Contenido del artículo
VI DIABETES MELLITUS
- Patogenia
- Metabolismo
- Diagnóstico y escrutinio
- Tratamiento de la DMID
- METAS DEL TRATAMIENTO PARA LA GLUCEMIA: DCCT
- Diseño del estudio
- Tratamiento intensivo y objetivos metabólicos del estudio
- Resultados del estudio
- Recomendaciones de tratamiento
- AUTOVIGILANCIA
- VIGILANCIA POR PARTE DEL MEDICO
- INSULINA
- DIETA
- EJERCICIO
- CETOACIDOSIS DIABETICA
- HIPOGLUCEMIA
- TRATAMIENTOS INNOVADORES Y EXPERIMENTALES DE LA DMID
- Tratamiento de la DMNID
- DIETA
- VIGILANCIA
- MEDICAMENTOS HIPOGLUCEMIANTES
- EJERCICIO
- TRATAMIENTO INNOVADOR Y EXPERIMENTAL PARA LA DMNID
- COMA HIPEROSMOLAR, HIPERGLUCEMICO, NO CETOSICO
- Complicaciones
DR. DAVID M. NATHAN
La diabetes mellitus es un trastorno de la regulación metabólica, principalmente del metabolismo de la glucosa, que se acompaña de complicaciones vasculares y neurológicas características a largo plazo. La diabetes tiene varias formas clínicas, cada una con distinta etiología, presentación clínica y evolución. Los estudios metabólicos, la posibilidad de radioinmunoensayos de insulina y glucagon y la aplicación de estrategias de biología molecular han permitido conocer más sobre la fisiopatología de este padecimiento y administrar un tratamiento racional para la enfermedad y sus complicaciones.
Al identificar la sustancia llamada en un principio isletina (i.e., insulina) en los islotes, como el agente activo para disminuir la glucosa, Benting y Best en Toronto y Paulesco en Rumania la usaron en animales diabéticos, lo que culminó en el tratamiento eficaz para Leonard Thompson el 11 de enero de 1922.1,2 La insulina controló una enfermedad que antes era siempre mortal. Sin embargo, las experiencias clínicas demostraron que aunque la insulina disminuía en forma dramática entre el 50 a 100 porciento de la mortalidad asociada con la cetoacidosis diabética, los pacientes tratados con insulina desarrollaban complicaciones inexorables a largo plazo, incluyendo ceguera, insuficiencia renal y enfermedad cardiovascular y periférica. La enfermedad aguda mortal se había transformado en un padecimiento crónico con morbilidad significativa y mortalidad prematura.
Durante los primeros 75 años de su uso, la insulina ha sido purificada, estableciéndose su secuencia de aminoácidos. La insulina se ha sintetizado por técnicas de fase sólida y, en forma más reciente, por modificación de la insulina porcina y por métodos recombinantes en bacterias o levaduras, lo que aumenta su disponibilidad en forma ilimitada. Las modificaciones a la insulina han originado diversas fórmulas que pueden emplearse en forma creativa para alcanzar niveles específicos de glucosa durante todo el día.3 Se están investigando análogos de la insulina, creados por sustitución de aminoácidos específicos, que sin duda constituirán nuevos agentes útiles para el tratamiento de la diabetes.4 El trasplante de páncreas es cada vez más exitoso,5 y en la actualidad se investiga sobre el trasplante de islotes aislados y el uso de equipos híbridos (i.e., islotes encapsulados en cámaras artificiales).6,7 El Estudio de Control de Diabetes y sus Complicaciones (DCCT, por sus siglas en inglés, n. del t.) demostró la eficacia del tratamiento intensivo al mantener el control de la glucosa y prevenir o retrasar las complicaciones de la diabetes a largo plazo. El DCCT estableció un nuevo estándar de tratamiento para la diabetes mellitus insulinodependiente (DMID) y, por extensión, para la diabetes mellitus no insulinodependiente (DMNID).8 También deben establecerse nuevos métodos de tratamiento intensivo que sean más cómodos para el paciente y tengan menos riesgo de causar hipoglucemia. El desarrollo de sensores de glucosa no invasores9 y bombas de insulina implantables10 ayudarán en este aspecto. En la actualidad se investigan nuevos tratamientos dirigidos a las complicaciones, independientemente del control de la glucemia, y otros estudios examinan la posibilidad de prevenir la DMID y la DMNID.
EPIDEMIOLOGIA
Todas las observaciones iniciales respecto a la diabetes se enfocaron en su expresión más dramática, la DMID, porque afecta a niños y tiene una evolución tórpida. La DMID y la otra forma clínica principal, la DMNID no se distinguieron en forma clara sino hasta hace poco tiempo. En 1936 se demostró por primera vez la existencia de dos formas clínicas de diabetes, la DMID (denominada entonces diabetes sensible a insulina) y la DMNID (denominada diabetes no sensible a insulina).11 Los estudios subsecuentes que han empleado bio y radioinmunoensayos para insulina demostraron la diferencia fisiopatológica fundamental de las dos formas clínicas principales. Los pacientes con DMID tienen una deficiencia absoluta de insulina causada por destrucción de los islotes,12 mientras que los pacientes con DMNID, que tienen un volumen de islotes casi normal, no secretan la insulina suficiente para satisfacer la mayor demanda causada por resistencia a la insulina.13
El National Diabetes Data Group (NDDG, Grupo nacional de datos sobre diabetes de los EUA, n. del t.) y la Organización Mundial de la Salud (OMS) emitieron en 1979 definiciones semejantes para las diferentes formas clínicas de la diabetes.14 Las definiciones se basaron en manifestaciones clínicas características y en la fisiopatología, y se apoyaron en marcadores inmunológicos y aspectos hereditarios [ver tabla 1]. Aunque las nuevas definiciones no tienen el criterio de edad que consistía antes en diabetes de inicio juvenil y de inicio en la edad adulta, la DMID suele ser una enfermedad de la infancia: la mayoría de los casos aparecen en pacientes menores de 20 años, y el inicio máximo es alrededor de la pubertad. Menos del 10 porciento de los casos de DMID aparecen en pacientes mayores de 50 años. Estos enfermos son delgados y tienen propensión a la cetosis, además de que comparten los marcadores genéticos e inmunológicos de los pacientes con DMID de aparición en la juventud. Los enfermos que desarrollan DMID como parte de un síndrome de deficiencia endócrina múltiple tipo II tienen en forma típica 30 o más años de edad al comienzo.15 La mayoría de los casos de DMNID ocurre en pacientes mayores de 40 años, un pequeño porcentaje de pacientes con DMNID desarrollan el padecimiento durante la tercera y la cuarta décadas de la vida. Estos pacientes son muy obesos, son hijos de padres con DMNID, son mujeres con historia de diabetes gestacional o son miembros de un grupo étnico o racial con prevalencia especialmente alta de DMNID [ver adelante, DMNID]. La Asociación Americana de Diabetes revisó en 1997 las normas para usar los niveles de glucosa en plasma en ayuno como criterio para el diagnóstico de diabetes [ver tabla 1].16
|
||||||||||||||||||
* Revisados por el Comité de Expertos de la Asociación Americana de Diabetes en 1997.16
|
DMID
La DMID es una enfermedad poco frecuente, que afecta solo a uno de cada 250 individuos en los Estados Unidos, en donde existen alrededor de 10,000 a 15,000 nuevos casos notificados cada año [ver tabla 2].17 La mayor prevalencia de la DMID se encuentra en el norte de Europa, en donde más de uno de cada 150 finlandeses desarrollan DMID para los15 años de edad. Datos recientes sugieren que, por motivos desconocidos, la incidencia de DMID está aumentado en Europa. Por otro lado, la DMID es menos frecuente (menos de la mitad que en la población caucásica) en los individuos de raza negra y oriental.
|
||||||||||||||||||||
* Derivado del Estudio Nacional de Salud y Nutrición,
usando los criterios del Grupo Nacional de Datos sobre Diabetes de los
EUA.14
|
DMNID
Comparado con la DMID, la DMNID es una enfermedad muy frecuente [ver tabla 2], con una prevalencia de 6.6 porciento en los Estados Unidos.18 En la segunda mitad del siglo XX, la DMNID se ha convertido en una de las enfermedades crónicas más frecuentes en los Estados Unidos y otros países industrializados.19 Existen más de 600,000 casos notificados cada año, y la prevalencia proyectada para la próxima década es de 10 porciento. Alrededor de la mitad de la población con DMNID no sabía de su padecimiento.18 El aumento en la prevalencia de la DMNID en los Estados Unidos se atribuye al incremento en la población de edad avanzada, que también es más obesa y sedentaria. La DMNID es especialmente común en ciertos grupos raciales y étnicos, como los nativos americanos, los nativos de Nauru en el Pacífico Sur y Mauritanía en el Océano Indico, los hispano-americanos, los asiáticos y los afro-americanos. La prevalencia de DMNID entre personas mayores de 65 años excede el 18 porciento y, comparado con los individuos de peso normal, la gente obesa (i.e., los que tienen un índice de masa corporal mayor de 30) tienen 10 a 20 veces más riesgo de DMNID.20 Casi el 90 porciento de los pacientes con DMNID son obesos. La obesidad abdominal (i.e., androide, opuesta a la ginecoide), que refleja aumento en la grasa visceral y puede medirse por un aumento en la relación cintura-cadera, puede constituir un factor de riesgo aún más importante que la obesidad.21 Por último, ciertas poblaciones étnicas aisladas, como los indios Pima de Gila River, Arizona, y los nativos Nauruan, tienen prevalencias de DMNID hasta de 50 porciento en la población adulta.22,23 Esta altísima prevalencia en poblaciones aisladas es consecuencia de cambios dietéticos, mayor prevalencia de obesidad y estilo de vida muy sedentario (en la reservación en el caso de los Pima y por la mejor economía en el caso de los nauruanos).
Aunque se han encontrado marcadores genéticos e inmunológicos para la DMID, el NDDG no encontró que fueran suficientemente sensibles ni específicos para definir a la DMID o distinguir entre la DMID y la DMNID. En forma semejante, las mediciones de la excreción de insulina o del péptido C no distinguen en forma fidedigna entre la DMID y la DMNID, y no se usan para diagnosticar ninguna de las dos. La identificación de un anticuerpo dirigido contra la descarboxilasa del ácido glutámico (anti-DAG) como marcador para la DMID24 y la estandarización del ensayo para anticuerpos contra células de los islotes (ACI)25 ofrece una mayor especificidad y sensibilidad para identificar a las parsonas con riesgo de DMID. Además, la búsqueda de una causa genética en la DMID ha permitido identificar varios loci en el cromosoma 6 que podrían ser responsables del componente genético de la DMID,26 y el uso de genes candidatos y estrategias genómicas ha permitido descartar muchas causas potenciales de la DMNID.
OTRAS FORMAS DE DIABETES
Además de las dos principales formas clínicas de diabetes, se conocen otras formas de diabetes con una etiología relativamente clara. La diabetes causada por la destrucción pancreática (v.gr., por pancreatitis recurrente, pancreatectomía o toxinas como el raticida Vacor) se denomina diabetes secundaria, lo mismo que la asociada con medicamentos que aumentan la resistencia a la insulina (v.gr., esteroides) o disminuyen la secreción de insulina (v.gr., fenitoína o bloqueadores adrenérgicos beta). La diabetes mellitus suele desarrollarse durante el tercer trimestre del embarazo, y la gran mayoría de estos casos tienen una fisiopatología semejante a la de la DMNID. La tolerancia a la glucosa se normaliza después del parto, pero el 30 al 50 porciento de las mujeres con diabetes gestacional desarrolla DMNID en un lapso de 10 años desde el diagnóstico inicial.27 Se ha identificado una forma relativamente rara de diabetes, en la que los individuos afectados tienen propensión a la cetosis y que se hereda como un rasgo autosómico dominante.28 Tiende a presentarse en adolescentes delgados y se conoce como diabetes de inicio del adulto en la juventud (MODY, por sus siglas en inglés, n. del t.). Se ha identificado una glucocinasa con una Km (constante de Michaelis) muy alta en algunas familias con MODY, pero no en todas, y esta podría ser la causa de la menor secreción de insulina.29 En familias principalmente japonesas se ha identificado otra forma rara de diabetes que puede asociarse con deficiencia a la insulina o recordar fenotípicamente a la DMNID y relacionarse con hipoacusia, otras alteraciones del sistema nervioso central y un ARN de transferencia mitocondrial mutante con herencia materna.30
Aunque la DMNID, la DMID y la diabetes gestacional constituyen la mayoría de los casos de diabetes en los Estados Unidos y otros países industrializados, la diabetes famélica puede ser la forma más frecuente en países con aportes limitados de alimentos. Se sabe que la desnutrición y las dietas con restrición de carbohidratos se asocian con una menor secreción de insulina.
ALTERACION EN LA TOLERANCIA A LA GLUCOSA
La definición de diabetes implica tanto la alteración en la glucemia como las complicaciones vasculares a largo plazo, características que comparten todas las formas de diabetes. En la población general la concentración de glucosa después de la administración de una carga de la misma (v.gr., la prueba de tolerancia con 75 g de glucosa por vía oral) tiene una distribución normal. Los valores que se consideran anormales pero que no se asocian con complicaciones microvasculares específicas a largo plazo representan la intolerancia a la glucosa. Los valores para diagnosticar intolerancia a la glucosa se basaron en datos de varios estudios epidemiológicos extensos [ver tabla 1]. El motivo de esta categoría diagnóstica separada es que los pacientes con intolerancia a la glucosa, aunque no tienen mayor riesgo de retinopatía o neuropatía, sí lo tienen de enfermedad macrovascular, en comparación con los individuos con tolerancia a la glucosa normal. Además, el 30 a 40 porciento de los pacientes con intolerancia a la glucosa desarrollan DMNID en un lapso de 10 años después del diagnóstico.31,32 En la actualidad se estudia la posible intervención en esta fase para prevenir el desarrollo subsecuente de DMNID.
Patogenia
DMID
La DMID se caracteriza por deficiencia absoluta de insulina, lo que hace al paciente dependiente de la insulina exógena para sobrevivir. Aunque la DMID tiene en forma característica un inicio agudo con síntomas de hiperglucemia (incluyendo poliuria, polidipsia, pérdida de peso, visión borrosa o todos) que preceden a la cetoacidosis o al diagnóstico por solo días o semanas, en la actualidad se sabe que la historia natural de la DMID tienen un periodo preclínico asintomático largo.33 Durante este periodo preclínico las células beta productoras de insulina se destruyen en forma progresiva [ver figura 1]. El páncreas normal contiene 1.0 a 1.5 millones de islotes, y alrededor del 80 porciento de las células de éstos son células beta secretoras de insulina. Aparece diabetes clínica cuando se han destruido más del 90 porciento de las células beta.

|
| Figura 1 |
| Fases del desarrollo de la DMID y volumen de células beta |
La destrucción autoinmune de las células beta se asocia con, y quizá es causada por infiltración linfocítica denominada insulinitis. Se han observado cambios característicos en los subgrupos de células T (v.gr., aumento en las células supresoras y disminución en las cooperadoras) que preceden a la destrucción de los islotes.34 Los pacientes que van a desarrollar DMID pueden identificarse por la aparición de ACI hasta 10 años antes del comienzo clínico de la diabetes.33 Estos anticuerpos no son citotóxicos y quizá indican la destrucción inmunológica. Aunque varios estudios han sugerido que la destrucción de los islotes tiene una progresión inexorable, otros datos sugieren que la presencia de ACI puede ser intermitente y que estos anticuerpos, sobre todo si están en títulos bajos, no predicen con exactitud el desarrollo de DMID.35 También se han identificado otros anticuerpos, dirigidos contra la insulina y la descarboxilasa del ácido glutámico, en etapas tempranas de la DMID.36 Cuando existen más de uno de los anticuerpos en asociación con menor secreción de insulina o con un tipo HLA que es característico de la DMID, el valor predictivo de la prueba aumenta.37 Se han identificado varios autoantígenos que pueden participar en la patogenia de la DMID, incluyendo insulina DAG65, DAG67, proteínas de calor de choque y otros antígenos de las células de los islotes.38 No es claro si las infecciones virales precipitan o causan DMID en personas susceptibles.
Genéica
Aunque es claro que la DMID es un padecimiento hereditario, el patrón de herencia escapa a las definiciones actuales. En un estudio de gemelos o triates monocigóticos (i.e., idénticos) entre los que existía DMID en solo uno de los hermanos, la enfermedad se desarrolló después en solo el 50 porciento de los hermanos restantes.39 Esta observación sugiere que existe una influencia ambiental que se agrega al componente hereditario de la DMID. El seguimiento de estos individuos demostró que los pacientes con DMID desarrollaron primero ACI y después disminución en la secreción de insulina en respuesta a la glucosa intravenosa.33 Las concentraciones en ayuno y posprandiales de glucosa en plasma de estos individuos no pudieron distinguirse de las personas no diabéticas sino hasta poco tiempo antes de la aparición de diabetes clínica.
