Hematología
⭳ Abrir artículo (PDF)1.2 MBEste artículo fue revisado respecto a la Edición 2/2000. Ver esa versión →
Contenido del artículo
IV HEMOGLOBINOPATIAS Y ANEMIAS HEMOLITICAS
- Desarrollo, estructura y fisiología del eritrocito
- HEMOGLOBINA
- CITOSOL NO HEMOGLOBINICO
- MEMBRANA PLASMATICA
- CONTROL DE LA HIDRATACION
- CAMBIOS DE FORMA
- PRINCIPIOS DE FLUJO SANGUINEO
- ENVEJECIMIENTO Y MUERTE CELULAR
- Características generales de las anemias hemolíticas
- Defectos en la membrana de los eritrocitos
- TRASTORNOS DEL METABOLISMO DEL SODIO Y DEL AGUA
- ALTERACIONES DE LAS PROTEINAS
- HEMOGLOBINURIA PAROXISTICA NOCTURNA
- Alteraciones del metabolismo del eritrocito
- DEFECTOS DE LA CAPACIDAD REDUCTORA
- DEFICIENCIA DE GLUCOSA-6-FOSFATO DESHIDROGENASA
- DEFECTOS EN LA GLUCOLISIS
- DEFECTOS EN EL METABOLISMO DE LOS NUCLEOSIDOS
- Trastornos de la hemoglobina
- CLASIFICACION DE LAS HEMOGLOBINOPATIAS
- ANEMIA DE CELULAS FALCIFORMES
- CRISIS FALCIFORMES
- Diagnóstico
- Otras consideraciones terapéuticas
- Anestesia general
- Pronóstico
- Tratamiento
- Consejo genético, anticoncepción y embarazo
- VARIANTES DE LA ENFERMEDAD FALCIFORME
- OTRAS HEMOGLOBINOPATIAS
- Enfermedad por hemoglobina C
- Hemoglobinopatías inestables
- Hemoglobina con afinidad anormal para el oxígeno
- Metahemoglobinemia
- TALASEMIAS
- Defectos extracorpusculares
- LESION MECANICA: HEMOLISIS MICROANGIOPATICA
- HEMOLISIS INMUNE
- Anemia hemolítica autoinmune
- Hemólisis inmune relacionada con medicamentos
- Hemólisis tardía de eritrocitos transfundidos
- Enfermedad por crioaglutininas
- Hemoglobinuria paroxística por frío
- HIPERESPLENISMO
- MEDICAMENTOS, TOXINAS, VENENOS Y AGENTES FISICOS COMO CAUSAS DE HEMOLISIS
- Fisiopatología
- Tratamiento
- Otras formas de hemólisis inducida por medicamentos
- Ataque enzimático a los eritrocitos
- Causas físicas de hemólisis
- Causas infecciosas de hemólisis
- HEMOLISIS RELACIONADA CON ENFERMEDAD HEPATICA
- OTRAS CAUSAS DE HEMOLISIS
IV HEMOGLOBINOPATIAS Y ANEMIAS HEMOLITICAS
DR. STANLEY L. SCHRIER
Las alteraciones de la membrana eritrocitaria son la vía final común que conduce a la hemólisis. Estas alteraciones de la membrana suelen indicar a los macrófagos reticuloendoteliales que retiren de la circulación los eritrocitos dañados. Sin embargo, en circunstancias extraordinarias el daño es tan grande que el contenido intracelular, incluyendo la hemoglobina, se libera en el plasma. Se conoce ya con sorprendente detalle la manera como se altera la membrana. Esta subsección describe varias enfermedades de la arquitectura de la membrana, variaciones genéticas de diversas proteínas del eritrocito, incluyendo la hemoglobina, y factores extracorpusculares que pueden causar acortamiento en la vida de los eritrocitos y muchas otras consecuencias serias.
Desarrollo, estructura y fisiología del eritrocito
Las células eritroides sufren cuatro o cinco divisiones celulares en la médula ósea y después expulsan su núcleo. Al madurar la célula enucleada disminuye la síntesis de hemoglobina y los reticulocitos pierden la mayoría de sus receptores de transferrina y penetran a la sangre periférica.
En la circulación los eritrocitos sobreviven alrededor de 4 meses. Durante este tiempo el eritrocito debe soportar tensiones mecánicas y metabólicas severas: se deforma para atravesar capilares con diámetros equivalentes a la mitad del suyo, resiste altas fuerzas de corte al cruzar las válvulas cardiacas, se aglomera con otros eritrocitos sin adherirse, y sobrevive a episodios repetidos de acidemia inducida por estasis y carencia de sustratos, y eliminación por macrófagos en las estructuras reticuloendoteliales. Debe también mantener un ambiente interno que proteja a la hemoglobina del ataque oxidativo y conservar la concentración óptima de 2,3-difosfoglicerato (2,3-DPG) que se necesita para la función de la hemoglobina.
HEMOGLOBINA
El eritrocito adulto normal contiene tres formas diferentes de hemoglobina (Hb): HbA (96 por ciento), HbA2 (2 a 3 por ciento) y Hb F (< 2 por ciento). La hemoglobina A normal se compone de 2 cadenas alfa y 2 cadenas beta (alfa2beta2), codificadas por cuatro genes en el cromosoma 16. Los genes beta y los otros genes no alfa, que codifican las cadenas gama de la HbF (hemoglobina fetal) y las cadenas delta de la HbA2, están en íntima relación mutua en el cromosoma 11. La concentración extraordinariamente alta de hemoglobina en el eritrocito, 33 a 35 g/dl (concentración media de hemoglobina corpuscular o CMHC) produce una solución viscosa.
CITOSOL NO HEMOGLOBINICO
Los eritrocitos utilizan la glucosa principalmente para mantener el poder reductor que protege a las células contra el ataque oxidativo, generar el 2,3-DPG requerido para modular la función de la hemoglobina, y para controlar el contenido de sal y agua de los eritrocitos a través de las acciones del trifosfato de adenosina (ATP) y de las trifosfatasas de adenosina (ATPasas) de transporte [ver tabla 1]. El contenido de agua y hemoglobina de los eritrocitos determinan el volumen corpuscular medio (VCM) y la CMHC.
MEMBRANA PLASMATICA
El eritrocito normal tiene forma discoide, un diámetro de 7 a 8 µm, un VCM de 85 a 90 femtolitros (fl) (1 fl =10-15 L), y una superficie de 140 µm2 [ver figura 1]. La forma bicóncava del eritrocito le permite introducirse a través de capilares tan angostos como de 3 µm de diámetro.
Los lípidos (fosfolípidos y colesterol) son responsables de alrededor del 50% del peso de la membrana de superficie. Los fosfolípidos se distribuyen de modo asimétrico en una bicapa, los que tienen carga positiva, como la esfingomielina y la fosfatidilcolina se localizan en la mitad externa, y los aminofosfolípidos con carga negativa, como la fosfatidilserina y la fosfatidiletanolamina, se encuentran sobre todo en la mitad interna de la bicapa. Esta asimetría en la distribución de fosfolípidos permite que pequeñas moléculas con carga puedan intercalarse en forma selectiva y causar expansión de la mitad externa o de la interna de la bicapa, produciendo equinocitos o estomatocitos. [ver figura 1].
Las proteínas de la membrana del eritrocito se clasifican con base en su movilidad en la electroforesis en gel de sodio dodecil sulfato- poliacrilamida (SDS-PAGE). Las proteínas integrales interactúan con la bicapa hidrofóbica de fosfolípidos [ver figura 2]. Las principales proteínas integrales de la membrana del eritrocito son las glucoforinas (que contienen la mayor parte del ácido siálico y portan los antígenos de grupo sanguíneo MNS) y la banda 3, que es el transportador aniónico y de bicarbonato.
Las proteínas periféricas se encuentran en la cara de la membrana que da al citosol. Las bandas 1 (alfa) y 2(beta) de la espectrina, una proteína periférica, interactúan para formar un heterodímero; enseguida se unen dos de estos heterodímeros para formar un heterotetrámero, la forma predominante de la espectrina. Por sí mismos, los heterotetrámeros contribuyen poco a la estabilidad, pero cuando están unidos en forma cruzada por oligómeros de la banda 5, o actina, en una interacción que está muy reforzada por la banda 4.1, el resultado es el citoesqueleto fuerte y elástico del eritrocito.
El citoesqueleto periférico está conectado a las proteínas integrales por medio de la banda 2.1 (anquirina), que une a la espectrina con la banda 3 [ver figura 2]. Una de las glucoforinas, la glucoforina C, se fija también a la banda 4.1. Se han identificado nuevas proteínas periféricas de la membrana, que existen en cantidades pequeñas y que son la aducina, la tropomiosina y la dematina, y todas parecen tener funciones como esqueleto de la membrana.1,2
Los carbohidratos contribuyen a la carga externa negativa de la membrana y en parte funcionan como antígenos de grupo sanguíneo. Algunos de estos glucolípidos se asocian con fosfatidilinositol para formar un ancla glucolípida llamada el ancla glucosil-fosfatidilinositol (GPI) [ver figura 3]. Estas anclas de GPI proporcionan sitios de sostén en la membrana para diversos tipos de proteína, como el factor acelerador de la degradación (DAF o CD55) y el inhibidor de la lisis reactiva de la membrana (MIRL o CD59), que sirve para controlar la acción del complemento [ver adelante, Hemoglobinuria paroxística nocturna].3
CONTROL DE LA HIDRATACION
El control del volumen del eritrocito tiene gran importancia fisiopatológica porque el contenido de agua y cationes de la célula determina su viscosidad intracelular y la relación entre el área de superficie y el volumen. El contenido de Na+ y K+ está determinado por una combinación de difusión pasiva y transporte activo, y este último mecanismo depende principalmente de la Na+-K+-ATPasa. El principal anión intracelular es el Cl-, que entra a la célula con gran permeabilidad a través de la banda 3, el principal transportador de aniones (también denominado AE1 o transportador de aniones-1). El cotransportador de K+ - Cl - origina el gradiente K+ - Cl - y es activado por edema de los eritrocitos y un pH intracelular bajo para causar una pérdida neta de K+ y Cl -. La Ca2+ATPasa bombea en forma activa Ca2+ al exterior, disminuyendo el contenido de Ca2+ libre en el citosol a menos de 0.1 µM, cuatro órdenes de magnitud menores que la concentración en plasma de 1 mM. El canal de Gardos, que extrae K+ en forma dependiente de Ca2+, tiene un papel importante en el control del volumen. El agua entra y sale a través de un canal de agua conocido como CHIP 28 (proteína de membrana integral que forma el canal de 28 kd) o acuaforina. Otros aniones intracelulares importantes son el 2,3-DPG y la hemoglobina, ninguno de los cuales atraviesa la membrana celular. Cuando la concentración de Ca2+ libre en el citosol aumenta a 0.3 µM, el canal se activa y causa una pérdida neta de K+. Si esta pérdida no se corrige, el eritrocito se deshidratará.4
CAMBIOS DE FORMA
La carencia de ATP, el acúmulo de ión calcio o el tratamiento con lisolecitina o con compuestos anfipáticos aniónicos, transforman el eritrocito normal o discocito en una célula espiculada crenada, equinocito o célula erizo [ver figura 1]. El calcio, solo o combinado con una proteína fijadora de calcio, la calmodulina, puede inducir modificaciones en la forma del equinocito. Si el proceso equinocítico persiste o progresa, las puntas del equinocito se fragmentan y se pierden componentes de la membrana, sobre todo de la banda 3 y fosfolípidos. Esta pérdida causa reducción en el área de superficie, con disminución en la relación superficie-volumen y formación de esferoequinocitos poco deformables.
PRINCIPIOS DE FLUJO SANGUINEO
Los principales determinantes del flujo sanguíneo son el hematócrito, la concentración plasmática de proteínas como fibrinógeno e inmunoglobulinas, que influyen en el grado de formación de rouleaux o agregación, la deformabilidad de los eritrocitos, el calibre de los vasos sanguíneos y la proporción de fuerzas de cizallamiento o de corte (la relación entre la velocidad de flujo y el radio del tubo). A las bajas fuerzas de cizallamiento que existen en las vénulas poscapilares los eritrocitos tienden a agruparse en masas asimétricas, con un aumento consecuente en la viscosidad y resistencia al flujo de la sangre.
ENVEJECIMIENTO Y MUERTE CELULAR
En la médula ósea el reticulocito pierde en forma progresiva su ARN residual durante un periodo de 4 días posteriores a la expulsión nuclear, y a partir de ese momento no puede ya realizar síntesis proteica. El cotransporte activo de K+-Cl- sirve para reducir su volumen. Con un ensamble proteico completo en la membrana, el eritrocito maduro entra a la circulación y sobrevive durante un periodo de 100 a 120 días.5 La muerte de los eritrocitos es un fenómeno que depende de la edad y puede relacionarse con fuerzas mecánicas y químicas que la célula encuentra en la circulación. A medida que envejece, el eritrocito pierde agua y superficie, disminuye la relación entre volumen y superficie y aumenta la CMHC, deteriorándose la deformabilidad de la célula. Además, la disminución de la actividad enzimática reduce la capacidad para soportar las presiones metabólicas. El envejecimiento puede manifestarse por cambios en la superficie del eritrocito, como pudiera ser una disminución en la densidad o tipo de carga superficial o la aparición de un neoantígeno de envejecimiento, quizá un grupo de banda 3 [ver figura 2], que se une a inmunoglobulinas específicas y componentes del complemento.6 A través de estos cambios el eritrocito viejo indica su incapacidad al sistema reticuloendotelial, lo que desencadena su eliminación por los macrófagos.
En condiciones fisiológicas, poco menos del 1 por ciento de los eritrocitos son destruidos diariamente y sustituidos por una cantidad prácticamente idéntica de nuevas células. Todos los días, un varón de 70 kg de peso con un volumen sanguíneo aproximado de 5 litros destruye y sustituye cerca de 50 ml de sangre total, que contiene aproximadamente 22 ml de eritrocitos. Como la hemoglobina equivale a un tercio del peso total de cada eritrocito, la reposición de estas células requiere la síntesis diaria de 7 g de hemoglobina. La médula ósea del adulto normal puede aumentar fácilmente su producción eritroide en 5 veces. Después de cualquier situación que ocasione anemia intensa y prolongada, la producción eritroide se puede elevar 7 u 8 veces. Sin embargo, el aporte de hierro representa un límite importante en la reposición de los eritrocitos: tres cuartas partes del hierro que se utiliza para la síntesis de eritrocitos en un día proviene de células que fueron destruidas el día anterior.
Características generales de las anemias hemolíticas
En términos generales, la severidad de una anemia está determinada por la velocidad de destrucción de los eritrocitos y por la capacidad de la médula ósea para producirlos. Cuando una persona tiene una médula normal el tiempo de vida de los eritrocitos puede disminuirse de 120 días a 20 días sin causar anemia o ictericia; sin embargo, en dichos casos se encontrará reticulocitosis importante.
La mayor parte de las formas de hemólisis son extravasculares, el sistema reticuloendotelial detecta las alteraciones de los eritrocitos en su membrana y los retira de la circulación. En circunstancias excepcionales, en las que el daño a los eritrocitos es devastador, como en algunas formas de lisis mediada por complemento, o en circunstancias en las que el sistema reticuloendotelial no puede eliminar el gran volumen de células dañadas, se desarrolla lisis intravascular y esto causa hemoglobinemia. La hemoglobina liberada hacia el plasma es degradada a dímeros alfa-beta, que se unen a la haptoglobina. Los complejos hemoglobina-haptoglobina son retirados por el sistema reticuloendotelial. Cuando se excede la capacidad de unión de la haptoglobina, los dímeros alfa-beta pasan al filtrado glomerular. Algunos de los dímeros alfa-beta se excretan directamente en la orina, produciendo hemoglobinuria, mientras que otros son captados y procesados por las células del túbulo renal. Varios días después de un episodio de hemólisis intravascular pueden excretarse células de los túbulos renales que contienen hierro. Es posible diagnosticar la hemosiderinuria identificando dichas células con tinción de azul de Prusia. La hemoglobina libre en el plasma se puede disociar en globina y hemina. La hemina puede unirse a la hemopexina y llegar a las células del túbulo renal en esa forma, o se puede unir a la albúmina plasmática, produciendo metemalbuminemia.
La hemólisis intravascular en un ejemplo notable de destrucción de los eritrocitos y puede producir anemia severa en forma aguda. Además, las partículas de la membrana eritrocítica liberada al plasma pueden actuar como estímulos potentes para el desarrollo de coagulación intravascular diseminada.
Cuando la hemólisis es crónica suelen desarrollarse cálculos de pigmento en la vesícula. Cuando un paciente que está compensando la hemólisis sufre una infección que altera en forma importante la actividad eritroide de la médula ósea,7 el nivel de hemoglobina puede disminuir en forma dramática, una condición denominada crisis aplástica. La hemólisis severa aguda también es causa de insuficiencia renal aguda y la hemólisis intravascular aguda puede provocar coagulación intravascular diseminada.
Las causas de hemólisis se clasifican como extracorpusculares o intracorpusculares. Las causas intracorpusculares, que son esencialmente defectos del eritrocito, incluyen alteraciones de la membrana celular, alteraciones metabólicas y trastornos de la estructura o biosíntesis de la hemoglobina. Las causas extracorpusculares representan elementos anormales dentro del lecho vascular del paciente que atacan y destruyen a los eritrocitos normales. Como los eritrocitos con defectos que causan hemólisis son anormales de manera intrínseca, siguen mostrando un tiempo de supervivencia característicamente acortado cuando se transfunden a receptores normales. De los defectos intracorpusculares el único que no es hereditario es la hemoglobinuria paroxística nocturna.
Defectos en la membrana de los eritrocitos
TRASTORNOS DEL METABOLISMO DEL SODIO Y DEL AGUA
Hidrocitosis (Estomatocitosis hereditaria)
La hidrocitosis es un trastorno hereditario que suele presentarse en etapas tempranas de la vida en forma de una anemia hemolítica parcialmente compensada, en ocasiones con bazo palpable. El VCM suele estar elevado. El frotis periférico demuestra estomatocitos [ver figura 4]. El flujo pasivo de Na+ y K+ aumenta mucho. La Na+-K+ATPasa es insuficiente y la concentración de cationes y el agua celular aumentan, provocando un incremento en el VCM y la disminución en la relación entre la superficie y el volumen. Algunos enfermos con hidrocitosis tienen un defecto en uno de los componentes de la membrana del eritrocito en la región de la banda 7. La esplenectomía puede mejorar la anemia. Los estomatocitos parecen adherirse en forma más ávida que los eritrocitos normales, un dato que puede explicar el reporte reciente de mayor frecuencia de eventos tromboembólicos.8
Xerocitosis
La xerocitosis, otro padecimiento hemolítico hereditario, se caracteriza por un defecto de membrana que origina pérdida de cationes y deshidratación celular. La salida de K+ excede la entrada de Na+, lo que ocasiona la pérdida de agua. Los pacientes presentan hemólisis variable compensada. La esplenomegalia no es una característica importante. El resultado del frotis periférico es variable, mostrando células en blanco de tiro, estomatocitos, equinocitos o los llamados budines de hemoglobina (i.e., hemoglobina acumulada alrededor de la célula). La CMH aumenta. Debido a que estas células rígidas son eliminadas en muchas partes del sistema reticuloendotelial, la esplenectomía no es muy útil.9
ALTERACIONES DE LAS PROTEINAS
Eliptocitosis hereditaria
Existen quizá 250 a 500 casos de eliptocitosis hereditaria por un millón de habitantes.10 En estos pacientes pueden observarse tres variedades morfológicas: (1) eliptocitosis hereditaria común, (2) eliptocitosis hereditaria esferocítica y (3) eliptocitosis hereditaria estomatocítica.10 La mayoría de los pacientes con eliptocitosis hereditaria común son heterocigotos para este trastorno autosómico dominante y solo tienen eritrocitos elípticos o, en el peor de los casos, hemólisis compensada. Los homocigotos pueden tener anemia hemolítica severa muy descompensada.
Al aplicar fuerzas de cizallamiento, los discocitos asumen una forma elíptica y cuando se elimina la fuerza recuperan su forma discoide. Se ha propuesto que los defectos de memebrana en la eliptocitosis hereditaria interfieren con la recuperación de la forma normal. Este defecto de membrana parece consistir en una lesión del citoesqueleto que afecta a la espectrina. En la mayoría de los pacientes los heterodímeros de espectrina no se asocian entre sí normalmente para formar tetrámeros u oligómeros [ver figura 2]. Otros defectos incluyen a la banda 4.1 o la interacción entre la anquirina y la banda 3. Las membranas de los eritrocitos de los pacientes con eliptocitosis hereditaria son invariablemente frágiles.
El diagnóstico se realiza en un paciente con hemólisis extravascular intracorpuscular con eliptocitos. Debe establecerse un patrón de herencia autosómico dominante porque la eliptocitosis puede observarse en la deficiencia severa de hierro, en los trastornos mieloproliferativos y mielodisplásicos y, en ocasiones, en las deficiencias de cobalamina y folato.10 La prueba de fragilidad osmótica suele ser normal. La esplenectomía ha sido útil en los pacientes con eliptocitosis hereditaria común.
Piropoiquilocitosis hereditaria
El síndrome de piropoiquilosis hereditaria (autosómico recesivo), una variedad de eliptocitosis hereditaria, produce hemólisis grave en niños pequeños. Es causado por una mutación anormal de alfa o beta espectrina. El frotis de sangre muestra microcitosis extrema y una extraordinaria variación en el tamaño y forma de los eritrocitos [ver figura 4]. La esplenectomía puede disminuir la magnitud de la hemólisis.
Esferocitosis hereditaria
La esferocitosis hereditaria se trasmite con carácter autosómico dominante y afecta alrededor de 220 personas por cada millón en todo el mundo. Se ha descrito una variante autosómica recesiva rara.
El dato característico de la enfermedad es la disminución en el área de superficie del eritrocito, que asume una forma microesferocítica y por lo tanto no puede deformarse lo suficiente para pasar a través de la vasculatura esplénica. Esto causa atrapamiento de los eritrocitos y hemólisis, con un aumento compensatorio en la eritropoyesis. Los defectos de la membrana ocasionan formación de vesículas en la misma ante condiciones de depleción metabólica. Estas vesículas de la membrana contienen fosfolípidos de la bicapa junto con proteínas transmembrana asociadas [ver figura 2]. Las lesiones moleculares subyacentes parecen consistir en deficiencia de la espectrina, la espectrina-anquirina, la banda 3 y la banda 4.210,11
Alrededor del 25 por ciento de los pacientes con esferocitosis hereditaria tienen hemólisis totalmente compensada sin anemia, y se diagnostican solo cuando un padecimiento concomitante, como una infección o embarazo, aumenta la velocidad de la hemólisis o reduce la capacidad compensatoria de la médula ósea.
Otros pacientes pueden sufrir anemia leve, cálculos de pigmentos biliares en la vesícula, úlceras en las piernas y ruptura esplénica. Las infecciones ordinarias del aparato respiratorio pueden precipitar crisis aplásticas, en especial la infección por parvovirus.7 El diagnóstico es sugerido por el predominio de microesferocitos en el frotis de sangre periférica [ver figura 4], una CMHC de 35 g/dl o mayor, reticulocitosis, ictericia leve, esplenomegalia y antecedentes familiares positivos. El diagnóstico se confirma por medio de una prueba de fragilidad osmótica con incubación de 24 horas. Una prueba de Coombs negativa en un paciente con antecedentes familiares positivos para esferocitosis hereditaria descarta el diagnóstico de anemia hemolítica autoinmune adquirida.
La esplenectomía erradica las manifestaciones clínicas del padecimiento, incluso las crisis aplásticas.
HEMOGLOBINURIA PAROXISTICA NOCTURNA
La hemoglobinuria paroxística nocturna (HPN) es un trastorno clonal de la hematopoyesis causada por una deficiencia en la proteína de anclaje de membrana fosfatidilinositol glican de clase A (PIG-A). Provoca muchas manifestaciones, incluyendo anemia hemolítica. Suele aparecer en la edad adulta y se debe a una mutación somática en las células tronco de la médula ósea.12
Los eritrocitos humanos normales modulan el ataque del complemento por lo menos por medio de tres proteínas que se fijan a la membrana: DAF, proteína fijadora de C8 (C8BP) y MIRL. Todas estas proteínas tienen deficiencia variable en los eritrocitos de los pacientes con HPN, con quimerismo variable en los diferentes pacientes. 12 En ausencia de la proteína de anclaje GPI, todas las proteínas que usan este anclaje de la membrana serán deficientes. La síntesis defectuosa de GPI afecta a las células tronco, por lo que los pacientes con HPN tienen neutropenia y trombocitopenia y pueden desarrollar pancitopenia, mielodisplasia o leucemia mieloide aguda.
Manifestaciones clínicas
Los episodios agudos de hemólisis intravascular se caracterizan por aparecer como complicaciones en pacientes con hemólisis crónica. En forma típica el paciente se percata de la hemoglobinuria al orinar después de dormir.14,15 Las oclusiones venosas recurrentes causan embolias pulmonares y trombosis de las venas hepática y mesentérica, quizá por liberación de factores trombogénicos o por activación de las plaquetas por complemento. En ocasiones se piensa erróneamente que estos pacientes con trombosis tienen trastornos psicosomáticos, pues se quejan de episodios de dolor abdominal y de espalda, sin presentar datos clínicos que indiquen la causa. En estos casos la anemia y la hemólisis asociada pueden ser muy leves, y los episodios de hemólisis no necesariamente se correlacionan con las crisis dolorosas.
Un estudio de 80 pacientes con HPN indicó que la supervivencia promedio fue de 10 años.14 La causa de la muerte relacionada con la HPN fue trombocitopenia, hemólisis, trombosis o anemia aplástica . Es interesante que el 15 por ciento de los pacientes presentó remisión espontánea. En este estudio las tromboembolias venosas recurrieron en sitios poco usuales y de gran riesgo, como la vena hepática (síndrome de Budd-Chiari), la vena cava inferior, las venas cerebrales y mesentéricas y el pulmón (embolia pulmonar).14 Ocurrió aplasia en cinco de los 80 pacientes. En casos raros puede ocurrir pérdida prolongada y severa de hierro como resultado de hemosiderinuria crónica, produciendo deficiencia de hierro; sin embargo, otros pacientes han desarrollado hemocromatosis asociada a transfusiones.15
Se ha desarrollado leucemia mieloide aguda durante la evolución de la HPN. En una serie la incidencia fue de solo tres casos en 80 pacientes, en otra, que incluyó 220 pacientes, la incidencia de síndromes mielodisplásicos fue de 5 por ciento y la de leucemia aguda de 1 por ciento.15
Diagnóstico
Se debe considerar el diagnóstico de HPN en cualquier caso de hemólisis crónica o episódica, o de tromboembolia venosa recurrente, en especial si el trombo ocurrió en un sitio como la vena cava inferior o el sistema porta-mesentérico, o si produjo un síndrome de Budd-Chiari. Las evidencias de hemólisis intravascular, como hemoglobinemia, disminución de la haptoglobina sérica, aumento de la metahemalbumina sérica, hemoglobinuria o hemosiderinuria, sugieren el diagnóstico. Un dato importante es la asociación de hipoplasia medular con hemólisis. Los eritrocitos de los pacientes con HPN no muestran alteraciones morfológicas. El diagnóstico con la prueba de Ham, que mide el grado de hemólisis en suero acidificado, es confiable, pero la prueba ha sido sustituida por pruebas específicas basadas en análisis de fluorescencia en células activadas (FACS) usando anticuerpos que evalúan en forma cuantitativa a las proteínas DAF y MIRL (CD59) en la superficie del eritrocito.16
Tratamiento
En la HPN la anemia puede ser tan severa (hemoglobina < 8 g/dl) que el paciente necesita transfusiones en forma regular.15 Por lo tanto, la elección del componente por transfundir es crucial. Se considera que la infusión de productos sanguíneos que contengan complemento puede aumentar la hemólisis. También es posible que la infusión de glóbulos bláncos (que generalmente están presentes en una unidad de paquete globular) del donador a un receptor HLA-inmunizado pueda provocar la reacción antígeno-anticuerpo que activa el complemento por la vía clásica. En tal caso, puede ser útil el uso de unidades especiales pobres en leucocitos.
Se puede intentar un tratamiento de prueba con prednisona (60 mg diarios con disminución rápida o 20 a 60 mg cada tercer día), que puede ayudar a reducir las necesidades de tranfusión al aliviar la anemia. La utilidad de la esplenectomía es muy dudosa. La cirugía es un problema en pacientes con HPN porque la estasis y el trauma agravan la hemólisis y las oclusiones venosas. Ante la necesidad de llevar a cabo una cirugía debe considerarse la anticoagulación profiláctica con warfarina en el periodo posoperatorio inmediato.
Los pacientes con HPN a menudo presentan deficiencia de hierro. Sin embargo, la simple administración de hierro para corregir este defecto agrava a menudo la hemólisis, puesto que el tratamiento con hierro produce una estirpe de células nuevas, muchas de las cuales son susceptibles a la lisis mediada por complemento. La transfusión previa puede ayudar a evitar este problema porque disminuye el impulso eritropoyético en la médula ósea.
La trombocitopenia por mala producción de plaquetas puede requerir transfusión de las mismas .14 El síndrome de Budd-Chiari y la trombosis de la vena cava inferior requieren de un diagnóstico pronto y tratamiento rápido con heparina, seguido de administración de warfarina a largo plazo. Si la heparinización es ineficaz puede usarse tratamiento trombolítico (v.gr., estreptocinasa). En un caso de síndrome de Budd-Chiari,17 se inició trombolisis con estreptocinasa sistémica con una dosis de impregnación de 250,000 unidades durante 40 minutos, seguido de una dosis de mantenimiento de 250,000 unidades en infusión IV continua por 12 horas. Los niños y los adolescentes con HPN y anemia aplástica pueden ser candidatos a trasplante alogénico de médula ósea.15
Alteraciones del metabolismo del eritrocito
DEFECTOS DE LA CAPACIDAD REDUCTORA
El glutatión reducido (GSH) y las coenzimas reducidas nicotinamida-adenina dinucleótido (NADH) y nicotinamida-adenina dinucléotido fosfato (NADPH) proveen la capacidad reductora del eritrocito [ver tabla 1]. Cuando las reservas eritrocíticas de estos materiales son inadecuadas, la hemoglobina y las proteínas asociadas a la membrana pueden oxidarse, causando la producción de cuerpos de Heinz, que consisten predominantemente en productos de degradación oxidativa de la hemoglobina. Estos cuerpos pueden verse con microscopía por contraste de fases o con microscopía ordinaria de luz después de teñir con metil violeta [ver figura 4]. Los eritrocitos que contienen cuerpos de Heinz son rígidos, por lo que el sistema reticuloendotelial los retira en forma selectiva de la circulación.
Defectos en la síntesis de glutatión
Las deficiencias de ciertas enzimas que participan en la síntesis de GSH causan hemólisis y ataques oxidativos a los eritrocitos [ver figura 5]. Varios informes han descrito familias cuyos miembros tienen síntesis imperceptible de GSH y sufren hemólisis asociada con producción de cuerpos de Heinz. La deficiencia de glutatión peroxidasa al parecer contribuye a la hemólisis en los recién nacidos.
DEFICIENCIA DE GLUCOSA-6-FOSFATO DESHIDROGENASA
La glucosa-6-fosfato deshidrogenasa (G6PD) es la primera enzima en la vía pentosa fosfato o hexosa monofosfato. Cataliza la conversión de NADP+ a NADPH, un agente reductor muy potente. El NDPH es un cofactor para la glutación reductasa, por lo que tiene un papel importante en la protección de la célula contra el ataque oxidativo. Por lo tanto, los eritrocitos deficientes en G6PD son susceptibles a la oxidación y, eventualmente, a la hemólisis.18,19
La deficiencia de G6PD es uno de los trastornos más comunes en el mundo; aproximadamente el 10 por ciento de los varones afroamericanos en los Estados Unidos están afectados, al igual que un gran número de africanos negros y algunos habitantes del litoral mediterráneo. Se supone que el trastorno alguna vez confirió cierta ventaja selectiva contra la malaria endémica, pero se desconoce el mecanismo de este efecto.
El gen que codifica la G6PD está en la banda q28 del cromosoma X; los hombres sólo poseen un gen para esta enzima y, por lo tanto, los afectados por el trastorno son hemicigotos. Las mujeres resultan afectadas con mucho menor frecuencia, ya que deben ser portadoras de dos genes G6PD defectuosos para manifestar una enfermedad de la misma severidad que los varones. Sin embargo, la expresión de un gen G6PD defectuoso no suele enmascararse completamente en las mujeres heterocigotas; de hecho, dichas mujeres muestran actividad muy variable de la G6PD. De acuerdo con la hipótesis de inactivación del cromosoma X, o hipótesis de Lyon-Beutler,19 las mujeres heterocigotas en el locus ligado al cromosoma X para el gen que codifica la G6PD tienen 2 líneas celulares, una que contiene un cromosoma X activo con un gen que codifica la producción de G6PD normal, y la otra que contiene un cromosoma X activo con un gen que codifica la producción de G6PD deficiente. El azar determina parcialmente la proporción de las dos líneas celulares, lo que a su vez controla la severidad clínica del defecto.
Existen tres tipos clínicos del padecimiento: el tipo I, que es la anemia hemolítica no esferocítica congénita crónica poco común, el tipo II, en el que la deficiencia de la enzima es severa pero la hemólisis tiende a ser episódica, y el tipo III, la variante más común, en la que la deficiencia enzimática es moderada y la hemólisis es causada por un evento oxidante. La clonación y secuencia del gen G6PD ha aclarado muchos aspectos complejos de la literatura que describía más de 300 variantes de la deficiencia de G6PD.19
Ocurre hemólisis en las personas con deficiencia de G6PD después de la exposición a un medicamento o sustancia que produce una tensión antioxidante. La ingestión o exposición a habas puede causar hemólisis intravascular devastadora (conocida como favismo) en los pacientes con deficiencia de G6PD, pero ocurre por lo general solo en los que tienen la variedad Mediterránea. Las habas contienen isouramilo y divicina, dos agentes reductores potentes. El peróxido de hidrógeno producido puede causar daño oxidativo importante. Al parecer, los oxidantes de las habas pueden también atacar la membrana y disminuir la barrera normal a la entrada de Ca2+. La combinación de niveles muy aumentados de Ca2+ y oxidación intracelular reduce el nivel de proteasas de eritrocitos. Estas proteasas protegen de modo ordinario contra el acúmulo de productos oxidados de la hemoglobina. El ataque oxidante a los tioles de la membrana causa oxidación de las proteínas de la membrana. Esto produce una célula rígida en la que la hemoglobina está confinada a una parte del citosol y la otra parte aparece como un fantasma claro (i.e., célula de media ampolla, o cruzada) [ver figura 6]. Estos defectos de memebrana provocan hemólisis extra e intravascular.18 También puede ocurrir hemólisis en los individuos con deficiencia de G6PD e infecciones severas, cetoacidosis diabética e insuficiencia renal. Es interesante que la deficiencia de G6PD produce, por mecanismos desconocidos, cierta protección contra la cardiopatía isquémica y varias enfermedades vasculares.20
La anemia hemolítica caracterizada por la aparición de cuerpos de Heinz después de la administración de ciertos medicamentos sugiere la posibilidad de deficiencia de G6PD [ver tabla 2]. El cloranfenicol y la quinina causan hemólisis en pacientes con la variedad Mediterránea de deficiencia de G6PD, pero no en pacientes con el tipo A-. La dapsona se usa cada vez más como profilaxis para la neumonía por Pneumocystis carinii en pacientes infectados por VIH. Por lo tanto, es importante buscar en los posibles receptores deficiencia de G6PD con las pruebas enzimáticas estándar porque la dapsona es capaz de inducir hemólisis de tipo oxidativo [ver tabla 2].
Otros trastornos que hay que considerar en el diagnóstico diferencial de la hemólisis oxidativa incluyen la hemoglobinopatía inestable, la enfermedad por hemoglobina M y las deficiencias de las enzimas esenciales para el metabolismo del glutatión. Una prueba de detección de G6PD o un ensayo enzimático directo suelen resolver la duda. Sin embargo, los pacientes con deficiencia de G6PD del tipo A- y reticulocitosis de rápida aparición pueden tener un nivel casi normal de G6PD. En dichos casos es mejor repetir los exámenes cuando la cuenta de reticulocitos vuelva a lo normal.
El evitar medicamentos que puedan producir hemólisis es crucial en el manejo. El favismo agudo requiere apoyo circulatorio, mantener un buen flujo sanguíneo renal y transfusiones con eritrocitos que no sean deficientes en G6PD. El médico también debe estar alerta por el posible inicio de coagulación intravascular diseminada.
DEFECTOS EN LA GLUCOLISIS
La serie de reacciones que constituyen la vía glucolítica [ver figura 5] generan diversos productos, incluyendo ATP, que tienen varias funciones esenciales en el metabolismo del eritrocito [ver tabla 1]. Existen defectos que afectan la vía glucolítica principal (vía de Embden-Meyerhof) y que generalmente interfieren con la producción de ATP.
La piruvato cinasa (PC) cataliza la formación de piruvato, reacción asociada con la síntesis de ATP. Después de la deficiencia de G6PD, la deficiencia de PC es la segunda de las enzimopatías hereditarias en orden de frecuencia. El síntoma inicial es la hemólisis de severidad variable, y se acompaña por ictericia leve y, en ocasiones, por esplenomegalia palpable. El examen de un frotis de sangre periférica suele mostrar eritrocitos normales, pero en algunos casos éstos presentan innumerables espículas. Pueden ocurrir crisis aplásticas.7
La hemólisis congénita no esferocítica plantea la posibilidad de deficiencia de PC. Un ensayo enzimático establece el diagnóstico. Se debe considerar la opción de esplenectomía en los pacientes que requieren transfusiones.
La deficiencia de glucosafosfato isomerasa es la tercera enzimopatía en frecuencia como causa de hemólisis. Otras son muy raras, pero se cuenta ya con pruebas de escrutinio y específicas para deficiencias de enzimas como hexocinasa, fosfofructosinasa, triosa fosfato isomerasa, fosfoglicerato cinasa y aldolasa.
