Contenido del artículo
XIV TRASTORNOS TROMBOTICOS
- Trombosis e inflamación vascular
- Estudio del paciente con trastornos trombóticos
- Estados hipercoagulables hereditarios
- DEFICIENCIA DE ANTITROMBINA III
- DEFICIENCIA DE PROTEINA C Y PROTEINA S
- FACTOR V DE LEIDEN
- MUTACION 20210A DEL GEN DE LA PROTROMBINA
- HIPERHOMOCISTEINEMIA
- LIPOPROTEINA(A)
- DISFIBRINOGENEMIA
- DISPLASMINOGENO Y FIBRINOLISIS ANORMAL
- DEFICIENCIA DE FACTOR XII
- NIVELES ELEVADOS DE FIBRINOGENO, FACTOR VII Y FACTOR VIII
- Estados hipercoagulables adquiridos
- SINDROME DE ANTICUERPOS ANTIFOSFOLIPIDO
- Epidemiología
- Fisiopatología
- Diagnóstico y cuadro clínico
- Evolución clínica y tratamiento
- Pérdidas fetales y síndrome de anticuerpos antifosfolípido
- TROMBOCITOPENIA Y TROMBOSIS INDUCIDAS POR HEPARINA
- NEOPLASIAS - SINDROME DE TROUSSEAU
- REACCIONES TROMBOTICAS POR ESTROGENOS
- Tratamiento de los pacientes con factores de riesgo para tromboembolia venosa
DR. LAWRENCE L. K. LEUNG
Trombosis e inflamación vascular
El proceso de trombosis es más que una coagulación sanguínea excesiva, y muchas veces debe considerarse como inflamación vascular. La triada clásica de Virchow identifica a tres elementos importantes en la fisiopatología de la trombosis, daño endotelial, reducción del flujo sanguíneo y desequilibrio entre los factores pro y anticoagulantes.
Las células endoteliales pueden activarse o lesionarse por diversos estímulos, incluyendo traumatismos mecánicos, endotoxinas y citocinas, proteasas, mediadores inflamatorios, depósito de complejos inmunes, radicales de oxígeno e hipoxia. Cada uno de estos estímulos endoteliales afecta múltiples facetas de las propiedades funcionales de la célula endotelial, cambiando al final su fenotipo funcional general de un estado antitrombótico natural a un estado protrombótico.
El endotelio vascular, en su localización única en la pared del vaso, es capaz de detectar y responder a las diferentes fuerzas mecánicas en la circulación sanguínea. Las fuerzas de cizalla causadas por la fuerza de fricción del flujo sanguíneo parecen ser especialmente importantes para regular la función endotelial. En las regiones de flujo lineal la circulación la sangre fluye en patrones laminares en forma pulsátil y regular. Este flujo laminar constante parece promover un fenotipo endotelial antitrombótico. En las áreas en que se altera el flujo laminar, como los sitios de bifurcación vascular o estenosis, el endotelio puede estar expuesto a cambios significativos en los gradientes de cizalla y a inversión del flujo, con lo que las células se activan y se vuelven protrombóticas.
Las causas del desequilibrio entre los factores procoagulantes y anticoagulantes pueden ser hereditarios o adquiridos. Algunos factores de la coagulación, como el factor VIII y el fibrinógeno, son reactantes de fase aguda y sus niveles en plasma aumentan en forma significativa en los estados de inflamación aguda, quizá confiriendo un estado protrombótico transitorio. Algunas deficiencias hereditarias de las proteínas anticoagulantes se asocian con trombosis recurrentes, y constituyen algunos de los estados clínicos de hipercoagulabilidad mejor comprendidos.
A pesar de la presencia de un estado de hipercoagulabilidad sistémica, la trombosis ocurre en forma local (v.gr., en las extremidades inferiores). Los factores locales que interactúan con la predisposición protrombótica sistémica pueden tener un papel determinante en la evolución clínica. Es posible que los mecanismos de control hemostático específicos del lecho vascular sean importantes en la compleja fisiopatología de la trombosis.
Estudio del paciente con trastornos trombóticos
Como se describió antes, la trombosis es la culminación de una serie compleja de eventos que provocan inflamación vascular. Son eventos desencadenantes comunes la cirugía, el traumatismo, el embarazo, las neoplasias, la inmovilización prolongada y la infección. El trastorno puede ser evidente o subclínico (i.e., pretrombótico). Cuando ocurre una trombosis en una persona aparentemente normal sin ningún evento desencadenante, en especial cuando la persona es joven, debe pensarse en un estado de hipercoagulabilidad.
La trombofilia, un estado hipercoagulable, se refiere a la predisposición clínica ad desarrollar trombosis recurrentes, por lo general sin factores de riesgo evidentes asociados [ver tabla 1]. El factor V de Leiden y la deficiencia de antitrombina-III (AT-III) se encuentran entre los estados hipercoagulables mejor caracterizados. Los estados hipercoagulables pueden ser hereditarios o adquiridos, asociados con diversos procesos fisiológicos y clínicos.
|
|
|
ASPECTOS CLINICOS PRINCIPALES EN LOS TRASTORNOS TROMBOTICOS
Los aspectos importantes a considerar en un paciente con un trastorno trombótico incluyen los siguientes (1) ¿Qué probabilidad hay de que la trombosis del paciente sea causada por un estado hipercoagulable? (2) ¿Qué tan extenso debe ser el estudio del paciente? (3) ¿Cuándo debe realizarse la evaluación? (4) ¿El paciente ha sido tratado en forma adecuada para la trombosis? (5) ¿Durante cuánto tiempo debe recibir anticoagulantes el paciente? Las respuestas a estas preguntas deben tomar en consideración la edad del paciente en el momento de la primera trombosis, la historia familiar y la historia médica por situaciones asociadas con riesgo alto de trombosis, además del sitio, tipo y severidad de la trombosis.
Edad en el momento de la primera trombosis
En un estudio retrospectivo reciente que incluyó a 150 familias con trombofilia hereditaria, la edad promedio en el momento de la primera trombosis fue de 35 a 40 años. Sin embargo, ocurrieron episodios hasta durante la segunda década de la vida si el paciente tenía más de un factor de riesgo hereditario.1
Presencia o ausencia de factores de riesgo trombótico coexistentes
En los pacientes con trombofilia hereditaria con frecuencia la trombosis coincide con un evento desencadenante, como embarazo, uso de anticonceptivos orales o cirugía.1 En ocasiones la trombosis puede ser espontánea e idiopática. Cuando ocurren trombosis espontáneas e idiopáticas en un paciente joven, debe considerarse la posibilidad de un estado hipercoagulable hereditario. Debe pensarse en un estado hipercoagulable adquirido cuando el paciente ha tenido ya embarazos o cirugías previas (en especial procedimientos ortopédicos) sin complicaciones trombóticas y acude por una trombosis de inicio reciente. Las condiciones probables en estos casos son el síndrome de anticuerpos antifosfolípido y el síndrome de Trousseau, causado por una neoplasia subyacente. El síndrome de anticuerpos antifosfolípido puede asociarse con una enfermedad autoinmune del tejido conectivo y en este caso deberán buscarse manifestaciones clínicas y serológicas. Las neoplasias asociadas al estado hipercoagulable pueden ser subclínicas, pero no suele ser útil realizar una investigación extensa para detectarlas. Es útil conocer la duración de la trombosis. Las trombosis recurrentes que tardan varios años en repetir es poco probable que se relacionen con cáncer porque éste suele ser aparente durante el primer año posterior al evento trombótico inicial [ver adelante, Neoplasias, síndrome de Trousseau].
Es obvio que la historia familiar de trombosis es sugestiva de un trastorno hereditario. Sin embargo, la historia familiar negativa no excluye un origen hereditario. La trombosis clínica suele ser la culminación de más de un factor de riesgo trombogénico, de los cuales solo uno puede ser irreversible y hereditario. En los pacientes con trombosis sintomática y estados hipercoagulables hereditarios bien documentados no es raro encontrar otros familiares con la misma deficiencia pero sin trombosis clínica.
Trombosis venosa contra trombosis arterial
El diagnóstico diferencial de la trombosis venosa y la trombosis arterial recurrentes es muy diferente. La mayoría de los estados hipercoagulables hereditarios, como la deficiencia de AT-III y la existencia de factor V de Leiden, se asocian con trombosis venosas, como las trombosis venosas profundas (TVP) de las extremidades inferiores. Rara vez causan trombosis arteriales como isquemia cerebral transitoria, eventos cerebrovasculares, isquemia digital e infarto al miocardio. Solo algunos estados hipercoagulables, como el síndrome antifosfolípido y la hiperhomocistinemia, se asocian con ambos tipos de trombosis.
La mayoría de las trombosis consisten en TVP de las extremidades inferiores. Un sitio atípico sugiere la existencia de un estado hipercoagulable subyacente. Las trombosis de venas hepáticas, venas mesentéricas, venas cerebrales o la necrosis cutánea por la administración de warfarina obligan a investigar esta posibilidad. Las trombosis espontáneas de las venas axilares también indican la presencia de un estado hipercoagulable subyacente; sin embargo, esta asociación ha sido motivo de controversia.1-2a
Evaluación del estado hipercoagulable
Con base en los factores anteriores, en un paciente que presenta un primer episodio de trombosis clínica debe decidirse qué evaluación se realizará para investigar un estado hipercoagulable. En general, si el primer episodio de una TVP en una extremidad inferior se asocia con un evento desencadenante reversible claramente identificable, no se requiere un estudio extenso para buscar un estado hipercoagulable. Por otro lado, si la trombosis parece ser idiopática u ocurre en un sitio atípico, está justificada la evaluación.
En el paciente con trombosis recurrente existe la posibilidad de un estado hipercoagulable (hereditario o adquirido). Sin embargo, cuando las trombosis recurrentes ocurren en solo una extremidad es importante distinguir si se trata de trombosis recurrentes o de complicaciones del síndrome posflebítico. La exacerbación aguda del síndrome posflebítico, con aumento en el edema y dolor de la extremidad, pueden ser difíciles de distinguir de la TVP aguda recurrente. La revisión de los estudios previos de Doppler puede ser muy útil al considerar estas posibilidades.
Debido a que no se cuenta con estudios de costo-eficacia y evolución, es difícil establecer lineamientos prácticos respecto a la extensión que debe tener la evaluación del estado hipercoagulable. En general, esto dependerá de la posibilidad de que exista el padecimiento y de la severidad de la trombosis. Por ejemplo, puede ser suficiente investigar el estado del factor V de Leiden, AT-III, proteína C, proteína S y anticoagulante lúpico en una mujer joven que desarrolla TVP en una vena femoral superficial mientras recibe anticonceptivos orales. Por otro lado, debe pensarse en un estado de hipercoagulabilidad adquirido en un anciano con una TVP espontánea e historia previa negativa de trombosis. Las posibilidades en este paciente deben incluir síndrome de anticuerpos antifosfolípico, deficiencia adquirida de AT-III (por síndrome nefrótico no detectado) o cáncer que causa un síndrome de Trousseau [ver tabla 2].
|
||||||||
*En estudios clínicos apropiados.
|
Momento de la evaluación También es importante el momento
adecuado para la evaluación. En el momento del diagnóstico
inicial de un evento trombótico se han consumido muchos inhibidores de
la cascada de la coagulación (v.gr., AT-III y proteína C). Por lo
tanto, la reducción en los niveles plasmáticos de estas
proteínas no pueden atribuirse a una deficiencia hereditaria. Por lo
general, es mejor posponer la evaluación hasta que se haya resuelto por
completo el episodio trombótico agudo, de preferencia algunas semanas
después de concluida la anticoagulación por vía oral. Sin
embargo, las pruebas para genotipos específicos (v.gr., factor V de
Leiden) pueden realizarse en cualquier momento. Las pruebas de anticuerpos
antifosfolípido deben hacerse en el momento del diagnóstico
porque la presencia de estos anticuerpos modificará el tratamiento
anticoagulante.
