 40 porciento cuando se practica en forma urgente.102 Cuando el sangrado se detiene y la cirugía se puede realizar en forma electiva, la mortalidad se reduce sustancialmente. Aunque las derivaciones portosistémicas no mejoran la supervivencia, sí reducen los episodios de hemorragia en forma satisfactoria. La complicación mayor de este tipo de cirugía es la insuficiencia hepática con encefalopatía intratable.103 El procedimiento quirúrgico de elección será aquel con el cual el cirujano esté más familiarizado. Aunque las derivaciones grandes son más efectivas para prevenir nuevas hemorragias, las pequeñas se relacionan con una menor incidencia de insuficiencia y encefalopatía hepática; sin embargo, estas derivaciones tienen una mayor tendencia a trombosarse, lo cual causará nuevos episodios de hemorragia. Una derivación esplenorrenal distal con devasculariza-ción selectiva gastroesofágica concomitante, provee una descompresión selectiva de las várices esofágicas al mismo tiempo que mantiene el flujo sanguíneo mesentérico al hígado. La mayoría de los estudios demostraron que la incidencia de encefalopatía severa se reducía significativa-mente en los dos o tres años posteriores a la cirugía cuando se utilizaba la derivación esplenorrenal distal; ello comparándola con los métodos convencionales de derivación.104-106. Sin embargo, el procedimiento tiene una gran dificultad técnica, y sólo el tiempo mostrará si tiene ventajas a largo plazo.
Se han hecho trabajos comparando las derivaciones portocava de urgencia con la escleroterapia en pacientes con hepatopatía alcohólica muy descompensada y hemorragia activa de várices esofágicas.107 Aunque los requerimientos transfusionales fueron inicialmente mayores en los enfermos sometidos a derivación, la recurrencia subsecuente de hemorragias fue mucho menor en esos pacientes que en los sometidos a escleroterapia. La incidencia de encefalopatía fue un poco mayor en los casos con derivación, pero los índices de supervivencia fueron semejantes para ambos grupos.
El propranolol produce reducciones sustanciales de la presión portal en pacientes con cirrosis, por lo que es de esperarse que disminuya el riesgo de hemorragia por várices esofágicas.108 En un estudio se encontró una reducción notable en los episodios de resangrado y mejoría en la supervivencia a dos años cuando se administraba propranolol en una dosis suficiente como para reducir la frecuencia cardiaca en reposo en un 25 porciento.109 Sin embargo, otro trabajo no corroboró dicha disminución en los episodios de hemorragia utilizando dosis similares y se encontró además que el bloqueo beta producido por el propranolol hacía sumamente díficil la reanimación de los enfermos con hemorragia.110 En sujetos susceptibles, el medicamento puede además inducir el desarrollo de encefalopatía hepática.111 Este medicamento parece ser menos eficaz que la escleroterapia para prevenir el sangrado recurrente por várices esofágicas.112 Tanto la escleroterapia como el propranolol tienen efectos adversos significativos, y la elección de uno u otro tratamiento dependerá de la tolerancia y cumplimiento por parte del paciente.
La colocación de una derivación portosistémica intrahepática a través de la vía transyugular (conocida como procedimiento TIPS por sus siglas en inglés, n. del t.) se ha convertido en una técnica muy aceptada para el tratamiento de las várices esofágicas sangrantes.113 Este procedimiento crea una derivación quirúrgica, pero evita las complicaciones de la cirugía mayor. Sus beneficios y complicaciones a largo plazo se desconocen todavía. Parece razonable dejar esta forma de tratamiento a las instituciones que puedan reunir una experiencia de magnitud considerable y para los pacientes que sean malos candidatos para cirugía y refractarios a la escleroterapia.
Debido a que el primer episodio de hemorragia por várices puede tener una gran morbimortalidad, ha existido gran interés en relación con el tratamiento profiláctico de las várices esofágicas. Las derivaciones portosistémicas profilácticas disminuyen el sangrado recurrente, pero no aumentan la supervivencia.114 En varias instituciones se han realizado estudios sobre escleroterapia profiláctica con resultados contradictorios.115-118 En el estudio más extenso, que se limitó a pacientes alcohólicos, este procedimiento fue incluso dañino.119 La experiencia con los antagonistas beta adrenérgicos propranolol y nadolol ha sido alentadora porque ambos medicamentos parecen prevenir el primer episodio de hemorragia y reducir la mortalidad asociada con hemorragia en los pacientes con várices esofágicas moderadas o graves.120 Si un paciente que se sabe tienen várices de gran tamaño está bien motivado y tolera el medicamento, debe considerarse administrarle antagonistas beta adrenérgicos. El mononitrato-5 de isosorbide, un nitrato de acción prolongada, puede también ayudar a prevenir la primera hemorragia por várices.121 Sin embargo, este medicamentos debe emplearse con precaución porque daña la función renal, en especial en pacientes con ascitis.122
El tratamiento de las várices esofágicas incluye también el manejo de sostén habitual, con infusión de líquidos intravenosos para restablecer el volumen plasmático y el balance de electrolitos y administración de sangre. Cuando las alteraciones de la coagulación son importantes deberá administrarse también vitamina K por vía subcutánea, plasma fresco congelado y plaquetas.
ASCITIS
La ascitis es una secuela habitual en muchas formas de cirrosis, y puede ser detectada cuando se encuentra matidez cambiante o una onda líquida en la exploración abdominal. En ocasiones la ascitis debe sospecharse en pacientes con derrame pleural derecho.123 Los factores que contribuyen a la formación de ascitis son la hipertensión porta, la disminución en la albúmina sérica con la consecuente pérdida de presión oncótica vascular e intersticial y la retención renal de sodio y agua.
Aunque la ascitis de origen infeccioso, pancreático o neoplásico no es frecuente, no deben olvidarse estas posibilidades porque el tratamiento y pronóstico son diferentes para cada situación.124 Para excluir esas posibilidades debe obtenerse una muestra de líquido de ascitis por medio de una aguja delgada. La apariencia macroscópica del líquido puede sugerir una etiología poco habitual: por ejemplo, el líquido turbio sugiere una etiología infecciosa, el hemorrágico, un tumor, y el líquido lechoso, obstrucción linfática. Los estudios de laboratorio del líquido deben incluir citología diferencial, determinaciones de proteínas y amilasa, citología y cultivo. En la ascitis trasudativa secundaria a cirrosis, la cuenta total de proteínas es menor a 2.5 g/dl, la cuenta leucocitaria es menor a 300/mm3, la proporción de granulocitos es menor al 30 porciento, la amilasa está en un nivel menor al sérico y tanto la citología como el cultivo son negativos. Alrededor del cinco porciento de los pacientes con ascitis atribuible a cirrosis tienen una concentración total de proteínas mayor a 2.5 g/dl.