DEFECTOS EN EL METABOLISMO DE LOS NUCLEOSIDOS
En la anemia hemolítica asociada con deficiencia de pirimidina 5'-nucleotidasa, persiste un puntilleo grueso basofílico en los eritrocitos maduros, supuestamente porque la deficiencia enzimática evita la degradación del ARN del reticulocito. Este acúmulo causa expansión de la reserva total de nucleótidos en los eritrocitos a cinco veces el normal. Los nucleótidos de pirimidina se acumulan y los de adenina disminuyen. El acúmulo y agregación de ribosomas no degradados contribuye también al puntilleo basofílico. La glucólisis se altera por un mecanismo aún no bien aclarado.
Trastornos de la hemoglobina
CLASIFICACION DE LAS HEMOGLOBINOPATIAS
Las hemoglobinopatías clínicamente importantes se clasifican en cinco categorías, con base en el defecto de fondo. Los defectos principales son los siguientes:
1. La hemoglobina tiende a convertirse en gel o cristalizarse (v.gr., anemia de células falciformes o enfermedad por hemoglobina C).
2. La hemoglobina es inestable (v.gr.,las anemias congénitas con cuerpos de Heinz).
3. La hemoglobina tiene propiedades anormales de captación de oxígeno (v.gr., el trastorno por hemoglobina Chesapeake).
4. La hemoglobina se oxida fácilmente a metahemoglobina (v.gr., metahemoglobinemia).
5. Las cadenas de hemoglobina se sintetizan en proporciones desiguales (v.gr., las talasemias).
ANEMIA DE CELULAS FALCIFORMES
La anemia de células falciformes es un padecimiento causado por la sustitución del aminoácido valina por glutamina en la sexta posición de la cadena beta de la hemoglobina. El gen se hereda con patrón autosómico recesivo y se encuentra en el 8 a 10 porciento de los afroamericanos en los Estados Unidos y en menor porcentaje en personas con ascendencia Mediterránea oriental, de la India o de Arabia Saudita. Los eritrocitos de individuos con el rasgo falciforme (HbAS) tienen una concentración de hemoglobina S menor del 50 por ciento, y con frecuencia menor del 30 por ciento. Con raras excepciones, el rasgo falciforme suele ser asintomático. La enfermedad se desarrolla en personas que son homocigotas para el gen falciforme (HbSS). En un paciente con anemia falciforme el 70 a 98 por ciento de la hemoglobina es de tipo S. Alrededor del 0.2 por ciento de los afroamericanos tienen anemia de células falciformes.
Varias teorías intentan explicar porqué la anemia de células falciformes se distribuye en todo el mundo y ha persistido en las poblaciones humanas. La coincidencia geográfica del gen falciforme y del paludismo falciparum endémico sugiere que la heterocigocidad falciforme confiere una ventaja protectora contra el paludismo.21 Tanto la resistencia inherente a la invasión como la mayor deformación de los eritrocitos parasitados interfieren con el crecimiento del parásito.21
Los análisis de endonucleasas de restricción indican que la mutación del gen de la anemia de células falciformes pudo aparecer en forma espontánea en por lo menos cinco sitios geográficos. Estas variantes son denominadas Senegal, Benin, Centroafricana (o Bantú), Saudi-Asiática, Camerún e India (que puede ser la misma que la Saudi-Asiática). Estas variantes son importantes desde el punto de vista clínico porque algunas se asocian con mayor producción de cadenas de globina gama (y por tanto niveles mayores de HbF) y otras se asocian con más frecuencia con el gen de la talasemia alfa-2 [ver adelante, Talasemias]. Cualquiera de estas asociaciones alivia ciertos aspectos del proceso de deformación falciforme.21
Fisiopatología
Dos características clínicas son las más importantes en la anemia de células falciformes: (1) la hemólisis crónica que es estable y moderadamente debilitante, y (2) las crisis vasoespásticas agudas y episódicas, que causan falla orgánica y son responsables de la mayor parte de la morbimortalidad de la enfermedad.
La HbS unida al oxígeno o al monóxido de carbono tiene una solubilidad casi normal. Sin embargo, cuando la molécula deja el oxígeno y cambia a su forma desoxi S, su solubilidad disminuye y tiende a polimerizarse en fibras tubulares largas, que inducen la deformación falciforme del eritrocito.22
Los análisis de cinética de la polimerización han aclarado mucho el conocimiento sobre la fisiopatología de la anemia de células falciformes. Puede considerarse que el polímero S de desoxihemoglobina está en equilibrio con las moléculas solubles circundantes de desoxihemoglobina S. Los factores que alteran este equilibrio afectan la magnitud de gelación de la hemoglobina. La reducción en la concentración de oxígeno u otros factores como la reducción en el pH o el aumento en la concentración de 2,3-DPG (ambos tienden a estabilizar la forma desoxi S) aumentan la gelación. Otro determinante importante de la polimerización en equilibrio es la concentración absoluta de la HbS y HbF. 22 La desoxihemoglobina S pura se polimeriza a 16 g/dl, una concentración muy por debajo de la CMHC habitual. La cinética de gelación es muy dependiente de la concentración de hemoglobina. Además, las células falciformes retienen un cotransportador activo K+-Cl- y tienen suficiente calcio intracelular para activar el canal de salida de Gardos23 [ver antes, Control de la hidratación y volumen]. Estos dos mecanismos actúan juntos para producir una población de eritrocitos falciformes muy densos con CMHC de hasta 50 g/dl. 23 La HbF inhibe la polimerización porque el residuo glutamina en la posición 87 de la cadena gama bloquea el contacto lateral de la fibra falciforme.23 Por lo tanto, los pacientes con valores altos de HbF, como los de la variante Saudi-Asiática de la anemia de células falciformes, tienen una enfermedad más leve [ver figura 7].21 Cuando estos factores, la hipoxemia y la CMHC, alcanzan un nivel crítico, ocurre polimerización después de un periodo de retraso variable.23
El retraso en el cambio de viscosidad puede representar el periodo durante el
cual los tetrámeros de desoxihemoglobina S se asocian en forma lenta
para formar un núcleo. Cuando el núcleo alcanza un tamaño
crítico, ocurre gelación rápida y casi explosiva al
fijarse los tetrámeros de desoxihemoglobina S libre al núcleo
para producir las largas fibras tubulares que se alínean para formar
estructuras tubulares paralelas que alteran las células y producen la
forma falciforme [ver figura 7].
La mayoría de las células en la circulación venosa no son falciformes. Sin embargo, ocurrirá este cambio si el tiempo de retraso para la polimerización se acorta a menos de 1 segundo o si los eritrocitos son atrapados en la microcirculación. Algunos eritrocitos contienen hemoglobina falciforme polimerizada incluso en la circulación arterial y el tiempo de retraso para la gelación es mucho más corto en estas células precondicionadas y más rígidas.
La sensibilidad extrema del cambio falciforme al ambiente local ha llamado la atención hacia los factores celulares. La hiperosmolaridad extrema de la médula renal (1,200 mOsm) deshidrata los eritrocitos y aumenta la CMHC. En consecuencia, puede ocurrir un cambio falciforme suficiente para abolir la capacidad de concentración de la médula renal incluso en pacientes que solo tienen rasgo falciforme. Los eritrocitos falciformes tienen una mayor tendencia a adherirse a las células endoteliales24 que los eritrocitos normales. El grado de adhesividad parece correlacionar con la severidad de los episodios vaso-oclusivos. Esto se atribuye a la desorganización de la bicapa de fosfolípidos de la membrana, al moverse la fosfatidilserina hacia el exterior, quizá aumentando las manifestaciones tromboembólicas de la enfermedad falciforme.25 En la anemia de células falciformes parece existir también aumento en las células endoteliales circulantes, que expresan en forma anormal factor tisular y pueden proporcionar una base adicional para la ocurrencia de tromboembolias.26
Otra manifestación del daño en la membrana en las células falciformes es el cambio falciforme irreversible, que se mantiene a pesar de que la célula sea reoxigenada.27 Estas poblaciones poco deformables y deshidratadas derivan directamente de una subpoblación de reticulocitos con concentraciones bajas de HbF28 y son eliminadas principalmente en el sistema reticuloendotelial. Ocurre hemólisis con anemia variable. La anemia se tolera bastante bien, quizá por un mayor aporte de oxígeno por la oxihemoglobina S.
CRISIS FALCIFORMES
En contraste con la anemia, la crisis falciforme es una complicación potencialmente mortal de la enfermedad falciforme. La crisis falciforme, una expresión dolorosa y notable de oclusión vascular, muestra características que dependen mucho de factores reológicos. La disminución del hematocrito en la anemia de células falciformes disminuye la viscosidad sanguínea y tiene efecto protector. Por el contrario, el nivel de fibrinógeno plasmático, característicamente elevado y que parece ser una manifestación de la respuesta de fase aguda que ocurre en muchos pacientes con anemia de células falciformes en fase estable,29 aumenta la agregación de los eritrocitos y la viscosidad, en particular ante las fuerzas de cizallamiento bajas que se encuentran en la microcirculación. La rigidez del eritrocito falciforme, que es causada por la polimerización de la desoxihemoglobina S y por la CMHC elevada, es el elemento crucial para elevar la viscosidad sanguínea y precipitar la oclusión vascular.
Si disminuye el pH o se eleva la CMHC, si existe enfermedad microvascular o se prolonga el tiempo de tránsito capilar por la adherencia de eritrocitos al endotelio, se formarán racimos de células falciformes rígidas que ocluirán la microcirculación. Estos bloqueos causan infartos isquémicos y una secuencia amplificadora de oclusión inducida por estasis que puede evolucionar hasta una crisis falciforme. Las circulaciones porta, en la que la tensión de oxígeno es baja, como las del hígado o riñón, tienen especial riesgo de oclusión. Se ha postulado que la trombosis también participa en la oclusión falciforme.
Se desconoce el evento iniciador de la crisis falciforme, y tampoco está claro por qué algunos pacientes tienen crisis severas y otros no. El dolor de la crisis falciforme puede ser causado por necrosis avascular de la médula ósea. Entre los factores de riesgo que predisponen a una crisis dolorosa se incluye una concentración de hemoglobina mayor de 8.5 g/dl, embarazo, clima frío, y una cuenta alta de reticulocitos.
La coherencia de otros genes influye en la gravedad de la anemia de células falciformes. La frecuencia de heterocigosidad para alfa-talasemia entre afroamericanos es de 30 por ciento.21 Las personas con alfa-talasemia tienen concentraciones más altas de hemoglobina, menores cuentas de reticulocitos, menor CMHC, menor VCM y menos eritrocitos densos que las personas que tienen solo anemia de células falciformes. Estos cambios, en especial la menor CMHC, deben beneficiar a los pacientes con los dos padecimientos, aunque la interacción es compleja. Los pacientes pueden tener mayor expectancia de vida y quizá menos úlceras en miembros inferiores, pero sufren más crisis y mayor necrosis aséptica del hueso.30 La combinación de deficiencia de G6PD y anemia de células falciformes no causa efectos benéficos ni dañinos.31
Diagnóstico
El aspecto clínico del paciente y un frotis de sangre que muestre células falciformes, células con aspecto de "flor de Nochebuena" y eritrocitos con cuerpos de Howell-Jolly son muy compatibles con anemia de células falciformes. Los cuerpos de Howell-Jolly representan restos citoplásmicos de cromatina nuclear que generalmente son retirados por la acción modeladora del bazo. Las cuentas de plaquetas y leucocitos suelen encontrarse elevadas. A menos que el enfermo se encuentre en crisis aplástica, lo que produciría ausencia total de normoblastos, la médula muestra hiperplasia eritroide. El diagnóstico se confirma realizando una "preparación falciforme": incubando una gota de sangre con solución fresca de metabisulfito de sodio al 2 por ciento y midiendo la proporción de células falciformes inmediatamente y una hora después. Para confirmar el diagnóstico, los equipos comerciales, como el Sickledex, se basan en la insolubilidad relativa de la hemoglobina S en soluciones amortiguadoras de fosfato 1.0 M. Sin embargo, la prueba definitiva para la anemia de células falciformes es la electroforesis de hemoglobina, que indica los porcentajes relativos de hemoglobina S y de hemoglobina F. Todas las pruebas mencionadas tienen la misma utilidad para examinar a los miembros de la familia en busca del rasgo de células falciformes. Los pacientes que son heterocigotos para los genes de la HbS y de la beta-talasemia pueden aparentar ser homocigotos para la HbS. Otras variedades de hemoglobina que causan transformación falciforme se observan con muy poca frecuencia. También pueden emplearse métodos basados en el estudio del ADN para determinar la alteración genética específica e identificar la subpoblación a la que pertenece el paciente.21
Otras consideraciones terapéuticas
Síntomas musculoesqueléticos Se presenta necrosis aséptica de la cabeza femoral (osteonecrosis) en alrededor del 10 por ciento de los pacientes, en especial en los que tienen también talasemia-alfa. La artroplastía ha resultado relativamente ineficaz, en parte por la presencia de hueso duro adyacente, que interfiere con la colocación de la prótesis, y por el mayor riesgo de infección.52
Enfermedad biliar La colelitiasis es una complicación común que ocurre en el 30 a 70 por ciento de los pacientes, algunos de los cuales desarrollan signos y síntomas de colecistitis.33 La frecuencia de colecistitis o de obstrucción del conducto biliar común es motivo de debate. En un estudio de 77 pacientes que se sabía tenían cálculos vesiculares de 1 a 10 años de evolución, solo cinco fueron sometidos a colecistectomía.34 Otro estudio encontró que de 604 procedimientos en la anemia de células falciformes, el 40 por ciento fueron por colecistitis y colelitiasis.33 Los pacientes que recibieron transfusiones hasta un nivel de hemoglobina de 10 g/dl antes de la cirugía tuvieron mejor evolución posoperatoria.33 Si va a realizarse una colecistectomía, debe esperarse hasta que pase la crisis dolorosa y el procedimiento debe practicarse por vía laparoscópica.33
Enfermedad hepática Las complicaciones hepáticas incluyen hepatopatía congestiva secundaria a insuficiencia cardiaca y hepatitis viral por las transfusiones frecuentes. La transformación falciforme en el hígado, órgano que extrae una gran cantidad de oxígeno de la sangre circulante, también puede producir hepatopatía. Es frecuente que los niveles séricos de bilirrubina excedan los 30 mg/dl en la colestasis intrahepática y las alteraciones en la coagulación pueden causar complicaciones hemorrágicas y muerte.
Enfermedad pulmonar Las complicaciones pulmonares agudas son una causa importante de morbimortalidad e incluyen infección local y oclusiones vasculares en el lecho arterial pulmonar. Es difícil la diferenciación entre las dos complicaciones. La oclusión vascular causada por un émbolo pulmonar puede ser imposible de distinguir de la oclusión in situ. Los problemas pulmonares agudos incluyen embolia grasa pulmonr por necrosis isquémica de la grasa de la médula.35 En un estudio de 1,722 episodios de dolor torácico agudo en 939 pacientes, los adultos en el estudio estaban afebriles pero tuvieron disnea, calosfríos y dolor torácico y en por lo menos una extremidad.36 El examen físico con frecuencia no mostró alteraciones torácicas. La tensión arterial de oxígeno (PaO2) estaba disminuida, en promedio en 71 mm Hg (en el 25 por ciento menor de 60 mm Hg). La tasa de muerte en adultos fue de 4.3 por ciento y fue precedida por un valor más bajo de hemoglobina, mayor cuenta de leucocitos y afección multilobar. La autopsia de 16 pacientes mostró que nueve tenían embolia pulmonar y embolia grasa y quizá el 20 por ciento tuvo infecciones bacterianas. Los infartos óseos de las vértebras torácicas contribuyeron en forma importante al dolor.37 Por lo general, el tratamiento debe incluir espirometría incentiva,37 búsqueda de infección y tratamiento con antimicrobianos, analgesia cuidadosa, remplazo agresivo de líquidos y consideración de lavado broncoalveolar para identificar la infección microbiana o los macrófagos llenos de lípidos típicos de la embolia grasa. Se requiere vigilancia cuidadosa que debe incluir medición periódica de la oxigenación y transfusiones cuando sea clínicamente necesario. Uno de los beneficios más importantes del tratamiento con hidroxiurea es su capacidad para disminuir la frecuencia del síndrome torácico agudo.38,39 Los niños pueden requerir profilaxis suplementaria con penicilina.40 Debido a que el uso rutinario de la vacuna neumocócica ha disminuido la incidencia de neumonía neumocócica en pacientes con anemia de células falciformes, Haemophilus influenzae es ahora responsable de un porcentaje relativamente mayor de casos de neumonía entre estos pacientes; de esta manera, la selección del antibiótico debe reflejar este cambio.
Complicaciones urogenitales La pérdida de agua, debida a la incapacidad para concentrar la orina, puede aumentar el proceso de transformación falciforme. El medio extremadamente hipertónico de la médula renal deshidrata los eritrocitos falciformes, induciendo transformación severa y destrucción de los vasos rectos. Se presenta hematuria y necrosis papilar. Estas complicaciones se observan especialmente en pacientes con el rasgo falciforme y en aquéllos que tienen enfermedad falciforme con hemoglobina C. El defecto en la capacidad de concentración renal parece depender de la cantidad de polímero de hemoglobina S en las células, por lo que es menos severa en los pacientes que tienen también variantes de talasemia alfa.41
Las complicaciones incluyen acidosis tubular renal, hipercalemia y proteinuria. El tratamiento con enalapril reduce la proteinuria, lo que sugiere la presencia de un componente de hipertensión capilar glomerular.42 La insuficiencia renal, con empeoramiento asociado de la anemia, contribuye a la muerte de alrededor de la quinta parte de los pacientes mayores de 40 años que tienen enfermedad falciforme homocigota.
El priapismo es una complicación muy dolorosa de la anemia de células falciformes y puede causar impotencia. Cuando las medidas conservadoras no producen mejoría en un periodo de 24 horas los autores recomiendan la exsanguineotransfusión de eritrocitos.
Complicaciones oculares Los principales problemas oculares que se asocian con la anemia de células falciformes son retinopatía, hemorragia del vítreo y neovascularización. Se recomienda el examen oftalmológico anual a estos pacientes. Se está investigando la eficacia de la fotocoagulación por láser en el tratamiento de las complicaciones oftalmológicas inducidas por la enfermedad.
Propensión a las infecciones Los pacientes con anemia de células falciformes son hipoesplénicos y tienen alteraciones del sistema del complemento. Una actividad opsonizadora deficiente contra Salmonella parece explicar que presenten propensión a desarrollar infecciones por este germen, incluyendo osteomielitis.
Trastornos neurológicos Las alteraciones neurológicas relacionadas incluyen eventos cerebrovasculares, hemorragia subaracnoidea y pérdidas funcionales aisladas compatibles con oclusión focal. Es probable que la patogenia de la oclusión de las arterias cerebrales de gran calibre sea diferente de los eventos oclusivos de la microcirculación que ocurren en los lechos capilares hipóxicos. El defecto subyacente parece ser el daño al endotelio vascular, seguido de proliferación extensa de la íntima y trombosis del lecho vascular lesionado.30 Un estudio multicéntrico reciente de 4,082 pacientes reveló una prevalencia de eventos cerebrovasculares (ECV) de 4 a 5 por ciento y una incidencia de 0.61 por 100 pacientes-año.43 De los ECV, el 54 por ciento fueron infartos, 34 por ciento hemorragias, 11 por ciento episodios de isquemia cerebral transitoria (ICT), y el 1 por ciento tenían datos tanto isquémicos como hemorrágicos. De los pacientes que sobrevivieron, la tasa de recurrencia de ECV fue de 14 por ciento. La mortalidad fue de 11 por ciento y virtualmente todos los pacientes que fallecieron tenían ECV hemorrágicos.
En un estudio prospectivo en el que se empleó la ultrasonografía transcraneal Doppler para identificar a los niños con riesgo de evento cerebrovascular, el tratamiento con atención estándar o transfusiones (para disminuir la concentración de HbS a < 30 por ciento) causó 10 ECV y un hematoma intracerebral en los 65 sujetos controles, mientras que el grupo transfundido tuvo solo 1 ECV (p < 0.002). El estudio se terminó antes de lo programado.44 El éxito del mismo destaca muchos aspectos serios sobre la necesidad de equipos ultrasonográficos para el manejo adecuado, la duración óptima del tratamiento de transfusión, las consecuencias inevitables de la hemocromatosis transfusional [ver adelante, beta-talasemia mayor (anemia de Cooley)], y la necesidad de administrar sangre específica de la etnia para minimizar las reacciones por alotransfusión, el deseo de los pacientes y familiares para aceptar el tratamiento transfusional y el papel del trasplante alogénico de médula ósea como una alternativa potencial.44,45
Crisis aplástica La crisis aplástica disminuye rápidamente los niveles de hemoglobina y hematocrito y produce reticulocitopenia, como sucede en cualquier estado hemolítico crónico. Se ha demostrado que el parvovirus causa crisis aplásticas,7 lo mismo que la necrosis de médula ósea.46
Anestesia general
La hipoxemia y la estasis vascular que pueden ocurrir durante la anestesia general aumenta la deformación falciforme y puede causar una crisis en el periodo posoperatorio. Un análisis de casi 4,000 pacientes asoció 12 muertes con 1,079 procedimientos. Ocurrieron más complicaciones después de la anestesia regional que de la general.47 Un programa de transfusión simple para aumentar el nivel de hemoglobina a 10 g/dl fue tan eficaz como los programas preoperatorios más agresivos para disminuir la tasa de complicaciones.48
Pronóstico
Un estudio de casi 4,000 pacientes ha proporcionado nueva información sobre la expectancia de vida y las causas de muerte en la enfermedad por células falciformes.49 Aunque se pensaba que la mayoría de los pacientes con anemia de células falciformes moría alrededor de los 20 años de edad, en la actualidad es claro que la edad promedio en el momento de la muerte es de 42 años para los hombres y 48 para las mujeres. Esta expectancia de vida representa una reducción de 25 a 30 años comparando con la población afroamericana general. De las causas de muerte identificadas, solo 18 por ciento fueron el resultado de falla orgánica, principalmente nefropatía, insuficiencia cardiaca congestiva o las consecuencias de los eventos cerebrovasculares crónicos. Treinta y tres por ciento de los pacientes murieron durante las crisis de dolor agudo, con frecuencia asociado con síndrome torácico agudo y menos común con eventos cerebrovasculares. La presencia de talasemia-alfa no tuvo un efecto mesurable. Los factores que predijeron mala evolución fueron una cuenta de leucocitos mayor de 15,000/µl, niveles bajos de hemoglobina F y afección orgánica manifestada por enfermedad renal, síndrome torácico agudo y eventos neurológicos.
Tratamiento
El manejo debe enfocarse al tratamiento y prevención de las crisis falciformes. El tratamiento conservador estándar de las crisis falciformes consiste en un examen apropiado, seguido de reposo, hidratación y analgesia. En aquellos pacientes en quienes se ha demostrado acidosis se debe inducir la alcalinización leve administrando solución de bicarbonato, que se prepara añadiendo un ámpula de bicarbonato de sodio a 1 L de solución glucosada al 5 por ciento, o solución salina medio normal. La solución de bicarbonato debe administrarse a una velocidad de 5 a 7 ml/kg/hora durante las primeras 4 horas, y después a una velocidad de 4 ml/kg/hr durante las siguientes 20 horas. La utilidad de la administración de O2 en pacientes con PaO2 normal y sin problemas cardiopulmonares no se ha demostrado.
Dolor El manejo de las crisis dolorosases la principal preocupación en el 10 al 20 por ciento de los pacientes con anemia de células falciformes. La necrosis avascular de la médula ósea produce dolor intenso que dura hasta 8 a 10 días. La necesidad de alivio del dolor causa en ocasiones habituación o adicción. Debido a que existen pocas maneras objetivas de evaluar las crisis falciformes, el médico puede no saber si la demanda de narcóticos es manifestación de un comportamiento de adicción.
Los pacientes con anemia de células falciformes deben recibir analgésicos orales para usarlos en casa en un intento por abortar las crisis de dolor en su inicio. Puede comenzarse con antinflamatorios no esteroides (AINE) como naproxen (500 mg v.o.) o ketorolaco (10 mg) y si no son útiles, éstos pueden ser seguidos de una combinación de analgésico-narcótico como acetaminofén e hidrocodona o aspirina y oxicodona. El uso de adyuvantes como la difenhidramina (50 mg v.o.) o loracepam (1 a 2 mg v.o.) puede calmar al paciente y quizá antagonizar las acciones de la histamina liberada.50 Si estas medidas, quizá repetidas cada 6 horas, no controlan el dolor, el paciente suele requerir tratamiento parenteral. La atención del paciente por su médico es preferible a confiar en un médico no conocido de un servicio de urgencias.50 El paciente requiere una evaluación rápida para investigar posible infección, síndrome torácico agudo, infarto óseo u otras complicaciones, y el dolor debe tratarse con 10 mg de morfina intravenosa junto con 50 mg de difenhidramina intramuscular cada 2 horas. o 4 mg de hidromorfona intramuscular junto con 50 mg de difenhidramina intramuscular cada 2 horas. Si no existe alivio del dolor o éste es inadecuado 30 minutos después de la primera dosis, puede administrarse el 50 por ciento de la dosis inicial de opioides, con vigilancia estrecha de la frecuencia respiratoria, en especial si llega a 10 respiraciones/min. Algunas unidades han usado analgesia controlada por el paciente con buenos resultados. Es importancia continuar la analgesia parenteral en intervalos regulares, proporcionando dosis mayores para eliminar el dolor. Es probable que el paciente requiera también un laxante y un antiemético, como proclorperacina (10 mg v.o o IM). Si el paciente responde, puede enviarse a casa con morfina de liberación controlada por vía oral. Si el dolor continúa por más de 8 a 12 horas es probable que deba ser internado para administrar dosis mayores de analgesia, líquidos parenterales y observación.50
Medidas generales para impedir la transformación falciforme El mayor conocimiento de la cinética de transformación falciforme sugiere algunos prospectos futuros para el tratamiento de la anemia de células falciformes. La disminución de la CMHC disminuye la gelación. Otro tratamiento consiste en bloquear la salida de K+ dependiente de Ca2+ (canal de Gardos) [ver antes, Control de la hidratación y el volumen].51 Un tratamiento semejante consiste en tratar a los pacientes con anemia de células falciformes con dosis grandes de pidolato de magnesio, que aumenta los niveles de Mg en los eritrocitos, inhibe el cotransportador de K+-Cl- y reduce el número de eritrocitos falciformes deshidratados y densos.52
Se han propuesto tratamientos que intentan corregir o compensar el defecto falciforme interviniendo a nivel del gen. La presencia de 20 a 30 por ciento de HbF en los eritrocitos falciformes retrasa la gelación en una proporción de 103 a 104, por lo que pudiera existir un mecanismo que activara los genes que controlan la síntesis de hemoglobina fetal y que disminuyera la gravedad de la enfermedad falciforme.49 La hidroxiurea produce un aumento en los reticulocitos F y en la HbF. Un estudio sobre hidroxiurea en fase III, con una dosis inicial de 15 mg/kg/día, mostró que los pacientes tratados con esta sustancia tuvieron menos crisis dolorosas, menos ingresos hospitalarios por crisis y menos episodios de síndrome torácico doloroso, requeriendo menos transfusiones que los pacientes que recibieron placebo.38 No se observó efecto sobre la frecuencia de muerte o eventos cerebrovasculares. El efecto benéfico se observó después de 8 semanas de tratamiento y se asoció con aumento en el VCM y la proporción de células F, junto con disminución en los neutrófilos y en la adhesión de los eritrocitos falciformes a las células endoteliales.53 También se realizan estudios con otros agentes, como el butirato, que puede aumentar la producción de cadenas-gama, aumentando así las concentraciones de HbF e interfiriendo con la gelación.54 Sin embargo, un estudio en el que se usó un tratamiento de 10 semanas de butirato de arginina por vía intravenosa no encontró beneficios hematológicos.55 El trasplante alogénico de médula ósea de un hermano compatible puede producir la curación o sustituir la anemia de células falciformes por solo un rasgo falciforme. En 22 pacientes seleccionados en forma cuidadosa, principalmente con eventos cerebrovasculares o episodios recurrentes de síndrome torácico agudo, el trasplante de médula ósea causó curación aparente en 15, con dos fallecimientos (9 por ciento); el resto de los pacientes tuvo complicaciones como fracaso del injerto.56
Papel del tratamiento transfusional por tiempo prolongado Se ha observado que el tratamiento transfusional prolongado evita los eventos cerebrovasculares (ver antes).44 Algunos investigadores han demostrado que las transfusiones preventivas pueden disminuir o eliminar las crisis dolorosas, los episodios de síndrome torácico agudo, la infección bacteriana o la hospitalización.57 Sin embargo, otros clínicos advierten sobre los peligros de la sobrecarga de hierro, la hepatitis por transfusión, los problemas con el acceso venoso y la aloinmunización de eritrocitos.37 Los estudios futuros podrán identificar mejor la utilidad del tratamiento transfusional crónico.
Consejo genético, anticoncepción y embarazo
Los elementos claves que deben considerarse al brindar consejo genético en pacientes con rasgo o enfermedad falciforme son: la morbilidad tan importante para niños y adultos con la enfermedad, el aumento del riesgo materno en el embarazo, y la frecuencia relativamente alta de muerte fetal. Los anticonceptivos orales también pueden representar un riesgo especial para las mujeres con anemia de células falciformes, porque producen un ligero aumento en la incidencia de evento cerebrovascular, tromboembolia venosa e infarto del miocardio. Sin embargo, los datos que han aparecido sobre la seguridad de los anticonceptivos orales que contienen menos de 50 µg de estrógenos sintéticos indican que las pacientes con enfermedad falciforme pueden tomar dichos medicamentos con seguridad razonable. Otra alternativa para algunas pacientes es el uso del implante anticonceptivo Norplant. En cualquier caso, el embarazo o el aborto en pacientes con enfermedad falciforme entrañan un riesgo elevado.58
Los peligros del embarazo en la enfermedad falciforme incluyen complicaciones pulmonares, mayor incidencia de infecciones de vías urinarias, hematuria, preclampsia y muerte materna. Al parecer, la hipoxia pélvica y la sobrecarga vascular asociadas con el embarazo causan una mayor transformación falciforme, con sus complicaciones. Las oclusiones vasculares en la placenta pueden causar muerte fetal y bajo peso al nacer.
Los clínicos experimentados tienen diferentes conceptos sobre la atención de la paciente embarazada con enfermedad falciforme. Algunos recomiendan solamente cuidado conservador meticuloso, mientras que otros indican transfusiones profilácticas. Un estudio controlado ha indicado que no existen ventajas con el uso de transfusiones profilácticas.58
Es posible que las parejas con enfermedad o rasgo falciforme quieran tener hijos a pesar de los riesgos fetales y maternos asociados. En Estados Unidos se presentan alrededor de 4,000 a 5,000 embarazos de este tipo al año.59 La muestra de vellosidades coriónicas (que proporciona ADN para análisis en el primer trimestre del embarazo), las técnicas de amplificación del ADN y las sondas que identifican el cambio específico de nucléotido en la anemia de células falciformes pueden proporcionar un diagnóstico prenatal relativamente seguro y muy confiable.59
VARIANTES DE LA ENFERMEDAD FALCIFORME
Rasgo falciforme
Por lo general los pacientes con el rasgo falciforme no presentan manifestaciones clínicas y llevan vidas normales y saludables. Se llegan a presentar algunas complicaciones: hipostenuria y hematuria renal, bacteriuria y pielonefritis durante el embarazo. Ocurre infarto esplénico cuando existe hipoxia y en altitudes elevadas, en especial en personas con anemia de células falciformes que no son de raza negra. También se ha identificado que el rasgo falciforme constituye un factor de riesgo para muerte súbita durante el entrenamiento militar básico,60 y la muerte ocurre por paro cardiaco inexplicable, golpe de calor, estrés por calor o rabdomiolisis. La mayor edad se ha correlacionado con mayor riesgo de muerte súbita. Sin embargo, estos eventos han ocurrido en condiciones muy extremas: actividad física muy extenuante en personas sin entrenamiento y en ocasiones en altitudes elevadas o con clima de calor extremo. Por lo general las personas con rasgo falciforme que están habituadas a realizar actividad física no tienen mayor riesgo de muerte súbita. Por ejemplo, entre los jugadores afroamericanos de futbol, los que tienen rasgo falciforme no tienen mayor riesgo de muerte súbita que otros jugadores.61 Las alternativas de tratamiento para la hematuria renal incluyen la administración de diuréticos, bicarbonato parenteral, transfusiones o ácido épsilon-aminocapropico.
Talasemia beta-falciforme
Cuando se combina el rasgo falciforme con un defecto en el gen de la talasemia beta se produce una enfermedad muy similar a la anemia de células falciformes. El gen de la talasemia beta reduce la velocidad de síntesis de la cadena ßa, lo que da como resultado que predominen las cadenas de ßs en pacientes con el rasgo falciforme. Los eritrocitos de los pacientes contienen cantidades variables de HbS, HbA, HbA2 y HbF. Cuando sólo existe una pequeña cantidad de hemoglobina A la enfermedad es tan grave como la anemia de células falciformes, pero en las variedades en las que la HbA constituye el 25 por ciento o más de la hemoglobina total y se produce menos HbS la enfermedad es leve. El diagnóstico se confirma si existe un nivel elevado de hemoglobina A2 en la electroforesis de hemoglobina, una historia familiar con el antecedente de talasemia y el gen falciforme. El tratamiento para la talasemia beta-falciforme es el mismo que se indica para la anemia de células falciformes.62
Enfermedad falciforme con hemoglobina C
En la enfermedad falciforme con hemoglobina C (HbSC) se forman cantidades casi iguales de hemoglobina S y C. Del 1 al 2 por ciento de la hemoglobina es HbF y también hay pequeñas cantidades de HbA2, pero no hay HbA. Los eritrocitos son microcíticos y la CMHC es alta, lo que facilita la cristalización y agregación de la HbC.63
Del 30 a 50 por ciento de los pacientes con este trastorno no están anémicos y sólo tienen reticulocitosis moderada. Los pacientes pueden no ser identificados hasta que el trastorno se manifiesta en forma de una crisis vaso oclusiva durante una cirugía, embarazo o urgencia médica.63 También ocurren esplenomegalia, retinopatía proliferativa y necrosis aséptica de los huesos largos. El frotis periférico [ver figura 8] contiene células falciformes irreversibles además de células en diana, estomatocitos y eritrocitos con depóstitos eccéntricos de hemoglobina, que probablemente representen agregados o cristales de hemoglobina C. El diagnóstico se confirma por electroforesis de hemoglobina.63 El tratamiento es el mismo que se indica en la anemia de células falciformes. Se administró hidroxiurea, 1,000 mg/día a seis pacientes con enfermedad de HbSC y presentaron aumento en el VCM, reducción en los llamados reticulocitos de estrés, incremento en la hemoglobina y quizá reducción en la densidad celular. Aunque no es definitivo, este pequeño estudio sugiere que la hidroxiurea es benéfica para los pacientes con enfermedad HbSC.64 La expectancia de vida es casi 20 años mayor que para los pacientes con enfermedad por HbSS.49
OTRAS HEMOGLOBINOPATIAS
Enfermedad por hemoglobina C
La molécula de hemoglobina C es a2ß26glu-lis, y la mutación del gen pareció originarse en un solo sitio en Africa Occidental, dentro de Burkina Faso.63 La presencia de esta hemoglobina casi no produce enfermedad en el estado heterocigoto, pero causa hemólisis moderada compensada y esplenomegalia palpable en el estado homocigoto.
La insolubilidad relativa de la hemoglobina C es responsable de los cambios patológicos que se asocian con su presencia. Es probable que la hemoglobina C interactúe con el cotransportador K+-Cl- para mantenerlo activo, en condiciones normales este cotransportador se inactiva en los eritrocitos después del periodo de reticulocito. Como consecuencia se presenta pérdida de K+, deshidratación celular con CMHC elevada y después agregación y cristalización de la hemoglobina C poco soluble.63 La insolubilidad relativa de la hemoglobina C causa rigidez de los eritrocitos, fragmentación y pérdida del material de la membrana; estos procesos producen los microesferocitos que se observan en el frotis de sangre periférica [ver figura 4].
Las células en diana, un dato morfológico importante, forman cerca del 80 por ciento de los eritrocitos. Se encuentran cristales de hemoglobina C en el estado de oxihemoglobina, que se disuelven cuanto los eritrocitos se desoxigenan. Esta propiedad parece explicar la ausencia de episodios vasculares oclusivos en pacientes con enfermedad por hemoglobina C.
El diagnóstico de enfermedad por hemoglobina C se basa en los datos del frotis de sangre periférica y en la ausencia de datos de deficiencia de hierro o talasemia, y se confirma por medio de electroforesis de hemoglobina. No se requiere tratamiento.
Enfermedad por hemoglobina E En la enfermedad por hemoglobina E la lisina es sustituida por ácido glutámico en la posición 26 de la cadena de la beta-globina. El rasgo de hemoglobina E se encuentra principalmente en el sureste de Asia. Se le ha prestado atención clínica en los Estados Unidos por la llegada de inmigrantes de esta región, en los que la incidencia del rasgo es de alrededor del 10 por ciento.
La hemoglobina E es inestable al oxidarse porque ocurre una mutación en el sitio de contacto alfa1beta1. El menor contenido de ßE es resultado de una menor producción de ARN mensajero (ARNm) ßE y de producción de ARNm ßE inestable.
Los pacientes heterocigotos para la hemoglobina E tienen valores de hemoglobina normales, microcitosis y no sufren esplenomegalia. La electroforesis revela que el 70 por ciento de la hemoglobina es HbA, el 25 por ciento HbE y el resto es HbA2 o HbF. En la electroforesis la HbE corre junto con la HbA2, y los laboratorios sin experiencia pueden reportarla como HbA2. La clave consiste en que la HbA2 nunca es más del 8 por ciento. Un reporte de laboratorio de concentración de HbA2 de 25 por ciento obliga a revisar los datos.
