Dermatología
⭳ Abrir artículo (PDF)656.1 KBEste artículo fue revisado respecto a la Edición 2/2000. Ver esa versión →
Contenido del artículo
IX ENFERMEDADES VESICULOBULOSAS
- Evaluación clínica general
- Pénfigo
- Penfigoide buloso
- PATOGENIA
- MANIFESTACIONES CLINICAS
- DATOS HISTOLOGICOS E INMUNOLOGICOS
- TRATAMIENTO
- PATOGENIA
- MANIFESTACIONES CLINICAS
- DATOS HISTOLOGICOS E INMUNOLOGICOS
- TRATAMIENTO
- Eritema multiforme
- Epidermolisis bulosa
- EPIDERMOLISIS BULOSA SIMPLE
- EPIDERMOLISIS BULOSA DE UNION
- EPIDERMOLISIS BULOSA DISTROFICA
- EPIDERMOLISIS BULOSA ADQUIRIDA
- Diagnóstico diferencial de las enfermedades vesiculobulosas
IX ENFERMEDADES VESICULOBULOSAS
DRA. ELIZABETH A. ABEL
DR. JEAN-CLAUDE BYSTRYN
Las enfermedades vesiculobulosas, que en número son más de 50, se caracterizan por la formación de ampollas llenas de líquido en la piel. Las ampollas menores de 0.5 cm se denominan vesículas y las mayores bulas. Las vesículas y las bulas son patrones de reacción de la piel a una lesión y pueden ser causadas por muy diversos padecimientos.
La mayoría de las enfermedades vesiculobulosas son de origen inmunológico o genético. Son causadas por reacciones autoinmunes a componentes de la piel, reacciones alérgicas a agentes externos en los que la piel es el principal órgano afectado, y por condiciones genéticas en las que algunos de los componentes de la piel están ausentes o son anormales. El denominador común es la falta de adhesión: una o más de las estructuras que mantienen junta a la piel se separan y aparece una cavidad llena de líquido. Las diversas enfermedades se clasifican por la estructura o estructuras afectadas y por el o los mecanismos por los que ocurre la falta de adhesión [ver tabla 1]. En esta subsección se analizan varias enfermedades vesiculobulosas dentro del contexto del enfoque diagnóstico general del paciente con lesiones ampollosas.
Evaluación clínica general
El diagnóstico se basa en los datos clínicos, histológicos e inmunológicos. Los datos clínicos con importancia diagnóstica incluyen:
1. La historia clínica. ¿El problema es agudo o crónico? ¿Se agrava con el sol o los traumatismos físicos?
2. La aparición de lesiones individuales [ver tabla 2]. ¿La lesión es una vesícula o una bula? ¿Es tensa, flácida o umbilicada? ¿La piel en la base es normal, tiene urticaria o cicatriz? ¿La vesícula está en medio de placas urticarianas o en la periferia? ¿Existe más de una bula en la misma placa?
3. Agrupación de las lesiones individuales. ¿Se encuentran estas en grupos (como ocurre en el herpes simple) o se distribuyen en forma aislada?
4. Sitios de afección. ¿Existen también lesiones en las mucosas? ¿Predominan en las superficies de flexión o extensión, en las palmas y plantas o en el dorso de manos y pies, en el cuero cabelludo, cara y tronco superior, o en áreas expuestas a traumatismos?
El dato histológico más importante es la capa de la piel en la que se forma la ampolla. Si la ampolla se forma en la epidermis, ¿lo hace inmediatamente sobre la capa de células basales o más arriba (por debajo del estrato córneo)? Si se forma en la zona de la membrana basal, ¿es dentro de la lámina lúcida o por debajo de la lámina densa? La localización precisa puede determinarse por procedimientos de inmunofluorescencia.
El hallazgo inmunológico más importante es la presencia o ausencia de anticuerpos anormales circulantes o fijos a los tejidos. Estos se detectan por técnicas de inmunofluorescencia: (1) inmunofluorescencia indirecta para detectar los anticuerpos circulantes y (2) inmunofluorescencia directa en la biopsia de piel para detectar anticuerpos fijos a los tejidos.
Pénfigo
DEFINICION Y PATOGENIA
El pénfigo se caracteriza por vesículas que se originan dentro de la epidermis y por pérdida de la cohesión de las células de la epidermis (acantolisis) que produce formación de hendiduras sobre la capa de células basales. Los autoanticuerpos dirigidos contra moléculas de adhesión causan que los queratinocitos epidérmicos se separen, provocando bulas intraepidérmicas. Existen dos tipos de pénfigo: profundo (v.gr., pénfigo vulgar) y superficial (v.gr., pénfigo foliáceo). Estos difieren en las capas epidérmicas que se lesionan, en las manifestaciones clínicas de las enfermedades y en las alteraciones inmunológicas asociadas.1 En las formas profundas las ampollas se forman inmediatamente por arriba de la capa de células basales y se asocian con autoanticuerpos contra desmogleina 3, una molécula de adhesión de queratinocitos. En las formas superficiales las bulas se forman inmediatamente por debajo del estrato córneo. Las formas superficiales del pénfigo se asocian con anticuerpos contra desmogleína 1.
MANIFESTACIONES CLINICAS
Pénfigo vulgar
El pénfigo vulgar es la forma más común de pénfigo. Puede desarrollarse a cualquier edad, pero suele ocurrir en personas entre 30 y 60 años de edad. El padecimiento tiende a afectar a personas con ascendencia mediterránea, pero puede ocurrir en cualquier etnia. El pénfigo es más común en personas con ciertos haplotipos HLA. La ocurrencia de la enfermedad en los familiares en primer grado, aunque rara, sugiere una susceptibilidad heredada transferida como un rasgo dominante. Sin embargo, se requieren otros factores desconocidos para la expresión de este trastorno en personas predispuestas.2 Los estudios de alelos de HLA de clase II en pacientes japoneses, así como en otros grupos étnicos, muestran asociación con el HLA-DRB1*04 y HLA-DRB1*14 en pacientes con pénfigo vulgar independientemente del grupo racial.3
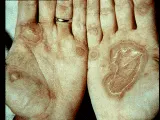
El pénfigo vulgar suele comenzar, aunque no siempre, con ulceraciones crónicas, dolorosas y que no cicatrizan en la cavidad oral [ver figura 1]. Rara vez se observan bulas porque se rompen con facilidad, dejando bases ulceradas. Las ulceraciones suelen ser múltiples, superficiales e irregulares. Puede afectarse cualquier parte de la mucosa oral, pero ocurre más en la mucosa labial, el paladar y la lengua. La presencia de ulceraciones múltiples permite distinguir a estas lesiones de tumores malignos ulcerados de la cavidad oral, que suelen ser únicos. El diagnóstico de pénfigo suele considerarse solo después de que las lesiones han durado semanas a meses.
También las lesiones en la piel pueden ser la manifestación inicial, comenzando con una ampolla pequeña y llena de líquido en piel que por lo demás parece normal. Las ampollas suelen ser flácidas porque la delgada epidermis suprayacente no suele tolerar mucha presión. Las bulas se rompen rápido, en varios días, y pueden no existir al examinar al paciente. En su lugar se observan erosiones superficiales, con bordes bien definidos y del tamaño de una moneda, con un collarín de pérdida de epidermis en la periferia. La parte superior del tronco, la espalda, el cuero cabelludo y la cara son sitios frecuentes de afección, pero las lesiones pueden ocurrir en cualquier parte del cuerpo. El problema progresa en semanas a meses [ver figura 2]. Las lesiones pueden pasar desapercibidas en las áreas periungueales (se manifiestan por inflamación paroniquia dolorosa y eritematosa), la faringe y laringe (dolor al deglutir y disfonía) y en la cavidad nasal (congestión nasal y descarga de moco con sangre, en especial al sonarse la nariz por la mañana).
Una característica de todas las formas activas de pénfigo es el signo de Nikolsky, en el que la aplicación de presión firme sobre piel de apariencia normal causa que la epidermis se separe de la dermis. El signo de Nikolsky se despierta con más facilidad en piel normal adyacente a una lesión activa.
Si no se trata las erosiones y bulas del pénfigo vulgar se diseminan en forma gradual, afectando áreas grandes de superficie y complicándose con infecciones severas y alteraciones metabólicas. Antes del advenimiento de los esteroides el pénfigo era casi invariablemente mortal, alrededor del 75 por ciento de los pacientes fallecían en un año.4 Sin embargo, al mejorar las técnicas de diagnóstico de formas más tempranas y leves de la enfermedad el pronóstico se ha modificado. Las formas leves pueden revertir en forma espontánea y en la mayoría de los casos puede limitarse la progresión de incluso las formas severas. Con el tratamiento (ver adelante) las lesiones sanan en forma normal sin cicatriz. La mayoría de los pacientes tratados por pénfigo entran en remisión parcial en 2 a 3 años y pueden mantenerse sin lesiones con dosis mínimas de esteroides (< 15 mg de prednisona al día). En un estudio longitudinal de evolución en 40 pacientes con pénfigo vulgar, el 45 por ciento tuvo remisión completa y a largo plazo después de 5 años y el 71 por ciento después de 10 años. Los pacientes en remisión permanecieron sin lesiones aún sin tratamiento.5 La hiperpigmentación que se asocia con frecuencia con el pénfigo suele resolverse después de varios meses.
Durante el embarazo el pénfigo parece asociarse con una mayor incidencia de parto prematuro y muerte fetal.6 Pueden aparecer lesiones de pénfigo en el neonato; sin embargo, se resuelven en forma espontánea en varias semanas.
Pénfigo foliáceo
El pénfigo foliáceo es la segunda forma en frecuencia de pénfigo. Suele comenzar con lesiones pequeñas (< 1 cm), costrosas y pruriginosas que recuerdan hojuelas de maíz sobre la parte superior del tronco y la cara. Las costras se retiran con facilidad, dejando lesiones crónicas superficiales.
El trastorno progresa en semanas a meses, con mayor número de lesiones en la parte superior del tronco, la cara y el cuero cabelludo. En los casos extensos se desarrollan lesiones en todo el cuerpo, que confluyen y pueden progresar hasta una eritrodermia exfoliativa. A diferencia de las formas profundas de pénfigo, en el pénfigo foliáceo la afección oral es muy rara.
El pronóstico del pénfigo foliáceo no tratado es más favorable que el del pénfigo vulgar. Las lesiones de pénfigo foliáceo no son tan profundas y existe menos posibilidad de infección, pérdida de líquido y alteraciones metabólicas. Aunque el pénfigo foliáceo es menos severo, las dosis de medicamento requeridas para su control son semejantes a las usadas para el pénfigo vulgar. Existen dos variantes clínicas, pénfigo eritematoso (también conocido como síndrome de Senear-Usher) y fogo salvagem (fuego salvaje, también conocido como pénfigo epidémico y pénfigo brasileño) [ver tabla 1].
