Infectología
⭳ Abrir artículo (PDF)805.7 KBEste artículo es idéntico en la Edición 2/2000.
Contenido del artículo
XXXIV INFECCIONES POR PROTOZOARIOS
- Toxoplasmosis
- SINDROMES CLINICOS
- Toxoplasmosis primaria
- Toxoplasmosis en el huésped inmunosuprimido
- Toxoplasmosis en pacientes con SIDA
- Toxoplasmosis congénita
- Toxoplasmosis ocular
- DIAGNOSTICO
- TRATAMIENTO
- Paludismo
- Babesiosis
- Infecciones por protozoarios intestinales
- GIARDIASIS
- AMIBIASIS
- COCCIDIOSIS
- INFECCION POR DIENTAMOEBA FRAGILIS
- INFECCION POR BLASTOCYSTIS HOMINIS
- MICROSPORIDIOSIS
- Infecciones por amibas de vida libre
- Leishmaniasis
- Tripanosomiasis
- TRIPANOSOMIASIS AFRICANA
XXXIV INFECCIONES POR PROTOZOARIOS
DR. PETER F. WELLER
Toxoplasmosis
Toxoplasma gondii, el agente casual de la toxoplasmosis, es un parásito intracelular obligado de distribución mundial. Infecta prácticamente a todos los mamíferos y aves, lo que le hace uno de los parásitos de mayor distribución en el mundo.1 En el ser humano la infección puede adquirirse por vía oral, transmisión transplacentaria, transfusiones sanguíneas o trasplantes de órganos. La infección por vía oral se debe a la ingestión de quistes de T. gonddi en alimentos mal cocinados o de los ooquistes del parásito contenidos en las heces de gato.1 En un brote pudo atribuirse la fuente a la ingestión de agua contaminada. Después de la ingestión los bradizoítos (de los quistes tisulares) o los esporozoítos (de los ooquistes) invaden las células circundantes y se convierten en taquizoítos, una forma de división rápida que puede diseminarse e invadir cualquier tipo de célula nucleada. Con el tiempo y el desarrollo de inmunidad, el parásito forma quistes tisulares que contienen muchos bradizoítos. Los quistes tisulares pueden permanecer viables por décadas sin causar enfermedad. Sin embargo, la pérdida de la inmunidad permite la reactivación de los quistes latentes y la generación de muchos taquizoítos invasores.
La toxoplasmosis es una infección muy frecuente, pero la mayoría de los casos, tanto congénitos como adquiridos, no dan manifestaciones clínicas y sólo se detectan por la presencia de anticuerpos. La prevalencia de la infección varía según los diferentes grupos étnicos y áreas geográficas; en la mayoría de los estudios se ha encontrado un aumento de la incidencia paralelo a la edad. En los Estados Unidos los estudios serológicos sugieren que el 3 a 30 por ciento de la población ha sufrido infección por Toxoplasma.
SINDROMES CLINICOS
La toxoplasmosis puede dividirse en cinco grandes síndromes clínicos: toxoplasmosis primaria, toxoplasmosis en el inmunosuprimido, toxoplasmosis del SIDA, toxoplasmosis congénita y toxoplasmosis ocular.
Toxoplasmosis primaria
La persona inmunocompetente suele presentar una afección subclínica al adquirir la infección primaria por Toxoplasma. La linfadenopatía no dolorosa es la manifestación más frecuente, puede ser localizada o generalizada y persistir por muchos meses. La linfadenopatía cervical aislada es el hallazgo más frecuente.2 La linfadenopatía puede ser el único signo de la infección y con menos frecuencia ocurren fiebre, malestar general, mialgias y ardor faríngeo; la fatiga y la debilidad pueden ser intensas. En la exploración física, además de la linfadenopatía, puede encontrarse faringitis, exantema maculopapular3 y, en un pequeño grupo de pacientes, hepatoesplenomegalia. No es raro encontrar linfocitos atípicos. El diagnóstico diferencial se hace con la mononucleosis infecciosa, la infección por citomegalovirus y el linfoma; en casos raros, la sarcoidosis, la enfermedad por rasguño de gato y otros procesos infecciosos pueden parecerse a la toxoplasmosis. Las pruebas serológicas dan la clave del diagnóstico.4
Aunque los síntomas de la linfadenopatía por Toxoplasma pueden persistir durante semanas o meses, casi siempre se autolimitan y no requieren tratamiento específico. En casos graves o persistentes, la pirimetamina y las sulfamidas son útiles. Estos mismos medicamentos pueden utilizarse en las complicaciones raras de la toxoplasmosis en el huésped normal, como la coriorretinitis, miocarditis, pericarditis, neumonitis, miositis y meningoencefalitis.
Toxoplasmosis en el huésped inmunosuprimido
En el huésped inmunosuprimido con inmunidad celular deficiente T. gondii puede causar enfermedad neurológica o diseminada grave. Por lo general el proceso se desarrolla a partir de una reactivación de una infección latente y no por una infección primaria. Los receptores seronegativos de órganos (en especial corazón), trasplantados de donadores seropositivos tienen especial riesgo,5,6 lo mismo que los pacientes con enfermedad de Hodgkin, leucemia de células peludas y otros trastornos malignos.7 En por lo menos el 50 por ciento de estos pacientes predominan las alteraciones neurológicas. El cuadro clínico es muy variable y puede semejar una encefalitis difusa, una meningoencefalitis o una lesión cerebral ocupativa.8 El líquido cefalorraquídeo muestra pleocitosis discreta de tipo linfocítico, con elevación de los niveles de proteínas y niveles normales de glucosa. Aparece neumonitis hasta en una tercera parte de los individuos inmunosuprimidas con infección por Toxoplasma.9 Hay fiebre y disnea, pero la tos está ausente y no se produce expectoración. La radiografía de tórax revela, en forma característica, infiltrados pulmonares bilaterales difusos. En algunos casos, el diagnóstico se establece al observar microorganismos en la muestra obtenida por lavado broncoalveolar. Otras manifestaciones de la toxoplasmosis diseminada en pacientes inmunosuprimidos incluyen miocarditis, pericarditis, peritonitis y linfadenitis. Muchos de estos individuos inmunosuprimidas presentan infecciones intercurrentes con otros patógenos oportunistas, sobre todo por herpesvirus y citomegalovirus.
Si la serología demuestra elevación de los títulos de anticuerpos, puede ser de ayuda diagnóstica, pero en estos enfermos los títulos son generalmente bajos y para establecer el diagnóstico se requiere una biopsia cerebral, pulmonar o de ganglio linfático. Se justifica el método diagnóstico agresivo, ya que el tratamiento con sulfonamidas y pirimetamina puede dar buenos resultados y la infección suele ser mortal si se deja sin tratamiento.
Toxoplasmosis en pacientes con SIDA
La toxoplasmosis es un problema muy grave en pacientes con síndrome de inmunodeficiencia adquirida (SIDA); del 3 al 40 por ciento de estos pacientes desarrollan una toxoplasmosis clínicamente aparente.10 Aunque los pacientes con SIDA pueden manifestar cualquiera de las variedades generalizadas de toxoplasmosis,11 la afectación del sistema nervioso central es la más frecuente. La mayoría de los casos de toxoplasmosis en pacientes con SIDA se deben a la reactivación de quistes latentes adquiridos antes de la infección por VIH. La reactivación es más probable cuando las cuentas de células T CD4+ caen a menos de 100 células /mm3. 10 Todas las personas infectadas por VIH que sean negativos a Toxoplasma requieren minimizar la exposición cocinando la carne a una temperatura interna de 150 'F, lavando sus manos después de tocar carne cruda o tierra, lavando frutas y verduras antes de comerlas y evitando el contacto con heces de gato o basura contaminada (o usando guantes al manejar estos materiales). 12
En pacientes con SIDA la toxoplasmosis se presenta con más frecuencia como una encefalitis necrosante. Los síntomas incluyen alteraciones focales (v. gr., hemiparesia, pérdida sensorial, alteraciones visuales, temblor, parálisis de pares craneales y crisis focales) y alteraciones neurológicas generalizadas (v. gr., cefalea, cambios de la personalidad, confusión, estupor o coma y convulsiones). Aunque la pleocitosis linfocítica del LCR apoya el diagnóstico de toxoplasmosis cerebral, la tomografia computada o la imagen por resonancia magnética es fundamental para el diagnóstico en la mayoría de los casos. Las lesiones nodulares únicas o múltiples son hallazgos típicos de la TC; cuando se administra un medio de contraste, más del 90 por ciento de estas lesiones nodulares aumentan de tamaño y tienen forma anular. En ocasiones los estudios del IRM revelan lesiones que no se pueden visualizar con la tomografía computada. 13 La tomografía de emisión de positrones (TEP) puede ser útil para distinguir las lesiones de la toxoplasmosis (que son hipometabólicas) de los linfomas (que son hipermetabólicos).14
Las pruebas de anticuerpos en suero son útiles para la detección de la toxoplasmosis cerebral en pacientes con SIDA; estas pruebas suelen ser positivas porque la mayoría de los casos de toxoplasmosis cerebral en estos pacientes surgen por la reactivación de una infección latente. Aunque las pruebas negativas sugieren un diagnóstico diferente, se han reportado muy pocos casos seronegativos. Sin embargo, las pruebas serológicas no siempre son definitivas para el diagnóstico de toxoplasmosis activa en pacientes con SIDA y los títulos de anticuerpos no alcanzan los altos niveles típicos de los pacientes inmunocompetentes con toxoplasmosis, ni tampoco existen anticuerpos IgM en los pacientes con SIDA.10 Se encuentran anticuerpos contra el Toxoplasma en el LCR en casi dos terceras partes de los pacientes con SIDA y toxoplasmosis cerebral, y su detección puede ayudar en el diagnóstico [ver adelante, diagnóstico]. 15
Otras causas de afectación cerebral en pacientes con SIDA, además de la toxoplasmosis, incluyen hongos, micobacterias, linfomas, sarcoma de Kaposi, tumores metastásicos, leucoencefalopatía multifocal y el VIH por sí mismo. A pesar del amplio diagnóstico diferencial de las lesiones cerebrales en los pacientes con SIDA, es preferible administrar tratamiento empírico para toxoplasmosis, a realizar una biopsia, si el cuadro clínico y radiológico es compatible con el diagnóstico y si los anticuerpos anti-toxoplasma séricos son positivos.16 Debido a que la toxoplasmosis es la causa más frecuente y tratable de lesiones cerebrales en los pacientes con SIDA, puede iniciarse un tratamiento empírico basado en la combinación de sulfonamidas y pirimetamina o clindamicina y pirimetamina. Si el diagnóstico es correcto se observará mejoría clínica y radiológica después de 1 a 2 semanas. Si los pacientes tienen poca respuesta al tratamiento, son seronegativos y si pertenecen a grupos con alto riesgo de tuberculosis (haitianos, africanos, adictos a drogas por vía intravenosa) se debe considerar seriamente realizar una biopsia.16
Toxoplasmosis congénita
La toxoplasmosis congénita aparece casi exclusivamente cuando la madre desarrolla una infección primaria durante el embarazo. La infección congénita casi nunca se desarrolla a partir de una toxoplasmosis latente adquirida antes del embarazo, y los pocos casos detectados ocurrieron en mujeres embarazadas con reactivación de la toxoplasmosis por tratamiento inmunosupresor o infección por VIH.17El riesgo de infección fetal depende de la época en que ocurra la infección materna, aumentando de 10 por ciento durante el primer trimestre a 60 por ciento durante el tercero.18 Las consecuencias de la infección fetal dependen también de cuando ocurra la infección: las infecciones adquiridas al principio de la gestación causan daño más severo.
El espectro clínico de la toxoplasmosis congénita varía mucho. Alrededor del 85 por ciento de los bebés infectados parecen normales al nacimiento; sin embargo, sin tratamiento el 85 por ciento desarrollará coriorretinitis, pérdida de la audición o retraso en el desarrollo.18,19 El espectro clínico de la toxoplasmosis congénita sintomática incluye muerte fetal, daño neurológico (calcificaciones cerebrales, convulsiones, retraso mental, hidrocefalia y microcefalia), coriorretinitis, fiebre, hepatoesplenomegalia y exantema. El diagnóstico diferencial se hace con otras infecciones congénitas por citomegalovirus, herpes simple, rubéola o sífilis. La toxoplasmosis congénita puede diagnosticarse por métodos serológicos por una reacción en cadena de la polimerasa (RCP) realizada en líquido amniótico,20 o al identificar al parásito en el tejido placentario o fetal. Los niños que llegan a sobrevivir pueden beneficiarse con el tratamiento a base de pirimetamina y sulfonamidas.21
La prevención de la toxoplasmosis congénita es de gran importancia. Las mujeres embarazadas deben evitar al máximo el contacto con gatos, en especial los gatos callejeros y los que se alimentan de carne cruda; es muy importante lavarse las manos después de tener contacto con gatos, y si se tienen en casa, otra persona debe encargarse de su aseo; también es recomendable que se laven todas las frutas y vegetales y se cocine adecuadamente la carne. Se recomienda la investigación serológica sistemática de todas las mujeres embarazadas.
Toxoplasmosis ocular
La toxoplasmosis puede causar el 30 por ciento de los casos de retinocoroiditis.22 Muchos casos se deben a reactivación de una infección congénita, por ello la toxoplasmosis ocular es más frecuente en niños mayores y adultos jóvenes. La retinocoroiditis también puede desarrollarse como manifestación de la infección primaria.23 Los síntomas más comunes son atribuibles a la disminución de la agudeza visual, pero en inflamaciones graves también se observa fotofobia y dolor ocular. Las lesiones típicas semejan racimos de exudados blandos de color blanco-amarillento que se observan en el polo posterior del ojo. Aunque el diagnóstico de esta variedad requiere demostración de anticuerpos contra toxoplasma, los títulos son por general bajos, puesto que la infección se adquirió varios años antes. Por lo tanto, en la mayoría de los casos la toxoplasmosis ocular puede diagnosticarse clínicamente reconociendo la morfología de los exudados. El diagnóstico diferencial incluye a la tuberculosis, la sarcoidosis, la sífilis, la histoplasmosis y la candidiasis.
DIAGNOSTICO
Las pruebas serológicas son la piedra angular en el diagnóstico de la toxoplasmosis. Aunque se dispone de muchas, las más utilizadas son la inmunofluorescencia indirecta y la prueba de Sabin y Felman. Los laboratorios de referencia emplean muchas otras pruebas. Todas ellas miden anticuerpos de la clase IgG y se vuelven positivas de 1 a 3 semanas después del comienzo de la infección. El diagnóstico de toxoplasmosis de adquisición reciente, especialmente importante durante el embarazo, y de la toxoplasmosis congénita en el recién nacido, requieren de la detección de anticuerpos anti-toxoplasma de IgM. Algunos estudios comerciales de IgM dan resultados falsos positivos y se asocian con detección de anticuerpos que persisten por más de 1 año, lo que pone en duda la confiabilidad de estas pruebas para detectar solo infecciones recientes.24 Los resultados positivos en las pruebas de IgM requerirán confirmación por pruebas alternativas.
Debido a que los quistes pueden persistir en los tejidos por años, la evidencia definitiva de toxoplasmosis activa a partir de la biopsia de un tejido requiere de la detección de taquizoitos. Estas formas no se observan con facilidad con las técnicas convencionales de patología y se requiere la aplicación de inmunofluorescencia o tinción de anticuerpos peroxidasa-antiperoxidasa. Los taquizoitos no suelen detectarse en la biopsia de ganglios linfáticos, aunque las características histopatológicas del ganglio pueden apoyar el diagnóstico.25 Los métodos de RCP son muy sensibles para detectar el organismo en el líquido amniótico en las infecciones congénitas, 20 pero ni la RCP ni el cultivo directo de los organismos a partir de la sangre o los tejidos se ha usado en el diagnóstico de otras formas de toxoplasmosis.
TRATAMIENTO
Toxoplasmosis primaria y asociada a SIDA
Aunque la mayoría de los pacientes con linfadenopatía por toxoplasma no requieren tratamiento, éste debe considerarse en los casos de enfermedad grave o prolongada. Los pacientes con coriorrenitis activa, afección del sistema nervioso central o toxoplasmosis diseminada sí deben recibir quimioterapia, así como todo paciente inmunosuprimido, incluyendo los pacientes con SIDA.
El tratamiento de elección consiste en una combinación de sulfonamidas (sulfadiazina o trisulfapirimidinas) y pirimetamina. 26 Deben evitarse otras sulfonamidas con pobre acción sobre T. gondii. En los pacientes adultos se administran 200 mg de pirimetamina el primer día, seguido de la dosis habitual de 50 a 75 mg/día. La sulfadiazina o trisulfapirimidinas se administran con una dosis de impregnación de 4 g, para continuar con dosis de mantenimiento de 1 a 2 gramos cuatro veces al día. El efecto tóxico más frecuente de la pirimetamina es la supresión de la médula ósea, por lo que debe vigilarse la cuenta de leucocitos, eritrocitos y plaquetas dos veces a la semana y administrar diario 10 a 15 mg de ácido folínico. El tratamiento combinado de pirimetamina y sulfonamida se administra por 3 a 6 semanas.
Para los pacientes con SIDA que no toleran las sulfonamidas y tienen toxoplasmosis del SNC ha sido eficaz la clindamicina, en dosis de 600 mg por vía oral o intravenosa cuatro veces al día, combinada con pirimetamina.27 También el atovaquone más pirimetamina es eficaz para los pacientes intolerantes a las sulfas.28
Los pacientes con SIDA tratados por toxoplasmosis y los que no sufren infección activa por toxoplasmosis pero que tienen serología positiva y cuentas de linfocitos T CD4+ menores de 100/mm3 tienen riesgo de reactivación y requieren de tratamiento supresor por tiempo prolongado. Existen diferentes esquemas para los pacientes con SIDA que se han recuperado de una toxoplasmosis. El primero consiste en sulfadiacina (500 a 1,000 mg cuatro veces al día) y pirimetamina (25 a 50 mg/día) más ácido folínico (5 mg/día).29 También la clindamicina (300 a 450 mg tres veces al día) es eficaz para los pacientes que no toleran las sulfonamidas.30 Algunos esquemas empleados para la prevención de la neumonía por Pneumocystis proporcionan profilaxis primaria para la toxoplasmosis, como el trimetoprim-sulfametoxazol, la dapsona-pirimetamina, el atovaquone y la pirimetamina sola para pacientes con intolerancia a las sulfas.31
Toxoplasmosis congénita
La toxoplasmosis de adquisición reciente durante el embarazo representa una dificultad terapéutica. La pirimetamina es teratogénica y debe evitarse durante el primer trimestre. La espiromicina, disponible en los Estados Unidos solo directamente a partir del fabricante (Rhone Poulenc Rorer), se administra en dosis de 3 a 4 g/día y puede disminuir el riesgo de infección transplacentaria.26 Si se demuestra infección in utero debe iniciarse tratamiento con pirimetamina y sulfadiacina. Puede considerarse un aborto terapéutico si la infección se adquirió en una etapa temprana del embarazo y usarse ultrasonido para detectar hidrocefalia, calcificaciones intracraneales y otros signos de daño fetal. Los niños que nacen con evidencia serológica o clínica de toxoplasmosis congénita deben tratarse durante un año con pirimetamina y sulfadiacina. 29
Toxoplasmosis ocular
La pirimetamina y las sulfonamidas son la base del tratamiento de la toxoplasmosis ocular, pero los resultados son impredecibles y las recaídas frecuentes. En los casos en los que existe riesgo de ceguera el paciente debe recibir esteroides en combinación con los antibióticos.
Paludismo
Cuatro especies de Plasmodium causan paludismo en el humano: P. falciparum, P. vivax, P. ovale y P. malarie [ver tabla 1]. P. vivax y P. falciparum son de los más prevalentes en el mundo y el paludismo falciparum es responsable de la mayoría de las muertes. El paludismo se trasmite por los mosquitos anófeles. Aunque la trasmisión del paludismo en los Estados Unidos es muy rara, se calcula que cada año ocurren en todo el mundo alrededor de 300 a 500 millones de casos, con tres millones de muertes.32,33 El paludismo ha reaparecido por múltiples factores, incluyendo resistencia del vector mosco a los insecticidas, incapacidad de los programas gubernamentales para controlar las poblaciones de moscos vectores, mayores oportunidades para el crecimiento de los vectores y mayor resistencia de los parásitos a los agentes quimioterápicos.
ETIOLOGIA Y EPIDEMIOLOGIA
El paludismo es endémico en muchos países. La mayoría del paludismo en los Estados Unidos es importado. En 1995 se reportaron a los Centros para el Control y Prevención de las Enfermedades (CDC) de los EUA 1,167 casos de paludismo, incluyendo seis muertes. De estos casos, 599 ocurrieron en civiles norteamericanos, 461 en civiles extranjeros y 12 en personal militar.32 P. vivax y P. falciparum causaron el 48 y 39 por ciento de las personas infectadas, respectivamente. Con menos frecuencia el paludismo se trasmite por transfusión sanguínea, al compartir agujas contaminadas, por trasplante de órganos y en infantes que son infectados en forma congénita al nacer de madres infectadas. 32,34 Además, debido a que en los Estados Unidos existen especies de mosquitos Anopheles que pueden trasmitir el paludismo, se han aislado casos de paludismo autóctono en norteamericanos picados por mosquitos que se alimentaron de personas con paludismo importado. 35 Un último medio de introducción del paludismo a los Estados Unidos es el llamado de aeropuerto, que surge cuando un mosco infectado entra al país en una aeronave de un área con paludismo y trasmite la infección en el área alrededor del aeropuerto.
PATOGENIA
Las infecciones se adquieren normalmente por la picadura de un mosco Anopheles hembra infectado, que introduce esporozoítos [ver figura 1]. En la fase preritrocítica de la infección las cuatro especies de Plasmodium infectan a los hepatocitos. Sin embargo, es notable que solo dos especies, P. vivax y P. ovale, causan infección persistente en el hígado. En la fase eritrocítica de la infección los merozoítos liberados por los hepatocitos infectados y después por los eritrocitos infectados interactúan con proteínas de membrana específicas de los eritrocitos y los invaden. (El antígeno sanguíneo Duffy es el receptor eritrocítico necesario para P. vivax. Los individuos que genéticamente no tienen antígeno sanguíneo Duffy, como muchos africanos occidentales y sus descendientes, son resistentes a esa especie de Plasmodium.) Dentro de las células infectadas los parásitos sufren un proceso de esquizogonia para formar nuevos merozoítos, que son liberados para reinfectar otros eritrocitos. Algunos pocos parásitos se diferencian en sus fases sexuales (gametocitos), capaces de infectar mosquitos. Los gametocitos masculinos y femeninos se transforman y reproducen en el intestino medio del mosco, causando la producción de nuevos esporozoítos que se localizan en las glándulas salivales de los mosquitos.
Debido a la capacidad de los parásitos para replicarse y aumentar su número dentro del humano infectado, pueden desarrollarse infecciones intensas a partir de un inóculo mínimo. Algunos viajeros han desarrollado paludismo después de ser picados por mosquitos durante una breve estancia en un aeropuerto de un área con paludismo y durante vuelos en aeronaves que se han detenido en zonas endémicas. En consecuencia, se justifica la profilaxis incluso cuando la exposición a los mosquitos vaya a ser muy breve.
Los pacientes permanecen asintomáticos durante el periodo entre la picadura del mosquito infectado y la fase eritrocítica de la infección, periodo que puede durar de 1 a 4 semanas. Debido a que la quimioprofilaxis no evita en realidad el paludismo sino que trata la infección en fase eritrocítica, debe continuarse durante 4 semanas después del regreso de una zona endémica. El no continuar la quimioprofilaxis semanal después del retorno del área endémica permite el desarrollo de la infección.
La infección palúdica despierta una respuesta inmunológica. El desarrollo de anticuerpos contra la especie más indolente y crónica del paludismo, P. malariae, puede provocar en años el inicio de un síndrome nefrótico mediado por complejos inmunes. Las infecciones repetitivas con otras especies de Plasmodium pueden causar una inmunidad parcialmente protectora que limita la severidad de la infección, aunque no la previene. Es importante mencionar que los residentes de las áreas con paludismo pierden su inmunidad relativa después de permanecer varios años en una región sin paludismo. Estas personas pueden ser muy susceptibles a la infección al regresar a la región endémica.
DIAGNOSTICO
El paludismo debe ser considerado como causa de cualquier enfermedad febril en personas provenientes de áreas endémicas o que han viajado o trabajado en esas regiones. Los receptores de transfusiones, trasplante de órganos y los neonatos de madres potencialmente infectadas también tienen riesgo. Por último, incluso en los residentes de Estados Unidos, debe pensarse en el diagnóstico en los pacientes con síndrome febril y un cuadro clínico compatible, por la posibilidad de la poco común transmisión autóctona.
Las infecciones con las cuatro especies de Plasmodium que causa paludismo en los humanos producen síndromes clínicos diferentes, en parte por las interacciones de cada especie con los eritrocitos. P. vivax infecta solo eritrocitos jóvenes, lo que ayuda a limitar la intensidad de la infección. También explica por qué los eritrocitos grandes son característicos de las infecciones por P. vivax en los frotis. Por el contrario, P. falciparum infecta eritrocitos de todas las edades. Esta capacidad del paludismo tipo falciparum, el mayor número de merozoítos producidos por esta especie y en especial la mayor propensión de los eritrocitos infectados para adherirse al endotelio de la microvasculatura, contribuyen a que el paludismo falciparum sea más severo que las otras formas. Los eritrocitos infectados con P. falciparum desarrollan protrusiones en la superficie que median la unión y adherencia a las células endoteliales en los capilares y las vénulas. Al secuestrarse los eritrocitos infectados en los vasos pequeños ocurre anoxia. Pueden ocurrir complicaciones severas, incluyendo paludismo cerebral y edema pulmonar [ver tabla 1].
Cuadro clínico
Los síntomas clínicos se desarrollan 1 a 4 semanas después de la infección y típicamente incluyen fiebre y calosfríos. Virtualmente todos los pacientes con paludismo agudo tienen episodios de fiebre. Al inicio la fiebre puede ser diaria, con el tiempo pueden desarrollarse los patrones típicos de cada tercer día (P. vivax, P. ovale, P. falciparum) o cada cuarto día (P. malariae). Los paroxismos de fiebre (hasta 41.5 'C [106.7 'F] y calosfríos (con o sin temblor) pueden ser irregulares, en especial en el paludismo falciparum. Otros síntomas incluyen cefalea, diaforesis, lumbalgia, mialgias, diarrea, náusea, vómito y tos. La constelación de síntomas posibles no es específica y puede sugerir otros diagnósticos.36 Con el tiempo se desarrollan anemia y esplenomegalia.
Debido a la capacidad de los eritrocitos infectados por plasmodio falciparum de causar bloqueo de la microvasculatura, en este tipo de paludismo puede desarrollarse afección orgánica potencialmente fatal. La afección cerebral puede ocasionar delirio, trastornos focales (v.gr., crisis convulsivas) y coma.37 La afección esplácnica produce náusea, vómito, diarrea, melena y dolor abdominal; este síndrome puede confundirse con una diarrea del viajero. El daño pulmonar produce edema y síndrome de insuficiencia respiratoria progresiva del adulto. Puede existir hipoglucemia severa. Un síndrome raro, la fiebre de agua negra, se produce por hemólisis intravascular masiva, lo que ocasiona hemoglobinuria e insuficiencia renal aguda.
Los organismos P. malariae pueden persistir en la sangre y provocan una infección indolente, incluso asintomática, durante años o aún décadas.38
Datos de laboratorio
En la mayoría de los pacientes la cuenta de leucocitos se encuentra dentro del rango normal. Se desarrolla anemia, pero esta puede no ser importante al inicio, en especial si el paciente está deshidratado. Puede ocurrir trombocitopenia y en ocasiones coagulación intravascular diseminada en el paludismo falciparum. Las enzimas hepáticas pueden aumentar.
El diagnóstico específico y de tipo depende de la identificación de los microorganismos en el frotis de sangre periférica teñido de manera adecuada. 32 Los frotis gruesos son más sensibles que los delgados, pero por las capas de células se requiere mayor experiencia para examinar la morfología. Los frotis deben tomarse diariamente durante varios días debido a la naturaleza cíclica de la parasitemia, lo que adquiere particular importancia si se sospecha infección por P. falciparaum, en la que las células infectadas pueden estar secuestradas en la microvasculatura. Las características morfológicas del parásito y de los eritrocitos infectados son útiles en la identificación de las especies, y en la distinción de Plasmodium del microorganismo muy similar, Babesia microti, que causa la babesiosis humana. Es muy importante identificar con rapidez las infecciones por P. falciparum por su rápida evolución cuando no se tratan apropiadamente.
La técnica de cubierta amortiguadora cuantitativa (QBC por sus siglas en inglés, n. del t.) es sensible para identificar la parasitemia por plasmodios, pero no determina la especie. Existen tiras de ensayo de captura de antígenos, que permiten a los clínicos detectar infecciones por P. falciparum, en especial cuando no se cuenta con laboratorios especializados.39 Las sondas de ADN, las pruebas de RCP y las pruebas serológicas no suelen estar muy disponibles para el diagnóstico. Por lo tanto, el frotis de sangre sigue siendo la prueba estándar para determinar, primero, si un paciente tiene paludismo y, segundo, si éste es causado por P. falciparum.
TRATAMIENTO
El tratamiento de los pacientes con paludismo agudo requiere tomar en cuenta la especie y, en el caso de paludismo falciparum, la posibilidad de que el parásito sea resistente a los antipalúdicos. La cloroquina es la base del tratamiento para todas las especies excepto para las cepas de P. falciparum resistentes al medicamento. Es curioso que el paludismo por P. falciparum resistente a cloroquina (PFRC) se ha extendido por todos los países en donde el paludismo por P. falciparum es endémico excepto en Haití, República Dominicana, las zonas de América Central al oeste del canal de Panamá, el Medio Oriente. Para las infecciones por P. falciparum adquiridas en países con cepas resistentes a paludismo deben utilizarse tratamientos alternativos. Se han identificado cepas resistentes a mefloquina en la frontera de Tailandia-Myanmar, y cepas aisladas de P. vivax resistente a cloroquina en Africa, America Central, Sudamérica y Asia.