El tratamiento de la ascitis no complicada secundaria a cirrosis debe ser directo.125 En primer lugar, deben suspenderse todos los medicamentos que inhiben la síntesis de prostaglandinas, ya que reducen la velocidad de filtración glomerular, la excreción urinaria basal de sodio y bloquean la respuesta natriurética a los diuréticos.126,127 La indometacina y la aspirina son los inhibidores de prostaglandinas más utilizados. Una vez que esos fármacos han sido suspendidos, debe instituirse la restricción de agua y sodio en la dieta. Aunque en medios hospitalarios puede mantenerse una restricción extrema de agua y sodio, tal medida no es generalmente necesaria, ya que no podrá ser mantenida en el hogar del enfermo. Una dieta en la que se limita el sodio a 2 g por día y el agua a 2,000 ml suele ser bien tolerada.128 Cuando la restricción de sodio y el reposo no inducen diuresis, el diurético de elección será la espironolactona.129 Setenta y cinco porciento de los sujetos hospitalizados por ascitis mejoran sustancialmente con el simple uso de espironolactona. La eficacia de este medicamento en cirróticos con ascitis contrasta con su débil efecto en quienes tienen retención hídrica de otro origen. Dado que la espironolactona inhibe la acción de la aldosterona, tiende a prevenir la excreción renal de potasio (efecto farmacológico deseable en la cirrosis). Sin embargo, por esta razón el medicamento no es recomendable para enfermos con insuficiencia renal. El uso a largo plazo de dosis altas produce ginecomastia en 20 a 30 porciento de los pacientes.
La espironolactona debe administrarse a una dosis inicial de 50 mg dos veces al día y puede incrementarse a 100 mg dos veces al día a los dos a tres días si no ha producido diuresis. Aunque la espironolactona puede administrarse hasta a 400 mg/día o más, el medicamento se tolera poco en estas dosis. Por lo tanto, si con 100 mg dos veces al día no se obtiene una diuresis adecuada, es mejor añadir furosemide, 40 mg por la mañana. La dosis de furosemide puede aumentarse cada día 40 mg (en una dosis por la mañana) hasta lograr la diuresis. La mayoría de los pacientes comienza a responder antes de que se alcancen dosis de 120 a 160 mg de furosemide y 200 mg de espironolactona. El objetivo es lograr una diuresis adecuada con las menores dosis posibles.
La diuresis máxima del líquido de ascitis no debe exceder los 1,000 ml/día.130 En base a ello, la pérdida ponderal en pacientes cirróticos no debe ser mayor a 0.5 a 1 kg/día. Una diuresis mayor conduce a la disminución peligrosa de los volúmenes intravascular e intracelular, especialmente en los individuos con edema periférico mínimo o ausente.
Entre las complicaciones del tratamiento diurético están las alteraciones electrolíticas severas, la encefalopatía, la uremia y la deshidratación. Estas son más comunes cuando la diuresis ha sido exagerada o cuando se ha continuado el tratamiento y el paciente ya estaba libre del exceso de líquido corporal.
En cerca del 5 porciento de los casos no se logrará obtener una respuesta diurética a pesar de utilizar dosis máximas o dicha respuesta se logrará sacrificando la función renal. En tales enfermos deberá considerarse la posibilidad de colocar una derivación peritoneovenosa de LeVeen.131 Dicha derivación hace pasar el líquido de ascitis del peritoneo a la vena yugular interna por medio de un tubo subcutáneo que tiene una válvula de una sola vía. Comparado con el tratamiento médico aislado, la derivación peritoneovenosa permite una resolución más rápida de la ascitis, estancias hospitalarias más breves y recurrencia más tardía de la ascitis. Además, la ascitis refractaria puede resolverse con la aplicación de dicha derivación, aunque el procedimiento no parece alterar la supervivencia. Debido a que la mayoría de los pacientes continúan requiriendo de diuréticos, aunque a dosis menores, lo más probable es que la derivación ejerza su efecto benéfico aumentando el flujo renal.
En los pacientes con derivación pueden desarrollarse complicaciones graves, e incluso mortales, incluyendo infección bacteriana del peritoneo, coagulación intravascular diseminada y ruptura de várices esofágicas. Además, la derivación puede trombosarse y requerir un remplazo. La derivación no debe ser colocada en los enfermos que puedan ser controlados con restricción dietética y medicamentos; tampoco debe utilizarse cuando no se espera que la ascitis tenga una duración prolongada, como es el caso en episodios de hepatitis alcohólica o después de una cirugía de derivación portocava. En general, no se debe considerar que un individuo tiene ascitis refractaria hasta que ésta persista por varios meses. Finalmente, no hay motivo para colocar una derivación peritoneovenosa a menos que el sujeto esté incapacitado seriamente por la ascitis crónica.
La paracentesis de grandes volúmenes se ha vuelto un procedimiento popular para el tratamiento de pacientes con ascitis. En un estudio se realizaron con seguridad paracentesis de 4 a 6 L/día, con estancias hospitalarias más cortas y menos complicaciones que con el tratamiento diurético convencional.132 Estudios subsecuentes han demostrado que la administración de 40 g de albúmina por vía intravenosa después de cada procedimiento evita la insuficiencia renal y la hiponatremia inducidas por la paracentesis.133 Por lo general los pacientes aceptan las paracentesis porque alivian sus molestias en forma importante. También proporcionan al médico líquido de ascitis para fines diagnósticos.
Se ha comparado la derivación peritoneovenosa con las paracentesis repetidas de grandes volúmenes para el tratamiento de la ascitis refractaria.134 Ambos procedimientos fueron eficaces para aliviar la ascitis. La derivación permitió un control a más largo plazo, pero con frecuencia tuvo que remplazarse y no proporcionó ventajas de supervivencia.
Se han realizado anastomosis portocava para el tratamiento de la ascitis refractaria, pero este procedimiento no puede ser recomendado mas que para casos extremos porque se asocia con una mortalidad y morbilidad muy elevadas. Por lo general es preferible recurrir a la derivación peritoneovenosa, que es más simple.
PERITONITIS BACTERIANA ESPONTANEA
Se desarrolla peritonitis bacteriana espontánea en el 10 a 25 porciento de los pacientes cirróticos que se vigilan en forma prospectiva por lo menos durante un año.135,136 Habitualmente la cirrosis se encuentra avanzada y activa, manifestada por encefalopatía hepática, várices esofágicas e ictericia marcada. La incidencia de peritonitis bacteriana espontánea es especialmente alta en pacientes con concentraciones de proteínas en el líquido de ascitis menores a 1.0 g/dl y de bilirrubinas séricas por arriba de 2.5 mg/dl. Estos hallazgos podrían explicar el mayor riesgo de infección del líquido de ascitis porque la actividad antibacteriana del líquido de ascitis, medida por actividad de opsoninas, es proporcional a la concentración de proteínas en el mismo. Se desconoce la patogenia exacta; se supone que existe siembra hematógena del líquido de ascitis, el cual funciona como un medio ideal de cultivo. La cirrosis indudablemente facilita el proceso permitiendo que los organismos entéricos penetren a la circulación venosa portal a través de las colaterales portosistémicas, evitando así el sistema reticuloendotelial del hígado.