Pénfigo relacionado con medicamentos
Tanto el pénfigo vulgar como el foliáceo puede ser inducido o desenmascarado (i.e., enfermedad latente enmascarada) por ciertos medicamentos. El pénfigo que continúa después de que el paciente usa un fármaco se denomina desenmascarado, mientras que las lesiones que desparecen pronto después de su supresión se conocen como inducidas. Aunque el pénfigo relacionado con fármacos no es frecuente, esta posibilidad debe excluirse en todos los pacientes con enfermedad de reciente inicio. Las alteraciones clínicas, histológicas,7 y de inmunofluorescencia8 del pénfigo inducido por fármacos son semejantes a las de la variedad idiopática. Sin embargo, el pénfigo causado por drogas que contienen un radical sulfihidrilo (fármacos tiol) es clínicamente distinto del pénfigo causado por fármacos no tiol. La presencia o ausencia del radical sulfhidrilo parece influir tanto en el tipo de pénfigo que se expresa como en el pronóstico de la condición inducida por el fármaco. Los fármacos tioles tienen más probabilidad de inducir pénfigo foliáceo, que revierte en forma espontánea con mayor probabilidad cuando se suspende el medicamento. Los medicamentos no tioles desencadenan más pénfigo vulgar, que puede persistir incluso después de suspender el fármaco. Los agentes implicados con más frecuencia son los fármacos tioles como la penicilamina y el captopril. Otros agentes responsables incluyen medicamentos que contienen azufre, como las penicilinas y las cefalosporinas. Estos sufren cambios metabólicos para formar grupos tioles y se denominan medicamentos tioles enmascarados. Los medicamentos no tioles que contienen grupos amida (v.gr., dipirona y enalapril) pueden provocar una enfermedad que es indistinguible del pénfigo vulgar que ocurre de modo espontáneo.8
DATOS HISTOLOGICOS E INMUNOLOGICOS
El diagnóstico debe confirmarse siempre por examen histopatológico y de inmunofluorescencia. Las biopsia de pénfigo y otras enfermedades bulosas deben realizarse en el borde de la lesión, de modo que se incluya piel adyacente no afectada. La acantolisis (separación de los queratinocitos entre sí) es la alteración fundamental en todas las formas de pénfigo.
Todas las formas de pénfigo se asocian con autoanticuerpos circulantes e intracelulares (IC) fijos a los tejidos que reaccionan contra antígenos en la superficie de los queratinocitos. La detección de estos autoanticuerpos es muy útil para establecer el diagnóstico, porque rara vez aparecen en otros padecimientos. Los autoanticuerpos IC circulantes se detectan por ensayos de inmunofluorescencia indirecta en el suero y los autoanticuerpos IC fijos a los tejidos se evidencian por inmunofluorescencia directa de la biopsia de piel. En ambos casos producen un patrón de fluorescencia semejante a encaje dentro de la epidermis. También pueden existir títulos bajos de autoanticuerpos IC en quemaduras, infecciones micóticas y reacciones alérgicas a fármacos. Los antígenos contra el ABO, que existen en alrededor del 5 por ciento de la población normal, son la causa más común de pruebas falsas positivas para autoanticuerpos IC. Existen autoanticuerpos IC fijos a los tejidos en las lesiones y en la piel normal adyacente de alrededor del 90 por ciento de los pacientes con pénfigo, y son más sensibles y específicos para el diagnóstico que los autoanticuerpos circulantes. Los autoanticuerpos más comunes son de IgG, pero también pueden depositarse de IgM e IgA (con o sin C3).
TRATAMIENTO
Tratamiento inicial
El tratamiento inicial está determinado por la extensión y velocidad de progresión de las lesiones. La enfermedad localizada y lentamente progresiva puede tratarse con inyecciones intralesionales de esteroides (acetónido de triamcinolona, 10 a 20 mg/ml) o administración tópica de esteroides de alta potencia. Las lesiones nuevas que continúan apareciendo en mayor número pueden controlarse en algunos casos con esteroides sistémicos en dosis bajas (prednsiona, 20 mg/día). Los pacientes con enfermedad extensa o rápidamente progresiva se tratan con dosis moderadamente altas de esteroides (prednisona 70 a 90 mg/día). Esta dosis se aumenta con rapidez cada 4 a 14 días en incrementos de 50 por ciento hasta que se controle la actividad de la enfermedad, lo que se evidencia por ausencia de lesiones nuevas y desaparición de dolor o prurito cutáneo. Si persiste la actividad de la enfermedad a pesar de las dosis altas de esteroides (> 120 a 160 mg/día de prednisona), deben considerarse alguna de las siguientes medidas para el control rápido:
1. Plasmaféresis, que se realiza tres veces por semana para retirar 1 a 2 L de plasma por procedimiento.9
2. Inmunoglobulina intravenosa (Ig IV), en dosis de 400 mg/kg/día por 5 días o en dosis mayores de 3 días.10 Recientemente se ha revisado el uso de Ig IV para el tratamiento de las enfermedades de la piel.11 Tanto con la Ig IV como con la plasmaféresis es importante administrar en forma concomitante un agente inmunosupresor como ciclofosfamida o azatioprina para minimizar el rebote en el nivel de anticuerpos,12 y para asegurar que el paciente esté respondiendo al tratamiento.
3. Tratamiento en pulsos con megadosis de metilprednisolona, en dosis de 1 g7día IV por 5 días.13
No se han evaluado aún estudios comparativos sobre la eficacia relativa de estos procedimientos. Con base en la experiencia limitada, la Ig IV puede preferirse en algunos casos porque tiene menos efectos adversos que otros procedimientos y se asocia con una tasa de respuesta muy alta. Una vez controlada la actividad de la enfermedad el paciente se mantiene con el tipo y dosis de medicamento requerido para establecer el control hasta que sanan alrededor del 80 por ciento de las lesiones. El tratamiento no debe disminuirse mientras sigan apareciendo lesiones nuevas.
Tratamiento adyuvante
El papel de los adyuvantes en el tratamiento del pénfigo sigue siendo tema de controversia. Debido a la falta de estudios controlados, no se sabe si los beneficios potenciales de los adyuvantes superan la toxicidad adicional.2 Las indicaciones para el tratamiento adyuvante incluye la presencia de contraindicaciones relativas para los esteroides sistémicos, desarrollo de efectos adversos serios de los esteroides y recaídas en la actividad de la enfermedad que hacen imposible disminuir las dosis de esteroides.5 Debido a que requieren 4 a 6 semanas para ser eficaces, los adyuvantes no se usan para controlar la enfermedad activa y de progresión rápida.
Los tratamientos adyuvantes para el pénfigo incluyen diversos agentes citotóxicos e inmunosupresores (v.gr., ciclofosfamida, azatioprina, ciclosporina, methotrexate y micofenolato mofetil14), dapsona, agentes antinflamatorios (v.gr., oro), antimaláricos y ciertos antibióticos (v.gr., tetraciclina y minociclina).
Penfigoide buloso
PATOGENIA
La causa inmediata del penfigoide buloso (PB o BP por sus siglas en inglés, n. del t.) parece ser una respuesta de autoanticuerpos a los antígenos de 180 kd (BP180) y de 230 kd (BP 230) de la zona de la membrana basal.15 La transferencia pasiva de estos anticuerpos a animales puede causar lesiones de la enfermedad,16 y en un estudio en humanos se encontró que los autoanticuerpos anti-BP180 constituyen un factor de mal pronóstico.17
MANIFESTACIONES CLINICAS
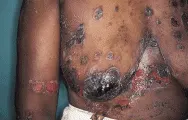
El PB es un padecimiento subepidérmico no cicatricial que se caracteriza por brotes recurrentes de ampollas grandes y tensas que se originan en bases urticarianas. Las lesiones suelen aparecer en el tronco y las zonas de flexión, en especial en cara interna de muslos y axilas. Las ampollas pueden variar en tamaño de algunos pocos milímetros a varios centímetros [ver figura 3]. Suelen estar llenas con un líquido claro, pero pueden ser hemorrágicas. Las erosiones son mucho menos frecuentes que en el pénfigo y el signo de Nikolsky es negativo. Un dato característico es que aparecen múltiples bulas a partir de placas urticarianas grandes (del tamaño de la palma o mayor) e irregulares. Esto contrasta con las bulas del eritema multiforme (ver adelante), en el eritema multiforme aparece una sola bula en el centro de una base urticariana más pequeña (del tamaño de una moneda).
En los brotes agudos de PB las bulas pueden originarse de piel con apariencia normal. Ocurren lesiones orales en el 10 a 25 por ciento de los pacientes, pero la afección ocular es rara. Sin tratamiento la enfermedad puede volverse muy extensa.
El PB es un padecimiento esporádico que ocurre principalmente en los ancianos, pero puede presentarse en cualquier edad y raza. Se ha reportado en un infante de 2 meses de edad.18 Entre los factores precipitantes se incluyen traumatismos, quemaduras, radiación ionizante, luz ultravioleta y ciertos fármacos. En un estudio de casos y controles de 116 casos, los neurolépticos y los diuréticos (en especial los antagonistas de la aldosterona) fueron más empleados en los pacientes con PB que en los sujetos controles.19 Aún existe controversia respecto a si el PB se asocia con mayor incidencia de cáncer;20 sin embargo, las correlaciones entre recaídas en la actividad de la enfermedad y recurrencia del cáncer subyacente sugieren esta asociación en pacientes individuales.
El PB se caracteriza por remisiones y recidivas espontáneas que pueden persistir por años. Incluso sin tratamiento, el PB suele ser autolimitado. Alrededor del 30 por ciento de los pacientes tienen remisión completa después de un promedio de 27 meses.21 A pesar de ello, la enfermedad es seria, en especial en ancianos.22 La mortalidad es baja en jóvenes, pero importante en ancianos. En un estudio de pacientes mayores de 68 años, casi la tercera parte falleció por la enfermedad o sus complicaciones (principalmente sepsis y enfermedad cardiovascular) en un lapso de 1 año.17
DATOS HISTOLOGICOS E INMUNOLOGICOS
La primera lesión en el PB es una ampolla que se origina en la lámina lúcida, entre la membrana basal de los queratinocitos y la lámina densa. Esta es seguida de pérdida de los filamentos de anclaje y hemidesmosomas. Desde el punto de vista histológico, existe infiltrado inflamatorio superficial y ampollas subepidérmicas sin queratinocitos necróticos. El infiltrado consiste principalmente en linfocitos e histiocitos, y es especialmente rico en eosinófilos. No ocurre cicatrización.
Alrededor del 70 a 80 por ciento de los pacientes con PB activo tienen anticuerpos circulantes a uno o más antígenos de la zona de la membrana basal. Según la inmunofluorescencia directa, los anticuerpos se depositan en un patrón lineal delgado y con la inmunoelectromicroscopía se observan dentro de la lámina lúcida. Por el contrario, los anticuerpos contra los antígenos de la membrana basal que existen en la piel de los pacientes con lupus eritematoso generalizado se depositan con un patrón granular.
Dos enfermedades ampollosas subepidérmicas menos frecuentes que están muy relacionadas al PB son el penfigoide cicatricial y el herpes gestacional [ver tabla 1]. El diagnóstico diferencial incluye también a la dermatitis herpetiforme y a la epidermolisis bulosa adquirida (ver adelante).
