Metabolismo
⭳ Abrir artículo (PDF)770.0 KBEste artículo es idéntico en la Edición 2/2000.
Contenido del artículo
VII BASES GENETICAS PARA EL CLINICO
VII BASES GENETICAS PARA EL CLINICO
DR. BRUCE R. KORF, PH.D.
Genoma humano y enfermedad
Un gen es una secuencia ordenada de cuatro bases de nucleótidos en una molécula de ácido desoxirribonucleico (ADN) que codifica un producto funcional específico (i.e., una proteína o molécula de ácido ribonucleico [ARN]). El desarrollo de un ser humano desde una sola célula hasta la fase adulta está dirigido por el genoma humano, un grupo de alrededor de 80,000 genes que codifican las estructuras para un número incluso mayor de proteínas. Estas proteínas catalizan reacciones químicas, proporcionan las estructuras básicas de las células y tejidos, efectúan comunicación inter e intracelular y dirigen la secuencia ordenada de la diferenciación celular. En la actualidad se realiza un enorme esfuerzo por identificar todos los genes humanos y determinar su secuencia de bases. El Proyecto del Genoma Humano tiene por objeto producir un adelanto revolucionario en el conocimiento de la biología humana. Al final, permitirá identificar en su totalidad los complementos de las proteínas expresadas en cualquier tejido en cualquier momento del desarrollo.
La complejidad del genoma humano lo hace vulnerable a sufrir disfunción con cambios incluso leves en el contenido de información. Estos cambios se denominan mutaciones. El papel de la herencia en la enfermedad humana comenzó a descubrirse a principios del siglo XX con el redescubrimiento de las leyes de la herencia de Mendel. Durante los últimos años el estudio de la enfermedad genética se había enfocado a algunos padecimientos raros causados por un solo gen, y en la mayor parte de los casos el conocimiento se limitaba a la descripción clínica y a los resultados del análisis bioquímico. Sin embargo, los grandes adelantos en genética molecular realizados en la segunda mitad del siglo XX, incluyendo el desarrollo de la tecnología de ADN recombinante en los años 70, ha hecho posible identificar los genes responsables de muchos padecimientos y comprender la patogenia de la enfermedad en términos celulares y moleculares precisos. Estos avances han permitido también el desarrollo de herramientas muy poderosas de diagnóstico y nuevos métodos de tratamiento. Además, en la actualidad es posible explorar la contribución genética a trastornos comunes como la enfermedad cardiovascular, la diabetes y el cáncer.
El médico requiere conocer cada vez más los patrones de trasmisión familiar de la enfermedad e identificar a los familiares con riesgo. La capacidad para interpretar los datos de las pruebas genéticas es una destreza cada vez más necesaria. Ningún área de la medicina está aislada de la influencia de los factores genéticos, y ningún médico puede no estar al tanto de la contribución de la genética a la salud y a la enfermedad.
Esta subsección se enfoca a la estructura y función del genoma humano y a la fisiopatología de los trastornos genéticos. Aunque el ámbito de la genética pertenece a la biología y el de las enfermedades genéticas a la medicina, existen temas comunes respecto a los mecanismos de la enfermedad que incluyen principalmente la expresión genética y su disrupción por mutaciones.
Estructura y función del genoma humano
El paradigma básico de la genética molecular, referido con frecuencia como el dogma central, es que la secuencia de aminoácidos de cada proteína está codificada por la secuencia de bases de su gen correspondiente.
ESTRUCTURA DEL ADN
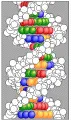
El descubrimiento de la estructura del ADN por Watson y Crick en 1953 inició la era moderna de la biología molecular. La molécula de ADN consiste en un par de tiras o cadenas de azúcar-fosfato con una base (adenina A, guanina G, citosina C o timidina T) unida a cada azúcar desoxirribosa [ver figuras 1 y 2]. Las dos cadenas están unidas entre sí por puentes de hidrógeno entre las bases, formando una doble hélice. La adenina y la guanina son purinas heterocíclicas, la citosina y la timina son pirimidinas más pequeñas, de un solo anillo. La adenina siempre hace par con la timina y la citosina con la guanina, de modo que el ancho de la doble hélice es constante, 2 nm. Existe un fosfato terminal fijo en la posición 5' del azúcar en un extremo de cada cadena y un grupo hidroxilo libre en la posición 3' del azúcar en el extremo opuesto. Esta polaridad 5' a 3' es opuesta en las dos cadenas, de modo que las cadenas que componen la doble hélice tienen orientación antiparalela.
Como reconocieron Watson y Crick, la estructura del ADN proporciona las bases
para su replicación. Durante el proceso de replicación las dos
cadenas se separan y cada una es copiada, sintetizándose una nueva
molécula en dirección 5' a 3' [ver figura 3]. El constante
apareamiento de la adenina con la timina y la guanina con la citosina asegura
que se mantiene la misma secuencia de bases en las dos moléculas hijas.
Cuando la replicación es completa, las dos moléculas de ADN (cada
una consistente de una cadena original y una recién sintetizada) se
separan y eventualmente se desplazan a las dos células hijas durante la
mitosis. Las moléculas de ARN se sintetizan en forma semejante, usando
una molécula de ADN como templete (ver adelante).
CODIGO Y EXPRESION GENETICA
En la década previa al descubrimiento de la estructura de doble hélice del ADN, Beadle y Tatum realizaron una serie de experimentos clásicos con el hongo Neurospora. Usando una cepa de Neurospora incapaz de sintetizar enzimas específicas por mutaciones genéticas, los investigadores demostraron que existe una corresponencia uno a uno entre el gen y la proteína específica. Esto originó la hipótesis denominada un gen-una enzima, y la noción de que la función de los genes consiste en codificar la estructura de las proteínas. Cuando se descubrió la estructura del ADN, fue claro que este código genético reside en la secuencia de bases del ADN.
El mayor conocimiento del código genético y el mecanismo por el que su información es transferida del ADN a la proteína fue la siguiente tarea importante en la biología molecular, y la mayoría de las investigaciones realizadas en la década siguiente a la publicación del trabajo de Watson y Crick estaban dedicadas a ello. El proceso básico consiste en que la secuencia de bases de un gen es copiada a una molécula de ARN conocida como ARN mensajero (ARNm) [ver figura 4]. El ARNm es exportado del núcleo al citoplasma, en donde se une a los ribosomas. Aquí se ensambla la proteína por la formación sucesiva de uniones peptídicas entre aminoácidos que son colocados en posición por moléculas de ARN de transferencia (ARNt) que reconocen la secuencia de bases del ARNm. En efecto, las moléculas de ARNt leen el código.
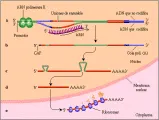
La transcripción se refiere a la copia de la secuencia de bases de un gen al ARN. El ARN es copiado de una de las dos cadenas de ADN, llamada la cadena de codificación. la base uracilo sustituye a la timina en el ARN, o se inserta adenina en el ARN opuesto a timina en el ADN y las guaninas y citosinas se insertan opuestas entre sí. La enzima ARN polimerasa II cataliza la transcripción de los genes que codifican proteínas, a diferencia de los genes que codifican las moléculas de ARN que funcionan sin traducirse a proteína (i.e., ARNt y ARN ribosomal [ARNr], un constituyente de los ribosomas). la transcripción se inicia al unirse la polimerasa a una secuencia de bases dentro de una región del gen denominada promotora. La regulación de la expresión de los genes está mediada por proteínas denominadas factores de transcripción, que se unen a secuencias específicas en la región promotora. La transcripción es estimulada por otras secuencias, denominadas facilitadoras, que pueden localizarse hacia arriba o abajo del promotor. Una secuencia específica de bases en el extremo del gen marca el sitio donde termina la transcripción.
