Inmunología
⭳ Abrir artículo (PDF)464.7 KBEste artículo fue revisado respecto a la Edición 2/2000. Ver esa versión →
Contenido del artículo
VI TOLERANCIA INMUNOLOGICA Y AUTOINMUNIDAD
- Tolerancia
- Enfermedades autoinmunes
- Mecanismos de autoinmunidad
- FALLA DE LA TOLERANCIA CENTRAL
- FALLA DE LA TOLERANCIA PERIFERICA
- MIMETISMO MOLECULAR
- ACTIVACION POLICLONAL DE LINFOCITOS
- REGULACION DEFICIENTE POR CELULAS T
- DEFECTOS EN LAS VIAS DE APOPTOSIS
- Susceptibilidad del huésped a las enfermedades autoinmunes
VI TOLERANCIA INMUNOLOGICA Y AUTOINMUNIDAD
DR. DWIGHT R. ROBINSON
Uno de los conceptos centrales de la inmunología lo constituye el concepto de que las reacciones autoinmunes son nocivas para el huésped. Alrededor de 1900, Ehrlich postuló que el sistema inmune adquiere un estado de tolerancia para los antígenos propios; además, propuso que el rompimiento de la tolerancia conduciría a la autodestrucción, estado que describió como horror autotóxico. Cerca de 50 años más tarde, Burnet indicó que las células inmunes autorreactivas son destruidas por los órganos linfoides en las etapas tempranas del desarrollo del individuo, lo que conduce a la tolerancia de los autoantígenos.
Las respuestas inmunológicas a los organismos extraños están estrictamente controladas para minimizar el daño a los tejidos propios. El sistema inmunológico se enfrenta a la enorme tarea de destruir los organismos extraños potencialmente dañinos sin lesionar los tejidos del huésped. La falla del sistema inmunológico para responder en forma adecuada a los organismos extraños puede causar infecciones graves y, por otro lado, las reacciones inmunológicas contra los tejidos del huésped pueden originar enfermedades autoinmunes. El mecanismo por el que el sistema inmunológico controla la especificidad e intensidad de estas reacciones es complejo y no se conoce bien, pero se han hecho importantes adelantos en el conocimiento de la autoinmunidad en los años recientes.
Tolerancia
La tolerancia puede definirse como un estado de no respuesta inmunológica a los antígenos. El reconocimiento antigénico tiene lugar en receptores específicos en las células T y B del sistema inmune. Estos encuentros pueden ocasionar activación o inhibición de los linfocitos, dependiendo de diversas condiciones en el momento del contacto. Puede adquirirse tolerancia ante antígenos extraños, y la autotolerancia es una forma especial que se aplica a los autoantígenos. La tolerancia es una consecuencia de uno de dos efectos sobre las células T y B: (1) deleción clonal, en la que el encuentro con el antígeno causa eliminación o muerte de los linfocitos específicos, o (2) anergia clonal, en la que los linfocitos no son destruidos sino que se vuelven no respondedores ante un antígeno específico.1-3
Burnet propuso la teoría de la selección clonal, la cual establece que en el sistema inmune existen clonas de células inmunes capaces de reaccionar genéticamente con todo inmunógeno potencial; de este modo, la producción de la respuesta inmune es el resultado de la amplificación y proliferación de clonas específicas. Burnet también postuló que la tolerancia puede ocurrir cuando los inmunocitos fetales se exponen y reconocen a un antígeno específico y que este proceso conduce a la deleción de dichas clonas. El apoyo al modelo de la eliminación clonal proviene de experimentos realizados por Billingham y Medawar; sus investigaciones demostraron que la infusión de células linfoides de una cepa murina (cepa B) a ratones recién nacidos de una segunda cepa (cepa A), permitía que la cepa A madura aceptara injertos de piel de los ratones de la cepa B.1
La tolerancia es inmunológicamente específica; puede inducirse hacia una estructura antigénica específica aunque se conserve la reactividad inmunológica contra otros antígenos. La tolerancia está mediada tanto por células T como B, por lo que afecta la inmunidad tanto humoral como celular. Los linfocitos inmaduros son más susceptibles a la inducción de tolerancia que los maduros. La tolerancia central se adquiere cuando los linfocitos inmaduros encuentran antígenos en sus órganos generadores, el timo (células T) y la médula ósea (células B). La tolerancia puede ser inducida en linfocitos maduros si éstos encuentran antígenos en tejidos fuera del timo o la médula ósea bajo ciertas condiciones especiales. A esto se le denomina tolerancia periférica. Tres factores generales pueden causar tolerancia periférica:
1. La forma del antígeno y su vía de administración. Los antígenos en dosis altas, administrados por vía oral y administrados en dosis bajas en forma repetida, pueden causar tolerancia. Los antígenos con alta afinidad por sus receptores antigénicos pueden también inducir tolerancia en lugar de activación.
2. El estado de la célula presentadora de antígeno (CPA). Las células presentadoras de antígeno, como los macrófagos, requieren coestimuladores, como la molécula de superficie B7, antes de que puedan activar a la célula T que reconoce al antígeno procesado que es presentado al receptor de la célula T por el antígeno leucocitario humano (HLA) en la superficie de la CPA. La exposición de las células T al antígeno en las CPA que no tienen B7 u otro estimulador puede causar tolerancia. Las células T que encuentran antígenos en las CPA en ausencia de coestimuladores permanecen tolerantes a estos antígenos, incluso si las células T se exponen después a CPA con antígenos en presencia de coestimuladores. Algunas CPA emiten también señales inhibitorias para la activación de la célula T.
3. La presencia de un subtipo de células T denominadas células T supresoras.
Los mecanismos anteriores se aplican a la inducción de tolerancia de células T, pero la tolerancia puede también ser ocasionada por las células B. En vista de que las respuestas inmunológicas, incluyendo la producción de anticuerpos, son dependientes de células T, el principal mecanismo de tolerancia de células B es la ausencia de la célula T cooperadora durante la presentación del antígeno a las células B. Las respuestas humorales o de células B a antígenos no proteicos (principalmente carbohidratos, como los antígenos de los grupos sanguíneos), son independientes de las células T. La tolerancia de las células B a antígenos T indepenientes puede ser inducida por ciertas formas de antígeno, antígenos en dosis muy altas y antígenos polivalentes.1-3
AUTOTOLERANCIA
La tolerancia del sistema inmunológico a los antígenos propios es indispensable para la salud. La ruptura de la autotolerancia es la base de las enfermedades autoinmunes. En las personas sanas existen antígenos potencialmente inmunogénicos en los tejidos del huésped que son expuestos al sistema inmune, y los tejidos propios no causan respuestas inmunológicas dañinas. La autotolerancia es adquirida, más que heredada, ya que todos los individuos heredan los mismos genes receptores de antígeno.
Durante la maduración de las células T llegan células tronco de la médula ósea hacia el timo, en donde los genes del receptor antigénico se recombinan para formar una variedad casi infinita de receptores antigénicos, algunos de los cuales son específicos para los autoantígenos. En una fase inmadura del desarrollo linfocitario, el encuentro con los antígenos causa tolerancia como resultado de anergia o deleción clonal, mientras que en una fase de mayor madurez se produce activación linfocitaria y expansión clonal. Las células Tinmaduras del timo y las células B inmaduras de la médula ósea se encuentran con autoantígens que son procesados y presentados a linfocitos que tienen receptores específicos para ellos. Los linfocitos que han desarrollado receptores que reconocen autoantígenos son eliminados del repertorio de receptores linfocíticos, y solo los linfocitos que no reconocen autoantígenos pueden dirigirse hacia los órganos linfoides periféricos. Por lo tanto, las clonas de linfocitos que reconocen autoantígenos son eliminadas (deleción clonal) o inactivadas (anergia clonal) en un proceso general denominado selección negativa.4
Las señales bioquímicas que determinan si las células sufren deleción o anergia se desconocen. Este proceso no es totalmente eficaz; algunas clonas autorreactivas escapan hacia los órganos linfoides periféricos, en donde pueden ser inactivadas por mecanismos que inducen tolerancia periférica (ver antes). La base más importante para la tolerancia a los antígenos proteicos es la tolerancia por parte de células T, debido a que la deleción o anergia de células T es insuficiente para prevenir la inmunidad celular y la activación de células B.1-4
Además, no se sabe porqué y cómo, las células T dirigidas contra los autoantígenos son seleccionadas en forma negativa, ni se sabe cómo otras células T que no son autorreactivas son seleccionadas en forma positiva y dejan el timo para poblar los órganos linfoides. Tampoco está clara la manera como se mantiene la autotolerancia al madurar la persona, ya que el timo parece atrofiarse en los adultos. Para las células T autorreactivas que escapan a la delección o inactivación en el timo, la tolerancia puede ser inducida en los órganos linfoides periféricos, por mecanismos que parecen ser semejantes a los de la inducción de tolerancia periférica a antígenos extraños (ver antes). Se ha postulado que la presentación de antígenos por CPA deficientes en coestimuladores es un mecanismo importante de tolerancia periférica. La presencia de inflamación puede inducir reacciones autoinmunes al activar CPA, ya que la activación puede inducir moléculas coestimuladoras.
La presencia de células T inhibidoras puede también mantener la tolerancia periférica. Por ejemplo, el subtipo de célula TH1 media las reacciones de hipersensibilidad tardía, pero la actividad de estas células puede ser inhibida por el subtipo TH2. Es posible que otras células T supresoras puedan también suprimir la autorreactividad inmunológica.1
Durante la maduración las células B pasan a través de un estado en el que expresan IgM, su primer receptor antigénico. Si la célula B se encuentra con autoantígenos en la médula ósea que reaccionan con sus receptores IgM en esta fase, sufre deleción o anergia clonal. Las células B inmaduras que no se encuentran con antígenos específicos continúan madurando a una fase más tardía en la que expresan receptores antigénicos tanto IgM como IgD. A continuación entran a los tejidos linfoides periféricos, en donde la exposición a antígenos causa su activación. Como en el caso de las células T, el grado de maduración de las células B determina si al encontrarse con el antígeno sufren anergia, deleción o activación clonal. Las células B más maduras localizadas en los órganos linfoides periféricos han sido seleccionadas de las autorreactivas, y por lo general se activan al exponerse a antígenos en presencia de células T.1-3
Enfermedades autoinmunes
Las enfermedades autoinmunes afectan a uno a dos porciento de la población, y se presentan en forma de trastornos muy diversos [ver tabla 1]. Sin embargo, es importante distinguir entre los fenómenos autoinmunes y las enfermedades autoinmunes.
Los fenómenos autoinmunes pueden desarrollarse en diversas enfermedades aunque tengan poco que ver con la patogenia de la enfermedad. Las enfermedades que causan lesión o muerte de los tejidos (v.gr., infarto del miocardio) pueden ocasionar la liberación de antígenos tisulares inmunogénicos que habían estado secuestrados y escondidos del sistema inmune. La respuesta inmune, incluyendo los anticuerpos producidos, es incidental y tiene poco que ver con la patología miocárdica causada por una oclusión coronaria.
En las enfermedades autoinmunes la autoinmunidad es un elemento importante en la patogenia de la enfermedad. Las enfermedades autoinmunes son causadas por ruptura de la tolerancia inmunológica a los autoantígenos. La pérdida de la autotolerancia puede deberse a uno o más de varios mecanismos, incluyendo la ausencia de eliminación de las clonas autorreactivas, la activación de las células anérgicas autorreactivas o la liberación de autoantígenos secuestrados que normalmente son inaccesibles al sistema inmunológico. Es claro que múltiples factores contribuyen al desarrollo de las enfermedades autoinmunes, incluyendo la constitución genética del huesped y la infección con ciertos microrganismos. Debido a la diversidad de mecanismos y factores del huésped que participan en las enfermedades autoinmunes, se piensa que son un gran número de padecimientos los que ocurren debido a la autoinmunidad [ver tabla 1].