Los ataques agudos producidos por todas las especies, excepto por P. falciparum resistente a cloroquina, se tratan con fosfato de cloroquina oral, con dosis inicial de 1 g (600 mg de cloroquina base), seguido de 500 mg (300 mg) a las 6 horas y después a las 24 y 48 horas.26 Para las infecciones por PFRC existen varios tratamientos alternativos.26 Puede combinarse sulfato de quinina (650 mg cada 8 horas por 3 a 7 días) con doxiciclina (100 mg bid por 7 días), pirimetamina-sulfadoxina (Fansidar, 3 tabletas en dosis única el último día de tratamiento con quinina), o clindamicina (900 mg tid por 5 días). Otras opciones incluyen mefloquina (1,250 mg en dosis única), halofantrine y atovaquone en combinación con proguanil o doxiciclina. 26
En los pacientes graves que no toleran el tratamiento oral se puede administrar gluconato de quinidina o dihidrocloruro de quinina por vía intravenosa para cualquier tipo de paludismo.26 La quinidina es eficaz y bien tolerada con las precauciones adecuadas, incluyendo vigilancia hemodinámica y de ECG. Se infunde una dosis de impregnación de 10 mg/kg (máximo de 600 mg) de quinidina en solución salina en forma lenta en 1 a 2 horas, seguido por infusión continua de 0.02 mg/kg/min. Debe continuarse el tratamiento parenteral hasta que pueda tolerarse el tratamiento oral, en la mayoría de los casos en 48 a 72 horas. Para el paludismo falciparum fulminante la exsanguíneo transfusión puede ayudar.40
Cada uno de los agentes mencionados antes es activo solo contra la fase eritrocítica de la infección. Debido a que P. vivax y P. ovale tienen fases hepáticas persistentes que pueden causar recaídas de paludismo después del uso de cloroquina [ver figura 1], debe administrarse después de la cloroquina un agente que sea eficaz sobre el ciclo exoeritrocítico. La primaquina es el único agente usado para erradicar la infección hepática; se usa en dosis de 26.3 mg (15 mg de medicamento base) por vía oral cada día durante 2 semanas. Debido a que la primaquina puede inducir hemólisis en personas con deficiencia de glucosa 6 fosfato deshidrogenasa (G6PD), debe investigarse esta posibilidad antes del tratamiento. Si se encuentra una deficiencia leve puede administrarse el medicamento en dosis de 79 mg (45 mg de base) por vía oral una vez por semana durante 8 semanas.
Prevención de los viajeros
La mayoría de los residentes en Estados Unidos no tienen inmunidad contra el paludismo y tienen riesgo de las consecuencias de morbimortalidad de la enfermedad, en especial de la forma falciparum, en los países en que ocurre la trasmisión. De los 591 civiles de los Estados Unidos que adquirieron paludismo en el extranjero en 1995, solo el 15.6% había seguido un esquema quimioprofiláctico recomendado por los CDC. 32 Es indispensable que los viajeros reciban la orientación adecuada para disminuir su riesgo de adquirir paludismo y que usen los esquemas adecuados.
Babesiosis
Babesia es un protozoario intraeritrocítico que produce una enfermedad muy parecida al paludismo. La mayoría de los casos de babesiosis se han observado en el Noreste de los Estados Unidos, pero han ocurrido casos también en la parte superior del Medio Oeste, la Costa Occidental y otras regiones de los Estados Unidos, Europa y otros sitios.41
ETIOLOGIA Y EPIDEMIOLOGIA
Se han detectado varias especies Babesia como causa de enfermedad humana. La primera fue B. divergens, un parásito del ganado que ha causado varias infecciones fatales en personas esplenectomizadas, en especial en Europa, aunque una especie relacionada, denominada MO1, provocó un caso fatal en un hombre esplenectomizado en Missouri.42 B. microti es la causa principal de babesiosis en los Estados Unidos. Este parásito del ratón de patas blancas se trasmite por garrapatas de venados y es prevalente en las islas de Massachusetts, New York y Rhode Island y en áreas focales en Connecticut, Wisconsin y Minnesota. El riesgo de babesiosis no aumenta por la esplenectomía, pero la enfermedad es más seria en personas sometidas a esta cirugía, que tienen infección por VIH o con algún otro trastorno inmunosupresor.41,43 Muchas personas infectadas tienen infecciones subclínicas, como lo evidencian los estudios serológicos en áreas endémicas. Debido a que las garrapatas ninfas son las más eficaces para trasmitir la infección, la mayoría de los casos se desarrollan entre mayo y agosto, cuando este tipo de garrapatas son más abundantes. 41
Otra forma de babesiosis se desarrolla a partir de la infección con un organismo encontrado en los estados a lo largo de la costa del Pacífico y que se denomina WA1. Aún se desconocen el vector y el reservorio de esta nueva especie de Babesia. Se han detectado infecciones en personas con esplenectomía y con menos frecuencia en huéspedes normales; los estudios serológicos en zonas rurales y semirrurales de California indican que puede desarrollarse infección subclínica por WA1 en hasta el 20% de la población.41
La babesiosis también puede adquirirse en forma perinatal y por transfusiones sanguíneas.41,44
DIAGNOSTICO
Manifestaciones clínicas
Muchas personas infectadas con B. microti o WA1 permanecen asintomáticas. En los que ocurren síntomas la enfermedad se desarrolla en forma gradual semanas después de la mordedura de la garrapata o la transfusión sanguínea. Ocurren malestar, anorexia y fatiga, seguidos de mialgias, fiebre y diaforesis. Son comunes la labilidad emocional, la depresión, náusea, vómito y cefalea. Los síntomas tienden a desaparecer en semanas, aunque la fatiga y el malestar pueden durar meses. En ocasiones existe esplenomegalia. Las infecciones pueden volverse fulminantes y persistentes en pacientes esplenectomizados, ancianos y pacientes infectados por VIH o con otros padecimientos inmunosupresores. Pueden desarrollarse hemólisis intravascular, hemoglobinuria e insuficiencia renal. Ocurren parasitemias hasta del 85%, y este grado de infección puede ser fatal, en especial en personas asplénicas.
Datos de laboratorio
Los datos de laboratorio pueden incluir anemia hemolítica, cuentas de leucocitos normales o bajas y pruebas de función hepática anormales. El diagnóstico se basa en la observación de formas en anillo o de los microrganismos intraeritrocíticos en un frotis de sangre periférica teñidos con Giemsa. En raras ocasiones es posible observar merozoítos extraeritrocíticos. A diferencia de los parásitos del paludismo, los organismos Babesia no producen pigmentos en los eritrocitos que parasitan. En algunos pacientes, una parasitemia del 5 por ciento o mayor, y la observación de formas en anillo en algunos eritrocitos, sugieren el diagnóstico de P. falciparum, pero la ausencia del pigmento y de gametocitos distingue la babesiosis del paludismo. Las pruebas serológicas son útiles para el diagnóstico de infecciones crónicas con B. microti y WA1 cuando no se detectan los parásitos en los frotis de sangre.
TRATAMIENTO
El tratamiento de elección en la babesiosis consiste en la combinación de clindamicina (1.2 g bid IV o 600 mg tid vo por 7 días) y quinina (650 mg tid vo por 7 días). 26 En una minoría de pacientes que mejoran desde el punto de vista sintomático persiste la parasitemia. 45 En ocasiones puede desarrollarse edema pulmonar después de iniciar el tratamiento para la babesiosis, igual que ocurre en el caso del paludismo. Las exsanguíneo transfusiones pueden ayudar en los casos muy graves. El vector garrapata de la babesiosis puede trasmitir en forma simultánea a Borrelia burgdorferi, el agente de la enfermedad de Lyme, y especies Ehrlichia, los agentes de la erhlichiosis. Estas coinfecciones pueden empeorar el curso clínico y justificar tratamiento específico.46
Infecciones por protozoarios intestinales
El tubo digestivo del hombre puede albergar varios protozoarios que parasitan al ser humano. Los protozoarios Entamoeba coli, Endolimax nana e Iodamoeba butschlii son patógenos y no requieren tratamiento. Los protozoarios intestinales que se han asociado con enfermedades son Giardia lamblia, Entamoeba histyolitica, coccidios, Balantidium coli y Dientamoeba fragilis.
GIARDIASIS
G. lamblia habita en el intestino proximal y su infección puede variar desde asintomática hasta malabsorción severa. G. lamblia y la criptosporidiosis son los parásitos intestinales patógenos más frecuentes en los Estados Unidos. 47
Etiología y epidemiología
Existen dos formas morfológicas del parásito: el trofozoíto y el quiste. El trofozoíto es un microrganismo con múltiples flagelos, en forma de pera, que mide 9 a 15 micras de largo, de 5 a 15 micras de ancho, y de 2 a 4 micras de espesor; tiene simetría bilateral y contiene dos núcleos prominentes. En su cara ventral tiene un disco adhesivo que le permite fijarse a la superficie mucosa del duodeno y el yeyuno. A su paso por el intestino el trofozoíto generalmente se enquista. Los quistes resultantes miden 8-12 por 7-10 micras, tienen una pared externa bien definida y cuando maduran contienen cuatro núcleos.
Los trotozoítos pueden encontrarse en el líquido duodenal y en las heces líquidas, pero generalmente están ausentes de las evacuaciones formadas. Los trofozoítos no resisten las condiciones del medio externo, pero en cambio los quistes son muy resistentes, pudiendo sobrevivir en el agua varios meses, y tienen mayor supervivencia cuando la temperatura del agua es menor. Aunque la infección puede adquirirse ingiriendo trofozoítos con suficiente cantidad de alimento que los proteja de la acidez del estómago, los quistes son los principales responsables de la infección en el hombre. En estudios experimentales se ha logrado infectar a voluntarios con la ingestión de tan sólo 10 quistes del parásito.
La infección, que se relaciona directamente con la excreción fecal del microrganismo, se disemina por vía fecal- oral, o bien a través de alimentos y agua contaminados. La trasmisión directa de persona a persona es la responsable de la alta prevalencia que se observa en ciertos grupos. En las instituciones en las que existe incontinencia fecal y mala higiene la giardiasis puede ser endémica. De manera análoga, la giardiasis puede ser causa de enfermedad intestinal en las guarderías.48El riego de la adquisición y trasmisión es muy alto entre los niños que aún no controlan esfínteres, éstos a su vez pueden ser la fuente de casos secundarios dentro de su familia. 48 La trasmisión de persona a persona también es responsable de la alta frecuencia de giardiasis entre los varones homosexuales promiscuos.48 Algunas prácticas sexuales, como el anilingus, permiten la trasmisión directa de los quistes infectantes.
La trasmisión por agua contaminada es la principal causa de giardiasis. Puesto que la filtración del agua a través de la tierra elimina los quistes de Giardia, el agua de pozos profundos suele ser segura, En comparación, el agua de 1a superficie, como el agua de los arroyos y fuentes de montaña y el agua estancada, puede estar contaminada con quistes de Giardia, que son resistentes al agua y a los niveles normales de cloración. El recuento de coliformes no es un índice confiable de la contaminación por Giardia. Además, los mamíferos acuáticos, como los castores, pueden infectarse y servir como fuente continua de contaminación del agua. Puede ser difícil reconocer un suministro de agua como el origen común de las giardiasis, debido a que la infección con frecuencia es asintomática.
G. lamblia tiene una distribución mundial. Los viajeros que visitan muchos países, no sólo subdesarrollados, pueden infectarse con el microrganismo.
Patogenia
Después de la ingestión y el paso por el estómago de los quistes de G. lamblia, se liberan en el intestino los trofozoítos, que proliferan por división binaria. Los trofozoítos localizados en el duodeno y yeyuno pueden adherirse a las microvellosidades del epitelio intestinal mediante su disco de succión ventral, vivir en la capa de moco que recubre el epitelio, moverse y permanecer en el contenido intestinal, o más raramente, invadir los tejidos adyacentes a través de las células epiteliales. Pueden desarrollarse cambios funcionales en la capacidad de absorción del intestino delgado. La actividad enzimática del borde epitelial en cepillo disminuye, produciéndose deficiencia de disacaridasas, incluyendo lactasa. Las biopsias de yeyuno en pacientes con giardiasis no suelen revelar alteraciones patológicas. 49 Los mecanismos fisiopatológicos que producen estas alteraciones funcionales son inciertos. Ambos tipos de alteraciones se normalizan con el tratamiento específico, aunque en algunos pacientes en forma lenta. La hipoclorhidria predispone a la infección. La giardiasis suele ser más grave en pacientes con fibrosis quística y con deficiencias de inmunoglobulinas, quizá por deficiencia en la IgA secretora.50
Diagnóstico
Manifestaciones clínicas Las manifestaciones clínicas de la giardiasis son variables. Un gran número de pacientes no sufre síntomas. De hecho, en un brote muy bien estudiado de giardiasis asociado con contaminación acuática, dos tercios de las personas infectadas permanecieron asintomáticas. Por el contrario, otro subgrupo de personas infectadas desarrolló una giardiasis aguda típica. Después de un periodo de incubación de 1 a 3 semanas se presenta diarrea acuosa, dolor abdominal tipo cólico (y con frecuencia molestia epigástrica), náusea (el vómito es menos frecuente) y síntomas generales. Puede existir fiebre. La excesiva producción de gas en el intestino produce flatulencia y eructos de olor sulfuroso. Son comunes la menor absorción de grasas y la esteatorrea. Las evacuaciones son grasosas, malolientes y flotan en el agua; es raro encontrar en ellas moco o sangre. Los síntomas son importantes durante una semana y disminuyen en intensidad en las siguientes semanas. Los datos clínicos que ayudan a identificar los casos de giardiasis en estudios epidemiológicos incluyen la duración de la enfermedad de 7 o más días con por lo menos dos de los siguientes seis síntomas: diarrea, flatulencia, heces fétidas, náusea, có1ico abdominal y fatiga excesiva.51
Una fase crónica de las giardiasis puede seguir a la fase aguda, o puede manifestarse sin el antecedente de una enfermedad aguda. La fase crónica se caracteriza por evacuaciones disminuidas de consistencia, aunque no necesariamente diarreicas, con aspecto graso. También se asocia con aumento de la cantidad de gas en el intestino, có1icos, borborigmos, flatulencia y distensión. La fiebre no es frecuente, pero sí pueden encontrarse malestar general, fatiga y depresión. La intolerancia a la lactosa, producida o exacerbada por la enfermedad, puede causar aumento de los síntomas al ingerir leche o sus productos. El curso clínico es variable, alternando períodos de remisión y exacerbación. En un número reducido de casos la persistencia de la infección puede asociarse con malabsorción moderada o grave y pérdida de peso.52
Muy raras veces, G. lamblia puede diseminarse del duodeno y yeyuno a las vías biliares y conductos pancreáticos. Se han comunicado casos de colecistitis, colangitis y hepatitis granulomatosa. También se han observado alteraciones en la función exócrina del páncreas, manifestada por secreción disminuida de tripsina y lipasa.
Estudios de laboratorio En la giardiasis no ocurren leucocitosis ni eosinofilia. La excreción de grasa en las heces está aumentada (ver antes) y los resultados de otras pruebas de laboratorio de malabsorción pueden ser anormales. Las series gastrointestinales no muestran cambios radiológicos significativos.
El diagnóstico definitivo de la infección requiere la identificación de los trofozoítos o quistes. El examen de las heces es habitualmente positivo en el cuadro agudo y la observación de series de tres muestras permite el diagnóstico en más de 90 por ciento de los casos. Los quistes, que pueden estar presentes en las heces líquidas o formadas son más resistentes y no requieren la búsqueda en fresco. Por el contrario cuando se buscan trofozoítos es importante analizar muestras en fresco, o conservadas en alcohol polivinílico o en una preparación de merthiolate- ioduro-formaldehido (MIF). Deben evitarse las sustancias que interfieren con la investigación en fresco, como el medio de contraste, los antiácidos y el aceite mineral. Los estudios inmunológicos detectan antígenos de giardia en las heces con mayor sensibilidad que una sola muestra de excremento.53
En la giardiasis crónica la frecuencia de detección del parásito en el excremento disminuye. Si se ha fracasado en tres muestras de heces, debe analizarse material duodenal obtenido por aspiración duodenoyeyunal. En forma alternativa, puede practicarse una biopsia intestinal. La detección de los trofozoítos en el material de la biopsia requiere de una búsqueda cuidadosa, el examen directo del frotis de mucosa en fresco puede facilitar la detección del parásito.
Diagnóstico diferencial
Debe pensarse en otros agentes infecciosos que causen gastroenteritis al evaluar a los pacientes con giardiasis aguda. Debido a que la mayoría de estos agentes producen un cuadro de corta duración, la persistencia de los síntomas después de una semana y los síntomas significativos de malabsorción (flatulencia, intolerancia a la lactosa, eructos) sugieren una giardiasis. La giardiasis crónica puede parecerse a otros padecimientos asociados con malabsorción.
Tratamiento
El metronidazol, aunque no aprobado por la Food and Drug Administration de los EUA, para el tratamiento de la giardiasis, es el principal agente empleado para tratar esta infección26 porque la quinacrina, el medicamento más eficaz, no se distribuye ya en los Estados Unidos. La dosis habitual es de 250 mg tres veces al día durante 5 días y en casos persistentes se han llegado a utilizar hasta 750 mg tres veces al día durante 10 días. Los efectos colaterales que se han observado son náusea, dolor de cabeza y sabor metálico en la boca; rara vez llega a oscurecerse la orina o a presentarse parestesias y mareos. El metronidazol tiene un efecto semejante al disulfiram, por lo que debe evitarse el consumo de alcohol durante el tratamiento. Aunque no está disponible en los Estados Unidos, el tinidazol (administrado en una sola dosis oral de 2 g) es muy eficaz.54
En los niños puede utilizarse una suspensión de furazolidona, que ha demostrado ser eficaz y bien tolerada.26 El tratamiento de la giardiasis durante el embarazo representa un verdadero reto. El metronidazol debe evitarse, aunque los estudios no han demostrado riesgos teratogénicos al usarlo durante el embarazo. 55 Si los síntomas de la giardiasis son mínimos el tratamiento puede esperar hasta el parto. Si hay molestias puede administrarse el aminoglucósico no absorbible paromomicina, 25 a 35 mg/kg/día por vía oral en tres dosis divididas por 7 días.26Este esquema puede proporcionar alivio por lo menos sintomático. Si la giardiasis durante el embarazo se asocia con deshidratación, malabsorción o síntomas severos está justificado el tratamiento con metronidazol. En cualquier paciente, la resolución de los síntomas de malabsorción pueden requerir de meses para que se regenere la mucosa intestinal funcional después del tratamiento antiparasitario eficaz.
La prevención de la giardiasis requiere atención hacia la higiene para evitar la trasmisión de persona a persona. No se han definido aún cuáles son los riesgos y los beneficios de tratar a los niños infectados asintomáticos que acuden a guarderías, pero está indicado el tratamiento de los portadores asintomáticos para evitar la diseminación de la enfermedad. La ebullición del agua o su calentamiento a 70'C [158' F] durante 10 minutos es suficiente para desinfectarla. Para los alpinistas y excursionistas, la iodinación del agua es más eficaz que la cloración, pero requiere de por lo menos 8 horas para que sea eficaz en un 99.9 por ciento. También son eficaces algunos filtros de agua de calidad para eliminar los quistes.
AMIBIASIS
La infección con Entamoeba histolytica es responsable de la amibiasis humana. La infección puede estar limitada al colon, con manifestaciones clínicas que varían desde el estado asintomático hasta la disentería grave, o puede afectar áreas extraintestinales, de las que el hígado es la más frecuente.
Etiología y epidemiología
E. histolytica puede distinguirse morfológicamente de las otras especies no patógenas de amibas intestinales, incluyendo a Escherichia coli y E. hartmanni. E. histolytica existe en dos formas: como trofozoíto y como quiste. El trofozoíto mide de 10 a 20 micras de diámetro, pero puede ser mayor en las evacuaciones disentéricas; es móvil y posee un núcleo único con citoplasma granular. Los trofozoítos se eliminan con las heces diarreicas, pero son poco resistentes a las condiciones del medio fuera del organismo. Por el contrario, el quiste que se forma en el colon es muy resistente a las condiciones del medio externo y a la acidez del estómago. Los quistes miden de 10 a 20 micras de diámetro, contienen de uno a cuatro núcleos pequeños, cariosomas centrales y un patrón de cromatina fina en la periferia.
No todas las cepas de E. histolytica son patógenas. Pueden aislarse cepas no patógenas de individuos asintomáticos, y son prevalentes entre los varones homosexuales promiscuos en los Estados Unidos e Inglaterra. Las cepas patógenas y no patógenas no pueden distinguirse por microscopía, excepto porque los trofozoítos patógenos con frecuencia fagocitan eritrocitos. Con base en datos bioquímicos, inmunológicos y genéticos se ha descrito a E. hystolitica. En la actualidad una nueva especie, E. dispar, representa las cepas no patógenas y aparentemente nunca es invasiva en los humanos, mientras que E. hystolitica incluye solo las cepas potencialmente patógenas.56 Debido a que no todas las cepas potencialmente patógenas de E. hystolitica producen enfermedad, sin duda otros procesos influyen en la virulencia de las amibas.
Se ha desarrollado un inmunoensayo para distinguir a E. hystolitica de E. dispar.57 En la actualidad no existe otro método disponible para distinguir en forma confiable entre ambos organismos [ver adelante, Diagnóstico].
Los quistes, que se eliminan por las heces humanas, son la principal fuente de infección para el hombre. La adquisición en la infección es producto de contaminación fecal - oral y puede ocurrir a través de agua y alimentos contaminados o de persona a persona. Esta última forma es la responsable de la alta prevalencia de la infección entre homosexuales promiscuos y en situaciones donde existe incontinencia fecal con malas medidas de higiene. La amibiasis es más común en los grupos socioeconómicos bajos, y se debe a las malas condiciones sanitarias y al hacinamiento. En los Estados Unidos se observan casos en personas que han regresado de viajes al extranjero o inmigrado de zonas en donde la amibiasis es endémica.
Patogenia
Los quistes ingeridos son transportados hasta el intestino, donde liberan los trofozoítos que se reproducen por fisión binaria dentro del colon. Los trofozoítos producen citólisis e invaden la pared del intestino, causando necrosis focal. Las úlceras que se producen tienen aspecto de cuello de botella, con la porción angosta a nivel de la mucosa y un cuerpo amplio en la submucosa. A menos que el daño clínico sea muy extenso, pueden observarse zonas de colon normal entre ellas. Las áreas que se afectan con mayor frecuencia son el rectosigmoides, el apéndice cecal, el colon transverso y descendente, y el íleon terminal.
La gravedad de la afección colónica en pacientes con amibiasis es muy variable y puede variar desde un padecimiento frecuente y leve o incluso despreciable hasta invasión difusa y extensa con necrosis. La mayoría de los pacientes que se infectan con E. histolytica sufren invasión importante del colon. Los factores determinantes de la gravedad se conocen poco y tal vez guarden relación con el tamaño del inóculo, la cepa de E. histolytica de que se trate, la flora colónica y el estado fisiológico y nutricional del huésped. Los esteroides y el embarazo disminuyen la resistencia del huésped a la invasión. Las complicaciones de la afección intestinal son hemorragia y peritonitis, produciéndose la última más por paso a través de la pared inflamada, que por una franca perforación del colon. La infección crónica produce una respuesta granulomatosa en el lugar afectado, más común en el ciego, que se manifiesta como una masa denominada ameboma. También puede desarrollarse estenosis del colon.
La amibiasis puede diseminarse por vía hematógena desde el colon para afectar cualquier parte del organismo. El hígado es el órgano más afectado, seguido en frecuencia por los pulmones, que se lesionan por diseminación transdiafragmática a partir del hígado. Los trofozoítos transportados por el sistema venoso porta producen necrosis en el hígado y la formación de abscesos. Alrededor del 90 por ciento de los abscesos se encuentran en el lóbulo derecho, en especial en la cara superior y anterior. Aunque los abscesos pueden ser múltiples, es más común encontrar un solo absceso que varía de pocos hasta 20 centímetros de diámetro. Los abscesos hepáticos amibianos son 7 a 9 veces más comunes en hombres que en mujeres. En pocas ocasiones un absceso puede desarrollarse en forma concomitante a una colitis amibiana. Dependiendo de la serie, el 50 a 70 por ciento de los pacientes con abscesos hepáticos amibianos no tienen historia de colitis amibiana. La ruptura de un absceso en el lóbulo derecho puede causar extensión hacia el tórax, produciendo un empiema amibiano, neumonía o una fístula broncopleural. La extensión hacia la cavidad peritoneal es menos común. La ruptura desde un absceso del lóbulo izquierdo puede invadir el pericardio, con frecuencia con consecuencias fatales.
Diagnóstico
Cuadro clínico La gran mayoría de los pacientes con amibiasis colónica no tiene síntomas. La excreción fecal de los quistes en estas personas es un hallazgo fortuito. En aquellos que sufren síntomas la enfermedad varía desde leves episodios de diarrea hasta una disentería grave. La primera forma de presentación es común y los pacientes no interrumpen sus actividades diarias, a pesar de la diarrea leve, que puede alternar con estreñimiento. El curso de la enfermedad puede ser remitente, con episodios sintomáticos intercalados con temporadas sin sintomatología. Es frecuente que el paciente se queje de malestar abdominal vago, flatulencia, tenesmo y dolor en la región sacra, pero los síntomas generales y la fiebre son raros, mucho menos frecuentes que en las formas bacterianas de gastroenteritis y colitis. Las evacuaciones se acompañan de moco y sangre. La exploración física del abdomen refleja la gravedad de la afección colónica. Puede haber dolor discreto en las zonas afectadas; en otros casos, el dolor intenso junto con fiebre alta indican afección grave del colon. 58 Los amebomas pueden palparse en ocasiones como masas dolorosas.
Los síntomas de presentación del paciente con un absceso hepático amibiano son malestar general, fatiga, anorexia, pérdida de peso, dolor abdominal y fiebre, con duración de una a varias semanas, aunque puede tener una evolución de meses. El dolor abdominal por lo general es constante, con localización en el cuadrante superior o hemitórax derechos. Algunas veces el paciente lo refiere en el hombro del mismo lado. Cuando se presenta como dolor pleurítico, acompañado de tos y disnea, puede interpretarse equivocadamente como una infección intrapulmonar. Un absceso en el lóbulo izquierdo produce dolor en el epigastrio o en el cuadrante superior izquierdo del abdomen. Al examen físico se encuentran hepatomegalia y dolor localizado al palpar la zona del absceso. La ictericia franca es rara Al examinar el tórax la base derecha se percute mate debido a la elevación del hemidiafragma y a la alta frecuencia de derrame pleural; con la auscultación pueden escucharse algunos estertores.
La amibiasis cutánea produce lesiones ulceradas o exofíticas, con localización frecuentes en el periné y los genitales. Estas se producen por invasión de los trofozoítos, como resultado de contaminación fecal o trasmisión sexual. Se diferencían de los procesos neoplásicos, la tuberculosis y la sífilis por la presencia de trofozoítos en los exudados y biopsias.
Estudios de laboratorio En la colitis amibiana sintomática es frecuente observar leucocitosis discreta a moderada. También es posible que se encuentre una anemia ligera y alteraciones en las pruebas de función hepática, sin que necesariamente indiquen formación de absceso. Las heces suelen contener sangre evidente u oculta, así como cristales de Charcot-Leyden, derivados de eosinófilos.
Cuando existe un absceso hepático amibiano se observa leucocitosis, anemia y elevación de la velocidad de sedimentación globular. En más de dos tercios de los pacientes la fosfatasa alcalina se eleva de una a cuatro veces por encima de sus valores normales. La elevación de las transaminasas se observa en menos del 50 por ciento de los pacientes; la bilirrubina se eleva poco, en general menos de 2.5 mg/dl. La radiografía del tórax muestra elevación del hemidiafragma derecho y derrame pleural. Las gamagrafías hepáticas revelan defectos de captación compatibles con lesiones ocupativas, el ultrasonido muestra la misma lesión, de características hipoecogénicas, de forma redonda u ovoide y es útil para detectar la diseminación transdiafragmática del absceso. La TAC o la IRM del abdomen también son útiles para detectar el absceso y valorar su extensión.
El diagnóstico definitivo de la amibiasis intestinal requiere la identificación en las heces de los quistes o trofozoítos de E. histolytica . Las evacuaciones diarreicas deben examinarse de inmediato en busca de trofozoítos, en cambio las heces formadas pueden examinarse directamente o después de someterlas a un proceso de concentración para búsqueda de quistes. Las heces se conservan en forma adecuada en alcohol polivinílico o en solución de MIF para su examen posterior. Es frecuente confundir los leucocitos fecales y las amibas no patógenas con E. histolytica, por lo que es recomendable realizar las tinciones adecuadas, y que el examen sea practicado por personal con experiencia. Los antimicrobianos, los catárticos, los antiácidos y el bario pueden interferir con el examen microscópico. Es recomendable examinar muestras de 3 o más días, ya que la excreción de los quistes varía día con día. Cerca de la mitad de los pacientes con colitis amibiana tiene lesiones en el rectosigmoides, por lo que los raspados, aspirados y biopsias de las lesiones mucosas tomados durante una rectosigmoidoscopía deben examinarse en busca de trofozoítos.
Se ha desarrollado una prueba de detección de un antígeno fecal para E. hystolitica, que se basa en un anticuerpo monoclonal contra una lectina específica en este organismo.57
Las pruebas serológicas para amibas son por lo general negativas en el paciente asintomático que excreta quistes, pero su positividad aumenta en forma paralela a la lesión colónica y al tiempo de evolución de la colitis. Estas pruebas pueden ayudar al diagnóstico de las colitis agudas y son muy útiles al evaluar la etiología de una colitis crónica. Al interpretar un resultado positivo, el clínico debe tomar en consideración que la positividad se mantiene durante meses o años después de una infección.
En el 90 a 95 por ciento de las infecciones extraintestinales existe seropositividad para anticuerpos contra amiba, y sus títulos se incrementan conforme aumenta el tiempo de evolución de la enfermedad. En el diagnóstico del absceso hepático amibiano, la combinación de una prueba serológica positiva, un cuadro clínico compatible y la observación de una lesión quística, es muy sugestiva del diagnóstico. En el caso de un absceso amibiano no es de utilidad la búsqueda del parásito en las heces. Rara vez es posible observar trofozoítos en el material aspirado de un absceso, cuyo aspecto se parece a la pasta de anchoas y es de apariencia achocolatada.
