Contenido del artículo
XI LINFOMAS MALIGNOS
- Generalidades
- Enfermedad de Hodgkin
- ETIOLOGIA Y EPIDEMIOLOGIA
- PATOLOGIA
- CARACTERISTICAS CLINICAS
- EVALUACION DIAGNOSTICA
- ESTADIFICACION Y PRONOSTICO
- TRATAMIENTO
- EFECTOS AGUDOS Y CRONICOS DEL TRATAMIENTO
- Linfoma no-Hodgkin
- PATOLOGIA Y CLASIFICACION
- EVALUACION DIAGNOSTICA
- ESTADIFICACION Y PRONOSTICO
- SUBTIPOS DE LINFOMA NO HODGKIN
- Linfoma folicular
- Linfoma linfocítico de células pequeñas
- Linfoma de células en manto
- Linfoma de la zona marginal
- Linfoma difuso de células grandes
- Linfoma de Burkitt y no Burkitt
- Linfoma linfoblástico
- Linfoma de células T periféricas
- Linfoma anaplásico primario de la piel
- LINFOMAS NO-HODGKIN EN EL SIDA
Generalidades
Con una incidencia de alrededor de 60,000 casos por año, los linfomas malignos representan el sexto tipo en frecuencia de cáncer en los Estados Unidos. Alrededor del 15 por ciento de los pacientes tienen linfoma de Hodgkin y el resto alguno de los linfomas de tipo no Hodgkin. La incidencia de linfoma no Hodgkin está aumentando, sobre todo en personas ancianas o con síndrome de inmunodeficiencia adquirida (SIDA). El enfoque multimodal para el diagnóstico y tratamiento de los linfomas malignos sirve como paradigma del tratamiento exitoso del cáncer. Los efectos a largo plazo del tratamiento anticanceroso pueden ser vigilados debido al mayor número de personas que sobreviven durante periodos prolongados. Los linfomas sirven también como un área de prueba para tratamientos novedosos, como la terapia biológica o los agentes que modulan la resistencia a los medicamentos.
Enfermedad de Hodgkin
ETIOLOGIA Y EPIDEMIOLOGIA
Existen evidencias que apoyan la participación de factores tanto ambientales como genéticos en la etiología de la enfermedad de Hodgkin. En los Estados Unidos y Europa occidental la enfermedad de Hodgkin es más frecuente en varones, y ocurre en una distribución bimodal de edades, con la incidencia máxima en la tercera década y una menor en la séptima década. Es más frecuente en miembros de familias pequeñas, primogénitos, personas criadas en un ambiente higiénico y personas con grados altos de educación. En países en vías de desarrollo la enfermedad de Hodgkin ocurre en la segunda década y predomina en varones. Este padecimiento es extremadamente raro en Japón.
Se ha observado un mayor riesgo en familiares de pacientes con enfermedad de Hodgkin, y este puede ser hasta 10 veces mayor en hermanos del mismo sexo. Se observa una concordancia significativa en los tipos HLA entre los familiares afectados. Las evidencias preliminares indican que algunos antígenos específicos de clase II se asocian con un mayor riesgo en la población, un hallazgo que puede relacionarse además con el subtipo histológico.1
Se ha postulado la asociación entre la infección por virus Epstein-Barr (VEB) y enfermedad de Hodgkin con base en múltiples líneas de evidencia, incluyendo una mayor incidencia de enfermedad de Hodgkin en individuos con antecedente de mononucleosis infecciosa y un estudio prospectivo y controlado que demostró títulos elevados de VEB en pacientes antes del diagnóstico de enfermedad de Hodgkin.2 Además, el genoma del virus Epstein-Barr puede identificarse por técnicas moleculares en las células malignas de alrededor del 50 por ciento de los pacientes con enfermedad de Hodgkin.3 La detección de proteína de membrana latente codificada por el VEB en la enfermedad de Hodgkin indica activación de por lo menos un gen del virus con potencial oncogénico. El genoma del VEB se detecta con más frecuencia en tejidos de enfermedad de Hodgkin en niños pequeños y pacientes que residen en países en vías de desarrollo, además de en enfermedad de Hodgkin subtipo celularidad mixta.
Las investigaciones recientes apoyan el concepto de que las células malignas en la enfermedad de Hodgkin se originan de células B de centros foliculares que sufren un evento de transformación.4 La producción resultante de diversas citocinas por las células de Hodgkin es la responsable de las características tisulares, los hallazgos serológicos, los síntomas constitucionales y la inmunodeficiencia asociada. Sin embargo, no es claro cuál es el evento transformador y la etiología de la enfermedad de Hodgkin sigue siendo un enigma. La apariencia tan diferente, las manifestaciones e historia natural de los subtipos histológicos de esta enfermedad sugiere que pudiera tener muchas causas probables, con interacciones complejas entre factores genéticos, ambientales y del sistema inmunológico de los individuos afectados.
PATOLOGIA
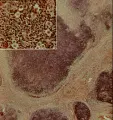
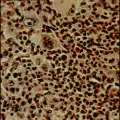
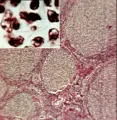
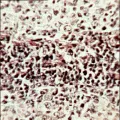
La célula de Reed-Sternberg, una célula gigante multinucleada, y sus variantes mononucleares se consideran las células malignas de la enfermedad de Hodgkin. El hecho de que estas células constituyen solo el 1 a 10 por ciento del número total de células en una muestra de patología ha contribuido a la dificultad para su caracterización. Para la nomenclatura histopatológica de la enfermedad de Hodgkin se usa mucho la clasificación de Rye [ver tabla 1]. Se han distinguido cuatro subtipos con características morfológicas muy variadas: predominio de linfocitos, esclerosis nodular, celularidad mixta y depleción linfocitaria. El subtipo de esclerosis nodular es el más frecuente, en especial en pacientes menores de 40 años, seguido de la celularidad mixta [ver figura 1]. El subtipo caracterizado por depleción linfocitaria es bastante raro y debe distinguirse de los linfomas no Hodgkin. Los nombres de los subtipos de enfermedad de Hodgkin describen el medio histológico en el que se encuentran las células de Reed-Sternberg y sus variantes, así como el grado de infiltración linfocitaria normal. Existen evidencias importantes de que las citocinas participan en el fondo histológico, que se caracteriza por células plasmáticas, eosinófilos, neutrófilos, linfocitos y, en el subtipo de esclerosis nodular, anchas bandas de colágena.
|
|
|
|
|
||||||||||
|
Los estudios inmunohistoquímicos en la enfermedad de Hodgkin revelan que las células de Reed-Sternberg se tiñen en forma positiva para el CD15 (un marcador de monocitos-macrófagos) y CD3 (un marcador de células activadas). La investigación molecular de la célula de Reed-Sternberg y sus variantes morfológicas se ha dificultado por el hecho de que estas células representan solo una pequeña porción del número total de células en el tejido. Sin embargo, en la actualidad se ha demostrado el linaje de célula B de las células de Reed-Sternberg por medio de análisis de una sola célula. Se ha encontrado una frecuencia excesiva de mutaciones dentro del gen de la región variable de la cadena pesada de la inmunoglobulina, lo que altera su función.5 Debido a que estas células no sufren apoptosis (muerte celular programada) a pesar de sus mutaciones, se ha postulado un evento transformador, como la infección por VEB.6 En un subgrupo de casos negativos para VEB se han encontrado mutaciones del gen p53.
Las células malignas del subtipo predominio de linfocitos tienen una apariencia única denominada en palomitas de maíz y expresión aberrante de antígenos. Se tiñen en forma positiva con anticuerpos contra células B y no con anticuerpos dirigidos contra CD15. Los estudios de células únicas indican que el tipo de predominio de linfocitos nodular se origina de la expansión clonal de células B de centros germinales con mutaciones importantes.7 Un grupo de estudio internacional ha descrito un nuevo subtipo de enfermedad de Hodgkin como "clásico, rico en linfocitos". Los tejidos de esta categoría provisional tienen algunas características morfológicas del tipo predominio de linfocitos, pero poseen datos inmunohistoquímicos de la enfermedad de Hodgkin típica descrita antes.8
CARACTERISTICAS CLINICAS
El dato de presentación más frecuente en la enfermedad de Hodgkin es el crecimiento no doloroso de los ganglios linfáticos, por lo general uno o más ganglios situados por arriba del diafragma. Es frecuente la adenopatía mediastinal, sobre todo en pacientes entre 15 y 35 años de edad que tienen el subtipo de esclerosis nodular. La afección subdiafragmática es menos común. En ocasiones la enfermedad de Hodgkin se manifiesta por tos, molestia torácica o síndrome de vena cava superior.
La enfermedad de Hodgkin se caracteriza por su diseminación ordenada de una región ganglionar a la contigua. La evolución de la enfermedad no tratada es muy variable. En muchos pacientes jóvenes puede ser indolente, con adenopatía que aumenta y disminuye de tamaño; sin embargo, en otros puede tener una evolución más agresiva. Con frecuencia el bazo y los ganglios linfáticos del tronco celiaco son los primeros sitios de enfermedad subdiafragmática, que se encuentra en el 20 a 30 por ciento de los pacientes con enfermedad que pareciera estar confinada al cuello, axila o tórax. Es típico que los ganglios paraórticos y pélvicos sean los siguientes en afectarse, y ocurre invasión hepática y de la médula ósea después de la afección del bazo. La enfermedad intratorácica es consecuencia de la diseminación hacia el parénquima pulmonar, el pericardio y la pared torácica desde la masa de adenopatía mediastinal. Las lesiones óseas, que son consecuencia de diseminación contigua de ganglios linfáticos o de diseminación hematógena, se manifiestan por cambios radiológicos osteoblásticos. Puede ocurrir compresión de la médula ósea por extensión directa de los ganglios crecidos o por enfermedad vertebral.
Algunos pacientes, en especial los que tienen enfermedad diseminada o con el subtipo de celularidad mixta o depleción linfocitaria, tienen síntomas generales, conocidos como síntomas B. Estos incluyen episodios nocturnos de diaforesis, fiebre y pérdida de peso. En ocasiones los pacientes presentan un patrón de fiebre de Pel-Ebstein, que consiste en episodios intermitentes de fiebres vespertinas que duran varios días y alternan con periodos afebriles. El prurito generalizado, que es muy incómodo y refractario al tratamiento con agentes antipruriginosos, puede ser un síntoma asociado. Otra característica es el dolor inducido en los sitios de adenopatía inmediatamente después de la ingestión de alcohol.