Las enfermedad autoinmune se clasifican como órgano específicas o sistémicas (i.e., no órgano específicas) con base en la localización principal del daño. Sin embargo, estas dos categorías, representan dos extremos; algunas enfermedades órgano específicas pueden afectar más de un órgano o producir efectos sistémicos, y algunas enfermedades sistémicas pueden dañar más un órgano que a otros. Por ejemplo, los pacientes con tiroiditis de Hashimoto, que se caracteriza por una reacción inflamatoria en la tiroides y por anticuerpos antitiroglobulina, también producen anticuerpos contra antígenos no tiroideos, como los presentes en las células gástricas parietales. Sin embargo, estos anticuerpos no suelen causar manifestaciones relacionadas con la mucosa gástrica, como anemia perniciosa. No obstante, la mayor parte de las enfermedades órgano específicas afectan solamente uno o unos cuantos órganos.
PATOGENIA DE LA LESION TISULAR
Tres mecanismos son los principales responsables de la inflamación y daño tisular en las enfermedades autoinmunes: lisis celular y liberación de moduladores inflamatorios estimulados por autoanticuerpos, enfermedad por complejos inmunes, e inmunidad modulada por células.
En el primer mecanismo, los autoanticuerpos circulantes reaccionan con antígenos modificados o no modificados sobre las superficies celulares. A continuación, los anticuerpos unidos estimulan la liberación de moduladores de la inflamación, activan la vía del complemento o activan células citotóxicas del sistema inmune que poseen receptores para la porción Fc de la IgG, como los macrófagos y una subpoblación de linfocitos granulares grandes conocidos como células K. Los últimos dos procesos provocan lisis celular.
En el segundo mecanismo, se forman complejos entre autoanticuerpos y antígenos en la circulación o en los líquidos intracelulares. Estos complejos inmunes se depositan después en varios tejidos, incluyendo glomérulos, articulaciones y vasos sanguíneos; en forma subsecuente fijan complemento y causan inflamación y daño tisular. El sitio del depósito se determina por las propiedades físicas del complejo inmune, como el tamaño y la carga.
En el tercer mecanismo, las células T sensibilizadas dañan a las células directamente o liberan linfocinas que amplifican las respuesta inflamatoria (v.gr., al aumentar la llegada de otras células inflamatorias). Aunque la lesión tisular causada por mecanismos modulados por células puede ser importante en las enfermedades autoinmunes, su papel no se ha esclarecido hasta la fecha.1,2
Mecanismos de autoinmunidad
Las enfermedades autoinmunes son causadas por alteraciones en la función de los linfocitos. Las alteraciones en la función de las células T pueden causar enfermedades a través de inmunidad mediada por células, y la actividad de las células T cooperadoras en la producción de anticuerpos puede contribuir a la formación de autoanticuerpos. El papel central de las células T cooperadoras en la enfermedad autoinmune se demustra por la asociación que tienen muchas de estas enfermedades con ciertas moléculas de HLA, ya que el reconocimiento de los antígenos procesados presentados por las moléculas de HLA es crítico para la activación de los linfocitos T cooperadores. La falla de uno o más pasos para mantener la tolerancia puede causar autoinmunidad.
FALLA DE LA TOLERANCIA CENTRAL
El proceso de selección negativa de las células T autorreactivas en el timo es el principal mecanismo de la autotolerancia, y la falla de esta selección negativa puede permitir que sobrevivan células T autorreactivas. Sin embargo, existen pocas evidencias directas de que este mecanismo sea responsable de la enfermedad humana autoinmune.
FALLA DE LA TOLERANCIA PERIFERICA
Un mecanismo de tolerancia periférica consiste en la presentación de un autoantígeno por una CPA que no tenga coestimuladores, como B7. En ciertas condiciones (v.gr., inflamación), las CPA pueden activarse para expresar coestimuladores, proporcionando así las condiciones necesarias para activar las células T específicas para autoantígenos. En un estudio, la interacción entre las moléculas accesorias B7 y CD28 fue bloqueada en ratones Nueva Zelanda por medio de la administración de una proteína de fusión que contenía el dominio extracelular CD28 unido a la porción Fc de la IgG. El bloqueo de esta interacción alivió la enfermedad autoinmune en estos ratones, que sirven como modelos del lupus eritematoso sistémico (LES) humano.5 En otro estudio, el tratamiento de los ratones con anticuerpos contra una proteína de 39 kd, la gp39, que es una molécula accesoria en las células T CD4+, evitó el desarrollo de artritis inducida por colágena tipo II.6 También se ha demostrado que la sobreproducción de la interleucina-2 (IL-2) impide la tolerancia en células T autorreactivas.
Otro mecanismo que se ha sugerido para superar la tolerancia periférica es la presencia de un antígeno con múltiples determinantes que contenga un epítope que parece un autoantígeno y otro que es extraño. Por ejemplo, un antígeno multivalente puede tener un epítope reconocido por una célula B. Debido a que las células B pueden ser también CPA, el mismo epítope antigénico u otro diferente puede ser procesado por la célula B y presentado en la molécula HLA sobre la superficie celular. Cuando el epítope presentado proviene de un autoantígeno, la célula T cooperadora lo reconoce como propio, y no proporciona la ayuda necesaria para la generación de clonas reactivas que proliferen en células B productoras de anticuerpos [ver figura 2]. Por otro lado, si el antígeno multideterminante contiene un epítope propio y uno extraño , la célula B puede reconocer el epítope propio y no reaccionar. Si el epítope extraño es procesado y presentado a las células T cooperadoras, las células reconocerán el antígeno como extraño y estimularán a las células B para que maduren en una célula que produzca anticuerpos contra el autoepítope. Esta hipótesis propone que las células B con receptores para antígenos propios pueden no reaccionar por tolerancia de las células T cooperadoras. Sin embargo, la misma célula B puede estimularse para producir anticuerpos si el antígeno contiene también un epítope antigénico extraño que sea presentado por las células B y reconocido por otras células T cooperadoras específicas para ese epítope extraño. Aunque no existen evidencias directas deque este mecanismo cause enfermedades autoinmunes, constituye una posible explicación para la hipótesis denominada mimetismo molecular.1,2
MIMETISMO MOLECULAR
Los antígenos extraños y los autoantígenos que comparten epítopes antigénicos similares o idénticos proporcionan la base para la hipótesis del mimetismo molecular. Existen varios ejemplos de bacterias y virus que contienen proteínas con secuencias cortas de aminoácidos que son idénticas a secuencias de aminoácidos en las proteínas del huésped. Se ha propuesto que estas secuencias de aminoácidos estructuralmente idénticas permiten que el organismo invasor induzca reacciones autoinmunes en el huésped. El huésped en cuestión debiera ser tolerante al epítope del microrganismo que es idéntico a un epítope propio. Muchos investigadores han propuesto que esta similitud causa autoinmunidad. Un posible mecanismo para la autoinmunidad basada en el mimetismo molecular es la presencia de antígenos complejos o multivalentes que presentan autoepítopes del huésped como si fueran epítopes extraños [ver figura 2]. Otra posibilidad es que las diferencias en los aminoácidos adyacentes a la secuencia idéntica de aminoácido sea considerada por el sistema inmunológico como semejante pero no idéntica (suficientemente parecida como para causar anticuerpos que tengan reacción cruzada con los epítopes, pero lo bastante diferente para que los autoepítopes evadan la tolerancia).
Se ha propuesto un ejemplo de mimetismo molecular en el síndrome de Reiter. Este consiste en uretritis o balanitis, artritis inflamatoria, conjuntivitis y/o dermatitis secundarias a la infección por enterobacterias patógenas (v.gr., organismos Shigella) o micoplasmas de adquisición venérea. El síndorme suele ocurrir en personas que portan el antígeno HLA-B27 del complejo principal de histocompatibilidad (CPH). Las cepas artritogénicas de Shigella contienen un plásmido de 2 Md (megadalton) que tiene una secuencia corta de aminoácidos idéntica a la secuencia de la región hipervariable del HLA-B27. Se ha sugerido que las infecciones porShigella en personas con el antígeno HLA-B27 causan síndrome de Reiter al inducir una reacción autoinmune con base en el mimetismo molecular entre estos epítopes, lo que causa reacciones autoinmunes contra las moléculas HLA-B27 del huésped.7 También la fiebre reumática representa una forma de mimetismo molecular. En este caso, la enfermedad reumática cardiaca puede ser causada por anticuerpos antiestreptococos que tienen reacción cruzada con antígenos miocárdicos.1
ACTIVACION POLICLONAL DE LINFOCITOS
Los linfocitos pueden ser activados por estímulos diferentes a los de los antígenos que se unen a sus receptores antigénicos. Las células B anérgicas autorreactivas pueden proliferar en respuesta a la endotoxina bacteriana, también llamada lipopolisacárido. De esta manera, ciertas infecciones bacterianas pueden estimular reacciones autoinmunes; sin embargo, no se ha demostrado que ninguna enfermedad humana conocida sea causada por este mecanismo.1
Los superantígenos bacterianos son una clase de activadores policlonales que actúan sobre ciertas células T al unirse a las regiones variables de las cadenas ß (regiones Vß) del receptor de antígeno de la célula T, en lugar de unirse al surco fijador de antígeno constituido por las cadenas a y ß de los receptores [ver figura 3]. Cada familia de receptores de células T que contienen Vß incluye a receptores para muchos antígenos que se unen en forma convencional dentro del surco fijador de antígenos entre las cadenas ay ß, debido a que la especificidad del receptor del antígeno está determinada por la recombinación de cada región Vß con diferentes regiones D (diversidad) y J (de unión) y con las mismas cadenas a. Por lo tanto, la activación de todas las células T dentro de una familia de receptores T que contienen Vß, como el Vß7+, activa células con un gran número de especificidades antigénicas, algunas de las cuales pueden ser autoantígenos. Por lo menos algunas de las manifestaciones clínicas de la enfermedad mediada por enterotoxina, como el síndrome de choque tóxico, pueden ser originadas por este mecanismo.1
Las enterotoxinas se unen a las moléculas clase II del CPH en las CPA. Las células T con una cierta región Vß reconocen al complejo superantígeno sobre la CPA, se unen a él y se activan. Las evidencias sugieren que algunas enfermedades autoinmunes pueden ser secundarias a la estimulación del superantígeno en las células T. En un estudio, los pacientes con diabetes mellitus insulinodependiente (DMID), una enfermedad en la que existe destrucción autoinmune de los islotes pancreáticos mediada por células T, tuvieron evidencia de un infiltrado de células T CD4+ rico en receptores celulares T de la familia Vß7 en el islote pancreático, lo que sugiere que el superantígeno participa en la patogenia de la destrucción de los islotes en la DMID.8 La participación del superantígeno implica a su vez a bacterias o virus, ya que los superantígenos suelen asociarse con microrganismos.9
REGULACION DEFICIENTE POR CELULAS T
Se ha postulado que el desequilibrio en las subpoblaciones de linfocitos puede ser el responsable de la autoinmunidad, aunque no existen evidencias directas de este mecanismo en alguna enfermedad autoinmune humana. Este desequilibrio puede incluir mayor actividad de células T supresoras o una alteración entre los subtipos TH1 y TH2 de las células T cooperadoras.
