Contenido del artículo
II FUNCION DEL ERITROCITO Y TRASTORNOS DEL METABOLISMO DEL HIERRO
- Función del eritrocito
- ESTRUCTURA DE LA HEMOGLOBINA
- FACTORES QUE AFECTAN LA CAPACIDAD DE LA HEMOGLOBINA PARA TRANSPORTAR AL OXIGENO
- TRANSPORTE DE OXIGENO
- TRANSPORTE DE BIOXIDO DE CARBONO
- Metabolismo del hierro
- BASES MOLECULARES DEL METABOLISMO DEL HIERRO
- Transferrina y apotransferrina
- Receptor de transferrina
- Ferritina
- Proteínas reguladoras del hierro
- Transportador divalente de metal-1
- Hefaestina
- HFE
- Ceruloplasmina
- ETH (Estimulador del transporte de hierro)
- APORTE Y ALMACEN DE HIERRO EN LA CELULA
- REGULACION DE LA CAPTACION DE HIERRO EN LA CELULA Y SU ALMACEN POR PROTEINAS REGULADORAS DE HIERRO
- PATRONES DE BALANCE Y METABOLISMO DEL HIERRO
- Deficiencia de hierro
- Exceso de hierro
DR. GARY M. BRITTENHAM
Las funciones principales del eritrocito son el transporte de oxígeno (O2) de los pulmones a los tejidos periféricos para su utilización y del bióxido de carbono (CO2) a los pulmones para su excreción.1 El eritrocito maduro dedica más del 95 por ciento de su proteína intracelular, la hemoglobina, a esta tarea. La hemoglobina, que es la molécula transportadora del oxígeno, se une a las moléculas de oxígeno en el ambiente de alta tensión de oxígeno de los alvéolos pulmonares y libera moléculas de oxígeno en los tejidos periféricos, en donde la tensión de oxígeno es baja. La hemoglobina también transporta óxido nítrico (NO), un vasodilatador potente que es liberado durante el tránsito arteriovenoso, aumentando el flujo sanguíneo y, por lo tanto, el transporte de oxígeno en el tejido hipóxico.2,3
El eritrocito tiene forma bicóncava y discoide, lo que optimiza su paso a través del sistema circulatorio y permite la aposición de los eritrocitos y las células parenquimatosas a través del delgado endotelio de los capilares, lo que facilita el intercambio de oxígeno y bióxido de carbono. Sin eritrocitos la sangre del plasma transportaría solo alrededor de 5 ml de O2/L, pero con una hemoglobina normal la sangre total puede transportar alrededor de 200 ml de O2/L.
La hemoglobina está compuesta de dos pares de cadenas polipeptídicas globinas diferentes con un grupo hem, la ferroprotoporfirina IX, unido en forma covalente a un sitio específico en cada cadena.4 El tetrámero forma una molécula esférica con peso molecular de 64,400. La principal hemoglobina adulta, la hemoglobina A, está formada por un par de cadenas a (cada una conteniendo 141 aminoácidos) y un par de cadenas b (cada una con 146 aminoácidos), y se expresa como a2b2 [ver figura 1].
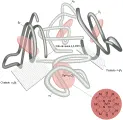
|
| Figura 1 |
| Molécula de hemoglobina |
La configuración de la hemoglobina cambia con la oxigenación y desoxigenación. La configuración desoxi de la hemoglobina se estabiliza por uniones de protones y por el 2,3-difosfoglicerato (2,3-DPG), un anión con carga importante. Con la oxigenación de una subunidad estos enlaces se rompen en forma secuencial y ocurre un cambio en la estructura terciaria que aumenta la afinidad para el oxígeno de las subunidades aún no oxigenadas. A este fenómeno se le denomina cooperatividad o interacción hem-hem. 5 Por el contrario, si el oxígeno es liberado de un hem en la oxihemoglobina, el resto de los sitios unidos sufren cambios que disminuyen su afinidad por el oxígeno, facilitando la liberación de esta molécula. Estos cambios conformacionales son también los responsables de la menor afinidad de la hemoglobina por el oxígeno al disminuir el pH. 1,4 Este efecto, denominado efecto Bohr, es fisiológicamente benéfico tanto en los pulmones, en donde la eliminación de bióxido de carbono aumenta el pH, incrementando la afinidad y captación de oxígeno, como en los tejidos, en donde la captación de bióxido de carbono reduce el pH, lo que reduce la afinidad por el oxígeno y facilita su liberación.
FACTORES QUE AFECTAN LA CAPACIDAD DE LA HEMOGLOBINA PARA TRANSPORTAR AL OXIGENO
Curva de disociación oxígeno-hemoglobina
La curva de disociación de oxígeno-hemoglobina [ver figura 2] es una gráfica del equilibrio entre el oxígeno y la hemoglobina a varias tensiones de oxígeno (PO2).1,5 A nivel del mar y con una presión parcial de oxígeno de alrededor de 90 mm Hg, la hemoglobina está saturada al 97 por ciento en los pulmones. Después de dejar el oxígeno en los tejidos, a una PO2 de alrededor de 40 mm Hg en la sangre venosa mixta, la saturación de la hemoglobina es de 75 por ciento. La P50, que es la presión parcial de oxígeno a la que la hemoglobina está saturada a la mitad, es una medida útil de la afinidad de la hemoglobina por el oxígeno: a mayor afinidad menor P50. En condiciones fisiológicas (i.e., temperatura de 37'C [98.6'F], pH de 7.40, 2,3-DPG de 5 mmol/L y tensión de bióxido de carbono [PCO2] de 40 mm Hg), la P50 de la sangre del adulto normal es de 26±1 mm Hg. La P50 disminuye (se desplaza a la izquierda) con el aumento en el pH, la reducción en el 2,3-DPG o la reducción en la temperatura.1,4

|
| Figura 2 |
| Curva de disociación de oxígeno-hemoglobina |
Efectos del 2,3-difosfoglicerato
El producto intermedio de la glucólisis 2,3-DPG está presente en los eritrocitos maduros en una concentración intracelular semejante a la hemoglobina, y es el regulador alostérico más importante de la afinidad por el oxígeno. En presencia de hipoxia aguda la concentración de 2,3-DPG aumenta en horas lo que desplaza la curva de disociación de oxígeno-hemoglobina a la derecha. El incremento en la concentración de 2,3-DPG facilita la entrega de oxígeno a los tejidos, pero a costa de disminuir su adquisición en los pulmones. Los aumentos en el 2,3-DPG pueden ayudar a adaptarse a corto plazo al estrés hipóxico si el aporte de oxígeno es abundante y la reserva cardiopulmonar robusta. A altitudes elevadas, cuando el sistema cardiovascular es incapaz de satisfacer en forma eficaz la mayor demanda circulatoria, o en otras circunstancias patológicas, el aumento en la cantidad de 2,3-DPG puede ser contraproducente.1
Varios otros factores fisiológicos funcionan en forma integral para proporcionar un aporte adecuado de oxígeno, incluyendo el volumen sanguíneo, la viscosidad, la función pulmonar y cardiaca y el flujo sanguíneo regional.4 La concentración de eritrocitos circulantes depende de la producción de eritropoyetina por el riñón y de la respuesta eritropoyética de la médula ósea. La hemoglobina transporta óxido nítrico, que se une al hierro hem y a la globina, ayudando a equilibrar el flujo sanguíneo regional y los requerimientos de oxígeno.2,3 En los tejidos periféricos, por ejemplo, los eritrocitos liberan óxido nítrico, que relaja la microvasculatura, mejora el flujo sanguíneo y aumenta el aporte de oxígeno.3
TRANSPORTE DE BIOXIDO DE CARBONO
La hemoglobina se une al bióxido de carbono después de dejar oxígeno en los tejidos. La mayoría del bióxido de carbono de los capilares tisulares es transportado a los pulmones como bicarbonato, y alrededor de un 10 por ciento es transportado como un complejo carbamino unido en forma reversible a los grupos N-terminales de las cadenas de globina. La desoxihemoglobina tiene mayor afinidad por el bióxido de carbono que la oxihemoglobina, lo que facilita su transporte desde los tejidos para eliminarse en el pulmón.1
Se han hecho progresos muy importantes en el conocimiento de los trastornos del metabolismo de hierro y para mejorar el diagnóstico y tratamiento tanto de la deficiencia como del exceso de este elemento. El hierro se usa para transportar y almacenar oxígeno, transportar electrones, catalizar reacciones en el metabolismo oxidativo y mantener el crecimiento y proliferación celulares. Cuando existe deficiencia de hierro el organismo es incapaz de sintetizar suficiente cantidad de hem, otros complejos hierro-porfirina, metaloenzimas u otros compuestos que contienen hierro para realizar las funciones normales. Con la sobrecarga de hierro, el exceso puede provocar reacciones nocivas, que causan daño progresivo y eventualmente letal a los órganos vitales.