Diagnóstico diferencial
En los casos de colitis amibiana leve, la duración y los síntomas pueden confundirse con un síndrome de colon irritable, con una diverticulitis o una enteritis regional. Cuando el cuadro disentérico es grave debemos distinguirlo de las infecciones por Shigella, Salmonella y Campylobacter, lo que puede lograrse mediante los cultivos apropiados y por la presencia de abundantes leucocitos en la materia fecal. La colitis ulcerosa y la enfermedad de Crohn deben considerarse en el diagnóstico diferencial de la colitis amibiana crónica. Las lesiones de la colitis amibiana y la colitis ulcerosa pueden tener un aspecto similar en la rectosigmoidoscopía y el enema con contraste, pero es muy importante distinguirlas porque los esteroides, que están indicados en la segunda, pueden agravar la colitis amibiana. Por esta razón las pruebas serológicas para amiba, y el examen de las heces y lesiones mucosas en busca de amibas son de gran importancia. La prueba terapéutica con metronidazol puede tener resultados benéficos en ambas enfermedades, por lo que no es de utilidad diagnóstica.
Un ameboma puede simular un adenocarcinoma o bien otros procesos granulomatosos. Una prueba serológica positiva para amiba y la resolución de la masa con el tratamiento con metronidazol apoyan el diagnóstico de ameboma. Si la respuesta con metronidazol no es completa, se recomienda practicar biopsia con el fin de descartar algún proceso asociado al ameboma.
El diagnóstico diferencial del absceso hepático amibiano se guía por dos observaciones: (1) una gamagrafía hepática que muestra una lesión ocupativa y (2) un ultrasonido, TC o IRM que demuestra la naturaleza quística de la lesión. Aunque los quistes hepáticos y los quistes equinocócicos no suelen asociarse con fiebre y los otros síntomas de una lesión amibiana, deben considerarse dentro del diagnóstico diferencial. También debe pensarse en abscesos piógenos. La serología positiva apoya una etiología amibiana. A diferencia de los quistes equinocócicos, los abscesos hepáticos amibianos rara vez se calcifican. Tanto el absceso hepático amibiano como el piógeno tienen buena respuesta al metronidazol, y por otro lado, los abscesos amibianos muy pocas veces se sobreinfectan con gérmenes piógenos. Aunque la aspiración del absceso no está indicada en el caso de que sea de etiología amibiana, esta medida puede ser necesaria para obtener material para cultivo y tinción de Gram cuando existen dudas diagnósticas.
Tratamiento
El tratamiento de elección para la amibiasis intestinal sintomática o extraintestinal es el metronidazol (750 mg vo tid por 10 días), que ha demostrado ser muy eficaz y constituye el tratamiento de elección.26 Ya se describieron en la sección de giardiasis los efectos colaterales y las precauciones al usarlo. Se han observado fracasos ocasionales en el tratamiento del absceso hepático. En caso de enfermedad intestinal, las recaídas son raras a menos que exista reinfección, por lo que se recomienda la vigilancia de los pacientes durante varios meses.
Se considera que el tratamiento farmacológico no es necesario para muchas personas asintomáticas con quistes porque con frecuencia tienen E. dispar no patógena. Si no es posible conocer la especie exacta de Entamoeba, los portadores de quistes deben tratarse solo si manejan alimentos o reciben esteroides; también en el caso de una epidemia de amibiasis.56 En los pacientes asintomáticos la concentración de metronidazol en la luz del colon puede ser inadecuada para erradicar las amibas, por lo que debe usarse uno de dos amebicidas intraluminales.26Puede emplearse paromomicina (25 a 35 mg/kg/día por 7 días) o como alternativa iodoquinol (650 mg vo, tid 20 días). La dosis no debe ser mayor por el riesgo de causar neuropatía óptica El iodoquinol está contraindicado en pacientes con neuropatía óptica o enfermedad tiroidea. Se recomienda la administración de un esquema de iodoquinol para las personas tratadas por amibiasis intestinal sintomática o extraintestinal.26
La aspiración terapéutica no está indicada en el tratamiento del absceso hepático amibiano, aunque la aspiración diagnóstica puede ser útil en ciertos casos [ver antes, Diagnóstico diferencial]. Está indicado el drenaje, que puede lograrse por aspiración cutánea, para las lesiones que no responden al tratamiento médico inicial o con peligro de ruptura inminente.
La prevención de la amibiasis se basa principalmente en la higiene personal, que evita la diseminación de persona a persona. En las comunidades donde la enfermedad es endémica, la previsión y el uso de sanitarios adecuados ha disminuido mucho su diseminación. Es conveniente evitar el consumo de vegetales que crecen en la tierra por la posibilidad de contaminación por heces humanas. El agua puede considerarse segura si se ha hervido o se ha tratado con tabletas de yodo.
COCCIDIOSIS
Los parásitos de la familia Coccidia se encuentran en el intestino de varios animales domésticos y salvajes, siendo parásitos unicelulares con ciclos de reproducción sexual y asexual que se llevan a cabo en las células del epitelio intestinal. De todos ellos, tres han demostrados ser patógenos en el humano, Isospora belli, Cryptosporidium y Cyclospora cayetanensis. El contacto con ganado infectado puede provocar la infección con Cryptosporidium, y el agua contaminada puede trasmitir I. belli, Cryptosporidium y C. cayetanensis.59 Los tres parásitos se han identificado como oportunistas en pacientes infectados por VIH.60
Isosporiasis
Diagnóstico La infección por I. belli se inicia en forma súbita. Los primeros síntomas son fiebre y malestar general, apareciendo después dolor abdominal, diarrea y pérdida de peso. En la mayoría de los casos el proceso se autolimita, aunque se han comunicado infecciones con duración de varios días hasta algunos meses. En estos casos, puede haber diarrea grave, esteatorrea y lesión hepática; sólo rara vez la infección provoca la muerte. En pacientes con SIDA la infección con I. belli causa diarrea acuosa crónica y pérdida de peso indistinguible de la enfermedad producida por Cryptosporidium.
El examen histológico de las lesiones mucosas muestra acortamiento de las vellosidades, hipertrofia de las criptas y un infiltrado compuesto por eosinófilos, linfocitos y células plasmáticas. Aunque la eosinofilia en sangre periférica es rara en las infecciones por protozoarios, sí se presenta en las coccidiosis. La presencia de ooquistes en las heces establece el diagnóstico, pero es difícil detectarlos en los exámenes de rutina; sin embargo, pueden demostrarse mediante tinción ácido resistente. Si el número de ooquistes en las heces es escaso, la incubación de las heces a temperatura ambiente y por espacio de 24 a 48 horas madura los quistes y facilita su detección; también se recomienda utilizar la técnica de concentración con sulfato de zinc. Los parásitos también pueden observarse en los aspirados y biopsias intestinales.
Tratamiento Las infecciones causadas por I. belli se tratan con trimetoprim-sulfametoxazol de doble potencia (160 mg de trimetoprim y 800 mg de sulfametoxazol) administrados por vía oral cuatro veces al día durante 10 días y después dos veces al día durante 3 semanas.26 Sin embargo, en los pacientes con SIDA son frecuentes las recaídas y el tratamiento inicial de 10 días debe continuarse con un tratamiento de sostén a largo plazo con una tableta de dosis doble de trimetoprim-sulfametoxazol tres veces a la semana o con la combinación de 25 mg de pirimetamina y 500 mg de sulfadoxina una vez a la semana. En los pacientes con intolerancia a las sulfas ha sido eficaz el tratamiento con pirimetamina (75 mg/día); puede administrarse una dosis de mantenimiento (50 a 75 mg/día) para prevenir las recaídas de las infecciones por I. belli. 60
Cryptosporidiosis
Cryptosporidium habita en el borde en cepillo de la mucosa intestinal y produce enterocolitis en individuos normales o inmunosuprimidos. La infección puede adquirirse al ingerir incluso menos de 100 ooquistes.61Se ha demostrado la trasmisión a través del agua para beber 62 y de albercas contaminadas. 47,63 La criptosporidiosis ha causado epidemias de diarrea en guarderías y puede adquirirse en viajes al extranejro. Debido a que los ooquistes fecales son infecciosos, puede ocurrir diseminación de la infección tanto nosocomial como en el hogar.
Diagnóstico En huéspedes normales la enfermedad comienza después de un periodo de incubación de cerca de una semana. Consiste en diarrea acuosa, no sanguinolenta, acompañada a veces por dolor abdominal, náusea, fiebre, anorexia y pérdida de peso. Los síntomas pueden persistir por 1 a 2 semanas y suelen autolimitarse sin tratamiento. Por el contrario, en los pacientes inmunosuprimidos la criptosporidiosis puede ser persistente y grave. En los pacientes infectados por VIH con cuentas de células T CD4+ mayores de 180/mm3 la criptosporidiosis puede ser autolimitada. Sin embargo, en los individuos más inmunosuprimidos, la diarrea secretora, que es crónica y profusa, puede no remitir. Cryptosporidia puede contribuir también a enfermedades hepatobiliares, como colecistitis, colangitis y estenosis papilar.
Se pueden preparar concentrados de ooquistes de Cryptosporidium mediante un tratamiento previo con la técnica de flotación de azúcar a partir de material fecal. Los ooquistes de Cryptosporidium pueden observarse en frotis de heces examinados al microscopio, ya sea con una preparación en fresco de lugol o después de teñir con un anticuerpo monoclonal o el agente modificado Kinyoun [ver figura 2].Además, las tinciones de fluorescencia directa y los inmunoensayos de muestras de heces pueden aumentar la sensibilidad diagnóstica.
Tratamiento En los pacientes inmunocompetentes la criptosporidiosis es
una enfermedad autolimitada y generalmente solo requiere tratamiento de apoyo.
Sin embargo, no se ha establecido una quimioterapia eficaz para los pacientes
inmunocomprometidos.26 Si se están
administrando fármacos inmunosupresores, la suspensión de
éstos puede resolver la diarrea. En algunos pacientes infectados por VIH
la paromomicina puede tener un beneficio parcial en el tratamiento de la
criptosporidiosis.26
Ciclosporidiosis
Cyclospora parece tener una distribución geográfica amplia y se ha descrito enfermedad atribuible a este protozoario en Estados Unidos, Latinoamérica, Africa, Europa y Asia.64 Los estudios epidemiológicos indican que el agua contaminada, 59 así como las frambuesas importadas, 65 pueden ser la fuente de infección. Se requieren más estudios para definir otros modos de trasmisión. El periodo de incubación es de alrededor de una semana. 59
Diagnóstico Muchos pacientes infectados han presentado diarrea, síntomas gripales, y síntomas comunes a otros patógenos del intestino delgado, incluyendo flatulencia y eructos. La enfermedad puede consistir en un episodio único o tener recurrencias, y son frecuentes la diarrea prolongado, la anorexia y los síntomas del tubo digestivo alto. La fatiga prolongada y la pérdida de peso también son comunes.
Las biopsias de intestino delgado permiten detectar el parásito en las células epiteliales y detectar inflamación yeyunal, que se asocia con aumento en el número de linfocitos intraepiteliales, atrofia de las vellosidades e hiperplasia de las criptas.143 No hay leucocitos ni sangre en las heces, lo que sugiere que la enfermedad se produce por mecanismos no invasores. El diagnóstico puede hacerse por detección de los ooquistes en el excremento que, como los ooquistes de Cryptosporidium, son aparentes con tinción ácido resistente. Aunque los ooquistes de Cyclospora, que miden entre 8 y 10 micras de diámetro, son más grandes que los de Cryptosporidium, se requiere tener cuidado para no confundir estos dos protozoarios. La microscopía fluorescente es un método rápido y sensible para detectar ooquistes autofluorescentes. 67 Pueden encontrarse ooquistes de Cyclospora en pacientes infectados por VIH.60
Tratamiento El trimetoprim-sulfametoxazol (160 mg de trimetoprim y 800 mg de sulfametoxazol) dos veces al día durante 7 días ha demostrado ser eficaz para la ciclosporidiasis. 26 Sin embargo, los pacientes infectados por VIH pueden requerir una dosis mayor (4 veces al día por 7 días) y un tratamiento de mantenimiento por tiempo prolongado (tres veces por semana). Como en el caso de la criptosporidiosis, debe pensarse en infección por Cyclospora en cualquier paciente con diarrea prolongada, anorexia y síntomas del tubo digestivo alto.
INFECCION POR DIENTAMOEBA FRAGILIS
A diferencia de las otras amibas, Dientamoeba fragilis no tiene una etapa de quistes. El trofozoíto no resiste la acidez del estómago, por lo que aún no se ha podido explicar cómo aparece la infección en el hombre. Es posible que los huevecillos del nemátodo Enterobius vermicularis acarreen los trofozoítos de Dientamoeba fragilis, puesto que es frecuente que ambas infecciones coexistan. D. fragilis produce una enfermedad caracterizada por dolor abdominal, anorexia y evacuaciones disminuidas de consistencia. Como en el caso de las infecciones por Isospora, pero no de otras infecciones por protozoarios, puede existir eosinofilia.68El estado de trofozoíto es muy frágil y difícil de detectar; para mejorar su detección, las muestras de excremento deben conservarse con fijador de alcohol polivinílico, fijador de acetato de sodio-ácido acético-formalina o líquido de Schaudinn, y deben examinarse después de su tinción permanente. La infección por D. fragilis puede tratarse con tetraciclina (500 mg qid durante 10 días), paromomicina (25 a 30 mg/kg por día en tres dosis durante 7 días) o iodoquinol (650 mg tid durante 20 días). 26
INFECCION POR BLASTOCYSTIS HOMINIS
Aunque antes se consideraba a Blastocystis hominis una levadura no patógena, en la actualidad algunos investigadores consideran que este microrganismo es un protozoario capaz de causar infección intestinal. Esta creencia parte del hallazgo de la presencia de grandes cantidades de microrganismos B. hominis en las heces del paciente con diarrea sin que se logre identificar otra causa. Sin embargo, otros investigadores han fracasado en confirmar estas observaciones,64 y no han encontrado concordancia entre el número de microrganismos en las heces, el grado de diarrea y la resolución de los síntomas después del tratamiento de la infección por Blastocystis. Mientras se resuelve la patogenicidad de B. hominis, los pacientes con diarrea que excretan este microrganismo en sus heces deben estudiarse para descartar infecciones parasitarias, bacterianas o virales. Si la diarrea es de suficiente magnitud para justificar tratamiento, se puede usar iodoquinol en dosis de 650 mg tres veces al día durante 20 días, o metronidazol en dosis de 750 mg tres veces al día por l0 días. 26
MICROSPORIDIOSIS
Los microsporidios, que son organismos intracelulares obligados formadores de esporas, pertenecen a un phylum distinto de protozoarios que incluye muchos géneros capaces de infectar a huéspedes vertebrados e invertebrados. Las infecciones humanas por microsporidios se han detectado sólo hasta hace poco y a la fecha se han identificado cuatro géneros (Enterocytozoon, Encephalitozoon, Pleistophora y Nosema) como causa de enfermedad en humanos, en especial en individuos infectados por VIH.70 Estos microsporidios se distinguen por su tamaño, morfología nuclear y modo de división, así como por el sitio de proliferación intracelular (los microsporidios se multiplican libres en el citoplasma a dentro de vacuolas unidas a la membrana). A pesar de la ubicuidad de estos organismos en otras especies, no se sabe la manera como se infectan los humanos. Debido a que casi todas las infecciones por microsporidios detectadas en humanos ocurren en huéspedes inmunosuprimidos, no se conoce la frecuencia con la que se infectan los pacientes inmunocompetentes o si en éstos últimos las infecciones son asintomáticas o autolimitadas.
El cuadro clínico de la enfermedad atribuible a los microsporidios es amplio y el estado de la enfermedad parece depender de la especie infectante y de la función inmunológica del huésped. Las infecciones causadas por microsporidios más frecuentes son las de Enterocitozoon bieneusi en pacientes infectados por VIH. Entre los pacientes con infección por VIH, cada vez se reconocen más infecciones intestinales por E. bieneusi y Encephalitozoon intestinalis (antes Septata intestinalis) que causan diarrea crónica.71,72 En estos pacientes E. bieneusi y E. intestinalis pueden infectar las vías biliares, causando colangitis. Las especies Encephalitozoon han provocado queratoconjuntivitis caracterizada por queratopatía epitelial con puntilleo burdo en pacientes infectados por VIH, mientras que las especies Vittaforma y Nosema se han relacionado con queratitis del estroma en algunos pacientes VIH seronegativos. Los microsporidios Encephalitozoon se han asociado también con peritonitis, hepatitis o infecciones nasales y sinusales en pacientes infectados por VIH y las especies Trachipleistophora y Pleistophora han causado miositis.
El tamaño tan pequeño de los microsporidios, que son organismos gram positivos con esporas maduras que miden 0.5 a 2 por 1 a 4 micras, dificulta la detección del parásito, y muchas infecciones han requerido de examen del tejido por microscopía electrónica para realizar el diagnóstico. Las esporas intracelulares pueden detectarse también por microscopía de luz si los tejidos se tiñen con hematoxilina-eosina, Giemsa o tinción de Gram. Los microsporidios intestinales parecen distribuirse en parches y pueden no detectarse en los tejidos biopsiados, pero un método basado en la tinción del cromotropo y los métodos de tinción de fluorocromo Uritex 2B facilitan la detección de las esporas en frotis de heces o de aspirado duodenal.
Para las infecciones intestinales con E. intestinalis, el albendazol oral es benéfico y curativo. Las infecciones por E. bieneusi responden menos, aunque los pacientes pueden presentar mejoría sintomática sin erradicación de la infección. 71,72 Para la queratoconjuntivitis causada por Encephalitozoon hellem se emplea el tratamiento tópico con suspensión de fumagilina.26
Infecciones por amibas de vida libre
Aunque algunas amibas como E. histolytica no pueden sobrevivir y replicarse fuera de los animales huéspedes, la mayor parte de las amibas son de vida libre en el suelo y el agua. Las amibas del género Naegleria y Acanthamoeba pueden causar meningitis aguda, meningoencefalitis aguda y meningoencefalitis granulomatosa crónica.73
Las especies Acanthamoeba se reconocen cada vez más como causa de queratitis74y, en un pequeño número de pacientes infectados por VIH, de enfermedad diseminada con manifestaciones cutáneas.75 Estos microrganismos han sido aislados del agua, polvo del aire, tuberías calientes y soluciones salinas empleadas para limpiar los lentes de contacto. Los factores relacionados con el desarrollo de queratitis amibiana incluyen la falta de desinfección de lentes de contacto, el antecedente de traumatismo corneal leve y la exposición a tierra o agua estancada. Las lesiones suelen ser crónicas y muy dolorosas y consisten en uveítis anterior variable, erosión epitelial, escleritis y queratitis infiltrativa del estroma, que a menudo tiene forma de anillo. Las lesiones son refractarias a los medicamentos antimicrobianos habituales y deben distinguirse de la queratitis producida por virus herpes simple. El diagnóstico se confirma por el examen microscópico de raspados corneales teñidos con Giemsa o tricrómico y mediante el uso de tinción indirecta con anticuerpos inmunofluorescentes de raspados corneales. Las amibas pueden cultivarse al inocular el tejido corneal en agar no nutritivo sembrado con E. coli.
La queratitis por Acanthamoeba puede ser tratada con esquemas tópicos con uno o varios fármacos como biguanida de polihexametileno, isetionato de propamidina y clotrimazol. 74
Leishmaniasis
Los protozoarios del género Leishmania, parásitos intracelulares obligados, producen una amplia gama de enfermedades en el hombre, que van desde la forma visceral generalizada, hasta las lesiones difusas o circunscritas de la piel y membranas mucosas. Cuatro especies de Leishmania parasitan al hombre: L. donavani, L. tropica, L. mexicana y L. braziliensis. Los síndromes clínicos que resultan de la infección dependen tanto del tropismo de la especie de protozoarios como de la respuesta inmunológica, principalmente celular, del huésped.
LEISHMANIASIS VISCERAL
Etiología y epidemiología
El complejo de especies L. donovani incluye varias especies (v.gr., L. infantum y L. chagasi). Estas especies causan la leishmaniasis visceral o kala-azar, que es endémica en varias zonas de la India, China, Centro y Sudamérica, Este y Oeste de Africa, y de los países que circundan el Mediterráneo. Los vectores son moscas del género Phlebotomus y la especie varía en las diferentes áreas. En la India no se conocen reservarios no humanos, pero en las otras regiones se ha observado la infección de otros mamíferos, como perros, zorros y roedores salvajes.
Patogenia
La mordedura del insecto introduce en el huésped promastigotes flagelados de L. donovani. Después de ser fagocitados por macrófagos del sistema reticuloendotelial, se transforman en amastigotes que se reproducen dentro de las células fagocíticas. Los amastigotes, que se liberan al morir la célula huésped, se diseminan por vía hematógena e infectan otras células reticuloendoteliales en el hígado, el bazo, los ganglios linfáticos, la médula ósea y la piel.
Diagnóstico
Cuadro clínico Los síntomas tienen un desarrollo gradual a lo largo de varios meses después de la infección; la enfermedad se caracteriza por debilidad, mareo, perdida de peso, diarrea y estreñimiento. Casi siempre hay fiebre, que presenta dos elevaciones diarias, y en algunas ocasiones se acompaña de calosfríos y diaforesis. Conforme la enfermedad progresa hay crecimiento del hígado y del bazo; este último llega a alcanzar la fosa iliaca. Cuando los macrófagos de la médula ósea son parasitados se desarrollan anemia y leucopenia. El paciente trombocitopénico sangra por las encías, la nariz y el tubo digestivo, en la piel pueden aparecen equímosis y petequias. La muerte es secundaria a infecciones bacterianas agregadas, anemia grave o hemorragia incontrolable. La infección latente se vuelve a manifestar durante la inmunopresión y la leishmaniasis visceral puede desarrollarse como infección oportunista en pacientes infectados por VIH. En estos pacientes los parásitos pueden localizarse en sitios no habituales, incluyendo la laringe y el tubo digestivo.
Estudios de laboratorio Los hallazgos de laboratorio que sugieren el diagnóstico de leishmaniasis visceral, cuando se observan en un paciente con fiebre, hepatosplenomegalia e historia de haber visitado las zonas endémicas, son los siguientes: anemia, leucopenia, trombocitopenia, hiperglobulinemia e hipoalbuminemia. El diagnóstico definitivo requiere del cultivo del microrganismo en medio de Novy-MacNeal-Nicolle (NNN) o algún otro medio específico, o bien la observación microscópica de los cuerpos de Leishman Donovan (amastigotes) en los frotis de tejido. En la mayoría de los casos el diagnóstico puede establecerse examinando aspirados de médula ósea. También es muy sensible para el diagnóstico la punción esplénica, pero tiene mucho riesgo. Otros lugares de donde puede obtenerse material para el diagnóstico son el hígado o los ganglios linfáticos afectados.
Tratamiento
El estibogluconato de sodio (antimonio pentavalente) es el tratamiento de elección para la mayoría de los casos de leishmaniasis, y está disponible en los EUA como Pentostam a través del Servicio de Medicamentos de los CDC en EUA (404-639-3670 en días hábiles y 404-639-2888 durante la noche y fines de semana) [ver cuadro, Información sobre infecciones por protozoarios en internet]. 26 Cuando el tratamiento inicial del kala-azar con estibogluconato de sodio fracasa, puede emplearse anfotericina B o pentamidina.26 El kala-azar resistente al estibogluconato de sodio puede también responder al tratamiento con anfotericina B liposomal.76
LEISHMANIASIS CUTANEA Y MUCOCUTANEA
Etiología y epidemiología
La leishmaniasis cutánea del Viejo Mundo es causada por tres especies de Leishmania que pertenecen al complejo L tropica: L. tropica existe en el Medio Oriente y en el litoral del Mediterráneo, L. major se encuentra en el Medio Oriente, Arabia, la antes Unión Soviética, India y la región Sub-Sahara de Africa, y L aethiopica se encuentra sobre todo en Etiopía y Kenya. Las moscas del género Phlebotomus son los vectores principales, aunque el contacto directo con una persona enferma puede también ser infectante. Las infecciones que son causadas por Leishmania pueden ser adquiridas por viajeros, así como por personal militar y otras personas que residan en áreas endémicas. El personal militar asignado al Medio Oriente puede adquirir leishmaniasis cutánea por L. major, así como infecciones viscerales por L. tropica.
La leishmaniasis del Nuevo Mundo se debe a infección por parásitos pertenecientes al grupo L. mexicana o L. viannia. El cuadro clínico varía de acuerdo con la naturaleza del microrganismo infectante, que a su vez es especifico de las diferentes regiones de América del Norte, del Centro y del Sur [ver tabla 2 ]. En las áreas de Centro y Sudamérica la infección por microrganismos del complejo L. mexicana produce una leishmaniasis cutánea. Se han informado algunos casos de leishmaniasis autóctona de este tipo en Texas. La infección con algunas de las cepas de L. viannia que son endémicas en varias áreas de Sudamérica, causa leishmaniasis cutánea, pero con el tiempo pueden evolucionar a una forma mucocutánea. La forma mucocutánea de esta leishmaniasis (espundia) afecta principalmente la mucosa nasal y de la orofaringe, y en algunos casos puede ser mortal. Todos estos parásitos de leishmania del Nuevo Mundo son también flebotomos, aunque el contacto directo con una persona enferma también puede trasmitir la infección. Varios mamíferos de vida salvaje son los reservorios naturales del microrganismo.
Patogenia
Tanto la leishmaniasis del Viejo Mundo como la del Nuevo Mundo se inician con la mordedura de un flebotomo, que inyecta los promastigotes al huésped. Entonces el microrganismo penetra en los macrófagos y en las células endoteliales, se convierte en amastigotes y se multiplica. En el sitio de la picadura se desarrolla una respuesta inflamatoria granulomatosa. Con la isquemia local la lesión se ulcera y la infección bacteriana del área necrótica extiende la ulceración.
En la leishmaniasis cutánea del Viejo Mundo el periodo de incubación dura semanas o meses, después de los cuales se forma una pápula en el lugar de la picadura. La lesión inicial puede resolverse espontáneamente, pero en general progresa hacia la ulceración, formándose una placa eritematosa circular, con diámetro de varios centímetros y borde elevado [ver figura 3]. La infección bacteriana agregada puede producir linfadenopatía regional. Las lesiones suelen ser únicas, pero las picaduras múltiples pueden producir varias lesiones concomitantes. La resolución de las lesiones es lenta y en ocasiones tarda más de un año.
Diagnóstico
Cuadro clínico La infección por L. mexicana produce una o varias lesiones localizadas en las áreas expuestas del organismo, como la cara y los pabellones auriculares. 77 Las úlceras cicatrizan en forma espontánea en un periodo de 6 meses, pero si se localizan en los pabellones auriculares pueden causar gran destrucción tisular. La infección por L. viannia provoca lesiones en la piel o mucosas que pueden ser múltiples y muy grandes, sobre todo cuando tienen infección agregada por bacterias. Las lesiones cutáneas causadas por L. braziliensis cicatrizan en forma espontánea con mucho menos frecuencia que las ocasionadas por L mexicana. 77 En ocasiones las lesiones se diseminan por los vasos linfáticos de los brazos o piernas, o se extienden en forma local y afectan las mucosas. Después de un periodo de meses o años pueden encontrarse lesiones metastásicas en las mucosas de nariz y boca. Las úlceras en estos lugares pueden erosionar el tabique nasal y el paladar duro y blando. Algunos pacientes mueren por desnutrición o infecciones bacterianas agregadas.
La leishmaniasis cutánea difusa se ha observado en Etiopía, Venezuela, Brasil y la República Dominicana. En estos casos el nódulo inicial no se ulcera, sino que aparecen múltiples nódulos diseminados en todo el cuerpo. En las lesiones los microrganismos son muy abundantes. Esta forma diseminada de leishmaniasis se asocia con un defecto en la inmunidad celular, similar al que se observa en la lepra lepromatosa.
Estudios de laboratorio El diagnóstico definitivo se basa en la observación de amastigotes en frotis de tejidos teñidos en forma adecuada [ver figura 4], obtenidos mediante la biopsia o raspado del borde de las úlceras. Otra alternativa diagnóstica consiste en cultivar las amastigotes en medio NNN, inoculándolo en forma directa con material de las úlceras. El uso de sondas de ADN basado en el ADN cinetoplástico del parásito ha permitido la detección de microrganismos que podrían pasar desapercibidos en la sección histológica o los cultivos. Así mismo, esta técnica constituye un método rápido para determinar si la especie es L mexicana o L. viannia. A diferencia de los amastigotes de L. tropica y L mexicana, que son abundantes en las lesiones y fáciles de cultivar, los amastigotes de L. viannia son escasos y difíciles de cultivar, sobre todo en el paciente con leishmaniasis monocutánea. Los microorganismos de las tres especies no pueden distinguirse por su morfología y cuando se desea el diagnóstico de la especie deben utilizarse estudios inmunológicos especializados, enzimáticos o del ácido nucleico del parásito. La prueba cutánea (intradermorreacción) con leishmanina es positiva en todos los casos, excepto en la leishmaniasis cutánea diseminada.
Tratamiento
Aunque los compuestos animoniales pentavalentes, estibogluconato de sodio [ver antes, Leishmaniasis visceral] y antimonato de meglumina (Glucantime), son los tratamientos de elección para la leishmaniasis cutánea del Nuevo y Viejo Mundo,26no se han establecido en definitiva los esquemas terapéuticos óptimos con éstos u otros agentes. Las diferentes especies y cepas geográficas de Leishmania responden en forma distinta al tratamiento y tienen diferente capacidad para causar enfermedad mucosa o cicatrizar en forma espontánea. Además, es difícil distinguir las especies infecciosas solo por clínica. Por lo tanto, no se ha logrado avanzar en el desarrollo de esquemas de tratamiento adecuados. La División de Enfermedades Parasitarias de los CDC de los EUA proporcionan orientación sobre el tratamiento de la leishmaniasis [ver cuadro, Información sobre infecciones por protozoarios en internet].
Tripanosomiasis
La infección por el protozoario flagelado Trypanosama cruzi produce la tripanosomiasis americana o enfermedad de Chagas, que tiene una forma aguda y otra crónica.78
Etiología y epidermiología
Dos fases de T. cruzi son capaces de infectar a los mamíferos, los tripanosomas, que se encuentran en la sangre, y los amastigotes, que se localizan dentro de las células infectadas. Varios géneros de chinches, triatomídeas o reduvídeos trasmiten el parásito; estos vectores se conocen comúnmente como chinches asesinas por ser predadoras de otros insectos u hociconas por su predilección por morder la cara de sus víctimas. Los reservorios no humanos son gatos, perros, ratas, mapaches y armadillos. El patrón de trasmisión de T. cruzi es diferente según la zona y depende de la especie de reduvídeo que prevalezca en la región, de los reservorios silvestres y domésticos y de las viviendas. Los insectos vectores habitan los orificios y nichos de las paredes, y los canceles de las puertas de las viviendas mal construidas. Los animales infectados que habitan en los alrededores de las viviendas rurales son los que trasmiten el agente a los vectores. Por las noches los vectores salen y se alimentan de la sangre de las personas que duermen, produciéndoles picaduras en las áreas expuestas, principalmente la cara. Durante el acto de chupar sangre, los insectos defecan, y sus heces contienen tripanosomas metacíclicos en estado infectante, que entran en el huésped a través de la herida producida por la picadura, por lesiones cutáneas, o atravesando la mucosa de las conjuntivas y los labios.