La exposición de los antígenos proteicos a las células linfoides en el intestino puede inducir tolerancia periférica que parece evitar las respuestas sistémicas inmunes a las proteínas de la dieta. La tolerancia por vía oral es inducida por lo menos por dos mecanismos. Uno es la supresión inmunológica por células T CD8+ específicas. Un segundo mecanismo es la supresión inmunológica por células T CD4+ del subtipo TH2, que secretan IL-4, IL-10 y factor de transformación del crecimiento-ß (FTC-ß). La tolerancia oral puede relacionarse con la capacidad de las células linfoides derivadas del intestino para inducir, ante la exposición al antígeno, un subtipo principal de células T CD4+ que secretan ciertas citocinas, incluyendo IL-4 y FTC-ß, que suprimen a las células TH1. Un estudio mostró que la tolerancia oral a la proteína básica de mielina suprime la encefalomielitis experimental autoinmune, un modelo animal que equivale a la esclerosis múltiple en humanos.10 Los resultados de estos estudios preliminares de tolerancia inducida por vía oral han sugerido que puede ser benéfico aplicar este mecanismo en la esclerosis múltiple y la artritis reumatoide.1,40
DEFECTOS EN LAS VIAS DE APOPTOSIS
La apoptosis, o muerte celular programada, es indispensable para la deleción de linfocitos autorreactivos. El proceso se inicia por una de varias interacciones ligando-receptor y continúa por la activación de endonucleasas como consecuencia de la generación de mensajeros intracelulares. La apoptosis también puede iniciarse por la ausencia de factores de crecimiento como la IL-2.
Es posible que el sistema Fas-Fas ligando sea importante en la enfermedad autoinmune. (Fas es una proteína de la superficie celular que media la apoptosis, mientras que el ligando Fas es una proteína de membrana tipo II relacionada con el factor de necrosis tumoral que se fija a Fas.) Las lesiones genéticas que causan alteraciones y disfunción de Fas o del ligando Fas se asocian con enfermedad autoinmune murina. En uno de estos modelos animales (la cepa MRL que porta en gen lpr [de linfoproliferación]) se acumulan gran número de linfocitos en los órganos linfoides como resultado de una apoptosis defectusa relacionada con una proteína Fas anormal. Este dato puede ser importante en el lupus humano, ya que se ha visto que los pacientes con LES tienen mayor producción de una forma soluble de molécula Fas causada por la delección de la porción transmembrana de la molécula. La proteína Fas circulante puede inhibir la interacción Fas-Fas ligando necesaria para la apoptosis normal y puede permitir la persistencia de células autorreactivas. Las lesiones en el sistema Fas y en otros sistemas requeridos para la apoptosis normal pueden contribuir a la patogenia de otras enfermedades autoinmunes. Varios medicamentos inmunosupresores y citotóxicos actúan a través de la inducción de apoptosis. La investigación relacionada con el sistema Fas-Fas ligando puede permitir el desarrollo de agentes que induzca una apoptosis más específica que la inducida por los agentes actuales.11,12
Susceptibilidad del huésped a las enfermedades autoinmunes
Los clínicos han observado desde hace mucho tiempo que los factores hereditarios parecen contribuir a la incidencia de las enfermedades autoinmunes. Varios estudios han demostrado una alta tasa de concordancia en gemelos monocigotos comparada con gemelos dicigotos. Sin embargo, la concordancia de varias enfermedades autoinmunes, incluyendo la DMID, el LES y la artritis reumatoide, es cercana al 15 a 30 porciento en la mayoría de los estudios. Por lo tanto, los factores genéticos son importantes en la susceptibilidad a las enfermedades autoinmunes, pero incluso las personas con genomas idénticos difieren en su susceptibilidad. La pregunta entonces es, ¿qué factores diferentes a los controlados por la genética pueden ser responsables de estos trastornos? Los factores ambientales podrían ser importantes, incluyendo a ciertas infecciones, pero también se ha sugerido que elementos estocásticos, no genéticos, relacionados con la maduración de los linfocitos, pueden influir en la susceptibilidad a la enfermedad. Por ejemplo, la recombinación de los segmentos genéticos para los receptores de las células T y B no se encuentra bajo control genético, sino que ocurre en forma aleatoria. Sin embargo, el reconocimiento antigénico por las células T requiere del sistema HLA, que está definido genéticamente.13
PAPEL DE LOS ANTIGENOS LEUCOCITARIOS HUMANOS
Se han definido muchas asociaciones entre las enfermedades autoinmunes y los antígenos del HLA. Estas varían desde asociaciones leves, con riesgos relativos de 1 o 2, hasta la asociación más estrecha, la de la espondilitis anquilosante con el HLA-B27, que confiere un riesgo relativo de 90 a 100 [ver tabla 2]. La interpretación de estas asociaciones sigue siendo difícil, y se han propuesto muchas explicaciones. Una posible explicación para la asociación de una enfermedad con un antígeno HLA es que el antígeno HLA puede ser más eficaz que otros para apoyar el reconocimiento de las células T de un autoantígeno. Otra posibilidad es que ciertos antígenos HLA sean menos eficaces que otros para apoyar la respuesta inmunológica contra un agente infeccioso, que puede iniciar la enfermedad autoinmune. Además, se ha postulado que un agente infeccioso puede utilizar un cierto antígeno HLA en la superficie de las células como un receptor para entrar a la célula, en la misma manera que el virus de inmunodeficiencia humana (VIH) utiliza a la molécula CD4 para la invasión celular.1,2,14
Algunas asociaciones entre los antígenos HLA y las enfermedades autoinmunes representan asociaciones estrechas entre otros antígenos HLA que se encuentran en desequilibrio de unión; esto es, dos alelos HLA están lo bastante cerca uno de otro en el cromosoma para heredarse juntos. Un ejemplo sucede en la DMID, que se asocia tanto con DR3 como con DR4. Sin embargo, la DMID se asocia en forma más estrecha con un alelo DQ, el DQ3.2, que se encuentra en desequilibrio de unión con DR3 y DR4. Aún más, cada antígeno HLA definido serológicamente, como el DR4, es una familia de alelos estructuralmente semejantes, aunque no idénticos. El análisis molecular de estos alelos ha revelado algunos requerimientos estructurales para las asociaciones con enfermedades autoinmunes. Por ejemplo, en por lo menos un grupo poblacional, las cadenas ß del DQ asociado a la DMID tienen uno de tres aminoácidos (alanina, valina o serina) cerca del sitio de ruptura de unión del péptido en la posición 57 del alelo, mientras que los alelos de las caenas ß del DQ que no se asocian con DMID tienen ácido aspártico en esa posición. Estas observaciones sugieren que la susceptibilidad a la DMID está determinada, en parte, por la estructura de la región de unión al péptido de la molécula DQ que determina la capacidad del alelo DQ para presentar ciertos antígenos a las células T.
Otro ejemplo de requerimientos estructurales discretos de las moléculas de clase II del HLA para la susceptibilidad a la enfermedad es la asociación de la artritis reumatoide con el antígeno DR4. Se ha determinado por análisis estructural de las moléculas individuales DR4 que ciertas secuencias de aminoácidos (en la región Vß cerca de los sitios de combinación con el antígeno y las regiones de contacto con células T de estas moléculas) se asocian con la artritis reumatoide, mientras que otras secuencias no tienen esta asociación. Además, la asociación entre artritis reumatoide y DR1 en otras poblaciones se basa en una semejante estructural de los aminoácidos entre el DR1 y el DR4.13
Es posible que el sistema HLA en sí pueda no ser responsable de la susceptibilidad genética a una enfermedad autoinmune y que otros genes que se encuentran en desequilibrio de unión con los genes HLA sean los responsables. Sin embargo, es probable que las moléculas HLA per se sean las directamente responsables en vista de su función para dirigir las respuestas inmunológicas. Las principales evidencias al respecto provienen de los trabajos de Taurog y colaboradores. Estos investigadores han mostrado que líneas transgénicas de ratas que expresan el HLA-B27 humano en forma espontánea desarrollan lesiones inflamatorias semejantes a las del síndrome de Reiter humano que, como la espondilitis anquilosante, se asocia en forma estrecha con el HLA-B27.15,16
Es claro, a partir de los análisis genéticos de las enfermedades autoinmunes murinas, que varios genes participan en la patogenia de una sola enfermedad.2 Las enfermedades autoinmunes humanas tienen la misma complejidad, y es posible que varios rasgos genéticos sean responsables de la susceptibilidad del huésped. Las infecciones con agentes virales o bacterianos parecen iniciar por lo menos algunas enfermedades autoinmunes, pero no se encuentran estos microrganismos en los tejidos lesionados en las etapas subsiguientes de la enfermedad. Existen varios mecanismos posibles por los que las infecciones pueden desencadenar autoinmunidad (ver antes). Por ejemplo, las infecciones pueden activar la detección inmune de células autorreactivas a través de la expresión de coestimuladores, como el B7, sobre las CPA o a través de la activación de la producción de citocinas. Las infecciones pueden exponer al sistema inmune a autoantígenos previamente secuestrados, a los cuales el sistema inmunológico no tiene tolerancia. La lesión tisular puede crear también neoantígenos que inciten reacciones inmunológicas. La exposición hormonal puede modificar la autoinmunidad, según indica la dependencia genética de muchas enfermedades autoinmunes. Por ejemplo, alrededor del 90 porciento de los casos de LES ocurren en mujeres, mientras que un porcentaje semejante de casos de espondilitis anquilosante se presentan en varones. No son claros los motivos para este sesgo en función del sexo en las enfermedades autoinmunes.1,2
PAPEL DE LOS AUTOANTICUERPOS
Con frecuencia existen autoanticuerpos en las enfermedades autoinmunes, pero puede ser difícil establecer que estos anticuerpos contribuyen en forma significativa a la patogenia de las enfermedades autoinmunes. Los autoanticuerpos que contribuyen con más probabilidad en la patogenia son los dirigidos contra antígenos de la superficie celular. Los autoanticuerpos dirigidos contra antígenos extracelulares pueden ser o no patogénicos. Por lo general se acepta que los anticuerpos no pueden reaccionar con antígenos en localizaciones intracelulares, pero las reacciones de los anticuerpos con antígenos intracelulares pueden contribuir a la enfermedad en ciertas circunstancias. Es posible que los antígenos intracelulares causen reacciones patógenas con autoanticuerpos si los antígenos son liberados y pueden fijarse a la superficie celular o a localizaciones extracelulares, o si los antígenos son traslocados a áreas accesibles de la superficie celular, ya sea que sean específicos o tengan reacción cruzada. Algunos ejemplos de enfermedades autoinmunes ilustran estos principios (ver adelante).17
Autoanticuerpos contra moléculas de la superficie celular
El pénfigo es una enfermedad cutánea bulosa relacionada con acantolisis, o pérdida de la adhesión de las células epidérmicas y que se asocia con anticuerpos contra los desmosomas (moléculas de la superficie celular epidérmica que participan en la adhesión intercelular). Varias líneas de evidencia apoyan el papel patogénico de estos autoanticuerpos en el pénfigo. Primero, es lógico suponer que la menor función de adhesión de los desmosomas epidérmicos puede causar acantolisis. Segundo, existen anticuerpos antidesmosomas en forma constante en los pacientes con pénfigo (y los niveles de anticuerpos correlacionan con la actividad de la enfermedad) y la eliminación de los anticuerpos séricos por plasmaféresis causa mejoría de los síntomas. Tercero, la transferencia pasiva de suero de pacientes con pénfigo vulgar causa lesiones semejantes en ratones. Cuatro, los recién nacidos de madres con pénfigo tienen una enfermedad transitoria causada por la trasmisión transplacentaria de los anticuerpos maternos.17
Casi todos los pacientes con miastenia gravis tienen autoanticuerpos contra receptores de acetilcolina (RAC) en el músculo esquelético [ver figura 4]. Sin embargo, el grado de bloqueo neuromuscular observado en esta enfermedad no siempre es paralelo a los niveles de anticuerpos anti-RAC. La inyección de anticuerpos contra RAC puede inducir debilidad miasténica en animales de experimentación. Los anticuerpos anti-RAC en los pacientes con miastenia gravis causan destrucción del RAC, pero rara vez bloquean el sitio de fijación de la acetilcolina. Los anticuerpos son policlonales y reaccionan con varios idiotipos en el RAC, así como con porciones no-RAC de la unión neuromuscular. No existe una buena correlación entre la severidad de la enfermedad y los niveles de anticuerpos anti-RAC; sin embargo, la respuesta de la enfermedad a la plasmaféresis sugiere que algunos, pero no todos, los anticuerpos, pueden ser responsables de la enfermedad.17,18
Los anticuerpos contra varios receptores hormonales pueden ocasionar endocrinopatías. Los niveles altos de anticuerpos contra el receptor periférico de insulina se asocian con DMID, quizá como resultado de la degradación de los receptores por mecanismos autoinmunes. En forma paradójica, los niveles bajos de anticuerpos pueden estimular al receptor de insulina al reproducir el efecto de la insulina, ocasionando hipoglucemia.