El cuadro típico de peritonitis bacteriana se manifiesta por la presencia de fiebre, leucocitosis periférica, dolor abdominal, peristalsis disminuida o ausente y rebote. No todos los enfermos tienen estos datos, y algunos no tendrán ni siquiera uno de ellos. Por ello, se debe realizar un estudio de líquido de ascitis siempre que las condiciones de un cirrótico empeoren súbitamente.
El líquido de ascitis se verá turbio debido a la leucocitosis y a la proliferación bacteriana. Son comunes las cifras de leucocitos superiores a 1,000 células/mm3 con más del 85 porciento de granulocitos.137 Prácticamente todos los casos tienen más de 300 leucocitos/mm3 y más de la mitad de estas células corresponderán a polimorfonucleares. Sin embargo, no todos los pacientes con leucocitosis en líquido de ascitis tienen peritonitis bacteriana. En la práctica es recomendable dar antibióticos a todos los individuos con un cuadro clínico sugerente de peritonitis y con una cifra de leucocitos en líquido de ascitis superior a 500 células/mm3.138 El criterio diagnóstico definitivo es la demostración de bacterias, ya sea por medio de la tinción de Gram (una cuarta parte de los casos) o por medio de cultivo. Para maximizar la detección de los organismos infecciosos responsables deben inyectarse 5 ml de líquido de ascitis en botellas de hemocultivo aeróbico y anaeróbico al lado de la cama del paciente. Dos terceras partes de los agentes etiológicos son enterales, siendo E. Coli y Klebsiella los más comunes. Los neumococos y estreptococos son responsables de hasta el 20 porciento de los casos. En cerca de la mitad de los pacientes el hemocultivo es positivo para el mismo microrganismo encontrado en el líquido de ascitis.
La cefotaxima, administrada por vía intravenosa en dosis de 2 g cada seis horas, es el tratamiento inicial apropiado para los episodios de peritonitis bacteriana espontánea.140 En los pacientes con insuficiencia renal la dosis se ajusta según sea necesario. Con este esquema se resuelve el 75 a 80 porciento de los episodios. Algunos requieren un cambio de esquema. Sin embargo, el 20 a 25 porciento de los pacientes fallecen antes de que la infección se resuelva. El tratamiento tiene la misma eficacia si se administra por cinco o por 10 días.141 Los pacientes deben ser vigilados en forma estrecha, reanalizando el líquido de ascitis por lo menos en una ocasión para segurarse que se está tratando la infección en forma adecuada. A pesar de un tratamiento óptimo, 40 a 60 porciento de estos enfermos fallece; la mitad como consecuencia directa de la peritonitis, mientras que el resto por otras complicaciones de la hepatopatía. El tratamiento a largo plazo con norfloxacina (400 mg/día) reduce la incidencia de peritonitis bacteriana espontánea recurrente atribuíble a bacterias aeróbicas gram negativas y debe considerarse en los pacientes con alto riesgo de recurrencia.142
SINDROME HEPATORRENAL
El síndrome hepatorrenal se define como una insuficiencia renal funcional relacionada con cirrosis bien establecida y habitualmente descompensada. Cuando aparece, el síndrome hepatorrenal generalmente tiene consecuencias mortales.143 El paciente típico suele tener ictericia profunda, está prácticamente moribundo y tendrá además ascitis a tensión, hipoalbuminemia e hipoprotrombinemia, y encefalopatía. Conforme la hepatopatía progresa, los volúmenes urinarios y la excreción de sodio disminuyen y la creatinina y nitrógeno de urea (BUN) séricos aumentan antes de la muerte. Bajo estas circunstancias, podemos decir que la insuficiencia renal es uno más de los incidentes relacionados con una hepatopatía severa. Sin embargo, en 10 a 20 porciento de los casos la hepatopatía puede estar razonablemente estable, por lo cual la insuficiencia renal aguda será la principal amenaza seria contra la vida del enfermo.
La patogenia del síndrome hepatorrenal no ha sido bien aclarada; tanto la reducción en el flujo sanguíneo renal como en el índice de filtración glomerular pueden preceder a la insuficiencia renal manifiesta incluso por varios meses. Paradójicamente, estas alteraciones ocurren combinadas con un aumento en el volumen plasmático.144 También ocurre un fenómeno llamado cortocircuito intrarrenal que se caracteriza por un incremento en el flujo a la médula renal a expensas de la corteza. Debido a que muchos pacientes presentan además hipotensión y no responden a agentes presores, se han implicado a falsos neuro-transmisores en la patogenia del síndrome hepatorrenal.
Aunque hay muchos padecimientos que pueden afectar al mismo tiempo al hígado y el riñón, la historia del enfermo con oliguria, retención importante de sal y hepatopatía severa generalmente limita las posibilidades diagnósticas a dos: síndrome hepatorrenal o azoemia prerrenal. Dado que estas dos alteraciones son indistinguibles desde el punto de vista clínico y de laboratorio, es indispensable que todos los enfermos sean tratados inicialmente como si tuvieran azoemia prerrenal.145 Debe suspenderse la medicación antidurética, cualquier pérdida sanguínea debe ser remplazada y el plasma se expandirá por medio de soluciones salinas o glucosadas. Estas medidas deben ser vigiladas estrechamente con la medición de la presión venosa central, y deben ser suspendidas si no se obtiene diuresis a pesar de que la presión venosa central haya ascendido sastisfactoriamente. Una vez que se ha descartado la presencia de azoemia prerrenal con dichas acciones, el tratamiento del síndrome hepatorrenal debe basarse en medidas de apoyo básicamente conservadoras. Aunque es poco frecuente, puede haber una corrección del síndrome cuando la hepatopatía mejora.
Se han informado casos de corrección del síndrome hepatorrenal después de la colocación de una derivación peritoneovenosa. Tal procedimiento debe considerarse en el pequeño grupo de pacientes en el que las manifestaciones de insuficiencia renal predominan sobre las de falla hepática; sin embargo no es raro que estos enfermos desarrollen complicaciones posiblemente mortales. Entre otras medidas que se han intentado sin algún beneficio están la derivación portocava de urgencia, los esteroides, la fenoxibenzamina, el metaraminol y la metildopa.
ENCEFALOPATIA HEPATICA
El diagnóstico de encefalopatía hepática se basa en la demostración de alteraciones mentales, asterixis y hedor hepático.146 Este último consiste en un olor desagradable, como de frutas fermentadas, del aliento. La asterixis se refiere a la flexión irregular de las extremidades, fácilmente demostrable pidiéndole al paciente que sostenga su brazo horizontalmente con las manos extendidas a la altura de la cintura. El movimiento de aleteo producido por la pérdida intermitente del tono extensor es un rasgo diferencial de la encefalopatía hepática, aunque de hecho el signo puede encontrarse también en la uremia y en las neumopatías severas. Un electroencefalograma con ondas lentas y aplanadas confirma el diagnóstico.