Aunque en los Estados Unidos existen los vectores reduvídeos, y también se han demostrado mamíferos infectados, parece ser que las condiciones generales de vivienda impiden que aparezca la trasmisión a los humanos. Sólo se han notificado algunos pocos casos autóctonos de enfermedad de Chagas en los Estados Unidos. 78 En las zonas rurales de Centro y Sudamérica las infecciones son frecuentes cuando las condiciones del medio propician el contacto entre el vector y el huésped humano. Además, la infección se puede trasmitir a través de transfusiones de sangre o sus productos, a través de la placenta y, en raros casos, por la ingestión de alimentos contaminados con deyecciones infectadas.
Patogenia
Se han descrito dos formas de enfermedad de Chagas. La forma aguda se desarrolla inmediatamente después de la infección, y afecta sobre todo a los niños de las áreas endémicas. 78 Después de algunos días de inóculo, aparece una lesión eritematosa indurada en el lugar de entrada del microrganismo que se denomina chagoma. Cuando el lugar de entrada es la conjuntiva ocurre una conjuntivitis unilateral, edema periocular y linfadenopatía preauricular que en conjunto constituyen el denominado signo de romaña. Después de dos semanas los tripanosomas aparecen en la sangre y a través de ella infectan las células, principalmente las de origen mesenquimatoso, donde se multiplican en su forma de amastigotes. La proliferación excesiva produce un seudoquiste celular, que al romperse libera tanto tripanosomas como amastigotes.
Diagnóstico
Cuadro clínico Durante esta fase aguda el huésped experimenta un cuadro de fiebre continua o intermitente, una erupción evanescente semejante a la del sarampión, hepatoesplenomegalia, linfadenopatía, edema sin fóvea en la cara y extremidades, nódulos subcutáneos dolorosos denominados chagomas hematógenos y, en los niños, diarrea. En los casos graves puede presentarse miocarditis y meningoencefalitis mortales. La fase aguda de la enfermedad es autolimitada, los síntomas desaparecen y los parásitos ya no pueden detectarse en la sangre del huésped.
La forma crónica de la enfermedad de Chagas se hace aparente después de una infección aguda o más frecuente subclínica. En general se observa en la segunda o tercera décadas de la vida y progresa en las décadas posteriores. Las complicaciones crónicas de la enfermedad de Chagas son producto de la destrucción de los ganglios del sistema nervioso autónomo, y de la miositis; la patogenia de tales lesiones se desconoce. El órgano que con mayor frecuencia se afecta es el corazón, 78 que sufre hipertrofia biventricular y miocarditis con infiltrado mononuclear. Los trastornos del ritmo que se asocian al daño cardiaco suelen ser bloqueo de rama derecha del haz de His, bloqueo completo o incompleto de la conducción aurículoventricular y presencia de contracciones ventriculares prematuras. Ha ocurrido muerte súbita en pacientes con enfermedad de Chagas, y también defunciones por complicación de insuficiencia cardiaca congestiva.
El tubo digestivo es el segundo lugar de afección en frecuencia.78 La enfermedad produce denervación, con los consiguientes trastornos de la motilidad y dilatación, siendo frecuente encontrar magaesófago y megacolon.
Las infecciones congénitas producen nacimientos prematuros, con niños que presentan hepatoesplenomegalia, distensión abdominal, cardiomegalia, megaesófago y meningoencefalitis. En los pacientes infectados por VIH la reactivación de la enfermedad de Chagas puede provocar masas cerebrales y, en pacientes con infecciones agudas, encefalitis necrosante.
Estudios de laboratorio En la fase aguda de la enfermedad de Chagas es usual encontrar leucocitosis superior a 18,000/mm 3 (con un 70 a 90 por ciento de linfocitos), además de detectarse el parásito en la sangre. Se han podido observar los parásitos en movimiento en exámenes de sangre fresca sin teñir; cuando los frotis se tiñen con Giemsa, los parásitos se observan con forma de una C mayúscula. Si los frotis no muestran parásitos, se recomienda centrifugar varios centímetros cúbicos de sangre, lisando previamente los eritrocitos, y realizando nuevos frotis con sedimento. Los microrganismos pueden cultivarse en muestras de sangre en medio NNN o en agar sangre. Otra alternativa es inocular a algún roedor de laboratorio con sangre problema y vigilar estrechamente la aparición de parasitemia. La técnica de laboratorio más sensible que existe es el xenodiagnóstico, que por desgracia no es accesible a todos los laboratorios. La técnica consiste en alimentar reduvídeos cultivados en laboratorio con la sangre del paciente, si la sangre ingerida por los insectos contiene tripanosomas los insectos se infectarán, lo que se detecta por examen subsecuente de las heces en busca de los parásitos. La sangre que contiene tripanosomas es un material infeccioso y debe manejarse con precaución. Los CDC pueden realizar, a solicitud del médico, una prueba de fijación de complemento que es de ayuda diagnóstica en la fase aguda.
En la fase crónica de la enfermedad de Chagas la radiografía de tórax puede mostrar cardiomegalia biventricular y datos de insuficiencia cardiaca congestiva. Es frecuente encontrar alteraciones electrocardiográficas. De los otros métodos, sólo el xenodiagnóstico es capaz de detectar la parasitemia. La prueba de fijación de complemento puede ser positiva, pero sólo indica una infección previa, y no establece una correlación entre los hallazgos clínicos y una enfermedad de Chagas activa.
Tratamiento
Aún no se ha establecido el tratamiento óptimo de la enfermedad de Chagas. El nifurtimox (disponible por medio de los CDC) elimina la parasitemia [ver cuadro, Información sobre infecciones por protozoarios en internet]. Se recomienda su administración en pacientes con fase aguda y en pacientes con fase crónica y parasitemia demostrada.26 Los efectos colaterales son frecuentes, incluyendo anemia hemolítica en pacientes con deficiencia de G6PD, neuritis periférica y psicosis. Las complicaciones cardiovasculares y gastrointestinales de la fase crónica se manejan con medicamentos, aunque en ocasiones se requiere cirugía para el megacolon.
TRIPANOSOMIASIS AFRICANA
Etiología y epidemiología
La tripanosomiasis africana o enfermedad del sueño, es una parasitosis aguda o crónica causada por hemoflagelados del género Trypanosoma. La enfermedad es endémica en un ancho cinturón que atraviesa Africa Ecuatorial. Dos variedades infectan al hombre: la tripanosomiasis del Africa Occidental se presenta en los bosques de la porción Occidental y Central de Africa, y es causada por T. brucei gambiense, un parásito que no tiene reservorio animal importante. En cambio, T. brucei rhodesiense, que produce la enfermedad del sueño de Africa Oriental, sí tiene reservorios animales, en especial de vida salvaje. Los visitantes de los parques nacionales tienen riesgo de adquirir este tipo de tripanosomiasis. 79 Ambas tripanosomiasis africanas son trasmitidas por varias especies de moscas tsetsé (del género Glossina ).
Diagnóstico
Cuadro clínico Después de un período de incubación que varía entre unos cuantos días a un par de semanas, aparece el chancro tripanosómico en el lugar de la picadura de la mosca tsetsé, localizado en áreas expuestas de la piel.79 El chancro en un principio es una pápula, que evoluciona en 2 semanas hacia un nódulo doloroso e inflamado que se resuelve espontáneamente. Aunque el chancro tripanosómico es común en pacientes no africanos, rara vez se observa en pacientes africanos.
Sigue después la fase hemolinfática de la enfermedad, en la que los protozoarios invaden el torrente sanguíneo y los ganglios linfáticos. Los síntomas que acompañan esta fase se desarrollan con lentitud en los africanos y consisten en fiebre, linfadenopatía, debilidad y cefalea. En cambio, en los no africanos el inicio es súbito y a menudo coincide con el desarrollo del chancro. En estos últimos llaman la atención los periodos febriles que duran de 1 a 7 días, y que recurren después de cortos periodos afebriles. A la fiebre se asocian otros síntomas: calosfríos, cefalea, malestar general y anorexia. La linfadenopatía, con ganglios blandos y no dolorosos, es más frecuente en la tripanosomiasis de Africa Occidental, e incluye los ganglios de las cadenas cervicales posteriores (signo de Winterbottom). También se presenta una erupción típica, que se observa mejor en los individuos de color claro, y aparece entre la sexta y octava semana de la infección, caracterizada por máculas eritematosas, circinadas y evanescentes, de predominio en el tronco.
La siguiente fase en la tripanosomiasis africana es la invasión del sistema nervioso central, produciendo una meningoencefalitis o meningomielitis difusa. En la tripanosomiasis del Africa Occidental, que tiene una evolución muy lenta, los síntomas de la enfermedad del sueño se desarrollan varios años después de la infección. La debilidad e indiferencia al medio evolucionan hacia el coma y la muerte por inanición o enfermedades intercurrentes. Por el contrario, en la tripanosomiasis del Africa Oriental la evolución es más rápida y la fase neurológica se presenta en forma temprana. Aún antes de que exista afección del sistema nervioso central es frecuente encontrar somnolencia, cambios de la personalidad y dificultad para concentrarse. En la tripanosomiasis del Africa Oriental puede haber pancarditis durante la fase aguda, que puede producir la muerte antes de la fase neurológica. Debido a que la tripanosomiasis del Africa Oriental puede adquirirse al visitar los parques y reservas de animales y es una enfermedad aguda y febril, es frecuente que se confunda con el paludismo. La enfermedad del sueño debe considerarse en el diagnóstico diferencial de cualquier proceso febril que aparece en el paciente que regresa de un área endémica.
Estudios de laboratorio El recuento leucocitario es normal, pero puede existir una mononucleosis de 50 al 70 por ciento. Los niveles séricos de IgM se elevan de 1 a 2 semanas después del comienzo de la parasitemia y pueden alcanzar hasta 7 veces los valores normales. El diagnóstico definitivo se hace por observación de los tripanosomas en la sangre, médula ósea, líquido de los ganglios infectados o líquido cefalorraquídeo previamente centrifugado. Los microrganismos teñidos con Wright, Giemsa o tinción de Leishman, pueden observarse en los frotis tomados del chancro hasta 48 horas antes de que aparezcan en la sangre. Si el examen en sangre periférica no es diagnóstico, los protozoarios pueden encontrarse en aspirados de ganglios linfáticos infectados, o en leucocitos del sobrenadante de una muestra de sangre centrifugada. Incluso en los casos en que no hay síntomas neurológicos, debe investigarse la enfermedad del sistema nervioso central mediante punción lumbar, y aun cuando no se observen los tripanosomas en el centrifugado del líquido cefalorraquídeo, la elevación del recuento celular, de las proteínas o de la fracción IgM, habla en favor de enfermedad neurológica.
Tratamiento El eflornitine ha demostrado ser eficaz para las infecciones por T. brucei gambiense, 2,23 incluso las que son refractarias al agente arsenical melarsoprol. El eflornitine es el tratamiento de elección para los estadios hemolinfático temprano y tardío del SNC de la tripanosomiasis de Africa Occidental. 26 Debido a que este medicamento puede causar anemia, leucopenia y trombocitopenia, deben realizarse citologías hemáticas dos veces por semana durante el tratamiento. El eflornitine no ha sido eficaz para las infecciones por T. brucei rhodesiense. El estadio temprano de la tripanosomiasis de Africa Oriental se trata con suramina, y el estadio de SNC con melarsoprol.26 Ambos medicamentos pueden solicitarse en los EUA en el Servicio de Medicamentos de los CDC [ver cuadro, Información sobre infecciones por protozoarios en internet].
Reconocimientos
Figura 1 Seward Hung.
Bibliografía
DR. PETER F. WELLER
Toxoplasmosis
Toxoplasma gondii, el agente casual de la toxoplasmosis, es un parásito intracelular obligado de distribución mundial. Infecta prácticamente a todos los mamíferos y aves, lo que le hace uno de los parásitos de mayor distribución en el mundo.1 En el ser humano la infección puede adquirirse por vía oral, transmisión transplacentaria, transfusiones sanguíneas o trasplantes de órganos. La infección por vía oral se debe a la ingestión de quistes de T. gonddi en alimentos mal cocinados o de los ooquistes del parásito contenidos en las heces de gato.1 En un brote pudo atribuirse la fuente a la ingestión de agua contaminada. Después de la ingestión los bradizoítos (de los quistes tisulares) o los esporozoítos (de los ooquistes) invaden las células circundantes y se convierten en taquizoítos, una forma de división rápida que puede diseminarse e invadir cualquier tipo de célula nucleada. Con el tiempo y el desarrollo de inmunidad, el parásito forma quistes tisulares que contienen muchos bradizoítos. Los quistes tisulares pueden permanecer viables por décadas sin causar enfermedad. Sin embargo, la pérdida de la inmunidad permite la reactivación de los quistes latentes y la generación de muchos taquizoítos invasores.
La toxoplasmosis es una infección muy frecuente, pero la mayoría de los casos, tanto congénitos como adquiridos, no dan manifestaciones clínicas y sólo se detectan por la presencia de anticuerpos. La prevalencia de la infección varía según los diferentes grupos étnicos y áreas geográficas; en la mayoría de los estudios se ha encontrado un aumento de la incidencia paralelo a la edad. En los Estados Unidos los estudios serológicos sugieren que el 3 a 30 por ciento de la población ha sufrido infección por Toxoplasma.
SINDROMES CLINICOS
La toxoplasmosis puede dividirse en cinco grandes síndromes clínicos: toxoplasmosis primaria, toxoplasmosis en el inmunosuprimido, toxoplasmosis del SIDA, toxoplasmosis congénita y toxoplasmosis ocular.
Toxoplasmosis primaria
La persona inmunocompetente suele presentar una afección subclínica al adquirir la infección primaria por Toxoplasma. La linfadenopatía no dolorosa es la manifestación más frecuente, puede ser localizada o generalizada y persistir por muchos meses. La linfadenopatía cervical aislada es el hallazgo más frecuente.2 La linfadenopatía puede ser el único signo de la infección y con menos frecuencia ocurren fiebre, malestar general, mialgias y ardor faríngeo; la fatiga y la debilidad pueden ser intensas. En la exploración física, además de la linfadenopatía, puede encontrarse faringitis, exantema maculopapular3 y, en un pequeño grupo de pacientes, hepatoesplenomegalia. No es raro encontrar linfocitos atípicos. El diagnóstico diferencial se hace con la mononucleosis infecciosa, la infección por citomegalovirus y el linfoma; en casos raros, la sarcoidosis, la enfermedad por rasguño de gato y otros procesos infecciosos pueden parecerse a la toxoplasmosis. Las pruebas serológicas dan la clave del diagnóstico.4
Aunque los síntomas de la linfadenopatía por Toxoplasma pueden persistir durante semanas o meses, casi siempre se autolimitan y no requieren tratamiento específico. En casos graves o persistentes, la pirimetamina y las sulfamidas son útiles. Estos mismos medicamentos pueden utilizarse en las complicaciones raras de la toxoplasmosis en el huésped normal, como la coriorretinitis, miocarditis, pericarditis, neumonitis, miositis y meningoencefalitis.
Toxoplasmosis en el huésped inmunosuprimido
En el huésped inmunosuprimido con inmunidad celular deficiente T. gondii puede causar enfermedad neurológica o diseminada grave. Por lo general el proceso se desarrolla a partir de una reactivación de una infección latente y no por una infección primaria. Los receptores seronegativos de órganos (en especial corazón), trasplantados de donadores seropositivos tienen especial riesgo,5,6 lo mismo que los pacientes con enfermedad de Hodgkin, leucemia de células peludas y otros trastornos malignos.7 En por lo menos el 50 por ciento de estos pacientes predominan las alteraciones neurológicas. El cuadro clínico es muy variable y puede semejar una encefalitis difusa, una meningoencefalitis o una lesión cerebral ocupativa.8 El líquido cefalorraquídeo muestra pleocitosis discreta de tipo linfocítico, con elevación de los niveles de proteínas y niveles normales de glucosa. Aparece neumonitis hasta en una tercera parte de los individuos inmunosuprimidas con infección por Toxoplasma.9 Hay fiebre y disnea, pero la tos está ausente y no se produce expectoración. La radiografía de tórax revela, en forma característica, infiltrados pulmonares bilaterales difusos. En algunos casos, el diagnóstico se establece al observar microorganismos en la muestra obtenida por lavado broncoalveolar. Otras manifestaciones de la toxoplasmosis diseminada en pacientes inmunosuprimidos incluyen miocarditis, pericarditis, peritonitis y linfadenitis. Muchos de estos individuos inmunosuprimidas presentan infecciones intercurrentes con otros patógenos oportunistas, sobre todo por herpesvirus y citomegalovirus.
Si la serología demuestra elevación de los títulos de anticuerpos, puede ser de ayuda diagnóstica, pero en estos enfermos los títulos son generalmente bajos y para establecer el diagnóstico se requiere una biopsia cerebral, pulmonar o de ganglio linfático. Se justifica el método diagnóstico agresivo, ya que el tratamiento con sulfonamidas y pirimetamina puede dar buenos resultados y la infección suele ser mortal si se deja sin tratamiento.
Toxoplasmosis en pacientes con SIDA
La toxoplasmosis es un problema muy grave en pacientes con síndrome de inmunodeficiencia adquirida (SIDA); del 3 al 40 por ciento de estos pacientes desarrollan una toxoplasmosis clínicamente aparente.10 Aunque los pacientes con SIDA pueden manifestar cualquiera de las variedades generalizadas de toxoplasmosis,11 la afectación del sistema nervioso central es la más frecuente. La mayoría de los casos de toxoplasmosis en pacientes con SIDA se deben a la reactivación de quistes latentes adquiridos antes de la infección por VIH. La reactivación es más probable cuando las cuentas de células T CD4+ caen a menos de 100 células /mm3. 10 Todas las personas infectadas por VIH que sean negativos a Toxoplasma requieren minimizar la exposición cocinando la carne a una temperatura interna de 150 'F, lavando sus manos después de tocar carne cruda o tierra, lavando frutas y verduras antes de comerlas y evitando el contacto con heces de gato o basura contaminada (o usando guantes al manejar estos materiales). 12
En pacientes con SIDA la toxoplasmosis se presenta con más frecuencia como una encefalitis necrosante. Los síntomas incluyen alteraciones focales (v. gr., hemiparesia, pérdida sensorial, alteraciones visuales, temblor, parálisis de pares craneales y crisis focales) y alteraciones neurológicas generalizadas (v. gr., cefalea, cambios de la personalidad, confusión, estupor o coma y convulsiones). Aunque la pleocitosis linfocítica del LCR apoya el diagnóstico de toxoplasmosis cerebral, la tomografia computada o la imagen por resonancia magnética es fundamental para el diagnóstico en la mayoría de los casos. Las lesiones nodulares únicas o múltiples son hallazgos típicos de la TC; cuando se administra un medio de contraste, más del 90 por ciento de estas lesiones nodulares aumentan de tamaño y tienen forma anular. En ocasiones los estudios del IRM revelan lesiones que no se pueden visualizar con la tomografía computada. 13 La tomografía de emisión de positrones (TEP) puede ser útil para distinguir las lesiones de la toxoplasmosis (que son hipometabólicas) de los linfomas (que son hipermetabólicos).14
Las pruebas de anticuerpos en suero son útiles para la detección de la toxoplasmosis cerebral en pacientes con SIDA; estas pruebas suelen ser positivas porque la mayoría de los casos de toxoplasmosis cerebral en estos pacientes surgen por la reactivación de una infección latente. Aunque las pruebas negativas sugieren un diagnóstico diferente, se han reportado muy pocos casos seronegativos. Sin embargo, las pruebas serológicas no siempre son definitivas para el diagnóstico de toxoplasmosis activa en pacientes con SIDA y los títulos de anticuerpos no alcanzan los altos niveles típicos de los pacientes inmunocompetentes con toxoplasmosis, ni tampoco existen anticuerpos IgM en los pacientes con SIDA.10 Se encuentran anticuerpos contra el Toxoplasma en el LCR en casi dos terceras partes de los pacientes con SIDA y toxoplasmosis cerebral, y su detección puede ayudar en el diagnóstico [ver adelante, diagnóstico]. 15
Otras causas de afectación cerebral en pacientes con SIDA, además de la toxoplasmosis, incluyen hongos, micobacterias, linfomas, sarcoma de Kaposi, tumores metastásicos, leucoencefalopatía multifocal y el VIH por sí mismo. A pesar del amplio diagnóstico diferencial de las lesiones cerebrales en los pacientes con SIDA, es preferible administrar tratamiento empírico para toxoplasmosis, a realizar una biopsia, si el cuadro clínico y radiológico es compatible con el diagnóstico y si los anticuerpos anti-toxoplasma séricos son positivos.16 Debido a que la toxoplasmosis es la causa más frecuente y tratable de lesiones cerebrales en los pacientes con SIDA, puede iniciarse un tratamiento empírico basado en la combinación de sulfonamidas y pirimetamina o clindamicina y pirimetamina. Si el diagnóstico es correcto se observará mejoría clínica y radiológica después de 1 a 2 semanas. Si los pacientes tienen poca respuesta al tratamiento, son seronegativos y si pertenecen a grupos con alto riesgo de tuberculosis (haitianos, africanos, adictos a drogas por vía intravenosa) se debe considerar seriamente realizar una biopsia.16
Toxoplasmosis congénita
La toxoplasmosis congénita aparece casi exclusivamente cuando la madre desarrolla una infección primaria durante el embarazo. La infección congénita casi nunca se desarrolla a partir de una toxoplasmosis latente adquirida antes del embarazo, y los pocos casos detectados ocurrieron en mujeres embarazadas con reactivación de la toxoplasmosis por tratamiento inmunosupresor o infección por VIH.17El riesgo de infección fetal depende de la época en que ocurra la infección materna, aumentando de 10 por ciento durante el primer trimestre a 60 por ciento durante el tercero.18 Las consecuencias de la infección fetal dependen también de cuando ocurra la infección: las infecciones adquiridas al principio de la gestación causan daño más severo.
El espectro clínico de la toxoplasmosis congénita varía mucho. Alrededor del 85 por ciento de los bebés infectados parecen normales al nacimiento; sin embargo, sin tratamiento el 85 por ciento desarrollará coriorretinitis, pérdida de la audición o retraso en el desarrollo.18,19 El espectro clínico de la toxoplasmosis congénita sintomática incluye muerte fetal, daño neurológico (calcificaciones cerebrales, convulsiones, retraso mental, hidrocefalia y microcefalia), coriorretinitis, fiebre, hepatoesplenomegalia y exantema. El diagnóstico diferencial se hace con otras infecciones congénitas por citomegalovirus, herpes simple, rubéola o sífilis. La toxoplasmosis congénita puede diagnosticarse por métodos serológicos por una reacción en cadena de la polimerasa (RCP) realizada en líquido amniótico,20 o al identificar al parásito en el tejido placentario o fetal. Los niños que llegan a sobrevivir pueden beneficiarse con el tratamiento a base de pirimetamina y sulfonamidas.21
La prevención de la toxoplasmosis congénita es de gran importancia. Las mujeres embarazadas deben evitar al máximo el contacto con gatos, en especial los gatos callejeros y los que se alimentan de carne cruda; es muy importante lavarse las manos después de tener contacto con gatos, y si se tienen en casa, otra persona debe encargarse de su aseo; también es recomendable que se laven todas las frutas y vegetales y se cocine adecuadamente la carne. Se recomienda la investigación serológica sistemática de todas las mujeres embarazadas.
Toxoplasmosis ocular
La toxoplasmosis puede causar el 30 por ciento de los casos de retinocoroiditis.22 Muchos casos se deben a reactivación de una infección congénita, por ello la toxoplasmosis ocular es más frecuente en niños mayores y adultos jóvenes. La retinocoroiditis también puede desarrollarse como manifestación de la infección primaria.23 Los síntomas más comunes son atribuibles a la disminución de la agudeza visual, pero en inflamaciones graves también se observa fotofobia y dolor ocular. Las lesiones típicas semejan racimos de exudados blandos de color blanco-amarillento que se observan en el polo posterior del ojo. Aunque el diagnóstico de esta variedad requiere demostración de anticuerpos contra toxoplasma, los títulos son por general bajos, puesto que la infección se adquirió varios años antes. Por lo tanto, en la mayoría de los casos la toxoplasmosis ocular puede diagnosticarse clínicamente reconociendo la morfología de los exudados. El diagnóstico diferencial incluye a la tuberculosis, la sarcoidosis, la sífilis, la histoplasmosis y la candidiasis.
DIAGNOSTICO
Las pruebas serológicas son la piedra angular en el diagnóstico de la toxoplasmosis. Aunque se dispone de muchas, las más utilizadas son la inmunofluorescencia indirecta y la prueba de Sabin y Felman. Los laboratorios de referencia emplean muchas otras pruebas. Todas ellas miden anticuerpos de la clase IgG y se vuelven positivas de 1 a 3 semanas después del comienzo de la infección. El diagnóstico de toxoplasmosis de adquisición reciente, especialmente importante durante el embarazo, y de la toxoplasmosis congénita en el recién nacido, requieren de la detección de anticuerpos anti-toxoplasma de IgM. Algunos estudios comerciales de IgM dan resultados falsos positivos y se asocian con detección de anticuerpos que persisten por más de 1 año, lo que pone en duda la confiabilidad de estas pruebas para detectar solo infecciones recientes.24 Los resultados positivos en las pruebas de IgM requerirán confirmación por pruebas alternativas.
Debido a que los quistes pueden persistir en los tejidos por años, la evidencia definitiva de toxoplasmosis activa a partir de la biopsia de un tejido requiere de la detección de taquizoitos. Estas formas no se observan con facilidad con las técnicas convencionales de patología y se requiere la aplicación de inmunofluorescencia o tinción de anticuerpos peroxidasa-antiperoxidasa. Los taquizoitos no suelen detectarse en la biopsia de ganglios linfáticos, aunque las características histopatológicas del ganglio pueden apoyar el diagnóstico.25 Los métodos de RCP son muy sensibles para detectar el organismo en el líquido amniótico en las infecciones congénitas, 20 pero ni la RCP ni el cultivo directo de los organismos a partir de la sangre o los tejidos se ha usado en el diagnóstico de otras formas de toxoplasmosis.
TRATAMIENTO
Toxoplasmosis primaria y asociada a SIDA
Aunque la mayoría de los pacientes con linfadenopatía por toxoplasma no requieren tratamiento, éste debe considerarse en los casos de enfermedad grave o prolongada. Los pacientes con coriorrenitis activa, afección del sistema nervioso central o toxoplasmosis diseminada sí deben recibir quimioterapia, así como todo paciente inmunosuprimido, incluyendo los pacientes con SIDA.
El tratamiento de elección consiste en una combinación de sulfonamidas (sulfadiazina o trisulfapirimidinas) y pirimetamina. 26 Deben evitarse otras sulfonamidas con pobre acción sobre T. gondii. En los pacientes adultos se administran 200 mg de pirimetamina el primer día, seguido de la dosis habitual de 50 a 75 mg/día. La sulfadiazina o trisulfapirimidinas se administran con una dosis de impregnación de 4 g, para continuar con dosis de mantenimiento de 1 a 2 gramos cuatro veces al día. El efecto tóxico más frecuente de la pirimetamina es la supresión de la médula ósea, por lo que debe vigilarse la cuenta de leucocitos, eritrocitos y plaquetas dos veces a la semana y administrar diario 10 a 15 mg de ácido folínico. El tratamiento combinado de pirimetamina y sulfonamida se administra por 3 a 6 semanas.
Para los pacientes con SIDA que no toleran las sulfonamidas y tienen toxoplasmosis del SNC ha sido eficaz la clindamicina, en dosis de 600 mg por vía oral o intravenosa cuatro veces al día, combinada con pirimetamina.27 También el atovaquone más pirimetamina es eficaz para los pacientes intolerantes a las sulfas.28
Los pacientes con SIDA tratados por toxoplasmosis y los que no sufren infección activa por toxoplasmosis pero que tienen serología positiva y cuentas de linfocitos T CD4+ menores de 100/mm3 tienen riesgo de reactivación y requieren de tratamiento supresor por tiempo prolongado. Existen diferentes esquemas para los pacientes con SIDA que se han recuperado de una toxoplasmosis. El primero consiste en sulfadiacina (500 a 1,000 mg cuatro veces al día) y pirimetamina (25 a 50 mg/día) más ácido folínico (5 mg/día).29 También la clindamicina (300 a 450 mg tres veces al día) es eficaz para los pacientes que no toleran las sulfonamidas.30 Algunos esquemas empleados para la prevención de la neumonía por Pneumocystis proporcionan profilaxis primaria para la toxoplasmosis, como el trimetoprim-sulfametoxazol, la dapsona-pirimetamina, el atovaquone y la pirimetamina sola para pacientes con intolerancia a las sulfas.31
Toxoplasmosis congénita
La toxoplasmosis de adquisición reciente durante el embarazo representa una dificultad terapéutica. La pirimetamina es teratogénica y debe evitarse durante el primer trimestre. La espiromicina, disponible en los Estados Unidos solo directamente a partir del fabricante (Rhone Poulenc Rorer), se administra en dosis de 3 a 4 g/día y puede disminuir el riesgo de infección transplacentaria.26 Si se demuestra infección in utero debe iniciarse tratamiento con pirimetamina y sulfadiacina. Puede considerarse un aborto terapéutico si la infección se adquirió en una etapa temprana del embarazo y usarse ultrasonido para detectar hidrocefalia, calcificaciones intracraneales y otros signos de daño fetal. Los niños que nacen con evidencia serológica o clínica de toxoplasmosis congénita deben tratarse durante un año con pirimetamina y sulfadiacina. 29
Toxoplasmosis ocular
La pirimetamina y las sulfonamidas son la base del tratamiento de la toxoplasmosis ocular, pero los resultados son impredecibles y las recaídas frecuentes. En los casos en los que existe riesgo de ceguera el paciente debe recibir esteroides en combinación con los antibióticos.