La enfermedad autoinmune de la tiroides se asocia con anticuerpos dirigidos contra tres antígenos: peroxidasa tiroidea microsomal, tiroglobulina y receptor tiroideo de la hormona estimulante de la tiroides (TSH). Los anticuerpos contra el receptor de TSH pueden tener la misma acción de esta hormona, ocasionando enfermedad de Graves. Otra enfermedad autoinmune de la tiroides, la tiroiditis de Hashimoto, se asocia con anticuerpos contra el receptor de TSH, pero su papel patogénico en esta enfermedad no es claro. Es menos frecuente que los anticuerpos contra el receptor de TSH puedan bloquear la acción de la TSH y causar hipotiroidismo. No se ha establecido el papel patogénico de las otras dos clases de autoanticuerpos antitiroideos.17
Autoanticuerpos contra moléculas extracelulares
El síndrome de anticuerpos antifosfolípido (SAF) consiste en trombosis recurrente, pérdidas fetales y trombocitopenia asociadas con anticuerpos contra cardiolipina u otros fosfolípidos de carga negativa, y alteraciones de ciertas pruebas de coagulación, conocidas como anticoagulante lúpico. El SAF puede ser primario, en ausencia de una enfermedad asociada, o secundario, casi siempre asociado a LES o sus variantes. No es claro si el anticuerpo antifosfolípido induce las trombosis en forma directa, aunque la asociación de estos anticuerpos con el SAF es muy estrecha. Los anticuerpos no se unen a los fosfolípidos aislados, sino a un complejo de fosfolípidos y un cofactor del plasma, la ß2-glicoproteína I. El anticoagulante lúpico se refiere a la capacidad de los anticuerpos antifosfolípidos para interferir con varias pruebas de la coagulación in vitro, pero in vivo estos anticuerpos promueven la trombosis, en lugar de inhibirla. Otros autoanticuerpos contra las moléculas extracelulares incluyen anticuerpos anti-insulina que pueden ser espontáneos o secundarios a la administración de insulina, así como anticuerpos contra la colágena tipo II que se asocian con la artritis reumatoide humana y la policondritis. Los anticuerpos contra colágena tipo II son casi siempre secundarios al daño en el cartílago, pero pueden contribuir a la inflamación en el cartílago de articulaciones y otras estructuras.
Autoanticuerpos contra moléculas intracelulares
Aunque los autoanticuerpos contra antígenos intracelulares se asocian mucho con enfermedades, suele ser difícil determinar si estos anticuerpos contribuyen a la patogenia de la enfermedad. La cirrosis biliar primaria (CBP) se asocia con forma estrecha con la presencia de anticuerpos antimitocondriales, y estos anticuerpos son muy específicos para la enfermedad. A pesar de la amplia distribución de las mitocondrias, la patología en la CBP se limita en forma casi exclusiva a los conductos biliares intrahepáticos. Los anticuerpos antimitoconcriales se dirigen contra un epítope en el complejo multienzimático piruvato deshidrogenasa. A pesar de la especificidad de estos anticuerpos para la CBP, parece muy poco probable que estos anticuerpos puedan causar la CBP, por la inaccesibilidad del antígeno al anticuerpo circulante.
Otro ejemplo de asociación muy específica de autoanticuerps con significado patogénico dudoso es el de la presencia de anticuerpos anti-aminoacil-ARNt sintetasa en la polimiositis. Estas enzimas están presentes en el ARN citoplásmico y son muy importantes para la síntesis proteica. Los niveles de anticuerpos contra sintetasas de ARNt correlacionan con la actividad de la enfermedad, pero la plasmaféresis es ineficaz. Existen autoanticuerpos contra varios componentes nucleares en diversas enfermedades autoinmunes reumatológicas, incluyendo el LES, la esclerodermia y el síndrome de Sjögren. Estos anticuerpos están dirigidos hacia ribonucleoproteínas nucleares pequeñas que participan en la separación del ARN mensajero, los componentes nucleares del centrómero y otras proteínas nucleares. Ninguno de estos anticuerpos han demostrado tener un papel definido en la patogenia de las enfermedades asociadas.17
Los autoanticuerpos pueden reaccionar con proteínas intracelulares que también aparecen en las superficies celulares o en localizaciones extracelulares. Ocurren anticuerpos contra las proteínas ribosomales P en el LES, sobre todo en pacientes con depresión o psicosis. Es interesante que estos anticuerpos reaccionan también con una proteína en la superficie celular que es semejante en tamaño al antígeno proteico ribosomal. No es claro si la proteína de superficie celular es la proteína ribosomal que ha sido transportada a la superficie celular o es otra proteína semejante. Sin embargo, la presencia del antígeno sobre la superficie celular sugiere que los anticuerpos contra proteínas ribosomales P pueden contribuir a las manifestaciones neurológicas del LES. En enfermos con LES y síndrome de Sjögren se presentan anticuerpos contra las pequeñas nucleoproteínas antigénicas SS-A (también llamada Ro), y SS-B (o La) lo mismo que en pacientes con lupus neonatal y bloqueo cardiaco congénito. Estos dos últimos transtornos del recién nacido se asocian con el paso trasplacentario de anticuerpos maternos. Se ha demostrado que los anticuerpos anti-Ro reaccionan con y alteran la permeabilidad del tejido cardiaco fetal, pero no el tejido cardiaco de los adultos.
Otro anticuerpo dirigido contra un antígeno intracelular que quizá contribuye a la enfermedad es el anticuerpo contra citoplasma de neutrófilos (ANCA). Este anticuerpo está dirigido contra la proteinasa-3, que existe en los lisosomas primarios de neutrófilos y monocitos. Al liberarse estos lisosomas durante la activación celular, su contenido se expone lo suficiente como para raccionar con los anticuerpos y las células inmunológicas. Desde el punto de vista experimental, los neutrófilos se activan al exponerse a citocinas y liberan sus gránulos y otras sustancias inflamatorias al exponerse a los ANCA. Se ha sugerido que en estos experimentos, la activación causa exposición de proteinasa-3 e imunización o formación de complejos inmunes con anticuerpos específicos, y que estos complejos pueden contribuir a la vasculitis y la lesión tisular. Los anticuerpos ANCA son específicos para la granulomatosis de Wegener y un pequeño número de enfermedades con patología renal semejante. La proteinasa-3 podría contribuir a la patología de estas enfermedades, ya sea por su movimiento hacia la superficie celular o por su liberación hacia el ambiente extracelular, con la subsecuente activación de los neutrófilos por los complejos inmunes. La activación de los neutrófilos puede entonces dañar las paredes vasculares y los tejidos circundantes.
Se observan anticuerpos contra ADN desnaturalizado o de una cadena en diversas enfermedades autoinmunes, pero su papel patogénico no es claro. Los anticuerpos contra el ADN nativo o de doble cadena son bastante específicos del LES, y es posible que participen en la enfermedad. En alguna ocasión se penso que complejos inmunes de ADN nativo-antiADN viajaban en la circulación y se depositaban en los tejidos, como en otras enfermedades por complejos inmunes, como la enfermedad del suero. Sin embargo, estudios posteriores sugieren un mecanismo diferente. El glomérulo normal contiene heparán sulfato, un polisacárido aniónico que puede formar complejos con histonas catiónicas, y se ha sugerido que estas moléculas unidas a histonas pueden a su vez captar al ADN en los complejos circulantes ADN-antiADN, fijando los complejos al glomérulo. Sin embargo, el papel de los complejos ADN-antiADN en el LES no se ha comprobado aún. La actividad de la enfermedad no siempre correlaciona con las concentraciones de anticuerpos anti-ADN, y la plasmaféresis ha sido ineficaz en la nefritis por LES.17
Los pacientes con DMID desarrollan autoanticuerpos contra varias proteínas de las células de los islotes pancreáticos. Entre los primeros anticuerpos encontrados antes del desarrollo de la diabetes se encuentran los anticuerpos contra la proteína de 64 kd presente tanto en el citoplasma como en las superficies celulares. Los pacientes con el síndrome diabético de hombre rígido, una enfermedad rara que consiste en diabetes, epilepsia y espasticidad muscular, desarrollan también anticuerpos contra el mismo antígeno de 64 kd. Este antígeno parece ser la decarboxilasa del ácido glutámico, una enzima que convierte el ácido glutámico al neurotrasmisor gama-aminobutírico en ciertas neuronas. La decarboxilasa del ácido glutámico se encuentra también en los islotes pancreáticos, aunque se desconoce su función en estas células. Una secuencia de 24 aminoácidos de esta enzima tiene homología estructural con la proteína P2-C de un picornavirus (el coxsackievirus B4), que se ha implicado como agente etiológico de la DMID. La plasmaféresis puede ser benéfica en el síndrome de hombre rígido, lo que sugiere que la decarboxilasa del ácido glutámico tiene un papel patogénico.18 El síndrome de hombre rígido se presenta también como un complejo paraneoplásico asociado con cáncer de mama. Sin embargo, en estos pacientes se desarrolla un anticuerpo contra la proteína neuronal de 128 kd que se concentra cerca de las sinapsis, más que contra la decarboxilasa del ácido glutámico.19
En el ratón diabético no obeso, un modelo para la DMID humana, se desarrolla una respuesta de células TH1 contra un epítope en la decarboxilasa del ácido glutámico al inicio de la insulitis; después se presenta reactividad de las células T a otros epítopes de la decarboxilasa del ácido glutámico, lo mismo que reactividad contra otros antígenos de las células de los islotes. La inducción de tolerancia de las células T a la decarboxilasa del ácido glutámico evita la autoinmunidad contra otros antígenos de las células de los islotes, lo mismo que la insulitis y la diabetes. Estos datos sugieren que la autoinmunidad contra una porción especial de la decarboxilasa del ácido glutámico es un factor importante en la patogenia de la DMID murina.20
Bibliografía
DR. DWIGHT R. ROBINSON
Uno de los conceptos centrales de la inmunología lo constituye el concepto de que las reacciones autoinmunes son nocivas para el huésped. Alrededor de 1900, Ehrlich postuló que el sistema inmune adquiere un estado de tolerancia para los antígenos propios; además, propuso que el rompimiento de la tolerancia conduciría a la autodestrucción, estado que describió como horror autotóxico. Cerca de 50 años más tarde, Burnet indicó que las células inmunes autorreactivas son destruidas por los órganos linfoides en las etapas tempranas del desarrollo del individuo, lo que conduce a la tolerancia de los autoantígenos.
Las respuestas inmunológicas a los organismos extraños están estrictamente controladas para minimizar el daño a los tejidos propios. El sistema inmunológico se enfrenta a la enorme tarea de destruir los organismos extraños potencialmente dañinos sin lesionar los tejidos del huésped. La falla del sistema inmunológico para responder en forma adecuada a los organismos extraños puede causar infecciones graves y, por otro lado, las reacciones inmunológicas contra los tejidos del huésped pueden originar enfermedades autoinmunes. El mecanismo por el que el sistema inmunológico controla la especificidad e intensidad de estas reacciones es complejo y no se conoce bien, pero se han hecho importantes adelantos en el conocimiento de la autoinmunidad en los años recientes.