TRATAMIENTO
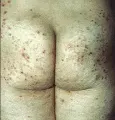
La dermatitis herpetiforme (DH) es una enfermedad vesiculobulosa rara caracterizada por vesículas pequeñas e intensamente pruriginosas que se agrupan en pequeños racimos y típicamente aparecen en la cara extensora de las extremidades y en los glúteos, cuero cabelludo y espalda. La condición parece ser un trastorno inmune y se asocia con depósitos granulares anormales de IgA en la zona de la membrana basal y con enteropatía sensible al gluten, semejante al esprue y asintomática. La enfermedad es crónica, con periodos de exacerbación y remisión. Las lesiones pueden desaparecer si los pacientes siguen una dieta estricta libre de gluten. La dermatosis lineal por IgA [ver tabla 1] es una enfermedad ampollosa subepidérmica que clínicamente se parece a la DH o al eritema multiforme (ver adelante).
PATOGENIA
Se desconoce la causa de la DH. Puede relacionarse con enfermedad celiaca sensible al gluten, ya existe asociación intensa entre ambos padecimientos y comparten bases genéticas similares (ambas se asocian con el HLA-B8 y HLA-DR3). Se piensa que la DH es causada por una respuesta inmune de IgA anormal contra un antígeno no identificado (quizá encontrado en el gluten) que tiene contacto con el intestino. Las lesiones de la piel pueden deberse a depósito de complejos inmunes contra este antígeno en la piel.
MANIFESTACIONES CLINICAS
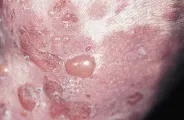
Las lesiones de la piel son polimórficas. Suelen comenzar como pápulas o vesículas urticarianas pequeñas, muy pruriginosas que se agrupan en un patrón herpetiforme [ver figura 4]. Rara vez se observan las vesículas u otras lesiones primarias porque sufren excoriación por el rascado. La distribución de las lesiones es característica: ocurren principalmente en los codos, rodillas, glúteos, área escapular y cuero cabelludo. En ocasiones las lesiones se distribuyen en todo el cuerpo. Tienden a aparecer en forma súbita y simétrica, en ocasiones después de la ingestión de cantidades grandes de glúten. Las lesiones cicatrizan dejando hiperpigmentación, y la cicatriz puede ser secundaria a rascado o infección secundaria. Es raro que se afecten las mucosas.
La enfermedad es dos veces más común en hombres que en mujeres. Afecta principalmente a personas entre 20 y 50 años. Puede asociarse con atrofia duodenal y yeyunal en parches que recuerda a la enteropatía sensible al gluten de la enfermedad celiaca.26,27 La enteropatía suele ser asintomática y, como la enfermedad celiaca, responde a restricción de gluten. Debido a que la enfermedad celiaca se asocia con linfoma gastrointestinal, existe preocupación de que lo mismo suceda con la DH. Sin embargo, aunque se han reportado linfomas del intestino delgado en la DH,28 la asociación parece ser rara.
DATOS HISTOLOGICOS E INMUNOLOGICOS
Para el diagnóstico se usan dos datos de laboratorio característicos en la DH. Primero, la enfermedad se caracteriza histológicamente por acúmulo de neutrófilos y eosinófilos en microabscesos en las puntas de las papilas dérmicas. En los casos más severos aparece edema, que puede progresar a vesículas subepidérmicas que aparecen justo por debajo de la lámina densa. Segundo, se encuentran depósitos granulares de IgA en la zona de la membrana basal en casi todos los pacientes. Estos suelen asociarse con depósitos granulares de C3 y en ocasiones de IgG e IgM. Cuando está sola, la IgA es uno de los marcadores diagnósticos más sensibles y específicos para la DH. Cuando se encuentra IgA con depósitos de IgG, IgM o C3, se agregan vasculitis por complejos inmunes y lupus eritematoso generalizado al diagnóstico diferencial. Aunque pueden ocurrir depósitos de IgA en la membrana basal en la enfermedad lineal por IgA,29 los depósitos en este padecimiento son lineales más que granulares. No existen anticuerpos circulantes contra componentes normales de la piel en la DH.
TRATAMIENTO
La DH responde en forma rápida y dramática a las sulfonas. La dapsona, en dosis de 100 a 200 mg/día, es el tratamiento que se usa con más frecuencia. Puede usarse sulfpiridina, en dosis de 1 a 3 g/día en dosis divididas (o sulfametoxipiridacina) en los pacientes que no pueden tolerar la dapsona. Las dosis de estos medicamentos se reducen en forma gradual hasta la menor cantidad que pueda suprimir el prurito y el desarrollo de nuevas lesiones. Como está indicado, los pacientes también responden a la dieta sin gluten, sin embargo, estas dietas son difíciles de seguir. A pesar de ello, incluso la reducción parcial en la ingesta de gluten causa menor requerimiento de sulfonas, por lo que siempre debe recomendarse la modificación de la dieta.
Eritema multiforme
El eritema multiforme es un padecimiento agudo, recurrente y autolimitado que afecta todos los grupos de edad y razas. Se caracteriza por erupción súbita de lesiones en racimos, que representan una reacción de hipersensibilidad mediada por células en la piel y las mucosas a diversos factores precipitantes, incluyendo agentes infecciosos y fármacos [ver tabla 3].30
MANIFESTACIONES CLINICAS
Las lesiones pueden ser localizadas o diseminadas y afectar tanto la piel como las mucosas. La erupción suele ser bilateral y simétrica, en las superficies extensoras de las extremidades y en las caras tanto dorsal como palmar de manos y pies. Las lesiones varían desde máculas y pápulas edematosas bien definidas, rojas a púrpura, hasta vesículas o bulas que pueden ulcerarse, formar costras, erosionarse e infectarse. Son características las lesiones en blanco de tiro, que consisten en una pápula o vesícula rodeada de una región de piel normal y un halo de eritema en la periferia [ver figura 5].
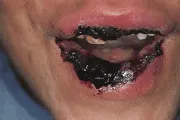
El síndrome de Stevens-Johnson es una forma severa de eritema multiforme que suele ser diseminado, fulminante y multisistémico [ver figura 6]. El síndrome puede acompañarse de fiebre alta, malestar, calosfríos, cefalea, taquicardia, taquipnea y postración. Las mucosas en la boca, ano y vagina contienen máculas redondas u ovales que forman vesículas, bulas y úlceras. Las lesiones oculares consisten en pápulas amarillas-grises bilaterales, que con frecuencia se ulceran e infectan de modo secundario, causando conjuntivitis. La afección ocular puede producir ceguera.
Algunos especialistas consideran que la necrolisis epidérmica tóxica (NET) es una forma de eritema multiforme, y suele ser una reacción a medicamentos (v.gr., ciertas sulfonamidas de acción prolongada [en especial trimetoprim-sulfametoxazol], anticonvulsivantes, barbitúricos y antinflamatorios no esteroides). La ausencia de reactantes inmunes dentro de los vasos sanguíneos de la piel y la escasez de inflamación dérmica ha hecho que otros investigadores consideren a la NET como una enfermedad independiente. En ocasiones el síndrome de piel escaldada por estafilococo, que se debe a toxinas producidas por Staphylococcus aureus,31 puede confundirse con la NET [ver tabla 1].
DATOS HISTOLOGICOS E INMUNOLOGICOS
Los datos histológicos cutáneos característicos del eritema multiforme incluyen edema subepidérmico, edema, formación de bulas, necrosis de célula epidérmicas y un infiltrado inflamatorio perivascular profundo. No existen datos inmunofluorescentes específicos, aunque la inmunofluorescencia directa puede mostrar depósitos granulares de C3 y fibrina en la unión dermoepidérmica y depósitos de IgM, C3 y fibrina en los vasos sanguíneos de la dermis. El eritema multiforme recurrente se asocia con frecuencia con infecciones por virus del herpes simple, como se reportó en el 71 por ciento en una serie de 65 pacientes con eritema multiforme. En estos pacientes pueden existir complejos inmunes circulantes contra el virus herpes simple.32
TRATAMIENTO
Las erupciones del eritema multiforme pueden recurrir en forma inesperada a pesar de las medidas preventivas. Por lo tanto, es importante identificar y eliminar las causas subyacentes. Los casos leves se tratan en forma sintomática con esteroides, antinflamatorios, antipruriginosos o preparaciones antibióticas tópicas. El aciclovir oral puede ser eficaz en la profilaxis del eritema multiforme posherpético. En los casos más severos está indicado el tratamiento con prednisona, 40 a 120 mg/día en dosis divididas. Si existe afección ocular debe solicitarse interconsulta inmediata al oftalmólogo.
Epidermolisis bulosa
La epidermolisis bulosa (EB) comprende un grupo de trastornos genéticos con una prevalencia aproximada de 1 por cada 500,000 personas. Existen más de 20 fenotipos diferentes de EB. Esta entidad patológica se caracteriza por la presencia de ampollas y erosiones secundarias a fricciones menores o traumatismos. Las vesículas pueden variar desde formas localizadas (p. ej., ampollas en palmas y plantas de los pies) a formas severas con desprendimiento generalizados de la piel y riesgo de infecciones graves, desequilibrio hidroelectrolítico, anemia y otras complicaciones mortales.
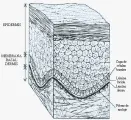
La EB se clasifica principalmente con base en el nivel ultraestructural en el que se desprende la piel dentro de la zona de la membrana basal [ver figura 7]. Los tres subtipos principales incluyen a la EB simple o epidermolítica (intradérmica), la EB de unión (dentro de la lámina lúcida) y la EB distrófica o dermolítica (bajo la lámina densa). El examen con microscopio electrónico localiza la capa en que ocurren las lesiones. Debido a que esta tecnología puede no estar siempre disponible, puede usarse el mapeo inmunofluorescente con anticuerpos para detectar componentes en las capas de la membrana basal, como el antígeno BP (células de la capa basal), la laminina (lámina lúcida) y colágena tipo IV (lámina densa).34 Puede hacerse diagnóstico prenatal por medio de sondas inmunohistoquímicas dirigidas contra componentes antigénicos de la membrana basal en la bipsia de piel del feto, como en el caso de EB de unión con atresia pilórica.35
EPIDERMOLISIS BULOSA SIMPLE
Existen por lo menos cinco formas de EB simple. El tipo más frecuente es una forma leve, autosómica dominante, que se inicia al nacimiento o poco tiempo después, con ampollas localizadas o generalizadas que generalmente no cicatrizan. Un segundo tipo es la enfermedad de Weber-Cockayne, que puede ser localizada o generalizada. En la forma localizada las ampollas aparecen en las zonas distales de las regiones palmares y plantares y comienzan durante la niñez o la adolescencia . En la forma generalizada, descrita por Koebner, la actividad de la enfermedad suele ser mayor en climas cálidos.
La variante denominada de Dowling-Meara (EB herpetiforme) es la forma menos frecuente de EB simple; se presenta como un proceso grave con ampollas generalizadas durante la infancia, que se parece a la EB de unión y a la variedad distrófica. La gravedad de este padecimiento disminuye con la edad.