Algunos loci HLA (DR3 y DR4) se asocian con mayor riesgo de desarrollar DMID, mientras que otros parecen ser protectores.40 Se ha encontrado que la sustitución de alanina, valina o serina por ácido aspártico en la posición 57 de la cadena § codificada por el locus HLA-DQ se relaciona en forma estrecha con mayor riesgo de desarrollar DMID.41 Todas las asociaciones genéticas se localizan en la región de del CPH de clase II en el cromosoma 6.
DMNID
Resistencia a la insulina en la DMNID
Aunque desde hace mucho tiempo se conoce la causa de la DMID, que es la destrucción de las células beta productoras de insulina, la causa de la DMNID continúa siendo motivo de controversia [ver figura 2]. En sus primeros estudios sobre concentraciones circulantes de insulina medidas por radioinmunoensayo, Berson y Yalow notaron concentraciones normales o incluso altas en pacientes diabéticos de edad avanzada, obesos y que no eran tratados con insulina.42,43 Apoyando estos datos, los estudios de necropsia de páncreas de pacientes que tenían DMNID demostraban solo disminución moderada en el volumen de islotes, comparado con los páncreas de individuos no diabéticos de la misma edad y peso.44 Para explicar la observación de aumento en la glucemia con concentraciones absolutas de insulina normales o aumentadas, se sugirió la posibilidad de resistencia a la insulina (i.e., disminución en la sensibilidad a la actividad de la insulina). Después de que se identificó que el primer paso en la acción de la insulina era la fijación de la misma a receptores de superficie de gran afinidad, estudios in vivo usando técnicas de fijación de insulina, demostraron una disminución modesta en los receptores de insulina en pacientes resistentes a la misma.45 El sitio principal de resistencia parecía ser distal al receptor (posreceptor). La identificación, aislamiento y clonación posteriores del receptor de insulina ha permitido a los investigadores estudiar su función en individuos no diabéticos y en pacientes con DMNID. En la DMNID no existen alteraciones aparentes ni la estructura del receptor de insulina ni en la tirosina cinasa que está integrada a su función. Sin embargo, la estimulación mediada por insulina de la tirosina cinasa y la autofosforilación están alteradas.46 Estos efectos son reversibles al mejorar la glucemia, y quizá no sean del todo responsables de la resistencia a la insulina.
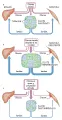
|
| Figura 2 |
| Patrones de secreción de insulina |
La principal secuela de la resistencia a la insulina es la menor captación de glucosa por el músculo. Esta alteración podría explicar los altos valores posprandiales de glucemia que se presentan en individuos con intolancia a la glucosa o con DMNID incipientes, porque la mayor parte del metabolismo de la glucosa en el posprandio se debe a captación muscular.48 La captación anormal de glucosa por el músculo puede ser secundaria a transportadores anormales de la misma, menor fosforilación de la glucosa, que es el paso que limita la velocidad en el metabolismo de la glucosa en el músculo, menor síntesis de glucógeno, glucogenolisis o gluconeogénesis relativamente no suprimida, o una combinación de estas condiciones. Por desgracia, la deficiencia de insulina y la hiperglucemia pueden causar muchos de estos trastornos, lo que dificulta la identificación del problema primario.
La célula beta dañada en la DMNID
Sin embargo, la demostración de resistencia a la insulina en muchos de (aunque no todos) los pacientes con DMNID no es suficiente para explicar la evolución de la intolerancia a la glucosa a diabetes franca. Primero, incluso en presencia de un menor transporte de glucosa y de resistencia a la insulina, el aumento en la salida hepática de glucosa que causa la hiperglucemia en ayuno característica de la DMNID sigue sin explicación. Segundo, aunque muchas personas son resistentes a la insulina, incluyendo a los obesos y a pacientes con síndrome de ovarios poliquísticos, acromegalia y síndrome de Cushing, solo una minoría de ellos desarrolla DMNID. Todos los defectos asociados con resistencia a la insulina pueden subsanarse con un mayor aporte de insulina. Por lo tanto, la mayoría de los pacientes con resistencia a la insulina no desarrollan diabetes porque pueden aumentar su secreción de insulina lo suficiente para superar la resistencia. Los pacientes que se descompensan y desarrollan DMNID son incapaces de aumentar su secreción de insulina lo suficiente o parecen cansarse de producir estos niveles aumentados, y desarrollan DMNID al disminuir la secreción de esta hormona.13 De hecho, aunque muchos estudios han demostrado hiperinsulinemia en pacientes con intolerancia a la glucosa, la disminución en la secreción de insulina acompaña siempre la transición a DMNID. Además, aunque la cantidad absoluta de insulina secretada puede aumentar antes de que se desarrolle diabetes clínica en el paciente, el 20 a 40 porciento de la insulina está constituida por proinsulina y productos de degradación de la proinsulina, lo que representa una separación incompleta del péptido conector (péptido C) de la proinsulina durante el proceso de postraducción. Por el contrario, la proinsulina corresponde a menos del 15 porciento de la insulina total en los no diabéticos. Debido a que la proinsulina y sus productos de degradación tienen solo el 10 porciento de la actividad de la insulina, la actividad real de la insulina en la prediabetes puede no estar aumentada.50
Al disminuir la secreción de insulina es frecuente encontrar pérdida de la primera fase de secreción de insulina.51 La secreción normal de insulina en respuesta a una administración rápida de glucosa intravenosa incluye liberación rápida de la hormona en los primeros uno a tres minutos y una liberación más prolongada que comienza 10 minutos después de la infusión I.V. [ver figura 3]. Se ha descrito también una tercera fase, que consiste en liberación muy lenta de insulina. La primera fase o espiga, del perfil espiga-meseta de secreción, se pierde no solo en la DMNID, sino también en las células beta dañadas de la DMID. Se ha inducido un fenómeno semejante en modelos en animales sometidos a pancreatectomía parcial.52 Por lo tanto, la pérdida de la primera fase de secreción es frecuente en las células beta limitadas o sometidas a estrés.

|
| Figura 3 |
| Fases de liberación de la insulina |
Una característica importante de la DMNID es que tanto la resistencia a la insulina como la secreción de la misma mejoran si se normaliza la glucemia basal (o de ayuno), independientemente del método para normalizarla. Los tratamientos con dieta, insulina o sulfonilureas han demostrado mejorar la secreción endógena de insulina.53-56 El efecto reversible de la hipoglucemia sobre la resistencia y secreción de la insulina ha sido denominado glucotoxicidad. Aunque el mecanismo por el que ocurre esto se desconoce, la glucotoxicidad puede explicar porqué la hiperglucemia engendra más hiperglucemia. Por último, un patrón específico de obesidad central, que refleja un mayor depósito visceral de grasa, está especialmente relacionado con el riesgo de desarrollar DMNID.20 La mayor liberación de ácidos grasos libres a partir de estas reservas grasas viscerales, con entrega directa al hígado y páncreas, parece explicar este metabolismo anormal de los lípidos y la resistencia a la insulina.
Genética
En vista de estas observaciones, la secreción anormal de insulina debe considerarse como integrada al desarrollo de la DMNID, e incluso un factor determinante de la evolución. Los estudios realizados en gemelos monocigóticos han demostrado una gran influencia genética en la DMNID, casi todos los gemelos idénticos de pacientes con DMNID desarrollan también el padecimiento.39 Los pacientes con alto riesgo para DMNID (v.gr., indios Pima e hijos de ambos padres con DMNID) tienen resistencia a la insulina antes de desarrollar intolerancia a la glucosa.57,58 Aunque varios estudios han demostrado una secreción de insulina normal o alta en esta etapa, otros han demostrado secreción intermitente anormal o disminuida.59-61 No se han identificado marcadores HLA específicos para DMNID.
Por lo tanto, la DMNID es un padecimiento hereditario en el que la resistencia y secreción anormal de insulina tienen funciones patogénicas importantes. La intolerancia a la glucosa precede al desarrollo de DMNID y se caracteriza por resistencia a la insulina, que causa hiperglucemia posprandial debido a menor captación de glucosa por el músculo. La resistencia a la insulina parece ser una característica hereditaria. Aunque el 30 al 50 porciento de los pacientes con intolerancia a la glucosa desarrollan después DMNID, el resto no lo hace. El factor crítico asociado con la progresión de intolerancia a la glucosa a DMNID es la reducción en la secreción de insulina. No se sabe si la secreción anormal de insulina es un trastorno adquirido o hereditario. Sin embargo, la posibilidad de que dos rasgos hereditarios independientes participen en una enfermedad tan frecuente como la DMNID parece ser remota. No se han identificado mutaciones específicas para la secreción de insulina o resistencia a la misma por medio de genes candidatos y estrategias genómicas, que expliquen la mayoría de los casos de DMNID. El tipo MODY es la única forma de DMNID con una mutación de un solo gen bien caracterizada, que proporciona explicación fisiopatológica para la intolerancia a la glucosa (aunque existe en solo alrededor de la mitad de las familias con MODY); las mutaciones en la glucocinasa causan secreción defectuosa de insulina.29
DIABETES SECUNDARIA
La diabetes secundaria a otras causas es relativamente rara y por lo general fácil de diagnósticar. Su fisiopatología también parece fácil de entender. Varias causas de diabetes secundaria, como la pancreatitis crónica o la pancreatectomía, se deben a pérdida de células beta y de la capacidad secretora de insulina. Debe perderse una gran parte de las células beta, y el daño pancreático debe ser importante, para que se desarrolle diabetes. Los medicamentos como los esteroides aumentan la resistencia a la insulina. La diabetes gestacional se desarrolla por la presencia de somatomamotropina placentaria (lactógeno placentario humano), que aumenta la resistencia a la insulina.
Metabolismo
DMID
La insulina es la principal hormona anabólica en los humanos. Cuando los requerimientos y el aporte de insulina están desequilibrados, se altera el metabolismo de la glucosa, lípidos y proteínas. La insulina causa sus muchísimos efectos celulares al unirse a receptores específicos de alta afinidad. Se ha aislado y purificado el receptor para la insulina, determinando y clonando su secuencia de ADN.62 El receptor de la insulina es un heterodímero con un dominio de fijación extracelular, un dominio transmembrana y un dominio intracelular, que tiene actividad de tirosina cinasa [ver figura 4]. La molécula de tirosina cinasa promueve la autofosforilación en diversos sitios después de que la insulina se fija al receptor. La fosforilación y la defosforilación tienen una función muy importante para mediar la actividad del receptor de la insulina. Se han identificado varios sustratos proximales del receptor activado, incluyendo los sustratos 1 y 2 del receptor de insulina, que se fijan y activan proteínas que median los efectos biológicos de la insulina.47
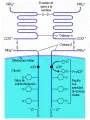
|
| Figura 4 |
| Receptor de la insulina |
Es evidente que en la DMID no existen alteraciones en el receptor de insulina ni en la acción de la hormona. La anomalía predominante es la deficiencia de insulina, con las siguientes consecuencias [ver figura 5]
|
|
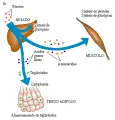
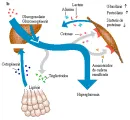
2. Aumento en la concentración de ácidos grasos libres por mayor lipólisis.
3. Aumento en la concentración de cetonas por cetogénesis hepática no limitada, a partir de los ácidos grasos libres.
4. Mayores niveles de triglicéridos por disminución en la lipoproteína lipasa, debida a mayor síntesis de lipoproteínas de muy baja densidad (LMBD) y a su menor depuración.
5. Aumento en los aminoácidos de cadena ramificada y disminución en la síntesis proteica.
6. Debido a que la glucosa actúa como un diurético osmótico, la hiperglucemia ocasiona deshidratación cuando la glucemia excede el nivel máximo de reab-sorción tubular renal.
Aunque la deficiencia de insulina es suficiente para causar todas estas alteraciones, el aumento en la concentración de hormonas contrarreguladoras como glucagon, epinefrina, norepinefrina, hormona del crecimiento y cortisol, exacerba muchas de las alteraciones metabólicas.63 En los pacientes con deficiencia de insulina el aumento en la concentración de glucagon y catecolaminas promueve la cetogénesis y la liberación de glucosa hepática. Puede desarrollarse cetoacidosis diabética en los pacientes sin que exista aumento en el glucagon, hormona del crecimiento o catecolaminas, pero esta complicación se desarrolla en forma más lenta que en los enfermos con niveles elevados de hormonas contrarreguladoras.64 Debido a que la cetogénesis es más sensible que la liberación de la glucosa hepática a la supresión de insulina, los niveles bajos de insulina se asocian con hiperglucemia, pero no con cetosis.
La destrucción completa de las células beta productoras de insulina de los islotes de Langherhans predispone a los pacientes con DMID a un control lábil de la glucosa. En ausencia de cantidades significativas de insulina endógena, el metabolismo de la glucosa depende por completo de la insulina exógena. Puede predecirse que la destrucción progresiva de las células beta, que en la actualidad se sabe constituye la evolución natural de la DMID, ocasionará inestabilidad metabólica al avanzar la enfermedad. En las etapas iniciales de la DMID no siempre existe inestabilidad metabólica. La primera manifestación de este fenómeno en los pacientes con DMID de inicio reciente es el llamado periodo de "luna de miel". Durante este periodo la insulina puede disminuirse o incluso suspenderse durante semanas o meses. Al parecer, los pacientes en los que se desarrolla este fenómeno aún tienen una función de células beta suficiente para poder sobrevivir sin insulina exógena. Con frecuencia el inicio clínico de la DMID se asocia, o es precipitado, por un cuadro gripal u otra enfermedad estresante que aumenta los requerimientos de insulina. Puede entonces requerirse de insulina exógena a pesar de la función de los islotes residuales. Una vez que la enfermedad se resuelve y la función de los islotes residuales mejora, y debido a que la hiperglucemia fue tratada con insulina exógena, puede seguir un periodo temporal en el que la insulina endógena es suficiente para satisfacer las demandas. Sin embargo, con el tiempo la masa de células beta disminuye, lo que llega a ocasionar deficiencia total de insulina. Además del periodo de luna de miel, se conocen otros dos ejemplos clínicos de estabilidad metabólica en la DMID. Los pacientes en los que la DMID se desarrolla a una edad más avanzada (i.e., al final de la adolescencia o en la tercera década de la vida) tienen en forma característica una evolución temprana más variable, y con frecuencia puede controlarse la glucemia con esquemas de una sola inyección diaria de insulina. Durante este periodo de estabilidad existe insulina endógena susceptible de estimulación, aunque a bajos niveles. A diferencia de los pacientes con DMID de inicio temprano, los pacientes en los que la enfermedad comienza a edad más avanzada suelen ser fáciles de tratar durante varios años. También puede lograrse un control metabólico más estable en los pacientes con DMID que son tratados desde etapas tempranas de la evolución con esquemas intensivos de insulina que tienen por objeto mantener la glucemia dentro del rango normal. Se ha demostrado que este tipo de tratamiento conserva la secreción endógena.65 Aunque la función de la célula declina con el tiempo, lo hace en forma más lenta que en los pacientes tratados de modo convencional.
Después de un periodo inicial de estabilidad metabólica que suele ser corto, aunque varía, la mayoría de los pacientes con DMID desarrollan deficiencia total de insulina y una inestabilidad metabólica característica [ver figura 6]. Por lo general, el control de la glucosa es muy variable en la DMID por el desequilibrio que existe entre los requerimientos de insulina y el aporte de la misma en la mayoría de los esquemas de tratamiento convencionales. Incluso con los esquemas intensivos que se basan en mediciones frecuentes de la glucemia para determinar la dosis y momento de las inyecciones de insulina, el control de la glucosa no es normal. La labilidad en las concentraciones de glucosa puede explicase por diversas variables que influyen en el control de la misma en la diabetes, incluyendo el estado prandial, el ejercicio, el estrés y el aporte de insulina. A su vez, numerosas variables influyen en la disponibilidad de la insulina [ver tabla 3].

|
| Figura 6 |
| Resultados de diversos esquemas de administración de insulina |
|
|
|
La expresión más dramática de la inestabilidad metabólica en la DMID es la cetoacidosis. Esta puede ser precipitada por interrupción del tratamiento con insulina o por un estado de estrés, con aumento en las concentraciones de hormona contrarreguladoras y un tratamiento con insulina inadecuado. La deficiencia de insulina y el glucagon aumentan la producción de cetonas a partir de los ácidos grasos libres formados por lipólisis. Esta cetogénesis y la liberación no limitada de glucosa del hígado causan hiperglucemia, con deshidratación secundaria y cetoacidosis.