La encefalopatía hepática puede ser clasificada a grandes rasgos en cuatro etapas: la primera consiste en agitación sin hallazgos físicos acompañantes; los sujetos en esta etapa suelen recibir por error sedantes que rápidamente profundizan la encefalopatía. En la segunda el paciente estará moderadamente confuso pero aún reactivo, pudiéndose detectar asterixis. En la tercera etapa, el enfermo estará estuporoso y prácticamente sin respuesta. En la cuarta, el individuo se encontrará en coma profundo. En la tercera y cuarta etapas puede no haber asterixis.
Es indudable que la causa de la encefalopatía es multifactorial. En el origen de este síndrome se ha implicado a las concentraciones elevadas de amonio en suero, a los ácidos grasos de cadena corta, a falsos neurotransmisores y a ciertos aminoácidos.147 Además puede existir en la insuficiencia hepática una sustancia circulante que tiene propiedades semejantes a los agonistas benzodiacepínicos y que pueden potencializar la acción del ácido g-aminobutírico, contribuyendo a la encefalopatía hepática. Algunos pacientes con encefalopatía parecen responder a la administración de un antagonista del receptor de benzodiacepinas,148 lo que apoya la hipótesis de que la sustancia semejante a las benzodiacepinas participa en este transtorno. A la potogenia contribuyen la derivación de sangre alrededor del hígado que ocurre como consecuencia de la hipertensión porta y el deterioro en la función hepática. También es cierto que el sistema nervioso central de los pacientes con cirrosis parece ser más sensible a los efectos sedantes de productos endógenos y de medicamentos exógenos.
En la mayoría de los casos es posible identificar algún factor presipitante de la encefalopatía hepática.149 Entre los eventos causales están la hemorragia gastrointestinal, las alteraciones electrolíticas, los transtornos ácido-base, la hipoxia, la retención de CO2, las infecciones y el uso inadecuado de diuréticos, sedantes y otros fármacos.
El aspecto más importante es la eliminación o corrección de la causa presipitante. Debe investigarse con cuidado sobre los antecedentes de ingesta de sedantes, que deberán suspenderse. Una vez que se han tomado dichas medidas, el tratamiento estándar de la encefalopatía hepática incluye restricción de las proteínas de la dieta y la administración de lactulosa y neomicina. El criterio habitual es limitar las proteínas a 40-60 g/día producirá desnutrición proteica.
El medicamento de elección es la lactulosa, disacárido que llega sin haber sido digerido al colon, en donde sufre degradación bacteriana a ácidos de dos o tres carbonos que reducen el pH intraluminal y producen diarrea; la dosis habitual es de 30 ml. tres o cuatro veces al día. No se conoce con certeza el mecanismo de acción de la lactulosa; aparentemente la reducción en el pH de las heces hace que el amonio sea reducido a su forma iónica ( NH4 ), que se absorbe poco y es excretada con la materia fecal.
Se ha demostrado que la lactulosa es tan eficaz como la neomicina en el tratamiento de la encefalopatía hepática crónica o recurrente. Cerca del 80 porciento de los casos responde a uno de estos medicamentos.150 En algunos pacientes la neomicina puede ser eficaz aun cuando la lactulosa no lo haya sido. La dosis habitual de neomicina es de 1 g. VO c/6 hrs., que puede ser aumentada hasta 12 g. por día. Aunque la absorción intestinal de neomicina es muy pobre, se han publicado casos de otoxicidad y daño renal; por ello, dado que la lactulosa es menos tóxica, debe ser intentada en primer lugar. Aunque desde el punto de vista teórico podría parecer que la neomicina interfiera con la lactulosa, la evidencia existente indica que ambos medicamentos pueden actuar en forma sinérgica.
Tanto la lactulosa como la neomicina son eficaces en la encefalopatía crónica o recurrente, sin embargo, sus efectos benéficos pueden no funcionar en la enfalopatía que acompaña a hepatopatías severas agudas. Algunos enfermos pueden desarrollar una encefalopatía crónica refractaria, que generalmente se debe a cortocircuitos portosistémicos.
LITIASIS VESICULAR
En la cirrosis se incrementa la incidencia de litiasis biliar, probablemente como resultado del aumento en la producción de bilirrubina secundaria a la anemia hemolítica crónica y al hiperesplenismo.151 Es por ello que siempre debe pensarse en la posibilidad de coledocolitiasis cuando encontremos a un paciente cirrótico con ictericia. Afortunadamente esta complicación es rara, porque el diagnóstico de dicha entidad puede ser sumamente difícil en un cirrótico descompensado. La ultrasonografía es un procedimiento simple que detecta con certeza la dilatación de los conductos biliares en un 90 porciento de los casos.152 Este recurso se debe usar con premura siempre que se sospeche litiasis. La colangiografía intravenosa no tiene ninguna utilidad, ya que no se puede visualizar el colédoco en un sujeto con cirrosis e ictericia. Si es necesario descartar la litiasis biliar en forma concluyente deberá pensarse en la necesidad de una colangiografía percutánea o en una colangiografía endoscópica retrógrada.153 A menos que la sospecha diagnóstica sea muy alta, siempre será recomendable evitar estos procedimientos.
ULCERA PEPTICA
Los pacientes cirróticos tienen una incidencia mayor de úlcera péptica que la población general. El diagnóstico deberá considerarse en todos los casos en los que exista dolor abdominal o hemorragia del aparato digestivo. Debido a la importancia de definir si el origen de una hemorragia son las várices esofágicas o una úlcera péptica, habitualmente es necesario realizar endoscopía en todo cirrótico que desarrolle hemorragia gastrointestinal de importancia.
HIPOXIA
La hipoxia es una complicación frecuente en enfermos con cirrosis avanzada, en los que no son raras las cifras de tensión de oxígeno (pO2) de 60 a 70 mm Hg. La ascitis dificulta la ventilación y puede haber angiomas pulmonares responsables de cortocircuitos sanguíneos de derecha a izquierda.154 Dado que muchos cirróticos fuman al mismo tiempo que beben, es común que haya cierto grado de neumopatía obstructiva complicando el cuadro.
DESNUTRICION
Casi todos los pacientes con enfermedad hepática avanzada tienen algún grado de desnutrición proteico-calórica. La desnutrición severa contribuye a la retención de sal y agua, a una respuesta inmunológica defectuosa y a recuperación más tardía de la función hepática. Debe realizarse una evaluación nutricional en todos los pacientes con hepatopatía avanzada, y administrarse suplementos nutricionales cuando estén indicados. Los suplementos a la nutrición pueden proporcionarse añadiendo fórmulas estándar enterales a la dieta, y en algunos pacientes graves pueden indicarse suplementos parenterales. Existen pocas evidencias de que las fórmulas enriquecidas con aminoácidos de cadena ramificada tengan alguna ventaja sobre las fórmulas con composición estándar de aminoácidos, y las primeras son bastante más costosas. Por lo general los suplementos son bien tolerados, por vía oral o parenteral. Se espera con ellos un retorno más rápido a un balance positivo de nitrógeno y mejoría en la función hepática. No se ha demostrado mejoría convincente en la supervivencia.155
CANCER PRIMARIO DEL HIGADO
El cáncer primario del hígado se desarrolla en cinco a 20 porciento de los pacientes con cirrosis. Si no es tratada, la enfermedad evoluciona hacia el deterioro con rapidez.