Los pacientes homocigotos para la HbE tienen anemia leve, con concentraciones de hemoglobina de alrededor de 12 a 13 g/dl, VCM bajo y eritrocitosis sin reticulocitosis, mostrando microcitos y células en diana. La electroforesis muestra solo HbE. No ocurre hemólisis crónica. Deben evitarse los medicamentos oxidantes, como la dapsona, tanto en heterocigotos como en homocigotos.
Ocurre un problema clínico grave cuando un paciente es heterocigoto doble para la HbE y para el rasgo de talasemia beta, porque la combinación produce el cuadro clínico de talasemia beta intermedia, con anemia severa, esplenomegalia y en ocasiones necesidad de transfusión de sangre e incluso médula ósea.65
Hemoglobinopatías inestables
Muchas variedades individuales constituyen las hemoglobinopatías inestables. La inestabilidad de la hemoglobina se origina de las sustituciones de aminoácidos que privan a la molécula de su grupo hem, alteran el sitio receptor del hem, debilitan la unión entre cadenas alfa y beta, o debilitan la estructura de la subunidad. El resultado es la ruptura y precipitación de la hemoglobina, en particular cuando se somete a un fenómeno oxidante. La hemoglobina precipitada forma cuerpos de Heinz, que se observan incluso en personas heterocigotas para la variante de hemoglobina inestable. Debido al efecto dañino de los cuerpos de Heinz sobre el eritrocito y su membrana, ocurre hemólisis importante incluso en el estado heterocigoto.
La presencia de hemólisis no esferocítica parcialmente compensada sugiere el diagnóstico de una hemoglobinopatía inestable. Se observan cuerpos de Heinz en los eritrocitos de pacientes que han sido sometidos a esplenectomía. Los eritrocitos de pacientes que no han sido esplenectomizados presentan cuerpos de Heinz al ser incubados con tinción azul brillante de cresilo. El diagnóstico diferencial de una hemoglobinopatía debe hacerse con la deficiencia de G6PD; por lo general puede descartarse este trastorno por medio de una prueba directa para buscar esta enzima.
El tratamiento incluye evitar el uso de medicamentos oxidantes. Se puede considerar la esplenectomía cuando la hemólisis es severa y no se compensa en forma adecuada.
Hemoglobina con afinidad anormal para el oxígeno
Se debe considerar la posibilidad de que exista una hemoglobina con mayor afinidad para el oxígeno en el diagnóstico diferencial de la eritrocitosis inexplicable, particularmente si hay asociación familiar . La electroforesis de hemoglobina puede demostrar el trastorno, pero en los casos sospechosos se prefiere tener como base para el diagnóstico la curva de disociación de la oxihemoglobina. La hemoglobina Chesapeake y la hemoglobina Rainier son ejemplos de tipos de hemoglobina con afinidad aumentada.
Los casos raros de hemoglobina con afinidad disminuida para el oxígeno, como la hemoglobina Kansas, representan mutaciones. Los pacientes con hemoglobinopatía con afinidad disminuida para el oxígeno pueden presentar cianosis debido a una mayor liberación del oxígeno.
Metahemoglobinemia
La metahemoglobina es un producto de oxidación de la hemoglobina cuya molécula de hierro está en forma férrica; de esta manera, la molécula no puede fijar oxígeno en forma reversible. En condiciones normales el 1 por ciento de la hemoglobina se encuentra en estado férrico, y entre el 0.5 y el 3 por ciento de la desoxihemoglobina se oxida en forma espontánea a metahemoglobina cada día. El poder reductor normal de los eritrocitos [ver tabla 1] mantiene el balance oxidación-reducción. El sistema enzimático que reduce el 95 por ciento de la metahemoglobina a hemoglobina incluye dos proteínas, la NADH-citocromo b5 reductasa y la citocromo b5, y también requiere de NADH. (El NADH es generado por la gliceraldehido fosfato deshidrogenasa [ver figura 5].) Como el nombre lo indica, la NADH-citocromo b5 reductasa, una enzima que contiene flavina, usa NADH para reducir a la citocromo b5. La citocromo b5 reducida reduce a la metahemoglobina.66,67 Se han descrito mutaciones nuevas en el gen afectado.68
En la forma enzimopénica hereditaria de metahemoglobinemia, los pacientes son homocigotos o heterocigotos dobles para la deficiencia de NADH-citocromo b5 reductasa. Estos enfermos tienen apariencia azul incluso cuando solo el 10 por ciento de su hemoglobina es metahemoglobina, pero no están enfermos y toleran con facilidad concentraciones de metahemoglobina hasta de 25 por ciento o más. Por el contrario, la presencia de alrededor de 5 g/dl o hemoblogina desoxigenada reducida causa cianosis. Los pacientes con esta forma de metahemoglobinemia no sufren hemólisis ni requieren tratamiento. La determinación de NADH-citocromo b5 reductasa, realizada en un laboratorio especial, puede establecer el diagnóstico. Si se desea puede usarse azul de metileno, en dosis de 100 a 300 mg/día por vía oral, aunque causa molestias urinarias.66 El azul de metileno transfiere electrones del NADPH a la metahemoglobina.
La otra forma hereditaria de metahemoglobinemia es causada por la hemoglobina M, de la que existen cuatro variantes poco frecuentes. Cada variante contiene una sustitución de aminoácido en el sitio receptor del hem, que permite que se formen enlaces estables entre el hierro hem y la cadena lateral de aminoácidos. Estas uniones mantienen a la hemoglobina en la forma Fe3+, incapaz de unir oxígeno e inaccesible a las enzimas reductoras. El trastorno se observa solo en los heterocigotos, en los que el 30 por ciento de la hemoglobina es anormal según la electroforesis. Se observa cianosis al nacimiento. La hemólisis es mínima y no se requiere tratamiento.
TALASEMIAS
Las talasemias tienen distribución mundial, y en muchas regiones estos trastornos son responsables de perturbaciones médicas, sociales y económicas importantes. La distribución mundial de las talasemias se superpone en forma estrecha con la distribución del paludismo, lo que indica que las formas heterocigotas de talasemia proporcionan protección contra esta enfermedad.69 Las técnicas de biología molecular han ayudado a dilucidar la fisiopatología de estos síndromes.69 En la actualidad, la mejor compresión de la base molecular de estos trastornos ha permitido que los investigadores realicen diagnósticos prenatales fidedignos. Usando estos datos, los padres pueden tomar decisiones razonables e informadas con respecto al resultado de embarazos en los cuales el feto se encuentre severamente afectado.
Fisiopatología
En un individuo normal, la síntesis de cadenas alfa y beta está meticulosamente coordinada para la producción de la HbA del adulto (a2b2). En comparación, los pacientes con talasemia suelen presentar alteraciones en la síntesis de cadenas normales de globina. No obstante, en ocasiones pueden presentarse síndromes parecidos a la talasemia como resultado de la producción disminuida de una cadena estructuralmente anormal.70 Debido a que una de las cadenas de globina existe en cantidad reducida, la cadena excedente se acumula en la célula precursora eritroide en desarrollo y se produce toxicidad. En consecuencia, las células eritroides mueren en la médula ósea, dando origen a una forma clásica de eritropoyesis ineficaz.
Las talasemias beta se caracterizan por menor producción de cadenas de globina beta, con acúmulo y agregación de cadenas alfa no pareadas. Estos agregados de cadenas alfa se precipitan, causando disminución de la síntesis de ATP, pérdida de potasio, disminución del ácido siálico superficial, y otras anormalidades de la superficie, incluyendo anticuerpos IgG dirigidos contra los residuos de galactosa, que pueden ser detectados por los macrófagos. Los eritrocitos afectados tienen forma anormal y son relativamente rígidos. La barrera de la membrana para el Ca2+ se rompe, lo que permite la entrada de este ión. Se encuentran acúmulos anormales de globina, quizá agregados de cadenas alfa, unidos al citoesqueleto de la membrana. Es posible que éstos sean responsables de la apoptosis observada en los precursores eritroides de la médula y de la mayor rigidez e inestabilidad de la membrana que se observa en los eritrocitos en sangre periférica. Estos agregados de globina alfa mantienen funcionando el cotransporte K+-Cl-, lo que provoca la deshidratación variable que se observa en las formas severas de talasemia beta.71 Las alteraciones destructivas de la membrana pueden ser causadas en parte por oxidación local desencadenada por el acúmulo de hemicromos en la membrana, como en la enfermedad de células falciformes. La reducción global en la síntesis de hemoglobina por la célula es responsable de la hipocromía y de la formación de células en diana.
Los pacientes con talasemia alfa muestran acúmulos en exceso de cadena beta que, si están presentes en cantidades suficientes, forman tetrámeros ß4 (HbH) [ver figura 8]. Dichos tetrámeros tienen una elevada afinidad por el oxígeno y son inestables, por lo que se agregan en situaciones de oxidación, como en las infecciones. Los acúmulos de globina beta se fijan también al esqueleto de la membrana eritrocitaria, pero producen lesiones diferentes que los agregados de globina alfa. En las talasemias alfa severas la eritropoyesis ineficaz es menor y la destrucción periférica de eritrocitos más intensa. Los eritrocitos en la talasemia alfa son rígidos, pero a diferencia de la talasemia beta grave, las membranas en la talasemia alfa severa son más estables de lo normal. También, a diferencia de la talasemia beta, los eritrocitos de la talasemia alfa están hidratados en exceso.71
Tanto las talasemias alfa como las beta se caracterizan por presentar grados variables de anemia. Esta variación se atribuye a grados diversos de eritropoyesis ineficaz y a la destrucción prematura de los eritrocitos circulantes. Cuando la anemia es grave la hipoxemia asociada induce una notable eritropoyesis compensadora que causa expansión de la cavidad medular, osteopenia y crecimiento de los órganos reticuloendoteliales; se pueden originar tumores en los sitios de actividad eritropoyética extramedular. La destrucción de eritroblastos y eritrocitos predispone a colelitiasis e ictericia obstructiva. Los pacientes con formas más graves de talasemia necesitan ser transfundidos con regularidad, lo que puede ocasionar finalmente sobrecarga de hierro y complicaciones tales como cardiopatía, hepatopatía y endocrinopatía.
Genética molecular
Los defectos que causan un desequilibrio en la síntesis de cadenas incluyen deleción de genes, anormalidades en la transcripción y en la traducción, e inestabilidad del ARNm que dirige la síntesis de globina, o de la globina misma. Los genes que controlan la síntesis de las cadenas alfa y no alfa de la hemoglobina se localizan en los cromosomas 11 y 16 [ver figura 8]. Durante la vida fetal y adulta no existen sustitutos para la cadena alfa de la hemoglobina, lo que tiene implicaciones clínicas importantes. En comparación, las cadenas delta y las gama suplementan la cadena beta normal en la vida adulta, y las cadenas gama (de las cuales hay dos variedades, que difieren solamente en un aminoácido) pueden servir como globina no alfa en la vida fetal y neonatal.73
Talasemia beta clínica
La síntesis deficiente de globina beta, característica de la talasemia beta, causa acúmulo de las cadenas alfa no complementarias. Esta disminución en la síntesis de cadenas beta puede ser consecuencia de un procedimiento nuclear anormal de transcripción de ARNm, de terminación prematura de la traducción, o de deleción genética. Un adelanto de importancia diagnóstica en la talasemia beta es el aumento compensatorio parcial de las cadenas delta y gama que produce aumento en los niveles de HbA2 (a2d2) o HbF (a2g2), respectivamente [ver figura 8]. Las variedades de talasemia beta producen tres síndromes clínicos: talasemia mayor beta, talasemia menor beta y talasemia intermedia.
Talasemia mayor beta (anemia de Cooley) La talasemia mayor beta se caracteriza por anemia severa que aparece en el primer año de vida. Se asocia con ictericia, hepatoesplenomegalia, expansión de la médula eritroide con cambios corporales secundarios incluyendo retraso en el crecimiento, y aumento en la susceptibilidad a las infecciones. La anemia es tan severa que se necesitan transfusiones en forma crónica. Estas ocasionan sobrecarga de hierro que, si no se trata, causa la muerte en la adolescencia por hemocromatosis cardiaca.
El diagnóstico no debe representar un problema; ninguna otra enfermedad se asemeja mucho a la anemia de Cooley. El frotis de sangre periférica muestra eritrocitos nucleados, eritrocitos hipocrómicos distorsionados y puntilleo basófilo, que representa acúmulos de ARN ribosomal [ver figura 7]. Las tinciones supravitales muestran acúmulos de cadenas alfa excedentes no complementarias.
La talasemia mayor beta es una enfermedad que se trasmite como una condición homocigota o heterocigota doble; ambos padres de un individuo afectado son portadores del rasgo de la talasemia. En la talasemia ßo, la variedad más grave, no se sintetizan cadenas beta, y solo se encuentra HbF y HbA2. La talasemia ß+ es menos severa, se caracteriza por cantidades pequeñas de cadenas beta y de HbA, además de HbF y HbA2. La talasemia delta beta es aún más moderada, y es ocasionada por la deleción de los genes que codifican la globina delta y beta. Esta mutación inhibe la producción de HbA2 y HbA, permitiendo solamente la síntesis de hemoglobina fetal.
El tratamiento consiste en transfusiones repetidas con el objeto de mantener la concentración de hemoglobina alrededor de 12 g/dl, evitando de esta manera complicaciones como insuficiencia cardiaca congestiva, sobrecarga de líquidos y deformidades esqueléticas. Por lo general se requiere esplenectomía para aumentar la supervivencia de los eritrocitos del paciente y de los transfundidos.74 Está indicada la vacunación con vacuna neumocócica por el riesgo de sepsis por neumococo después de la esplenectomía. La sobrecarga de hierro debe tratarse por medio de la administración de infusiones subcutáneas de desferroxamina, un agente quelante, antes de que ocurra exceso de hierro. La desferroxamina subcutánea, en dosis de 50 mg/kg/día puede lograr eliminación de 50 a 200 mg de hierro/día, pero solo si se infunde en forma continua durante 8 a 12 horas.75 Este tratamiento no solo evita la disfunción ventricular izquierda, sino que revierte las alteraciones ya establecidas.76 Los efectos benéficos de la quelación de hierro han mejorado el pronóstico de las personas con anemia de Cooley:76 ya no es inevitable que los pacientes mueran en la segunda década de la vida por arritmias e insuficiencia ventricular izquierda. Con el tratamiento actual con desferroxamina, el 61 por ciento de los pacientes nacidos antes de 1976 no han tenido enfermedad cardiovascular. Los pacientes que cumplen el esquema de tratamiento, en los que el valor de ferritina casi siempre es menor de 2,500 ng/ml tienen una tasa de supervivencia de 91 por ciento después de 15 años.77 Se ha probado un quelante oral de hierro, el deferiprone, comparando con la desferroxamina parenteral, pero no ha demostrado un control adecuado de la carga de hierro al medir los niveles de hierro hepático pre y postratamiento.78 Sin embargo, se recomienda que no se descarte su uso hasta contar con más datos.79
Se ha realizado trasplante de médula ósea usando hermanos HLA compatibles. Más de 600 pacientes han sido sometidos a este tipo de procedimiento con donadores normales o que tenían rasgo de talasemia beta.80 Algunos pacientes con hemoglobina E-talasemia beta tienen un fenotipo tan severo como el de la talasemia mayor beta y requieren el mismo tratamiento, incluyendo trasplante alogénico de médula ósea.65 La experiencia con el trasplante de células tronco de cordón umbilical es más limitado.81 Dependiendo de la condición del paciente en el momento del trasplante, la tasa de mortalidad relacionada al mismo fue de 5 a 19 por ciento, con una tasa de curación de 54 a 90 porciento.82 Otros enfoques para el tratamiento de la talasemia beta severa están aún en fase experimental.54,55,83,84
Talasemia beta menor (rasgo de talasemia beta) Por lo común, los pacientes con talasemia menor son heterocigotos para una mutación beta globina, y pueden o no presentar anemia leve. El frotis de sangre periférica muestra hipocromía notable y microcitos con puntilleo basófilo. En ocasiones se encuentra esplenomegalia.
El nivel de HbA2 se eleva por arriba del 5 por ciento en 90 por ciento de los pacientes y el nivel de HbF se eleva por arriba del 2 por ciento en el 50 por ciento de los casos. Este aumento en la hemoglobina fetal ocurre en proporciones variables en los eritrocitos (distribución heterocelular), como lo muestra la tinción de Kleihuaer Betke. Los pacientes con niveles más elevados de HbF tienen anemia menos grave. Los heterocigotos para la talasemia delta beta producen cantidades aumentadas de HbF con cantidades normales de HbA2.
Se debe descartar la anemia por deficiencia de hierro en el diagnóstico diferencial del rasgo de la talasemia beta. Por lo general es fácil distinguir los dos trastornos. Ambas enfermedades se asocian con hipocromia y microcitosis, pero la deficiencia de hierro produce hipoproliferación de eritrocitos, mientras que la talasemia menor causa sólo una reducción mínima en su número. Con un nivel de hemoglobina de 9 g/dl, los pacientes con deficiencia de hierro tienen una cifra de eritrocitos cercana a 3 millones/µl, mientras que los pacientes con el rasgo de talasemia tienen cifras de eritrocitos de alrededor de 5 millones /µl. La cifra de eritrocitos, relativamente normal en la talasemia, se utiliza como base para todas las fórmulas que usan índices para distinguir el rasgo de talasemia de la deficiencia de hierro. Si persisten dudas acerca del diagnóstico pueden usarse las mediciones del hierro sérico y de la capacidad de fijación de hierro o del nivel sérico de ferritina con el fin de distinguir estos trastornos. Sin embargo, es importante recordar que un paciente con el rasgo de talasemia también puede tener deficiencia de hierro como consecuencia de hemorragias vaginales, gastrointestinales o ambas.
Talasemia intermedia Como el término lo implica, la talasemia intermedia se caracteriza por manifestaciones clínicas de severidad moderada. Los pacientes con este síndrome tienen anemia importante, con niveles de hemoglobina tan disminuidos como de 6-7 g/dl, y muestran grados variables de hepatoesplenomegalia, pero es usual que no requieran de transfusiones en forma regular. No obstante, durante las infecciones u otras agresiones eritropoyéticas pueden necesitarse transfusiones en forma temporal.
Variedades parecidas a la talasemia beta La hemoglobinopatía asociada con la hemoglobina Lepore representa otra variedad de talasemia beta. Los pacientes que son homocigotos para este trastorno presentan anemia de Cooley o talasemia intermedia, y sus eritrocitos contienen sólo hemoglobina Lepore y HbF.70
Persistencia hereditaria de la hemoglobina fetal Los eritrocitos de pacientes que son heterocigotos para la persistencia hereditaria de hemoglobina fetal (PHHF) contienen alrededor del 50 por ciento de HbF, mientras que los homocigotos tienen el 100 por ciento de dicha hemoglobina. Hasta hace poco tiempo se suponía que los pacientes con PHHF se encontraban en buenas condiciones de salud y que presentaban anemia mínima o no tenían. Sin embargo, desde entonces se han descrito algunas variedades clínicas de la PHHF en las que hay anemia notable.
Talasemia alfa
Dos diferencias principales sirven para distinguir los genes de globina alfa y beta. Primero, no hay sustitutos fetales, neonatales o adultos para los genes alfa, y segundo, sólo hay dos genes de globina beta, pero cuatro de globina alfa, dos en cada cromosoma 16 [ver figura 8]. El genotipo de la globina alfa normal se designa con aa/ aa. Los pacientes portadores de la variedad talasemia alfa 1 muestran una deleción de dos genes de la cadena alfa del mismo cromosoma y por lo tanto, tienen el haplotipo--/ o alfa0. Esta deleción es común entre pacientes asiáticos. Los pacientes que tienen la variedad de talasemia alfa 2 han perdido un gen alfa en un cromosoma, y muestran el haplotipo -a / o alfa+. Aunque esta mutación es particularmente frecuente en negros, también se observa en poblaciones asiáticas y mediterráneas. Se han reconocido cinco síndromes clínicamente distintos: la hemoglobina Barts, o hidropesía fetal (--/--), la enfermedad por hemoglobina H (--/-a), la talasemia alfa 1 heterocigota (--/aa), la talasemia alfa 2 homocigota (-a /-a), y el síndrome del portador silencioso (-a / aa).
Hemoglobina de Barts Los niños con el síndrome de hemoglobina Barts son homocigotos para la talasemia alfa 1 (--/--), por lo que no producen cadenas alfa. Las cadenas gama no pareadas producen tetrámeros g4 (hemoglobina de Barts). Todos los niños con esta enfermedad nacen hidrópicos. Es común que los padres sean heterocigotos para la talasemia alfa 1 (--/aa).
Enfermedad por hemoglobina H El cuadro clínico de la enfermedad debida a hemoglobina H es el de una anemia hemolítica leve que se presenta en pacientes de origen asiático, del Medio Oriente o del Mediterráneo. Por lo general puede detectarse HbH que se precipita, al teñir con azul brillante de cresilo, en los eritrocitos recién extraídos del paciente. Los mecanismos moleculares que originan la enfermedad por hemoglobina H son heterogéneos. Algunos pacientes tienen un genotipo que sugiere la deleción de tres genes alfa, como sería el caso si el paciente fuera doblemente heterocigoto para la talasemia alfa 1 (--/) y talasemia alfa 2 (-a/), dando un genotipo
--/-a. La esplenomegalia es común. Los pacientes no suelen requerir de transfusiones regulares, pero puede necesitarse apoyo temporal cuando sufren infecciones u otros eventos oxidativos estresantes que causen precipitación de la HbH inestable y mayor hemólisis.
Talasemia alfa heterocigota La talasemia alfa 1 heterocigota (--/aa), genotipo común entre asiáticos, se caracteriza por anemia leve o ausente y la presencia en el frotis de sangre periférica de eritrocitos notablemente hipocrómicos y microcíticos. Los pacientes homocigotos para la talasemia alfa 2 (-a/-a), un genotipo frecuente en la raza negra, no tienen dos genes alfa, y las manifestaciones clínicas de este genotipo recuerdan a las de los pacientes heterocigotos para la talasemia alfa 1. El estado heterocigoto de la talasemia alfa 2 (-a/aa) no se manifiesta clínicamente y por ese motivo representa el síndrome del portador silencioso.
Síndrome parecido a la talasemia alfa La hemoglobina Constant Spring (hemoglobina CS) es una hemoglobina estructuralmente anormal común en algunas poblaciones asiáticas. El gen de la globina alfa contiene una mutación en el codón de terminación que da como resultado la síntesis de una globina alfa que contiene 31 aminoácidos adicionales. Los pacientes heterocigotos con este defecto tienen un cuadro clínico similar al de los pacientes homocigotos para la talasemia alfa 2. Los pacientes homocigotos para la hemoglobina CS tienden a presentar manifestaciones clínicas un poco más graves que los pacientes heterocigotos para la talasemia alfa 1.
Diagnóstico
Las mediciones de HbF y HbA2 que ayudan en el diagnóstico de las talasemias beta están fácilmente disponibles en los laboratorios clínicos. En comparación, las pruebas necesarias para diagnosticar las talasemias alfa son bastante sofisticadas y se llevan a cabo en instituciones destinadas a la investigación de las talasemias. Los elementos diagnósticos utilizados en los pacientes en los cuales se sospecha talasemia alfa incluyen historia clínica, evaluación del frotis de sangre, cálculo de índices, tinción con azul brillante de cresilo y estudios familiares. En la práctica el rasgo de la talasemia alfa se diagnostica basándose en el hallazgo de microcitosis en los pacientes sin deficiencia de hierro que tienen niveles normales de HbA2 y HbF.
El diagnóstico de talasemia alfa o beta debe sospecharse cuando se asocia un VCM menor de 75 fl con una cuenta de eritrocitos mayor de 5 millones de células/µl. Un paciente con estos dos datos tiene una posibilidad de 85 por ciento de tener un síndrome de talasemia.85 En un estudio, el diagnóstico de talasemia no se consideró como opción en alrededor de la mitad de los pacientes con la enfermedad.
Consejo genético y diagnóstico prenatal
Los padres que han tenido un mortinato hidrópico o un niño con anemia de Cooley se niegan, y con justificación, a repetir la experiencia. Los adultos que han crecido en familias con talasemias y que se sabe que son heterocigotos para talasemia, están con frecuencia ansiosos de recibir consejo genético cuando inician sus propias familias. El consejo genético incluye el estudio de los futuros padres con base en exámenes diagnósticos rutinarios y estudios familiares. Además, los adelantos en la genética molecular, como el análisis de endonucleasas de restricción, permiten hacer diagnósticos prenatales precisos en el caso de las talasemias. Durante el primer trimestre, la muestra de vellosidades coriónicas combinada con el uso de marcadores de polimorfismo de ADN y sondas sintéticas de oligonucleótidos, puede proporcionar el diagnóstico definitivo en alrededor del 80 por ciento de los casos de talasemia beta.86,87
Defectos extracorpusculares
Los eritrocitos pueden dañarse por traumatismos, anticuerpos, fármacos y toxinas. Debe pensarse en un defecto extracorpuscular siempre que se presente hemólisis en los pacientes que tienen una historia personal o familiar negativa de anemia.
LESION MECANICA: HEMOLISIS MICROANGIOPATICA
La hemólisis microangiopática se caracteriza por la presencia de eritrocitos atípicos y fragmentados en un frotis de sangre periférica (v.gr., esquistocitos o células en casco), y por signos de hemólisis intra y extravascular.
Fisiopatología
El eritrocito normal puede soportar grados moderados de deformación y torcedura, pero se desintegra cuando se somete a fuerzas excesivas de estiramiento o cizallamiento. Se ha observado que ocurren tensiones de esta magnitud en las turbulencias producidas por válvulas aórticas deformes, por fístulas arteriovenosas, por comunicaciones interventriculares, o por las prótesis valvulares antiguas.
Se cree que ocurre coagulación intravascular localizada, en la cual bandas de fibrina cruzan la luz arteriolar, en las arteriolas que irrigan tejidos inflamados o neoplásicos. El estudio de modelos in vitro muestra que dichas bandas de fibrina desprenden fragmentos de los eritrocitos, cuyas membranas se sellan rápidamente. Sin embargo, parte del contenido del eritrocito se fuga, produciendo grados variables de hemólisis intravascular. Los eritrocitos alterados son retirados por el sistema reticuloendotelial.
Diagnóstico
La hemólisis, junto con los hallazgos típicos en el frotis de sangre, indican el diagnóstico [ver figura 7]. Si la angiopatía es extensa puede ocurrir trombocitopenia y coagulación intravascular diseminada. Las causas incluyen turbulencias hemodinámicas, vasculitis, hemangiomas gigantes, púrpura trombocitopénica, cáncer metastásico,89 ciertas infecciones (en especial meningococemia y enfermedades por rickettsias, incluyendo infecciones por hantavirus), síndrome urémico-hemolítico, coagulación intravascular diseminada, fármacos (cocaína, ciclosporina, mitomicina y tacrolimus [FK506]), e incluso catéteres subclavios.89,90
Tratamiento
La atención primaria se debe enfocar hacia la enfermedad principal. Los pacientes pueden desarrollar deficiencia de hierro y requerir tratamiento. Los suplementos en caso de reservas disminuidas de folato pueden estimular la eritropoyesis. En raras ocasiones la anemia causada por una válvula aórtica protésica vieja es lo suficientemente grave para requerir remplazo valvular. La plasmaféresis constituye un tratamiento eficaz para la púrpura trombocitopénica trombótica.
Hemoglobinuria por la marcha La hemoglobinuria por la marcha, trastorno parecido a la hemólisis microangiopática, ocurre por lo común en personas jóvenes después de correr o de tocar bongos durante tiempo prolongado. Se piensa que el trauma severo y repetitivo a los pies y manos destruye los eritrocitos que circulan en los vasos de plantas y palmas. El paciente presenta orina de color rojo que se aclara en 1 día o menos después de la actividad. El diagnóstico se confirma al demostrar hemoglobinemia y hemoglobinuria pasajeras sin anemia, alteraciones en el frotis de sangre, ni reticulocitosis. El uso de zapatos con plantilla y el evitar las superficies pavimentadas puede evitar recurrencias en personas que deciden seguir corriendo.
HEMOLISIS INMUNE
Un ejemplo clásico de hemólisis inmune ( pero no autoinmune) es la incompatiblidad materno fetal en el locus Rh D, en la que la madre D negativa, después de estar en contacto con eritrocitos D positivos, puede producir un anticuerpo IgG anti-D; el anticuerpo cruza la barrera placentaria y ataca y destruye los eritrocitos fetales. El feto presenta ictericia y eritrocitos esferocíticos.
Los eritrocitos fetales, ahora cubiertos con el anticuerpo IgG anti-D, se fijan a los macrófagos y monocitos fetales que contienen receptores para la porción Fc de estas moléculas de IgG. La digestión macrofágica de porciones de la membrana del eritrocito causa pérdida de áreas importantes de la superficie. Los esferocitos rígidos retornan a la circulación y son atrapados por el sistema reticuloendotelial fetal, en especial en el bazo. El anticuerpo IgG es más activo a 37°C, por lo común no puede activar en forma extensa la vía del complemento y no puede aglutinar eritrocitos atacados que están suspendidos en solución salina.
La prueba de antiglobulina directa de Coombs [ver figura 9a]. se utiliza clínicamente para detectar IgG que está cubriendo eritrocitos. Esta prueba es negativa en la madre, cuyos eritrocitos carecen de antígeno D y por lo tanto no están cubiertos por anticuerpo anti-D. La prueba de Coombs indirecta [ver figura 9b], que detecta la presencia de anticuerpos libres en el suero, es positiva en la madre porque tiene anticuerpos anti-D en su suero. En comparación, el feto tiene una prueba de Coombs directa fuertemente positiva, porque sus eritrocitos (con antígenos D) están cubiertos con anticuerpo anti-D materno. La prueba de Coombs indirecta del feto puede ser positiva o negativa, dependiendo de la cantidad de anticuerpos anti-D transferidos por la madre, de la avidez del anti-D por los eritrocitos D positivos fetales, y de la disponibilidad de sitios D en los eritrocitos fetales.
Estos anticuerpos se describen como calientes (porque tienen actividad máxima a 37 'C, por lo general IgG1 o IgG3) o fríos (con actividad máxima a 5 'C, por lo general IgM). Los anticuerpos también se han clasificado como completos (i.e., capaces de aglutinar eritrocitos suspendidos en solución salina) e incompletos (i.e., incapaces de hacerlo) y su detección requiere de técnicas como la prueba de antiglobulina directa de Coombs [ver figura 9a] o del tratamiento enzimático de los eritrocitos.91 Los autoanticuerpos calientes suelen ser incompletos, mientras que las aglutininas frías, por lo general IgM, suelen ser completas.92
Anemia hemolítica autoinmune
La anemia hemolítica autoinmune suele ser un trastorno agudo caracterizado por hemólisis extravascular. La hemólisis intravascular es rara e indica una velocidad extremadamente rápida de destrucción de los eritrocitos o que se han excedido los mecanismos de eliminación extravascular.
Fisiopatología En la anemia hemolítica autoinmune los autoanticuerpos suelen dirigirse contra componentes importantes del eritrocito (v.gr., antígeno Rh, antígeno Kell,91 glucoforina A).93 En forma alternativa, los eritrocitos del paciente están sensibilizados tanto con un anticuerpo IgG como con un componente del complemento, por lo general C3d. En otras circunstancias parece ser que el complemento se fija al eritrocito por un anticuerpo IgM que después se desprende. En ocasiones los eritrocitos pueden mostrar solo componentes del complmento sin que se detecte IgG por la prueba de Coombs. La fijación del complemento en estas circunstancias se puede explicar por la presencia continua de IgG en un nivel por debajo del detectable por medio de la prueba usual de antiglobulina directa; en forma alternativa, pudiera ser que una IgG que fija complemento se hubiera unido antes a la célula, pero fue lavada por el procedimiento de la prueba.92
La severidad de la hemólisis correlaciona con el número y la clase de moléculas IgG unida a la superficie del eritrocito. Los eritrocitos cubiertos con anticuerpo se fijan a los macrófagos a través de receptores (FcRI, FcRII o FcRIII) a través de la porción Fc del anticuerpo. La unión firme de los eritrocitos a los macrófagos es seguida de fagocitosis y lisis.91 Concentraciones relativamente bajas de IgG1 fija a los eritrocitos producen una prueba positiva de antiglobulina directa de Coombs sin evidencia de hemólisis (alrededor de 1,000 moléculas por eritrocito), mientras que concentraciones mucho más altas de autoanticuerpos IgG1 por célula se asocian con evidencias de hemólisis.91 La presencia combinada de IgG y de fracciones del complemento puede aumentar también la severidad de la hemólisis.
Los eritrocitos sensibilizados sólo por IgG suelen ser eliminados por el bazo, mientras que los eritrocitos sensibilizados por IgG y complemento o solo complemento son destruidos por el hígado, porque las células de Kupffer portan receptores específicos para el componente C3b del complemento.
Diagnóstico diferencial Se ha descrito una variedad idiopática y una secundaria a otros padecimientos. Estos incluyen lupus eritematoso generalizado, linfoma no-Hodgkin (en especial leucemia linfocítica crónica), enfermedad de Hodgkin, cáncer, mieloma, quiste dermoide, infección por VIH, linfadenopatía angioinmunoblástica con disproteinemia y colitis ulcerativa crónica.
Manifestaciones clínicas Los pacientes pueden estar desde asintomáticos hasta tener una enfermedad severa. Durante los exámenes de detección en bancos de sangre, o antes de la donación de sangre pueden descubrirse pacientes con prueba de Coombs positiva. Generalmente se puede demostrar que dichas personas tienen complemento o la combinación de complemento e IgG (casi siempre IgG1 o IgG4) en sus eritrocitos, pero por lo común no presentan hemólisis. En el otro extremo, un episodio hemolítico agudo puede disminuir el hematocrito de 45 a 15 por ciento en 2 días. Los pacientes con este tipo de episodios tienen fatiga severa y síntomas cardiorrespiratorios, aunados a ictericia, linfadenopatía y hepatoesplenomegalia.
Diagnóstico En los casos severos los frotis de sangre periférica muestran macrocitosis, policromatofilia, esferocitosis variable y autoaglutinación de eritrocitos. En casos severos la cuenta plaquetaria también está disminuida (síndrome de Evans) y puede haber leucopenia. La tercera parte de los pacientes tiene reticulocitopenia al inicio,94 debido a que la respuesta de reticulocitos puede tardar algunos días en desarrollarse. La prueba directa de Coombs será positiva. Cualquiera o todos estos datos pueden estar ausentes en la enfermedad leve.
Se debe determinar si hay complemento, IgG, o ambos en los eritrocitos por medio del uso de los reactivos de Coombs que están dirigidos específicamente contra la IgG o componentes del complemento. En algunas ocasiones se sospecha una anemia hemolítica autoinmune, pero la prueba directa de Coombs es repetidamente negativa. En estos casos el nivel de autoanticuerpo puede estar por debajo del límite de detección para autoanticuerpos muy activos, como los de la subclase IgG3 o el autoanticuerpo puede ser de tipo IgA o IgM.91
A los pacientes con evidencia de anemia hemolítica se les debe investigar una enfermedad autoinmune (v.gr., lupus eritematoso genealizado) y otras formas de hemólisis, como hemoglobinuria paroxística nocturna, enfermedad por crioaglutininas y hemoglobinuria paroxística por frío.
Tratamiento Los pacientes con enfermedad muy leve no requieren tratamiento inmediato. El tratamiento debe estar encaminado a disminuir la producción de autoanticuerpos y a reducir el ataque por los macrófagos que desencadena la eliminación de los eritrocitos por el sistema reticuloendotelial. El tratamiento inicial consiste en la administración de 60 a100 mg de prednisona al día en dosis divididas. Este tratamiento suele producir una disminución lenta en la fijación de anticuerpos a los eritrocitos y se cree que interfiere con el ataque fagocítico a los eritrocitos ya cubiertos. Una buena respuesta al uso de corticosteroides, indicada por una elevación en la cuenta reticulocitos y una mejoría en los valores sanguíneos, puede ser aparente en 1 o 2 días. Se recomiendan la administración de 1 mg de ácido fólico al día.
Después de la respuesta inicial al tratamiento, que suele ser muy satisfactoria, se permite que la hemoglobina y la cuenta de reticulocitos vuelvan a lo normal. Entonces se repite la prueba de Coombs para determinar si existe disminución en el título de anticuerpos, y en caso positivo comienza a disminuirse la dosis de prednisona de modo cuidadoso. Cerca de la quinta parte de los pacientes se mantienen en buen estado en forma indefinida, pero la mayoría presenta una enfermedad crónica y agresiva que puede producir recaídas repentinas con anemia súbita. La dosis de prednisona se puede regular con base en el nivel de hemoglobina, la cuenta de reticulocitos (su elevación indica persistencia de la hemólisis), y el título de la prueba de Coombs directa. Se ha recurrido a la administración de prednisona en días alternos para minimizar los efectos secundarios de los esteroides. Si el paciente no responde al tratamiento estándar con prednisona, se usa dexametasona en dosis altas (v.gr., 40 mg/día vo por 4 días consecutivos en ciclos de 28 días95 como para la púrpura trombocitopénica idiopática crónica refractaria.
Si la dosis de esteroides necesaria para el tratamiento crónico produce morbilidad importante, el autor ha decidido en forma empírica realizar esplenectomía o administrar agentes inmunosupresores. La medición del secuestro esplénico de eritrocitos marcados con Cr51 no predice en forma confiable el éxito de la esplenectomía. La esplenectomía rara vez causa remisión prolongada, pero es útil para ahorrar esteroides. Después de la esplenectomía, una dosis pequeña de prednisona (5 ó 10 mg al día), puede estabilizar la concentración de hemoglobina.
Se pueden administrar agentes inmunosupresores como la azatioprina o la ciclofosfamida como alternativa a la esplenectomía. Sin embargo, no existe evidencia confiable que pueda servir como base para elegir entre los dos agentes. En los pacientes con enfermedad muy agresiva la ciclosporina ha sido útil.96 La azatioprina se debe iniciar en una dosis de 100 a 200 mg al día, vigilando cuidadosamente la citología hemática para evitar reticulocitopenia o neutropenia. La ciclofosfamida se inicia en dosis de 100 a 200 mg al día, vigilando la citología hemática y la orina. Debido a que la ciclofosfamida puede causar leucemia mielocítica aguda o síndrome mielodisplásico relacionados al tratamiento, su uso es limitado.