Tolerancia
La tolerancia puede definirse como un estado de no respuesta inmunológica a los antígenos. El reconocimiento antigénico tiene lugar en receptores específicos en las células T y B del sistema inmune. Estos encuentros pueden ocasionar activación o inhibición de los linfocitos, dependiendo de diversas condiciones en el momento del contacto. Puede adquirirse tolerancia ante antígenos extraños, y la autotolerancia es una forma especial que se aplica a los autoantígenos. La tolerancia es una consecuencia de uno de dos efectos sobre las células T y B: (1) deleción clonal, en la que el encuentro con el antígeno causa eliminación o muerte de los linfocitos específicos, o (2) anergia clonal, en la que los linfocitos no son destruidos sino que se vuelven no respondedores ante un antígeno específico.1-3
Burnet propuso la teoría de la selección clonal, la cual establece que en el sistema inmune existen clonas de células inmunes capaces de reaccionar genéticamente con todo inmunógeno potencial; de este modo, la producción de la respuesta inmune es el resultado de la amplificación y proliferación de clonas específicas. Burnet también postuló que la tolerancia puede ocurrir cuando los inmunocitos fetales se exponen y reconocen a un antígeno específico y que este proceso conduce a la deleción de dichas clonas. El apoyo al modelo de la eliminación clonal proviene de experimentos realizados por Billingham y Medawar; sus investigaciones demostraron que la infusión de células linfoides de una cepa murina (cepa B) a ratones recién nacidos de una segunda cepa (cepa A), permitía que la cepa A madura aceptara injertos de piel de los ratones de la cepa B.1
La tolerancia es inmunológicamente específica; puede inducirse hacia una estructura antigénica específica aunque se conserve la reactividad inmunológica contra otros antígenos. La tolerancia está mediada tanto por células T como B, por lo que afecta la inmunidad tanto humoral como celular. Los linfocitos inmaduros son más susceptibles a la inducción de tolerancia que los maduros. La tolerancia central se adquiere cuando los linfocitos inmaduros encuentran antígenos en sus órganos generadores, el timo (células T) y la médula ósea (células B). La tolerancia puede ser inducida en linfocitos maduros si éstos encuentran antígenos en tejidos fuera del timo o la médula ósea bajo ciertas condiciones especiales. A esto se le denomina tolerancia periférica. Tres factores generales pueden causar tolerancia periférica:
1. La forma del antígeno y su vía de administración. Los antígenos en dosis altas, administrados por vía oral y administrados en dosis bajas en forma repetida, pueden causar tolerancia. Los antígenos con alta afinidad por sus receptores antigénicos pueden también inducir tolerancia en lugar de activación.
2. El estado de la célula presentadora de antígeno (CPA). Las células presentadoras de antígeno, como los macrófagos, requieren coestimuladores, como la molécula de superficie B7, antes de que puedan activar a la célula T que reconoce al antígeno procesado que es presentado al receptor de la célula T por el antígeno leucocitario humano (HLA) en la superficie de la CPA. La exposición de las células T al antígeno en las CPA que no tienen B7 u otro estimulador puede causar tolerancia. Las células T que encuentran antígenos en las CPA en ausencia de coestimuladores permanecen tolerantes a estos antígenos, incluso si las células T se exponen después a CPA con antígenos en presencia de coestimuladores. Algunas CPA emiten también señales inhibitorias para la activación de la célula T.
3. La presencia de un subtipo de células T denominadas células T supresoras.
Los mecanismos anteriores se aplican a la inducción de tolerancia de células T, pero la tolerancia puede también ser ocasionada por las células B. En vista de que las respuestas inmunológicas, incluyendo la producción de anticuerpos, son dependientes de células T, el principal mecanismo de tolerancia de células B es la ausencia de la célula T cooperadora durante la presentación del antígeno a las células B. Las respuestas humorales o de células B a antígenos no proteicos (principalmente carbohidratos, como los antígenos de los grupos sanguíneos), son independientes de las células T. La tolerancia de las células B a antígenos T indepenientes puede ser inducida por ciertas formas de antígeno, antígenos en dosis muy altas y antígenos polivalentes.1-3
AUTOTOLERANCIA
La tolerancia del sistema inmunológico a los antígenos propios es indispensable para la salud. La ruptura de la autotolerancia es la base de las enfermedades autoinmunes. En las personas sanas existen antígenos potencialmente inmunogénicos en los tejidos del huésped que son expuestos al sistema inmune, y los tejidos propios no causan respuestas inmunológicas dañinas. La autotolerancia es adquirida, más que heredada, ya que todos los individuos heredan los mismos genes receptores de antígeno.
Durante la maduración de las células T llegan células tronco de la médula ósea hacia el timo, en donde los genes del receptor antigénico se recombinan para formar una variedad casi infinita de receptores antigénicos, algunos de los cuales son específicos para los autoantígenos. En una fase inmadura del desarrollo linfocitario, el encuentro con los antígenos causa tolerancia como resultado de anergia o deleción clonal, mientras que en una fase de mayor madurez se produce activación linfocitaria y expansión clonal. Las células Tinmaduras del timo y las células B inmaduras de la médula ósea se encuentran con autoantígens que son procesados y presentados a linfocitos que tienen receptores específicos para ellos. Los linfocitos que han desarrollado receptores que reconocen autoantígenos son eliminados del repertorio de receptores linfocíticos, y solo los linfocitos que no reconocen autoantígenos pueden dirigirse hacia los órganos linfoides periféricos. Por lo tanto, las clonas de linfocitos que reconocen autoantígenos son eliminadas (deleción clonal) o inactivadas (anergia clonal) en un proceso general denominado selección negativa.4
Las señales bioquímicas que determinan si las células sufren deleción o anergia se desconocen. Este proceso no es totalmente eficaz; algunas clonas autorreactivas escapan hacia los órganos linfoides periféricos, en donde pueden ser inactivadas por mecanismos que inducen tolerancia periférica (ver antes). La base más importante para la tolerancia a los antígenos proteicos es la tolerancia por parte de células T, debido a que la deleción o anergia de células T es insuficiente para prevenir la inmunidad celular y la activación de células B.1-4
Además, no se sabe porqué y cómo, las células T dirigidas contra los autoantígenos son seleccionadas en forma negativa, ni se sabe cómo otras células T que no son autorreactivas son seleccionadas en forma positiva y dejan el timo para poblar los órganos linfoides. Tampoco está clara la manera como se mantiene la autotolerancia al madurar la persona, ya que el timo parece atrofiarse en los adultos. Para las células T autorreactivas que escapan a la delección o inactivación en el timo, la tolerancia puede ser inducida en los órganos linfoides periféricos, por mecanismos que parecen ser semejantes a los de la inducción de tolerancia periférica a antígenos extraños (ver antes). Se ha postulado que la presentación de antígenos por CPA deficientes en coestimuladores es un mecanismo importante de tolerancia periférica. La presencia de inflamación puede inducir reacciones autoinmunes al activar CPA, ya que la activación puede inducir moléculas coestimuladoras.
La presencia de células T inhibidoras puede también mantener la tolerancia periférica. Por ejemplo, el subtipo de célula TH1 media las reacciones de hipersensibilidad tardía, pero la actividad de estas células puede ser inhibida por el subtipo TH2. Es posible que otras células T supresoras puedan también suprimir la autorreactividad inmunológica.1
Durante la maduración las células B pasan a través de un estado en el que expresan IgM, su primer receptor antigénico. Si la célula B se encuentra con autoantígenos en la médula ósea que reaccionan con sus receptores IgM en esta fase, sufre deleción o anergia clonal. Las células B inmaduras que no se encuentran con antígenos específicos continúan madurando a una fase más tardía en la que expresan receptores antigénicos tanto IgM como IgD. A continuación entran a los tejidos linfoides periféricos, en donde la exposición a antígenos causa su activación. Como en el caso de las células T, el grado de maduración de las células B determina si al encontrarse con el antígeno sufren anergia, deleción o activación clonal. Las células B más maduras localizadas en los órganos linfoides periféricos han sido seleccionadas de las autorreactivas, y por lo general se activan al exponerse a antígenos en presencia de células T.1-3
Enfermedades autoinmunes
Las enfermedades autoinmunes afectan a uno a dos porciento de la población, y se presentan en forma de trastornos muy diversos [ver tabla 1]. Sin embargo, es importante distinguir entre los fenómenos autoinmunes y las enfermedades autoinmunes.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Los fenómenos autoinmunes pueden desarrollarse en diversas enfermedades aunque tengan poco que ver con la patogenia de la enfermedad. Las enfermedades que causan lesión o muerte de los tejidos (v.gr., infarto del miocardio) pueden ocasionar la liberación de antígenos tisulares inmunogénicos que habían estado secuestrados y escondidos del sistema inmune. La respuesta inmune, incluyendo los anticuerpos producidos, es incidental y tiene poco que ver con la patología miocárdica causada por una oclusión coronaria.
En las enfermedades autoinmunes la autoinmunidad es un elemento importante en la patogenia de la enfermedad. Las enfermedades autoinmunes son causadas por ruptura de la tolerancia inmunológica a los autoantígenos. La pérdida de la autotolerancia puede deberse a uno o más de varios mecanismos, incluyendo la ausencia de eliminación de las clonas autorreactivas, la activación de las células anérgicas autorreactivas o la liberación de autoantígenos secuestrados que normalmente son inaccesibles al sistema inmunológico. Es claro que múltiples factores contribuyen al desarrollo de las enfermedades autoinmunes, incluyendo la constitución genética del huesped y la infección con ciertos microrganismos. Debido a la diversidad de mecanismos y factores del huésped que participan en las enfermedades autoinmunes, se piensa que son un gran número de padecimientos los que ocurren debido a la autoinmunidad [ver tabla 1].
Las enfermedad autoinmune se clasifican como órgano específicas o sistémicas (i.e., no órgano específicas) con base en la localización principal del daño. Sin embargo, estas dos categorías, representan dos extremos; algunas enfermedades órgano específicas pueden afectar más de un órgano o producir efectos sistémicos, y algunas enfermedades sistémicas pueden dañar más un órgano que a otros. Por ejemplo, los pacientes con tiroiditis de Hashimoto, que se caracteriza por una reacción inflamatoria en la tiroides y por anticuerpos antitiroglobulina, también producen anticuerpos contra antígenos no tiroideos, como los presentes en las células gástricas parietales. Sin embargo, estos anticuerpos no suelen causar manifestaciones relacionadas con la mucosa gástrica, como anemia perniciosa. No obstante, la mayor parte de las enfermedades órgano específicas afectan solamente uno o unos cuantos órganos.
PATOGENIA DE LA LESION TISULAR
Tres mecanismos son los principales responsables de la inflamación y daño tisular en las enfermedades autoinmunes: lisis celular y liberación de moduladores inflamatorios estimulados por autoanticuerpos, enfermedad por complejos inmunes, e inmunidad modulada por células.
En el primer mecanismo, los autoanticuerpos circulantes reaccionan con antígenos modificados o no modificados sobre las superficies celulares. A continuación, los anticuerpos unidos estimulan la liberación de moduladores de la inflamación, activan la vía del complemento o activan células citotóxicas del sistema inmune que poseen receptores para la porción Fc de la IgG, como los macrófagos y una subpoblación de linfocitos granulares grandes conocidos como células K. Los últimos dos procesos provocan lisis celular.
En el segundo mecanismo, se forman complejos entre autoanticuerpos y antígenos en la circulación o en los líquidos intracelulares. Estos complejos inmunes se depositan después en varios tejidos, incluyendo glomérulos, articulaciones y vasos sanguíneos; en forma subsecuente fijan complemento y causan inflamación y daño tisular. El sitio del depósito se determina por las propiedades físicas del complejo inmune, como el tamaño y la carga.