Trasplante
La gran disponibilidad del trasplante de hígado ha mejorado en forma importante el diagnóstico de casi todas las formas de hepatopatía terminal.156,157 Debido a que los porcentajes de supervivencia a uno y cinco años llegan a ser de 90 y 70 porciento, respectivamente, en varias instituciones, esta forma de tratamiento es muy aceptada. (Algunos pacientes han vivido más de 20 años después de un trasplante de hígado). Las únicas excepciones para ser candidato a este procedimiento son la presencia de otras enfermedades debilitantes y la mayoría de los padecimientos neoplásicos del hígado. La principal limitante de este procedimiento es la disponibilidad de órganos.
- Menghini G: OIte-second biopsy of the liver-problems of its clinical application. N Engl J Med 283:582, 1970
- Reynolds TB, Ito S, Iwatsuki S: Measurement of portal pressure and its clinical application. Am J Med 49:649,1970
- Viamonte MJr, Warren WD, Fomon JJ: Liver panangiography in the assessment of portal hypertension in liver cirrhosis. Radiol Clin North Am 8:147,1970
- Kreel L: Radiology of the portal system. Gut 11:620, 1970
- Trujillo NP: Peritoneoscopy and guided biopsy in the diagnosis of intraabdominal disease. Gastroenterology 71:1083,1976
- Lieber CS: Pathogenesis and early diagnosis of alcoholic liver injury. N Engl J Med 298:888,1978
- Rubin E, Lieber CS: Fatty liver, alcoholic hepatitis and cirrhosis produced by alcohol in primates. N Engl J Med 290:128, 1974
- Lieber CS: Liver adaptation and injury in alcoholism. N Engl J Med 288:356,1973
- Rubin E, Lieber CS: Alcohol-induced hepatic injury in nonalcoholic volunteers. N Engl J Med 278:869,1968
- Reynolds TB, Hidemura R, Michel H, et al: Portal hypertension without cirrhosis in alcoholic liver disease. Ann Intern Med 70:497,1969
- Helman RA, Temko MH, Nye SW, et al: Alcoholic hepatitis-natural history and evaluation of prednisolone therapy. Ann Intern Med 74:311,1971
- Powell WJ Jr, Klatskin G: Duration of survival in patients with Laennec's cirrhosis: influence of alcohol withdrawal and possible effects of recent changes in general management of the disease. Am J Med 44:406, 1968
- Mendenhall CL, Anderson S, Garcia-Pont P, et al: Short-term and long-term survival in patients with alcoholic hepatitis treated with oxandrolone and prednisolone. N Engl J Med 311:1464, 1984
- Mendenhall CL, Moritz TE, Roselle GA, et al: A study of oral nutritional support with oxandrolone in malnourished patients with alcoholic hepatitis: results of a Department of Veterans Affairs cooperative study. Hepatology 17:564,1993
- Porter HP, Simon FR, Pope CE, et al: Corticosteroid therapy in severe alcoholic hepatitis. N Engl J Med 284:1350, 1971
- Campra JL, Hamlin EM, Kirshbaum RJ, et al: Prednisone therapy of acute alcoholic hepatitis: report of a controlled trial. Ann Intem Med 79:625, 1973
- Lesesne HR, Bozymski EM, Fallon HJ: Treatment of alcoholic hepatitis with encephalopathy: comparison of prednisolone with caloric supplements. Gastroenterology 74:169,1978
- Maddrey WC, Boitnott JK, Bedine MS, et al: Corticosteroid therapy of alcoholic hepatitis. Gastroenterology 75:193, 1978
- Depew W, Boyer T, Omata M, et al: Double-blindcontrolled trial of prednisolone therapy in patients with severe acute alcoholic hepatitis and spontaneous encephalopathy. Gastroenterology 78:524,1980
- Carithers RL Jr, Herlong HF, Diehl AM, et al: Methylprednisolone therapy in patients with severe alcoholic hepatitis. Ann Intern Med 110:685, 1989
- Abdi W, Millan JC, Mezey E: Sampling variability on percutaneous liver biopsy. Arch Intern Med 139:667,1979
- Davis GL, Czaja AJ: Prolonged steroid therapy for severe chronic active liver disease (CALD): a diminishing return? Gastroenterology 78:1l53, 1980
- Sherlock S, Scheuer PJ: The presentation and diagnosis of loopatients with primary biliary cirrhosis. N Engl J Med 289:674,1973
- James SP, Hoofnagle JH, Strober W, et al: Primary biliary cirrhosis: a model autoimmune disease. Ann Intern Med 99:500,1983
- Underhill J, Donaldson P, Bray G, et al: Susceptibility to primary biliary cirrhosis is associated with the HLA-DR8-DQB1.0402 haplotype. Hepatology 16:1404,1992
- Hodgson SF, Dickson ER, Wahner HW, et al: Bone loss and reduced osteoblast function in primary biliary cirrhosis. Ann Intem Med 103:855, 1985
- RollJ, Boyer JL, Barry D, et al: The prognostic importance of clinical and histologic features in asymptomatic and symptomatic primary biliary cirrhosis. N Engl J Med 308:1,1983
- Beswick DR, Klatskin G, Boyer JL: Asymptomatic primary biliary cirrhosis: a progress report on long-term follow-up and natural history .Gastroenterology 89:267, 1985
- Balasubramaniam K, Grambsch PM, Wiesner RH, et al: Diffiinished survival in asymptomatic primary biliary cirrhosis: a prospective study. Gastroenterology 98:1567,1990
- Mitchison HC, Bassendine MF, Malcolm AJ, et al: A pilot, doubleblind, controlled 1-year trial of prednisolone treatment in primarybiliary cirrhosis: hepatic improvement but greater bone loss.Hepatology 10:420,1989
- Heathcote J, Ross A, Sherlock S: A prospective controlled trial of azathioprine in primary biliary cirrhosis. Gastroenterology 70:656, 1976
- Christensen E, Neuberger J, Crowe J, et al: Beneficial effect of azathioprine and prediction of prognosis in primary biliary cirrhosis: final results of an intrnational trial. Gastroenterology 89:1084,1985
- Epstein O, Jain S, Lee RG, et al: D-Penicillamine treatment improves survival in primary biliary cirrhosis. Lancet 1:1275,1981
- Matloff DS, Alpert E, Resnick R, et al: A prospective trial of D-penicillamine in primary biliary cirrhosis. N Engl J Med 306:319,1982
- Dickson ER, Fleming TR, Wiesner RH, et al: Trial of penicillamine in advanced primary biliary cirrhosis. N Engl J Med 312:10l1, 1985
- Bodenheimer HC Jr, Schaffner F, Stemlieb I, et al: A prospective clinical trial of D-penicillamine in the treatment of primary biliary cirrhosis. Hepatology 5:1139, 1985
- Neuberger J, Christensen E, Portmann B, et al: Double blind controlled trial of D-penicillamine in patients with primary biliary cirrhosis. Gut 26:l14, 1985