DMNID
Las consecuencias metabólicas de la deficiencia parcial de insulina (o resistencia a la insulina con cantidad inadecuada de ésta) pueden predecirse con facilidad. La hiperglucemia ocasiona deshidratación y un estado hiperosmolar. A diferencia de los pacientes con DMID, los enfermos con DMNID son resistentes a la cetoacidosis porque la concentración de insulina existente suele ser suficiente para evitar la cetogénesis. Sin embargo, en condiciones de estrés intenso (v.gr., choque séptico o infarto del miocardio), estos pacientes pueden desarrollar cetoacidosis. Con más frecuencia los pacientes ancianos con DMNID tienen riesgo de sufrir coma hiperglucémico hiperosmolar no cetósico.66 Esta urgencia metabólica se desarrolla en pacientes con DMNID que tienen cada vez más hiperglucemia por estrés, administración de esteroides o alimentación por sonda. Al aumentar la concentración de glucosa en estos pacientes, la falta de reposición de las pérdidas de agua libre por deterioro del mecanismo de la sed o del estado mental, o por falta de acceso a líquidos, causa mayor hiperglucemia, hiperosmolaridad y, al final, depresión del estado mental.
Por lo general los niveles de glucosa en la DMNID son mucho menos lábiles que en la DMID. La glucemia tiende a ser menor antes de las comidas y aumenta después de ellas [ver figura 6]. El aumento en la glucemia posprandial en los pacientes con intolerancia a la glucosa es causado por una menor captación de la misma en el músculo asociada a resistencia a la insulina. Con el desarrollo de la DMNID, el aumento en la liberación de glucosa hepática ocasiona hiperglucemia en ayuno y contribuye en forma significativa a la hiperglucemia posprandial.67 La concentración de glucosa en ayuno y las determinaciones de glucemia tienden a ser estables y reproducibles en el paciente con DMNID no sometido a estrés.68,69 Otras alteraciones metabólicas que se asocian con frecuencia a la DMNID incluyen aumento en la concentración de LMBD y triglicéridos, partículas de lipoproteína de baja densidad (LBD) densas y pequeñas, que son especialmente aterogénicas, y menor concentración de colesterol de lipoproteínas de alta densidad (LAD).70
Diagnóstico y escrutinio
Por lo general, la presentación clínica de la DMID no deja lugar a dudas. No se requieren pruebas diagnósticas especiales excepto la medición de la glucemia y, dependiendo del cuadro clínico, de cetonas y electrolitos en orina y suero. Aunque existen en el comercio pruebas de escrutinio que miden ACI, la sensibilidad de estas pruebas es menor al 75 porciento, incluso en circunstancias ideales.35 Además, es indispensable que las pruebas para una enfermedad con prevalencia baja sean muy específicas. Los estudios han sugerido que la medición combinada de anticuerpos ACI y de la respuesta de insulina ante una prueba de tolerancia a la glucosa por vía intravenosa aumenta la especificidad.37 Sin embargo, para que se justifique este tipo de programa de escrutinio debe mejorar el estado de salud al identificar a la enfermedad en su estado preclínico. Aunque se está investigando el uso de inmunoterapia profiláctica [ver adelante, Tratamientos novedosos y experimentales de la DMID], en la actualidad no se ha demostrado un beneficio claro, y los programas de escrutinio para la DMID no se recomiendan.
Comparando con los pacientes con DMID, los enfermos con DMNID presentan pocos o ningún síntoma, y el diagnóstico puede requerir la medición de la glucemia en ayuno o la realización de una prueba de tolerancia a la glucosa. En 1977, el Comité de Expertos de la Asociación Americana de Diabetes emitió nuevas normas para el diagnóstico del padecimiento. Estas se basaron en estudios epidemiológicos que correlacionaron los niveles de glucosa en plasma con la prevalencia de retinopatía diabética. El comité recomienda que el nivel de glucemia en ayuno usado como criterio para DMNID se reduzca de 140 mg/dl a mayor de 126 mg/dl.16 Además, el comité recomienda que se evite la prueba de tolerancia a la glucosa, excepto en casos en que se sospeche diabetes gestacional, y que los adultos mayores de 45 años se realicen una prueba de glucemia en ayuno por lo menos cada tres años para investigar diabetes [ver tabla 1]. Aunque hasta el 50 porciento de la población con DMNID no saben de su padecimiento, los beneficios del diagnóstico temprano no han sido comprobados, y es difícil justificar el escrutinio de grandes grupos de población por medio de una prueba de tolerancia a la glucosa.71
Tratamiento de la DMID
La mayoría del tratamiento de la diabetes mellitus depende del propio paciente. De todas las enfermedades crónicas, la diabetes resalta porque su manejo incluye una serie de adaptaciones de estilo de vida. Además de los medicamentos, la DMID requiere de autovigilancia y de atención a todos los aspectos de la vida diaria, desde la dieta hasta el ejercicio.
METAS DEL TRATAMIENTO PARA LA GLUCEMIA: DCCT
Los objetivos específicos del tratamiento incluyen mantener un crecimiento, desarrollo y peso corporal normales, evitar la hiperglucemia sintomática, la hipoglucemia clínica y la cetoacidosis, y detectar y tratar en forma pronta las complicaciones. Los objetivos del tratamiento para la glucemia fueron motivo de grandes controversias hasta que los resultados del DCCT demostraron que el tratamiento intensivo dirigido a mantener los niveles normales de glucosa tenía un efecto benéfico importante en el desarrollo y progresión de las complicaciones diabéticas a largo plazo.8
El debate respecto a la relación entre el control metabólico y la presencia de complicaciones a largo plazo en la diabetes mellitus comenzó en los primero 10 años después de la introducción del tratamiento con insulina.72-74 Durante las primeras décadas de uso de la insulina los clínicos comenzaron a observar complicaciones microvasculares y neurológicas.75,76 Estas complicaciones a largo plazo, incluyendo retinopatía, neuropatía y nefropatía, no se habían observado en la era previa a la insulina porque los pacientes con DMID no sobrevivían lo suficiente como para que ocurrieran. Aunque muchas teorías intentaron explicar la presencia de estas complicaciones, la hipótesis más popular sugirió como causa un control insuficiente de la glucosa. La llamada hipótesis de la glucosa fue apoyada por estudios realizados en modelos animales de diabetes y por estudios epidemiológicos.69,77,78 Aunque los partidarios de la hipótesis de la glucosa apoyaban la importancia del denominado control metabólico estricto, no existían datos clínicos definitivos que apoyaran la eficacia de ningún método especial de tratamiento para prevenir el desarrollo o enlentecer la progresión de las complicaciones. Además, no existió un consenso respecto a qué tipo de tratamiento establece y mantiene niveles normales o casi normales de glucemia durante el tiempo.
A finales de los años 70 se contó con los medios para lograr un control casi normal y a largo plazo de la glucosa, como la autovigilancia de la glucemia y los esquemas intensivos de insulina.79 Al mismo tiempo se desarrollaron métodos objetivos para medir la glucemia a largo plazo y evaluar las complicaciones.80 Estos avances hicieron posible diseñar y realizar experimentos clínicos para aclarar el debate.
Diseño del estudio
El DCCT, iniciado en 1983, se diseñó para establecer si el manejo intensivo de la diabetes con objeto de lograr niveles de glucemia lo más cercanos al rango no diabético afectaría el desarrollo (prevención primaria) o la progresión (prevención secundaria) de las complicaciones a largo plazo de la DMID.8
Se incluyeron dos grupos de pacientes en el estudio. Los pacientes en prevención pirmaria tenían de 13 a 39 años de edad, diabetes por uno a cinco años y no evidencia de retinopatía o nefropatía. Los pacientes del grupo de intervención secundaria tenían edades semejantes, pero DMID de hasta 15 años de evolución y por lo menos un microaneurisma y excreción de albúmina de menos de 200 mg/24 hr.
Tratamiento intensivo y objetivos metabólicos del estudio
Las dos cohortes del estudio se distribuyeron de modo aleatorio para recibir tratamiento convencional, que consistió en una o más inyecciones diarias de insulina y vigilancia diaria de la glucemia, o tratamiento intensivo. El objetivo clínico del tratamiento convencional consistió en prevenir cualquier síntoma de hiperglucemia o hipoglucemia, pero no se especificaron metas numéricas de glucemia. Los objetivos del tratamiento intensivo consistieron en controlar la glucemia lo más cerca posible al rango no diabético, incluyendo concentraciones preprandiales de glucemia de 70 a 120 mg/dl y concentraciones posprandiales máximas de menos de 180 mg/dl, para lograr niveles de hemoglobina A1c (HbA1c) dentro del rango no diabético (<6.05 porciento). Para lograr estas metas los pacientes que fueron asignados al tratamiento intensivo se autoadministraron tres o más inyecciones de insulina diario o usaron una bomba de insulina. El tratamiento fue guiado por la autovigilancia frecuente de la glucemia [ver tabla 4]. El tratamiento con insulina se ajustó en respuesta a los cambios en los niveles de glucemia, la dieta y el ejercicio.
|
||||||||||||||||||||||||||||
* Rango no diabético, 4.0-6.05%
|
Resultados del estudio
Durante el seguimiento promedio de 6.5 años (rango de tres a nueve años), el tratamiento intensivo produjo niveles promedio de HbA1c que fueron alrededor de dos porciento menores de los causados con el tratamiento convencional (siete y nueve porciento, respectivamente). Sin embargo, el tratamiento intensivo no redujo los niveles promedio de HbA1c a rangos no diabéticos.8 Comparado con el tratamiento convencional, el tratamiento intensivo redujo el riesgo de desarrollo y progresión de la retinopatía diabética, nefropatía y neuropatía en aproximadamente 40 a 76 porciento. Al parecer los efectos benéficos del tratamiento intensivo aumentaron con el tiempo.
Aunado a los efectos saludables del tratamiento intensivo sobre las complicaciones a largo plazo, existió un incremento cercano al triple en la incidencia de hipoglucemia severa, que se definió como cualquier episodio que requirió de ayuda para ser tratado. La incidencia de hipoglucemia que causó coma o crisis convulsivas o que requirió de tratamiento en un servicio de urgencias también aumentó al doble o triple. Las reacciones más severas, como las que causaron crisis convulsivas o coma, fueron relativamente raras (16 episodios por 100 paciente-años en el grupo de tratamiento intensivo y cinco episodios por 100 paciente-años en el grupo de tratamiento convencional). Otros efectos adversos que acompañaron al grupo en tratamiento intensivo incluyeron un mayor riesgo de aumento de peso y de infecciones del catéter en los pacientes que usaron una bomba de insulina. En conjunto, ninguno de estos efectos adversos causó morbilidad o mortalidad significativas. No ocurrieron eventos macrovasculares o muertes por hipoglucemia. Además, la mayor frecuencia de hipoglucemia con el tratamiento intensivo no tuvo efectos adversos sobre la función neurocognitiva medida por pruebas frecuentes. Por último, a pesar de las demanas del tratamiento intensivo, la calidad de vida, determinada por reportes de los mismos pacientes, no fue diferente entre los dos grupos de tratamiento.8
Recomendaciones de tratamiento
El grupo de investigación del DCCT, la Asociación Americana para la Diabetes y otros grupos han concluido que los efectos del tratamiento intensivo, importantes y constantes, para reducir el riesgo de complicaciones a largo plazo, superan el considerable esfuerzo y los costos asociados con este tratamiento, además del mayor riesgo de hipoglucemia. En la actualidad se considera que el tratamiento intensivo es el de elección para la mayoría de los pacientes con DMID. Siendo realistas, no todos los pacientes serán capaces de seguirlo; sin embargo, la meta para todos los enfermos debe ser reducir la glucemia lo más cercano al rango normal y con la mayor seguridad posible según las condiciones de vida. Para los pacientes que no pueden seguir el tratamiento intensivo, existen tratamientos de intensidad variable [ver tabla 4].
Durante más de una década el tratamiento intensivo ha sido obligatorio para las mujeres con DMID que desean concebir o están embarazadas, ya que este tratamiento ha demostrado que mejora la evolución neonatal.81-83 El manejo intensivo de la paciente embarazada con DMID es especialmente difícil por los cambiantes requerimientos de insulina durante este periodo, y solo debe intentarse por médicos con experiencia.
Aún quedan muchas preguntas y retos que aclarar después del DCCT. Primero, si los pacientes con DMID que no están dentro de un protocolo de investigación pueden seguir, y hasta qué grado, el tratamiento intensivo. Segundo, se requieren mucho más recursos para aplicar el tratamiento intensivo. Es necesario determinar si será posible obtener estos recursos fuera del campo de la investigación. Deben establecerse equipos de expertos para ayudar a implementar y manejar el tratamiento intensivo. Es necesario eliminar barreras económicas para hacer que el tratamiento sea lo más accesible posible. Tercero, de acuerdo con una interpretación estricta de los resultados del estudio, fue el tratamiento intensivo y no el alcance de la normoglucemia lo que proporcionó el beneficio al prevenir el desarrollo y retrasar la progresión de la diabetes mellitus. No es claro si todos los métodos de tratamiento diseñados para alcanzar la normoglucemia tienen efectos semejantes. Es probable que el logro de la normoglucemia haya sido la principal razón por la que el tratamiento intensivo fue eficaz, y es posible que métodos de tratamiento semejante, como el basado en un páncreas artificial (bombas implantables con sensores de glucosa internos), proporcione beneficios parecidos, cuando estén disponibles.
Por otro lado, el trasplante de páncreas, que es otro medio de alcanzar la normoglucemia, introduce variables como la inmunosupresión, que pueden afectar la evolución. Este tratamiento debe evaluarse para determinar su relación costo-beneficio. Por último, no se incluyó en el DCCT ningún pacientes con DMNID, la forma más frecuente de diabetes, por lo que no se sabe si los resultados del DCCT en enfermos con DMID pueden aplicarse a pacientes con DMNID.84 La semejanza entre las complicaciones de la DMID y la DMNID, así como la relación semejante entre la progresión de la retinopatía y los niveles de glucemia crónicos en ambos padecimientos sugieren que el tratamiento intensivo puede también ser eficaz en la DMNID. Sin embargo, no existen datos directos para apoyar esta hipótesis. Ningún estudio a largo plazo ha establecido los riesgos relativos de los diferentes tratamientos diseñados a mantener la normoglucemia en la DMNID. Las dosis altas de insulina que se requieren al final para tratar a muchos pacientes con DMNID pueden ser aterogénicas, una preocupación que ha reducido el entusiasmo por implementar en forma más amplia el tratamiento intensivo en pacientes con esta enfermedad.
AUTOVIGILANCIA
Durante la década pasada aumento el interés de una vigilancia más exacta del estado metabólico por el propio paciente, y se han desarrollado más equipos para ayudarles a realizar esta vigilancia. Los pacientes deben determinar ellos mismos sus concentraciones de glucosa en sangre por los siguientes motivos:
1. La hipoglucemia es el principal efecto colateral del tratamiento con insulina, y se requiere una vigilancia oportuna para detectar, prevenir y tratar esta complicación.
2. El desarrollo de los nuevos esquemas de administración de insulina, que semejan en forma más estrecha a la fisiología normal, depende de la retroalimentación frecuente con los niveles de glucemia, lo que solo se logra solo por medio de la autovigilancia.
3. Por último, los resultados de la autovigilancia proporcionan información que ayuda a educar al paciente diabético sobre su enfermedad y sobre el efecto que tienen el ejercicio, las variaciones en la dieta, la enfermedad y otros factores, en sus síntomas y en el control de la glucosa.
Los perfiles de glucemia en los pacientes con DMID son muy lábiles, y las pruebas de glucosa en orina no proporcionan información exacta sobre las concentraciones en sangre.85 Además, las consecuencias de la hipoglucemia pueden ser graves. Por lo tanto, es evidente la necesidad de contar con un método para medir con frecuencia la glucemia en el hogar, en la escuela o en el trabajo. Se han desarrollado tiras reactivas y sistemas de medición de gran precisión y exactitud.86
La frecuencia de la vigilancia en la DMID depende del riesgo de hipoglucemia y de los objetivos del tratamiento en cada individuo. Como mínimo, los pacientes deben ser capaces de medir su glucemia cuando cambian la dosis de insulina, dieta o actividad, o cuando la hipoglucemia pudiera ser especialmente peligrosa (i.e., antes de un viaje en automóvil). Además, la autovigilancia durante las enfermedades puede ayudar a los pacientes a ajustar la dosis de insulina. Mientras más complejo sea el esquema de tratamiento, los pacientes deberán checar su nivel de glucosa con más frecuencia, dependiendo del esquema específico y del horario de alimentos y actividades diarios. Por lo general la medición se realiza en ayuno y antes de la cena o de acostarse. Los esquemas intensivos requieren, como mínimo, una medición antes de las comidas y antes de acostarse, y las dosis de insulina se ajustan según los resultados.
