Oncología
⭳ Abrir artículo (PDF)444.7 KBEste artículo es idéntico en la Edición 2/2000.
Contenido del artículo
XV LEUCEMIAS LINFOIDES CRONICAS Y TRASTORNOS DE LAS CELULAS PLASMATICAS
- Leucemia linfocítica crónica
- EPIDEMIOLOGIA
- FISIOPATOLOGIA
- PATOGENIA
- DIAGNOSTICO DE LEUCEMIA LINFOCITICA CRONICA
- ESTADIFICACION Y FACTORES PRONOSTICOS
- EVOLUCION CLINICA Y COMPLICACIONES
- TRATAMIENTO PARA LA LEUCEMIA LINFOCITICA CRONICA
- TRATAMIENTO DE LA ENFERMEDAD CON RECAIDAS O REFRACTARIA
- Leucemia prolinfocítica
- Mieloma múltiple
- EPIDEMIOLOGIA
- PATOGENIA
- INMUNOLOGIA
- DIAGNOSTICO DEL MIELOMA MULTIPLE
- ESTADIFICACION Y FACTORES PRONOSTICOS
- TRATAMIENTO DEL MIELOMA MULTIPLE
- Amiloidosis
XV LEUCEMIAS LINFOIDES CRONICAS Y TRASTORNOS DE LAS CELULAS PLASMATICAS
DR. BRUCE D. CHESON
Las leucemias linfoides crónicas son un grupo de trastornos linfoides de tipo clonal relativamente indolentes, principalmente derivados de las células B y que incluyen a la leucemia linfocítica crónica (LLC), la leucemia prolinfocítica (LPL), la leucemia de células peludas (LCP) y la fase leucémica de los linfomas no Hodgkin (LNH).
Las discrasias de las células plasmáticas se caracterizan por acúmulo de células plasmáticas malignas en la médula ósea, hueso o tejidos blandos. Su nombre refleja el hallazgo en la biopsia de hueso de múltiples tumores de células plasmáticas, en lugar de una sola masa. Estas enfermedades, incluyen al mieloma múltiple (MM), el plasmacitoma extramedular y la amiloidosis. La macroglobulinemia de Waldenström, caracterizada por una gamopatía IgM, linfadenopatía y hepatoesplenomegalia, se incluía antes junto con estos trastornos, pero en la actualidad se clasifica en forma más apropiada como un linfoma no Hodgkin indolente linfoplasmocitoide.1
Leucemia linfocítica crónica
EPIDEMIOLOGIA
La LLC es la forma más común de leucemia en adultos del mundo occidental. La incidencia anual de esta enfermedad parece estar disminuyendo en los Estados Unidos, de 10,000 nuevos casos por año hace una década a alrededor de 7,300 casos en 1998.2 Esta reducción puede reflejar, en parte, una clasificación más apropiada de otras neoplasias linfoides que antes se diagnosticaban mal como LLC. La edad media del diagnóstico es de 62 años, y solo 10 a 15 por ciento de los casos se diagnostican antes de los 50 años. La LLC parece ser más común en varones que en mujeres y es más frecuente en los judíos de ascendencia rusa o europea oriental que en la población general. Ocurre con frecuencia similar en negros y blancos. La LLC es poco común en Japón, China y otros países asiáticos.
FISIOPATOLOGIA
La etiología de la LLC se desconoce y no existen factores de riesgo conocidos, incluyendo radiación ionizante, químicos y fármacos.3-5 Se han reportado familias con casos múltiples de LLC y varios pares de gemelos afectados, con mayor frecuencia de padecimientos linfoproliferativos en los familiares en primer grado de los pacientes con LLC.6 También se han reportado cónyuges con LLC.6
PATOGENIA
La LLC es una expansión clonal de linfocitos de apariencia madura. Aunque la morfología de las células B normales y de la LLC es parecida, existen diferencias inmunológicas y de marcadores moleculares ente las dos poblaciones celulares. Tanto las células de LLC como las normales expresan antígenos de linaje B (v.gr., CD19 y CD20); sin embargo, las células de la LLC también expresan antígenos de activación (v.gr., CD23) y CD5. Las células B CD5+ no son únicas de la LLC, también pueden ser una fuente de anticuerpos y su número aumenta en trastornos autoinmunes como el lupus eritematoso generalizado y la artritis reumatoide, en la púrpura trombocitopénica inmune y después de un trasplante alogénico de médula ósea.
A pesar de ello, existen numerosas diferencias funcionales, inmunológicas y moleculares entre la célula B de la LLC y la célula B CD5+ benigna, incluyendo propiedades de adhesión, transformación por el virus de Epstein-Barr, proliferación en respuesta al estímulo con diversos mitógenos y transducción de señales.7,8
Citogenética
Pueden detectarse alteraciones citogenéticas adquiridas en alrededor del 50 por ciento de los pacientes con LLC por el uso de técnicas de bandeo convencionales,9 y estas alteraciones son más comunes en los estados avanzados de la enfermedad. Se pensaba que la trisomía 12, que ocurre en el 30 a 50 por ciento de los casos, era la alteración citogenética más común. Sin embargo, ensayos más sensibles, como la hibridización in situ con fluorescencia (HISF), han revelado defectos en más del 70 por ciento de los casos,10 con deleciones de 13q en más de la mitad de los pacientes, seguido de deleciones de 11q en 15 a 20 por ciento de los casos y trisomía 12 en el 15 a 20 por ciento. Más de la tercera parte de los pacientes tienen alteraciones complejas.
Biología molecular
No se ha implicado a una sola alteración genética en la patogenia de la LLC. Sin embargo, las células de LLC se caracterizan por un defecto en la apoptosis, o muerte programada. Existe expresión exagerada del gen bcl-2 en más del 70 por ciento de los casos, incluso en ausencia del rearreglo del cromosoma t(14;18).11 La relación entre el gen antiapoptósico bcl-2 y el gen proapoptótico BAX aumenta en las células de la LLC, lo que favorece la supervivencia celular. Por técnicas citogenéticas convencionales se han identificado deleciones que afectan a 13q34 en hasta el 70 por ciento de los casos; este defecto, que antes se pensaba que estaba en el sitio del gen supresor del retinoblastoma, ha demostrado ser telomérico a esa región.12 La región de pérdida contiene un gen supresor recién detectado denominado DBM (alterado en las neoplasias de células B). Existe una relación aparente entre la proteína antiapoptósica Mcl-1 y la resistencia a la quimioterapia.13
DIAGNOSTICO DE LEUCEMIA LINFOCITICA CRONICA
Manifestaciones clínicas
Los pacientes con LLC suelen estar asintomáticos al momento del diagnóstico y este se realiza en forma incidental al encontrar linfocitosis durante una evaluación de rutina. El examen fisico inicial es normal en el 20 a 30 por ciento de los pacientes y existe linfadenopatía, hepatoesplenomegalia o ambos en el 40 a 50 por ciento de los casos. Sin embargo, al progresar la enfermedad la adenopatía generalizada y la esplenomegalia son datos frecuentes.
Las infecciones son una complicación frecuente y pueden ser recurrentes, siendo los patógenos más comunes Staphylococcus, Streptococcus, otras bacterias que requieren opsonización y los herpesvirus.
Puede ocurrir un resultado positivo de antiglobulina Coombs en hasta el 20 por ciento de los casos con LLC, aunque la hemólisis clínica es aparente en solo la mitad de estos casos. La frecuencia de trombocitopenia inmune parece ser de alrededor del 2 por ciento. La anemia o la trombocitopenia autoinmunes suelen responder al tratamiento con esteroides. Si el paciente no mejora con esteroides pueden usarse otras alternativas, incluyendo inmunoglobulina en dosis altas y esplenectomía.14
La aplasia pura de células rojas ocurre en menos del 5 por ciento de los casos de LLC. Las opciones de tratamiento incluyen esteroides, ciclosporina con o sin esteroides concomitantes y quimioterapia sistémica para la LLC.15
Datos de laboratorio
Debe pensarse en el diagnóstico de LLC si existe aumento sostenido en el número de linfocitos pequeños de apariencia madura (i.e., a > 5,000/ml) circulantes en sangre periférica que no se explica por otros padecimientos médicos. El aspirado de médula ósea y la biopsia revelan infiltración de por lo menos 30 por ciento de linfocitos. Rara vez se requiere evaluar la médula ósea para confirmar el diagnóstico, pero el estudio proporciona importante información pronóstica y se requiere para evaluar la respuesta al tratamiento.16
La inmunofenotipificación ayuda a distinguir la LLC de otras leucemias linfoides crónicas, como la LPL, LCP y sus variantes, el LNH en fase leucémica (v.gr., linfomas linfoplasmacíticos, LNH de zona marginal, incluyendo el linfoma esplénico con linfocitos velloso y LNH de células en manto), y leucemia de células plasmáticas [ver figura 1 y tabla 1].1,16 Los linfocitos de la LLC son células B monoclonales que expresan inmunoglobulinas de superficie en bajo nivel y el antígeno CD5 junto con los antígenos normales de células B CD19, CD20 y CD23. El LNH en mano, otro trastorno de células B CD5+, puede distinguirse de la LLC por su intensa expresión de inmunoglobulinas de superficie y la falta de CD23.
ESTADIFICACION Y FACTORES PRONOSTICOS
La LLC es un trastorno clínicamente heterogéneo, algunos pacientes viven por décadas sin requerir tratamiento, mientras que otros mueren en meses después del diagnóstico. El primer sistema de estadificación que se adoptó en forma amplia fue la clasificación de cinco estadios de Rai [ver tabla 2]. Este sistema se simplificó después a tres estadios: riesgo bajo (estadio 0), riesgo intermedio (estadios I y II) y riesgo alto (estadios III y IV). En algunas partes de Europa se usa más la clasificación de Binet17 [ver tabla 3]. Aunque el sistema de Binet no identifica a los pacientes con estadio 0 de Rai, los dos sistemas tienen el mismo valor predictivo. Entre el 60 al 70 por ciento de los pacientes con LLC pertenecen a un grupo de riesgo bajo o intermedio.
Aunque la LLC tiende a ocurrir en pacientes mayores de 60 años, el 10 a 15 por ciento son menores de 50. Para cada estadio, los pacientes más jóvenes tienen una supervivencia promedio semejante a la de los mayores cuando se corrigen las causas de muerte no relacionadas a la LLC.18
Los factores de riesgo incluyen sexo masculino, raza negra, mal estado funcional y edad avanzada.19,20 El tiempo de duplicación de los linfocitos tiene mayor valor predictivo que el número absoluto de linfocitos circulantes.21 La beta2-microglobulina sérica, el CD23 soluble, 22-24 y los receptores solubles de interleucina-225 también predicen la evolución.
Las alteraciones citogenéticas simples se asocian con mayor supervivencia en comparación con las alteraciones citogenéticas complejas. Los pacientes con alteraciones 14q tienen especial peor pronóstico, mientras que la evolución de los enfermos con alteraciones 13q es semejante a la de los pacientes con cariotipo normal. No se recomiendan los estudios citogenéticos como parte de la evaluación de rutina del paciente con LLC porque son costosos, difíciles de realizar y, lo más importante, no dirigen las decisiones terapéuticas.
La relación de proteínas bcl-2 a BAX en las células de LLC favorece a la bcl-2 y correlaciona con resistencia clínica a la quimioterapia y a la evolución clínica.26,27 Las deleciones de p53 predicen una respuesta pobre al tratamiento con fludarabine o pentostatin.28
EVOLUCION CLINICA Y COMPLICACIONES
Condiciones de progresión
En alrededor del 10 a 15 por ciento de los pacientes con LLC se desarrolla una enfermedad linfoproliferativa más agresiva, con más frecuencia el síndrome de Richter, que es un linfoma agresivo de células grandes que ocurre en alrededor del 5 por ciento de los enfermos.29 Los pacientes característicamente presentan mayor linfadenopatía, hepatoesplenomegalia, fiebre, dolor abdominal, pérdida de peso, anemia progresiva y trombocitopenia, con elevación rápida en la cuenta de linfocitos en sangre periférica. La respuesta de los pacientes con síndrome de Richter al tratamiento sistémico es mala, con poca supervivencia.
La segunda transformación en frecuencia es la LPL. Otras incluyen leucemia linfocítica aguda, leucemia de células plasmáticas, mieloma múltiple y enfermedad de Hodgkin.
Neoplasias secundarias
Las neoplasias secundarias parecen ocurrir con más frecuencia en pacientes con LLC y se relacionan tanto con los defectos inmunes asociados a la enfermedad como con las consecuencias del tratamiento. Estas neoplasias afectan principalmente a la piel, próstata y tubo digestivo.30-33
Alteraciones inmunológicas
En la LLC se han reportado diversos defectos inmunológicos. Las células B de la LLC producen cantidades disminuidas de inmunoglobulinas normales en respuesta a los estímulos antigénicos y existen alteraciones cuantitativas y cualitativas de células B normales, células T y células asesinas naturales, reducción en el número y función de las células T normales y menor activación del complemento.34,35
TRATAMIENTO PARA LA LEUCEMIA LINFOCITICA CRONICA
El pronóstico de los pacientes con LLC se relaciona con la fase de la enfermedad. Los pacientes con una enfermedad en estadio temprano pueden vivir una vida normal; sin embargo, la evolución no ha mejorado en forma significativa durante las últimas décadas para quienes tienen enfermedad avanzada. Debido a que los tratamientos disponibles en la actualidad no son curativos, es importante el momento en que se inicia el tratamiento. No existe ventaja en la supervivencia para los pacientes con enfermedad en fase temprana que se tratan al hacer el diagnóstico. El Grupo Cooperativo Francés sobre LLC realizó dos estudios de pacientes con enfermedad en fase A de Binet. En el primero los pacientes fueron distribuidos para recibir tratamiento diario con clorambucil por vía oral u observación. En el segundo los pacientes recibieron clorambucil intermitente más prednisona o no se les dió tratamiento. Ningún estudio detectó ventajas con la intervención temprana y en el primer estudio existió mayor número de tumores sólidos secundarios fatales,33 quizá relacionados con el clorambucil. Por lo tanto, el tratamiento debe iniciarse solo cuando esté indicado por uno o más síntomas relacionados con la enfermedad (v.gr., fiebre, calosfríos, pérdida de peso o fatiga importante), falla progresiva de la médula ósea con anemia, trombocitopenia o ambos, anemia o trombocitopenia autoinmunes, hepatoesplenomegalia o linfadenopatía masivas o progresivas, o infecciones recurrentes.35 Aunque no existe un número absoluto de linfocitos que indique la necesidad de tratamiento, el tiempo de duplicación rápida de los mismos (< 6 meses) puede justificar el inicio del tratamiento.21
Agentes aislados
El medicamento estándar para la LLC ha sido el clorambucil. Cuando se administra este fármaco a dosis de 6 a 14 mg por vía oral diario por 4 a 8 semanas o en dosis más altas, pulsos intermitentes de 20 mg/m2 por vía oral cada 3 semanas o 30 a 40 mg/m2 una vez al mes, ocurre regresión de la enfermedad en los ganglios linfáticos, hígado y bazo, y normalización de las cuentas de células en sangre periférica en alrededor de la mitad de los pacientes sin tratamiento previo, aunque es rara la desaparición del padecimiento.36-38 La ciclofosfamida parece tener una actividad semejante a la del clorambucil, pero se usa solo cuando el clorambucil no es bien tolerado.