BASES MOLECULARES DEL METABOLISMO DEL HIERRO
Transferrina y apotransferrina
La transferrina transporta el hierro a los eritrocitos en desarrollo y a otras células del organismo, en especial las que están en crecimiento o proliferación.6 La apotransferrina, que es la transferrina sin el hierro unido, es una glucoproteína bilobular de una sola cadena con dos lóbulos semejantes en el extremo N-terminal y el carboxi (C) terminal. Debido a que un solo ión férrico puede unirse en cada lóbulo, existen cuatro formas de la molécula: (1) apotransferrina, (2) transferrina monomérica, con un átomo de hierro unido al lóbulo N-terminal (FeNTf), (3) transferrina monoférrica, con un átomo de hierro unido al lóbulo C-terminal (FeCTf), y (4) transferrina diférrica (Fe2Tf). En los humanos, el hepatocito es la fuente de origen de casi toda la apotransferrina circulante. Después de dejar el hierro a las células, la apotransferrina regresa con rapidez al plasma para retomar su función con transportadora de hierro.6 La saturación de la transferrina es la proporción de sitios disponibles para unión de hierro en la transferrina que están ocupados por átomos de hierro, expresado en porcentaje.
Los receptores de transferrina se localizan en la superficie de la membrana de todas las células nucleadas y proporcionan la única vía fisiológica de entrada para el hierro unido a transferrina. El número de receptores de transferrina expresados en la superficie celular es el principal determinante del aporte de hierro a la célula. Por ello, el número de receptores de transferrina es más alto en las células de la médula eritroide, el hígado y la placenta.7 El receptor de transferrina ayuda a liberar el hierro de la transferrina para su uso en la célula.8 Desde el punto de vista estructural el receptor de transferrina consiste en dos subunidades transmembrana glucoproteicas idénticas unidas por un puente disulfuro. Cada subunidad puede fijar una molécula de transferrina, de modo que el receptor puede aceptar dos moléculas. La eficacia con la que el receptor de transferrina puede entregar el hierro unido a la transferrina depende del contenido de hierro en esta molécula. Por lo tanto, el receptor dimérico puede permitir la entrada de cuatro átomos de hierro si cada transferrina es diférrica o solo dos átomos de hierro si cada una es monoférrica.
Casi todas las células contienen ferritina, que funciona tanto como un sitio de almacén seguro para hierro como una reserva accesible que ha sido adquirida por la célula ante la existencia en exceso a sus necesidades metabólicas inmediatas. En consecuencia, las mayores cantidades de ferritina se encuentran en las células dedicadas al almacén de hierro (i.e., macrófagos y hepatocitos) y en las células con los mayores requerimientos para la síntesis de compuestos que contienen hierro (i.e., células eritroides en desarrollo). La ferritina hepática tiene una vida media de alrededor de 60 horas.9
La apoferritina, que es la ferritina sin el hierro unido, es una concha esférica con un peso molecular de 440,000. Cada molécula puede almacenar hasta 4,500 átomos de hierro en el interior en un centro polinuclear de hidrofosfato férrico.9 La apoferritina está formada por 24 subunidades oblongas que se denominan como H (pesadas) y L (ligeras). Las moléculas de ferritina con mayor proporción de subunidades H parecen ser más activas en el metabolismo del hierro, y las que tienen más subunidades L se usan para el almacenamiento a largo plazo de este elemento.9
Proteínas reguladoras del hierro
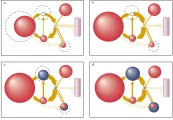
Las proteínas reguladoras del hierro PRH-1 y PRH-2 permiten al hierro autorregular su disponibilidad intracelular [ver figura 4]. Funcionan al unirse a elementos que responden al hierro (ERH) en el ARN mensajero (ARNm).10,11 Los ERH funcionales se localizan en la región no trasducida (RNT) 3' del ARNm para la transferrina y en la región no traducida 5' del ARNm para la ferritina, en la forma eritroide específica de la sintetasa de ácido d-aminolevulínico (eAAL) y en la aconitasa mitocondrial.
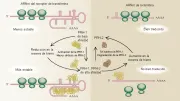
|
| Figura 4 |
| Receptor de transferrina y ferritina |
Transportador divalente de metal-1
El DMT1,12 antes llamado transportador divalente de cationes (DCT1 por sus siglas en inglés, n. del t.) o proteína asociada a macrófagos de resistencia natural-2 (Nrampo2), 13 mueve el hierro de la luz del tubo digestivo hacia la célula de absorción de la parte superior del intestino (enterocito). El transportador de hierro intestinal, localizado en la superficie apical del enterocito duodenal, es una glucoproteína de membrana con 12 dominios putativos de membrana y un amplio rango de sustratos que incluyen no sólo Fe2+, sino también Zn2+, Mn2+, Co2+, Cd2+, Cu2+, Ni2+ y Pb2+ [ver figura 5 ]. 12 El DMT1 también se presenta en las vesículas endosómicas de los precursores hematopoyéticos, los macrófagos y otras células, y media el paso de hierro de la transferrina dentro del endosoma al citoplasma de la célula para su utilización o almacén. 14,15
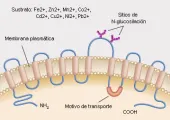
|
| Figura 5 |
| Transportador divalente de metales-1 |
Hefaestina
La hefaestina es una proteína transmembrana homóloga a la ceruloplasmina recién identificada con localización perinuclear dentro del enterocito intestinal.16 Esta proteína multicobre parece ser necesaria para la salida del hierro del enterocito intestinal y hacia la circulación sistémica. La homología con la ceruloplasmina sugiere que la hefaestina funciona como una ferroxidasa intracelular, pero no se ha determinado su mecanismo de acción exacto.
La HFE, que es la proteína que está defectuosa en la mayoría de los pacientes con hemocromatosis hereditaria (ver adelante), es estructuralmente similar a las proteínas del complejo principal de histocompatibilidad (CPH) de clase I.17 Aún se desconocen el papel exacto de la HFE en el metabolismo del hierro y la forma como las mutaciones en la HFE causan la mayor absorción de hierro que se observa en la hemocromatosis hereditaria, pero se han hecho varias observaciones potencialmente importantes. Por ejemplo, la HFE recién sintetizada forma un complejo 1:1 con la b2-microglobulina, que puede a su vez formar un complejo estable con el receptor de transferrina y al parecer disminuir la afinidad del receptor de transferrina para la transferrina.18-20 También, la HFE se expresa en forma abundante en las células de las criptas de la mucosa duodenal,21 y algunas evidencias sugieren que los niveles de HFE y DMT1 se relacionan en forma recíproca en las células intestinales.22
La ceruloplasmina es una glucoproteína sérica a2 y una oxidasa multicobre que transporta el 95 por ciento del cobre que se encuentra en el plasma. Cada molécula puede fijar hasta seis átomos de cobre. Las personas con deficiencia hereditaria de la ceruloplasmina desarrollan una forma de sobrecarga de hierro (ver adelante), 23 lo que indica que esta proteína tiene algún papel en el metabolismo humano del hierro, aunque se desconoce aún la función exacta. Los estudios recientes han sugerido que la ceruloplasmina aumenta la captación celular de hierro independiente de transferrina, un papel que puede depender en parte de la capacidad de esta proteína para catalizar la conversión del ión ferroso a férrico.24
ETH (Estimulador del transporte de hierro)
Se ha identificado un aparente estimulador del transporte de hierro (ETH). Esta proteína parece aumentar la captación celular del elemento, aunque su función exacta y relación con otras proteínas del metabolismo del hierro se desconocen. 25
APORTE Y ALMACEN DE HIERRO EN LA CELULA
La transferrina, el receptor de transferrina y la ferritina actúan juntos para proporcionar hierro a la célula [ver figura 4]. 8 Al inicio dos moléculas de transferrina (monoférrica o diférrica) se unen a un receptor de transferrina sobre la superficie celular. Es posible que la HFE tenga algún papel para determinar la afinidad del receptor de la transferrina por esta proteína.19 El complejo hierro-transferrina-receptor de transferrina-HFE se agrega con otros ocmplejos en una zona cubierta con clatrin, que es traslocada después al interior de la célula como una vesícula encubierta. Después de que el clatrin es separado, la vesícula sin cubierta se fusionan con otras vesículas para formar un endosoma multivesicular. El endosoma se acidifica hasta un pH de 5.6 por una bomba de protones mientras viaja en el interior de la célula. Con la caída en el pH, el receptor de transferrina sufre cambios de conformación que ayudan a liberar el hierro de la transferrina mono o diférrica.8 El hierro disociado es entonces transportado a través de la membrana endosómica por medio del DMT1 para ser utilizado en la síntesis de compuestos que contengan hierro o para su almacén en la ferritina citoplásmica.14 En el endosoma a un pH ácido, la apotransferrina sin hierro se une en forma ávida al receptor de transferrina,8 y el complejo es transportado de regreso a la membrana celular en el endosoma. Por el contrario, la unión de alta afinidad entre la HFE y el receptor de transferrina se pierde si el pH es ácido.20 Aunque algunos endosomas se desvían hacia el aparato de Golgi para su resialilación y otros son reparados, la mayoría de los endosomas llegan en forma directa a la superficie celular y se fusionan con la membrana. El complejo apotransferrina-receptor de transferrina se expone entonces al pH de 7.4 del plasma, lo que causa pérdida de la afinidad de la apotransferrina por el receptor. La apotransferrina entra al reservorio circulante en el plasma y el receptor de transferrina, ahora unido aparentemente en forma estrecha a la HFE, está de nuevo disponible para unir a una transferrina que lleve hierro.