Cuando concentraciones demasiado altas de glucosa indican el riesgo de cetoacidosis secundaria a una enfermedad agregada, a la administración inadecuada de insulina o a ambas, deberá investigarse la presencia de cetonas en la orina del paciente. Los pacientes deben ser instruidos para que consulten a su médico si encuentran más que trazas de cetonas en orina. También deben medirse las cetonas si el paciente tiene náusea o vómito, porque estos síntomas pueden indicar cetoacidosis.
VIGILANCIA POR PARTE DEL MEDICO
Es evidente la utilidad de medir la concentración de proteínas glucosiladas (que son el producto de la glucosa y de los grupos amino circulantes y proteínas de la membrana) como un índice de los niveles crónicos de glucosa.80 La concentración de una proteína glucosilada revela la concentración promedio de glucosa en sangre durante la mitad de la vida media de la proteína [ver figura 7]. Por lo tanto, la concentración de hemoglobina glucosilada (HbA1 o HbA1c, dependiendo del método específico que se use) indica el promedio de la glucemia durante alrededor de 60 días, porque la vida media promedio del eritrocito es de 120 días. La concentración de albúmina glucosilada o de otra proteína sérica (v.gr., fructosamina) indica la glucemia promedio en un periodo de 20 días. Estas mediciones proporcionan una manera objetiva de evaluar la glucemia a largo plazo y evaluar la eficacia de las intervenciones terapéuticas. En por lo menos un estudio, los pacientes en los que se midió la HbA1c tuvieron un mejor estado general, con menos hiperglucemia y menos hospitalizaciones que un grupo seleccionado en forma aleatoria en el que ni los pacientes ni el médico conocieron el valor de la HbA1c.87 Para el tratamiento clínico de la DMID es suficiente medir la HbA1c tres o cuatro veces al año. Las mediciones que reflejan la glucemia promedio en periodos más cortos, como la medición de fructosamina, deben repetirse con más frecuencia, aunque su papel en el tratamiento de la diabetes no está bien establecido.
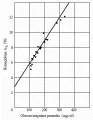
|
| Figura 7 |
| Relación entre hemoglobina glucosilada y glucemia |
INSULINA
La insulina está disponible en varias especies, fórmulas y grados de purificación. Por lo general, todos los tipos de insulina que existen en los Estados Unidos son muy purificadas y contienen menos de 50 ppm de proinsulina. Las insulinas porcina y bovina difieren de la insulina humana por uno y tres aminoácidos, respectivamente. A pesar de las diferencias en la secuencia de aminoácidos y de los anticuerpos contra insulina generados por la insulina bovina, porcina o por combinaciones de ambas, éstas tienen una potencia semejante a la de la insulina humana y muy pocas veces causan problemas de tipo inmunológico. En los raros casos de resistencia a la insulina mediada por anticuerpos o de alergia local o sistémica a la insulina, es eficaz la desensibilización humana o el cambio a una insulina porcina muy purificada o a insulina humana. La insulina porcina muy purificada contiene menos de10 ppm de proinsulina, y la insulina humana solo es necesaria en los pacientes que tienen complicaciones inmunológicas después de recibir insulinas de origen animal menos purificadas y por ello más antigénicas. Además, los pacientes que usan insulina en forma intermitente (i.e., las pacientes con diabetes gestacional) y que pueden tener mayor riesgo de complicaciones inmunológicas, deben ser tratados con insulina porcina purificada o insulina humana. La insulina humana se sintetiza por la modificación de la insulina porcina o por métodos recombinantes en Escherichia coli o levaduras. Aunque muy pocos pacientes diabéticos tienen indicaciones absolutas para el uso de insulina humana (o una contraindicación absoluta para el uso de insulinas de origen animal), los fabricantes de insulina desean modificar la producción de insulina animal a humana por razones económicas
Las fórmulas de insulina pueden clasificarse como de acción rápida (i.e., insulina regular o zinc cristalina [IZC]), de acción intermedia (i.e., insulina lenta o protamina neutral Hagerdon [NPH]) o de acción prolongada (i.e., insulina Ultralenta). Una nueva insulina humana recombinante de acción rápida (lispro o Humalog), creada por inversión de los aminoácidos 28 y 29 de la cadena B, no se polimeriza tan rápido como la IZC. En lugar de hexámeros, la mayoría de la insulina se encuentra en forma monomérica y se absorbe y elimina con mayor rapidez.4 Cada fórmula tiene diferente perfil de acción [ver tabla 5]. Sin embargo, la variabilidad en la absorción de la insulina entre las diferentes personas, e incluso en un mismo individuo, hace que estos perfiles no sean exactos. Además, por lo general las insulinas humanas tienen un inicio de acción más rápido y una duración más corta que los de las insulinas animales correspondientes. Existen en el mercado varias combinaciones fijas (v.gr., 70 porciento de NPH y 30 porciento de insulina regular). Estas son útiles en muy pocas ocasiones en la DMID porque las fórmulas con relación fija no son eficaces para los requerimientos tan cambiantes de los pacientes; sin embargo, sí pueden ser apropiadas en la DMNID. Las fórmulas pueden combinarse en un esquema diario para proporcionar un aporte de insulina que se parezca a la fisiología normal [ver figura 8]. Sin embargo, los esquemas más complejos requieren inyecciones y pruebas de glucosa más frecuentes para determinar la dosis a administrar. Los pacientes tratados con esquemas intensivos deben recibir un algoritmo individual para ayudarlos a seleccionar la dosis correcta de insulina regular para una determinada glucemia, comida y rutina de ejercicio. Debido a que el inicio de la acción de la insulina regular subcutánea es tardío, los pacientes deben administrarse la insulina regular preprandial entre 30 y 60 minutos antes de los alimentos. En este sentido, la insulina lispro es más adecuada porque puede administrarse 10 a 15 minutos antes de cada comida.

|
| Figura 8 |
| Esquemas de administración de insulina |
|
|||||||||||||||||||||||||
* Todas disponibles en fórmulas bovinas, porcina, mixta y humana en los Estados Unidos, excepto por la lispro y Ultralenta, que están disponibles solo como insulina humana (recombinante). Las fórmulas humanas, en especial la NPH y la Ultralenta, tienen un inicio de acción más rápido y duración más corta que las insulinas de origen animal correspondientes. |
Se han desarrollado equipos de administración de insulina para facilitar la administración frecuente de insulina regular en los tratamientos intensivos. Las jeringas mecánicas, los inyectores con propulsión de aire, las bombas externas con baterías y las bombas implantables son todos eficaces para estos esquemas, y permiten lograr una glucemia casi normal.8,10,88 Pueden obtenerse resultados metabólicos semejantes con tres o más inyecciones diarias.89,90 El principal riesgo de todos los esquemas intensivos es la frecuencia del doble o triple en los episodios de hipoglucemia grave.91 (Un estudio que usó bombas implantables que administraban insulina por vía intravenosa o intraperitoneal sugirió que en estos esquemas la hipoglucemia puede no ser un problema tan frecuente como en los esquemas en que se administra por vía subcutánea.92) Además, los equipos que usan catéteres subcutáneos pueden ocasionar inflamación o infecciones en el sitio de entrada del catéter.
DIETA
El tratamiento dietético es un componente crucial en la atención del diabético. Como en el caso de la vigilancia y del tratamiento con insulina, el grado de intensidad del tratamiento debe basarse en el estilo de vida del paciente, su motivación y los objetivos globales de la atención de la diabetes [ver tabla 4]. El enfoque moderno para el tratamiento dietético de la DMID enfatiza el adaptar la insulina a la dieta y no al revés. Para facilitar el equilibrio entre las dosis de insulina y las comidas y para evitar la hipoglucemia, el paciente con DMID debe comer alimentos semejantes en forma regular. Se recomienda una dieta alta en carbohidratos (45 a 50 porciento de las calorías) baja en grasas (< 30 porciento de las calorías) y baja en colesterol (< 300 mg).93 Enfoques más sofisticados incluyen listas de intercambio para ayudar al paciente a mantener un contenido de carbohidratos semejantes en todas las comidas. Debido a que los carbohidratos ingeridos se digieren a velocidades diferentes y tienen efectos distintos sobre los niveles de glucemia, se han desarrollado índices glucémicos para usarse en los esquemas de tratamiento más estrictos. Estos índices señalan los efectos que los diferentes alimentos que contienen carbohidratos ejercen sobre el nivel de glucosa.94 Debido a la importancia de la dieta en el tratamiento de la DMID, debe existir un dietista dentro del equipo de atención para la salud.
EJERCICIO
Otro componente del cuidado en la diabetes es el ejercicio, importante para conservar la condición cardiovascular y la sensibilidad a la insulina. Sin embargo, los pacientes deben ser instruidos a ajustar sus alimentos, su ingesta de insulina o ambos, inmediatamente después, o incluso seis a 12 horas después de realizar ejercicio [ver adelante, Hipoglucemia]. Los deportes de alto impacto pueden ser muy peligrosos para los pacientes con retinopatía avanzada o con neuropatía periférica, que tienen mayor riesgo de traumatismos en los pies.
CETOACIDOSIS DIABETICA
La cetoacidosis diabética (CAD) es poco frecuente, con 10 a 15 casos por cada 1,000 pacientes-años. La mayoría de los casos ocurren después de que se ha diagnosticado la diabetes, pocas veces la cetoacidosis es la primera manifestación de la enfermedad.95 Con la disponibilidad de la autovigilancia de la glucemia y de las cetonas en orina, la mayoría de los pacientes son capaces de detectar y detener la evolución de la CAD; el tratamiento pronto y agresivo con insulina extra y líquidos puede lograrlo. Desde cierto punto de vista, esta complicación representa casi siempre deficiencias en la educación del paciente o incapacidad del mismo para cumplir con un esquema terapéutico determinado.
Aunque la mortalidad asociada con la CAD debe ser de cero, un estudio realizado en Rhode Island demostró una mortalidad de casi el 10 porciento.95 A pesar de esta estadística, la CAD debe considerarse una urgencia metabólica susceptible de tratamiento y no fatal, y debe realizarse una investigación clínica cuidadosa en cualquier caso que termine en defunción. Los fallecimientos casi siempre se presentan cuando la CAD es la primera manifestación de la DMID en niños y no se diagnostica a tiempo, o cuando es precipitada por una enfermedad grave, como sepsis o infarto del miocardio en adultos con DMID. En los niños puede ocurrir edema cerebral fatal, aunque no es frecuente , y éste suele desarrollarse seis a 12 horas después de iniciado el tratamiento de la CAD. El cuadro clínico incluye signos y síntomas de deshidratación (secundaria a diuresis osmótica y vómito), respiración de Kussmaul (para aumentar la espiración de ácidos volátiles), náusea y vómito (quizá debidas a la alteración en el metabolismo de las prostaglandinas y por gastroparesia y reducción de la motilidad intestinal), y depresión del estado de conciencia [ver tabla 6]. La glucemia es alta, casi siempre entre 350 y 700 mg/dl, y es típico que exista una brecha aniónica aumentada, acompañada por elevación en la concentración de cuerpos cetónicos (acetoacetato, beta-hdroxibutirato y acetona) en orina y suero. Aunque el potasio y el fosfato pueden ser normales o incluso altos al principio, la hidratación y la corrección de la acidosis disminuyen en forma invariable su concentración sérica, desenmascarando una depleción corporal de potasio importante. Otros factores puede alterar los valores de laboratorio habituales. El tratamiento con insulina administrado antes por el paciente, la inanición profunda o el embarazo, pueden ocasionar la llamada CAD euglucémica, en la que la concentración de glucosa es menor de 250 mg/dl.
|
||||
* La creatinina aumenta por la deshidratación y porque
las cetonas interfieren con el estudio habitual de la creatinina.
|
El tratamiento fundamental incluye remplazo de la pérdida de volumen y de electrolitos (en especial de potasio), administración de insulina, y diagnóstico y tratamiento del padecimiento médico asociado [ver tabla 7]. Desde su introducción en 1974, el tratamiento con insulina intravenosa denominado de microdosis, ha sido el más usado para la CAD.96 Aunque no es obligatorio que el tratamiento se administre en una unidad de cuidados intensivos, sí debe contarse con facilidades para administrar la insulina y medir en forma frecuente los signos vitales, la glucosa y los electrolitos. Durante la obnulación o el estado de coma la vía aérea del paciente debe protegerse para prevenir la aspiración. Se requiere una línea intravenosa de gran calibre para administrar los líquidos intravenosos. Un paciente de 70 kg con CAD tiene una deficiencia de volumen promedio de 5 a 8 L, una deficiencia aproximada de cloruro de sodio de 350 a 600 mEq, y una deficiencia de potasio entre 200 y 400 mEq. Por lo tanto, es indispensable corregir estas deficiencias por vía intravenosa. Al corregir la brecha aniónica, con frecuencia se agrega una acidosis metabólica hiperclorémica.97
|
||||||||||||||||||||||
|
Aunque la CAD puede tratarse en forma satisfactoria con insulina administrada por vía intramuscular o incluso subcutánea, la administración intravenosa es mucho más predecible y causa menos hipoglucemia o hipocalemia. El remplazo de bicarbonato y de fosfato continúan siendo tema de controversia. No existen estudios que apoyen el uso de bicarbonato,98 aunque la mayoría de los textos continúan aconsejando administrarlo si el pH arterial es menor de 7.05. La depleción grave de fosfato (fosfato < 2.0 mEq/L) puede causar rabdomiolisis y alteraciones diafragmáticas. También ocasiona depleción del 2,3-difosfoglicerato, con disociación anormal de la hemoglobina y el oxígeno. Aunque la administración de fosfato puede revertir todas estas alteraciones, la importancia del tratatamiento de remplazo con fosfato es dudosa porque estudios clínicos controlados no han demostrado que esta medida mejore la evolución de los pacientes con CAD.99
HIPOGLUCEMIA
La hipoglucemia es más frecuente que la cetoacidosis diabética y puede ser igualmente peligrosa. La hipoglucemia clínica puede variar desde síntomas molestos que se asocian con concentraciones bajas de glucosa (< 55 mg/dl) hasta confusión, crisis convulsivas o pérdida de la conciencia (durante la hipoglucemia grave). Los síntomas que con más frecuencia caracterizan a la hipoglucemia no grave son de tipo adrenérgico e incluyen temblor, diaforesis y taquicardia. Es obvio que la hipoglucemia grave puede tener consecuencias desastrosas, dependiendo de las circunstancias en que ocurra.
El glucagon y la epinefrina son las principales hormonas contrarreguladoras que se secretan en respuesta a la hipoglucemia y que restablecen los niveles de glucosa al estimular la salida de glucosa hepática cuando disminuyen los niveles de insulina.100 Además, la secreción de catecolaminas, que suele acompañar a la hipoglucemia, puede alertar al paciente con síntomas simpaticoadrenales a tratar el episodio. Aunque menos importantes, el cortisol, la hormona del crecimiento y la tiroxina también participan en el mantenimiento de la concentración de glucosa. Los pacientes suelen reconocer con rapidez la hipoglucemia y pueden tratarse en forma eficaz usando un carbohidrato de absorción rápida. Alrededor de 15 g de carbohidrato son suficientes para normalizar la glucemia [ver tabla 8]. El uso de carbohidratos complejos y de alimentos con alto contenido de grasas, como el chocolate, puede retrasar la digestión y la absorción de azúcares, y no es adecuado para el tratamiento de la hipoglucemia. Si un paciente no puede deglutir o cooperar, deberá administrarse dextrosa intravenosa o glucagon intramuscular o subcutáneo. La administración de 25 ml (i.e., media ampolleta) de dextrosa al 50 porciento (12.5 g) como un bolo I.V. casi siempre es suficiente para revertir la hipoglucemia clínca. Los infantes y niños pueden requerir incluso una dosis menor. El equipo de inyección de glucagon contiene 1 mg de este medicamento, que puede aumentar la glucemia en 15 a 30 minutos. El glucagon puede causar náusea, vómito y cefalea, en especial en los niños.
|
||||
* Todos proporcionan alrededor de 15 g de carbohidratos
|
Por lo general, los pacientes con hipoglucemia grave responden con rapidez al tratamiento, aunque los enfermos en estado postictal o en coma prolongado pueden requerir varios días para recuperar su función mental y cognoscitiva normal. Las deficiencias neurológicas permanentes después de un episodio de hipoglucemia son muy poco frecuentes. En vista de las consecuencias potenciales de la hipoglucemia prolongada, ésta debe tratarse de inmediato y, ante la duda diagnóstica, iniciar tratamiento. Los pacientes deben ser enseñados a tratarse ellos mismos cuando sospechen que tienen hipoglucemia y no puedan confirmarlo por medio de una medición de glucosa en sangre.