Frecuencia y riesgo relativo de la tromboembolia venosa
La frecuencia de varios estados hipercoagulables en pacientes no seleccionados que presentan trombosis venosa varía de 1 a 25 por ciento [ver tabla 3]. Los pacientes con trombosis sintomática con frecuencia tienen más de un factor de riesgo, que puede tener un efecto sinérgico para aumentar el riesgo de trombosis. Por ejemplo, los pacientes con dos factores de riesgo (factor V de Leiden y uso de anticonceptivos orales) tienen un incremento de 35 veces en el riesgo relativo de tromboembolia venosa en comparación con la población general.
|
||||||||||||||||||||||||
*La incidencia de tromboembolia venosa en la población normal
se calcula que es de 0.008 por ciento por año (0.03 por ciento por
año en pacientes que reciben anticonceptivos orales).
|
Estados hipercoagulables hereditarios
DEFICIENCIA DE ANTITROMBINA III
La frecuencia de deficiencia de AT-III hereditaria sintomática en la población general se ha calculado en una de cada 2,000 personas.3 Los dos tipos de deficiencia de AT-III se trasmiten con patrón autosómico dominante. El tipo I es causado por una deficiencia cuantitativa de AT-III, medida por ensayos antigénicos y funcionales. En la deficiencia de AT-III de tipo I se han caracterizado gran cantidad de mutaciones moleculares, incluyendo deleciones parciales del gen y sustituciones de un solo nucléotido, que causan mutaciones sin sentido o con sentido equivocado, lo que provoca señales de detención prematura en el proceso de traducción de la proteína. La deficiencia de tipo II es causada por un defecto cualitativo de la AT-III en el que los niveles plasmáticos del antígeno son normales. El defecto suele consistir en un cambio de un solo nucleótido que provoca mutaciones sin sentido, dando origen a una proteína disfuncional. Puede ocurrir deficiencia adquirida de AT-III después de la administración de heparina intravenosa por más de 3 días, después del tratamiento con asparaginasa y en pacientes con coagulación intravascular diseminada (CID), hepatopatía severa o síndrome nefrótico.
Fisiopatología y cuadro clínico
La deficiencia de AT-III altera la inactivación de los factores de la coagulación Xa y la trombina, lo que aumenta la trombosis. La AT-III también inactiva a los factores XIIa y XIa al formar un complejo estoiquiométrico con cada uno de ellos (por el mismo mecanismo por el que inactiva a la trombina y al factor Xa). La AT-III existe en cantidades suficientes en el plasma como para inactivar toda la trombina formada en un volumen plasmático determinado, pero lo hace en forma muy lenta, a menos que se active por el heparán sulfato de la superficie de la célula endotelial o por la heparina administrada Los pacientes con deficiencia hereditaria de AT-III tienen evidencia de activación continua del factor X y generación de trombina (lo que se demuestra por el aumento en los niveles en plasma de fragmento F1.2 de la protrombina).
Los pacientes con deficiencia de AT-III tienen mayor incidencia de trombosis venosas, por lo general desencadenada por un factor específico como embarazo, cirugía, infección, inmovilización o traumatismo. Esta asociación sugiere que la sobreposición de un estímulo protrombótico en un estado hipercoagulable causa trombosis clínica. Los pacientes suelen sufrir TVP de miembros inferiores, embolia pulmonar y, en ocasiones, trombosis de las venas mesentéricas. No existen evidencias convincentes que sugieran que la deficiencia de AT-III aumenta el riesgo de trombosis arterial.4 Los pacientes afectados suelen tener historia familiar de trombosis recurrentes, que por lo general comienzan en la juventud y suelen asociarse con cirugía o traumatismos. El embarazo y el uso de anticonceptivos orales aumenta el riesgo de trombosis en los pacientes con deficiencia de AT-III. La tendencia a desarrollar trombosis aumenta con la edad, para los 50 años sólo el 10 por ciento de estos pacientes no ha tenido síntomas.
Los pacientes con deficiencia de AT-III tienen una reducción sorprendentemente modesta en la proteína, y sus valores medidos por bioensayo e inmunoensayo varían de 25 a 60 por ciento del normal en el caso de deficiencia tipo I. La deficiencia homocigota es muy rara, quizá porque la condición es incompatible con el desarrollo fetal normal. Debe determinarse en nivel de AT-III por una prueba funcional y no una antigénica de modo que se evalúen ambos tipos de deficiencia.
El estudio de familias grandes con deficiencia de AT-III indica que la profilaxis con anticoagulación no está indicada en los portadores asintomáticos de la deficiencia.5 Estos portadores asintomáticos deben recibir anticoagulación profiláctica si están expuestos a situaciones que se sabe aumentan el riesgo trombogénico, como la cirugía abdominal.5 Sin embargo, una vez que ha ocurrido un evento trombótico es probable que los pacientes requieren tratamiento de por vida con warfarina. Los episodios agudos de trombosis deben tratarse con heparina. Debido a que la deficiencia de AT-III puede hacer que la heparina sea relativamente ineficaz, el médico debe estar alerta ante esta resistencia, que se manifiesta por prolongación mínima del tiempo parcial de tromboplastina (TPT) después de la administración de dosis terapéuticas de este medicamento.
Si ocurre resistencia a la heparina debe administrarse heparina más concentrados purificados de AT-III o plasma fresco congelado. La AT-III tiene una vida media de alrededor de 60 horas. Estas preparaciones pueden usarse para proteger al paciente con deficiencia de AT-III durante una cirugía o parto y deben ser suficientes para normalizar el nivel de AT-III al 100 por ciento, dependiendo del nivel basal de AT-III. El nivel de AT-III debe checarse para repetir la infusión a las 24 horas de modo que se mantenga un nivel normal de AT-III durante 5 a 7 días después del parto o cirugía. El embarazo en las pacientes con deficiencia de AT-III es difícil de manejar. Debe iniciarse la combinación de heparina subcutánea (en dosis de 15,000 unidades cada 12 horas) y AT-III al comenzar el trabajo de parto y continuarse durante el alumbramiento.6 La warfarina es el tratamiento base a largo plazo para los pacientes con deficiencia de AT-III y tromboembolia.
DEFICIENCIA DE PROTEINA C Y PROTEINA S
Fisiopatología y cuadro clínico
La deficiencia o defecto en la proteína C o la proteína S causa pérdida de la capacidad para inactivar el factor VIIIa y el factor Va excesivos, los dos principales cofactores que regulan la amplificación de la cascada de coagulación. Los niveles de proteína C son bajos en los pacientes con CID y hepatopatía, quizá porque la activación de la hemostasia consume este factor.
La deficiencia homocigota de proteína C provoca trombosis letal en la infancia. Es probable que la deficiencia heterocigota ocurra con una prevalencia de uno por 200 a 300 habitantes. La manifestación trombótica de la deficiencia de proteína C puede expresarse en forma autosómica recesiva o autosómica dominante. Muchos pacientes con deficiencia heterocigota y otras personas con niveles normales bajos de proteína C no tienen historia de trombosis.7 Sin embargo, otros enfermos con deficiencia heterocigota tienen una tendencia definitiva a desarrollar trombosis venosas incluso a pesar de niveles de proteínas C en 40 a 50 por ciento del normal. Esta variabilidad fenotípica sugiere interacciones multigénicas y apoya la hipótesis de que la trombosis clínica en estos pacientes puede deberse a la combinación de deficiencia de proteína C y una o más mutaciones protrombóticas.8 Al parecer la trombosis venosa cerebral explica los casos de infarto cerebral hemorrágico que ocurren en adultos jóvenes con deficiencia de proteína C.
La deficiencia de proteína S también causa trombosis venosa, incluyendo trombosis venosa mesentérica. El embarazo y el uso de anticonceptivos orales disminuye el nivel de proteína S, lo que puede explicar algunos casos de tromboembolia que ocurren en estas circunstancias.9 También ocurre deficiencia adquirida de proteína S en pacientes con síndrome nefrótico, causada por pérdida de la proteína en la orina.10 Se han reportado casos de deficiencia de proteína S con necrosis de la piel inducida por warfaina,11aunque no se conoce la fisiopatología de este fenómeno. Debido a que la proteína S tiene una vida media mucho más larga que la proteína C (alrededor de 60 horas para la proteína S contra 6 horas para la proteína C), la proteína S no debe afectarse por la warfarina del mismo modo.
En la actualidad se cuenta con estudios funcionales y antigénicos de proteína C y proteína S en casi todos los laboratorios de coagulación. Los estudios funcionales son preferibles ara el diagnóstico. Es importante medir la proteína S libre porque algunos pacientes con niveles bajos de proteína S libre tienen niveles normales o limítrofe de proteína S total. Los ensayos de coagulación para proteína C y S pueden mostrar niveles falsos bajos cuando existe mutación del factor V de Leiden.12
Los anticoagulantes orales, como la warfarina, son el tratamiento de elección para prevenir las trombosis, a pesar de que disminuyen aún más los niveles de proteína C. Debido a que la vida media de la proteína C es de solo 6 a 7 horas, mucho más corta que la de la protrombina y el factor X, al iniciar el tratamiento con warfarina en un paciente con deficiencia de proteína C ocurre un periodo de mayor hipercoagulabilidad. Es por esto que se administra al inicio heparina en forma concomitante, que puede suspenderse después. La necrosis de la piel inducida por warfarina es una complicación rara del tratamiento anticoagulante.
De la población caucásica en general, alrededor del 5 por ciento es heterocigota para el factor V de Leiden. Este defecto está prácticamente ausente en otros grupos étnicos.18 El factor V de Leiden se refiere a la resistencia a los efectos anticoagulantes de la proteína C activada (PCA) causada por una mutación en el factor V. El defecto se trasmite como un rasgo autosómico dominante y en la actualidad se considera como el estado hipercoagulable hereditario más común. La prevalencia de factor V de Leiden en pacientes con trombofilia es hasta de 20 a 50 por ciento.14,15 En un estudio grande de cohortes de pacientes no seleccionados con un primer episodio de TVP sintomática, se encontró factor V de Leiden en el 16 por ciento de los pacientes.16 En las mujeres que desarrollan trombosis asociadas al uso de anticonceptivos orales la frecuencia de factor V de Leiden es de alrededor del 23 por ciento.17 El riesgo relativo de TVP en un paciente con mutación homocigota de factor V de Leiden (con incidencia calculada de 0.5 a 1 por ciento por año) es de alrededor de 80 veces mayor que el de una persona normal.18 El riesgo de trombosis en portadores de factor V de Leiden se calcula en 4 a 8 veces más que en no portadores y el riesgo relativo aumenta más de 30 veces si la presencia de este factor se combina con el uso de anticonceptivos orales. Sin embargo, el riesgo absoluto de trombosis es bajo.19 Los portadores de factor V de Leiden tienen un riesgo relativo de trombosis de 3.3.20 En algunas familias se ha reportado la asociación de factor V de Leiden con deficiencias de proteína C, proteína S o AT-III.21-23 En general, aunque el factor V de Leiden es muy prevalente, es un factor de riesgo relativamente débil para trombosis.