La evolución clínica de la enfermedad de Hodgkin difiere de acuerdo con la edad, geografía e integridad del sistema inmunológico. El subtipo de predominio linfocitario tiene un curso especialmente indolente. Los pacientes típicamente presentan ganglios linfáticos periféricos por arriba o debajo del diafragma y la adenopatía mediastinal es poco común. Los síntomas generales son raros. Los pacientes ancianos, los que pertenecen a países en vías de desarrollo y los enfermos con infección por virus de inmunodeficiencia humana (VIH) tienen más probabilidad de sufrir enfermedad diseminada y síntomas generales en el momento del diagnóstico. El subtipo de celularidad mixta es la variable más frecuente observada en este grupo. Los pacientes jóvenes de los Estados Unidos y de Europa occidental presentan con mayor frecuencia enfermedad limitada de subtipo esclerosis nodular.
EVALUACION DIAGNOSTICA
El propósito de la evaluación diagnóstica es investigar la extensión (estadio) de la enfermedad de Hodgkin, y se usa para establecer el pronóstico y el tratamiento. La prueba diagnóstica aislada más útil para un paciente con probable linfoma es una biopsia de ganglio linfático realizada y evaluada en forma adecuada. Debe revisarse la historia del paciente en busca de síntomas generales. Aunque deben considerarse otras causas de adenopatía, como enfermedad por rasguño de gato o infección por VIH, estos diagnósticos pueden excluirse por medio de una revisión patológica correcta, incluyendo estudios inmunohistoquímicos.
El examen físico debe incluir la evaluación cuidadosa de todas las regiones linfáticas para evaluar la extensión de la linfadenopatía periférica. También está indicado revisar la pared torácica, palpar la columna vertebral en busca de dolor y examinar el abdomen para detectar hepatoesplenomegalia.
Siempre está indicado realizar una radiografía de tórax de escrutinio. La mayoría de los pacientes, incluso los que tienen una radiografía normal, deben someterse a una TC del tórax, esta es crucial para planear la radioterapia definitiva porque permite examinar el mediastino y la cavidad intratorácica con detalle, siendo muy valiosa para definir la extensión de la enfermedad y su diseminacipón a sitios extranodales. Por lo general se solicita una TC de abdomen y pelvis para visualizar el tronco celiaco y detectar la presencia de linfadenopatía retroperitoneal y pélvica. La afección mesentérica no es frecuente en la enfermedad de Hodgkin. La linfangiografía de las extremidades inferiores permite estudiar los ganglios linfáticos con más detalle, y su exactitud diagnóstica es alrededor de 10 por ciento mayor que la de la TC. Además, el medio de contraste se mantiene dentro de los ganglios opacificados durante 1 a 2 años. Sin embargo, en muchos centros médicos no se realizan linfangiografías porque el número de técnicos y radiólogos capaces de realizar e interpretar este estudio ha disminuido. Se ha incorporado la imagen por resonancia magnética (IRM) al protocolo diagnóstico con un éxito variable. La IRM es más sensible que la TC para valorar la enfermedad de la pared torácica, pero se ha encontrado que es poco útil para analizar la afección abdominal. Al parecer el galio, que es captado en forma ávida por el tejido afectado, es útil para evaluar la respuesta al tratamiento. Varios estudios sugieren que el fracaso para transformar un gamagrama positivo con galio en un gamagrama negativo después del tratamiento indica una alta probabilidad de persistencia de la enfermedad de Hodgkin. La tomografía de emisión de positrones (TEP) distingue al tejido neoplásico del normal con base en sus características metabólicas. En la actualidad existen estudios en progreso para determinar la utilidad de la TEP para estadificar la enfermedad o medir la respuesta al tratamiento.
Un problema importante con todas las técnicas radiológicas es la imposibilidad para diagnosticar la afección esplénica. Se ha empleado la laparotomía exploradora como un medio para investigar la diseminación subdiafragmática de la enfermedad de Hodgkin, lo que ha permitido también un mayor conocimiento de la historia natural de este padecimiento. A pesar de los importantes adelantos en las técnicas de imagenología, la laparotomía sigue siendo el método más definitivo para detectar enfermedad nodal, hepática o esplénica ocultas. Sin embargo, en la actualidad su uso se restringe a situaciones en las que los pacientes recibirán quimioterapia. El procedimiento incluye esplenectomía; muestras y extirpación de los ganglios celiacos, del hilio esplénico, de la porta hepatis y paraórticos (poniendo especial atención en las áreas que parecen sospechosas en los estudios radiológicos); muestras del hígado por medio de biopsia en cuña y con aguja bajo visión directa, y biopsia abierta de la médula ósea de la cresta iliaca. La radiografía abdominal durante o después de la laparotomía puede ayudar a extirpar los ganglios sospechosos que se opacifiquen con aceite etiodizado después de la linfangiografía. En este momento se emplea la laparotomía en forma más limitada porque la quimioterapia se incluye en muchas circunstancias clínicas, y porque los estudios retrospectivos proporcionan una base para determinar la probabilidad de enfermedad abdominal según las diferentes presentaciones clínicas.9 Un estudio prospectivo demostró que la laparotomía no tuvo un impacto significativo en la supervivencia o el éxito del tratamiento en pacientes con un pronóstico favorable y que fueron distribuidos en forma aleatoria para someterse a estadificación con laparotomía o a estadificación solo clínica.10 El riesgo de enfermedad subdiafragmática oculta puede calcularse en función del sexo, edad, velocidad de sedimentación globular (VSG), número de sitios ganglionares afectados y subtipo histológico. La biopsia de médula ósea se incluye en la estadificación rutinaria de los pacientes que tienen enfermedad en un estadio avanzado, síntomas generales o ambos.
Los estudios de laboratorio incluyen una citología hemática completa y determinación de química sérica y VSG. La alteración hematológica más frecuente es la anemia normocítica normocrómica, que suele asociarse con enfermedad extensa y sintomática. En pocas ocasiones se observan citopenias secundarias a hiperesplenismo o a enfermedad de la médula ósea. Pueden ocurrir elevaciones discretas de la fosfatasa alcalina en pacientes con enfermedad limitada y los niveles más altos se asocian con afección hepática, ósea o de la médula ósea. La hipercalcemia es una manifestación paraneoplásica poco común. Es frecuente que la VSG se eleve en la enfermedad de Hodgkin y varios estudios han demostrado que el grado de elevación tiene un significado pronóstico en los pacientes con enfermedad temprana.
ESTADIFICACION Y PRONOSTICO
Mientras que el estadio clínico se refiere a la extensión de la enfermedad según lo determinan las biopsias diagnósticas y los estudios radiológicos, el término de estadio patológico suele indicar que se realizó una laparotomía. En 1971 se desarrolló el sistema de estadificación de Ann Arbor para la enfermedad de Hodgkin [ver tabla 2], en un momento cuando el principal método de tratamiento era la radioterapia.11 La enfermedad en estadio I se limita a un solo grupo de ganglios linfáticos, el estadio II refleja enfermedad en múltiples regiones ganglionares en un solo lado del diafragma y el estadio III se refiere a linfadenopatía diseminada en ambos lados del diafragma. Los pacientes con enfermedad diseminada a sitios extranodales se clasifican en estadio IV. La extensión de un sitio extranodal a partir de ganglios linfáticos contiguos, en una región anatómica que pueda abarcarse por un solo puerto de radioterapia, se considera como lesión E en lugar de estadio IV. Debido al mayor uso actual de la quimioterapia y al conocimiento de otras variables pronósticas se han propuesto varias modificaciones para este sistema de estadificación. En 1990 se propuso la modificación de Cotswold para los criterios de Ann Arbor [ver tabla 2].12 Esta modificación incorporó el significado pronóstico de la masa tumoral, sobre todo en el mediastino, y los subtipos de estadificación del estadio IIIA.
|
|||||||||||||||
* Todos los casos subclasifican para designar la ausencia (A) o presencia (B) de síntomas generales, como fiebre significativa
(>38°C; 100.4°F), diaforesis nocturna y pérdida de peso inexplicable que exceda el 10% del peso corporal normal en los 6 meses
previos.
|
El análisis de un extenso grupo de datos internacionales reveló siete factores adversos, además del estadio, que pueden predecir la evolución en la enfermedad de Hodgkin avanzada13 [ver tabla 3]. Cada factor contribuyó a una reducción en el número de pacientes vivos y libre de enfermedad de Hodgkin 5 años después del tratamiento. Los factores fueron edad mayor de 45 años, enfermedad en estadio IV, sexo masculino, anemia, hipoalbuminemia, linfocitopenia y neutrofilia. Estos factores se están considerando ya en los estudios clínicos nacionales de enfermedad de Hodgkin avanzada.
|
||||||||||||||||||||||||||||
Nota:Los factores de riesgo adversos son edad >-45 años, enfermedad en estadio
|
TRATAMIENTO
Una clasificación histopatológica que se acepta en forma general, la estadificación exacta, los mejores esquemas de radioterapia y la existencia de agentes quimioterapéuticos más eficaces, contribuyen al alto porcentaje de curación de la enfermedad de Hodgkin. El éxito dependerá de que la atención sea administrada por un equipo multidisciplinario con experiencia en histopatología, radiología diagnóstica, cirugía, radioterapia y oncología médica. Los nuevos esquemas de tratamiento intentan mantener tasas altas de curación con menos complicaciones a largo plazo.
Tratamiento primario
El tratamiento inicial de la enfermedad de Hodgkin depende de la extensión de la enfermedad, que está determinada por el estadio y los factores pronósticos. Históricamente, la radioterapia era el tratamiento preferido, reservándose la quimioterapia para la enfermedad avanzada o los fracasos de la radioterapia. Sin embargo, el conocimiento de los efectos tardíos de la radioterapia, incluyendo un mayor riesgo de tumores sólidos y enfermedad cardiaca, y el desarrollo de combinaciones de quimioterapia menos tóxicas han originado nuevos enfoques terapéuticos. Los tratamientos actuales tienen por objeto mantener tasas altas de curación y disminuir la toxicidad aguda y crónica.