- Bodenheimer H Jr, Schaffner F, Pezzulo J: A randomized doubleblind trial of colchicine in primary biliary cirrhosis (abstr). Hepatology 5:968,1985
- Kaplan MM, Alling DW, Wolfe HJ, et al: Colchicine is effective in the treatment of primary biliary cirrhosis (abstr). Hepatology 5:967, 1985
- Wiesner RH, Ludwig J, Lindor KD, et al: A controlled trial of cyclosporine in the treatment of primary biliary cirrhosis. N Engl J Med 322:1419,1990
- Lombard M, Portmann B, Neuberger J, et al: Cyclosporin A treatment in primary biliary clrrhosis: results of a long-term placebo controlled trial. Gastroenterology 104:519,1993
- Poupon RE, Balkau B, Eschwege E, et al: A multi-center, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC Study Group. N Engl J Med 324:1548,1991
- Ghent CN, Carruthers SG: Treatment of pruritus in primary biliary cirrhosis with rifampin: results of adouble-blind, crossover, randomized trial. Gastroenterology 94:488,1988
- Reed JS, Meredith SC, Nemchausky BA, et al: Bone disease in primary biliary cirrhosis: reversal of osteomalacia with oral 25-hydroxyvitamin D. Gastroenterology 78:512, 1980
- Matloff DS, Kaplan MM, Neer RM, et al: Osteoporosis in primary biliary cirrhosis: effects of 25-hydroxyvitamin D3 treatment. Gastroenterology 83:97,1982
- Finch SC, Finch CA: Idiopathic hemochromatosis, an iron storage disease. Medicine (Baltimore) 34:381, 1955
- Eciwards CQ, Griffen LM, Dadone MM, et al: Mapping the locus for hereditary hemochromatosis: localization between HLA-B and HLA-A. Am J Hum Genet 38:805,1986
- Edwards CQ: Early detection of hereditary hemochromatosis (editorial). Ann Intem Med 101:707,1984
- Stocks AE, Powell LW: Pituitary function in idiopathic haemochromatosis and cirrhosis of the liver. Lancet 2:298,1972
- Serum-ferritin (editorial). Lancet 1:533, 1979
- Howard JM, Ghent CN, Carey LS, et al: Diagnostic efficacy of hepatic computed tomography in the detection of body iron overload. Gastroenterology 84:209,1983
- Johnson CD: Magnetic resonance imaging of the liver: current clinical applications. Mayo Clin Proc 68:147,1993
- Edwards CQ, Griffen LM, Goldgar D, et al: Prevalence of hemochromatosis among 11,065 presumably healthy blood donors. N Engl J Med 318:1355, 1988
- Crawford DH, Halliday JW , summers KM, et al: Concordance of iron storage in siblings with genetic hemochromatosis: evidence for a predominantly genetic effect on iron storage. Hepatology 17:833,1993
- Kent G, Popper H: Liver biopsy in diagnosis of hemochromatosis. Am J Med 44:837,1968
- Ludwig J, Batts KP, Moyer TP , et al: Liver biopsy diagnosis of homozygous hemochromatosis: a diagnostic algorithm. Mayo Clin Proc 68:263,1993
- Williams R, smith PM, spicer EJF, et al: Venesection therapy in idiopathic haemochromatosis-an analysis of 40 treated and 18 untreated patients. Q J Med 38:1, 1969
- Niederau C, Fischer R, Sonnenberg A, et al: survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N Engl J Med 313:1256,1985
- Propper RD, Cooper B, Rufo RR, et al: Continuous subcutaneous administration of deferoxamine in patients with iron overload. N Engl J Med 297:418, 1977
- Strickland GT, Leu M: Wilson's disease--clinical and laboratory manifestations in 40 patients. Medicine (Baltimore) 54:113, 1975
- Chelly J, Monaco AP: Cloning the Wilson disease gene. Nature Genetics 5:317,1993
- Walshe JM: Wilson's disease presenting with features of hepatic dysfunction: a clinical analysis of eighty-seven patients. Q J Med 70:253, 1989
- Carpenter TO, Carnes DL, Anast Cs: Hypoparathyroidism in Wilson's disease. N Engl J Med 309:873, 1983
- Martins da Costa C, Baldwin D, Portrnann B, et al: Value of urinary copper excretion after penicillamine challenge in the diagnosis of Wilson's disease. Hepatology 15:609,1992
- Deiss A, Lynch RE, Lee GR, et al: Long-term therapy of Wilson's disease. Ann Intern Med 75:57, 1971
- Brewer GJ, Hill GM, Prasad AS, et al: Oral zinc therapy for Wilson's disease. Ann Intern Med 99:314,1983
- Erickson S, Carlson J, Velez R: Risk of cirrhosis and primary liver cancer in alpha1-antitrypsin deficiency. N Engl J Med 314:736,1986
- PalmerPE, WolfeHJ,GherardiGJ: Hepaticchanges inadultal-antitrypsin deficiency. Gastroenterology 65:284,1973
- Alpha1-antitrypsin, Z, and the liver (editorial). Lancet 340:402,1992
- Propst T, Propst A, Dietze O, et al: High prevalence of viral infection in adults with homozygous and heterozygous alpha1-antitrypsin deficiency and chronic liver disease. Ann Intern Med 117:641,1992
- Warren Ks, Rebou[varsigma]as G, Baptista AG: Ammonia metabolism and hepatic coma in hepatosplenic schistosomiasis: patients studied before and after portacaval shunt. Ann Intern Med 62:ll13, 1965
- Jordan P: Factors affecting results of chemotherapy in schistosomiasis. Ann NY Acad Sci 160:602, 1969
- Holzbach RT, Wieland RG, Lieber Cs, et al: Hepatic lipid in morbid obesity: assessment at and subsequent to jejunoileal bypass. N Engl J Med 290:296, 1974
- Andrassy RJ, Haff RC, Lobritz RW: Liver failure after jejunoileal shunt. Arch surg 110:332, 1975
- McGill DB, Humpherys SR, Baggenstoss AH, et al: Cirrhosis and death after jejunoileal shunt. Gastroenterology 63:872, 1972
- Hamilton DL, Vest TK, Brown Bs, et al: Liver injury with alcoholiclike hyalin after gastroplasty for morbid obesity. Gastroenterology 85:722, 1983
- Adler M, Schaffner F: Fatty liver hepatitis and cirrhosis in obese patients. Am J Med 67:8l1, 1979
- Miller DJ, Ishimaru H, Klatskin G: Non-alcoholic liver disease mimicking alcoholic hepatitis and cirrhosis (abstr). Gastroenterology 77:A27,1979
- Powell EE, Cooksley WG, Hanson R, et al: The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology 11:74, 1990
- Mitchell MC, Boitnott JK, Kaufman S, et al: Budd-Chiari syndrome: etiology, diagnosis and management. Medicine (Baltimore) 61:199, 1982
- Morris Js, Schmid M, Newman S, et al: Arsenic and non-cirrhotic portal hypertension. Gastroenterology 66:86,1974
- Russell RM, Boyer JL, BagherisA, et al: Hepatic injury from chronic hypervitaminosis A resulting in portal hypertension and ascites. N Engl J Med 291:435, 1974