Las manifestaciones clínicas del factor V de Leiden son semejantes a las de las deficiencias de AT-III, proteína C y proteína S (principalmente trombosis venosa). Sin embargo, la principal manifestación trombótica en estos casos suele ocurrir más tarde que en otros estados trombofílicos hereditarios. En varones aparentemente sanos y mayores de 60 años que sufren un primer episodio de trombosis venosa, alrededor del 25 por ciento son portadores del factor V de Leiden.24
Alrededor del 5 por ciento de los casos asociados con resistencia hereditaria a la PCA se atribuyen a otras mutaciones y defectos.25-27 Ciertas condiciones (v.gr., aumento del factor VIII, embarazo, uso de anticonceptivos orales y anticoagulante lúpico) pueden causar un fenotipo de resistencia a la PCA.28 El fenotipo que no es causado por factor V de Leiden puede constituir un factor de riesgo para eventos cerebrovasculares29,30 y trombosis venosas.31,32 El riesgo global de trombosis venosas por resistencia a la PCA es semejante o menor al implicado por el factor V de Leiden.31
Fisiopatología y cuadro clínico
La resistencia a los efectos anticoagulantes de la PCA es causada por una mutación específica en el factor V (factor V de Leiden o factor V R506Q) que se debe a la sustitución de un solo nucleótido que provoca la sustitución de arginina con glutamina en la posición 506.33 La arginina 506 se localiza en uno de los dos principales sitios de ruptura de la PCA sobre el factor V activado. El factor V de Leiden activado tiene una actividad procoagulante normal, pero su degradación por la PCA es alrededor de 10 veces más lenta que la del factor V activado (factor Va) normal. Este cambio provoca mayor generación de trombina.34 Además, evidencias recientes sugieren que el factor V (pero no el factor Va), junto con la proteína S, sirve como un cofactor de la PCA en la inhibición del complejo factor VIIIa/factor IXa y que el factor V de Leiden tiene una mala función como cofactor de la PCA [ver figura 1].
|
| Figura 1 |
| Factor V de Leiden |
Diagnóstico
El diagnóstico del factor V de Leiden puede hacerse en forma rápida y con precisión con pruebas simples basadas en el ADN. Estas pruebas permiten hacer el diagnóstico en pacientes que han recibido tratamiento anticoagulante con warfarina y en los que tienen anticuerpos antifosfolípidos coexistentes. Debido a que el fenotipo de resistencia a la PCA no es causado solo por la mutación V de Leiden, puede ser adecuado completar el diagnóstico con una prueba de resistencia a la PCA en casos seleccionados.
El tratamiento del factor V de Leiden es similar al de las deficiencias de AT-III, proteína C y proteína S. Los pacientes con un primer episodio de trombosis venosa deben recibir tratamiento con anticoagulación por 6 meses. Se administrará anticoagulación profiláctica en situaciones que se sabe provocan trombosis, como cirugía. Debe considerarse la anticoagulación a largo plazo en los pacientes con trombosis recurrente.35 En las mujeres jóvenes que se sabe son portadoras de factor V de Leiden el uso de anticonceptivos orales aumenta el riesgo relativo de trombosis (que de cualquier modo es bajo). El escrutinio e rutina en familiares de pacientes con factor V de Leiden no parece tener buena relación costo-eficacia.20,36
MUTACION 20210A DEL GEN DE LA PROTROMBINA
Una mutación G a A en el nucleótido en la posición 20210 de la región 3' no traducida del gen de la protrombina se asocia con mayor incidencia de trombosis venosa. El riesgo relativo de trombosis en pacientes con esta mutación es de 2.8, similar al riesgo relativo en pacientes con factor V de Leiden.37 La prevalencia de la mutación en personas sanas es de alrededor del 2.3 por ciento. La mutación puede encontrarse en hasta el 18 por ciento de los pacientes con trombosis e historia familiar de trombosis. La presentación más común es la TVP de las extremidades inferiores. La combinación de uso de anticonceptivos orales y mutación del gen de protrombina se asocia con un incremento de alrededor de 150 veces más en el riesgo de trombosis venosa cerebral en mujeres jóvenes.37a
La hiperhomocisteinemia puede dividirse en tres clases: severa (concentración en plasma de homocisteína > 100 µml/L), moderada (25 a 100 µmol/L) y leve (16 a 24 µmol/L). La hiperhomocisteinemia severa suele ser causada por una deficiencia homocigota de la enzima cistationina [beta]-sintetasa. Las personas afectadas tienen retraso mental, luxación del cristalino, alteraciones esqueléticas y enfermedad trombótica arterial y venosa severa prematura.38 La hiperhomocisteinemia leve o moderada ocurre en los pacientes con defectos hereditarios o adquiridos en la vía metabólica de la homocisteína. La deficiencia heterocigota en la cistationina b-sintetasa es muy común en la población general, con una frecuencia del 0.3 a 1.4 por ciento.38 Ocurre con frecuencia un defecto en la vía de la remetilación causado por un mutante termolábil de la enzima metileno-tetrahidrofolato reductasa (MTHFR) que tiene alrededor del 50 por ciento de la actividad de la enzima normal. El estado homocigoto tiene una prevalencia del 5 por ciento en la población general.39 Las causas comunes de hiperhomocisteinemia adquirida incluyen deficiencias dietéticas de cobalamina, folato o piridoxina (los cofactores esenciales para la vía metabólica de la homocisteína). Un estudio prospectivo reciente mostró que la hiperhomocisteinemia leve es muy común en los ancianos, pesar de concentraciones normales de vitamina en suero.40
La hiperhomocisteinemia leve a moderada se asocia con enfermedad cerebrovascular, enfermedad coronaria y enfermedad vascular periférica en personas menores de 55 años,41 y con estenosis de la arteria carotídea en los ancianos.42 Se ha encontrado en el 10 por ciento de los pacientes con un primer episodio de TVP.43 En un estudio prospectivo reciente, se encontró una relación gradual entre los niveles aumentados de homocisteína en el plasma y la mortalidad en pacientes con enfermedad coronaria.44
Fisiopatología y cuadro clínico
En condiciones normales la homocisteína deriva de la metionina por un proceso de transmetilación. La homocisteína puede entonces ser remetilada a metionina por dos vías o convertirse a cisteína por transulfuración [ver figura 2]. Una vía de remetilación es catalizada por la metionina sintetasa, en la que el grupo metil es donado por la 5-metiltretrahidrofolato y con cobalamina (vitamina B12) actuando como cofactor. La otra vía de remetilación incluye a la betaína como donador del metilo. En el proceso de transulfuración la homocisteína se convierte por primera vez a cistationina, catalizada por la enzima cistationina b-sintetasa y con piridoxina como cofactor.
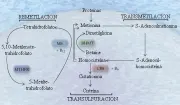
|
| Figura 2 |
| Metabolismo de la homocisteína |
La homocisteína es un aminoácido muy reactivo que contiene un grupo sulfhidrilo libre. Puede promover la oxidación de las lipoproteínas de baja densidad (LBD), del colesterol y al parecer es tóxico para el endotelio vascular.45,46 También puede inhibir la expresión de la trombomodulina y la activación de la proteína C47 y suprime la expresión de heparán sulfato en el endotelio.48 Todos estos efectos causan hipercoagulabilidad. Recientemente se ha demostrado que la homocisteína aumenta la unión de la lipoproteína(a), una lipoproteína aterogénica, a la fibrina, lo que puede establecer un enlace entre la hiperhomocistinemia, la trombosis y la ateroesclerosis prematura [ver adelante, Lipoproteína(a)].49 El daño vascular causado por los niveles altos de homocisteína causan trombosis arterial y venosa, y quiza ateroesclerosis acelerada.
Debe sospecharse hiperhomocisteinemia en casos de enfermedad trombótica arterial y venosa, incluyendo afección cerebrovascular, enfermedad arterial periférica y TVP, en especial en personas jóvenes. La homocisteína del plasma existe en forma libre y unida a proteínas, y generalmente se mide y reporta como homocisteína total en plasma (rango normal de 6 a 15 µmol/L). El diagnóstico de hiperhomocisteinemia suele hacerse midiendo el nivel en plasma después de una noche de ayuno. Sin embargo, debido a que hasta el 40 por ciento de los pacientes con hiperhomocisteinemia pueden tener una concentración normal en ayuno, debe considerarse realizar una prueba de carga con metionina cuando esté indicado.50 Se administra L-metionina (100 mg/kg) por vía oral al paciente después de una noche de ayuno y se toman muestras a las 4, 6 y 8 horas para medir el nivel de homocisteína en plasma. Las percentilas 90 para las muestras a las 4 y 6 horas son de 41.3 y 58.8 µmol/L, respectivamente.50,51 La percentila 95 para el nivel de 8 horas es de 35.8 µmol/L en la mujer y de 38.9 µmol/L en el varón.52 Deben medirse también los niveles de folato y vitamina B12 en el plasma para excluir hiperhomocisteinemia causada por deficiencias de folato o B12. También se cuenta con una prueba de genotipificación de ADN para la enzima MTHFR en laboratorios comerciales.
Los pacientes deben ser tratados diario con un esquema por vía oral de piridoxina (250 mg) y ácido fólico (5 mg), que debe normalizar los niveles elevados de homocisteína en la mayoría de los pacientes.53 Los enfermos deben recibir suplementos de vitamina B12 si existe deficiencia. En los pacientes con niveles altos de homocisteína en el plasma puede ser prudente medir la homocisteína después del tratamiento con piridoxina y folato para asegurarse de que el tratamiento fue eficaz. En ocasiones la betaína (3 g vo bid) es eficaz en pacientes con hiperhomocisteinemia resistente a piridoxina y folato. No se sabe si la corrección de la hiperhomocisteinemia por estas medidas tiene beneficio clínico.
La lipoproteína(a) [Lp(a)] constituye un factor de riesgo independiente para la trombosis de las arterias coronarias.54 Un estudio reciente, prospectivo, de casos y controles, asoció los niveles elevados en plasma de Lp(a) con un incremento alrededor del triple en el riesgo de enfermedad coronaria en varones.55 La asociación entre la Lp(a) alta y los eventos cerebrovasculares isquémicos en adultos jóvenes es motivo de controversia.56,57 El rango de distribución de la Lp(a) en la población general es estrecho; la percentila 95 se calcula en un rango de 25 a 30 mg/dl.58
La clase Lp(a) de las lipoproteínas se forma por el ensamble de partículas de LBD y apoproteína(a), una proteína que tiene cierta semejanza estructural con el plasminógeno y la apoproteína(a) compite con el plasminógeno por su sitio de unión en la célula endotelial, desplazándolo y disminuyendo así la generación de plasmina en la superficie de la célula endotelial.59 Por lo tanto, las concentraciones elevadas en plasma de Lp(a) pueden suprimir la respuesta endotelial fibrinolítica. La Lp(a) se encuentra en la íntima de los vasos ateroescleróticos humanos y los ratones transgénicos que expresan Lp(a) desarrollan ateroesclerosis extensa.60 En la actualidad pueden determinarse los niveles de Lp(a) en laboratorios comerciales, y debe considerarse realizar esta prueba en pacientes jóvenes con trombosis arterial. Debido a que los niveles altos de colesterol de LBD parecen aumentar los factores de riesgo asociados con la Lp(a) alta, deben usarse medidas de dieta, ejercicio y fármacos estándar en los pacientes con niveles altos de colesterol de LBD.61 En estudios pequeños, las dosis altas de niacina (2 a 4 g vo al día) y el tamoxifén (20 mg al día) han disminuido los niveles elevados de Lp(a) en 30 a 40 por ciento.62,63 Con frecuencia las dosis altas de niacina provocan rubor y cefalea, pero estos efectos desagradables pueden disminuirse si la niacina se inicia a dosis bajas (v.gr., 300 mg al día) y después se aumenta la dosis en forma gradual. Debe vigilarse la función hepática en forma periódica.
Se han definido alrededor de 300 fibrinógenos anormales (disfibrinógenos) y en la disfibrinogenemia se han detectado alrededor de 85 defectos estructurales. Estos defectos casi siempre se caracterizan por alteración funcional en la liberación del fibrinopéptido A y en la polimerización de la fibrina, y con menos frecuencia por unión y activación defectuosa del plasminógeno. Alrededor de la mitad de las mutaciones del fibrinógeno no se asocian con ningún síntoma clínico. Ocurre hemorragia leve o trombosis recurrente con la misma frecuencia en el resto de los casos.64 En casos raros coexiste hemorragia y trombosis en un solo paciente. La disfibrinogenemia adquirida puede asociarse con carcinoma hepatocelular o hepatopatía crónica. La evaluación general de laboratorio suele mostrar discrepancia entre los niveles antígenos y los niveles funcionales del fibrinógeno porque la mayoría de los disfibrinógenos tienen una función de coagulación subóptima, con prolongación del tiempo de trombina (TT) y del tiempo de reptilasa (TR). Los fibrinógenos anormales forman coágulos de fibrina que son resistentes a la lisis del coágulo. La identificación precisa del defecto estructural requiere de un esfuerzo importante en laboratorios de investigación. Un paciente con trombosis recurrente causada por disfibrinogenemia debe ser manejado en la misma manera que otros pacientes con trombofilia.