Los esteroides son menos activos que los agentes alquilantes en la LLC y deben reservarse para los pacientes con complicaciones autoinmunes por el riesgo de infecciones bacterianas, virales y micóticas, diabetes y osteoporosis.
Análogos de purina
Fludarabine El fludarabine ha demostrado ser el agente más activo en el tratamiento de la LLC. Se estudió en forma inicial en pacientes que no respondieron o que tuvieron recaídas durante el tratamiento con agentes alquilantes.39-42 Debido a la dificultad para comparar los resultados de los diferentes tratamientos, el Grupo de Trabajo sobre LLC patrocinado por el Instituto Nacional de Cáncer de los EUA estandarizó los criterios de elegibilidad, respuesta y toxicidad. Con el uso de estas normales se lograron remisiones completas clínicas y hematológicas en el 3 a 13 por ciento de los pacientes, con una respuesta global de 40 a 50 por ciento. Los resultados variaron por factores como la edad del paciente, el estadio y la capacidad funcional. El tiempo promedio de progresión fue de alrededor de 18 meses para los pacientes refractarios a agentes alquilantes y de 17 meses para pacientes que tuvieron recaídas después de una respuesta previa. La supervivencia promedio fue de 29 meses para los pacientes con recaídas y de 9 meses para los refractarios a tratamiento.
Cuando se usó fludarabine como tratamiento inicial la tasa de respuesta completa fue de alrededor de 30 por ciento, con una tasa de respuesta global de 70 por ciento.38,43,44 Los estudios aleatorios han demostrado tasas de respuesta y tiempos para la progresión más altos con fludarabine que con los agentes alquilantes y los esquemas basados en antraciclinas, aunque no se ha observado ventaja en la supervivencia.38,43 El tiempo promedio para la progresión en pacientes sin tratamiento previo fue de 32 meses, con supervivencia promedio mayor de 5 años. El fludarabine es una opción de elección eficaz para la LLC.
Los principales efectos tóxicos por este medicamento con el esquema actual de 25 mg/m2 IV durante 5 días consecutivos una vez por mes son la mielosupresión moderada y la inmunosupresión severa, con neurotoxicidad ocasional, en especial con dosis más altas que las recomendadas.45,46 Las cuentas de linfocitos, en especial de células CD4, disminuye en semanas y no se normaliza durante un año o más después de suspendido el tratamiento.44,46 No se recomienda el uso de antibióticos profilácticos porque sería imposible cubrir todo el espectro de patógenos potenciales, a pesar del costo y toxicidad potencial de los medicamentos adicionales.
Otros agentes Tanto el cladribine como la 2'-deoxidoformicina (pentostatin) son activos en la LLC, pero se usan con menos frecuencia que el fludarabine por sus tasas de respuesta relativamente menores, remisiones menos duraderas y mayor costo.47,48
Quimioterapia combinada
Los esquemas combinados usados con más frecuencia en LLC incluyen clorambucil más prednisona (CP) y ciclofosfamida, vincristina y prednisona (CVP). Las tasas de respuesta reportadas con los esquemas CP y CVP en pacientes sin tratamiento previo con enfermedad en fase avanzada varían de menos de 10 a más de 60 por ciento, dependiendo en gran parte de los criterios usados para evaluar la respuesta. No es común lograr desaparición de la enfermedad evidente y la supervivencia promedio es menor de 2 años.36,49-51
El tratamiento combinado no es claramente superior al de un solo fármaco. Los estudios iniciales, incluyeron pocos casos, sugieren que la prednisona puede aumentar la tasa de respuesta al clorambucil, pero no aumentar la supervivencia.49,50 Los esquemas más intensos, como el CHOP (ciclofosfamida, doxorrubicina [hidroxidaunomicina], vincristina [Oncovin] y prednisona), no son claramente mejores que los menos intensos.36,51-53 En la actualidad se desarrollan combinaciones que incluyen fludarabine.
TRATAMIENTO DE LA ENFERMEDAD CON RECAIDAS O REFRACTARIA
Enfoques estándar
Los pacientes con una recurrencia asintomática no suelen tratarse sino hasta que esté indicado el tratamiento por los mismos criterios que al inicio, en un intento paliativo. El fludarabine es el agente de salvamento estándar para pacientes que no han respondido a los agentes alquilantes. Los pacientes que son refractarios a fludarabine tienen menos probabilidad de responder a agentes alquilantes.
Tratamiento con dosis altas y apoyo de células tronco
La experiencia con el trasplante alogénico de la médula ósea en la LLC es limitada, en parte porque la edad promedio de los pacientes al momento del diagnóstico es mayor de 60 años.54-56 Alrededor de la mitad de los pacientes que reciben un trasplante de médula ósea pueden permanecer sin enfermedad por periodos prolongados; sin embargo, no es claro si están curados. Por desgracia, la mortalidad relacionada al tratamiento es de 25 a 50 por ciento.
El trasplante autólogo de células tronco tiene una función dudosa en los pacientes con LLC.56,57
Tratamiento biológico
La mayor experiencia con un agente biológico ha sido con interferón alfa. Las respuestas tienden a ser breves, sin influencia en la evolución. Los estudios preliminares con el anticuerpo monoclonal anti-CD52 CAMPATH han sido promisorios tanto en LLC como el LPL.58,59 El anticuerpo C2B8 anti CD20 (rituximab), que tiene actividad considerable en los linfomas foliculares, no parece tener la misma eficacia en la LLC.
Tratamiento de sostén
Factores de crecimiento mieloide No se ha demostrado si los factores de crecimiento mieloide pueden proteger contra la mielosupresión inducida por la quimioterapia, por lo que no se incluyen en el tratamiento estándar.
Inmunoglobulinas intravenosas La mayoría de los pacientes con LLC sufren hipogamaglobulinemia, que es más común en las fases avanzadas de la LLC y que empeora al progresar la enfermedad. Desde el punto de vista histórico, los patógenos más comunes en la LLC son los que requieren de opsonización antes de ser destruidos. Por desgracia, las inmunoglobulinas intravenosas en dosis altas no protegen a los pacientes de diversas infecciones bacterianas, virales o micóticas. No influyen en la supervivencia global y no tienen buena relación costo eficacia.60,61 Este tratamiento debe reservarse para pacientes con infecciones bacterianas demostradas de repetición.
Eritropoyetina La anemia en la LLC no es consecuencia de producción deficiente de eritropoyetina, sino más bien consecuencia de sustitución de la médula ósea, secuestro esplénico o hemólisis. Sin embargo, la eritropoyetina puede reducir la necesidad de transfusión en pacientes seleccionados y puede intentarse una prueba con este agente en los pacientes con LLC y anemia que no tienen otra causa obvia y corregible de la anemia.62
Esplenectomía
La esplenectomía puede tener un papel importante en el tratamiento paliativo de los pacientes con LLC que no se han beneficiado del tratamiento sistémico.14 La trombocitopenia responde en grado variable en casi todos los pacientes. Cuando se realiza este procedimiento por un cirujano experimentado la mortalidad relacionada es menor al 10 por ciento.
Leucemia prolinfocítica
La LPL representa la forma más común de transformación agresiva en los pacientes con LLC. Los pacientes presentan deterioro clínico, con frecuencia con síndrome consuncional. Comparado con los pacientes cuya LPL ha evolucionado de una LLC, los que tienen LPL de novo tienden a ser de mayor edad (70 o más años de edad) que los que han sufrido transformación (alrededor de 60 años), tienen linfocitosis más importante, esplenomegalia masiva y no moderada y casi nunca linfadenopatía importante. El frotis de sangre periférica revela una población dual de células, algunas son linfocitos pequeños característicos de la LLC y otras son más grandes con nucleolos prominentes. La LPL puede presentarse como una enfermedad de novo; en cualquier caso, se caracteriza por una cuenta de leucocitos alta, con por lo menos el 55 por ciento de prolinfocitos circulantes. En los pacientes con LPL primaria se ha encontrado rearreglo cromosómico t(6;12). Las alteraciones de p53, que ocurren en más de la mitad de los pacientes pueden causar resistencia a la quimioterapia.
Se han publicado resultados preliminares promisorios con los análogos de purina y el anticuerpo monoclonal CAMPATH.
Leucemia de células peludas
La LCP es una neoplasia de células B poco común que ocurre en alrededor de 500 pacientes nuevos cada año en los Estados Unidos. La enfermedad es más común en personas mayores, predominando los hombres en relación con las mujeres. Aunque los pacientes con LCP pueden no tener síntomas al inicio, suelen presentar manifestaciones de menor producción de la médula ósea, como infecciones, sangrado y debilidad. En casos raros los pacientes sufren un trastorno que recuerda a una vasculitis. El examen físico es notable por la esplenomegalia que con frecuencia es masiva y la hepatomegalia asociada; con menos frecuencia ocurre linfadenopatía con esplenomegalia. La evaluación de laboratorio revela pancitopenia, que aumenta la posibilidad de anemia aplástica, leucemia aguda, síndrome mielodisplásico o LCP. Puede requerirse una biopsia de médula ósea para hacer el diagnóstico porque el aspirado suele taparse por las células peludas, y también existe fibrosis. Las células malignas son de origen B, expresando CD19, CD20 y el antígeno monocito CD11c. Es posible que el marcador más específico sea Bly-7 (CD103).
La LCP suele ser un trastorno indolente y el 10 por ciento de los pacientes puede nunca requerir tratamiento. El tratamiento está indicado si existe esplenomegalia masiva o progresiva, empeoramiento de las cuentas celulares, infecciones recurrentes, más de 20,000 células peludas/ml de sangre periférica o linfadenopatía importante. Hasta hace 15 años la esplenectomía era el tratamiento estándar para la LCP porque mejora los síntomas relacionados con la esplenomegalia y en la mayoría de los casos las cuentas de sangre periférica. Sin embargo, el procedimiento no afecta la enfermedad en sí. En la actualidad se realizan menos esplenectomías por la posibilidad de tratamientos sistémicos eficaces. El interferón alfa fue el primer agente que logró respuestas significativas en pacientes con LCP. La mayoría de los pacientes sufren mejoría clínica y hematológica, aunque en solo 10 por ciento de los mismos la respuesta es completa e invariablemente la enfermedad recurre después de suspender el tratamiento. El interferón alfa se asocia con síntomas de tipo catarral (fiebre, mialgias y malestar) en casi todos los pacientes. Otros efectos tóxicos incluyen erupción, alteraciones en el sitio de aplicación y síntomas gastrointestinales como náusea, vómito y anorexia.
Los análogos de purina han revolucionado el tratamiento de los pacientes con LCP. En un estudio realizado en Norteamérica, pacientes con LCP sin tratamiento previo recibieron interferón alfa o pentostatin, se observaron tasas de respuesta completa en alrededor del 11 por ciento de los pacientes tratados con interferón alfa, comparado con 76 por ciento en los que recibieron pentostatin, con una diferencia significativa en la duración de la respuesta a favor del pentostatin.63 Recientemente se ha demostrado que el cladribine induce respuestas en 80 a más del 90 por ciento de los pacientes, con 65 a 85 por ciento de remisiones completas.64 Solo 25 a 30 por ciento de los pacientes recaen después del seguimiento prolongado. En muchos casos la recaída se caracteriza solo por aumento en las células peludas de la médula ósea, sin indicación de tratamiento. La mayoría de los pacientes que requieren tratamiento logran una segunda respuesta duradera. Los resultados con pentostatin y cladribine son equivalentes y el medicamento de elección dependerá de la preferencia del médico.
Mieloma múltiple
EPIDEMIOLOGIA
El MM es poco común en personas menores de 40 años, su incidencia aumenta con rapidez después de los 50 años, con igual frecuencia en hombres y mujeres. Aunque las personas de raza negra se afectan más que los blancos, la evolución en los dos grupos es semejante, dependiendo del estadio.65 Se calcula que existieron alrededor de 14,000 nuevos casos en 1998 en los Estados Unidos.2 No se ha identificado relación entre el MM y el estado socioeconómico. La incidencia de esta enfermedad parece ser menor en chinos y otros grupos asiáticos. La etiología del MM no es clara. Se ha implicado a la exposición a radiación ionizante, pinturas, solventes y pesticidas, pero nunca ha podido establecerse una relación firme.
PATOGENIA
El MM se caracteriza por acúmulo de células plasmáticas en la médula ósea y, con menos frecuencia, en los tejidos blandos u órganos. La célula maligna no es la célula plasmática madura, sino una célula previa en la vía de diferenciación de la célula B, después del rearreglo de los genes de las cadenas pesadas. Estas células tienen la apariencia inmadura de un plasmablasto, pueden ser identificadas en la sangre periférica de los pacientes con MM y se considera que son precursoras de las células malignas que se encuentran en la médula ósea. Expresan una variedad diferente de moléculas de adhesión (v.gr., CD11b),66 que parecen permitir que las células B circulantes se fijen a las células del estroma de la médula ósea no malignas que producen citocinas capaces de apoyar el crecimiento de las células de mieloma, como IL-6. La IL-6 actúa como un factor de crecimiento autócrino y parácrino en el MM, estimulando así el crecimiento celular y evitando la apoptosis. Se ha sugerido que los herpesvirus (herpesvirus asociado al sarcoma de Kaposi o el virus del herpes simple-8) tienen algún papel en la patogenia.67
Los análisis citogenéticos son difíciles de realizar en el MM. Sin embargo, estudios recientes que han usado HISF han demostrado aneuploidía en alrededor del 70 por ciento de los casos.68 Las alteraciones más frecuentes afectan los cromosomas 13 (13q-) y 14 (14q+). Estos defectos pueden causar menor apoptosis y resistencia subsecuente al tratamiento. Se han identificado mutaciones de p53 en los pacientes con MM que presentan recaídas, estas parecen correlacionar con mala evolución.