Enfermedad limitada Durante varias décadas la radioterapia ha sido el tratamiento de elección para los pacientes con enfermedad favorable y limitada, que se define como una presentación sin una masa de gran tamaño, supradiafragmática y sin afección extranodal o solo con afección extranodal en un sitio, en un paciente asintomático.14 Debido a que la enfermedad de Hodgkin se disemina en forma ordenada a través de los grupos ganglionares adyacentes, se administra radioterapia con megavoltaje dirigida a los ganglios afectados y los no afectados de regiones contiguas. En pacientes estadificados por laparotomía, dosis diarias de radiación de 180 a 200 cGy, hasta un total de 3,500 a 4,400 cGy, produjeron tasas altas (> 80 por ciento) de supervivencia libre de recidivas. Para fines terapéuticos el organismo se divide en campos. El tratamiento de radiación linfoide subtotal se dirige a ciertas áreas localizadas, que incluyen el manto (ganglios cervicales, supraclaviculares, infraclaviculares, axilares, mediastinales e hiliares) y el campo abdominal (ganglios celiacos, porta hepatis, del hilio esplénico y paraórticos, así como el bazo si no se ha extirpado). Sin embargo, los efectos tardíos de la radioterapia extensa han originado nuevos enfoques terapéuticos en los que se emplea radiación, en combinación con quimioterapia, en forma más selectiva y a menor dosis.
Con objeto de disminuir los efectos tardíos, se emplean ahora esquemas de quimioterapia menos tóxica, como VBM (vinblastina, bleomicina y methotrexate), combinado con radioterapia dirigida a campos más limitados.15 Se han reportado altas tasas de curación usando esquemas breves (dos a cuatro ciclos) de quimioterapia estándar, como ABVD (doxorrubicina [Adriamicina], bleomicina, vinblastina, dacarbazina) y radioterapia en campos limitados.16 A diferencia de los estudios de radioterapia, en estos estudios no se realizó estadificación patológica. Los estudios clínicos que se realizan en la actualidad determinarán la duración óptima de la quimioterapia y la dosis de radioterapia. Además, varios grupos están evaluando la quimioterapia sola en la enfermedad de Hodgkin limitada y favorable (sin datos pronósticos adversos).
La quimioterapia combinada, con o sin radioterapia, es el tratamiento de elección para la enfermedad de Hodgkin estadio IIB. Los pacientes con enfermedad de Hodgkin limitada que se caracteriza por una masa mediastinal voluminosa y afección de múltiples sitios extranodales suelen tratarse con quimioterapia combinada y radioterapia. La enfermedad mediastinal voluminosa se define como una masa mayor de la tercera parte del diámetro intratorácico máximo en una tele de tórax de pie posteroanterior. La selección del esquema de quimioterapia dependerá de la toxicidad asociada y de la cantidad de radiación cardiaca requerida. La combinación de ABVD y radioterapia fue superior a la combinación de MOPP (metotrexate, vincristina [Oncovin], procarbacina y prednisona) y radioterapia en un estudio aleatorio extenso. Las modalidades terapéuticas combinadas causan un porcentaje de curación mayor al 70 o 75 por ciento en pacientes con enfermedad limitada y no favorable.
Enfermedad avanzada La quimioterapia combinada está indicada para los pacientes con enfermedad en estadio III o IV. La enfermedad en estadio IIIA es curable en muchas ocasiones con diversos esquemas de quimioterapia [ver tabla 4]. Estudios comparativos17-23 han demostrado que el esquema ABVD es el más tolerado, tiene la mayor tasa de cumplimiento y la menor toxicidad a corto y largo plazo. Estos estudios demuestran que puede lograrse una supervivencia libre de recaídas del 65 al 70 por ciento en pacientes con las presentaciones menos favorables de enfermedad de Hodgkin. En la actualidad se investigan en estudios clínicos nuevas combinaciones farmacológicas, como la Stanford V.24,25
|
||||||||||||||||||
|
De los factores pronósticos en la enfermedad de Hodgkin, la edad del paciente es muy significativa. La mala evolución en los pacientes mayores se atribuye tanto a una respuesta menos favorable al tratamiento en comparación con los más jóvenes como a un tratamiento inadecuado.26,27 La administración de dosis óptimas de quimioterapia puede beneficiar a esta población. Los estudios intergrupo en los Estados Unidos emplearán a los factores pronósticos dentro de los criterios de inclusión. Por ejemplo, los pacientes con un pronóstico menos favorable (tres o más factores adversos) serán distribuidos a ABVD o ABVD seguido de quimioterapia de dosis intensivas y trasplante autólogo de células hematopoyéticas tronco,13 y los que tengan un pronóstico más favorable (menos de dos factores adversos) recibirán ABVD o Stanford V solo o con radioterapia.
Tratamiento para las recaídas
Los pacientes que sufren recaídas después del tratamiento primario de radiación pueden tener una evolución posterior libre de recaídas cuando son tratados con alguno de los esquemas de quimioterapia descritos para la enfermedad no favorable (i.e., MOPP, ABVD o esquemas híbridos). Las recaídas después de la quimioterapia combinada o de los tratamientos con modalidades mixtas representan un reto importante. Si la duración de la remisión inicial es mayor de 12 meses, el retratamiento con quimioterapia, sea con la misma combinación o con una combinación que no tenga resistencia cruzada, permite una supervivencia libre de recidivas de alrededor del 50 por ciento a los 5 años.28,29 Los pacientes que no alcanzan la remisión completa con la quimioterapia inicial o que recaen antes de pasado un año de la quimioterapia inicial tienen un pronóstico menos favorable.28-30 En los pacientes menores de 60 años muchos clínicos indican el tratamiento en dosis altas (quimioterapia o quimioterapia combinada con radioterapia), facilitada por trasplante autólogo de células tronco.31 Los siguientes factores predicen una evolución favorable con el trasplante autólogo de células hematopoyéticas tronco: ausencia de síntomas generales, ausencia de enfermedad extranodal al recaer y persistencia de sensibilidad al tratamiento convencional.31 Como en el caso del tratamiento convencional, los pacientes que progresan durante el tratamiento primario de inducción, que tienen enfermedad persistente o que progresan en 6 a 12 meses después del tratamiento tienen menos probabilidad de curarse con el trasplante. Con base en estos factores pronósticos, la supervivencia libre de enfermedad varía de 20 a más de 80 por ciento a 2 a 5 años después de la terapia con dosis altas combinada con trasplante de células tronco. Sin embargo, debido a la naturaleza indolente de la enfermedad de Hodgkin recurrente en algunos pacientes, la tasa absoluta de curación después del tratamiento con dosis intensivas no se conoce con claridad.
El seguimiento de los pacientes retratados con quimioterapia por enfermedad de Hodgkin recurrente ha revelado que ocurren muchas defunciones por efectos secundarios tardíos. Al final, la elección del tratamiento secundario, como en el caso del tratamiento primario, debe basarse en la eficacia y la toxicidad aguda y crónica de los esquemas.
EFECTOS AGUDOS Y CRONICOS DEL TRATAMIENTO
Los efectos agudos de la radioterapia dependen del campo, de las fracciones y de la dosis total. El tratamiento subtotal es bien tolerado, con pocos efectos agudos. Debido a que es probable que la toxicidad tardía por radioterapia no se presente sino hasta 2 o más décadas después, hasta ahora se está conociendo el perfil tóxico completo. Se ha detectado un aumento de 7 veces en el riesgo de tumores sólidos específicos, del pulmón, estómago, tejidos blandos, piel y mama.32,33 El cáncer de mama se asocia con radioterapia torácica y axilar y se observa un riesgo relativo aumentado en las pacientes tratadas antes de los 30 años de edad.32 Las adolescentes tienen mayor riesgo. Se ha reportado también mayor mortalidad por afección cardiaca.32,33 Los factores técnicos tienen un papel importante en la toxicidad cardiaca. Las dosis de radiación de menos de 3,000 cGy no parecen implicar mayor riesgo de complicaciones cardiacas diferentes a la enfermedad arterial coronaria.34,35 Por desgracia, los largos periodos de latencia hacen difícil definir las complejas relaciones entre dosis total, fracciones factores técnicos. Sin embargo, datos recientes de la Universidad de Stanford sugieren que el exceso de mortalidad relacionada a la radioterapia en pacientes con enfermedad de Hodgkin ha disminuido en los últimos 16 años, quizá como resultado de modificaciones en la técnica aunado al tratamiento combinado.36
Los efectos colaterales de la quimioterapia se relacionan con las dosis acumuladas de los fármacos individuales en un esquema de combinación [ver tabla 4]. Los efectos adversos agudos de la mayoría de los esquemas incluyen náusea y vómito. Por fortuna, los nuevos antieméticos son muy eficaces. Ocurre mielosupresión en todos los esquemas de quimioterapia eficaces, pero pueden usarse factores de crecimiento hematopoyético para disminuir la incidencia y duración de la neutropenia febril. La alopecia temporal sigue siendo un problema con algunos esquemas. La leucemia y la esterilidad se encuentran entre los efectos tardíos más temidos, ambos se relacionan con las dosis acumuladas de agentes alquilantes como la mostaza nitrogenada y la procarbazina en el esquema MOPP.37-39 Ocurre esterilidad en el 90 por ciento o más de los varones tratados con un esquema completo de MOPP. En las mujeres tratadas con este esquema, las mayores de 25 años tienen un riesgo de 80 por ciento de esterilidad y menopausia temprana, pero las de menor edad tienen más probabilidad de conservar sus periodos menstruales y tener hijos. La administración de 2 a 4 ciclos de MOPP, como en los esquemas alternantes e híbridos, se asocia con menor incidencia de esterilidad, aunque persiste cierto riesgo. No existe un mayor riesgo de defectos al nacimiento en los hijos de los pacientes tratados con radioterapia, quimioterapia o ambos.
La incidencia de mielodisplasia o de leucemia aguda después de 6 ciclos de MOPP alcanza una meseta de 1 a 3 por ciento a los 10 años. Aunque existen algunas controversias respecto al mayor riesgo de leucemia por las modalidades combinadas de tratamiento, un gran estudio de población demostró que no había mayor riesgo cuando se empleaba radioterapia y MOPP.37 El riesgo de leucemia aguda disminuye, aunque no se elimina, por el uso de 2 a 4 ciclos de MOPP.