El ARNm se sintetiza en dirección 5' a 3'. Pronto después del inicio de la transcripción, la primera base del ARNm se une en forma covalente al residuo 7-metil-guanosina. La función de esta porción no se comprende del todo, pero parece ser importante para mantener la estabilidad del ARNm, sacarlo del núcleo e iniciar la traducción. El ARNm de longitud completa consiste en una secuencia no codificadora (conocida como la región 5' no traducida), la secuencia codificadora y una región 3' no traducida. Después de la transcripción la mayoría de los ARNm se modifican por la adición de 100 a 200 residuos de adenina, la cola poli (A). Una enzima rompe el ARNm en una secuencia AAUAAA en la región no traducida 3' y se agregan bases adenina sucesivas alrededor de 20 pares de bases hacia abajo de la señal AAUAAA. La cola sirve para mantener la estabilidad del ARNm y facilitar su salida del núcleo, y también parece participar en el inicio de la traducción.
Antes de salir, debe ocurrir un paso importante en el procesamiento del ARNm, la separación de los intrones. A diferencia de los genes procariocíticos, en los que existe una correspondencia uno a uno entre la secuencia de bases y la secuencia de aminoácidos de una proteína, la secuencia de codificación de los genes eucariotes, denominados exones, está interrumpida por secuencias no codificadoras denominadas intrones. Al inicio se transcribe toda la longitud del gen, incluyendo los intrones y los exones. Después se extraen los intrones y los exones se unen entre sí por medio de un complejo proceso mediado por enzimas que tiene lugar en el núcleo. Por último, el ARNm maduro es estraído al citoplasma para su traducción a proteína. La función de los intrones se desconoce, aunque se especula que pueden ser remanentes de partes arrastradas del genoma durante la evolución. La secuencia de bases de los exones tiende a estar muy conservada, pero las secuencias de base de los intrones no están sujetas a presión de selección, excepto las secuencias de bases al final de los intrones y en los sitios internos requeridos para su separación normal. Estos sitios son vulnerables a sufrir mutaciones que pueden alterar el proceso de separación (ver adelante).
El número de intrones en los genes varía mucho. Por ejemplo, el gen de la beta-globina incluye dos intrones y tres exones. El gen de la distrofina, una proteína del citoesqueleto muscular que se afecta en la distrofia muscular de Duchenne contiene 79 exones.1 Por lo general los intrones son mucho más largos que los exones, lo que indica que una porción significativa del ADN en el genoma no codifica información genética. Los tamaños de los genes varían con el número de exones e intrones desde algunos miles a varios cientos de miles de pares de bases. El gen más grande conocido es el de la distrofina, que incluye más de 2.5 millones de pares de bases.
El proceso de separación no produce un ARNm idéntico para cada gen en todos los tipos de células. Un transcrito puede dividirse en forma diferente en células diferentes y en momentos diferentes del desarrollo. Por lo tanto, en algunas células un exón específico es separado del ARNm junto con los intrones, en otras este exón es incluído en el ARNm. Además, pueden existir secuencias promotoras localizadas dentro de los genes que son reconocidas como sitios de inicio de la transcripción en ciertos tipos celulares, pero no en otros. Estos dos fenómenos permiten que una sola unidad de transcripción codifique diferentes formas de la misma proteína, aumentando en forma sustancial la diversidad de proteínas codificadas en el genoma. Este es el motivo por el que alrededor de 80,000 genes codifican para un número mucho mayor de proteínas.
La traducción, el proceso de ensamble de la proteína, tiene lugar en los ribosomas del citoplasma. La proteína es ensamblada como una cadena de aminoácidos, que se agregan uno por uno. Cada aminoácido es especificado por un triplete de bases en el ARNm (un codón) que corresponde a un triplete en el gen. El primer aminoácido siempre es metionina, codificado por el codón AUG (en algunas proteínas esta metionina es eliminada después). Existen 61 posibles codones que especifican 20 aminoácidos, de modo que muchos aminoácidos son determinados por más de un codón, y se dice que el código genético es degenerado [ver tabla 1]. La información del gen es traducido cuando cada codón en el ARNm correspondiente se une a la moléducla de ARNt que contiene una tripleta complementaria de bases (un anticodón). Cada ARNt está unido en forma covalente a un aminoácido específico. La maquinaria enzimática del ribosoma crea una unión peptídica entre los aminoácidos adyacentes atraídos hacia la cercanía del ribosoma al unirse los ARNt respectivos al ARNm. Los dobleces y modificación (v.gr., glucosilación) de la proteína comienzan incluso antes de que se termine la traducción. El codón final en una secuencia codificadora es un codón de final, sea UGA, UAA o UAG, que indica el final de la traducción y permite que la proteína sea liberada del ribosoma.
Es importante reconocer que una parte sustancial de los tres billones de pares de bases que constituyen el genoma humano no codifican. Además de los intrones, existen otros segmentos importantes del genoma que no codifican proteínas o ARN. Muchos de estos segmentos incluyen secuencias que se repiten muchas veces, y algunas tienen un papel estructural en la organización del genoma en sí. Las repeticiones de secuencias simples pueden contener miles de copias y tienden a acumularse en regiones cercanas a los centrómeros del cromosoma (ver adelante). Otras secuencias repetidas están dispersas en el genoma, y su función se desconoce.
GENOMA MITOCONDRIAL
Además de los genes en el núcleo celular, existe ADN fuera del núcleo, en la mitocondria. Cada mitocondria contiene múltiples copias de una molécula circular de ADN de doble cadena formada por 16,569 pares de bases.2 Aunque la mayoría de las proteínas en las mitocondrias son codificadas por el genoma nuclear, el genoma mitocondrial incluye las secuencias de codificación para 13 subunidades de proteínas que participan en la fosforilación oxidativa, un grupo completo de ARNt mitocondriales y el ARNr mitocondrial. El ADN mitocondrial se transcribe dentro de la mitocondria (usando una ARN polimerasa codificada por los genes nucleares) y es traducida en la mitocondria (usando ARNt y ARNr mitocondrial pero proteínas ribosomales codificadas por genes nucleares). Los genes mitocondriales, como los genes procarióticos de los que derivan, no tienen intrones. Las mutaciones en el ADN mitocondrial pueden alterar la fosforilación oxidativa, causando trastornos del metabolismo energético.
Todo el complemento del ADN mitocondrial en el huevo fertilizado es contribución del oocito, las mitocondrias de los espermatozoides se pierden en el momento de la fertilización. Los rasgos genéticos codificados en el ADN mitocondrial son, por lo tanto, trasmitidos de la madre a todos sus hijos (herencia materna). Este patrón de trasmisión se observa en trastornos energéticos que son resultado de mutaciones en el ADN mitocondrial.
Otra propiedad de la herencia del ADN mitocondrial importante en relación con la patogenia de los padecimientos mitocondriales consiste en la segregación de las mitocondrias en el momento de la mitosis.3 Cada mitocondria contiene múltiples copias de ADN y existen muchas mitocondrias en cada célula. Las mitocondrias se reparten al azar en las dos células hijas durante la mitosis, en contraste con la segregación ordenada de los genes del genoma nuclear. Cuando surge una mutación del ADN mitocondrial, suele ocurrir en una célula que contiene una mezcla de ADN mutante y nativo. La segregación pasiva de las mitocondrias durante la mitosis causa una distribución aleatoria de las moléculas de ADN mutante y nativo en las células hijas. Por azar algunas células pueden recibir un mayor número de mitocondrias con la mutación, mientras que otras pueden recibir muy pocas. Esto produce heteroplasmía, esto es, que la proporción de ADN mitocondrial mutado y nativo en los diversos tejidos es diferente, lo que puede causar diversos efectos clínicos en los miembros de la misma familia, a pesar de que compartan una mutación mitocondrial.