Uno de los propósitos de la autodeterminación de la glucemia es que los pacientes puedan detectar y tratar los episodios de hipoglucemia, aunque no se ha demostrado que esta automedición sea eficaz para disminuir la frecuencia de los episodios. Sin embargo, la opinión general es que los pacientes con riesgo de hipoglucemia deben vigilar sus niveles de glucosa para ajustar mejor sus esquemas y evitar esta complicación.101
Varios factores contribuyen al desarrollo de hipoglucemia grave [ver tabla 9]. Los episodios de hipoglucemia que ocurren durante periodos de estrés, ejercicio o sueño, o en el caso de uso de alcohol o drogas ilícitas, pueden no causar síntomas característicos. Por lo tanto, en ocasiones no se administra tratamiento, lo que empeora la hipoglucemia. Algunos factores de riesgo, como el uso de alcohol, pueden tener efectos múltiples que precipitan, prolongan o empeoran la gravedad de la hipoglucemia. Por ejemplo, el alcohol altera el juicio, inhibe la liberación de glucosa del hígado (retrasando la recuperación) y puede aumentar la secreción de insulina. Los pacientes que reconocen la hipoglucemia incipiente pero que no responden en forma adecuada (v.gr., esperando para comer en un restaurante o continuando la conducción de su automóvil a pasar de los síntomas) pueden tener también mayor riesgo de hipoglucemia grave. Por último, debido a que el glucagon y la epinefrina son las principales defensas contra la hipoglucemia prolongada, su ausencia facilita que los episodios de hipoglucemia sean más duraderos y severos por dos mecanismos: (1) sin glucagon ni epinefrina disminuye la liberación de glucosa hepática ante la hipoglucemia, y (2) en ausencia de epinefrina los síntomas hipoglucémicos se modifican, lo que dificulta su detección y el tratamiento del episodio. Con frecuencia la respuesta del glucagón ante la hipoglucemia disminuye después de cinco años de evolución de la DMID.100,102 En ausencia de la respuesta de glucagon, la secreción de epinefrina permite una contrarregulación adecuada; sin embargo, ésta puede perderse también, en ocasiones por asociación con otras neuropatías autonómicas. Por lo general, muchos pacientes pierden su capacidad de contrarregulación después de 10 años de evolución de la DMID. Aún más, se ha observado un menor umbral de glucosa para la liberación de glucagon y epinefrina en respuesta a la hipoglucemia en los pacientes que reciben tratamiento intensivo con insulina.103 Mientras menor sea el nivel de glucosa necesario para estimular la concentrarregulación, se angosta el margen de seguridad del tratamiento. Por ejemplo, si los primeros síntomas de hipoglucemia ocurren cuando la glucemia es tan baja como 35 mg/dl, y el síntoma consiste en confusión, puede interferir con el autotratamiento. La predilección de la hipoglucemia en estos casos puede ser secundaria a hipoglucemia previa, un periodo de hipoglucemia sin tratamiento puede restablecer la contrarregulación.104
|
|
|
TRATAMIENTOS INNOVADORES Y EXPERIMENTALES DE LA DMID
Al evolucionar el conocimiento sobre la fisiopatología de la DMID también se han modificado los tratamientos experimentales para esta enfermedad. Aunque no se ha desarrollado un páncreas artificial verdadero por la falta de un sensor confiable de glucosa que ajuste en forma automática la liberación de insulina, se están investigando varias bombas implantables.10 Estos equipos se basan en la actualidad en la automedición del nivel de glucosa para elegir las dosis de insulina.
El trasplante total de páncreas tiene en la actualidad mucho mayor éxito para establecer la normoglucemia que en el pasado, pero requiere de cirugía mayor y de inmunosupresión.6 Por este motivo la mayoría de los programas de trasplante reservan este procedimiento para pacientes con DMID que serán sometidos a trasplante concomitante de riñón y que, por lo tanto, serán operados e inmunosuprimidos. Aunque es más probable que las complicaciones de la diabetes puedan evitarse en los pacientes jóvenes, el trasplante total de páncreas no es adecuado para ellos por los riesgos asociados de la cirugía mayor y de la inmunosupresión a largo plazo.
Debido a las limitaciones del trasplante total del órgano, los investigadores han intentado el trasplante de los islotes, que no requiere de una cirugía mayor. Además, la capacidad para inmunomodular los islotes aislados (enmascarando o retirando los antígenos de la superficie celular) puede permitir el trasplante con muy poca o ninguna inmunosupresión.105 En forma alternativa, los islotes pueden colocarse en tubos semipermeables que permiten a la glucosa e insulina entrar y salir, pero que aíslan a los islotes de los linfocitos y macrófagos.7 Los trasplantes de islotes han tenido éxito en modelos animales y en número muy limitado de humanos.105,106
Por último, debido a que en la actualidad se comprende mejor la inmunopatogenia de la DMID y porque ésta puede detectarse en estados preclínicos, en teoría es posible interrumpir su desarrollo por medio de inmunoterapia. Varios estudios han usado con éxito fármacos inmunosupresores, incluyendo azatioprina, prednisona o ciclosporina, para prevenir la destrucción progresiva de los islotes en pacientes con DMID que se identificaron en etapas clínicas tempranas. Por desgracia, la mayoría de los pacientes han perdido ya la función de los islotes en el momento del diagnóstico, y la suspensión de la inmunoterapia puede causar deterioro en un periodo corto de tiempo.107 Además, la toxicidad de los medicamentos hace que sea problemático mantener el tratamiento a largo plazo. Sin embargo, la identificación de la DMID preclínica y el desarrollo de estrategias de inmunoterapia más específicas y menos tóxicas hace que la prevención de la DMID sea una posibilidad. Un estudio multicéntrico extenso está examinando si el tratamiento con insulina, administrado por vía subcutánea, intravenosa u oral, retrasa el desarrollo de la DMID en personas identificadas en fase preclínica.
Tratamiento de la DMNID
Los objetivos clínicos del tratamiento de la DMNID son semejantes a los de la DMID. Al igual que en la DMID, deben balancearse los costos y riesgos del tratamiento y los beneficios a largo plazo en la DMNID. Por desgracia, los beneficios a largo plazo y riesgo del control estricto en la DMNID no han sido tan bien estudiados como en la DMID. Un estudio semejante al DCCT relativamente pequeño en pacientes con DMNID ha demostrado que el tratamiento intensivo es benéfico para las complicaciones a largo plazo.108 El estudio se realizó en pacientes japoneses muy delgados y que requerían dosis muy pequeñas de insulina, lo que hace difícil extrapolar los resultados a la población no japonesa con DMNID. Aunque es probable que los beneficios del tratamiento intensivo en la DMNID sean parecidos a los de la DMID, los costos específicos y riesgos para la DMNID ciertamente son diferentes. Por ejemplo, muchos pacientes con DMNID pueden tratarse con dieta, un tratamiento inocuo con una buena relación costo-eficacia y que cuando es exitoso disminuye muchos riesgos cardiovasculares, además de mejorar la tolerancia a la glucosa. Siempre debe intentarse lograr la euglucemia con un bajo costo y riesgo. Cuando se requieren tratamientos más complejos para lograr la normoglucemia en la DMNID la relación costo-beneficio debe analizarse con más cuidado y, como en el caso de la DMID, los intentos por lograr el control metabólico dependerán de factores individuales del paciente. Aunque por lo general puede lograrse la normoglucemia en la DMNID con esquemas menos complejos y con menos riesgo de hipoglucemia que en la DMID, la mayoría de los pacientes con DMNID que no son tratados en forma intensiva tienen riesgo claro de complicaciones a largo plazo.
DIETA
La dieta constituye el principal tratamiento en la DMNID. Ningún tratamiento farmacológico puede igualar la relación riesgo-beneficio del tratamiento dietético. La disminución del peso es el principal objetivo de la dieta en más del 85 porciento de los pacientes con DMNID que son obesos. Debido al mayor riesgo de enfermedad vascular coronaria en la DMNID, es importante que la dieta contenga una cantidad prudente de lípidos y poco colesterol. Las dietas hipocalóricas deben estructurarse en tres comidas al día y ser realistas. Con frecuencia no es posible alcanzar el peso corporal ideal, pero por fortuna la pérdida moderada de peso (v.gr., 5 a 10 kg) puede amilorar o curar la diabetes.109 Aunque las muchas dietas que contienen suplementos líquidos, extractos de toronja, fórmulas especiales de vitamina y patrocinadores famosos pueden ser atractivas para el paciente, no existe evidencia de que estos métodos costosos sean más útiles que las dietas que consisten en pequeñas porciones balanceadas de alimento. De hecho, aunque cualquier dieta hipocalórica puede reducir el peso corporal, la transición de suplementos y fórmulas a alimentos normales casi siempre ocasiona aumento en el peso. Los suplementos con pocas proteínas, con periodos de ayuno, en especial los que usan suplementos proteicos líquidos de poca calidad (casi siempre extraídos de productos de caballo) pueden ocasionar arritmias y muerte súbita. Las dietas con muy pocas calorías (< 800 kcal/día) causan pérdida de peso en los muy obesos, pero pueden acompañarse de complicaciones significativas, por lo que deben ser vigiladas por un médico capacitado. Los procedimientos quirúrgicos, como la plicación gástrica con derivación del intestino delgado, puede ser muy eficaz, pero pueden acompañarse de morbimortalidad y por lo tanto se reservan para pacientes con obesidad severa.110
Entre los beneficios esperados de la pérdida de peso se incluyen la menor hiperglucemia (o normoglucemia), la disminución en la presión arterial y en la concentración de lípidos, y la reducción del riesgo cardiovascular y de enfermedades articulares. Estos beneficios se logran casi sin ningún costo ni riesgo para el paciente. Por supuesto, el problema es que los beneficios no se observan con mucha frecuencia porque la gran mayoría de los pacientes obesos que realizan una dieta con éxito inicial aumentan de nuevo el peso perdido en uno a dos años.111 Cada intento sucesivo por perder peso se asocia con mayor dificultad. Aunque los pacientes diabéticos tienen más ventajas al perder peso que las personas no diabéticas, parece costarles más trabajo reducir su peso corporal.112
VIGILANCIA
La vigilancia de los pacientes con DMNID es menos demandante que la de los pacientes con DMID. Los perfiles de glucemia de los pacientes con DMNID son relativamente estables y el riesgo de hipoglucemia es bajo porque por lo menos una tercera parte de los enfermos son tratados solo con dieta. Además, comparado con los pacientes con DMID, los enfermos con DMNID tratados con sulfonilureas y con insulina tienden a tener menos riesgo de episodios de hipoglucemia. Por último, el riesgo de cetoacidosis es muy bajo, y no se requieren pruebas de cetonas en orina. Los pacientes con DMNID tratados con insulina y los tratados con sulfonilureas que tienen riesgo de hipoglucemia deben vigilar su glucemia en sangre, en especial cuando se inicia el tratamiento farmacológico o se ajustan las dosis. Después de que se determina la dosis adecuada, la estabilidad de la glucemia en sangre hace que no sean necesarias las pruebas de glucemia frecuentes. Algunos pacientes con DMNID son tratados con esquemas de insulina complejos (v.gr., dos inyecciones mixtas o separadas) y requieren de una autovigilancia más frecuente [ver tabla 4]. Por el contrario, los pacientes con DMNID tratados sólo con dieta no necesitan vigilar su glucemia en forma rutinaria.113
En la DMNID las correlaciones entre la glucemia en ayuno en varios días, entre la glucemia en ayuno y la HbA1c, y entre los niveles de HbA1c en diferentes periodos, son relativamente altas (r > 0.80).68,69 Esta estabilidad glucémica hace más fácil la vigilancia en los pacientes con DMNID que en los que sufren DMID.114Las mediciones de laboratorio de la glucosa o de la HbA1c, tomadas cada tres a seis meses, suelen proporcionar un índice exacto de la glucemia crónica en los pacientes con DMNID estable. Si se presenta algún cambio en la dieta, ejercicio, esquema de medicamentos o estado de salud, se requerirá una vigilancia más estrecha.
MEDICAMENTOS HIPOGLUCEMIANTES
Los medicamentos hipoglucemiantes son necesarios en los pacientes con DMNID en quienes falla el tratamiento dietético. La falla de la dieta casi siempre significa que el paciente no es capaz de alcanzar o mantener una pérdida de peso adecuada para amilorar la diabetes. Antes de comenzar el tratamiento farmacológico deben compararse los riesgos y costos de los medicamentos con los beneficios que se esperan. No existen datos que confirmen que la reducción de los niveles de glucosa puede prevenir o disminuir las complicaciones a largo plazo. Sin embargo, ciertos criterios, como los síntomas de hiperglucemia no controlada por dieta o la hiperglucemia tan grave para causar deshidratación, justifican el uso de tratamiento farmacológico [ver tabla 10]. Todos los tratamientos farmacológicos son más efectivos si se asocian con atención continua a la dieta.
|
|
|
Una vez que el médico analiza la necesidad de los medicamentos con el paciente, debe elegir el fármaco indicado, ya que cada uno tiene sus ventajas y desventajas [ver tabla 11].
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
* Ver tabla 12 para los rasgos de dosis específicos para las sulfonilureas de primera y segunda generación.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Las sulfonilureas son medicamentos que se administran por vía oral y que actúan principalmente al estimular la secreción endógena de insulina.115 Sus efectos hipoglucemiantes se observaron por primera vez como un efecto colateral al investigar una sulfonamida. Aunque las sulfonilureas disminuyen la resistencia a la insulina, este efecto parece participar muy poco, si es que algo, en la reducción de los niveles de glucemia. En promedio, las sulfonilureas disminuyen los niveles de HbA1c en uno a dos porciento. Las sulfonilureas no tienen efecto ni función en el tratamiento de la DMID, y no suelen ser eficaces en pacientes con DMNID no obesos. En los Estados Unidos existen varias sulfonilureas disponibles [ver tabla 12]. Aunque más potentes al compararlos en miligramos, los agentes de segunda generación no son más eficaces que los fármacos de primera generación en sus dosis máximas. Hasta el 20 porciento de los pacientes con DMNID que comienzan el tratamiento con sulfonilureas no logran una respuesta hipoglucémica adecuada a dosis máximas (a estos pacientes se les considera fracasos primarios), y cada año, el 10 a 15 porciento adicional de pacientes que inicialmente respondieron a estos medicamentos, fallan (fracasos secundarios). Muchas de las interacciones farmacológicas y los efectos adversos de las sulfonilureas de primera generación no ocurren o se reducen con los agentes de segunda generación. Aunque la hipoglucemia secundaria a sulfonilureas no es frecuente, suele ser más prolongada y peligrosa que la secundaria a insulina. Se ha observado mayor frecuencia de hipoglucemia en pacientes que recibieron dosis iniciales de sulfonilureas de segunda generación, y debe tenerse cuidado para indicar dosis bajas al inicio, que pueden aumentarse según se requiera. El tratamiento con sulfonilureas suele asociarse con incremento de peso. Por último, el Programa Multicéntrico del Grupo Universitario de Diabetes ha demostrado mayor riesgo de mortalidad cardiovascular asociada con la tolbutamida,116 un efecto que puede ser común para todos estos medicamentos.
|
|||||||||||||||||||||||||||||||||||
* Si la dosis máxima de un agente oral es ineficaz, es poco probable que el cambio a otra sulfonilurea dé mejor resultado. |
|||||||||||||||||||||||||||||||||||
La biguanida metformin es una adición relativamente nueva a los medicamentos disponibles en los Estados Unidos para el tratamiento de la DMNID, aunque se ha usado en Canadá y Europa por más de 20 años.117 El metformin tiene un mecanismo de acción diferente al de las sulfonilureas, disminuyendo principalmente la salida de glucosa del hígado. A diferencia de las sulfonilureas, el metformin es eficaz en pacientes tanto obesos como no obesos y no se asocia con hipoglucemia o aumento de peso. Su potencia es semejante a la de las sulfonilureas, disminuyendo la HbA1c glucosilada en uno a dos porciento. Los efectos adversos más comunes del metformin son gastrointestinales, causa distensión abdominal, molestia o incluso diarrea en alrededor del 10 porciento de los pacientes. Este efecto adverso puede disminuirse aumentando la dosis en forma lenta. La acidosis láctica, un efecto potencialmente fatal que ocurrió con más frecuencia con la biguanida fenformin, es rara con el metformin (tres episodios por 100,000 paciente-años). El riesgo de acidosis lática está aumentado en los pacienes con menor velocidad de la filtración glomerular (creatinina sérica > 1.4 mg/dl en las mujeres y > 1.5 mg/dl en los varones) y con falla circulatoria (v.gr., insuficiencia cardiaca congestiva o choque). Debido a las consecuencias potencialmente fatales de la acidosis láctica, los pacientes deben vigilarse y seguirse en forma cuidadosa. El metformin puede usarse como monoterapia o en combinación con sulfonilureas en pacientes en los que fracase el tratamiento con sulfonilureas.118
Otro agente relativamente nuevo de administración por vía oral que puede usarse para tratar la DMID y la DMNID es el inhibidor de la a-glucosidasa, acarbosa. Este medicamento no absorbible actúa como la fibra dietética, diminuyendo la velocidad de absorción de los carbohidratos al inhibir en forma competitiva a las enzimas de la pared intestinal que digieren a los polisacáridos en monosacáridos absorbibles. El resultado es un menor incremento de la glucemia en el periodo posprandial. La acarbosa es un poco menos potente que las sulfonilureas y el metformin, disminuyendo la HbA1c en 0.5 a 1.5 porciento. Sin embargo, es eficaz en todos los pacientes diabéticos, incluyendo DMID, y puede usarse como monoterapia o en combinación con alguna de los otros medicamentos hipoglucemiantes disponibles.119 Cuando se usó como monoterapia, la acarbosa no se asoció con hipoglucemia o aumento de peso. La llegada de polisacáridos sin digerir al intestino grueso causa aumento en la formación de gas intestinal, lo que disminuye la tolerancia al medicamento en algunos pacientes.