- Dahl MGC, Gregory MM, Scheuer PJ: Methotrexate hepatotoxicity in psoriasis-comparison of different dose regimens. Br Med J 1:654,1972
- Stern RC, stevens DP, Boat TF, et al: symptomatic hepatic disease in cystic fibrosis: incidence, course and outcome of portal systemic shunting. Gastroenterology 70:645,1976
- Thaler MM, Gellis 55: studies in neonatal hepatitis and biliary atresia: m. Progression and regression of cirrhosis in biliary atresia. Am J Dis Child 116:271,1968
- Kerr DNs, Okonkwo S, Choa RG: Congenital hepatic fibrosis: the long-term prognosis. Gut 19:514,1978
- Talente GM, Coleman RA, Alter C, et al: Glycogen storage disease in adults. Ann Intem Med 120:218,1994
- Baker LA, smith C, Lieberman G: The natural history of esophageal varices: a study of 115 cirrhotic patients in whom varices were diagnosed prior to bleeding. Am J Med 26:228, 1959
- Lebrec D, De Fleury P, Rueff B, et al: Portal hypertension, size of esophageal varices, and risk of gastrointestinal bleeding in alcoholic cirrhosis. Gastroenterology 79:1139,1980
- Smith JL, Graham DY: Variceal hemorrhage: a critical evaluation of survival analysis. Gastroenterology 82:968,1982
- Terblanche J, Bornman PC, Kahn D, et al: Failure of repeated injection sclerotherapy to improve long-term survival after oesophageal variceal bleeding: a five-year prospective controlled clinical trial. Lancet 2:1328, 1983
- Sclerotherapy after first variceal hemorrhage in cirrhosis: a randomized multicenter trial. The Copenhagen Esophageal Varices Sclerotherapy Project. N Engl J Med 311:1594,1984
- Westaby D, MacDougall BR, Williams R: Improved survival following injection sclerotherapy for esophageal varices: final analysis of a controlled trial. Hepatology 5:827, 1985
- Ayres SJ, Goff JS, Warren GH: Endoscopic sclerotherapy for bleeding esophageal varices: effects and complications. Ann Intern Med 98:900,1983
- Stiegmann GV, Goff JS, Michaletz-Onody P A, et al: Endoscopic sclerotherapy as compared with endoscopic ligation for bleeding esophageal varices. N Engl J Med 326:1527,1992
- Gimson AES, Ramage JK, Panos MZ, et al: Randomised trial of variceal banding ligation versus injection sclerotherapy for bleeding oesophageal varices. Lancet 342:391,1993
- Laine L, EI-Newihi HM, Migikovsky B, et al: Endoscopic ligation compared with sclerotherapy for the treatment of bleeding esophageal varices. Ann Intern Med 119:1, 1993
- Sarin SK, Lahoti D, Saxena SP, et al: Prevalence, classification and natural history of gastric varices: a long-term follow-up study in 568 portal hypertension patients. Hepatology 16:1343, 1992
- Pitcher JL: safety and effectiveness of the modified Sengstaken Blakemore tube: a prospective study. Gastroenterology 61:291,1971
- Fogel MR, Knauer CM, Andres LL, et al: Continuous intravenous vasopressin in active upper gastrointestinal bleeding: a placebocontrolled trial. Ann Intern Med 96:565,1982
- Benner KG, Keeffe EB, Keller FS, et al: Clinical outcome after percutaneous transhepatic obliteration of esophageal varices. Gastroenterology 85:146,1983
- Malt RA: Portasystemic venous shunts (pts 1 and 2). N Engl J Med 295:24,80,1976
- Jackson FC, Perrin EB, Felix WR, et al: A clinical investigation of the portacaval shunt: V. survival analysis of the therapeutic operation. Ann surg 174:672,1971
- Galambos JT, Warren WD, Rudman D, et al: Selective and total shunts in the treatment ofbleeding varices. N EnglJ Med 295:1089, 1976
- Conn HO, Resnick RH, Grace ND, et al: Distal splenorenal shunt vs. portal-systernic shunt: current status of a controlled trial. Hepatology 1:151,1981
- Langer B, Taylor BR, Mackenzie DR, et al: Further report of a prospective randomized trial comparing distal splenorenal shunt with end-to-side portacaval shunt: an analysis of encephalopathy, survival, and quality of life. Gastroenterology 88:424,1985
- Cello JP, Grendell JH, Crass RA, et al: Endoscopic sclerotherapy versus portacaval shunt in patients with severe cirrhosis and variceal hemorrhage. N Engl J Med 3l1:1589, 1984
- LeBrec D, Hillon P, Múnoz C, et al: The effect of propranolol on portal hypertension in patients with drrhosis: a hemodynamic study. Hepatology 2:523,1982
- LeBrec D, Poynard T, Bemuau J, et al: A randomized controlled study of propranolol for prevention of recurrent gastrointestinal bleeding in patients with cirrhosis: a final report. Hepatology 4:355, 1984
- Burroughs AK, Jenkins WJ, sherlock S, et al: Controlled trial of propranolol for the prevention of recurrent variceal hemorrhage in patients with cirrhosis. N Engl J Med 309:1539,1983
- Tarver D, Walt RP , Dunk AH, et al: Precipitation of hepatic encephalopathy by propranolol in cirrhosis. Br Med J 287:585, 1983
- Dasarathy S, Dwivedi M, Bhargava DK, et al: A prospective randomized trial comparing repeated endoscopic sclerotherapy and propranolol in decompensated (child class B and C) drrhotic patients. Hepatology 16:89,1992
- Conn HO: Transjugular intrahepatic portal-systemic shunts: the state of the art. Hepatology 17:148, 1993
- Conn HO, Lindenmuth WW, May CJ, et al: Prophylactic portacaval anastomosis: a tale oftwo studies. Medidne (Baltimore) 51:27,1972
- Paquet KJ: Prophylactic endoscopic sclerosing treatrnent of the esophageal wall in varices-a prospective controlled randomized trial. Endoscopy 14:4,1982
- Witzel L, Wolbergs E, Merki H: Prophylactic endoscopic sclerotherapy of oesophageal varices: a prospective controlled study. Lancet 1:773, 1985
- Santangelo WC, Dueno Ml, Estes BL, et al: Prophylactic sclerotherapy of large esophageal varices. N Engl J Med 318:814,1988
- Piai G, Cipolletta L, Claar M, et al: Prophylactic sclerotherapy of high-risk esophageal varices: results of a multicentric prospective controlled trial. Hepatology 8:1495,1988
- Prophylactic sclerotherapy for esophageal varices in men with alcoholic liver disease: a randomized, single-blind, multicenter clinical trial. The Veterans Affairs Cooperative Variceal Sclerotherapy Group. N Engl J Med 324:1779,1991