DISPLASMINOGENO Y FIBRINOLISIS ANORMAL
En casos raros plasminógenos anormales (displasminógenos) que son defectuosos en su activación a la plasmina se asocian con trombosis. Los pacientes con este trastorno pueden tener un nivel bajo de plasminógeno en los ensayos funcionales del plasma.65 Se han reportado niveles aumentados del inhibidor del activador del plasminógeno-1 (IAP-1) y menor actividad fibrinolítica en plasma en pacientes con preclampsia.66 La alteración adquirida en la actividad fibrinolítica puede asociarse con trombosis posoperatoria.67 Sin embargo, se requieren más estudios para establecer el papel de la fibrinolisis anormal en la trombosis clínica recurrente.68 Existen ensayos antigénicos para el activador tisular del plasminógeno (t-PA) y el IAP-1 en algunos laboratorios comerciales, pero los ensayos funcionales específicos se realizan solo en laboratorios de experimentación.
La deficiencia de factor XII no produce un trastorno hemorrágico, a pesar de la prolongación del TPT activado (TPTa). Paradójicamente, los pacientes homocigotos con deficiencia de factor XII pueden desarrollar tromboembolia. El factor XII puede participar en la activación por contacto de la fibrinolisis y este proceso estará defectuoso en los pacientes con deficiencia de factor XII. El tratamiento es semejante al de otros padecimientos tromboembólicos.69
NIVELES ELEVADOS DE FIBRINOGENO, FACTOR VII Y FACTOR VIII
El nivel elevado de fibrinógeno en plasma constituye un factor de riesgo independiente para la enfermedad arterial coronaria.70 El incremento en el nivel de factor VII también se asocia con el desarrollo de cardiopatía.71 Un factor VIII por arriba del 150 por ciento de lo normal se asocia con un incremento de alrededor de 5 veces en el riesgo de un primer episodio de TVP.72,73 Se requieren estudios adicionales para establecer la utilidad clínica de estos parámetros en los pacientes con trombosis.
Estados hipercoagulables adquiridos
SINDROME DE ANTICUERPOS ANTIFOSFOLIPIDO
El síndrome de anticuerpos antifosfolípido es causado por autoanticuerpos a proteínas asociadas con fosfolípidos cargados en forma negativa. Los términos antifosfolípidos y anticardiolipina se emplean como sinónimos. El término anticoagulante lúpico o parecido a lupus se refiere a un inhibidor que se identificó por primera vez en pacientes con lupus eritematoso. Parece correlacionar con una reacción de Wassermann para sífilis falsa positiva. A pesar del término "lúpico", muchos pacientes con estos inhibidores no tienen una enfermedad de tejido conectivo subyacente.
El síndrome de anticuerpos antifosfolípido ocurre en forma secundaria al lupus eritematoso generalizado y, con menos frecuencia a artritis reumatoide, arteritis de la temporal y otros trastornos del tejido conectivo. También se asocia con infecciones por VIH-1 y hepatitis C, trastornos linfoproliferativos y ciertos fármacos (v.gr., fenotiacinas y procainamida). Cuando no se asocia con ningún factor de riesgo identificable el síndrome se considera primario.
Las dos proteínas blanco más comunes para los anticuerpos antifosfolípidos parecen ser la b2-glicoproteína I (b2-GPI) y la protrombina. La b2-GPI es una proteína plasmática que fija fosfolípidos aniónicos con gran afinidad. Tiene una función anticoagulante débil in vitro. La b2-GPI puede inducir un cambio en la cardiolipina de su forma habitual bilaminar a una forma hexagonal que es muy inmunogénica.74 La prueba inmunosorbente ligada a enzimas (ELISA) para anticuerpos anticardiolipina suele detectar anticuerpos dirigidos contra este complejo cardiolipina/b2-GPI. Se han purificado anticuerpos que reaccionan específicamente con la protrombina pero no con la trombina. Estos anticuerpos purificados pueden aumentar la unión de la protrombina a la superficie de las células endoteliales en cultivo.75 Es posible que también participen otros complejos blanco proteínas-fosfolípidos. Se encuentran anticuerpos antifosfatidiletanolamina en muchos pacientes con síndrome de anticuerpos antifosfolípido y algunos de estos anticuerpos inhiben la función de la proteína C activada.76 Se han detectado también anticuerpos contra heparina y heparán sulfato, que inhiben la neutralización dependiente de heparina de la trombina por la AT-III.77 Debido a la heterogeneidad de los anticuerpos antifosfolípido, parece ser que múltiples mecanismos participan en la patogenia de la trombosis en este síndrome.
Ocurren eventos trombóticos en alrededor del 30 por ciento de los pacientes con anticuerpos antifosfolípido (incidencia global de 2.5 eventos por 100 pacientes año).79 Ocurre trombosis venosa en alrededor de dos terceras partes de los pacientes y trombosis arterial en un tercio. Las trombosis venosas suelen manifestarse como TVP de las extremidades inferiores, que en ocasiones se asocia con embolia pulmonar. También pueden ocurrir tromboflebitis superficial, trombosis de la vena de la retina y trombosis de las venas hepáticas (síndrome de Budd-Chiari) [ver tabla 4].
|
||||
GPL- unidades fosfolípido IgG MPL- unidades fosfolípido IgM |
Los eventos cerebrovasculares trombóticos y los episodios de isquemia
cerebral transitoria son manifestaciones importantes de las trombosis
arteriales. Las complicaciones obstétricas incluyen pérdidas
fetales recurrentes (ver adelante). Otras manifestaciones son anemia
hemolítica autoinmune, trombocitopenia y livedo reticularis.
Los anticuerpos antifosfolípido se detectan por ELISA (ver antes) o ensayos de coagulación. Debe pensarse en este diagnóstico en cualquier paciente que presente una trombosis arterial o venosa idiopática. Los criterios generales para el diagnóstico de anticoagulante lúpico son (1) prolongación de por lo menos una prueba de coagulación dependiente de fosfolípidos, (2) demostración de que la prolongación es causada por un inhibidor y no por deficiencia de un factor de coagulación y (3) confirmación de que el inhibidor depende de fosfolípidos.
Las pruebas de coagulación usadas con más frecuencia son el TPTa y el tiempo de veneno de víbora de Rusell diluido (TVVR). Los reactivos en el TPTa tienen sensibilidad variable al anticoagulante lúpico y están influenciados por la concentración en el plasma de ciertos factores de la coagulación (v.gr., factor VIII). Por lo tanto, la sensibilidad de esta prueba no es alta. Por lo menos debe realizarse otra prueba de coagulación. El TVVR diluido es mucho más sensible que el TPTa, pero es una prueba manual y no siempre está disponible. Cuando están disponibles, otras pruebas son útiles, como el tiempo de coagulación con caolín y la prueba de inhibición tisular de tromboplastina. La presencia de un inhibidor requiere de un estudio de mezcla para demostrar que no corrige con el plasma normal. La corrección de la prolongación adicionando un fosfolípido en la forma de plaquetas o un fosfolípido en fase hexagonal confirmará el diagnóstico. En los casos dudosos pueden realizarse ensayos de factores de la coagulación. La presencia de un anticoagulante lúpico causará deficiencia funcional de varios factores de la coagulación dependientes de fosfolípidos, y no de un factor en particular.
El diagnóstico de anticuerpos anticardiolipina, que generalmente se realiza por ELISA, se reporta como anticardiolipina de IgG (en unidades de antifosfolípido IgG [GPL]) y anticardiolipina de IgM (en unidades de antifosfolípido IgM [MPL]). La prevalencia de aumento en los niveles de anticardiolipina IgG e IgM en la población normal es de alrededor del 5 por ciento, y menos del 2 por ciento de las pruebas sigue siendo anormal al repetirlas.79 Los títulos altos de anticuerpos anticardiolipina IgG (> 33 GPL) se asocian con un incremento de alrededor del 5 por ciento en el riesgo trombótico general.79,80 No se ha establecido la importancia de los títulos bajos de anticuerpos IgG (< 20 GPL), anticuerpos IgM aislados o anticuerpos IgA.81,82 Deben solicitarse tanto ensayos funcionales como antigénicos en la evaluación del paciente porque estas dos pruebas no corresponden del todo.
Ciertas infecciones y exposición a fármacos pueden causar aparición transitoria de anticuerpos antifosfolípidos, que desaparecen al resolverse la infección o suspenderse el medicamento [ver tabla 5]. Por lo tanto, las pruebas de laboratorio deben repetirse por lo menos una vez (6 semanas después de la primera vez) para confirmar el diagnóstico. Por el contrario, alrededor del 20 por ciento de los pacientes con títulos bajos de anticardiolipinas de IgG tendrán títulos altos al repetir la prueba. Siempre está justificado repetir el estudio en pacientes con trombosis nuevas o recurrentes.81
|
|
|
Evolución clínica y tratamiento
Las recomendaciones terapéuticas actuales para el síndrome e anticuerpos antifosfolípido se basan casi en forma exclusiva en estudios observacionales que apoyan la asociación entre estos anticuerpos y la trombosis, en especial la trombosis recurrente.78-82 En el tratamiento agudo de la TVP en pacientes con síndrome antifosfolípido la vigilancia de la heparina regular por vía intravenosa puede complicarse porque el anticoagulante lúpico prolonga el TPTa. El uso de heparina de bajo peso molecular (HBPM) evita este problema porque no se requiere graduación de la dosis ni monitoreo. El paciente debe tratarse con HBPM y warfarina en la forma habitual, con un traslape de 5 días entre ambos fármacos.
Los análisis retrospectivos muestran que los pacientes con síndrome de anticuerpos antifosfolípido e historia de trombosis tienen alta tasa de recurrencias de trombosis (rango de 50 a 70 por ciento) si no reciben tratamiento intensivo con warfarina.83,84 El sitio del primer evento trombótico (i.e., arterial o venoso) tiende a predecir el sitio de las recurrencias.84 El mejor resultado se logra con tratamiento intensivo con warfarina, intentando mantener una relación internacional normalizada (INR) mayor a 3.0, aunque esto se asocia con un riesgo significativo de hemorragia. La aspirina no proporciona un beneficio adicional y aumenta el riesgo de sangrado.85 El autor mantiene el INR entre 3.0 y 3.5. Sin embargo, debido al mayor riesgo de sangrado, algunos clínicos experimentados emplean un límite de INR de 2.5 en lo que se obtienen los resultados de los estudios prospectivos.86
La duración óptima del tratamiento con anticoagulación oral no se ha establecido del todo. Las trombosis venosas recurrentes y los eventos cerebrovasculares en pacientes con síndrome antifosfolípido suelen justificar el tratamiento a largo plazo con warfarina. En un paciente con un primer episodio de TVP que tiene anticuerpos antifosfolípido está indicado el tratamiento con warfarina por lo menos durante 6 meses, y quizá de por vida.87 Es necesario tomar en cuenta la severidad del episodio trombótico específico, la coexistencia de cualquier factor de riesgo trombótico y los riesgos del tratamiento con anticoagulación oral a largo plazo. Debe mencionarse que el anticoagulante lúpico en ocasiones puede aumentar el tiempo de protombina y, asu vez, el INR, complicando la vigilancia del tratamiento con warfarina.88 Cuando aumentan el TP y el INR es útil emplear una tromboplastina reactiva insensible al anticoagulante lúpico. En los pacientes asintomáticos con anticuerpos anticardiolipina o anticoagulante lúpico pero sin historia de trombosis no se requiere anticoagulación.
Pérdidas fetales y síndrome de anticuerpos antifosfolípido
Varios estudios prospectivos confirman la asociación entre las pérdidas fetales recurrentes y los anticuerpos antifosfolípido. Deben medirse estos anticuerpos en las pacientes con historia de pérdidas del segundo o tercer trimestre sin explicación, muerte fetal, inicio temprano de preclampsia severa y retraso en el crecimiento intrauterino.89 Por el contrario, los anticuerpos antifosfolípido no se asocian con pérdidas tempranas esporádicas,90 que suelen deberse a alteraciones genéticas del feto. La relación entre anticuerpos antifosfolípido con infertilidad es incierta hasta el momento.