Estructura y tinción de los cromosomas
Aunque los genes están colocados en un orden lineal a lo largo de la cadena de ADN, el genoma humano se divide en 23 pares de moléculas de ADN denominadas cromosomas. Cada cromosoma consiste en una sola cadena continua de ADN que incluye varios miles de genes. En el núcleo el ADN existe como una estructura muy organizada acoplada con múltiples proteínas. La unidad básica de organización es el nucleosoma. El nucleosoma consiste en un centro de ocho proteínas histonas (dos copias cada una de H2A, H2B, H3 y H4) alrededor de las cuales se envuelve el ADN (con una longitud de alrededor de146 pares de bases). Los nucleosomas adyacentes están conectados como cuentas en un cordel por un ADN de unión de hasta 80 pares de bases de longitud. La fibra de cromatina que se forma tiene alrededor de 11 nm de grosor. Esta se dobla a su vez en una estructura semejante a un solenoide unida con histona H1 y otras proteínas no histonas. Esta estructura se desdobla durante la transcripción. Durante la división celular la cromatina se vuelve muy compacta y enrollada, condensándose para formar la estructura conocida como cromosoma mitótico.
La célula nucleada somática humana normal contiene 46 cromosomas, 23 heredados de cada padre. Existen 22 pares de cromosomas no sexuales, conocidos como autosomas, y dos cromosomas sexuales, un cromosoma X y uno Y en los varones y dos cromosomas X en las mujeres. La célula germinal contiene un grupo haploide de autosomas y uno de los cromosomas sexuales (un X en los oocitos y un X o un Y en los espermatozoides) Se usa una nomenclatura estándar para describir los cromosomas y las alteraciones cromosómicas.4 El cariotipo masculino normal se describe como 46,XY y el femenino normal como 46,XX.
Los cromosomas se observan con más facilidad en el estado muy compacto que ocurre durante la división celular. Las dos cadenas replicadas de ADN aparecen como dos cromátides unidas junto al centrómero. El centrómero divide al cromosoma en un brazo corto (p, por petit) y uno largo (q). La región centromérica contiene un bloque de material que se tiñe en forma muy intensa conocida como heterocromatina centromérica. Esta área consiste de muchas secuencias repetidas que no son genéticamente activas. Los extremos de los cromosomas se conocen como telómeros. Los telómeros incluyen secuencias especializadas que se requieren para la replicación del cromosoma.
La identificación de los cromosomas se facilita por patrones de bandeo específicos que se descubren por medio de protocolos especiales de tinción. Estos incluyen pretratamientos, como digestión con proteasa, antes de la tinción con Giemsa o colorante fluorescente. Cada par homólogo de cromosomas tiene un tamaño y forma característicos que se aprecia como una estructura fina reproducible y que se tiñe [ver figura 6]. Recientemente, la técnica de hibridización fluorescente in situ (FISH, por sus siglas en inglés, n. del t.) ha hecho posible teñir secuencias específicas del ADN en un cromosoma (ver adelante).
El análisis clínico del número y estructura de los cromosomas suele realizarse en las células en división. Estas células son principalmente linfocitos de sangre periférica que se estimulan para dividirse en cultivo por medio de lectina fitohemaglutinina. Los fibroblastos de piel, las células de líquido amniótico, las células de las vellosidades coriónicas o las células tumorales con división espontánea pueden también usarse para el análisis cromosómico.
La división de las células somáticas ocurre por el proceso de mitosis [ver figura 7]. La replicación del ADN ocurre durante la fase S (de síntesis) del ciclo celular, varias horas antes de que se inicie la división celular. La mitosis comienza con la disolución del núcleo y el comienzo de la condensación de los cromosomas (profase). Los cromosomas se alinean entre los dos polos del huso mitótico durante la metafase. Las cromátides se separan durante la anafase y se forma un núcleo en cada célula hija durante la telofase. La división del citoplasma por medio del proceso de citocinesis completa la mitosis.
Las células germinales sufren un proceso de división especial denominado meiosis, en la que las células germinales primordiales diploides son reducidas a un estado haploide [ver figura 7]. La replicación cromosómica ocurre antes de la meiosis. Durante la profase de la primera división meiótica los cromosomas homólogos se aparean (sinapsis) y pueden intercambiar segmentos a través del proceso de recombinación (entrecruzamiento). Las copias mezcladas de genes específicos sin cambiar el orden genético global del cromosoma producen un incremento sustancial en la diversidad genética. La meiosis ocurre en un periodo de varios días en los espermatocitos. Sin embargo, los oocitos comienzan la meiosis durante la vida embrionaria y permanecen en la primera división meiótica de la profase hasta la ovulación (i.e., durante muchos años).
La primera división meiótica provoca la separación de los cromosomas homólogos en dos células hijas. Estas células sufren otra división completa, sin síntesis de ADN, para producir cuatro células germinales haploides. En el varón, estas cuatros células son los espermatozoides. En la mujer, la primera división meiótica produce un oocito primario y un primer cuerpo polar. La segunda división meiótica del oocito se completa solo después de la fertilización y produce un segundo cuerpo polar además del pronúcleo del oocito.
Se conoce como no disyunción al hecho de que las dos cromátides no pasen en forma independiente a las células hijas durante la mitosis o la meiosis. Esto provoca la producción de una célula con una copia extra de un cromosoma y una sin la copia. La fertilización de una célula germinal con un cromosoma extra da lugar a una trisomía, mientras que la fertilización de una célula germinal con un cromosoma ausente origina una monosomía. Las células monosómicas para algún autosoma generalmente no son viables, pero sí lo son las trisomías de los cromosomas 13, 18 y 21. Si ocurre no disyunción durante la mitosis durante el desarrollo embrionario, existirá una mezcla de células normales y trisómicas en el embrión (suponiendo que las células monosómicas no son viables). A esto se conoce como mosaicismo. Los efectos clínicos del mosaicismo suelen ser menos severos que los de las trisomías presentes en todas las células. Para algunas trisomías autosómicas, como la trisomía 8, el embrión es viable solo si la trisomía se presenta en forma de mosaico.
La causa de la nodisyunción se desconoce. Ejemplos poco frecuentes de asociación familiar de personas con trisomía sugieren la existencia de factores genéticos predisponentes.5 El único riesgo bien documentado para la nodisyunción es la edad materna. La frecuencia de no disyunción claramente aumenta en función de la edad materna, con un incremento constante a partir de la mitad de los 30 años.6 Los estudios sobre trisomía 21 indican que la nodisyunción ocurre con más frecuencia en la primera división meiótica materna.7 Esto puede ser una consecuencia del periodo tan prolongado de la meiosis I en la mujer. Si la sinapsis de un par de cromosomas homólogos se rompe durante este periodo, los dos cromosomas pueden separarse en forma aleatoria hacia las células hijas, causando no disyunción. Estudios sobre los efectos de la edad paterna no han revelado mas que, si acaso, una correlación muy débil con la no disyunción.8 Debido a la relación entre la edad materna y la frecuencia de trisomía, es práctica común ofrecer diagnóstico prenatal para detectar trisomías fetales a las mujeres que tendrán 35 o más años de edad en el momento del parto.
Proyecto genoma humano
Durante la mayor parte de este siglo, los genes se conocieron principalmente porque causaban fenotipos anormales. Con excepción de los genes de las moléculas de globina y algunas enzimas, las secuencias de los genes humanos y las proteínas que codifican seguían siendo un misterio. Además, el catálogo de rasgos genéticos humanos contenía solo algunos pocos miles de los alrededor de 80,000 genes que constituyen el genoma. El Proyecto genoma humano ha sido posible gracias al desarrollo de tecnologías que permiten aislar segmentos individuales del genoma y estudiarlos con detalle. En esta sección se revisan en forma breve estos adelantos técnicos dentro del contexto de su aplicación al estudio del genoma humano y la información que se está acumulado sobre la huella digital genética para el organismo humano.
