Neurología
⭳ Abrir artículo (PDF)628.4 KBEste artículo fue revisado respecto a la Edición 2/2000. Ver esa versión →
Contenido del artículo
VI NEOPLASIAS DEL SISTEMA NERVIOSO CENTRAL
- Tumores primarios del sistema nervioso
- EPIDEMIOLOGIA
- CLASIFICACION
- ETIOLOGIA
- FISIOPATOLOGIA
- SINTOMAS Y SIGNOS
- DIAGNOSTICO
- TRATAMIENTO
- PRONOSTICO
- TUMORES ESPECIFICOS
- Metástasis al sistema nervioso central
- Complicaciones no metastasicas del cáncer
VI NEOPLASIAS DEL SISTEMA NERVIOSO CENTRAL
DR. JEROME B. POSNER
Las neoplasias pueden dañar al sistema nervioso en tres formas: las células del sistema nervioso central o periférico pueden volverse cancerígenas,1-3 tumores localizados fuera del sistema nervioso (i.e., neoplasias sistémicas) pueden dar metástasis a estructuras neurales,3,4 y cánceres sistémicos pueden causar disfunción neurológica de modo indirecto3,4 [ver tabla 1]. Este capítulo se enfoca a los tumores primarios y metastásicos del sistena nervioso, aunque se mencionan algunos efectos no metastásicos del cáncer sobre el sistema nervioso.
Tumores primarios del sistema nervioso
EPIDEMIOLOGIA
Pueden desarrollarse tumores tanto en el cerebro como en la médula espinal, en especial en las células de la glía, las meninges y las estructuras asociadas, como la hipófisis y la glándula pineal. En 1994 la Asociación Americana contra el Cáncer predijo que existirían para fines del año siguiente 17,500 nuevos casos de cánceres en el cerebro y otros sitios del SNC (9,600 en varones y 7,900 en mujeres).5 Esta incidencia es mayor del doble que la de la enfermedad de Hodgkin y más de la mitad de la del melanoma. En los niños, los tumores cerebrales son los tumores sólidos más frecuentes, y ocupan en ese grupo de edad el segundo lugar (después de la leucemia) como causa global de cáncer. Debido a que las neoplasias del SNC son más frecuentes y más graves que las del sistema nervioso periférico, la discusión que sigue se enfoca sobre todo a las primeras.
Los meningiomas y los adenomas hipofisiarios son más frecuentes en mujeres6 y en afroamericanos.7 Por el contrario, los gliomas son más comunes en varones y blancos. Los gliomas de bajo grado de malignidad, como los astrocitomas, son más frecuentes en jóvenes que en ancianos, mientras que los de alto grado de malignidad, como los astrocitomas anaplásicos y el glioma multiforme, son más comunes en ancianos.
Las evidencias sugieren que la incidencia de los tumores primarios del SNC va en aumento.8,9 Este incremento es especialmente notable en la incidencia del linfoma primario del SNC (LPSNC) tanto en pacientes inmunosuprimidos (sobre todo los infectados por el virus de inmunodeficiencia humana [VIH]) como en huéspedes inmunocompetentes.8 Aunque antes era un tumor raro del SNC que se identificaba solo por autopsia, el LPSNC debe tenerse muy en cuenta en la actualidad para el diagnóstico y tratamiento de los tumores cerebrales.10 Es motivo de controversia si también está aumentando la incidencia de los tumores gliales, los estudios no invasores de neuroimagen han mejorado en forma sustancial la identificación de estos tumores, en especial en los ancianos en los que antes se hacía el diagnóstico erróneo de infartos cerebrales o demencia.11
En 1994 los cánceres del SNC fueron la causa de muerte de alrededor de 12,600 personas. En las mujeres la mortalidad causada por estas neoplasias fue más o menos la misma que la ocasionada por el cáncer de ovario. Los tumores cerebrales, que son los más temidos porque ocasionan incapacidad grave como parálisis, crisis convulsivas, demencia y disfunción del lenguaje, son la principal causa de muerte por cáncer en personas menores de 35 años.
CLASIFICACION
La Organización Mundial de la Salud ha clasificado a los tumores del sistema nervioso central y periférico por su origen celular12 [ver tabla 2]. Aproximadamente el 70 porciento de estos tumores primarios del SNC se originan en el cerebro o en la médula espinal. De estos, alrededor del 75 porciento son de origen neuroepitelial, y ocurren sobre todo en las células de la glía (por lo general astrocitos) o sus precursores. Estos tumores se subdividen por criterios histológicos en alto o bajo grado de malignidad, la atipia nuclear, las figuras mitóticas, la proliferación endotelial y la necrosis identifican a los de grado alto. Si existen tres o cuatro de estas características, el pronóstico es peor
Aunque los tumores cerebrales y de la médula espinal suelen clasificarse como benignos o malignos, es preferible su clasificación por grado histológico. Estos tumores rara vez son verdaderamente benignos porque la cirugía casi nunca es curativa, y tampoco son realmente malignos porque casi no dan metástasis. La cirugía fracasa porque incluso los tumores histológicamente benignos infiltran el tejido normal, lo que impide su resección total por el riesgo de destruir funciones cerebrales vitales.13 Incluso las cirugías radicales, como la hemisferectomía, han fallado para evitar la recurrencia tumoral en pacientes con glioblastoma. Además, ciertos tumores, como el meduloblastoma, el ependimoma, el glioblastoma y el LPSNC, se diseminan a las leptomeninges o a sitios distantes dentro del neuroeje. Sin embargo, muy rara vez se encuentran fuera del SNC.
ETIOLOGIA
Factores ambientales de riesgo
Existen pocas asociaciones inequívocas entre el ambiente y la ocurrencia de tumores cerebrales.14 La exposición a la radiación ionizante es el factor de riesgo mejor establecido.15 Se ha demostrado que la radiación en dosis bajas al cráneo, que en algún tiempo se usó como tratamiento para la tiña de la cabeza, ocasiona una incidencia de 10 veces más meningiomas, muchos de los cuales son anaplásicos o malignos, y un aumento de 3 veces en la incidenca de tumores gliales.16 La radiación en dosis altas para cánceres intra y extracraneanos, incluyendo la radiación profiláctica para la leucemia, aumenta la incidencia de gliomas y sarcomas.17 Sin embargo, las radiografías dentales no parecen constituir un factor de riesgo.14
El traumatismo craneano puede ser un factor de riesgo ambiental para el desarrollo de tumores gliales y otros tumores cerebrales.18 La infección por VIH es un factor de riesgo para el desarrollo de LPSNC y quizá de gliomas.19 Se ha sugerido que la exposición a la radiación electromagnética, los colorantes para el cabello, los pesticidas, el formaldehido u otras sustancias de uso industrial o laboral causan tumores cerebrales, pero esta hipótesis no ha sido confirmada.14 Si en realidad estas sustancias constituyen factores de riesgo, causan solo una proporción pequeña de los tumores cerebrales.
Factores genéticos de riesgo
Algunas enfermedades hereditarias predisponen al desarrollo de tumores del SNC [ver tabla 3].20 La neurofibromatosis se asocia con gliomas en hasta el 14 porciento de los pacientes que tienen neurofibromatosis tipo 1 (NF-1 o enfermedad de von Recklinghausen) y con schwannomas, meningiomas y, menos común, ependimoma, en pacientes con neurofibromatosis tipo 2 (NF-2). El gen supresor de tumores NF-1 ya ha sido clonado,21,22 y se ha reportado un posible gen supresor de tumores para la NF-2.23,24
Las alteraciones genéticas adquiridas (i.e., no familares) se asocian también con tumores del SNC. Estas alteraciones incluyen la pérdida o mutación de un gen supresor de tumores, como el p53 o el gen del retinoblastoma (Rb), y la amplificación y rearreglo de los oncogenes. Muchos oncogenes codifican para factores de crecimiento o receptores de factores de crecimiento que pueden autoestimular células tumorales (estimulación autócrina) o células cercanas (estimulación parácrina).
Existen diversas alteraciones genéticas asociadas con los tumores cerebrales primarios más frecuentes [ver tabla 4]. Las alteraciones más comunes identificadas en pacientes con tumores gliales son las mutaciones en el p53, que ocurren en el 40 porciento de los pacientes con astrocitoma y con aproximadamente la misma frecuencia en pacientes con astrocitoma anaplásico o glioblastoma multiforme,25-27 y la expresión exagerada del receptor del factor de crecimiento epidérmico, en forma normal o mutada, que ocurre en alrededor del 40 porciento de los pacientes con glioblastoma multiforme.28-30 También se han implicado al factor de crecimiento derivado de plaquetas31,32 y a los factores de crecimiento de fibroblastos.33 No han podido identificarse secuencias específicas de alteraciones genéticas en la progresión de un astrocitoma de bajo grado a un glioblastoma multiforme. En consecuencia, algunos investigadores han propuesto que el glioblastoma multiforme puede originarse de novo o por una serie de pasos desde el astrocitoma de bajo grado de malignidad.34
FISIOPATOLOGIA
La fisiopatología de los tumores del SNC explica porqué incluso los tumores relativamente pequeños causan síntomas, mientras que los tumores sistémicos con frecuencia no.4 Primero, los tumores pequeños pero con localización estratégica, dañan vías vitales del cerebro y la médula espinal, ocasionando disfunción importante. Lesiones semejantes en órganos más homogéneos, como el pulmón o el hígado, deben destruir grandes áreas del órgano para que aparezcan síntomas. Segundo, debido a que el cerebro y la médula espinal se encuentran contenidos en la duramadre y el hueso, existe poco espacio para que crezca una neoplasia sin comprimir el tejido normal [ver figura 1]. Tercero, los vasos tumorales no poseen una barrera hemato-encefálica normal. Por pequeñas hendiduras pasan proteínas y otras sustancias nocivas que pueden entrar al tumor y difundir al tejido normal circundante, ocasionando edema y mayor compresión de las estructuras normales [ver figura 1]. La eliminación del edema es muy lenta porque el cerebro no tiene linfáticos.
Aunque la mayoría de los tumores pequeños causa síntomas, ocurren excepciones: los tumores de crecimiento lento, en especial los que se encuentran en áreas cerebrales silenciosas, pueden no causar síntomas hasta que son muy grandes. Incluso el glioblastoma multiforme produce en ocasiones menos síntomas de lo esperado por el tamaño de la lesión y el grado de desplazamiento observado en los estudios de imagen.
Los síntomas del SNC son causados por tres mecanismos:
1.El tumor invade, irrita y remplaza el tejido normal. Este mecanismo es especialmente característico de los gliomas infiltrantes de bajo grado de malignidad, pero es raro en los tumores metastásicos.
2.Tanto el tumor como el edema comprimen el tejido normal y sus vasos sanguíneos, produciendo isquemia.
3.Los tumores del tercero, cuarto ventrículo o las leptomenínges, bloquean la circulación del líquido cefalorraquídeo, causando hidrocefalia.
Los efectos de la invasión tumoral, el edema peritumoral y la hidrocefalia se combinan para herniar las estructuras cerebrales normales por debajo de la hoz cerebral, a través de la tienda del cerebelo o a través del foramen magno [ver figura 1].
SINTOMAS Y SIGNOS
Los pacientes con tumores cerebrales pueden tener síntomas generalizados causados por presión intracraneal difusa, síntomas focales ocasionados por isquemia y compresión en el sitio del tumor,35 o síntomas de falsa localización por el desplazamiento de estructuras cerebrales.36 Los síntomas generalizados o de falsa localización se presentan más en tumores de crecimiento lento localizados en el lóbulo frontal (relativamente silente), mientras que ocurren síntomas focales con tumores relativamente menores en áreas funcionales más importantes, como el área motora y el tallo cerebral, o en la médula espinal.
Síntomas y signos generalizados
La cefalea, el síntoma más frecuente de hipertensión intracraneana, es el primer síntoma en alrededor del 40 porciento de los pacientes con un tumor cerebral.37 Es más común como primer síntoma de un tumor en los pacientes con antecedente de cefalea no tumoral que en los que no tienen este antecedente. La mayoría de las cefaleas asociadas a tumores son inespecíficas; sin embargo, debe sospecharse un tumor cuando el dolor existe al despertar el paciente y desaparece en el lapso de una hora, cuando la cefalea comienza en una persona de edad media o anciana que antes no sufría esta molestia, o cuando se presenta un cambio súbito en las características o intensidad de la cefalea en un paciente con cefaleas crónicas. Cuando la cefalea es localizada, es un indicador confiable del lado de la cabeza que contiene el tumor, aunque no marca su localización precisa. Por ejemplo, una cefalea frontal derecha indica que el tumor está del lado derecho, pero no que el tumor es frontal, puede ser occipital o incluso cerebeloso.
El vómito, con o sin náusea previa, en especial al despertar, es un síntoma común de tumor cerebral en niños, pero menos frecuente en adultos. La cefalea aguda seguida de inmediato por vómito es característica de un tumor e indica hipertensión intracraneana. Por el contrario, la cefalea más prolongada seguida de vómito varias horas después es típica de migraña. El papiledema, que ocurre con frecuencia en niños, pero es menos común en pacientes de mayor edad, es un signo de hipertensión intracraneana.38 Suele ser asintomático, pero puede causar episodios temporales de ceguera.
Los cambios mentales asociados con los tumores cerebrales comienzan con irritabilidad y progresan hacia apatía. Los pacientes duermen más tiempo, parecen preocupados al despertar y tienen dificultad para iniciar alguna actividad, incluso una conversación; sin embargo, si se les habla suelen responder en forma adecuada. Es frecuente que se acuda a un psiquiatra por lo que se piensa es un cuadro de depresión, antes de sospechar un tumor cerebral.
Los tumores cerebrales pueden asociarse con síntomas episódicos que incluyen cefalea, pérdida visual, alteraciones de la conciencia y en ocasiones debilidad temporal de las extremidades. Estos episodios suelen ser precipitados al incorporarse desde la posición de decúbito, al toser o estornudar, y se asocian con ondas de meseta,39,40 incrementos súbitos en una presión intracraneal ya elevada que duran de cinco a 20 minutos. Estos síntomas no corresponden a crisis convulsivas, y mejoran con la reducción de la presión intracraneana, pero no con tratamiento anticonvulsivante.
Síntomas y signos focales
Las crisis convulsivas son los signos focales más frecuentes de un tumor cerebral. Son especialmente comunes en pacientes que tienen tumores que comprimen la corteza (como los meningiomas) o se originan cerca del área motora o del lóbulo temporal, ambas zonas epileptógenas. Las crisis focales causadas por focos frontales o temporales suelen causar síntomas de comportamiento o emocionales que pueden confundirse con ataques de pánico o trastornos psicológicos. Las convulsiones generalizadas son también de inicio focal, originándose en un foco de descarga asintomático. Dependiendo de la velocidad de crecimiento del tumor, las crisis convulsivas pueden presentarse meses a años antes de que se desarrollen otros síntomas. Cualquier paciente con crisis convulsivas focales o generalizadas que comienzan en la edad adulta debe someterse a una evaluación diagnóstica para investigar un tumor cerebral (ver adelante).
Otros síntomas y signos focales de un tumor cerebral dependen del sitio de la lesión. Estos son los mismos que los causados por una infección, un episodio cerebrovascular y otras enfermedades estructurales del cerebro o la médula espinal.
Síntomas de localización falsa
Los síntomas de localización falsa se deben al desplazamiento de estructuras cerebrales. La diplopia puede ser causada por desplazamiento o compresión del sexto par craneal en la base cerebral. La hemianopsia se explica por herniación tentorial que comprime a la arteria cerebral posterior. También pueden ocurrir otras parálisis de nervios craneales asociadas con desplazamiento de las estructuras del tallo cerebral.36
DIAGNOSTICO
Debe sospecharse una neoplasia del SNC en todos los adultos con crisis convulsivas de inicio reciente y en todos los pacientes con papiledema o nuevos signos motores focales o sensoriales. En estos pacientes se justifica realizar una imagen por resonancia magnética (IRM) con inyección de medio de contraste, como gadolinio, para determinar si existe ruptura de la barrera hematoencefálica y su localización.41 Los enfermos con cambios en la personalidad o el comportamiento deben ser evaluados con una IRM cuando los síntomas psiquiátricos son atípicos o se acompañan de somnolencia, apatía o pérdida de la memoria. Los pacientes con cefalea solo requieren IRM cuando ésta es de inicio reciente, ha cambiado en características o no responde a tratamiento. Una IRM negativa descarta la posibilidad de un tumor como causa de los síntomas o signos del paciente, no tiene utilidad repetir la prueba.