Al parecer las pérdidas fetales recurrentes en pacientes con síndrome de anticuerpos antifosfolípido se explican por daño a la placenta por los anticuerpos. La circulación materna en la interfase con la placenta está con contacto con el sinciciotrofoblasto, que puede ser más susceptible al daño que las células endoteliales. Aunque las células trofoblásticas pueden tener muchas propiedades semejantes a las de las células endoteliales,91 aún no se determina si poseen todos los mecanismos antitrombóticos de las segundas. La fosfatidilserina puede tener mayor exposición en los trofoblastos, lo que puede promover la formación del complejo protrombinasa y trombina. La anexina V (también denominada proteína anticoagulante placentaria-1), que en condiciones normales se encuentra en la placenta y que se une a la fosfatidilserina con gran afinidad, puede tener un efecto anticoagulante protector. Recientemente se ha reportado que los anticuerpos antifosfolípidos reducen los niveles de anexina V y aceleran la coagulación del plasma en cultivos de trofoblastos y células endoteliales. Por lo tanto, los anticuerpos antifosfolípido pueden ser un factor importante en la patogenia de la trombosis placentaria.92
El tratamiento de la mujer embarazada con síndrome de anticuerpos antifosfolípido es difícil por la asociación del síndrome con trombosis y el mayor riesgo de hemorragia asociado al tratamiento antitrombótico. En un estudio reciente, prospectivo, aleatorio y controlado con placebo, se demostró que la combinación de prednisona y aspirina era ineficaz para promover nacimientos vivos; de hecho, esta combinación pareció aumentar el riesgo de prematurez.93 Por otro lado, dos estudios prospectivos recientes demostraron que la heparina y la aspirina en dosis bajas (81 mg/día) permiten una evolución significativamente mejor de los embarazos que la aspirina sola en dosis bajas, logrando nacimientos vivos en el 70 a 80 por ciento de los casos.94,95 Aún más, la heparina en dosis bajas (administrada al inicio en dosis de 5,000 unidades por vía subcutánea cada 12 horas y ajustada para mantener el TPTa en el límite superior del rango normal) parece ser tan eficaz como las dosis altas de heparina combinadas con aspirina en dosis bajas.96 El tratamiento debe comenzar en cuanto se confirme el embarazo. Es preferible emplear HBPM porque este tipo de heparina puede administrarse una o dos veces al día y reduce el riesgo de osteopenia y trombocitopenia inducida por heparina (ver adelante). La HBPM se ha empleado con seguridad en mujeres embarazadas, pero no se han realizado estudios prospectivos y controlados que comparen la eficacia de la HBPM con la heparina regular en estas pacientes.
TROMBOCITOPENIA Y TROMBOSIS INDUCIDAS POR HEPARINA
Alrededor del 1 por ciento de los pacientes que reciben heparina porcina y el 5 por ciento de los que reciben heparina bovina sufren trombocitopenia 5 a 10 días después de que comienza su administración.77 Ocurre trombocitopenia de inicio más rápido en los pacientes que han recibido heparina en los 3 a 4 meses previos. La trombocitopenia suele ser leve, con cuentas de plaquetas que van de 50 a 60,000/µl, y los pacientes rara vez tienen hemorragia significativa. Un subgrupo de pacientes con trombocitopenia inducida por heparina (TIH) progresa a un trastorno muy peligroso e incluso fatal caracterizado por trombosis venosas y arteriales.
La patogenia de la TIH se atribuye a la presenica de anticuerpo IgG que reconoce un complejo de heparina y factor 4 plaquetario (F4P) [ver figura 3].98 El F4P es una proteína catiónica que se encuentra en los gránulos a de las plaquetas y que, cuando se libera, se une a la molécula de heparina (con carga negativa) con gran afinidad. El anticuerpo IgG se une al complejo F4P-heparina en las membranas de las plaquetas, formando un complejo ternario que a su vez se une al receptor FcgRII de la membrana plaquetaria. Esta unión activa a las plaquetas, causando mayor liberación de F4P y la formación del complejo F4P-heparina. Las plaquetas cubiertas con complejos inmunes son eliminadas con rapidez por el sistema retículoendotelial, causando así la trombocitopenia. Las complicaciones trombóticas en la TIH se deben a la activación de las plaquetas por los complejos inmunes, lo que provoca la formación de micropartículas de plaquetas y mayor generación de trombina.99 El F4P también se une a los glucosaminoglucanos sulfatados semejantes a heparina (v.gr., heparán sulfato) sobre la superficie de las células endoteliales. Las evidencias in vitro indican que el anticuerpo en la TIH es capaz de unirse a las células endoteliales, lo que las activa y predispone a la trombosis.100
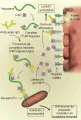
|
| Figura 3 |
| Trombocitopenia inducida por heparina |
Cuadro clínico
La TVP es el evento que con frecuencia lleva al diagnóstico de TIH. El trastorno puede complicarse por gangrena de la extremidad, en especial en el caso del tratamiento con warfarina sin una cobertura anticoagulante alternativa concomitante (ver adelante). La trombosis arterial incluye isquemia de la extremidad, eventos cerebrovasculares, infarto al miocardio y, con menos frecuencia, trombosis mesentérica y de la arteria renal. Algunos pacientes tienen datos de laboratorio que apoyan el diagnóstico de CID. Al inicio se pensó que el riesgo de trombosis asociada a la TIH era bajo; sin embargo, en un estudio clínico extenso reciente el riesgo de trombosis fue mayor al 50 por ciento en un grupo de pacientes ortopédicos que desarrollaron TIH durante el posoperatorio como resultado de la profilaxis con heparina.99
La prueba más empleada para diagnosticar TIH es la agregación de plaquetas inducida por heparina en presencia de suero del paciente. Sin embargo, la sensibilidad de esta prueba puede ser tan baja como del 50 por ciento.101 En ocasiones el suero del paciente puede causar agregación espontánea de las plaquetas del donador en ausencia de heparina, haciendo imposible la interpretación correcta de los resultados. Se ha desarrollado una prueba de ELISA para detectar anticuerpos en el suero de los pacientes reactivos al complejo F4P-heparina. Sin embargo, debido a que la TIH puede complicarse con problemas trombóticos serios, el diagnóstico de TIH debe basarse principalmente en los datos clínicos y el tratamiento deberá iniciarse aún antes de la confirmación por laboratorio.
El tratamiento de la TIH consiste en suspender el uso de heparina de inmediato e iniciar un tratamiento anticoagulante alternativo. Es importante suspender todos los tipos de administración de heparina: existen reportes anecdóticos de TIH causada por las cantidades mínimas de heparina que se usaban en las líneas arteriales.
Si el paciente ha recibido warfarina durante 4 a 5 días y el INR ha alcanzado niveles terapéuticos adecuados, el clínico puede usar solo warfarina y vigilar al paciente en forma cuidadosa. Sin embargo, si el paciente ha recibido warfarina durante menos de 4 días o tiene evidencia de una complicación trombótica deberá emplearse un anticoagulante alternativo.
Si el paciente tiene solo trombocitopenia leve, sin evidencia de trombosis, puede ser adecuado suspender la heparina y vigilar al paciente en forma cuidadosa sin un tratamiento anticoagulante alternativo. Sin embargo, el autor suele tratar el estado hipercoagulable subyacente con un anticoagulante alternativo. Este enfoque agresivo está apoyado por un estudio reciente retrospectivo de cohortes en el que alrededor de la mitad de los pacientes con TIH aislada al inicio desarrollaron trombosis en los primeros 30 días después de suspendida la heparina.97
Aunque la HBPM es mucho menos inmunogénica que la heparina estándar para causar TIH,99 no puede usarse como un sustituto seguro cuando un paciente desarrolla TIH por la heparina estándar. La HBPM y la heparina estándar tienen una extensa reactividad cruzada (> 90 por ciento) en términos de reconocimiento por anticuerpos. La HBPM no es una buena opción en los pacientes con TIH.
El danaparoide es un anticoagulante autorizado recientemente para la profilaxis de las TVP. El medicamento puede usarse también en pacientes con TIH. Es un compuesto heparinoide de bajo peso molecular formado de heparán sulfato, dermatán sulfato y condroitín sulfato. El efecto anticoagulante del danaparoide está mediado por una combinación de AT-III, cofactor II de heparina y un mecanismo celular endotelial no definido que actúa para hacer al danaparoide un inhibidor más selectivo del factor Xa que la HBPM. La relación anti-Xa-antitrombina del danaparoide es 28:1, lo que es alrededor de 10 veces mayor que para la HBPM. El danaparoide se vigila por una prueba anti-Xa, no con TPTa, y tiene escasa reacción cruzada in vitro (alrededor del 10 por ciento) con el anticuerpo de la TIH.102 El nivel anti-Xa se mantiene entre 0.5 y 0.8 U/ml.
El danaparoide es eficaz para tratar las TVP agudas y la TIH (en dosis de 2,000 unidades anti-Xa en bolo IV, seguido de 2,000 unidades por vía subcutánea bid).103 También se ha empleado con éxito en mujeres embarazadas con TIH.104 Las desventajas potenciales del danaparoide son su costo y su vida media larga (25 horas), además de que no tiene antídoto. Cuando ocurre hemorragia por la administración de danaparoide, ésta puede ser difícil de revertir. Al igual que la HBPM, el danaparoide se elimina por vía renal. En los pacientes con insuficiencia renal deben medirse los niveles anti-Xa.
El hirudin, un péptido de 65 aminoácidos extraído de la glándula salival de la sanguijuela medicinal (Hirudo medicinalis), es un inhibidor directo y potente de la trombina. El hirudin se une en forma directa al sitio activo de la trombina, independientemente de la AT-III. No es neutralizado por el F4P. La función anticoagulante del hirudin es vigilada por medio de TPTa.
El lepirudin es una forma recombinante de hirudin que recientemente se autorizó para emplearse en el tratamiento de la TIH. En dos estudios clínicos prospectivos, el uso de lepirudin redujo las complicaciones trombóticas serias en alrededor del 20 por ciento en los pacientes que recibieron el fármaco (comparado con una tasa de alrededor del 40 por ciento en los sujetos controles históricos).104,105 El lepirudin se administró en bolo IV (0.4 mg/kg) seguido de una infusión continua de 0.15 mg/kg/h. Los pacientes fueron vigilados para mantener un TPTa de 1.5 a 3 veces el normal por 2 a 10 días, según estuviera indicado. Los pacientes presentaron incremento en sangrados leves (en sitios de punción, epistaxis y hematuria) pero no hemorragia intracraneal. Es importante mencionar que el 40 por ciento de los pacientes desarrolló anticuerpos antihirudin. No existen anticuerpos antihirudin y en realidad éstos pueden aumentar la potencia del fármaco, quizá al retrasar su eliminación, otro motivo para vigilar los niveles del TPTa. Según estudios de cardiología intervencionista, el riesgo de hemorragia aumenta en forma sustancial con el uso concomitante de trombolíticos, por lo que no es aconsejable usar estos agentes en combinación. No existe un antídoto eficaz para el lepirudin, pero éste tiene una vida media más corta que el danaparoide (alrededor de 1.3 contra 25 horas). En la actualidad están en desarrollo otros inhibidores directos de la trombina con posibles aplicaciones para la TIH.
NEOPLASIAS - SINDROME DE TROUSSEAU
Algunos pacientes con cáncer (con frecuencia tumores sólidos ocultos del páncreas, ovario, hígado, cerebro, colon, pulmón o mama) tienen mayor propensión a desarrollar trombosis arterial recurrente, trombosis venosa, tromboembolia y síndrome de Trousseau.106 En un estudio prospectivo reciente de pacientes que presentaron TVP idiopática sintomática, se desarrolló cáncer en alrededor del 8 por ciento de los pacientes durante un periodo de seguimiento de 2 años (relación de momios de 2.3). En los pacientes con trombosis recurrente la incidencia de cáncer fue aún mayor (17 por ciento, con relación de momios de 4.3).107 Estos pacientes desarrollaron una endocarditis trombótica no bacteriana en la que se depositó fibrina estéril en la válvula mitral. El coágulo de fibrina puede embolizar para causar isquemia digital, isquemia cerebral transitoria y eventos cerebrovasculares. La trombosis venosa suele manifestarse como tromboflebitis migratoria superficial o TVP de las extremidades inferiores.