ADELANTOS TECNOLOGICOS
La base para el estudio de los genes humanos a nivel molecular es la tecnología de ADN recombinante. Esta se usa para aislar fragmentos de ADN, cuyo tamaño varía de algunos cientos a más de un millón de pares de bases. Los fragmentos se recombinan con vectores de ADN que son capaces de tener replicación autónoma dentro de las células procarióticas. Esto permite obtener fragmentos en grandes cantidades para su análisis, incluyendo la determinación de secuencias nucléotidas.
La creación de moléculas recombinantes depende especialmente de un grupo de endonucleasas de restricción, que son enzimas que reconocen y rompen el ADN en sitios específicos (i.e., definidos por una secuencia especial de bases). Los sitios de reconocimiento con secuencia específica suelen consistir en cuatro a ocho nucleótidos y suelen manifestar simetría palindrómica [ver figura 8]. Incluso un solo cambio de base en el sitio de reconocimiento puede impedir la ruptura; por lo tanto, una endonucleasa de restricción específica cortará un segmento determinado de ADN en forma muy reproducible. En muchos casos las endonucleasas de restricción dejan fragmentos libres sin sus correspondientes pares de bases en los extremos de la molécula de ADN. Por lo tanto, dos fragmentos cortados con la misma enzima pueden formar moléculas híbridas que pueden unirse en forma covalente con una ADN ligasa. Las endonucleasas de restricción se aislan de las células bacterianas, en donde su función consiste en restringir el crecimiento viral al romper el ADN viral cuando éste entra en la célula. Estas enzimas reciben nombres cortos que corresponden a las bacterias de las que derivan (v.gr., EcoRI, que deriva de Escherichia coli).
Un experimento básico de clonación del ADN consiste en generar fragmentos de ADN para ser clonados y ligar estos fragmentos a un vector de ADN [ver figura 9]. Se usan diferentes vectores, dependiendo del tamaño del ADN que va a clonarse. Los plásmidos bacterianos acomodan pequeños fragmentos de ADN, hasta 10,000 pares de bases (10kb). Los vectores bacteriófagos pueden clonar fragmentos tan largos como de 23 kb. Los cósmidos, que no se encuentran en la naturaleza, son vectores construidos que pueden comportarse como híbridos de los plásmidos y bacteriófagos y que pueden acomodar fragmentos de alrededor de 50 kb. Los cromosomas artificales de levaduras tienen la mayor capacidad de clonación, varios cientos de miles a un millón de pares de bases.
Pueden usarse varias fuentes de ADN para obtener el material para la clonación génica. El ADN genómico total puede romperse en pequeños fragmentos en forma mecánica o por medio de endonucleasas de restricción. Los fragmentos se insertan en forma aleatoria en un vector de clonación. Entonces se genera una librería genómica, en la que cada colonia bacteriana que resulta incluye un fragmento separado de ADN genómico. Otra fuente común de material para clonación es el llamado ADN complementario (ADNc). El ADNc se produce usando la enzima transcriptasa inversa para copiar el ARN a ADN. La transcriptasa inversa se aisla de virus tumorales ARN, en donde copia el genoma viral ARN en una doble cadena de ADN. Las secuencias del ADNc también pueden clonarse. Sin embargo, la biblioteca del ADNc no representa el ADN genómico total, sino solo los fragmentos que se expresan como ARN en un tipo celular determinado. Si el ADNc se forma a partir de un ARNm totalmente procesado, la secuencia clonada incluirá solo la secuencia codificadora y no los intrones. Las bibliotecas de ADNc son útiles para determinar la secuencia de codificación de un gen e identificar los transcritos que se elaboraron en un tipo celular específico.
El ADN clonado puede analizarse de diferentes maneras. Se han desarrollado técnicas químicas para la determinación rápida de la secuencia de bases. El enfoque más empleado, desarrollado por Sanger, Nicken y Coulson,9 se practica en la actualidad con un aparato automático capaz de secuenciar miles de bases por día.
El ADN clonado, en una sola cadena, se unirá a una molécula de ADN o ARN que incluya una secuencia de bases complementaria. A esta unión se le conoce como hibridización. Por lo tanto, un fragmento clonado puede ser marcado con radioactividad o fluorescencia y usarse como una sonda para buscar secuencias homólogas en una muestra de ARN o de ADN de cualquier origen. El análisis de Southern blot es una técnica que consiste en separar los fragmentos de ADN de acuerdo por su tamaño por electroforesis, inmovilizando los diferentes segmentos en una membrana e hibridizándolos con un fragmento clonado y marcado.10 El análisis de Northern blot usa el mismo principio pero con ARN unidos a la membrana en lugar de ADN. La FISH usa el mismo principio, pero el ADN dentro de una célula o en un cromosoma es tratado sobre un portaobjetos de vidrio.
Otra técnica importante es la reacción en cadena de la polimerasa, que se usa para amplificar la cantidad de un fragmento de ADN por medio de ciclos repetidos de síntesis enzimática de ADN [ver figura 10]. Un par de pequeños oligonucleótidos sintéticos (20 a 30 bases) (denominados cebadores) se unen a las cadenas opuestas del fragmento de ADN blanco. Estos cebadores sirven como puntos de inicio para la replicación del ADN. El proceso incluye 20 a 30 ciclos de unión sucesiva de cebadores, su replicación y separación de los productos en cadenas simples. En cada ciclo los productos del ciclo previo sirven como un templete, causando un incremento exponencial en la cantidad de ADN formado por los cebadores. Esto permite la amplificación a gran escala del segmento de ADN, que entonces puede ser estudiado.
MAPEO GENETICO
El esfuerzo para localizar cada uno de los genes humanos ha requerido la construcción de un mapa de alta resolución del genoma [ver figura 11]. Para este propósito se han generado fragmentos clonados del ADN para usarse como marcadores genéticos. Estos fragmentos incluyen segmentos polimórficos, secuencias de bases que varían de persona a persona. Estas secuencias se detectaron primero como fragmentos polimórficos de longitud restringida (FPLR),11 porque los cambios en la secuencia de bases alteran la capacidad de la endonucleasa de restricción para cortarlos en un sitio determinado. Los FPLR fueron analizados cortando el ADN por medio de una endonucleasa de restricción apropiada y realizando un estudio de Southern blot con una sonda para la región del polimorfismo. Se identificaron alelos como bandas de diferentes tamaños en el ensayo. Otro tipo de polimorfismo que se usa con más frecuencia en la actualidad en los estudios de mapeo genético es la repetición simple de la secuencia polimórfica. Es frecuente encontrar fragmentos de repeticiones simples, como el dinucleótido CACACA...., dentro y alrededor de los genes, y el número exacto de repeticiones suele ser polimórfica. Los alelos pueden detectarse usando RCP para amplificar un segmento de la región que contiene la repetición y después midiendo el tamaño de los productos por electroforesis.