REGULACION DE LA CAPTACION DE HIERRO EN LA CELULA Y SU ALMACEN POR PROTEINAS REGULADORAS DE HIERRO
De una manera coordinada, las PRH controlan la captación intracelular y el almacén del hierro al controlar la traducción en la síntesis del receptor de transferrina y de la ferritina [ver figura 4].10,11 Las PRH1 y PRH2 responden en forma semejante a cambios en la disponibilidad intracelular de hierro, aunque responden a través de vías regulatorias diferentes. La síntesis del receptor de transferrina está controlada por la estabilidad del ARNm del receptor de transferrina citoplásmico, mientras que la síntesis de ferritina depende de la traducción del ARNm de ferritina sin que cambie la cantidad de ARNm de ferritina en el citoplasma. Como consecuencia, los cambios en la cantidad de PRH tienen efectos opuestos sobre la producción del receptor de transferrina y de ferritina. Si cae el nivel de hierro intracelular, la cantidad de PRH de alta afinidad aumenta. A mayor cantidad de PRH la unión a los ERH en la RNT3' del ARNm del receptor de transferrina estabiliza al ARNm, aumentando la producción del receptor de transferrina y la disponibilidad de hierro intracelular. La unión de las PRH al RNT5' del ARNm de la ferritina detiene la síntesis de la proteína, disminuyendo el almacén de hierro y aumentando la disponibilidad de hierro intracelular. Por el contrario, el aumento en el hierro intracelular disminuye la cantidad de PRH de alta afinidad, disminuyendo la captación de hierro por la célula al reducir la síntesis de la proteína del receptor de transferrina y aumenta el almacén de hierro al incrementar la síntesis de la proteína ferritina.
PATRONES DE BALANCE Y METABOLISMO DEL HIERRO
La concentración del hierro en el organismo está cuidadosamente controlada y se mantiene en condiciones normales en alrededor de 40 mg Fe/kg en la mujer y 50 mg Fe/kg en el varón.26,27 El balance del hierro es el resultado de la diferencia entre la cantidad de hierro captada por el organismo y la cantidad perdida [ver figura 3]. Debido a que los humanos son incapaces de excretar el exceso de hierro, el equilibrio de este elemento está regulado de modo fisiológico por el control en su absorción. Los dos principales factores que influyen en la absorción del hierro son la cantidad de hierro almacenado y la magnitud de la eritropoyesis.28 Si las reservas de hierro aumentan la absorción se reduce, si las reservas disminuyen la absorción aumenta. La absorción aumenta con la mayor actividad eritropoyética, en especial cuando es ineficaz. La principal vía de flujo interno de hierro [ver figura 3] es un flujo unidireccional de la transferrina plasmática al eritrón (el eritrón consiste en la totalidad de eritrocitos circulantes y sus precursores en la médula ósea), al sistema monocito macrófago y después de regreso a la transferrina.26,27 La mayoría del hierro del organismo se localiza en el eritrón. El eritrón usa alrededor del 80 por ciento del hierro que pasa a través del compartimiento de la transferrina cada día. En condiciones normales este proceso es muy eficaz y se adquieren o pierden menos de 0.05 por ciento del hierro corporal total por día.
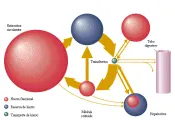
|
| Figura 3 |
| Aporte y almacén del hierro corporal en el organismo |
El eritrón adquiere el hierro a través de la transferrina por un receptor específico localizado en la membrana de superficie de las células eritroides inmaduras. La mayoría del hierro se usa para la síntesis de hemoglobina. Pequeñas cantidades se almacenan en la ferritina, ingresan a enzimas de las células eritroides inmaduras que contienen hierro o se pierden en los productos de hematopoyesis ineficaz. Los eritrocitos viejos son fagocitados por macrófagos especializados en el bazo, médula ósea e hígado, que después regresan la mayor parte del hierro al compartimiento de la transferrina, en donde el ciclo comienza de nuevo. La fagocitosis de los eritrocitos dañados o viejos explica casi la totalidad del hierro almacenado que se encuentra en condiciones normales en los macrófagos del hígado, médula ósea y bazo. Por el contrario, las células parenquimatosas del hígado pueden intercambiar hierro con la transferrina del plasma.
El hierro se requiere no solo para restablecer las pérdidas fisiológicas y satisfacer las necesidades de crecimiento y embarazo, sino también para sustituir las pérdidas patológicas. La deficiencia de hierro indica condiciones en las que el hierro corporal está reducido por un incremento sostenido en los requerimientos que son superiores al aporte.28
La pérdida diaria fisiológica de hierro en el varón es un poco menor de 1.0 mg/día, y en la mujer normal que menstrúa de 1.5 mg/día.26 Pueden identificarse estadios secuenciales en la reducción del hierro corporal [ver figura 6 y tabla 1]. La reducción en las reservas de hierro sin un cambio en la cantidad de compuestos de hierro funcionales se denomina como reducción en la reserva de hierro. Cuando las reservas se agotan, se dice que los pacientes tienen depleción de hierro. La mayor disminución en el hierro corporal causa menor producción de hemoglobina y de otros compuestos funcionales que contienen hierro, estadio denominado eritropoyesis deficiente en hierro. La reducción aún mayor en el hierro corporal produce anemia por deficiencia de hierro. En todo el mundo, la deficiencia de hierro es la causa más común de anemia.29
|
| Figura 6 |
| Reservas y distribución del hierro |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
* Puede ser normal en algunos pacientes |
Casi la tercera parte de la población mundial de alrededor de seis billones tiene anemia, y la anemia por deficiencia de hierro causa o contribuye a la anemia en por lo menos 500 millones de personas.
La pérdida de sangre es la causa más común de aumento en el requerimiento de hierro que origina deficiencia del mismo [ver tabla 2].29 En los hombres y las mujeres posmenopáusicas, la deficiencia de hierro casi siempre es resultado de pérdida gastrointestinal. En las mujeres menstruando la pérdida genitourinaria suele ser la causa del aumento en los requerimientos de hierro. Los anticonceptivos orales pueden disminuir la pérdida menstrual de sangre, pero los dispositivos intrauterinos la aumentan. Deben considerarse también otras causas de hemorragia genitourinaria o respiratoria [ver tabla 2]. Para los donadores de sangre, cada donación significa una pérdida de 200 a 250 mg de hierro. Durante los periodos de crecimiento en la infancia, niñez y adolescencia, los requerimientos de hierro pueden superar el aporte a partir de las dietas y reservas.30 El embarazo aumenta las necesidades de hierro y, sin hierro suplementario, causa una pérdida neta equivalente a alrededor de 1,200 a 1,500 ml de sangre.30 Después del parto, si el niño es alimentado al seno materno, la lactación aumenta los requerimientos de hierro en alrededor de 0.5 a 1.0 mg por día.
|
||
|
El aporte insuficiente de hierro puede contribuir al desarrollo de deficiencia
de hierro. En los niños y mujeres con requerimientos altos de hierro,
las dietas que contienen cantidades inadecuadas de hierro biodisponible pueden
aumentar el riesgo de deficiencia. En los niños mayores, varones y
mujeres posmenopáusicas, el aporte escaso de hierro en la dieta casi
nunca es el único factor responsable de la deficiencia, por lo que deben
buscarse otros factores etiológicos, en especial pérdida de
sangre. La menor absorción de hierro es una causa poco común de
deficiencia. En algunos pacientes la malabsorción intestinal de hierro
es solo un aspecto más de la malabsorción generalizada [ver
tabla 2]. La cirugía gástrica, en especial la
resección gástrica parcial o total o la gastroenterostomía
para derivación del duodeno, puede causar deficiencia de hierro. Aunque
la absorción de hierro en la dieta puede ser pobre, las sales
terapéuticas se absorben bien, y la deficiencia puede corregirse con
facilidad.