La ABVD se asocia con una incidencia de esterilidad en ambos sexos de10 a 20 por ciento, que es mucho menor que la asociada con el esquema MOPP.40,41 En raras ocasiones se ha reportado leucemia aguda en los pacientes tratados con ABVD. Aunque los estudios no han publicado un aumento importante en los riesgos de enfermedad cardiopulmonar, las dos principales preocupaciones con este esquema son la posibilidad tardía de enfermedad cardiaca, que se relaciona con el uso de doxorrubicina, y de enfermedad pulmonar, que se relaciona con la bleomicina. Los niños parecen tener mayor riesgo tanto de toxicidad por bleomicina como de toxicidad por doxorrubicina.42,43 Aunque la combinación de ABVD y radioterapia se ha asociado en forma anecdótica con toxicidad pulmonar severa en adultos, esta no se ha reportado en estudios aleatorios grandes.18
La toxicidad aguda de los nuevos esquemas de tratamiento para la enfermedad de Hodgkin, como el Stanford V, son semejantes a la de otras combinaciones de quimioterapia: náusea y vómito, alopecia temporal y mielosupresión. Aún es demasiado pronto para describir sus efectos tardíos.
Linfoma no-Hodgkin
PATOLOGIA Y CLASIFICACION
Los linfomas no-Hodgkin han sido clasificados sobre todo desde el punto de vista morfológico. En 1994 un grupo internacional de estudio del linfoma propuso la clasificación europea-americana revisada para el linfoma.44 Posteriormente un grupo internacional de patólogos, con apoyo de médicos clínicos, desarrolló una clasificación del linfoma para la Organización Mundial de la Salud. La nueva clasificación divide a los linfomas no Hodgkin de acuerdo con la célula de origen (usando anticuerpos que reconocen antígenos de células B y T), las manifestaciones clínicas y los datos morfológicos. Otros marcadores inmunohistoquímicos, datos citogenéticos y caracterización genotípica de los rearreglos de receptores de las células B y T pueden ayudar en el diagnóstico. El significado clínico y la posibilidad de reproducción de la clasificación propuesta se ha corroborado en un estudio internacional.45
Clasificación histopatológica
Diversos patólogos expertos desarrollaron en forma independiente esquemas de clasificación para los linfomas no Hodgkin. Para facilitar la traducción de un esquema de clasificación a otro, el Instituto Nacional del Cáncer patrocinó el desarrollo de una Fórmula de Trabajo [ver tabla 5], en la que los linfomas se clasifican por sus características morfológicas, sobre todo por la presencia o ausencia de un patrón folicular y por la descripción de las células malignas.46 El grado bajo indica una supervivencia promedio mayor, y el grado alto una supervivencia más breve.
|
||||||||||
|
La Fórmula de Trabajo se usó en forma amplia en la práctica clínica en los Estados Unidos, mientras que la clasificación de Kiel modificada por Lennert fue más popular en Europa. La clasificación de la Organización Mundial de la Salud incluye varios linfomas que se han descrito en forma más exacta o que representan nuevas entidades. Entre estos linfomas se encuentran el linfoma de células en manto, el linfoma de tejido linfoide asociado a mucosas (MALT), el linfoma de células B monocitoides, el linfoma anaplásico K-1 y el linfoma de células T periféricas [ver tabla 6].
|
||||
+ Limitado a linfomas, los subtipos en itálicas son los más comunes.
|
Los linfomas con arquitectura mixta, con elementos tanto foliculares como difusos, son frecuentes. Hasta el 20 o 30 por ciento de los pacientes tienen más de un subtipo histológico si se realizan varias biopsias para el diagnóstico. Es muy común el patrón discordante que se caracteriza por diferentes subtipos histológicos en tejidos diversos, en especial un patrón difuso de células grandes en los ganglios linfáticos y un patrón de células pequeñas hendidas en la médula ósea. El linfoma compuesto, que se caracteriza por más de un subtipo histológico en una sola biopsia, no es frecuente. Los linfomas foliculares no-Hodgkin tienen una gran propensión a sufrir, con el tiempo, transformación histológica a linfomas de mayor grado.
Inmunohistoquímica
En general los linfomas no-Hodgkin corresponden a subpoblaciones de células B y T que están activadas o en proliferación. La mayoría se originan de células B. Su origen monoclonal está indicado por la expresión de cadenas ligeras, que se restringe a cadenas kappa o lambda. Los linfomas de células B se clasifican a su vez en expansiones de células malignas del centro germinal, zona del manto o zona marginal de los ganglios linfáticos normales. Los anticuerpos monoclonales dirigidos contra determinantes antigénicos específicos son parte integral del diagnóstico diferencial. Además, la detección inmunohistoquímica de productos genéticos, como bcl-2, una proteína que inhibe la apoptosis, y bcl-1, una proteína que promueve la progresión a través del ciclo celular, se ha vuelto de rutina en el análisis patológico del linfoma.
Menos del 20 por ciento de los casos de linfoma maligno en los Estados Unidos se originan de células T. Los linfomas de células T pueden clasificarse en forma amplia con base en su expresión antigénica como de origen precursor (tímico) o maduro (periférico). Desde el punto de vista clínico estas clasificaciones se traducen en linfoma linfoblástico precursor o en el grupo heterogéneo de linfomas de células T periféricas. El patrón de expresión de los antígenos de superficie celular en los linfomas no-Hodgkin de células T no recuerda los fenotipos de las células T normales en forma tan constante como en el caso de los linfomas no-Hodgkin de células B.
La relación entre la expresión de marcadores de superficie y el pronóstico en los linfomas no-Hodgkin ha sido examinada en varios estudios. Estos marcadores de superficie incluyen marcadores de líneas celulares T o B, antígenos asociados con células normales activadas o en proliferación, expresión de bcl-2, antígenos de histocompatibilidad, moléculas de adhesión celular y linfocitos infiltrantes de tumores.
Análisis molecular
El rearreglo de genes somáticos es un aspecto integral de la diferenciación de las células T y B. Los genes de las regiones variable y constante de las cadenas ligeras y pesadas de las inmunoglobulinas no son continuos en el ADN de las células B de línea germinal, y se combinan por medio de rearreglos somáticos para producir una molécula de anticuerpo funcional. El gen del receptor de la célula T tiene una estructura semejante a la de la molécula de inmunoglobulina en relación a que los segmentos no continuos de este gen sufren rearreglos somáticos en etapas tempranas del desarrollo de las células T. El análisis de hibridización del ADN usando sondas adecuadas permite reconocer una banda de movimiento electroforético que sirve como indicador de una población monoclonal de células de linfoma. En todos los subtipos clínicos de linfoma no-Hodgkin se han observado rearreglos genéticos, y el análisis de hibridización de ADN puede servir para confirmar el diagnóstico de un linfoma que puede ser ambiguo desde el punto de vista histológico o fenotípico.
En la mayoría de los linfomas no-Hodgkin de células B se han detectado alteraciones cariotípicas. Las traslocaciones más prevalentes incluyen: t(8;14), t(14;18) y t(11;14). Cada traslocación afecta al locus genético de la cadena pesada de la inmunoglobulina en el cromosoma 14 (14q32). La identificación y clonación de los puntos de ruptura ha identificado a 8q24 como myc, un oncogén bien caracterizado que tiene asociación evidente con las neoplasias. Las regiones 18q21 y 11q13 han sido designadas como los sitios de los oncogenes putativos asociados a linfoma de células B bcl-2 y bcl-1, respectivamente. La proximidad de estos oncogenes al gen de la inmunoglobulina causa desrregulación y mayor expresión del producto del gen. Alrededor de la tercera parte de los linfomas difusos de células grandes demuestran traslocación de 3q27, que ha sido designada como bcl-6.47 Las traslocaciones de 3q27 incluyen diversas parejas cromosómicas, incluyendo el cromosoma 14.
El oncogén bcl-2, que está presente en la mayoría de los linfomas foliculares, codifica una proteína de membrana que bloquea la apoptosis cuando se expresa en forma exagerada.48 Se ha propuesto que la expresión o ausencia de la traslocación bcl-2, evaluada por el muy sensible método de reacción en cadena de la polimerasa (detección de una célula de linfoma en una población de un millón de células) indica si existe un estado de remisión en los pacientes con linfomas caracterizados por esta traslocación. Esto se basa en reportes de pacientes tratados con quimioterapia combinada sola o seguida de tratamiento mieloablativo y trasplante.49,50 Sin embargo, se han encontrado células que contienen t(14;18) en la sangre de pacientes con linfoma que han tenido remisión continua por muchos años y en las amígdalas y linfocitos de sangre periférica de personas normales.
En la mayoría de las neoplasias humanas se han detectado mutaciones en el gen supresor de tumores p53. Se ha reportado que existe relación entre las mutaciones en el gen p53 y un pronóstico no favorable en los linfomas no Hodgkin agresivos.51
EVALUACION DIAGNOSTICA
Como en la enfermedad de Hodgkin, la prueba diagnóstica aislada más útil en el linfoma no-Hodgkin es una biopsia realizada y evaluada en forma correcta. La historia clínica debe dirigirse hacia la detección de síntomas relacionados tanto con afección extranodal como sistémicos. El exámen físico debe incluir la evaluación cuidadosa de todas las regiones ganglionares (incluyendo el anillo de Waldeyer) y de hepatoesplenomegalia, y debe estar dirigida por la historia del paciente (por ejemplo, por la presencia de dolor en sitios específicos o de lesiones cutáneas).
La evaluación radiológica es crucial para el diagnóstico y estadificación adecuados. Además de una radiografía de tórax de escrutinio, es necesario realizar TC del tórax, abdomen y pelvis. La adenopatía mesentérica, que no ocurre con frecuencia en la enfermedad de Hodgkin, sí es común en los linfomas no-Hodgkin. La necesidad de estudios adicionales, como estudios gastrointestinales con medio de contraste o IRM del cerebro o de la columna, depende de las manifestaciones clínicas y del subtipo histológico.
Los estudios de laboratorio incluyen una biometría hemática completa y la determinación de químicas séricas. El parámetro más significativo es la lactato deshidrogenasa sérica, que es un factor predictor importante de la evolución. La punción lumbar está indicada en pacientes seleccionados que se considera tienen un alto riesgo de afección del sistema nervioso central. También se realiza biopsia de médula ósea.
ESTADIFICACION Y PRONOSTICO
El sistema de estadificación de Ann Arbor para enfermedad de Hodgkin se aplica mucho a los linfomas no-Hodgkin, aunque existen ciertas deficiencias importantes por la gran heterogeneidad de los linfomas no Hodgkin, la presencia de afección extranodal en la mayoría de los pacientes y la mala correlación entre el estadio por Ann Arbor y el pronóstico.