La IRM identifica tumores que la tomografía computada no detecta, y distingue a los tumores de las malformaciones arteriovenosas. Con excepción de la biopsia (ver adelante), no se requieren otros estudios de laboratorio. La IRM sugiere también la histología del tumor: los gliomas de bajo grado de malignidad suelen no tener reforzamiento con el medio de contraste, y su imagen se caracteriza por ser hiperintensa en T2 y normal o hipointensa en T1 (T2 se refiere a tiempo de relajación giro-giro o transverso, y T1 a tiempo de relajación giro-reja, o longitudinal). El área hiperintensa en la imagen T2 incluye tanto al tumor como al edema asociado. Por el contrario, los gliomas de alto grado de malignidad suelen tener reforzamiento con el medio de contraste, la imagen T1 muestra un anillo reforzado de borde y grosor irregulares que rodea a un centro hipointenso, y la imagen T2 muestra solo hiperintensidad [ver figura 2a]. Aunque el reforzamiento no corresponde a todo el margen infiltrante, permite una aproximación clínica sobre el volumen del tumor. Las metástasis tienen un borde regular y esférico [ver figura 2b]. Ocurren más en forma múltiple: el 50 porciento de los pacientes con metástasis tienen lesiones múltiples, comparando con solo el cinco porciento de los pacientes con gliomas. De los enfermos con LPSNC, el 40 porciento tiene tumores múltiples, que se localizan en forma periventricular, mostrando por lo general reforzamiento difuso con el medio de contraste, márgenes poco delimitados al comparar con los gliomas y las metástasis, y menos edema circundante que en el caso de otros tumores [ver figura 2c].
Aunque la IRM puede sugerir el diagnóstico histológico, solo
la biopsia es definitiva. Una excepción a esta regla ocurre en el LPSNC,
en el que la punción lumbar o la vitrectomía (cuando existe
afección ocular) pueden demostrar las células malignas. En los
otros tumores la realización de una punción lumbar se asocia con
riesgo de herniación cerebral,42 y con beneficio casi
nulo.
TRATAMIENTO
El tratamiento de los pacientes con neoplasias del SNC depende del tipo y localización del tumor y del estado general del paciente. Sin embargo, existen ciertos principios generales que se aplican a todos los casos de neoplasias del SNC.2-4
Esteroides
Los esteroides alivian en forma dramática los síntomas de los tumores cerebrales y de la médula al disminuir la presión intracraneana y el edema que rodea a las lesiones. La mejoría sintomática comienza en minutos y los pacientes suelen estar asintomáticos a las 24 a 48 horas después de su administración. Los mecanismos por los que los esteroides tienen estos efectos no son muy claros; sin embargo, muchos investigadores afirman que estos medicamentos disminuyen el flujo de agentes hidrosolubles a través de la barrera hematoencefálica dañada. Es posible que también tengan el efecto no buscado de inhibir la entrada de agentes quimioterápicos al tumor.
Los esteroides están indicados en todos los pacientes sintomáticos con tumores cerebrales y medulares, con excepción de los que tienen LPSNC. Por lo general se administra dexametasona, en dosis de 16 mg/día. Debido a sus efectos linfolíticos, pueden ocasionar necrosis tumoral, impidiendo el diagnóstico definitivo. Por lo tanto, su administración debe retrasarse cuando se sospeche LPSNC hasta después de realizada la biopsia.
Una vez comenzados, la administración del esteroide se continúa hasta que los síntomas del paciente se alivien y disminuya la presión intracraneana. Después se reduce la dosis en forma paulatina hasta lograr la mínima compatible con una buena función neurológica. Con frecuencia pueden suspenderse por completo.
Cirugía
Aunque la cirugía para pacientes con tumores cerebrales o de la médula espinal rara vez es curativa, este es el tratamiento más importante en pacientes con tumores accesibles que no son LPSNC. La cirugía establece el diagnóstico, alivia la presión intracraneana y mejora los síntomas y el control de las crisis convulsivas. Además, el uso de técnicas quirúrgicas modernas en manos de un cirujano experimentado y la administración de esteroides para evitar el edema posoperatorio, reduce el riesgo de empeoramiento de la función neurológica. Aunque existe cierta controversia respecto al papel de la cirugía en pacientes con gliomas, la mayoría de las evidencias indican que la extirpación quirúrgica del tumor mejora tanto la duración de la supervivencia como la calidad de vida.43-45 Si el tumor muestra reforzamiento con medio de contraste antes de la cirugía, la realización de una IRM con medio de contraste tres días después de la resección predice la magnitud del tumor residual y ayuda a establecer el pronóstico.44 Los cálculos de la extensión de la resección por parte del cirujano suelen ser inexactos.
Radioterapia
El tratamiento con radioterapia posoperatoria prolonga la duración de la supervivencia y mejora la calidad de vida en pacientes con tumores de alto grado de malignidad (i.e., astrocitomas anaplásicos o gliomas), pero su papel en los pacientes con tumores de bajo grado, en especial asintomáticos, no se ha definido. Después de la resección total de los astrocitomas pilocíticos no se requiere radioterapia. En los pacientes asintomáticos con astrocitomas de bajo grado u oligodendrogliomas, la radioterapia puede retrasarse en forma segura hasta que se presenten síntomas. Los gliomas cerebrales y de la médula espinal deben tratarse con radiación en dosis altas (5,500 a 6,000 cGy [5,500 a 6,000 rads] en fracciones de 180 a 200 cGy [180 a 200 rads]) administrados en un campo limitado que incluya al tumor y a sus zonas circundantes.46 En algunos pacientes puede ser útil aumentar la radioterapia convencional con braquiterapia (en la que la fuente de radiación se implanta dentro del tumor)46,47 o radiocirugía estereotáctica (en la que se dirigen dosis altas de radiación ionizante en múltiples haces angostos hacia una localización intracraneana precisa por medio de estereotaxia).46 Aún no se ha determinado del todo el papel exacto de estas técnicas.
Quimioterapia
La respuesta de la mayoría de los gliomas a la quimioterapia es limitada, pero la adición de nitrosureas o procarbacina al tratamiento aumenta la supervivencia de algunos pacientes con gliomas de alto grado de malignidad.48 Es imposible predecir qué pacientes responderan a la quimioterapia, por lo que todos los pacientes con gliomas de alto grado deben tratarse con una combinación de radioterapia y una nitrosurea u otro agente quimioterápico liposoluble.
Otras modalidades terapéuticas
Ni la inmunoterapia ni el tratamiento genético están indicados en la actualidad para los tumores cerebrales o de la médula spinal.49 Sin embargo, en la actualidad estas modalidades son motivo de gran investigación, y quizá en un futuro demuestren ser benéficas.
PRONOSTICO
Varios factores determinan el pronóstico de los pacientes con tumores cerebrales primarios.50,51 El grado histológico es un factor pronóstico importante. Por ejemplo, la supervivencia promedio de los pacientes con glioblastoma multiforme es un poco mayor de un año, y la tasa de supervivencia a cinco años es menor del 10 porciento.52 Los pacientes con astrocitoma anaplásico tienen una supervivencia promedio de dos a tres años, y los que tienen astrocitomas, de cinco a 10 años. Sin embargo, los enfermos con astrocitoma pilocítico tienen una esperanza de vida normal.
La edad es otro factor importante para determinar el pronóstico, y los pacientes más jóvenes sobreviven más tiempo que los de mayor edad con tumores similares histológicamente. En el caso del glioblastoma multiforme, el origen del tumor tiene también importancia pronóstica: un paciente con un glioblastoma que se origina de un astrocitoma de bajo grado de malignidad puede tener mejor pronóstico que un paciente con un glioblastoma que se origina de novo.53,54 Otros factores pronósticos incluyen la extensión de la cirugía (la resección más completa mejora la supervivencia),43-45 la dosis de radiación administrada (a mayor dosis mayor supervivencia),46 y la administración de quimioterapia (los agentes quimioterapéuticos mejoran la supervivencia en el 20 a 30 porciento de los pacientes).48
TUMORES ESPECIFICOS
Meningiomas
Los meningiomas se originan de la duramadre y de las vellosidades aracnoideas de los espacios intracraneal y espinal. Se localizan a lo largo de la superficie dorsal y base del cerebro, la hoz cerebral y el borde esfenoidal, así como en los ventrículos laterales y el canal medular torácico. Son más comunes en mujeres que en hombres. Poseen receptores para progesterona y, con menos frecuencia, para estrógenos.55 Las mujeres con cáncer de mama pueden tener mayor riesgo de meningiomas.56
Las alteraciones genéticas que se han implicado en el desarrollo de los meningiomas incluyen la pérdida del brazo largo del cromosoma 22, que es la más frecuente,57,58 del brazo corto del cromosoma 14 y, más raro, del brazo corto de los cromosomas 1, 7 o Y.59-62 Los meningiomas pueden ser mútiples y con frecuencia crecen con lentitud, causando síntomas por compresión cerebral, no por invasión. Los síntomas comunes de presentación de los meningiomas craneales son crisis convulsivas y parálisis de pares craneales, los de los meningiomas medulares incluyen paraparesias graduales y en ocasiones no dolorosas.
Los pacientes con meningiomas pueden diagnosticarse con facilidad por medio de una IRM con contraste. Los meningiomas son isodensos en la imagen T1, muestran reforzamiento uniforme con el medio de contraste y se distinguen con claridad del cerebro o la médula espinal. En ocasiones puede identificarse un área de fijación dural elongada, o pueden aparecer en forma de placa, causando engrosamiento difuso de la duramadre.
Los pacientes con meningiomas pueden ser curados con la extirpación quirúrgica completa del tumor o tumores. Sin embargo, muchos meningiomas, en especial los localizados en los senos cavernosos y en la base del cerebro, no pueden resecarse en su totalidad. Además, algunos meningiomas, en especial los que son anaplásicos, recurren a pesar de una resección aparentemente completa. La radioterapia puede controlar estas recidivas. No se ha demostrado que el uso de agentes quimioterápicos u hormonales55 sea benéfico. Debido a la lentitud de su crecimiento, los meningiomas pequeños o asintomáticos pueden vigilarse en lugar de resecarse, en especial en ancianos.
Schwannomas
Los schawnnomas (también llamados tumores de células de Schwann o neurilemomas) pueden presentarse en el cráneo o en el sistema nervioso periférico. Los neuromas del acústico, los schwannomas más frecuentes, ocurren en la porción vestibular del octavo par craneal, cerca o en el interior del canal auditivo interno, en donde las células de Schwann remplazan a la oligodendroglia como productores de mielina. Por este motivo debe realizarse una IRM del octavo par craneal en cualquier paciente que desarrolle sordera nerviosa unilateral inexplicable, en especial cuando el estímulo del canal semicircular del lado ipsilateral por medio del cambio en la temperatura de la membrana timpánica (respuesta calórica) no cause nistagmus.
Los schwannomas suelen crecer con lentitud. Al inicio causan falla vestibular unilateral asintomática, y después ocurre hipoacusia unilateral sintomática. Los tumores no tratados pueden crecer mucho, comprimiendo los nervios trigeminal y facial, así como el cerebelo. Estos tumores suelen asociarse con pérdida del brazo largo del cromosoma 22.63 La presencia de neuromas acústicos bilaterales sugieren que el paciente tiene también NF-2. Los enfermos con hipoacusia unilateral inexplicable, tinnitus unilateral o vértigo asociado a una respuesta calórica ausente en un lado, deben someterse a una evaluación más profunda porque el diagnóstico temprano por medio de pruebas de audición e IRM permite al cirujano extirpar el tumor por microcirugía, lo que conserva la función del nervio facial y en ocasiones la audición. Algunos investigadores están a favor de la radiocirugía estereotáctica en pacientes con neuromas del acústico pequeños.64
Tumores hipofisiarios
Los tumores hipofisiarios suelen manifestarse por síntomas endocrinológicos. Los macroadenomas pueden comprimir el diafragma selar, causando cefalea bitemporal o bifrontal. Cuando rompen el diafragma y comprimen el quiasma óptico, causan hemianopsia bitemporal. La masa se extiende hacia los lados hacia los senos cavernosos, ocasionando diplopia al comprimir los nervios oculares. La pérdida visual y la diplopia son agudas cuando el tumor sangra o se infarta (apoplejía hipofisiaria). Los pacientes con tumores hipofisiarios pueden tratarse por medio de resección transesfenoidal, radioterapia o, en los pacientes con prolactinomas, con hormonoterapia.65 La mayoría de los pacientes con tumores que afectan al quiasma óptico se tratan en forma quirúrgica.
Tumores de la región pineal
El pineocitoma y el pineoblastoma son tumores agresivos que suelen ocurrir en niños, se originan de la glándula pineal y con frecuencia se diseminan a las leptomeninges.66 Por lo general, los tumores de la región pineal se originan de estructuras de células germinales e incluyen los germinomas, los tumores del saco vitelino y los coriocarcinomas. Los tumores de la región pineal comprimen el tallo cerebral, causando paresia de la mirada hacia arriba, disfunción pupilar, e hidrocefalia por compresión del acueducto de Silvio. Los pacientes refieren primero cefalea y después desarrollan papiledema por aumento de la presión intracraneana causada por hidrocefalia, la visión doble y la ataxia por compresión del tallo cerebral superior pueden ser también síntomas tempranos.
Los coriocarcinomas y los tumores del saco vitelino se caracterizan por marcadores bioquímicos en el LCR67 (gonadotrofina coriónica humana-§ y a-fetoproteína, respectivamente). Los germinomas no producen estos marcadores. Debe realizarse un diagnóstico definitivo porque esto tiene implicaciones pronósticas importantes: los pacientes con germinomas pueden curarse por radioterapia o una combinación de quimio y radioterapia, mientras que los otros tumores son incurables.66
Linfoma primario del sistema nervioso central
Los LPSNC se originan de células B que han entrado al SNC desde la sangre. El LPSNC difiere del linfoma sistémico en que, a pesar de que es localizado, rara vez se cura.10 Además, cuando el linfoma sistémico da metástasis al SNC, suele afectar las leptomeninges, mientras que el LPSNC se presenta casi siempre como una masa cerebral. El LPSNC puede iniciar también en el ojo, la médula espinal o las leptomeninges.
Puede presentarse cefalea o hidrocefalia en el caso de los tumores de leptomeninges. Si la afección es ocular, la primera molestia puede ser dificultad visual que sugiere uveítis o vitreítis. Debido a que las lesiones ocupativas en el cerebro suelen ser profundas, los pacientes pueden iniciar con cambios en el comportamiento o personalidad, y con menos frecuencia sufren crisis convulsivas. El LPSNC crece con rapidez y se disemina por todo el cerebro y la médula espinal, pero rara vez da metástasis fuera del SNC. Son comunes las lesiones múltiples.
El diagnóstico de LPSNC puede hacerse por vitrectomía (en casos de afección ocular), examen del LCR (cuando el tumor se encuentra en las leptomeninges), o por biopsia con aguja del cerebro. En todos los casos el diagnóstico se realiza al identificar linfocitos malignos (por lo general células B), la mayoría de los cuales son células grandes, hendidas, de tipo inmunoblástico. Los pacientes no tratados mueren varios meses después del diagnóstico. El tratamiento consiste en quimioterapia sistémica e intratecal seguida de radioterapia. Comparado con la radioterapia sola, este tratamiento aumenta el tiempo promedio de recaída de 12 a 40 meses,68 y algunos pacientes se curan. Los pacientes con LPSNC y VIH positivo también se tratan con esta esquema, aunque muchos pacientes inmunosuprimidos no pueden tolerar las dosis altas de quimioterapia.