Paludismo
Cuatro especies de Plasmodium causan paludismo en el humano: P. falciparum, P. vivax, P. ovale y P. malarie [ver tabla 1]. P. vivax y P. falciparum son de los más prevalentes en el mundo y el paludismo falciparum es responsable de la mayoría de las muertes. El paludismo se trasmite por los mosquitos anófeles. Aunque la trasmisión del paludismo en los Estados Unidos es muy rara, se calcula que cada año ocurren en todo el mundo alrededor de 300 a 500 millones de casos, con tres millones de muertes.32,33 El paludismo ha reaparecido por múltiples factores, incluyendo resistencia del vector mosco a los insecticidas, incapacidad de los programas gubernamentales para controlar las poblaciones de moscos vectores, mayores oportunidades para el crecimiento de los vectores y mayor resistencia de los parásitos a los agentes quimioterápicos.
|
||||||||||||||||||||||||||||||||||||
|
ETIOLOGIA Y EPIDEMIOLOGIA
El paludismo es endémico en muchos países. La mayoría del paludismo en los Estados Unidos es importado. En 1995 se reportaron a los Centros para el Control y Prevención de las Enfermedades (CDC) de los EUA 1,167 casos de paludismo, incluyendo seis muertes. De estos casos, 599 ocurrieron en civiles norteamericanos, 461 en civiles extranjeros y 12 en personal militar.32 P. vivax y P. falciparum causaron el 48 y 39 por ciento de las personas infectadas, respectivamente. Con menos frecuencia el paludismo se trasmite por transfusión sanguínea, al compartir agujas contaminadas, por trasplante de órganos y en infantes que son infectados en forma congénita al nacer de madres infectadas. 32,34 Además, debido a que en los Estados Unidos existen especies de mosquitos Anopheles que pueden trasmitir el paludismo, se han aislado casos de paludismo autóctono en norteamericanos picados por mosquitos que se alimentaron de personas con paludismo importado. 35 Un último medio de introducción del paludismo a los Estados Unidos es el llamado de aeropuerto, que surge cuando un mosco infectado entra al país en una aeronave de un área con paludismo y trasmite la infección en el área alrededor del aeropuerto.
PATOGENIA
Las infecciones se adquieren normalmente por la picadura de un mosco Anopheles hembra infectado, que introduce esporozoítos [ver figura 1]. En la fase preritrocítica de la infección las cuatro especies de Plasmodium infectan a los hepatocitos. Sin embargo, es notable que solo dos especies, P. vivax y P. ovale, causan infección persistente en el hígado. En la fase eritrocítica de la infección los merozoítos liberados por los hepatocitos infectados y después por los eritrocitos infectados interactúan con proteínas de membrana específicas de los eritrocitos y los invaden. (El antígeno sanguíneo Duffy es el receptor eritrocítico necesario para P. vivax. Los individuos que genéticamente no tienen antígeno sanguíneo Duffy, como muchos africanos occidentales y sus descendientes, son resistentes a esa especie de Plasmodium.) Dentro de las células infectadas los parásitos sufren un proceso de esquizogonia para formar nuevos merozoítos, que son liberados para reinfectar otros eritrocitos. Algunos pocos parásitos se diferencian en sus fases sexuales (gametocitos), capaces de infectar mosquitos. Los gametocitos masculinos y femeninos se transforman y reproducen en el intestino medio del mosco, causando la producción de nuevos esporozoítos que se localizan en las glándulas salivales de los mosquitos.
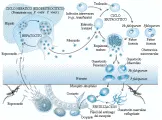
|
| Figura 1 |
| Ciclo vital de Plasmodium |
Debido a la capacidad de los parásitos para replicarse y aumentar su número dentro del humano infectado, pueden desarrollarse infecciones intensas a partir de un inóculo mínimo. Algunos viajeros han desarrollado paludismo después de ser picados por mosquitos durante una breve estancia en un aeropuerto de un área con paludismo y durante vuelos en aeronaves que se han detenido en zonas endémicas. En consecuencia, se justifica la profilaxis incluso cuando la exposición a los mosquitos vaya a ser muy breve.
Los pacientes permanecen asintomáticos durante el periodo entre la picadura del mosquito infectado y la fase eritrocítica de la infección, periodo que puede durar de 1 a 4 semanas. Debido a que la quimioprofilaxis no evita en realidad el paludismo sino que trata la infección en fase eritrocítica, debe continuarse durante 4 semanas después del regreso de una zona endémica. El no continuar la quimioprofilaxis semanal después del retorno del área endémica permite el desarrollo de la infección.
La infección palúdica despierta una respuesta inmunológica. El desarrollo de anticuerpos contra la especie más indolente y crónica del paludismo, P. malariae, puede provocar en años el inicio de un síndrome nefrótico mediado por complejos inmunes. Las infecciones repetitivas con otras especies de Plasmodium pueden causar una inmunidad parcialmente protectora que limita la severidad de la infección, aunque no la previene. Es importante mencionar que los residentes de las áreas con paludismo pierden su inmunidad relativa después de permanecer varios años en una región sin paludismo. Estas personas pueden ser muy susceptibles a la infección al regresar a la región endémica.
DIAGNOSTICO
El paludismo debe ser considerado como causa de cualquier enfermedad febril en personas provenientes de áreas endémicas o que han viajado o trabajado en esas regiones. Los receptores de transfusiones, trasplante de órganos y los neonatos de madres potencialmente infectadas también tienen riesgo. Por último, incluso en los residentes de Estados Unidos, debe pensarse en el diagnóstico en los pacientes con síndrome febril y un cuadro clínico compatible, por la posibilidad de la poco común transmisión autóctona.
Las infecciones con las cuatro especies de Plasmodium que causa paludismo en los humanos producen síndromes clínicos diferentes, en parte por las interacciones de cada especie con los eritrocitos. P. vivax infecta solo eritrocitos jóvenes, lo que ayuda a limitar la intensidad de la infección. También explica por qué los eritrocitos grandes son característicos de las infecciones por P. vivax en los frotis. Por el contrario, P. falciparum infecta eritrocitos de todas las edades. Esta capacidad del paludismo tipo falciparum, el mayor número de merozoítos producidos por esta especie y en especial la mayor propensión de los eritrocitos infectados para adherirse al endotelio de la microvasculatura, contribuyen a que el paludismo falciparum sea más severo que las otras formas. Los eritrocitos infectados con P. falciparum desarrollan protrusiones en la superficie que median la unión y adherencia a las células endoteliales en los capilares y las vénulas. Al secuestrarse los eritrocitos infectados en los vasos pequeños ocurre anoxia. Pueden ocurrir complicaciones severas, incluyendo paludismo cerebral y edema pulmonar [ver tabla 1].
Cuadro clínico
Los síntomas clínicos se desarrollan 1 a 4 semanas después de la infección y típicamente incluyen fiebre y calosfríos. Virtualmente todos los pacientes con paludismo agudo tienen episodios de fiebre. Al inicio la fiebre puede ser diaria, con el tiempo pueden desarrollarse los patrones típicos de cada tercer día (P. vivax, P. ovale, P. falciparum) o cada cuarto día (P. malariae). Los paroxismos de fiebre (hasta 41.5 'C [106.7 'F] y calosfríos (con o sin temblor) pueden ser irregulares, en especial en el paludismo falciparum. Otros síntomas incluyen cefalea, diaforesis, lumbalgia, mialgias, diarrea, náusea, vómito y tos. La constelación de síntomas posibles no es específica y puede sugerir otros diagnósticos.36 Con el tiempo se desarrollan anemia y esplenomegalia.
Debido a la capacidad de los eritrocitos infectados por plasmodio falciparum de causar bloqueo de la microvasculatura, en este tipo de paludismo puede desarrollarse afección orgánica potencialmente fatal. La afección cerebral puede ocasionar delirio, trastornos focales (v.gr., crisis convulsivas) y coma.37 La afección esplácnica produce náusea, vómito, diarrea, melena y dolor abdominal; este síndrome puede confundirse con una diarrea del viajero. El daño pulmonar produce edema y síndrome de insuficiencia respiratoria progresiva del adulto. Puede existir hipoglucemia severa. Un síndrome raro, la fiebre de agua negra, se produce por hemólisis intravascular masiva, lo que ocasiona hemoglobinuria e insuficiencia renal aguda.
Los organismos P. malariae pueden persistir en la sangre y provocan una infección indolente, incluso asintomática, durante años o aún décadas.38
Datos de laboratorio
En la mayoría de los pacientes la cuenta de leucocitos se encuentra dentro del rango normal. Se desarrolla anemia, pero esta puede no ser importante al inicio, en especial si el paciente está deshidratado. Puede ocurrir trombocitopenia y en ocasiones coagulación intravascular diseminada en el paludismo falciparum. Las enzimas hepáticas pueden aumentar.
El diagnóstico específico y de tipo depende de la identificación de los microorganismos en el frotis de sangre periférica teñido de manera adecuada. 32 Los frotis gruesos son más sensibles que los delgados, pero por las capas de células se requiere mayor experiencia para examinar la morfología. Los frotis deben tomarse diariamente durante varios días debido a la naturaleza cíclica de la parasitemia, lo que adquiere particular importancia si se sospecha infección por P. falciparaum, en la que las células infectadas pueden estar secuestradas en la microvasculatura. Las características morfológicas del parásito y de los eritrocitos infectados son útiles en la identificación de las especies, y en la distinción de Plasmodium del microorganismo muy similar, Babesia microti, que causa la babesiosis humana. Es muy importante identificar con rapidez las infecciones por P. falciparum por su rápida evolución cuando no se tratan apropiadamente.
La técnica de cubierta amortiguadora cuantitativa (QBC por sus siglas en inglés, n. del t.) es sensible para identificar la parasitemia por plasmodios, pero no determina la especie. Existen tiras de ensayo de captura de antígenos, que permiten a los clínicos detectar infecciones por P. falciparum, en especial cuando no se cuenta con laboratorios especializados.39 Las sondas de ADN, las pruebas de RCP y las pruebas serológicas no suelen estar muy disponibles para el diagnóstico. Por lo tanto, el frotis de sangre sigue siendo la prueba estándar para determinar, primero, si un paciente tiene paludismo y, segundo, si éste es causado por P. falciparum.
TRATAMIENTO
El tratamiento de los pacientes con paludismo agudo requiere tomar en cuenta la especie y, en el caso de paludismo falciparum, la posibilidad de que el parásito sea resistente a los antipalúdicos. La cloroquina es la base del tratamiento para todas las especies excepto para las cepas de P. falciparum resistentes al medicamento. Es curioso que el paludismo por P. falciparum resistente a cloroquina (PFRC) se ha extendido por todos los países en donde el paludismo por P. falciparum es endémico excepto en Haití, República Dominicana, las zonas de América Central al oeste del canal de Panamá, el Medio Oriente. Para las infecciones por P. falciparum adquiridas en países con cepas resistentes a paludismo deben utilizarse tratamientos alternativos. Se han identificado cepas resistentes a mefloquina en la frontera de Tailandia-Myanmar, y cepas aisladas de P. vivax resistente a cloroquina en Africa, America Central, Sudamérica y Asia.
Los ataques agudos producidos por todas las especies, excepto por P. falciparum resistente a cloroquina, se tratan con fosfato de cloroquina oral, con dosis inicial de 1 g (600 mg de cloroquina base), seguido de 500 mg (300 mg) a las 6 horas y después a las 24 y 48 horas.26 Para las infecciones por PFRC existen varios tratamientos alternativos.26 Puede combinarse sulfato de quinina (650 mg cada 8 horas por 3 a 7 días) con doxiciclina (100 mg bid por 7 días), pirimetamina-sulfadoxina (Fansidar, 3 tabletas en dosis única el último día de tratamiento con quinina), o clindamicina (900 mg tid por 5 días). Otras opciones incluyen mefloquina (1,250 mg en dosis única), halofantrine y atovaquone en combinación con proguanil o doxiciclina. 26
En los pacientes graves que no toleran el tratamiento oral se puede administrar gluconato de quinidina o dihidrocloruro de quinina por vía intravenosa para cualquier tipo de paludismo.26 La quinidina es eficaz y bien tolerada con las precauciones adecuadas, incluyendo vigilancia hemodinámica y de ECG. Se infunde una dosis de impregnación de 10 mg/kg (máximo de 600 mg) de quinidina en solución salina en forma lenta en 1 a 2 horas, seguido por infusión continua de 0.02 mg/kg/min. Debe continuarse el tratamiento parenteral hasta que pueda tolerarse el tratamiento oral, en la mayoría de los casos en 48 a 72 horas. Para el paludismo falciparum fulminante la exsanguíneo transfusión puede ayudar.40
Cada uno de los agentes mencionados antes es activo solo contra la fase eritrocítica de la infección. Debido a que P. vivax y P. ovale tienen fases hepáticas persistentes que pueden causar recaídas de paludismo después del uso de cloroquina [ver figura 1], debe administrarse después de la cloroquina un agente que sea eficaz sobre el ciclo exoeritrocítico. La primaquina es el único agente usado para erradicar la infección hepática; se usa en dosis de 26.3 mg (15 mg de medicamento base) por vía oral cada día durante 2 semanas. Debido a que la primaquina puede inducir hemólisis en personas con deficiencia de glucosa 6 fosfato deshidrogenasa (G6PD), debe investigarse esta posibilidad antes del tratamiento. Si se encuentra una deficiencia leve puede administrarse el medicamento en dosis de 79 mg (45 mg de base) por vía oral una vez por semana durante 8 semanas.
Prevención de los viajeros
La mayoría de los residentes en Estados Unidos no tienen inmunidad contra el paludismo y tienen riesgo de las consecuencias de morbimortalidad de la enfermedad, en especial de la forma falciparum, en los países en que ocurre la trasmisión. De los 591 civiles de los Estados Unidos que adquirieron paludismo en el extranjero en 1995, solo el 15.6% había seguido un esquema quimioprofiláctico recomendado por los CDC. 32 Es indispensable que los viajeros reciban la orientación adecuada para disminuir su riesgo de adquirir paludismo y que usen los esquemas adecuados.
Babesiosis
Babesia es un protozoario intraeritrocítico que produce una enfermedad muy parecida al paludismo. La mayoría de los casos de babesiosis se han observado en el Noreste de los Estados Unidos, pero han ocurrido casos también en la parte superior del Medio Oeste, la Costa Occidental y otras regiones de los Estados Unidos, Europa y otros sitios.41
ETIOLOGIA Y EPIDEMIOLOGIA
Se han detectado varias especies Babesia como causa de enfermedad humana. La primera fue B. divergens, un parásito del ganado que ha causado varias infecciones fatales en personas esplenectomizadas, en especial en Europa, aunque una especie relacionada, denominada MO1, provocó un caso fatal en un hombre esplenectomizado en Missouri.42 B. microti es la causa principal de babesiosis en los Estados Unidos. Este parásito del ratón de patas blancas se trasmite por garrapatas de venados y es prevalente en las islas de Massachusetts, New York y Rhode Island y en áreas focales en Connecticut, Wisconsin y Minnesota. El riesgo de babesiosis no aumenta por la esplenectomía, pero la enfermedad es más seria en personas sometidas a esta cirugía, que tienen infección por VIH o con algún otro trastorno inmunosupresor.41,43 Muchas personas infectadas tienen infecciones subclínicas, como lo evidencian los estudios serológicos en áreas endémicas. Debido a que las garrapatas ninfas son las más eficaces para trasmitir la infección, la mayoría de los casos se desarrollan entre mayo y agosto, cuando este tipo de garrapatas son más abundantes. 41
Otra forma de babesiosis se desarrolla a partir de la infección con un organismo encontrado en los estados a lo largo de la costa del Pacífico y que se denomina WA1. Aún se desconocen el vector y el reservorio de esta nueva especie de Babesia. Se han detectado infecciones en personas con esplenectomía y con menos frecuencia en huéspedes normales; los estudios serológicos en zonas rurales y semirrurales de California indican que puede desarrollarse infección subclínica por WA1 en hasta el 20% de la población.41
La babesiosis también puede adquirirse en forma perinatal y por transfusiones sanguíneas.41,44
DIAGNOSTICO
Manifestaciones clínicas
Muchas personas infectadas con B. microti o WA1 permanecen asintomáticas. En los que ocurren síntomas la enfermedad se desarrolla en forma gradual semanas después de la mordedura de la garrapata o la transfusión sanguínea. Ocurren malestar, anorexia y fatiga, seguidos de mialgias, fiebre y diaforesis. Son comunes la labilidad emocional, la depresión, náusea, vómito y cefalea. Los síntomas tienden a desaparecer en semanas, aunque la fatiga y el malestar pueden durar meses. En ocasiones existe esplenomegalia. Las infecciones pueden volverse fulminantes y persistentes en pacientes esplenectomizados, ancianos y pacientes infectados por VIH o con otros padecimientos inmunosupresores. Pueden desarrollarse hemólisis intravascular, hemoglobinuria e insuficiencia renal. Ocurren parasitemias hasta del 85%, y este grado de infección puede ser fatal, en especial en personas asplénicas.
Datos de laboratorio
Los datos de laboratorio pueden incluir anemia hemolítica, cuentas de leucocitos normales o bajas y pruebas de función hepática anormales. El diagnóstico se basa en la observación de formas en anillo o de los microrganismos intraeritrocíticos en un frotis de sangre periférica teñidos con Giemsa. En raras ocasiones es posible observar merozoítos extraeritrocíticos. A diferencia de los parásitos del paludismo, los organismos Babesia no producen pigmentos en los eritrocitos que parasitan. En algunos pacientes, una parasitemia del 5 por ciento o mayor, y la observación de formas en anillo en algunos eritrocitos, sugieren el diagnóstico de P. falciparum, pero la ausencia del pigmento y de gametocitos distingue la babesiosis del paludismo. Las pruebas serológicas son útiles para el diagnóstico de infecciones crónicas con B. microti y WA1 cuando no se detectan los parásitos en los frotis de sangre.
TRATAMIENTO
El tratamiento de elección en la babesiosis consiste en la combinación de clindamicina (1.2 g bid IV o 600 mg tid vo por 7 días) y quinina (650 mg tid vo por 7 días). 26 En una minoría de pacientes que mejoran desde el punto de vista sintomático persiste la parasitemia. 45 En ocasiones puede desarrollarse edema pulmonar después de iniciar el tratamiento para la babesiosis, igual que ocurre en el caso del paludismo. Las exsanguíneo transfusiones pueden ayudar en los casos muy graves. El vector garrapata de la babesiosis puede trasmitir en forma simultánea a Borrelia burgdorferi, el agente de la enfermedad de Lyme, y especies Ehrlichia, los agentes de la erhlichiosis. Estas coinfecciones pueden empeorar el curso clínico y justificar tratamiento específico.46
Infecciones por protozoarios intestinales
El tubo digestivo del hombre puede albergar varios protozoarios que parasitan al ser humano. Los protozoarios Entamoeba coli, Endolimax nana e Iodamoeba butschlii son patógenos y no requieren tratamiento. Los protozoarios intestinales que se han asociado con enfermedades son Giardia lamblia, Entamoeba histyolitica, coccidios, Balantidium coli y Dientamoeba fragilis.
GIARDIASIS
G. lamblia habita en el intestino proximal y su infección puede variar desde asintomática hasta malabsorción severa. G. lamblia y la criptosporidiosis son los parásitos intestinales patógenos más frecuentes en los Estados Unidos. 47
Etiología y epidemiología
Existen dos formas morfológicas del parásito: el trofozoíto y el quiste. El trofozoíto es un microrganismo con múltiples flagelos, en forma de pera, que mide 9 a 15 micras de largo, de 5 a 15 micras de ancho, y de 2 a 4 micras de espesor; tiene simetría bilateral y contiene dos núcleos prominentes. En su cara ventral tiene un disco adhesivo que le permite fijarse a la superficie mucosa del duodeno y el yeyuno. A su paso por el intestino el trofozoíto generalmente se enquista. Los quistes resultantes miden 8-12 por 7-10 micras, tienen una pared externa bien definida y cuando maduran contienen cuatro núcleos.
Los trotozoítos pueden encontrarse en el líquido duodenal y en las heces líquidas, pero generalmente están ausentes de las evacuaciones formadas. Los trofozoítos no resisten las condiciones del medio externo, pero en cambio los quistes son muy resistentes, pudiendo sobrevivir en el agua varios meses, y tienen mayor supervivencia cuando la temperatura del agua es menor. Aunque la infección puede adquirirse ingiriendo trofozoítos con suficiente cantidad de alimento que los proteja de la acidez del estómago, los quistes son los principales responsables de la infección en el hombre. En estudios experimentales se ha logrado infectar a voluntarios con la ingestión de tan sólo 10 quistes del parásito.
La infección, que se relaciona directamente con la excreción fecal del microrganismo, se disemina por vía fecal- oral, o bien a través de alimentos y agua contaminados. La trasmisión directa de persona a persona es la responsable de la alta prevalencia que se observa en ciertos grupos. En las instituciones en las que existe incontinencia fecal y mala higiene la giardiasis puede ser endémica. De manera análoga, la giardiasis puede ser causa de enfermedad intestinal en las guarderías.48El riego de la adquisición y trasmisión es muy alto entre los niños que aún no controlan esfínteres, éstos a su vez pueden ser la fuente de casos secundarios dentro de su familia. 48 La trasmisión de persona a persona también es responsable de la alta frecuencia de giardiasis entre los varones homosexuales promiscuos.48 Algunas prácticas sexuales, como el anilingus, permiten la trasmisión directa de los quistes infectantes.
La trasmisión por agua contaminada es la principal causa de giardiasis. Puesto que la filtración del agua a través de la tierra elimina los quistes de Giardia, el agua de pozos profundos suele ser segura, En comparación, el agua de 1a superficie, como el agua de los arroyos y fuentes de montaña y el agua estancada, puede estar contaminada con quistes de Giardia, que son resistentes al agua y a los niveles normales de cloración. El recuento de coliformes no es un índice confiable de la contaminación por Giardia. Además, los mamíferos acuáticos, como los castores, pueden infectarse y servir como fuente continua de contaminación del agua. Puede ser difícil reconocer un suministro de agua como el origen común de las giardiasis, debido a que la infección con frecuencia es asintomática.
G. lamblia tiene una distribución mundial. Los viajeros que visitan muchos países, no sólo subdesarrollados, pueden infectarse con el microrganismo.
Patogenia
Después de la ingestión y el paso por el estómago de los quistes de G. lamblia, se liberan en el intestino los trofozoítos, que proliferan por división binaria. Los trofozoítos localizados en el duodeno y yeyuno pueden adherirse a las microvellosidades del epitelio intestinal mediante su disco de succión ventral, vivir en la capa de moco que recubre el epitelio, moverse y permanecer en el contenido intestinal, o más raramente, invadir los tejidos adyacentes a través de las células epiteliales. Pueden desarrollarse cambios funcionales en la capacidad de absorción del intestino delgado. La actividad enzimática del borde epitelial en cepillo disminuye, produciéndose deficiencia de disacaridasas, incluyendo lactasa. Las biopsias de yeyuno en pacientes con giardiasis no suelen revelar alteraciones patológicas. 49 Los mecanismos fisiopatológicos que producen estas alteraciones funcionales son inciertos. Ambos tipos de alteraciones se normalizan con el tratamiento específico, aunque en algunos pacientes en forma lenta. La hipoclorhidria predispone a la infección. La giardiasis suele ser más grave en pacientes con fibrosis quística y con deficiencias de inmunoglobulinas, quizá por deficiencia en la IgA secretora.50
Diagnóstico
Manifestaciones clínicas Las manifestaciones clínicas de la giardiasis son variables. Un gran número de pacientes no sufre síntomas. De hecho, en un brote muy bien estudiado de giardiasis asociado con contaminación acuática, dos tercios de las personas infectadas permanecieron asintomáticas. Por el contrario, otro subgrupo de personas infectadas desarrolló una giardiasis aguda típica. Después de un periodo de incubación de 1 a 3 semanas se presenta diarrea acuosa, dolor abdominal tipo cólico (y con frecuencia molestia epigástrica), náusea (el vómito es menos frecuente) y síntomas generales. Puede existir fiebre. La excesiva producción de gas en el intestino produce flatulencia y eructos de olor sulfuroso. Son comunes la menor absorción de grasas y la esteatorrea. Las evacuaciones son grasosas, malolientes y flotan en el agua; es raro encontrar en ellas moco o sangre. Los síntomas son importantes durante una semana y disminuyen en intensidad en las siguientes semanas. Los datos clínicos que ayudan a identificar los casos de giardiasis en estudios epidemiológicos incluyen la duración de la enfermedad de 7 o más días con por lo menos dos de los siguientes seis síntomas: diarrea, flatulencia, heces fétidas, náusea, có1ico abdominal y fatiga excesiva.51
Una fase crónica de las giardiasis puede seguir a la fase aguda, o puede manifestarse sin el antecedente de una enfermedad aguda. La fase crónica se caracteriza por evacuaciones disminuidas de consistencia, aunque no necesariamente diarreicas, con aspecto graso. También se asocia con aumento de la cantidad de gas en el intestino, có1icos, borborigmos, flatulencia y distensión. La fiebre no es frecuente, pero sí pueden encontrarse malestar general, fatiga y depresión. La intolerancia a la lactosa, producida o exacerbada por la enfermedad, puede causar aumento de los síntomas al ingerir leche o sus productos. El curso clínico es variable, alternando períodos de remisión y exacerbación. En un número reducido de casos la persistencia de la infección puede asociarse con malabsorción moderada o grave y pérdida de peso.52
Muy raras veces, G. lamblia puede diseminarse del duodeno y yeyuno a las vías biliares y conductos pancreáticos. Se han comunicado casos de colecistitis, colangitis y hepatitis granulomatosa. También se han observado alteraciones en la función exócrina del páncreas, manifestada por secreción disminuida de tripsina y lipasa.
Estudios de laboratorio En la giardiasis no ocurren leucocitosis ni eosinofilia. La excreción de grasa en las heces está aumentada (ver antes) y los resultados de otras pruebas de laboratorio de malabsorción pueden ser anormales. Las series gastrointestinales no muestran cambios radiológicos significativos.
El diagnóstico definitivo de la infección requiere la identificación de los trofozoítos o quistes. El examen de las heces es habitualmente positivo en el cuadro agudo y la observación de series de tres muestras permite el diagnóstico en más de 90 por ciento de los casos. Los quistes, que pueden estar presentes en las heces líquidas o formadas son más resistentes y no requieren la búsqueda en fresco. Por el contrario cuando se buscan trofozoítos es importante analizar muestras en fresco, o conservadas en alcohol polivinílico o en una preparación de merthiolate- ioduro-formaldehido (MIF). Deben evitarse las sustancias que interfieren con la investigación en fresco, como el medio de contraste, los antiácidos y el aceite mineral. Los estudios inmunológicos detectan antígenos de giardia en las heces con mayor sensibilidad que una sola muestra de excremento.53
En la giardiasis crónica la frecuencia de detección del parásito en el excremento disminuye. Si se ha fracasado en tres muestras de heces, debe analizarse material duodenal obtenido por aspiración duodenoyeyunal. En forma alternativa, puede practicarse una biopsia intestinal. La detección de los trofozoítos en el material de la biopsia requiere de una búsqueda cuidadosa, el examen directo del frotis de mucosa en fresco puede facilitar la detección del parásito.
Diagnóstico diferencial
Debe pensarse en otros agentes infecciosos que causen gastroenteritis al evaluar a los pacientes con giardiasis aguda. Debido a que la mayoría de estos agentes producen un cuadro de corta duración, la persistencia de los síntomas después de una semana y los síntomas significativos de malabsorción (flatulencia, intolerancia a la lactosa, eructos) sugieren una giardiasis. La giardiasis crónica puede parecerse a otros padecimientos asociados con malabsorción.
Tratamiento
El metronidazol, aunque no aprobado por la Food and Drug Administration de los EUA, para el tratamiento de la giardiasis, es el principal agente empleado para tratar esta infección26 porque la quinacrina, el medicamento más eficaz, no se distribuye ya en los Estados Unidos. La dosis habitual es de 250 mg tres veces al día durante 5 días y en casos persistentes se han llegado a utilizar hasta 750 mg tres veces al día durante 10 días. Los efectos colaterales que se han observado son náusea, dolor de cabeza y sabor metálico en la boca; rara vez llega a oscurecerse la orina o a presentarse parestesias y mareos. El metronidazol tiene un efecto semejante al disulfiram, por lo que debe evitarse el consumo de alcohol durante el tratamiento. Aunque no está disponible en los Estados Unidos, el tinidazol (administrado en una sola dosis oral de 2 g) es muy eficaz.54
En los niños puede utilizarse una suspensión de furazolidona, que ha demostrado ser eficaz y bien tolerada.26 El tratamiento de la giardiasis durante el embarazo representa un verdadero reto. El metronidazol debe evitarse, aunque los estudios no han demostrado riesgos teratogénicos al usarlo durante el embarazo. 55 Si los síntomas de la giardiasis son mínimos el tratamiento puede esperar hasta el parto. Si hay molestias puede administrarse el aminoglucósico no absorbible paromomicina, 25 a 35 mg/kg/día por vía oral en tres dosis divididas por 7 días.26Este esquema puede proporcionar alivio por lo menos sintomático. Si la giardiasis durante el embarazo se asocia con deshidratación, malabsorción o síntomas severos está justificado el tratamiento con metronidazol. En cualquier paciente, la resolución de los síntomas de malabsorción pueden requerir de meses para que se regenere la mucosa intestinal funcional después del tratamiento antiparasitario eficaz.
La prevención de la giardiasis requiere atención hacia la higiene para evitar la trasmisión de persona a persona. No se han definido aún cuáles son los riesgos y los beneficios de tratar a los niños infectados asintomáticos que acuden a guarderías, pero está indicado el tratamiento de los portadores asintomáticos para evitar la diseminación de la enfermedad. La ebullición del agua o su calentamiento a 70'C [158' F] durante 10 minutos es suficiente para desinfectarla. Para los alpinistas y excursionistas, la iodinación del agua es más eficaz que la cloración, pero requiere de por lo menos 8 horas para que sea eficaz en un 99.9 por ciento. También son eficaces algunos filtros de agua de calidad para eliminar los quistes.
AMIBIASIS
La infección con Entamoeba histolytica es responsable de la amibiasis humana. La infección puede estar limitada al colon, con manifestaciones clínicas que varían desde el estado asintomático hasta la disentería grave, o puede afectar áreas extraintestinales, de las que el hígado es la más frecuente.
Etiología y epidemiología
E. histolytica puede distinguirse morfológicamente de las otras especies no patógenas de amibas intestinales, incluyendo a Escherichia coli y E. hartmanni. E. histolytica existe en dos formas: como trofozoíto y como quiste. El trofozoíto mide de 10 a 20 micras de diámetro, pero puede ser mayor en las evacuaciones disentéricas; es móvil y posee un núcleo único con citoplasma granular. Los trofozoítos se eliminan con las heces diarreicas, pero son poco resistentes a las condiciones del medio fuera del organismo. Por el contrario, el quiste que se forma en el colon es muy resistente a las condiciones del medio externo y a la acidez del estómago. Los quistes miden de 10 a 20 micras de diámetro, contienen de uno a cuatro núcleos pequeños, cariosomas centrales y un patrón de cromatina fina en la periferia.