Esta falta de exactitud en el sistema de estadificación de Ann Arbor ha originado un gran esfuerzo internacional por lograr un consenso en relación con los factores pronósticos en los linfomas difusos de células grandes.52 Los grupos de riesgo se determinan de acuerdo con la presencia o ausencia de los siguientes factores adversos de riesgo: edad mayor de 60 años, enfermedad avanzada (estadio Ann Arbor III o IV), pobre condición funcional, niveles anormales de lactato deshidrogenasa, y 2 o más sitios de afección extranodal [ver tabla 7]. El estadio, el estado funcional y la concentración de lactato deshidrogenasa determinan el pronóstico en los pacientes menores de 60 años, y estos factores han sido ya incorporados a los nuevos programas de tratamiento para todos los pacientes con mal pronóstico. Es importante recordar que estos factores clínicos indican factores pronósticos biológicos, algunos de los cuales ya se mencionaron antes. Varios estudios han indicado que algunos de estos factores biológicos, especialmente el linaje de células T y la mayor expresión de bcl-2, son de importancia pronóstica además del uso del índice internacional.53-55
|
||||||||
Nota: Factores adversos de riesgo: edad mayor de 60 años, estadío de Ann Arbor III-IV, dos o más sitios de enfermedad extranodal, estado funcional del paciente >-2, lactato deshidrogenasa (DHL) > aumentada. |
Los factores pronósticos para otros subtipos histológicos de linfoma no han sido estudiados en forma tan extensa, pero el índice internacional se aplica a la mayoría de los subtipos de linfoma no Hodgkin [ver tabla 8].
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
SUBTIPOS DE LINFOMA NO HODGKIN
Linfoma folicular
Grados I y II (células hendidas pequeñas y mixto de células hendidas pequeñas y células grandes) El linfoma folicular fue el segundo subtipo histológico más prevalente en el estudio de clasificación internacional [ver figura 1].45 Por lo general el linfoma folicular de grado bajo se manifiesta como una adenopatía difusa asintomática en un individuo de edad media. En la mayoría de los pacientes se presenta afección de la médula ósea. Es frecuente que las adenopatías tengan una evolución oscilante, y en el 20 a 30 por ciento de los pacientes se ha observado regresión espontánea de la enfermedad objetiva.56 Existe un riesgo real de transformación a un grado histológico más agresivo, que aumenta en forma constante después del diagnóstico y alcanza un 40 a 50 por ciento entre los 8 y 10 años de evolución.
El tratamiento habitual incluye la administración de agentes alquilantes como la ciclofosfamida y la prednisona, que suelen lograr un porcentaje global de respuesta mayor del 80 por ciento. Aunque el 50 a 75 por ciento de los pacientes logran la llamada remisión completa, los linfomas foliculares de bajo grado se caracterizan por un patrón continuo de recaídas con un tiempo promedio entre el tratamiento y la recaída de 1.5 a 3 años. En la actualidad estos linfomas no se consideran curables con tratamiento convencional. El análisis molecular de la traslocación bcl-2 sugiere que muy pocos, si es que alguno, de estos pacientes logran la remisión completa con el tratamiento estándar.57 Debido a que los linfomas foliculares de bajo grado no Hodgkin tienen un comportamiento indolente, aparecen hacia la mitad de la vida o en edad avanzada y no son curables, en algunos pacientes seleccionados se ha propuesto observar y diferir el tratamiento.56 En la actualidad se investigan métodos para modificar la historia natural de los linfomas foliculares de bajo grado no Hodgkin, y éstos incluyen el uso de interferón con quimioterapia combinada, una estrategia que ha producido resultados preliminares promisorios. En varios centros se estudia el uso de la quimioterapia en dosis altas combinada con radioterapia y trasplante autólogo de células hematopoyéticas tronco como parte del tratamiento inicial o como tratamiento para las recidivas. Aunque se logran remisiones más prolongadas con este método, no se ha definido en estos estudios no controlados que la supervivencia sea mayor. En esquemas de fármacos múltiples se está evaluando el análogo de nucléosidos fludarabina, que es muy activo en el linfoma de bajo grado.58,59
Los anticuerpos monoclonales contra el antígeno de células B CD20 han demostrado tener actividad antitumoral y el anticuerpo quimérico (murino-humano) rituximab ha sido autorizado para usarse en linfomas de bajo grado con base en estudios de fase I y II que sugieren una tasa de respuesta de alrededor del 50 por ciento pero que no implican cambio en la supervivencia.60 El objetivo de múltiples estudios clínicos en progreso consiste en incorporar este anticuerpo a las estrategias primarias de manejo, combinándolo con terapias citotóxicas. La conjugación de un anticuerpo anti-CD20 a los radioisótopos yodo-131 e itrio-90 representa una estrategia promisoria porque los linfomas foliculares son especialmente sensibles a la radiación.61,62 Por último, el idiotipo (determinante molecular de la región variable de la inmunoglobulina de superficie) de la célula B del linfoma puede funcionar como marcador específico del tumor. Se han inducido respuestas inmunes contra el idiotipo por medio de vacunas dirigidas.63 Estas vacunas contra linfomas se preparan aislando la proteína idiotipo de los tejidos del linfoma, conjugándola in vitro con una proteína transportadora y mezclándola con un adyuvante inmunológico. Nuevas estrategias incluyen la exposición del idiotipo a células dendríticas ex vivo para una presentación más eficaz al sistema inmune del huésped.64
Grado III (linfoma de células grandes) Como otros linfomas foliculares, el subtipo de células grandes suele caracterizarse al inicio por adenopatía difusa y asintomática. La afección de la médula ósea es menos prevalente. El tratamiento con quimioterapia combinada que contenga doxorrubicina puede causar una remisión inicial prolongada. La supervivencia promedio es de 6 a 8 años. Desde el punto de vista histológico se caracteriza por su presentación discordante con linfoma folicular de grado bajo y coexistencia con linfoma difuso de células grandes.65
Linfoma linfocítico de células pequeñas
El linfoma linfocítico de células pequeñas es un padecimiento indolente muy relacionado con la leucemia linfocítica crónica. Suele presentarse en pacientes de edad avanzada con adenopatía difusa y afección esplénica y de la médula ósea. En una proporción importante de los pacientes ocurre progresión a la fase leucémica. El linfoma linfocítico de células pequeñas se trata en forma muy semejante a los linfomas foliculares de grado bajo y con resultados parecidos. Los análogos de nucleósidos representan una alternativa terapéutica nueva e importante para estos linfomas, en algunos estudios las tasas de respuesta han sido superiores a las alcanzadas con agentes alquilantes. Es importante distinguir el linfoma linfocítico de células pequeñas del linfoma de células en manto porque su historia natural es muy distinta [ver tabla 8].
Linfoma de células en manto
El linfoma de células en manto, antes conocido como linfoma linfocítico intermedio, se compone principalmente de células linfoides pequeñas con bordes nucleares irregulares, es más común en varones de edad avanzada y se presenta con adenopatía y hepatoesplenomegalia. Un subtipo diferente de pacientes tiene afección de la médula ósea y de sangre periférica. Se ha asociado un pronóstico menos favorable con la afección hepática, edad avanzada, patrón histológico difuso y expresión aumentada de p53. Una variante blástica que se caracteriza por rasgos morfológicos distintivos, alta frecuencia de mitosis, hiperploidía y mutaciones de p53 es especialmente agresiva. La traslocación t(11;14) se observa en más del 50 por ciento de los casos sometidos a análisis citogenético. Sin embargo, un anticuerpo monoclonal dirigido contra la ciclina D1 (bcl-1) tiñe los tejidos embebidos en parafina y este anticuerpo se emplea en la actualidad en los análisis estándar de patología para confirmar el diagnóstico de linfoma de células en manto. Por lo tanto, el diagnóstico se basa en criterios mofológicos con confirmación por tinción de bcl-1. La inmunohistoquímica para p53 y la tasa de proliferación pueden proporcionar información pronóstica adicional. El linfoma de células en manto típicamente es más refractario al tratamiento que los linfomas de grado bajo, la supervivencia promedio en pacientes con linfoma de células en manto es de 3 a 5 años.66 Los agentes alquilantes y la prednisona han sido la base del tratamiento. Este es difícil porque el linfoma de células en manto parece ser incurable incluso con quimioterapia agresiva. Los datos respecto a remisiones prolongadas o curación con tratamiento en dosis altas y trasplante autólogo de células hematopoyéticas tronco son conflictivos. En un reporte con seguimiento prolongado, se observó un patrón continuo de recaída en los pacientes que fueron sometidos a trasplante durante la primera remisión o remisiones subsecuentes. 67
Linfoma de la zona marginal
Linfoma del tejido linfoide asociado a mucosas Los linfomas MALT pueden originarse en el estómago, pulmones, piel, glándulas parótidas, tiroides, mamas y otros sitios extranodales, en donde se caracterizan por alinearse con células epiteliales. 68 Antes del uso de técnicas para determinar la clonalidad y definir sus datos histológicos, los linfomas MALT se clasificaban como seudolinfomas. Desde el punto de vista clínico, estos linfomas tienden a ser indolentes y permanecen localizados por periodos prolongados. Los linfomas MALT tienen propensión para afectar múltiples sitios extranodales, mientras que otros linfomas de grado bajo característicamente afectan los ganglios linfáticos periféricos. Recientemente se ha establecido una asociación casi constante entre los linfomas gástricos MALT y la infección por Helicobacter pylori. Se ha reportado resolución de las características clínicas e histológicas del linfoma en una alta proporción de pacientes con enfermedad de IE [ver tabla 2] tratada con antibióticos eficaces contra H. pylori.69 Hasta la fecha estas respuestas han sido duraderas. Sin embargo, un linfoma MALT indolente puede coexistir con un MALT de alto grado, de modo que se recomienda que un oncólogo sea quien trate esta enfermedad. Además, se ha notado aumento en la incidencia de tumores sólidos secundarios en pacientes con linfoma MALT.