EPIDERMOLISIS BULOSA DE UNION
La EB de unión, incluye un grupo de procesos autosómicos recesivos que se caracterizan por una disminución en el número e hipoplasia de los hemidesmosomas, demostrado por microscopía electrónica, y separación a nivel de la lámina lúcida. Con frecuencia se afectan las mucosas y se observan uñas distróficas. La forma más grave, denominada EB letalis, se presenta a los pocos días del nacimiento o durante los primeros meses y tienen una mortalidad muy alta. Estos pacientes tienen una elevada incidencia de paro respiratorio debido a la afección de la laringe y la tráquea. Las formas menos graves de la EB de unión consisten en ampollas severas generalizadas al nacimiento que mejoran gradualmente. Se puede desarrollar estenosis esofágica.
EPIDERMOLISIS BULOSA DISTROFICA
Hay dos formas de EB distrófica de tipo hereditario, que se trasmite de forma autosómica dominante. La EB distrófica hiperplásica (síndrome de Cockayne-Touraine) se presenta en etapas tempranas de la vida con formación de ampollas de contenido serohemático predominantemente en las superficies extensoras de las extremidades inferiores y asociadas con uñas distróficas. El tipo albopapuloide de EB distrófica se caracteriza por la presencia de pápulas blancas que se desarrollan durante la adolescencia en el tronco o las extremidades; sin embargo, las ampollas están presentes en el periodo perinatal. En ambas formas el estudio ultraestructural revela separación por debajo de la membrana basal, con alteración en las fibrillas de anclaje o disminución en su número.
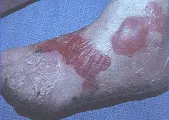
Las formas recesivas de EB distrófica se presentan en el periodo neonatal con ampollas graves de contenido serohemático, localizada en zonas de traumatismos o de forma generalizada. La formación de milia es poco frecuente, pero puede causar cicatrices en las lesiones. Otras complicaciones son: dentición anormal, pérdida o distrofia de las uñas, fusión digital, contracturas en flexión y estenosis esofágica [ver figura 8]. Ocurre también retraso en el crecimiento, desnutrición y anemia crónica. Estos pacientes tienen un riesgo elevado de carcinoma epidermoide, con una incidencia elevada de metástasis y muerte.
Se puede hacer el diagnóstico de esta entidad patológica antes del nacimiento mediante fetoscopía con toma de biopsias de la piel. Desde el punto de vista ultraestructural, el tejido muestra formación de ampollas dermolíticas. Una alternativa cuando se sospecha EB distrófica recesiva son las pruebas bioquímicas para detectar aumento de la expresión de colagenasa en los fibroblastos fetales.36
El tratamiento de apoyo está dirigido a curar las heridas y prevenir las complicaciones. Los cuidados diarios de la piel incluyen la aplicación de compresas húmedas, ungüentos con antibióticos y compresas no adherentes como mallas de gasa fina (N-terface). Es esencial un método multidisciplinario que incluya el consejo genético, psiquiátrico o psicológico y sistemas de apoyo para el paciente y su familia, en especial para las formas graves de la enfermedad.
La Dystrophic Epidermolysis Bulosa Research Association of America (http://www.debra.org) ha establecido un registro nacional para obtener datos epidemiológicos y determinar los aspectos económicos y sociales de la EB, así como llevar un registro de los pacientes que quieran participar en diversos protocolos de investigación.53 Para una mayor información puede comunicarse con DEBRA, en la ciudad de Nueva York, en el teléfono 995-2220 (212).
EPIDERMOLISIS BULOSA ADQUIRIDA
La aparición de ampollas en adultos en quienes no existe una base genética causante de epidermólisis bulosa se denomina epidermólisis bulosa adquirida o epidermólisis bulosa acquisita (EBA). Pueden demostrarse por inmunohistología anticuerpos IgG contra la membrana basal, tanto circulantes como fijos a los tejidos. Las ampollas se desarrollan por debajo de la epidermis y curan dejando como secuela cicatrices atróficas y malformaciones. Dichas ampollas están casi siempre limitadas a las extremidades, en áreas expuestas a traumatismos mecánicos. La EBA puede estar asociada con lesiones orales y distrofia ungueal. Pueden estar asociadas alteraciones subyacentes de origen maligno, autoinmune e inflamatorio. La presencia de colitis ulcerosa y enfermedad de Crohn en cerca del 30 por ciento de los casos sugiere que la EBA debería ser incluida entre las manifestaciones extraintestinales de la enfermedad inflamatoria del intestino.37
El diagnóstico se hace excluyendo otras enfermedades bulosas, en especial PB (ver antes). La microscopía inmunoelectrónica puede ser recurso adicional para establecer el diagnóstico, aunque la técnica no siempre está disponible.55 La inmunofluorescencia directa con técnicas para separar la lámina lúcida ayuda en el diagnóstico diferencial. Con este método se observan los anticuerpos de IgG en el lado dérmico en la EBA y en la epidermis en el penfigoide.38 Se ha identificado que el antígeno en la EBA es el extremo globular carboxilo de la procolágena tipo VII.39
Diagnóstico diferencial de las enfermedades vesiculobulosas
Se han analizado las principales formas de padecimientos bulosos de origen autoinmune o hereditario. El diagnóstico diferencial incluye diversas otras condiciones en las que las vesículas o bulas son menos comunes o aparecen en forma secundaria a otros procesos patológicos. Una erupción fija a drogas puede causar bulas localizadas que aparezcan después de la ingestión de algún medicamento en particular . La dermatitis eccematosa produce vesículas espongióticas por edema intercelular. Este se manifiesta clínicamente por grandes bulas en la dermatitis alérgica por contacto aguda debida a hiedra o roble venenosos. En ocasiones el lupus eritematoso generalizado produce bulas al causar degeneración de las células basales.
Una erupción bulosa en el dorso de las manos y otros sitios expuestos al sol en pacientes sometidos a hemodiálisis por tiempo prolongado puede parecerse a la porfiria cutánea tarda.40 Los niveles de porfirina suelen estar en límites normales. Las bulas intraepidérmicas o subepidérmicas, principalmente en las extremidades, pueden ser un signo cutáneo de diabetes mellitus.41 Las infeciones bacterianas de la piel, como el impétigo, pueden asociarse con formación subcorneal de bulas.
También deben incluirse en el diagnóstico diferencial varias infecciones virales, incluyendo varicela, herpes simple y herpes zoster .
DRA. ELIZABETH A. ABEL
DR. JEAN-CLAUDE BYSTRYN
Las enfermedades vesiculobulosas, que en número son más de 50, se caracterizan por la formación de ampollas llenas de líquido en la piel. Las ampollas menores de 0.5 cm se denominan vesículas y las mayores bulas. Las vesículas y las bulas son patrones de reacción de la piel a una lesión y pueden ser causadas por muy diversos padecimientos.
La mayoría de las enfermedades vesiculobulosas son de origen inmunológico o genético. Son causadas por reacciones autoinmunes a componentes de la piel, reacciones alérgicas a agentes externos en los que la piel es el principal órgano afectado, y por condiciones genéticas en las que algunos de los componentes de la piel están ausentes o son anormales. El denominador común es la falta de adhesión: una o más de las estructuras que mantienen junta a la piel se separan y aparece una cavidad llena de líquido. Las diversas enfermedades se clasifican por la estructura o estructuras afectadas y por el o los mecanismos por los que ocurre la falta de adhesión [ver tabla 1]. En esta subsección se analizan varias enfermedades vesiculobulosas dentro del contexto del enfoque diagnóstico general del paciente con lesiones ampollosas.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
ZMB-zona de membrana basal IC-intercelular IF-inmunofluorescencia Ig IV-inmunoblogulina intravenosa |
Evaluación clínica general
El diagnóstico se basa en los datos clínicos, histológicos e inmunológicos. Los datos clínicos con importancia diagnóstica incluyen:
1. La historia clínica. ¿El problema es agudo o crónico? ¿Se agrava con el sol o los traumatismos físicos?
2. La aparición de lesiones individuales [ver tabla 2]. ¿La lesión es una vesícula o una bula? ¿Es tensa, flácida o umbilicada? ¿La piel en la base es normal, tiene urticaria o cicatriz? ¿La vesícula está en medio de placas urticarianas o en la periferia? ¿Existe más de una bula en la misma placa?
3. Agrupación de las lesiones individuales. ¿Se encuentran estas en grupos (como ocurre en el herpes simple) o se distribuyen en forma aislada?
4. Sitios de afección. ¿Existen también lesiones en las mucosas? ¿Predominan en las superficies de flexión o extensión, en las palmas y plantas o en el dorso de manos y pies, en el cuero cabelludo, cara y tronco superior, o en áreas expuestas a traumatismos?
|
|||||||||||||||||||||||||||||||||||||||||
|
El dato histológico más importante es la capa de la piel en la que se forma la ampolla. Si la ampolla se forma en la epidermis, ¿lo hace inmediatamente sobre la capa de células basales o más arriba (por debajo del estrato córneo)? Si se forma en la zona de la membrana basal, ¿es dentro de la lámina lúcida o por debajo de la lámina densa? La localización precisa puede determinarse por procedimientos de inmunofluorescencia.
El hallazgo inmunológico más importante es la presencia o ausencia de anticuerpos anormales circulantes o fijos a los tejidos. Estos se detectan por técnicas de inmunofluorescencia: (1) inmunofluorescencia indirecta para detectar los anticuerpos circulantes y (2) inmunofluorescencia directa en la biopsia de piel para detectar anticuerpos fijos a los tejidos.
Pénfigo
DEFINICION Y PATOGENIA
El pénfigo se caracteriza por vesículas que se originan dentro de la epidermis y por pérdida de la cohesión de las células de la epidermis (acantolisis) que produce formación de hendiduras sobre la capa de células basales. Los autoanticuerpos dirigidos contra moléculas de adhesión causan que los queratinocitos epidérmicos se separen, provocando bulas intraepidérmicas. Existen dos tipos de pénfigo: profundo (v.gr., pénfigo vulgar) y superficial (v.gr., pénfigo foliáceo). Estos difieren en las capas epidérmicas que se lesionan, en las manifestaciones clínicas de las enfermedades y en las alteraciones inmunológicas asociadas.1 En las formas profundas las ampollas se forman inmediatamente por arriba de la capa de células basales y se asocian con autoanticuerpos contra desmogleina 3, una molécula de adhesión de queratinocitos. En las formas superficiales las bulas se forman inmediatamente por debajo del estrato córneo. Las formas superficiales del pénfigo se asocian con anticuerpos contra desmogleína 1.