Estas variantes en las secuencias de bases pueden servir como simples rasgos mendelianos y permitir que los alelos sean rastreados dentro de una familia para determinar si los alelos se segregan junto con un rasgo o trastorno genético. Si un marcador en particular se localiza cerca del rasgo que será mapeado, se segregará unto con el rasgo en una familia, y se dirá que el rasgo y el marcador están ligados entre sí. El entrecruzamiento entre el marcador y el locus del rasgo altera esta segregación. Mientras más cerca entre sí se encuentren en marcador y el rasgo, menor será la probabilidad de que se separen durante la recombinación genética. La distancia genética entre el rasgo y el marcador se mide como una función de la frecuencia de recombinación. La herramienta estadística empleada con más frecuencia es la calificación de lod (lod es el acrónimo para logaritmo de momios). Los datos provenientes de una familia se usan para calcular los momios relativos de que el marcador y el rasgo que se están mapeando estén ligados entre sí o no. La relación de momios se convierte a un logaritmo para facilitar el acúmulo de datos de múltiples familias al sumar los logaritmos en lugar de multiplicar la relación de momios. Por conveniencia, el marcador y el rasgo están ligados entre sí si la calificación lod es mayor de 3 (i.e., 1,000:1 momios favoreciendo la unión contra la no unión). Se excluye la unión si la calificación de lod es menor de -2 (i.e., 100:1 momios favoreciendo la no unión sobre la unión). En la actualidad el mapeo genético incluye marcadores polimórficos espaciados alrededor de cada 1 cM (centimorgan, que corresponde a la longitud del ADN entre marcadores dentro de los que existe un porciento de probabilidad de entrecruzamiento).12 Esta densidad es suficiente para asegurar que virtualmente cualquier rasgo genético puede ser mapeado si se cuenta con un ADN del pedigree de tamaño adecuado.
La identificación de los genes individuales requiere la correlación del mapa genético con un mapa físico del ADN, que es un mapa de las localizaciones de los marcadores identificables (v.gr., sitios de corte por las endonucleasas de restricción o marcadores de repetición CA).13 El mapa físico consiste en segmentos muy grandes del genoma clonados en vectores como los cromosomas artificiales de levaduras. El mapa físico se alínea con el mapa genético localizando los marcadores polimórficos a lo largo del ADN clonado. El mapa físico permite al investigador relacionar los segmentos clonados del ADN con un locus de interés que se ha colocado en el mapa genético. Para encontrar el gen en sí se requiere investigar las secuencias expresadas a lo largo del ADN clonado en la región y después examinar las secuencas buscando las propiedades esperadas en el gen blanco.14 En los casos en que se encuentran los genes responsables de la enfermedad, el proceso incluye buscar mutaciones en los genes de las personas afectados.
Durante la década pasada se han identificado varios genes con base en su localización dentro del genoma,15 lo que se conoce como genética reversa o, en forma más apropiada, clonación posicional. Esta técnica ha hecho posible clonar genes de trastornos cuya base bioquímica se desconoce. La identificación de los genes de padecimientos como la distrofia muscular de Duchenne,16 la fibrosis quística,17 y la neurofibromatosis tipo 118,19 por este método, ha facilitado el mayor conocimiento de su patogenia y, en consecuencia, de las observaciones clínicas. Antes de su descubrimiento por clonación posicional se ignoraba la existencia de las proteínas que participan en estos trastornos.
EL PROYECTO DEL GENOMA HUMANO Y LA BIOLOGIA HUMANA
El Proyecto del genoma humano va a la vanguardia de una revolución en el conocimiento de la biología humana que no dejará de abarcar a ningún área de la medicina. La identificación de todos los genes humanos permitirá estudiar en detalle la secuencia de los eventos responsables del desarrollo normal y revelará los mecanismos que ocasionan alteraciones en el desarrollo. También aclarará la patogenia de los trastornos genéticos. En la actualidad ya se están desarrollando nuevos métodos de diagnóstico basados en la detección de mutaciones en el ADN. El conocimiento de los mecanismos celulares de la enfermedad impulsará nuevos métodos de tratamiento que pueden incluir manipulación directa de genes, como la reparación de los genes defectuosos.
Fisiopatología genética
La variación en la secuencia de bases entre una persona y otra es la base de la diversidad genética en la población. La composición genética de un locus genético en particular se conoce como genotipo, y las manifestaciones físicas que resultan de la expresión de un gen en particular constituyen el fenotipo. Algunos cambios de bases no afectan la secuencia codificadora de las proteínas, o tienen un impacto mínimo en la estructura y función proteica. Otros cambios de bases pueden abolir por completo la síntesis de una proteína o alterar en forma sustancial sus propiedades químicas. Los efectos fenotípicos son también variables, siendo desde no detectables hasta manifestaciones de un trastorno genético severo o incluso mortal. La mayoría del conocimiento sobre la patología genética deriva del estudio de cambios en genes individuales que causan fenotipos obvios y severos o de cambios que pueden alterar grandes segmentos cromosómicos. Aunque clínicamente importantes, los trastornos estrictamente genéticos son poco frecuentes. Los factores genéticos contribuyen a la etiología de padecimientos mucho más comunes, pero estas enfermedades suelen ser resultado de la acción conjunta entre múltiples genes y factores ambientales. En esta sección se revisarán los mecanismos por los que los cambios genéticos influyen en la expresión genética y tienen efectos patológicos.
MUTACION
Un cambio en la estructura de un gen o un grupo de genes se conoce como mutación. El más sutil de estos cambios en el ADN es la alteración de una sola base, que sin embargo puede tener importantes efectos sobre la expresión de la proteína correspondiente. El extremo opuesto incluye cambios que afectan a grandes bloques de genes o cromosomas enteros.
Los cambios cromosómicos incluyen poliploidía (uno o más juegos extra completos de cromosomas), aneuploidía (ganancia o pérdida de uno o más cromosomas individuales), traslocaciones (intercambio de segmentos entre dos o más cromosomas) y deleción, duplicación, inserción o inversión dentro de un solo cromosoma. Algunos cambios cromosómicos alteran la estructura del cromosoma pero no causan ganancia o pérdida del material genético. Pueden no tener efectos fenotípicos y existen como variantes en la población general. En casos raros el sitio de ruptura de un cromosoma para un rearreglo balanceado puede interrumpir un gen, causando el fenotipo de un trastorno de un solo gen. Por ejemplo, algunas personas con distrofia muscular de Duchenne ligada al X tienen rearreglos en el cromosoma X que alteran el gen de la distrofina.20
En la mayor parte de los casos, las ganancias o pérdidas de grandes bloques de material cromosómico son letales y producen un aborto. Solo una minoría de los infantes llega a término, aunque con alteraciones importantes del desarrollo tanto físico como neurológico. Las alteraciones cromosómicoas ejercen sus efectos fenotípicos al modificar el balance en el nivel de expresión genética. Al tener tres copias de un gen existen niveles más altos de transcripción, lo que, por lo menos en algunos genes, altera la función. Se han hecho intentos por mapear los efectos fenotípicos asociados con ganancia o pérdida de regiones cromosómicas específicas. Incluso los fenotipos complejos como el del síndrome de Down, que se debe a la trisomía 21, se han analizado de esta forma.21 Al completar el mapa genético de cada cromosoma, se identificarán los genes asociados con los fenotipos particulares y podrán estudiarse los mecanismos por los que los genes causan los problemas del desarrollo.
Las deleciones cromosómicas que son visibles al microscopio incluyen más de un millón de pares de base, lo que equivale a docenas de genes. Con FISH pueden detectarse deleciones sutiles que no se observan con microscopía de luz.22 Estas deleciones pueden producir fenotipos característicos conocidos como síndromes de microdeleción. Muchos de estos síndromes se atribuyen a los efectos acumulativos de deleción de los genes contiguos. Por ejemplo, en el síndrome WAGR (por tumor de Wilms, aniridia, alteraciones genitourinarias o gonadoblastoma y retraso mental), cada componente del síndrome es resultado de la deleción de un gen en particular.23 Pueden ocurrir diversas manifestaciones del síndrome dependiendo de la magnitud de una deleción en particular. Otros síndromes puede ser también resultado de la deleción de un solo gen. Por ejemplo, en algunos casos el fenotipo complejo del síndrome de Alagille (asociado con atresia biliar intrahepática, estenosis pulmonar y anomalías faciales) es causado por deleción parcial del cromosoma 20, y en otros casos la enfermedad ocurre por mutaciones dentro de un solo gen en esta región cromosómica.24-26
El estudio de las deleciones cromosómicas ha revelado un mecanismo importante de la regulación genética, conocido como impresión genómica, en el que existe expresión desequilibrada de la copia materna y paterna de un gen [ver figura 12].27 Solo un subgrupo de genes sufren impresión genómica. En éstos solo se expresa la copia materna o paterna, y la huella o impresión puede cambiar en cada generación. La deleción de un gen de este tipo causará diversos efectos fenotípicos, dependiendo cual copia se pierda. La pérdida de la secuencia expresada puede tener efectos fenotípicos importantes, mientras que la deleción de la secuencia no expresada puede no tener consecuencias hasta que el gen pasa a la siguiente generación, cuando quizá deberá expresarse.