Linfoma monocitoide de células B En la nueva clasificación de los linfomas se considera que el linfoma monocitoide de células B es la contraparte nodal del linfoma MALT. Los linfomas monocitoides de células B ocurren en pacientes femeninas de edad avanzada y suelen presentarse con crecimiento no masivo y asintomático de los ganglios linfáticos periféricos y afección de los ganglios paraparótidos o intraparótidos.70 Existe una fuerte asociación con el síndrome de Sjögren y otros padecimientos del tejido conjuntivo. Aunque la afección esplénica y de la médula ósea son frecuentes en otros linfomas de bajo grado, esta no es común en los linfomas monocitoides de células B. Puede ocurrir transformación a un linfoma de grado medio o alto. Debido a la falta de certeza en relación con el diagnóstico y al curso muy indolente de la enfermedad, muchos pacientes reportados en la literatura no recibieron nunca tratamiento antineoplásico. Se recomienda un manejo semejante al de otros linfomas de grado bajo.
Linfoma difuso de células grandes
El linfoma difuso de células grandes fue el subtipo más prevalente de linfoma no-Hodgkin en el estudio internacional descrito antes. La enfermedad afecta una gran variedad de pacientes, variando de niños a ancianos. Las formas de presentación se distribuyen en forma equitativa entre enfermedad limitada (estadios I a II) y enfermedad avanzada, de acuerdo con el sistema de estadificación de Ann Arbor. Alrededor de la tercera parte de los casos presentan afección extranodal, sobre todo la cabeza y el cuello, el estómago, la piel, el hueso, los testículos y el sistema nervioso. Como se mencionó antes, el pronóstico se relaciona con el estadio de la enfermedad y con otros factores [ver tabla 5].
El tratamiento inicial en todos los estadios es la quimioterapia. Los pacientes en estadio I y II tienen una supervivencia libre de recidivas alta (más del 85 por ciento para el estadio I y 70 a 80 por ciento para el estadio II).71,72 La consolidación de la respuesta inducida por la quimioterapia por medio de radioterapia logró mejores resultados en un estudio clínico aleatorio y extenso de pacientes con enfermedad limitada.71 En los pacientes con enfermedad avanzada los porcentajes de supervivencia libre de recidivas varían de menos del 20 por ciento a más del 50 por ciento con la quimioterapia combinada. Además de la quimioterapia, está indicada la profilaxis contra la afección del sistema nervioso central para algunos casos de linfoma difuso de células grandes, como cuando existe afección de la médula ósea (sobre todo con células circulantes), enfermedad epidural, linfoma testicular bilateral e invasión de senos paranasales.
La búsqueda de la quimioterapia combinada óptima para los linfomas difusos de células grandes ha ocupado un papel central en la investigación clínica durante más de dos décadas. Un estudio multi-institucional dirigido por el Southwestern Oncology Group probó cuatro esquemas principales. A pesar de los indicios preliminares de superioridad de los nuevos esquemas en estudios pilotos de una sola institución, ninguno de estos esquemas demostró ser mejor que el CHOP (ciclofosfamida, doxorrubidina [hidroxidaunomicina], vincristina [Oncovin], prednisona) cuando se evaluó en el estudio multi-institucional; además, los nuevos esquemas se asociaron con mayor toxicidad.67 Aunque este estudio define al CHOP como el tratamiento de elección para el linfoma difuso de células grandes, es claro que se requieren tratamientos más eficaces para los pacientes con mal pronóstico.
La disponibilidad de factores de crecimiento hematopoyético y, en menor grado, la introducción de antieméticos muy eficaces, hace posible administrar fármacos en dosis mayores, una modificación que se ha asociado con mejoría de la evolución en varios estudios retrospectivos. La reducción de los efectos mielotóxicos graves de la quimioterapia por medio de rescate con células tronco y factores de crecimiento permite aumentar la intensidad de los esquemas terapéuticos. Debido a que el tratamiento en dosis altas y el autotrasplante es superior a la quimioterapia de salvamento en las recaídas del linfoma de células grande difuso sensible a quimioterapia, existe gran interés en extender este tipo de manejo a pacientes sin tratamiento previo y con alto riesgo de fracaso terapéutico.73 Los análisis retrospectivos de pacientes con riesgo algo que fueron sometidos a trasplante durante la primera remisión completa mostraron un beneficio significativo del trasplante en dos estudios clínicos.74,75 Por el contrario, un estudio aleatorio y prospectivo de este subgrupo no encontró beneficio.76 Debido a estos datos contradictorios, aún está en duda el papel de la intensificación de la dosis en el paciente en su primera remisión. Un enfoque novedoso de intensificación de dosis, el uso de agentes quimioterápicos secuenciales en su dosis máxima tolerada, mostró resultados promisorios en un pequeño estudio aleatorio realizado en Italia.77
El linfoma de células grandes puede manifestarse en cualquiera de varios sitios extranodales, en especial el tubo digestivo y la cabeza y cuello. Se han observado manifestaciones únicas en sitios seleccionados, como el sistema nervioso central, los senos paranasales y los testículos.
El linfoma tímico de células B merece atención especial como una entidad clinicopatológica distinta compuesta de células grandes y con frecuencia asociada a esclerosis. En el momento del diagnóstico el timo siempre está afectado. Los pacientes, que casi siempre son mujeres, tienen una masa mediastinal grande, localmente invasiva y que puede causar compromiso de las vías respiratorias o síndrome de vena cava superior. Puede existir enfermedad extranodal en el momento del diagnóstico o si existe recaída. El tratamiento agresivo con quimio y radioterapia permite tasas de curación que son semejantes a las obtenidas en pacientes con linfoma difuso de células grandes.45
Dos de los subtipos histológicos en la Fórmula de Trabajo, el linfoma de células pequeñas hendidas y el linfoma difuso mixto, de células pequeñas hendidas y grandes, no se reconocen en forma independiente en la clasificación de la Organización Mundial de la Salud. Con los mejores métodos diagnósticos y el reconocimiento morfológico la mayoría de los linfomas colocados en la actualidad en estas categorías podrían reclasificarse.
Linfoma de Burkitt y no Burkitt
Las características patológicas monomórficas distinguen al linfoma de Burkitt, una enfermedad que ocurre en población joven y que tiene alta incidencia de afección al intestino y médula ósea. En comparación con casos estudiados en Africa, en donde la enfermedad es endémica, los pacientes de los Estados Unidos tienen una menor incidencia de afección aislada de la mandíbula y de asociación con el virus de Epstein-Barr. El pronóstico se relaciona con la masa tumoral. Los tratamientos agresivos y en dosis altas son curativos en la mayoría de los pacientes pediátricos, incluyendo los que tienen afección al SNC. Son componentes importantes del tratamiento la ciclofosfamida en dosis altas, el metotrexate, la citarabina y la profilaxis intensiva al SNC. Los adultos que presentan una masa tumoral importante y aumento en la concentración de deshidrogenasa láctica tienen mal pronóstico. Se ha reportado que el tratamiento pediátrico ha sido útil en adultos. 78,79 La enfermedad caracterizada por una gran masa tumoral puede asociarse con un síndrome hipermetabólico que empeora con el tratamiento, cuando el tumor sufre lisis súbita. Esto puede causar hipercalemia que pone en peligro la vida, hiperfosfatemia, hipocalcemia y tetania. Además de la hidratación vigorosa y la diuresis, puede requerirse hemodiálisis para controlar la hipercalemia.
En algunas instituciones se trata al linfoma parecido al Burkitt de la misma manera que al linfoma de Burkitt, mientras que en otras se administra el tratamiento del linfoma difuso de células grandes.
Linfoma linfoblástico
Un linfoma linfoblástico idéntico desde el punto de vista citológico a la leucemia linfoblástica aguda ocurre con más frecuencia en varones adolescentes o adultos jóvenes. Los datos de presentación incluyen una masa mediastinal y adenopatía periférica; puede existir afección de la médula ósea y del SNC en el momento del diagnóstico o durante la evolución de la enfermedad. Los estudios en adultos y niños indican que el tratamiento apropiado para la leucemia aguda, que consiste en quimioterapia combinada y profilaxis para el SNC, da los mejores resultados.80 Los pacientes con mal pronóstico incluyen los que tienen enfermedad en estadio IV (en especial con afección a la médula ósea o del SNC), los mayores de 40 años y los que tienen concentraciones aumentadas de deshidrogenasa láctica.
Linfoma de células T periféricas
Se han logrado grandes adelantos en la última década sobre el conocimiento de los linfomas de células T periféricas, muchos de los cuales se consideraban no clasificables o no se reconocían como neoplasias al usar la Fórmula de Trabajo. Los linfomas de células T periféricas son muy diversos en sus características morfológicas y clínicas, que incluyen una gran variedad de fenómenos paraneoplásicos. Gran parte de la dificultad para caracterizar a los linfomas de células T se basa en su diversidad y su relativa poca frecuencia. En el proyecto de Clasificación Internacional, estos linfomas de células T periféricas, junto con los linfomas de células en manto, tuvieron el pronóstico menos favorable entre todos los linfomas no Hodgkin.45
En la clasificación actual se describen diversas entidades individuales, con características clínicas y patológicas distintivas [ver tabla 8]. Los linfomas cutáneos de células T, que incluyen a las micosis fungoides y al síndrome de Sézary (una forma de micosis fungoide) se analizan en otro capítulo.
Los linfomas nasales T/NK típicamente afectan las vías respiratorias superiores. Desde el punto de vista histórico, las presentaciones aerodigestivas superiores se denominaron enfermedad destructiva de la línea media idiopática o reticulosis polimórfica. Existe un padecimiento clínico muy agresivo que puede afectar otros sitios anatómicos.
La linfadenopatía angioinmunoblástica (LAIB) es una entidad clínica definida que se caracteriza por erupción cutánea, síntomas generales, linfadenopatía generalizada, hepatoesplenomegalia y anemia hemolítica autoinmune. Suele afectar individuos de edad avanzada y puede desarrollarse pronto después de la exposición a un agente viral o un fármaco. El pronóstico es malo y la supervivencia promedio es menor de 2 años. Aunque algunos patólogos han intentado distinguir el cuadro histológico denominado premaligno en este padecimiento de un linfoma franco de células T, la distinción es difícil porque la histología es muy semejante y la mayoría de los pacientes con LAIB tienen poblaciones clonales de células T. Los pacientes más jóvenes con lesiones muy proliferativas son candidatos para quimioterapia combinada agresiva, mientras que los pacientes de mayor edad con enfermedad relativamente hipocelular pueden tratarse con esteroides solos o en combinación con ciclofosfamida.