MANIFESTACIONES CLINICAS
Pénfigo vulgar
El pénfigo vulgar es la forma más común de pénfigo. Puede desarrollarse a cualquier edad, pero suele ocurrir en personas entre 30 y 60 años de edad. El padecimiento tiende a afectar a personas con ascendencia mediterránea, pero puede ocurrir en cualquier etnia. El pénfigo es más común en personas con ciertos haplotipos HLA. La ocurrencia de la enfermedad en los familiares en primer grado, aunque rara, sugiere una susceptibilidad heredada transferida como un rasgo dominante. Sin embargo, se requieren otros factores desconocidos para la expresión de este trastorno en personas predispuestas.2 Los estudios de alelos de HLA de clase II en pacientes japoneses, así como en otros grupos étnicos, muestran asociación con el HLA-DRB1*04 y HLA-DRB1*14 en pacientes con pénfigo vulgar independientemente del grupo racial.3
El pénfigo vulgar suele comenzar, aunque no siempre, con ulceraciones crónicas, dolorosas y que no cicatrizan en la cavidad oral [ver figura 1]. Rara vez se observan bulas porque se rompen con facilidad, dejando bases ulceradas. Las ulceraciones suelen ser múltiples, superficiales e irregulares. Puede afectarse cualquier parte de la mucosa oral, pero ocurre más en la mucosa labial, el paladar y la lengua. La presencia de ulceraciones múltiples permite distinguir a estas lesiones de tumores malignos ulcerados de la cavidad oral, que suelen ser únicos. El diagnóstico de pénfigo suele considerarse solo después de que las lesiones han durado semanas a meses.
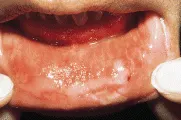
|
| Figura 1 |
| Pénfigo vulgar generalizado |
También las lesiones en la piel pueden ser la manifestación inicial, comenzando con una ampolla pequeña y llena de líquido en piel que por lo demás parece normal. Las ampollas suelen ser flácidas porque la delgada epidermis suprayacente no suele tolerar mucha presión. Las bulas se rompen rápido, en varios días, y pueden no existir al examinar al paciente. En su lugar se observan erosiones superficiales, con bordes bien definidos y del tamaño de una moneda, con un collarín de pérdida de epidermis en la periferia. La parte superior del tronco, la espalda, el cuero cabelludo y la cara son sitios frecuentes de afección, pero las lesiones pueden ocurrir en cualquier parte del cuerpo. El problema progresa en semanas a meses [ver figura 2]. Las lesiones pueden pasar desapercibidas en las áreas periungueales (se manifiestan por inflamación paroniquia dolorosa y eritematosa), la faringe y laringe (dolor al deglutir y disfonía) y en la cavidad nasal (congestión nasal y descarga de moco con sangre, en especial al sonarse la nariz por la mañana).
|
| Figura 2 |
| Pénfigo vulgar |
Una característica de todas las formas activas de pénfigo es el signo de Nikolsky, en el que la aplicación de presión firme sobre piel de apariencia normal causa que la epidermis se separe de la dermis. El signo de Nikolsky se despierta con más facilidad en piel normal adyacente a una lesión activa.
Si no se trata las erosiones y bulas del pénfigo vulgar se diseminan en forma gradual, afectando áreas grandes de superficie y complicándose con infecciones severas y alteraciones metabólicas. Antes del advenimiento de los esteroides el pénfigo era casi invariablemente mortal, alrededor del 75 por ciento de los pacientes fallecían en un año.4 Sin embargo, al mejorar las técnicas de diagnóstico de formas más tempranas y leves de la enfermedad el pronóstico se ha modificado. Las formas leves pueden revertir en forma espontánea y en la mayoría de los casos puede limitarse la progresión de incluso las formas severas. Con el tratamiento (ver adelante) las lesiones sanan en forma normal sin cicatriz. La mayoría de los pacientes tratados por pénfigo entran en remisión parcial en 2 a 3 años y pueden mantenerse sin lesiones con dosis mínimas de esteroides (< 15 mg de prednisona al día). En un estudio longitudinal de evolución en 40 pacientes con pénfigo vulgar, el 45 por ciento tuvo remisión completa y a largo plazo después de 5 años y el 71 por ciento después de 10 años. Los pacientes en remisión permanecieron sin lesiones aún sin tratamiento.5 La hiperpigmentación que se asocia con frecuencia con el pénfigo suele resolverse después de varios meses.
Durante el embarazo el pénfigo parece asociarse con una mayor incidencia de parto prematuro y muerte fetal.6 Pueden aparecer lesiones de pénfigo en el neonato; sin embargo, se resuelven en forma espontánea en varias semanas.
Pénfigo foliáceo
El pénfigo foliáceo es la segunda forma en frecuencia de pénfigo. Suele comenzar con lesiones pequeñas (< 1 cm), costrosas y pruriginosas que recuerdan hojuelas de maíz sobre la parte superior del tronco y la cara. Las costras se retiran con facilidad, dejando lesiones crónicas superficiales.
El trastorno progresa en semanas a meses, con mayor número de lesiones en la parte superior del tronco, la cara y el cuero cabelludo. En los casos extensos se desarrollan lesiones en todo el cuerpo, que confluyen y pueden progresar hasta una eritrodermia exfoliativa. A diferencia de las formas profundas de pénfigo, en el pénfigo foliáceo la afección oral es muy rara.
El pronóstico del pénfigo foliáceo no tratado es más favorable que el del pénfigo vulgar. Las lesiones de pénfigo foliáceo no son tan profundas y existe menos posibilidad de infección, pérdida de líquido y alteraciones metabólicas. Aunque el pénfigo foliáceo es menos severo, las dosis de medicamento requeridas para su control son semejantes a las usadas para el pénfigo vulgar. Existen dos variantes clínicas, pénfigo eritematoso (también conocido como síndrome de Senear-Usher) y fogo salvagem (fuego salvaje, también conocido como pénfigo epidémico y pénfigo brasileño) [ver tabla 1].
Pénfigo relacionado con medicamentos
Tanto el pénfigo vulgar como el foliáceo puede ser inducido o desenmascarado (i.e., enfermedad latente enmascarada) por ciertos medicamentos. El pénfigo que continúa después de que el paciente usa un fármaco se denomina desenmascarado, mientras que las lesiones que desparecen pronto después de su supresión se conocen como inducidas. Aunque el pénfigo relacionado con fármacos no es frecuente, esta posibilidad debe excluirse en todos los pacientes con enfermedad de reciente inicio. Las alteraciones clínicas, histológicas,7 y de inmunofluorescencia8 del pénfigo inducido por fármacos son semejantes a las de la variedad idiopática. Sin embargo, el pénfigo causado por drogas que contienen un radical sulfihidrilo (fármacos tiol) es clínicamente distinto del pénfigo causado por fármacos no tiol. La presencia o ausencia del radical sulfhidrilo parece influir tanto en el tipo de pénfigo que se expresa como en el pronóstico de la condición inducida por el fármaco. Los fármacos tioles tienen más probabilidad de inducir pénfigo foliáceo, que revierte en forma espontánea con mayor probabilidad cuando se suspende el medicamento. Los medicamentos no tioles desencadenan más pénfigo vulgar, que puede persistir incluso después de suspender el fármaco. Los agentes implicados con más frecuencia son los fármacos tioles como la penicilamina y el captopril. Otros agentes responsables incluyen medicamentos que contienen azufre, como las penicilinas y las cefalosporinas. Estos sufren cambios metabólicos para formar grupos tioles y se denominan medicamentos tioles enmascarados. Los medicamentos no tioles que contienen grupos amida (v.gr., dipirona y enalapril) pueden provocar una enfermedad que es indistinguible del pénfigo vulgar que ocurre de modo espontáneo.8
DATOS HISTOLOGICOS E INMUNOLOGICOS
El diagnóstico debe confirmarse siempre por examen histopatológico y de inmunofluorescencia. Las biopsia de pénfigo y otras enfermedades bulosas deben realizarse en el borde de la lesión, de modo que se incluya piel adyacente no afectada. La acantolisis (separación de los queratinocitos entre sí) es la alteración fundamental en todas las formas de pénfigo.
Todas las formas de pénfigo se asocian con autoanticuerpos circulantes e intracelulares (IC) fijos a los tejidos que reaccionan contra antígenos en la superficie de los queratinocitos. La detección de estos autoanticuerpos es muy útil para establecer el diagnóstico, porque rara vez aparecen en otros padecimientos. Los autoanticuerpos IC circulantes se detectan por ensayos de inmunofluorescencia indirecta en el suero y los autoanticuerpos IC fijos a los tejidos se evidencian por inmunofluorescencia directa de la biopsia de piel. En ambos casos producen un patrón de fluorescencia semejante a encaje dentro de la epidermis. También pueden existir títulos bajos de autoanticuerpos IC en quemaduras, infecciones micóticas y reacciones alérgicas a fármacos. Los antígenos contra el ABO, que existen en alrededor del 5 por ciento de la población normal, son la causa más común de pruebas falsas positivas para autoanticuerpos IC. Existen autoanticuerpos IC fijos a los tejidos en las lesiones y en la piel normal adyacente de alrededor del 90 por ciento de los pacientes con pénfigo, y son más sensibles y específicos para el diagnóstico que los autoanticuerpos circulantes. Los autoanticuerpos más comunes son de IgG, pero también pueden depositarse de IgM e IgA (con o sin C3).
TRATAMIENTO
Tratamiento inicial
El tratamiento inicial está determinado por la extensión y velocidad de progresión de las lesiones. La enfermedad localizada y lentamente progresiva puede tratarse con inyecciones intralesionales de esteroides (acetónido de triamcinolona, 10 a 20 mg/ml) o administración tópica de esteroides de alta potencia. Las lesiones nuevas que continúan apareciendo en mayor número pueden controlarse en algunos casos con esteroides sistémicos en dosis bajas (prednsiona, 20 mg/día). Los pacientes con enfermedad extensa o rápidamente progresiva se tratan con dosis moderadamente altas de esteroides (prednisona 70 a 90 mg/día). Esta dosis se aumenta con rapidez cada 4 a 14 días en incrementos de 50 por ciento hasta que se controle la actividad de la enfermedad, lo que se evidencia por ausencia de lesiones nuevas y desaparición de dolor o prurito cutáneo. Si persiste la actividad de la enfermedad a pesar de las dosis altas de esteroides (> 120 a 160 mg/día de prednisona), deben considerarse alguna de las siguientes medidas para el control rápido:
1. Plasmaféresis, que se realiza tres veces por semana para retirar 1 a 2 L de plasma por procedimiento.9
2. Inmunoglobulina intravenosa (Ig IV), en dosis de 400 mg/kg/día por 5 días o en dosis mayores de 3 días.10 Recientemente se ha revisado el uso de Ig IV para el tratamiento de las enfermedades de la piel.11 Tanto con la Ig IV como con la plasmaféresis es importante administrar en forma concomitante un agente inmunosupresor como ciclofosfamida o azatioprina para minimizar el rebote en el nivel de anticuerpos,12 y para asegurar que el paciente esté respondiendo al tratamiento.
3. Tratamiento en pulsos con megadosis de metilprednisolona, en dosis de 1 g7día IV por 5 días.13
No se han evaluado aún estudios comparativos sobre la eficacia relativa de estos procedimientos. Con base en la experiencia limitada, la Ig IV puede preferirse en algunos casos porque tiene menos efectos adversos que otros procedimientos y se asocia con una tasa de respuesta muy alta. Una vez controlada la actividad de la enfermedad el paciente se mantiene con el tipo y dosis de medicamento requerido para establecer el control hasta que sanan alrededor del 80 por ciento de las lesiones. El tratamiento no debe disminuirse mientras sigan apareciendo lesiones nuevas.