Oligodendrogliomas
Los oligodendrogliomas de bajo grado de malignidad y anaplásicos recuerdan a sus equivalentes astrocitomas (i.e., astrocitomas y astrocitomas anaplásicos) en muchas cosas, excepto en que crecen con más lentitud y son más sensibles a la quimioterapia.69 En los pacientes asintomáticos, incluso los oligodendrogliomas de bajo grado de malignidad y gran tamaño pueden no requerir tratamiento. Los pacientes con oligodendrogliomas de bajo grado y anaplásicos y que están sintomáticos deben tratarse con quimioterapia seguida de radioterapia.70
Meduloblastomas
Los meduloblastomas, tumores muy agresivos que ocurren con más frecuencia en niños, se originan de células neuroectodérmicas en la vermis del cerebelo.71,72 Son síntomas tempranos de estos tumores la ataxia troncal, la cefalea, la náusea y el vómito (estos últimos causados por hidrocefalia temprana). Son signos frecuentes tanto el papiledema como la hidrocefalia. Tanto en niños como en adultos el tumor invade las leptomeninges. El tratamiento consiste en extirpación quirúrgica seguida de radiación a todo el neuroeje. En los pacientes con riesgo alto también se administra quimioterapia.72 Es posible lograr la curación en el 50 a 60 porciento de los pacientes con meduloblastoma.
Tumores de la médula espinal
Los tumores de la médula espinal pueden ser epidurales, subdurales, extra o intramedulares.73 Su localización no puede determinarse por clínica, pero sí por medio de IRM, lo que obvia la necesidad de la mielografía, que es un estudio invasor. Los tumores de la médula espinal se caracterizan por mielopatía, que suele consistir en debilidad bilateral, reflejos hiperactivos, signos de Babinski y pérdida de la sensación por debajo de la lesión. Puede aparecer dolor local y en ocasiones dolor radicular semanas a meses antes del desarrollo de la mielopatía. Por lo general se conservan las funciones vesical e intestinal hasta una fase tardía de la enfermedad.
Los tumores epidurales suelen ser metástasis (ver adelante). Por lo general los tumores subdurales son meningiomas benignos o schwannomas. En ocasiones las metástasis leptomeníngeas crecen suficiente como para causar una masa subdural. Los signos y síntomas de los meningiomas y schwannomas subdurales se desarrollan en forma lenta, y puede no existir dolor, en especial en los pacientes con meningiomas.
El tratamiento de los meningiomas y los schwannomas es la cirugía.
Los tumores intramedulares incluyen los ependimomas, los astrocitomas y las metástasis. Los pacientes con ependimomas de la médula espinal suelen curarse con la extirpación quirúrgica de los tumores.74 Los pacientes con ependimomas que se originan por debajo de la médula, en la cola de caballo, pueden también curarse si se logra extirpar por completo el tumor. Los astrocitomas de la médula espinal no pueden resecarse en su totalidad, y los pacientes con este tipo de tumores se tratan con radioterapia.
Metástasis al sistema nervioso central
Las metástasis al sistema nervioso central son más frecuentes que los tumores primarios. Cada año ocurren metástasis sintomáticas al cerebro, médula espinal o sistema nervioso periférico en alrededor de 80,000 pacientes.3,4 Además, con la disponibilidad de mejores agentes quimioterápicos sistémicos, las metástasis al sistema nervioso central se están volviendo más comunes.
Las metástasis afectan al cerebro por diseminación hematógena directa y a la médula espinal por compresión epidural. Los nervios craneales y espinales se alteran por diseminación a las leptomenínges o por compresión directa por metástasis óseas o de ganglios linfáticos. Las metástasis cerebrales pueden ser el dato de presentación hasta en el 10 porciento de los pacientes con cáncer pulmonar que por otro lado están asintomáticos. Por el contrario, los gamagramas cerebrales realizados de rutina a pacientes con cáncer pulmonar de diagnóstico reciente muestran lesiones metastásicas en el cinco a 10 porciento.76 La compresión de la médula espinal es el primer signo de cáncer en hasta el 50 porciento de los pacientes internados en hospitales generales y en alrededor el ocho porciento de los internados en hospitales oncológicos.4,73
Los tumores que dan metástasis cerebrales con frecuencia incluyen a los canceres de pulmón, mama y melanoma maligno. Los canceres de útero, ovario y próstata rara vez dan metastasis cerebrales. La compresión epidural es una complicación frecuente de los canceres de próstata, mama y pulmón. Las metastasis leptomeníngeas complican al cancer de mama, la leucemia, los linfomas y el cancer pulmonar de células pequeñas.
METASTASIS CEREBRALES
Los signos y síntomas de las metastasis cerebrales son las mismas que las de los tumores cerebrales primarios. La IRM suele mostrar una o mas lesiones esféricas dentro de la sustancia blanca que tienen reforzamiento con contraste y estan rodeadas por edema. Con las lesiones pequeñas el reforzamiento con contraste puede ser uniforme, en las de mayor tamaño existe un borde simétrico de reforzamiento que rodea una zona central isointensa. La IRM no puede distinguir en forma inequívoca estas lesiones de los abscesos cerebrales o los tumores primarios. Las metastasis durales son semejantes a los meningiomas, pero suelen causar mas edema cerebral. En ciertas situaciones clínicas (i.e., lesiones múltiples en el cerebro que se desarrollan en un paciente con diagnóstico de un cancer activo) la IRM es diagnóstica. Si la imagen es atípica o si el paciente no tiene aún diagnóstico de cancer, puede requerirse una biopsia.4 En una serie de posibles metastasis cerebrales únicas, la biopsia reveló un 11 porciento de error; varias de estas lesiones fueron benignas.77
Los esteroides se emplean de la misma manera para las metastasis cerebrales que para los tumores cerebrales primarios (ver antes). En un paciente cuyo cancer sistémico esta controlado, las metastasis cerebrales accesibles y únicas deben extirparse.77,78 La mayoría de los médicos estan a favor de la radioterapia cerebral después de la cirugía, pero la eficacia de este tratamiento no se ha demostrado por medio de estudios controlados.4 Los pacientes con tumores metastasicos múltiples o quirúrgicamente inaccesibles se tratan con radiación cerebral total para erradicar tanto el tumor visible como las micrometastasis que pueden estar diseminadas en todo el cerebro. Aunque la radiocirugía estereotactica puede controlar las metastasis menores de 3 cm de diametro, su papel preciso no se ha definido aún.79
Los tumores cerebrales metastasicos que pueden identificarse en la IRM con contraste y el cancer del mismo tipo en el resto del organismo responden a la quimioterapia en el mismo grado. El motivo de esta respuesta en las metastasis es la ruptura de la barrera hematoencefalica, que permite que los agentes hidrosolubles penetren al cerebro. Esta observación no es contraria al concepto de que el cerebro actúa como una especie de santuario para metastasis pequeñas, que no muestran reforzamiento con contraste, porque estas no se exponen a la quimioterapia hidrosoluble. Es frecuente que se presenten recaídas aisladas al SNC en pacientes con cancer de mama periféricamente controlado.
METASTASIS A LA MEDULA ESPINAL
Las metastasis a los cuerpos vertebrales causan síntomas al comprimir la médula espinal.4,73 La compresión epidural de la médula por un tumor metastasico constituye una urgencia neurológica. El tratamiento consiste en dexametasona en dosis altas, 100 mg en bolo intravenoso, seguido de 100 mg/día en dosis divididas durante tres días, con reducción rapida de la dosis.4,80,81 El tratamiento mas definitivo incluye radioterapia con o sin descompresión quirúrgica. Los procedimientos quirúrgicos incluyen laminectomía y extirpación de un cuerpo vertebral. Las evidencias actuales no demuestran que la descompresión quirúrgica sea mejor que la radioterapia.4,82
Los pacientes que caminan, cuando son tratados, suelen permanecer así después del tratamiento. Los que no, pueden recuperar la capacidad para caminar, aunque los que estan paraplégicos rara vez se recuperan.
METASTASIS A LAS LEPTOMENINGES
Al igual que la incidencia de las metastasis cerebrales, la incidencia de metastasis a las leptomeninges parece estar aumentando. El diagnóstico se establece por identificación citológica de células malignas en el LCR, por IRM con contraste de las leptomeninges cerebrales o medulares, o por la presencia de una concentración excesiva de marcadores tumorales en el LCR cuando se compara con el suero.4,83 Los tumores que se originan de un linfoma no Hodgkin o de una leucemia suelen responder a la administración de medicamentos como el methotrexate o la citarabina. la radioterapia a areas asintomaticas causa paliación importante, y algunos tumores sólidos responden. La quimioterapia sistémica puede tener la misma eficacia que en el resto del organismo por la ruptura de la barrera hematoencefalica por neovascularización del tumor.83
Complicaciones no metastasicas del cáncer
Los canceres pueden afectar de modo importante al sistema nervioso incluso cuando no dan metastasis a éste [ver tabla 1]. Las complicaciones no metastasicas de los tumores que se analizan en esta sección incluyen a los síndromes paraneoplasicos y a los efectos adversos del tratamiento anticanceroso.
SINDROMES PARANEOPLASICOS
Los síndromes paraneoplasicos,4,84 también denominados efectos remotos del cancer sobre el sistema nervioso,85 son alteraciones neurológicas causadas por el cancer pero que no pueden atribuirse a metastasis o a un trastorno infeccioso, vascular, nutricional, tóxico o metabólico identificable. Se piensa que la mayoría de los síndromes paraneoplasicos son resultado de una respuesta inmune montada contra un antígeno que se expresa tanto en el cancer como en el sistema nervioso. La respuesta inmune suele controlar el crecimiento del cancer, pero daña la parte del sistema nervioso que expresa el mismo antígeno o antígenos semejantes. El mejor ejemplo de este mecanismo ocurre en el síndrome miasténico de Eaton-Lambert (SMEL), en el que se desarrolla una respuesta inmune humonal contra las proteínas de los canales del calcio en el cancer pulmonar de células pequeñas. Este anticuerpo se fija a los canales del calcio en el lado presinaptico de las sinapsis colinÉrgicas, bloqueando la captura de calcio necesaria para la liberación de acetilcolina. El resultado es debilidad muscular.
Los síndromes paraneoplasicos pueden afectar cualquier estructura en el SNC o el sistema nervioso periférico y causar síntomas que son semejantes a los de síndromes autoinmunes no asociados con cancer. Los síntomas neurológicos suelen preceder a la identificación del cancer, ya que éste puede ser demasiado pequeño para detectarse. Varias características clínicas sugieren un síndrome paraneoplasico:
1.El trastorno neurológico suele tener una evolución rapida que evoluciona en un periodo de semanas a meses y después se estabiliza.
2.La incidencia de ciertos síndromes clínicos (v.gr., SMEL)es sustancialmente mas alta en pacientes con cáncer que en la población general [ver tabla 5].
3.En algunos pacientes con un síndrome clínico determinado, los anticuerpos séricos específicos reaccionan contra el sistema nervioso y contra el cáncer. La presencia de estos autoanticuerpos identifica el trastorno clínico como paraneoplassico y sugiere el tipo de neoplasia responsable. Estos autoanticuerpos incluyen los anti-Yo, encontrados en pacientes con degeneración cerebelosa paraneoplasica asociada con canceres ginecológicos;86 los anticuerpos contra glucoproteína asociada a mielina, que se observan en pacientes con neuropatía periférica asociada con linfoma y enfermedad de Waldenström;87 los anticuerpos anti-Hu, encontrados en pacientes con neuropatía sensorial o encefalomielitis asociadas a cancer de pulmón de células pequeñas;88 y los anticuerpos contra el antígeno de la retinopatía asociada al cancer.89 Existen pruebas comerciales para la detección de la mayor parte de estos antígenos.
Degeneración cerebelosa paraneoplasica
La degeneración cerebelosa paraneoplasica se caracteriza por el desarrollo rapido de disfunción cerebelosa bilateral, por lo general simétrica, incluyendo ataxia de brazos y piernas y disartria severa. Son frecuentes el vértigo, la diplopia y el nistagmus.86 Los datos del LCR incluyen pleocitosis y aumento en la concentración de proteínas y de IgG. La enfermedad, que puede asociarse con cualquier cáncer, suele preceder a su diagnóstico por algunas semanas o hasta tres años. Evoluciona durante un periodo de semanas a meses, causando gran incapacidad. En raras ocasiones el trastorno mejora al tratar el tumor. La IRM muestra atrofia cerebelosa, sobre todo si se realiza varios meses después de comenzada la enfermedad. Se encuentran anticuerpos contra las células de Purkinje en el suero de algunos pacientes, en especial en los que tienen cancer de ovario.
Los cambios patológicos suelen consistir en la pérdida de células de Purkinje en todo el cerebelo. Pueden encontrarse acúmulos de linfocitos alrededor de los vasos sanguíneos, sobre todo en los núcleos cerebelosos profundos. Desde el punto de vista clínico, la degeneración cerebelosa paraneoplasica puede distinguirse de las metastasis cerebelosas por la simetría de sus signos y la ausencia de hipertensión intracraneana. Difiere de la degeneración alcohólica en que la disartria y la ataxia de las extremidades superiores son características prominentes de la forma neoplasica, y casi siempre son leves o no existen en la forma alcohólica. Difieren de los tipos hereditarios de degeneración cerebelosa en que estos últimos rara vez tienen una evolución tan rapida.
Opsoclonus paraneoplasico
El opsoclonus paraneoplasico (movimientos oculares incontrolables, caóticos, conjugados y espontaneos) se asocia con ataxia cerebelosa y mioclonus del tronco y las extremidades. Ocurre sobre todo en niños como un efecto remoto del neuroblastoma, y también en adultos con diversos canceres.90 En los niños los síntomas neurológicos responden al tratamiento con esteroides y al manejo antitumoral.
Neuropatía sensorial paraneoplasica
La neuropatía sensorial paraneoplasica se caracteriza por pérdida de la sensación con conservación relativa de la función motora. La enfermedad suele preceder a la aparición del cancer, típicamente del pulmonar de células pequeñas (y en este caso se conoce como síndrome anti-Hu88), y progresa durante varios meses, dejando al paciente con incapacidad moderada o severa. Es común la pleocitosis inicial del LCR, que desaparece en algunas semanas. Los datos patológicos incluyen destrucción de los ganglios del hasta dorsal y de las columnas posteriores de la médula espinal, infiltrado linfocítico perivascular y degeneración walleriana de los nervios sensoriales. Muchos pacientes tienen también cambios inflamatorios y degenerativos en el cerebro y la médula.
Síndrome miasténico de Eaton-Lambert
El SMEL se caracteriza por debilidad muscular proximal con ptosis y xerostomía; en los varones puede ocurrir impotencia.91 El examen físico revela que el paciente tiene mayor fuerza de lo esperado por sus molestias y que cuando realiza un esfuerzo sostenido la fuerza aumenta durante varios segundos. Los reflejos tendinosos profundos rotuliano y aquíleo estan ausentes, pero en ocasiones reaparecen después del ejercicio. El diagnóstico se realiza por medio de estudios eléctricos; en los pacientes con SMEL la trasmisión neuromuscular aumenta con la estimulación repetitiva, que es lo contrario a lo que ocurre en los enfermos con miastenia gravis.91 Son tratamientos útiles la plasmaféresis y la erradicación del cancer subyacente.
NEUROTOXICIDAD CAUSADA POR AGENTES QUIMIOTERAPEUTICOS
La quimioterapia anticancer puede ocasionar diversos efectos neurotóxicos4,92 [ver tabla 6]. Sobresalen entre estos efectos adversos la neuropatía periférica y la encefalopatía. Cuando ocurre disfunción neurologica en pacientes que reciben quimioterapia, incluso en los que reciben medicamentos considerados no neurotóxicos, debe pensarse en la posibilidad de neurotoxicidad , consultando en busca de datos precedentes similares.93,94
Neuropatía periférica
Los alcaloides de la vinca, en especial la vincristina, afectan a los microtúbulos, causando una neuropatía periférica sensorimotora.4,92 Virtualmente todos los pacientes tratados con vincristina refieren parestesias en las puntas de los dedos de las manos y en ocasiones de los pies. Los reflejos aquíleos desaparecen pronto. Algunos enfermos pueden desarrollar debilidad motora que ocurre en una distribución distal difusa o en nervios craneales o periféricos individuales. También puede ocurrir neuropatía autonómica. Estos síntomas son reversibles.