En el tercer mecanismo, las células T sensibilizadas dañan a las células directamente o liberan linfocinas que amplifican las respuesta inflamatoria (v.gr., al aumentar la llegada de otras células inflamatorias). Aunque la lesión tisular causada por mecanismos modulados por células puede ser importante en las enfermedades autoinmunes, su papel no se ha esclarecido hasta la fecha.1,2
Mecanismos de autoinmunidad
Las enfermedades autoinmunes son causadas por alteraciones en la función de los linfocitos. Las alteraciones en la función de las células T pueden causar enfermedades a través de inmunidad mediada por células, y la actividad de las células T cooperadoras en la producción de anticuerpos puede contribuir a la formación de autoanticuerpos. El papel central de las células T cooperadoras en la enfermedad autoinmune se demustra por la asociación que tienen muchas de estas enfermedades con ciertas moléculas de HLA, ya que el reconocimiento de los antígenos procesados presentados por las moléculas de HLA es crítico para la activación de los linfocitos T cooperadores. La falla de uno o más pasos para mantener la tolerancia puede causar autoinmunidad.
FALLA DE LA TOLERANCIA CENTRAL
El proceso de selección negativa de las células T autorreactivas en el timo es el principal mecanismo de la autotolerancia, y la falla de esta selección negativa puede permitir que sobrevivan células T autorreactivas. Sin embargo, existen pocas evidencias directas de que este mecanismo sea responsable de la enfermedad humana autoinmune.
FALLA DE LA TOLERANCIA PERIFERICA
Un mecanismo de tolerancia periférica consiste en la presentación de un autoantígeno por una CPA que no tenga coestimuladores, como B7. En ciertas condiciones (v.gr., inflamación), las CPA pueden activarse para expresar coestimuladores, proporcionando así las condiciones necesarias para activar las células T específicas para autoantígenos. En un estudio, la interacción entre las moléculas accesorias B7 y CD28 fue bloqueada en ratones Nueva Zelanda por medio de la administración de una proteína de fusión que contenía el dominio extracelular CD28 unido a la porción Fc de la IgG. El bloqueo de esta interacción alivió la enfermedad autoinmune en estos ratones, que sirven como modelos del lupus eritematoso sistémico (LES) humano.5 En otro estudio, el tratamiento de los ratones con anticuerpos contra una proteína de 39 kd, la gp39, que es una molécula accesoria en las células T CD4+, evitó el desarrollo de artritis inducida por colágena tipo II.6 También se ha demostrado que la sobreproducción de la interleucina-2 (IL-2) impide la tolerancia en células T autorreactivas.
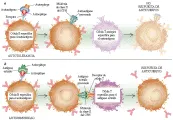
Otro mecanismo que se ha sugerido para superar la tolerancia periférica es la presencia de un antígeno con múltiples determinantes que contenga un epítope que parece un autoantígeno y otro que es extraño. Por ejemplo, un antígeno multivalente puede tener un epítope reconocido por una célula B. Debido a que las células B pueden ser también CPA, el mismo epítope antigénico u otro diferente puede ser procesado por la célula B y presentado en la molécula HLA sobre la superficie celular. Cuando el epítope presentado proviene de un autoantígeno, la célula T cooperadora lo reconoce como propio, y no proporciona la ayuda necesaria para la generación de clonas reactivas que proliferen en células B productoras de anticuerpos [ver figura 2]. Por otro lado, si el antígeno multideterminante contiene un epítope propio y uno extraño , la célula B puede reconocer el epítope propio y no reaccionar. Si el epítope extraño es procesado y presentado a las células T cooperadoras, las células reconocerán el antígeno como extraño y estimularán a las células B para que maduren en una célula que produzca anticuerpos contra el autoepítope. Esta hipótesis propone que las células B con receptores para antígenos propios pueden no reaccionar por tolerancia de las células T cooperadoras. Sin embargo, la misma célula B puede estimularse para producir anticuerpos si el antígeno contiene también un epítope antigénico extraño que sea presentado por las células B y reconocido por otras células T cooperadoras específicas para ese epítope extraño. Aunque no existen evidencias directas deque este mecanismo cause enfermedades autoinmunes, constituye una posible explicación para la hipótesis denominada mimetismo molecular.1,2
|
| Figura 2 |
| Epítopes antigénicos |
MIMETISMO MOLECULAR
Los antígenos extraños y los autoantígenos que comparten epítopes antigénicos similares o idénticos proporcionan la base para la hipótesis del mimetismo molecular. Existen varios ejemplos de bacterias y virus que contienen proteínas con secuencias cortas de aminoácidos que son idénticas a secuencias de aminoácidos en las proteínas del huésped. Se ha propuesto que estas secuencias de aminoácidos estructuralmente idénticas permiten que el organismo invasor induzca reacciones autoinmunes en el huésped. El huésped en cuestión debiera ser tolerante al epítope del microrganismo que es idéntico a un epítope propio. Muchos investigadores han propuesto que esta similitud causa autoinmunidad. Un posible mecanismo para la autoinmunidad basada en el mimetismo molecular es la presencia de antígenos complejos o multivalentes que presentan autoepítopes del huésped como si fueran epítopes extraños [ver figura 2]. Otra posibilidad es que las diferencias en los aminoácidos adyacentes a la secuencia idéntica de aminoácido sea considerada por el sistema inmunológico como semejante pero no idéntica (suficientemente parecida como para causar anticuerpos que tengan reacción cruzada con los epítopes, pero lo bastante diferente para que los autoepítopes evadan la tolerancia).
Se ha propuesto un ejemplo de mimetismo molecular en el síndrome de Reiter. Este consiste en uretritis o balanitis, artritis inflamatoria, conjuntivitis y/o dermatitis secundarias a la infección por enterobacterias patógenas (v.gr., organismos Shigella) o micoplasmas de adquisición venérea. El síndorme suele ocurrir en personas que portan el antígeno HLA-B27 del complejo principal de histocompatibilidad (CPH). Las cepas artritogénicas de Shigella contienen un plásmido de 2 Md (megadalton) que tiene una secuencia corta de aminoácidos idéntica a la secuencia de la región hipervariable del HLA-B27. Se ha sugerido que las infecciones porShigella en personas con el antígeno HLA-B27 causan síndrome de Reiter al inducir una reacción autoinmune con base en el mimetismo molecular entre estos epítopes, lo que causa reacciones autoinmunes contra las moléculas HLA-B27 del huésped.7 También la fiebre reumática representa una forma de mimetismo molecular. En este caso, la enfermedad reumática cardiaca puede ser causada por anticuerpos antiestreptococos que tienen reacción cruzada con antígenos miocárdicos.1
ACTIVACION POLICLONAL DE LINFOCITOS
Los linfocitos pueden ser activados por estímulos diferentes a los de los antígenos que se unen a sus receptores antigénicos. Las células B anérgicas autorreactivas pueden proliferar en respuesta a la endotoxina bacteriana, también llamada lipopolisacárido. De esta manera, ciertas infecciones bacterianas pueden estimular reacciones autoinmunes; sin embargo, no se ha demostrado que ninguna enfermedad humana conocida sea causada por este mecanismo.1
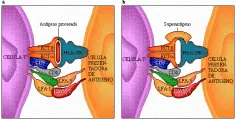
Los superantígenos bacterianos son una clase de activadores policlonales que actúan sobre ciertas células T al unirse a las regiones variables de las cadenas ß (regiones Vß) del receptor de antígeno de la célula T, en lugar de unirse al surco fijador de antígeno constituido por las cadenas a y ß de los receptores [ver figura 3]. Cada familia de receptores de células T que contienen Vß incluye a receptores para muchos antígenos que se unen en forma convencional dentro del surco fijador de antígenos entre las cadenas ay ß, debido a que la especificidad del receptor del antígeno está determinada por la recombinación de cada región Vß con diferentes regiones D (diversidad) y J (de unión) y con las mismas cadenas a. Por lo tanto, la activación de todas las células T dentro de una familia de receptores T que contienen Vß, como el Vß7+, activa células con un gran número de especificidades antigénicas, algunas de las cuales pueden ser autoantígenos. Por lo menos algunas de las manifestaciones clínicas de la enfermedad mediada por enterotoxina, como el síndrome de choque tóxico, pueden ser originadas por este mecanismo.1
|
| Figura 3 |
| Procesamiento del epítope antigénico |
Las enterotoxinas se unen a las moléculas clase II del CPH en las CPA. Las células T con una cierta región Vß reconocen al complejo superantígeno sobre la CPA, se unen a él y se activan. Las evidencias sugieren que algunas enfermedades autoinmunes pueden ser secundarias a la estimulación del superantígeno en las células T. En un estudio, los pacientes con diabetes mellitus insulinodependiente (DMID), una enfermedad en la que existe destrucción autoinmune de los islotes pancreáticos mediada por células T, tuvieron evidencia de un infiltrado de células T CD4+ rico en receptores celulares T de la familia Vß7 en el islote pancreático, lo que sugiere que el superantígeno participa en la patogenia de la destrucción de los islotes en la DMID.8 La participación del superantígeno implica a su vez a bacterias o virus, ya que los superantígenos suelen asociarse con microrganismos.9
REGULACION DEFICIENTE POR CELULAS T
Se ha postulado que el desequilibrio en las subpoblaciones de linfocitos puede ser el responsable de la autoinmunidad, aunque no existen evidencias directas de este mecanismo en alguna enfermedad autoinmune humana. Este desequilibrio puede incluir mayor actividad de células T supresoras o una alteración entre los subtipos TH1 y TH2 de las células T cooperadoras.
La exposición de los antígenos proteicos a las células linfoides en el intestino puede inducir tolerancia periférica que parece evitar las respuestas sistémicas inmunes a las proteínas de la dieta. La tolerancia por vía oral es inducida por lo menos por dos mecanismos. Uno es la supresión inmunológica por células T CD8+ específicas. Un segundo mecanismo es la supresión inmunológica por células T CD4+ del subtipo TH2, que secretan IL-4, IL-10 y factor de transformación del crecimiento-ß (FTC-ß). La tolerancia oral puede relacionarse con la capacidad de las células linfoides derivadas del intestino para inducir, ante la exposición al antígeno, un subtipo principal de células T CD4+ que secretan ciertas citocinas, incluyendo IL-4 y FTC-ß, que suprimen a las células TH1. Un estudio mostró que la tolerancia oral a la proteína básica de mielina suprime la encefalomielitis experimental autoinmune, un modelo animal que equivale a la esclerosis múltiple en humanos.10 Los resultados de estos estudios preliminares de tolerancia inducida por vía oral han sugerido que puede ser benéfico aplicar este mecanismo en la esclerosis múltiple y la artritis reumatoide.1,40
DEFECTOS EN LAS VIAS DE APOPTOSIS
La apoptosis, o muerte celular programada, es indispensable para la deleción de linfocitos autorreactivos. El proceso se inicia por una de varias interacciones ligando-receptor y continúa por la activación de endonucleasas como consecuencia de la generación de mensajeros intracelulares. La apoptosis también puede iniciarse por la ausencia de factores de crecimiento como la IL-2.