La leucemia/linfoma de células T del adulto, es una enfermedad agresiva caracterizada por adenopatía generalizada, hipercalcemia, hipergamaglobulinemia policlonal, lesiones óseas líticas y células muy anormales en la médula ósea y la sangre periférica.
Linfoma anaplásico de células grandes (células T y nulas)
Los linfomas anaplásicos de células grandes constituyen un grupo heterogéneo de neoplasias caracterizadas por la proliferación de células muy atípicas que expresan el antígeno CD30. El término de linfoma anaplásico Ki-1 deriva del anticuerpo monoclonal Ki-1 que reacciona con el CD30. La inmunotipificación confirma el diagnóstico y permite diferenciar a estos trastornos de las neoplasias epiteliales. Se ha identificado una traslocación cromosómica recurrente, t(2;5)(p23;q35), que causa una proteína de fusión NPM-ALK, en los linfomas anaplásicos Ki-1. La expresión de la proteína ALK resultante, que está característicamente limitada al tejido del sistema nervioso central, en las muestras de linfoma, define un subtipo favorable de linfoma anaplásico.81 Estos pacientes con frecuencia son niños o adultos jóvenes que tienen alta probabilidad de curación con la quimioterapia combinada estándar. El cuadro clínico de los linfomas anaplásicos de células grandes puede ser indistinguible de los linfomas difusos agresivos típicos. Se observa enfermedad extranodal que afecta al tubo digestivo, pulmones, tejidos blandos, hueso y bazo. Es probable que la expresión de la proteína ALK se vuelva una parte indispensable del proceso diagnóstico.
Linfoma anaplásico primario de la piel
Este tipo de linfoma tiene un pronóstico favorable, aunque se caracteriza por recaídas crónicas limitadas a la piel. La papulomatosis linfomatoide, un proceso linfomatoso que se resuelve solo, y los linfomas anaplásicos de células grandes limitados a la piel son clínicamente semejantes y se traslapan desde el punto de vista morfológico. Debido a su naturaleza indolente y de recurrencias crónicas, estos linfomas pueden manejarse con tratamiento local.82
LINFOMAS NO-HODGKIN EN EL SIDA
Los linfomas no-Hodgkin ocurren con mayor frecuencia en la población con riesgo de SIDA. Existen varias características clínicas y patológicas que distinguen a los linfomas no-Hodgkin en los pacientes con SIDA. El linfoma relacionado al SIDA suele ser avanzado, y 65 a 98 por ciento de los pacientes tienen enfermedad extranodal, que con frecuencia afecta el aparato digestivo, el hígado y el SNC. Desde el punto de vista de patología, estos linfomas son difusos de linaje B, Burkitt o semejantes a Burkitt. A pesar del uso de factores de crecimiento hematopoyético y medicamentos antirretrovirales, la administración de la quimioterapia combinada se complica por las citopenias e infecciones oportunistas relacionadas al SIDA. El pronóstico es malo, con una supervivencia promedio de menor de 1 año, y la menor longevidad se relaciona con frecuencia con la severidad de la inmunodeficiencia subyacente.
- Klitz W, Aldrich CL, Fildes N, et al: Localization of predisposition to Hodgkin disease in the HLA class II region. Am J Hum Genet 54:497, 1994
- Mueller N, Evans A, Harris NL, et al: Hodgkin's disease and Epstein-Barr virus: altered antibody pattern before diagnosis. N Engl J Med 320:689, 1989
- Weiss LM, Chen YY, Liu XF, et al: Epstein-Barr virus and Hodgkin's disease: a correlative in situ hybridization and polymerase chain reaction study. Am J Pathol 139:1259, 1991
- Küppers R, Rajewsky K: The origin of Hodgkin and Reed/Sternberg cells in Hodgkin's disease. Annu Rev Immunol 16:471, 1998
- Kanzler H, Kuppers R, Hansmann ML, et al: Hodgkin and Reed-Sternberg cells in Hodgkin's disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med 184:1495, 1996
- Stein H, Hummel M, Marafioti T, et al: Molecular biology of Hodgkin's disease. Cancer Surv 30:107, 1997
- Marafioti T, Hummel M, Anagnostopoulos I, et al: Origin of nodular lymphocyte-predominant Hodgkin's disease from a clonal expansion of highly mutated germinal-center B cells. N Engl J Med 337:453, 1997
- Sextro M, Diehl V, Franklin J, et al: Lymphocyte predominant Hodgkin's disease-a workshop report. European Task Force on Lymphoma. Ann Oncol 7 (suppl 4):61, 1996
- Leibenhaut MH, Hoppe RT, Efron B, et al: Prognostic indicators of laparotomy findings in clinical stage I-II supradiaphragmatic Hodgkin's disease. J Clin Oncol 7:81, 1989
- Carde P, Hagenbeek A, Hayat M, et al: Clinical staging versus laparotomy and combined modality with MOPP versus ABVD in early-stage Hodgkin's disease: the H6 twin randomized trials from the European Organization for Research and Treatment of Cancer Lymphoma Cooperative Group. J Clin Oncol 11:2258, 1993
- Carbone P, Kaplan H, Musshoff K: Report of the committee on the Hodgkin's disease staging. Cancer Res 31:1860, 1971
- Lister TA, Crowther D, Sutcliffe SB, et al: Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin's disease: Cotswolds meeting. J Clin Oncol 7:1630, 1989
- Hasenclever D, Diehl V: A prognostic score for advanced Hodgkin's disease. International Prognostic Factors Project on Advanced Hodgkin's Disease. N Engl J Med 339:1506, 1998
- Mauch PM: Controversies in the management of early stage Hodgkin's disease. Blood 83:318, 1994
- Horning SJ, Hoppe RT, Mason J, et al: Stanford-Kaiser Permanente G1 study for clinical stage I to IIA Hodgkin's disease: subtotal lymphoid irradiation versus vinblastine, methotrexate, and bleomycin chemotherapy and regional irradiation. J Clin Oncol 15:1736, 1997
- Connors JM, Reece DE, Diehl V, et al: Evaluation and treatment of early stage Hodgkin's lymphoma. Hematology 1:274, 1998
- Bonfante V, Santoro A, Viviani S, et al: ABVD in the treatment of Hodgkin's disease. Semin Oncol 19(suppl 5):38, 1992
- Canellos GP, Anderson JR, Propert KJ, et al: Chemotherapy of advanced Hodgkin's disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med 327:1478, 1992
- Connors JM, Klimo P, Adams G, et al: Treatment of advanced Hodgkin's disease with chemotherapy-comparison of MOPP/ABV hybrid regimen with alternating courses of MOPP and ABVD: a report from the National Cancer Institute of Canada clinical trials group. J Clin Oncol 15:1638, 1997
- Viviani S, Bonadonna G, Santoro A, et al: Alternating versus hybrid MOPP and ABVD combinations in advanced Hodgkin's disease: ten-year results. J Clin Oncol 14:1421, 1996
- Duggan D, Petroni G, Johnson J, et al: MOPP/ABV vs ABVD for advanced Hodgkin's disease: a preliminary report of CALGB 8952 (with SWOG, ECOG, NCIC). Proc Am Soc Clin Oncol 16:12a, 1997
- Diehl V, Franklin J, Hasenclever D, et al: BEACOPP, a new dose-escalated and accelerated regimen, is at least as effective as COPP/ABVD in patients with advanced-stage Hodgkin's lymphoma: interim report from a trial of the German Hodgkin's Lymphoma Study Group. J Clin Oncol 16:3810, 1998
- Tesch H, Diehl V, Lathan B, et al: Moderate dose escalation for advanced stage Hodgkin's disease using the bleomycin, etoposide, Adriamycin, cyclophosphamide, vincristine, procarbazine, and prednisone scheme and adjuvant radiotherapy: a study of the German Hodgkin's Lymphoma Study Group. Blood 92:4560, 1998
- Bartlett NL, Rosenberg SA, Hoppe RT, et al: Brief chemotherapy, Stanford V, and adjuvant radiotherapy for bulky or advanced-stage Hodgkin's disease: a preliminary report. J Clin Oncol 13:1080, 1995
-
Horning SJ, Rosenberg SA, Hoppe RT, et al: Brief chemotherapy
(Stanford V) and adjuvant radiotherapy for bulky or advanced Hodgkin's
disease: an update. Ann Oncol 7 (suppl 4):105, 1996
-
Kennedy BJ, Loeb VJ, Peterson V, et al: Survival in
Hodgkin's disease by stage and age. Med Pediatr Oncol 20:100, 1992
-
Enblad G, Glimelius B, Sundstrom C: Treatment outcome
in Hodgkin's disease in patients above the age of 60: a population-based
study. Ann Oncol 2:297, 1991
-
Lohri A, Barnett M, Fairey RN, et al: Outcome of treatment
of first relapse of Hodgkin's disease after primary chemotherapy: identification
of risk factors from the British Columbia experience 1970 to 1988. Blood
77:2292, 1991
-
Viviani S, Santoro A, Negretti E, et al: Salvage chemotherapy
in Hodgkin's disease: results in patients relapsing more than twelve months
after first complete remission. Ann Oncol 1:123, 1990
-
Yuen AR, Rosenberg SA, Hoppe RT, et al: Comparison
between conventional salvage therapy and high-dose therapy with autografting
for recurrent or refractory Hodgkin's disease. Blood 89:814, 1997
-
Horning SJ, Chao NJ, Negrin RS, et al: High-dose therapy
and autologous hematopoietic progenitor cell transplantation for recurrent
or refractory Hodgkin's disease: analysis of the Stanford University results
and prognostic indices. Blood 89:801, 1997
-
Hancock SL, Tucker MA, Hoppe RT. Breast cancer after
treatment of Hodgkin's disease. J Natl Cancer Inst 85:25, 1993
-
Tucker MA, Coleman CN, Cox RS, et al: Risk of second
cancers after treatment for Hodgkin's disease. N Engl J Med 318:76, 1988
-
Hancock SL, Tucker MA, Hoppe RT: Factors affecting
late mortality from heart disease after treatment of Hodgkin's disease.