El cisplatino causa una neuropatía sensorial de fibras largas al dañar los ganglios de las astas dorsales.94 Este trastornos se relacionan con la dosis y ocurren en pacientes que reciben mas de 450 mg/m2. Los síntomas suelen comenzar varias semanas después de terminado el tratamiento con cisplatino y progresan durante varios meses. La pérdida de la sensación propioceptiva puede ser tan severa como para impedir que el paciente camine o use sus manos. Las sensaciones a piquetes y temperatura son normales, lo mismo que la fuerza motora. Algunos pacientes se recuperan.
Las dosis de paclitaxel (Taxol) que son mayores de 250 mg/m2 causan una neuropatía predominantemente sensorial que se caracteriza por parestesias en manos y pies y que puede asociarse con ataxia sensorial. La debilidad muscular proximal puede complicar el cuadro. La neuropatía puede resolverse incluso si se continúa el medicamento, y pocas veces constituye un problema incapacitante.
Incluso las dosis estandar de esteroides (prednisona en dosis de 60 a 100 mg/día, o su equivalente) causan miopatía que se caracteriza por debilidad de los músculos flexores del cuello y de la porción proximal de las extremidades.4,92 El primer signo de esta neurotoxicidad es la dificultad para levantarse de sillas bajas o del inodoro sin sostenerse de los brazos. Después ocurre dificultad para subir escaleras. Pueden afectarse los músculos respiratorios. La miopatía casi siempre se resuelve cuando se suspenden los esteroides.
Encefalopatía
Muchos agentes quimioterapicos causan encefalopatía4,92 [ver tabla 6]. La ciclofosfamida en dosis altas puede asociarse con delirio agudo. Las dosis altas de citarabina ocasionan delirio agdo o degeneración cerebelosa aguda, ambos reversibles. El methotrexate en dosis elevadas o las dosis estandar de medicamentos intratecales pueden ocasionar encefalopatía aguda reversible. Sin embargo, el uso prolongado de methotrexate, en especial aunado a radioterapia, puede causar encefalopatía crónica caracterizada por demencia progresiva.
NEUROTOXICIDAD CAUSADA POR RADIOTERAPIA
Los efectos neurotóxicos de la radioterapia (ver adelante) pueden afectar cualquier porción del SNC o periférico y ocurrir en forma aguda o tardía, varias semanas a años después de la radiación [ver tabla 7]. El grado de neurotoxicidad esta determinado por la dosis total de radiación, el tamaño de cada fracción, el tiempo total en el que se recibe la dosis, el volumen de tejido nervioso que se irradíe y el tiempo transcurrido desde el tratamiento.4,95 Otros factores, como la enfermedad neruológica subyacente (v.gr., un tumor o edema cerebrales), la cirugía previa, el uso concomitante de quimioterapicos y la susceptibilidad individual (en especial a methotrexate), hacen imposible definir con precisión la dosis segura para cada paciente. Sin embargo, algunos lineamientos permiten al radio-oncólogo calcular dosis que sean seguras.
Encefalopatía aguda
Ocurre encefalopatía aguda en pacientes que tienen hipertensión intracraneana, en especial en los que han recibido fracciones grandes de radiación sin profilaxis con esteroides. Inmediatamente después del primer tratamiento, los pacientes susceptibles sufren cefalea, nausea y vómito, somnolencia, fiebre y en ocasiones empeoramiento de los signos neurológicos. Es raro, pero puede ocurrir herniación cerebral y muerte. La encefalopatía aguda, que responde a los esteroides, parece ser causada por aumento en la presión intracraneana, edema cerebral por alteración de la barrera hemato-encefalica inducida por la radiación, o por ambos factores. No ocurre empeoramiento agudo de los síntomas neurológicos después de la radiación a la médula espinal.
Encefalopatía subaguda o mielopatía
La encepalopatía subaguda o mielopatía aparece seis a 16 semanas después del tratamiento y persiste por días a semanas. Estos síndromes subagudos por radiación parecen ser secundarios a desmielinización, que puede ser causada por daño a la oligodendroglia por la radiación.
En los niños la encefalopatía subaguda, también conocida como síndrome de somnolencia posradiación, ocurre con frecuencia después de la radiación profilactica del cerebro en casos de leucemia.92,95 El trastorno se caracteriza por somnolencia asociada a cefalea, nausea y vómito, y en ocasiones, a fiebre. La electroencefalografía muestra ondas lentas, sin signos focales. En adultos el síndrome suele presentarse después de la radiación craneal de tumores cerebrales y se caracteriza por letargo y empeoramiento de los signos neurológicos focales. Tanto en niños como en adultos el trastorno responde a esteroides, y si no se trata suele resolverse en forma espontanea.
Ocurre mielopatía subaguda después de la radiación del cuello o porción superior del tórax. Esta se caracteriza por el signo de Lhermitte, una sensación de choque eléctrico que se irradía a varias partes del cuerpo cuando se flexiona el cuello. Los síntomas se resuelven en forma espontanea.
Lesión cerebral tardía
La lesión tardía por radiación aparece meses a años después del tratamiento y puede afectar cualquier parte del sistema nervioso. En el cerebro pueden ocurrir dos síndromes clínicos: lesión difusa o daño local por radiación. La lesión difusa puede ser causada por radiación cerebral total en pacientes que no tienen tumores cerebrales (i.e., en los que reciben radiación profilactica para el cáncer de pulmón de células pequeñas) o en algunos pacientes con tumores primarios o metastásicos. El trastorno se caracteriza por demencia y ausencia de signos focales. La IRM muestra atrofia cerebral, y los cambios patológicos son inespecíficos. No existe tratamiento.
El daño focal por radiación afecta a pacientes que reciben radiación cerebral focal para el tratamiento de neoplasias extracraneales o radiación cerebral total para el manejo de neoplasias intracraneales. Los signos neurológicos sugieren que existe una masa e incluyen cefalea, crisis convulsivas focales o generalizadas y hemiparesia. La IRM revela una masa hipointensa que en ocasiones muestra reforzamiento con el medio de contraste. Las características neuropatológicas de la lesión consisten en necrosis coagulativa en la sustancia blanca, alteraciones vasculares (telangiectasias, necrosis fibrinoide y formación de trombos) y proliferación glial con astrocitos bizarros multinucleados. Los datos clínicos y de IRM en los pacientes con daño focal por radiación no pueden distinguirse de los de los pacientes con tumores cerebrales, y el diagnóstico solo se realiza por biopsia. Los esteroides pueden mejorar los síntomas; si fallan, el tratamiento consiste en la extirpación quirúrgica de la masa.
Mielopatía o neuropatía tardía
La mielopatía tardía suele comenzar con el síndrome de Brown-Séquard (debilidad y pérdida de la propiocepción en las extremidades de un lado, y pÉrdida de la sensación de dolor y temperatura del otro), con desarrollo posterior de paralisis progresiva, cambios sensoriales y en ocasiones dolor. Los pacientes pueden tener una respuesta temporal a los esteroides, con freno del proceso. Sin embargo, casi siempre se desarrolla paraplegia o cuadriplegia. Los cambios patológicos incluyen necrosis de la médula espinal.
La neuropatía tardía puede afectar cualquier nervio craneal o periférico. Los trastornos comunes incluyen ceguera, causada por neuropatía óptica, y paralisis de una extremidad superior, ocasionada por plexopatía braquial después del tratamiento para el cáncer de pulmón o mama. Los mecanismos patogénicos suelen ser fibrosis e isquemia del plexo. No existe tratamiento para esta complicación.
Tumores inducidos por radiación
Pueden aparecer meningiomas, sarcomas o, con menos frecuencia gliomas, años a décadas después de la radiación craneal y asociarse incluso con dosis bajas de radioterapia. También pueden desarrollarse tumores malignos o atípicos de las vainas nerviosas después de la irradiación de los plexos braquial, cervical o lumbar. El SNC puede lesionarse cuando la radiación altera estructuras extraneurales. La radioterapia acelera la ateroesclerosis, y pueden ocurrir infartos cerebrales asociados con oclusión de la arteria carótida muchos años después de la radiación del cuello. La radioterapia puede causar disfunción endocrinológica (hipofisiaria, tiroidea o paratiroidea) y asociarse con signos neurológicos. El hipotiroidismo secundario a la radiación puede causar también encefalopatía.95
Bibliografía
DR. JEROME B. POSNER
Las neoplasias pueden dañar al sistema nervioso en tres formas: las células del sistema nervioso central o periférico pueden volverse cancerígenas,1-3 tumores localizados fuera del sistema nervioso (i.e., neoplasias sistémicas) pueden dar metástasis a estructuras neurales,3,4 y cánceres sistémicos pueden causar disfunción neurológica de modo indirecto3,4 [ver tabla 1]. Este capítulo se enfoca a los tumores primarios y metastásicos del sistena nervioso, aunque se mencionan algunos efectos no metastásicos del cáncer sobre el sistema nervioso.
|
||||||||
|
Tumores primarios del sistema nervioso
EPIDEMIOLOGIA
Pueden desarrollarse tumores tanto en el cerebro como en la médula espinal, en especial en las células de la glía, las meninges y las estructuras asociadas, como la hipófisis y la glándula pineal. En 1994 la Asociación Americana contra el Cáncer predijo que existirían para fines del año siguiente 17,500 nuevos casos de cánceres en el cerebro y otros sitios del SNC (9,600 en varones y 7,900 en mujeres).5 Esta incidencia es mayor del doble que la de la enfermedad de Hodgkin y más de la mitad de la del melanoma. En los niños, los tumores cerebrales son los tumores sólidos más frecuentes, y ocupan en ese grupo de edad el segundo lugar (después de la leucemia) como causa global de cáncer. Debido a que las neoplasias del SNC son más frecuentes y más graves que las del sistema nervioso periférico, la discusión que sigue se enfoca sobre todo a las primeras.
Los meningiomas y los adenomas hipofisiarios son más frecuentes en mujeres6 y en afroamericanos.7 Por el contrario, los gliomas son más comunes en varones y blancos. Los gliomas de bajo grado de malignidad, como los astrocitomas, son más frecuentes en jóvenes que en ancianos, mientras que los de alto grado de malignidad, como los astrocitomas anaplásicos y el glioma multiforme, son más comunes en ancianos.
Las evidencias sugieren que la incidencia de los tumores primarios del SNC va en aumento.8,9 Este incremento es especialmente notable en la incidencia del linfoma primario del SNC (LPSNC) tanto en pacientes inmunosuprimidos (sobre todo los infectados por el virus de inmunodeficiencia humana [VIH]) como en huéspedes inmunocompetentes.8 Aunque antes era un tumor raro del SNC que se identificaba solo por autopsia, el LPSNC debe tenerse muy en cuenta en la actualidad para el diagnóstico y tratamiento de los tumores cerebrales.10 Es motivo de controversia si también está aumentando la incidencia de los tumores gliales, los estudios no invasores de neuroimagen han mejorado en forma sustancial la identificación de estos tumores, en especial en los ancianos en los que antes se hacía el diagnóstico erróneo de infartos cerebrales o demencia.11
En 1994 los cánceres del SNC fueron la causa de muerte de alrededor de 12,600 personas. En las mujeres la mortalidad causada por estas neoplasias fue más o menos la misma que la ocasionada por el cáncer de ovario. Los tumores cerebrales, que son los más temidos porque ocasionan incapacidad grave como parálisis, crisis convulsivas, demencia y disfunción del lenguaje, son la principal causa de muerte por cáncer en personas menores de 35 años.
CLASIFICACION
La Organización Mundial de la Salud ha clasificado a los tumores del sistema nervioso central y periférico por su origen celular12 [ver tabla 2]. Aproximadamente el 70 porciento de estos tumores primarios del SNC se originan en el cerebro o en la médula espinal. De estos, alrededor del 75 porciento son de origen neuroepitelial, y ocurren sobre todo en las células de la glía (por lo general astrocitos) o sus precursores. Estos tumores se subdividen por criterios histológicos en alto o bajo grado de malignidad, la atipia nuclear, las figuras mitóticas, la proliferación endotelial y la necrosis identifican a los de grado alto. Si existen tres o cuatro de estas características, el pronóstico es peor
|
|||||||||||||||||||||||||||||||||||||||
* Suele ocurrir en niños.
|
Aunque los tumores cerebrales y de la médula espinal suelen clasificarse como benignos o malignos, es preferible su clasificación por grado histológico. Estos tumores rara vez son verdaderamente benignos porque la cirugía casi nunca es curativa, y tampoco son realmente malignos porque casi no dan metástasis. La cirugía fracasa porque incluso los tumores histológicamente benignos infiltran el tejido normal, lo que impide su resección total por el riesgo de destruir funciones cerebrales vitales.13 Incluso las cirugías radicales, como la hemisferectomía, han fallado para evitar la recurrencia tumoral en pacientes con glioblastoma. Además, ciertos tumores, como el meduloblastoma, el ependimoma, el glioblastoma y el LPSNC, se diseminan a las leptomeninges o a sitios distantes dentro del neuroeje. Sin embargo, muy rara vez se encuentran fuera del SNC.
ETIOLOGIA
Factores ambientales de riesgo
Existen pocas asociaciones inequívocas entre el ambiente y la ocurrencia de tumores cerebrales.14 La exposición a la radiación ionizante es el factor de riesgo mejor establecido.15 Se ha demostrado que la radiación en dosis bajas al cráneo, que en algún tiempo se usó como tratamiento para la tiña de la cabeza, ocasiona una incidencia de 10 veces más meningiomas, muchos de los cuales son anaplásicos o malignos, y un aumento de 3 veces en la incidenca de tumores gliales.16 La radiación en dosis altas para cánceres intra y extracraneanos, incluyendo la radiación profiláctica para la leucemia, aumenta la incidencia de gliomas y sarcomas.17 Sin embargo, las radiografías dentales no parecen constituir un factor de riesgo.14
El traumatismo craneano puede ser un factor de riesgo ambiental para el desarrollo de tumores gliales y otros tumores cerebrales.18 La infección por VIH es un factor de riesgo para el desarrollo de LPSNC y quizá de gliomas.19 Se ha sugerido que la exposición a la radiación electromagnética, los colorantes para el cabello, los pesticidas, el formaldehido u otras sustancias de uso industrial o laboral causan tumores cerebrales, pero esta hipótesis no ha sido confirmada.14 Si en realidad estas sustancias constituyen factores de riesgo, causan solo una proporción pequeña de los tumores cerebrales.
Factores genéticos de riesgo
Algunas enfermedades hereditarias predisponen al desarrollo de tumores del SNC [ver tabla 3].20 La neurofibromatosis se asocia con gliomas en hasta el 14 porciento de los pacientes que tienen neurofibromatosis tipo 1 (NF-1 o enfermedad de von Recklinghausen) y con schwannomas, meningiomas y, menos común, ependimoma, en pacientes con neurofibromatosis tipo 2 (NF-2). El gen supresor de tumores NF-1 ya ha sido clonado,21,22 y se ha reportado un posible gen supresor de tumores para la NF-2.23,24
|
|||||||||||||||||||||||||||||||||||||||||||||||||
AD-autosómico dominante AR-autosómico recesivo |
Las alteraciones genéticas adquiridas (i.e., no familares) se asocian también con tumores del SNC. Estas alteraciones incluyen la pérdida o mutación de un gen supresor de tumores, como el p53 o el gen del retinoblastoma (Rb), y la amplificación y rearreglo de los oncogenes. Muchos oncogenes codifican para factores de crecimiento o receptores de factores de crecimiento que pueden autoestimular células tumorales (estimulación autócrina) o células cercanas (estimulación parácrina).