Las dosis de azatioprina o ciclofosfamida deben ajustarse para reducir la cuenta de leucocitos a alrededor de 3,000/mm3. La mejoría ocurre en un lapso de 3 a 4 semanas. Cuando existe respuesta al tratamiento se puede reducir la dosis de prednisona; en este momento es necesario comprobar de nuevo el nivel de hemoglobina, cuenta de reticulocitos y títulos de la prueba de Coombs para determinar la dosis mínima de tratamiento requerida. Se han usado con éxito dosis elevadas de IgG intravenosa para tratar la anemia hemolítica autoinmune, en forma análoga a su uso en la púrpura trombocitopénica idiopática. Sin embargo, no existen estudios controlados a gran escala. En un reporte, un tercio de los pacientes tuvieron una respuesta temporal y se requirieron dosis mayores a las empleadas para púrpura trombocitopénica idiopática.97,98
El paciente con anemia sintomática requiere transfusión de eritrocitos. En estos casos no es una sorpresa que el banco de sangre informe incompatibilidad. Muchos bancos de sangre hacen en forma regular una prueba de antiglobulina directa en los eritrocitos del receptor. Los pacientes que tienen anticuerpos libres en el suero presentarán reactividad muy amplia y extensa contra los donadores de eritrocitos, y producirán pruebas cruzadas incompatibles cuando se examinan con el reactivo para antiglogulinas. Si se requieren transfusiones para apoyar las funciones cardiorrespiratorias y del sistema nervioso central, se recomienda una consulta pronta con el servicio de medicina de transfusión.92
Hemólisis inmune relacionada con medicamentos
Ciertos casos de hemólisis inmune que son clínicamente indistinguibles de la anemia hemolítica autoinmune son inducidos por la administración de medicamentos. Existen dos variables, la anemia de tipo hapteno y la hemolítica autoinmune.99
Tipo hapteno Medicamentos como las penicilinas y cefalosporinas se unen firmemente a la membrana del eritrocito. En circunstancias excepcionales en las que se requieren dosis masivas del medicamento (v.gr., más de 10 millones de unidades de penicilina al día), el medicamento unido a las proteínas puede actuar como un hapteno y causar una respuesta inmune. Al parecer se produce un anticuerpo que está dirigido contra el complejo medicamento-eritrocito.100,101 El medicamento puede fijarse de modo firme o débil al eritrocito y producir un neoantígeno que genera una respuesta inmune.99
La prueba de Coombs directa es positiva con agentes anti-IgG. Si se prueba el suero del paciente contra células normales (prueba de Coombs indirecta) no se presenta reacción a menos que el medicamento agresor se añada primero a los eritrocitos normales. El suspender o cambiar el medicamento es eficaz para eliminar la hemólisis, ya que el anticuerpo tiende a ser muy específico.
Anemia hemolítica autoinmune inducida por medicamentos La metildopa es el ejemplo clásico de un medicamento que causa anemia hemolítica autoinmune. Otros son la levodopa, el ácido mefenámico y quizá la procainamida. La administración de dichos medicamentos desencadena una prueba de Coombs directa positiva con reactivos anti-IgG en el 15 al 20 por ciento de los pacientes tratados, pero se presenta hemólisis en menos del 1 por ciento. Cuando el anticuerpo se separa se observa que es un anticuerpo IgG clásico dirigido contra los componentes Rh. El mecanismo de la hemólisis es idéntico al de la anemia hemolítica autoinmune.102
Se ha reportado que el AINE diclofenaco sódico causa anemia hemolítica aguda devastante, con evidencia de hemólisis intra y extravascular asociada a estado de choque, falla orgánica e incluso coagulación intravascular diseminada (CID).103 Los pacientes desarrollan tanto autoanticuerpos contra eritrocitos como anticuerpos dependientes del fármaco. Se piensa que el diclofenaco sódico se une a la superficie del eritrocito, formando neoantígenos que provocan la generación de autoanticuerpos verdderos, así como anticuerpos dependientes del fármaco. La prueba directa de Coombs es positiva tanto con reactantes IgG como C3d. Ocurre reactividad de anticuerpo adicional si se agregan metabolitos del diclofenaco sódico obtenidos de la orina de los pacientes tratados con el medicamento. El tratamiento consiste en reconocer la causa, suspender el medicamento y dar manejo de apoyo durante varios días hasta que cese el proceso.103
Hemólisis tardía de eritrocitos transfundidos
La sangre se tipifica sólo para los antígenos ABO y Rh-D, pero existen otros antígenos presentes en los eritrocitos. Por ello, un paciente que es trasfundido en forma múltiple puede desarrollar, en un periodo de 1 a 2 semanas, una respuesta de anticuerpos a uno o más de estos antígenos. Los agresores usuales son los antígenos Kell, Duffy, Kidd, y antígenos Rh diferentes al D. Puede presentarse una hemólisis aguda autolimitada, que por lo común es extravascular. Los datos primordiales son el antecedente de transfusión previa, esferosis en el frotis en sangre periférica, una prueba de Coombs directa positiva y la aparición reciente de un anticuerpo en el suero del paciente (prueba indirecta de Coombs positiva). En general no se requiere tratamiento, pero las transfusiones subsecuentes deben cruzarse con el suero del paciente. Aparece un problema semejante con el trasplante de médula ósea y de otros tejidos.104
Enfermedad por crioaglutininas
La enfermedad por crioaglutininas tiene varios tipos. Una variedad poco común afecta adultos jóvenes y suele estar precedida por una infección por Mycoplasma pneumoniae o mononucleosis infecciosa; sin embargo, se han informado varios casos de esta variedad relacionados con paludismo crónico por Plasmodium falciparum. Una forma más común afecta a individuos de alrededor de 60 años de edad y puede presentarse como enfermedad idiopática por crioaglutininas, como pródromo a un trastorno linfoproliferativo o inmunoproliferativo, o en asociación a un padecimiento linfoproliferativo ya establecido.105
Fisiopatología y manifestaciones clínicas Serológicamente, el trastorno se caracteriza por la presencia en el suero de títulos elevados (más de 1:1,000 y por lo común más de 1:10,000) de aglutininas IgM. Estos anticuerpos son más activos a 4'C, son capaces de activar la secuencia del complemento y están dirigidos contra el sistema antigénico Ii de los eritrocitos. Al parecer la IgM reacciona con los eritrocitos que circulan en la sangre de menor temperatura de la nariz, oídos y espinillas. El anticuerpo fija complemento y posteriormente se disocia de los eritrocitos al llegar a zonas más templadas del organismo. En la variedad posinfecciosa de este trastorno, la crioaglutinina IgM es policlonal y de vida corta. A la inversa, en la variedad idiopática de enfermedad por crioaglutininas o en los casos asociados con macroglobulinemia de Waldenström, leucemia linfocítica crónica u otros linfomas, la IgM es monoclonal. Esta consiste casi en su totalidad de cadenas ligeras kappa en los pacientes con enfermedad idiopática por crioaglutininas o macroglobulinemia de Waldenström, pero en los pacientes con linfoma la IgM está formada sobre todo de cadenas ligeras lambda. En ocasiones la IgM se detecta como un pico de proteína M en la electroforesis de proteínas séricas.
En la variante posMycoplasma, los micoplasmas parecen unirse a la superficie del eritrocito en el sitio antigénico Ii. Esta interacción receptor-ligando causa la presentación del antígeno I en una forma inmunogénica.106 Listeria monocytogenes contiene al antígeno I,105 lo que apoya la teoría de que algunos agentes infecciosos estimulan las crioaglutininas que ocurren en forma natural y que causan la enfermedad por crioaglutininas posinfecciosa.
El síndrome clínico de la enfermedad por crioaglutininas es muy variable, y algunos pacientes sólo muestran títulos bajos de crioaglutininas sin síntomas o historia de neumonía reciente. En otros con anemia hemolítica autoinmune caliente y fría la hemólisis asociada tiende a ser severa y crónica. Los eritrocitos de estos pacientes están cubiertos con IgG y componentes del complemento, mientras que sus sueros contienen un título relativamente bajo de crioaglutininas que actúan a 30'C y que incluso pueden actuar a los 37'C.
Diagnóstico La presencia de anemia hemolítica con signos y síntomas acrales sugiere el diagnóstico. Puede ser difícil tomar una muestra de sangre, y la sangre puede aglutinarse a la vista en una jeringa fría y en el frotis. Los contadores automáticos de células sanguíneas pueden contar los eritrocitos aglutinados como eritrocitos independientes y en consecuencia informar valores absurdamente altos para el VCM y la CMHC.
Por lo general el banco de sangre detecta una crioaglutinina con actividad amplia. La prueba de Coombs directa es positiva con reactivos anticomplemento pero no con reactivos anti-IgG.
Los datos que apoyan el diagnóstico de enfermedad idiopática por crioaglutininas incluyen los siguientes: títulos elevados de crioaglutinina IgM con reactividad termica amplia91 y especificidad a I (que reaccionan con eritrocitos de adultos pero no con eritrocitos de cordón umbilical), composición pura de cadenas ligeras kappa, en forma ocasional elevación absoluta de la IgM sérica, y un patrón de proteína M en la electroforesis de proteínas. Debe intentar detectarse un posible linfoma u otro padecimiento subyacente. Por el contrario, las crioaglutininas pos-Mycoplasma y pos-mononucleosis infecciosa son policlonales y los anticuerpos pos-mononucleosis infecciosa están dirigidas contra los antígenos i (eritrocitos de cordón umbilical).
Tratamiento La variedad pos-Mycoplasma o pos-mononucleosis infecciosa suele ser leve, autolimitada y no requiere tratamiento específico. Los pacientes con la variedad idiopática que tienen síntomas acrales deben cambiar su forma de vida, trasladándose a un sitio con clima más cálido o manteniendo cubiertos sus oídos, nariz, manos y pies durante la temporada fría. Pueden requerirse transfusiones de concentrado globular en los pacientes con anemia severa, lo que amerita pruebas cruzadas cuidadosas y calentar la sangre para minimizar la aglutinación por frío.
La esplenectomía y los esteroides no suelen controlar la hemólisis asociada con la enfermedad por crioaglutininas. Se supone que los eritrocitos cubiertos por complemento son retirados de la circulación en grado importante por macrófágos hepáticos más que esplénicos, y las células que producen IgM son relativamente insensibles a los efectos de los esteroides. Sin embargo, en ocasiones la administración de esteroides en dosis elevadas (v.gr.,100 mg de prednisona al día), ha reducido la velocidad de hemólisis en pacientes con títulos relativamente bajos de crioaglutininas. En la variante relativamente rara causada por crioaglutininas de IgG los esteroides y la esplenectomía pueden ser benéficos. No se observan buenos resultados con el uso de penicilina u otros agentes reductores que contienen grupos sulfhidrilo. La exsanguinotransfusión y la plasmaféresis parecen ser tratamientos lógicos para la crisis aguda, pero se necesitan más estudios clínicos para evaluar estas técnicas. El interferón alfa, en dosis de 3 millones U/m2 tres veces por semana, parece producir una reducción impresionante en el título de crioaglutininas, con reducción en la proteína M monoclonal en suero y en los síntomas acrales durante un periodo de un mes.91
Hemoglobinuria paroxística por frío
Los pacientes con el raro transtorno de hemoglobinuria paroxística por frío tienen signos y síntomas de hemólisis intravascular inducidos por el frío. La hemólisis parece estar asociada con la presencia de un anticuerpo sérico IgG que está dirigido contra el sistema P de los eritrocitos. El anticuerpo IgG se demuestra mejor por prueba de Donath-Landsteiner: se mezcla el suero con los eritrocitos del propio paciente o con eritrocitos de una persona normal y se enfría la mezcla a 4'C. Si está presente el anticuerpo de IgG asociado con este trastorno, producirá hemólisis después de calentar a 37'C. En el pasado la hemoglobinuria paroxística por frío se observaba generalmente como una complicación de la sífilis, pero en la actualidad se asocia a infecciones virales y a linfoma no Hodgkin.107
HIPERESPLENISMO
Los trastornos hiperesplénicos comprenden un grupo diverso de entidades clínicas que comparten las características comunes de esplenomegalia y hemólisis. La esplenomegalia y la hemólisis se presentan en muchos trastornos, incluyendo cirrosis hepática con esplenomegalia congestiva, enfermedad de Gaucher, linfoma, trastornos del tejido conjuntivo, síndrome de Felty, sarcoidosis, tuberculosis y otras enfermedades infecciosas.
Fisiopatología
La estructura singular del bazo es responsable de varias de las características fisiopatológicas del hiperesplenismo. Las arteriolas esplénicas tienen algunas ramas directas que van a los sinusoides, pero la mayoría de las arteriolas terminales se abren hacia los cordones esplénicos. Las células sanguíneas pasan desde los cordones hacia la pulpa a través de hendiduras en las paredes sinusoidales, que tienen dimensiones de entre 1 y 3 µm.108 Las células sanguíneas pasan entre los espacios longitudinales, que están cubiertos con fibras reticulares, y entre las células de la adventicia que se localizan fuera del sinusoide. Los macrófagos y las células endoteliales cubren la parte interna del sinusoide, y de esta manera existe contacto íntimo repetido entre las células sanguíneas y estos macrófagos.
El flujo sanguíneo en el bazo es lento. El pH y pO2 del eritrocito disminuyen, se consume glucosa y se altera el metabolismo de la célula. Puede aumentar el hematocrito, elevando más la viscosidad y la resistencia al flujo. Como consecuencia, las células sanguíneas son expuestas a tensiones metabólicas y mecánicas en presencia de macrófagos y otros leucocitos que tienen capacidad de reconocer el daño en la membrana celular. Al envejecer los eritrocitos, los fagocitos les quitan las áreas de superficie defectuosa, transformando a los eritrocitos bicóncavos en esferocitos rígidos o fragmentos de eritrocitos; estas partículas son atrapadas y después retiradas por el sistema reticuloendotelial. Un bazo grande tiene un flujo sanguíneo mayor de lo normal y expone una importante proporción de células sanguíneas a sus actividades de filtro. De esta manera, el problema en el hiperesplenismo es en esencia cuantitativo. Puede originarse un círculo vicioso en los pacientes que presentan hemólisis porque la hemólisis misma causa esplenomegalia.
Diagnóstico
Si el bazo no es palpable, pero se tiene la sospecha clínica de esplenomegalia, puede ser útil realizar un ultrasonido o un gamagrama con radioisótopos. Debido a que un bazo grande afecta, además de los eritrocitos a otras células sanguíneas, el paciente puede tener pancitopenia. A menos que el trastorno de fondo se relacione específicamente con la médula ósea, ésta suele encontrarse hiperplásica en el hiperesplenismo, a causa de la regeneración rápida de todas las líneas celulares afectadas. El frotis de sangre periférica no confirma el diagnóstico de hiperesplenismo.
Tratamiento
Si el hiperesplenismo está produciendo complicaciones clínicamente importantes y el tratamiento de la enfermedad principal no causa disminución del bazo, puede ser necesaria la esplenectomía. Sin embargo, la anemia no necesariamente debe atribuirse al hiperesplenismo, independientemente del tamaño del bazo. La hemodilución es otro mecanismo posible. Los pacientes con esplenomegalia masiva que tienen valores bajos de hematocrito y hemoglobina pueden tener volúmenes normales de eritrocitos según la técnica de Cr51. La esplenomegalia masiva produce un aumento en el volumen plasmático que causa una hemodilución extraordinaria. Es más, los bazos muy grandes pueden contener una reserva de eritrocitos que constituyen hasta el 25 por ciento del volumen total de eritrocitos; en comparación, los bazos normales no tienen reserva de eritrocitos. En pacientes con esplenomegalia que tienen una verdadera disminución en el volumen de eritrocitos el trastorno principal puede actuar reduciendo la producción de eritrocitos más que por destrucción acelerada. Por lo tanto, es conveniente llevar a cabo una determinación del volumen de eritrocitos antes de hacer el diagnóstico de hiperesplenismo.
MEDICAMENTOS, TOXINAS, VENENOS Y AGENTES FISICOS COMO CAUSAS DE HEMOLISIS
Fisiopatología
Ataque oxidativo Medicamentos como dapsona, sulfasalazina, fenacetina, perclorato de sodio, nitroglicerina, fenazopiridina, primaquina,66 y agentes como el paraquat y los análogos de la vitamina K, pueden introducirse en la hendidura fijadora de oxígeno de la hemoglobina . Por medio de esta acción esos medicamentos pueden generar radicales libres oxidantes como superóxido, radicales hidroxilo libres y peróxido. Si los mecanismos reductores del eritrocito son superados [ver tabla I], la hemoglobina es oxidada para formar cuerpos de Heinz y metahemoglobina. También se produce sulfahemoglobina por el ataque oxidativo. Esta molécula contiene un átomo de azufre en el anillo de porfirina, lo que le da un color azul-verdoso. No se conoce bien el origen del átomo de azufre, pero su presencia en el anillo hem deteriora el transporte de oxígeno.109 A pesar de ello, la sulfahemoglobina es una causa rara de cianosis.148 La membrana del eritrocito también puede estar sujeta al ataque oxidativo. Las células dañadas son retiradas por el sistema reticuloendotelial. La hemólisis suele ser extravascular, aunque no siempre, y pueden observarse cuerpos de Heinz en un frotis de sangre con tinción especial. Se eleva el nivel de la metahemoglobina. (Una cantidad tan pequeña como 1.5 g/dl de metahemoglobina o 0.5 g/dl de sulfahemoglobina puede producir datos físicos de cianosis. Por el contrario, se requieren 5 g/dl de desoxihemoglobina reducida para producir una cianosis comparable.66) El frotis puede mostrar las células con las muescas o ampollas típicas del ataque oxidativo a los eritrocitos [ver figura 6].
Los nitritos pueden oxidar la hemoglobina a metahemoglobina. En consecuencia, el uso recreativo de butil e isobutil nitritos como estimulantes, psicodélicos y afrodisiácos ha causado problemas clínicos. Cuando se inhalan en las cantidades habituales, estos agentes pueden producir un incremento leve a moderado en la metahemoglobina, elevando su concentración del nivel normal de 1 a 2 por ciento hasta el 20 por ciento. La inhalación más abundantes o la ingestión de estos compuestos induce metahemoglobinemia severa, caracterizada por concentraciones que alcanzan el 62 por ciento. Debido a que la metahemoglobina no transporta oxígeno, estos niveles elevados se asocian con manifestaciones de hipoxia tisular como cefalea, disnea, letargo y estupor. El examen físico demuestra taquicardia, hipotensión postural y cianosis, la sangre venosa es de color café-púrpura.110
Al parecer el daño oxidativo severo hace que la hemoglobina se agrupe en un lado del eritrocito, dejando un 'medio fantasma' cubierto por membrana plasmática en el resto de la célula. Estos hemifantasmas se pueden detectar en la sangre periférica. La destrucción oxidativa severa se asocia con aumento en los niveles de metahemoglobina y disminución en los niveles de glutatión reducido, y puede ser mortal si no se trata.
El diagnóstico se basa en el antecedente de exposición a un medicamento oxidante o a otra toxina, junto con la presencia de datos característicos en el frotis de sangre periférica y de elevación en la metahemoglobina.
Tratamiento
El tratamiento consiste en identificar y suspender el agente agresor. Los pacientes con metahemoglobinemia severa deben tratarse inmediatamente con una dosis de 1 a 2 mg/kg de azul de metileno; éste se administra por vía intravenosa en una solución de 1 g/dl en un periodo de 5 minutos. En presencia de la enzima eritrocítica NADPH-metahemoglobina reductasa y de cantidades adecuadas del donador de electrones NADPH [ver tabla I], el azul de metileno es reducido rápidamente a leuco-azul de metileno. Este producto reduce a su vez en forma rápida a la metahemoglobina a hemoglobina. Por lo tanto la cianosis revierte y el paciente adquiere un tono rosado de la piel inmediatamente después de la administración. Sin embargo, varias horas después el paciente puede ponerse otra vez cianótico, al parecer porque los nitratos liberados de los tejidos vuelven a entrar a la sangre periférica en ese momento. La readministración de azul de metileno, en una dosis de 1 mg/kg por vía intravenosa en un periodo de 5 minutos, debe restablecer los niveles de hemoglobina.
El éxito del tratamiento con azul de metileno depende de la presencia de suministros adecuados de NADPH. Los pacientes que tienen alteraciones de la vía de la pentosa fosfato [ver figura 5], como deficiencia de G6PD, no responderán a este tratamiento y deben recibir exsanguinotransfusiones de urgencia.110
Los pacientes con concentraciones muy altas de metahemoglobina (de por lo menos 60 por ciento) o los que tienen muchos hemifantasmas en el frotis deben someterse a exsanguineotransfusión, quizá con hemodiálisis.66,110
Otras formas de hemólisis inducida por medicamentos
Fisiopatología La exposición al plomo da como resultado un cuadro clínico que consiste en encefalopatía hipertensiva, neuropatía y anemia hemolítica que se caracteriza por puntilleo basófilo irregular en los eritrocitos. El mecanismo de la hemólisis inducida por plomo es complejo porque el metal tiene varias acciones: bloquea la síntesis del hem y causa de esta manera un almacenamiento de protoporfinas en el eritrocito, produce una deficiencia de pirimidina 5'-nucleotidasa111 y ataca los fosfolípidos de la membrana del eritrocito, causando fuga de potasio e interfiriendo con la actividad de la ATPasa de Na+-K+.
Tratamiento La detección de la intoxicación por plomo se lleva a cabo midiendo el nivel de protoporfirina eritrocítica libre. El diagnóstico se confirma al medir los niveles de plomo en sangre y orina. Se puede considerar el uso de un agente quelante, como edetato disódico del calcio (Ca Na2 EDTA), después de suspender la exposición al plomo. El tratamiento se inicia con 0.5 a 1.0 g de Ca Na2 EDTA, administrados por vía intravenosa en un periodo de 6 a 8 horas. El compuesto se administra diario por 5 días, y luego se administran 0.5 g de Ca Na2 EDTA como bolo intravenoso o inyección intramuscular, cada tercer día durante 2 semanas, mientras se lleva un control de los niveles urinarios de plomo. Una alternativa a la administración de Ca Na2 EDTA, después de los primeros 5 días, es el tratamiento con penicilamina oral de la siguiente manera: los primeros 7 días, 1 g/día; la siguiente semana se suspende el medicamento, y durante los 7 últimos días del régimen se repite el ciclo y se mide el nivel de plomo en la orina. Un estudio recomienda administrar 500 mg de penicilamina por día y continuar esta dosis durante 60 días después de que el paciente está asintomático.112
Ataque enzimático a los eritrocitos
Ejemplos clásicos de enzimas que atacan a los eritrocitos son el veneno de serpientes o las lecitinasas de los clostridios (fosfolipasa C). Estas enzimas literalmente atacan a los fosfolípidos de la bicapa de la membrana y producen fragmentación de los eritrocitos, esferocitosis y evidencia de hemólisis intravascular y extravascular. Puede haber coagulación intravascular diseminada y estado de choque. La identificación pronta y el tratamiento del trastorno primario son decisivos, al igual que el tratamiento de sostén.
Causas físicas de hemólisis
El casi ahogamiento en agua dulce y la administración accidental de agua estéril puede causar hemólisis intravascular por lisis osmótica. En dichos casos los eritrocitos se hinchan y se vuelven esferoidales. El casi ahogamiento en agua salada puede inducir hemólisis al desecar los eritrocitos. Las quemaduras causan desnaturalización mediada por temperatura de los polipéptidos de la membrana del eritrocito, provocando hemólisis.
Causas infecciosas de hemólisis
Las infecciones producen hemólisis por varios mecanismos. Las infecciones por Mycoplasma pneumoniae y la mononucleosis infecciosa pueden causar hemólisis por crioaglutininas. La infección por H. influenzae tipo b puede provocar hemólisis, el principal factor de virulencia de este organismo, polirribosa ribosil fosfato (PRRP) permite que el organismo escape a la fagocitosis. El PRRP liberado a la circulación se fija a los eritrocitos, y la unión consecuente de aticuerpos anti-PRRP causa hemólisis dependiente del complemento.113 Los pacientes infectados por VIH o citomegalovirus (CMV) pueden tener anemia hemolítica autoinmune (ver antes).114
La sepsis por clostridios puede ser devastadora y la aparición de hemoglobina libre en plasma o de hemoglobinuria sugiere una infección fatal. Las especies Clostridium son capaces de crecer en forma súbita y explosiva, y pueden liberar muchas enzimas, incluyendo fosfolipasas y proteasas, que digieren a los eritrocitos, causando hemólisis intravascular.
Algunas infecciones pueden causar esplenomegalia y hemólisis por hiperesplenismo. La meningococcemia o la septicemia severa por gram negativos pueden producir CID y hemólisis microangiopática. Algunos agentes infecciosos, tales como Plasmodium, que causa el paludismo, parasitan y destruyen directamente los eritrocitos. Los eritrocitos autólogos no parasitados, se eliminan con más rapidez de la habitual, e incluso los eritrocitos normales son eliminados con rapidez de pacientes cuyos eritrocitos están parasitados por paludismo.115 Este dato sugiere que el sistema reticuloendotelial es hiperactivo durante el paludismo y, quizá también en otras infecciones.
Cada vez se diagnóstica con más frecuencia a la babesiosis, en especial en el área de Nueva Inglaterra, enfermedad causada por un parásito que invade los eritrocitos y que se trasmite de su reservorio roedor por la misma garrapata ixodes que porta la enfermedad de Lyme y la erlichiosis humana. Los individuos inmunosuprimidos, como los infectados por VIH, tienen más riesgo de sufrir infecciones crónicas y severas. El diagnóstico se realiza por frotis de sangre periférica, pero los nuevos métodos de reacción en cadena de la polimerasa son más sensibles.116
HEMOLISIS RELACIONADA CON ENFERMEDAD HEPATICA
Varios mecanismos, incluyendo la hemólisis, pueden producir anemia en pacientes con hepatopatías. El aspecto inflamatorio de un trastorno de fondo (como en la hepatitis viral) puede causar un defecto en la producción. La ingestión de alcohol bloquea también la producción de eritrocitos. La deficiencia de folato en los pacientes con cirrosis alcohólica puede dar como resultado eritropoyesis ineficaz megaloblástica clásica. La gastritis alcohólica puede causar hemorragias severas. Las reservas de hierro pueden agotarse en los vagabundos alcohólicos que venden su sangre con demasiada frecuencia. El paciente cirrótico puede tener esplenomegalia congestiva con hemólisis hiperesplénica. También son comunes los macrocitos (con o sin deficiencia de B12 o folato) y las células en diana (causadas por elevación de colesterol).
Equinocitosis y acantocitosis
En los pacientes con hepatopatía es frecuente observar equinocitosis cuando se examinan los eritrocitos en una preparación húmeda o por microscopía electrónica, aunque los frotis estándar pocas veces muestran estos equinocitos.117 Cuando los eritrocitos normales se incuban en el plasma de pacientes con equinocitosis, las células se vuelven equinocíticas con rapidez.
Los equinocitos pueden sufrir un cambio de forma a células en erizo (acantocitos). La acantositosis o hemólisis de células en erizo es causada por alteraciones en los lípidos de la membrana de los eritrocitos, con disminución en la síntesis de fosfolípidos, que causa disminución en la relación entre fosfolípidos y colesterol.118 Estos cambios en la membrana acortan la vida de los eritrocitos.
OTRAS CAUSAS DE HEMOLISIS
Acúmulo de cobre
Pocas veces la enfermedad de Wilson, trastorno metabólico asociado con depósitos excesivos de cobre, se detecta por un episodio de hemólisis grave y agudo. Se piensa que la liberación de cobre al suero y su entrada subsecuente al eritrocito es el mecanismo hemolítico principal. Además de afectar los niveles de hexocinasa, el cobre intracelular parece causar la formación de radicales de oxígeno que reaccionan con los componentes de la membrana y los oxidan. Aunque no se ha informado ningún tratamiento con buenos resultados, puede administrarse penicilamina en una dosis de 2 a 4 g vo diarios en un esfuerzo por reducir el nivel de cobre libre. Si el ataque oxidativo es un factor importante, la administración de 1,000 a 2,000 UI de vitamina E (alfa-tocoferol) diarios, durante varios días, puede ser de utilidad.
Derivación cardiopulmonar
Después de la derivación cardiopulmonar puede presentarse leucopenia transitoria en algunos pacientes. Se piensa que esta leucopenia es causada por la activación del complemento, lo que origina C3a y C5a . También aumenta la hemoglobina libre en plasma, probablemente por hemólisis causada por el depósito del complejo de ataque C5b-C9 sobre la superficie del eritrocito.119
Bibliografía
DR. STANLEY L. SCHRIER
Las alteraciones de la membrana eritrocitaria son la vía final común que conduce a la hemólisis. Estas alteraciones de la membrana suelen indicar a los macrófagos reticuloendoteliales que retiren de la circulación los eritrocitos dañados. Sin embargo, en circunstancias extraordinarias el daño es tan grande que el contenido intracelular, incluyendo la hemoglobina, se libera en el plasma. Se conoce ya con sorprendente detalle la manera como se altera la membrana. Esta subsección describe varias enfermedades de la arquitectura de la membrana, variaciones genéticas de diversas proteínas del eritrocito, incluyendo la hemoglobina, y factores extracorpusculares que pueden causar acortamiento en la vida de los eritrocitos y muchas otras consecuencias serias.
Desarrollo, estructura y fisiología del eritrocito
Las células eritroides sufren cuatro o cinco divisiones celulares en la médula ósea y después expulsan su núcleo. Al madurar la célula enucleada disminuye la síntesis de hemoglobina y los reticulocitos pierden la mayoría de sus receptores de transferrina y penetran a la sangre periférica.
En la circulación los eritrocitos sobreviven alrededor de 4 meses. Durante este tiempo el eritrocito debe soportar tensiones mecánicas y metabólicas severas: se deforma para atravesar capilares con diámetros equivalentes a la mitad del suyo, resiste altas fuerzas de corte al cruzar las válvulas cardiacas, se aglomera con otros eritrocitos sin adherirse, y sobrevive a episodios repetidos de acidemia inducida por estasis y carencia de sustratos, y eliminación por macrófagos en las estructuras reticuloendoteliales. Debe también mantener un ambiente interno que proteja a la hemoglobina del ataque oxidativo y conservar la concentración óptima de 2,3-difosfoglicerato (2,3-DPG) que se necesita para la función de la hemoglobina.
HEMOGLOBINA
El eritrocito adulto normal contiene tres formas diferentes de hemoglobina (Hb): HbA (96 por ciento), HbA2 (2 a 3 por ciento) y Hb F (< 2 por ciento). La hemoglobina A normal se compone de 2 cadenas alfa y 2 cadenas beta (alfa2beta2), codificadas por cuatro genes en el cromosoma 16. Los genes beta y los otros genes no alfa, que codifican las cadenas gama de la HbF (hemoglobina fetal) y las cadenas delta de la HbA2, están en íntima relación mutua en el cromosoma 11. La concentración extraordinariamente alta de hemoglobina en el eritrocito, 33 a 35 g/dl (concentración media de hemoglobina corpuscular o CMHC) produce una solución viscosa.
CITOSOL NO HEMOGLOBINICO
Los eritrocitos utilizan la glucosa principalmente para mantener el poder reductor que protege a las células contra el ataque oxidativo, generar el 2,3-DPG requerido para modular la función de la hemoglobina, y para controlar el contenido de sal y agua de los eritrocitos a través de las acciones del trifosfato de adenosina (ATP) y de las trifosfatasas de adenosina (ATPasas) de transporte [ver tabla 1]. El contenido de agua y hemoglobina de los eritrocitos determinan el volumen corpuscular medio (VCM) y la CMHC.
|
|||||||||||||
ATP- Trifosfato de adenosina, 2,3-DPG-2,3 -difosfoglicerato, NADH-dinucleotido de nicotinamida-adenina fosfato reducido |
MEMBRANA PLASMATICA
El eritrocito normal tiene forma discoide, un diámetro de 7 a 8 µm, un VCM de 85 a 90 femtolitros (fl) (1 fl =10-15 L), y una superficie de 140 µm2 [ver figura 1]. La forma bicóncava del eritrocito le permite introducirse a través de capilares tan angostos como de 3 µm de diámetro.
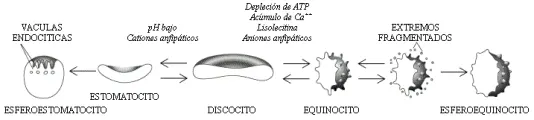
|
| Figura 1 |
| Cambios en la forma del eritrocito |
Los lípidos (fosfolípidos y colesterol) son responsables de alrededor del 50% del peso de la membrana de superficie. Los fosfolípidos se distribuyen de modo asimétrico en una bicapa, los que tienen carga positiva, como la esfingomielina y la fosfatidilcolina se localizan en la mitad externa, y los aminofosfolípidos con carga negativa, como la fosfatidilserina y la fosfatidiletanolamina, se encuentran sobre todo en la mitad interna de la bicapa. Esta asimetría en la distribución de fosfolípidos permite que pequeñas moléculas con carga puedan intercalarse en forma selectiva y causar expansión de la mitad externa o de la interna de la bicapa, produciendo equinocitos o estomatocitos. [ver figura 1].
Las proteínas de la membrana del eritrocito se clasifican con base en su movilidad en la electroforesis en gel de sodio dodecil sulfato- poliacrilamida (SDS-PAGE). Las proteínas integrales interactúan con la bicapa hidrofóbica de fosfolípidos [ver figura 2]. Las principales proteínas integrales de la membrana del eritrocito son las glucoforinas (que contienen la mayor parte del ácido siálico y portan los antígenos de grupo sanguíneo MNS) y la banda 3, que es el transportador aniónico y de bicarbonato.
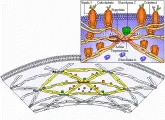
|
| Figura 2 |
| Proteínas de la membrana del eritrocito |
Las proteínas periféricas se encuentran en la cara de la membrana que da al citosol. Las bandas 1 (alfa) y 2(beta) de la espectrina, una proteína periférica, interactúan para formar un heterodímero; enseguida se unen dos de estos heterodímeros para formar un heterotetrámero, la forma predominante de la espectrina. Por sí mismos, los heterotetrámeros contribuyen poco a la estabilidad, pero cuando están unidos en forma cruzada por oligómeros de la banda 5, o actina, en una interacción que está muy reforzada por la banda 4.1, el resultado es el citoesqueleto fuerte y elástico del eritrocito.
El citoesqueleto periférico está conectado a las proteínas integrales por medio de la banda 2.1 (anquirina), que une a la espectrina con la banda 3 [ver figura 2]. Una de las glucoforinas, la glucoforina C, se fija también a la banda 4.1. Se han identificado nuevas proteínas periféricas de la membrana, que existen en cantidades pequeñas y que son la aducina, la tropomiosina y la dematina, y todas parecen tener funciones como esqueleto de la membrana.1,2
Los carbohidratos contribuyen a la carga externa negativa de la membrana y en parte funcionan como antígenos de grupo sanguíneo. Algunos de estos glucolípidos se asocian con fosfatidilinositol para formar un ancla glucolípida llamada el ancla glucosil-fosfatidilinositol (GPI) [ver figura 3]. Estas anclas de GPI proporcionan sitios de sostén en la membrana para diversos tipos de proteína, como el factor acelerador de la degradación (DAF o CD55) y el inhibidor de la lisis reactiva de la membrana (MIRL o CD59), que sirve para controlar la acción del complemento [ver adelante, Hemoglobinuria paroxística nocturna].3
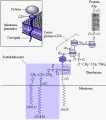
|
| Figura 3 |
| Molécula glucosil-fosfatidilinositol |
CONTROL DE LA HIDRATACION
El control del volumen del eritrocito tiene gran importancia fisiopatológica porque el contenido de agua y cationes de la célula determina su viscosidad intracelular y la relación entre el área de superficie y el volumen. El contenido de Na+ y K+ está determinado por una combinación de difusión pasiva y transporte activo, y este último mecanismo depende principalmente de la Na+-K+-ATPasa. El principal anión intracelular es el Cl-, que entra a la célula con gran permeabilidad a través de la banda 3, el principal transportador de aniones (también denominado AE1 o transportador de aniones-1). El cotransportador de K+ - Cl - origina el gradiente K+ - Cl - y es activado por edema de los eritrocitos y un pH intracelular bajo para causar una pérdida neta de K+ y Cl -. La Ca2+ATPasa bombea en forma activa Ca2+ al exterior, disminuyendo el contenido de Ca2+ libre en el citosol a menos de 0.1 µM, cuatro órdenes de magnitud menores que la concentración en plasma de 1 mM. El canal de Gardos, que extrae K+ en forma dependiente de Ca2+, tiene un papel importante en el control del volumen. El agua entra y sale a través de un canal de agua conocido como CHIP 28 (proteína de membrana integral que forma el canal de 28 kd) o acuaforina. Otros aniones intracelulares importantes son el 2,3-DPG y la hemoglobina, ninguno de los cuales atraviesa la membrana celular. Cuando la concentración de Ca2+ libre en el citosol aumenta a 0.3 µM, el canal se activa y causa una pérdida neta de K+. Si esta pérdida no se corrige, el eritrocito se deshidratará.4
CAMBIOS DE FORMA
La carencia de ATP, el acúmulo de ión calcio o el tratamiento con lisolecitina o con compuestos anfipáticos aniónicos, transforman el eritrocito normal o discocito en una célula espiculada crenada, equinocito o célula erizo [ver figura 1]. El calcio, solo o combinado con una proteína fijadora de calcio, la calmodulina, puede inducir modificaciones en la forma del equinocito. Si el proceso equinocítico persiste o progresa, las puntas del equinocito se fragmentan y se pierden componentes de la membrana, sobre todo de la banda 3 y fosfolípidos. Esta pérdida causa reducción en el área de superficie, con disminución en la relación superficie-volumen y formación de esferoequinocitos poco deformables.