El riesgo de deficiencia de hierro es especialmente alto cuando los requerimientos aumentan y el aporte es inadecuado. Por ejemplo, los infantes alimentados con leche de vaca suelen tener deficiencia de hierro por la combinación de mayor pérdida por hemorragia gastrointestinal inducida por la leche de vaca y por las pequeñas cantidades de hierro biodisponible que existen en ésta.30 Las mujeres con requerimientos elevados de hierro por la menstruación suelen consumir dietas que tienen poco hierro biodisponible y con inhibidores de la absorción de hierro, como el calcio.
Desde el punto de vista clínico los pacientes con deficiencia de hierro pueden estar asintomáticos y detectarse solo por resultados anormales en las pruebas de laboratorio. Otros pacientes pueden solicitar atención médica por las manifestaciones del trastorno subyacente que causó la deficiencia de hierro, pero pueden no tener datos relacionados con esta última. Un tercer grupo de enfermos puede sufrir signos y síntomas comunes a todas las anemias, como debilidad, mareo, fatiga fácil, palidez, irritabilidad y otras molestias indefinidas e inespecíficas. La deficiencia de hierro puede asociarse también con signos y síntomas que no se relacionan con anemia, como estomatitis angular, glositis, estenosis o pliegues esofágicos poscricoideos y atrofia gástrica. Por último, algunos pacientes presentan uno o más de los pocos signos y síntomas que se piensa son muy específicos de la deficiencia de hierro (escleras azules, coiloniquia y pagofagia). La pagofagia, o pica con hielo, parece ser un síntoma muy específico de deficiencia de hierro y desaparece poco después de que se inicia el tratamiento.29 Otros tipos de pica pueden acompañar a la deficiencia de hierro, pero ninguno es tan específico como la pagofagia. Las consecuencias no hematológicas de la falta de hierro incluyen menor tolerancia al ejercicio y desempeño laboral y deterioro en la inmunidad y resistencia a la infección. En los niños la deficiencia de hierro afecta en forma adversa al crecimiento, el desarrollo motor y las funciones cognoscitivas y de comportamiento. Estas alteraciones pueden no ser reversibles con el tratamiento posterior.30,31
La anemia por deficiencia de hierro es la única anemia microcítica hipocrómica asociada con ausencia en las reservas de hierro. En las otras anemias microcíticas hipocrómicas las reservas de hierro en la médula son normales o están aumentadas. Pueden usarse medidas indirectas del hierro corporal para identificar una secuencia característica de cambios que ocurren al disminuir el hierro corporal desde los niveles de reserva normal hasta los encontrados en la anemia por deficiencia de hierro [ver figura 6 y tabla 1]. La concentración de ferritina en plasma es la prueba más útil para la detección de la deficiencia de hierro porque las concentraciones de ferritina en plasma disminuyen al reducirse las reservas corporales.29 Una concentración de ferritina en plasma por debajo de 12 µg/L es virtualmente diagnóstica de ausencia de reservas de hierro. Por el contrario, la concentración normal de ferritina en plasma no confirma la presencia de reservas de hierro porque esta proteína puede aumentar en forma independiente por infecciones, inflamación, enfermedad hepática, neoplasias y otros trastornos. En estos casos la medición de la concentración del receptor de transferrina en plasma proporciona una manera de distinguir entre la anemia por deficiencia de hierro y la anemia asociada con trastornos inflamatorios crónicos.32 La saturación de transferrina en plasma está disminuida tanto en la deficiencia de hierro como en los estados infecciosos e inflamatorios, y es de poca ayuda práctica para distinguir entre estos trastornos. Aunque rara vez se realiza examen de la médula ósea solo para evaluar el estado del hierro, casi siempre puede verificarse el estado de deficiencia de hierro por la evaluación directa de las reservas de este elemento en la médula. Si no existen reservas de hierro se establece diagnóstico de deficiencia de hierro, si se encuentra hemosiderina, se excluye esta posibilidad. Además, en la deficiencia de hierro no existen sideroblastos en la médula, o estos están muy disminuidos, constituyendo menos del 10 por ciento de los normoblastos. Una prueba terapéutica con hierro puede ser un medio eficaz de establecer el diagnóstico de deficiencia de hierro.
El tratamiento para la anemia por deficiencia de hierro consiste en corregir tanto la deficiencia de hemoglobina como reemplazar las reservas de hierro. Para casi todos los pacientes el hierro oral es el tratamiento de elección.29 El tratamiento con hierro oral es eficaz, seguro y económico. Debido al riesgo de reacciones adversas locales y sistémicas, el hierro parenteral solo debe usarse en el pequeño número de pacientes que no pueden absorber o tolerar el hierro oral o que tienen requerimientos de hierro que no pueden satisfacerse por el tratamiento oral debido a hemorragia crónica no controlable. En la anemia severa por deficiencia de hierro puede requerirse transfusión de eritrocitos para evitar isquemia cardiaca o cerebral. En pocas ocasiones se requieren también para los pacientes en los que la velocidad de pérdida crónica de hierro excede la velocidad a la que puede reemplazarse el hierro con tratamiento parenteral. Aunque la mayoría de los pacientes toman el hierro oral sin dificultad, el 10 a 20 por ciento sufre efectos colaterales, principalmente de tipo gastrointestinal.
La deficiencia de hierro casi siempre puede tratarse en forma eficaz, y el tratamiento oral y parenteral da resultados semejantes [ver cuadro, Tratamiento de sustitución de hierro].29 Ocurre mejoría de los síntomas en los primeros días de tratamiento. En la anemia no complicada la respuesta hematológica inicial, que consiste en reticulocitosis leve, comienza 3 a 5 días después del inicio del tratamiento, alcanza un máximo a los 8 a 10 días y disminuye después. Después de la primera semana la concentración de hemoglobina comienza a aumentar y suele ser normal alrededor de 6 semanas después. La microcitosis puede no resolverse por completo hasta después de 4 meses. Si la deficiencia de hierro se trata con hierro oral en dosis de 200 mg/día o menos, la concentración de ferritina en plasma permanece en menos de 12 µg/L hasta que la anemia se corrija y después aumenta en forma gradual al repletarse las reservas de hierro. Si la respuesta al tratamiento con hierro no es completa y característica, estará indicado reevaluar al paciente. Uno de los problemas más comunes es confundir la anemia de una enfermedad crónica con anemia por deficiencia de hierro. La recuperación puede ser más lenta cuando coexisten padecimientos, incluyendo otras deficiencias nutricionales, enfermedad hepática o renal, infecciones, inflamación o neoplasias, o pérdida de sangre oculta continua. Si el paciente fue tratado con hierro oral, deberán revisarse la forma y dosis de hierro, la constancia al tratamiento y la posibilidad de malabsorción.