Tratamiento adyuvante
El papel de los adyuvantes en el tratamiento del pénfigo sigue siendo tema de controversia. Debido a la falta de estudios controlados, no se sabe si los beneficios potenciales de los adyuvantes superan la toxicidad adicional.2 Las indicaciones para el tratamiento adyuvante incluye la presencia de contraindicaciones relativas para los esteroides sistémicos, desarrollo de efectos adversos serios de los esteroides y recaídas en la actividad de la enfermedad que hacen imposible disminuir las dosis de esteroides.5 Debido a que requieren 4 a 6 semanas para ser eficaces, los adyuvantes no se usan para controlar la enfermedad activa y de progresión rápida.
Los tratamientos adyuvantes para el pénfigo incluyen diversos agentes citotóxicos e inmunosupresores (v.gr., ciclofosfamida, azatioprina, ciclosporina, methotrexate y micofenolato mofetil14), dapsona, agentes antinflamatorios (v.gr., oro), antimaláricos y ciertos antibióticos (v.gr., tetraciclina y minociclina).
Penfigoide buloso
PATOGENIA
La causa inmediata del penfigoide buloso (PB o BP por sus siglas en inglés, n. del t.) parece ser una respuesta de autoanticuerpos a los antígenos de 180 kd (BP180) y de 230 kd (BP 230) de la zona de la membrana basal.15 La transferencia pasiva de estos anticuerpos a animales puede causar lesiones de la enfermedad,16 y en un estudio en humanos se encontró que los autoanticuerpos anti-BP180 constituyen un factor de mal pronóstico.17
MANIFESTACIONES CLINICAS
El PB es un padecimiento subepidérmico no cicatricial que se caracteriza por brotes recurrentes de ampollas grandes y tensas que se originan en bases urticarianas. Las lesiones suelen aparecer en el tronco y las zonas de flexión, en especial en cara interna de muslos y axilas. Las ampollas pueden variar en tamaño de algunos pocos milímetros a varios centímetros [ver figura 3]. Suelen estar llenas con un líquido claro, pero pueden ser hemorrágicas. Las erosiones son mucho menos frecuentes que en el pénfigo y el signo de Nikolsky es negativo. Un dato característico es que aparecen múltiples bulas a partir de placas urticarianas grandes (del tamaño de la palma o mayor) e irregulares. Esto contrasta con las bulas del eritema multiforme (ver adelante), en el eritema multiforme aparece una sola bula en el centro de una base urticariana más pequeña (del tamaño de una moneda).
|
| Figura 3 |
| Penfigoide buloso |
En los brotes agudos de PB las bulas pueden originarse de piel con apariencia normal. Ocurren lesiones orales en el 10 a 25 por ciento de los pacientes, pero la afección ocular es rara. Sin tratamiento la enfermedad puede volverse muy extensa.
El PB es un padecimiento esporádico que ocurre principalmente en los ancianos, pero puede presentarse en cualquier edad y raza. Se ha reportado en un infante de 2 meses de edad.18 Entre los factores precipitantes se incluyen traumatismos, quemaduras, radiación ionizante, luz ultravioleta y ciertos fármacos. En un estudio de casos y controles de 116 casos, los neurolépticos y los diuréticos (en especial los antagonistas de la aldosterona) fueron más empleados en los pacientes con PB que en los sujetos controles.19 Aún existe controversia respecto a si el PB se asocia con mayor incidencia de cáncer;20 sin embargo, las correlaciones entre recaídas en la actividad de la enfermedad y recurrencia del cáncer subyacente sugieren esta asociación en pacientes individuales.
El PB se caracteriza por remisiones y recidivas espontáneas que pueden persistir por años. Incluso sin tratamiento, el PB suele ser autolimitado. Alrededor del 30 por ciento de los pacientes tienen remisión completa después de un promedio de 27 meses.21 A pesar de ello, la enfermedad es seria, en especial en ancianos.22 La mortalidad es baja en jóvenes, pero importante en ancianos. En un estudio de pacientes mayores de 68 años, casi la tercera parte falleció por la enfermedad o sus complicaciones (principalmente sepsis y enfermedad cardiovascular) en un lapso de 1 año.17
DATOS HISTOLOGICOS E INMUNOLOGICOS
La primera lesión en el PB es una ampolla que se origina en la lámina lúcida, entre la membrana basal de los queratinocitos y la lámina densa. Esta es seguida de pérdida de los filamentos de anclaje y hemidesmosomas. Desde el punto de vista histológico, existe infiltrado inflamatorio superficial y ampollas subepidérmicas sin queratinocitos necróticos. El infiltrado consiste principalmente en linfocitos e histiocitos, y es especialmente rico en eosinófilos. No ocurre cicatrización.
Alrededor del 70 a 80 por ciento de los pacientes con PB activo tienen anticuerpos circulantes a uno o más antígenos de la zona de la membrana basal. Según la inmunofluorescencia directa, los anticuerpos se depositan en un patrón lineal delgado y con la inmunoelectromicroscopía se observan dentro de la lámina lúcida. Por el contrario, los anticuerpos contra los antígenos de la membrana basal que existen en la piel de los pacientes con lupus eritematoso generalizado se depositan con un patrón granular.
Dos enfermedades ampollosas subepidérmicas menos frecuentes que están muy relacionadas al PB son el penfigoide cicatricial y el herpes gestacional [ver tabla 1]. El diagnóstico diferencial incluye también a la dermatitis herpetiforme y a la epidermolisis bulosa adquirida (ver adelante).
TRATAMIENTO
La dermatitis herpetiforme (DH) es una enfermedad vesiculobulosa rara caracterizada por vesículas pequeñas e intensamente pruriginosas que se agrupan en pequeños racimos y típicamente aparecen en la cara extensora de las extremidades y en los glúteos, cuero cabelludo y espalda. La condición parece ser un trastorno inmune y se asocia con depósitos granulares anormales de IgA en la zona de la membrana basal y con enteropatía sensible al gluten, semejante al esprue y asintomática. La enfermedad es crónica, con periodos de exacerbación y remisión. Las lesiones pueden desaparecer si los pacientes siguen una dieta estricta libre de gluten. La dermatosis lineal por IgA [ver tabla 1] es una enfermedad ampollosa subepidérmica que clínicamente se parece a la DH o al eritema multiforme (ver adelante).
PATOGENIA
Se desconoce la causa de la DH. Puede relacionarse con enfermedad celiaca sensible al gluten, ya existe asociación intensa entre ambos padecimientos y comparten bases genéticas similares (ambas se asocian con el HLA-B8 y HLA-DR3). Se piensa que la DH es causada por una respuesta inmune de IgA anormal contra un antígeno no identificado (quizá encontrado en el gluten) que tiene contacto con el intestino. Las lesiones de la piel pueden deberse a depósito de complejos inmunes contra este antígeno en la piel.
MANIFESTACIONES CLINICAS
Las lesiones de la piel son polimórficas. Suelen comenzar como pápulas o vesículas urticarianas pequeñas, muy pruriginosas que se agrupan en un patrón herpetiforme [ver figura 4]. Rara vez se observan las vesículas u otras lesiones primarias porque sufren excoriación por el rascado. La distribución de las lesiones es característica: ocurren principalmente en los codos, rodillas, glúteos, área escapular y cuero cabelludo. En ocasiones las lesiones se distribuyen en todo el cuerpo. Tienden a aparecer en forma súbita y simétrica, en ocasiones después de la ingestión de cantidades grandes de glúten. Las lesiones cicatrizan dejando hiperpigmentación, y la cicatriz puede ser secundaria a rascado o infección secundaria. Es raro que se afecten las mucosas.
|
| Figura 4 |
| Dermatitis herpetiforme |
La enfermedad es dos veces más común en hombres que en mujeres. Afecta principalmente a personas entre 20 y 50 años. Puede asociarse con atrofia duodenal y yeyunal en parches que recuerda a la enteropatía sensible al gluten de la enfermedad celiaca.26,27 La enteropatía suele ser asintomática y, como la enfermedad celiaca, responde a restricción de gluten. Debido a que la enfermedad celiaca se asocia con linfoma gastrointestinal, existe preocupación de que lo mismo suceda con la DH. Sin embargo, aunque se han reportado linfomas del intestino delgado en la DH,28 la asociación parece ser rara.
DATOS HISTOLOGICOS E INMUNOLOGICOS
Para el diagnóstico se usan dos datos de laboratorio característicos en la DH. Primero, la enfermedad se caracteriza histológicamente por acúmulo de neutrófilos y eosinófilos en microabscesos en las puntas de las papilas dérmicas. En los casos más severos aparece edema, que puede progresar a vesículas subepidérmicas que aparecen justo por debajo de la lámina densa. Segundo, se encuentran depósitos granulares de IgA en la zona de la membrana basal en casi todos los pacientes. Estos suelen asociarse con depósitos granulares de C3 y en ocasiones de IgG e IgM. Cuando está sola, la IgA es uno de los marcadores diagnósticos más sensibles y específicos para la DH. Cuando se encuentra IgA con depósitos de IgG, IgM o C3, se agregan vasculitis por complejos inmunes y lupus eritematoso generalizado al diagnóstico diferencial. Aunque pueden ocurrir depósitos de IgA en la membrana basal en la enfermedad lineal por IgA,29 los depósitos en este padecimiento son lineales más que granulares. No existen anticuerpos circulantes contra componentes normales de la piel en la DH.
TRATAMIENTO
La DH responde en forma rápida y dramática a las sulfonas. La dapsona, en dosis de 100 a 200 mg/día, es el tratamiento que se usa con más frecuencia. Puede usarse sulfpiridina, en dosis de 1 a 3 g/día en dosis divididas (o sulfametoxipiridacina) en los pacientes que no pueden tolerar la dapsona. Las dosis de estos medicamentos se reducen en forma gradual hasta la menor cantidad que pueda suprimir el prurito y el desarrollo de nuevas lesiones. Como está indicado, los pacientes también responden a la dieta sin gluten, sin embargo, estas dietas son difíciles de seguir. A pesar de ello, incluso la reducción parcial en la ingesta de gluten causa menor requerimiento de sulfonas, por lo que siempre debe recomendarse la modificación de la dieta.