PRINCIPIOS DE FLUJO SANGUINEO
Los principales determinantes del flujo sanguíneo son el hematócrito, la concentración plasmática de proteínas como fibrinógeno e inmunoglobulinas, que influyen en el grado de formación de rouleaux o agregación, la deformabilidad de los eritrocitos, el calibre de los vasos sanguíneos y la proporción de fuerzas de cizallamiento o de corte (la relación entre la velocidad de flujo y el radio del tubo). A las bajas fuerzas de cizallamiento que existen en las vénulas poscapilares los eritrocitos tienden a agruparse en masas asimétricas, con un aumento consecuente en la viscosidad y resistencia al flujo de la sangre.
ENVEJECIMIENTO Y MUERTE CELULAR
En la médula ósea el reticulocito pierde en forma progresiva su ARN residual durante un periodo de 4 días posteriores a la expulsión nuclear, y a partir de ese momento no puede ya realizar síntesis proteica. El cotransporte activo de K+-Cl- sirve para reducir su volumen. Con un ensamble proteico completo en la membrana, el eritrocito maduro entra a la circulación y sobrevive durante un periodo de 100 a 120 días.5 La muerte de los eritrocitos es un fenómeno que depende de la edad y puede relacionarse con fuerzas mecánicas y químicas que la célula encuentra en la circulación. A medida que envejece, el eritrocito pierde agua y superficie, disminuye la relación entre volumen y superficie y aumenta la CMHC, deteriorándose la deformabilidad de la célula. Además, la disminución de la actividad enzimática reduce la capacidad para soportar las presiones metabólicas. El envejecimiento puede manifestarse por cambios en la superficie del eritrocito, como pudiera ser una disminución en la densidad o tipo de carga superficial o la aparición de un neoantígeno de envejecimiento, quizá un grupo de banda 3 [ver figura 2], que se une a inmunoglobulinas específicas y componentes del complemento.6 A través de estos cambios el eritrocito viejo indica su incapacidad al sistema reticuloendotelial, lo que desencadena su eliminación por los macrófagos.
En condiciones fisiológicas, poco menos del 1 por ciento de los eritrocitos son destruidos diariamente y sustituidos por una cantidad prácticamente idéntica de nuevas células. Todos los días, un varón de 70 kg de peso con un volumen sanguíneo aproximado de 5 litros destruye y sustituye cerca de 50 ml de sangre total, que contiene aproximadamente 22 ml de eritrocitos. Como la hemoglobina equivale a un tercio del peso total de cada eritrocito, la reposición de estas células requiere la síntesis diaria de 7 g de hemoglobina. La médula ósea del adulto normal puede aumentar fácilmente su producción eritroide en 5 veces. Después de cualquier situación que ocasione anemia intensa y prolongada, la producción eritroide se puede elevar 7 u 8 veces. Sin embargo, el aporte de hierro representa un límite importante en la reposición de los eritrocitos: tres cuartas partes del hierro que se utiliza para la síntesis de eritrocitos en un día proviene de células que fueron destruidas el día anterior.
Características generales de las anemias hemolíticas
En términos generales, la severidad de una anemia está determinada por la velocidad de destrucción de los eritrocitos y por la capacidad de la médula ósea para producirlos. Cuando una persona tiene una médula normal el tiempo de vida de los eritrocitos puede disminuirse de 120 días a 20 días sin causar anemia o ictericia; sin embargo, en dichos casos se encontrará reticulocitosis importante.
La mayor parte de las formas de hemólisis son extravasculares, el sistema reticuloendotelial detecta las alteraciones de los eritrocitos en su membrana y los retira de la circulación. En circunstancias excepcionales, en las que el daño a los eritrocitos es devastador, como en algunas formas de lisis mediada por complemento, o en circunstancias en las que el sistema reticuloendotelial no puede eliminar el gran volumen de células dañadas, se desarrolla lisis intravascular y esto causa hemoglobinemia. La hemoglobina liberada hacia el plasma es degradada a dímeros alfa-beta, que se unen a la haptoglobina. Los complejos hemoglobina-haptoglobina son retirados por el sistema reticuloendotelial. Cuando se excede la capacidad de unión de la haptoglobina, los dímeros alfa-beta pasan al filtrado glomerular. Algunos de los dímeros alfa-beta se excretan directamente en la orina, produciendo hemoglobinuria, mientras que otros son captados y procesados por las células del túbulo renal. Varios días después de un episodio de hemólisis intravascular pueden excretarse células de los túbulos renales que contienen hierro. Es posible diagnosticar la hemosiderinuria identificando dichas células con tinción de azul de Prusia. La hemoglobina libre en el plasma se puede disociar en globina y hemina. La hemina puede unirse a la hemopexina y llegar a las células del túbulo renal en esa forma, o se puede unir a la albúmina plasmática, produciendo metemalbuminemia.
La hemólisis intravascular en un ejemplo notable de destrucción de los eritrocitos y puede producir anemia severa en forma aguda. Además, las partículas de la membrana eritrocítica liberada al plasma pueden actuar como estímulos potentes para el desarrollo de coagulación intravascular diseminada.
Cuando la hemólisis es crónica suelen desarrollarse cálculos de pigmento en la vesícula. Cuando un paciente que está compensando la hemólisis sufre una infección que altera en forma importante la actividad eritroide de la médula ósea,7 el nivel de hemoglobina puede disminuir en forma dramática, una condición denominada crisis aplástica. La hemólisis severa aguda también es causa de insuficiencia renal aguda y la hemólisis intravascular aguda puede provocar coagulación intravascular diseminada.
Las causas de hemólisis se clasifican como extracorpusculares o intracorpusculares. Las causas intracorpusculares, que son esencialmente defectos del eritrocito, incluyen alteraciones de la membrana celular, alteraciones metabólicas y trastornos de la estructura o biosíntesis de la hemoglobina. Las causas extracorpusculares representan elementos anormales dentro del lecho vascular del paciente que atacan y destruyen a los eritrocitos normales. Como los eritrocitos con defectos que causan hemólisis son anormales de manera intrínseca, siguen mostrando un tiempo de supervivencia característicamente acortado cuando se transfunden a receptores normales. De los defectos intracorpusculares el único que no es hereditario es la hemoglobinuria paroxística nocturna.
Defectos en la membrana de los eritrocitos
TRASTORNOS DEL METABOLISMO DEL SODIO Y DEL AGUA
Hidrocitosis (Estomatocitosis hereditaria)
La hidrocitosis es un trastorno hereditario que suele presentarse en etapas tempranas de la vida en forma de una anemia hemolítica parcialmente compensada, en ocasiones con bazo palpable. El VCM suele estar elevado. El frotis periférico demuestra estomatocitos [ver figura 4]. El flujo pasivo de Na+ y K+ aumenta mucho. La Na+-K+ATPasa es insuficiente y la concentración de cationes y el agua celular aumentan, provocando un incremento en el VCM y la disminución en la relación entre la superficie y el volumen. Algunos enfermos con hidrocitosis tienen un defecto en uno de los componentes de la membrana del eritrocito en la región de la banda 7. La esplenectomía puede mejorar la anemia. Los estomatocitos parecen adherirse en forma más ávida que los eritrocitos normales, un dato que puede explicar el reporte reciente de mayor frecuencia de eventos tromboembólicos.8
|
|
|
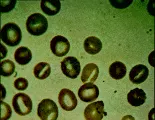

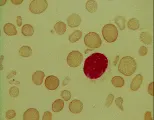
La xerocitosis, otro padecimiento hemolítico hereditario, se caracteriza por un defecto de membrana que origina pérdida de cationes y deshidratación celular. La salida de K+ excede la entrada de Na+, lo que ocasiona la pérdida de agua. Los pacientes presentan hemólisis variable compensada. La esplenomegalia no es una característica importante. El resultado del frotis periférico es variable, mostrando células en blanco de tiro, estomatocitos, equinocitos o los llamados budines de hemoglobina (i.e., hemoglobina acumulada alrededor de la célula). La CMH aumenta. Debido a que estas células rígidas son eliminadas en muchas partes del sistema reticuloendotelial, la esplenectomía no es muy útil.9
ALTERACIONES DE LAS PROTEINAS
Eliptocitosis hereditaria
Existen quizá 250 a 500 casos de eliptocitosis hereditaria por un millón de habitantes.10 En estos pacientes pueden observarse tres variedades morfológicas: (1) eliptocitosis hereditaria común, (2) eliptocitosis hereditaria esferocítica y (3) eliptocitosis hereditaria estomatocítica.10 La mayoría de los pacientes con eliptocitosis hereditaria común son heterocigotos para este trastorno autosómico dominante y solo tienen eritrocitos elípticos o, en el peor de los casos, hemólisis compensada. Los homocigotos pueden tener anemia hemolítica severa muy descompensada.
Al aplicar fuerzas de cizallamiento, los discocitos asumen una forma elíptica y cuando se elimina la fuerza recuperan su forma discoide. Se ha propuesto que los defectos de memebrana en la eliptocitosis hereditaria interfieren con la recuperación de la forma normal. Este defecto de membrana parece consistir en una lesión del citoesqueleto que afecta a la espectrina. En la mayoría de los pacientes los heterodímeros de espectrina no se asocian entre sí normalmente para formar tetrámeros u oligómeros [ver figura 2]. Otros defectos incluyen a la banda 4.1 o la interacción entre la anquirina y la banda 3. Las membranas de los eritrocitos de los pacientes con eliptocitosis hereditaria son invariablemente frágiles.
El diagnóstico se realiza en un paciente con hemólisis extravascular intracorpuscular con eliptocitos. Debe establecerse un patrón de herencia autosómico dominante porque la eliptocitosis puede observarse en la deficiencia severa de hierro, en los trastornos mieloproliferativos y mielodisplásicos y, en ocasiones, en las deficiencias de cobalamina y folato.10 La prueba de fragilidad osmótica suele ser normal. La esplenectomía ha sido útil en los pacientes con eliptocitosis hereditaria común.
Piropoiquilocitosis hereditaria
El síndrome de piropoiquilosis hereditaria (autosómico recesivo), una variedad de eliptocitosis hereditaria, produce hemólisis grave en niños pequeños. Es causado por una mutación anormal de alfa o beta espectrina. El frotis de sangre muestra microcitosis extrema y una extraordinaria variación en el tamaño y forma de los eritrocitos [ver figura 4]. La esplenectomía puede disminuir la magnitud de la hemólisis.
Esferocitosis hereditaria
La esferocitosis hereditaria se trasmite con carácter autosómico dominante y afecta alrededor de 220 personas por cada millón en todo el mundo. Se ha descrito una variante autosómica recesiva rara.
El dato característico de la enfermedad es la disminución en el área de superficie del eritrocito, que asume una forma microesferocítica y por lo tanto no puede deformarse lo suficiente para pasar a través de la vasculatura esplénica. Esto causa atrapamiento de los eritrocitos y hemólisis, con un aumento compensatorio en la eritropoyesis. Los defectos de la membrana ocasionan formación de vesículas en la misma ante condiciones de depleción metabólica. Estas vesículas de la membrana contienen fosfolípidos de la bicapa junto con proteínas transmembrana asociadas [ver figura 2]. Las lesiones moleculares subyacentes parecen consistir en deficiencia de la espectrina, la espectrina-anquirina, la banda 3 y la banda 4.210,11
Alrededor del 25 por ciento de los pacientes con esferocitosis hereditaria tienen hemólisis totalmente compensada sin anemia, y se diagnostican solo cuando un padecimiento concomitante, como una infección o embarazo, aumenta la velocidad de la hemólisis o reduce la capacidad compensatoria de la médula ósea.
Otros pacientes pueden sufrir anemia leve, cálculos de pigmentos biliares en la vesícula, úlceras en las piernas y ruptura esplénica. Las infecciones ordinarias del aparato respiratorio pueden precipitar crisis aplásticas, en especial la infección por parvovirus.7 El diagnóstico es sugerido por el predominio de microesferocitos en el frotis de sangre periférica [ver figura 4], una CMHC de 35 g/dl o mayor, reticulocitosis, ictericia leve, esplenomegalia y antecedentes familiares positivos. El diagnóstico se confirma por medio de una prueba de fragilidad osmótica con incubación de 24 horas. Una prueba de Coombs negativa en un paciente con antecedentes familiares positivos para esferocitosis hereditaria descarta el diagnóstico de anemia hemolítica autoinmune adquirida.
La esplenectomía erradica las manifestaciones clínicas del padecimiento, incluso las crisis aplásticas.
HEMOGLOBINURIA PAROXISTICA NOCTURNA
La hemoglobinuria paroxística nocturna (HPN) es un trastorno clonal de la hematopoyesis causada por una deficiencia en la proteína de anclaje de membrana fosfatidilinositol glican de clase A (PIG-A). Provoca muchas manifestaciones, incluyendo anemia hemolítica. Suele aparecer en la edad adulta y se debe a una mutación somática en las células tronco de la médula ósea.12
Los eritrocitos humanos normales modulan el ataque del complemento por lo menos por medio de tres proteínas que se fijan a la membrana: DAF, proteína fijadora de C8 (C8BP) y MIRL. Todas estas proteínas tienen deficiencia variable en los eritrocitos de los pacientes con HPN, con quimerismo variable en los diferentes pacientes. 12 En ausencia de la proteína de anclaje GPI, todas las proteínas que usan este anclaje de la membrana serán deficientes. La síntesis defectuosa de GPI afecta a las células tronco, por lo que los pacientes con HPN tienen neutropenia y trombocitopenia y pueden desarrollar pancitopenia, mielodisplasia o leucemia mieloide aguda.
Manifestaciones clínicas
Los episodios agudos de hemólisis intravascular se caracterizan por aparecer como complicaciones en pacientes con hemólisis crónica. En forma típica el paciente se percata de la hemoglobinuria al orinar después de dormir.14,15 Las oclusiones venosas recurrentes causan embolias pulmonares y trombosis de las venas hepática y mesentérica, quizá por liberación de factores trombogénicos o por activación de las plaquetas por complemento. En ocasiones se piensa erróneamente que estos pacientes con trombosis tienen trastornos psicosomáticos, pues se quejan de episodios de dolor abdominal y de espalda, sin presentar datos clínicos que indiquen la causa. En estos casos la anemia y la hemólisis asociada pueden ser muy leves, y los episodios de hemólisis no necesariamente se correlacionan con las crisis dolorosas.
Un estudio de 80 pacientes con HPN indicó que la supervivencia promedio fue de 10 años.14 La causa de la muerte relacionada con la HPN fue trombocitopenia, hemólisis, trombosis o anemia aplástica . Es interesante que el 15 por ciento de los pacientes presentó remisión espontánea. En este estudio las tromboembolias venosas recurrieron en sitios poco usuales y de gran riesgo, como la vena hepática (síndrome de Budd-Chiari), la vena cava inferior, las venas cerebrales y mesentéricas y el pulmón (embolia pulmonar).14 Ocurrió aplasia en cinco de los 80 pacientes. En casos raros puede ocurrir pérdida prolongada y severa de hierro como resultado de hemosiderinuria crónica, produciendo deficiencia de hierro; sin embargo, otros pacientes han desarrollado hemocromatosis asociada a transfusiones.15
Se ha desarrollado leucemia mieloide aguda durante la evolución de la HPN. En una serie la incidencia fue de solo tres casos en 80 pacientes, en otra, que incluyó 220 pacientes, la incidencia de síndromes mielodisplásicos fue de 5 por ciento y la de leucemia aguda de 1 por ciento.15
Diagnóstico
Se debe considerar el diagnóstico de HPN en cualquier caso de hemólisis crónica o episódica, o de tromboembolia venosa recurrente, en especial si el trombo ocurrió en un sitio como la vena cava inferior o el sistema porta-mesentérico, o si produjo un síndrome de Budd-Chiari. Las evidencias de hemólisis intravascular, como hemoglobinemia, disminución de la haptoglobina sérica, aumento de la metahemalbumina sérica, hemoglobinuria o hemosiderinuria, sugieren el diagnóstico. Un dato importante es la asociación de hipoplasia medular con hemólisis. Los eritrocitos de los pacientes con HPN no muestran alteraciones morfológicas. El diagnóstico con la prueba de Ham, que mide el grado de hemólisis en suero acidificado, es confiable, pero la prueba ha sido sustituida por pruebas específicas basadas en análisis de fluorescencia en células activadas (FACS) usando anticuerpos que evalúan en forma cuantitativa a las proteínas DAF y MIRL (CD59) en la superficie del eritrocito.16
Tratamiento
En la HPN la anemia puede ser tan severa (hemoglobina < 8 g/dl) que el paciente necesita transfusiones en forma regular.15 Por lo tanto, la elección del componente por transfundir es crucial. Se considera que la infusión de productos sanguíneos que contengan complemento puede aumentar la hemólisis. También es posible que la infusión de glóbulos bláncos (que generalmente están presentes en una unidad de paquete globular) del donador a un receptor HLA-inmunizado pueda provocar la reacción antígeno-anticuerpo que activa el complemento por la vía clásica. En tal caso, puede ser útil el uso de unidades especiales pobres en leucocitos.
Se puede intentar un tratamiento de prueba con prednisona (60 mg diarios con disminución rápida o 20 a 60 mg cada tercer día), que puede ayudar a reducir las necesidades de tranfusión al aliviar la anemia. La utilidad de la esplenectomía es muy dudosa. La cirugía es un problema en pacientes con HPN porque la estasis y el trauma agravan la hemólisis y las oclusiones venosas. Ante la necesidad de llevar a cabo una cirugía debe considerarse la anticoagulación profiláctica con warfarina en el periodo posoperatorio inmediato.
Los pacientes con HPN a menudo presentan deficiencia de hierro. Sin embargo, la simple administración de hierro para corregir este defecto agrava a menudo la hemólisis, puesto que el tratamiento con hierro produce una estirpe de células nuevas, muchas de las cuales son susceptibles a la lisis mediada por complemento. La transfusión previa puede ayudar a evitar este problema porque disminuye el impulso eritropoyético en la médula ósea.
La trombocitopenia por mala producción de plaquetas puede requerir transfusión de las mismas .14 El síndrome de Budd-Chiari y la trombosis de la vena cava inferior requieren de un diagnóstico pronto y tratamiento rápido con heparina, seguido de administración de warfarina a largo plazo. Si la heparinización es ineficaz puede usarse tratamiento trombolítico (v.gr., estreptocinasa). En un caso de síndrome de Budd-Chiari,17 se inició trombolisis con estreptocinasa sistémica con una dosis de impregnación de 250,000 unidades durante 40 minutos, seguido de una dosis de mantenimiento de 250,000 unidades en infusión IV continua por 12 horas. Los niños y los adolescentes con HPN y anemia aplástica pueden ser candidatos a trasplante alogénico de médula ósea.15
Alteraciones del metabolismo del eritrocito
DEFECTOS DE LA CAPACIDAD REDUCTORA
El glutatión reducido (GSH) y las coenzimas reducidas nicotinamida-adenina dinucleótido (NADH) y nicotinamida-adenina dinucléotido fosfato (NADPH) proveen la capacidad reductora del eritrocito [ver tabla 1]. Cuando las reservas eritrocíticas de estos materiales son inadecuadas, la hemoglobina y las proteínas asociadas a la membrana pueden oxidarse, causando la producción de cuerpos de Heinz, que consisten predominantemente en productos de degradación oxidativa de la hemoglobina. Estos cuerpos pueden verse con microscopía por contraste de fases o con microscopía ordinaria de luz después de teñir con metil violeta [ver figura 4]. Los eritrocitos que contienen cuerpos de Heinz son rígidos, por lo que el sistema reticuloendotelial los retira en forma selectiva de la circulación.
Defectos en la síntesis de glutatión
Las deficiencias de ciertas enzimas que participan en la síntesis de GSH causan hemólisis y ataques oxidativos a los eritrocitos [ver figura 5]. Varios informes han descrito familias cuyos miembros tienen síntesis imperceptible de GSH y sufren hemólisis asociada con producción de cuerpos de Heinz. La deficiencia de glutatión peroxidasa al parecer contribuye a la hemólisis en los recién nacidos.
|
| Figura 5 |
| Metabolismo de la glucosa en el eritrocito |
DEFICIENCIA DE GLUCOSA-6-FOSFATO DESHIDROGENASA
La glucosa-6-fosfato deshidrogenasa (G6PD) es la primera enzima en la vía pentosa fosfato o hexosa monofosfato. Cataliza la conversión de NADP+ a NADPH, un agente reductor muy potente. El NDPH es un cofactor para la glutación reductasa, por lo que tiene un papel importante en la protección de la célula contra el ataque oxidativo. Por lo tanto, los eritrocitos deficientes en G6PD son susceptibles a la oxidación y, eventualmente, a la hemólisis.18,19
La deficiencia de G6PD es uno de los trastornos más comunes en el mundo; aproximadamente el 10 por ciento de los varones afroamericanos en los Estados Unidos están afectados, al igual que un gran número de africanos negros y algunos habitantes del litoral mediterráneo. Se supone que el trastorno alguna vez confirió cierta ventaja selectiva contra la malaria endémica, pero se desconoce el mecanismo de este efecto.
El gen que codifica la G6PD está en la banda q28 del cromosoma X; los hombres sólo poseen un gen para esta enzima y, por lo tanto, los afectados por el trastorno son hemicigotos. Las mujeres resultan afectadas con mucho menor frecuencia, ya que deben ser portadoras de dos genes G6PD defectuosos para manifestar una enfermedad de la misma severidad que los varones. Sin embargo, la expresión de un gen G6PD defectuoso no suele enmascararse completamente en las mujeres heterocigotas; de hecho, dichas mujeres muestran actividad muy variable de la G6PD. De acuerdo con la hipótesis de inactivación del cromosoma X, o hipótesis de Lyon-Beutler,19 las mujeres heterocigotas en el locus ligado al cromosoma X para el gen que codifica la G6PD tienen 2 líneas celulares, una que contiene un cromosoma X activo con un gen que codifica la producción de G6PD normal, y la otra que contiene un cromosoma X activo con un gen que codifica la producción de G6PD deficiente. El azar determina parcialmente la proporción de las dos líneas celulares, lo que a su vez controla la severidad clínica del defecto.
Existen tres tipos clínicos del padecimiento: el tipo I, que es la anemia hemolítica no esferocítica congénita crónica poco común, el tipo II, en el que la deficiencia de la enzima es severa pero la hemólisis tiende a ser episódica, y el tipo III, la variante más común, en la que la deficiencia enzimática es moderada y la hemólisis es causada por un evento oxidante. La clonación y secuencia del gen G6PD ha aclarado muchos aspectos complejos de la literatura que describía más de 300 variantes de la deficiencia de G6PD.19
Ocurre hemólisis en las personas con deficiencia de G6PD después de la exposición a un medicamento o sustancia que produce una tensión antioxidante. La ingestión o exposición a habas puede causar hemólisis intravascular devastadora (conocida como favismo) en los pacientes con deficiencia de G6PD, pero ocurre por lo general solo en los que tienen la variedad Mediterránea. Las habas contienen isouramilo y divicina, dos agentes reductores potentes. El peróxido de hidrógeno producido puede causar daño oxidativo importante. Al parecer, los oxidantes de las habas pueden también atacar la membrana y disminuir la barrera normal a la entrada de Ca2+. La combinación de niveles muy aumentados de Ca2+ y oxidación intracelular reduce el nivel de proteasas de eritrocitos. Estas proteasas protegen de modo ordinario contra el acúmulo de productos oxidados de la hemoglobina. El ataque oxidante a los tioles de la membrana causa oxidación de las proteínas de la membrana. Esto produce una célula rígida en la que la hemoglobina está confinada a una parte del citosol y la otra parte aparece como un fantasma claro (i.e., célula de media ampolla, o cruzada) [ver figura 6]. Estos defectos de memebrana provocan hemólisis extra e intravascular.18 También puede ocurrir hemólisis en los individuos con deficiencia de G6PD e infecciones severas, cetoacidosis diabética e insuficiencia renal. Es interesante que la deficiencia de G6PD produce, por mecanismos desconocidos, cierta protección contra la cardiopatía isquémica y varias enfermedades vasculares.20
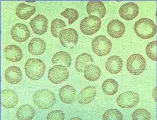
|
| Figura 6 |
| Eritrocitos en hemiámpula |
La anemia hemolítica caracterizada por la aparición de cuerpos de Heinz después de la administración de ciertos medicamentos sugiere la posibilidad de deficiencia de G6PD [ver tabla 2]. El cloranfenicol y la quinina causan hemólisis en pacientes con la variedad Mediterránea de deficiencia de G6PD, pero no en pacientes con el tipo A-. La dapsona se usa cada vez más como profilaxis para la neumonía por Pneumocystis carinii en pacientes infectados por VIH. Por lo tanto, es importante buscar en los posibles receptores deficiencia de G6PD con las pruebas enzimáticas estándar porque la dapsona es capaz de inducir hemólisis de tipo oxidativo [ver tabla 2].
|
|
||||||||||||||
|
Otros trastornos que hay que considerar en el diagnóstico diferencial de la hemólisis oxidativa incluyen la hemoglobinopatía inestable, la enfermedad por hemoglobina M y las deficiencias de las enzimas esenciales para el metabolismo del glutatión. Una prueba de detección de G6PD o un ensayo enzimático directo suelen resolver la duda. Sin embargo, los pacientes con deficiencia de G6PD del tipo A- y reticulocitosis de rápida aparición pueden tener un nivel casi normal de G6PD. En dichos casos es mejor repetir los exámenes cuando la cuenta de reticulocitos vuelva a lo normal.
El evitar medicamentos que puedan producir hemólisis es crucial en el manejo. El favismo agudo requiere apoyo circulatorio, mantener un buen flujo sanguíneo renal y transfusiones con eritrocitos que no sean deficientes en G6PD. El médico también debe estar alerta por el posible inicio de coagulación intravascular diseminada.
DEFECTOS EN LA GLUCOLISIS
La serie de reacciones que constituyen la vía glucolítica [ver figura 5] generan diversos productos, incluyendo ATP, que tienen varias funciones esenciales en el metabolismo del eritrocito [ver tabla 1]. Existen defectos que afectan la vía glucolítica principal (vía de Embden-Meyerhof) y que generalmente interfieren con la producción de ATP.
La piruvato cinasa (PC) cataliza la formación de piruvato, reacción asociada con la síntesis de ATP. Después de la deficiencia de G6PD, la deficiencia de PC es la segunda de las enzimopatías hereditarias en orden de frecuencia. El síntoma inicial es la hemólisis de severidad variable, y se acompaña por ictericia leve y, en ocasiones, por esplenomegalia palpable. El examen de un frotis de sangre periférica suele mostrar eritrocitos normales, pero en algunos casos éstos presentan innumerables espículas. Pueden ocurrir crisis aplásticas.7
La hemólisis congénita no esferocítica plantea la posibilidad de deficiencia de PC. Un ensayo enzimático establece el diagnóstico. Se debe considerar la opción de esplenectomía en los pacientes que requieren transfusiones.
La deficiencia de glucosafosfato isomerasa es la tercera enzimopatía en frecuencia como causa de hemólisis. Otras son muy raras, pero se cuenta ya con pruebas de escrutinio y específicas para deficiencias de enzimas como hexocinasa, fosfofructosinasa, triosa fosfato isomerasa, fosfoglicerato cinasa y aldolasa.
DEFECTOS EN EL METABOLISMO DE LOS NUCLEOSIDOS
En la anemia hemolítica asociada con deficiencia de pirimidina 5'-nucleotidasa, persiste un puntilleo grueso basofílico en los eritrocitos maduros, supuestamente porque la deficiencia enzimática evita la degradación del ARN del reticulocito. Este acúmulo causa expansión de la reserva total de nucleótidos en los eritrocitos a cinco veces el normal. Los nucleótidos de pirimidina se acumulan y los de adenina disminuyen. El acúmulo y agregación de ribosomas no degradados contribuye también al puntilleo basofílico. La glucólisis se altera por un mecanismo aún no bien aclarado.
Trastornos de la hemoglobina
CLASIFICACION DE LAS HEMOGLOBINOPATIAS
Las hemoglobinopatías clínicamente importantes se clasifican en cinco categorías, con base en el defecto de fondo. Los defectos principales son los siguientes:
1. La hemoglobina tiende a convertirse en gel o cristalizarse (v.gr., anemia de células falciformes o enfermedad por hemoglobina C).
2. La hemoglobina es inestable (v.gr.,las anemias congénitas con cuerpos de Heinz).
3. La hemoglobina tiene propiedades anormales de captación de oxígeno (v.gr., el trastorno por hemoglobina Chesapeake).
4. La hemoglobina se oxida fácilmente a metahemoglobina (v.gr., metahemoglobinemia).
5. Las cadenas de hemoglobina se sintetizan en proporciones desiguales (v.gr., las talasemias).
ANEMIA DE CELULAS FALCIFORMES
La anemia de células falciformes es un padecimiento causado por la sustitución del aminoácido valina por glutamina en la sexta posición de la cadena beta de la hemoglobina. El gen se hereda con patrón autosómico recesivo y se encuentra en el 8 a 10 porciento de los afroamericanos en los Estados Unidos y en menor porcentaje en personas con ascendencia Mediterránea oriental, de la India o de Arabia Saudita. Los eritrocitos de individuos con el rasgo falciforme (HbAS) tienen una concentración de hemoglobina S menor del 50 por ciento, y con frecuencia menor del 30 por ciento. Con raras excepciones, el rasgo falciforme suele ser asintomático. La enfermedad se desarrolla en personas que son homocigotas para el gen falciforme (HbSS). En un paciente con anemia falciforme el 70 a 98 por ciento de la hemoglobina es de tipo S. Alrededor del 0.2 por ciento de los afroamericanos tienen anemia de células falciformes.
Varias teorías intentan explicar porqué la anemia de células falciformes se distribuye en todo el mundo y ha persistido en las poblaciones humanas. La coincidencia geográfica del gen falciforme y del paludismo falciparum endémico sugiere que la heterocigocidad falciforme confiere una ventaja protectora contra el paludismo.21 Tanto la resistencia inherente a la invasión como la mayor deformación de los eritrocitos parasitados interfieren con el crecimiento del parásito.21
Los análisis de endonucleasas de restricción indican que la mutación del gen de la anemia de células falciformes pudo aparecer en forma espontánea en por lo menos cinco sitios geográficos. Estas variantes son denominadas Senegal, Benin, Centroafricana (o Bantú), Saudi-Asiática, Camerún e India (que puede ser la misma que la Saudi-Asiática). Estas variantes son importantes desde el punto de vista clínico porque algunas se asocian con mayor producción de cadenas de globina gama (y por tanto niveles mayores de HbF) y otras se asocian con más frecuencia con el gen de la talasemia alfa-2 [ver adelante, Talasemias]. Cualquiera de estas asociaciones alivia ciertos aspectos del proceso de deformación falciforme.21
Fisiopatología
Dos características clínicas son las más importantes en la anemia de células falciformes: (1) la hemólisis crónica que es estable y moderadamente debilitante, y (2) las crisis vasoespásticas agudas y episódicas, que causan falla orgánica y son responsables de la mayor parte de la morbimortalidad de la enfermedad.
La HbS unida al oxígeno o al monóxido de carbono tiene una solubilidad casi normal. Sin embargo, cuando la molécula deja el oxígeno y cambia a su forma desoxi S, su solubilidad disminuye y tiende a polimerizarse en fibras tubulares largas, que inducen la deformación falciforme del eritrocito.22
Los análisis de cinética de la polimerización han aclarado mucho el conocimiento sobre la fisiopatología de la anemia de células falciformes. Puede considerarse que el polímero S de desoxihemoglobina está en equilibrio con las moléculas solubles circundantes de desoxihemoglobina S. Los factores que alteran este equilibrio afectan la magnitud de gelación de la hemoglobina. La reducción en la concentración de oxígeno u otros factores como la reducción en el pH o el aumento en la concentración de 2,3-DPG (ambos tienden a estabilizar la forma desoxi S) aumentan la gelación. Otro determinante importante de la polimerización en equilibrio es la concentración absoluta de la HbS y HbF. 22 La desoxihemoglobina S pura se polimeriza a 16 g/dl, una concentración muy por debajo de la CMHC habitual. La cinética de gelación es muy dependiente de la concentración de hemoglobina. Además, las células falciformes retienen un cotransportador activo K+-Cl- y tienen suficiente calcio intracelular para activar el canal de salida de Gardos23 [ver antes, Control de la hidratación y volumen]. Estos dos mecanismos actúan juntos para producir una población de eritrocitos falciformes muy densos con CMHC de hasta 50 g/dl. 23 La HbF inhibe la polimerización porque el residuo glutamina en la posición 87 de la cadena gama bloquea el contacto lateral de la fibra falciforme.23 Por lo tanto, los pacientes con valores altos de HbF, como los de la variante Saudi-Asiática de la anemia de células falciformes, tienen una enfermedad más leve [ver figura 7].21 Cuando estos factores, la hipoxemia y la CMHC, alcanzan un nivel crítico, ocurre polimerización después de un periodo de retraso variable.23
|
|
|
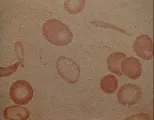
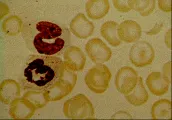

La mayoría de las células en la circulación venosa no son falciformes. Sin embargo, ocurrirá este cambio si el tiempo de retraso para la polimerización se acorta a menos de 1 segundo o si los eritrocitos son atrapados en la microcirculación. Algunos eritrocitos contienen hemoglobina falciforme polimerizada incluso en la circulación arterial y el tiempo de retraso para la gelación es mucho más corto en estas células precondicionadas y más rígidas.
La sensibilidad extrema del cambio falciforme al ambiente local ha llamado la atención hacia los factores celulares. La hiperosmolaridad extrema de la médula renal (1,200 mOsm) deshidrata los eritrocitos y aumenta la CMHC. En consecuencia, puede ocurrir un cambio falciforme suficiente para abolir la capacidad de concentración de la médula renal incluso en pacientes que solo tienen rasgo falciforme. Los eritrocitos falciformes tienen una mayor tendencia a adherirse a las células endoteliales24 que los eritrocitos normales. El grado de adhesividad parece correlacionar con la severidad de los episodios vaso-oclusivos. Esto se atribuye a la desorganización de la bicapa de fosfolípidos de la membrana, al moverse la fosfatidilserina hacia el exterior, quizá aumentando las manifestaciones tromboembólicas de la enfermedad falciforme.25 En la anemia de células falciformes parece existir también aumento en las células endoteliales circulantes, que expresan en forma anormal factor tisular y pueden proporcionar una base adicional para la ocurrencia de tromboembolias.26
Otra manifestación del daño en la membrana en las células falciformes es el cambio falciforme irreversible, que se mantiene a pesar de que la célula sea reoxigenada.27 Estas poblaciones poco deformables y deshidratadas derivan directamente de una subpoblación de reticulocitos con concentraciones bajas de HbF28 y son eliminadas principalmente en el sistema reticuloendotelial. Ocurre hemólisis con anemia variable. La anemia se tolera bastante bien, quizá por un mayor aporte de oxígeno por la oxihemoglobina S.
CRISIS FALCIFORMES
En contraste con la anemia, la crisis falciforme es una complicación potencialmente mortal de la enfermedad falciforme. La crisis falciforme, una expresión dolorosa y notable de oclusión vascular, muestra características que dependen mucho de factores reológicos. La disminución del hematocrito en la anemia de células falciformes disminuye la viscosidad sanguínea y tiene efecto protector. Por el contrario, el nivel de fibrinógeno plasmático, característicamente elevado y que parece ser una manifestación de la respuesta de fase aguda que ocurre en muchos pacientes con anemia de células falciformes en fase estable,29 aumenta la agregación de los eritrocitos y la viscosidad, en particular ante las fuerzas de cizallamiento bajas que se encuentran en la microcirculación. La rigidez del eritrocito falciforme, que es causada por la polimerización de la desoxihemoglobina S y por la CMHC elevada, es el elemento crucial para elevar la viscosidad sanguínea y precipitar la oclusión vascular.
Si disminuye el pH o se eleva la CMHC, si existe enfermedad microvascular o se prolonga el tiempo de tránsito capilar por la adherencia de eritrocitos al endotelio, se formarán racimos de células falciformes rígidas que ocluirán la microcirculación. Estos bloqueos causan infartos isquémicos y una secuencia amplificadora de oclusión inducida por estasis que puede evolucionar hasta una crisis falciforme. Las circulaciones porta, en la que la tensión de oxígeno es baja, como las del hígado o riñón, tienen especial riesgo de oclusión. Se ha postulado que la trombosis también participa en la oclusión falciforme.
Se desconoce el evento iniciador de la crisis falciforme, y tampoco está claro por qué algunos pacientes tienen crisis severas y otros no. El dolor de la crisis falciforme puede ser causado por necrosis avascular de la médula ósea. Entre los factores de riesgo que predisponen a una crisis dolorosa se incluye una concentración de hemoglobina mayor de 8.5 g/dl, embarazo, clima frío, y una cuenta alta de reticulocitos.