|
|
|
El exceso de hierro se origina de un aumento constante en el aporte de hierro en relación con los requerimientos, y causa cambios característicos en la función, transporte y almacén del hierro [ver figura 6 y tabla 1]. La cantidad de hierro corporal suele controlarse en condiciones normales por regulación en la absorción dietética de hierro. Se desarrolla exceso de hierro en condiciones que modifican o superan la regulación de la absorción del ión en el intestino [ver tabla 3]. Debido a que los humanos no tienen una forma fisiológica de eliminar el exceso de hierro, cualquier incremento persistente en la ingesta puede eventualmente causar exceso de hierro. Independientemente de la causa del acúmulo del hierro, cuando la cantidad del mismo excede la capacidad del organismo para secuestrar el exceso se desarrolla daño tisular potencialmente letal. Las características precisas de las consecuencias patológicas de la sobrecarga de hierro son resultado, en parte, de la magnitud de la carga de hierro corporal, la velocidad a la que ha ocurrido el aumento, la distribución específica del exceso del ión entre los sitios de almacén en macrófagos, los depósitos, potencialmente más dañinos, en células parenquimatosas, y la coexistencia de condiciones que pueden disminuir (v.gr., deficiencia de ácido ascórbico) o pueden empeorar (v.gr., uso de alcohol o hepatitis) la evolución. Las consecuencias más comunes de la sobrecarga de hierro incluyen enfermedad hepática, enfermedad pancreática asociada con diabetes mellitus, trastornos endócrinos asociados con insuficiencia gonadal, disfunción cardiaca, artropatía y, en ocasiones, alteraciones neurológicas y psicológicas.
|
|
* Puede tener un componente genético |
Puede ocurrir mayor absorción de hierro por un control anormal en su
absorción aún cuando la dieta contenga cantidades normales de
hierro biodisponible, como en la hemocromatosis hereditaria o por una dieta con
tanto exceso de hierro biodisponible que supera los mecanismos de control, como
en el exceso dietético de hierro que ocurre en africanos. Las anemias
con sobrecarga de hierro son un grupo heterogéneo de padecimientos que
tienen en común una hiperplasia eritroide marcada asociada con
eritropoyesis ineficaz. La sobrecarga de hierro puede deberse también a
transfusiones (ver adelante).
La hemocromatosis hereditaria, un padecimiento autosómico recesivo, es el padecimiento genético más común en personas con ascendencia del norte de Europa. En los Estados Unidos hasta el 10 por ciento de la población es heterocigota para esta condición, y se cree que el estado homocigoto afecta hasta al 0.5 por ciento de la población, aunque está muy subdiagnosticada.33
El descubrimiento del gen responsable de la mayoría de los casos de hemocromatosis hereditaria ha revolucionado tanto el conocimiento como el diagnóstico de este padecimiento.17 Un defecto en la proteína HFE causa alteración en el control de la absorción de hierro, con un aumento inapropiado en su captación y crecimiento progresivo de las reservas corporales de hierro. Al inicio la sobrecarga tiene principalmente un patrón de depósito parenquimatoso, acúmulándose primero en los hepatocitos y después en el páncreas, corazón y otros órganos.34 Característicamente el hierro en los macrófagos de la médula ósea puede ser normal o estar incluso disminuido a pesar del depósito parenquimatoso severo [ver figura 6 y tabla 1].
Las mutaciones sin sentido en el gen HFE son responsables de alrededor del 85 por ciento de los casos de hemocromatosis hereditaria en los Estados Unidos, en otras áreas del mundo el porcentaje varía de alrededor de 60 a 100 por ciento.35 En un estudio, la mutación más prevalente (homocigota en el 83 por ciento de los pacientes) fue una sustitución Cys282Tyr. Una segunda mutación, His63Asp, existió en los pacientes que eran heterocigotos compuestos para la sustitución Cys282Tyr.17 Recientemente se identificó una tercera mutación, Ser65Cys.36 Es importante mencionar que en los Estados Unidos el 10 a 15 por ciento de los pacientes con sobrecarga primaria de hierro no tienen ninguna de estas tres mutaciones, pero que clínicamente son indistinguibles de los pacientes que sí las portan. La mutación Cys282Tyr interrumpe la unión de la HFE a la b2-microglobulina, lo que evita casi por completo la asociación de la HFE mutante con el receptor de transferrina.19 Los efectos de las mutaciones His63Asp y Ser65Cys están menos bien caracterizados. Los datos disponibles sugieren que la patogenia de la hemocromatosis hereditaria puede relacionarse con alteración en la vía de la transferrina-receptor de la transferrina, pero se desconoce el mecanismo exacto por el que este cambio aumenta la absorción del hierro de la dieta.
Manifestaciones clínicas En los homocigotos que presentan hemocromatosis hereditaria en la edad media o después, la tétrada clásica de signos clínicos consiste en enfermedad hepática, diabetes mellitus, pigmentación cutánea y falla gonadal.37-39 Se desarrolla insuficiencia cardiaca en alrededor del 10 a 15 por ciento de los homocigotos no tratados. Las reservas de hierro corporal suelen haber aumentado de la cantidad normal de 1 g o menos a 15 o 20 g o más cuando ocurren los síntomas de daño parenquimatoso, por lo general en la edad media o tardía.38 Los aumentos adicionales en el hierro corporal pueden ser fatales, aunque algunos pacientes son capaces de tolerar una reserva total de hierro de hasta 40 a 50 g. Los factores ambientales, incluyendo el contenido de hierro de la dieta y el uso de alcohol, pueden influir mucho en la velocidad y severidad del daño orgánico. En los heterocigotos la hemocromatosis hereditaria se expresa en forma incompleta, y solo alrededor del 25 por ciento de estas personas sufren un aumento leve en las reservas de hierro, con cantidades totales de 4 o 5 g y sin presentar otras manifestaciones de la enfermedad.37,38
Escrutinio y pruebas diagnósticas Debido a que la detección y tratamiento tempranos pueden evitar las manifestaciones de la enfermedad, se recomienda en todos los pacientes el escrutinio de rutina para hemocromatosis.40 La medición de la concentración de hierro en plasma, saturación de transferrina y concentración de ferritina proporciona la mejor manera de realizar escrutinio para la hemocromatosis hereditaria, aunque debe reconocerse que la concentración de ferritina puede ser normal en un pequeño número de pacientes con esta enfermedad.41 La genotipificación puede emplearse como prueba confirmatoria en un paciente con sospecha o datos clínicos de hemocromatosis o que ha tenido una saturación de transferrina, nivel de ferritina o ambos elevados.
La caracterización de las mutacciones del gen HFE fue seguida con rapidez por el desarrollo de una prueba de genotipificación diagnóstica.35 El papel diagnóstico exacto de esta prueba depende en parte de la población a examinar porque el porcentaje de pacientes con hemocromatosis hereditaria que tienen mutaciones HFE varía del 60 a 100 por ciento en diferentes poblaciones.33 Además, se han identificado varias personas que son homocigotas para la mutación Cys282Tyr pero que no tienen sobrecarga de hierro.37,42,43 El número de pacientes en los Estados Unidos que no son homocigotos para la mutación Cys282Tyr pero que satisfacen los criterios clínicos para hemocromatosis hereditaia es tan grande que el solo confiar en la pruebas genéticas podría dejar sin diagnóstico a muchos individuos afectados. Un enfoque más práctico consiste en realizar un escrutinio inicial para sobrecarga de hierro con pruebas fenotípicas.33,37
En los estudios de pedigree la genotipificación debe sustituir a la tipificación del HLA al evaluar a los hermanos de un homocigoto Cys282Tyr. Además, la genotipificación de la esposa de un homocigoto Cys282Tyr es una estrategia con buena relación costo eficacia que lleva a una investigación más selectiva de los hijos para buscar el gen de la hemocromatosis.42
Recientemente existen estudios que sugieren que el escrutinio inicial para la hemocromatosis hereditaria debe consistir en la saturación de transferrina, seguido de la búsqueda de la mutación Cys282Tyr en pacientes con una saturación de transferrina mayor de 40 por ciento y, al identificar a los homocigotos, la evaluación de la carga de hierro por medio de medición de la ferritina en suero o de biopsia de hígado.37,44 En los pacientes con hepatopatía y sospecha de sobrecarga de hierro el escrutinio genotípico puede identificar a los homocigotos Cys282Tyr, que de otro modo pasarían desapercibidos.45 La biopsia de hígado permite el diagnóstico definitivo de hemocromatosis hereditaria, independientemente del genotipo.34,36
La evaluación debe incluir la determinación cuantitativa de la concentración del hierro no hem, la evaluación histoquímica del patrón de depósito de hierro y la evaluación patológica de la lesión tisular. El cálculo del índice hepático de hierro (la concentración de hierro en el hígado [expresada como µmol Fe/g de peso seco de hígado] dividido entre la edad del paciente [expresada en años]) puede ser útil para distinguir de los homocigotos con hemocromatosis hereditaria de los heterocigotos o de los pacientes con aumento en el hierro corporal asociado con hepatopatía crónica (por lo general alcohólica).37 La biopsia hepática es especialmente útil para evaluar la magnitud del daño hepático y para detectar el desarrollo de fibrosis o cirrosis.