El síndrome de Prader-Willi y el síndrome de Angelman son dos trastornos causados por cambios en la expresión de genes impresos.28 Ambos síndromes causan deterioro del desarrollo, pero por lo demás son muy diferentes. Los signos y síntomas del síndrome de Prader-Willi incluyen una apariencia facial característica y un trastorno de la alimentación que causa obesidad. En el síndrome de Angelman existen características dismórficas, movimientos mal coordinados y crisis convulsivas. Ambos síndromes se asocian con deleciones en la región proximal del brazo largo del cromosoma 15; la deleción de las secuencias paternas causa el síndrome de Prader-Willi, mientras que la deleción de las secuencias maternas produce el síndrome de Angelman.29 Un solo gen es responsable de muchos de los efectos del síndrome de Angelman,30,31 pero el gen o genes responsables del síndrome de Prader-Willi no ha sido identificado aún.
Algunos casos de síndrome de Prader-Willi o de Angelman no son causados por deleción sino por un fenómeno conocido como disomía unipaterna. En estos casos las dos copias del cromosoma 15 se heredan del mismo padre. Debido a que existen genes impresos tanto paternos como maternos en el cromosoma 15, las dos copias del cromosoma 15 que derivan del mismo padre provocan la falta de expresión de algunos genes impresos y, en consecuencia, el síndrome de Prader-Willi o de Angelman. Ocurre disomía unipaterna cuando un cromosoma trisómico sufre un evento secundario de no disyunción, restableciendo la disomía para el cromosoma pero produciendo una disomía unipaterna si las dos copias derivan del mismo padre. No todos los cromosomas tienen genes impresos, pero los que los tienen originan alteraciones fenotípicas cuando ocurre disomía unipaterna.32
Las mutaciones dentro de genes individuales se analizan mejor desde el punto de vista del impacto sobre la función del gen [ver figura 13]. La mutación genética puede causar reducción o ausencia de la transcripción, producción de un producto truncado, duplicación o deleción de aminoácidos en la proteína o sustitución de un aminoácido por otro dentro de la proteína. Las mutaciones pueden ocurrir dentro de los exones o los intrones. Las mutaciones en los intrones causan efectos fenotípicos al alterar la separación del ARN.
Las mutaciones en la región promotora causan niveles aberrantes de transcripción genética. Es posible que el promotor se pierda o que los cambios de una sola base inhiban la unión de la polimerasa de ARN o de las moléculas reguladoras. Las mutaciones en el promotor son responsables de algunos casos de talasemia beta: los cambios en la secuencia ocasionan una transcripción deficiente del gen de beta-globina.33 Las mutaciones del promotor pueden también causar expresión constitutiva de un gen. Estas mutaciones se han demotrado en procariocitos. La activación de un gen por alteración en la secuencia promotora es un mecanismo importante de algunos cambios genéticos somáticos que originan neoplasias (ver adelante).
Las mutaciones que crean nuevos codones de terminación o alteran el marco de lectura de la transcripción provocan la producción de un producto truncado. Los nuevos codones de terminación pueden originarse por el cambio de una sola base en una tripleta. Por ejemplo, la tripleta CGA codifica al aminoácido arginina. El cambio de C a T produce un codón de terminación TGA. Este es un tipo especialmente frecuente de mutación. Las bases de citosina seguidas por guanina (dinucleótidos CpG) suelen estar metiladas , y la deaminación de la citosina metilada origina timina.
El desplazamiento del marco de lectura se produce por la inserción de material en el exón o deleción de material de un exón en un múltiplo diferente a tres bases. La alteración del marco de lectura produce una secuencia sin sentido que inevitablemente genera un codón de terminación. La inserción, deleción o duplicación de una o dos base produce desplazamiento del marco de lectura. Esto puede ocurrir también cuando todos los exones o grupos de exones sufren deleción o se duplican. Los exones no necesariamente terminan en la tercera posición de un codón, pero el marco de lectura se conserva mientras el siguiente exón continúe en la posición en que se quedó el exón previo.34 Cuando un exón o grupo de ellos sufre deleción, los dos exones que limitan la porción perdida serán yuxtapuestos en el ARNm. Si el marco de lectura se conserva, la proteína será más corta, pero por lo demás se traducirá en su totalidad. Si el marco de lectura no se conserva, se producirá una proteína truncada. Ocurre un proceso semejante si un exón o grupo de exones se duplica, la yuxtaposición de los exones en el ARNm puede o no conservar el mismo marco de lectura. Las mutaciones de los intrones pueden causar que se brinquen exones (ver adelante) y provocar desplazamiento del marco de lectura por el mismo mecanismo.
Las inserciones o deleciones de múltiples aminoácidos o las sustituciones de los mismos pueden causar una proteína estructuralmente aberrante. Sin embargo, debido a la degeneración del códico genético, algunas sustituciones de base no cambian del todo la secuencia de aminoácidos; a éstas se les denomina mutaciones silenciosas. Otras sustituciones de base causan la incorporación de aminoácidos con propiedades químicas semejantes a los nativos, por lo que no existe impacto, o este es mínimo, sobre la estructura o función de la proteína; éstas se denominan mutaciones conservadoras. Ni las mutaciones silenciosas ni las conservadoras están sujetas a presión selectiva y pueden existir como variantes de secuencias en la población general (polimorfismos). Los cambios de base que causan sustitución de aminoácidos con diferentes propiedades químicas pueden alterar en forma importante las propiedades de una proteína (v.gr., un aminoácido con carga, como la arginina, es sustituido por uno no polar, como la leucina). Con algunas sustituciones de aminoácidos la proteína se vuelve inestable y es degradada en la célula. Con otras se produce una proteína intacta pero sin actividad enzimática o sin que pueda realizar un papel estructural normal.
Las mutaciones dentro de los intrones pueden tener consecuencias al interferir con la formación del ARN. Aunque la secuencia de bases exacta de un intrón no está muy conservada, si se conservan un grupo de bases en los extremos 5' y 3' (donador y aceptor del empalme, respectivamente), ya que esta es la región dentro del intrón necesaria para el empalme normal. La región donadora siempre comienza con la secuencia GT, seguida de un grupo más variable pero generalmente conservado de bases (una secuencia de consenso). La región aceptora termina con AG, precedida de una secuencia de consenso. Las mutaciones en la región donadora o aceptora pueden causar que todo un exón sea excluido en la transcripción al ARNm (exón brincado) o pueden ocasionar la inserción de cierto material del intrón en la molécula madura de ARNm. Esto último ocurre si se presenta un donador o aceptor denominado críptico dentro del intrón. Esta es una secuencia que puede servir como donadora o aceptora si la secuencia real se altera. El brinco de un exón tiene el mismo impacto que su deleción. La inserción de un intrón suele causar desplazamiento del marco de lectura o producir un codón de terminación que provoca el fin prematuro de la traducción.