La causa subyacente del síndrome de Trousseau es una forma compensada y crónica de CID. Los procoagulantes activados generados en la CID aumentan la trombosis. En un estudio histológico reciente, las tinciones inmunoquímicas de cortes de tumores que con frecuencia se asocian con síndrome de Trousseau mostraron la presencia de factor tisular en la superficie tumoral.108,109 Se ha purificado un procoagulante tumoral, identificado como una proteasa de cisteína, de algunos adenocarcinomas y células leucémicas que puede activar la cascada de la coagulación al activar en forma directa el factor X.110,111 La interacción de las células tumorales con monocitos, plaquetas y células endoteliales puede generar también citocinas inflamatorias e inducir actividad procoagulante endotelial y monocítica, exacerbando la trombosis.
Los datos de laboratorio en el síndrome de Trousseau muestran evidencia de CID crónica de bajo grado (i.e., niveles anormalmente altos o bajos de fibrinógeno y plaquetas y niveles altos de dímero D). El TP y el TPT generalmente no están prolongados. Es rara la CID en estos pacientes.
Siempre surge la duda de qué tan agresivo debe ser el abordaje diagnóstico de un cáncer subyacente en un paciente con una TVP idiopática. Los estudios no han demostrado que las evaluaciones muy intensas tengan beneficio y una buena relación costo-eficacia.112 En un estudio reciente de cohortes, los diagnósticos de cáncer después de un evento trombótico fueron mayores durante los primeros 6 meses de seguimiento y disminuyeron con rapidez a niveles normales después del primer año. Además, el 40 por ciento de los pacientes con diagnóstico de cáncer en el primer año tenían metástasis a distancia en el momento del diagnóstico. No es claro si un diagnóstico más temprano, después del evento trombótico, hubiera cambiado el pronóstico de estos pacientes. Los investigadores concluyeron que no se justifica un enfoque agresivo para investigar un cáncer oculto en estos pacientes.113
En la actualidad es prudente realizar una historia clínica cuidadosa, un examen físico, radiografía de tórax, citología hemática y química sanguínea de rutina. Algunos especialistas han recomendado pruebas múltiples de sangre oculta en heces, antígeno prostático específico en los varones y mamografía y ultrasonido pélvico en las mujeres.114,115 Debe realizarse una vigilancia cuidadosa y se harán las pruebas indicadas según los resultados de la evaluación inicial.116
La clave para el manejo del síndrome de Trosseau consiste en diagnosticar y tratar el tumor subyacente. Por desgracia, los tumores suelen manifestarse en forma explosiva en los pacientes con este síndrome y pueden no responder a los tratamientos habituales. La warfarina, en dosis suficiente para mantener el INR de 2 a 3, suele no ser eficaz para el tratamiento de las trombosis en estos pacientes. Los enfermos con síndrome de Trousseau suelen requerir la administración a largo plazo de heparina en dosis completas (sea estándar o HBPM). Si un paciente está recibiendo quimioterapia para tratar el cáncer subyacente puede esperarse una exacerbación de la CID asociada con la lisis del tumor por que se requerirá mayor dosis de heparina.
REACCIONES TROMBOTICAS POR ESTROGENOS
Los anticonceptivos orales aumentan el riesgo de enfermedad tromboembólica alrededor de cuatro veces.117 Estudios epidemiológicos recientes indican que los anticonceptivos que contienen progestágenos de tercera generación tienen un riesgo del doble de trombosis comparado con los fármacos de segunda generación. Por este motivo, se prefieren los medicamentos de segunda generación para quienes emplean por primera vez anticonceptivos orales.118 El riesgo de tromboembolia venosa desaparece al suspender estos medicamentos.
El riesgo de tromboembolia venosa es alrededor de tres veces mayor en mujeres pomenopáusicas que reciben sustitución estrogénica en comparación con las que no reciben estrógenos. Sin embargo, el riesgo absoluto es bajo, calculado en alrededor de 3.2 por 10,000 pacientes-año. Por lo tanto, la sustitución estrogénica no está contraindicada en los pacientes que buscan los beneficios potenciales relacionados a la osteoporosis y la enfermedad coronaria.119,120
La explicación para la asociación entre el uso de estrógenos y la enfermedad tromboembólica se desconoce. Se han observado cambios en los niveles plasmáticos de gran número de proteínas que participan en la coagulación y la fibrinolisis, incluyendo reducción en la proteína S y la AT-III y aumento en el plasminógeno. Sin embargo, los cambios suelen ser modestos y no parecen ser los responsables del mayor riesgo de trombosis.
Tratamiento de los pacientes con factores de riesgo para tromboembolia venosa
TRATAMIENTO AGUDO DE LA TROMBOEMBOLIA VENOSA
El manejo agudo de un episodio inicial de TVP o embolia pulmonar en los pacientes con factores de riesgo posibles o demostrados para trombosis es el mismo que para el resto de los pacientes. En cuanto se realiza el diagnóstico el paciente debe ser anticoagulado con heparina. La HBPM por vía subcutánea se ha convertido en el fármaco de elección por su facilidad de uso y por el hecho de que no requiere la medición frecuente del TPT para ajustar la dosis. Los estudios aleatorios han demostrado que la HBPM es segura y eficaz para el tratamiento en casa.121,122
Después de iniciar la heparina los pacientes comienzan con warfarina en forma concomitante. Los medicamentos deben traslaparse por lo menos 5 días, incluso si el INR alcanza un rango terapeútico (i.e., 2 a 3) antes de ese tiempo. La prolongación inicial del TP después de la administración de warfarina puede atribuirse principalmente a la depleción factor VII funcional. El efecto antitrombótico real de la warfarina ocurre después, cuando el factor X y la protrombina disminuyen a menos del 20 por ciento. Puede ocurrir propagación de la trombosis si la heparina se suspende en forma prematura.
El INR debe mantenerse entre 2 y 3 en la mayoría de los pacientes. Sin embargo, en pacientes con síndrome de anticuerpos antifosfolípido suele recomendarse un tratamiento anticoagulante más agresivo (i.e., INR 3.0 a 3.5) (ver adelante).
El tratamiento con warfarina suele suspenderse por 3 a 6 meses. En un estudio reciente y prospectivo de tratamiento con anticoagulación oral en pacientes con un primer episodio de tromboembolia venosa, fue adecuado el tratamiento durante 6 semanas para los pacientes con factores de riesgo reversibles (v.gr., cirugía, traumatismo, inmovilización temporal y uso de anticonceptivos). Por otro lado, el tratamiento anticoagulante por vía oral durante 6 meses fue claramente superior para los pacientes con tromboembolia venosa idiopática (que se supone tienen factores de riesgo intrínsecos).123 Sin embargo, la tasa de recurrencia fue alta, alrededor de 12 por ciento a 2 años. Con base en esta evidencia, está indicado administrar por lo menos 6 meses de anticoagulación oral en un paciente con un primer episodio de TVP idiopática o embolia pulmonar. Durante 3 a 4 semanas después de suspender la warfarina los pacientes deben ser evaluados en busca de factores de riesgo de hipercoagulabilidad (v.gr., deficiencia de AT-III, proteína C y proteína S, anticuerpos antifosfolípidos y resistencia a la proteína C activada). La genotipificación con ADN para el factor V de Leiden puede realizarse en el momento del diagnóstico inicial.
El tratamiento posterior dependerá de los resultados del estado hipercoagulable. También es importante evaluar otros factores de riesgo. Las deficiencias heterocigotas de AT-III, proteína C y proteína S suelen considerarse factores de riesgo más importantes que el factor V de Leiden. La deficiencia de AT-III probablemente implique el mayor riesgo. La tasa de recurrencia en pacientes con síndrome antifosfolípido es tan alta como 50 a 70 por ciento en algunos estudios.
Dos estudios prospectivos ayudaron a evaluar el riesgo y beneficio del tratamiento a largo de la tromboembolia venosa. En un estudio, los pacientes fueron asignados en forma aleatoria para recibir tratamiento anticoagulante por 6 meses o en forma indefinida después de un segundo episodio de TVP o embolia pulmonar.124 Por el diseño del estudio se excluyó a los enfermos con deficiencias de AT-III, proteína C y proteína S. Después de 4 años de seguimiento el grupo asignado a 6 meses de tratamiento tuvo una tasa de recurrencia mucho más alta (20.7 por ciento) que el grupo con tratamiento continuo (2.6 por ciento). No fue sorprendente que existieron más sangrados en este último grupo (8.6 por ciento contra 2.7 por ciento, con una incidencia anual de hemorragias importantes de 2.4 por ciento por año).
En otro estudio, los pacientes con un primer episodio de tromboembolia venosa idiopática fueron distribuidos en forma aleatoria para recibir placebo o tratamiento anticoagulante oral por 2 años después de completar 3 meses de tratamiento anticoagulante.125 El grupo placebo tuvo una tasa de recurrencia de alrededor del 27 por ciento por paciente-año y el grupo con warfarina tuvo una tasa de recurrencia de 1.3 por ciento por paciente-año. El grupo con warfarina tuvo una tasa de hemorragias importantes de 3.8 por ciento por paciente-año. Cuando se analizaron los factores de riesgo para recurrencia, ni el factor V de Leiden ni la mutación 20210A de la protrombina tuvieron un efecto significativo. Solo el anticoagulante lúpico se asoció en forma significativa con las recurrencias.125
Estos estudios demuestran que el tratamiento con anticoagulación oral durante 3 meses es inadecuado en los pacientes con tromboembolia venosa idiopática. Sin embargo, aún no se ha definido la duración óptima del tratamiento anticoagulante oral a largo plazo. Es importante mencionar que no existen diferencias significativas en la mortalidad entre los pacientes que reciben tratamiento prolongado o los que reciben esquemas más breves.
Hasta el momento no existen evidencias que sugieran que el tratamiento anticoagulante profiláctico mejora la supervivencia global. Estudios históricos de familias con deficiencias de AT-III y proteína C no muestran mayor mortalidad que la de la población general.126,127
El riesgo de recurrencia determina el tratamiento a largo plazo de la tromboembolia [ver tabla 6].128,129 Antes de iniciar tratamiento de anticoagulación por tiempo prolongado en un paciente con riesgo alto el clínico debe considerar también el peligro de que el paciente sangre. Debido a que suele ser difícil distinguir entre la TVP recurrente y las exacerbaciones de un síndrome posflebítico subyacente, en ocasiones es adecuado realizar un ultrasonido duplex de las extremidades inferiores después de la resolución de un episodio de TVP. El estudio servirá como base para una comparación futura.
|
||||||||
INR- relación normalizada internacional |
Figuras 1 a 3 Dr. Rajeev Doshi.