Existen diversas alteraciones genéticas asociadas con los tumores cerebrales primarios más frecuentes [ver tabla 4]. Las alteraciones más comunes identificadas en pacientes con tumores gliales son las mutaciones en el p53, que ocurren en el 40 porciento de los pacientes con astrocitoma y con aproximadamente la misma frecuencia en pacientes con astrocitoma anaplásico o glioblastoma multiforme,25-27 y la expresión exagerada del receptor del factor de crecimiento epidérmico, en forma normal o mutada, que ocurre en alrededor del 40 porciento de los pacientes con glioblastoma multiforme.28-30 También se han implicado al factor de crecimiento derivado de plaquetas31,32 y a los factores de crecimiento de fibroblastos.33 No han podido identificarse secuencias específicas de alteraciones genéticas en la progresión de un astrocitoma de bajo grado a un glioblastoma multiforme. En consecuencia, algunos investigadores han propuesto que el glioblastoma multiforme puede originarse de novo o por una serie de pasos desde el astrocitoma de bajo grado de malignidad.34
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
PH-pérdida de la heterocigocidad RFCE-receptor del factor de crecimiento epidérmico RFCDP-a-receptor del factor de crecimiento derivado de plaquetas a FRLP-fragmento de restricción de longitud polimórfica |
FISIOPATOLOGIA
La fisiopatología de los tumores del SNC explica porqué incluso los tumores relativamente pequeños causan síntomas, mientras que los tumores sistémicos con frecuencia no.4 Primero, los tumores pequeños pero con localización estratégica, dañan vías vitales del cerebro y la médula espinal, ocasionando disfunción importante. Lesiones semejantes en órganos más homogéneos, como el pulmón o el hígado, deben destruir grandes áreas del órgano para que aparezcan síntomas. Segundo, debido a que el cerebro y la médula espinal se encuentran contenidos en la duramadre y el hueso, existe poco espacio para que crezca una neoplasia sin comprimir el tejido normal [ver figura 1]. Tercero, los vasos tumorales no poseen una barrera hemato-encefálica normal. Por pequeñas hendiduras pasan proteínas y otras sustancias nocivas que pueden entrar al tumor y difundir al tejido normal circundante, ocasionando edema y mayor compresión de las estructuras normales [ver figura 1]. La eliminación del edema es muy lenta porque el cerebro no tiene linfáticos.
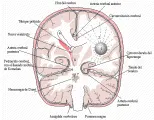
|
| Figura 1 |
| Tumor en el hemisferio cerebral |
Aunque la mayoría de los tumores pequeños causa síntomas, ocurren excepciones: los tumores de crecimiento lento, en especial los que se encuentran en áreas cerebrales silenciosas, pueden no causar síntomas hasta que son muy grandes. Incluso el glioblastoma multiforme produce en ocasiones menos síntomas de lo esperado por el tamaño de la lesión y el grado de desplazamiento observado en los estudios de imagen.
Los síntomas del SNC son causados por tres mecanismos:
1.El tumor invade, irrita y remplaza el tejido normal. Este mecanismo es especialmente característico de los gliomas infiltrantes de bajo grado de malignidad, pero es raro en los tumores metastásicos.
2.Tanto el tumor como el edema comprimen el tejido normal y sus vasos sanguíneos, produciendo isquemia.
3.Los tumores del tercero, cuarto ventrículo o las leptomenínges, bloquean la circulación del líquido cefalorraquídeo, causando hidrocefalia.
Los efectos de la invasión tumoral, el edema peritumoral y la hidrocefalia se combinan para herniar las estructuras cerebrales normales por debajo de la hoz cerebral, a través de la tienda del cerebelo o a través del foramen magno [ver figura 1].
SINTOMAS Y SIGNOS
Los pacientes con tumores cerebrales pueden tener síntomas generalizados causados por presión intracraneal difusa, síntomas focales ocasionados por isquemia y compresión en el sitio del tumor,35 o síntomas de falsa localización por el desplazamiento de estructuras cerebrales.36 Los síntomas generalizados o de falsa localización se presentan más en tumores de crecimiento lento localizados en el lóbulo frontal (relativamente silente), mientras que ocurren síntomas focales con tumores relativamente menores en áreas funcionales más importantes, como el área motora y el tallo cerebral, o en la médula espinal.
Síntomas y signos generalizados
La cefalea, el síntoma más frecuente de hipertensión intracraneana, es el primer síntoma en alrededor del 40 porciento de los pacientes con un tumor cerebral.37 Es más común como primer síntoma de un tumor en los pacientes con antecedente de cefalea no tumoral que en los que no tienen este antecedente. La mayoría de las cefaleas asociadas a tumores son inespecíficas; sin embargo, debe sospecharse un tumor cuando el dolor existe al despertar el paciente y desaparece en el lapso de una hora, cuando la cefalea comienza en una persona de edad media o anciana que antes no sufría esta molestia, o cuando se presenta un cambio súbito en las características o intensidad de la cefalea en un paciente con cefaleas crónicas. Cuando la cefalea es localizada, es un indicador confiable del lado de la cabeza que contiene el tumor, aunque no marca su localización precisa. Por ejemplo, una cefalea frontal derecha indica que el tumor está del lado derecho, pero no que el tumor es frontal, puede ser occipital o incluso cerebeloso.
El vómito, con o sin náusea previa, en especial al despertar, es un síntoma común de tumor cerebral en niños, pero menos frecuente en adultos. La cefalea aguda seguida de inmediato por vómito es característica de un tumor e indica hipertensión intracraneana. Por el contrario, la cefalea más prolongada seguida de vómito varias horas después es típica de migraña. El papiledema, que ocurre con frecuencia en niños, pero es menos común en pacientes de mayor edad, es un signo de hipertensión intracraneana.38 Suele ser asintomático, pero puede causar episodios temporales de ceguera.
Los cambios mentales asociados con los tumores cerebrales comienzan con irritabilidad y progresan hacia apatía. Los pacientes duermen más tiempo, parecen preocupados al despertar y tienen dificultad para iniciar alguna actividad, incluso una conversación; sin embargo, si se les habla suelen responder en forma adecuada. Es frecuente que se acuda a un psiquiatra por lo que se piensa es un cuadro de depresión, antes de sospechar un tumor cerebral.
Los tumores cerebrales pueden asociarse con síntomas episódicos que incluyen cefalea, pérdida visual, alteraciones de la conciencia y en ocasiones debilidad temporal de las extremidades. Estos episodios suelen ser precipitados al incorporarse desde la posición de decúbito, al toser o estornudar, y se asocian con ondas de meseta,39,40 incrementos súbitos en una presión intracraneal ya elevada que duran de cinco a 20 minutos. Estos síntomas no corresponden a crisis convulsivas, y mejoran con la reducción de la presión intracraneana, pero no con tratamiento anticonvulsivante.
Síntomas y signos focales
Las crisis convulsivas son los signos focales más frecuentes de un tumor cerebral. Son especialmente comunes en pacientes que tienen tumores que comprimen la corteza (como los meningiomas) o se originan cerca del área motora o del lóbulo temporal, ambas zonas epileptógenas. Las crisis focales causadas por focos frontales o temporales suelen causar síntomas de comportamiento o emocionales que pueden confundirse con ataques de pánico o trastornos psicológicos. Las convulsiones generalizadas son también de inicio focal, originándose en un foco de descarga asintomático. Dependiendo de la velocidad de crecimiento del tumor, las crisis convulsivas pueden presentarse meses a años antes de que se desarrollen otros síntomas. Cualquier paciente con crisis convulsivas focales o generalizadas que comienzan en la edad adulta debe someterse a una evaluación diagnóstica para investigar un tumor cerebral (ver adelante).
Otros síntomas y signos focales de un tumor cerebral dependen del sitio de la lesión. Estos son los mismos que los causados por una infección, un episodio cerebrovascular y otras enfermedades estructurales del cerebro o la médula espinal.
Síntomas de localización falsa
Los síntomas de localización falsa se deben al desplazamiento de estructuras cerebrales. La diplopia puede ser causada por desplazamiento o compresión del sexto par craneal en la base cerebral. La hemianopsia se explica por herniación tentorial que comprime a la arteria cerebral posterior. También pueden ocurrir otras parálisis de nervios craneales asociadas con desplazamiento de las estructuras del tallo cerebral.36
DIAGNOSTICO
Debe sospecharse una neoplasia del SNC en todos los adultos con crisis convulsivas de inicio reciente y en todos los pacientes con papiledema o nuevos signos motores focales o sensoriales. En estos pacientes se justifica realizar una imagen por resonancia magnética (IRM) con inyección de medio de contraste, como gadolinio, para determinar si existe ruptura de la barrera hematoencefálica y su localización.41 Los enfermos con cambios en la personalidad o el comportamiento deben ser evaluados con una IRM cuando los síntomas psiquiátricos son atípicos o se acompañan de somnolencia, apatía o pérdida de la memoria. Los pacientes con cefalea solo requieren IRM cuando ésta es de inicio reciente, ha cambiado en características o no responde a tratamiento. Una IRM negativa descarta la posibilidad de un tumor como causa de los síntomas o signos del paciente, no tiene utilidad repetir la prueba.
La IRM identifica tumores que la tomografía computada no detecta, y distingue a los tumores de las malformaciones arteriovenosas. Con excepción de la biopsia (ver adelante), no se requieren otros estudios de laboratorio. La IRM sugiere también la histología del tumor: los gliomas de bajo grado de malignidad suelen no tener reforzamiento con el medio de contraste, y su imagen se caracteriza por ser hiperintensa en T2 y normal o hipointensa en T1 (T2 se refiere a tiempo de relajación giro-giro o transverso, y T1 a tiempo de relajación giro-reja, o longitudinal). El área hiperintensa en la imagen T2 incluye tanto al tumor como al edema asociado. Por el contrario, los gliomas de alto grado de malignidad suelen tener reforzamiento con el medio de contraste, la imagen T1 muestra un anillo reforzado de borde y grosor irregulares que rodea a un centro hipointenso, y la imagen T2 muestra solo hiperintensidad [ver figura 2a]. Aunque el reforzamiento no corresponde a todo el margen infiltrante, permite una aproximación clínica sobre el volumen del tumor. Las metástasis tienen un borde regular y esférico [ver figura 2b]. Ocurren más en forma múltiple: el 50 porciento de los pacientes con metástasis tienen lesiones múltiples, comparando con solo el cinco porciento de los pacientes con gliomas. De los enfermos con LPSNC, el 40 porciento tiene tumores múltiples, que se localizan en forma periventricular, mostrando por lo general reforzamiento difuso con el medio de contraste, márgenes poco delimitados al comparar con los gliomas y las metástasis, y menos edema circundante que en el caso de otros tumores [ver figura 2c].
|
|
|
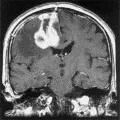
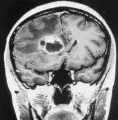
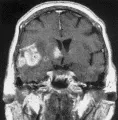
TRATAMIENTO
El tratamiento de los pacientes con neoplasias del SNC depende del tipo y localización del tumor y del estado general del paciente. Sin embargo, existen ciertos principios generales que se aplican a todos los casos de neoplasias del SNC.2-4
Esteroides
Los esteroides alivian en forma dramática los síntomas de los tumores cerebrales y de la médula al disminuir la presión intracraneana y el edema que rodea a las lesiones. La mejoría sintomática comienza en minutos y los pacientes suelen estar asintomáticos a las 24 a 48 horas después de su administración. Los mecanismos por los que los esteroides tienen estos efectos no son muy claros; sin embargo, muchos investigadores afirman que estos medicamentos disminuyen el flujo de agentes hidrosolubles a través de la barrera hematoencefálica dañada. Es posible que también tengan el efecto no buscado de inhibir la entrada de agentes quimioterápicos al tumor.
Los esteroides están indicados en todos los pacientes sintomáticos con tumores cerebrales y medulares, con excepción de los que tienen LPSNC. Por lo general se administra dexametasona, en dosis de 16 mg/día. Debido a sus efectos linfolíticos, pueden ocasionar necrosis tumoral, impidiendo el diagnóstico definitivo. Por lo tanto, su administración debe retrasarse cuando se sospeche LPSNC hasta después de realizada la biopsia.
Una vez comenzados, la administración del esteroide se continúa hasta que los síntomas del paciente se alivien y disminuya la presión intracraneana. Después se reduce la dosis en forma paulatina hasta lograr la mínima compatible con una buena función neurológica. Con frecuencia pueden suspenderse por completo.
Cirugía
Aunque la cirugía para pacientes con tumores cerebrales o de la médula espinal rara vez es curativa, este es el tratamiento más importante en pacientes con tumores accesibles que no son LPSNC. La cirugía establece el diagnóstico, alivia la presión intracraneana y mejora los síntomas y el control de las crisis convulsivas. Además, el uso de técnicas quirúrgicas modernas en manos de un cirujano experimentado y la administración de esteroides para evitar el edema posoperatorio, reduce el riesgo de empeoramiento de la función neurológica. Aunque existe cierta controversia respecto al papel de la cirugía en pacientes con gliomas, la mayoría de las evidencias indican que la extirpación quirúrgica del tumor mejora tanto la duración de la supervivencia como la calidad de vida.43-45 Si el tumor muestra reforzamiento con medio de contraste antes de la cirugía, la realización de una IRM con medio de contraste tres días después de la resección predice la magnitud del tumor residual y ayuda a establecer el pronóstico.44 Los cálculos de la extensión de la resección por parte del cirujano suelen ser inexactos.
Radioterapia
El tratamiento con radioterapia posoperatoria prolonga la duración de la supervivencia y mejora la calidad de vida en pacientes con tumores de alto grado de malignidad (i.e., astrocitomas anaplásicos o gliomas), pero su papel en los pacientes con tumores de bajo grado, en especial asintomáticos, no se ha definido. Después de la resección total de los astrocitomas pilocíticos no se requiere radioterapia. En los pacientes asintomáticos con astrocitomas de bajo grado u oligodendrogliomas, la radioterapia puede retrasarse en forma segura hasta que se presenten síntomas. Los gliomas cerebrales y de la médula espinal deben tratarse con radiación en dosis altas (5,500 a 6,000 cGy [5,500 a 6,000 rads] en fracciones de 180 a 200 cGy [180 a 200 rads]) administrados en un campo limitado que incluya al tumor y a sus zonas circundantes.46 En algunos pacientes puede ser útil aumentar la radioterapia convencional con braquiterapia (en la que la fuente de radiación se implanta dentro del tumor)46,47 o radiocirugía estereotáctica (en la que se dirigen dosis altas de radiación ionizante en múltiples haces angostos hacia una localización intracraneana precisa por medio de estereotaxia).46 Aún no se ha determinado del todo el papel exacto de estas técnicas.
Quimioterapia
La respuesta de la mayoría de los gliomas a la quimioterapia es limitada, pero la adición de nitrosureas o procarbacina al tratamiento aumenta la supervivencia de algunos pacientes con gliomas de alto grado de malignidad.48 Es imposible predecir qué pacientes responderan a la quimioterapia, por lo que todos los pacientes con gliomas de alto grado deben tratarse con una combinación de radioterapia y una nitrosurea u otro agente quimioterápico liposoluble.
Otras modalidades terapéuticas
Ni la inmunoterapia ni el tratamiento genético están indicados en la actualidad para los tumores cerebrales o de la médula spinal.49 Sin embargo, en la actualidad estas modalidades son motivo de gran investigación, y quizá en un futuro demuestren ser benéficas.
PRONOSTICO
Varios factores determinan el pronóstico de los pacientes con tumores cerebrales primarios.50,51 El grado histológico es un factor pronóstico importante. Por ejemplo, la supervivencia promedio de los pacientes con glioblastoma multiforme es un poco mayor de un año, y la tasa de supervivencia a cinco años es menor del 10 porciento.52 Los pacientes con astrocitoma anaplásico tienen una supervivencia promedio de dos a tres años, y los que tienen astrocitomas, de cinco a 10 años. Sin embargo, los enfermos con astrocitoma pilocítico tienen una esperanza de vida normal.