No todas las cepas de E. histolytica son patógenas. Pueden aislarse cepas no patógenas de individuos asintomáticos, y son prevalentes entre los varones homosexuales promiscuos en los Estados Unidos e Inglaterra. Las cepas patógenas y no patógenas no pueden distinguirse por microscopía, excepto porque los trofozoítos patógenos con frecuencia fagocitan eritrocitos. Con base en datos bioquímicos, inmunológicos y genéticos se ha descrito a E. hystolitica. En la actualidad una nueva especie, E. dispar, representa las cepas no patógenas y aparentemente nunca es invasiva en los humanos, mientras que E. hystolitica incluye solo las cepas potencialmente patógenas.56 Debido a que no todas las cepas potencialmente patógenas de E. hystolitica producen enfermedad, sin duda otros procesos influyen en la virulencia de las amibas.
Se ha desarrollado un inmunoensayo para distinguir a E. hystolitica de E. dispar.57 En la actualidad no existe otro método disponible para distinguir en forma confiable entre ambos organismos [ver adelante, Diagnóstico].
Los quistes, que se eliminan por las heces humanas, son la principal fuente de infección para el hombre. La adquisición en la infección es producto de contaminación fecal - oral y puede ocurrir a través de agua y alimentos contaminados o de persona a persona. Esta última forma es la responsable de la alta prevalencia de la infección entre homosexuales promiscuos y en situaciones donde existe incontinencia fecal con malas medidas de higiene. La amibiasis es más común en los grupos socioeconómicos bajos, y se debe a las malas condiciones sanitarias y al hacinamiento. En los Estados Unidos se observan casos en personas que han regresado de viajes al extranjero o inmigrado de zonas en donde la amibiasis es endémica.
Patogenia
Los quistes ingeridos son transportados hasta el intestino, donde liberan los trofozoítos que se reproducen por fisión binaria dentro del colon. Los trofozoítos producen citólisis e invaden la pared del intestino, causando necrosis focal. Las úlceras que se producen tienen aspecto de cuello de botella, con la porción angosta a nivel de la mucosa y un cuerpo amplio en la submucosa. A menos que el daño clínico sea muy extenso, pueden observarse zonas de colon normal entre ellas. Las áreas que se afectan con mayor frecuencia son el rectosigmoides, el apéndice cecal, el colon transverso y descendente, y el íleon terminal.
La gravedad de la afección colónica en pacientes con amibiasis es muy variable y puede variar desde un padecimiento frecuente y leve o incluso despreciable hasta invasión difusa y extensa con necrosis. La mayoría de los pacientes que se infectan con E. histolytica sufren invasión importante del colon. Los factores determinantes de la gravedad se conocen poco y tal vez guarden relación con el tamaño del inóculo, la cepa de E. histolytica de que se trate, la flora colónica y el estado fisiológico y nutricional del huésped. Los esteroides y el embarazo disminuyen la resistencia del huésped a la invasión. Las complicaciones de la afección intestinal son hemorragia y peritonitis, produciéndose la última más por paso a través de la pared inflamada, que por una franca perforación del colon. La infección crónica produce una respuesta granulomatosa en el lugar afectado, más común en el ciego, que se manifiesta como una masa denominada ameboma. También puede desarrollarse estenosis del colon.
La amibiasis puede diseminarse por vía hematógena desde el colon para afectar cualquier parte del organismo. El hígado es el órgano más afectado, seguido en frecuencia por los pulmones, que se lesionan por diseminación transdiafragmática a partir del hígado. Los trofozoítos transportados por el sistema venoso porta producen necrosis en el hígado y la formación de abscesos. Alrededor del 90 por ciento de los abscesos se encuentran en el lóbulo derecho, en especial en la cara superior y anterior. Aunque los abscesos pueden ser múltiples, es más común encontrar un solo absceso que varía de pocos hasta 20 centímetros de diámetro. Los abscesos hepáticos amibianos son 7 a 9 veces más comunes en hombres que en mujeres. En pocas ocasiones un absceso puede desarrollarse en forma concomitante a una colitis amibiana. Dependiendo de la serie, el 50 a 70 por ciento de los pacientes con abscesos hepáticos amibianos no tienen historia de colitis amibiana. La ruptura de un absceso en el lóbulo derecho puede causar extensión hacia el tórax, produciendo un empiema amibiano, neumonía o una fístula broncopleural. La extensión hacia la cavidad peritoneal es menos común. La ruptura desde un absceso del lóbulo izquierdo puede invadir el pericardio, con frecuencia con consecuencias fatales.
Diagnóstico
Cuadro clínico La gran mayoría de los pacientes con amibiasis colónica no tiene síntomas. La excreción fecal de los quistes en estas personas es un hallazgo fortuito. En aquellos que sufren síntomas la enfermedad varía desde leves episodios de diarrea hasta una disentería grave. La primera forma de presentación es común y los pacientes no interrumpen sus actividades diarias, a pesar de la diarrea leve, que puede alternar con estreñimiento. El curso de la enfermedad puede ser remitente, con episodios sintomáticos intercalados con temporadas sin sintomatología. Es frecuente que el paciente se queje de malestar abdominal vago, flatulencia, tenesmo y dolor en la región sacra, pero los síntomas generales y la fiebre son raros, mucho menos frecuentes que en las formas bacterianas de gastroenteritis y colitis. Las evacuaciones se acompañan de moco y sangre. La exploración física del abdomen refleja la gravedad de la afección colónica. Puede haber dolor discreto en las zonas afectadas; en otros casos, el dolor intenso junto con fiebre alta indican afección grave del colon. 58 Los amebomas pueden palparse en ocasiones como masas dolorosas.
Los síntomas de presentación del paciente con un absceso hepático amibiano son malestar general, fatiga, anorexia, pérdida de peso, dolor abdominal y fiebre, con duración de una a varias semanas, aunque puede tener una evolución de meses. El dolor abdominal por lo general es constante, con localización en el cuadrante superior o hemitórax derechos. Algunas veces el paciente lo refiere en el hombro del mismo lado. Cuando se presenta como dolor pleurítico, acompañado de tos y disnea, puede interpretarse equivocadamente como una infección intrapulmonar. Un absceso en el lóbulo izquierdo produce dolor en el epigastrio o en el cuadrante superior izquierdo del abdomen. Al examen físico se encuentran hepatomegalia y dolor localizado al palpar la zona del absceso. La ictericia franca es rara Al examinar el tórax la base derecha se percute mate debido a la elevación del hemidiafragma y a la alta frecuencia de derrame pleural; con la auscultación pueden escucharse algunos estertores.
La amibiasis cutánea produce lesiones ulceradas o exofíticas, con localización frecuentes en el periné y los genitales. Estas se producen por invasión de los trofozoítos, como resultado de contaminación fecal o trasmisión sexual. Se diferencían de los procesos neoplásicos, la tuberculosis y la sífilis por la presencia de trofozoítos en los exudados y biopsias.
Estudios de laboratorio En la colitis amibiana sintomática es frecuente observar leucocitosis discreta a moderada. También es posible que se encuentre una anemia ligera y alteraciones en las pruebas de función hepática, sin que necesariamente indiquen formación de absceso. Las heces suelen contener sangre evidente u oculta, así como cristales de Charcot-Leyden, derivados de eosinófilos.
Cuando existe un absceso hepático amibiano se observa leucocitosis, anemia y elevación de la velocidad de sedimentación globular. En más de dos tercios de los pacientes la fosfatasa alcalina se eleva de una a cuatro veces por encima de sus valores normales. La elevación de las transaminasas se observa en menos del 50 por ciento de los pacientes; la bilirrubina se eleva poco, en general menos de 2.5 mg/dl. La radiografía del tórax muestra elevación del hemidiafragma derecho y derrame pleural. Las gamagrafías hepáticas revelan defectos de captación compatibles con lesiones ocupativas, el ultrasonido muestra la misma lesión, de características hipoecogénicas, de forma redonda u ovoide y es útil para detectar la diseminación transdiafragmática del absceso. La TAC o la IRM del abdomen también son útiles para detectar el absceso y valorar su extensión.
El diagnóstico definitivo de la amibiasis intestinal requiere la identificación en las heces de los quistes o trofozoítos de E. histolytica . Las evacuaciones diarreicas deben examinarse de inmediato en busca de trofozoítos, en cambio las heces formadas pueden examinarse directamente o después de someterlas a un proceso de concentración para búsqueda de quistes. Las heces se conservan en forma adecuada en alcohol polivinílico o en solución de MIF para su examen posterior. Es frecuente confundir los leucocitos fecales y las amibas no patógenas con E. histolytica, por lo que es recomendable realizar las tinciones adecuadas, y que el examen sea practicado por personal con experiencia. Los antimicrobianos, los catárticos, los antiácidos y el bario pueden interferir con el examen microscópico. Es recomendable examinar muestras de 3 o más días, ya que la excreción de los quistes varía día con día. Cerca de la mitad de los pacientes con colitis amibiana tiene lesiones en el rectosigmoides, por lo que los raspados, aspirados y biopsias de las lesiones mucosas tomados durante una rectosigmoidoscopía deben examinarse en busca de trofozoítos.
Se ha desarrollado una prueba de detección de un antígeno fecal para E. hystolitica, que se basa en un anticuerpo monoclonal contra una lectina específica en este organismo.57
Las pruebas serológicas para amibas son por lo general negativas en el paciente asintomático que excreta quistes, pero su positividad aumenta en forma paralela a la lesión colónica y al tiempo de evolución de la colitis. Estas pruebas pueden ayudar al diagnóstico de las colitis agudas y son muy útiles al evaluar la etiología de una colitis crónica. Al interpretar un resultado positivo, el clínico debe tomar en consideración que la positividad se mantiene durante meses o años después de una infección.
En el 90 a 95 por ciento de las infecciones extraintestinales existe seropositividad para anticuerpos contra amiba, y sus títulos se incrementan conforme aumenta el tiempo de evolución de la enfermedad. En el diagnóstico del absceso hepático amibiano, la combinación de una prueba serológica positiva, un cuadro clínico compatible y la observación de una lesión quística, es muy sugestiva del diagnóstico. En el caso de un absceso amibiano no es de utilidad la búsqueda del parásito en las heces. Rara vez es posible observar trofozoítos en el material aspirado de un absceso, cuyo aspecto se parece a la pasta de anchoas y es de apariencia achocolatada.
Diagnóstico diferencial
En los casos de colitis amibiana leve, la duración y los síntomas pueden confundirse con un síndrome de colon irritable, con una diverticulitis o una enteritis regional. Cuando el cuadro disentérico es grave debemos distinguirlo de las infecciones por Shigella, Salmonella y Campylobacter, lo que puede lograrse mediante los cultivos apropiados y por la presencia de abundantes leucocitos en la materia fecal. La colitis ulcerosa y la enfermedad de Crohn deben considerarse en el diagnóstico diferencial de la colitis amibiana crónica. Las lesiones de la colitis amibiana y la colitis ulcerosa pueden tener un aspecto similar en la rectosigmoidoscopía y el enema con contraste, pero es muy importante distinguirlas porque los esteroides, que están indicados en la segunda, pueden agravar la colitis amibiana. Por esta razón las pruebas serológicas para amiba, y el examen de las heces y lesiones mucosas en busca de amibas son de gran importancia. La prueba terapéutica con metronidazol puede tener resultados benéficos en ambas enfermedades, por lo que no es de utilidad diagnóstica.
Un ameboma puede simular un adenocarcinoma o bien otros procesos granulomatosos. Una prueba serológica positiva para amiba y la resolución de la masa con el tratamiento con metronidazol apoyan el diagnóstico de ameboma. Si la respuesta con metronidazol no es completa, se recomienda practicar biopsia con el fin de descartar algún proceso asociado al ameboma.
El diagnóstico diferencial del absceso hepático amibiano se guía por dos observaciones: (1) una gamagrafía hepática que muestra una lesión ocupativa y (2) un ultrasonido, TC o IRM que demuestra la naturaleza quística de la lesión. Aunque los quistes hepáticos y los quistes equinocócicos no suelen asociarse con fiebre y los otros síntomas de una lesión amibiana, deben considerarse dentro del diagnóstico diferencial. También debe pensarse en abscesos piógenos. La serología positiva apoya una etiología amibiana. A diferencia de los quistes equinocócicos, los abscesos hepáticos amibianos rara vez se calcifican. Tanto el absceso hepático amibiano como el piógeno tienen buena respuesta al metronidazol, y por otro lado, los abscesos amibianos muy pocas veces se sobreinfectan con gérmenes piógenos. Aunque la aspiración del absceso no está indicada en el caso de que sea de etiología amibiana, esta medida puede ser necesaria para obtener material para cultivo y tinción de Gram cuando existen dudas diagnósticas.
Tratamiento
El tratamiento de elección para la amibiasis intestinal sintomática o extraintestinal es el metronidazol (750 mg vo tid por 10 días), que ha demostrado ser muy eficaz y constituye el tratamiento de elección.26 Ya se describieron en la sección de giardiasis los efectos colaterales y las precauciones al usarlo. Se han observado fracasos ocasionales en el tratamiento del absceso hepático. En caso de enfermedad intestinal, las recaídas son raras a menos que exista reinfección, por lo que se recomienda la vigilancia de los pacientes durante varios meses.
Se considera que el tratamiento farmacológico no es necesario para muchas personas asintomáticas con quistes porque con frecuencia tienen E. dispar no patógena. Si no es posible conocer la especie exacta de Entamoeba, los portadores de quistes deben tratarse solo si manejan alimentos o reciben esteroides; también en el caso de una epidemia de amibiasis.56 En los pacientes asintomáticos la concentración de metronidazol en la luz del colon puede ser inadecuada para erradicar las amibas, por lo que debe usarse uno de dos amebicidas intraluminales.26Puede emplearse paromomicina (25 a 35 mg/kg/día por 7 días) o como alternativa iodoquinol (650 mg vo, tid 20 días). La dosis no debe ser mayor por el riesgo de causar neuropatía óptica El iodoquinol está contraindicado en pacientes con neuropatía óptica o enfermedad tiroidea. Se recomienda la administración de un esquema de iodoquinol para las personas tratadas por amibiasis intestinal sintomática o extraintestinal.26
La aspiración terapéutica no está indicada en el tratamiento del absceso hepático amibiano, aunque la aspiración diagnóstica puede ser útil en ciertos casos [ver antes, Diagnóstico diferencial]. Está indicado el drenaje, que puede lograrse por aspiración cutánea, para las lesiones que no responden al tratamiento médico inicial o con peligro de ruptura inminente.
La prevención de la amibiasis se basa principalmente en la higiene personal, que evita la diseminación de persona a persona. En las comunidades donde la enfermedad es endémica, la previsión y el uso de sanitarios adecuados ha disminuido mucho su diseminación. Es conveniente evitar el consumo de vegetales que crecen en la tierra por la posibilidad de contaminación por heces humanas. El agua puede considerarse segura si se ha hervido o se ha tratado con tabletas de yodo.
COCCIDIOSIS
Los parásitos de la familia Coccidia se encuentran en el intestino de varios animales domésticos y salvajes, siendo parásitos unicelulares con ciclos de reproducción sexual y asexual que se llevan a cabo en las células del epitelio intestinal. De todos ellos, tres han demostrados ser patógenos en el humano, Isospora belli, Cryptosporidium y Cyclospora cayetanensis. El contacto con ganado infectado puede provocar la infección con Cryptosporidium, y el agua contaminada puede trasmitir I. belli, Cryptosporidium y C. cayetanensis.59 Los tres parásitos se han identificado como oportunistas en pacientes infectados por VIH.60
Isosporiasis
Diagnóstico La infección por I. belli se inicia en forma súbita. Los primeros síntomas son fiebre y malestar general, apareciendo después dolor abdominal, diarrea y pérdida de peso. En la mayoría de los casos el proceso se autolimita, aunque se han comunicado infecciones con duración de varios días hasta algunos meses. En estos casos, puede haber diarrea grave, esteatorrea y lesión hepática; sólo rara vez la infección provoca la muerte. En pacientes con SIDA la infección con I. belli causa diarrea acuosa crónica y pérdida de peso indistinguible de la enfermedad producida por Cryptosporidium.
El examen histológico de las lesiones mucosas muestra acortamiento de las vellosidades, hipertrofia de las criptas y un infiltrado compuesto por eosinófilos, linfocitos y células plasmáticas. Aunque la eosinofilia en sangre periférica es rara en las infecciones por protozoarios, sí se presenta en las coccidiosis. La presencia de ooquistes en las heces establece el diagnóstico, pero es difícil detectarlos en los exámenes de rutina; sin embargo, pueden demostrarse mediante tinción ácido resistente. Si el número de ooquistes en las heces es escaso, la incubación de las heces a temperatura ambiente y por espacio de 24 a 48 horas madura los quistes y facilita su detección; también se recomienda utilizar la técnica de concentración con sulfato de zinc. Los parásitos también pueden observarse en los aspirados y biopsias intestinales.
Tratamiento Las infecciones causadas por I. belli se tratan con trimetoprim-sulfametoxazol de doble potencia (160 mg de trimetoprim y 800 mg de sulfametoxazol) administrados por vía oral cuatro veces al día durante 10 días y después dos veces al día durante 3 semanas.26 Sin embargo, en los pacientes con SIDA son frecuentes las recaídas y el tratamiento inicial de 10 días debe continuarse con un tratamiento de sostén a largo plazo con una tableta de dosis doble de trimetoprim-sulfametoxazol tres veces a la semana o con la combinación de 25 mg de pirimetamina y 500 mg de sulfadoxina una vez a la semana. En los pacientes con intolerancia a las sulfas ha sido eficaz el tratamiento con pirimetamina (75 mg/día); puede administrarse una dosis de mantenimiento (50 a 75 mg/día) para prevenir las recaídas de las infecciones por I. belli. 60
Cryptosporidiosis
Cryptosporidium habita en el borde en cepillo de la mucosa intestinal y produce enterocolitis en individuos normales o inmunosuprimidos. La infección puede adquirirse al ingerir incluso menos de 100 ooquistes.61Se ha demostrado la trasmisión a través del agua para beber 62 y de albercas contaminadas. 47,63 La criptosporidiosis ha causado epidemias de diarrea en guarderías y puede adquirirse en viajes al extranejro. Debido a que los ooquistes fecales son infecciosos, puede ocurrir diseminación de la infección tanto nosocomial como en el hogar.
Diagnóstico En huéspedes normales la enfermedad comienza después de un periodo de incubación de cerca de una semana. Consiste en diarrea acuosa, no sanguinolenta, acompañada a veces por dolor abdominal, náusea, fiebre, anorexia y pérdida de peso. Los síntomas pueden persistir por 1 a 2 semanas y suelen autolimitarse sin tratamiento. Por el contrario, en los pacientes inmunosuprimidos la criptosporidiosis puede ser persistente y grave. En los pacientes infectados por VIH con cuentas de células T CD4+ mayores de 180/mm3 la criptosporidiosis puede ser autolimitada. Sin embargo, en los individuos más inmunosuprimidos, la diarrea secretora, que es crónica y profusa, puede no remitir. Cryptosporidia puede contribuir también a enfermedades hepatobiliares, como colecistitis, colangitis y estenosis papilar.
Se pueden preparar concentrados de ooquistes de Cryptosporidium mediante un tratamiento previo con la técnica de flotación de azúcar a partir de material fecal. Los ooquistes de Cryptosporidium pueden observarse en frotis de heces examinados al microscopio, ya sea con una preparación en fresco de lugol o después de teñir con un anticuerpo monoclonal o el agente modificado Kinyoun [ver figura 2].Además, las tinciones de fluorescencia directa y los inmunoensayos de muestras de heces pueden aumentar la sensibilidad diagnóstica.
|
|


Ciclosporidiosis
Cyclospora parece tener una distribución geográfica amplia y se ha descrito enfermedad atribuible a este protozoario en Estados Unidos, Latinoamérica, Africa, Europa y Asia.64 Los estudios epidemiológicos indican que el agua contaminada, 59 así como las frambuesas importadas, 65 pueden ser la fuente de infección. Se requieren más estudios para definir otros modos de trasmisión. El periodo de incubación es de alrededor de una semana. 59
Diagnóstico Muchos pacientes infectados han presentado diarrea, síntomas gripales, y síntomas comunes a otros patógenos del intestino delgado, incluyendo flatulencia y eructos. La enfermedad puede consistir en un episodio único o tener recurrencias, y son frecuentes la diarrea prolongado, la anorexia y los síntomas del tubo digestivo alto. La fatiga prolongada y la pérdida de peso también son comunes.
Las biopsias de intestino delgado permiten detectar el parásito en las células epiteliales y detectar inflamación yeyunal, que se asocia con aumento en el número de linfocitos intraepiteliales, atrofia de las vellosidades e hiperplasia de las criptas.143 No hay leucocitos ni sangre en las heces, lo que sugiere que la enfermedad se produce por mecanismos no invasores. El diagnóstico puede hacerse por detección de los ooquistes en el excremento que, como los ooquistes de Cryptosporidium, son aparentes con tinción ácido resistente. Aunque los ooquistes de Cyclospora, que miden entre 8 y 10 micras de diámetro, son más grandes que los de Cryptosporidium, se requiere tener cuidado para no confundir estos dos protozoarios. La microscopía fluorescente es un método rápido y sensible para detectar ooquistes autofluorescentes. 67 Pueden encontrarse ooquistes de Cyclospora en pacientes infectados por VIH.60
Tratamiento El trimetoprim-sulfametoxazol (160 mg de trimetoprim y 800 mg de sulfametoxazol) dos veces al día durante 7 días ha demostrado ser eficaz para la ciclosporidiasis. 26 Sin embargo, los pacientes infectados por VIH pueden requerir una dosis mayor (4 veces al día por 7 días) y un tratamiento de mantenimiento por tiempo prolongado (tres veces por semana). Como en el caso de la criptosporidiosis, debe pensarse en infección por Cyclospora en cualquier paciente con diarrea prolongada, anorexia y síntomas del tubo digestivo alto.
INFECCION POR DIENTAMOEBA FRAGILIS
A diferencia de las otras amibas, Dientamoeba fragilis no tiene una etapa de quistes. El trofozoíto no resiste la acidez del estómago, por lo que aún no se ha podido explicar cómo aparece la infección en el hombre. Es posible que los huevecillos del nemátodo Enterobius vermicularis acarreen los trofozoítos de Dientamoeba fragilis, puesto que es frecuente que ambas infecciones coexistan. D. fragilis produce una enfermedad caracterizada por dolor abdominal, anorexia y evacuaciones disminuidas de consistencia. Como en el caso de las infecciones por Isospora, pero no de otras infecciones por protozoarios, puede existir eosinofilia.68El estado de trofozoíto es muy frágil y difícil de detectar; para mejorar su detección, las muestras de excremento deben conservarse con fijador de alcohol polivinílico, fijador de acetato de sodio-ácido acético-formalina o líquido de Schaudinn, y deben examinarse después de su tinción permanente. La infección por D. fragilis puede tratarse con tetraciclina (500 mg qid durante 10 días), paromomicina (25 a 30 mg/kg por día en tres dosis durante 7 días) o iodoquinol (650 mg tid durante 20 días). 26
INFECCION POR BLASTOCYSTIS HOMINIS
Aunque antes se consideraba a Blastocystis hominis una levadura no patógena, en la actualidad algunos investigadores consideran que este microrganismo es un protozoario capaz de causar infección intestinal. Esta creencia parte del hallazgo de la presencia de grandes cantidades de microrganismos B. hominis en las heces del paciente con diarrea sin que se logre identificar otra causa. Sin embargo, otros investigadores han fracasado en confirmar estas observaciones,64 y no han encontrado concordancia entre el número de microrganismos en las heces, el grado de diarrea y la resolución de los síntomas después del tratamiento de la infección por Blastocystis. Mientras se resuelve la patogenicidad de B. hominis, los pacientes con diarrea que excretan este microrganismo en sus heces deben estudiarse para descartar infecciones parasitarias, bacterianas o virales. Si la diarrea es de suficiente magnitud para justificar tratamiento, se puede usar iodoquinol en dosis de 650 mg tres veces al día durante 20 días, o metronidazol en dosis de 750 mg tres veces al día por l0 días. 26
MICROSPORIDIOSIS
Los microsporidios, que son organismos intracelulares obligados formadores de esporas, pertenecen a un phylum distinto de protozoarios que incluye muchos géneros capaces de infectar a huéspedes vertebrados e invertebrados. Las infecciones humanas por microsporidios se han detectado sólo hasta hace poco y a la fecha se han identificado cuatro géneros (Enterocytozoon, Encephalitozoon, Pleistophora y Nosema) como causa de enfermedad en humanos, en especial en individuos infectados por VIH.70 Estos microsporidios se distinguen por su tamaño, morfología nuclear y modo de división, así como por el sitio de proliferación intracelular (los microsporidios se multiplican libres en el citoplasma a dentro de vacuolas unidas a la membrana). A pesar de la ubicuidad de estos organismos en otras especies, no se sabe la manera como se infectan los humanos. Debido a que casi todas las infecciones por microsporidios detectadas en humanos ocurren en huéspedes inmunosuprimidos, no se conoce la frecuencia con la que se infectan los pacientes inmunocompetentes o si en éstos últimos las infecciones son asintomáticas o autolimitadas.
El cuadro clínico de la enfermedad atribuible a los microsporidios es amplio y el estado de la enfermedad parece depender de la especie infectante y de la función inmunológica del huésped. Las infecciones causadas por microsporidios más frecuentes son las de Enterocitozoon bieneusi en pacientes infectados por VIH. Entre los pacientes con infección por VIH, cada vez se reconocen más infecciones intestinales por E. bieneusi y Encephalitozoon intestinalis (antes Septata intestinalis) que causan diarrea crónica.71,72 En estos pacientes E. bieneusi y E. intestinalis pueden infectar las vías biliares, causando colangitis. Las especies Encephalitozoon han provocado queratoconjuntivitis caracterizada por queratopatía epitelial con puntilleo burdo en pacientes infectados por VIH, mientras que las especies Vittaforma y Nosema se han relacionado con queratitis del estroma en algunos pacientes VIH seronegativos. Los microsporidios Encephalitozoon se han asociado también con peritonitis, hepatitis o infecciones nasales y sinusales en pacientes infectados por VIH y las especies Trachipleistophora y Pleistophora han causado miositis.
El tamaño tan pequeño de los microsporidios, que son organismos gram positivos con esporas maduras que miden 0.5 a 2 por 1 a 4 micras, dificulta la detección del parásito, y muchas infecciones han requerido de examen del tejido por microscopía electrónica para realizar el diagnóstico. Las esporas intracelulares pueden detectarse también por microscopía de luz si los tejidos se tiñen con hematoxilina-eosina, Giemsa o tinción de Gram. Los microsporidios intestinales parecen distribuirse en parches y pueden no detectarse en los tejidos biopsiados, pero un método basado en la tinción del cromotropo y los métodos de tinción de fluorocromo Uritex 2B facilitan la detección de las esporas en frotis de heces o de aspirado duodenal.
Para las infecciones intestinales con E. intestinalis, el albendazol oral es benéfico y curativo. Las infecciones por E. bieneusi responden menos, aunque los pacientes pueden presentar mejoría sintomática sin erradicación de la infección. 71,72 Para la queratoconjuntivitis causada por Encephalitozoon hellem se emplea el tratamiento tópico con suspensión de fumagilina.26
Infecciones por amibas de vida libre
Aunque algunas amibas como E. histolytica no pueden sobrevivir y replicarse fuera de los animales huéspedes, la mayor parte de las amibas son de vida libre en el suelo y el agua. Las amibas del género Naegleria y Acanthamoeba pueden causar meningitis aguda, meningoencefalitis aguda y meningoencefalitis granulomatosa crónica.73
Las especies Acanthamoeba se reconocen cada vez más como causa de queratitis74y, en un pequeño número de pacientes infectados por VIH, de enfermedad diseminada con manifestaciones cutáneas.75 Estos microrganismos han sido aislados del agua, polvo del aire, tuberías calientes y soluciones salinas empleadas para limpiar los lentes de contacto. Los factores relacionados con el desarrollo de queratitis amibiana incluyen la falta de desinfección de lentes de contacto, el antecedente de traumatismo corneal leve y la exposición a tierra o agua estancada. Las lesiones suelen ser crónicas y muy dolorosas y consisten en uveítis anterior variable, erosión epitelial, escleritis y queratitis infiltrativa del estroma, que a menudo tiene forma de anillo. Las lesiones son refractarias a los medicamentos antimicrobianos habituales y deben distinguirse de la queratitis producida por virus herpes simple. El diagnóstico se confirma por el examen microscópico de raspados corneales teñidos con Giemsa o tricrómico y mediante el uso de tinción indirecta con anticuerpos inmunofluorescentes de raspados corneales. Las amibas pueden cultivarse al inocular el tejido corneal en agar no nutritivo sembrado con E. coli.
La queratitis por Acanthamoeba puede ser tratada con esquemas tópicos con uno o varios fármacos como biguanida de polihexametileno, isetionato de propamidina y clotrimazol. 74
Leishmaniasis
Los protozoarios del género Leishmania, parásitos intracelulares obligados, producen una amplia gama de enfermedades en el hombre, que van desde la forma visceral generalizada, hasta las lesiones difusas o circunscritas de la piel y membranas mucosas. Cuatro especies de Leishmania parasitan al hombre: L. donavani, L. tropica, L. mexicana y L. braziliensis. Los síndromes clínicos que resultan de la infección dependen tanto del tropismo de la especie de protozoarios como de la respuesta inmunológica, principalmente celular, del huésped.
LEISHMANIASIS VISCERAL
Etiología y epidemiología
El complejo de especies L. donovani incluye varias especies (v.gr., L. infantum y L. chagasi). Estas especies causan la leishmaniasis visceral o kala-azar, que es endémica en varias zonas de la India, China, Centro y Sudamérica, Este y Oeste de Africa, y de los países que circundan el Mediterráneo. Los vectores son moscas del género Phlebotomus y la especie varía en las diferentes áreas. En la India no se conocen reservarios no humanos, pero en las otras regiones se ha observado la infección de otros mamíferos, como perros, zorros y roedores salvajes.