El tratamiento de elección para la hemocromatosis hereditaria consiste en flebotomías para reducir y mantener el hierro corporal en niveles normales o casi normales. En los pacientes con hemocromatosis hereditaria e insuficiencia cardiaca se ha sugerido el uso de flebotomía y tratamiento quelante. El tratamiento con flebotomías debe iniciarse en cuanto se hace el diagnóstico de estado homocigoto para la enfermedad, su postergación solo aumenta el riesgo de daño orgánico por exceso de hierro. El programa de flebotomía debe eliminar 500 ml de sangre (conteniendo 200 a 250 mg de hierro) una vez por semana o, en pacientes con exceso importante, dos veces por semana, hasta que el paciente tenga deficiencia de hierro. Antes de cada flebotomía debe medirse el hematocrito o la hemoglobina. Al principio éstos deben disminuir alrededor de 10 por ciento de sus valores iniciales, pero después pueden aumentar al aumentar la velocidad de la eritropoyesis para satisfacer las demandas de la flebotomía. Debe medirse en forma regular la ferritina, el hierro y la saturación de transferrina, para vigilar el progreso en la eliminación del hierro. Al eliminar hierro la concentración de ferritina en plasma disminuirá en forma progresiva, pero el hierro y la saturación de transferrina permanecerán elevados hasta que las reservas estén casi agotadas. Por último, cuando se ha eliminado todo el hierro acumulado la ferritina disminuirá a menos de 12 µg/L, el hierro y la saturación de transferrina caerán y la concentración de hemoglobina disminuirá a menos de 10 g/dl durante 2 semanas sin otra flebotomía. Para ilustrar qué tanto tiempo puede requerirse el tratamiento, si la reserva inicial de hierro es de 25 g, su eliminación total con flebotomías semanales puede necesitar de 2 años o más. Después de haber terminado la eliminación del hierro se requiere un programa de por vida de flebotomías de mantenimiento para evitar su reacúmulo. Típicamente se necesitan flebotomías de 500 ml cada 3 a 4 meses. El objetivo de la fletobomía de mantenimiento debe ser mantener una saturación de transferrina normal con una concentración de ferritina en plasma de menos de alrededor de 50 µg/L.
Si el tratamiento con flebotomía elimina la carga de hierro antes que se desarrolle cirrosis la esperanza de vida para el paciente es normal.47 Si por el contrario ya existe cirrosis, el riesgo de carcinoma hepatocelular aumenta en más de 200 veces. En la hemocromatosis hereditaria se desarrollan hepatomas casi en forma exclusiva en los pacientes con cirrosis hepática, siendo la causa de muerte en el 20 a 30 por ciento de estos pacientes, incluso después de la eliminación exitosa del exceso de hierro. Casi siempre está indicado el tratamiento con flebotomías para los pacientes con hemocromatosis hereditaria, incluso si ya existe cirrosis o daño orgánico, porque puede detenerse la progresión de la enfermedad e incluso mejorar parte de la disfunción orgánica.
En los pacientes con anemias con exceso de hierro puede desarrollarse sobrecarga severa de hierro como resultado de una mayor absorción gastrointestinal del mismo.48 Cualquier transfusión de eritrocitos que reciban estos pacientes contribuye al acúmulo del hierro. Las anemias con sobrecarga de hierro incluyen a la anemia diseritropoyética congénita, la deficiencia de piruvato cinasa, la talasemia mayor (anemia de Cooley) y la talasemia intermedia, la talasemia-a con hemoglobina E, diversas formas de anemia sideroblástica, algunas anemias mielodispásicas y varios otros trastornos anémicos en los que se altera la incorporación del hierro a la hemoglobina.48 Debido a que la magnitud de la eritropoyesis ineficaz, y no la severidad de la anemia, parece determinar la velocidad de acúmulo de hierro, puede desarrollarse sobrecarga severa de hierro en pacientes con anemia solo leve. Las manifestaciones clínicas y la patología que puede desarrollarse en pacientes con anemias con exceso de hierro es semejante a las de la hemocromatosis hereditaria, incluyendo enfermedad hepática, diabetes mellitus, trastornos endócrinos y disfunción cardiaca.
OTRAS FORMAS DE SOBRECARGA DE HIERRO POR MAYOR ABSORCION
Algunos pacientes con hepatopatía crónica, incluyendo los que tienen cirrosis alcohólica y con derivación portocava, pueden sufrir grados leves o moderados de exceso de hierro como resultado de mayor absorción del hierro de la dieta.48 Los mecanismos responsables de la mayor captación gastrointestinal de hierro no se han identificado, aunque se han propuesto como factores etiológicos a la eritropoyesis ineficaz y a la hiperferremia asociada con alteraciones sideroblásticas y del folato inducidas por alcohol. Las reservas corporales de hierro están aumentadas solo un poco, típicamente a 2 a 4 g. El depósito de hierro predomina en las células de Kupffer, más que en los hepatocitos, como en la hemocromatosis hereditaria. Como ya se mencionó, la distinción entre la sobrecarga de hierro en los pacientes con hepatopatía crónica y la de los homocigotos para la hemocromatosis hereditaria casi siempre puede hacerse por determinación cuantitativa de hierro en una muestra de hígado obtenida por biopsia calculando el índice de hierro hepático.
Los pacientes sintomáticos con porfiria cutánea tarda, una porfiria hepática, suelen tener un aumento modesto en el hierro corporal que casi siempre se debe a mayor absorción gastrointestinal.8 La aceruloplasminemia es un trastorno autosómico recesivo del metabolismo del hierro que causa acúmulo del mismo tanto en el hígado como en el cerebro, manifestándose como un padecimiento neurológico en las etapas tardías de la vida.23 Se han descubierto otros defectos congénitos raros asociados con sobrecarga de hierro. Por ejemplo, en los pacientes con atransferrinemia prácticamente no existe transferrina.48 La absorción gastrointestinal de hierro está aumentada, pero debido a que no existen mecanismos fisiológicos para su transporte, casi nada del hierro absorbido puede ser captado por las células eritroides en desarrollo. El hierro absorbido se acumula entonces en el hígado, páncreas, corazón, tiroides y riñones, con casi nada de hierro depositado en la médula ósea o el bazo. Se ha descrito exceso dietético de hierro en africanos en por lo menos 9 países en la región sub-Sahara de Africa en asociación con una ingesta muy abundante de hierro por una bebida fermentada de maíz preparada en casa en tambos de acero.49 El acúmulo de hierro puede ser tan importante como el encontrado en la hemocromatosis hereditaria, y puede desarrollarse enfermedad hepática (con cirrosis y hepatoma), daño pancreático (con diabetes mellitus), trastornos endócrinos y disfunción cardiaca. Aunque durante mucho tiempo se consideró que la mayor ingesta en la dieta era la única causa de la mayor absorción de hierro en este padecimiento, los análisis de pedigree han sugerido que puede participar un componente genético que es común en poblaciones con ancestros africanos,50 aunque aún se investiga la relación con la sobrecarga asociada a ingesta dietética. Sin duda la ingestión medicinal de hierro puede contribuir al acúmulo del mismo en los pacientes con estos padecimientos, pero aún no se tiene certeza del grado en el que el hierro administrado por vía oral puede aumentar las reservas corporales de hierro en los individuos normales. Aunque algunos reportes de casos han descrito exceso de hierro en pacientes que toman hierro medicinal por periodos prolongados, no puede excluirse en estos pacientes la participación potencial de un alelo no reconocido para la hemocromatosis hereditaria.