JAMA 270:1949, 1993
-
Boivin JF, Hutchison GB, Lubin JH, et al: Coronary
artery disease mortality in patients treated for Hodgkin's disease. Cancer
69:1241, 1992
-
Hancock SL, Hoppe RT: Long-term complications of treatment
and causes of mortality after Hodgkin's disease. Semin Radiat Oncol 6:225,
1996
-
Kaldor JM, Day NE, Clarke EA, et al: Leukemia following
Hodgkin's disease. N Engl J Med 322:7, 1990
-
Horning SJ, Hoppe RT, Kaplan HS, et al: Female reproductive
potential after treatment for Hodgkin's disease. N Engl J Med 304:1377,
1981
-
Chapman RM, Sutcliffe SB, Malpas JS: Male gonadal dysfunction
in Hodgkin's disease: a prospective study. JAMA 245:1323, 1981
-
Anselmo AP, Cartoni C, Bellantuono P, et al: Risk of
infertility in patients with Hodgkin's disease treated with ABVD vs MOPP
vs ABVD/MOPP. Haematologica 75:155, 1990
-
Viviani S, Santoro A, Ragni G, et al: Gonadal toxicity
after combination chemotherapy for Hodgkin's disease: comparative results
of MOPP vs ABVD. Eur J Cancer Clin Oncol 21:601, 1985
-
Fryer CJ, Hutchinson RJ, Krailo M, et al: Efficacy
and toxicity of 12 courses of ABVD chemotherapy followed by low-dose regional
radiation in advanced Hodgkin's disease in children: a report from the
Children's Cancer Study Group. J Clin Oncol 8:1971, 1990
-
Lipshultz SE, Lipsitz SR, Mone SM, et al: Female sex
and drug dose as risk factors for late cardiotoxic effects of doxorubicin
therapy for childhood cancer. N Engl J Med 332: 1738, 1995
-
Harris NL, Jaffe ES, Stein H, et al: A revised European-American
classification of lymphoid neoplasms: a proposal from the International
Lymphoma Study Group. Blood 84:1361, 1994
-
The Non-Hodgkin's Lymphoma Classification Project: A clinical
evaluation of the International Lymphoma Study Group classification of
non-Hodgkin's lymphoma. Blood 89:3909, 1997
-
The Non-Hodgkin's Lymphoma Pathologic Classification
Project: National Cancer Institute sponsored study of classifications of
non-Hodgkin's lymphomas: summary and description of a working formulation
for clinical usage. Cancer 49:2112, 1982
-
Ye BH, Lista F, Lo Coco F, et al: Alterations of a
zinc finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science
262:747, 1993
-
Hockenbery D, Nunez G, Milliman C, et al: Bcl-2 is
an inner mitochondrial membrane protein that blocks programmed cell death.
Nature 348:334, 1990
-
Gribben JG, Neuberg D, Freedman AS, et al: Detection
by polymerase chain reaction of residual cells with the bcl-2 translocation
is associated with increased risk of relapse after autologous bone marrow
transplantation for B-cell lymphoma. Blood 81:3449, 1993
-
López-Guillermo A, Cabanillas F, McLaughlin
P, et al: The clinical significance of molecular response in indolent follicular
lymphomas. Blood 91:2955, 1998
-
Ichikawa A, Kinoshita T, Watanabe T, et al: Mutations
of the p53 gene as a prognostic factor in aggressive B-cell lymphoma.
N Engl J Med 337:529, 1997
-
The International Non-Hodgkin's Lymphoma Prognostic
Factors Project: A predictive model for aggressive non-Hodgkin's lymphoma.
N Engl J Med 329:987, 1993
-
Gascoyne RD, Adomat SA, Krajewski S, et al: Prognostic
significance of Bcl-2 protein expression and bcl-2 gene rearrangement
in diffuse aggressive non-Hodgkin's lymphoma. Blood 90:244, 1997
-
Hermine O, Haioun C, Lepage E, et al: Prognostic significance
of bcl-2 protein expression in aggressive non-Hodgkin's lymphoma. Groupe
d'Etude des Lymphomes de l'Adulte (GELA). Blood 87:265, 1996
-
Gisselbrecht C, Gaulard P, Lepage E, et al: Prognostic
significance of T-cell phenotype in aggressive non-Hodgkin's lymphomas.
Groupe d'Etudes des Lymphomes de l'Adulte (GELA). Blood 92:76, 1998
-
Horning SJ, Rosenberg SA: The natural history of initially
untreated low-grade non-Hodgkin's lymphomas. N Engl J Med 311:1471, 1984
-
Gribben JG, Freedman AS, Woo SD, et al: All advanced
stage non-Hodgkin's lymphomas with a polymerase chain reaction amplifiable
breakpoint of bcl-2 have residual cells containing the bcl-2 rearrangement
at evaluation and after treatment. Blood 78:3275, 1991
-
McLaughlin P, Hagemeister FB, Romaguera JE, et al:
Fludarabine, mitoxantrone, and dexamethasone: an effective new regimen
for indolent lymphoma. J Clin Oncol 14:1262, 1996
-
Hochster H, Oken M, Winter J, et al: Prolonged time
to progression in patients with low-grade lymphoma treated with cyclophosphamide
and fludarabine (ECOG 1491). Proc Am Soc Clin Oncol 17:17a, 1998
-
McLaughlin P, Grillo-López AJ, Link BK, et al:
Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent
lymphoma: half of patients respond to a four-dose treatment program. J
Clin Oncol 16:2825, 1998
-
Kaminski MS, Zasadny KR, Francis IR, et al: Iodine-131-anti-B1
radioimmunotherapy for B-cell lymphoma. J Clin Oncol 14:1974, 1996
-
Knox SJ, Goris ML, Trisler K: Yttrium-90-labeled anti-CD20
monoclonal antibody therapy of recurrent B-cell lymphoma. Clin Cancer Res
2:457, 1996
-
Kwak LW, Campbell MJ, Czerwinski DK, et al: Induction
of immune responses in patients with B-cell lymphoma against the surface-immunoglobulin
idiotype expressed by their tumors. N Engl J Med 327:1209, 1992
-
Hsu FJ, Benike C, Fagnoni F, et al: Vaccination of
patients with B-cell lymphoma using autologous antigen-pulsed dendritic
cells. Nat Med 2:52, 1996
-
Bartlett NL, Rizeq M, Dorfman RF, et al: Follicular large-cell
lymphoma: intermediate or low grade? J Clin Oncol 12:1349, 1994
-
Fisher RI, Dahlberg S, Nathwani BN, et al: A clinical
analysis of two indolent lymphoma entities: mantle cell lymphoma and marginal
zone lymphoma (including the mucosa-associated lymphoid tissue and monocytoid
B-cell subcategories): a Southwest Oncology Group study. Blood 85:1075,
1995
-
Freedman AS, Neuberg D, Gribben JG, et al: High-dose chemoradiotherapy
and anti- B-cell monoclonal antibody-purged autologous bone marrow transplantation
in mantle-cell lymphoma: no evidence for long-term remission. J Clin Oncol
16:13, 1998
-
Isaacson PG: Lymphomas of mucosa-associated lymphoid
tissue (MALT). Histopathology 16:617, 1990
-
Neubauer A, Thiede C, Morgner A, et al: Cure of Helicobacter
pylori infection and duration of remission of low-grade gastric mucosa-associated
lymphoid tissue lymphoma. J Natl Cancer Inst 89:1350, 1997
-
Ngan BY, Warnke RA, Wilson M, et al: Monocytoid B-cell
lymphoma: a study of 36 cases. Hum Pathol 22:409, 1991
-
Miller TP, Dahlberg S, Cassady JR, et al: Chemotherapy
alone compared with chemotherapy plus radiotherapy for localized intermediate-
and high-grade non-Hodgkin's lymphoma. N Engl J Med 339:21, 1998
-
Glick JH, Kim K, Earle J, et al: An ECOG randomized
phase III trial of CHOP vs CHOP + radiotherapy for intermediate grade early
stage non-Hodgkin's lymphoma. Proc Am Soc Clin Oncol 14:391, 1995
-
Philip T, Guglielmi C, Hagenbeek A, et al: Autologous
bone marrow transplantation as compared with salvage chemotherapy in relapses
of chemotherapy-sensitive non-Hodgkin's lymphoma. N Engl J Med 333:1540,
1995
-
Haioun C, Lepage E, Gisselbrecht C, et al: Benefit
of autologous bone marrow transplantation over sequential chemotherapy
in poor-risk aggressive non-Hodgkin's lymphoma: updated results of the
prospective study LNH87-2. Groupe d'Etude des Lymphomes de l'Adulte. J
Clin Oncol 15:1131, 1997
-
Santini G, Salvagno L, Leoni P, et al: VACOP-B versus
VACOP-B plus autologous bone marrow transplantation for advanced diffuse
non-Hodgkin's lymphoma: results of a prospective randomized trial by the
non-Hodgkin's Lymphoma Cooperative Study Group. J Clin Oncol 16:2796, 1998
-
Reyes F, Lepage D, Morel P, et al: Failure of first-line
inductive high-dose chemotherapy in poor-risk patients with aggressive
lymphoma: updated results of the randomized LNH93-3 study. Blood 90 (suppl
I):594a, 1997
-
Gianni AM, Bregni M, Siena S, et al: High-dose chemotherapy
and autologous bone marrow transplantation compared with MACOP-B in aggressive
B-cell lymphoma. N Engl J Med 336:1290, 1997
-
Magrath I, Adde M, Shad A, et al: Adults and children
with small non-cleaved-cell lymphoma have a similar excellent outcome when
treated with the same chemotherapy regimen. J Clin Oncol 14:925, 1996
-
Soussain C, Patte C, Ostronoff M, et al: Small noncleaved
cell lymphoma and leukemia in adults: a retrospective study of 65 adults
treated with the LMB pediatric protocols. Blood 85:664, 1995
-
Bouabdallah R, Xerri L, Bardou VJ, et al: Role of induction
chemotherapy and bone marrow transplantation in adult lymphoblastic lymphoma:
a report on 62 patients from a single center. Ann Oncol 9:619, 1998
-
Benharroch D, Meguerian-Bedoyan Z, Lamant L, et al:
ALK-positive lymphoma: a single disease with a broad spectrum of morphology.
Blood 91:2076,1998
-
Paulli M, Berti E, Rosso R, et al: CD30/Ki-1-positive
lymphoproliferative disorders of the skin-clinicopathologic correlation
and statistical analysis of 86 cases: a multicentric study from the European
Organization for Research and Treatment of Cancer Cutaneous Lymphoma Project
Group. J Clin Oncol 13:1343, 1995