Eritema multiforme
El eritema multiforme es un padecimiento agudo, recurrente y autolimitado que afecta todos los grupos de edad y razas. Se caracteriza por erupción súbita de lesiones en racimos, que representan una reacción de hipersensibilidad mediada por células en la piel y las mucosas a diversos factores precipitantes, incluyendo agentes infecciosos y fármacos [ver tabla 3].30
|
||||||||||||||||||||||||||||||||||||||
|
MANIFESTACIONES CLINICAS
Las lesiones pueden ser localizadas o diseminadas y afectar tanto la piel como las mucosas. La erupción suele ser bilateral y simétrica, en las superficies extensoras de las extremidades y en las caras tanto dorsal como palmar de manos y pies. Las lesiones varían desde máculas y pápulas edematosas bien definidas, rojas a púrpura, hasta vesículas o bulas que pueden ulcerarse, formar costras, erosionarse e infectarse. Son características las lesiones en blanco de tiro, que consisten en una pápula o vesícula rodeada de una región de piel normal y un halo de eritema en la periferia [ver figura 5].
|
| Figura 5 |
| Eritema multiforme |
El síndrome de Stevens-Johnson es una forma severa de eritema multiforme que suele ser diseminado, fulminante y multisistémico [ver figura 6]. El síndrome puede acompañarse de fiebre alta, malestar, calosfríos, cefalea, taquicardia, taquipnea y postración. Las mucosas en la boca, ano y vagina contienen máculas redondas u ovales que forman vesículas, bulas y úlceras. Las lesiones oculares consisten en pápulas amarillas-grises bilaterales, que con frecuencia se ulceran e infectan de modo secundario, causando conjuntivitis. La afección ocular puede producir ceguera.
|
| Figura 6 |
| Síndrome de Stevens-Johnson |
Algunos especialistas consideran que la necrolisis epidérmica tóxica (NET) es una forma de eritema multiforme, y suele ser una reacción a medicamentos (v.gr., ciertas sulfonamidas de acción prolongada [en especial trimetoprim-sulfametoxazol], anticonvulsivantes, barbitúricos y antinflamatorios no esteroides). La ausencia de reactantes inmunes dentro de los vasos sanguíneos de la piel y la escasez de inflamación dérmica ha hecho que otros investigadores consideren a la NET como una enfermedad independiente. En ocasiones el síndrome de piel escaldada por estafilococo, que se debe a toxinas producidas por Staphylococcus aureus,31 puede confundirse con la NET [ver tabla 1].
DATOS HISTOLOGICOS E INMUNOLOGICOS
Los datos histológicos cutáneos característicos del eritema multiforme incluyen edema subepidérmico, edema, formación de bulas, necrosis de célula epidérmicas y un infiltrado inflamatorio perivascular profundo. No existen datos inmunofluorescentes específicos, aunque la inmunofluorescencia directa puede mostrar depósitos granulares de C3 y fibrina en la unión dermoepidérmica y depósitos de IgM, C3 y fibrina en los vasos sanguíneos de la dermis. El eritema multiforme recurrente se asocia con frecuencia con infecciones por virus del herpes simple, como se reportó en el 71 por ciento en una serie de 65 pacientes con eritema multiforme. En estos pacientes pueden existir complejos inmunes circulantes contra el virus herpes simple.32
TRATAMIENTO
Las erupciones del eritema multiforme pueden recurrir en forma inesperada a pesar de las medidas preventivas. Por lo tanto, es importante identificar y eliminar las causas subyacentes. Los casos leves se tratan en forma sintomática con esteroides, antinflamatorios, antipruriginosos o preparaciones antibióticas tópicas. El aciclovir oral puede ser eficaz en la profilaxis del eritema multiforme posherpético. En los casos más severos está indicado el tratamiento con prednisona, 40 a 120 mg/día en dosis divididas. Si existe afección ocular debe solicitarse interconsulta inmediata al oftalmólogo.
Epidermolisis bulosa
La epidermolisis bulosa (EB) comprende un grupo de trastornos genéticos con una prevalencia aproximada de 1 por cada 500,000 personas. Existen más de 20 fenotipos diferentes de EB. Esta entidad patológica se caracteriza por la presencia de ampollas y erosiones secundarias a fricciones menores o traumatismos. Las vesículas pueden variar desde formas localizadas (p. ej., ampollas en palmas y plantas de los pies) a formas severas con desprendimiento generalizados de la piel y riesgo de infecciones graves, desequilibrio hidroelectrolítico, anemia y otras complicaciones mortales.
La EB se clasifica principalmente con base en el nivel ultraestructural en el que se desprende la piel dentro de la zona de la membrana basal [ver figura 7]. Los tres subtipos principales incluyen a la EB simple o epidermolítica (intradérmica), la EB de unión (dentro de la lámina lúcida) y la EB distrófica o dermolítica (bajo la lámina densa). El examen con microscopio electrónico localiza la capa en que ocurren las lesiones. Debido a que esta tecnología puede no estar siempre disponible, puede usarse el mapeo inmunofluorescente con anticuerpos para detectar componentes en las capas de la membrana basal, como el antígeno BP (células de la capa basal), la laminina (lámina lúcida) y colágena tipo IV (lámina densa).34 Puede hacerse diagnóstico prenatal por medio de sondas inmunohistoquímicas dirigidas contra componentes antigénicos de la membrana basal en la bipsia de piel del feto, como en el caso de EB de unión con atresia pilórica.35
|
| Figura 7 |
| Epidermolisis bulosa |
EPIDERMOLISIS BULOSA SIMPLE
Existen por lo menos cinco formas de EB simple. El tipo más frecuente es una forma leve, autosómica dominante, que se inicia al nacimiento o poco tiempo después, con ampollas localizadas o generalizadas que generalmente no cicatrizan. Un segundo tipo es la enfermedad de Weber-Cockayne, que puede ser localizada o generalizada. En la forma localizada las ampollas aparecen en las zonas distales de las regiones palmares y plantares y comienzan durante la niñez o la adolescencia . En la forma generalizada, descrita por Koebner, la actividad de la enfermedad suele ser mayor en climas cálidos.
La variante denominada de Dowling-Meara (EB herpetiforme) es la forma menos frecuente de EB simple; se presenta como un proceso grave con ampollas generalizadas durante la infancia, que se parece a la EB de unión y a la variedad distrófica. La gravedad de este padecimiento disminuye con la edad.
EPIDERMOLISIS BULOSA DE UNION
La EB de unión, incluye un grupo de procesos autosómicos recesivos que se caracterizan por una disminución en el número e hipoplasia de los hemidesmosomas, demostrado por microscopía electrónica, y separación a nivel de la lámina lúcida. Con frecuencia se afectan las mucosas y se observan uñas distróficas. La forma más grave, denominada EB letalis, se presenta a los pocos días del nacimiento o durante los primeros meses y tienen una mortalidad muy alta. Estos pacientes tienen una elevada incidencia de paro respiratorio debido a la afección de la laringe y la tráquea. Las formas menos graves de la EB de unión consisten en ampollas severas generalizadas al nacimiento que mejoran gradualmente. Se puede desarrollar estenosis esofágica.
EPIDERMOLISIS BULOSA DISTROFICA
Hay dos formas de EB distrófica de tipo hereditario, que se trasmite de forma autosómica dominante. La EB distrófica hiperplásica (síndrome de Cockayne-Touraine) se presenta en etapas tempranas de la vida con formación de ampollas de contenido serohemático predominantemente en las superficies extensoras de las extremidades inferiores y asociadas con uñas distróficas. El tipo albopapuloide de EB distrófica se caracteriza por la presencia de pápulas blancas que se desarrollan durante la adolescencia en el tronco o las extremidades; sin embargo, las ampollas están presentes en el periodo perinatal. En ambas formas el estudio ultraestructural revela separación por debajo de la membrana basal, con alteración en las fibrillas de anclaje o disminución en su número.
Las formas recesivas de EB distrófica se presentan en el periodo neonatal con ampollas graves de contenido serohemático, localizada en zonas de traumatismos o de forma generalizada. La formación de milia es poco frecuente, pero puede causar cicatrices en las lesiones. Otras complicaciones son: dentición anormal, pérdida o distrofia de las uñas, fusión digital, contracturas en flexión y estenosis esofágica [ver figura 8]. Ocurre también retraso en el crecimiento, desnutrición y anemia crónica. Estos pacientes tienen un riesgo elevado de carcinoma epidermoide, con una incidencia elevada de metástasis y muerte.
|
| Figura 8 |
| Epidermolisis bulosa distrófica recesiva |
Se puede hacer el diagnóstico de esta entidad patológica antes del nacimiento mediante fetoscopía con toma de biopsias de la piel. Desde el punto de vista ultraestructural, el tejido muestra formación de ampollas dermolíticas. Una alternativa cuando se sospecha EB distrófica recesiva son las pruebas bioquímicas para detectar aumento de la expresión de colagenasa en los fibroblastos fetales.36
El tratamiento de apoyo está dirigido a curar las heridas y prevenir las complicaciones. Los cuidados diarios de la piel incluyen la aplicación de compresas húmedas, ungüentos con antibióticos y compresas no adherentes como mallas de gasa fina (N-terface). Es esencial un método multidisciplinario que incluya el consejo genético, psiquiátrico o psicológico y sistemas de apoyo para el paciente y su familia, en especial para las formas graves de la enfermedad.
La Dystrophic Epidermolysis Bulosa Research Association of America (http://www.debra.org) ha establecido un registro nacional para obtener datos epidemiológicos y determinar los aspectos económicos y sociales de la EB, así como llevar un registro de los pacientes que quieran participar en diversos protocolos de investigación.53 Para una mayor información puede comunicarse con DEBRA, en la ciudad de Nueva York, en el teléfono 995-2220 (212).
EPIDERMOLISIS BULOSA ADQUIRIDA
La aparición de ampollas en adultos en quienes no existe una base genética causante de epidermólisis bulosa se denomina epidermólisis bulosa adquirida o epidermólisis bulosa acquisita (EBA). Pueden demostrarse por inmunohistología anticuerpos IgG contra la membrana basal, tanto circulantes como fijos a los tejidos. Las ampollas se desarrollan por debajo de la epidermis y curan dejando como secuela cicatrices atróficas y malformaciones. Dichas ampollas están casi siempre limitadas a las extremidades, en áreas expuestas a traumatismos mecánicos. La EBA puede estar asociada con lesiones orales y distrofia ungueal. Pueden estar asociadas alteraciones subyacentes de origen maligno, autoinmune e inflamatorio. La presencia de colitis ulcerosa y enfermedad de Crohn en cerca del 30 por ciento de los casos sugiere que la EBA debería ser incluida entre las manifestaciones extraintestinales de la enfermedad inflamatoria del intestino.37
El diagnóstico se hace excluyendo otras enfermedades bulosas, en especial PB (ver antes). La microscopía inmunoelectrónica puede ser recurso adicional para establecer el diagnóstico, aunque la técnica no siempre está disponible.55 La inmunofluorescencia directa con técnicas para separar la lámina lúcida ayuda en el diagnóstico diferencial. Con este método se observan los anticuerpos de IgG en el lado dérmico en la EBA y en la epidermis en el penfigoide.38 Se ha identificado que el antígeno en la EBA es el extremo globular carboxilo de la procolágena tipo VII.39
Diagnóstico diferencial de las enfermedades vesiculobulosas
Se han analizado las principales formas de padecimientos bulosos de origen autoinmune o hereditario. El diagnóstico diferencial incluye diversas otras condiciones en las que las vesículas o bulas son menos comunes o aparecen en forma secundaria a otros procesos patológicos. Una erupción fija a drogas puede causar bulas localizadas que aparezcan después de la ingestión de algún medicamento en particular . La dermatitis eccematosa produce vesículas espongióticas por edema intercelular. Este se manifiesta clínicamente por grandes bulas en la dermatitis alérgica por contacto aguda debida a hiedra o roble venenosos. En ocasiones el lupus eritematoso generalizado produce bulas al causar degeneración de las células basales.