En los años recientes se ha descubierto un mecanismo adicional de mutación genética. Este es la expansión de repeticiones de tripletas, que es responsable de varios trastornos neurológicos.35 Esta mutación ocurre en genes que incluyen segmentos de una tripleta de bases que se repite múltiples veces. La expansión de la repetición puede ocurrir en la región 5' no traducida, como en el gen FNR1, que participa en el síndrome del X frágil (retraso mental ligado al X), en la región 3' no traducida, como en el gen de la distrofia miotónica, en la secuencia codificada, como en el gen de la enfermedad de Huntington, o en un intrón, como en el gen de la ataxia de Friederich. El número exacto de copias de la tripleta puede diferir de persona a persona en la población, pero suele ser estable de generación en generación. La expansión del número de repeticiones por arriba de un umbral causa la expresión genética anormal. Los trastornos de repetición de tripletas sufren un fenómeno conocido como anticipación, en el que el padecimiento se vuelve más severo o tiene un inicio más temprano de una generación a la siguiente.36 La tripleta expandida es inestable y, por lo tanto, puede expanderse de generación en generación. La severidad clínica del padecimiento tiende a relacionarse con el tamaño de la región expandida. Hasta el momento todos los padecimientos identificados que resultan de la expansión repetida de tripletas afectan principalmente el sistema nervioso. Sin embargo, aún debe establecerse el rango completo de efectos fenotípicos de estos trastornos.
FISIOPATOLOGIA DE LA ENFERMEDAD GENETICA
Las consecuencias fisiológicas de alterar la cantidad o calidad de una proteína específica dependen de la función normal de la misma. Debido a que se han identificado más de decenas de miles de proteínas diferentes, cada vez se aclara más el repertorio completo de la función proteica.
En vista de que los humanos son organismos diploides, cada individuo tiene dos copias de cada gen (excepto para los genes de los cromosomas X y Y en los varones). Las dos copias de un gen en particular se denominan alelos. Con respecto a un cambio particular de secuencia de bases los dos alelos pueden ser idénticos, caso en el que el individuo se denomina homocigoto, o pueden diferir, caso en el que se le considera heterocigoto. En algunos casos ambos alelos tienen mutaciones, pero estas son diferentes. A esto se le conoce como herocigocidad compuesta.
Los primeros trastornos genéticos humanos fueron descrito por Archibald Garrod en la primera década del siglo XX. Los errores congénitos del metabolismo (término acuñado por Garrod) son causados por deficiencias enzimáticas que resultan de la mutación de los genes que codifican las enzimas o los cofactores que catalizan las reacciones químicas. Los efectos fenotípicos son consecuencia de la deficiencia de la producción de la reacción, el acúmulo de sustratos o ambos. La mayoría de los errores congénitos del metabolismo se heredan en forma recesiva, esto es, ambos alelos deben ser mutados para producir el efecto fenotípico. Esto se debe a la naturaleza catalítica de las enzimas. Debido a que solo pequeñas cantidades de proteína activa son suficientes para producir una reacción adecuada, los portadores heterocigotos de una mutación enzimática suelen ser fenotípicamente normales. Virtualmente cualquier tipo de mutación, sea que produzca deficiencia del producto o altere la secuencia de aminoácidos de una enzima, puede causar un error congénito del metabolismo.
Las mutaciones de los genes que codifican las proteínas estructurales pueden causar efectos fenotípicos en un estado heterocigoto y se dice que se heredan en forma dominante. Las proteínas estructurales son las que se secretan hacia la matriz extracelular, como la colágena, la elastina y la fibrilina, así como las que tienen un papel estructural dentro de la célula, como la globina. La mayoría de las proteínas estructurales participan en interacciones complejas proteína-proteína. Con frecuencia ocurren dos tipos de mutaciones en los padecimientos asociados con las proteínas estructurales. El primer tipo consiste en mutaciones que causan deficiencia del producto, como deleciones, mutaciones en el promotor y mutaciones que causan un péptido truncado no funcional. Por ejemplo, las deleciones provocan algunos tipos de talasemia, en especial las talasemias-alfa. Las mutaciones con deficiencia del producto suelen ocasionar un fenotipo leve en los heterocigotos. La deficiencia de una proteína estructural puede debilitar un tejido o disminuir el estado funcional del sistema, pero si cierta proteína normal se produce a partir del otro alelo el fenotipo será relativamente leve.
Por el contrario, las mutaciones que alteran la secuencia de aminoácidos de una proteína estructural suelen tener un efecto más severo, incluso en el estado heterocigoto, porque pueden alterar las interacciones proteína-proteína. Puede producirse una proteína estructural mutante en cantidades normales y afectar la integridad tisular. Estas mutaciones se conocen como dominantes negativas. Incluyen, por ejemplo, las mutaciones de la colágena que causan la osteogénesis imperfecta (huesos brillantes con fracturas múltiples). La colágena tipo I es una triple hélice con dos cadenas alfa (I)1 y una alfa (I)2. La mutación de un alelo para una cadena alfa (I)1 causa que tres cuartas partes de las moléculas de colágena tipo I sean anormales, lo que ocasiona una forma severa de osteogénesis imperfecta.37
Una tercera clase de trastornos es causada por mutación de genes que codifican proteínas que participan en las señales intra o extracelulares. Estas incluyen hormonas peptídicas, receptores de membrana, canales iónicos, proteínas del citoesqueleto, la cascada de proteínas que envía las señales recibidas en la membrana celular y factores de transcripción. Esta clase heterogénea de proteínas está sujeta a una amplia variedad de mutaciones. La alteración en la secuencia de aminoácidos puede ocasionar reducción en la función o activación constitutiva. Un ejemplo interesante es el gen ret, que codifica una tirosina cinasa de membrana.38 Algunas mutaciones del ret ocasionan activación constitutiva del receptor, lo que origina división celular aberrante en ausencia del ligando. Este tipo de mutación causa que la proteína ret funcione como un oncogén (un gen cuya actividad contribuye a las propiedades neoplásicas de una célula canderosa) y causa el trastorno de herencia dominante conocido como neoplasia endócrina múltiple tipo II. Por el contrario, un grupo diferente de mutaciones del ret causa inactivación del receptor. Cuando ambos alelos han mutado ocurre diferenciación aberrante de las células de la cresta neural en el intestino y se presenta la enfermedad de Hirschprung.
Los diferentes efectos producidos por las mutaciones en el gen ret ilustran el principio de la heterogeneidad genética. La heterogeneidad en el locus génico se refiere a mutaciones de diferentes genes que originan un fenotipo similar, con frecuencia indistinguible. El trastorno esclerosis tuberosa (asociado con displasia cortical, tumores renales, máculas hipopigmentadas y rabdomiomas cardiacos) puede ser causado por mutaciones en alguno de dos loci (TSC1 o TSC2).39 La heterogeneidad del locus génico puede reflejar el hecho de que múltiples proteínas participan en vías comunes del metabolismo celular. La otra forma de heterogeneidad genética es la heterogeneidad alélica. Aquí diferentes tipos de mutaciones del mismo gen causan uno o más fenotipos diferentes. La fibrosis quística, asociada con secreciones espesas, enfermedad pulmonar crónica y malabsorción) es causada por mutación en el gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR, por sus siglas en inglés, n. del t.). Una deleción de tres pares de bases denominada ?F508 es responsable de alrededor del 70 porciento de las mutaciones CFTR en la población blanca, y más de 600 mutaciones diferentes causan el 30 porciento restante de los casos.40 El caso del gen ret muestra que trastornos completamente diferentes pueden ser causados por mutaciones diferentes del mismo gen. Los mismo es verdad para las mutaciones de los genes del receptor de factor de crecimiento de fibroblastos.41 Diferentes mutaciones del gen que codifica al receptor 2 del factor de crecimiento de fibroblastos (FGFR2, por sus siglas en inglés, n. del t.) pueden originar el síndrome de Apert (asociado con craniosinostosis y sindactilia), el síndrome de Crouzon (asociado con craniosinostosis y órbitas poco profundas) o el síndrome de Pfeiffer (asociado con craniosinostosis, pulgares anchos y primeros ortejos grandes). El síndrome de Pfeiffer puede ser causado también por mutaciones en el FGFR1. El descubrimiento de las bases genéticas para los trastornos clínicos con frecuencia pone en duda los esquemas antiguos de clasificación clínica y demuestra que fenotipos que se superponen pueden ser causados por mutaciones de genes que codifican proteínas con funciones que también se superponen.