Es posible que el sistema Fas-Fas ligando sea importante en la enfermedad autoinmune. (Fas es una proteína de la superficie celular que media la apoptosis, mientras que el ligando Fas es una proteína de membrana tipo II relacionada con el factor de necrosis tumoral que se fija a Fas.) Las lesiones genéticas que causan alteraciones y disfunción de Fas o del ligando Fas se asocian con enfermedad autoinmune murina. En uno de estos modelos animales (la cepa MRL que porta en gen lpr [de linfoproliferación]) se acumulan gran número de linfocitos en los órganos linfoides como resultado de una apoptosis defectusa relacionada con una proteína Fas anormal. Este dato puede ser importante en el lupus humano, ya que se ha visto que los pacientes con LES tienen mayor producción de una forma soluble de molécula Fas causada por la delección de la porción transmembrana de la molécula. La proteína Fas circulante puede inhibir la interacción Fas-Fas ligando necesaria para la apoptosis normal y puede permitir la persistencia de células autorreactivas. Las lesiones en el sistema Fas y en otros sistemas requeridos para la apoptosis normal pueden contribuir a la patogenia de otras enfermedades autoinmunes. Varios medicamentos inmunosupresores y citotóxicos actúan a través de la inducción de apoptosis. La investigación relacionada con el sistema Fas-Fas ligando puede permitir el desarrollo de agentes que induzca una apoptosis más específica que la inducida por los agentes actuales.11,12
Susceptibilidad del huésped a las enfermedades autoinmunes
Los clínicos han observado desde hace mucho tiempo que los factores hereditarios parecen contribuir a la incidencia de las enfermedades autoinmunes. Varios estudios han demostrado una alta tasa de concordancia en gemelos monocigotos comparada con gemelos dicigotos. Sin embargo, la concordancia de varias enfermedades autoinmunes, incluyendo la DMID, el LES y la artritis reumatoide, es cercana al 15 a 30 porciento en la mayoría de los estudios. Por lo tanto, los factores genéticos son importantes en la susceptibilidad a las enfermedades autoinmunes, pero incluso las personas con genomas idénticos difieren en su susceptibilidad. La pregunta entonces es, ¿qué factores diferentes a los controlados por la genética pueden ser responsables de estos trastornos? Los factores ambientales podrían ser importantes, incluyendo a ciertas infecciones, pero también se ha sugerido que elementos estocásticos, no genéticos, relacionados con la maduración de los linfocitos, pueden influir en la susceptibilidad a la enfermedad. Por ejemplo, la recombinación de los segmentos genéticos para los receptores de las células T y B no se encuentra bajo control genético, sino que ocurre en forma aleatoria. Sin embargo, el reconocimiento antigénico por las células T requiere del sistema HLA, que está definido genéticamente.13
PAPEL DE LOS ANTIGENOS LEUCOCITARIOS HUMANOS
Se han definido muchas asociaciones entre las enfermedades autoinmunes y los antígenos del HLA. Estas varían desde asociaciones leves, con riesgos relativos de 1 o 2, hasta la asociación más estrecha, la de la espondilitis anquilosante con el HLA-B27, que confiere un riesgo relativo de 90 a 100 [ver tabla 2]. La interpretación de estas asociaciones sigue siendo difícil, y se han propuesto muchas explicaciones. Una posible explicación para la asociación de una enfermedad con un antígeno HLA es que el antígeno HLA puede ser más eficaz que otros para apoyar el reconocimiento de las células T de un autoantígeno. Otra posibilidad es que ciertos antígenos HLA sean menos eficaces que otros para apoyar la respuesta inmunológica contra un agente infeccioso, que puede iniciar la enfermedad autoinmune. Además, se ha postulado que un agente infeccioso puede utilizar un cierto antígeno HLA en la superficie de las células como un receptor para entrar a la célula, en la misma manera que el virus de inmunodeficiencia humana (VIH) utiliza a la molécula CD4 para la invasión celular.1,2,14
|
||||||||||||||||||||||||
* Posibilidad de que personas con uno o más alelos HLA desarrollen una enfermedad, comparado con personas que no tienen estos alelos. |
Algunas asociaciones entre los antígenos HLA y las enfermedades autoinmunes representan asociaciones estrechas entre otros antígenos HLA que se encuentran en desequilibrio de unión; esto es, dos alelos HLA están lo bastante cerca uno de otro en el cromosoma para heredarse juntos. Un ejemplo sucede en la DMID, que se asocia tanto con DR3 como con DR4. Sin embargo, la DMID se asocia en forma más estrecha con un alelo DQ, el DQ3.2, que se encuentra en desequilibrio de unión con DR3 y DR4. Aún más, cada antígeno HLA definido serológicamente, como el DR4, es una familia de alelos estructuralmente semejantes, aunque no idénticos. El análisis molecular de estos alelos ha revelado algunos requerimientos estructurales para las asociaciones con enfermedades autoinmunes. Por ejemplo, en por lo menos un grupo poblacional, las cadenas ß del DQ asociado a la DMID tienen uno de tres aminoácidos (alanina, valina o serina) cerca del sitio de ruptura de unión del péptido en la posición 57 del alelo, mientras que los alelos de las caenas ß del DQ que no se asocian con DMID tienen ácido aspártico en esa posición. Estas observaciones sugieren que la susceptibilidad a la DMID está determinada, en parte, por la estructura de la región de unión al péptido de la molécula DQ que determina la capacidad del alelo DQ para presentar ciertos antígenos a las células T.
Otro ejemplo de requerimientos estructurales discretos de las moléculas de clase II del HLA para la susceptibilidad a la enfermedad es la asociación de la artritis reumatoide con el antígeno DR4. Se ha determinado por análisis estructural de las moléculas individuales DR4 que ciertas secuencias de aminoácidos (en la región Vß cerca de los sitios de combinación con el antígeno y las regiones de contacto con células T de estas moléculas) se asocian con la artritis reumatoide, mientras que otras secuencias no tienen esta asociación. Además, la asociación entre artritis reumatoide y DR1 en otras poblaciones se basa en una semejante estructural de los aminoácidos entre el DR1 y el DR4.13
Es posible que el sistema HLA en sí pueda no ser responsable de la susceptibilidad genética a una enfermedad autoinmune y que otros genes que se encuentran en desequilibrio de unión con los genes HLA sean los responsables. Sin embargo, es probable que las moléculas HLA per se sean las directamente responsables en vista de su función para dirigir las respuestas inmunológicas. Las principales evidencias al respecto provienen de los trabajos de Taurog y colaboradores. Estos investigadores han mostrado que líneas transgénicas de ratas que expresan el HLA-B27 humano en forma espontánea desarrollan lesiones inflamatorias semejantes a las del síndrome de Reiter humano que, como la espondilitis anquilosante, se asocia en forma estrecha con el HLA-B27.15,16
Es claro, a partir de los análisis genéticos de las enfermedades autoinmunes murinas, que varios genes participan en la patogenia de una sola enfermedad.2 Las enfermedades autoinmunes humanas tienen la misma complejidad, y es posible que varios rasgos genéticos sean responsables de la susceptibilidad del huésped. Las infecciones con agentes virales o bacterianos parecen iniciar por lo menos algunas enfermedades autoinmunes, pero no se encuentran estos microrganismos en los tejidos lesionados en las etapas subsiguientes de la enfermedad. Existen varios mecanismos posibles por los que las infecciones pueden desencadenar autoinmunidad (ver antes). Por ejemplo, las infecciones pueden activar la detección inmune de células autorreactivas a través de la expresión de coestimuladores, como el B7, sobre las CPA o a través de la activación de la producción de citocinas. Las infecciones pueden exponer al sistema inmune a autoantígenos previamente secuestrados, a los cuales el sistema inmunológico no tiene tolerancia. La lesión tisular puede crear también neoantígenos que inciten reacciones inmunológicas. La exposición hormonal puede modificar la autoinmunidad, según indica la dependencia genética de muchas enfermedades autoinmunes. Por ejemplo, alrededor del 90 porciento de los casos de LES ocurren en mujeres, mientras que un porcentaje semejante de casos de espondilitis anquilosante se presentan en varones. No son claros los motivos para este sesgo en función del sexo en las enfermedades autoinmunes.1,2
PAPEL DE LOS AUTOANTICUERPOS
Con frecuencia existen autoanticuerpos en las enfermedades autoinmunes, pero puede ser difícil establecer que estos anticuerpos contribuyen en forma significativa a la patogenia de las enfermedades autoinmunes. Los autoanticuerpos que contribuyen con más probabilidad en la patogenia son los dirigidos contra antígenos de la superficie celular. Los autoanticuerpos dirigidos contra antígenos extracelulares pueden ser o no patogénicos. Por lo general se acepta que los anticuerpos no pueden reaccionar con antígenos en localizaciones intracelulares, pero las reacciones de los anticuerpos con antígenos intracelulares pueden contribuir a la enfermedad en ciertas circunstancias. Es posible que los antígenos intracelulares causen reacciones patógenas con autoanticuerpos si los antígenos son liberados y pueden fijarse a la superficie celular o a localizaciones extracelulares, o si los antígenos son traslocados a áreas accesibles de la superficie celular, ya sea que sean específicos o tengan reacción cruzada. Algunos ejemplos de enfermedades autoinmunes ilustran estos principios (ver adelante).17
Autoanticuerpos contra moléculas de la superficie celular
El pénfigo es una enfermedad cutánea bulosa relacionada con acantolisis, o pérdida de la adhesión de las células epidérmicas y que se asocia con anticuerpos contra los desmosomas (moléculas de la superficie celular epidérmica que participan en la adhesión intercelular). Varias líneas de evidencia apoyan el papel patogénico de estos autoanticuerpos en el pénfigo. Primero, es lógico suponer que la menor función de adhesión de los desmosomas epidérmicos puede causar acantolisis. Segundo, existen anticuerpos antidesmosomas en forma constante en los pacientes con pénfigo (y los niveles de anticuerpos correlacionan con la actividad de la enfermedad) y la eliminación de los anticuerpos séricos por plasmaféresis causa mejoría de los síntomas. Tercero, la transferencia pasiva de suero de pacientes con pénfigo vulgar causa lesiones semejantes en ratones. Cuatro, los recién nacidos de madres con pénfigo tienen una enfermedad transitoria causada por la trasmisión transplacentaria de los anticuerpos maternos.17
Casi todos los pacientes con miastenia gravis tienen autoanticuerpos contra receptores de acetilcolina (RAC) en el músculo esquelético [ver figura 4]. Sin embargo, el grado de bloqueo neuromuscular observado en esta enfermedad no siempre es paralelo a los niveles de anticuerpos anti-RAC. La inyección de anticuerpos contra RAC puede inducir debilidad miasténica en animales de experimentación. Los anticuerpos anti-RAC en los pacientes con miastenia gravis causan destrucción del RAC, pero rara vez bloquean el sitio de fijación de la acetilcolina. Los anticuerpos son policlonales y reaccionan con varios idiotipos en el RAC, así como con porciones no-RAC de la unión neuromuscular. No existe una buena correlación entre la severidad de la enfermedad y los niveles de anticuerpos anti-RAC; sin embargo, la respuesta de la enfermedad a la plasmaféresis sugiere que algunos, pero no todos, los anticuerpos, pueden ser responsables de la enfermedad.17,18
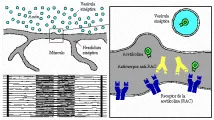
|
| Figura 4 |
| Unión neuromuscular normal |
Los anticuerpos contra varios receptores hormonales pueden ocasionar endocrinopatías. Los niveles altos de anticuerpos contra el receptor periférico de insulina se asocian con DMID, quizá como resultado de la degradación de los receptores por mecanismos autoinmunes. En forma paradójica, los niveles bajos de anticuerpos pueden estimular al receptor de insulina al reproducir el efecto de la insulina, ocasionando hipoglucemia.