La coherencia de otros genes influye en la gravedad de la anemia de células falciformes. La frecuencia de heterocigosidad para alfa-talasemia entre afroamericanos es de 30 por ciento.21 Las personas con alfa-talasemia tienen concentraciones más altas de hemoglobina, menores cuentas de reticulocitos, menor CMHC, menor VCM y menos eritrocitos densos que las personas que tienen solo anemia de células falciformes. Estos cambios, en especial la menor CMHC, deben beneficiar a los pacientes con los dos padecimientos, aunque la interacción es compleja. Los pacientes pueden tener mayor expectancia de vida y quizá menos úlceras en miembros inferiores, pero sufren más crisis y mayor necrosis aséptica del hueso.30 La combinación de deficiencia de G6PD y anemia de células falciformes no causa efectos benéficos ni dañinos.31
Diagnóstico
El aspecto clínico del paciente y un frotis de sangre que muestre células falciformes, células con aspecto de "flor de Nochebuena" y eritrocitos con cuerpos de Howell-Jolly son muy compatibles con anemia de células falciformes. Los cuerpos de Howell-Jolly representan restos citoplásmicos de cromatina nuclear que generalmente son retirados por la acción modeladora del bazo. Las cuentas de plaquetas y leucocitos suelen encontrarse elevadas. A menos que el enfermo se encuentre en crisis aplástica, lo que produciría ausencia total de normoblastos, la médula muestra hiperplasia eritroide. El diagnóstico se confirma realizando una "preparación falciforme": incubando una gota de sangre con solución fresca de metabisulfito de sodio al 2 por ciento y midiendo la proporción de células falciformes inmediatamente y una hora después. Para confirmar el diagnóstico, los equipos comerciales, como el Sickledex, se basan en la insolubilidad relativa de la hemoglobina S en soluciones amortiguadoras de fosfato 1.0 M. Sin embargo, la prueba definitiva para la anemia de células falciformes es la electroforesis de hemoglobina, que indica los porcentajes relativos de hemoglobina S y de hemoglobina F. Todas las pruebas mencionadas tienen la misma utilidad para examinar a los miembros de la familia en busca del rasgo de células falciformes. Los pacientes que son heterocigotos para los genes de la HbS y de la beta-talasemia pueden aparentar ser homocigotos para la HbS. Otras variedades de hemoglobina que causan transformación falciforme se observan con muy poca frecuencia. También pueden emplearse métodos basados en el estudio del ADN para determinar la alteración genética específica e identificar la subpoblación a la que pertenece el paciente.21
Otras consideraciones terapéuticas
Síntomas musculoesqueléticos Se presenta necrosis aséptica de la cabeza femoral (osteonecrosis) en alrededor del 10 por ciento de los pacientes, en especial en los que tienen también talasemia-alfa. La artroplastía ha resultado relativamente ineficaz, en parte por la presencia de hueso duro adyacente, que interfiere con la colocación de la prótesis, y por el mayor riesgo de infección.52
Enfermedad biliar La colelitiasis es una complicación común que ocurre en el 30 a 70 por ciento de los pacientes, algunos de los cuales desarrollan signos y síntomas de colecistitis.33 La frecuencia de colecistitis o de obstrucción del conducto biliar común es motivo de debate. En un estudio de 77 pacientes que se sabía tenían cálculos vesiculares de 1 a 10 años de evolución, solo cinco fueron sometidos a colecistectomía.34 Otro estudio encontró que de 604 procedimientos en la anemia de células falciformes, el 40 por ciento fueron por colecistitis y colelitiasis.33 Los pacientes que recibieron transfusiones hasta un nivel de hemoglobina de 10 g/dl antes de la cirugía tuvieron mejor evolución posoperatoria.33 Si va a realizarse una colecistectomía, debe esperarse hasta que pase la crisis dolorosa y el procedimiento debe practicarse por vía laparoscópica.33
Enfermedad hepática Las complicaciones hepáticas incluyen hepatopatía congestiva secundaria a insuficiencia cardiaca y hepatitis viral por las transfusiones frecuentes. La transformación falciforme en el hígado, órgano que extrae una gran cantidad de oxígeno de la sangre circulante, también puede producir hepatopatía. Es frecuente que los niveles séricos de bilirrubina excedan los 30 mg/dl en la colestasis intrahepática y las alteraciones en la coagulación pueden causar complicaciones hemorrágicas y muerte.
Enfermedad pulmonar Las complicaciones pulmonares agudas son una causa importante de morbimortalidad e incluyen infección local y oclusiones vasculares en el lecho arterial pulmonar. Es difícil la diferenciación entre las dos complicaciones. La oclusión vascular causada por un émbolo pulmonar puede ser imposible de distinguir de la oclusión in situ. Los problemas pulmonares agudos incluyen embolia grasa pulmonr por necrosis isquémica de la grasa de la médula.35 En un estudio de 1,722 episodios de dolor torácico agudo en 939 pacientes, los adultos en el estudio estaban afebriles pero tuvieron disnea, calosfríos y dolor torácico y en por lo menos una extremidad.36 El examen físico con frecuencia no mostró alteraciones torácicas. La tensión arterial de oxígeno (PaO2) estaba disminuida, en promedio en 71 mm Hg (en el 25 por ciento menor de 60 mm Hg). La tasa de muerte en adultos fue de 4.3 por ciento y fue precedida por un valor más bajo de hemoglobina, mayor cuenta de leucocitos y afección multilobar. La autopsia de 16 pacientes mostró que nueve tenían embolia pulmonar y embolia grasa y quizá el 20 por ciento tuvo infecciones bacterianas. Los infartos óseos de las vértebras torácicas contribuyeron en forma importante al dolor.37 Por lo general, el tratamiento debe incluir espirometría incentiva,37 búsqueda de infección y tratamiento con antimicrobianos, analgesia cuidadosa, remplazo agresivo de líquidos y consideración de lavado broncoalveolar para identificar la infección microbiana o los macrófagos llenos de lípidos típicos de la embolia grasa. Se requiere vigilancia cuidadosa que debe incluir medición periódica de la oxigenación y transfusiones cuando sea clínicamente necesario. Uno de los beneficios más importantes del tratamiento con hidroxiurea es su capacidad para disminuir la frecuencia del síndrome torácico agudo.38,39 Los niños pueden requerir profilaxis suplementaria con penicilina.40 Debido a que el uso rutinario de la vacuna neumocócica ha disminuido la incidencia de neumonía neumocócica en pacientes con anemia de células falciformes, Haemophilus influenzae es ahora responsable de un porcentaje relativamente mayor de casos de neumonía entre estos pacientes; de esta manera, la selección del antibiótico debe reflejar este cambio.
Complicaciones urogenitales La pérdida de agua, debida a la incapacidad para concentrar la orina, puede aumentar el proceso de transformación falciforme. El medio extremadamente hipertónico de la médula renal deshidrata los eritrocitos falciformes, induciendo transformación severa y destrucción de los vasos rectos. Se presenta hematuria y necrosis papilar. Estas complicaciones se observan especialmente en pacientes con el rasgo falciforme y en aquéllos que tienen enfermedad falciforme con hemoglobina C. El defecto en la capacidad de concentración renal parece depender de la cantidad de polímero de hemoglobina S en las células, por lo que es menos severa en los pacientes que tienen también variantes de talasemia alfa.41
Las complicaciones incluyen acidosis tubular renal, hipercalemia y proteinuria. El tratamiento con enalapril reduce la proteinuria, lo que sugiere la presencia de un componente de hipertensión capilar glomerular.42 La insuficiencia renal, con empeoramiento asociado de la anemia, contribuye a la muerte de alrededor de la quinta parte de los pacientes mayores de 40 años que tienen enfermedad falciforme homocigota.
El priapismo es una complicación muy dolorosa de la anemia de células falciformes y puede causar impotencia. Cuando las medidas conservadoras no producen mejoría en un periodo de 24 horas los autores recomiendan la exsanguineotransfusión de eritrocitos.
Complicaciones oculares Los principales problemas oculares que se asocian con la anemia de células falciformes son retinopatía, hemorragia del vítreo y neovascularización. Se recomienda el examen oftalmológico anual a estos pacientes. Se está investigando la eficacia de la fotocoagulación por láser en el tratamiento de las complicaciones oftalmológicas inducidas por la enfermedad.
Propensión a las infecciones Los pacientes con anemia de células falciformes son hipoesplénicos y tienen alteraciones del sistema del complemento. Una actividad opsonizadora deficiente contra Salmonella parece explicar que presenten propensión a desarrollar infecciones por este germen, incluyendo osteomielitis.
Trastornos neurológicos Las alteraciones neurológicas relacionadas incluyen eventos cerebrovasculares, hemorragia subaracnoidea y pérdidas funcionales aisladas compatibles con oclusión focal. Es probable que la patogenia de la oclusión de las arterias cerebrales de gran calibre sea diferente de los eventos oclusivos de la microcirculación que ocurren en los lechos capilares hipóxicos. El defecto subyacente parece ser el daño al endotelio vascular, seguido de proliferación extensa de la íntima y trombosis del lecho vascular lesionado.30 Un estudio multicéntrico reciente de 4,082 pacientes reveló una prevalencia de eventos cerebrovasculares (ECV) de 4 a 5 por ciento y una incidencia de 0.61 por 100 pacientes-año.43 De los ECV, el 54 por ciento fueron infartos, 34 por ciento hemorragias, 11 por ciento episodios de isquemia cerebral transitoria (ICT), y el 1 por ciento tenían datos tanto isquémicos como hemorrágicos. De los pacientes que sobrevivieron, la tasa de recurrencia de ECV fue de 14 por ciento. La mortalidad fue de 11 por ciento y virtualmente todos los pacientes que fallecieron tenían ECV hemorrágicos.
En un estudio prospectivo en el que se empleó la ultrasonografía transcraneal Doppler para identificar a los niños con riesgo de evento cerebrovascular, el tratamiento con atención estándar o transfusiones (para disminuir la concentración de HbS a < 30 por ciento) causó 10 ECV y un hematoma intracerebral en los 65 sujetos controles, mientras que el grupo transfundido tuvo solo 1 ECV (p < 0.002). El estudio se terminó antes de lo programado.44 El éxito del mismo destaca muchos aspectos serios sobre la necesidad de equipos ultrasonográficos para el manejo adecuado, la duración óptima del tratamiento de transfusión, las consecuencias inevitables de la hemocromatosis transfusional [ver adelante, beta-talasemia mayor (anemia de Cooley)], y la necesidad de administrar sangre específica de la etnia para minimizar las reacciones por alotransfusión, el deseo de los pacientes y familiares para aceptar el tratamiento transfusional y el papel del trasplante alogénico de médula ósea como una alternativa potencial.44,45
Crisis aplástica La crisis aplástica disminuye rápidamente los niveles de hemoglobina y hematocrito y produce reticulocitopenia, como sucede en cualquier estado hemolítico crónico. Se ha demostrado que el parvovirus causa crisis aplásticas,7 lo mismo que la necrosis de médula ósea.46
Anestesia general
La hipoxemia y la estasis vascular que pueden ocurrir durante la anestesia general aumenta la deformación falciforme y puede causar una crisis en el periodo posoperatorio. Un análisis de casi 4,000 pacientes asoció 12 muertes con 1,079 procedimientos. Ocurrieron más complicaciones después de la anestesia regional que de la general.47 Un programa de transfusión simple para aumentar el nivel de hemoglobina a 10 g/dl fue tan eficaz como los programas preoperatorios más agresivos para disminuir la tasa de complicaciones.48
Pronóstico
Un estudio de casi 4,000 pacientes ha proporcionado nueva información sobre la expectancia de vida y las causas de muerte en la enfermedad por células falciformes.49 Aunque se pensaba que la mayoría de los pacientes con anemia de células falciformes moría alrededor de los 20 años de edad, en la actualidad es claro que la edad promedio en el momento de la muerte es de 42 años para los hombres y 48 para las mujeres. Esta expectancia de vida representa una reducción de 25 a 30 años comparando con la población afroamericana general. De las causas de muerte identificadas, solo 18 por ciento fueron el resultado de falla orgánica, principalmente nefropatía, insuficiencia cardiaca congestiva o las consecuencias de los eventos cerebrovasculares crónicos. Treinta y tres por ciento de los pacientes murieron durante las crisis de dolor agudo, con frecuencia asociado con síndrome torácico agudo y menos común con eventos cerebrovasculares. La presencia de talasemia-alfa no tuvo un efecto mesurable. Los factores que predijeron mala evolución fueron una cuenta de leucocitos mayor de 15,000/µl, niveles bajos de hemoglobina F y afección orgánica manifestada por enfermedad renal, síndrome torácico agudo y eventos neurológicos.
Tratamiento
El manejo debe enfocarse al tratamiento y prevención de las crisis falciformes. El tratamiento conservador estándar de las crisis falciformes consiste en un examen apropiado, seguido de reposo, hidratación y analgesia. En aquellos pacientes en quienes se ha demostrado acidosis se debe inducir la alcalinización leve administrando solución de bicarbonato, que se prepara añadiendo un ámpula de bicarbonato de sodio a 1 L de solución glucosada al 5 por ciento, o solución salina medio normal. La solución de bicarbonato debe administrarse a una velocidad de 5 a 7 ml/kg/hora durante las primeras 4 horas, y después a una velocidad de 4 ml/kg/hr durante las siguientes 20 horas. La utilidad de la administración de O2 en pacientes con PaO2 normal y sin problemas cardiopulmonares no se ha demostrado.
Dolor El manejo de las crisis dolorosases la principal preocupación en el 10 al 20 por ciento de los pacientes con anemia de células falciformes. La necrosis avascular de la médula ósea produce dolor intenso que dura hasta 8 a 10 días. La necesidad de alivio del dolor causa en ocasiones habituación o adicción. Debido a que existen pocas maneras objetivas de evaluar las crisis falciformes, el médico puede no saber si la demanda de narcóticos es manifestación de un comportamiento de adicción.
Los pacientes con anemia de células falciformes deben recibir analgésicos orales para usarlos en casa en un intento por abortar las crisis de dolor en su inicio. Puede comenzarse con antinflamatorios no esteroides (AINE) como naproxen (500 mg v.o.) o ketorolaco (10 mg) y si no son útiles, éstos pueden ser seguidos de una combinación de analgésico-narcótico como acetaminofén e hidrocodona o aspirina y oxicodona. El uso de adyuvantes como la difenhidramina (50 mg v.o.) o loracepam (1 a 2 mg v.o.) puede calmar al paciente y quizá antagonizar las acciones de la histamina liberada.50 Si estas medidas, quizá repetidas cada 6 horas, no controlan el dolor, el paciente suele requerir tratamiento parenteral. La atención del paciente por su médico es preferible a confiar en un médico no conocido de un servicio de urgencias.50 El paciente requiere una evaluación rápida para investigar posible infección, síndrome torácico agudo, infarto óseo u otras complicaciones, y el dolor debe tratarse con 10 mg de morfina intravenosa junto con 50 mg de difenhidramina intramuscular cada 2 horas. o 4 mg de hidromorfona intramuscular junto con 50 mg de difenhidramina intramuscular cada 2 horas. Si no existe alivio del dolor o éste es inadecuado 30 minutos después de la primera dosis, puede administrarse el 50 por ciento de la dosis inicial de opioides, con vigilancia estrecha de la frecuencia respiratoria, en especial si llega a 10 respiraciones/min. Algunas unidades han usado analgesia controlada por el paciente con buenos resultados. Es importancia continuar la analgesia parenteral en intervalos regulares, proporcionando dosis mayores para eliminar el dolor. Es probable que el paciente requiera también un laxante y un antiemético, como proclorperacina (10 mg v.o o IM). Si el paciente responde, puede enviarse a casa con morfina de liberación controlada por vía oral. Si el dolor continúa por más de 8 a 12 horas es probable que deba ser internado para administrar dosis mayores de analgesia, líquidos parenterales y observación.50
Medidas generales para impedir la transformación falciforme El mayor conocimiento de la cinética de transformación falciforme sugiere algunos prospectos futuros para el tratamiento de la anemia de células falciformes. La disminución de la CMHC disminuye la gelación. Otro tratamiento consiste en bloquear la salida de K+ dependiente de Ca2+ (canal de Gardos) [ver antes, Control de la hidratación y el volumen].51 Un tratamiento semejante consiste en tratar a los pacientes con anemia de células falciformes con dosis grandes de pidolato de magnesio, que aumenta los niveles de Mg en los eritrocitos, inhibe el cotransportador de K+-Cl- y reduce el número de eritrocitos falciformes deshidratados y densos.52
Se han propuesto tratamientos que intentan corregir o compensar el defecto falciforme interviniendo a nivel del gen. La presencia de 20 a 30 por ciento de HbF en los eritrocitos falciformes retrasa la gelación en una proporción de 103 a 104, por lo que pudiera existir un mecanismo que activara los genes que controlan la síntesis de hemoglobina fetal y que disminuyera la gravedad de la enfermedad falciforme.49 La hidroxiurea produce un aumento en los reticulocitos F y en la HbF. Un estudio sobre hidroxiurea en fase III, con una dosis inicial de 15 mg/kg/día, mostró que los pacientes tratados con esta sustancia tuvieron menos crisis dolorosas, menos ingresos hospitalarios por crisis y menos episodios de síndrome torácico doloroso, requeriendo menos transfusiones que los pacientes que recibieron placebo.38 No se observó efecto sobre la frecuencia de muerte o eventos cerebrovasculares. El efecto benéfico se observó después de 8 semanas de tratamiento y se asoció con aumento en el VCM y la proporción de células F, junto con disminución en los neutrófilos y en la adhesión de los eritrocitos falciformes a las células endoteliales.53 También se realizan estudios con otros agentes, como el butirato, que puede aumentar la producción de cadenas-gama, aumentando así las concentraciones de HbF e interfiriendo con la gelación.54 Sin embargo, un estudio en el que se usó un tratamiento de 10 semanas de butirato de arginina por vía intravenosa no encontró beneficios hematológicos.55 El trasplante alogénico de médula ósea de un hermano compatible puede producir la curación o sustituir la anemia de células falciformes por solo un rasgo falciforme. En 22 pacientes seleccionados en forma cuidadosa, principalmente con eventos cerebrovasculares o episodios recurrentes de síndrome torácico agudo, el trasplante de médula ósea causó curación aparente en 15, con dos fallecimientos (9 por ciento); el resto de los pacientes tuvo complicaciones como fracaso del injerto.56
Papel del tratamiento transfusional por tiempo prolongado Se ha observado que el tratamiento transfusional prolongado evita los eventos cerebrovasculares (ver antes).44 Algunos investigadores han demostrado que las transfusiones preventivas pueden disminuir o eliminar las crisis dolorosas, los episodios de síndrome torácico agudo, la infección bacteriana o la hospitalización.57 Sin embargo, otros clínicos advierten sobre los peligros de la sobrecarga de hierro, la hepatitis por transfusión, los problemas con el acceso venoso y la aloinmunización de eritrocitos.37 Los estudios futuros podrán identificar mejor la utilidad del tratamiento transfusional crónico.
Consejo genético, anticoncepción y embarazo
Los elementos claves que deben considerarse al brindar consejo genético en pacientes con rasgo o enfermedad falciforme son: la morbilidad tan importante para niños y adultos con la enfermedad, el aumento del riesgo materno en el embarazo, y la frecuencia relativamente alta de muerte fetal. Los anticonceptivos orales también pueden representar un riesgo especial para las mujeres con anemia de células falciformes, porque producen un ligero aumento en la incidencia de evento cerebrovascular, tromboembolia venosa e infarto del miocardio. Sin embargo, los datos que han aparecido sobre la seguridad de los anticonceptivos orales que contienen menos de 50 µg de estrógenos sintéticos indican que las pacientes con enfermedad falciforme pueden tomar dichos medicamentos con seguridad razonable. Otra alternativa para algunas pacientes es el uso del implante anticonceptivo Norplant. En cualquier caso, el embarazo o el aborto en pacientes con enfermedad falciforme entrañan un riesgo elevado.58
Los peligros del embarazo en la enfermedad falciforme incluyen complicaciones pulmonares, mayor incidencia de infecciones de vías urinarias, hematuria, preclampsia y muerte materna. Al parecer, la hipoxia pélvica y la sobrecarga vascular asociadas con el embarazo causan una mayor transformación falciforme, con sus complicaciones. Las oclusiones vasculares en la placenta pueden causar muerte fetal y bajo peso al nacer.
Los clínicos experimentados tienen diferentes conceptos sobre la atención de la paciente embarazada con enfermedad falciforme. Algunos recomiendan solamente cuidado conservador meticuloso, mientras que otros indican transfusiones profilácticas. Un estudio controlado ha indicado que no existen ventajas con el uso de transfusiones profilácticas.58
Es posible que las parejas con enfermedad o rasgo falciforme quieran tener hijos a pesar de los riesgos fetales y maternos asociados. En Estados Unidos se presentan alrededor de 4,000 a 5,000 embarazos de este tipo al año.59 La muestra de vellosidades coriónicas (que proporciona ADN para análisis en el primer trimestre del embarazo), las técnicas de amplificación del ADN y las sondas que identifican el cambio específico de nucléotido en la anemia de células falciformes pueden proporcionar un diagnóstico prenatal relativamente seguro y muy confiable.59
VARIANTES DE LA ENFERMEDAD FALCIFORME
Rasgo falciforme
Por lo general los pacientes con el rasgo falciforme no presentan manifestaciones clínicas y llevan vidas normales y saludables. Se llegan a presentar algunas complicaciones: hipostenuria y hematuria renal, bacteriuria y pielonefritis durante el embarazo. Ocurre infarto esplénico cuando existe hipoxia y en altitudes elevadas, en especial en personas con anemia de células falciformes que no son de raza negra. También se ha identificado que el rasgo falciforme constituye un factor de riesgo para muerte súbita durante el entrenamiento militar básico,60 y la muerte ocurre por paro cardiaco inexplicable, golpe de calor, estrés por calor o rabdomiolisis. La mayor edad se ha correlacionado con mayor riesgo de muerte súbita. Sin embargo, estos eventos han ocurrido en condiciones muy extremas: actividad física muy extenuante en personas sin entrenamiento y en ocasiones en altitudes elevadas o con clima de calor extremo. Por lo general las personas con rasgo falciforme que están habituadas a realizar actividad física no tienen mayor riesgo de muerte súbita. Por ejemplo, entre los jugadores afroamericanos de futbol, los que tienen rasgo falciforme no tienen mayor riesgo de muerte súbita que otros jugadores.61 Las alternativas de tratamiento para la hematuria renal incluyen la administración de diuréticos, bicarbonato parenteral, transfusiones o ácido épsilon-aminocapropico.
Talasemia beta-falciforme
Cuando se combina el rasgo falciforme con un defecto en el gen de la talasemia beta se produce una enfermedad muy similar a la anemia de células falciformes. El gen de la talasemia beta reduce la velocidad de síntesis de la cadena ßa, lo que da como resultado que predominen las cadenas de ßs en pacientes con el rasgo falciforme. Los eritrocitos de los pacientes contienen cantidades variables de HbS, HbA, HbA2 y HbF. Cuando sólo existe una pequeña cantidad de hemoglobina A la enfermedad es tan grave como la anemia de células falciformes, pero en las variedades en las que la HbA constituye el 25 por ciento o más de la hemoglobina total y se produce menos HbS la enfermedad es leve. El diagnóstico se confirma si existe un nivel elevado de hemoglobina A2 en la electroforesis de hemoglobina, una historia familiar con el antecedente de talasemia y el gen falciforme. El tratamiento para la talasemia beta-falciforme es el mismo que se indica para la anemia de células falciformes.62
Enfermedad falciforme con hemoglobina C
En la enfermedad falciforme con hemoglobina C (HbSC) se forman cantidades casi iguales de hemoglobina S y C. Del 1 al 2 por ciento de la hemoglobina es HbF y también hay pequeñas cantidades de HbA2, pero no hay HbA. Los eritrocitos son microcíticos y la CMHC es alta, lo que facilita la cristalización y agregación de la HbC.63
Del 30 a 50 por ciento de los pacientes con este trastorno no están anémicos y sólo tienen reticulocitosis moderada. Los pacientes pueden no ser identificados hasta que el trastorno se manifiesta en forma de una crisis vaso oclusiva durante una cirugía, embarazo o urgencia médica.63 También ocurren esplenomegalia, retinopatía proliferativa y necrosis aséptica de los huesos largos. El frotis periférico [ver figura 8] contiene células falciformes irreversibles además de células en diana, estomatocitos y eritrocitos con depóstitos eccéntricos de hemoglobina, que probablemente representen agregados o cristales de hemoglobina C. El diagnóstico se confirma por electroforesis de hemoglobina.63 El tratamiento es el mismo que se indica en la anemia de células falciformes. Se administró hidroxiurea, 1,000 mg/día a seis pacientes con enfermedad de HbSC y presentaron aumento en el VCM, reducción en los llamados reticulocitos de estrés, incremento en la hemoglobina y quizá reducción en la densidad celular. Aunque no es definitivo, este pequeño estudio sugiere que la hidroxiurea es benéfica para los pacientes con enfermedad HbSC.64 La expectancia de vida es casi 20 años mayor que para los pacientes con enfermedad por HbSS.49
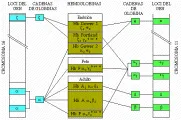
|
| Figura 8 |
| Codificación de las cadenas de globina |
OTRAS HEMOGLOBINOPATIAS
Enfermedad por hemoglobina C
La molécula de hemoglobina C es a2ß26glu-lis, y la mutación del gen pareció originarse en un solo sitio en Africa Occidental, dentro de Burkina Faso.63 La presencia de esta hemoglobina casi no produce enfermedad en el estado heterocigoto, pero causa hemólisis moderada compensada y esplenomegalia palpable en el estado homocigoto.
La insolubilidad relativa de la hemoglobina C es responsable de los cambios patológicos que se asocian con su presencia. Es probable que la hemoglobina C interactúe con el cotransportador K+-Cl- para mantenerlo activo, en condiciones normales este cotransportador se inactiva en los eritrocitos después del periodo de reticulocito. Como consecuencia se presenta pérdida de K+, deshidratación celular con CMHC elevada y después agregación y cristalización de la hemoglobina C poco soluble.63 La insolubilidad relativa de la hemoglobina C causa rigidez de los eritrocitos, fragmentación y pérdida del material de la membrana; estos procesos producen los microesferocitos que se observan en el frotis de sangre periférica [ver figura 4].
Las células en diana, un dato morfológico importante, forman cerca del 80 por ciento de los eritrocitos. Se encuentran cristales de hemoglobina C en el estado de oxihemoglobina, que se disuelven cuanto los eritrocitos se desoxigenan. Esta propiedad parece explicar la ausencia de episodios vasculares oclusivos en pacientes con enfermedad por hemoglobina C.
El diagnóstico de enfermedad por hemoglobina C se basa en los datos del frotis de sangre periférica y en la ausencia de datos de deficiencia de hierro o talasemia, y se confirma por medio de electroforesis de hemoglobina. No se requiere tratamiento.
Enfermedad por hemoglobina E En la enfermedad por hemoglobina E la lisina es sustituida por ácido glutámico en la posición 26 de la cadena de la beta-globina. El rasgo de hemoglobina E se encuentra principalmente en el sureste de Asia. Se le ha prestado atención clínica en los Estados Unidos por la llegada de inmigrantes de esta región, en los que la incidencia del rasgo es de alrededor del 10 por ciento.
La hemoglobina E es inestable al oxidarse porque ocurre una mutación en el sitio de contacto alfa1beta1. El menor contenido de ßE es resultado de una menor producción de ARN mensajero (ARNm) ßE y de producción de ARNm ßE inestable.
Los pacientes heterocigotos para la hemoglobina E tienen valores de hemoglobina normales, microcitosis y no sufren esplenomegalia. La electroforesis revela que el 70 por ciento de la hemoglobina es HbA, el 25 por ciento HbE y el resto es HbA2 o HbF. En la electroforesis la HbE corre junto con la HbA2, y los laboratorios sin experiencia pueden reportarla como HbA2. La clave consiste en que la HbA2 nunca es más del 8 por ciento. Un reporte de laboratorio de concentración de HbA2 de 25 por ciento obliga a revisar los datos.
Los pacientes homocigotos para la HbE tienen anemia leve, con concentraciones de hemoglobina de alrededor de 12 a 13 g/dl, VCM bajo y eritrocitosis sin reticulocitosis, mostrando microcitos y células en diana. La electroforesis muestra solo HbE. No ocurre hemólisis crónica. Deben evitarse los medicamentos oxidantes, como la dapsona, tanto en heterocigotos como en homocigotos.
Ocurre un problema clínico grave cuando un paciente es heterocigoto doble para la HbE y para el rasgo de talasemia beta, porque la combinación produce el cuadro clínico de talasemia beta intermedia, con anemia severa, esplenomegalia y en ocasiones necesidad de transfusión de sangre e incluso médula ósea.65
Hemoglobinopatías inestables
Muchas variedades individuales constituyen las hemoglobinopatías inestables. La inestabilidad de la hemoglobina se origina de las sustituciones de aminoácidos que privan a la molécula de su grupo hem, alteran el sitio receptor del hem, debilitan la unión entre cadenas alfa y beta, o debilitan la estructura de la subunidad. El resultado es la ruptura y precipitación de la hemoglobina, en particular cuando se somete a un fenómeno oxidante. La hemoglobina precipitada forma cuerpos de Heinz, que se observan incluso en personas heterocigotas para la variante de hemoglobina inestable. Debido al efecto dañino de los cuerpos de Heinz sobre el eritrocito y su membrana, ocurre hemólisis importante incluso en el estado heterocigoto.
La presencia de hemólisis no esferocítica parcialmente compensada sugiere el diagnóstico de una hemoglobinopatía inestable. Se observan cuerpos de Heinz en los eritrocitos de pacientes que han sido sometidos a esplenectomía. Los eritrocitos de pacientes que no han sido esplenectomizados presentan cuerpos de Heinz al ser incubados con tinción azul brillante de cresilo. El diagnóstico diferencial de una hemoglobinopatía debe hacerse con la deficiencia de G6PD; por lo general puede descartarse este trastorno por medio de una prueba directa para buscar esta enzima.
El tratamiento incluye evitar el uso de medicamentos oxidantes. Se puede considerar la esplenectomía cuando la hemólisis es severa y no se compensa en forma adecuada.
Hemoglobina con afinidad anormal para el oxígeno
Se debe considerar la posibilidad de que exista una hemoglobina con mayor afinidad para el oxígeno en el diagnóstico diferencial de la eritrocitosis inexplicable, particularmente si hay asociación familiar . La electroforesis de hemoglobina puede demostrar el trastorno, pero en los casos sospechosos se prefiere tener como base para el diagnóstico la curva de disociación de la oxihemoglobina. La hemoglobina Chesapeake y la hemoglobina Rainier son ejemplos de tipos de hemoglobina con afinidad aumentada.
Los casos raros de hemoglobina con afinidad disminuida para el oxígeno, como la hemoglobina Kansas, representan mutaciones. Los pacientes con hemoglobinopatía con afinidad disminuida para el oxígeno pueden presentar cianosis debido a una mayor liberación del oxígeno.
Metahemoglobinemia
La metahemoglobina es un producto de oxidación de la hemoglobina cuya molécula de hierro está en forma férrica; de esta manera, la molécula no puede fijar oxígeno en forma reversible. En condiciones normales el 1 por ciento de la hemoglobina se encuentra en estado férrico, y entre el 0.5 y el 3 por ciento de la desoxihemoglobina se oxida en forma espontánea a metahemoglobina cada día. El poder reductor normal de los eritrocitos [ver tabla 1] mantiene el balance oxidación-reducción. El sistema enzimático que reduce el 95 por ciento de la metahemoglobina a hemoglobina incluye dos proteínas, la NADH-citocromo b5 reductasa y la citocromo b5, y también requiere de NADH. (El NADH es generado por la gliceraldehido fosfato deshidrogenasa [ver figura 5].) Como el nombre lo indica, la NADH-citocromo b5 reductasa, una enzima que contiene flavina, usa NADH para reducir a la citocromo b5. La citocromo b5 reducida reduce a la metahemoglobina.66,67 Se han descrito mutaciones nuevas en el gen afectado.68
En la forma enzimopénica hereditaria de metahemoglobinemia, los pacientes son homocigotos o heterocigotos dobles para la deficiencia de NADH-citocromo b5 reductasa. Estos enfermos tienen apariencia azul incluso cuando solo el 10 por ciento de su hemoglobina es metahemoglobina, pero no están enfermos y toleran con facilidad concentraciones de metahemoglobina hasta de 25 por ciento o más. Por el contrario, la presencia de alrededor de 5 g/dl o hemoblogina desoxigenada reducida causa cianosis. Los pacientes con esta forma de metahemoglobinemia no sufren hemólisis ni requieren tratamiento. La determinación de NADH-citocromo b5 reductasa, realizada en un laboratorio especial, puede establecer el diagnóstico. Si se desea puede usarse azul de metileno, en dosis de 100 a 300 mg/día por vía oral, aunque causa molestias urinarias.66 El azul de metileno transfiere electrones del NADPH a la metahemoglobina.
La otra forma hereditaria de metahemoglobinemia es causada por la hemoglobina M, de la que existen cuatro variantes poco frecuentes. Cada variante contiene una sustitución de aminoácido en el sitio receptor del hem, que permite que se formen enlaces estables entre el hierro hem y la cadena lateral de aminoácidos. Estas uniones mantienen a la hemoglobina en la forma Fe3+, incapaz de unir oxígeno e inaccesible a las enzimas reductoras. El trastorno se observa solo en los heterocigotos, en los que el 30 por ciento de la hemoglobina es anormal según la electroforesis. Se observa cianosis al nacimiento. La hemólisis es mínima y no se requiere tratamiento.
TALASEMIAS
Las talasemias tienen distribución mundial, y en muchas regiones estos trastornos son responsables de perturbaciones médicas, sociales y económicas importantes. La distribución mundial de las talasemias se superpone en forma estrecha con la distribución del paludismo, lo que indica que las formas heterocigotas de talasemia proporcionan protección contra esta enfermedad.69 Las técnicas de biología molecular han ayudado a dilucidar la fisiopatología de estos síndromes.69 En la actualidad, la mejor compresión de la base molecular de estos trastornos ha permitido que los investigadores realicen diagnósticos prenatales fidedignos. Usando estos datos, los padres pueden tomar decisiones razonables e informadas con respecto al resultado de embarazos en los cuales el feto se encuentre severamente afectado.
Fisiopatología
En un individuo normal, la síntesis de cadenas alfa y beta está meticulosamente coordinada para la producción de la HbA del adulto (a2b2). En comparación, los pacientes con talasemia suelen presentar alteraciones en la síntesis de cadenas normales de globina. No obstante, en ocasiones pueden presentarse síndromes parecidos a la talasemia como resultado de la producción disminuida de una cadena estructuralmente anormal.70 Debido a que una de las cadenas de globina existe en cantidad reducida, la cadena excedente se acumula en la célula precursora eritroide en desarrollo y se produce toxicidad. En consecuencia, las células eritroides mueren en la médula ósea, dando origen a una forma clásica de eritropoyesis ineficaz.
Las talasemias beta se caracterizan por menor producción de cadenas de globina beta, con acúmulo y agregación de cadenas alfa no pareadas. Estos agregados de cadenas alfa se precipitan, causando disminución de la síntesis de ATP, pérdida de potasio, disminución del ácido siálico superficial, y otras anormalidades de la superficie, incluyendo anticuerpos IgG dirigidos contra los residuos de galactosa, que pueden ser detectados por los macrófagos. Los eritrocitos afectados tienen forma anormal y son relativamente rígidos. La barrera de la membrana para el Ca2+ se rompe, lo que permite la entrada de este ión. Se encuentran acúmulos anormales de globina, quizá agregados de cadenas alfa, unidos al citoesqueleto de la membrana. Es posible que éstos sean responsables de la apoptosis observada en los precursores eritroides de la médula y de la mayor rigidez e inestabilidad de la membrana que se observa en los eritrocitos en sangre periférica. Estos agregados de globina alfa mantienen funcionando el cotransporte K+-Cl-, lo que provoca la deshidratación variable que se observa en las formas severas de talasemia beta.71 Las alteraciones destructivas de la membrana pueden ser causadas en parte por oxidación local desencadenada por el acúmulo de hemicromos en la membrana, como en la enfermedad de células falciformes. La reducción global en la síntesis de hemoglobina por la célula es responsable de la hipocromía y de la formación de células en diana.
Los pacientes con talasemia alfa muestran acúmulos en exceso de cadena beta que, si están presentes en cantidades suficientes, forman tetrámeros ß4 (HbH) [ver figura 8]. Dichos tetrámeros tienen una elevada afinidad por el oxígeno y son inestables, por lo que se agregan en situaciones de oxidación, como en las infecciones. Los acúmulos de globina beta se fijan también al esqueleto de la membrana eritrocitaria, pero producen lesiones diferentes que los agregados de globina alfa. En las talasemias alfa severas la eritropoyesis ineficaz es menor y la destrucción periférica de eritrocitos más intensa. Los eritrocitos en la talasemia alfa son rígidos, pero a diferencia de la talasemia beta grave, las membranas en la talasemia alfa severa son más estables de lo normal. También, a diferencia de la talasemia beta, los eritrocitos de la talasemia alfa están hidratados en exceso.71
Tanto las talasemias alfa como las beta se caracterizan por presentar grados variables de anemia. Esta variación se atribuye a grados diversos de eritropoyesis ineficaz y a la destrucción prematura de los eritrocitos circulantes. Cuando la anemia es grave la hipoxemia asociada induce una notable eritropoyesis compensadora que causa expansión de la cavidad medular, osteopenia y crecimiento de los órganos reticuloendoteliales; se pueden originar tumores en los sitios de actividad eritropoyética extramedular. La destrucción de eritroblastos y eritrocitos predispone a colelitiasis e ictericia obstructiva. Los pacientes con formas más graves de talasemia necesitan ser transfundidos con regularidad, lo que puede ocasionar finalmente sobrecarga de hierro y complicaciones tales como cardiopatía, hepatopatía y endocrinopatía.
Genética molecular
Los defectos que causan un desequilibrio en la síntesis de cadenas incluyen deleción de genes, anormalidades en la transcripción y en la traducción, e inestabilidad del ARNm que dirige la síntesis de globina, o de la globina misma. Los genes que controlan la síntesis de las cadenas alfa y no alfa de la hemoglobina se localizan en los cromosomas 11 y 16 [ver figura 8]. Durante la vida fetal y adulta no existen sustitutos para la cadena alfa de la hemoglobina, lo que tiene implicaciones clínicas importantes. En comparación, las cadenas delta y las gama suplementan la cadena beta normal en la vida adulta, y las cadenas gama (de las cuales hay dos variedades, que difieren solamente en un aminoácido) pueden servir como globina no alfa en la vida fetal y neonatal.73
Talasemia beta clínica
La síntesis deficiente de globina beta, característica de la talasemia beta, causa acúmulo de las cadenas alfa no complementarias. Esta disminución en la síntesis de cadenas beta puede ser consecuencia de un procedimiento nuclear anormal de transcripción de ARNm, de terminación prematura de la traducción, o de deleción genética. Un adelanto de importancia diagnóstica en la talasemia beta es el aumento compensatorio parcial de las cadenas delta y gama que produce aumento en los niveles de HbA2 (a2d2) o HbF (a2g2), respectivamente [ver figura 8]. Las variedades de talasemia beta producen tres síndromes clínicos: talasemia mayor beta, talasemia menor beta y talasemia intermedia.