- Poynard T, Cales P, Pasta L, et al: Beta-adrenergic-antagonist drugs in the prevention of gastrointestinal bleeding in patients with cirrhosis and esophageal varices: an analysis of data and prognostic factors in 589 patients from four randomized clinical trials. Francoltalian Multicenter study Group. N Engl J Med 324:1532, 1991
- Angelico M, Carli L, Piat C, et al: Isosorbide-5-mononitrate versus propranolol in the prevention of first bleeding in cirrhosis. Gastroenterology 104:1460,1993
- Salmeron JM, Ruiz del Arbol L, Gines A, et al: Renal effects of acute isosorbide-5-mononitrate administration in cirrhosis. Hepatology 17:800,1993
- Rubinstein D, Mclnnes lE, Dudley FJ: Hepatic hydrothorax in the absence of clinical asdtes: diagnosis and management. Gastroenterlogy 88:188, 1985
- Cabot RC: The causes of ascites: a study of five thousand cases. Am J Med Sd 143:1,1912
- Runyon BA: Care of patients with ascites. N Engl J Med 330:337,1994
- Mirouze D, Zipser RD, Reynolds TB: Effect of inhibitors of prostaglandin synthesis on induced diuresis in cirrhosis. Hepatology 3:50,1983
- Wong F, Massie D, Hsu P, et al: lndomethacin-induced renal dysfunction in patients with well-compensated cirrhosis. Gastroenterology 104:869,1993
- Reynolds TB, Lieberman FL, Goodman AR: Advantages of treatment of asdtes without sodium restriction and without complete removal of excess fluid. Gut 19:549,1978
- Gregory PB, Broekelschen PH, Hill MD, et al: Complications of diuresis in the alcoholic patient with ascites: a controlled trial. Gastroenterology 73:534,1977
- Shear L, Ching S, Gabuzda GJ: Compartrnentalization of ascites and edema in patients with hepatic cirrhosis. N Engl J Med 282:1391,1970
- Stanley MM, Ochi S, Lee KK, et al: Peritoneovenous shunting as compared with medical treatrnent in patients with alcoholic cirrhosis and massive ascites. Veterans Administration Cooperative study on Treatment of Alcoholic Cirrhosis with Ascites. N Engl J Med 321:1632,1989
- Gines P, Arroyo V, Quintero E, et al: Comparison of paracentesis and diuretics in the treatment of cirrhotics with tense ascites: results of a randomized study. Gastroenterology 93:234,1987
- Gines P, Tito L, Arroyo V, et al: Randomized comparative study of therapeutic paracentesis with and without intravenous albumin in cirrhosis. Gastroenterology 94:1493, 1988
- Gines P, Arroyo V, Vargas V, et al: Paracentesis with intravenous infusion of albumin as compared with peritoneovenous shunting in cirrhosis with refractory ascites. N Engl J Med 325:829,1991
- Llach J, Rimola A, Navasa M, et al: Incidence and predictive factors of first episode of spontaneous bacterial peritonitis in cirrhosis with ascites: relevance of ascitic fluid protein concentration. Hepatology 16:724,1992
- Andreu M, Sola R, sitges-Serra A, et al: Risk factors for spontaneous bacterial peritonitis in cirrhotic patients with ascites. Gastroenterology 104:1133,1993
- Weinstein MP, Iannini PB, stratton CW, et al: Spontaneous bacterial peritonitis: a review of 28 cases with emphasis on improved survival and factors influencing prognosis. Am J Med 64:592,1978
- Conn HO: spontaneous bacterial peritonitis: multiple revisitations (editorial). Gastroenterology 70:455,1976
- Runyon BA, Umland ET, Merlin T: Inoculation of blood culture bottles with ascitic fluid. Arch Intern Med 147:73,1987
- Toledo C, Salmeron JM, Rimola A, et al: Spontaneous bacterial peritonitis in cirrhosis: predictive factors of infection resolution and survival in patients treated with cefotaxime. Hepatology 17:251, 1993
- Runyon BA, McHutchison JG, Antillon MR, et al: short-course versus long-course antibiotic treatment of spontaneous bacterial peritonitis: a randomized controlled study of 100 patients. Gastroenterology 100:1737,1991
- Gines P, Rimola A, Planas R, et al: Norfloxacin prevents spontaneous bacterial peritonitis recurrence in cirrhosis: results of a doubleblind, placebo-controlled trial. Hepatology 12:716,1990
- Baldus WP, Feichter RN, summerskill WH: The kidney in cirrhosis: I. Clinical and biochemical features of azotemia in hepatic failure. Ann Intern Med 60:353,1964
- McCloy RM, Baldus WP , Tauxe WN, et al: Plasma volume and renal circulatory function in cirrhosis. Ann Intern Med 66:307,1967
- Conn HO: A rational approach to the hepatorenal syndrome. Gas-troenterology 65:321, 1973
- Adams RD, Foley JM: The neurological disorder assodated with liver disease. Metabolic and Toxic Diseases of the Nervous system. Williams & Wilkins, Baltimore, 1953, p 198
- Fraser CL, Arieff Al: Hepatic encephalopathy. N Engl J Med 313:865, 1985
- Pomier-Layrargues G, Giguere JF, Lavoie J, et al: Flumazenil in cirrhotic patients in hepatic coma: a randomized double-blind placebo-controlled crossover trial. epatology 19:32,1994
- Stormont JM, Mackie JE, Davidson CS: Observations on antibiotics in the treatrnent of hepatic coma and on factors contributing to prognosis. N Engl J Med 259:l145, 1958
- Conn HO, Leevy CM, Vlahcevic ZR, et al: Comparison of lactulose and neomycin in the treatrnent of chronic portal-systemic encephalopathy: a double-blind controlled trial. Gastroenterology 72:573,1977
- Nicholas P, Rinaudo P A, Conn HO: Increased incidence of choleli-thiasis in Laennec's cirrhosis: a postmortem evaluation of pathogenesis. Gastroenterology 63:l12, 1972
- Lapis JL, Orlando RC, Mittelstaedt CA, et al: Ultrasonography in the diagnosis of obstructive jaundice. Ann Intem Med 89:61,1978
- Elias E, Hamlyn AN, Jain S, et al: A randomized trial of percutaneous transhepatic cholangiography with the Chiba needle versus endoscopic retrograde cholangiography for bile duct visualization in jaundice. Gastroenterology 71:439, 1976
- Robin ED, Hom B, Goris ML, et al: Detection, quantitation and pathophysiology of lung "spiders." Trans Assoc Am Physicians 88:202, 1975
- Nompleggi DJ, Bonkovsky HL: Nutritional supplementation in chronic liver disease: an analytical review. Hepatology 19:518, 1994
- Starzl TE, Demetris AJ, Van Thiel D: Liver transplantation (first of two parts). N Engl J Med 321:1014,1989
- Starzl TE, Demetris AJ, Van Thiel D: Liver transplantation (second of two parts). N Engl J Med 321:1092,1989