Una erupción bulosa en el dorso de las manos y otros sitios expuestos al sol en pacientes sometidos a hemodiálisis por tiempo prolongado puede parecerse a la porfiria cutánea tarda.40 Los niveles de porfirina suelen estar en límites normales. Las bulas intraepidérmicas o subepidérmicas, principalmente en las extremidades, pueden ser un signo cutáneo de diabetes mellitus.41 Las infeciones bacterianas de la piel, como el impétigo, pueden asociarse con formación subcorneal de bulas.
También deben incluirse en el diagnóstico diferencial varias infecciones virales, incluyendo varicela, herpes simple y herpes zoster .
Bibliografìa
- Baxter DL: Vesiculobullous diseases: introduction. Clinical Dermatology, Vol 2, Subsection 6-1. Demis DJ, Dobson RL, McGuire J, Eds. Harper & Row, Pubs, Inc, Hagerstown, Maryland, 1980
- Korman NJ, Eyre RW, Klaus-Kovtun V, et al: Demonstration of an adhering-junction molecule (plakoglobin) in the autoantigens of pemphigus foliaceus and pemphigus vulgaris. N Engl J Med 321:631, 1989 [PMID 2770792]
- Anhalt GJ, Labib RS, Voorhees JJ, et al: Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med 306:1190, 1982
- Hu C-H, Michel B, Schiltz JR: Epidermal acantholysis induced in vitro by pemphigus autoantibody: an ultrastructural study. Am J Pathol 90:345, 1978
- Wasserstrum N, Laros RK Jr: Transplacental transmission of pemphigus. JAMA 249:1480, 1983
- Jablonska S, Chorzelski TP, Beutner EH, et al: Utilization of immunofluorescence in the diagnosis of bullous diseases, lupus erythematosus and certain other dermatoses. Int J Dermatol 14:83, 1975
- Yung CW, Hambrick GW: D-Penicillamine-induced pemphigus syndrome. J Am Acad Dermatol 6:317, 1982
- Stanley JR, Yaar M, Hawley-Nelson P, et al: Pemphigus antibodies identify a cell surface glycoprotein synthesized by human and mouse keratinocytes. J Clin Invest 70:281, 1982
- Lever WF, Schaumburg-Lever G: Treatment of pemphigus vulgaris: results obtained in 84 patients between 1961 and 1982. Arch Dermatol 120:44, 1984 [PMID 6691714]
- Jordon RE: Pemphigus. Dermatology in General Medicine, 2nd ed. Fitzpatrick TB, Eisen AZ, Wolff K, et al, Eds. McGraw-Hill Book Co, New York, 1979, p 310
- Bystryn J-C: Adjuvant therapy of pemphigus. Arch Dermatol 120:941, 1984
- Lever WF: Methotrexate and prednisone in pemphigus vulgaris. Arch Dermatol 106:491, 1972
- Burton JL, Greaves MW: Azathioprine for pemphigus and pemphi-goid-a 4 year follow-up.Br J Dermatol 91:103, 1974 [PMID 4850720]
- Aberer W, Wolff-Schreiner EC, Stingl G, et al: Azathioprine in the treatment of pemphigus vulgaris: a long-term follow-up. J Am Acad Dermatol 16:527, 1987 [PMID 3819096]
- Thomas I: Gold therapy and its indications in dermatology: a review. J Am Acad Dermatol 16:845, 1987
- Penneys NS: Gold therapy: dermatologic uses and toxicities. J Am Acad Dermatol 1:315, 1979
- Barthelemy H, Frappaz A, Cambazard F, et al: Treatment of nine cases of pemphigus vulgaris with cyclosporine. J Am Acad Dermatol 18:1262, 1988 [PMID 3385040]
- Tan-Lim R, Bystryn J-C: Effect of plasmapheresis therapy on circulating levels of pemphigus antibodies. J Am Acad Dermatol 22:35, 1990 [PMID 2298963]
- Rook AH, Jegosothy BV, Heald P, et al: Extracorporeal photochemotherapy for drug-resistant pemphigus vulgaris. Ann Intern Med 112:303, 1990 [PMID 2297209]
- Pearson RW, O'Donoghue M, Kaplan SJ: Pemphigus vegetans: its relationship to eosinophilic spongiosis and favorable response to dapsone. Arch Dermatol 116:65, 1980 [PMID 7352766]
- Provost TT: Pemphigus. N Engl J Med 306:1224, 1982
- Diaz LA, Sampaio SA, Rivitti EA, et al: Endemic pemphigus foliaceus (fogo selvagem): I. Clinical features and immunopathology. J Am Acad Dermatol 20:647, 1989
- Rock B, Martins CR, Theofilopoulos AN, et al: The pathogenic effect of IgG4 autoantibodies in endemic pemphigus foliaceus (fogo selvagem). N Engl J Med 320:1463, 1989 [PMID 2654636]
- Anhalt GJ, Kim SC, Stanley JR, et al: Paraneoplastic pemphigus: an autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med 323:1729, 1990 [PMID 2247105]
- Fullerton SH, Woodley DT, Smoller BR, et al: Paraneoplastic pemphigus with autoantibody deposition in bronchial epithelium after autologous bone marrow transplantation. JAMA 267:1500, 1992 [PMID 1538540]
- Oursler JR, Ariss-Abdo L, Labib RS, et al: Autoantibodies against desmoplakins: a component of the immune response against epithelia and tumor in paraneoplastic pemphigus. Clin Res 39:195A, 1991
- Stanley JR, Woodley DT, Katz SI: Identification and partial characterization of pemphigoid antigen extracted from normal human skin. J Invest Dermatol 82:108, 1984 [PMID 6361166]
- Mueller S, Klaus-Kovtun V, Stanley JR: A 230-kd basic protein is the major bullous pemphigoid antigen. J Invest Dermatol 92:33, 1989 [PMID 2462597]
- Thornfeldt CR, Menkes AW: Bullous pemphigoid controlled by tetracycline. J Am Acad Dermatol 16:305, 1987
- Korman N: Bullous pemphigoid. J Am Acad Dermatol 16:907, 1987
- Leenutaphong V, von Kries R, Plewig G: Localized cicatricial pemphigoid (Brunsting-Perry): electron microscopic study. J Am Acad Dermatol 21:1089, 1989 [PMID 2681294]
- Lawley TJ, Hall RP, Fauci AS, et al: Defective Fc-receptor functions associated with the HLA-B8/DRw3 haplotype: studies in patients with dermatitis herpetiformis and normal subjects. N Engl J Med 304:185, 1981 [PMID 6969363]
- Brow JR, Parker F, Weinstein WM, et al: The small intestinal mucosa in dermatitis herpetiformis: I. Severity and distribution of the small intestinal lesion and associated malabsorption. Gastroenterology 60:355, 1971
- Katz SI, Hall RP, Lawley TJ, et al: Dermatitis herpetiformis: the skin and the gut. Ann Intern Med 93:857, 1980 [PMID 7447195]
- Jenkins D, Lynde CW, Stewart WD: Histiocytic lymphoma occurring in a patient with dermatitis herpetiformis. J Am Acad Dermatol 9:252, 1983 [PMID 6350384]
- Prost C, De Leca AC, Combemale P, et al: Diagnosis of adult linear IgA dermatosis by immunoelectronmicroscopy in 16 patients with linear IgA deposits. J Invest Dermatol 92:39, 1989
- Lang PG Jr: Sulfones and sulfonamides in dermatology today. J Am Acad Dermatol 1:479, 1979
- Lamkin BC: Dermatitis herpetiformis: its relationship to gastrointestinal disease. Cutis 28:302, 1981
- Tonnesen MG, Soter NA: Erythema multiforme. J Am Acad Dermatol 1:357, 1979 [PMID 512083]
- Howland WW, Golitz LE, Weston WL, et al: Erythema multiforme: clinical, histopathologic, and immunologic study. J Am Acad Dermatol 10:438, 1984 [PMID 6725656]
- Kazmierowski JA, Peizner DS, Wuepper KD: Herpes simplex antigen in immune complexes of patients with erythema multiforme: presence following recurrent herpes simplex infection. JAMA 247:2547, 1982 [PMID 6279897]
- Orton PW, Huff JC, Tonnesen MG, et al: Detection of a herpes simplex viral antigen in skin lesions of erythema multiforme. Ann Intern Med 101:48, 1984 [PMID 6329053]
- Green JA, Spruance SL, Wenerstrom G: Post-herpetic erythema multiforme prevented with prophylactic oral acyclovir. Ann Intern Med 102:632, 1985 [PMID 3985514]
- Orfanos CE: Oral retinoids-present status. Br J Dermatol 103:473, 1980
- Pearson RW: Clinicopathologic types of epidermolysis bullosa and their nondermatological complications. Arch Dermatol 124:718, 1988
- Tabas M, Gibbons S, Bauer EA: The mechanobullous diseases. Dermatol Clin 5:123, 1987 [PMID 3549073]
- Fine J-D: Epidermolysis bullosa: clinical aspects, pathology, and recent advances in research. Int J Dermatol 25:143, 1986
- Hintner H, Stingl G, Schuler G, et al: Immunofluorescence mapping of antigenic determinants within the dermal-epidermal junction in the mechanobullous diseases. J Invest Dermatol 76:113, 1981
- Savolainen E-R, Kero M, Pihlajaniemi T, et al: Deficiency of galactosylhydroxylysyl glucosyltransferase, an enzyme of collagen synthesis, in a family with dominant epidermolysis bullosa simplex. N Engl J Med 304:197, 1981
- Bauer EA, Ludman MD, Goldberg JD, et al: Antenatal diagnosis of recessive dystrophic epidermolysis bullosa: collagenase expression in cultured fibroblasts as a biochemical marker. J Invest Dermatol 87:597, 1986 [PMID 3021861]
- Cooper TW, Bauer EA: Therapeutic efficacy of phenytoin in recessive dystrophic epidermolysis. Arch Dermatol 120:490, 1984 [PMID 6703753
- Haber RM, Hanna W, Ramsay CA, et al: Hereditary epidermolysis bullosa. J Am Acad Dermatol 13:252, 1985
- Carter DM: Announcement of the National Epidermolysis Bullosa Registry (letter). J Am Acad Dermatol 16:1058, 1987
- Raab B, Fretzin DF, Bronson DM, et al: Epidermolysis bullosa acquisita and inflammatory bowel disease. JAMA 250:1746, 1983 [PMID 6887488]
- Nieboer C, Boorsma DM, Woerdeman MJ, et al: Epidermolysis bullosa acquisita: immunofluorescence, electron microscopic and immunoelectron microscopic studies in four patients. Br J Dermatol 102:383, 1980 [PMID 6992836]
- Woodley DT, Briggaman RA, O'Keefe EJ, et al: Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med 310:1007, 1984 [PMID 6369131]
- Goldsman CI, Taylor JS: Porphyria cutanea tarda and bullous dermatoses associated with chronic renal failure: a review. Cleve Clin Q 50:151, 1983 [PMID 6357544]
- Bernstein JE, Medenica M, Soltani K, et al: Bullous eruption of diabetes mellitus. Arch Dermatol 115:324, 1979 [PMID 373635]