Patogenia
La mordedura del insecto introduce en el huésped promastigotes flagelados de L. donovani. Después de ser fagocitados por macrófagos del sistema reticuloendotelial, se transforman en amastigotes que se reproducen dentro de las células fagocíticas. Los amastigotes, que se liberan al morir la célula huésped, se diseminan por vía hematógena e infectan otras células reticuloendoteliales en el hígado, el bazo, los ganglios linfáticos, la médula ósea y la piel.
Diagnóstico
Cuadro clínico Los síntomas tienen un desarrollo gradual a lo largo de varios meses después de la infección; la enfermedad se caracteriza por debilidad, mareo, perdida de peso, diarrea y estreñimiento. Casi siempre hay fiebre, que presenta dos elevaciones diarias, y en algunas ocasiones se acompaña de calosfríos y diaforesis. Conforme la enfermedad progresa hay crecimiento del hígado y del bazo; este último llega a alcanzar la fosa iliaca. Cuando los macrófagos de la médula ósea son parasitados se desarrollan anemia y leucopenia. El paciente trombocitopénico sangra por las encías, la nariz y el tubo digestivo, en la piel pueden aparecen equímosis y petequias. La muerte es secundaria a infecciones bacterianas agregadas, anemia grave o hemorragia incontrolable. La infección latente se vuelve a manifestar durante la inmunopresión y la leishmaniasis visceral puede desarrollarse como infección oportunista en pacientes infectados por VIH. En estos pacientes los parásitos pueden localizarse en sitios no habituales, incluyendo la laringe y el tubo digestivo.
Estudios de laboratorio Los hallazgos de laboratorio que sugieren el diagnóstico de leishmaniasis visceral, cuando se observan en un paciente con fiebre, hepatosplenomegalia e historia de haber visitado las zonas endémicas, son los siguientes: anemia, leucopenia, trombocitopenia, hiperglobulinemia e hipoalbuminemia. El diagnóstico definitivo requiere del cultivo del microrganismo en medio de Novy-MacNeal-Nicolle (NNN) o algún otro medio específico, o bien la observación microscópica de los cuerpos de Leishman Donovan (amastigotes) en los frotis de tejido. En la mayoría de los casos el diagnóstico puede establecerse examinando aspirados de médula ósea. También es muy sensible para el diagnóstico la punción esplénica, pero tiene mucho riesgo. Otros lugares de donde puede obtenerse material para el diagnóstico son el hígado o los ganglios linfáticos afectados.
Tratamiento
El estibogluconato de sodio (antimonio pentavalente) es el tratamiento de elección para la mayoría de los casos de leishmaniasis, y está disponible en los EUA como Pentostam a través del Servicio de Medicamentos de los CDC en EUA (404-639-3670 en días hábiles y 404-639-2888 durante la noche y fines de semana) [ver cuadro, Información sobre infecciones por protozoarios en internet]. 26 Cuando el tratamiento inicial del kala-azar con estibogluconato de sodio fracasa, puede emplearse anfotericina B o pentamidina.26 El kala-azar resistente al estibogluconato de sodio puede también responder al tratamiento con anfotericina B liposomal.76
LEISHMANIASIS CUTANEA Y MUCOCUTANEA
Etiología y epidemiología
La leishmaniasis cutánea del Viejo Mundo es causada por tres especies de Leishmania que pertenecen al complejo L tropica: L. tropica existe en el Medio Oriente y en el litoral del Mediterráneo, L. major se encuentra en el Medio Oriente, Arabia, la antes Unión Soviética, India y la región Sub-Sahara de Africa, y L aethiopica se encuentra sobre todo en Etiopía y Kenya. Las moscas del género Phlebotomus son los vectores principales, aunque el contacto directo con una persona enferma puede también ser infectante. Las infecciones que son causadas por Leishmania pueden ser adquiridas por viajeros, así como por personal militar y otras personas que residan en áreas endémicas. El personal militar asignado al Medio Oriente puede adquirir leishmaniasis cutánea por L. major, así como infecciones viscerales por L. tropica.
La leishmaniasis del Nuevo Mundo se debe a infección por parásitos pertenecientes al grupo L. mexicana o L. viannia. El cuadro clínico varía de acuerdo con la naturaleza del microrganismo infectante, que a su vez es especifico de las diferentes regiones de América del Norte, del Centro y del Sur [ver tabla 2 ]. En las áreas de Centro y Sudamérica la infección por microrganismos del complejo L. mexicana produce una leishmaniasis cutánea. Se han informado algunos casos de leishmaniasis autóctona de este tipo en Texas. La infección con algunas de las cepas de L. viannia que son endémicas en varias áreas de Sudamérica, causa leishmaniasis cutánea, pero con el tiempo pueden evolucionar a una forma mucocutánea. La forma mucocutánea de esta leishmaniasis (espundia) afecta principalmente la mucosa nasal y de la orofaringe, y en algunos casos puede ser mortal. Todos estos parásitos de leishmania del Nuevo Mundo son también flebotomos, aunque el contacto directo con una persona enferma también puede trasmitir la infección. Varios mamíferos de vida salvaje son los reservorios naturales del microrganismo.
|
||||||||||||||||||||||||||||||||||||
|
Patogenia
Tanto la leishmaniasis del Viejo Mundo como la del Nuevo Mundo se inician con la mordedura de un flebotomo, que inyecta los promastigotes al huésped. Entonces el microrganismo penetra en los macrófagos y en las células endoteliales, se convierte en amastigotes y se multiplica. En el sitio de la picadura se desarrolla una respuesta inflamatoria granulomatosa. Con la isquemia local la lesión se ulcera y la infección bacteriana del área necrótica extiende la ulceración.
En la leishmaniasis cutánea del Viejo Mundo el periodo de incubación dura semanas o meses, después de los cuales se forma una pápula en el lugar de la picadura. La lesión inicial puede resolverse espontáneamente, pero en general progresa hacia la ulceración, formándose una placa eritematosa circular, con diámetro de varios centímetros y borde elevado [ver figura 3]. La infección bacteriana agregada puede producir linfadenopatía regional. Las lesiones suelen ser únicas, pero las picaduras múltiples pueden producir varias lesiones concomitantes. La resolución de las lesiones es lenta y en ocasiones tarda más de un año.
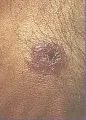
|
| Figura 3 |
| Leishmaniasis: lesión cutánea |
Diagnóstico
Cuadro clínico La infección por L. mexicana produce una o varias lesiones localizadas en las áreas expuestas del organismo, como la cara y los pabellones auriculares. 77 Las úlceras cicatrizan en forma espontánea en un periodo de 6 meses, pero si se localizan en los pabellones auriculares pueden causar gran destrucción tisular. La infección por L. viannia provoca lesiones en la piel o mucosas que pueden ser múltiples y muy grandes, sobre todo cuando tienen infección agregada por bacterias. Las lesiones cutáneas causadas por L. braziliensis cicatrizan en forma espontánea con mucho menos frecuencia que las ocasionadas por L mexicana. 77 En ocasiones las lesiones se diseminan por los vasos linfáticos de los brazos o piernas, o se extienden en forma local y afectan las mucosas. Después de un periodo de meses o años pueden encontrarse lesiones metastásicas en las mucosas de nariz y boca. Las úlceras en estos lugares pueden erosionar el tabique nasal y el paladar duro y blando. Algunos pacientes mueren por desnutrición o infecciones bacterianas agregadas.
La leishmaniasis cutánea difusa se ha observado en Etiopía, Venezuela, Brasil y la República Dominicana. En estos casos el nódulo inicial no se ulcera, sino que aparecen múltiples nódulos diseminados en todo el cuerpo. En las lesiones los microrganismos son muy abundantes. Esta forma diseminada de leishmaniasis se asocia con un defecto en la inmunidad celular, similar al que se observa en la lepra lepromatosa.
Estudios de laboratorio El diagnóstico definitivo se basa en la observación de amastigotes en frotis de tejidos teñidos en forma adecuada [ver figura 4], obtenidos mediante la biopsia o raspado del borde de las úlceras. Otra alternativa diagnóstica consiste en cultivar las amastigotes en medio NNN, inoculándolo en forma directa con material de las úlceras. El uso de sondas de ADN basado en el ADN cinetoplástico del parásito ha permitido la detección de microrganismos que podrían pasar desapercibidos en la sección histológica o los cultivos. Así mismo, esta técnica constituye un método rápido para determinar si la especie es L mexicana o L. viannia. A diferencia de los amastigotes de L. tropica y L mexicana, que son abundantes en las lesiones y fáciles de cultivar, los amastigotes de L. viannia son escasos y difíciles de cultivar, sobre todo en el paciente con leishmaniasis monocutánea. Los microorganismos de las tres especies no pueden distinguirse por su morfología y cuando se desea el diagnóstico de la especie deben utilizarse estudios inmunológicos especializados, enzimáticos o del ácido nucleico del parásito. La prueba cutánea (intradermorreacción) con leishmanina es positiva en todos los casos, excepto en la leishmaniasis cutánea diseminada.
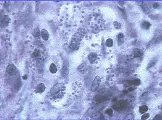
|
| Figura 4 |
| Amastigotes |
Tratamiento
Aunque los compuestos animoniales pentavalentes, estibogluconato de sodio [ver antes, Leishmaniasis visceral] y antimonato de meglumina (Glucantime), son los tratamientos de elección para la leishmaniasis cutánea del Nuevo y Viejo Mundo,26no se han establecido en definitiva los esquemas terapéuticos óptimos con éstos u otros agentes. Las diferentes especies y cepas geográficas de Leishmania responden en forma distinta al tratamiento y tienen diferente capacidad para causar enfermedad mucosa o cicatrizar en forma espontánea. Además, es difícil distinguir las especies infecciosas solo por clínica. Por lo tanto, no se ha logrado avanzar en el desarrollo de esquemas de tratamiento adecuados. La División de Enfermedades Parasitarias de los CDC de los EUA proporcionan orientación sobre el tratamiento de la leishmaniasis [ver cuadro, Información sobre infecciones por protozoarios en internet].
Tripanosomiasis
La infección por el protozoario flagelado Trypanosama cruzi produce la tripanosomiasis americana o enfermedad de Chagas, que tiene una forma aguda y otra crónica.78
Etiología y epidermiología
Dos fases de T. cruzi son capaces de infectar a los mamíferos, los tripanosomas, que se encuentran en la sangre, y los amastigotes, que se localizan dentro de las células infectadas. Varios géneros de chinches, triatomídeas o reduvídeos trasmiten el parásito; estos vectores se conocen comúnmente como chinches asesinas por ser predadoras de otros insectos u hociconas por su predilección por morder la cara de sus víctimas. Los reservorios no humanos son gatos, perros, ratas, mapaches y armadillos. El patrón de trasmisión de T. cruzi es diferente según la zona y depende de la especie de reduvídeo que prevalezca en la región, de los reservorios silvestres y domésticos y de las viviendas. Los insectos vectores habitan los orificios y nichos de las paredes, y los canceles de las puertas de las viviendas mal construidas. Los animales infectados que habitan en los alrededores de las viviendas rurales son los que trasmiten el agente a los vectores. Por las noches los vectores salen y se alimentan de la sangre de las personas que duermen, produciéndoles picaduras en las áreas expuestas, principalmente la cara. Durante el acto de chupar sangre, los insectos defecan, y sus heces contienen tripanosomas metacíclicos en estado infectante, que entran en el huésped a través de la herida producida por la picadura, por lesiones cutáneas, o atravesando la mucosa de las conjuntivas y los labios.
Aunque en los Estados Unidos existen los vectores reduvídeos, y también se han demostrado mamíferos infectados, parece ser que las condiciones generales de vivienda impiden que aparezca la trasmisión a los humanos. Sólo se han notificado algunos pocos casos autóctonos de enfermedad de Chagas en los Estados Unidos. 78 En las zonas rurales de Centro y Sudamérica las infecciones son frecuentes cuando las condiciones del medio propician el contacto entre el vector y el huésped humano. Además, la infección se puede trasmitir a través de transfusiones de sangre o sus productos, a través de la placenta y, en raros casos, por la ingestión de alimentos contaminados con deyecciones infectadas.
Patogenia
Se han descrito dos formas de enfermedad de Chagas. La forma aguda se desarrolla inmediatamente después de la infección, y afecta sobre todo a los niños de las áreas endémicas. 78 Después de algunos días de inóculo, aparece una lesión eritematosa indurada en el lugar de entrada del microrganismo que se denomina chagoma. Cuando el lugar de entrada es la conjuntiva ocurre una conjuntivitis unilateral, edema periocular y linfadenopatía preauricular que en conjunto constituyen el denominado signo de romaña. Después de dos semanas los tripanosomas aparecen en la sangre y a través de ella infectan las células, principalmente las de origen mesenquimatoso, donde se multiplican en su forma de amastigotes. La proliferación excesiva produce un seudoquiste celular, que al romperse libera tanto tripanosomas como amastigotes.
Diagnóstico
Cuadro clínico Durante esta fase aguda el huésped experimenta un cuadro de fiebre continua o intermitente, una erupción evanescente semejante a la del sarampión, hepatoesplenomegalia, linfadenopatía, edema sin fóvea en la cara y extremidades, nódulos subcutáneos dolorosos denominados chagomas hematógenos y, en los niños, diarrea. En los casos graves puede presentarse miocarditis y meningoencefalitis mortales. La fase aguda de la enfermedad es autolimitada, los síntomas desaparecen y los parásitos ya no pueden detectarse en la sangre del huésped.
La forma crónica de la enfermedad de Chagas se hace aparente después de una infección aguda o más frecuente subclínica. En general se observa en la segunda o tercera décadas de la vida y progresa en las décadas posteriores. Las complicaciones crónicas de la enfermedad de Chagas son producto de la destrucción de los ganglios del sistema nervioso autónomo, y de la miositis; la patogenia de tales lesiones se desconoce. El órgano que con mayor frecuencia se afecta es el corazón, 78 que sufre hipertrofia biventricular y miocarditis con infiltrado mononuclear. Los trastornos del ritmo que se asocian al daño cardiaco suelen ser bloqueo de rama derecha del haz de His, bloqueo completo o incompleto de la conducción aurículoventricular y presencia de contracciones ventriculares prematuras. Ha ocurrido muerte súbita en pacientes con enfermedad de Chagas, y también defunciones por complicación de insuficiencia cardiaca congestiva.
El tubo digestivo es el segundo lugar de afección en frecuencia.78 La enfermedad produce denervación, con los consiguientes trastornos de la motilidad y dilatación, siendo frecuente encontrar magaesófago y megacolon.
Las infecciones congénitas producen nacimientos prematuros, con niños que presentan hepatoesplenomegalia, distensión abdominal, cardiomegalia, megaesófago y meningoencefalitis. En los pacientes infectados por VIH la reactivación de la enfermedad de Chagas puede provocar masas cerebrales y, en pacientes con infecciones agudas, encefalitis necrosante.
Estudios de laboratorio En la fase aguda de la enfermedad de Chagas es usual encontrar leucocitosis superior a 18,000/mm 3 (con un 70 a 90 por ciento de linfocitos), además de detectarse el parásito en la sangre. Se han podido observar los parásitos en movimiento en exámenes de sangre fresca sin teñir; cuando los frotis se tiñen con Giemsa, los parásitos se observan con forma de una C mayúscula. Si los frotis no muestran parásitos, se recomienda centrifugar varios centímetros cúbicos de sangre, lisando previamente los eritrocitos, y realizando nuevos frotis con sedimento. Los microrganismos pueden cultivarse en muestras de sangre en medio NNN o en agar sangre. Otra alternativa es inocular a algún roedor de laboratorio con sangre problema y vigilar estrechamente la aparición de parasitemia. La técnica de laboratorio más sensible que existe es el xenodiagnóstico, que por desgracia no es accesible a todos los laboratorios. La técnica consiste en alimentar reduvídeos cultivados en laboratorio con la sangre del paciente, si la sangre ingerida por los insectos contiene tripanosomas los insectos se infectarán, lo que se detecta por examen subsecuente de las heces en busca de los parásitos. La sangre que contiene tripanosomas es un material infeccioso y debe manejarse con precaución. Los CDC pueden realizar, a solicitud del médico, una prueba de fijación de complemento que es de ayuda diagnóstica en la fase aguda.
En la fase crónica de la enfermedad de Chagas la radiografía de tórax puede mostrar cardiomegalia biventricular y datos de insuficiencia cardiaca congestiva. Es frecuente encontrar alteraciones electrocardiográficas. De los otros métodos, sólo el xenodiagnóstico es capaz de detectar la parasitemia. La prueba de fijación de complemento puede ser positiva, pero sólo indica una infección previa, y no establece una correlación entre los hallazgos clínicos y una enfermedad de Chagas activa.
Tratamiento
Aún no se ha establecido el tratamiento óptimo de la enfermedad de Chagas. El nifurtimox (disponible por medio de los CDC) elimina la parasitemia [ver cuadro, Información sobre infecciones por protozoarios en internet]. Se recomienda su administración en pacientes con fase aguda y en pacientes con fase crónica y parasitemia demostrada.26 Los efectos colaterales son frecuentes, incluyendo anemia hemolítica en pacientes con deficiencia de G6PD, neuritis periférica y psicosis. Las complicaciones cardiovasculares y gastrointestinales de la fase crónica se manejan con medicamentos, aunque en ocasiones se requiere cirugía para el megacolon.
TRIPANOSOMIASIS AFRICANA
Etiología y epidemiología
La tripanosomiasis africana o enfermedad del sueño, es una parasitosis aguda o crónica causada por hemoflagelados del género Trypanosoma. La enfermedad es endémica en un ancho cinturón que atraviesa Africa Ecuatorial. Dos variedades infectan al hombre: la tripanosomiasis del Africa Occidental se presenta en los bosques de la porción Occidental y Central de Africa, y es causada por T. brucei gambiense, un parásito que no tiene reservorio animal importante. En cambio, T. brucei rhodesiense, que produce la enfermedad del sueño de Africa Oriental, sí tiene reservorios animales, en especial de vida salvaje. Los visitantes de los parques nacionales tienen riesgo de adquirir este tipo de tripanosomiasis. 79 Ambas tripanosomiasis africanas son trasmitidas por varias especies de moscas tsetsé (del género Glossina ).
Diagnóstico
Cuadro clínico Después de un período de incubación que varía entre unos cuantos días a un par de semanas, aparece el chancro tripanosómico en el lugar de la picadura de la mosca tsetsé, localizado en áreas expuestas de la piel.79 El chancro en un principio es una pápula, que evoluciona en 2 semanas hacia un nódulo doloroso e inflamado que se resuelve espontáneamente. Aunque el chancro tripanosómico es común en pacientes no africanos, rara vez se observa en pacientes africanos.
Sigue después la fase hemolinfática de la enfermedad, en la que los protozoarios invaden el torrente sanguíneo y los ganglios linfáticos. Los síntomas que acompañan esta fase se desarrollan con lentitud en los africanos y consisten en fiebre, linfadenopatía, debilidad y cefalea. En cambio, en los no africanos el inicio es súbito y a menudo coincide con el desarrollo del chancro. En estos últimos llaman la atención los periodos febriles que duran de 1 a 7 días, y que recurren después de cortos periodos afebriles. A la fiebre se asocian otros síntomas: calosfríos, cefalea, malestar general y anorexia. La linfadenopatía, con ganglios blandos y no dolorosos, es más frecuente en la tripanosomiasis de Africa Occidental, e incluye los ganglios de las cadenas cervicales posteriores (signo de Winterbottom). También se presenta una erupción típica, que se observa mejor en los individuos de color claro, y aparece entre la sexta y octava semana de la infección, caracterizada por máculas eritematosas, circinadas y evanescentes, de predominio en el tronco.
La siguiente fase en la tripanosomiasis africana es la invasión del sistema nervioso central, produciendo una meningoencefalitis o meningomielitis difusa. En la tripanosomiasis del Africa Occidental, que tiene una evolución muy lenta, los síntomas de la enfermedad del sueño se desarrollan varios años después de la infección. La debilidad e indiferencia al medio evolucionan hacia el coma y la muerte por inanición o enfermedades intercurrentes. Por el contrario, en la tripanosomiasis del Africa Oriental la evolución es más rápida y la fase neurológica se presenta en forma temprana. Aún antes de que exista afección del sistema nervioso central es frecuente encontrar somnolencia, cambios de la personalidad y dificultad para concentrarse. En la tripanosomiasis del Africa Oriental puede haber pancarditis durante la fase aguda, que puede producir la muerte antes de la fase neurológica. Debido a que la tripanosomiasis del Africa Oriental puede adquirirse al visitar los parques y reservas de animales y es una enfermedad aguda y febril, es frecuente que se confunda con el paludismo. La enfermedad del sueño debe considerarse en el diagnóstico diferencial de cualquier proceso febril que aparece en el paciente que regresa de un área endémica.
Estudios de laboratorio El recuento leucocitario es normal, pero puede existir una mononucleosis de 50 al 70 por ciento. Los niveles séricos de IgM se elevan de 1 a 2 semanas después del comienzo de la parasitemia y pueden alcanzar hasta 7 veces los valores normales. El diagnóstico definitivo se hace por observación de los tripanosomas en la sangre, médula ósea, líquido de los ganglios infectados o líquido cefalorraquídeo previamente centrifugado. Los microrganismos teñidos con Wright, Giemsa o tinción de Leishman, pueden observarse en los frotis tomados del chancro hasta 48 horas antes de que aparezcan en la sangre. Si el examen en sangre periférica no es diagnóstico, los protozoarios pueden encontrarse en aspirados de ganglios linfáticos infectados, o en leucocitos del sobrenadante de una muestra de sangre centrifugada. Incluso en los casos en que no hay síntomas neurológicos, debe investigarse la enfermedad del sistema nervioso central mediante punción lumbar, y aun cuando no se observen los tripanosomas en el centrifugado del líquido cefalorraquídeo, la elevación del recuento celular, de las proteínas o de la fracción IgM, habla en favor de enfermedad neurológica.
Tratamiento El eflornitine ha demostrado ser eficaz para las infecciones por T. brucei gambiense, 2,23 incluso las que son refractarias al agente arsenical melarsoprol. El eflornitine es el tratamiento de elección para los estadios hemolinfático temprano y tardío del SNC de la tripanosomiasis de Africa Occidental. 26 Debido a que este medicamento puede causar anemia, leucopenia y trombocitopenia, deben realizarse citologías hemáticas dos veces por semana durante el tratamiento. El eflornitine no ha sido eficaz para las infecciones por T. brucei rhodesiense. El estadio temprano de la tripanosomiasis de Africa Oriental se trata con suramina, y el estadio de SNC con melarsoprol.26 Ambos medicamentos pueden solicitarse en los EUA en el Servicio de Medicamentos de los CDC [ver cuadro, Información sobre infecciones por protozoarios en internet].
Figura 1 Seward Hung.