La edad es otro factor importante para determinar el pronóstico, y los pacientes más jóvenes sobreviven más tiempo que los de mayor edad con tumores similares histológicamente. En el caso del glioblastoma multiforme, el origen del tumor tiene también importancia pronóstica: un paciente con un glioblastoma que se origina de un astrocitoma de bajo grado de malignidad puede tener mejor pronóstico que un paciente con un glioblastoma que se origina de novo.53,54 Otros factores pronósticos incluyen la extensión de la cirugía (la resección más completa mejora la supervivencia),43-45 la dosis de radiación administrada (a mayor dosis mayor supervivencia),46 y la administración de quimioterapia (los agentes quimioterapéuticos mejoran la supervivencia en el 20 a 30 porciento de los pacientes).48
TUMORES ESPECIFICOS
Meningiomas
Los meningiomas se originan de la duramadre y de las vellosidades aracnoideas de los espacios intracraneal y espinal. Se localizan a lo largo de la superficie dorsal y base del cerebro, la hoz cerebral y el borde esfenoidal, así como en los ventrículos laterales y el canal medular torácico. Son más comunes en mujeres que en hombres. Poseen receptores para progesterona y, con menos frecuencia, para estrógenos.55 Las mujeres con cáncer de mama pueden tener mayor riesgo de meningiomas.56
Las alteraciones genéticas que se han implicado en el desarrollo de los meningiomas incluyen la pérdida del brazo largo del cromosoma 22, que es la más frecuente,57,58 del brazo corto del cromosoma 14 y, más raro, del brazo corto de los cromosomas 1, 7 o Y.59-62 Los meningiomas pueden ser mútiples y con frecuencia crecen con lentitud, causando síntomas por compresión cerebral, no por invasión. Los síntomas comunes de presentación de los meningiomas craneales son crisis convulsivas y parálisis de pares craneales, los de los meningiomas medulares incluyen paraparesias graduales y en ocasiones no dolorosas.
Los pacientes con meningiomas pueden diagnosticarse con facilidad por medio de una IRM con contraste. Los meningiomas son isodensos en la imagen T1, muestran reforzamiento uniforme con el medio de contraste y se distinguen con claridad del cerebro o la médula espinal. En ocasiones puede identificarse un área de fijación dural elongada, o pueden aparecer en forma de placa, causando engrosamiento difuso de la duramadre.
Los pacientes con meningiomas pueden ser curados con la extirpación quirúrgica completa del tumor o tumores. Sin embargo, muchos meningiomas, en especial los localizados en los senos cavernosos y en la base del cerebro, no pueden resecarse en su totalidad. Además, algunos meningiomas, en especial los que son anaplásicos, recurren a pesar de una resección aparentemente completa. La radioterapia puede controlar estas recidivas. No se ha demostrado que el uso de agentes quimioterápicos u hormonales55 sea benéfico. Debido a la lentitud de su crecimiento, los meningiomas pequeños o asintomáticos pueden vigilarse en lugar de resecarse, en especial en ancianos.
Schwannomas
Los schawnnomas (también llamados tumores de células de Schwann o neurilemomas) pueden presentarse en el cráneo o en el sistema nervioso periférico. Los neuromas del acústico, los schwannomas más frecuentes, ocurren en la porción vestibular del octavo par craneal, cerca o en el interior del canal auditivo interno, en donde las células de Schwann remplazan a la oligodendroglia como productores de mielina. Por este motivo debe realizarse una IRM del octavo par craneal en cualquier paciente que desarrolle sordera nerviosa unilateral inexplicable, en especial cuando el estímulo del canal semicircular del lado ipsilateral por medio del cambio en la temperatura de la membrana timpánica (respuesta calórica) no cause nistagmus.
Los schwannomas suelen crecer con lentitud. Al inicio causan falla vestibular unilateral asintomática, y después ocurre hipoacusia unilateral sintomática. Los tumores no tratados pueden crecer mucho, comprimiendo los nervios trigeminal y facial, así como el cerebelo. Estos tumores suelen asociarse con pérdida del brazo largo del cromosoma 22.63 La presencia de neuromas acústicos bilaterales sugieren que el paciente tiene también NF-2. Los enfermos con hipoacusia unilateral inexplicable, tinnitus unilateral o vértigo asociado a una respuesta calórica ausente en un lado, deben someterse a una evaluación más profunda porque el diagnóstico temprano por medio de pruebas de audición e IRM permite al cirujano extirpar el tumor por microcirugía, lo que conserva la función del nervio facial y en ocasiones la audición. Algunos investigadores están a favor de la radiocirugía estereotáctica en pacientes con neuromas del acústico pequeños.64
Tumores hipofisiarios
Los tumores hipofisiarios suelen manifestarse por síntomas endocrinológicos. Los macroadenomas pueden comprimir el diafragma selar, causando cefalea bitemporal o bifrontal. Cuando rompen el diafragma y comprimen el quiasma óptico, causan hemianopsia bitemporal. La masa se extiende hacia los lados hacia los senos cavernosos, ocasionando diplopia al comprimir los nervios oculares. La pérdida visual y la diplopia son agudas cuando el tumor sangra o se infarta (apoplejía hipofisiaria). Los pacientes con tumores hipofisiarios pueden tratarse por medio de resección transesfenoidal, radioterapia o, en los pacientes con prolactinomas, con hormonoterapia.65 La mayoría de los pacientes con tumores que afectan al quiasma óptico se tratan en forma quirúrgica.
Tumores de la región pineal
El pineocitoma y el pineoblastoma son tumores agresivos que suelen ocurrir en niños, se originan de la glándula pineal y con frecuencia se diseminan a las leptomeninges.66 Por lo general, los tumores de la región pineal se originan de estructuras de células germinales e incluyen los germinomas, los tumores del saco vitelino y los coriocarcinomas. Los tumores de la región pineal comprimen el tallo cerebral, causando paresia de la mirada hacia arriba, disfunción pupilar, e hidrocefalia por compresión del acueducto de Silvio. Los pacientes refieren primero cefalea y después desarrollan papiledema por aumento de la presión intracraneana causada por hidrocefalia, la visión doble y la ataxia por compresión del tallo cerebral superior pueden ser también síntomas tempranos.
Los coriocarcinomas y los tumores del saco vitelino se caracterizan por marcadores bioquímicos en el LCR67 (gonadotrofina coriónica humana-§ y a-fetoproteína, respectivamente). Los germinomas no producen estos marcadores. Debe realizarse un diagnóstico definitivo porque esto tiene implicaciones pronósticas importantes: los pacientes con germinomas pueden curarse por radioterapia o una combinación de quimio y radioterapia, mientras que los otros tumores son incurables.66
Linfoma primario del sistema nervioso central
Los LPSNC se originan de células B que han entrado al SNC desde la sangre. El LPSNC difiere del linfoma sistémico en que, a pesar de que es localizado, rara vez se cura.10 Además, cuando el linfoma sistémico da metástasis al SNC, suele afectar las leptomeninges, mientras que el LPSNC se presenta casi siempre como una masa cerebral. El LPSNC puede iniciar también en el ojo, la médula espinal o las leptomeninges.
Puede presentarse cefalea o hidrocefalia en el caso de los tumores de leptomeninges. Si la afección es ocular, la primera molestia puede ser dificultad visual que sugiere uveítis o vitreítis. Debido a que las lesiones ocupativas en el cerebro suelen ser profundas, los pacientes pueden iniciar con cambios en el comportamiento o personalidad, y con menos frecuencia sufren crisis convulsivas. El LPSNC crece con rapidez y se disemina por todo el cerebro y la médula espinal, pero rara vez da metástasis fuera del SNC. Son comunes las lesiones múltiples.
El diagnóstico de LPSNC puede hacerse por vitrectomía (en casos de afección ocular), examen del LCR (cuando el tumor se encuentra en las leptomeninges), o por biopsia con aguja del cerebro. En todos los casos el diagnóstico se realiza al identificar linfocitos malignos (por lo general células B), la mayoría de los cuales son células grandes, hendidas, de tipo inmunoblástico. Los pacientes no tratados mueren varios meses después del diagnóstico. El tratamiento consiste en quimioterapia sistémica e intratecal seguida de radioterapia. Comparado con la radioterapia sola, este tratamiento aumenta el tiempo promedio de recaída de 12 a 40 meses,68 y algunos pacientes se curan. Los pacientes con LPSNC y VIH positivo también se tratan con esta esquema, aunque muchos pacientes inmunosuprimidos no pueden tolerar las dosis altas de quimioterapia.
Oligodendrogliomas
Los oligodendrogliomas de bajo grado de malignidad y anaplásicos recuerdan a sus equivalentes astrocitomas (i.e., astrocitomas y astrocitomas anaplásicos) en muchas cosas, excepto en que crecen con más lentitud y son más sensibles a la quimioterapia.69 En los pacientes asintomáticos, incluso los oligodendrogliomas de bajo grado de malignidad y gran tamaño pueden no requerir tratamiento. Los pacientes con oligodendrogliomas de bajo grado y anaplásicos y que están sintomáticos deben tratarse con quimioterapia seguida de radioterapia.70
Meduloblastomas
Los meduloblastomas, tumores muy agresivos que ocurren con más frecuencia en niños, se originan de células neuroectodérmicas en la vermis del cerebelo.71,72 Son síntomas tempranos de estos tumores la ataxia troncal, la cefalea, la náusea y el vómito (estos últimos causados por hidrocefalia temprana). Son signos frecuentes tanto el papiledema como la hidrocefalia. Tanto en niños como en adultos el tumor invade las leptomeninges. El tratamiento consiste en extirpación quirúrgica seguida de radiación a todo el neuroeje. En los pacientes con riesgo alto también se administra quimioterapia.72 Es posible lograr la curación en el 50 a 60 porciento de los pacientes con meduloblastoma.
Tumores de la médula espinal
Los tumores de la médula espinal pueden ser epidurales, subdurales, extra o intramedulares.73 Su localización no puede determinarse por clínica, pero sí por medio de IRM, lo que obvia la necesidad de la mielografía, que es un estudio invasor. Los tumores de la médula espinal se caracterizan por mielopatía, que suele consistir en debilidad bilateral, reflejos hiperactivos, signos de Babinski y pérdida de la sensación por debajo de la lesión. Puede aparecer dolor local y en ocasiones dolor radicular semanas a meses antes del desarrollo de la mielopatía. Por lo general se conservan las funciones vesical e intestinal hasta una fase tardía de la enfermedad.
Los tumores epidurales suelen ser metástasis (ver adelante). Por lo general los tumores subdurales son meningiomas benignos o schwannomas. En ocasiones las metástasis leptomeníngeas crecen suficiente como para causar una masa subdural. Los signos y síntomas de los meningiomas y schwannomas subdurales se desarrollan en forma lenta, y puede no existir dolor, en especial en los pacientes con meningiomas.
El tratamiento de los meningiomas y los schwannomas es la cirugía.
Los tumores intramedulares incluyen los ependimomas, los astrocitomas y las metástasis. Los pacientes con ependimomas de la médula espinal suelen curarse con la extirpación quirúrgica de los tumores.74 Los pacientes con ependimomas que se originan por debajo de la médula, en la cola de caballo, pueden también curarse si se logra extirpar por completo el tumor. Los astrocitomas de la médula espinal no pueden resecarse en su totalidad, y los pacientes con este tipo de tumores se tratan con radioterapia.
Metástasis al sistema nervioso central
Las metástasis al sistema nervioso central son más frecuentes que los tumores primarios. Cada año ocurren metástasis sintomáticas al cerebro, médula espinal o sistema nervioso periférico en alrededor de 80,000 pacientes.3,4 Además, con la disponibilidad de mejores agentes quimioterápicos sistémicos, las metástasis al sistema nervioso central se están volviendo más comunes.
Las metástasis afectan al cerebro por diseminación hematógena directa y a la médula espinal por compresión epidural. Los nervios craneales y espinales se alteran por diseminación a las leptomenínges o por compresión directa por metástasis óseas o de ganglios linfáticos. Las metástasis cerebrales pueden ser el dato de presentación hasta en el 10 porciento de los pacientes con cáncer pulmonar que por otro lado están asintomáticos. Por el contrario, los gamagramas cerebrales realizados de rutina a pacientes con cáncer pulmonar de diagnóstico reciente muestran lesiones metastásicas en el cinco a 10 porciento.76 La compresión de la médula espinal es el primer signo de cáncer en hasta el 50 porciento de los pacientes internados en hospitales generales y en alrededor el ocho porciento de los internados en hospitales oncológicos.4,73
Los tumores que dan metástasis cerebrales con frecuencia incluyen a los canceres de pulmón, mama y melanoma maligno. Los canceres de útero, ovario y próstata rara vez dan metastasis cerebrales. La compresión epidural es una complicación frecuente de los canceres de próstata, mama y pulmón. Las metastasis leptomeníngeas complican al cancer de mama, la leucemia, los linfomas y el cancer pulmonar de células pequeñas.
METASTASIS CEREBRALES
Los signos y síntomas de las metastasis cerebrales son las mismas que las de los tumores cerebrales primarios. La IRM suele mostrar una o mas lesiones esféricas dentro de la sustancia blanca que tienen reforzamiento con contraste y estan rodeadas por edema. Con las lesiones pequeñas el reforzamiento con contraste puede ser uniforme, en las de mayor tamaño existe un borde simétrico de reforzamiento que rodea una zona central isointensa. La IRM no puede distinguir en forma inequívoca estas lesiones de los abscesos cerebrales o los tumores primarios. Las metastasis durales son semejantes a los meningiomas, pero suelen causar mas edema cerebral. En ciertas situaciones clínicas (i.e., lesiones múltiples en el cerebro que se desarrollan en un paciente con diagnóstico de un cancer activo) la IRM es diagnóstica. Si la imagen es atípica o si el paciente no tiene aún diagnóstico de cancer, puede requerirse una biopsia.4 En una serie de posibles metastasis cerebrales únicas, la biopsia reveló un 11 porciento de error; varias de estas lesiones fueron benignas.77
Los esteroides se emplean de la misma manera para las metastasis cerebrales que para los tumores cerebrales primarios (ver antes). En un paciente cuyo cancer sistémico esta controlado, las metastasis cerebrales accesibles y únicas deben extirparse.77,78 La mayoría de los médicos estan a favor de la radioterapia cerebral después de la cirugía, pero la eficacia de este tratamiento no se ha demostrado por medio de estudios controlados.4 Los pacientes con tumores metastasicos múltiples o quirúrgicamente inaccesibles se tratan con radiación cerebral total para erradicar tanto el tumor visible como las micrometastasis que pueden estar diseminadas en todo el cerebro. Aunque la radiocirugía estereotactica puede controlar las metastasis menores de 3 cm de diametro, su papel preciso no se ha definido aún.79
Los tumores cerebrales metastasicos que pueden identificarse en la IRM con contraste y el cancer del mismo tipo en el resto del organismo responden a la quimioterapia en el mismo grado. El motivo de esta respuesta en las metastasis es la ruptura de la barrera hematoencefalica, que permite que los agentes hidrosolubles penetren al cerebro. Esta observación no es contraria al concepto de que el cerebro actúa como una especie de santuario para metastasis pequeñas, que no muestran reforzamiento con contraste, porque estas no se exponen a la quimioterapia hidrosoluble. Es frecuente que se presenten recaídas aisladas al SNC en pacientes con cancer de mama periféricamente controlado.
METASTASIS A LA MEDULA ESPINAL
Las metastasis a los cuerpos vertebrales causan síntomas al comprimir la médula espinal.4,73 La compresión epidural de la médula por un tumor metastasico constituye una urgencia neurológica. El tratamiento consiste en dexametasona en dosis altas, 100 mg en bolo intravenoso, seguido de 100 mg/día en dosis divididas durante tres días, con reducción rapida de la dosis.4,80,81 El tratamiento mas definitivo incluye radioterapia con o sin descompresión quirúrgica. Los procedimientos quirúrgicos incluyen laminectomía y extirpación de un cuerpo vertebral. Las evidencias actuales no demuestran que la descompresión quirúrgica sea mejor que la radioterapia.4,82
Los pacientes que caminan, cuando son tratados, suelen permanecer así después del tratamiento. Los que no, pueden recuperar la capacidad para caminar, aunque los que estan paraplégicos rara vez se recuperan.
METASTASIS A LAS LEPTOMENINGES
Al igual que la incidencia de las metastasis cerebrales, la incidencia de metastasis a las leptomeninges parece estar aumentando. El diagnóstico se establece por identificación citológica de células malignas en el LCR, por IRM con contraste de las leptomeninges cerebrales o medulares, o por la presencia de una concentración excesiva de marcadores tumorales en el LCR cuando se compara con el suero.4,83 Los tumores que se originan de un linfoma no Hodgkin o de una leucemia suelen responder a la administración de medicamentos como el methotrexate o la citarabina. la radioterapia a areas asintomaticas causa paliación importante, y algunos tumores sólidos responden. La quimioterapia sistémica puede tener la misma eficacia que en el resto del organismo por la ruptura de la barrera hematoencefalica por neovascularización del tumor.83
Complicaciones no metastasicas del cáncer
Los canceres pueden afectar de modo importante al sistema nervioso incluso cuando no dan metastasis a éste [ver tabla 1]. Las complicaciones no metastasicas de los tumores que se analizan en esta sección incluyen a los síndromes paraneoplasicos y a los efectos adversos del tratamiento anticanceroso.