Bibliografía
- Martinelli I, Mannucci PM, De Stefano V, et al: Different risks of thrombosis in four coagulation defects associated with inherited thrombophilia: a study of 150 families. Blood 92:2353, 1998
- Martinelli I, Cattaneo M, Panzeri D, et al: Risk factors for deep venous thrombosis of the upper extremities. Ann Intern Med 126:707, 1997
| 2a. | Prandoni P, Polistena P, Bernardi E, et al: Upper-extremity deep vein thrombosis: risk factors, diagnosis, and complications. Arch Intern Med 157:57, 1997 |
- Abilgaard U: Antithrombin and related inhibitors of coagulation. Recent Advances in Blood Coagulation. Poller L, Ed. Churchill Livingstone, Edinburgh, Scotland, 1981, p 151
- De Steano V, Leone G, Mastrangelo S, et al: Clinical manifestations and management of inherited thrombophilia: retrospective analysis and follow-up after diagnosis of 238 patients with congenital deficiency of antithrombin III, protein C, protein S. Thromb Haemost 72:352, 1994
- Demers C, Ginsberg JS, Hirsh J, et al: Thrombosis in antithrombin-III-deficient persons: report of a large kindred and literature review. Ann Intern Med 116:754, 1992
- Menache D, O'Malley JP, Schorr JB, et al: Evaluation of the safety, recovery, half-life, and clinical efficacy of antithrombin III (human) in patients with hereditary antithrombin III deficiency. Blood 75:33, 1990
- Miletich J, Sherman L, Broze G Jr: Absence of thrombosis in subjects with heterozygous protein C deficiency. N Engl J Med 317:991, 1987
- Miletich JP, Prescott SM, White R, et al: Inherited predisposition to thrombosis. Cell 72:477, 1993
- Boerger LM, Morris PC, Thurnau GR, et al: Oral contraceptives and gender affect protein S status. Blood 69:692, 1987
- Vigano-D'Angelo S, D'Angelo A, Kaufman CE Jr, et al: Protein S deficiency occurs in the nephrotic syndrome. Ann Intern Med 107:42, 1987 [PMID 2954500 ]
- Sallah S, Abdallah JM, Gagnon GA: Recurrent warfarin-induced skin necrosis in kindreds with protein-S deficiency. Haemostasis 28:25, 1998 [PMID 10047610 ]
- Faioni EM, Franchi F, Asti D, et al: Resistance to activated protein C in nine thrombophilic families: interference in a protein S functional assay. Thromb Haemost 70:1067, 1993 [PMID 8165605 ]
- Rees DC, Cox M, Clegg JB: World distribution of factor V Leiden. Lancet 346:1133, 1995 [PMID 7475606 ]
- Svensson PJ, Dahlback B: Resistance to activated protein C as a basis for venous thrombosis. N Engl J Med 330:517, 1994 [PMID 8302317 ]
- Koster T, Rosendaal FR, de Ronde H, et al: Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden Thrombophilia Study. Lancet 342:1503, 1993 [PMID 7902898 ]
- Simioni P, Prandoni P, Lensing AWA, et al: The risk of recurrent venous thromboembolism in patients with an Arg506ÆGln mutation in the gene for factor V (factor V Leiden). N Engl J Med 336:399, 1997 [PMID 9010145 ]
- Vandenbroucke JP, Koster T, Briet E, et al: Increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet 344:1453, 1994 [PMID 7968118 ]
- Rosendaal FR, Koster T, Vandenbroucke JP, et al: High-risk of thrombosis in patients homozygous for factor V Leiden (APC-resistance). Blood 85:1504, 1995 [PMID 7888671 ]
- Middledorp S, Henkens CMA, Koopman MMW, et al: The incidence of venous thromboembolism in family members of patients with factor V Leiden mutation and venous thrombosis. Ann Intern Med 128:15, 1998
- Rodeghiero F, Tosetto A: Activated protein C resistance and factor V Leiden mutation are independent risk factors for venous thromboembolism. Ann Intern Med 130:643, 1999 [PMID 10215560 ]
- Koeleman BPC, Reitsma PH, Allaart RC, et al: Activated protein C resistance as an additional risk factor for thrombosis in protein C-deficient families. Blood 84:1031, 1994
- Zoller B, Berntsdotter A, Garcia de Frutos P, et al: Resistance to activated protein C resistance as an additional genetic risk factor in hereditary deficiency of protein S. Blood 85: 3518, 1995 [PMID 7780138 ]
- van Boven HH, Reitsma PH, Rosendaal FR, et al: Factor V Leiden (FV R506Q) in families with inherited antithrombin deficiency. Thromb Haemost 75:417, 1996 [PMID 8701400 ]
- Ridker PM, Hennekens CH, Lindpaintner K, et al: Mutation in the gene coding for coagulation factor V and the risk for myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 332:912, 1995 [PMID 7877648]
- Williamson D, Brown K, Luddington R, et al: Factor V Cambridge: a new mutation (Arg306CÆ Thr) associated with resistance to activated protein C. Blood 91:1140, 1998 [çPMID 9454742 ]
- Chan WP, Lee CK, Kwong YL, et al: A novel mutation of Arg306 of factor V gene in Hong Kong Chinese. Blood 91:1135, 1998 [PMID 9454741 ]
- Zoller B, Svensson PJ, He X, et al: Identification of the same factor V gene mutation in 47 out of 50 thrombosis-prone families with inherited resistance to activated protein C. J Clin Invest 94:2521, 1994 [PMID 7989612 ]
- Bertina RM: Laboratory diagnosis of resistance to activated protein C (APC-resistance). Thromb Haemost 78:478, 1997
- van der Bom JG, Bots ML, Haverkate F, et al: Reduced response to protein C is associated with increased risk for cerebral vascular disease. Ann Intern Med 125:265, 1996 [PMID 8678388 ]
- Fisher M, Fernandez JA, Ameriso SF, et al: Activated protein C resistance in ischemic stroke not due to factor V arginine506Æ glutamine mutation. Stroke 27:1163, 1996 [PMID 8685921 ]
- Rodeghiero F, Tosetto A: Activated protein C resistance and factor V Leiden mutation are independent risk factors for venous thromboembolism. Ann Intern Med 130:643, 1999 [PMID 10215560 ]
- de Visser MCH, Rosendaal FR, Bertina RM: A reduced sensitivity to activated protein C in the absence of factor V Leiden increases the risk of venous thrombosis. Blood 93:1271, 1999
- Bertina RM, Koeleman BPC, Koster T, et al: Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 369:64, 1994 [PMID 8164741 ]
- Kalafatis M, Bertina RM, Rand MD, et al: Characterization of the molecular defect in factor V R506Q. J Biol Chem 270:4053, 1995 [PMID 7876154 ]
- Dahlback B: Resistance to activated protein C caused by the factor V R506Q mutation is a common risk factor for venous thrombosis. Thromb Haemost 78:483, 1997
- Dahlback B: Are we ready for factor V Leiden screening? Lancet 347:1346, 1996
- Poort SR, Rosendaal FR, Reitsma PH, et al: A common genetic variation in the 3¢-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood 88:3698, 1996 [PMID 8916933 ]
| 37a. | Martinelli I, Sacchi E, Landi G, et al:High risk of cerebral-vein thrombosis in carriers of a prothrombin-gene mutation and in users of oral contraceptives. NEngl J Med 338: 1793, 1998 |
- Mudd SH, Levy HL, Skovby F: Disorders of transulfuration. The Metabolic Basis of Inherited Disease. Scriver CR, Beaudet AL, Sly WS, et al, Eds. McGraw-Hill, New York, 1989, p 693
- Malinow MR: Homocyst(e)ine and arterial occlusive diseases. J Intern Med 236:603, 1994
- Naurath HJ, Joosten E, Riezier R, et al: Effects of vitamin B12, folate, and vitamin B6 supplements in elderly people with normal serum vitamin concentrations. Lancet 346:85, 1995 [PMID 7603218 ]
- Clarke R, Daly L, Robinson K, et al: Hyperhomocysteinemia: An independent risk factor for vascular disease. N Engl J Med 324:1149, 1991 [PMID 2011158 ]
- Selhub J, Jacques PF, Bostom AG, et al: Association between plasma homocysteine concentrations and extracranial carotid-artery stenosis. N Engl J Med 332:286, 1995 [PMID 7816063 ]
- Den Heijer M, Koster T, Blom HJ, et al: Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N Engl J Med 334:759, 1996 [PMID 8592549 ]
- Nygard O, Nordrehaug JE, Refsum H, et al: Plasma homocysteine levels and mortality in patients with coronary artery disease. N Engl J Med 337:230, 1997 [PMID 9227928 ]
- Harker LA, Harlan JM, Ross R: Effect of sulfinpyrazone on homocysteine-induced endothelial injury and arteriosclerosis in baboons. Circ Res 53:731, 1983 [PMID 6640860 ]
- Starkebaum G, Harlan JM: Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. J Clin Invest 77:1370, 1986 [PMID 3514679 ]
- Lentz SR, Sadler JE: Inhibition of thrombomodulin surface expression and protein C activation by the thrombogenic agent homocysteine. J Clin Invest 88:1906, 1991 [PMID 1661291 ]
- Nishinaga M, Ozawa T, Shimada K: Homocysteine, a thrombogenic agent, suppresses anticoagulant heparan sulfate expression in cultured porcine aortic endothelial cells. J Clin Invest 92:1281, 1993
- Harpel PC, Chang VT, Borth W: Homocysteine and other sulfhydryl compounds enhance the binding of lipoprotein(a) to fibrin: a potential biochemical link between thrombosis, atherogenesis, and sulfhydryl compound metabolism. Proc Natl Acad Sci USA 89: 10193, 1992 [PMID 1438209 ]
- Bostom AG, Jacques PF, Nadeau MR, et al: Post-methionine load hyperhomocysteinemia in persons with normal fasting total plasma homocysteine: initial results from The NHLBI Family Heart Study. Arteriosclerosis 116:147, 1995
- Den Heijer M, Blom HJ, Gerrits WBJ, et al: Is hyperhomocysteinemia a risk factor for recurrent venous thrombosis? Lancet 345:882, 1995
- Fermo I, D'Angelo SV, Paroni R, et al: Prevalence of moderate hyperhomocysteinemia in patients with early-onset venous and arterial occlusive disease. Ann Intern Med 123: 747, 1995 [PMID 7574192 ]
- Franken DG, Boers GHJ, Blom HJ, et al: Treatment of mild hyperhomocysteinemia in vascular disease patients. Arterioscler Thromb Vasc Biol 14:465, 1994
- Bostom A, Cupples A, Jenner J, et al: Elevated plasma lipoprotein(a) and coronary heart disease in men aged 55 years and younger. JAMA 276:544, 1996 [PMID 8709403 ]
- Wild SH, Fortmann SP, Marcovina SM: A prospective case-control study of lipoprotein(a) levels and Apo(a) size and risk of coronary heart disease in Stanford five-city project participants. Arterioscler Thromb Vasc Biol 17:239, 1997 [PMID 9081676 ]
- Nagayama M, Shinohara Y, Nagayama T: Lipoprotein(a) and ischemic cerebrovascular disease in young adults. Stroke 25:74, 1994 [PMID 8266386 ]
- Ridker PM, Stampfer MJ, Hennekens CH: Plasma concentration of lipoprotein(a) and the risk of future stroke. JAMA 273:1269, 1995 [PMID 7715039 ]
- Lipoprotein(a) concentrations and apolipoprotein(a) phenotypes in Caucasians and African-Americans. The CARDIA study. Arterioscler Thromb 13:1037, 1993
- Hajjar KA, Gavish D, Breslow JL, et al: Lipoprotein (a) modulation of endothelial cell surface fibrinolysis and its potential role in atherosclerosis. Nature 339:303, 1989 [PMID 2524666 ]
- Callow MJ, Verstuyft J, Tangirala R, et al: Atherogenesis in transgenic mice with human apolipoprotein B and lipoprotein(a). J Clin Invest 96:1639, 1995 [PMID 7657833 ]
- Maher VM, Brown BG, Marcovina SM, et al: Effects of lowering elevated LDL cholesterol on the cardiovascular risk of lipoprotein(a). JAMA 274:1771, 1995 [PMID 7500507 ]
- Gurakar A, Hoeg JM, Kostner G, et al: Levels of lipoprotein(a) decline with neomycin and niacin treatment. Atherosclerosis 57:293, 1985 [PMID 2935163 ]
- Shewmon DA, Stock JL, Rosen CJ, et al: Tamoxifen and estrogen lower circulating lipoprotein(a) concentrations in healthy postmenopausal women. Arterioscler Thromb 14: 1586, 1994 [PMID 7522547 ]
- Martinez J: Congenital dysfibrinogenemia. Curr Opin Hematol 4:357, 1997