INMUNOLOGIA
El MM se caracteriza por la producción exagerada de inmunoglobulina monoclonal. La frecuencia de los diversos tipos de inmunoglobulinas del MM es paralela a la concentración de estas Igs: IgG en el 60 a 70 por ciento, IgA en el 20 por ciento y cadenas ligeras en solo el 15 por ciento. Se han reportado algunos casos de IgD e IgE, y alrededor del 1 por ciento de los pacientes son no secretores. Un segundo hallazgo son los niveles disminuidos de inmunoglobulinas residuales normales.
Las proteínas de Bence Jones (cadenas ligeras urinarias) se detectan en la orina con antisuero contra las cadenas ligeras sin un componente anti-cadenas pesadas.
DIAGNOSTICO DEL MIELOMA MULTIPLE
El diagnóstico de MM suele hacerse en forma incidental cuando se detecta una mayor concentración de proteína en suero en la evaluación de rutina o al evaluar a un paciente por anemia, disfunción renal o hipercalcemia asintomática.
Gamopatía monoclonal de significado no determinado
Ocurre gamopatía monoclonal de significado no determinado (GMSND) en alrededor del 1 por ciento de la población general y en el 3 por ciento de las personas mayores de 70 años; la IgG es la proteína aumentada con más frecuencia. Las características del MM que lo distinguen de la GMSND incluyen concentraciones mayores de proteína M en suero y orina, aumento en el porcentaje de células plasmáticas en la médula ósea, aumento en el índice de marcación de células plasmáticas, presencia de lesiones óseas líticas y presencia de células plasmáticas circulantes en la sangre periférica. Después de un seguimiento prolongado, pueden identificarse cuatro grupos de pacientes con GMSND: (1) los que tienen gamopatía monoclonal limitada, (2) los que tienen concentración e inmunoglobulina sérica mayor de 3 g/dl pero no requieren ningún tratamiento (10 por ciento), (3) los que mueren sin mieloma múltiple u otra enfermedad relacionada (47 por ciento) y (4) aquellos en los que se desarrolla mieloma, amiloidosis, macroglobulinemia u otro trastorno linfoproliferativo (24 por ciento de los cuales son mielomas). Ocurre progresión a una neoplasia en 8 a 10 años. Por desgracia no existen estudios de laboratorio que predigan con exactitud qué pacientes con GMSND progresarán a MM.
Cuadro clínico
El diagnóstico de MM requiere de por lo menos uno de los siguientes criterios mayores: plasmocitoma, médula ósea con por lo menos el 30 por ciento de células plasmáticas y proteína M en la electroforesis que tenga niveles de IgG mayores de 3.5 g/dl, IgA mayor de 2.0 g/dl o excreción de cadenas ligeras en la orina mayor de 1 g/24 h. También se requieren uno o más de los siguientes criterios menores: plasmacitosis en la médula ósea mayor de 10 por ciento, proteína M en el suero u orina pero menor de los niveles mencionados en los criterios mayores, lesiones óseas líticas y reducción de las inmunoglobulinas residuales.
El mieloma múltiple puede afectar muchos órganos, incluyendo los huesos, riñones, nervios, corazón e hígado [ver tabla 4].
Plasmacitomas Los plasmacitomas son tumores sólidos formados por células plasmáticas y que pueden ocurrir en los tejidos blandos (plasmacitomas extramedulares). Se presentan como masas palpables pero con frecuencia causan compresión de la médula espinal. Estos tumores pueden curarse con radioterapia local. Los pacientes con plasmacitomas que ocurren en el hueso pueden responder al inicio a la radioterapia, pero la mitad progresarán a MM franco, dos tercios lo harán antes de 3 años después del diagnóstico. La persistencia de proteína M después de la radioterapia se asocia con mayor probabilidad de evolución a MM.
Enfermedad ósea La enfermedad ósea es una de las características más comunes y diagnósticas del MM. Aunque las lesiones líticas son típicas [ver figura 2], la osteopenia es la forma más común de afección ósea en el mieloma. También se ha reportado enfermedad osteoesclerótica. Es notable el aumento en la actividad osteoclástica en los huesos afectados, y se cree que se relaciona con el llamado factor activador de osteoclastos, que es una manifestación de los efectos del factor de necrosis tumoral, la IL-1 y la IL-6.
La radioterapia para las lesiones óseas dolorosas en un paciente con MM recién diagnósticado debe diferirse lo más posible hasta que pueda evaluarse la eficacia del tratamiento sistémico porque la radioterapia puede impedir la administración de dosis adecuadas de quimioterapia. Son excepciones a esto la compresión de la médula espinal y las fracturas potenciales en huesos que cargan peso y que requieren de intervención inmediata. Para los pacientes con recaídas la radioterapia local puede proporcionar un beneficio paliativo importante.
Las fracturas por compresión vertebral durante el curso del tratamiento sistémico no justifican siempre la administración de radiación porque estas lesiones suelen ser causadas por osteopenia, no necesariamente representan progresión de la enfermedad y no responden siempre a la radiación.
El pamidronato es un bifosfonato de segunda generación que ha sido estudiado en un estudio clínico controlado con placebo en pacientes no tratados antes y en pacientes con recaídas y MM en estadio III.69 El fármaco proporciona protección significativa contra las complicaciones esqueléticas (medidas por el tiempo que transcurre antes del primer evento esquelético), parece mejorar la calidad de vida y quizá proporciona ventajas para la supervivencia.70 Como resultado, en la actualidad se recomienda administrar bifosfonatos a todos los pacientes con evidencia de afección ósea, aunque algunos clínicos recomiendan este agente en todos los pacientes con MM.
Anemia Ocurre anemia en la mayoría de los pacientes con MM, en especial al progresar la enfermedad, como consecuencia de sustitución de la médula ósea, insuficiencia renal y anemia asociada a la enfermedad crónica. En los pacientes con MM que tienen niveles séricos inapropiadamente bajos de eritropoyetina, esta sustancia puede disminuir la necesidad de transfusión de paquetes eritrocitarios.71
Hipercalcemia Alrededor del 10 a 20 por ciento de los pacientes con MM presentan hipercalcemia. Los niveles de calcio en suero, corregidos según la albúmina, pueden usarse para identificar a los pacientes con riesgo de síntomas relacionados a la hipercalcemia. Las paraproteínas pueden en ocasiones fijar calcio, dando un nivel elevado falso; por lo tanto, debe medirse el calcio libre (ionizado) en el paciente asintomático con un nivel elevado de calcio en suero.
Los pacientes sin tratamiento previo y que están asintomáticos o tienen solo síntomas leves relacionados con hipercalcemia leve o moderada deben ser tratados con quimioterapia sistémica, incluyendo esteroides. Los pacientes con niveles altos de calcio sérico o síntomas como letargo, confusión y constipación requieren de hidratación con solución salina. Los bifosfonatos son útiles para disminuir la concentración de calcio sérico, lo mismo que la mitramicina y la calcitonina.
Insuficiencia renal La insuficiencia renal es común en el MM y puede deberse a varios mecanismos. Los túbulos pueden obstruirse por la proteína del mieloma (riñón de mieloma) o los riñones pueden infiltrarse por proteína amiloide. El mayor riesgo de infecciones crea predisposición para pielonefritis. Otras causas incluyen hipercalcemia, nefropatía por uratos, síndrome nefrótico, hiperviscosidad, infiltración de los riñones por células plasmáticas y uropatía obstructiva. Los pacientes con sospecha de MM y función renal anormal deben mantenerse bien hidratados y no deben someterse a procedimientos que deshidraten como tomografía computada con medio de contraste o urografía excretora, lo que puede precipitar insuficiencia renal aguda en el paciente con daño renal subclínico. La plasmaféresis puede aliviar la insuficiencia renal aguda. Pueden ser útiles la diálisis e incluso el trasplante renal.
Hiperviscosidad Los signos y síntomas de hiperviscosidad, como disnea, confusión y dolor torácico, son poco comunes en pacientes con MM por IgG, pero ocurren con concentraciones de proteína en suero muy altas. La hiperviscosidad se presenta con más frecuencia en pacientes con MM de tipo IgA por la mayor polimerización de esta paraproteína. El tratamiento debe dirigirse a tratar el MM subyacente. Puede considerarse la realización de plasmaféresis si se requiere reducción rápida de la paraproteína para aliviar los síntomas.
Enfermedades infecciosas Los pacientes con mieloma tienen mayor riesgo de infecciones bacterianas que ponen en peligro la vida, incluso en presencia de números normales de granulocitos circulantes. Esta alteración se relaciona con la menor síntesis de inmunoglobulinas normales y una activación del complemento defectuosa. Al inicio del padecimiento los pacientes pueden infectarse con organismos encapsulados que requieren de opsonización (v.gr., Streptococcus pneumoniae, Haemophilus influenzae y Staphylococcus), mientras que el uso de quimioterapia aumenta la posibilidad de infecciones por gram negativos.
Es poco probable que la vacuna polivalente antineumocócica sea eficaz porque los pacientes responden mal a los antígenos bacterianos y tienen otros defectos bactericidas que no son corregidos por la vacunación. Ni las inmunoglobulinas intravenosas ni los antibióticos profilácticos tienen buena relación costo eficacia o protegen de modo uniforme, y deben reservarse para los pacientes con infecciones bacterianas recurrentes demostradas.
Síntomas neurológicos La compresión de la médula es un dato de presentación común del plasmacitoma y se considera una urgencia oncológica que requiere descompresión y radioterapia. Las paraproteinemias pueden asociarse con otros síntomas neurológicos, como el síndrome de POEMS. POEMS es el acrónimo de una serie de datos que incluyen polineuropatía (generalmente sensorial), organomegalia (v.gr., hepatomegalia), endocrinopatía (v.gr., diabetes, amenorrea y ginecomastia), proteína M y cambios cutáneos (v.gr., hiperpigmentación). Otras características son la linfadenopatía y las lesiones óseas osteoescleróticas.
ESTADIFICACION Y FACTORES PRONOSTICOS
Una vez hecho el diagnóstico de MM la evaluación inicial debe incluir una biometría hemática completa, así como aspiración y biopsia de la médula ósea. La electroforesis sérica y la cuantificación de inmunoglobulinas determinan la concentración de la paraproteína, que correlaciona con la masa tumoral. La inmunoelectroforesis define el subtipo de inmunoglobulina. La electroforesis urinaria detectará cadenas ligeras, cuando existen. Se realizará una investigación radiológica ósea para identificar los sitios de afección y las fracturas potenciales. Los gamagramas óseos con radionúclidos son de poco valor para evaluar el MM.
El cuadro clínico de los subtipos de MM varía mucho. Por ejemplo, los pacientes que secretan solo cadenas ligeras pueden presentar insuficiencia renal progresiva, al inicio sin mieloma franco. Los pacientes con mieloma por IgD tienen una evolución más agresiva, caracterizada por alta frecuencia de insuficiencia renal, plasmacitomas y amiloidosis, y tienen supervivencia más corta que los pacientes con MM de IgG a pesar de tener concentraciones bajas de paraproteína.72 El mieloma de IgE, que es raro, parece asociarse con mayor frecuencia de leucemia de células plasmáticas.
Los pacientes suelen estadificarse de acuerdo con la clasificación de Durie y Salmon73 [ver tabla 5], que correlaciona bien con la masa tumoral y el pronóstico. El estadio está determinado por el nivel de proteína M, el número de lesiones óseas líticas, la concentración de hemoglobina y el calcio sérico (corregido según la albúmina). Los pacientes después se subdividen en clases A y B con base en la concentración de creatinina sérica.
La supervivencia promedio es de alrededor de 5 años para los pacientes con enfermedad en estadio IA, pero de 15 meses para los que están en estadio IIA. Sin embargo, el sistema de estadificación no tiene unformidad confiable para predecir la evolución y ha sido sustituido por marcadores biológicos de mayor importancia, principalmente la beta2-microglobulina, el índice de marcación de células plasmáticas, y las alteraciones citogenéticas, así como la deshidrogenasa láctica, el Ki-67, los niveles séricos de IL-6, la proteína C reactiva y la presencia de células plasmáticas circulantes.68,74-78 La citogenética no es parte de la evaluación estándar porque las pruebas son costosas y no influyen en el tratamiento. La imagen por resonancia magnética puede ser útil en la evaluación de la afección de la médula ósea y como indicador pronóstico.79
TRATAMIENTO DEL MIELOMA MULTIPLE
Indicaciones de tratamiento
Una proporción pequeña de pacientes se clasifican como con MM latente según las siguientes características: proteína M en el suero menor de 3.0 g/dl, menos del 10 por ciento de infiltración de células plasmáticas en la médula ósea, no anemia, niveles de calcio y creatinina normales en suero, ausencia de lesiones líticas y un índice de marcaje de células plasmáticas bajo. Estos pacientes pueden permanecer estables durante varios años sin tratamiento.
Debe considerarse el tratamiento del MM en estadios I y II para los pacientes con enfermedad progresiva, que incluye un aumento en el nivel de proteína M en el suero, orina o ambos, reducción en el nivel sérico de hemoglobina, falla progresiva de la médula ósea con neutropenia, trombocitopenia o ambos, aumento en la concentración de calcio, creatinina o ácido úrico séricos, lesiones óseas líticas múltiples, dolor óseo, infecciones recurrentes y plasmacitoma extramedular. Los pacientes con enfermedad en fase III generalmente tienen síntomas u alguna otra alteración hematólogica o química en el momento de su evaluación que justifica el tratamiento.
Tratamiento estándar
Una vez que se ha tomado la decisión de que el tratamiento está indicado existen algunas opciones terapéuticas eficaces. Los agentes aislados que son activos incluyen a los esteroides, los agentes alquilantes y el interferón alfa. Por lo general se usa melfalán con prednisona (MP) por la alta tasa de respuesta cuando se administran ambos fármacos. El esquema MP se administra cada 3 a 4 semanas durante 6 a 9 meses, y el tratamiento se continúa cuando existe respuesta. Lo más frecuente es que la concentración de la paraproteína disminuya a un nivel estable (fase de meseta) y continuar el tratamiento después de este punto no prolonga la supervivencia y sí aumenta las complicaciones (v.gr., infecciones). Otros agentes alquilantes también son activos pero no tienen ventajas sobre el melfalán. Algunos medicamentos usados para tratar a los pacientes con MM, como las antraciclinas, tienen utilidad limitada como agentes aislados.