El tratamiento más novedoso para la DMNID es el tiazolidinedione troglitazone, un agente insulinomimético. En dosis de 200 a 800 mg al día, el troglitazone disminuye la HbA1c en 0.5 a 1.0 porciento y mejora los niveles de triglicéridos con menos insulina y sin aumento significativo de peso u otros efectos adversos serios.120
La insulina es el agente hipoglucemiante más eficaz y, debido a que la dosis no tiene un límite superior, casi siempre puede encontrarse una dosis eficaz. En la DMNID la dosis total de insulina puede ser más importante que el perfil de su administración, a diferencia de la DMID, en la que es crucial seguir un esquema de administración fisiológico. Generalmente se requieren dosis altas (50 a más de 100 U/día) para alcanzar niveles normales de glucemia en los pacientes obesos con DMNID.54,55,121-123 La insulina puede administrarse en fórmula intermedia o de acción prolongada en la mañana o al momento de acostarse, en los esquemas llamados mixtos (con insulina de acción rápida e intermedia) antes del desayuno o antes del desayuno y la cena, o en fórmulas con combinaciones fijas. Aunque algunos pacientes con DMNID, como los que no tienen sobrepeso, los que tienen hiperglucemia severa con pérdida de peso o cetosis, o los que tienen niveles muy altos de triglicéridos, deben iniciar con insulina como tratamiento de elección, la mayoría de los médicos reservan la insulina para cuando han fallado los agentes orales más débiles. Debido a que la mayoría de los pacientes con DMNID que inician agentes orales eventualmente requieren insulina para mantener una glucemia casi normal, los agentes orales deben considerarse como una medida temporal.
La resistencia a usar insulina con más frecuencia o en forma más agresiva, por los médicos y pacientes, parece residir en la necesidad de inyectarse y el temor a los efectos adversos, incluyendo la hipoglucemia y el aumento de peso. De hecho, incluso cuando se usa en dosis altas, el tratamiento con insulina rara vez causa hipoglucemia severa en pacientes con DMNID.122,123 Además, la mayoría de los estudios que han comparado la inyección de insulina con el tratamiento oral con una sulfonilurea en pacientes con DMNID demuestran niveles de cumplimiento semejantes.122,124 Es obvio que deben tomarse en cuenta las preferencias del paciente e individualizar la elección de los agentes terapéuticos.
Se ha propuesto el tratamiento combinado para el manejo de la DMNID, por lo general para cuando falla la monoterapia. Una de las estrategias propuestas combina las sulfonilureas y la insulina. Al usar el efecto ahorrador de insulina de las sulfonilureas, este tratamiento logra concentraciones de glucemia semejantes con dosis de insulina cinco a 20 porciento menores que si se usara insulina sola.125 Aunque pueden lograrse los mismos resultados metabólicos con dosis de insulina más altas, algunos investigadores tienen la hipótesis de que los niveles (o dosis) más altos de insulina pueden ser aterogénicos, por lo que deben evitarse. La asociación entre la concentración de insulina y la enfermedad coronaria se estableció en varios estudios amplios de población que incluyeron a los pacientes con diabetes, y existen pocos datos que sugieran que el disminuir las concentraciones de insulina en los pacientes con DMNID mejore la evolución. Además, aunque las dosis de insulina pueden disminuirse con la terapia combinada, las concentraciones de insulina no son significativamente diferentes que con la administración solo de insulina. Por lo tanto, debido al mayor costo, posibilidad de interacciones medicamentosas, efectos colaterales y complicaciones, y por el menor cumplimiento cuando se usan los dos medicamentos, no se recomienda el tratamiento combinado en forma rutinaria.
Pueden usarse en forma eficaz otras combinaciones, incluyendo metformin y sulfonilureas118 o acarbosa y cualquiera de los otros agentes hipoglucemiantes,119 aunque con mayor costo y riesgo de hipoglucemia que con la insulina sola.
EJERCICIO
Las prescripciones de ejercicio para los pacientes con DMNID tienen por objeto disminuir el peso corporal, aumentar la sensibilidad a la insulina y mejorar la condición cardiovascular. Debido a que los pacientes con DMNID tienen mayor riesgo de enfermedad cardiovascular, los enfermos que comienzan un nuevo esquema de ejercicio deben someterse a un estudio electrocardiográfico y, si es necesario, a una prueba de estrés con ejercicio. Al igual que los pacientes con DMID y neuropatía periférica, los enfermos con DMNID y neuropatía, pies insensibles o circulación limitada tienen mayor riesgo de sufrir traumatismos en los pies durante los deportes de alto impacto.
TRATAMIENTO INNOVADOR Y EXPERIMENTAL PARA LA DMNID
Se han desarrollado varios principios terapéuticos novedosos para tratar la DMNID. El péptido-I-(7-37) semejante a glucagon (GLP-I-[7-37), un polipéptido natural que es procesado por las células intestinales L a partir de la molécula de preproglucagon, ha demostrado tener potentes efectos sobre la secreción y síntesis de insulina. Además, el GLP-I(7-37) inhibe la secreción de glucagon y quizá retrasa el vaciamiento gástrico, contribuyendo a su efecto reductor de la glucemia. Su administración intravenosa o subcutánea mejora la hiperglucemia en pacientes con DMNID sin riesgo de hipoglucemia.126 Por último, se ha iniciado un estudio multicéntrico extenso, el Programa de Prevención de Diabetes, para determinar si es posible prevenir o retrasar la DMNID en voluntarios con riesgo alto por tener una tolerancia a la glucosa deteriorada.
COMA HIPEROSMOLAR, HIPERGLUCEMICO, NO CETOSICO
El coma hiperosmolar, hiperglucémico, no cetósico se asocia con hiperglucemia grave (> 600 mg/dl), pero por definición no con cetosis o cetoacidosis. La depleción de volumen es importante (con una deficiencia promedio de líquido de 25 porciento del agua corporal total) y la osmolaridad sérica es mayor de 350 mOsm/kg. La combinación de una enfermedad, medicamentos o tratamiento nutricional que exacerbe en forma aguda la hiperglucemia (v.gr., tratamiento esteroideo o alimentación por sonda con soluciones de carbohidratos concentrados) con la falla o incapacidad para sustituir las pérdidas de líquidos, precipita deshidratación grave, obnubilación y, al final, estado de coma. Es necesario identificar y tratar los efectos precipitantes, incluyendo las infecciones. La deshidratación hiperosmolar se trata con solución salina hipotónica (al medio normal) para remplazar tanto los solutos como el agua libre. La infusión de insulina en dosis bajas es eficaz para disminuir la concentración de glucosa. A diferencia de la cetoacidosis diabética, la mortalidad por el coma hiperosmolar, hiperglucémico, no cetósico puede ser hasta del 50 porciento, lo que quizá se explique por la edad de las personas afectadas y por la gravedad de los factores precipitantes. Después de que se trata el episodio inicial con frecuencia los pacientes no requieren ya insulina.
Complicaciones
Todas las formas de diabetes mellitus se acompañan de un espectro semejante de complicaciones microvasculares y neurológicas. Las complicaciones como la retinopatía y la nefropatía son específicas de la diabetes; por el contrario, las complicaciones macrovasculares, incluyendo la enfermedad cardiaca, vascular periférica y cerebrovascular, ocurren también en población no diabética. Sin embargo, el riesgo de enfermedades macrovasculares en la población diabética es mayor que en la población general.
El desarrollo de complicaciones a largo plazo en el paciente diabético transforma una enfermedad metabólica relativamente benigna, aunque difícil de manejar, en un padecimiento devastador con gran morbimortalidad. La evolución clínica de los pacientes con DMID suele incluir alteraciones visuales o ceguera, insuficiencia renal y trastornos vasculares [ver tabla 13]. El riesgo de sufrir estas complicaciones se reduce 35 a 76 porciento con el tratamiento intensivo de la diabetes [ver antes, Metas del tratamiento para la glucemia: DCCT].
|
||||
|
Aunque la incidencia de las complicaciones específicas puede variar un poco en cada tipo de diabetes, su expresión clínica es idéntica. La causa del gran número de complicaciones no se conoce. Sin embargo, debido a que ocurren en formas de diabetes que son diferentes desde el punto de vista genético pero que tienen un trastorno metabólico en común, es posible que alguna de las características metabólicas de la diabetes sea la que ocasione las complicaciones. No se sabe si la hiperglucemia per se causa estas complicaciones, quizá por glucosilación de las proteínas de la membrana o por otros mecanismos. Estudios intervencionistas realizados en modelos animales apoyan el papel central de la hiperglucemia para causar complicaciones específicas de la diabetes (la llamada hipótesis de la glucosa), y aún más los resultados del DDCT. Un estudio semejante al DCCT realizado en japoneses apoya un beneficio semejante del tratamiento intensivo sobre las complicaciones a largo plazo de la DMNID.108
Se han identificado otros factores de riesgo para las complicaciones de la diabetes además de la hiperglucemia. El más poderoso es la duración de la enfermedad. La hipertensión es un factor de riesgo para la nefropatía, y el embarazo (en la DMID) es un factor de riesgo para la progresión de la retinopatía.127,128 Por último, puede existir un componente genético que incluya el riesgo de desarrollar complicaciones a largo plazo.
RETINOPATIA
Si la evolución de la diabetes es la suficiente, se desarrolla retinopatía en casi todos los pacientes con DMID y en la mayoría de los que tienen DMNID. Después de 15 años de evolución con tratamiento convencional, más del 95 porciento de los enfermos con DMID tienen retinopatía clínica.127 La forma más frecuente de retinopatía es la no proliferativa (también llamada retinopatía de fondo). Comienza con la pérdida de los pericitos, las células que apoyan a los vasos de la retina y progresa para formar microaneurismas caracteríscos que son visibles por oftalmoscopía directa [ver figura 9a]. Los microaneurismas, que miden 50 a 100 µm de diámetro, pueden ocurrir en cualquier parte de la retina, pero tienden a acumularse cerca de la mácula, la porción de la retina responsable de la visión central y de la agudeza visual. Se forman hemorragias puntiformes (o de punto y mancha) cuando los microaneurismas sangran. La salida de suero ocasiona la formación de exudados duros. Estas lesiones suelen ser benignas hasta que ocurren cerca de la mácula y en número suficiente para causar edema macular, que puede lesionar la visión central y la agudeza visual.
|
| Figura 9a |
| Retinopatía diabética |
Al progresar la retinopatía los capilares terminales se obstruyen y la retina sufre isquemia. Aparecen infartos de la capa nerviosa como exudados suaves (los llamados exudados algodonosos). En respuesta a la isquemia proliferan vasos de la superficie de la retina y hacia el vítreo, lo que explica el nombre de retinopatía proliferativa [ver figura 9b]. Los nuevos vasos son muy frágiles y pueden sangrar dentro del vítreo, causando pérdida temporal de la visión hasta que la sangre se reabsorbe. Los vasos proliferantes que son mayores de un cuarto del diámetro del disco óptico y que ocurren dentro de un diámetro de disco alrededor del mismo tienen más posibilidades de sangrar. Cuando la sangre del vítreo se reabsorbe se forman cicatrices, y la tracción sobre la retina puede ocasionar desprendimiento de la misma, con pérdida importante y permanente de la visión.
|
| Figura 9b |
| Retinopatía diabética |
Se ha demostrado que el tratamiento con laser para la retinopatía proliferativa y el edema macular conserva la visión.130,131 El papel del internista y del oftalmólogo consiste en detectar y tratar los casos de retinopatía que requieren tratamiento con laser antes de que ocurra daño irreversible. Aunque la fotografía de fondo de ojo es la forma más sensible para detectar retinopatía, los oftalmólogos e incluso los internistas bien entrenados pueden detectarla.132 Es preferible realizar el examen con la pupila dilatada. Debido a que la retinopatía casi nunca ocurre antes de cinco años de evolución de la DMID, los estudios oftalmológicos periódicos deben iniciarse hasta ese momento. En vista de que la duración de la DMNID es mucho más díficil de establecer con seguridad, se recomienda realizar exámenes anuales, comenzando en el momento del diagnóstico. Se sabe que el embarazo es un factor de riesgo para la progresión de la retinopatía en la DMID, y se recomiendan los estudios oculares frecuentes durante este periodo. Por último, los pacientes con cicatrización y detritos importantes del vítreo y desprendimiento de la retina pueden ser sometidos a vitrectomía. Durante este procedimiento de alto riesgo se extirpan las cicatrices fibroproliferativas y si es posible se intenta pegar de nuevo la retina, remplazando el vítreo lleno de detritos con solución salina. En algunos casos seleccionados la vitrectomía puede restablecer la visión.133
NEFROPATIA
La nefropatía es la complicación diabética que se asocia con mayor mortalidad. Mientras que la gran mayoría de los pacientes diabéticos desarrollan retinopatía, solo el 35 a 45 porciento de los enfermos con DMID y menos del 20 porciento de los que tienen DMNID sufren nefropatía clínica.134,135 Se ha identificado ya el mecanismo de progresión de la nefropatía. Esta comienza con excreción ligera de albúmina o microalbuminuria (concentración urinaria de albúmina de 30 a 300 mg/24 hr), tan pronto como a los cinco años de evolución de la DMID. Esta llamada nefropatía incipiente progresa a macroalbuminuria (i.e., niveles de albúmina en orina > 300 mg/24 hr medidos por medio de una tira reactiva de proteinuria positiva o por recolección de orina de 24 hr) con hipertensión asociada después de 12 a 20 años de duración de la DMID. Al final se desarrolla síndrome nefrótico y disminución en la velocidad de filtración glomerular (VFG), que progresa a insuficiencia renal terminal (IRT) [ver figura 10].136,137 A diferencia del riesgo de retinopatía, el riesgo de nefropatía no continúa aumentando con la duración de la enfermedad.127 Si no existe proteinuria por cinta reactiva (que es el indicador más confiable de nefropatía diabética), después de 25 a 30 años de duración de la diabetes, el riesgo de desarrollar nefropatía comienza a disminuir. La duración de la diabetes y quizá la historia familiar de hipertensión son factores de riesgo de nefropatía,138 mientras que el control de la glucemia no se ha identificado en forma clara como un factor de riesgo. El control de hipertensión y el uso de dietas bajas en proteínas pueden disminuir la velocidad de deterioro de la VFG en las etapas tardías de la nefropatía.139-141 El tratamiento intensivo de la diabetes disminuye la progresión y quizá previene el desarrollo de microalbuminuria y proteinuria positiva con cinta reactiva (concentración de albumina en orina > 300 mg/24 hr).8,145,146 No se sabe si el tratamiento intensivo de la diabetes afecta la progresión de la microalbuminuria a IRT.

|
| Figura 10 |
| Progreso de la nefropatía en la DMID |
Los pacientes diabéticos con IRT pueden ser tratados con trasplante, hemodiálisis o diálisis peritoneal. La hemodiálisis se asocia con un mayor riesgo de eventos cardiovasculares y de hemorragia retiniana por las grandes fluctuaciones en la presión arterial y el uso de anticoagulantes. Por lo tanto, el trasplante y la diálisis peritoneal son los tratamientos preferidos.