SINDROMES PARANEOPLASICOS
Los síndromes paraneoplasicos,4,84 también denominados efectos remotos del cancer sobre el sistema nervioso,85 son alteraciones neurológicas causadas por el cancer pero que no pueden atribuirse a metastasis o a un trastorno infeccioso, vascular, nutricional, tóxico o metabólico identificable. Se piensa que la mayoría de los síndromes paraneoplasicos son resultado de una respuesta inmune montada contra un antígeno que se expresa tanto en el cancer como en el sistema nervioso. La respuesta inmune suele controlar el crecimiento del cancer, pero daña la parte del sistema nervioso que expresa el mismo antígeno o antígenos semejantes. El mejor ejemplo de este mecanismo ocurre en el síndrome miasténico de Eaton-Lambert (SMEL), en el que se desarrolla una respuesta inmune humonal contra las proteínas de los canales del calcio en el cancer pulmonar de células pequeñas. Este anticuerpo se fija a los canales del calcio en el lado presinaptico de las sinapsis colinÉrgicas, bloqueando la captura de calcio necesaria para la liberación de acetilcolina. El resultado es debilidad muscular.
Los síndromes paraneoplasicos pueden afectar cualquier estructura en el SNC o el sistema nervioso periférico y causar síntomas que son semejantes a los de síndromes autoinmunes no asociados con cancer. Los síntomas neurológicos suelen preceder a la identificación del cancer, ya que éste puede ser demasiado pequeño para detectarse. Varias características clínicas sugieren un síndrome paraneoplasico:
1.El trastorno neurológico suele tener una evolución rapida que evoluciona en un periodo de semanas a meses y después se estabiliza.
2.La incidencia de ciertos síndromes clínicos (v.gr., SMEL)es sustancialmente mas alta en pacientes con cáncer que en la población general [ver tabla 5].
|
||||||||||||||||||||
|
3.En algunos pacientes con un síndrome clínico determinado, los anticuerpos séricos específicos reaccionan contra el sistema nervioso y contra el cáncer. La presencia de estos autoanticuerpos identifica el trastorno clínico como paraneoplassico y sugiere el tipo de neoplasia responsable. Estos autoanticuerpos incluyen los anti-Yo, encontrados en pacientes con degeneración cerebelosa paraneoplasica asociada con canceres ginecológicos;86 los anticuerpos contra glucoproteína asociada a mielina, que se observan en pacientes con neuropatía periférica asociada con linfoma y enfermedad de Waldenström;87 los anticuerpos anti-Hu, encontrados en pacientes con neuropatía sensorial o encefalomielitis asociadas a cancer de pulmón de células pequeñas;88 y los anticuerpos contra el antígeno de la retinopatía asociada al cancer.89 Existen pruebas comerciales para la detección de la mayor parte de estos antígenos.
Degeneración cerebelosa paraneoplasica
La degeneración cerebelosa paraneoplasica se caracteriza por el desarrollo rapido de disfunción cerebelosa bilateral, por lo general simétrica, incluyendo ataxia de brazos y piernas y disartria severa. Son frecuentes el vértigo, la diplopia y el nistagmus.86 Los datos del LCR incluyen pleocitosis y aumento en la concentración de proteínas y de IgG. La enfermedad, que puede asociarse con cualquier cáncer, suele preceder a su diagnóstico por algunas semanas o hasta tres años. Evoluciona durante un periodo de semanas a meses, causando gran incapacidad. En raras ocasiones el trastorno mejora al tratar el tumor. La IRM muestra atrofia cerebelosa, sobre todo si se realiza varios meses después de comenzada la enfermedad. Se encuentran anticuerpos contra las células de Purkinje en el suero de algunos pacientes, en especial en los que tienen cancer de ovario.
Los cambios patológicos suelen consistir en la pérdida de células de Purkinje en todo el cerebelo. Pueden encontrarse acúmulos de linfocitos alrededor de los vasos sanguíneos, sobre todo en los núcleos cerebelosos profundos. Desde el punto de vista clínico, la degeneración cerebelosa paraneoplasica puede distinguirse de las metastasis cerebelosas por la simetría de sus signos y la ausencia de hipertensión intracraneana. Difiere de la degeneración alcohólica en que la disartria y la ataxia de las extremidades superiores son características prominentes de la forma neoplasica, y casi siempre son leves o no existen en la forma alcohólica. Difieren de los tipos hereditarios de degeneración cerebelosa en que estos últimos rara vez tienen una evolución tan rapida.
Opsoclonus paraneoplasico
El opsoclonus paraneoplasico (movimientos oculares incontrolables, caóticos, conjugados y espontaneos) se asocia con ataxia cerebelosa y mioclonus del tronco y las extremidades. Ocurre sobre todo en niños como un efecto remoto del neuroblastoma, y también en adultos con diversos canceres.90 En los niños los síntomas neurológicos responden al tratamiento con esteroides y al manejo antitumoral.
Neuropatía sensorial paraneoplasica
La neuropatía sensorial paraneoplasica se caracteriza por pérdida de la sensación con conservación relativa de la función motora. La enfermedad suele preceder a la aparición del cancer, típicamente del pulmonar de células pequeñas (y en este caso se conoce como síndrome anti-Hu88), y progresa durante varios meses, dejando al paciente con incapacidad moderada o severa. Es común la pleocitosis inicial del LCR, que desaparece en algunas semanas. Los datos patológicos incluyen destrucción de los ganglios del hasta dorsal y de las columnas posteriores de la médula espinal, infiltrado linfocítico perivascular y degeneración walleriana de los nervios sensoriales. Muchos pacientes tienen también cambios inflamatorios y degenerativos en el cerebro y la médula.
Síndrome miasténico de Eaton-Lambert
El SMEL se caracteriza por debilidad muscular proximal con ptosis y xerostomía; en los varones puede ocurrir impotencia.91 El examen físico revela que el paciente tiene mayor fuerza de lo esperado por sus molestias y que cuando realiza un esfuerzo sostenido la fuerza aumenta durante varios segundos. Los reflejos tendinosos profundos rotuliano y aquíleo estan ausentes, pero en ocasiones reaparecen después del ejercicio. El diagnóstico se realiza por medio de estudios eléctricos; en los pacientes con SMEL la trasmisión neuromuscular aumenta con la estimulación repetitiva, que es lo contrario a lo que ocurre en los enfermos con miastenia gravis.91 Son tratamientos útiles la plasmaféresis y la erradicación del cancer subyacente.
NEUROTOXICIDAD CAUSADA POR AGENTES QUIMIOTERAPEUTICOS
La quimioterapia anticancer puede ocasionar diversos efectos neurotóxicos4,92 [ver tabla 6]. Sobresalen entre estos efectos adversos la neuropatía periférica y la encefalopatía. Cuando ocurre disfunción neurologica en pacientes que reciben quimioterapia, incluso en los que reciben medicamentos considerados no neurotóxicos, debe pensarse en la posibilidad de neurotoxicidad , consultando en busca de datos precedentes similares.93,94
|
||
* Causas comunes de estas complicaciones IV-intravenoso IT-intratecal IA-intrarterial |
Neuropatía periférica
Los alcaloides de la vinca, en especial la vincristina, afectan a los microtúbulos, causando una neuropatía periférica sensorimotora.4,92 Virtualmente todos los pacientes tratados con vincristina refieren parestesias en las puntas de los dedos de las manos y en ocasiones de los pies. Los reflejos aquíleos desaparecen pronto. Algunos enfermos pueden desarrollar debilidad motora que ocurre en una distribución distal difusa o en nervios craneales o periféricos individuales. También puede ocurrir neuropatía autonómica. Estos síntomas son reversibles.
El cisplatino causa una neuropatía sensorial de fibras largas al dañar los ganglios de las astas dorsales.94 Este trastornos se relacionan con la dosis y ocurren en pacientes que reciben mas de 450 mg/m2. Los síntomas suelen comenzar varias semanas después de terminado el tratamiento con cisplatino y progresan durante varios meses. La pérdida de la sensación propioceptiva puede ser tan severa como para impedir que el paciente camine o use sus manos. Las sensaciones a piquetes y temperatura son normales, lo mismo que la fuerza motora. Algunos pacientes se recuperan.
Las dosis de paclitaxel (Taxol) que son mayores de 250 mg/m2 causan una neuropatía predominantemente sensorial que se caracteriza por parestesias en manos y pies y que puede asociarse con ataxia sensorial. La debilidad muscular proximal puede complicar el cuadro. La neuropatía puede resolverse incluso si se continúa el medicamento, y pocas veces constituye un problema incapacitante.
Incluso las dosis estandar de esteroides (prednisona en dosis de 60 a 100 mg/día, o su equivalente) causan miopatía que se caracteriza por debilidad de los músculos flexores del cuello y de la porción proximal de las extremidades.4,92 El primer signo de esta neurotoxicidad es la dificultad para levantarse de sillas bajas o del inodoro sin sostenerse de los brazos. Después ocurre dificultad para subir escaleras. Pueden afectarse los músculos respiratorios. La miopatía casi siempre se resuelve cuando se suspenden los esteroides.
Encefalopatía
Muchos agentes quimioterapicos causan encefalopatía4,92 [ver tabla 6]. La ciclofosfamida en dosis altas puede asociarse con delirio agudo. Las dosis altas de citarabina ocasionan delirio agdo o degeneración cerebelosa aguda, ambos reversibles. El methotrexate en dosis elevadas o las dosis estandar de medicamentos intratecales pueden ocasionar encefalopatía aguda reversible. Sin embargo, el uso prolongado de methotrexate, en especial aunado a radioterapia, puede causar encefalopatía crónica caracterizada por demencia progresiva.
NEUROTOXICIDAD CAUSADA POR RADIOTERAPIA
Los efectos neurotóxicos de la radioterapia (ver adelante) pueden afectar cualquier porción del SNC o periférico y ocurrir en forma aguda o tardía, varias semanas a años después de la radiación [ver tabla 7]. El grado de neurotoxicidad esta determinado por la dosis total de radiación, el tamaño de cada fracción, el tiempo total en el que se recibe la dosis, el volumen de tejido nervioso que se irradíe y el tiempo transcurrido desde el tratamiento.4,95 Otros factores, como la enfermedad neruológica subyacente (v.gr., un tumor o edema cerebrales), la cirugía previa, el uso concomitante de quimioterapicos y la susceptibilidad individual (en especial a methotrexate), hacen imposible definir con precisión la dosis segura para cada paciente. Sin embargo, algunos lineamientos permiten al radio-oncólogo calcular dosis que sean seguras.
|
||||||||||||
|
||||||||||||
Encefalopatía aguda
Ocurre encefalopatía aguda en pacientes que tienen hipertensión intracraneana, en especial en los que han recibido fracciones grandes de radiación sin profilaxis con esteroides. Inmediatamente después del primer tratamiento, los pacientes susceptibles sufren cefalea, nausea y vómito, somnolencia, fiebre y en ocasiones empeoramiento de los signos neurológicos. Es raro, pero puede ocurrir herniación cerebral y muerte. La encefalopatía aguda, que responde a los esteroides, parece ser causada por aumento en la presión intracraneana, edema cerebral por alteración de la barrera hemato-encefalica inducida por la radiación, o por ambos factores. No ocurre empeoramiento agudo de los síntomas neurológicos después de la radiación a la médula espinal.
Encefalopatía subaguda o mielopatía
La encepalopatía subaguda o mielopatía aparece seis a 16 semanas después del tratamiento y persiste por días a semanas. Estos síndromes subagudos por radiación parecen ser secundarios a desmielinización, que puede ser causada por daño a la oligodendroglia por la radiación.
En los niños la encefalopatía subaguda, también conocida como síndrome de somnolencia posradiación, ocurre con frecuencia después de la radiación profilactica del cerebro en casos de leucemia.92,95 El trastorno se caracteriza por somnolencia asociada a cefalea, nausea y vómito, y en ocasiones, a fiebre. La electroencefalografía muestra ondas lentas, sin signos focales. En adultos el síndrome suele presentarse después de la radiación craneal de tumores cerebrales y se caracteriza por letargo y empeoramiento de los signos neurológicos focales. Tanto en niños como en adultos el trastorno responde a esteroides, y si no se trata suele resolverse en forma espontanea.
Ocurre mielopatía subaguda después de la radiación del cuello o porción superior del tórax. Esta se caracteriza por el signo de Lhermitte, una sensación de choque eléctrico que se irradía a varias partes del cuerpo cuando se flexiona el cuello. Los síntomas se resuelven en forma espontanea.
Lesión cerebral tardía
La lesión tardía por radiación aparece meses a años después del tratamiento y puede afectar cualquier parte del sistema nervioso. En el cerebro pueden ocurrir dos síndromes clínicos: lesión difusa o daño local por radiación. La lesión difusa puede ser causada por radiación cerebral total en pacientes que no tienen tumores cerebrales (i.e., en los que reciben radiación profilactica para el cáncer de pulmón de células pequeñas) o en algunos pacientes con tumores primarios o metastásicos. El trastorno se caracteriza por demencia y ausencia de signos focales. La IRM muestra atrofia cerebral, y los cambios patológicos son inespecíficos. No existe tratamiento.
El daño focal por radiación afecta a pacientes que reciben radiación cerebral focal para el tratamiento de neoplasias extracraneales o radiación cerebral total para el manejo de neoplasias intracraneales. Los signos neurológicos sugieren que existe una masa e incluyen cefalea, crisis convulsivas focales o generalizadas y hemiparesia. La IRM revela una masa hipointensa que en ocasiones muestra reforzamiento con el medio de contraste. Las características neuropatológicas de la lesión consisten en necrosis coagulativa en la sustancia blanca, alteraciones vasculares (telangiectasias, necrosis fibrinoide y formación de trombos) y proliferación glial con astrocitos bizarros multinucleados. Los datos clínicos y de IRM en los pacientes con daño focal por radiación no pueden distinguirse de los de los pacientes con tumores cerebrales, y el diagnóstico solo se realiza por biopsia. Los esteroides pueden mejorar los síntomas; si fallan, el tratamiento consiste en la extirpación quirúrgica de la masa.
Mielopatía o neuropatía tardía
La mielopatía tardía suele comenzar con el síndrome de Brown-Séquard (debilidad y pérdida de la propiocepción en las extremidades de un lado, y pÉrdida de la sensación de dolor y temperatura del otro), con desarrollo posterior de paralisis progresiva, cambios sensoriales y en ocasiones dolor. Los pacientes pueden tener una respuesta temporal a los esteroides, con freno del proceso. Sin embargo, casi siempre se desarrolla paraplegia o cuadriplegia. Los cambios patológicos incluyen necrosis de la médula espinal.
La neuropatía tardía puede afectar cualquier nervio craneal o periférico. Los trastornos comunes incluyen ceguera, causada por neuropatía óptica, y paralisis de una extremidad superior, ocasionada por plexopatía braquial después del tratamiento para el cáncer de pulmón o mama. Los mecanismos patogénicos suelen ser fibrosis e isquemia del plexo. No existe tratamiento para esta complicación.