La enfermedad autoinmune de la tiroides se asocia con anticuerpos dirigidos contra tres antígenos: peroxidasa tiroidea microsomal, tiroglobulina y receptor tiroideo de la hormona estimulante de la tiroides (TSH). Los anticuerpos contra el receptor de TSH pueden tener la misma acción de esta hormona, ocasionando enfermedad de Graves. Otra enfermedad autoinmune de la tiroides, la tiroiditis de Hashimoto, se asocia con anticuerpos contra el receptor de TSH, pero su papel patogénico en esta enfermedad no es claro. Es menos frecuente que los anticuerpos contra el receptor de TSH puedan bloquear la acción de la TSH y causar hipotiroidismo. No se ha establecido el papel patogénico de las otras dos clases de autoanticuerpos antitiroideos.17
Autoanticuerpos contra moléculas extracelulares
El síndrome de anticuerpos antifosfolípido (SAF) consiste en trombosis recurrente, pérdidas fetales y trombocitopenia asociadas con anticuerpos contra cardiolipina u otros fosfolípidos de carga negativa, y alteraciones de ciertas pruebas de coagulación, conocidas como anticoagulante lúpico. El SAF puede ser primario, en ausencia de una enfermedad asociada, o secundario, casi siempre asociado a LES o sus variantes. No es claro si el anticuerpo antifosfolípido induce las trombosis en forma directa, aunque la asociación de estos anticuerpos con el SAF es muy estrecha. Los anticuerpos no se unen a los fosfolípidos aislados, sino a un complejo de fosfolípidos y un cofactor del plasma, la ß2-glicoproteína I. El anticoagulante lúpico se refiere a la capacidad de los anticuerpos antifosfolípidos para interferir con varias pruebas de la coagulación in vitro, pero in vivo estos anticuerpos promueven la trombosis, en lugar de inhibirla. Otros autoanticuerpos contra las moléculas extracelulares incluyen anticuerpos anti-insulina que pueden ser espontáneos o secundarios a la administración de insulina, así como anticuerpos contra la colágena tipo II que se asocian con la artritis reumatoide humana y la policondritis. Los anticuerpos contra colágena tipo II son casi siempre secundarios al daño en el cartílago, pero pueden contribuir a la inflamación en el cartílago de articulaciones y otras estructuras.
Autoanticuerpos contra moléculas intracelulares
Aunque los autoanticuerpos contra antígenos intracelulares se asocian mucho con enfermedades, suele ser difícil determinar si estos anticuerpos contribuyen a la patogenia de la enfermedad. La cirrosis biliar primaria (CBP) se asocia con forma estrecha con la presencia de anticuerpos antimitocondriales, y estos anticuerpos son muy específicos para la enfermedad. A pesar de la amplia distribución de las mitocondrias, la patología en la CBP se limita en forma casi exclusiva a los conductos biliares intrahepáticos. Los anticuerpos antimitoconcriales se dirigen contra un epítope en el complejo multienzimático piruvato deshidrogenasa. A pesar de la especificidad de estos anticuerpos para la CBP, parece muy poco probable que estos anticuerpos puedan causar la CBP, por la inaccesibilidad del antígeno al anticuerpo circulante.
Otro ejemplo de asociación muy específica de autoanticuerps con significado patogénico dudoso es el de la presencia de anticuerpos anti-aminoacil-ARNt sintetasa en la polimiositis. Estas enzimas están presentes en el ARN citoplásmico y son muy importantes para la síntesis proteica. Los niveles de anticuerpos contra sintetasas de ARNt correlacionan con la actividad de la enfermedad, pero la plasmaféresis es ineficaz. Existen autoanticuerpos contra varios componentes nucleares en diversas enfermedades autoinmunes reumatológicas, incluyendo el LES, la esclerodermia y el síndrome de Sjögren. Estos anticuerpos están dirigidos hacia ribonucleoproteínas nucleares pequeñas que participan en la separación del ARN mensajero, los componentes nucleares del centrómero y otras proteínas nucleares. Ninguno de estos anticuerpos han demostrado tener un papel definido en la patogenia de las enfermedades asociadas.17
Los autoanticuerpos pueden reaccionar con proteínas intracelulares que también aparecen en las superficies celulares o en localizaciones extracelulares. Ocurren anticuerpos contra las proteínas ribosomales P en el LES, sobre todo en pacientes con depresión o psicosis. Es interesante que estos anticuerpos reaccionan también con una proteína en la superficie celular que es semejante en tamaño al antígeno proteico ribosomal. No es claro si la proteína de superficie celular es la proteína ribosomal que ha sido transportada a la superficie celular o es otra proteína semejante. Sin embargo, la presencia del antígeno sobre la superficie celular sugiere que los anticuerpos contra proteínas ribosomales P pueden contribuir a las manifestaciones neurológicas del LES. En enfermos con LES y síndrome de Sjögren se presentan anticuerpos contra las pequeñas nucleoproteínas antigénicas SS-A (también llamada Ro), y SS-B (o La) lo mismo que en pacientes con lupus neonatal y bloqueo cardiaco congénito. Estos dos últimos transtornos del recién nacido se asocian con el paso trasplacentario de anticuerpos maternos. Se ha demostrado que los anticuerpos anti-Ro reaccionan con y alteran la permeabilidad del tejido cardiaco fetal, pero no el tejido cardiaco de los adultos.
Otro anticuerpo dirigido contra un antígeno intracelular que quizá contribuye a la enfermedad es el anticuerpo contra citoplasma de neutrófilos (ANCA). Este anticuerpo está dirigido contra la proteinasa-3, que existe en los lisosomas primarios de neutrófilos y monocitos. Al liberarse estos lisosomas durante la activación celular, su contenido se expone lo suficiente como para raccionar con los anticuerpos y las células inmunológicas. Desde el punto de vista experimental, los neutrófilos se activan al exponerse a citocinas y liberan sus gránulos y otras sustancias inflamatorias al exponerse a los ANCA. Se ha sugerido que en estos experimentos, la activación causa exposición de proteinasa-3 e imunización o formación de complejos inmunes con anticuerpos específicos, y que estos complejos pueden contribuir a la vasculitis y la lesión tisular. Los anticuerpos ANCA son específicos para la granulomatosis de Wegener y un pequeño número de enfermedades con patología renal semejante. La proteinasa-3 podría contribuir a la patología de estas enfermedades, ya sea por su movimiento hacia la superficie celular o por su liberación hacia el ambiente extracelular, con la subsecuente activación de los neutrófilos por los complejos inmunes. La activación de los neutrófilos puede entonces dañar las paredes vasculares y los tejidos circundantes.
Se observan anticuerpos contra ADN desnaturalizado o de una cadena en diversas enfermedades autoinmunes, pero su papel patogénico no es claro. Los anticuerpos contra el ADN nativo o de doble cadena son bastante específicos del LES, y es posible que participen en la enfermedad. En alguna ocasión se penso que complejos inmunes de ADN nativo-antiADN viajaban en la circulación y se depositaban en los tejidos, como en otras enfermedades por complejos inmunes, como la enfermedad del suero. Sin embargo, estudios posteriores sugieren un mecanismo diferente. El glomérulo normal contiene heparán sulfato, un polisacárido aniónico que puede formar complejos con histonas catiónicas, y se ha sugerido que estas moléculas unidas a histonas pueden a su vez captar al ADN en los complejos circulantes ADN-antiADN, fijando los complejos al glomérulo. Sin embargo, el papel de los complejos ADN-antiADN en el LES no se ha comprobado aún. La actividad de la enfermedad no siempre correlaciona con las concentraciones de anticuerpos anti-ADN, y la plasmaféresis ha sido ineficaz en la nefritis por LES.17
Los pacientes con DMID desarrollan autoanticuerpos contra varias proteínas de las células de los islotes pancreáticos. Entre los primeros anticuerpos encontrados antes del desarrollo de la diabetes se encuentran los anticuerpos contra la proteína de 64 kd presente tanto en el citoplasma como en las superficies celulares. Los pacientes con el síndrome diabético de hombre rígido, una enfermedad rara que consiste en diabetes, epilepsia y espasticidad muscular, desarrollan también anticuerpos contra el mismo antígeno de 64 kd. Este antígeno parece ser la decarboxilasa del ácido glutámico, una enzima que convierte el ácido glutámico al neurotrasmisor gama-aminobutírico en ciertas neuronas. La decarboxilasa del ácido glutámico se encuentra también en los islotes pancreáticos, aunque se desconoce su función en estas células. Una secuencia de 24 aminoácidos de esta enzima tiene homología estructural con la proteína P2-C de un picornavirus (el coxsackievirus B4), que se ha implicado como agente etiológico de la DMID. La plasmaféresis puede ser benéfica en el síndrome de hombre rígido, lo que sugiere que la decarboxilasa del ácido glutámico tiene un papel patogénico.18 El síndrome de hombre rígido se presenta también como un complejo paraneoplásico asociado con cáncer de mama. Sin embargo, en estos pacientes se desarrolla un anticuerpo contra la proteína neuronal de 128 kd que se concentra cerca de las sinapsis, más que contra la decarboxilasa del ácido glutámico.19
En el ratón diabético no obeso, un modelo para la DMID humana, se desarrolla una respuesta de células TH1 contra un epítope en la decarboxilasa del ácido glutámico al inicio de la insulitis; después se presenta reactividad de las células T a otros epítopes de la decarboxilasa del ácido glutámico, lo mismo que reactividad contra otros antígenos de las células de los islotes. La inducción de tolerancia de las células T a la decarboxilasa del ácido glutámico evita la autoinmunidad contra otros antígenos de las células de los islotes, lo mismo que la insulitis y la diabetes. Estos datos sugieren que la autoinmunidad contra una porción especial de la decarboxilasa del ácido glutámico es un factor importante en la patogenia de la DMID murina.20
- Abbas AK, Lichtman AH, Pober JS: Cellular and Molecular Immunology , 2nd ed. WB Saunders Co, Philadelphia, 1994
- Steinberg AD: Mechanisms of disordered immune regulation. Basic and Clinical Immunology , 8th ed. Stites DP, Terr Al, Parslow TG, Eds. Appleton & Lange, Norwalk, Connecticut, 1994, p 380
- Nossal GJV: Immunologic tolerance: collaboration between antigen and lymphokines. Science 245:147,1989
- Shortman K, Scollay R: Death in the thymus. Nature 372:44,1994
- Finck BK, Unsley PS, Wofsy D: Treatment of murine lupus with CTLA41g. Science 265:1225, 1994
- Durie FH, Fava RA, Foy TM, et al: Prevention of collagen-induced arthritis with an antibody to gp39, the ligand for CD40. Science 261:1328, 1993
- Stieglitz H, Lipsky P: Association between reactive arthritis and antecedent infection with ShigeIla flexneri carrying a 2-Md plasmid and encoding an HLA-B27 mimetic epitope. Arthritis Rheum 36:1387,1993
- Conrad B, Weidmann E, Trucco G, et al: Evidence for superantigen involvement in insulin-dependent diabetes mellitus aetiology .Nature 371:351, 1994
- Torres BA, Griggs ND, Johnson HM: Bacterial and retroviral superantigens share a common binding region on class II MHC antigens. Nature 364:152, 1993 .
- Chen Y, Kuchroo VK, Inobe J-i, et al: Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science 265:1237,1994
- MountzJD, Wu J,ChengJ,etal: Autoimmune disease: a problem of defective apoptosis. Arthritis Rheum 37:1415, 1994
- Takahashi T, Tanaka M, Brannan CI et al: Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell 76:969, 1994
- Kotzin BL: Twins and T-cell responses. Nature 364:187,1993
- Schwartz BD: Infectious agents, immunity , and rheumatic diseases. Arthritis Rheum 33:457,1990
- Breban M, Hammer RE, Richardson JA, et al: Transfer of the inflammatory disease of HLA-B27 transgenic rats by bone marrow engraftment. J Exp Med 178:16JJ7, 1993
- Taurog JD, Maika SD, Simmons W A, et al: Susceptibility to inf1ammatory disease in HLA-B27 transgenic rat lines correlates with the level of B27 expression. J mmunoI150:4168, 1993
- Nanarstek Y, Plotz PH: The role of autoantibodies in autoimmune disease. Annu Rev Immunol 11:79, 1993
- Protti MP, Manfredi AA, Horton RB, et al: Myasthenia gravis: recognition of a human autoantigen at the molecular level. Immunol Today 14:363,1993
- Folli F, Solimena M, Cofiell R, et al: Autoantibodies to a 128-kd synaptic protein in three women with the stiff-man syndrome and breast cancer. N Engl J Med 328:546,1993
- Kaufman DL,Clare-SalzlerM, Tian J, et al: Spontaneousloss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-depen-dent diabetes. Nature 366:69,1993