- Joiner KA, Dubremetz JF: Toxoplasma gandii: a protozoan for the nineties. Infect Immun 61:1169, 1993 [PMID 8454321]
- Teutsch SM, Juranek DD, Sulzer A, et al: Epidemic toxoplasmosis associated with infected cats. N Engl J Med 300:695, 1979 [PMID 763300]
- Benenson MW, Takafuji ET, Lemon SM, et al: Oocyst-transmitted toxoplasmosis associated with ingestion of contaminated water. N Engl J Med 307:666, 1982 [PMID 7110216]
- Krick JA, Remington JS: Current concepts in parasitology: toxoplasmosis in the adult-an overview. N Engl J Med 298:550, 1978 [PMID 342953]
- Stray-Pedersen B: A prospective study of acquired toxoplasmosis among 8,043 pregnant women in the Oslo area. Am J Obstet Gynecol 136:399, 1980
- Beach PG: Prevalence of antibodies to Toxoplasma gondii in pregnant women in Oregon. J Infect Dis 140:780, 1979
- Guerina NG, Hsu H, Meissner C, et al: Neonatal serologic screening and early treatment for congenital Toxoplasma gondii infection. N Engl J Med 330:1858, 1994 [PMID 7818637]
- Wong S, Remington JS: Toxoplasmosis in pregnancy. Clin Infect Dis 18:853, 1994 [PMID 8086543]
- Hohlfeld P, Daffos F, Costa J, et al: Prenatal diagnosis of congenital toxoplasmosis with a polymerase-chain-reaction test on amniotic fluid. N Engl J Med 331:695, 1994 [PMID 8058075]
- McAuley J, Boyer KM, Patel D, et al: Early longitudinal evaluations of treated infants and children and untreated historical patients with congenital toxoplasmosis: the Chicago Collaborative Treatment Trial. Clin Infect Dis 18:38, 1994 [PMID 8054436]
- McCabe R, Remington JS: Toxoplasmosis: the time has come (editorial). N Engl J Med 318:313, 1988
- Daffos F, Forestier F, Capella-Pavlovsky M, et al: Prenatal management of 746 pregnancies at risk for congenital toxoplasmosis. N Engl J Med 318:271, 1988 [PMID 3336419]
- Nussenblatt RB, Belfort R Jr: Ocular toxoplasmosis. JAMA 271:304, 1994 [PMID 8295291]
- McCabe RE, Brooks RG, Dorfman RF, et al: Clinical spectrum in 107 cases of toxoplasmic lymphadenopathy. Rev Infect Dis 9:754, 1987 [PMID 3326123]
- Mawhorter SD, Effron D, Blinkhorn R, et al: Cutaneous manifestations of toxoplasmosis. Clin Infect Dis 14:1084, 1992 [PMID 1600010]
- Montoya JG, Remington JS: Studies on the serodiagnosis of toxoplasmic lymphadenitis. Clin Infect Dis 20:781, 1995 [PMID 7795074]
- Luft BJ, Remington JS: Acute Toxoplasma infection among family members of patients with acute lymphadenopathic toxoplasmosis. Arch Intern Med 144:53, 1984 [PMID 6691774]
- Hunter CA, Remington JS: Immunopathogenesis of toxoplasmic encephalitis. J Infect Dis 170:1057, 1994
- Luft BJ, Naot Y, Araujo FG, et al: Primary and reactivated toxoplasma infection in patients with cardiac transplants: clinical spectrum and problems in diagnosis in a defined population. Ann Intern Med 99:27, 1983 [PMID 6344718]
- Israelski DM, Remington JS: Toxoplasmosis in patients with cancer. Clin Infect Dis 17(suppl2):S423, 1993
- Pomeroy C, Filice GA: Pulmonary toxoplasmosis: a review. Clin Infect Dis 14: 863, 1992 [PMID 1576281]
- Shepp DH, Hackman RC, Conley FK, et al: Toxoplasma gondii reactivation identified by detection of parasitemia in tissue culture. Ann Intern Med 103:218, 1985 [PMID 3893254]
- Hofflin JM, Remington JS: Tissue culture isolation of Toxoplasma from blood of a patient with AIDS. Arch Intern Med 145:925, 1985 [PMID 3994469]
- Luft BJ, Remington JS: Toxoplasmic encephalitis in AIDS. Clin Infect Dis 15: 221, 1992
- Rabaud C, May T, Amiel C, et al: Extracerebral toxoplasmosis in patients infected with HIV: a French national study. Medicine Baltimore) 73:306, 1994 [PMID 7984082]
- Luft BJ, Brooks RG, Conley FK, et al: Toxoplasmic encephalitis in patients with acquired immune deficiency syndrome. JAMA 252:913, 1994
- Wallace MR, Rossetti RJ, Olson PE: Cats and toxoplasmosis risk in HIV-infected adults. JAMA 269:76, 1993 [PMID 8416410]
- Richards FO Jr, Kovacs JA, Luft BJ: Preventing toxoplasmic encephalitis in persons infected with human immunodeficiency virus. Clin Infect Dis 21 (suppl 1):S49, 1995
- Milligan SA, Katz MS, Craven PC, et al: Toxoplasmosis presenting as panhypopituitarism in a patient with the acquired immune deficiency syndrome. Am J Med 77:760, 1984
- Knobel H, Guelar A, Graus F, et al: Toxoplasmic encephalitis with normal CT scan and pathologic MRI. Am J Med 99:220, 1995 [PMID 7625428]
- Pierce MA, Johnson MD, Maciunas RJ, et al: Evaluating contrast-enhancing brain lesions in patients with AIDS by using positron emission tomography. Ann Intern Med 123:594, 1995
- Potasman I, Resnick L, Luft BJ, et al: Intrathecal production of antibodies against Toxoplasma gondii in patients with toxoplasmic encephalitis and the acquired immunodeficiency syndrome (AIDS). Ann Intern Med 108:49, 1988 [PMID 3337515]
- Shaw GM, Harper ME, Hahn BH: HTLV-III infection in brains of children and adults with AIDS encephalopathy. Science 227:177, 1985 [PMID 2981429]
- Cohn JA, McMeeking A, Cohen W, et al: Evaluation of the policy of empiric treatment of suspected Toxoplasma encephalitis in patients with the acquired immunodeficiency syndrome. Am J Med 86:521, 1989 [PMID 2712059]
- Araujo FG, Remington JS: Antigenemia in recently acquired acute toxoplasmosis. J Infect Dis 141:144, 1990
- Rougier D, Ambroise-Thomas P: Detection of toxoplasmic immunity by multipuncture skin test with excretory-secretory antigen. Lancet 2:121, 1985 [PMID 2862318]
- Lane HC, Laughon BE, Falloon J: Recent advances in the management of AIDS-related opportunistic infections. Ann Intern Med 120:945, 1994 [PMID 7909657]
- Wynn RF, Leen CLS, Brettle RP: Azithromycin for cerebral toxoplasmosis in AIDS. Lancet 341:243, 1993 [PMID 8093526]
- Kovacs JA: Efficacy of atovaquone in treatment of toxoplasmosis in patients with AIDS. The NIAID-Clinical Center Intramural AIDS Program. Lancet 340:637, 1992
- Morris JT, Kelly JW: Effective treatment of cerebral toxoplasmosis with doxycycline. Am J Med 93:107,1992 [PMID 1626560]
- The TE Study Group: Assessment of therapy for Toxoplasma encephalitis. Am J Med 82:907, 1987
- Leport C, Raffi F, Matheron S, et al: Treatment of central nervous system toxoplasmosis with pyrimethamine/sulfadiazine combination in 35 patients with the acquired immunodeficiency syndrome: efficacy of long-term continuous therapy. Am J Med 84:94, 1988 [PMID 3337134]
- Rolston KVI, Hoy J: Role of clindamycin in the treatment of central nervous system toxoplasmosis. Am J Med 83:551, 1987
- Dannemann BR, Israelski DM, Remington JS: Treatment of toxoplasmic encephalitis with intravenous clindamycin. Arch Intern Med 148:2477, 1988 [PMID 3190380]
- Dannemann B, McCutchan JA, Israelski D, et al: Treatment of toxoplasmic encephalitis in patients with AIDS: a randomized trial comparing pyrimethamine plus clindamycin to pyrimethamine plus sulfadiazine. Ann Intern Med 116:33, 1992 [PMID 1727093]
- Luft BJ, Harner R, Korzun AH, et al: Toxoplasmic encephalitis in patients with the acquired immunodeficiency syndrome. N Engl J Med 329:995, 1993 [PMID 8366923]
- Podzamczer D, Miro JM, Bolao F, et al: Twice-weekly maintenance therapy with sulfadiazine- pyrimethamine to prevent recurrent toxoplasmic encephalitis in patients with AIDS. Ann Intern Med 123:175, 1995 [PMID 7598298]
- Rolston KVI: Clindamycin in cerebral toxoplamosis (letter). Am J Med 85:284, 1988
- Caumes E, Bocquet H, Guermonprez G, et al: Adverse cutaneous reactions to pyrimethamine/sulfadiazine and pyrimethamine/clindamycin in patients with AIDS and toxoplasmic encephalitis. Clin Infect Dis 21:656, 1995 [PMID 8527561]
- Murphy K, Groarke M, O'Shaughnessy E, et al: Pyrimethamine alone as long-term suppressive therapy in cerebral toxoplasmosis. Am J Med 96:95, 1994 [PMID 8304371]
- Bozzette SA, Finkelstein DM, Spector SA, et al: A randomized trial of three antipneumocystis agents in patients with advanced immunodeficiency infection. N Engl J Med 332:693, 1995
- Opravil M, Hirschel B, Lazzarin A, et al: Once-weekly administration of dapsone/pyrimethamine vs. aerosolized pentamidine as combined prophylaxis for Pneumocystis carinii pneumonia and toxoplasmic encephalitis in human immunodeficiency virus-infected patients. Clin Infect Dis 20:531, 1995
- Ruebush TK II, Breman JG, Kaiser RL, et al: Selective primary health care: XXIV. Malaria. Rev Infect Dis 8:454, 1986
- Wyler DJ: Malaria: overview and update. Clin Infect Dis 16:449, 1993
- Zucker JR, Barber AM, Paxton LA, et al: Malaria surveillance-United States, 1992. MMWR CDC Surveill Summ 44(5):1, 1995
- Guerrero IC, Weniger BC, Schultz MG: Transfusion malaria in the United States, 1972-1981. Ann Intern Med 99:221, 1983 [PMID 6349456]
- Hulbert TV: Congenital malaria in the United States: report of a case and review. Clin Infect Dis 14:922, 1992
- Thompson RG, Harper IA: Malaria in returning travellers (letter). Br Med J 1:1468, 1977 [PMID 861694]
- Singal M, Shaw PK, Lindsay RC, et al: An outbreak of introduced malaria in California possibly involving secondary transmission. Am J Trop Med Hyg 26:1, 1977 [PMID 320891]
- Transmission of Plasmodium vivax malaria-San Diego County, California, 1988 and 1989. MMWR 39:91, 1990
- Brook JH, Genese CA, Bloland PB, et al: Brief report: malaria probably locally acquired in New Jersey. N Engl J Med 331:22, 1994 [PMID 8202097]
- Layton M, Parise ME, Campbell CC, et al: Mosquito-transmitted malaria in New York City, 1993. Lancet 346:729, 1995 [PMID 7658873]
- Lobel HO, Campbell CC, Schwartz IK, et al: Recent trends in the importation of malaria caused by Plasmodium falciparum into the United States from Africa. J Infect Dis 152:613, 1985
- Svenson JE, MacLean JD, Gyorkos TW, et al: Imported malaria: clinical presentation and examination of symptomatic travelers. Arch Intern Med 155:861, 1995 [PMID 7717795]
- White NJ, Warrell DA, Chanthavanich P, et al: Severe hypoglycemia and hyperinsulinemia in falciparum malaria. N Engl Med 309:61, 1983
- McLaughlin GL, Ruth JL, Jablonski E, et al: Use of enzyme-linked synthetic DNA in diagnosis of falciparun malaria. Lancet 1:714, 1987 [PMID 2882132]
- Sethabutr O, Brown AE, Panyim S, et al: Detection of Plasmodium falciparum by polymerase chain reaction in a field study. J Infect Dis 166:145, 1992 [PMID 1607686]
- Beadle C, Long GW, Weiss WR, et al: Diagnosis of malaria by detection of Plasmodium falciparum HRP-2 antigen with a rapid dipstick antigen-capture assay. Lancet 343:564, 1994 [PMID 7906328]
- Tracey KJ: TNF and Mae West or: death from too much of a good thing. Lancet 345:75, 1995
- Wyler DJ: Malaria chemoprophylaxis for the traveler. N Engl J Med 329:31, 1993
- Drugs for parasitic infections. Med Lett Drugs Ther 32:23, 1990
- Treatment with quinidine gluconate of persons with severe Plasmodium falciparum infection: discontinuation of parenteral quinine from CDC Drug Service. MMWR 40(RR-4):21, 1991
- Phillips RE, Warrell DA, White NJ, et al: Intravenous quinidine for the treatment of severe falciparum malaria: clinical and pharmacokinetic studies. N Engl J Med 312:1273, 1985 [PMID 3887162]
- Miller KD, Greenberg AE, Campbell CC: Treatment of severe malaria in the United States with a continuous infusion of quinidine gluconate and exchange transfusion. N Engl J Med 321:65, 1989
- Hoffman SL, Rustama D, Punjabi NH, et al: High-dose dexamethasone in quinine-treated patients with cerebral malaria: a double-blind, placebo-controlled trial. J Infect Dis 158:325, 1988 [PMID 3042874]
- Warrell DA, Looareesuwan S, Warrell MJ, et al: Dexamethasone proves deleterious in cerebral malaria: a double blind trial in 100 atients. N Engl J Med 306: 313, 1982 [PMID 7033788]
- Phillips P, Nantel S, Benny WB: Exchange transfusion as an adjunct to the treatment of severe falciparum malaria: case report and review. Rev Infect Dis 12:1100, 1990 [PMID 2267486]
- Gordeuk V, Thuma P, Brittenham G, et al: Effect of iron chelation therapy on recovery from deep coma in children with cerebral malaria. N Engl J Med 327:1473, 1992 [PMID 1406879]
- Nosten F, ter Kuile FO, Luxemburger C, et al: Cardiac effects of antimalarial treatment with halofantrine. Lancet 341:1054, 1993 [PMID 8096959]
- Hien TT, White NJ: Qinghaosu. Lancet 341:603, 1993 [PMID 8094838]
- Watt G, Shanks GD, Edstein MD, et al: Ciprofloxacin treatment of drug-resistant falciparum malaria. J Infect Dis 164:602, 1991 [PMID 1869847]
- McClean KL, Hitchman D, Shafran SD: Norfloxacin is inferior to chloroquine for falciparum malaria in northwestern Zambia: a comparative clinical trial. J Infect Dis 165:904, 1992
- Sadig ST, Glasgow KW, Drakeley CJ, et al: Effects of azithromycin on malariometic indices in the Gambia. Lancet 346:881, 1995
- Shmuklarsky MJ, Boudreau EF, Pang LW, et al: Failure of doxycycline as a causal prophylactic agent against Plasmodium falciparum malaria in healthy nonimmune volunteers. Ann Intern Med 120:294, 1994 [PMID 8291822]
- Pruthi RK, Marshall WF, Wiltsie JC, et al: Human babesiosis. Mayo Clin Proc 70:853, 1995 [PMID 7643639]
- Persing DH, Herwaldt BL, Glaser C, et al: Infection with a babesia-like organism in Northern California. N Engl J Med 332:298, 1995 [PMID 7816065]
- Weiss LM, Wittner M, Wasserman S, et al: Efficacy of azithromycin for treating Babesia microti infection in the hamster model. J Infect Dis 168:1289, 1993 [PMID 8228366]
- Kappus KD, Lundgren RG Jr, Juranek DD, et al: Intestinal parasitism in the United States: update on a continuing problem. Am J Trop Med Hyg 50:705, 1994 [PMID 8024063]
- Overturf GD: Endemic giardiasis in the United States-role of the daycare center. Clin Infect Dis 18:764, 1994
- Moldwin RM: Sexually transmitted protozoal infections: Trichomonas vaginalis, Entamoeba histolytica, and Giardia lamblia. Urol Clin North Am 19:93, 1992
- Mintz ED, Hudson-Wragg M, Mshar P, et al: Foodborne giardiasis in a corporate office setting. J Infect Dis 167:250, 1993 [PMID 8418177]
- Quick R, Paugh K, Addiss D, et al: Restaurant-associated outbreak of giardiasis. J Infect Dis 166:673, 1992
- Chute CG, Smith RP, Baron JA: Risk factors for endemic giardiasis. Am J Public Health 77:585, 1987
- Moore AC, Herwaldt BL, Craun GF, et al: Surveillance for waterborne disease outbreaks-United States, 1991-1992. MMWR CDC Surveill Summ 42:1, 1993 [PMID 8232179]
- Dykes AC, Juranek DD, Lorenz RA, et al: Municipal waterborne giardiasis: an epidemiologic investigation: beavers implicated as a possible reservoir. Ann Intern Med 92:165, 1980
- Brodsky RE, Spencer HC Jr, Schultz MG: Giardiasis in American travelers to the Soviet Union. J Infect Dis 130:319, 1974 [PMID 4413575]
- Nash TE, Herrington DA, Losonsky GA, et al: Experimental human infections with Giardia lamblia. J Infect Dis 156:974, 1987 [PMID 3680997]
- Clyne CA, Eliopoulos GM: Fever and urticaria in acute giardiasis. Arch Intern Med 149:939, 1989
- Hopkins RS, Juranek DD: Acute giardiasis: an improved clinical case definition for epidemiologic studies. Am J Epidemiol 133:402, 1991 [PMID 1994703]
- Lengerich EJ, Addiss DG, Juranek DD: Severe giardiasis in the United States. Clin Infect Dis 18:760, 1994
- Gunasekaran TS, Hassall E: Giardiasis mimicking inflammatory bowel disease. J Pediatr 120:424, 1992
- Zimmerman SK, Needham CA: Comparison of conventional stool concentration and preserved-smear methods with Merifluor Cryptosporidium/Giardia Direct Immunofluorescence Assay and ProSpecT Giardia EZ Microplate Assay for detection of Giardia lamblia J Clin Microbiol 33:1942, 1995
- Alles AJ, Waldron MA, Sierra LS, et al: Prospective comparison of direct immunofluorescence and conventional staining methods for detection of Giardia and Cryptosporidium spp. in human fecal specimens. J Clin Microbiol 33:1632, 1995
- Drugs for parasitic infections. Med Lett Drugs Ther 37:99, 1995
- Craft JC, Murphy T, Nelson JD: Furazolidone and quinacrine: comparative study of therapy for giardiasis in children. Am J Dis Child 135:164, 1981 [PMID 7468550]
- Burtin P, Taddio A, Ariburnu O, et al: Safety of metronidazole in pregnancy: a meta-analysis. Am J Obstet Gynecol 172:525, 1995 [PMID 7856680]
- Addiss DG, Juranek DD, Spencer HC: Treatment of children with asymptomatic and nondiarrheal Giardia infection. Pediatr Infect Dis J 10:843, 1991 [PMID 1823546]
- Pickering LK, Morrow AL: Commentary. Pediatr Infect Dis J 10:846, 1991
- Ongerth JE, Johnson RL, Macdonald SC, et al: Backcountry water treatment to prevent giardiasis. Am J Public Health 79:1633, 1989 [PMID 2817191]
- Petri WA Jr, Clark CG, Diamond LS: Host-parasite relationships in amebiasis: conference report. J Infect Dis 169:483, 1994 [PMID 7908923]
- Adams EB, MacLeod IN: Invasive amebiasis: I. Amebic dysentery and its complications. Medicine (Baltimore) 56:315, 1977
- Chun D, Chandrasoma P, Kiyabu M: Fulminant amebic colitis: a morphologic study of four cases. Dis Colon Rectum 37:535, 1994 [PMID 8200230]
- Adams EB, MacLeod IN: Invasive amebiasis: II. Amebic liver abscess and its complications. Medicine (Baltimore) 56:325, 1977
- Peters RS, Gitlin N, Libke RD: Amebic liver abscess. Annu Rev Med 32:161, 1981 [PMID 7013659]
- Katzenstein D, Rickerson V, Braude A: New concepts of amebic liver abscess derived from hepatic imaging, serodiagnosis, and hepatic enzymes in 67 consecutive cases in San Diego. Medicine (Baltimore) 61:237, 1982 [PMID 6806561]
- Gomersall LN, Currie J, Jeffrey R: Amoebiasis: a rare cause of cardiac tamponade. Br Heart J 71:368, 1994 [PMID 8198889]
- Veliath AJ, Bansal R, Sankaran V, et al: Genital amebiasis. Int J Gynaecol Obstet 25:249, 1987
- Ralls PW, Barnes PF, Radin DR, et al: Sonographic features of amebic and pyogenic liver abscesses: a blinded comparison. AJR Am J Roentgenol 149:499, 1987 [PMID 3303877]
- Radin DR, Ralls PW, Colletti PM, et al: CT of amebic liver abscess. AJR Am J Roentgenol 150:1297, 1988
- Ralls PW, Henley DS, Colletti PM, et al: Amebic liver abscess: MR imaging. Radiology 165:801, 1987 [PMID 3317504]
- Elizondo G, Weissleder R, Stark DD, et al: Amebic liver abscess: diagnosis and treatment evaluation with MR imaging. Radiology 165:795, 1987 [PMID 2891154]
- McAuley JB, Herwaldt BL, Stokes SL, et al: Diloxanide furoate for treating asymptomatic Entamoeba histolytica cyst passers: 14 years' experience in the United States. Clin Infect Dis 15:464, 1992 [PMID 1520794]
- Van Allan RJ, Katz MD, Johnson MB, et al: Uncomplicated amebic liver abscess: prospective evaluation of percutaneous therapeutic aspiration. Radiology 183:827, 1992 [PMID 1584941]
- Ralls PW, Barnes PF, Johnson MB, et al: Medical treatment of hepatic amebic abscess: rare need for percutaneous drainage. Radiology 165:805, 1987 [PMID 3317505]
- Singh JP, Kashyap A: A comparative evaluation of percutaneous cathe ter drainage for resistant amebic liver abscesses. Am J Surg 158:58, 1989 [PMID 2662790]
- DeHovitz JA, Pape JW, Boncy M, et al: Clinical manifestations and therapy of Isospora belli infection in patients with the acquired immunodeficiency syndrome. N Engl J Med 315:87, 1986 [PMID 3487730]
- Pape JW, Verdier R-I, Johnson WD Jr: Treatment and prophylaxis of Isospora belli infection in patients with the acquired immunodeficiency syndrome. N Engl J Med 320:1044, 1989
- Weiss LM, Perlman DC, Sherman J, et al: Isospora belli infection: treatment with pyrimethamine. Ann Intern Med 109:474, 1988 [PMID 3261956]
- MacKenzie WR, Hoxie NJ, Proctor ME, et al: A massive outbreak in Milwaukee of Cryptosporidium infection transmitted through the public water supply. N Engl J Med 331:161, 1994
- McAnulty JM, Fleming DW, Gonzalez AH: A community-wide outbreak of cryptosporidiosis associated with swimming at a wave pool. JAMA 272:1597, 1994 [PMID 7966870]
- Jokipii L, Jokipii AMM: Timing of symptoms and oocyst excretion in human cryptosporidiosis. N Engl J Med 315:1643, 1986 [PMID 2431315]
- Koch KL, Phillips DJ, Aber RC, et al: Cryptosporidiosis in hospital personnel: evidence for person-to-person transmission. Ann Intern Med 102:593, 1985 [PMID 3885814]
- Newman RD, Zu SX, Wuhib T, et al: Household epidemiology of Cryptosporidium paroum infection in an urban community in northeast Brazil. Ann Intern Med 120:500, 1994 [PMID 8311373]
- DuPont HL, Chappell CL, Sterling CR, et al: The infectivity of Cryptosporidium paroum in healthy volunteers. Cryptosporidium parvum N Engl J Med 332:855, 1995
- Current WL, Reese NC, Ernst JV, et al: Human cryptosporidiosis in immunocompetent and immunodeficient persons: studies of an outbreak and experimental transmission. N Engl J Med 308:1252, 1983
- Wolfson JS, Richter JM, Waldron MA, et al: Cryptosporidiosis in immunocompetent patients. N Engl J Med 312:1278, 1985 [PMID 4039408]
- Flanigan T, Whalen C, Turner J, et al: Cryptosporidium infection and CD4 counts. Ann Intern Med 116:840, 1992 [PMID 1348918]
- Navin TR, Juranek DD: Cryptosporidiosis: clinical, epidemiologic, and parasitologic review. Rev Infect Dis 6:313, 1984 [PMID 6377439]
- Ma P, Villanueva TG, Kaufman D, et al: Respiratory cryptosporidiosis in the acquired immune deficiency syndrome: use of modified cold Kinyoun and Hemacolor stains for rapid diagnoses. JAMA 252:1298, 1994
- Teixidor HS, Godwin TA, Ramirez EA: Cryptosporidiosis of the biliary tract in AIDS. Radiology 180:51, 1991 142. Kehl KS, Cicirello H, Havens PL: Comparison of four different methods for detection of Cryptosporidium species. J Clin Microbiol 33:416, 1995
- Kehl KS, Cidrello H, Havens PL: Comparison of four different methods for detection of Cryptosporidium species. J Clin Microbiol 33:416, 1995 [PMID 7536216]
- White AC Jr, Chappell CL, Hayat CS, et al: Paromomycin for crypt osporidiosis in AIDS: a prospective, double-blind trial. J Infect Dis 170:419, 1994 [PMID 8035029]
- Ortega YR, Sterling CR, Gilman RH, et al: Cyclospora species-a new protozoan pathogen of humans. N Engl J Med 328:1308, 1993 [PMID 8469253]
- Hoge CW, Shlim DR, Rajah R, et al: Epidemiology of diarrhoeal illness associated with coccidian-like organism among travellers and foreign residents in Nepal. Lancet 341:1175, 1993
- Huang P, Weber JT, Sosin DM, et al: The first reported outbreak of diarrheal illness associated with Cyclospora in the United States. Ann Intern Med 123:409, 1995 [PMID 7639439]
- Bendall RP, Lucas S, Moody A, et al: Diarrhoea associated with Cyanobacterium -like bodies: a new coccidian enteritis of man. Lancet 341:590, 1993 [PMID 8094829]
- Pape JW, Verdier RI, Boncy M, et al: Cyclospora infection in adults infected with HIV: clinical manifestations, treatment, and prophylaxis. Ann Intern Med 121:654, 1994
- Hoge CW, Shlim DR, Ghimire M, et al: Placebo-controlled trial of co-trimoxazole for Cyclospora infections among travellers and foreign residents in Nepal. Lancet 345:691, 1995
- Frenkel JK: Sarcosporidiosis. Hunter's Tropical Medicine, 6th ed. Strickland GT, Ed. WB Saunders Co, Philadelphia, 1984, p 611
- Beaver PC, Gadgil RK, Morera P: Sarcocystis in man: a review and report of five cases. Am J Trop Med Hyg 28:819, 1979 [PMID 114067]
- Bunyaratvej S, Bunyawongwiroj P, Nitiyanant P: Human intestinal sarcosporidiosis: report of six cases. Am J Trop Med Hyg 31:36, 1982 [PMID 6800273]
- Walzer PD, Judson FN, Murphy KB, et al: Balantidiasis outbreak in Truk. Am J Trop Med Hyg 22:33, 1973
- Swartzwelder JC: Balantidiasis. Am J Dig Dis 17:173, 1950
- Yang J, Scholten TH: Dientamoeba fragilis: a review with notes on its epidemiology, pathogenicity, mode of transmission, and diagnosis. Am J Trop Med Hyg 26:16, 1977
- Preiss U, Ockert G, Broemme S, et al: On the clinical importance of Dientamoeba fragilis infections in childhood. J Hyg Epidemiol Microbiol Immunol 35:27, 1991 [PMID 1880405]
- Spencer MJ, Garcia LS, Chapin MR: Dientamoeba fragilis: an intestinal pathogen in children? Am J Dis Child 133:390, 1979
- Grendon JH, Digiacomo RF, Frost FJ: Dientamoeba fragilis detection methods and prevalence: a survey of state public health laboratories. Public Health Rep 106:322, 1991
- Vannatta JB, Adamson D, Mullican K: Blastocystis hominis infection presenting as recurrent diarrhea. Ann Intern Med 102:495, 1985 [PMID 4038860]
- Markell EK, Udkow MP: Blastocystis hominis: pathogen or fellow traveler? Am J Trop Med Hyg 35:1023, 1986
- Grossman I, Weiss LM, Simon D, et al: Blastocystis hominis in hospital employees. Am J Gastroenterol 87:729, 1992 [PMID 1590309]
- Weiss LM: . . . and now microsporidiosis (editorial). Ann Intern Med 123:954, 1995
- Weber R, Bryan RT, Owen RL, et al: Improved light-microscopical detection of microsporidia spores in stool and duodenal aspirates. N Engl J Med 326:161, 1992 [PMID 1370122]
- Molina JM, Oksenhendler E, Beauvais B, et al: Disseminated microsporidiosis due to Septata intestinalis in patients with AIDS: clinical features and response to albendazole therapy. J Infect Dis 171:245, 1995 [PMID 7798674]
- Weber R, Sauer B, Spycher MA, et al: Detection of Septata intestinalis in stool specimens and coprodiagnostic monitoring of successful treatment with albendazole. Clin Infect Dis 19:342, 1994
- Dieterich DT, Lew EA, Kotler DP, et al: Treatment with albendazole for intestinal disease due to Enterocytozoon bieneusi in patients with AIDS. J Infect Dis 169:178, 1994 [PMID 8277179]
- Orenstein JM, Chiang J, Steinberg W, et al: Intestinal microsporidiosis as a cause of diarrhea in human immunodeficiency virus-infected patients: a report of 20 cases. Hum Pathol 21:475, 1990
- Molina J-M, Sarfati C, Beauvais B, et al: Intestinal microsporidiosis in human immunodeficiency virus-infected patients with chronic unexplained diarrhea: prevalence and clinical and biologic features. J Infect Dis 167:217, 1993
- Pol S, Romana CA, Richard S, et al: Microsporidia infection in patients with the human immunodeficiency virus and unexplained cholangitis. N Engl J Med 328:95, 1993 [PMID 8416439]
- Willson R, Harrington R, Stewart B, et al: Human immunodeficiency virus 1-associated necrotizing cholangitis caused by infection with Septata intestinalis. Gastroenterology 108:247, 1995 [PMID 7806048]
- Weber R, Kuster H, Keller R, et al: Pulmonary and intestinal microsporidiosis in a patient with the acquired immunodeficiency syndrome. Am Rev Respir Dis 146:1603, 1992 [PMID 1456583]
- Didier ES, Didier PJ, Friedberg DN, et al: Isolation and characterization of a new human microsporidian, Encephalitozoon hellem (n. sp.), from three AIDS patients with keratoconjunctivitis. J Infect Dis 163:617, 1991
- Terada S, Reddy KR, Jeffers LJ, et al: Microsporidian hepatitis in the acquired immunodeficiency syndrome. Ann Intern Med 107:61, 1987 [PMID 3109297]
- Zender HO, Arrigoni E, Eckert J, et al: A case of Encephalitozoon cuniculi peritonitis in a patient with AIDS. Am J Clin Pathol 92:352, 1989 [PMID 2505609]
- Lacey CJ, Clarke AM, Fraser P, et al: Chronic microsporidian infection of the nasal mucosae, sinuses and conjunctivae in HIV disease. Genitourin Med 68:179, 1992 [PMID 1607196]
- DeGirolami PC, Ezratty CR, Desai G, et al: Diagnosis of intestinal microsporidiosis by examination of stool and duodenal aspirate with Weber's modified trichrome and Uvitex 2B strains. J Clin Microbiol 33:805, 1995 [PMID 7540626]
- Diesenhouse MC, Wilson LA, Corrent GF, et al: Treatment of microsporidial keratoconjunctivitis with topical fumagillin. Am J Ophthalmol 115:293, 1993 [PMID 8117342]
- Liu LX, Weller PF: An update on antiparasitic drugs. N Engl J Med (in press)
- John DT, Howard MJ: Seasonal distribution of pathogenic free-living amebae in Oklahoma waters. Parasitol Res 81:193, 1995 [PMID 7770424]
- Stehr-Green JK, Bailey TM, Visvesvara GS: The epidemiology of Acanthamoeba keratitis in the United States. Am J Ophthalmol 107:331, 1989 [PMID 2929702]
- D'Aversa G, Stern GA, Driebe WT Jr: Diagnosis and successful medical treatment of Acanthamoeba keratitis. Arch Ophthalmol 113:1120, 1995 [PMID 7661744]
- Radford CF, Bacon AS, Dart JK, et al: Risk factors for Acanthamoeba keratitis in contact lens users: a case-control study. BMJ 310:1567, 1995 [PMID 7787645]
- Sison JP, Kemper CA, Loveless M, et al: Disseminated Acanthamoeba infection in patients with AIDS: case reports and review. Clin Infect Dis 20:1207, 1995 [PMID 7620001]
- Moore MB, McCulley JP: Acanthamoeba keratitis associated with contact lenses: six consecutive cases of successful management. Br J Ophthalmol 73:271, 1989 [PMID 2540794]
- Ishibashi Y, Matsumoto Y, Kabata T, et al: Oral itraconazole and topical miconazole with debridement for Acanthamoeba keratitis. Am J Ophthalmol 109:121, 1990 [PMID 2154105]
- Larkin DF, Kilvington S, Dart JK: Treatment of Acanthamoeba keratitis with polyhexamethylene biguanide. Ophthalmology 99:185, 1992 [PMID 1553206]
- Andrade TM, Carvalho EM, Rocha H: Bacterial infections in patients with visceral leishmaniasis. J Infect Dis 162:1354, 1990 [PMID 2230265]
- Rosenthal E, Marty P, Poizot-Martin I, et al: Visceral leishmaniasis in 50 HIV-1 infected patients. Int Conf AIDS 10:160 (abstr.), 1994
- Laguna F, Garcia-Samaniego J, Soriano V, et al: Gastrointestinal leishmaniasis in human immunodeficiency virus-infected patients: report of five cases and review. Clin Infect Dis 19:48, 1994
- Siddig M, Ghalib H, Shillington DC, et al: Visceral leishmaniasis in the Sudan: comparative parasitological methods of diagnosis. Trans R Soc Trop Med Hyg 82:66, 1988 [PMID 3176153]
- Mishra M, Biswas UK, Jha DN, et al: Amphotericin versus pentamidine in antimony-unresponsive kala-azar. Lancet 340:1256, 1992 [PMID 1359322]
- Badaro R, Falcoff E, Badaro FS, et al: Treatment of visceral leishmaniasis with pentavalent antimony and interferon gamma. N Engl J Med 322:16, 1990 [PMID 2104665]
- Davidson RN, Croft SL, Scott A, et al: Liposomal amphotericin B in drug-resistant visceral leishmaniasis. Lancet 337:1061, 1991 [PMID 1673494]
- Wali JP, Aggarwal P, Gupta U, et al: Ketoconazole in the treatment of antimony- and pentamidine-resistant kala-azar (letter). J Infect Dis 166:215, 1992 [PMID 1607703]
- Magill AJ, Grogl M, Gasser RA Jr, et al: Visceral infection caused by Leishmania tropica in veterans of Operation Desert Storm. N Engl J Med 328:1383, 1993 [PMID 8292114]
- Norton SA, Frankenburg S, Klaus SN: Cutaneous leishmaniasis acquired during military service in the Middle East. Arch Dermatol 128:83, 1992 [PMID 1739291]
- Furner BB: Cutaneous leishmaniasis in Texas: report of a case and review of the literature. J Am Acad Dermatol 23:368, 1990
- Herwaldt BL, Arana BA, Navin TR: The natural history of cutaneous leishmaniasis in Guatemala. J Infect Dis 165:518, 1992 [PMID 1538157]
- Navin TR, Arana FE, de Merida AM, et al: Cutaneous leishmaniasis in Guatemala: comparison of diagnostic methods. Am J Trop Med Hyg 42:36, 1990 [PMID 2301704]
- Grogl M, Thomason TN, Franke ED: Drug resistance in leishmaniasis: its implication in systemic chemotherapy of cutaneous and mucocutaneous disease. Am J Trop Med Hyg 47:117, 1992 [PMID 1322070]
- Martinez S, Marr JJ: Allopurinol in the treatment of American cutaneous leishmaniasis. N Engl J Med 326:741, 1992 [PMID 1738379]
- Guderian RH, Chico ME, Rogers MD, et al: Placebo controlled treatment of Ecuadorian cutaneous leishmaniasis. Am J Trop Med Hyg 45:92, 1991 [PMID 1651060]
- Navin TR, Arana BA, Arana FE, et al: Placebo-controlled clinical trial of meglumine antimonate (Glucantime) vs localized controlled heat in the treatment of cutaneous leishmaniasis in Guatemala. Am J Trop Med Hyg 42:43, 1990 [PMID 2405727]
- Navin TR, Arana BA, Arana FE, et al: Placebo-controlled clinical trial of sodium stibogluconate (Pentostam) versus ketoconazole for treating cutaneous leishmaniasis in Guatemala. J Infect Dis 165:528, 1992 [PMID 1311351]
- Kirchhoff LV: American trypanosomiasis (Chagas' disease)-a tropical disease now in the United States. N Engl J Med 329:639, 1993
- Hagar JM, Rahimtoola SH: Chagas' heart disease in the United States. N Engl J Med 325:763, 1991
- Gluckstein D, Ciferri F, Ruskin J: Chagas' disease: another cause of cerebral mass in the acquired immunodeficiency syndrome. Am J Med 92:429, 1992 [PMID 1558089]
- Oddo D, Casanova M, Acuna G, et al: Acute Chagas' disease (Trypanosomiasis americana) in acquired immunodeficiency syndrome: report of two cases. Hum Pathol 23:41, 1992
- Cattand P, deRaadt P: Laboratory diagnosis of trypanosomiasis. Clin Lab Med 11:899, 1991
- Gear JHS, Miller GB: The clinical manifestations of Rhodesian trypanosomiasis: an account of cases contracted in the Okavango swamps of Botswana. Am J Trop Med Hyg 35:1146, 1986
- Foulkes JR: Human trypanosomiasis in Africa. Br Med J 283:1172, 1981
- Van Nieuwenhove S: Advances in sleeping sickness therapy. Ann Soc Belg Med Trop 72(suppl 1):39, 1992