NEUROPATIA
La neuropatía en la diabetes tiene manifestaciones muy diversas. La más frecuente es la neuropatía sensorimotora periférica simétrica, que se presenta como adormicimiento u hormigueo en los ortejos o pies. Los síntomas suelen ser poco molestos y no requieren tratamiento específico. Con frecuencia los pacientes mencionan que estos síntomas desaparecen con el tiempo, porque al empeorar la neuropatía las parestesias y disestesias son sustituidas por hipoestesia y anestesia. El pie insensible se vuelve vulnerable a los traumatismos, y las úlceras en pies neuropáticos son causa frecuente de hospitalización y amputación. En raros casos la neuropatía periférica es dolorosa, con disestesias que interfieren con el sueño y la actividad. Los antidepresivos tricíclicos, la fenitoína y la carbamacepina, suelen tener alguna eficacia en estos casos. Se ha demostrado que la capsaicina, un tratamiento tópico que depleta la sustancia P, neutrotrasmisor modulador del dolor a través de las fibras nocipeptivas tipo C, tiene alguna eficacia.147 Deben evitarse los narcóticos porque la posibilidad de adicción es alta. El DCCT demostró un beneficio importante del tratamiento intensivo en la neuropatía diabética.8 Para los pacientes cuyos pies tienen riesgo de ulceración, se ha demostrado que la educación para cuidar e inspeccionar los pies disminuye los traumatismos y las hospitalizaciones.148
Entre otras formas de neuropatía periférica se incluyen las mononeuropatías, los síndromes de atrapamiento y la neuropatía autonómica. Las mononeuropatías, que por lo general son asimétricas, afectan nervios craneales o periféricos y se piensa que son secundarias a oclusión vascular, con infartos nerviosos subsecuentes. Los síndromes de atrapamiento, como el síndrome del túnel carpiano, ocurren con más frecuencia en diabéticos que en no diabéticos. Por último, la neuropatía autonómica puede afectar los sistemas gastrointestinal, cardiovascular y genitourinario. Los síndromes clínicos incluyen gastroparesia diabética, caracterizada por retraso en el vaciamiento gástrico, así como los siguientes síntomas: saciedad temprana, sensación de plenitud, náusea y vómito. La diarrea diabética se manifiesta por diarrea e incontinencia nocturnas que alternan con constipación. La neuropatía produce taquicardia en reposo con hipotensión postural. La neuropatía genitourinaria provoca impotencia e hipotonía, o atonía de la vejiga, lo que produce incontinencia por rebosamiento. La gastroparesia se trata en forma eficaz con metoclopramida, y la diarrea diabética con clonidina;149,150 la impotencia puede manejarse con inyecciones de vasodilatadores en el pene o con prótesis implantables o equipos de vacío.151,152
ENFERMEDAD CARDIOVASCULAR
La enfermedad cardiovascular no es específica de la diabetes. Sin embargo, el riesgo de enfermedad cardiaca aumenta dos a casi 10 veces en la DMNID.153 El riesgo relativo de enfermedad cardiovascular en las mujeres diabéticas (comparadas con las no diabéticas) es incluso mayor que para los varones, porque la diabetes anula el efecto protector que normalmente confieren los estrógenos. Existen numerosos factores de riesgo para la enfemedad cardiovascular asociadad con DMNID, incluyendo obesidad, hipertensión, dislipidemia (reducción de LAD y LMBD altas con LBD pequeñas densas) e hiperglucemia. Un estudio de supervivencia de la cohorte del Estudio original de Framinghan ha demostrado que la concentración de HbA1c se asocia con la ocurrencia de enfermedad cardiaca en las mujeres, independientemente de los otros factores de riesgo conocidos.154 El desarrollo de nefropatía tanto en la DMID como en la DMNID constituye un riesgo especial para mayor enfermedad coronaria.
La incidencia de enfermedad vascular periférica y cerebrovascular también está aumentada en pacientes con diabetes, y estos trastornos originan una gran morbimortalidad entre los pacientes diabéticos. La combinación de neuropatía periférica y enfermedad vascular periférica provoca un aumento de 40 veces en el riesgo de amputaciones en la población diabética en relación con la población general.
Figuras 1, 3, 6, 8 y 10 Janet Betries.
Figuras 2, 4, 5 y 9 Andy Christie.
Tabla 1 Adaptada de "Classification and Diagnosis of Diabetes Mellitus and Other Categories of Glucose Intolerance", por la National Diabetes Date Group, en iabetes 28: 1039, 1979. Utilizada con autorización.
Tabla 2 Datos de intolerancia a la glucosa derivados de "Prevalence of Diabetes and Impaired Glucose Tolerance and Plasma Glucose Levels in U.S. Population Aged 20-74 Yr" por M.I. Harris, W.C. Hadeen, W.C. Knowler y cols, en Diabetes 36: 523, 1987. Utilizado con autorización.
Bibliografìa
-
Banting FG, Best CH, Collip JB, et al: Pancreatic extracts
in the treatment of diabetes mellitus. Can Med Assoc J 12:141, 1922
-
Burrow GN, Hazlett BE, Phillips MJ: A case of diabetes
mellitus. N Engl J Med 306:340, 1982 [PMID
7033790]
-
Zinman B: The physiologic replacement of insulin. N
Engl J Med 321:363, 1989
-
Howey DC, Bowsher RR, Brunelle RL, et al: [Lys(B28),
Pro(B29)]-Human insulin. Diabetes 43:396, 1994 [PMID
8314011]
-
Nathan DM, Fogel H, Norman D, et al: Long-term metabolic
and quality of life results with pancreatic/renal transplantation in insulin-dependent
diabetes mellitus. Transplantation 52:85, 1991 [PMID
1858159]
-
Robertson AP: Pancreatic and islet transplantation
for diabetes-cures or curiosities? N Engl J Med 327:1861, 1992
-
Lanza RP, Sullivan SJ, Chick WL: Islet transplantation
with immunoisolation. Diabetes 41:1503, 1992 [PMID
1446791]
-
The effect of intensive treatment of diabetes on the
development and progression of long-term complications in insulin-dependent
diabetes mellitus. Diabetes Control and Complications Trial Research Group.
N Engl J Med 329:977, 1993
-
Gough DA, Armour JC: Development of the implant glucose
sensor-what are the prospects and why is it taking so long? Diabetes 44:1005,
1995
-
Selam J-L, Micossi P, Dunn FL, et al: Clinical trial
of programmable implantable insulin pump for type I diabetes. Diabetes
Care 15:877, 1992
-
Himsworth HP: Diabetes mellitus: its differentiation
into insulin-sensitive and insulin insensitive types. Lancet 1:117, 1936
-
Eisenbarth GS: Type I diabetes mellitus: a chronic
autoimmune disease. N Engl J Med 314:1360, 1986
-
Leahy JL: Natural history of b-cell
dysfunction in NIDDM. Diabetes Care 13:992, 1990
-
National Diabetes Data Group: Classification and diagnosis
of diabetes mellitus and other categories of glucose intolerance. Diabetes
28:1039, 1979
-
Neufield M, Maclaren NK, Blizzard RM: Two types of
autoimmune Addison's disease associated with different polyglandular autoimmune
(PGA) syndromes. Medicine (Baltimore) 60:355, 1981
-
Report of the Expert Committee on the diagnosis and
classification of diabetes mellitus. Expert Committee on the Diagnosis
and Classification of Diabetes Mellitus. Diabetes Care 20:1183, 1997
-
LaPorte R, Matsushima M, Chang YF: Prevalence and incidence
of insulin-dependent diabetes. Diabetes in America, 2nd ed. Harris M, Ed.
National Institutes of Health, Bethesda, Maryland, NIH Publication No 95-1468,
1995, p 37 [see
Web link]
-
Harris MI, Hadden WC, Knowler WC, et al: Prevalence
of diabetes and impaired glucose tolerance and plasma glucose levels in
U.S. population aged 20-74 yr. Diabetes 36:523, 1987
-
King H, Zimmet P: Trends in the incidence and prevalence
of diabetes: non-insulin dependent diabetes mellitus. World Health Stat
Q 41:190, 1988 [PMID
2466380]
-
Bennett PH, Bogardus C, Tuomilehto J, et al: Epidemiology
and natural history of NIDDM: nonobese and obese. International Textbook
of Diabetes. Alberti KGMM, DeFronzo RA, Keen H, et al, Eds. John Wiley
& Sons, Ltd, Chichester, England, 1992, p 148
-
Hartz AJ, Rupley DC, Rimm AA: The association of girth
measurements with disease in 32,856 women. Am J Epidemiol 119:71, 1984
[PMID
6691337]
-
Savage PJ, Bennett PH, Senter RG, et al: High prevalence
of diabetes in young Pima Indians: evidence of phenotypic variation in
a genetically isolated population. Diabetes 28:937, 1979 [PMID
478185]
-
Zimmet P, Taft P, Guinea A, et al: The high prevalence
of diabetes mellitus on a Central Pacific island. Diabetologia 13:111,
1977 [PMID
852640]
-
Rowley MJ, MacKay IR, Chen Q-Y, et al: Antibodies to
glutamic acid decarboxylase discriminate major types of diabetes mellitus.
Diabetes 41:548, 1992 [PMID
1607079]
-
Greenbaum CJ, Palmer JP, Nagataki S, et al: Participating
labs improved specificity of ICA assays in the Fourth International Immunology
of Diabetes Exchange Workshop. Diabetes 41:1570, 1992 [PMID
1446798]
-
Davies JL, Kawaguchi Y, Bennett ST, et al: A genome-wide
search for human type 1 diabetes susceptibility genes. Nature 371:130,
1994 [PMID
8072542]
-
Oats JN, Beischer NA, Grant PT: The emergence of diabetes
and impaired glucose tolerance in women who had gestational diabetes. Gestational
Diabetes. Weiss PA, Coustan DR, Eds. Springer-Verlag, New York, 1988, p
199
-
Tattersall RB: Mild familial diabetes with dominant
inheritance. Q J Med 43:339, 1974
-
Froguel P, Vaxillaire M, Sun F, et al: Close linkage
of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent
diabetes mellitus. Nature 356:162, 1992 [PMID
1545870]
-
Kadowaki T, Kadowaki H, Mori Y, et al: A subtype of
diabetes mellitus associated with a mutation of mitochondrial DNA. N Engl
J Med 330:962, 1994
-
Jarrett JJ, McCartney P, Keen H: The Bedford survey:
ten year mortality rates in newly diagnosed diabetics, borderline diabetics
and normoglycaemic controls and risk indices for coronary heart disease
in borderline diabetics. Diabetologia 22:79, 1982
-
Fuller JH, McCartney P, Jarrett RJ, et al: Hyperglycaemia
and coronary heart disease: the Whitehall Study. J Chron Dis 32:721, 1979
-
Ziegler AG, Herskowitz RD, Jackson RA, et al: Predicting
type I diabetes. Diabetes Care 13:762, 1990 [PMID
2201499]
-
Faustman D, Eisenbarth G, Daley J, et al: Abnormal
T-lymphocyte subsets in type I diabetes. Diabetes 38:1462, 1989 [PMID
2533573]
-
Palmer JP, McCulloch DK: Prediction and prevention
of IDDM-1991. Diabetes 40:943, 1991 [PMID
1860558]
-
Baekkeskov S, Aanstoot H-J, Christgau S, et al: Identification
of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing
enzyme glutamic acid decarboxylase. Nature 347:151, 1990 [PMID
1697648]
-
Verge CF, Gianani R, Kawasaki E, et al: Prediction
of type I diabetes in first-degree relatives using a combination of insulin,
GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes 45:926, 1996 [PMID
8666144]
-
Roep BO: T-cell responses to autoantigens in IDDM:
the search for the Holy Grail. Diabetes 45:1147, 1996
-
Barnett AH, Eff C, Leslie RDG, et al: Diabetes in identical
twins: a study of 200 pairs. Diabetologia 20:87, 1981 [PMID
7193616]
-
Thomson G, Robinson WP, Kuhner MK: Genetic heterogeneity,
modes of inheritance, and risk estimates for a joint study of Caucasians
with insulin-dependent diabetes mellitus. Am J Hum Genet 43:799, 1988 [PMID
3057885]
-
Baisch JM, Weeks T, Giles R, et al: Analysis of HLA-DQ
genotypes and susceptibility in insulin-dependent diabetes mellitus. N
Engl J Med 322:1836, 1990 [PMID
2348836]
-
Yalow RS, Berson SA: Plasma insulin concentrations
in nondiabetic and early diabetic subjects: determinations by a new sensitive
immuno-assay technique. Diabetes 9:254, 1960
-
Yalow RS, Berson SA: Immunoassay of endogenous plasma
insulin in man. J Clin Invest 39:1157, 1960
-
Rahier J, Goebbels RM, Henquin JC: Cellular composition
of the human diabetic pancreas. Diabetologia 24:366, 1983 [PMID
6347784]
-
Reaven GM, Bernstein R, Davis B, et al: Nonketotic
diabetes mellitus: insulin deficiency or insulin resistance? Am J Med 60:80,
1976
-
Moller DE, Flier JS: Insulin resistance-mechanisms,
syndromes, and implications. N Engl J Med 325:938, 1991 [PMID
1881419]
-
Sun XJ, Rothenberg P, Kahn CR, et al: Structure of
the insulin receptor substrate IRS-1 defines a unique signal transduction
protein. Nature 352:73, 1991 [PMID
1648180]
-
DeFronzo RA: The triumvirate: b-cell,
muscle, liver: a collusion responsible for NIDDM. Diabetes 37:667, 1988
-
Brunzell JD, Robertson RP, Lerner RL, et al: Relationships
between fasting plasma glucose levels and insulin secretion during intravenous
glucose tolerance tests. J Clin Endocrinol Metab 42:222, 1976 [PMID
1262429]
-
Yoshioka N, Kuzuya T, Matsuda A, et al: Effects of
dietary treatment on serum insulin and proinsulin response in newly diagnosed
NIDDM. Diabetes 38:262, 1989 [PMID
2644145]
-
Cerasi E, Luft R: The plasma insulin response to glucose
infusion in healthy subjects and in diabetes mellitus. Acta Endocrinol
55:278, 1967 [PMID
5338206]
-
Leahy JL, Bonner-Weir S, Weir GC: Abnormal glucose
regulation of insulin secretion in models of reduced B-cell mass. Diabetes
33:667, 1984 [PMID
6203798]
-
Turner RC, Holman RR: Beta cell function during insulin
or chlorpropamide treatment of maturity-onset diabetes mellitus. Diabetes
27(suppl 1):241, 1978
-
Andrews WJ, Vasquez B, Nagulesparan M, et al: Insulin
therapy in obese, non-insulin-dependent diabetes induces improvements in
insulin action and secretion that are maintained for two weeks after insulin
withdrawal. Diabetes 33:634, 1984 [PMID
6376219]
-
Garvey WT, Olefsky JM, Griffin J, et al: The effect
of insulin treatment on insulin secretion and insulin action in type II
diabetes mellitus. Diabetes 34:222, 1985 [PMID
3882489]
-
Blackshear PJ, Shulman GI, Roussell AM, et al: Metabolic
response to three years of continuous, basal rate intravenous insulin infusion
in type II diabetic patients. J Clin Endocrinol Metab 61:753, 1985 [PMID
3897260]
-
Lillioja S, Mott DM, Howard BV, et al: Impaired glucose
tolerance as a disorder of insulin action: longitudinal and cross-sectional
studies in Pima Indians. N Engl J Med 318:1217, 1988 [PMID
3283552]
-
Warram JH, Martin BC, Krolewski AS, et al: Slow glucose
removal rate and hyperinsulinemia precede the development of type II diabetes
in the offspring of diabetic parents. Ann Intern Med 113:909, 1990 [PMID
2240915]
-
O'Rahilly S, Turner RC, Matthews DR: Impaired pulsatile
secretion of insulin in relatives of patients with non-insulin-dependent
diabetes. N Engl J Med 318:1225, 1988 [PMID
3283553]
-
Eriksson J, Franssila-Kallunki A, Ekstrand A, et al:
Early metabolic defects in persons at increased risk for non-insulin-dependent
diabetes mellitus. N Engl J Med 321:337, 1989 [PMID
2664520]
-
Pimenta W, Korytkowski M, Mitrakou A, et al: Pancreatic
beta-cell dysfunction as the primary genetic lesion in NIDDM: evidence
from studies in normal glucose-tolerant individuals with a first-degree
NIDDM relative. JAMA 273:1855, 1995 [PMID
7776502]
-
Ullrich A, Bell JR, Chen EY, et al: Human insulin receptor
and its relationship to the tyrosine kinase family of oncogenes. Nature
313:756, 1985 [PMID
2983222]
-
Foster DW, McGarry JD: The metabolic derangements and
treatment of diabetic ketoacidosis. N Engl J Med 309:159, 1983 [PMID
6408476]
-
Gerich JE, Lorenzi M, Bier DM, et al: Prevention of
human diabetic ketoacidosis by somatostatin: evidence for an essential
role of glucagon. N Engl J Med 292:985, 1975 [PMID
804137]
-
Effects of age, duration and treatment of insulin-dependent
diabetes mellitus on residual b-cell function:
observations during eligibility testing for the Diabetes Control and Complications
Trial (DCCT). The DCCT Research Group. J Clin Endocrinol Metab 65:30, 1987
-
Podolsky S: Hyperosmolar nonketotic coma in the elderly
diabetic. Med Clin North Am 62:815, 1978
-
Mitrakou A, Kelley D, Veneman T, et al: Contribution
of abnormal muscle and liver glucose metabolism to postprandial hyperglycemia
in NIDDM. Diabetes 39:1381, 1990 [PMID
2121568]
-
Holman RR, Turner RC: The basal plasma glucose: a simple
relevant index of maturity-onset diabetes. Clin Endocrinol 14:279, 1980
-
Nathan DM, Singer DE, Godine JE, et al: Retinopathy
in older type II diabetics: association with glucose control. Diabetes
35:797, 1986 [PMID
3721064]
-
Walden CE, Knopp RH, Wahl PW, et al: Sex differences
in the effect of diabetes mellitus on lipoprotein triglyceride and cholesterol
concentrations. N Engl J Med 311:953, 1984 [PMID
6472421]
-
Singer DE, Samet JH, Coley CM, et al: Screening for
diabetes mellitus. Ann Intern Med 109:639, 1988 [PMID
3138934]
-
Cahill GF Jr, Etzwiler LD, Freinkel N: "Control" and
diabetes (editorial). N Engl J Med 294:1004, 1976 [PMID
1256496]
-
Siperstein MD, Foster DW, Knowles HC, et al: Control
of blood glucose and diabetic vascular disease (editorial). N Engl J Med
296:1060, 1977
-
Ingelfinger FJ: Debates on diabetes. N Engl J Med 296:1228,
1977
-
Ballantyne AJ, Loewenstein A: The pathology of diabetic
retinopathy. Trans Ophthalmol UK 63:95, 1943
-
Kimmelstiel P, Wilson C: Intercapillary lesions in
the glomeruli of the kidney. Am J Pathol 12:83, 1936
-
Engerman R, Bloodworth JM Jr, Nelson S: Relationship
of microvascular disease in diabetes to metabolic control. Diabetes 26:760,
1977
-
Klein R, Klein BE, Moss SE, et al: The Wisconsin epidemiologic
study of diabetic retinopathy: II. Prevalence and risk of diabetic retinopathy
when age at diagnosis is less than 30 years. Arch Ophthalmol 102:520, 1984
-
Nathan DM: Modern management of insulin-dependent diabetes
mellitus. Med Clin North Am 72:1365, 1988
-
Nathan DM, Singer DE, Hurxthal K, et al: The clinical
information value of the glycosylated hemoglobin assay. N Engl J Med 310:341,
1984
-
Management of diabetes mellitus in pregnancy. ACOG
Technical Bulletin 92:1, 1986
-
Fuhrmann K, Reiher H, Semmler K, et al: Prevention
of congenital malformations in infants of insulin-dependent diabetic mothers.