Talasemia mayor beta (anemia de Cooley) La talasemia mayor beta se caracteriza por anemia severa que aparece en el primer año de vida. Se asocia con ictericia, hepatoesplenomegalia, expansión de la médula eritroide con cambios corporales secundarios incluyendo retraso en el crecimiento, y aumento en la susceptibilidad a las infecciones. La anemia es tan severa que se necesitan transfusiones en forma crónica. Estas ocasionan sobrecarga de hierro que, si no se trata, causa la muerte en la adolescencia por hemocromatosis cardiaca.
El diagnóstico no debe representar un problema; ninguna otra enfermedad se asemeja mucho a la anemia de Cooley. El frotis de sangre periférica muestra eritrocitos nucleados, eritrocitos hipocrómicos distorsionados y puntilleo basófilo, que representa acúmulos de ARN ribosomal [ver figura 7]. Las tinciones supravitales muestran acúmulos de cadenas alfa excedentes no complementarias.
La talasemia mayor beta es una enfermedad que se trasmite como una condición homocigota o heterocigota doble; ambos padres de un individuo afectado son portadores del rasgo de la talasemia. En la talasemia ßo, la variedad más grave, no se sintetizan cadenas beta, y solo se encuentra HbF y HbA2. La talasemia ß+ es menos severa, se caracteriza por cantidades pequeñas de cadenas beta y de HbA, además de HbF y HbA2. La talasemia delta beta es aún más moderada, y es ocasionada por la deleción de los genes que codifican la globina delta y beta. Esta mutación inhibe la producción de HbA2 y HbA, permitiendo solamente la síntesis de hemoglobina fetal.
El tratamiento consiste en transfusiones repetidas con el objeto de mantener la concentración de hemoglobina alrededor de 12 g/dl, evitando de esta manera complicaciones como insuficiencia cardiaca congestiva, sobrecarga de líquidos y deformidades esqueléticas. Por lo general se requiere esplenectomía para aumentar la supervivencia de los eritrocitos del paciente y de los transfundidos.74 Está indicada la vacunación con vacuna neumocócica por el riesgo de sepsis por neumococo después de la esplenectomía. La sobrecarga de hierro debe tratarse por medio de la administración de infusiones subcutáneas de desferroxamina, un agente quelante, antes de que ocurra exceso de hierro. La desferroxamina subcutánea, en dosis de 50 mg/kg/día puede lograr eliminación de 50 a 200 mg de hierro/día, pero solo si se infunde en forma continua durante 8 a 12 horas.75 Este tratamiento no solo evita la disfunción ventricular izquierda, sino que revierte las alteraciones ya establecidas.76 Los efectos benéficos de la quelación de hierro han mejorado el pronóstico de las personas con anemia de Cooley:76 ya no es inevitable que los pacientes mueran en la segunda década de la vida por arritmias e insuficiencia ventricular izquierda. Con el tratamiento actual con desferroxamina, el 61 por ciento de los pacientes nacidos antes de 1976 no han tenido enfermedad cardiovascular. Los pacientes que cumplen el esquema de tratamiento, en los que el valor de ferritina casi siempre es menor de 2,500 ng/ml tienen una tasa de supervivencia de 91 por ciento después de 15 años.77 Se ha probado un quelante oral de hierro, el deferiprone, comparando con la desferroxamina parenteral, pero no ha demostrado un control adecuado de la carga de hierro al medir los niveles de hierro hepático pre y postratamiento.78 Sin embargo, se recomienda que no se descarte su uso hasta contar con más datos.79
Se ha realizado trasplante de médula ósea usando hermanos HLA compatibles. Más de 600 pacientes han sido sometidos a este tipo de procedimiento con donadores normales o que tenían rasgo de talasemia beta.80 Algunos pacientes con hemoglobina E-talasemia beta tienen un fenotipo tan severo como el de la talasemia mayor beta y requieren el mismo tratamiento, incluyendo trasplante alogénico de médula ósea.65 La experiencia con el trasplante de células tronco de cordón umbilical es más limitado.81 Dependiendo de la condición del paciente en el momento del trasplante, la tasa de mortalidad relacionada al mismo fue de 5 a 19 por ciento, con una tasa de curación de 54 a 90 porciento.82 Otros enfoques para el tratamiento de la talasemia beta severa están aún en fase experimental.54,55,83,84
Talasemia beta menor (rasgo de talasemia beta) Por lo común, los pacientes con talasemia menor son heterocigotos para una mutación beta globina, y pueden o no presentar anemia leve. El frotis de sangre periférica muestra hipocromía notable y microcitos con puntilleo basófilo. En ocasiones se encuentra esplenomegalia.
El nivel de HbA2 se eleva por arriba del 5 por ciento en 90 por ciento de los pacientes y el nivel de HbF se eleva por arriba del 2 por ciento en el 50 por ciento de los casos. Este aumento en la hemoglobina fetal ocurre en proporciones variables en los eritrocitos (distribución heterocelular), como lo muestra la tinción de Kleihuaer Betke. Los pacientes con niveles más elevados de HbF tienen anemia menos grave. Los heterocigotos para la talasemia delta beta producen cantidades aumentadas de HbF con cantidades normales de HbA2.
Se debe descartar la anemia por deficiencia de hierro en el diagnóstico diferencial del rasgo de la talasemia beta. Por lo general es fácil distinguir los dos trastornos. Ambas enfermedades se asocian con hipocromia y microcitosis, pero la deficiencia de hierro produce hipoproliferación de eritrocitos, mientras que la talasemia menor causa sólo una reducción mínima en su número. Con un nivel de hemoglobina de 9 g/dl, los pacientes con deficiencia de hierro tienen una cifra de eritrocitos cercana a 3 millones/µl, mientras que los pacientes con el rasgo de talasemia tienen cifras de eritrocitos de alrededor de 5 millones /µl. La cifra de eritrocitos, relativamente normal en la talasemia, se utiliza como base para todas las fórmulas que usan índices para distinguir el rasgo de talasemia de la deficiencia de hierro. Si persisten dudas acerca del diagnóstico pueden usarse las mediciones del hierro sérico y de la capacidad de fijación de hierro o del nivel sérico de ferritina con el fin de distinguir estos trastornos. Sin embargo, es importante recordar que un paciente con el rasgo de talasemia también puede tener deficiencia de hierro como consecuencia de hemorragias vaginales, gastrointestinales o ambas.
Talasemia intermedia Como el término lo implica, la talasemia intermedia se caracteriza por manifestaciones clínicas de severidad moderada. Los pacientes con este síndrome tienen anemia importante, con niveles de hemoglobina tan disminuidos como de 6-7 g/dl, y muestran grados variables de hepatoesplenomegalia, pero es usual que no requieran de transfusiones en forma regular. No obstante, durante las infecciones u otras agresiones eritropoyéticas pueden necesitarse transfusiones en forma temporal.
Variedades parecidas a la talasemia beta La hemoglobinopatía asociada con la hemoglobina Lepore representa otra variedad de talasemia beta. Los pacientes que son homocigotos para este trastorno presentan anemia de Cooley o talasemia intermedia, y sus eritrocitos contienen sólo hemoglobina Lepore y HbF.70
Persistencia hereditaria de la hemoglobina fetal Los eritrocitos de pacientes que son heterocigotos para la persistencia hereditaria de hemoglobina fetal (PHHF) contienen alrededor del 50 por ciento de HbF, mientras que los homocigotos tienen el 100 por ciento de dicha hemoglobina. Hasta hace poco tiempo se suponía que los pacientes con PHHF se encontraban en buenas condiciones de salud y que presentaban anemia mínima o no tenían. Sin embargo, desde entonces se han descrito algunas variedades clínicas de la PHHF en las que hay anemia notable.
Talasemia alfa
Dos diferencias principales sirven para distinguir los genes de globina alfa y beta. Primero, no hay sustitutos fetales, neonatales o adultos para los genes alfa, y segundo, sólo hay dos genes de globina beta, pero cuatro de globina alfa, dos en cada cromosoma 16 [ver figura 8]. El genotipo de la globina alfa normal se designa con aa/ aa. Los pacientes portadores de la variedad talasemia alfa 1 muestran una deleción de dos genes de la cadena alfa del mismo cromosoma y por lo tanto, tienen el haplotipo--/ o alfa0. Esta deleción es común entre pacientes asiáticos. Los pacientes que tienen la variedad de talasemia alfa 2 han perdido un gen alfa en un cromosoma, y muestran el haplotipo -a / o alfa+. Aunque esta mutación es particularmente frecuente en negros, también se observa en poblaciones asiáticas y mediterráneas. Se han reconocido cinco síndromes clínicamente distintos: la hemoglobina Barts, o hidropesía fetal (--/--), la enfermedad por hemoglobina H (--/-a), la talasemia alfa 1 heterocigota (--/aa), la talasemia alfa 2 homocigota (-a /-a), y el síndrome del portador silencioso (-a / aa).
Hemoglobina de Barts Los niños con el síndrome de hemoglobina Barts son homocigotos para la talasemia alfa 1 (--/--), por lo que no producen cadenas alfa. Las cadenas gama no pareadas producen tetrámeros g4 (hemoglobina de Barts). Todos los niños con esta enfermedad nacen hidrópicos. Es común que los padres sean heterocigotos para la talasemia alfa 1 (--/aa).
Enfermedad por hemoglobina H El cuadro clínico de la enfermedad debida a hemoglobina H es el de una anemia hemolítica leve que se presenta en pacientes de origen asiático, del Medio Oriente o del Mediterráneo. Por lo general puede detectarse HbH que se precipita, al teñir con azul brillante de cresilo, en los eritrocitos recién extraídos del paciente. Los mecanismos moleculares que originan la enfermedad por hemoglobina H son heterogéneos. Algunos pacientes tienen un genotipo que sugiere la deleción de tres genes alfa, como sería el caso si el paciente fuera doblemente heterocigoto para la talasemia alfa 1 (--/) y talasemia alfa 2 (-a/), dando un genotipo
--/-a. La esplenomegalia es común. Los pacientes no suelen requerir de transfusiones regulares, pero puede necesitarse apoyo temporal cuando sufren infecciones u otros eventos oxidativos estresantes que causen precipitación de la HbH inestable y mayor hemólisis.
Talasemia alfa heterocigota La talasemia alfa 1 heterocigota (--/aa), genotipo común entre asiáticos, se caracteriza por anemia leve o ausente y la presencia en el frotis de sangre periférica de eritrocitos notablemente hipocrómicos y microcíticos. Los pacientes homocigotos para la talasemia alfa 2 (-a/-a), un genotipo frecuente en la raza negra, no tienen dos genes alfa, y las manifestaciones clínicas de este genotipo recuerdan a las de los pacientes heterocigotos para la talasemia alfa 1. El estado heterocigoto de la talasemia alfa 2 (-a/aa) no se manifiesta clínicamente y por ese motivo representa el síndrome del portador silencioso.
Síndrome parecido a la talasemia alfa La hemoglobina Constant Spring (hemoglobina CS) es una hemoglobina estructuralmente anormal común en algunas poblaciones asiáticas. El gen de la globina alfa contiene una mutación en el codón de terminación que da como resultado la síntesis de una globina alfa que contiene 31 aminoácidos adicionales. Los pacientes heterocigotos con este defecto tienen un cuadro clínico similar al de los pacientes homocigotos para la talasemia alfa 2. Los pacientes homocigotos para la hemoglobina CS tienden a presentar manifestaciones clínicas un poco más graves que los pacientes heterocigotos para la talasemia alfa 1.
Diagnóstico
Las mediciones de HbF y HbA2 que ayudan en el diagnóstico de las talasemias beta están fácilmente disponibles en los laboratorios clínicos. En comparación, las pruebas necesarias para diagnosticar las talasemias alfa son bastante sofisticadas y se llevan a cabo en instituciones destinadas a la investigación de las talasemias. Los elementos diagnósticos utilizados en los pacientes en los cuales se sospecha talasemia alfa incluyen historia clínica, evaluación del frotis de sangre, cálculo de índices, tinción con azul brillante de cresilo y estudios familiares. En la práctica el rasgo de la talasemia alfa se diagnostica basándose en el hallazgo de microcitosis en los pacientes sin deficiencia de hierro que tienen niveles normales de HbA2 y HbF.
El diagnóstico de talasemia alfa o beta debe sospecharse cuando se asocia un VCM menor de 75 fl con una cuenta de eritrocitos mayor de 5 millones de células/µl. Un paciente con estos dos datos tiene una posibilidad de 85 por ciento de tener un síndrome de talasemia.85 En un estudio, el diagnóstico de talasemia no se consideró como opción en alrededor de la mitad de los pacientes con la enfermedad.
Consejo genético y diagnóstico prenatal
Los padres que han tenido un mortinato hidrópico o un niño con anemia de Cooley se niegan, y con justificación, a repetir la experiencia. Los adultos que han crecido en familias con talasemias y que se sabe que son heterocigotos para talasemia, están con frecuencia ansiosos de recibir consejo genético cuando inician sus propias familias. El consejo genético incluye el estudio de los futuros padres con base en exámenes diagnósticos rutinarios y estudios familiares. Además, los adelantos en la genética molecular, como el análisis de endonucleasas de restricción, permiten hacer diagnósticos prenatales precisos en el caso de las talasemias. Durante el primer trimestre, la muestra de vellosidades coriónicas combinada con el uso de marcadores de polimorfismo de ADN y sondas sintéticas de oligonucleótidos, puede proporcionar el diagnóstico definitivo en alrededor del 80 por ciento de los casos de talasemia beta.86,87
Defectos extracorpusculares
Los eritrocitos pueden dañarse por traumatismos, anticuerpos, fármacos y toxinas. Debe pensarse en un defecto extracorpuscular siempre que se presente hemólisis en los pacientes que tienen una historia personal o familiar negativa de anemia.
LESION MECANICA: HEMOLISIS MICROANGIOPATICA
La hemólisis microangiopática se caracteriza por la presencia de eritrocitos atípicos y fragmentados en un frotis de sangre periférica (v.gr., esquistocitos o células en casco), y por signos de hemólisis intra y extravascular.
Fisiopatología
El eritrocito normal puede soportar grados moderados de deformación y torcedura, pero se desintegra cuando se somete a fuerzas excesivas de estiramiento o cizallamiento. Se ha observado que ocurren tensiones de esta magnitud en las turbulencias producidas por válvulas aórticas deformes, por fístulas arteriovenosas, por comunicaciones interventriculares, o por las prótesis valvulares antiguas.
Se cree que ocurre coagulación intravascular localizada, en la cual bandas de fibrina cruzan la luz arteriolar, en las arteriolas que irrigan tejidos inflamados o neoplásicos. El estudio de modelos in vitro muestra que dichas bandas de fibrina desprenden fragmentos de los eritrocitos, cuyas membranas se sellan rápidamente. Sin embargo, parte del contenido del eritrocito se fuga, produciendo grados variables de hemólisis intravascular. Los eritrocitos alterados son retirados por el sistema reticuloendotelial.
Diagnóstico
La hemólisis, junto con los hallazgos típicos en el frotis de sangre, indican el diagnóstico [ver figura 7]. Si la angiopatía es extensa puede ocurrir trombocitopenia y coagulación intravascular diseminada. Las causas incluyen turbulencias hemodinámicas, vasculitis, hemangiomas gigantes, púrpura trombocitopénica, cáncer metastásico,89 ciertas infecciones (en especial meningococemia y enfermedades por rickettsias, incluyendo infecciones por hantavirus), síndrome urémico-hemolítico, coagulación intravascular diseminada, fármacos (cocaína, ciclosporina, mitomicina y tacrolimus [FK506]), e incluso catéteres subclavios.89,90
Tratamiento
La atención primaria se debe enfocar hacia la enfermedad principal. Los pacientes pueden desarrollar deficiencia de hierro y requerir tratamiento. Los suplementos en caso de reservas disminuidas de folato pueden estimular la eritropoyesis. En raras ocasiones la anemia causada por una válvula aórtica protésica vieja es lo suficientemente grave para requerir remplazo valvular. La plasmaféresis constituye un tratamiento eficaz para la púrpura trombocitopénica trombótica.
Hemoglobinuria por la marcha La hemoglobinuria por la marcha, trastorno parecido a la hemólisis microangiopática, ocurre por lo común en personas jóvenes después de correr o de tocar bongos durante tiempo prolongado. Se piensa que el trauma severo y repetitivo a los pies y manos destruye los eritrocitos que circulan en los vasos de plantas y palmas. El paciente presenta orina de color rojo que se aclara en 1 día o menos después de la actividad. El diagnóstico se confirma al demostrar hemoglobinemia y hemoglobinuria pasajeras sin anemia, alteraciones en el frotis de sangre, ni reticulocitosis. El uso de zapatos con plantilla y el evitar las superficies pavimentadas puede evitar recurrencias en personas que deciden seguir corriendo.
HEMOLISIS INMUNE
Un ejemplo clásico de hemólisis inmune ( pero no autoinmune) es la incompatiblidad materno fetal en el locus Rh D, en la que la madre D negativa, después de estar en contacto con eritrocitos D positivos, puede producir un anticuerpo IgG anti-D; el anticuerpo cruza la barrera placentaria y ataca y destruye los eritrocitos fetales. El feto presenta ictericia y eritrocitos esferocíticos.
Los eritrocitos fetales, ahora cubiertos con el anticuerpo IgG anti-D, se fijan a los macrófagos y monocitos fetales que contienen receptores para la porción Fc de estas moléculas de IgG. La digestión macrofágica de porciones de la membrana del eritrocito causa pérdida de áreas importantes de la superficie. Los esferocitos rígidos retornan a la circulación y son atrapados por el sistema reticuloendotelial fetal, en especial en el bazo. El anticuerpo IgG es más activo a 37°C, por lo común no puede activar en forma extensa la vía del complemento y no puede aglutinar eritrocitos atacados que están suspendidos en solución salina.
La prueba de antiglobulina directa de Coombs [ver figura 9a]. se utiliza clínicamente para detectar IgG que está cubriendo eritrocitos. Esta prueba es negativa en la madre, cuyos eritrocitos carecen de antígeno D y por lo tanto no están cubiertos por anticuerpo anti-D. La prueba de Coombs indirecta [ver figura 9b], que detecta la presencia de anticuerpos libres en el suero, es positiva en la madre porque tiene anticuerpos anti-D en su suero. En comparación, el feto tiene una prueba de Coombs directa fuertemente positiva, porque sus eritrocitos (con antígenos D) están cubiertos con anticuerpo anti-D materno. La prueba de Coombs indirecta del feto puede ser positiva o negativa, dependiendo de la cantidad de anticuerpos anti-D transferidos por la madre, de la avidez del anti-D por los eritrocitos D positivos fetales, y de la disponibilidad de sitios D en los eritrocitos fetales.
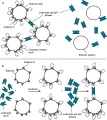
|
| Figura 9 |
| Prueba de Coombs |
Estos anticuerpos se describen como calientes (porque tienen actividad máxima a 37 'C, por lo general IgG1 o IgG3) o fríos (con actividad máxima a 5 'C, por lo general IgM). Los anticuerpos también se han clasificado como completos (i.e., capaces de aglutinar eritrocitos suspendidos en solución salina) e incompletos (i.e., incapaces de hacerlo) y su detección requiere de técnicas como la prueba de antiglobulina directa de Coombs [ver figura 9a] o del tratamiento enzimático de los eritrocitos.91 Los autoanticuerpos calientes suelen ser incompletos, mientras que las aglutininas frías, por lo general IgM, suelen ser completas.92
Anemia hemolítica autoinmune
La anemia hemolítica autoinmune suele ser un trastorno agudo caracterizado por hemólisis extravascular. La hemólisis intravascular es rara e indica una velocidad extremadamente rápida de destrucción de los eritrocitos o que se han excedido los mecanismos de eliminación extravascular.
Fisiopatología En la anemia hemolítica autoinmune los autoanticuerpos suelen dirigirse contra componentes importantes del eritrocito (v.gr., antígeno Rh, antígeno Kell,91 glucoforina A).93 En forma alternativa, los eritrocitos del paciente están sensibilizados tanto con un anticuerpo IgG como con un componente del complemento, por lo general C3d. En otras circunstancias parece ser que el complemento se fija al eritrocito por un anticuerpo IgM que después se desprende. En ocasiones los eritrocitos pueden mostrar solo componentes del complmento sin que se detecte IgG por la prueba de Coombs. La fijación del complemento en estas circunstancias se puede explicar por la presencia continua de IgG en un nivel por debajo del detectable por medio de la prueba usual de antiglobulina directa; en forma alternativa, pudiera ser que una IgG que fija complemento se hubiera unido antes a la célula, pero fue lavada por el procedimiento de la prueba.92
La severidad de la hemólisis correlaciona con el número y la clase de moléculas IgG unida a la superficie del eritrocito. Los eritrocitos cubiertos con anticuerpo se fijan a los macrófagos a través de receptores (FcRI, FcRII o FcRIII) a través de la porción Fc del anticuerpo. La unión firme de los eritrocitos a los macrófagos es seguida de fagocitosis y lisis.91 Concentraciones relativamente bajas de IgG1 fija a los eritrocitos producen una prueba positiva de antiglobulina directa de Coombs sin evidencia de hemólisis (alrededor de 1,000 moléculas por eritrocito), mientras que concentraciones mucho más altas de autoanticuerpos IgG1 por célula se asocian con evidencias de hemólisis.91 La presencia combinada de IgG y de fracciones del complemento puede aumentar también la severidad de la hemólisis.
Los eritrocitos sensibilizados sólo por IgG suelen ser eliminados por el bazo, mientras que los eritrocitos sensibilizados por IgG y complemento o solo complemento son destruidos por el hígado, porque las células de Kupffer portan receptores específicos para el componente C3b del complemento.
Diagnóstico diferencial Se ha descrito una variedad idiopática y una secundaria a otros padecimientos. Estos incluyen lupus eritematoso generalizado, linfoma no-Hodgkin (en especial leucemia linfocítica crónica), enfermedad de Hodgkin, cáncer, mieloma, quiste dermoide, infección por VIH, linfadenopatía angioinmunoblástica con disproteinemia y colitis ulcerativa crónica.
Manifestaciones clínicas Los pacientes pueden estar desde asintomáticos hasta tener una enfermedad severa. Durante los exámenes de detección en bancos de sangre, o antes de la donación de sangre pueden descubrirse pacientes con prueba de Coombs positiva. Generalmente se puede demostrar que dichas personas tienen complemento o la combinación de complemento e IgG (casi siempre IgG1 o IgG4) en sus eritrocitos, pero por lo común no presentan hemólisis. En el otro extremo, un episodio hemolítico agudo puede disminuir el hematocrito de 45 a 15 por ciento en 2 días. Los pacientes con este tipo de episodios tienen fatiga severa y síntomas cardiorrespiratorios, aunados a ictericia, linfadenopatía y hepatoesplenomegalia.
Diagnóstico En los casos severos los frotis de sangre periférica muestran macrocitosis, policromatofilia, esferocitosis variable y autoaglutinación de eritrocitos. En casos severos la cuenta plaquetaria también está disminuida (síndrome de Evans) y puede haber leucopenia. La tercera parte de los pacientes tiene reticulocitopenia al inicio,94 debido a que la respuesta de reticulocitos puede tardar algunos días en desarrollarse. La prueba directa de Coombs será positiva. Cualquiera o todos estos datos pueden estar ausentes en la enfermedad leve.
Se debe determinar si hay complemento, IgG, o ambos en los eritrocitos por medio del uso de los reactivos de Coombs que están dirigidos específicamente contra la IgG o componentes del complemento. En algunas ocasiones se sospecha una anemia hemolítica autoinmune, pero la prueba directa de Coombs es repetidamente negativa. En estos casos el nivel de autoanticuerpo puede estar por debajo del límite de detección para autoanticuerpos muy activos, como los de la subclase IgG3 o el autoanticuerpo puede ser de tipo IgA o IgM.91
A los pacientes con evidencia de anemia hemolítica se les debe investigar una enfermedad autoinmune (v.gr., lupus eritematoso genealizado) y otras formas de hemólisis, como hemoglobinuria paroxística nocturna, enfermedad por crioaglutininas y hemoglobinuria paroxística por frío.
Tratamiento Los pacientes con enfermedad muy leve no requieren tratamiento inmediato. El tratamiento debe estar encaminado a disminuir la producción de autoanticuerpos y a reducir el ataque por los macrófagos que desencadena la eliminación de los eritrocitos por el sistema reticuloendotelial. El tratamiento inicial consiste en la administración de 60 a100 mg de prednisona al día en dosis divididas. Este tratamiento suele producir una disminución lenta en la fijación de anticuerpos a los eritrocitos y se cree que interfiere con el ataque fagocítico a los eritrocitos ya cubiertos. Una buena respuesta al uso de corticosteroides, indicada por una elevación en la cuenta reticulocitos y una mejoría en los valores sanguíneos, puede ser aparente en 1 o 2 días. Se recomiendan la administración de 1 mg de ácido fólico al día.
Después de la respuesta inicial al tratamiento, que suele ser muy satisfactoria, se permite que la hemoglobina y la cuenta de reticulocitos vuelvan a lo normal. Entonces se repite la prueba de Coombs para determinar si existe disminución en el título de anticuerpos, y en caso positivo comienza a disminuirse la dosis de prednisona de modo cuidadoso. Cerca de la quinta parte de los pacientes se mantienen en buen estado en forma indefinida, pero la mayoría presenta una enfermedad crónica y agresiva que puede producir recaídas repentinas con anemia súbita. La dosis de prednisona se puede regular con base en el nivel de hemoglobina, la cuenta de reticulocitos (su elevación indica persistencia de la hemólisis), y el título de la prueba de Coombs directa. Se ha recurrido a la administración de prednisona en días alternos para minimizar los efectos secundarios de los esteroides. Si el paciente no responde al tratamiento estándar con prednisona, se usa dexametasona en dosis altas (v.gr., 40 mg/día vo por 4 días consecutivos en ciclos de 28 días95 como para la púrpura trombocitopénica idiopática crónica refractaria.
Si la dosis de esteroides necesaria para el tratamiento crónico produce morbilidad importante, el autor ha decidido en forma empírica realizar esplenectomía o administrar agentes inmunosupresores. La medición del secuestro esplénico de eritrocitos marcados con Cr51 no predice en forma confiable el éxito de la esplenectomía. La esplenectomía rara vez causa remisión prolongada, pero es útil para ahorrar esteroides. Después de la esplenectomía, una dosis pequeña de prednisona (5 ó 10 mg al día), puede estabilizar la concentración de hemoglobina.
Se pueden administrar agentes inmunosupresores como la azatioprina o la ciclofosfamida como alternativa a la esplenectomía. Sin embargo, no existe evidencia confiable que pueda servir como base para elegir entre los dos agentes. En los pacientes con enfermedad muy agresiva la ciclosporina ha sido útil.96 La azatioprina se debe iniciar en una dosis de 100 a 200 mg al día, vigilando cuidadosamente la citología hemática para evitar reticulocitopenia o neutropenia. La ciclofosfamida se inicia en dosis de 100 a 200 mg al día, vigilando la citología hemática y la orina. Debido a que la ciclofosfamida puede causar leucemia mielocítica aguda o síndrome mielodisplásico relacionados al tratamiento, su uso es limitado.
Las dosis de azatioprina o ciclofosfamida deben ajustarse para reducir la cuenta de leucocitos a alrededor de 3,000/mm3. La mejoría ocurre en un lapso de 3 a 4 semanas. Cuando existe respuesta al tratamiento se puede reducir la dosis de prednisona; en este momento es necesario comprobar de nuevo el nivel de hemoglobina, cuenta de reticulocitos y títulos de la prueba de Coombs para determinar la dosis mínima de tratamiento requerida. Se han usado con éxito dosis elevadas de IgG intravenosa para tratar la anemia hemolítica autoinmune, en forma análoga a su uso en la púrpura trombocitopénica idiopática. Sin embargo, no existen estudios controlados a gran escala. En un reporte, un tercio de los pacientes tuvieron una respuesta temporal y se requirieron dosis mayores a las empleadas para púrpura trombocitopénica idiopática.97,98
El paciente con anemia sintomática requiere transfusión de eritrocitos. En estos casos no es una sorpresa que el banco de sangre informe incompatibilidad. Muchos bancos de sangre hacen en forma regular una prueba de antiglobulina directa en los eritrocitos del receptor. Los pacientes que tienen anticuerpos libres en el suero presentarán reactividad muy amplia y extensa contra los donadores de eritrocitos, y producirán pruebas cruzadas incompatibles cuando se examinan con el reactivo para antiglogulinas. Si se requieren transfusiones para apoyar las funciones cardiorrespiratorias y del sistema nervioso central, se recomienda una consulta pronta con el servicio de medicina de transfusión.92
Hemólisis inmune relacionada con medicamentos
Ciertos casos de hemólisis inmune que son clínicamente indistinguibles de la anemia hemolítica autoinmune son inducidos por la administración de medicamentos. Existen dos variables, la anemia de tipo hapteno y la hemolítica autoinmune.99
Tipo hapteno Medicamentos como las penicilinas y cefalosporinas se unen firmemente a la membrana del eritrocito. En circunstancias excepcionales en las que se requieren dosis masivas del medicamento (v.gr., más de 10 millones de unidades de penicilina al día), el medicamento unido a las proteínas puede actuar como un hapteno y causar una respuesta inmune. Al parecer se produce un anticuerpo que está dirigido contra el complejo medicamento-eritrocito.100,101 El medicamento puede fijarse de modo firme o débil al eritrocito y producir un neoantígeno que genera una respuesta inmune.99
La prueba de Coombs directa es positiva con agentes anti-IgG. Si se prueba el suero del paciente contra células normales (prueba de Coombs indirecta) no se presenta reacción a menos que el medicamento agresor se añada primero a los eritrocitos normales. El suspender o cambiar el medicamento es eficaz para eliminar la hemólisis, ya que el anticuerpo tiende a ser muy específico.
Anemia hemolítica autoinmune inducida por medicamentos La metildopa es el ejemplo clásico de un medicamento que causa anemia hemolítica autoinmune. Otros son la levodopa, el ácido mefenámico y quizá la procainamida. La administración de dichos medicamentos desencadena una prueba de Coombs directa positiva con reactivos anti-IgG en el 15 al 20 por ciento de los pacientes tratados, pero se presenta hemólisis en menos del 1 por ciento. Cuando el anticuerpo se separa se observa que es un anticuerpo IgG clásico dirigido contra los componentes Rh. El mecanismo de la hemólisis es idéntico al de la anemia hemolítica autoinmune.102
Se ha reportado que el AINE diclofenaco sódico causa anemia hemolítica aguda devastante, con evidencia de hemólisis intra y extravascular asociada a estado de choque, falla orgánica e incluso coagulación intravascular diseminada (CID).103 Los pacientes desarrollan tanto autoanticuerpos contra eritrocitos como anticuerpos dependientes del fármaco. Se piensa que el diclofenaco sódico se une a la superficie del eritrocito, formando neoantígenos que provocan la generación de autoanticuerpos verdderos, así como anticuerpos dependientes del fármaco. La prueba directa de Coombs es positiva tanto con reactantes IgG como C3d. Ocurre reactividad de anticuerpo adicional si se agregan metabolitos del diclofenaco sódico obtenidos de la orina de los pacientes tratados con el medicamento. El tratamiento consiste en reconocer la causa, suspender el medicamento y dar manejo de apoyo durante varios días hasta que cese el proceso.103
Hemólisis tardía de eritrocitos transfundidos
La sangre se tipifica sólo para los antígenos ABO y Rh-D, pero existen otros antígenos presentes en los eritrocitos. Por ello, un paciente que es trasfundido en forma múltiple puede desarrollar, en un periodo de 1 a 2 semanas, una respuesta de anticuerpos a uno o más de estos antígenos. Los agresores usuales son los antígenos Kell, Duffy, Kidd, y antígenos Rh diferentes al D. Puede presentarse una hemólisis aguda autolimitada, que por lo común es extravascular. Los datos primordiales son el antecedente de transfusión previa, esferosis en el frotis en sangre periférica, una prueba de Coombs directa positiva y la aparición reciente de un anticuerpo en el suero del paciente (prueba indirecta de Coombs positiva). En general no se requiere tratamiento, pero las transfusiones subsecuentes deben cruzarse con el suero del paciente. Aparece un problema semejante con el trasplante de médula ósea y de otros tejidos.104
Enfermedad por crioaglutininas
La enfermedad por crioaglutininas tiene varios tipos. Una variedad poco común afecta adultos jóvenes y suele estar precedida por una infección por Mycoplasma pneumoniae o mononucleosis infecciosa; sin embargo, se han informado varios casos de esta variedad relacionados con paludismo crónico por Plasmodium falciparum. Una forma más común afecta a individuos de alrededor de 60 años de edad y puede presentarse como enfermedad idiopática por crioaglutininas, como pródromo a un trastorno linfoproliferativo o inmunoproliferativo, o en asociación a un padecimiento linfoproliferativo ya establecido.105
Fisiopatología y manifestaciones clínicas Serológicamente, el trastorno se caracteriza por la presencia en el suero de títulos elevados (más de 1:1,000 y por lo común más de 1:10,000) de aglutininas IgM. Estos anticuerpos son más activos a 4'C, son capaces de activar la secuencia del complemento y están dirigidos contra el sistema antigénico Ii de los eritrocitos. Al parecer la IgM reacciona con los eritrocitos que circulan en la sangre de menor temperatura de la nariz, oídos y espinillas. El anticuerpo fija complemento y posteriormente se disocia de los eritrocitos al llegar a zonas más templadas del organismo. En la variedad posinfecciosa de este trastorno, la crioaglutinina IgM es policlonal y de vida corta. A la inversa, en la variedad idiopática de enfermedad por crioaglutininas o en los casos asociados con macroglobulinemia de Waldenström, leucemia linfocítica crónica u otros linfomas, la IgM es monoclonal. Esta consiste casi en su totalidad de cadenas ligeras kappa en los pacientes con enfermedad idiopática por crioaglutininas o macroglobulinemia de Waldenström, pero en los pacientes con linfoma la IgM está formada sobre todo de cadenas ligeras lambda. En ocasiones la IgM se detecta como un pico de proteína M en la electroforesis de proteínas séricas.
En la variante posMycoplasma, los micoplasmas parecen unirse a la superficie del eritrocito en el sitio antigénico Ii. Esta interacción receptor-ligando causa la presentación del antígeno I en una forma inmunogénica.106 Listeria monocytogenes contiene al antígeno I,105 lo que apoya la teoría de que algunos agentes infecciosos estimulan las crioaglutininas que ocurren en forma natural y que causan la enfermedad por crioaglutininas posinfecciosa.
El síndrome clínico de la enfermedad por crioaglutininas es muy variable, y algunos pacientes sólo muestran títulos bajos de crioaglutininas sin síntomas o historia de neumonía reciente. En otros con anemia hemolítica autoinmune caliente y fría la hemólisis asociada tiende a ser severa y crónica. Los eritrocitos de estos pacientes están cubiertos con IgG y componentes del complemento, mientras que sus sueros contienen un título relativamente bajo de crioaglutininas que actúan a 30'C y que incluso pueden actuar a los 37'C.
Diagnóstico La presencia de anemia hemolítica con signos y síntomas acrales sugiere el diagnóstico. Puede ser difícil tomar una muestra de sangre, y la sangre puede aglutinarse a la vista en una jeringa fría y en el frotis. Los contadores automáticos de células sanguíneas pueden contar los eritrocitos aglutinados como eritrocitos independientes y en consecuencia informar valores absurdamente altos para el VCM y la CMHC.
Por lo general el banco de sangre detecta una crioaglutinina con actividad amplia. La prueba de Coombs directa es positiva con reactivos anticomplemento pero no con reactivos anti-IgG.
Los datos que apoyan el diagnóstico de enfermedad idiopática por crioaglutininas incluyen los siguientes: títulos elevados de crioaglutinina IgM con reactividad termica amplia91 y especificidad a I (que reaccionan con eritrocitos de adultos pero no con eritrocitos de cordón umbilical), composición pura de cadenas ligeras kappa, en forma ocasional elevación absoluta de la IgM sérica, y un patrón de proteína M en la electroforesis de proteínas. Debe intentar detectarse un posible linfoma u otro padecimiento subyacente. Por el contrario, las crioaglutininas pos-Mycoplasma y pos-mononucleosis infecciosa son policlonales y los anticuerpos pos-mononucleosis infecciosa están dirigidas contra los antígenos i (eritrocitos de cordón umbilical).
Tratamiento La variedad pos-Mycoplasma o pos-mononucleosis infecciosa suele ser leve, autolimitada y no requiere tratamiento específico. Los pacientes con la variedad idiopática que tienen síntomas acrales deben cambiar su forma de vida, trasladándose a un sitio con clima más cálido o manteniendo cubiertos sus oídos, nariz, manos y pies durante la temporada fría. Pueden requerirse transfusiones de concentrado globular en los pacientes con anemia severa, lo que amerita pruebas cruzadas cuidadosas y calentar la sangre para minimizar la aglutinación por frío.