Los esteroides (sea prednisona o dexametasona) pueden disminuir la proteína M en hasta el 40 por ciento de los pacientes no tratados antes y en el 20 a 40 por ciento de los pacientes que no han respondido al tratamiento inicial. Los esteroides también son una opción terapéutica para los pacientes ancianos y para los que tienen mal estado funcional.
La respuesta clínica suele asociarse con reducción del dolor óseo y normalización de la creatinina, el calcio y la hemoglobina. La supervivencia promedio de los pacientes con MM tratados con melfalán y prednisona es de 2.5 a 3 años y solo el 5 a 10 por ciento de los pacientes sobrevive 10 años o más.
Los esquemas de quimioterapia combinada (v.gr., vincristina, carmustina, melfalán, ciclofosfamida y prednisona; vincristina, carmustina, doxorrubicina y prednisona; o vincristina, ciclofosfamida, doxorrubicina y prednisona) no han demostrado mayor eficacia que la MP. El esquema VAD (vincristina, doxorrubicina [Adriamicina] y desametasona) como esquema inicial es menos tóxico para la población de células tronco que la MP y puede ser preferible para los pacientes que se consideran candidatos a trasplante autólogo de células tronco.
El interferón alfa ha sido uno de los agentes biológicos más estudiados. Tiene actividad como agente aislado, pero aún es motivo de controversia si ayuda al efecto de la quimioterapia durante la inducción o el tratamiento de mantenimiento. En un estudio realizado por Mandelli y colaboradores,80 pacientes que respondieron al esquema MP fueron distribuidos para recibir mantenimiento con interferón alfa o solo ser vigilados. Se observó una aparente prolongación del tiempo antes de la progresión y de la supervivencia con el interferón alfa, pero que desapareció al prolongar el tiempo de seguimiento. La mayoría de los otros datos publicados no han demostrado la utilidad del interferón alfa. Considerando la toxicidad asociada, el costo del agente y su beneficio clínico limitado, en la actualidad no se recomienda el interferón alfa como un componente habitual del tratamiento del MM.
Enfermedad con recaídas o refractaria
Las recaídas después de la respuesta inicial al tratamiento es común en el MM. Es poco frecuente que vuelva a haber respuesta a MP y ésta es de menos duración que con el tratamiento inicial, además de que se asocia con mayor mielosupresión. El esquema de salvamento estándar consiste en VAD, una infusión continua de 4 días de vincristina y doxorrubicina a través de un catéter y dexametasona por vía oral, que se repite los días 9 a 17 y en ciclos de cada 21 a 28 días. Se logran respuestas en la tercera parte de los pacientes resistentes y en casi la mitad de los que han sufrido una recaída.
Trasplante de células tronco El trasplante alogénico de médula ósea (TAMO) no se ha usado en forma muy frecuente en el MM porque los pacientes tienden a ser ancianos. Este método terapéutico induce remisiones en el 40 a 50 por ciento de los pacientes, y el 25 a 30 por ciento permanece vivo después de alrededor de 7 años. Sin embargo, pueden ocurrir recaídas después de varios años, aumentando la duda de si algún paciente en realidad se cura. Debido a la alta tasa de morbimortalidad asociada al procedimiento, el TAMO suele reservarse para los pacientes más jóvenes (< 50 años) sin respuesta o con enfermedad progresiva.
En los casos de recaída o refractario se han realizado estudios de quimioterapia en dosis altas con trasplante alogénico de células tronco. La frecuencia y duración de la respuesta puede predecirse por el nivel de deshidrogenasa láctica, la beta2-microglobulina, el estado funcional, la edad (>50 años) y la respuesta al tratamiento previo. Para los pacientes con buen perfil de riesgo (i.e., los que no tienen factores de mal pronóstico o solo uno), la tasa de respuesta completa es de 20 por ciento, con un periodo promedio de supervivencia sin recaídas de 1.5 años y una supervivencia global de 3.5 años. Cuando existen dos a tres factores de alto riesgo los resultados son sustancialmente peores, con un periodo promedio de supervivencia sin recaídas de 7 meses y una supervivencia global de 20 meses. Si existen cuatro más factores de alto riesgo la duración promedio de la respuesta es de 3 meses y la supervivencia global de 5 meses.
Recientemente algunos pacientes recibieron este tratamiento en etapas tempranas de la enfermedad. En un estudio realizado en Francia,81 pacientes sin tratamiento previo menores de 65 años recibieron dosis estándar de quimioterapia o quimioterapia en dosis alta con trasplante autólogo de células tronco. Existió una tasa de respuesta del 81 por ciento en el grupo con dosis altas, incluyendo 22 por ciento de remisiones completas y un 16 por ciento de remisiones parciales muy buenas, comparado con 57, 5 y 9 por ciento, respectivamente, en el grupo con tratamiento estándar. Estas diferencias fueron muy significativas. Sin embargo, la supervivencia a 6 años libre de eventos en el grupo trasplantado fue de solo 24 por ciento. En la actualidad se realiza un estudio aleatorio norteamericano extenso que, en combinación con el estudio francés, caracterizará mejor la función de la quimioterapia con apoyo autólogo o alogénico de células tronco.
Amiloidosis
La amiloidosis se caracteriza por el depósito de fibrillas de amiloide en varios tejidos.82 Puede ocurrir como secundaria a un padecimiento inflamatorio crónico (amiloide A o AA) o como una condición primaria (amiloide asociado a cadenas ligeras, o AL) en el caso de neoplasias de células B. En el AL las fibrillas están formadas por fragmentos de las cadenas ligeras de las inmunoglobulinas, con proliferación clonal de las células plasmáticas de la médula ósea y proteinuria de Bence Jones. La incidencia de amiloidosis primaria en los Estados Unidos es de 5.1 a 12.8 casos por millón de personas-año. Esta enfermedad afecta principalmente el corazón en el 90 por ciento de los casos y también el hígado, bazo, riñón, lengua y sistema nervioso. El diagnóstico se realiza por biopsia del órgano afectado o por una muestra de orina teñida con rojo Congo.
El tratamiento del amiloide AL se dirige tanto al órgano afectado como al padecimiento subyacente. Pocos medicamentos tienen actividad contra la amiloidosis. Un estudio aleatorio en el que se comparó MP con colchicina mostró mayor supervivencia en pacientes que recibieron el esquema MP.83 La supervivencia promedio es de 18 meses. Se ha reportado que la quimioterapia en dosis altas con apoyo de células tronco ha sido útil en algunos pacientes84, pero el beneficio clínico no es muy claro.
DR. BRUCE D. CHESON
Las leucemias linfoides crónicas son un grupo de trastornos linfoides de tipo clonal relativamente indolentes, principalmente derivados de las células B y que incluyen a la leucemia linfocítica crónica (LLC), la leucemia prolinfocítica (LPL), la leucemia de células peludas (LCP) y la fase leucémica de los linfomas no Hodgkin (LNH).
Las discrasias de las células plasmáticas se caracterizan por acúmulo de células plasmáticas malignas en la médula ósea, hueso o tejidos blandos. Su nombre refleja el hallazgo en la biopsia de hueso de múltiples tumores de células plasmáticas, en lugar de una sola masa. Estas enfermedades, incluyen al mieloma múltiple (MM), el plasmacitoma extramedular y la amiloidosis. La macroglobulinemia de Waldenström, caracterizada por una gamopatía IgM, linfadenopatía y hepatoesplenomegalia, se incluía antes junto con estos trastornos, pero en la actualidad se clasifica en forma más apropiada como un linfoma no Hodgkin indolente linfoplasmocitoide.1
Leucemia linfocítica crónica
EPIDEMIOLOGIA
La LLC es la forma más común de leucemia en adultos del mundo occidental. La incidencia anual de esta enfermedad parece estar disminuyendo en los Estados Unidos, de 10,000 nuevos casos por año hace una década a alrededor de 7,300 casos en 1998.2 Esta reducción puede reflejar, en parte, una clasificación más apropiada de otras neoplasias linfoides que antes se diagnosticaban mal como LLC. La edad media del diagnóstico es de 62 años, y solo 10 a 15 por ciento de los casos se diagnostican antes de los 50 años. La LLC parece ser más común en varones que en mujeres y es más frecuente en los judíos de ascendencia rusa o europea oriental que en la población general. Ocurre con frecuencia similar en negros y blancos. La LLC es poco común en Japón, China y otros países asiáticos.
FISIOPATOLOGIA
La etiología de la LLC se desconoce y no existen factores de riesgo conocidos, incluyendo radiación ionizante, químicos y fármacos.3-5 Se han reportado familias con casos múltiples de LLC y varios pares de gemelos afectados, con mayor frecuencia de padecimientos linfoproliferativos en los familiares en primer grado de los pacientes con LLC.6 También se han reportado cónyuges con LLC.6
PATOGENIA
La LLC es una expansión clonal de linfocitos de apariencia madura. Aunque la morfología de las células B normales y de la LLC es parecida, existen diferencias inmunológicas y de marcadores moleculares ente las dos poblaciones celulares. Tanto las células de LLC como las normales expresan antígenos de linaje B (v.gr., CD19 y CD20); sin embargo, las células de la LLC también expresan antígenos de activación (v.gr., CD23) y CD5. Las células B CD5+ no son únicas de la LLC, también pueden ser una fuente de anticuerpos y su número aumenta en trastornos autoinmunes como el lupus eritematoso generalizado y la artritis reumatoide, en la púrpura trombocitopénica inmune y después de un trasplante alogénico de médula ósea.
A pesar de ello, existen numerosas diferencias funcionales, inmunológicas y moleculares entre la célula B de la LLC y la célula B CD5+ benigna, incluyendo propiedades de adhesión, transformación por el virus de Epstein-Barr, proliferación en respuesta al estímulo con diversos mitógenos y transducción de señales.7,8
Citogenética
Pueden detectarse alteraciones citogenéticas adquiridas en alrededor del 50 por ciento de los pacientes con LLC por el uso de técnicas de bandeo convencionales,9 y estas alteraciones son más comunes en los estados avanzados de la enfermedad. Se pensaba que la trisomía 12, que ocurre en el 30 a 50 por ciento de los casos, era la alteración citogenética más común. Sin embargo, ensayos más sensibles, como la hibridización in situ con fluorescencia (HISF), han revelado defectos en más del 70 por ciento de los casos,10 con deleciones de 13q en más de la mitad de los pacientes, seguido de deleciones de 11q en 15 a 20 por ciento de los casos y trisomía 12 en el 15 a 20 por ciento. Más de la tercera parte de los pacientes tienen alteraciones complejas.
Biología molecular
No se ha implicado a una sola alteración genética en la patogenia de la LLC. Sin embargo, las células de LLC se caracterizan por un defecto en la apoptosis, o muerte programada. Existe expresión exagerada del gen bcl-2 en más del 70 por ciento de los casos, incluso en ausencia del rearreglo del cromosoma t(14;18).11 La relación entre el gen antiapoptósico bcl-2 y el gen proapoptótico BAX aumenta en las células de la LLC, lo que favorece la supervivencia celular. Por técnicas citogenéticas convencionales se han identificado deleciones que afectan a 13q34 en hasta el 70 por ciento de los casos; este defecto, que antes se pensaba que estaba en el sitio del gen supresor del retinoblastoma, ha demostrado ser telomérico a esa región.12 La región de pérdida contiene un gen supresor recién detectado denominado DBM (alterado en las neoplasias de células B). Existe una relación aparente entre la proteína antiapoptósica Mcl-1 y la resistencia a la quimioterapia.13
DIAGNOSTICO DE LEUCEMIA LINFOCITICA CRONICA
Manifestaciones clínicas
Los pacientes con LLC suelen estar asintomáticos al momento del diagnóstico y este se realiza en forma incidental al encontrar linfocitosis durante una evaluación de rutina. El examen fisico inicial es normal en el 20 a 30 por ciento de los pacientes y existe linfadenopatía, hepatoesplenomegalia o ambos en el 40 a 50 por ciento de los casos. Sin embargo, al progresar la enfermedad la adenopatía generalizada y la esplenomegalia son datos frecuentes.
Las infecciones son una complicación frecuente y pueden ser recurrentes, siendo los patógenos más comunes Staphylococcus, Streptococcus, otras bacterias que requieren opsonización y los herpesvirus.
Puede ocurrir un resultado positivo de antiglobulina Coombs en hasta el 20 por ciento de los casos con LLC, aunque la hemólisis clínica es aparente en solo la mitad de estos casos. La frecuencia de trombocitopenia inmune parece ser de alrededor del 2 por ciento. La anemia o la trombocitopenia autoinmunes suelen responder al tratamiento con esteroides. Si el paciente no mejora con esteroides pueden usarse otras alternativas, incluyendo inmunoglobulina en dosis altas y esplenectomía.14
La aplasia pura de células rojas ocurre en menos del 5 por ciento de los casos de LLC. Las opciones de tratamiento incluyen esteroides, ciclosporina con o sin esteroides concomitantes y quimioterapia sistémica para la LLC.15
Datos de laboratorio
Debe pensarse en el diagnóstico de LLC si existe aumento sostenido en el número de linfocitos pequeños de apariencia madura (i.e., a > 5,000/ml) circulantes en sangre periférica que no se explica por otros padecimientos médicos. El aspirado de médula ósea y la biopsia revelan infiltración de por lo menos 30 por ciento de linfocitos. Rara vez se requiere evaluar la médula ósea para confirmar el diagnóstico, pero el estudio proporciona importante información pronóstica y se requiere para evaluar la respuesta al tratamiento.16
La inmunofenotipificación ayuda a distinguir la LLC de otras leucemias linfoides crónicas, como la LPL, LCP y sus variantes, el LNH en fase leucémica (v.gr., linfomas linfoplasmacíticos, LNH de zona marginal, incluyendo el linfoma esplénico con linfocitos velloso y LNH de células en manto), y leucemia de células plasmáticas [ver figura 1 y tabla 1].1,16 Los linfocitos de la LLC son células B monoclonales que expresan inmunoglobulinas de superficie en bajo nivel y el antígeno CD5 junto con los antígenos normales de células B CD19, CD20 y CD23. El LNH en mano, otro trastorno de células B CD5+, puede distinguirse de la LLC por su intensa expresión de inmunoglobulinas de superficie y la falta de CD23.
|
|
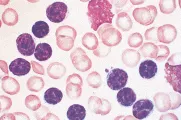
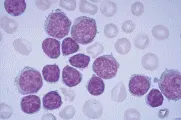
|
|
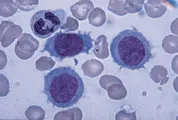
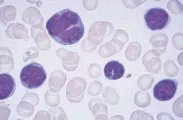
|
|||||||||||||||
Igs-inmunoglobulina de superficie |
ESTADIFICACION Y FACTORES PRONOSTICOS
La LLC es un trastorno clínicamente heterogéneo, algunos pacientes viven por décadas sin requerir tratamiento, mientras que otros mueren en meses después del diagnóstico. El primer sistema de estadificación que se adoptó en forma amplia fue la clasificación de cinco estadios de Rai [ver tabla 2]. Este sistema se simplificó después a tres estadios: riesgo bajo (estadio 0), riesgo intermedio (estadios I y II) y riesgo alto (estadios III y IV). En algunas partes de Europa se usa más la clasificación de Binet17 [ver tabla 3]. Aunque el sistema de Binet no identifica a los pacientes con estadio 0 de Rai, los dos sistemas tienen el mismo valor predictivo. Entre el 60 al 70 por ciento de los pacientes con LLC pertenecen a un grupo de riesgo bajo o intermedio.
|
||||||||||||||||
|
|
||||||||||||
|
Aunque la LLC tiende a ocurrir en pacientes mayores de 60 años, el 10 a 15 por ciento son menores de 50. Para cada estadio, los pacientes más jóvenes tienen una supervivencia promedio semejante a la de los mayores cuando se corrigen las causas de muerte no relacionadas a la LLC.18
Los factores de riesgo incluyen sexo masculino, raza negra, mal estado funcional y edad avanzada.19,20 El tiempo de duplicación de los linfocitos tiene mayor valor predictivo que el número absoluto de linfocitos circulantes.21 La beta2-microglobulina sérica, el CD23 soluble, 22-24 y los receptores solubles de interleucina-225 también predicen la evolución.