Diabetes Care 6:219, 1983
-
Jovanovic L, Druzin M, Peterson CM: Effect of euglycemia
on the outcome of pregnancy in insulin-dependent diabetic women as compared
with normal control subjects. Am J Med 71:921, 1981 [PMID
7032287]
-
Nathan DM: Inferences and implications: do results
from the Diabetes Control and Complications Trial apply in NIDDM? Diabetes
Care 18:251, 1995
-
Malone JI, Rosenbloom AL, Grgic A, et al: The role
of urine sugar in diabetic management. Am J Dis Child 130:1324, 1976 [PMID
998574]
-
Frishman D, Ardito DM, Graham SM: Performance of glucose
monitors. Lab Med 23:179, 1992
-
Larsen ML, Horder M, Mogensen EF: Effect of long-term
monitoring of glycosylated hemoglobin levels in insulin-dependent diabetes
mellitus. N Engl J Med 323:1021, 1990 [PMID
2215560]
-
Halle J-P, Lambert J, Lindmayer I, et al: Twice-daily
mixed regular and NPH insulin injections with new jet injector versus conventional
syringes: pharmacokinetics of insulin absorption. Diabetes Care 9:279,
1986
-
Rizza RA, Gerich JE, Haymond MW, et al: Control of
blood sugar in insulin-dependent diabetes: comparison of an artificial
endocrine pancreas, continuous subcutaneous insulin infusion, and intensified
conventional insulin therapy. N Engl J Med 303:1313, 1980 [PMID
7001229]
-
Nathan DM, Lou P, Avruch J: Intensive conventional
and insulin pump therapies in adult type I diabetes: a crossover study.
Ann Intern Med 97:31, 1982 [PMID
7046554]
-
Epidemiology of severe hypoglycemia in the Diabetes
Control and Complications Trial. The DCCT Research Group. Am J Med 90:450,
1991
-
Nathan DM, Dunn FL, Bruch J, et al: Postprandial insulin
profiles with implantable pump therapy may explain decreased frequency
of severe hypoglycemia, compared with intensive subcutaneous regimens,
in insulin-dependent diabetes mellitus patients. Am J Med 100:412, 1996
[PMID
8610727]
-
Nutritional recommendations and principles for individuals
with diabetes mellitus: 1986 position statement. American Diabetes Association.
Diabetes Care 10:126, 1987
-
Jenkins DJA, Wolever TMS, Wong GS, et al: Glycemic
responses to foods: possible differences between insulin-dependent and
noninsulin-dependent diabetics. Am J Clin Nutr 40:971, 1984
-
Faich GA, Fishbein HA, Ellis SE: The epidemiology of
diabetic acidosis: a population-based study. Am J Epidemiol 117:551, 1983
[PMID
6405612]
-
Page M, Alberti KGMM, Greenwood R, et al: Treatment
of diabetic coma with continuous low-dose infusion of insulin. Br Med J
2:687, 1974 [PMID
4855253]
-
Adrogue HJ, Wilson H, Boyd AE, et al: Plasma acid-base
patterns in diabetic ketoacidosis. N Engl J Med 307:1603, 1982 [PMID
6815530]
-
Morris LR, Murphy MB, Kitabchi AE: Bicarbonate therapy
in severe diabetic ketoacidosis. Ann Intern Med 105:836, 1986 [PMID
3096181]
-
Keller U, Berger W: Prevention of hypophosphatemia
by phosphate infusion during treatment of diabetic ketoacidosis and hyperosmolar
coma. Diabetes 29:87, 1980 [PMID
6766411]
-
Cryer PE, Gerich JE: Glucose counterregulation, hypoglycemia,
and intensive insulin therapy in diabetes mellitus. N Engl J Med 313:232,
1985 [PMID
2861565]
-
Consensus statement on self-monitoring of blood glucose.
American Diabetes Association. Diabetes Care 10:95, 1987
-
Bolli G, De Feo P, Compagnucci P, et al: Abnormal glucose
counterregulation in insulin-dependent diabetes mellitus: interaction of
anti-insulin antibodies and impaired glucagon and epinephrine secretion.
Diabetes 32:134, 1983 [PMID
6337896]
-
Amiel SA, Tamborlane WV, Simonson DC, et al: Defective
glucose counterregulation after strict glycemic control of insulin-dependent
diabetes mellitus. N Engl J Med 316:1376, 1987 [PMID
3553949]
-
Cranston I, Lomas J, Maran A, et al: Restoration of
hypoglycaemia awareness in patients with long-duration insulin-dependent
diabetes. Lancet 344:283, 1994 [PMID
7914259]
-
Faustman D, Coe C: Prevention of xenograft rejection
by masking donor HLA class I antigens. Science 252:1700, 1991 [PMID
1710828]
-
Scharp DW, Lacy PE, Santiago JV, et al: Insulin independence
after islet transplantation into type I diabetic patient. Diabetes 39:515,
1990 [PMID
2108071]
-
Cyclosporin-induced remission of IDDM after early intervention.
The Canadian-European Randomized Control Trial Group. Diabetes 37:1514,
1988
-
Ohkubo Y, Kishikawa H, Araki E, et al: Intensive insulin
therapy prevents the progression of diabetic microvascular complications
in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized
prospective 6-year study. Diabetes Res Clin Pract 28:103, 1995 [PMID
7587918]
-
Hadden DR, Montgomery DAD, Skelly RJ, et al: Maturity
onset diabetes mellitus: response to intensive dietary management. Br Med
J 3:276, 1975 [PMID
1148773]
-
Long SD, O'Brien K, MacDonald KG Jr, et al: Weight
loss in severely obese subjects prevents the progression of impaired glucose
tolerance to type II diabetes: a longitudinal interventional study. Diabetes
Care 17:372, 1994 [PMID
8062602]
-
Wing RR, Jeffery RW: Outpatient treatments of obesity:
a comparison of methodology and clinical results. Int J Obes 3:261, 1979
[PMID
395116]
-
Wing RR, Marcus MD, Epstein LH, et al: Type II diabetic
subjects lose less weight than their overweight nondiabetic spouses. Diabetes
Care 10:563, 1987
-
Allen BT, Delong ER, Feussner JR: Impact of glucose
self-monitoring on non-insulin-treated patients with type II diabetes mellitus.
Diabetes Care 13:1044, 1990
-
Singer DE, Coley CM, Samet JH, et al: Tests of glycemia
in diabetes mellitus: their use in establishing diagnosis and in treatment.
Ann Intern Med 110:125, 1989 [PMID
2642375]
-
Groop LC: Sulfonylureas in NIDDM. Diabetes Care 15:737,
1992
-
A study of the effects of hypoglycemic agents on vascular
complications in patients with adult onset diabetes: supplementary report
on nonfatal events in patients treated with tolbutamide. University Group
Diabetes Program. Diabetes 25:1129, 1976
-
Bailey CJ: Biguanides and NIDDM. Diabetes Care 15:755,
1992
-
DeFronzo RA, Goodman AM: Efficacy of metformin in patients
with non-insulin-dependent diabetes mellitus. N Engl J Med 333:541, 1995
[PMID
7623902]
-
Chiasson JL, Josse RG, Hunt JA, et al: The efficacy
of acarbose in the treatment of patients with non-insulin-dependent diabetes
mellitus: a multicenter controlled clinical trial. Ann Intern Med 121:928,
1994 [PMID
7734015]
-
Iwamoto Y, Kosaka K, Kuzuya T, et al: Effects of troglitazone,
a new hypoglycemic agent in patients with NIDDM poorly controlled by diet
therapy. Diabetes Care 19:151, 1996 [PMID
8718436]
-
Glaser B, Leibovich G, Nesher R, et al: Improved beta-cell
function after intensive insulin treatment in severe non-insulin-dependent
diabetes. Acta Endocrinol 118:365, 1988 [PMID
3293339]
-
Nathan DM, Roussell A, Godine JE: Glyburide or insulin
for metabolic control in non-insulin-dependent diabetes mellitus: a randomized
double-blind study. Ann Intern Med 108:334, 1988 [PMID
3124685]
-
Colwell JA: The feasibility of intensive insulin management
in non-insulin-dependent diabetes mellitus: implications of the Veterans
Affairs Cooperative Study on Glycemic Control and Complications in NIDDM.
Ann Intern Med 124:131, 1996
-
Diehl AK, Sugarek NJ, Bauer RL: Medication compliance
in non-insulin-dependent diabetes: a randomized comparison of chlorpropamide
and insulin. Diabetes Care 8:219, 1985 [PMID
3891264]
-
Peters AL, Davidson MB: Insulin plus a sulfonylurea
agent for treating type 2 diabetes. Ann Intern Med 115:45, 1991 [PMID
1828656]
-
Nathan DM, Schreiber E, Fogel H, et al: Insulinotropic
action of glucagon-like peptide-I-(7-37) in diabetic and nondiabetic subjects.
Diabetes Care 15:270, 1992 [PMID
1547685]
-
Krolewski AS, Warram JH, Rand LI, et al: Epidemiologic
approach to the etiology of type I diabetes mellitus and its complications.
N Engl J Med 317:1390, 1987 [PMID
3317040]
-
Klein BEK, Moss SE, Klein R: Effect of pregnancy on
progression of diabetic retinopathy. Diabetes Care 13:34, 1990
-
Palmberg P, Smith M, Waltman S, et al: The natural
history of retinopathy in insulin-dependent juvenile-onset diabetes. Ophthalmology
88:613, 1981 [PMID
7267029]
-
Aiello LP, Avery RL, Arrig PG, et al: Vascular endothelial
growth factor in ocular fluid of patients with diabetic retinopathy and
other retinal disorders. N Engl J Med 331:1480, 1994 [PMID
7526212]
-
Photocoagulation treatment of proliferative diabetic
retinopathy: clinical application of Diabetic Retinopathy Study (DRS) findings,
DRS report number 8. The Diabetic Retinopathy Study Research Group. Ophthalmology
88:583, 1981
-
Early photocoagulation for diabetic retinopathy: ETDRS
report number 9. Early Treatment Diabetic Retinopathy Study Research Group.
Ophthalmology 98:766, 1991
-
Nathan DM, Fogel HA, Godine JE, et al: Role of diabetologist
in evaluating diabetic retinopathy. Diabetes Care 14:26, 1991 [PMID
1991432]
-
Early vitrectomy for severe vitreous hemorrhage in
diabetic retinopathy. Diabetic Retinopathy Vitrectomy Study Group. Arch
Ophthalmol 103:1644, 1985
-
Andersen AR, Christiansen JS, Andersen JK, et al: Diabetic
nephropathy in type 1 (insulin-dependent) diabetes: an epidemiological
study. Diabetologia 25:496, 1982
-
Humphrey LL, Ballard DJ, Frohnert PP, et al: Chronic
renal failure in non-insulin-dependent diabetes mellitus: a population-based
study in Rochester, Minnesota. Ann Intern Med 111:788, 1989 [PMID
2817626]
-
Viberti GC, Jarrett RJ, Mahmud U, et al: Microalbuminuria
as a predictor of clinical nephropathy in insulin-dependent diabetes mellitus.
Lancet 1:1430, 1982 [PMID
6123720]
-
Krolewski AS, Canessa M, Warram JH, et al: Predisposition
to hypertension and susceptibility to renal disease in insulin-dependent
diabetes mellitus. N Engl J Med 318:140, 1988 [PMID
3336401]
-
Cohen D, Dodds R, Viberti G: Effect of protein restriction
in insulin-dependent diabetics at risk of nephropathy. Br Med J 294:295,
1987
-
Mogensen CE: Long-term antihypertensive treatment inhibits
progression of diabetic nephropathy. Br Med J 285:685, 1982
-
Vasquez B, Flock EV, Savage PJ, et al: Sustained reduction
of proteinuria in type 2 (non-insulin-dependent) diabetes following diet-induced
reduction of hyperglycaemia. Diabetologia 26:127, 1984 [PMID
6714534]
-
Marre M, Chatellier G, Leblanc H, et al: Prevention
of diabetic nephropathy with enalapril in normotensive diabetics with microalbuminuria.
BMJ 297:1092, 1988 [PMID
2848604]
-
Lewis EJ, Hunsicker LG, Bain RP, et al: The effect
of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N
Engl J Med 329:1456, 1993 [PMID
8413456]
-
Sano T, Kawamura T, Matsumae H, et al: Effects of long-term
enalapril treatment on persistent micro-albuminuria in well-controlled
hypertensive and normotensive NIDDM patients. Diabetes Care 17:420, 1994
[PMID
8062609]
-
Feldt-Rasmussen B, Mathiesen ER, Deckert T: Effect
of two years of strict metabolic control on progression of incipient nephropathy
in insulin-dependent diabetes. Lancet 2:1300, 1986 [PMID
2878175]
-
Reichard P, Rosenqvist U: Nephropathy is delayed by
intensified insulin treatment in patients with insulin-dependent diabetes
mellitus and retinopathy. J Intern Med 226:81, 1989 [PMID
2671247]
-
Donofrio PD, Walker FO, Hunt VP, et al: Treatment of
painful diabetic neuropathy with topical capsaicin: a multicenter, double-blind,
vehicle controlled study. Arch Intern Med 151:2225, 1991
-
Kitzelman DK, Slemenda CW, Langefeld CD, et al: Reduction
of lower extremity clinical abnormalities in patients with non-insulin-dependent
diabetes mellitus: a randomized, controlled trial. Ann Intern Med 119:36,
1993
-
Snape WJ, Battle WM, Schwartz SS, et al: Metoclopramide
to treat gastroparesis due to diabetes mellitus. Ann Intern Med 96:444,
1982 [PMID
7065559]
-
Fedorak RN, Field M, Chang EB: Treatment of diabetic
diarrhea with clonidine. Ann Intern Med 102:197, 1985 [PMID
3966758]
-
. Chen J, Godschalk M, Katz PG, et al: The lowest effective
dose of prostaglandin E1 as treatment for erectile dysfunction.
J Urol 153:80, 1995 [PMID
7966797]
-
Scott FB, Fishman IJ, Light JK: An inflatable penile
prosthesis for treatment of diabetic impotence. Ann Intern Med 92:340,
1980 [PMID
7356228]
-
Kannel WB, McGee DL: Diabetes and cardiovascular disease:
the Framing ham Study. JAMA 241:2035, 1979 [PMID
430798]
-
Singer DE, Nathan DM, Anderson KM, et al: Association
of HbA1c with prevalent cardiovascular disease in the original
cohort of the Framingham Heart Study. Diabetes 41:202, 1992 [PMID
1733810]