La esplenectomía y los esteroides no suelen controlar la hemólisis asociada con la enfermedad por crioaglutininas. Se supone que los eritrocitos cubiertos por complemento son retirados de la circulación en grado importante por macrófágos hepáticos más que esplénicos, y las células que producen IgM son relativamente insensibles a los efectos de los esteroides. Sin embargo, en ocasiones la administración de esteroides en dosis elevadas (v.gr.,100 mg de prednisona al día), ha reducido la velocidad de hemólisis en pacientes con títulos relativamente bajos de crioaglutininas. En la variante relativamente rara causada por crioaglutininas de IgG los esteroides y la esplenectomía pueden ser benéficos. No se observan buenos resultados con el uso de penicilina u otros agentes reductores que contienen grupos sulfhidrilo. La exsanguinotransfusión y la plasmaféresis parecen ser tratamientos lógicos para la crisis aguda, pero se necesitan más estudios clínicos para evaluar estas técnicas. El interferón alfa, en dosis de 3 millones U/m2 tres veces por semana, parece producir una reducción impresionante en el título de crioaglutininas, con reducción en la proteína M monoclonal en suero y en los síntomas acrales durante un periodo de un mes.91
Hemoglobinuria paroxística por frío
Los pacientes con el raro transtorno de hemoglobinuria paroxística por frío tienen signos y síntomas de hemólisis intravascular inducidos por el frío. La hemólisis parece estar asociada con la presencia de un anticuerpo sérico IgG que está dirigido contra el sistema P de los eritrocitos. El anticuerpo IgG se demuestra mejor por prueba de Donath-Landsteiner: se mezcla el suero con los eritrocitos del propio paciente o con eritrocitos de una persona normal y se enfría la mezcla a 4'C. Si está presente el anticuerpo de IgG asociado con este trastorno, producirá hemólisis después de calentar a 37'C. En el pasado la hemoglobinuria paroxística por frío se observaba generalmente como una complicación de la sífilis, pero en la actualidad se asocia a infecciones virales y a linfoma no Hodgkin.107
HIPERESPLENISMO
Los trastornos hiperesplénicos comprenden un grupo diverso de entidades clínicas que comparten las características comunes de esplenomegalia y hemólisis. La esplenomegalia y la hemólisis se presentan en muchos trastornos, incluyendo cirrosis hepática con esplenomegalia congestiva, enfermedad de Gaucher, linfoma, trastornos del tejido conjuntivo, síndrome de Felty, sarcoidosis, tuberculosis y otras enfermedades infecciosas.
Fisiopatología
La estructura singular del bazo es responsable de varias de las características fisiopatológicas del hiperesplenismo. Las arteriolas esplénicas tienen algunas ramas directas que van a los sinusoides, pero la mayoría de las arteriolas terminales se abren hacia los cordones esplénicos. Las células sanguíneas pasan desde los cordones hacia la pulpa a través de hendiduras en las paredes sinusoidales, que tienen dimensiones de entre 1 y 3 µm.108 Las células sanguíneas pasan entre los espacios longitudinales, que están cubiertos con fibras reticulares, y entre las células de la adventicia que se localizan fuera del sinusoide. Los macrófagos y las células endoteliales cubren la parte interna del sinusoide, y de esta manera existe contacto íntimo repetido entre las células sanguíneas y estos macrófagos.
El flujo sanguíneo en el bazo es lento. El pH y pO2 del eritrocito disminuyen, se consume glucosa y se altera el metabolismo de la célula. Puede aumentar el hematocrito, elevando más la viscosidad y la resistencia al flujo. Como consecuencia, las células sanguíneas son expuestas a tensiones metabólicas y mecánicas en presencia de macrófagos y otros leucocitos que tienen capacidad de reconocer el daño en la membrana celular. Al envejecer los eritrocitos, los fagocitos les quitan las áreas de superficie defectuosa, transformando a los eritrocitos bicóncavos en esferocitos rígidos o fragmentos de eritrocitos; estas partículas son atrapadas y después retiradas por el sistema reticuloendotelial. Un bazo grande tiene un flujo sanguíneo mayor de lo normal y expone una importante proporción de células sanguíneas a sus actividades de filtro. De esta manera, el problema en el hiperesplenismo es en esencia cuantitativo. Puede originarse un círculo vicioso en los pacientes que presentan hemólisis porque la hemólisis misma causa esplenomegalia.
Diagnóstico
Si el bazo no es palpable, pero se tiene la sospecha clínica de esplenomegalia, puede ser útil realizar un ultrasonido o un gamagrama con radioisótopos. Debido a que un bazo grande afecta, además de los eritrocitos a otras células sanguíneas, el paciente puede tener pancitopenia. A menos que el trastorno de fondo se relacione específicamente con la médula ósea, ésta suele encontrarse hiperplásica en el hiperesplenismo, a causa de la regeneración rápida de todas las líneas celulares afectadas. El frotis de sangre periférica no confirma el diagnóstico de hiperesplenismo.
Tratamiento
Si el hiperesplenismo está produciendo complicaciones clínicamente importantes y el tratamiento de la enfermedad principal no causa disminución del bazo, puede ser necesaria la esplenectomía. Sin embargo, la anemia no necesariamente debe atribuirse al hiperesplenismo, independientemente del tamaño del bazo. La hemodilución es otro mecanismo posible. Los pacientes con esplenomegalia masiva que tienen valores bajos de hematocrito y hemoglobina pueden tener volúmenes normales de eritrocitos según la técnica de Cr51. La esplenomegalia masiva produce un aumento en el volumen plasmático que causa una hemodilución extraordinaria. Es más, los bazos muy grandes pueden contener una reserva de eritrocitos que constituyen hasta el 25 por ciento del volumen total de eritrocitos; en comparación, los bazos normales no tienen reserva de eritrocitos. En pacientes con esplenomegalia que tienen una verdadera disminución en el volumen de eritrocitos el trastorno principal puede actuar reduciendo la producción de eritrocitos más que por destrucción acelerada. Por lo tanto, es conveniente llevar a cabo una determinación del volumen de eritrocitos antes de hacer el diagnóstico de hiperesplenismo.
MEDICAMENTOS, TOXINAS, VENENOS Y AGENTES FISICOS COMO CAUSAS DE HEMOLISIS
Fisiopatología
Ataque oxidativo Medicamentos como dapsona, sulfasalazina, fenacetina, perclorato de sodio, nitroglicerina, fenazopiridina, primaquina,66 y agentes como el paraquat y los análogos de la vitamina K, pueden introducirse en la hendidura fijadora de oxígeno de la hemoglobina . Por medio de esta acción esos medicamentos pueden generar radicales libres oxidantes como superóxido, radicales hidroxilo libres y peróxido. Si los mecanismos reductores del eritrocito son superados [ver tabla I], la hemoglobina es oxidada para formar cuerpos de Heinz y metahemoglobina. También se produce sulfahemoglobina por el ataque oxidativo. Esta molécula contiene un átomo de azufre en el anillo de porfirina, lo que le da un color azul-verdoso. No se conoce bien el origen del átomo de azufre, pero su presencia en el anillo hem deteriora el transporte de oxígeno.109 A pesar de ello, la sulfahemoglobina es una causa rara de cianosis.148 La membrana del eritrocito también puede estar sujeta al ataque oxidativo. Las células dañadas son retiradas por el sistema reticuloendotelial. La hemólisis suele ser extravascular, aunque no siempre, y pueden observarse cuerpos de Heinz en un frotis de sangre con tinción especial. Se eleva el nivel de la metahemoglobina. (Una cantidad tan pequeña como 1.5 g/dl de metahemoglobina o 0.5 g/dl de sulfahemoglobina puede producir datos físicos de cianosis. Por el contrario, se requieren 5 g/dl de desoxihemoglobina reducida para producir una cianosis comparable.66) El frotis puede mostrar las células con las muescas o ampollas típicas del ataque oxidativo a los eritrocitos [ver figura 6].
Los nitritos pueden oxidar la hemoglobina a metahemoglobina. En consecuencia, el uso recreativo de butil e isobutil nitritos como estimulantes, psicodélicos y afrodisiácos ha causado problemas clínicos. Cuando se inhalan en las cantidades habituales, estos agentes pueden producir un incremento leve a moderado en la metahemoglobina, elevando su concentración del nivel normal de 1 a 2 por ciento hasta el 20 por ciento. La inhalación más abundantes o la ingestión de estos compuestos induce metahemoglobinemia severa, caracterizada por concentraciones que alcanzan el 62 por ciento. Debido a que la metahemoglobina no transporta oxígeno, estos niveles elevados se asocian con manifestaciones de hipoxia tisular como cefalea, disnea, letargo y estupor. El examen físico demuestra taquicardia, hipotensión postural y cianosis, la sangre venosa es de color café-púrpura.110
Al parecer el daño oxidativo severo hace que la hemoglobina se agrupe en un lado del eritrocito, dejando un 'medio fantasma' cubierto por membrana plasmática en el resto de la célula. Estos hemifantasmas se pueden detectar en la sangre periférica. La destrucción oxidativa severa se asocia con aumento en los niveles de metahemoglobina y disminución en los niveles de glutatión reducido, y puede ser mortal si no se trata.
El diagnóstico se basa en el antecedente de exposición a un medicamento oxidante o a otra toxina, junto con la presencia de datos característicos en el frotis de sangre periférica y de elevación en la metahemoglobina.
Tratamiento
El tratamiento consiste en identificar y suspender el agente agresor. Los pacientes con metahemoglobinemia severa deben tratarse inmediatamente con una dosis de 1 a 2 mg/kg de azul de metileno; éste se administra por vía intravenosa en una solución de 1 g/dl en un periodo de 5 minutos. En presencia de la enzima eritrocítica NADPH-metahemoglobina reductasa y de cantidades adecuadas del donador de electrones NADPH [ver tabla I], el azul de metileno es reducido rápidamente a leuco-azul de metileno. Este producto reduce a su vez en forma rápida a la metahemoglobina a hemoglobina. Por lo tanto la cianosis revierte y el paciente adquiere un tono rosado de la piel inmediatamente después de la administración. Sin embargo, varias horas después el paciente puede ponerse otra vez cianótico, al parecer porque los nitratos liberados de los tejidos vuelven a entrar a la sangre periférica en ese momento. La readministración de azul de metileno, en una dosis de 1 mg/kg por vía intravenosa en un periodo de 5 minutos, debe restablecer los niveles de hemoglobina.
El éxito del tratamiento con azul de metileno depende de la presencia de suministros adecuados de NADPH. Los pacientes que tienen alteraciones de la vía de la pentosa fosfato [ver figura 5], como deficiencia de G6PD, no responderán a este tratamiento y deben recibir exsanguinotransfusiones de urgencia.110
Los pacientes con concentraciones muy altas de metahemoglobina (de por lo menos 60 por ciento) o los que tienen muchos hemifantasmas en el frotis deben someterse a exsanguineotransfusión, quizá con hemodiálisis.66,110
Otras formas de hemólisis inducida por medicamentos
Fisiopatología La exposición al plomo da como resultado un cuadro clínico que consiste en encefalopatía hipertensiva, neuropatía y anemia hemolítica que se caracteriza por puntilleo basófilo irregular en los eritrocitos. El mecanismo de la hemólisis inducida por plomo es complejo porque el metal tiene varias acciones: bloquea la síntesis del hem y causa de esta manera un almacenamiento de protoporfinas en el eritrocito, produce una deficiencia de pirimidina 5'-nucleotidasa111 y ataca los fosfolípidos de la membrana del eritrocito, causando fuga de potasio e interfiriendo con la actividad de la ATPasa de Na+-K+.
Tratamiento La detección de la intoxicación por plomo se lleva a cabo midiendo el nivel de protoporfirina eritrocítica libre. El diagnóstico se confirma al medir los niveles de plomo en sangre y orina. Se puede considerar el uso de un agente quelante, como edetato disódico del calcio (Ca Na2 EDTA), después de suspender la exposición al plomo. El tratamiento se inicia con 0.5 a 1.0 g de Ca Na2 EDTA, administrados por vía intravenosa en un periodo de 6 a 8 horas. El compuesto se administra diario por 5 días, y luego se administran 0.5 g de Ca Na2 EDTA como bolo intravenoso o inyección intramuscular, cada tercer día durante 2 semanas, mientras se lleva un control de los niveles urinarios de plomo. Una alternativa a la administración de Ca Na2 EDTA, después de los primeros 5 días, es el tratamiento con penicilamina oral de la siguiente manera: los primeros 7 días, 1 g/día; la siguiente semana se suspende el medicamento, y durante los 7 últimos días del régimen se repite el ciclo y se mide el nivel de plomo en la orina. Un estudio recomienda administrar 500 mg de penicilamina por día y continuar esta dosis durante 60 días después de que el paciente está asintomático.112
Ataque enzimático a los eritrocitos
Ejemplos clásicos de enzimas que atacan a los eritrocitos son el veneno de serpientes o las lecitinasas de los clostridios (fosfolipasa C). Estas enzimas literalmente atacan a los fosfolípidos de la bicapa de la membrana y producen fragmentación de los eritrocitos, esferocitosis y evidencia de hemólisis intravascular y extravascular. Puede haber coagulación intravascular diseminada y estado de choque. La identificación pronta y el tratamiento del trastorno primario son decisivos, al igual que el tratamiento de sostén.
Causas físicas de hemólisis
El casi ahogamiento en agua dulce y la administración accidental de agua estéril puede causar hemólisis intravascular por lisis osmótica. En dichos casos los eritrocitos se hinchan y se vuelven esferoidales. El casi ahogamiento en agua salada puede inducir hemólisis al desecar los eritrocitos. Las quemaduras causan desnaturalización mediada por temperatura de los polipéptidos de la membrana del eritrocito, provocando hemólisis.
Causas infecciosas de hemólisis
Las infecciones producen hemólisis por varios mecanismos. Las infecciones por Mycoplasma pneumoniae y la mononucleosis infecciosa pueden causar hemólisis por crioaglutininas. La infección por H. influenzae tipo b puede provocar hemólisis, el principal factor de virulencia de este organismo, polirribosa ribosil fosfato (PRRP) permite que el organismo escape a la fagocitosis. El PRRP liberado a la circulación se fija a los eritrocitos, y la unión consecuente de aticuerpos anti-PRRP causa hemólisis dependiente del complemento.113 Los pacientes infectados por VIH o citomegalovirus (CMV) pueden tener anemia hemolítica autoinmune (ver antes).114
La sepsis por clostridios puede ser devastadora y la aparición de hemoglobina libre en plasma o de hemoglobinuria sugiere una infección fatal. Las especies Clostridium son capaces de crecer en forma súbita y explosiva, y pueden liberar muchas enzimas, incluyendo fosfolipasas y proteasas, que digieren a los eritrocitos, causando hemólisis intravascular.
Algunas infecciones pueden causar esplenomegalia y hemólisis por hiperesplenismo. La meningococcemia o la septicemia severa por gram negativos pueden producir CID y hemólisis microangiopática. Algunos agentes infecciosos, tales como Plasmodium, que causa el paludismo, parasitan y destruyen directamente los eritrocitos. Los eritrocitos autólogos no parasitados, se eliminan con más rapidez de la habitual, e incluso los eritrocitos normales son eliminados con rapidez de pacientes cuyos eritrocitos están parasitados por paludismo.115 Este dato sugiere que el sistema reticuloendotelial es hiperactivo durante el paludismo y, quizá también en otras infecciones.
Cada vez se diagnóstica con más frecuencia a la babesiosis, en especial en el área de Nueva Inglaterra, enfermedad causada por un parásito que invade los eritrocitos y que se trasmite de su reservorio roedor por la misma garrapata ixodes que porta la enfermedad de Lyme y la erlichiosis humana. Los individuos inmunosuprimidos, como los infectados por VIH, tienen más riesgo de sufrir infecciones crónicas y severas. El diagnóstico se realiza por frotis de sangre periférica, pero los nuevos métodos de reacción en cadena de la polimerasa son más sensibles.116
HEMOLISIS RELACIONADA CON ENFERMEDAD HEPATICA
Varios mecanismos, incluyendo la hemólisis, pueden producir anemia en pacientes con hepatopatías. El aspecto inflamatorio de un trastorno de fondo (como en la hepatitis viral) puede causar un defecto en la producción. La ingestión de alcohol bloquea también la producción de eritrocitos. La deficiencia de folato en los pacientes con cirrosis alcohólica puede dar como resultado eritropoyesis ineficaz megaloblástica clásica. La gastritis alcohólica puede causar hemorragias severas. Las reservas de hierro pueden agotarse en los vagabundos alcohólicos que venden su sangre con demasiada frecuencia. El paciente cirrótico puede tener esplenomegalia congestiva con hemólisis hiperesplénica. También son comunes los macrocitos (con o sin deficiencia de B12 o folato) y las células en diana (causadas por elevación de colesterol).
Equinocitosis y acantocitosis
En los pacientes con hepatopatía es frecuente observar equinocitosis cuando se examinan los eritrocitos en una preparación húmeda o por microscopía electrónica, aunque los frotis estándar pocas veces muestran estos equinocitos.117 Cuando los eritrocitos normales se incuban en el plasma de pacientes con equinocitosis, las células se vuelven equinocíticas con rapidez.
Los equinocitos pueden sufrir un cambio de forma a células en erizo (acantocitos). La acantositosis o hemólisis de células en erizo es causada por alteraciones en los lípidos de la membrana de los eritrocitos, con disminución en la síntesis de fosfolípidos, que causa disminución en la relación entre fosfolípidos y colesterol.118 Estos cambios en la membrana acortan la vida de los eritrocitos.
OTRAS CAUSAS DE HEMOLISIS
Acúmulo de cobre
Pocas veces la enfermedad de Wilson, trastorno metabólico asociado con depósitos excesivos de cobre, se detecta por un episodio de hemólisis grave y agudo. Se piensa que la liberación de cobre al suero y su entrada subsecuente al eritrocito es el mecanismo hemolítico principal. Además de afectar los niveles de hexocinasa, el cobre intracelular parece causar la formación de radicales de oxígeno que reaccionan con los componentes de la membrana y los oxidan. Aunque no se ha informado ningún tratamiento con buenos resultados, puede administrarse penicilamina en una dosis de 2 a 4 g vo diarios en un esfuerzo por reducir el nivel de cobre libre. Si el ataque oxidativo es un factor importante, la administración de 1,000 a 2,000 UI de vitamina E (alfa-tocoferol) diarios, durante varios días, puede ser de utilidad.
Derivación cardiopulmonar
Después de la derivación cardiopulmonar puede presentarse leucopenia transitoria en algunos pacientes. Se piensa que esta leucopenia es causada por la activación del complemento, lo que origina C3a y C5a . También aumenta la hemoglobina libre en plasma, probablemente por hemólisis causada por el depósito del complejo de ataque C5b-C9 sobre la superficie del eritrocito.119
Bibliografía
-
Liu S-C, Derick LH: Molecular anatomy of the red blood cell
membrane skeleton: structure-function relationships. Semin Hematol 29:231,
1992
-
Cohen CM, Gascard P: Regulation and post-translational modification
of erythrocyte membrane and membrane-skeletal proteins. Semin Hematol 29:244,
1992
-
Schwartz RS: PIG-A: the target gene in paroxysmal nocturnal
hemoglobinuria (editorial). N Engl J Med 330:283, 1994
-
Canessa M: Red cell volume–related ion transport systems
in hemoglobinopathies. Hematol Oncol Clin North Am 5:495, 1991
-
Hanspal M, Palek J: Biogenesis of normal and abnormal red
blood cell membrane skeleton. Semin Hematol 29:305, 1992
-
Turrini F, Mannu F, Arese P, et al: Characterization of the
autologous antibodies that opsonize erythrocytes with clustered integral
membrane proteins. Blood 81:3146, 1993
-
Potter CG, Potter AC, Hatton CSR, et al: Variation of erythroid
and myeloid precursors in the marrow and peripheral blood of volunteer
subjects infected with human parvovirus (B19). J Clin Invest 79:1486, 1987
-
Smith BD, Segel GB: Abnormal erythrocyte endothelial adherence
in hereditary stomatocytosis. Blood 89:3451, 1997
-
Vives Corrons JL, Besson I, Aymerich M, et al: Hereditary
xerocytosis: a report of six unrelated Spanish families with leaky red
cell syndrome and increased heat stability of the erythrocyte membrane.
Br J Haematol 90:817, 1995
-
Palek J, Jarolim P: Clinical expression and laboratory detection
of red blood cell membrane protein mutations. Semin Hematol 30:249, 1993
-
Dhermy D, Galand C, Bournier O, et al: Heterogenous band
3 deficiency in hereditary spherocytosis related to different band 3 gene
defects. Br J Haematol 98:32, 1997
-
Rosse WF: Hematopoiesis and the defect in paroxysmal hemoglobinuria
(editorial). J Clin Invest 100:953, 1997
-
Ohashi H, Hotta T, Ichikawa A, et al: Peripheral blood cells
are predominantly chimeric of affected and normal cells in patients with
paroxysmal nocturnal hemoglobinuria: simultaneous investigation on clonality
and expression of glycophosphatidylinositol-anchored proteins. Blood 83:853,
1994
-
Hillmen P, Lewis SM, Bessler M, et al: Natural history of
paroxysmal nocturnal hemoglobinuria. N Engl J Med 333:1253, 1995
-
Socie G, Marie J-Y, de Gramont A, et al: Paroxysmal nocturnal
haemoglobinuria: long-term follow-up and prognostic factors. Lancet 348:573,
1996
-
Hall SE, Rosse WF: The use of monoclonal antibodies and flow
cytometry in the diagnosis of paroxysmal nocturnal hemoglobinuria. Blood
87:5332, 1996
-
Sholar PW, Bell WR: Thrombolytic therapy for inferior
vena cava thrombosis in paroxysmal nocturnal hemoglobinuria. Ann Intern
Med 103:539, 1985
-
Arese P, De Flora A: Pathophysiology of hemolysis in glucose-6-phosphate
dehydrogenase deficiency. Semin Hematol 27:1, 1990
-
Beutler E: G6PD deficiency. Blood 84:3613, 1994
-
Cocco P, Todde P, Fornera S, et al: Mortality in a cohort
of men expressing the glucose-6-phosphate dehydrogenase deficiency. Blood
91:706, 1998
-
Nagel RL, Ranney HM: Genetic epidemiology of structural mutations
of the b-globin gene. Semin Hematol 27:342,
1990
-
Rodgers GP, Noguchi CT, Schecter AN: Sickle cell anemia.
Scientific American Science & Medicine 1(4):48, 1994
-
Bunn HF: Pathogenesis and treatment of sickle cell disease.
N Engl J Med 337:762, 1997
-
Hebbel RP: Adhesive interactions of sickle erythrocytes with
endothelium. J Clin Invest 99:2561, 1997
-
Kuypers FA, Lewis RA, Hua M, et al: Detection of altered
membrane phospholipid asymmetry in subpopulations of human red blood cells
using fluorescently labeled annexin V. Blood 87:1179, 1996
-
Solovey A, Gui L, Key NS, et al: Tissue factor expression
by endothelial cells in sickle cell anemia. J Clin Invest 101:1899, 1998
-
Hebbel RP: Beyond hemoglobin polymerization: the red blood
cell membrane and sickle disease pathophysiology. Blood 77:214, 1991
-
Bookchin RM, Ortiz OE, Lew VL: Evidence for a direct reticulocyte
origin of dense red cells in sickle cell anemia. J Clin Invest 87:113,
1991
-
Singhal A, Doherty JF, Raynes JG: Is there an acute-phase
response in steady-state sickle cell disease? Lancet 341:651, 1993
-
Francis RB, Johnson CS: Vascular occlusion in sickle cell
disease: current concepts and unanswered questions. Blood 77:1405, 1991
-
Steinberg MH, West MS, Gallagher D, et al: Effects of glucose-6-phosphate
dehydrogenase deficiency upon sickle cell anemia. Blood 71:748, 1988
-
Milner PF, Kraus AP, Sebes JI, et al: Sickle cell disease
as a cause of osteonecrosis of the femoral head. N Engl J Med 325:1476,
1991
-
Haberkern CM, Neumayr LD, Orringer EP, et al: Cholecystectomy
in sickle cell anemia patients: perioperative outcome of 364 cases from
national preoperative transfusion study. Blood 89:1533, 1997
-
Serjeant GR: Sickle-cell disease. Lancet 350:725, 1997
-
Vichinsky E, Williams R, Das M, et al: Pulmonary fat
embolism: a distinct cause of severe acute chest syndrome in sickle cell
anemia. Blood 83:3107, 1994
-
Vichinsky EP, Styles LA, Colangelo LH, et al: Acute chest
syndrome in sickle cell disease: clinical presentation and course. Blood
89:1787, 1997
-
Bellet PS, Kalinyak KA, Shukla R, et al: Incentive
spirometry to prevent acute pulmonary complications in sickle cell diseases.
N Engl J Med 333:699, 1995
-
Charache S, Terrin ML, Moore RD, et al: Effect of hydroxyurea
on the frequency of painful crises in sickle cell anemia. N Engl J Med
332:1317, 1995
-
Styles LA, Schalkwijk CG, Aarsman AJ, et al: Phospholipase
A2 levels in acute chest syndrome of sickle cell disease. Blood
87: 2573, 1996
-
Gaston MH, Verter JI, Woods G, et al: Prophylaxis with oral
penicillin in children with sickle cell anemia: a randomized trial. N Engl
J Med 314:1593, 1986
-
Gupta AK, Kirchner KA, Nicholson R, et al: Effects
of a-thalassemia and sickle polymerization tendency
on the urine-concentrating defect of individuals with sickle cell trait.
J Clin Invest 88:1963, 1991
-
Falk RJ, Scheinman J, Phillips G, et al: Prevalence
and pathologic features of sickle cell nephropathy and response to inhibition
of angiotensin-converting enzyme. N Engl J Med 326:910, 1992
-
Ohene-Frempong K, Weiner SJ, Sleeper LA, et al: Cerebrovascular
accidents in sickle cell disease: rates and risk factors. Blood 91:288,
1998
-
Adams RJ, McKie VC, Hsu L, et al: Prevention of a first stroke
by transfusions in children with sickle cell anemia and abnormal results
on transcranial Doppler ultrasonography. N Engl J Med 339:5, 1998
-
Cohen AR: Sickle cell disease: new treatments, new
questions. N Engl J Med 339:42, 1998
-
Serjeant GR, Serjeant BE, Thomas PW, et al: Human parvovirus
infection in homozygous sickle cell disease. Lancet 341:1237, 1993
-
Koshy M, Weiner SJ, Miller ST, et al: Surgery and anesthesia
in sickle cell disease. Blood 86:3676, 1995
-
Vichinsky EP, Haberkern CM, Neumayr L, et al: A comparison
of conservative and aggressive transfusion regimens in the perioperative
management of sickle cell disease. N Engl J Med 333:206, 1995
-
Platt OS, Brambilla DJ, Rosse WF, et al: Mortality in sickle
cell disease: life expectancy and risk factors for early death. N Engl
J Med 330:1639, 1994
-
Ballas SK: Neurobiology and treatment of pain. Sickle Cell
Disease: Basic Principles and Clinical Practice. Embury SH, Hebbel RP,
Mohandas N, et al, Eds. Raven Press, New York, 1994, p 745
-
Brugnara C, de Franceschi L, Alper SL: Inhibition of Ca2+-dependent
K+ transport and cell dehydration in sickle erythrocytes by
clotrimazole and other imidazole derivatives. J Clin Invest 92:520, 1993
-
De Franceschi L, Bachir D, Galacteros F, et al: Oral magnesium
supplements reduce erythrocyte dehydration in patients with sickle cell
disease. J Clin Invest 100:1847, 1997
-
Bridges KR, Barabino GD, Brugnara C, et al: A multiparameter
analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy.
Blood 88:4701, 1996
-
Perrine SP, Ginder GD, Faller DV, et al: A short-term trial
of butyrate to stimulate fetal-globin-gene expression in the b-globin
disorders. N Engl J Med 328:81, 1993
-
Sher GD, Ginder GD, Little J, et al: Extended therapy
with intravenous arginine butyrate in patients with b-hemoglobinopathies.
N Engl J Med 332:1606, 1995
-
Walters MC, Patience M, Leisenring W, et al: Bone marrow
transplantation for sickle cell disease. N Engl J Med 335:369, 1996
-
Vichinsky EP: Understanding the morbidity of sickle cell
disease (correspondence). Br J Haematol 99:974, 1997
-
Koshy M, Burd L: Management of pregnancy in sickle cell syndromes.
Hematol Oncol Clin North Am 5:585, 191
-
Embury SH: Prenatal diagnosis. Sickle Cell Disease:
Basic Principles and Clinical Practice. Embury SH, Hebbel RP, Mohandas
N, et al, Eds. Raven Press, New York, 1994, p 485
-
Kark JA, Posey DM, Schumacher HR, et al: Sickle-cell trait
as a risk factor for sudden death in physical training. N Engl J Med 317:781,
1987
-
Sullivan LW: The risks of sickle-cell trait: caution
and common sense. N Engl J Med 317:830, 1987
-
Voskaridou E, Kalotychou V, Loukopoulos D: Clinical and laboratory
effects of long-term administration of hydroxyurea to patients with sickle-cell/b-thalassaemia.
Br J Haematol 89:479, 1995
-
Nagel RL, Lawrence C: The distinct pathobiology of sickle
cell-hemoglobin C disease. Hematol Oncol Clin North Am 5:433, 1991
-
Steinberg MH, Nagel RL, Brugnara C: Cellular effects of hydroxyurea
in Hb SC disease. Br J Haematol 98:838, 1997
-
Rees DC, Styles L, Vichinsky EP, et al: The hemoglobin
E syndromes. Ann NY Acad Sci 850:334, 1998
-
Jaffe ER: Methemoglobinemia in the differential diagnosis
of cyanosis. Hosp Pract (Off Ed) 20(12):92, 1985
-
Charache S: Methemoglobinemia—sleuthing for a new cause
(editorial). N Engl J Med 314:776, 1986
-
Manabe J, Arya R, Sumimoto H, et al: Two novel mutations
in the reduced nicotinamide adenine dinucleotide (NADH)–cytochrome b5 reductase
gene of a patient with generalized type, hereditary methemoglobinemia.
Blood 88:3208, 1996
-
Weatherall DJ, Clegg JB: Thalassemia: a global public health
problem. Nat Med 2: 847, 1996
-
Adams JG III, Coleman MB: Structural hemoglobin variants
that produce the phenotype of thalassemia. Semin Hematol 27:229, 1990
-
Schrier SL: Thalassemia: pathophysiology of red cell changes.
Annu Rev Med 45:211, 1994
-
Chui DHK, Waye JS: Hydrops fetalis caused by a-thalassemia:
an emerging health care problem. Blood 91:2213, 1998
-
Sadelain M: Genetic treatment of the haemoglobinopathies:
recombinations and new combinations. Br J Haematol 98:247, 1997
-
Piomelli S: The management of patients with Cooley's anemia:
transfusions and splenectomy. Semin Hematol 32:262, 1995
-
Hoffbrand AV: Oral iron chelation. Semin Hematol 33:1, 1996
-
Brittenham GM, Griffith PM, Nienhuis AW, et al: Efficacy
of deferoxamine in preventing complications of iron overload in patients
with thalassemia major. N Engl J Med 331:567, 1994
-
Olivieri NF, Nathan DG, MacMillan JH, et al: Survival in
medically treated patients with homozygous b-thalassemia.
N Engl J Med 331:574, 1994
-
Olivieri NF, Brittenham GM, McLaren CE, et al: Long-term
safety and effectiveness of iron-chelation therapy with deferiprone for
thalassemia major. N Engl J Med 339:417, 1998
-
Kowdley KV, Kaplan MM: Iron-chelation therapy with oral deferiprone:
toxicity or lack of efficacy (editorial)? N Engl J Med 339:468, 1998
-
Winterbourne CC: Oxidative denaturation in congenital
hemolytic anemias: the unstable hemoglobins. Semin Haematol 27:41, 1990
-
Issaragrisil S, Visuthisakchai S, Suvatte V, et al: Brief
report: transplantation of cord-blood stem cells into a patient with severe
thalassemia. N Engl J Med 332:367, 1995
-
Lucarelli G, Galimberti M, Giardini C, et al: Bone marrow
transplantation in thalassemia. Ann NY Acad Sci 850:270, 1998
-
Rodgers GP, Rachmilewitz EA: Novel treatment options in the
severe b-globin disorders. Br J Haematol 91:263,
1995
-
Olivieri NF: Reactivation of fetal hemoglobin in patients
with b-thalassemia. Semin Hematol 33:24, 1996
-
Hansen RM, Hanson G, Anderson T: Failure to suspect and diagnose
thalassemic syndromes: interpretation of RBC indices by the nonhematologist.
Arch Intern Med 145: 93, 1985
-
Kazazian HH Jr: The thalassemia syndromes: molecular basis
and prenatal diagnosis in 1990. Semin Hematol 27:209, 1990
-
Dover GJ, Valle D: Therapy for b-thalassemia:
a paradigm for the treatment of genetic disorders. N Engl J Med 331:609,
1994
-
Ross CN, Reuter H, Scott D, et al: Microangiopathic haemolytic
anaemia and systemic vasculitis. Br J Rheumatol 35:377, 1996
-
Rytting M, Worth L, Jaffe N: Hemolytic disorders associated
with cancer. Hematol Oncol Clin North Am 10:365, 1996
-
Mach-Pascual S, Samii K, Beris P, et al: Microangiopathic
hemolytic anemia complicating FK 506 (tacrolimus) therapy. Am J Hematol
52:310, 1996
-
Engelfriet CP, Overbeeke MAM, von dem Borne AEGK: Autoimmune
hemolytic anemia. Semin Hematol 29:3, 1992
-
Jefferies LC: Transfusion therapy in autoimmune hemolytic
anemia. Hematol Oncol Clin North Am 8:1087, 1994
-
Leddy JP, Falany JL, Kissel GE, et al: Erythrocyte membrane
proteins reactive with human (warm-reacting) anti–red cell autoantibodies.
J Clin Invest 91:1672, 1993
-
Liesveld JL, Rowe JM, Lichtman MA: Variability of the erythropoietic
response in autoimmune hemolytic anemia: analysis of 109 cases. Blood 69:820,
1987
-
Meyer O, Stahl D, Beckhove P, et al: Pulsed high-dose dexamethasone
in chronic autoimmune haemolytic anaemia of warm type. Br J Haematol 98:860,
1997
-
Emilia G, Messora C, Longo G, et al: Long-term salvage treatment
by cyclosporin in refractory autoimmune haematological disorders. Br J
Haematol 93:341, 1996
-
Collins PW, Newland AC: Treatment modalities of autoimmune
blood disorders. Semin Hematol 29:64, 1992
-
Flores G, Cunningham-Rundles C, Newland AC, et al: Efficacy
of intravenous immunoglobulin in the treatment of autoimmune hemolytic
anemia: results in 73 patients. Am J Hematol 44:237, 1993
-
Salama A, Mueller-Eckhardt C: Immune-mediated blood cell
dyscrasias related to drugs. Semin Hematol 29:54, 1992
-
Garratty G: Immune cytopenia associated with antibiotics.
Transfus Med Rev 7:255, 1993
-
Kopicky JA, Packman CH: The mechanisms of sulfonylurea-induced
immune hemolysis: case report and review of the literature. Am J Hematol
23:283, 1986
-
Petz LD: Drug-induced autoimmune hemolytic anemia. Transfus
Med Rev 7:242, 1993
-
Salama A, Kroll H, Wittmann G, et al: Diclofenac-induced
immune haemolytic anaemia: simultaneous occurrence of red blood cell autoantibodies
and drug-dependent antibodies. Br J Haematol 95:640, 1996
-
Ramsey G: Red cell antibodies arising from solid organ transplants.
Transfusion 31:76, 1991
-
Silberstein LE: B-cell origin of cold agglutinins. Adv Exp
Med Biol 347:193, 1994
-
Loomes LM, Uemura K, Childs RA, et al: Erythrocyte receptors
for Mycoplasma pneumoniae are sialylated oligosaccharides of Ii
antigen type. Nature 307:560, 1984
-
Sharara AI, Hillsley RE, Wax TD, et al: Paroxysmal cold hemoglobinuria
associated with non-Hodgkin's lymphoma. South Med J 87:397, 1994
-
Rosse WF: The spleen as a filter. N Engl J Med 317:704, 1987
-
Lu HC, Shih RD, Marcus S, et al: Pseudomethemoglobinemia:
a case and review of sulfhemoglobinemia. Arch Pediatr Adolesc Med 152:803,
1998
-
Coleman MD, Coleman NA: Drug-induced methaemoglobinaemia:
treatment issues. Drug Saf 14:394, 1996
-
Valentine WN, Paglia DE, Fink K, et al: Lead poisoning: association
with hemolytic anemia, basophilic stippling, erythrocyte pyrimidine 5'-nucleotidase
deficiency, and intraerythrocytic accumulation of pyrimidines. J Clin Invest
58:926, 1976
-
Carton JA, Maradona JA, Arribas JM: Acute-subacute
lead poisoning: clinical findings and comparative study of diagnostic tests.
Arch Intern Med 147:697, 1987
-
Shurin SB, Anderson P, Zollinger J, et al: Pathophysiology
of hemolysis in infections with Hemophilus influenzae type b. J
Clin Invest 77:1340, 1986
-
Van Spronsen DJ, Breed WPM: Cytomegalovirus-induced thrombocytopenia
and haemolysis in an immunocompetent adult. Br J Haematol 92:218, 1996
-
Looareesuwan S, Merry AH, Phillips RE, et al: Reduced erythrocyte
survival following clearance of malarial parasitaemia in Thai patients.
Br J Haematol 67:473, 1987
-
Krause PJ, Spielman A, Telford SR III, et al: Persistent
parasitemia after acute babesiosis. N Engl J Med 339:160, 1998
-
Owen JS, Brown DJC, Harry DS, et al: Erythrocyte echinocytosis
in liver disease. J Clin Invest 76:2275, 1985
-
Allen DW, Manning N: Cholesterol-loading of membranes of
normal erythrocytes inhibits phospholipid repair and arachidonoyl-CoA:1-palmitoyl-sn-glycero-3-phosphocholine
acyl transferase: a model of spur cell anemia. Blood 87:3489, 1996
-
Salama A, Hugo F, Heinrich D, et al: Deposition of
terminal C5b-complement complexes on erythrocytes and leukocytes during
cardiopulmonary bypass. N Engl J Med 318:408, 1988
-
Schrier SL: The red cell membrane and its abnormalities.
Recent Advances in Haematology. Hoffbrand AV, Ed. Churchill Livingstone,
Edinburgh, 1982
-
Rosse WF: The glycolipid anchor of membrane surface proteins.
Semin Hematol 30:219,1993
-
Swartz RS: PIG-A: the target gene in paroxysmal nocturnal
hemoglobinuria. N Engl J Med 330:283,1994
- Weatherall DJ: The molecular genetics of haemoglobin—the thalassemias. Recent Advances in Haematology. Hoffbrand AV, Ed. Churchill Livingstone, Edinburgh, 1982