Las alteraciones citogenéticas simples se asocian con mayor supervivencia en comparación con las alteraciones citogenéticas complejas. Los pacientes con alteraciones 14q tienen especial peor pronóstico, mientras que la evolución de los enfermos con alteraciones 13q es semejante a la de los pacientes con cariotipo normal. No se recomiendan los estudios citogenéticos como parte de la evaluación de rutina del paciente con LLC porque son costosos, difíciles de realizar y, lo más importante, no dirigen las decisiones terapéuticas.
La relación de proteínas bcl-2 a BAX en las células de LLC favorece a la bcl-2 y correlaciona con resistencia clínica a la quimioterapia y a la evolución clínica.26,27 Las deleciones de p53 predicen una respuesta pobre al tratamiento con fludarabine o pentostatin.28
EVOLUCION CLINICA Y COMPLICACIONES
Condiciones de progresión
En alrededor del 10 a 15 por ciento de los pacientes con LLC se desarrolla una enfermedad linfoproliferativa más agresiva, con más frecuencia el síndrome de Richter, que es un linfoma agresivo de células grandes que ocurre en alrededor del 5 por ciento de los enfermos.29 Los pacientes característicamente presentan mayor linfadenopatía, hepatoesplenomegalia, fiebre, dolor abdominal, pérdida de peso, anemia progresiva y trombocitopenia, con elevación rápida en la cuenta de linfocitos en sangre periférica. La respuesta de los pacientes con síndrome de Richter al tratamiento sistémico es mala, con poca supervivencia.
La segunda transformación en frecuencia es la LPL. Otras incluyen leucemia linfocítica aguda, leucemia de células plasmáticas, mieloma múltiple y enfermedad de Hodgkin.
Neoplasias secundarias
Las neoplasias secundarias parecen ocurrir con más frecuencia en pacientes con LLC y se relacionan tanto con los defectos inmunes asociados a la enfermedad como con las consecuencias del tratamiento. Estas neoplasias afectan principalmente a la piel, próstata y tubo digestivo.30-33
Alteraciones inmunológicas
En la LLC se han reportado diversos defectos inmunológicos. Las células B de la LLC producen cantidades disminuidas de inmunoglobulinas normales en respuesta a los estímulos antigénicos y existen alteraciones cuantitativas y cualitativas de células B normales, células T y células asesinas naturales, reducción en el número y función de las células T normales y menor activación del complemento.34,35
TRATAMIENTO PARA LA LEUCEMIA LINFOCITICA CRONICA
El pronóstico de los pacientes con LLC se relaciona con la fase de la enfermedad. Los pacientes con una enfermedad en estadio temprano pueden vivir una vida normal; sin embargo, la evolución no ha mejorado en forma significativa durante las últimas décadas para quienes tienen enfermedad avanzada. Debido a que los tratamientos disponibles en la actualidad no son curativos, es importante el momento en que se inicia el tratamiento. No existe ventaja en la supervivencia para los pacientes con enfermedad en fase temprana que se tratan al hacer el diagnóstico. El Grupo Cooperativo Francés sobre LLC realizó dos estudios de pacientes con enfermedad en fase A de Binet. En el primero los pacientes fueron distribuidos para recibir tratamiento diario con clorambucil por vía oral u observación. En el segundo los pacientes recibieron clorambucil intermitente más prednisona o no se les dió tratamiento. Ningún estudio detectó ventajas con la intervención temprana y en el primer estudio existió mayor número de tumores sólidos secundarios fatales,33 quizá relacionados con el clorambucil. Por lo tanto, el tratamiento debe iniciarse solo cuando esté indicado por uno o más síntomas relacionados con la enfermedad (v.gr., fiebre, calosfríos, pérdida de peso o fatiga importante), falla progresiva de la médula ósea con anemia, trombocitopenia o ambos, anemia o trombocitopenia autoinmunes, hepatoesplenomegalia o linfadenopatía masivas o progresivas, o infecciones recurrentes.35 Aunque no existe un número absoluto de linfocitos que indique la necesidad de tratamiento, el tiempo de duplicación rápida de los mismos (< 6 meses) puede justificar el inicio del tratamiento.21
Agentes aislados
El medicamento estándar para la LLC ha sido el clorambucil. Cuando se administra este fármaco a dosis de 6 a 14 mg por vía oral diario por 4 a 8 semanas o en dosis más altas, pulsos intermitentes de 20 mg/m2 por vía oral cada 3 semanas o 30 a 40 mg/m2 una vez al mes, ocurre regresión de la enfermedad en los ganglios linfáticos, hígado y bazo, y normalización de las cuentas de células en sangre periférica en alrededor de la mitad de los pacientes sin tratamiento previo, aunque es rara la desaparición del padecimiento.36-38 La ciclofosfamida parece tener una actividad semejante a la del clorambucil, pero se usa solo cuando el clorambucil no es bien tolerado.
Los esteroides son menos activos que los agentes alquilantes en la LLC y deben reservarse para los pacientes con complicaciones autoinmunes por el riesgo de infecciones bacterianas, virales y micóticas, diabetes y osteoporosis.
Análogos de purina
Fludarabine El fludarabine ha demostrado ser el agente más activo en el tratamiento de la LLC. Se estudió en forma inicial en pacientes que no respondieron o que tuvieron recaídas durante el tratamiento con agentes alquilantes.39-42 Debido a la dificultad para comparar los resultados de los diferentes tratamientos, el Grupo de Trabajo sobre LLC patrocinado por el Instituto Nacional de Cáncer de los EUA estandarizó los criterios de elegibilidad, respuesta y toxicidad. Con el uso de estas normales se lograron remisiones completas clínicas y hematológicas en el 3 a 13 por ciento de los pacientes, con una respuesta global de 40 a 50 por ciento. Los resultados variaron por factores como la edad del paciente, el estadio y la capacidad funcional. El tiempo promedio de progresión fue de alrededor de 18 meses para los pacientes refractarios a agentes alquilantes y de 17 meses para pacientes que tuvieron recaídas después de una respuesta previa. La supervivencia promedio fue de 29 meses para los pacientes con recaídas y de 9 meses para los refractarios a tratamiento.
Cuando se usó fludarabine como tratamiento inicial la tasa de respuesta completa fue de alrededor de 30 por ciento, con una tasa de respuesta global de 70 por ciento.38,43,44 Los estudios aleatorios han demostrado tasas de respuesta y tiempos para la progresión más altos con fludarabine que con los agentes alquilantes y los esquemas basados en antraciclinas, aunque no se ha observado ventaja en la supervivencia.38,43 El tiempo promedio para la progresión en pacientes sin tratamiento previo fue de 32 meses, con supervivencia promedio mayor de 5 años. El fludarabine es una opción de elección eficaz para la LLC.
Los principales efectos tóxicos por este medicamento con el esquema actual de 25 mg/m2 IV durante 5 días consecutivos una vez por mes son la mielosupresión moderada y la inmunosupresión severa, con neurotoxicidad ocasional, en especial con dosis más altas que las recomendadas.45,46 Las cuentas de linfocitos, en especial de células CD4, disminuye en semanas y no se normaliza durante un año o más después de suspendido el tratamiento.44,46 No se recomienda el uso de antibióticos profilácticos porque sería imposible cubrir todo el espectro de patógenos potenciales, a pesar del costo y toxicidad potencial de los medicamentos adicionales.
Otros agentes Tanto el cladribine como la 2'-deoxidoformicina (pentostatin) son activos en la LLC, pero se usan con menos frecuencia que el fludarabine por sus tasas de respuesta relativamente menores, remisiones menos duraderas y mayor costo.47,48
Quimioterapia combinada
Los esquemas combinados usados con más frecuencia en LLC incluyen clorambucil más prednisona (CP) y ciclofosfamida, vincristina y prednisona (CVP). Las tasas de respuesta reportadas con los esquemas CP y CVP en pacientes sin tratamiento previo con enfermedad en fase avanzada varían de menos de 10 a más de 60 por ciento, dependiendo en gran parte de los criterios usados para evaluar la respuesta. No es común lograr desaparición de la enfermedad evidente y la supervivencia promedio es menor de 2 años.36,49-51
El tratamiento combinado no es claramente superior al de un solo fármaco. Los estudios iniciales, incluyeron pocos casos, sugieren que la prednisona puede aumentar la tasa de respuesta al clorambucil, pero no aumentar la supervivencia.49,50 Los esquemas más intensos, como el CHOP (ciclofosfamida, doxorrubicina [hidroxidaunomicina], vincristina [Oncovin] y prednisona), no son claramente mejores que los menos intensos.36,51-53 En la actualidad se desarrollan combinaciones que incluyen fludarabine.
TRATAMIENTO DE LA ENFERMEDAD CON RECAIDAS O REFRACTARIA
Enfoques estándar
Los pacientes con una recurrencia asintomática no suelen tratarse sino hasta que esté indicado el tratamiento por los mismos criterios que al inicio, en un intento paliativo. El fludarabine es el agente de salvamento estándar para pacientes que no han respondido a los agentes alquilantes. Los pacientes que son refractarios a fludarabine tienen menos probabilidad de responder a agentes alquilantes.
Tratamiento con dosis altas y apoyo de células tronco
La experiencia con el trasplante alogénico de la médula ósea en la LLC es limitada, en parte porque la edad promedio de los pacientes al momento del diagnóstico es mayor de 60 años.54-56 Alrededor de la mitad de los pacientes que reciben un trasplante de médula ósea pueden permanecer sin enfermedad por periodos prolongados; sin embargo, no es claro si están curados. Por desgracia, la mortalidad relacionada al tratamiento es de 25 a 50 por ciento.
El trasplante autólogo de células tronco tiene una función dudosa en los pacientes con LLC.56,57
Tratamiento biológico
La mayor experiencia con un agente biológico ha sido con interferón alfa. Las respuestas tienden a ser breves, sin influencia en la evolución. Los estudios preliminares con el anticuerpo monoclonal anti-CD52 CAMPATH han sido promisorios tanto en LLC como el LPL.58,59 El anticuerpo C2B8 anti CD20 (rituximab), que tiene actividad considerable en los linfomas foliculares, no parece tener la misma eficacia en la LLC.
Tratamiento de sostén
Factores de crecimiento mieloide No se ha demostrado si los factores de crecimiento mieloide pueden proteger contra la mielosupresión inducida por la quimioterapia, por lo que no se incluyen en el tratamiento estándar.
Inmunoglobulinas intravenosas La mayoría de los pacientes con LLC sufren hipogamaglobulinemia, que es más común en las fases avanzadas de la LLC y que empeora al progresar la enfermedad. Desde el punto de vista histórico, los patógenos más comunes en la LLC son los que requieren de opsonización antes de ser destruidos. Por desgracia, las inmunoglobulinas intravenosas en dosis altas no protegen a los pacientes de diversas infecciones bacterianas, virales o micóticas. No influyen en la supervivencia global y no tienen buena relación costo eficacia.60,61 Este tratamiento debe reservarse para pacientes con infecciones bacterianas demostradas de repetición.
Eritropoyetina La anemia en la LLC no es consecuencia de producción deficiente de eritropoyetina, sino más bien consecuencia de sustitución de la médula ósea, secuestro esplénico o hemólisis. Sin embargo, la eritropoyetina puede reducir la necesidad de transfusión en pacientes seleccionados y puede intentarse una prueba con este agente en los pacientes con LLC y anemia que no tienen otra causa obvia y corregible de la anemia.62
Esplenectomía
La esplenectomía puede tener un papel importante en el tratamiento paliativo de los pacientes con LLC que no se han beneficiado del tratamiento sistémico.14 La trombocitopenia responde en grado variable en casi todos los pacientes. Cuando se realiza este procedimiento por un cirujano experimentado la mortalidad relacionada es menor al 10 por ciento.
Leucemia prolinfocítica
La LPL representa la forma más común de transformación agresiva en los pacientes con LLC. Los pacientes presentan deterioro clínico, con frecuencia con síndrome consuncional. Comparado con los pacientes cuya LPL ha evolucionado de una LLC, los que tienen LPL de novo tienden a ser de mayor edad (70 o más años de edad) que los que han sufrido transformación (alrededor de 60 años), tienen linfocitosis más importante, esplenomegalia masiva y no moderada y casi nunca linfadenopatía importante. El frotis de sangre periférica revela una población dual de células, algunas son linfocitos pequeños característicos de la LLC y otras son más grandes con nucleolos prominentes. La LPL puede presentarse como una enfermedad de novo; en cualquier caso, se caracteriza por una cuenta de leucocitos alta, con por lo menos el 55 por ciento de prolinfocitos circulantes. En los pacientes con LPL primaria se ha encontrado rearreglo cromosómico t(6;12). Las alteraciones de p53, que ocurren en más de la mitad de los pacientes pueden causar resistencia a la quimioterapia.
Se han publicado resultados preliminares promisorios con los análogos de purina y el anticuerpo monoclonal CAMPATH.