- Aoki N, Moroi M, Sakata Y, et al: Abnormal plasminogen: a hereditary molecular abnormality found in a patient with recurrent thrombosis. J Clin Invest 61:1186, 1978 [PMID 659588 ]
- Estelles A, Gilabert J, Aznar J, et al: Changes in the plasma levels of type 1 and type 2 plasminogen activators in normal pregnancy and in patients with severe preeclampsia. Blood 74:1332, 1989 [PMID 2504307 ]
- Prins MH, Hirsh J: A critical review of the evidence supporting a relationship between impaired fibrinolytic activity and venous thromboembolism. Arch Intern Med 151: 1721, 1991 [PMID 1888237 ]
- Wiman B: Plasminogen activator inhibitor-1 (PAI-1) in plasma: its role in thrombotic disease. Thromb Haemost 74:71, 1995
- Rodeghiero F, Castaman G, Ruggeri M, et al: Thrombosis in subjects with homozygous and heterozygous factor XII deficiency. Thromb Haemost 67:590, 1992 [PMID 1519222 ]
- Meade TW: Fibrinogen in ischemic heart disease. Eur Heart J 16(suppl):31, 1995
- Meade TW, Mellows S, Brozovic M, et al: Haemostatic function and ischaemic heart disease: principal results of the Northwick Park Heart Study. Lancet 2:533, 1986 [PMID 2875280 ]
- Koster T, Blann AD, Briet E, et al: Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 345:152, 1995 [PMID 7823669 ]
- O'Donnell J, Tuddenham EGD, Manning R, et al: High prevalence of elevated factor VIII levels in patients referred for thrombophilia screening: role of increased synthesis and relationship to the acute phase reaction. Thromb Haemost 77:825, 1997 [PMID 9184386 ]
- Rauch J, Janoff AS: Role of antibodies in understanding the interactions between anti-phospholipid antibodies and phospholipids. Phospholipid-Binding Antibodies. N Harris, T Exner, GRV Hughes, et al, Eds. CRC Press, Boca Raton, Florida, 1991, p 108
- Rao LVM, Hoang AD, Rapaport SI: Mechanisms and effects of the binding of lupus anticoagulant IgG and prothrombin to surface phospholipid. Blood 88:4173, 1996
- Smirnov MD, Triplett DT, Comp PC, et al: On the role of phosphatidylethanolamine in the inhibition of activated protein C activity by antiphospholipid antibodies. J Clin Invest 95:309, 1995 [PMID 7814631 ]
- Shibata S, Harpel PC, Gharavi A, et al: Autoantibodies to heparin from patients with antiphospholipid antibody syndrome inhibit formation of antithrombin III-thrombin complexes. Blood 83:2532, 1994 [PMID 8167338 ]
- Finazzi G, Brancaccio V, Ciavarella N, et al: Natural history and risk factors for thrombosis in 360 patients with antiphospholipid antibodies: a four-year prospective study from the Italian registry. Am J Med 100:530, 1996 [PMID 8644765 ]
- Vila P, Hernandez MC, Lopez-Fernadez MF, et al: Prevalence, follow-up and clinical significance of the anticardiolipin antibodies in normal subjects. Thromb Haemost 72:209, 1994 [PMID 7831653 ]
- Ginsburg KS, Liang MH, Newcomer L, et al: Anticardiolipin antibodies and the risk for ischemic stroke and venous thrombosis. Ann Intern Med 117:997, 1992 [PMID 1443986 ]
- Silver RM, Porter TF, van Leeuween I, et al: Anticardiolipin antibodies: clinical consequences of "low titers." Obstet Gynecol 87:494, 1996
- Selva-O'Callaghan A, Ordi-Ros J, Monegal-Ferran F, et al: IgA anticardiolipin antibodies: relation with other antiphospholipid antibodies and clinical significance. Thromb Haemost 79:282, 1998 [PMID 9493576 ]
- Khamashta MA, Cuardrado MJ, Mujic F, et al: The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med 332:993, 1995 [PMID 7885428 ]
- Rosove MH, Brewer MC: Antiphospholipid thrombosis: clinical course after the first thrombotic event in 70 patients. Ann Intern Med 117:303, 1992 [PMID 1637025 ]
- Palareti G, Leali N, Coccheri S, et al: Bleeding complications of oral anticoagulant treatment: an inception-cohort, prospective collaborative study (ISCOAT). Lancet 348:423, 1996 [PMID 8709780 ]
- Greaves M: Antiphospholipid antibodies and thrombosis. Lancet 353:1348, 1999
- Kearon C, Gent M, Hirsh J, et al: A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 340:901, 1999 [PMID 10089183 ]
- Moll S, Ortel TL: Monitoring warfarin therapy in patients with lupus anticoagulants. Ann Intern Med 127:177, 1997 [PMID 9245222 ]
- Kutteh WH, Rote NS, Silver R: Antiphospholipid antibodies and reproduction: the antiphospholipid antibody syndrome. Am J Reprod Immunol 41:133, 1999 [PMID 10102085 ]
- Infante-Rivard C, David M, Gauthier R, et al: Lupus anticoagulants, anticardiolipin antibodies, and fetal loss: a case-controlled study. N Engl J Med 325:1063, 1991 [PMID 1909770 ]
- Buttery LD, McCarthy A, Springall DR, et al: Endothelial nitric oxide synthetase in the human placenta: regional distribution and proposed regulatory role in the feto-maternal interface. Placenta 15:257, 1994 [PMID 7520584 ]
- Rand JH, Wu X-X, Andree HAM, et al: Pregnancy loss in the antiphospholipid-antibody syndrome-a possible thrombogenic mechanism. N Engl J Med 337:154, 1997 [PMID 9219701 ]
- Laskin CA, Bombardier C, Hannah ME, et al: Prednisone and aspirin in women with autoantibodies and unexplained recurrent fetal loss. N Engl J Med 337:148, 1997 [PMID 9219700 ]
- Kutteh WH: Antiphospholipid antibody-associated recurrent pregnancy loss: Treatment with heparin and low-dose aspirin is superior to low-dose aspirin alone. Am J Obstet Gynecol 174:1584, 1996
- Rai R, Cohen H, Dave M, et al: Randomized, controlled trial of aspirin and aspirin plus heparin in pregnant women with recurrent miscarriage asssociated with phospholipid antibodies. BMJ 314:253, 1997 [PMID 9022487 ]
- Kutteh WH, Ermel LD: A clinical trial for the treatment of antiphospholipid antibody-associated recurrent pregnancy loss with lower dose heparin and aspirin. Am J Reprod Immunol 35:402, 1996 [PMID 8739461 ]
- Warkentin TE: Heparin-induced thrombocytopenia: a ten-year retrospective. Ann Rev Med 50:129, 1999
- Amiral J, Bridey F, Dreyfus M, et al: Platelet factor 4 complexed to heparin is the target for antibodies generated in heparin-induced thrombocytopenia. Thromb Haemost 68:95, 1992 [PMID 1514184 ]
- Warkentin TE, Levine MN, Hirsh J, et al: Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med 332:1330, 1995 [PMID 7715641 ]
- Visentin GP, Ford SE, Scott JP, et al: Antibodies from patients with heparin-induced thrombocytopenia/thrombosis are specific for platelet factor 4 complexed with heparin or bound to endothelial cells. J Clin Invest 93:81, 1994 [PMID 8282825 ]
- Greinacher A, Amiral J, Dummel V, et al: Laboratory diagnosis of heparin-associated thrombocytopenia and comparison of platelet aggregation test, and platelet factor 4/heparin enzyme linked immunosorbent assay. Transfusion 34:381, 1994 [PMID 8191560 ]
- Magnani HN: Orgaran (danaparoid sodium) use in the syndrome of heparin-induced thrombocytopenia. Platelets 8:74, 1997
- de Valk HW, Banga JD, Wester JWJ, et al: Comparing subcutaneous danaparoid with intravenous unfractionated heparin for the treatment of venous thromboembolism. Ann Intern Med 123:1, 1995 [PMID 7539233 ]
- Greinacher A, Volpol H, Potzsch B: Recombinant hirudin in the treatment of patients with heparin-induced thrombocytopenia (HIT). Blood 88(suppl):281a, 1996
- Schiele F, Vuillemenot A, Kramarz P, et al: Use of recombinant hirudin as antithrombotic treatment in patients with heparin-induced thrombocytopenia. Am J Hematol 50:20, 1995 [PMID 7668220 ]
- Callander N, Rapaport SI: Trousseau's syndrome. West J Med 158:364, 1993 [PMID 8317122 ]
- Prandoni P, Lensing AWA, Buller HR, et al: Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med 327:1128, 1992 [PMID 1528208 ]
- Callander NS, Varki N, Rao LVM: Immunohistochemical identification of tissue factor in solid tumors. Cancer 70:1194, 1992 [PMID 1381270 ]
- Contrino J, Hair G, Kreutzer DL, Rickles FR: In situ detection of tissue factor in vascular endothelial cells: correlation with malignant phenotype of human breast disease. Nat Med 2:209, 1996 [PMID 8574967 ]
- Falanga A, Gordon SG: Isolation and characterization of cancer procoagulant: a cysteine proteinase from malignant tissue. Biochemistry 24:5558, 1985 [PMID 3935163 ]
- Falanga A, Consonni R, Marchetti M, et al: Cancer procoagulant and tissue factor are differentially modulated by all-trans-retinoic acid in acute promyelocytic leukemic cells. Blood 92:143, 1998 [PMID 9639510 ]
- Prins MH, Hettiarachchi RJK, Lensing AWA, et al: Newly diagnosed malignancy in patients with venous thromboembolism: search or wait and see? Thromb Haemost 78:121, 1997
- Sorensen HT, Mellemkjaer L, Steffensen FH, et al: The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med 338:1169, 1998 [PMID 9554856 ]
- Silverstein RL, Nachman RL: Cancer and clotting: Trousseau's warning. N Engl J Med 327:1163, 1992 [PMID 1308758 ]
- Buller H, Ten Cate JW: Primary venous thromboembolism and cancer screening. N Engl J Med 338:1221, 1998 [PMID 9554864 ]
- Cornuz J, Pearson SD, Creager MA, et al: Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 125:785, 1996 [PMID 8928984 ]
- Vandenbroucke JP, Koster T, Briet E, et al: Increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet 344:1453, 1994 [PMID 7968118 ]
- Helmerhorst FM, Bloemenkamp KWM, Rosendaal FR, et al: Oral contraceptives and thrombotic disease: risk of venous thromboembolism. Thromb Haemost 78:327, 1997 [PMID 9198174 ]
- Jick H, Derby LE, Myers MW, et al: Risk of hospital admission for idiopathic venous thromboembolism among users of postmenopausal oestrogens. Lancet 348:981, 1996 [PMID 8855853 ]
- Daly E, Vessey MP, Hawkins MM, et al: Risk of venous thromboembolism in users of hormone replacement therapy. Lancet 348:977, 1996 [PMID 8855852 ]
- Levine M, Gent M, Hirsh J, et al: A comparison of low-molecular-weight heparin administration primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 334:677, 1996 [PMID 8594425 ]
- Koopman MMW, Prandoni P, Piovella F, et al: Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. N Engl J Med 334:682, 1996 [PMID 8663633 ]
- Schulman S, Rhedin AS, Lindmarker P, et al: A comparison of six weeks with six months of oral anticoagulant therapy after a first episode of venous thromboembolism. N Engl J Med 332:1661, 1995 [PMID 7760866 ]
- Schulman S, Granqvist S, Holmstrom M, et al: The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. N Engl J Med 336:393, 1997 [PMID 9010144 ]
- Kearon C, Gent M, Hirsh J, et al: A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 340:901, 1999 [PMID 10089183 ]
- Rosendaal FR, Heijboer H, Briet E, et al: Mortality in hereditary antithrombin-III deficiency: 1830 to 1989. Lancet 337:260, 1991 [PMID 1671110 ]
- Allaart CF, Rosendaal FR, Noteboom WM, et al: Survival in families with hereditary protein C deficiency, 1820 to 1993. BMJ 311:910, 1995 [PMID 7580547 ]
- Bauer KA: Management of patients with hereditary defects predisposing to thrombosis including pregnant women. Thromb Haemost 74:94, 1995
- De Stefano V, Finazzi G, Mannucci PM: Inherited thrombophilia: pathogenesis, clinical syndromes, and management. Blood 87:3531, 1996 [PMID 8611675 ]
- Triplett DA: Protean clinical presentation of antiphospholipid-protein antibodies (APA). Thromb Haemost 74:329, 1995
- Aster RH: Heparin-induced thrombocytopenia and thrombosis. N Engl J Med 332:1374, 1995