Tumores inducidos por radiación
Pueden aparecer meningiomas, sarcomas o, con menos frecuencia gliomas, años a décadas después de la radiación craneal y asociarse incluso con dosis bajas de radioterapia. También pueden desarrollarse tumores malignos o atípicos de las vainas nerviosas después de la irradiación de los plexos braquial, cervical o lumbar. El SNC puede lesionarse cuando la radiación altera estructuras extraneurales. La radioterapia acelera la ateroesclerosis, y pueden ocurrir infartos cerebrales asociados con oclusión de la arteria carótida muchos años después de la radiación del cuello. La radioterapia puede causar disfunción endocrinológica (hipofisiaria, tiroidea o paratiroidea) y asociarse con signos neurológicos. El hipotiroidismo secundario a la radiación puede causar también encefalopatía.95
- Brain Tumors: AComprehensiveText. Morailtz RA,WalshJW,Eds: Marcel Dekker, Inc, New York, 1994
- Astrocytomas: Diagnosis, Treatment, and Biology. Black PM, Schoene WC, Lampson LA, Eds. Blackwell Scientific Publications, Boston, 1993
- Neur0-0ncology: Primary Tumors and Neurological Complications of Cancer. Twijnstra A, Keyser A, Ongerboer de Visser BW, Eds. Elsevier, Amsterdam, 1993
- Posner JB: Neurologic Complications of Cancer. FA Davis, Philadelphia (in press)
- Boring CC, Squires TS, Tong T, et al: Cancer statistics, 1994. CA Cancer J Clin 44:7, 1994
- Longstreth WT Jr, Dennis LK, McGuire VM, et al: Epidemiology of intracranial meningioma. Cancer 72:639,1993
- Fan K-J, Pezeshkpour GH: Ethnic distribution of primary central nervous system tumorsin Washington, DC, 1971 to 1985.J Natl Med Assoc 84:858, 1992
- Devesa 55, Fears T: Non-Hodgkin's lymphoma time trends: United States and international data. Cancer Res 52:5432s, 1992
- Davis DL,Ahlbom A,Hoel D, etal: Is braincancermortalityincreasing in industrial countries? Am J Ind Med 19:421,1991
- DeAngelis LM: Primary central nervous system lymphoma: a new clinical challenge. Neurology 4l1:619, 1991
- Boyle P, Maisonneuve P, Saracci R, et al: Is the increased incidence of primary malignant brain tumors in the elderly real? J Natl Cancer Inst 82:1594,1990
- Kleihue5 P, Burger PC, Scheithauer BW: Histological.Typing of Tumours of the Central Nervous System, 2nd ed. Springer-Verlag, Berlin, 1993
- Kelly PJ, Daumas-Duport C, Scheithauer BW: Stereotactic histologic correlations of computed tomography- and magnetic resonance imaging-defined abnormalities in patients with glial neoplasms. Mayo Clin Proc 62:450,1987
- Wrensch M, Bondy ML, Wiencke J, et al: Environmental risk factors for primary malignant brain tumors: a review. J NeurooncoI17:47, 1993
- Bem5tein M, Laperriere N: Radiation-induced tumors of the nervous system. Radiation Injury to the Nervous System. Gutin PH, Leibel SA, Sheline GE, Eds. Raven Press, New York, 1991, p 455
- Ron E, Modan B, Boice JD Jr, et al: Tumors of the brain and nervous system after radiotherapy in childhood. N Engl J Med 319:1033, 1988
- HawkinsMM, DraperGJ, KingstonJE: Incidenceofsecond primary tumours among childhood cancer 5urvivors. Br J Cancer 56:339, 1987
- Hochberg F, Toniolo P, Cole P: Head trauma and seizures as risk factors of glioblastoma. Neurology 34:l151, 1984
- Moulignier A, Mikol J, Pialoux G, et al: Cerebral glial tumors and human immunodeficiency Virus-1 infection: more than a coincidental association. Cancer 74:686, 1994
- Bondy M, Wiencke J, Wrensch M, et al: Genetics of primary brain tumors: a review. J Neurooncol18:69 , 1993
- Wallace MR, Marchuk DA, Andersen LB, et al: Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NFl patients. Science 249:181, 1990
- Cawthon RM, Wei55 R, Xu G, et al: A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutation. CeI162:193, 1990
- Trofatter JA, MacCollinMM, Rutter JL, et al: A novel moesin-,ezrin-,radixin-like gene is a candidate for the neuro-fibromatosis 2 tumor suppressor. CeI172:791, 1993
- Rouleau GA, Merel P, Lutchman M, et al: Alteration in a new gene encoding a putative membrane-organizing protein causes neurofibromatosis type 2. Nature 363:515, 1993
- Von Deimling A, Eibl RH, Ohgaki H, et al: p53 mutations are assodated with 17p allelicloss in grade II and grade III astrocytoma. Cancer Res 52:2987,1992
- Frankel RH, Bayona W, KoslowM, et al: p53 mutations in human malignant gliomas: comparison of loss of heterozygosity with mu- tation frequency. Cancer Res 52:1427,1992
- Louis DN, von Deimling A, Chung RY, et al: Comparative 5tudy of p53 gene and protein alterations in human a5trocytic tumors. J Neuropathol Exp NeuroI 52:31, 1993
- AgO5ti R, Leuthold M, Gullick W, et al: Expression of the epidermal growth factor receptor in astrocytic tumors is specifically assodated with glioblastoma multiforme. Virchows Arch A Pathol Anat HistopathoI420:321, 1992
- Ek5trand AJ, Sugawa N, James CD, et al: Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of 5equences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci USA 89:4309,1992
- Von Deimling A, Loui5 DN, von Ammon K, et al: Association of epidermal growth factor receptor gene amplification with 1055 of chromosome 10 in human gliobla5torna multiforme. J Neurosurg 77:295,1992
- We5termark B, Ni5ter M, Heldin C-H: Growth factors and oncogenes in human malignant glioma. Neurol Clin 3:785,1985
- Nister M, Heldin C-H, Westermark B: Clonal variation in the production of a platelet-derived growth factor-Iike protein and expression of corresponding receptors in a human malignant glioma. Cancer Res 46:332,1986
- Takahashi JA, Mori H, Fukumoto M, et al: Gene expression of fibroblast growth factors inhuman gliomas and meningiomas: demonstration of cellular source of basic fibroblast growth factor mRNA and peptide in tumor tissues. Proc Natl Acad Sd USA 87:5710,1990
- Von Deimling A, von Ammon K, Schoenfeld D, et al: Subsets of glioblastoma multiforme defined by molecular genetic analysis. Brain PathoI 3:19, 1993
- Kraemer DL, Bullard DE: Clinical presentation of the brain tumor patient. Brain Tumors: A Comprehensive Text. Morantz RA, Wal5h JW, Eds. Marcel, Dekker, Inc, New York, 1994, p 183
- Gassel MM: False localiiing 5ign5. Arch NeuroI 4:526, 1961
- Forsyth PA, Posner JB: Headaches in patients with brain tumors: a study of ll1 patients. Neurology 43:1678, 1993
- Van Crevel H: Papilloedema, CSF pressure, and CSF flow in cerebral tumours. J Neurol Neurosurg Psychiatry 42:493,1979
- Lyons MK, Meyer FB: Cerebrospinal fluid physiology and the management of increased intracranial pressure. Mayo Clin Proc 65:684, 1990
- Rottenberg DA, Posner JB: lntracranial pressure control. Anesthesia and Neurosurgery. Cottrell JE, Tumdorf H, Eds. CV Mosby Co, St Louis, 1980, p 89 .
- Edelman RR, Warach S: Magnetic resonance imaging (pt 1). N Engl J Med 328:708,1993
- Marton Kl, Gean AD: The spinal tap: a new look at an old test. Ann lntem Med 104:840,1986
- Berger MS: Malignant astrocytomas: surgical aspects. Semin Oncol 21:172,1994
- Albert FK, Forsting M, Sartor K, et al: Early postoperative magnetic resonance imaging after resection of malignant glioma: objective evaluation of residual tumor and its influence on regrowth and prognosis. Neurosurgery 34:45,1994
- Berger MS, Deliganis A V, Dobbins J, et al: The effect of extent of resection on recurrence in patients with low grade cerebral hemi- sphere gliomas. Cancer 74:1784, 1994
- Leibel SA, Scott CB, Loeffler JS: Contemporary approaches to the treatment of malignant gliomas with radiation therapy. Semin Oncol 21:198,1994
- Sneed PK, Larson DA, Gutin PH: Brachytherapy and hyperthermia for malignant astrocytomas. Semin Oncol21:186, 1994
- Lesser GJ, Grossman S: The chemotherapy of high-grade as-trocytomas. Semin Oncol21:220, 1994
- Jaeckle KA: lmrnunotherapy of malignant gliomas. Semin Oncol 21:249,1994
- The Medical Research Council Brain Tumour Working Party: Prognostic factors for high-grade malignant glioma: development of a prognostic index. J Neurooncol9:47, 1990
- HuttonJL, Srt\ith DF, Sandemann D, et al: Development ofprognostic index for primary supratentorial intracerebral tumours. J Neurol Neurosurg Psychiatry 55:271, 1992
- Salcman M, Scholtz H, Kaplan RS, et al: Long-term survival in patients with malignant astrocytoma. Neurosurgery 34:213, 1994
- Winger MJ, Macdonald DR, Caimcross JG: Supratentorial anaplastic gliomas in adults: the prognostic importance of extent of resection and prior low-grade glioma. J Neurosurg 71:487,1989
- Rogers LR, LewinJ, Estes ML, et al: Treatment response and survival in cerebral anaplastic glioma patients with prior low-grade glioma (abstr). Neurology 43:A249, 1993
- Grunberg SM: Role of antiprogestational therapy for meningiomas. Hum Reprod 9(suppl1):202, 1994
- Jacobs DH, Holmes FF, McFarlane MJ: Meningiomas are not significantly associated with breast cancer. Arch Neurol49:753, 1992
- Atkinson L, Schmidek HH: Genetic aspects of meningiomas. Meningiomas and Their Surgical Management. Schmidek HH, Ed. WB SaundersCo, Philadelphia, 1991, p 42
- Zankl H, Zang KD: Cytological and cytogenetical studies on brain tumors: 4. Identification of the missing G chromosome in human meningiomas as no.22 by fluorescence technique. Humangenetik 14:167,1972
- Al Saadi A, Latimer F, Madercic M, et al: Cytogenetic studies of human tumors and their clinical significance: II. Meningioma. Cancer GenetCytogenet 26:127,1987
- Rey JA, Bello MJ, de Campos JM, et al: lncidence and origin of dicentric chromosomes in cultured meningiomas. Cancer Genet Cytogenet 35:55,1988
- Poulsgard L, Ronne M, Schroder HD: Cytogenetic studies of 19 meningiornas and their clinical significance: I. Anticancer Res 9:109, 1989
- Poulsgard L, Schroder Hp, Ronne M: Cytogenetic studies of 11 meningiomas and their clinical significance: II. Anticancer Res 10:535,1990
- Seizinger BR, Rouleau G, Ozelius LJ, et al: Common pathogenetic mechanism for three tumor types in bilateral acoustic neu- rofibromatosis. Science 236:317,1987
- Lunsford LD, Kondziolka D, Flickinger JC: Stereotactic radiosurgery for benign intracranial tumors. Clin Neurosurg 40:.475,1993
- Klibanski A, Zervas NT: Diagnosis and management of hormonsecreting pituitary adenomas. N Engl J Med 324:822, 1991
- Schild SE, Scheithauer BW, Schomberg PJ, et al: Pineal parenchymal tumors: clinical, pathologic, and therapeutic aspects. Cancer 72:870, 1993
- Horowitz MB, Hall WA: Central nervous system germinomas: a review. Arch NeuroI 48:652, 1991
- DeAngelis LM, Yahalom J, Thaler HT, et al: Combined modality therapy for primary CNS Iymphoma. J Clin OncoI10:635, 1992
- Cairncross JG, Macdonald DR: Chemotherapy for oIigodendroglioma: progress report. Arch NeuroI48:225, 1991
- Mason WP, DeAnge1is LM: Procarbazine, CCNU , vincristine (PCV) chemotherapy (CT) for benign oIigodendrog1ioma (abstr). NeuroI- ogy 44(suppI2):A262, 1994
- Carrie C, Lasset C, Blay JY, et al: Medulloblastoma in adults: survivaI and prognostic factors. Radiother OncoI29:301, 1993
- Mosijczuk AD, Nigro MA, Thomas PR, et al: Preradiation chemotherapy in advanced medulloblastoma: a Pediatric Oncology Group pilot study. Cancer 72:2755, 1993
- Byme TN, Waxman SG: SpinaI Cord Compression: Diagnosis and PrincipIes of Management. FA Davis Co, Philadelphia, 1990
- Waldron JN, Laperrieré NJ, Jaakkimainen L, et al: SpinaI cord ependymomas: a retrospective analysis of 59 cases. Int J Radiat OncoI BioI Phys 27:223, 1993
- Hulshof MC, Menten J, Dito JJ, et al: Treatment results in primary intraspinaI g1iomas. Radiother OncoI29:294, 1993
- Jacobs L, KinkeI WR, Vincent RG: "Silent" brain metastasis from Iung carcinoma determined by computerized tomography. Arch NeuroI34:690, 1977
- Patchell RA, Tibbs PA, Walsh JW, et al: A randomized triaI of surgery in the treatment of single metastases to the brain. N EngI J Med 322:494,1990
- Noordijk EM, Vecht CJ, Naaxma-Reiche H, et al: The choice of treatment of single brain metastasis should be based on extracraniaI tumor activity and age. Int J Radiat OncoI BioI Phys 29:711, 1994
- Loeffler JS, Alexander E III: The role of stereotactic radiosurgery in the management of intracraniaI tumors. Oncology (Wil1iston Park) 4:21,1990
- Greenberg HS, Kim J-H, Posner JB: EpiduraI spinaI cord compression from metastatic tumor: results with a new treatment protocoI. Ann NeuroI 8:361, 1980
- Sorensen S, Helweg-LarsenS, MouridsenH,et al: Effectofhigh-dose dexamethasone in carcinomatous metastatic spinaI cord compression treated with radiotherapy: a randomised triaI. Eur J Cancer 30A:22,1994
- Grant R, Papadopoulos SM, Sandler HM, et al: Metastatic epiduraI spinaI cord compression: current concepts and treatment. J Neu-rooncoI19:79,1994
- SiegaI T, Lossos A, Pfeffer MR: LeptomeningeaI metastases: analysis of 31 patients with sustained off-therapy response following com-bined-moda1ity therapy. Neurology 44:1463,1994
- Posner JB: Paraneoplastic syndromes involving the nervous system. Neurology and General Medicine: The NeurologicaI Aspecrs of MedicaI Disorders, 2nd ed. Aminoff MJ, Ed. Churchill Livingstone, Inc, New York, 1995, p 401
- The Remote Effects of Cancer on the Nervous System. Brain L, Norris FH Jr, Eds. Grune & Stratton, New York, 1965
- Peterson K, Rosenblum MK, Kotanides H, et al: Paraneoplastic cerebellar degeneration: I. A clinicaI analysis of 55 anti- Yo antibody positive patients. Neurology 42:1931,1992
- Nobile-Orazio E, Marmiro1i P, Baldini L, et al: PeripheraI neuropa- thy in macroglobulinemia: incidence and antigen-specificity of M proteins. Neurology 37:1506, 1987
- Dalmau J, Graus F, Rosenblum MK, et al: Anti-Hu associated paraneoplastic encephalomye1itis/sensory neuronopathy: a clinicaI study of 71 patients. Medicine (Baltimore) 71:59, 92
- ThirkilI CE, Fitzgerald P, Sergott RC, et a\: Cancer-associated retinopathy (CAR syndrome) with antibodies reacting with retinaI, optic nerve, and cancer cells. N EngI J Med 321:1589,1989
- Luque FA, Fumeaux HM, Ferziger R, et al: Anti-Ri: an antibody associated with paraneop1astic opsoclonus and breast cancer. Ann NeuroI29:241, 1991
- O'N~I JH, Murray NM,~ewsom-Davis J: The Lambert-Eaton myasthenic syndrome: a review of 50 cases. Brain 111:577,
- Delattre J-Y, Posner JB: NeurologicaI comp1ications of chemotherapy and radiation therapy. Neurology and General Medicine: The NeurologicaI Aspects ofMedicaI Disorders, 2nd ed. Aminoff MJ, Ed. ChurchilI Livingstone, Inc, New York, 1995, p 421
- Toxicity of Chemotherapy (entire issue). Semin OncoI19:453, 1992
- Neurological Adverse Reactions to Anticancer Drugs. Hildebrand J, Ed. Springer-Verlag, Berlin, 1990
- Radiation Injury to the Nervous System. Gutin PH, Leibel SA, ~ Sheline GE, Eds. Raven Press, New York, 1991