EXCESO DE HIERRO POR TRANSFUSIONES Y ADMINISTRACION PARENTERAL
Un programa adecuado de transfusiones puede mantener la vida en pacientes con anemia severa crónica y refractaria, pero el tratamiento transfusional aislado produce acúmulo progresivo por el hierro contenido en los eritrocitos transfundidos.51 El acúmulo de hierro por transfusiones ocurre al inicio sobre todo en los macrófagos, seguido de redistribución a los tejidos parenquimatosos [ver figura 6]. En los pacientes con anemias congénitas severas, como la talasemia mayor y el síndrome de Blackfan-Diamond, las transfusiones regulares pueden prevenir la muerte por anemia en la infancia y permitir un crecimiento y desarrollo normal durante la niñez. Las anemias adquiridas dependientes de transfusión, como la anemia aplástica, la aplasia pura de eritrocitos, los trastornos hipoplásicos o mielodisplásicos y otros padecimientos, pueden causar exceso de hierro corporal. Si las anemias dependientes de transfusión incluyen hiperplasia eritroide con eritropoyesis ineficaz, puede agregarse mayor absorción gastrointestinal de hierro para empeorar el acúmulo. En estos casos debe minimizarse la captación de hierro de la dieta por medio de un programa adecuado de transfusiones. Aunque no dependen de transfusiones, los pacientes con trastornos falciformes, como la anemia de células falciformes y la talasemia-b con células falciformes, pueden adquirir una cantidad considerable de hierro por transfusiones crónicas para prevenir eventos cerebrovasculares,52 crisis dolorosas y otras complicaciones recurrentes. Debido a que el humano no tiene sistemas fisiológicos para eliminar el exceso de hierro, el hierro contenido en los eritrocitos transfundidos se acumula en forma progresiva y llega a dañar al hígado, corazón, páncreas y otros órganos. Suele ocurrir la muerte por insuficiencia cardiaca. En los pacientes más jóvenes la carga de hierro causa falla en el crecimiento y, en la adolescencia, retraso o ausencia de la maduración sexual. El hierro medicinal parenteral puede aumentar el exceso de hierro en los pacientes con anemias microcíticas refractarias que son diagnosticados en forma equívoca como con deficiencia de hierro.
Con cada unidad transfundida de eritrocitos se agregan a la reserva corporal alrededor de 200 a 250 mg de hierro. La mayoría de los pacientes dependientes de transfusión requieren de 200 a 300 ml/kg/año de sangre. Por ejemplo, un adulto de 70 kg requiere alrededor de dos a tres unidades de sangre cada 3 o 4 semanas, lo que agrega de 6 a 10 g de hierro por año. La severidad de la toxicidad por hierro parece relacionarse con la magnitud de la carga de hierro en el organismo. Casi todos los pacientes que han sido tratados solo con transfusión y han recibido 100 o más unidades de sangre (alrededor de 20 a 25 g de hierro) han desarrollado depósitos de hierro en el corazón, con frecuencia asociados con signos de daño hepático, pancreático y endócrino.51 Para los pacientes que son dependientes de transfusión o tienen anemia severa, la única manera de superar la limitación fisiológica que evita la eliminación de hierro es con el tratamiento con un agente quelante capaz de formar complejos con el hierro y permitir su excreción. El único agente quelante disponible en la actualidad para uso clínico es un sideróforo bacteriano que comenzó a usarse hace tres décadas, la desferroxamina B, un ácido trihidroxámico producido por Streptomyces pilosus. En la actualidad existen estudios clínicos que han demostrado la eficacia de la quelación de hierro como un enfoque terapéutico para su exceso, demostrando que la quelación regular de este elemento puede disminuir el exceso corporal, reducir la disfunción orgánica y mejorar la supervivencia.51,53 Aunque en la actualidad se han reconocido diversos efectos adversos tóxicos, en especial con el tratamiento intensivo, la desferroxamina es un medicamento muy seguro, incluso cuando se ha usado prácticamente de por vida en algunos pacientes.
El tratamiento de quelación del hierro debe comenzarse pronto para evitar el acúmulo de cantidades tóxicas de hierro en los tejidos susceptibles y para mantener las reservas corporales de hierro en concentraciones asociadas con un riesgo bajo de muerte temprana y complicaciones clínicas. Mientras más se retrase el tratamiento quelante mayor será el riesgo de toxicidad por hierro. Debido a que la desferroxamina se absorbe poco después de la administración oral y se elimina con tanta rapidez de la circulación, debe administrarse en forma de infusión intravenosa o subcutánea durante 9 a 12 horas cada día por lo menos 5 días de la semana para que sea útil en el tratamiento de pacientes con sobrecarga transfusional de hierro. En los pacientes con talasemia mayor y otras anemias refractarias congénitas que son dependientes de transfusión desde la infancia temprana, es mejor comenzar el tratamiento quelante después de 10 a 20 transfusiones, por lo general alrededor de los 3 a 4 años de edad, y administrar infusión subcutánea lenta en dosis no mayores de 25 mg/kg de peso/día para minimizar el riesgo de retraso en el crecimiento.51 En los pacientes ancianos y adultos con anemias refractarias adquiridas que requieren de transfusión regular y en los que tienen enfermedad de células falciformes que son transfundidos en forma crónica para prevenir complicaciones también parece prudente el tratamiento temprano, comenzando después de la transfusión de 10 a 20 unidades de sangre. La dosis habitual de desferroxamina en estos pacientes no debe ser mayor de 50 mg/kg/día, administrados en 9 a 12 horas por infusión subcutánea lenta por lo menos 5 días de la semana. En algunos pacientes que no son capaces de tolerar el dolor y molestia local de la infusión subcutánea o que requieren de la reducción rápida del exceso de hierro puede administrarse desferroxamina intravenosa a través de un reservorio implantable con acceso venoso.51 Puede ser difícil cumplir con estos esquemas casi diarios de infusión prolongada subcutánea o intravenosa, y la falta de constancia es el principal obstáculo para que el tratamiento sea eficaz. La administración de ácido ascórbico puede aumentar la excreción de hierro inducida por desferroxamina, pero se asocia con el riesgo de redistribución interna de hierro desde sitios de almacén relativamente benignos en los macrófagos hasta reservorios potencialmente tóxicos en las células parenquimatosas. Aunque la evidencia es anecdótica, las dosis grandes de ácido ascórbico deben considerarse como peligrosas en los pacientes con sobrecarga de hierro. Por lo general la desferroxamina es segura y no tóxica en el paciente con exceso de hierro, pero se han reportado complicaciones sistémicas,51 incluyendo reacciones anafilactoides alérgicas, complicaciones infecciosas, trastornos visuales, disfunción auditiva, y retraso del crecimiento. Como resultado, debe incluirse la evaluación regular de toxicidad medicamentosa en el tratamiento de cualquier paciente que reciba desferroxamina, incluyendo audiometrías anuales, examen de retina y evaluación del crecimiento en niños y adolescentes. El riesgo de muchas de estas complicaciones puede reducirse ajustando la dosis de desferroxamina según la magnitud del exceso de hierro. En la actualidad se están desarrollando agentes quelantes activos por vía oral, pero ninguno está clínicamente disponible aún.51,54
OTRAS FORMAS DE EXCESO DE HIERRO
Se ha detectado exceso de hierro perinatal en asociación con algunas
alteraciones metabólicas raras del neonato, incluyendo tirosinemia
hereditaria (hipermetioninemia), síndrome cerebrohepatorrenal o
síndrome de Zellweger, y hemocromatosis perinatal, también
conocida como hemocromatosis neonatal o enfermedad por almacenamiento de hierro
neonatal. El secuestro de hierro en otros trastornos raros produce varios
patrones de depósito de hierro localizado en la hemosiderosis pulmonar
idiopática, la hemosiderosis renal y la enfermedad de
Hallervorden-Spatz.48 La hiperferritinemia autosómico
dominante con cataratas congénitas es un trastorno recién
detectado del metabolismo del hierro,55 en el que los familiares
afectados presentan cataratas nucleares bilaterales de inicio temprano y
elevación moderada en la concentración de ferritina en plasma
(alrededor de 1,000 a 2,500 mg/L). Los dos trastornos se heredan en forma
autosómico dominante. La cantidad de hierro corporal en estos pacientes
es normal, pero con frecuencia se sospecha exceso por el aumento en la
concentración de ferritina.