Leucemia de células peludas
La LCP es una neoplasia de células B poco común que ocurre en alrededor de 500 pacientes nuevos cada año en los Estados Unidos. La enfermedad es más común en personas mayores, predominando los hombres en relación con las mujeres. Aunque los pacientes con LCP pueden no tener síntomas al inicio, suelen presentar manifestaciones de menor producción de la médula ósea, como infecciones, sangrado y debilidad. En casos raros los pacientes sufren un trastorno que recuerda a una vasculitis. El examen físico es notable por la esplenomegalia que con frecuencia es masiva y la hepatomegalia asociada; con menos frecuencia ocurre linfadenopatía con esplenomegalia. La evaluación de laboratorio revela pancitopenia, que aumenta la posibilidad de anemia aplástica, leucemia aguda, síndrome mielodisplásico o LCP. Puede requerirse una biopsia de médula ósea para hacer el diagnóstico porque el aspirado suele taparse por las células peludas, y también existe fibrosis. Las células malignas son de origen B, expresando CD19, CD20 y el antígeno monocito CD11c. Es posible que el marcador más específico sea Bly-7 (CD103).
La LCP suele ser un trastorno indolente y el 10 por ciento de los pacientes puede nunca requerir tratamiento. El tratamiento está indicado si existe esplenomegalia masiva o progresiva, empeoramiento de las cuentas celulares, infecciones recurrentes, más de 20,000 células peludas/ml de sangre periférica o linfadenopatía importante. Hasta hace 15 años la esplenectomía era el tratamiento estándar para la LCP porque mejora los síntomas relacionados con la esplenomegalia y en la mayoría de los casos las cuentas de sangre periférica. Sin embargo, el procedimiento no afecta la enfermedad en sí. En la actualidad se realizan menos esplenectomías por la posibilidad de tratamientos sistémicos eficaces. El interferón alfa fue el primer agente que logró respuestas significativas en pacientes con LCP. La mayoría de los pacientes sufren mejoría clínica y hematológica, aunque en solo 10 por ciento de los mismos la respuesta es completa e invariablemente la enfermedad recurre después de suspender el tratamiento. El interferón alfa se asocia con síntomas de tipo catarral (fiebre, mialgias y malestar) en casi todos los pacientes. Otros efectos tóxicos incluyen erupción, alteraciones en el sitio de aplicación y síntomas gastrointestinales como náusea, vómito y anorexia.
Los análogos de purina han revolucionado el tratamiento de los pacientes con LCP. En un estudio realizado en Norteamérica, pacientes con LCP sin tratamiento previo recibieron interferón alfa o pentostatin, se observaron tasas de respuesta completa en alrededor del 11 por ciento de los pacientes tratados con interferón alfa, comparado con 76 por ciento en los que recibieron pentostatin, con una diferencia significativa en la duración de la respuesta a favor del pentostatin.63 Recientemente se ha demostrado que el cladribine induce respuestas en 80 a más del 90 por ciento de los pacientes, con 65 a 85 por ciento de remisiones completas.64 Solo 25 a 30 por ciento de los pacientes recaen después del seguimiento prolongado. En muchos casos la recaída se caracteriza solo por aumento en las células peludas de la médula ósea, sin indicación de tratamiento. La mayoría de los pacientes que requieren tratamiento logran una segunda respuesta duradera. Los resultados con pentostatin y cladribine son equivalentes y el medicamento de elección dependerá de la preferencia del médico.
Mieloma múltiple
EPIDEMIOLOGIA
El MM es poco común en personas menores de 40 años, su incidencia aumenta con rapidez después de los 50 años, con igual frecuencia en hombres y mujeres. Aunque las personas de raza negra se afectan más que los blancos, la evolución en los dos grupos es semejante, dependiendo del estadio.65 Se calcula que existieron alrededor de 14,000 nuevos casos en 1998 en los Estados Unidos.2 No se ha identificado relación entre el MM y el estado socioeconómico. La incidencia de esta enfermedad parece ser menor en chinos y otros grupos asiáticos. La etiología del MM no es clara. Se ha implicado a la exposición a radiación ionizante, pinturas, solventes y pesticidas, pero nunca ha podido establecerse una relación firme.
PATOGENIA
El MM se caracteriza por acúmulo de células plasmáticas en la médula ósea y, con menos frecuencia, en los tejidos blandos u órganos. La célula maligna no es la célula plasmática madura, sino una célula previa en la vía de diferenciación de la célula B, después del rearreglo de los genes de las cadenas pesadas. Estas células tienen la apariencia inmadura de un plasmablasto, pueden ser identificadas en la sangre periférica de los pacientes con MM y se considera que son precursoras de las células malignas que se encuentran en la médula ósea. Expresan una variedad diferente de moléculas de adhesión (v.gr., CD11b),66 que parecen permitir que las células B circulantes se fijen a las células del estroma de la médula ósea no malignas que producen citocinas capaces de apoyar el crecimiento de las células de mieloma, como IL-6. La IL-6 actúa como un factor de crecimiento autócrino y parácrino en el MM, estimulando así el crecimiento celular y evitando la apoptosis. Se ha sugerido que los herpesvirus (herpesvirus asociado al sarcoma de Kaposi o el virus del herpes simple-8) tienen algún papel en la patogenia.67
Los análisis citogenéticos son difíciles de realizar en el MM. Sin embargo, estudios recientes que han usado HISF han demostrado aneuploidía en alrededor del 70 por ciento de los casos.68 Las alteraciones más frecuentes afectan los cromosomas 13 (13q-) y 14 (14q+). Estos defectos pueden causar menor apoptosis y resistencia subsecuente al tratamiento. Se han identificado mutaciones de p53 en los pacientes con MM que presentan recaídas, estas parecen correlacionar con mala evolución.
INMUNOLOGIA
El MM se caracteriza por la producción exagerada de inmunoglobulina monoclonal. La frecuencia de los diversos tipos de inmunoglobulinas del MM es paralela a la concentración de estas Igs: IgG en el 60 a 70 por ciento, IgA en el 20 por ciento y cadenas ligeras en solo el 15 por ciento. Se han reportado algunos casos de IgD e IgE, y alrededor del 1 por ciento de los pacientes son no secretores. Un segundo hallazgo son los niveles disminuidos de inmunoglobulinas residuales normales.
Las proteínas de Bence Jones (cadenas ligeras urinarias) se detectan en la orina con antisuero contra las cadenas ligeras sin un componente anti-cadenas pesadas.
DIAGNOSTICO DEL MIELOMA MULTIPLE
El diagnóstico de MM suele hacerse en forma incidental cuando se detecta una mayor concentración de proteína en suero en la evaluación de rutina o al evaluar a un paciente por anemia, disfunción renal o hipercalcemia asintomática.
Gamopatía monoclonal de significado no determinado
Ocurre gamopatía monoclonal de significado no determinado (GMSND) en alrededor del 1 por ciento de la población general y en el 3 por ciento de las personas mayores de 70 años; la IgG es la proteína aumentada con más frecuencia. Las características del MM que lo distinguen de la GMSND incluyen concentraciones mayores de proteína M en suero y orina, aumento en el porcentaje de células plasmáticas en la médula ósea, aumento en el índice de marcación de células plasmáticas, presencia de lesiones óseas líticas y presencia de células plasmáticas circulantes en la sangre periférica. Después de un seguimiento prolongado, pueden identificarse cuatro grupos de pacientes con GMSND: (1) los que tienen gamopatía monoclonal limitada, (2) los que tienen concentración e inmunoglobulina sérica mayor de 3 g/dl pero no requieren ningún tratamiento (10 por ciento), (3) los que mueren sin mieloma múltiple u otra enfermedad relacionada (47 por ciento) y (4) aquellos en los que se desarrolla mieloma, amiloidosis, macroglobulinemia u otro trastorno linfoproliferativo (24 por ciento de los cuales son mielomas). Ocurre progresión a una neoplasia en 8 a 10 años. Por desgracia no existen estudios de laboratorio que predigan con exactitud qué pacientes con GMSND progresarán a MM.
Cuadro clínico
El diagnóstico de MM requiere de por lo menos uno de los siguientes criterios mayores: plasmocitoma, médula ósea con por lo menos el 30 por ciento de células plasmáticas y proteína M en la electroforesis que tenga niveles de IgG mayores de 3.5 g/dl, IgA mayor de 2.0 g/dl o excreción de cadenas ligeras en la orina mayor de 1 g/24 h. También se requieren uno o más de los siguientes criterios menores: plasmacitosis en la médula ósea mayor de 10 por ciento, proteína M en el suero u orina pero menor de los niveles mencionados en los criterios mayores, lesiones óseas líticas y reducción de las inmunoglobulinas residuales.
El mieloma múltiple puede afectar muchos órganos, incluyendo los huesos, riñones, nervios, corazón e hígado [ver tabla 4].
|
||||||||||||||||||||
|
Plasmacitomas Los plasmacitomas son tumores sólidos formados por células plasmáticas y que pueden ocurrir en los tejidos blandos (plasmacitomas extramedulares). Se presentan como masas palpables pero con frecuencia causan compresión de la médula espinal. Estos tumores pueden curarse con radioterapia local. Los pacientes con plasmacitomas que ocurren en el hueso pueden responder al inicio a la radioterapia, pero la mitad progresarán a MM franco, dos tercios lo harán antes de 3 años después del diagnóstico. La persistencia de proteína M después de la radioterapia se asocia con mayor probabilidad de evolución a MM.
Enfermedad ósea La enfermedad ósea es una de las características más comunes y diagnósticas del MM. Aunque las lesiones líticas son típicas [ver figura 2], la osteopenia es la forma más común de afección ósea en el mieloma. También se ha reportado enfermedad osteoesclerótica. Es notable el aumento en la actividad osteoclástica en los huesos afectados, y se cree que se relaciona con el llamado factor activador de osteoclastos, que es una manifestación de los efectos del factor de necrosis tumoral, la IL-1 y la IL-6.
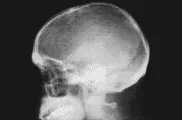
|
| Figura 2 |
| Mieloma múltiple: radiografía |
La radioterapia para las lesiones óseas dolorosas en un paciente con MM recién diagnósticado debe diferirse lo más posible hasta que pueda evaluarse la eficacia del tratamiento sistémico porque la radioterapia puede impedir la administración de dosis adecuadas de quimioterapia. Son excepciones a esto la compresión de la médula espinal y las fracturas potenciales en huesos que cargan peso y que requieren de intervención inmediata. Para los pacientes con recaídas la radioterapia local puede proporcionar un beneficio paliativo importante.
Las fracturas por compresión vertebral durante el curso del tratamiento sistémico no justifican siempre la administración de radiación porque estas lesiones suelen ser causadas por osteopenia, no necesariamente representan progresión de la enfermedad y no responden siempre a la radiación.
El pamidronato es un bifosfonato de segunda generación que ha sido estudiado en un estudio clínico controlado con placebo en pacientes no tratados antes y en pacientes con recaídas y MM en estadio III.69 El fármaco proporciona protección significativa contra las complicaciones esqueléticas (medidas por el tiempo que transcurre antes del primer evento esquelético), parece mejorar la calidad de vida y quizá proporciona ventajas para la supervivencia.70 Como resultado, en la actualidad se recomienda administrar bifosfonatos a todos los pacientes con evidencia de afección ósea, aunque algunos clínicos recomiendan este agente en todos los pacientes con MM.
Anemia Ocurre anemia en la mayoría de los pacientes con MM, en especial al progresar la enfermedad, como consecuencia de sustitución de la médula ósea, insuficiencia renal y anemia asociada a la enfermedad crónica. En los pacientes con MM que tienen niveles séricos inapropiadamente bajos de eritropoyetina, esta sustancia puede disminuir la necesidad de transfusión de paquetes eritrocitarios.71
Hipercalcemia Alrededor del 10 a 20 por ciento de los pacientes con MM presentan hipercalcemia. Los niveles de calcio en suero, corregidos según la albúmina, pueden usarse para identificar a los pacientes con riesgo de síntomas relacionados a la hipercalcemia. Las paraproteínas pueden en ocasiones fijar calcio, dando un nivel elevado falso; por lo tanto, debe medirse el calcio libre (ionizado) en el paciente asintomático con un nivel elevado de calcio en suero.
Los pacientes sin tratamiento previo y que están asintomáticos o tienen solo síntomas leves relacionados con hipercalcemia leve o moderada deben ser tratados con quimioterapia sistémica, incluyendo esteroides. Los pacientes con niveles altos de calcio sérico o síntomas como letargo, confusión y constipación requieren de hidratación con solución salina. Los bifosfonatos son útiles para disminuir la concentración de calcio sérico, lo mismo que la mitramicina y la calcitonina.
Insuficiencia renal La insuficiencia renal es común en el MM y puede deberse a varios mecanismos. Los túbulos pueden obstruirse por la proteína del mieloma (riñón de mieloma) o los riñones pueden infiltrarse por proteína amiloide. El mayor riesgo de infecciones crea predisposición para pielonefritis. Otras causas incluyen hipercalcemia, nefropatía por uratos, síndrome nefrótico, hiperviscosidad, infiltración de los riñones por células plasmáticas y uropatía obstructiva. Los pacientes con sospecha de MM y función renal anormal deben mantenerse bien hidratados y no deben someterse a procedimientos que deshidraten como tomografía computada con medio de contraste o urografía excretora, lo que puede precipitar insuficiencia renal aguda en el paciente con daño renal subclínico. La plasmaféresis puede aliviar la insuficiencia renal aguda. Pueden ser útiles la diálisis e incluso el trasplante renal.
Hiperviscosidad Los signos y síntomas de hiperviscosidad, como disnea, confusión y dolor torácico, son poco comunes en pacientes con MM por IgG, pero ocurren con concentraciones de proteína en suero muy altas. La hiperviscosidad se presenta con más frecuencia en pacientes con MM de tipo IgA por la mayor polimerización de esta paraproteína. El tratamiento debe dirigirse a tratar el MM subyacente. Puede considerarse la realización de plasmaféresis si se requiere reducción rápida de la paraproteína para aliviar los síntomas.
Enfermedades infecciosas Los pacientes con mieloma tienen mayor riesgo de infecciones bacterianas que ponen en peligro la vida, incluso en presencia de números normales de granulocitos circulantes. Esta alteración se relaciona con la menor síntesis de inmunoglobulinas normales y una activación del complemento defectuosa. Al inicio del padecimiento los pacientes pueden infectarse con organismos encapsulados que requieren de opsonización (v.gr., Streptococcus pneumoniae, Haemophilus influenzae y Staphylococcus), mientras que el uso de quimioterapia aumenta la posibilidad de infecciones por gram negativos.