Bibliografía
- Hsia CC: Respiratory function of hemoglobin. N Engl J Med 338:239-247, 1998
- Jia L, Bonaventura C, Bonaventura J, et al: S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature 380:221, 1996 [PMID 8637569 ]
- Stamler JS, Jia L, Eu JP, et al: Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science 276:2034, 1997 [PMID 9197264 ]
- Bunn HF, Forget BF: Hemoglobin: Molecular, Genetic and Clinical Aspects. WB Saunders Co, Philadelphia, 1986
- Perutz MF, Wilkinson AJ, Paoli M, et al: The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Annu Rev Biophys Biomol Struct 27:1, 1998 [PMID 9646860 ]
- Ponka P, Beaumont C, Richardson DR: Function and regulation of transferrin and ferritin. Semin Hematol 35:35, 1998 [PMID 9460808 ]
- Levy JE, Jin O, Fujiwara Y, et al: Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet 21:396, 1999 [PMID 10192390 ]
- Aisen P: Transferrin, the transferrin receptor, and the uptake of iron by cells. Met Ions Biol Syst 35:585, 1998
- Harrison PM, Arosio P: The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1275:161, 1996 [PMID 8695634 ]
- Hentze MW, Kuhn LC: Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc Natl Acad Sci USA 93:8175, 1996 [PMID 8710843 ]
- Rouault T, Klausner R: Regulation of iron metabolism in eukaryotes. Curr Top Cell Regul 35:1, 1997 [PMID 9192174 ]
- Gunshin H, Mackenzie B, Berger UV, et al: Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388:482, 1997 [PMID 9242408 ]
- Fleming MD, Trenor CC 3rd, Su MA, et al: Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet 16:383, 1997 [PMID 9241278]
- Fleming MD, Romano MA, Su MA, et al: Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci USA 95:1148, 1998 [PMID 9448300]
- Gruenheid S, Canonne-Hergaux F, Gauthier S, et al: The iron transport protein NRAMP2 is an integral membrane glycoprotein that colocalizes with transferrin in recycling endosomes. J Exp Med 189:831, 1999 [PMID 10049947]
- Vulpe CD, Kuo YM, Murphy TL, et al: Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 21:195, 1999 [PMID 9988272 ]
- Feder JN, Gnirke A, Thomas W, et al: A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 13:399, 1996 [PMID 8696333 ]
- Feder JN, Tsuchihashi Z, Irrinki A, et al: The hemochromatosis founder mutation in HLA-H disrupts beta2-microglobulin interaction and cell surface expression. J Biol Chem 272:14025, 1997 [PMID 9162021 ]
- Feder JN, Penny DM, Irrinki A, et al: The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci USA 95:1472, 1998 [PMID 9465039 ]
- Lebron JA, Bennett MJ, Vaughn DE, et al: Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell 93:111, 1998 [PMID 9546397 ]
- Waheed A, Parkkila S, Saarnio J, et al: Association of HFE protein with transferrin receptor in crypt enterocytes of human duodenum. Proc Natl Acad Sci USA 96:1579, 1999 [PMID 9990067 ]
- Han O, Fleet JC, Wood RJ: Reciprocal regulation of HFE and Nramp2 gene expression by iron in human intestinal cells. J Nutr 129:98, 1999 [PMID 9915882 ]
- Harris ZL, Takahashi Y, Miyajima H, et al: Aceruloplasminemia: molecular characterization of this disorder of iron metabolism. Proc Natl Acad Sci USA 92:2539, 1995 [PMID 7708681 ]
- Attieh ZK, Mukhopadhyay CK, Seshadri V, et al: Ceruloplasmin ferroxidase activity stimulates cellular iron uptake by a trivalent cation-specific transport mechanism. J Biol Chem 274:1116, 1999 [PMID 9873059 ]
- Yu J, Wessling-Resnick M: Structural and functional analysis of SFT, a stimulator of Fe Transport. J Biol Chem 273:21380, 1998 [PMID 9694900 ]
- Bothwell TH, Charlton RW, Cook JD, et al: Iron Metabolism in Man. Blackwell Scientific Publications, Oxford, England, 1979
- Brittenham GM: Iron in the red cell cycle. Iron Metabolism in Health and Disease. Brock J, Pippard M, Halliday J, et al, eds. Academic Press, London, 1994, p 31
- Finch C: Regulators of iron balance in humans. Blood 84:1697, 1994
- Cook JD: Iron-deficiency anaemia. Baillieres Clin Haematol 7:787, 1994
- Recommendations to prevent and control iron deficiency in the United States. MMWR Morb Mortal Wkly Rep 47(RR-3):1, 1998 [see Web Link]|http://www.cdc.gov/epo/mmwr/preview/mmwrhtml/00051880.htm||
- de Andraca I, Castillo M, Walter T: Psychomotor development and behavior in iron-deficient anemic infants. Nutr Rev 55:125, 1997 [PMID 9197132 ]
- Cook JD, Baynes RD, Skikne BS: The physiological significance of circulating transferrin receptors. Adv Exp Med Biol 352:119, 1994 [PMID 7832041 ]
- Cogswell ME, McDonnell SM, Khoury MJ, et al: Iron overload, public health, and genetics: evaluating the evidence for hemochromatosis screening. Ann Intern Med 129:971, 1998 [PMID 9867750 ]
- Edwards CQ, Griffen LM, Ajioka RS, et al: Screening for hemochromatosis: phenotype versus genotype. Semin Hematol 35:72, 1998 [PMID 9460810 ]
- Burke W, Thomson E, Khoury MJ, et al: Hereditary hemochromatosis: gene discovery and its implications for population-based screening. JAMA 280:172, 1998 [PMID 9669792 ]
- Mura C, Raguenes O, Ferec C: HFE mutations analysis in 711 hemochromatosis probands: evidence for S65C implication in mild form of hemochromatosis. Blood 93:2502, 1999 [PMID 10194428 ]
- Crawford DH, Jazwinska EC, Cullen LM, et al: Expression of HLA-linked hemochromatosis in subjects homozygous or heterozygous for the C282Y mutation. Gastroenterology 114:1003, 1998 [PMID 9558290 ]
- Niederau C, Stremmel W, Strohmeyer GW: Clinical spectrum and management of haemochromatosis. Baillieres Clin Haematol 7:881, 1994 [PMID 7881158 ]
- Adams PC, Deugnier Y, Moirand R, et al: The relationship between iron overload, clinical symptoms, and age in 410 patients with genetic hemochromatosis. Hepatology 25:162, 1997 [PMID 8985284 ]
- Phatuk PD, Guzman G, Woll JE, et al: Cost-effectiveness of screening for hereditary hemochromatosis. Arch Intern Med 154:769, 1994
- Feller ER, Pout A, Wands JR, et al: Familial hemochromatosis: physiologic studies in the precirrhotic stage of the disease. N Engl J Med 296:1422, 1977 [PMID 194151 ]
- Adams PC, Chakrabarti S: Genotypic/phenotypic correlations in genetic hemochromatosis: evolution of diagnostic criteria. Gastroenterology 114:319, 1998 [PMID 9453492 ]
- Brissot P, Moirand R, Jouanolle AM, et al: A genotypic study of 217 unrelated probands diagnosed as "genetic hemochromatosis" on "classical" phenotypic criteria. J Hepatol 30:588, 1999 [PMID 10207799 ]
- Olynyk JK: Hereditary haemochromatosis: diagnosis and management in the gene era. Liver 19:73, 1999
- Bacon BR, Olynyk JK, Brunt EM, et al: HFE genotype in patients with hemochromatosis and other liver diseases. Ann Intern Med 130:953, 1999 [PMID 10383365 ]
- Angelucci E, Baronciani D, Lucarelli G, et al: Needle liver biopsy in thalassaemia: analyses of diagnostic accuracy and safety in 1184 consecutive biopsies. Br J Haematol 89:757, 1995 [PMID 7772512 ]
- Niederau C, Fischer R, Purschel A, et al: Long-term survival in patients with hereditary hemochromatosis. Gastroenterology 110:1107, 1996 [PMID 8613000 ]
- Bottomley SS: Secondary iron overload disorders. Semin Hematol 35:77, 1998
- Moyo VM, Mandishona E, Hasstedt SJ, et al: Evidence of genetic transmission in African iron overload. Blood 91:1076, 1998 [PMID 9446671 ]
- Wurapa RK, Gordeuk VR, Brittenham GM, et al: Primary iron overload in African Americans. Am J Med 101:9, 1996 [PMID 8686721 ]
- Olivieri NF, Brittenham GM: Iron-chelating therapy and the treatment of thalassemia. Blood 89:739, 1997 [PMID 9028304 ]
- Adams RJ, McKie VC, Hsu L, et al: Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med 339:5, 1998 [PMID 9647873 ]
- Brittenham GM, Griffith PM, Nienhuis AW, et al: Efficacy of deferoxamine in preventing complications of iron overload in patients with thalassemia major. N Engl J Med 331:567, 1994 [PMID 8047080 ]
- Bergeron RJ, Wiegand J, Brittenham GM: HBED: the continuing development of a potential alternative to deferoxamine for iron-chelating therapy. Blood 93:370, 1999 [PMID 9864183 ]
- Beaumont C, Leneuve P, Devaux I, et al: Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinaemia and cataract. Nat Genet 11:444, 1995 [PMID 7493028 ]