Es poco probable que la vacuna polivalente antineumocócica sea eficaz porque los pacientes responden mal a los antígenos bacterianos y tienen otros defectos bactericidas que no son corregidos por la vacunación. Ni las inmunoglobulinas intravenosas ni los antibióticos profilácticos tienen buena relación costo eficacia o protegen de modo uniforme, y deben reservarse para los pacientes con infecciones bacterianas recurrentes demostradas.
Síntomas neurológicos La compresión de la médula es un dato de presentación común del plasmacitoma y se considera una urgencia oncológica que requiere descompresión y radioterapia. Las paraproteinemias pueden asociarse con otros síntomas neurológicos, como el síndrome de POEMS. POEMS es el acrónimo de una serie de datos que incluyen polineuropatía (generalmente sensorial), organomegalia (v.gr., hepatomegalia), endocrinopatía (v.gr., diabetes, amenorrea y ginecomastia), proteína M y cambios cutáneos (v.gr., hiperpigmentación). Otras características son la linfadenopatía y las lesiones óseas osteoescleróticas.
ESTADIFICACION Y FACTORES PRONOSTICOS
Una vez hecho el diagnóstico de MM la evaluación inicial debe incluir una biometría hemática completa, así como aspiración y biopsia de la médula ósea. La electroforesis sérica y la cuantificación de inmunoglobulinas determinan la concentración de la paraproteína, que correlaciona con la masa tumoral. La inmunoelectroforesis define el subtipo de inmunoglobulina. La electroforesis urinaria detectará cadenas ligeras, cuando existen. Se realizará una investigación radiológica ósea para identificar los sitios de afección y las fracturas potenciales. Los gamagramas óseos con radionúclidos son de poco valor para evaluar el MM.
El cuadro clínico de los subtipos de MM varía mucho. Por ejemplo, los pacientes que secretan solo cadenas ligeras pueden presentar insuficiencia renal progresiva, al inicio sin mieloma franco. Los pacientes con mieloma por IgD tienen una evolución más agresiva, caracterizada por alta frecuencia de insuficiencia renal, plasmacitomas y amiloidosis, y tienen supervivencia más corta que los pacientes con MM de IgG a pesar de tener concentraciones bajas de paraproteína.72 El mieloma de IgE, que es raro, parece asociarse con mayor frecuencia de leucemia de células plasmáticas.
Los pacientes suelen estadificarse de acuerdo con la clasificación de Durie y Salmon73 [ver tabla 5], que correlaciona bien con la masa tumoral y el pronóstico. El estadio está determinado por el nivel de proteína M, el número de lesiones óseas líticas, la concentración de hemoglobina y el calcio sérico (corregido según la albúmina). Los pacientes después se subdividen en clases A y B con base en la concentración de creatinina sérica.
|
|
|
La supervivencia promedio es de alrededor de 5 años para los pacientes con enfermedad en estadio IA, pero de 15 meses para los que están en estadio IIA. Sin embargo, el sistema de estadificación no tiene unformidad confiable para predecir la evolución y ha sido sustituido por marcadores biológicos de mayor importancia, principalmente la beta2-microglobulina, el índice de marcación de células plasmáticas, y las alteraciones citogenéticas, así como la deshidrogenasa láctica, el Ki-67, los niveles séricos de IL-6, la proteína C reactiva y la presencia de células plasmáticas circulantes.68,74-78 La citogenética no es parte de la evaluación estándar porque las pruebas son costosas y no influyen en el tratamiento. La imagen por resonancia magnética puede ser útil en la evaluación de la afección de la médula ósea y como indicador pronóstico.79
TRATAMIENTO DEL MIELOMA MULTIPLE
Indicaciones de tratamiento
Una proporción pequeña de pacientes se clasifican como con MM latente según las siguientes características: proteína M en el suero menor de 3.0 g/dl, menos del 10 por ciento de infiltración de células plasmáticas en la médula ósea, no anemia, niveles de calcio y creatinina normales en suero, ausencia de lesiones líticas y un índice de marcaje de células plasmáticas bajo. Estos pacientes pueden permanecer estables durante varios años sin tratamiento.
Debe considerarse el tratamiento del MM en estadios I y II para los pacientes con enfermedad progresiva, que incluye un aumento en el nivel de proteína M en el suero, orina o ambos, reducción en el nivel sérico de hemoglobina, falla progresiva de la médula ósea con neutropenia, trombocitopenia o ambos, aumento en la concentración de calcio, creatinina o ácido úrico séricos, lesiones óseas líticas múltiples, dolor óseo, infecciones recurrentes y plasmacitoma extramedular. Los pacientes con enfermedad en fase III generalmente tienen síntomas u alguna otra alteración hematólogica o química en el momento de su evaluación que justifica el tratamiento.
Tratamiento estándar
Una vez que se ha tomado la decisión de que el tratamiento está indicado existen algunas opciones terapéuticas eficaces. Los agentes aislados que son activos incluyen a los esteroides, los agentes alquilantes y el interferón alfa. Por lo general se usa melfalán con prednisona (MP) por la alta tasa de respuesta cuando se administran ambos fármacos. El esquema MP se administra cada 3 a 4 semanas durante 6 a 9 meses, y el tratamiento se continúa cuando existe respuesta. Lo más frecuente es que la concentración de la paraproteína disminuya a un nivel estable (fase de meseta) y continuar el tratamiento después de este punto no prolonga la supervivencia y sí aumenta las complicaciones (v.gr., infecciones). Otros agentes alquilantes también son activos pero no tienen ventajas sobre el melfalán. Algunos medicamentos usados para tratar a los pacientes con MM, como las antraciclinas, tienen utilidad limitada como agentes aislados.
Los esteroides (sea prednisona o dexametasona) pueden disminuir la proteína M en hasta el 40 por ciento de los pacientes no tratados antes y en el 20 a 40 por ciento de los pacientes que no han respondido al tratamiento inicial. Los esteroides también son una opción terapéutica para los pacientes ancianos y para los que tienen mal estado funcional.
La respuesta clínica suele asociarse con reducción del dolor óseo y normalización de la creatinina, el calcio y la hemoglobina. La supervivencia promedio de los pacientes con MM tratados con melfalán y prednisona es de 2.5 a 3 años y solo el 5 a 10 por ciento de los pacientes sobrevive 10 años o más.
Los esquemas de quimioterapia combinada (v.gr., vincristina, carmustina, melfalán, ciclofosfamida y prednisona; vincristina, carmustina, doxorrubicina y prednisona; o vincristina, ciclofosfamida, doxorrubicina y prednisona) no han demostrado mayor eficacia que la MP. El esquema VAD (vincristina, doxorrubicina [Adriamicina] y desametasona) como esquema inicial es menos tóxico para la población de células tronco que la MP y puede ser preferible para los pacientes que se consideran candidatos a trasplante autólogo de células tronco.
El interferón alfa ha sido uno de los agentes biológicos más estudiados. Tiene actividad como agente aislado, pero aún es motivo de controversia si ayuda al efecto de la quimioterapia durante la inducción o el tratamiento de mantenimiento. En un estudio realizado por Mandelli y colaboradores,80 pacientes que respondieron al esquema MP fueron distribuidos para recibir mantenimiento con interferón alfa o solo ser vigilados. Se observó una aparente prolongación del tiempo antes de la progresión y de la supervivencia con el interferón alfa, pero que desapareció al prolongar el tiempo de seguimiento. La mayoría de los otros datos publicados no han demostrado la utilidad del interferón alfa. Considerando la toxicidad asociada, el costo del agente y su beneficio clínico limitado, en la actualidad no se recomienda el interferón alfa como un componente habitual del tratamiento del MM.
Enfermedad con recaídas o refractaria
Las recaídas después de la respuesta inicial al tratamiento es común en el MM. Es poco frecuente que vuelva a haber respuesta a MP y ésta es de menos duración que con el tratamiento inicial, además de que se asocia con mayor mielosupresión. El esquema de salvamento estándar consiste en VAD, una infusión continua de 4 días de vincristina y doxorrubicina a través de un catéter y dexametasona por vía oral, que se repite los días 9 a 17 y en ciclos de cada 21 a 28 días. Se logran respuestas en la tercera parte de los pacientes resistentes y en casi la mitad de los que han sufrido una recaída.
Trasplante de células tronco El trasplante alogénico de médula ósea (TAMO) no se ha usado en forma muy frecuente en el MM porque los pacientes tienden a ser ancianos. Este método terapéutico induce remisiones en el 40 a 50 por ciento de los pacientes, y el 25 a 30 por ciento permanece vivo después de alrededor de 7 años. Sin embargo, pueden ocurrir recaídas después de varios años, aumentando la duda de si algún paciente en realidad se cura. Debido a la alta tasa de morbimortalidad asociada al procedimiento, el TAMO suele reservarse para los pacientes más jóvenes (< 50 años) sin respuesta o con enfermedad progresiva.
En los casos de recaída o refractario se han realizado estudios de quimioterapia en dosis altas con trasplante alogénico de células tronco. La frecuencia y duración de la respuesta puede predecirse por el nivel de deshidrogenasa láctica, la beta2-microglobulina, el estado funcional, la edad (>50 años) y la respuesta al tratamiento previo. Para los pacientes con buen perfil de riesgo (i.e., los que no tienen factores de mal pronóstico o solo uno), la tasa de respuesta completa es de 20 por ciento, con un periodo promedio de supervivencia sin recaídas de 1.5 años y una supervivencia global de 3.5 años. Cuando existen dos a tres factores de alto riesgo los resultados son sustancialmente peores, con un periodo promedio de supervivencia sin recaídas de 7 meses y una supervivencia global de 20 meses. Si existen cuatro más factores de alto riesgo la duración promedio de la respuesta es de 3 meses y la supervivencia global de 5 meses.
Recientemente algunos pacientes recibieron este tratamiento en etapas tempranas de la enfermedad. En un estudio realizado en Francia,81 pacientes sin tratamiento previo menores de 65 años recibieron dosis estándar de quimioterapia o quimioterapia en dosis alta con trasplante autólogo de células tronco. Existió una tasa de respuesta del 81 por ciento en el grupo con dosis altas, incluyendo 22 por ciento de remisiones completas y un 16 por ciento de remisiones parciales muy buenas, comparado con 57, 5 y 9 por ciento, respectivamente, en el grupo con tratamiento estándar. Estas diferencias fueron muy significativas. Sin embargo, la supervivencia a 6 años libre de eventos en el grupo trasplantado fue de solo 24 por ciento. En la actualidad se realiza un estudio aleatorio norteamericano extenso que, en combinación con el estudio francés, caracterizará mejor la función de la quimioterapia con apoyo autólogo o alogénico de células tronco.
Amiloidosis
La amiloidosis se caracteriza por el depósito de fibrillas de amiloide en varios tejidos.82 Puede ocurrir como secundaria a un padecimiento inflamatorio crónico (amiloide A o AA) o como una condición primaria (amiloide asociado a cadenas ligeras, o AL) en el caso de neoplasias de células B. En el AL las fibrillas están formadas por fragmentos de las cadenas ligeras de las inmunoglobulinas, con proliferación clonal de las células plasmáticas de la médula ósea y proteinuria de Bence Jones. La incidencia de amiloidosis primaria en los Estados Unidos es de 5.1 a 12.8 casos por millón de personas-año. Esta enfermedad afecta principalmente el corazón en el 90 por ciento de los casos y también el hígado, bazo, riñón, lengua y sistema nervioso. El diagnóstico se realiza por biopsia del órgano afectado o por una muestra de orina teñida con rojo Congo.
El tratamiento del amiloide AL se dirige tanto al órgano afectado como al padecimiento subyacente. Pocos medicamentos tienen actividad contra la amiloidosis. Un estudio aleatorio en el que se comparó MP con colchicina mostró mayor supervivencia en pacientes que recibieron el esquema MP.83 La supervivencia promedio es de 18 meses. Se ha reportado que la quimioterapia en dosis altas con apoyo de células tronco ha sido útil en algunos pacientes84, pero el beneficio clínico no es muy claro.
Bibliografía
- Harris NL, Jaffe ES, Stein H, et al: A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 84:1361, 1994 8068936
- Landis SH, Murray T, Bolden S, et al: Cancer statistics. CA Cancer J Clin 48:6, 1998 9449931
- Malone KE, Koepsell TD, Daling JR, et al: Chronic lymphocytic leukemia in relation to chemical exposures. Am J Epidemiol 130:1152, 1989 2589308
- Zahm SH, Weisenburger DD, Babbitt PA, et al: Use of hair coloring products and the risk of lymphoma, multiple myeloma, and chronic lymphocytic leukemia. Am J Public Health 82:990, 1992 1609918
- Inskip PD, Kleinerman RA, Stovall M, et al: Leukemia, lymphoma, and multiple myeloma after pelvic radiotherapy for benign disease. Radiat Res 135:108, 1993 8327655
- Cuttner J: Increased incidence of hematologic malignancies in first-degree relatives of patients with chronic lymphocytic leukemia. Cancer Invest 10:103, 1992
- Calligaris-Cappio F: B-chronic lymphocytic leukemia: a malignancy of anti-self B cells. Blood 87:2615, 1996
- Johnson TA, Rassenti LZ, Kipps TJ: Ig VH1 genes expressed in B cell chronic lymphocytic leukemia exhibit distinctive molecular features. J Immunol 158:235, 1997 8977195
- Juliusson G, Oscier DG, Fitchett M, et al: Prognostic subgroups in B-cell chronic lymphocytic leukemia defined by specific chromosomal abnormalities. N Engl J Med 323:720, 1990 2201915
- Stilgenbauer S, Döhner K, Bentz M, et al: Molecular cytogenetic analysis of B-cell chronic lymphocytic leukemia. Ann Hematol 76:101, 1998 9619726
- Hanada M, Delia D, Aiello A, et al: bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood 82:1820, 1993 8104532
- Bullrich F, Veronese ML, Kitada S, et al: Minimal region of loss at 13q14 in B-cell chronic lymphocytic leukemia. Blood 88:3109, 1996 8874210
- Kitada S, Andersen J, Akar S, et al: Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with in vitro and in vivo chemoresponses. Blood 91:3379, 1998 9558396
- Seymour JF, Cusack JD, Lerner SA, et al: Case/control study of the role of splenectomy in chronic lymphocytic leukemia. J Clin Oncol 15:52, 1997 8996124
- Chikkappa G, Pasquale D, Zarrabi MH, et al: Cyclosporine and prednisone therapy for pure red cell aplasia in patients with chronic lymphocytic leukemia. Am J Hematol 41:5, 1992 1503099
- Cheson BD, Bennett JM, Grever M, et al: National Cancer Institute–sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood 87:4990, 1996 8652811
- Binet JL, Auquier A, Dighiero G, et al: A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 48:198, 1981 7237385
- Montserrat E, Gomis F, Vallespí T, et al: Presenting features and prognosis of chronic lymphocytic leukemia in younger adults. Blood 78:1545, 1991 1884021
- Lee JS, Dixon DO, Kantarjian HM, et al: Prognosis of chronic lymphocytic leukemia: a multivariate regression analysis of 325 untreated patients. Blood 69:929, 1987 3814821
- Catovsky D, Fooks J, Richards S: Prognostic factors in chronic lymphocytic leukaemia: the importance of age, sex and response to treatment in survival: a report from the MRC CLL 1 trial. Br J Haematol 72:141, 1989 2757960
- Montserrat E, Sanchez-Bisono J, Viñolas N, et al: Lymphocyte doubling time in chronic lymphocytic leukaemia: analysis of its prognostic significance. Br J Haematol 62:567, 1986 3954968
- Reinish W, Willheim M, Hilgarth M, et al: Soluble CD23 reliably reflects disease activity in B-cell chronic lymphocytic leukemia. J Clin Oncol 12:2146, 1994
- Sarfati M, Chevret S, Chastang C, et al: Prognostic importance of serum soluble CD23 level in chronic lymphocytic leukemia. Blood 88:4259, 1996 8943862
- Molica S, Levato D, Dell'Olio M, et al: Cellular expression and serum circulating levels of CD23 in B-cell chronic lymphocytic leukemia: implications for prognosis. Haematologica 81:428, 1996 8952156
- Semenzato G, Foa R, Agostini C, et al: High serum levels of soluble interleukin 2 receptor in patients with B chronic lymphocytic leukemia. Blood 70:396, 1987 3111560
- Robertson LE, Plunkett W, McConnell K, et al: Bcl-2 expression in chronic lymphocytic leukemia and its correlation with the induction of apoptosis and clinical outcome. Leukemia 10:456, 1996 8642861
- Pepper C, Hoy T, Bentley DP: Bcl-2/Bax ratios in chronic lymphocytic leukaemia and their correlation with in vitro apoptosis and clinical resistance. Br J Cancer 76:935, 1997 9328155
- Döhner H, Fischer K, Bentz M, et al: p53 gene deletion predicts for poor survival and non-response to therapy with purine analogs in chronic B-cell leukemias. Blood 85:1580, 1995
- Robertson LE, Pugh W, O'Brien S, et al: Richter's syndrome: a report on 39 patients. J Clin Oncol 11:1985, 1993 8410123
- Molica S, Alberti A: Second neoplasms in chronic lymphocytic leukemia: analysis of incidence as a function of the length of follow-up. Haematologica 74:481, 1989 2511118
- Travis LB, Curtis RE, Hankey BF, et al: Second cancers in patients with chronic lymphocytic leukemia. J Natl Cancer Inst 84:1422, 1992 1512794
- Cheson BD, Vena D, Freidlin B, et al: The risk of secondary malignancies following purine analog therapy of chronic lymphocytic leukemia (CLL) and hairy cell leukemia (HCL). Proc ASCO 16:15a (abstr 52), 1997
- Dighiero G, Maloum K, Desablens B, et al: Chlorambucil in indolent chronic lymphocytic leukemia. French Cooperative Group on Chronic Lymphocytic Leukemia. N Engl J Med 338:1506, 1998
- Heath ME, Cheson BD: Defective complement activity in chronic lymphocytic leukemia. Am J Hematol 19:63, 1985 3845763
- Cheson BD, Bennett JM, Rai KR, et al: Guidelines for clinical protocols for chronic lymphocytic leukemia: report of the NCI-sponsored Working Group. Am J Hematol 29: 152, 1988 3189311
- Treatment of chronic lymphocytic leukemia: a preliminary report of Spanish (PETHEMA) trials. Spanish Cooperative Group. Leuk Lymphoma 5:89, 1991
- Molica S, Alberti A: Recombinant alpha-2a interferon in treatment of B-chronic lymphocytic leukemia: a preliminary report with emphasis on previously untreated patients in early stage of disease. Haematologica 75:75, 1990 2338291
- Rai KR, Peterson B, Elias L, et al: A randomized comparison of fludarabine and chlorambucil for patients with previously untreated chronic lymphocytic leukemia: a CALGB, SWOG, CTC/NCI-C and ECOG inter-group study. Blood 88:141a (abstr 552), 1996
- Grever MR, Kopecky KJ, Coltman CA, et al: Fludarabine monophosphate: a potentially useful agent in chronic lymphocytic leukemia. Nouv Rev Fr Hematol 30:457, 1988
- Keating MJ, Kantarjian H, Talpaz M, et al: Fludarabine: a new agent with major activity against chronic lymphocytic leukemia. Blood 74:19, 1989 2473795
- O'Brien S, Kantarjian H, Beran M, et al: Results of fludarabine and prednisone therapy in 264 patients with chronic lymphocytic leukemia with multivariate analysis-derived prognostic model for response to treatment. Blood 82:1695, 1993
- Sorensen JM, Vena D, Fallavollita A, et al: Treatment of refractory chronic lymphocytic leukemia with fludarabine phosphate via the group C mechanism of the National Cancer Institute: 5-year follow-up report. J Clin Oncol 15:458, 1997 9053466
- Multicentre prospective randomised trial of fludarabine versus cyclophosphamide, doxorubicin, and prednisone (CAP) for treatment of advanced-stage chronic lymphocytic leukaemia. The French Cooperative Group on CLL. Lancet 347:1432, 1996
- Keating MJ, O'Brien S, Lerner S, et al: Long-term follow-up of patients with chronic lymphocytic leukemia (CLL) receiving fludarabine regimens as initial therapy. Blood 92:1165,1998 9694704
- Cheson BD, Vena D, Foss F, et al: Neurotoxicity of purine analogs: a review. J Clin Oncol 12:2216, 1994 7931492
- Cheson BD: Immunologic and immunosuppressive complications of purine analogue therapy. J Clin Oncol 13:2431, 1995
- Saven A, Carrera CJ, Carson DA, et al: 2-Chlorodeoxyadenosine treatment of refractory chronic lymphocytic leukemia. Leuk Lymphoma 5:133, 1991
- Dillman RO, Mick R, McIntyre OR: Pentostatin in chronic lymphocytic leukemia: a phase II trial of Cancer and Leukemia Group B. J Clin Oncol 7:433, 1989 2784491
- Montserrat E, Alcala A, Parody R, et al: Treatment of chronic lymphocytic leukemia in advanced stages: a randomized trial comparing chlorambucil plus prednisone versus cyclophosphamide, vincristine, and prednisone. Cancer 56:2369, 1985 3899346
- Raphael B, Andersen JW, Silber R, et al: Comparison of chlorambucil and prednisone versus cyclophosphamide, vincristine, and prednisone as initial treatment for chronic lymphocytic leukemia: long-term follow-up of an Eastern Cooperative Oncology Group randomized clinical trial. J Clin Oncol 9:770, 1991 2016618
- Hansen MM, Andersen E, Birgens H, et al: CHOP versus chlorambucil + prednisolone in chronic lymphocytic leukemia. Leuk Lymphoma 5:97, 1991
- Montserrat E, Alcala A, Alonso C, et al: A randomized trial comparing chlorambucil plus prednisone vs cyclophosphamide, melphalan, and prednisone in the treatment of chronic lymphocytic leukemia stages B and C. Nouv Rev Fr Hematol 30:429, 1988
- Kimby E, Millstedt H: Chlorambucil/prednisone versus CHOP in symptomatic chronic lymphocytic leukemias of B-cell type: a randomized trial. Leuk Lymphoma 5:93, 1991
- Rabinowe SN, Soiffer RJ, Gribben JG, et al: Autologous and allogeneic bone marrow transplantation for poor prognosis patients with B-cell chronic lymphocytic leukemia. Blood 82:1366, 1993 7688995
- 55. Michallet M, Archimbaud E, Bandini G, et al: HLA-identical sibling bone marrow transplantation in younger patients with chronic lymphocytic leukemia. Ann Intern Med 124:311, 1996 8554226
- Khouri IF, Przepiorka D, van Besien K, et al: Allogeneic blood or marrow transplantation for chronic lymphocytic leukaemia: timing of transplantation and potential effect of fludarabine on acute graft-versus-host disease. Br J Haematol 97:466, 1997 9163617
- Khouri IF, Keating MJ, Vriesendorp HM, et al: Autologous and allogeneic bone marrow transplantation for chronic lymphocytic leukemia: preliminary results. J Clin Oncol 12:748, 1994 8151318
- Pawson R, Dyer MJS, Barge R, et al: Treatment of T-cell prolymphocytic leukemia with human CD52 antibody. J Clin Oncol 15:2667, 1997 9215839
- Österborg A, Dyer MJS, Bunjes D, et al: Phase II multicenter study of human CD52 antibody in previously treated chronic lymphocytic leukemia. J Clin Oncol 15:1567, 1997
- Intravenous immunoglobulin for the prevention of infection in chronic lymphocytic leukemia: a randomized, controlled clinical trial. Cooperative Group for the Study of Immunoglobulin in Chronic Lymphocytic Leukemia. N Engl J Med 319:902, 1988
- Weeks JC, Tierney MR, Weinstein MC: Cost effectiveness of prophylactic intravenous immune globulin in chronic lymphocytic leukemia. N Engl J Med 325:81, 1991 1904989
- Rose E, Rai K, Revicki D, et al: Clinical and health status assessments in anemic chronic lymphocytic leukemia (CLL) patients treated with epoietin alfa (EPO). Blood 84:526a (abstr 2091), 1994
- Grever M, Kopecky K, Foucar MK, et al: A randomized comparison of pentostatin vs alpha-interferon in previously untreated patients with hairy cell leukemia: an intergroup study. J Clin Oncol 13:974, 1995 7707126
- Saven A, Burian C, Koziol JA, et al: Long-term follow-up of patients with hairy cell leukemia after cladribine treatment. Blood 92:1918, 1998 9731048
- Modiano MR, Villar-Werstler P, Crowley J, et al: Evaluation of race as a prognostic factor in multiple myeloma: an ancillary of Southwest Oncology Group study 8229. J Clin Oncol 14:974, 1996. 8622048
- Vidriales MB, Anderson KC: Adhesion of multiple myeloma cells to the bone marrow microenvironment: implications for future therapeutic strategies. Mol Med Today 2:425, 1996 8897437
- Rettig MB, Ma HJ, Vescio RA, et al: Kaposi's sarcoma–associated herpesvirus infection of bone marrow dendritic cells from multiple myeloma patients. Science 276:1851, 1997 9188529
- Perez-Simón JA, Garcia-Sanz R, Tabernero MD, et al: Prognostic value of numerical chromosome aberrations in multiple myeloma: a FISH analysis of 15 different chromosomes. Blood 91:3366, 1998
-
Berenson JR, Lichtenstein A, Porter L, et al: Efficacy
of pamidronate in reducing skeletal events in patients with advanced multiple
myeloma. N Engl J Med 334:488, 1996 8559201
-
Berenson JR, Lichtenstein A, Porter L, et al: Long-term
pamidronate treatment of advanced multiple myeloma patients reduces skeletal
events. Myeloma Aredia Study Group. J Clin Oncol 16:593, 1998
-
Österborg A, Boogaerts MA, Cimino R, et al: Recombinant
human erythropoietin in transfusion-dependent anemic patients with multiple
myeloma and non-Hodgkin's lymphoma: a randomized multicenter study. Blood
87:2675, 1996
-
Bladé J, Lust JA, Kyle RA: Immunoglobulin D
multiple myeloma: presenting features, response to therapy, and survival
in a series of 53 cases. J Clin Oncol 12:2398 , 1994
-
Durie BGM, Salmon SE: A clinical staging system for
multiple myeloma. Cancer 36: 842, 1975
-
Dimopoulos MA, Barlogie B, Smith TL, et al: High serum
lactate dehydrogenase level as a marker for drug resistance and short survival
in multiple myeloma. Ann Intern Med 115:931, 1991 1952489
-
Greipp PR, Lust JA, O'Fallon M, et al: Plasma cell
labeling index and b2-microglobulin predict
survival independent of thymidine kinase and C-reactive protein in multiple
myeloma. Blood 81:3382, 1993 8507875
-
Ballester OF, Moscinski LC, Lyman GH, et al: High levels
of interleukin-6 are associated with low tumor burden and low growth fraction
in multiple myeloma. Blood 83: 1903, 1994 8142657
-
Witzig TE, Gertz MA, Lust JA, et al: Peripheral blood
monoclonal plasma cells as a predictor of survival in patients with multiple
myeloma. Blood 88:1780, 1996 8781435
-
Kay NE, Lwong T, Kyle RA, et al: Circulating blood
B cells in multiple myeloma: analysis and relationship to circulating clonal
cells and clinical parameters in a cohort of patients entered on the Eastern
Cooperative Oncology Group phase III E9486 clinical trial. Blood 90:340,
1997 9207470
-
Moulopoulos LA, Dimopoulos MA, Smith TL, et al: Prognostic
significance of magnetic resonance imaging in patients with asymptomatic
multiple myeloma. J Clin Oncol 13:251, 1995 7799027
-
Mandelli F, Avvisati G, Amadori S, et al: Maintenance
treatment with recombinant interferon alfa-2b in patients with multiple
myeloma responding to conventional induction chemotherapy. N Engl J Med
322:1430, 1990 2184356
-
Attal M, Harousseau J-L, Stoppa A-M, et al: A prospective,
randomized trial of autologous bone marrow transplantation and chemotherapy
in multiple myeloma. N Engl J Med 335:91, 1996
-
Falk RH, Comenzo RL, Skinner M: The systemic amyloidoses.
N Engl J Med 337:898, 1997 9302305
-
Kyle RA, Gertz MA, Greipp PR, et al: A trial of three
regimens for primary amyloidosis: colchicine alone, melphalan and prednisone,
and melphalan, prednisone, and colchicine. N Engl J Med 336:1202, 1997
9110907
- Comenzo RL, Vosburgh E, Falk RH, et al: Dose-intensive melphalan with blood stem-cell support for the treatment of AL (amyloid light-chain) amyloidosis: survival and responses in 25 patients. Blood 91:3662, 1998 9573002