La importancia médica de la mutación genética se aplica no solo a los trastornos hereditarios, sino también a mutaciones somáticas que se adquieren después de la concepción. Es probable que se desconozca el rango completo de los efectos fenotípicos causados por las mutaciones somáticas, pero mucho se ha aprendido sobre mutaciones que originan las propiedades fenotípicas de las células neoplásicas. Los genes cuyos productos contribuyen a las neoplasias se conocen como oncogenes.42 Los oncogenes dominantes son activados por mutaciones que afectan su intensidad de transcripción, estabilidad proteica o actividad biológica. Algunos mecanismos de la activación de los oncogenes no ocurren en los genes relacionados con las enfermedades hereditarias. Estos mecanismos incluyen traslocaciones que yuxtaponen oncogenes con promotores activos y amplificación del gen, en la que un gen o grupo de genes se replica decenas a cientos de veces en una célula.
Los oncogenes recesivos también se conocen como genes supresores de tumores. Estos genes ejercen su efecto solo cuando han mutado ambas copias, causando ausencia del producto. Las mutaciones de los genes supresores de tumores suelen ser deleciones o mutaciones truncadas que eliminan la actividad de la proteína. La pérdida de la actividad de un gen supresor de tumores puede influir en la etiología de un cáncer. Las personas que nacen con una mutación heterocigota tienen un mayor riesgo de cáncer porque el paso que limita hacia la aparición de la neoplasia será la pérdida de la copia restante del gen. En algunos casos las mutaciones heterocigotas pueden asociarse con efectos fenotípicos, principalmente alteraciones congénitas.
Trastornos genéticos en la población
Todos los humanos comparten la carga de la enfermedad genética, pero el espectro de padecimientos no es el mismo en todas las poblaciones. El estudio de las influencias sobre la frecuencia de la enfermedad genética en las diferentes poblaciones es el tema de la genética de población. Estas influencias incluyen mutación, selección, migración y el fenómeno conocido como el efecto de fundador (ver adelante).
Algunos genes están sometidos a una tasa relativamente alta de mutaciones nuevas. Por ejemplo, la prevalencia de neurofibromatosis tipo 1 es de alrededor de uno por 3,500 personas y el 50 porciento de los casos se debe a una mutación nueva.43 Los trastornos que se originan de una mutación nueva se distribuyen en todo el mundo, sin predilección racial o étnica. Aunque la frecuencia elevada de mutaciones debe originar un aumento gradual en la frecuencia del padecimiento, se llega a alcanzar un equilibrio entre los nuevos casos que se originan por mutación y la pérdida de alelos mutantes de la población como resultado de la selección (v.gr., muertes tempranas, infertilidad o menor capacidad para reproducirse).
Si la tasa de mutación es baja, el trastorno genético será poco frecuente, pero las fuerzas de selección o el efecto de fundador pueden causar una frecuencia aberrantemente alta en poblaciones específicas. En algunas circunstancias la selección puede favorecer a un alelo mutante sobre el alelo nativo (normal). El mejor ejemplo lo constituyen las mutaciones de la globina responsables de la anemia de células falciformes o de la talasemia. La heterocigosidad para las variantes de la globina tienden a conferir una resistencia relativa al paludismo.33 Esto preserva las mutaciones en estado heterocigoto en ciertas partes del mundo en donde el paludismo es prevalente. La ventaja de los heterocigotos también se ha propuesto como un mecanismo para mantener las mutaciones de la fibrosis quística, en este caso para mantener una resistencia relativa a la pérdida de líquido causada por la toxina del cólera.44
El efecto de fundador es otra manera por la que mutaciones específicas son confinadas a poblaciones específicas. Este ocurre cuando un pequeño número de individuos fundan una población nueva que permanece relativamente aislada. Los alelos recesivos que son raros en la población de origen pueden volverse más prevalentes en los fundadores de la nueva población. Se piensa que este es el mecanismo responsable de la prevalencia de alelos para enfermedad de Tay-Sachs, de Canavan y de Gaucher en los judíos Ashkenazi. El efecto de fundador puede explicar también porqué mutaciones de globina específicas (v.gr., celulas falciformes en Africa y beta-talasemia en la región del Mediterráneo) predominan en diferentes regiones del llamado cinturón del paludismo.
El reconocimiento de la predilección étnica para la enfermedad genética es importante en la práctica clínica. Las frecuencias de portadores para las enfermedades recesivas pueden variar mucho en las diferentes poblaciones. Las personas cuyos antecedentes étnicos les confieren un mayor riesgo de ser portadores de un trastorno específico deben someterse a escrutinio para determinar el riesgo de tener un hijo afectado. En muchos de estos padecimientos es posible realizar diagnóstico prenatal.
Conclusión
El siglo XX será recordado en la historia de la medicina como la era en la que se comprendieron las bases moleculares para el desarrollo normal y la función celular. Los cambios en la integridad del programa genético, sean heredados o adquiridos somáticamente, son la raíz o contribuyen en virtualmente todos los aspectos de la patología humana. Al aclararse la estructura y función del genoma humano, podrá contarse con las herramientas para definir la patogenia molecular de esencialmente todos los aspectos de la enfermedad. Será tarea de las siguientes generaciones de científicos médicos el integrar esta información en un cuadro de función normal y anormal, y es seguro suponer que la práctica médica se modificará en mucho al surgir nuevos conocimientos.
Reconocimientos
Figuras 1 a 3 George Kelvin.
Figuras 4,5,7,9,11,12 y 13 Dimitry Schidlovsky.
Figura 6 Micrografía cortesía del Dr. Jorge J. Yunis, Department of Laboratory Medicine and Pathology, University of Minnesota Medical School.
Figura 8 Talar Agasyan.
Figura 10 Tom Moore.
DR. BRUCE R. KORF, PH.D.
Genoma humano y enfermedad
Un gen es una secuencia ordenada de cuatro bases de nucleótidos en una molécula de ácido desoxirribonucleico (ADN) que codifica un producto funcional específico (i.e., una proteína o molécula de ácido ribonucleico [ARN]). El desarrollo de un ser humano desde una sola célula hasta la fase adulta está dirigido por el genoma humano, un grupo de alrededor de 80,000 genes que codifican las estructuras para un número incluso mayor de proteínas. Estas proteínas catalizan reacciones químicas, proporcionan las estructuras básicas de las células y tejidos, efectúan comunicación inter e intracelular y dirigen la secuencia ordenada de la diferenciación celular. En la actualidad se realiza un enorme esfuerzo por identificar todos los genes humanos y determinar su secuencia de bases. El Proyecto del Genoma Humano tiene por objeto producir un adelanto revolucionario en el conocimiento de la biología humana. Al final, permitirá identificar en su totalidad los complementos de las proteínas expresadas en cualquier tejido en cualquier momento del desarrollo.
La complejidad del genoma humano lo hace vulnerable a sufrir disfunción con cambios incluso leves en el contenido de información. Estos cambios se denominan mutaciones. El papel de la herencia en la enfermedad humana comenzó a descubrirse a principios del siglo XX con el redescubrimiento de las leyes de la herencia de Mendel. Durante los últimos años el estudio de la enfermedad genética se había enfocado a algunos padecimientos raros causados por un solo gen, y en la mayor parte de los casos el conocimiento se limitaba a la descripción clínica y a los resultados del análisis bioquímico. Sin embargo, los grandes adelantos en genética molecular realizados en la segunda mitad del siglo XX, incluyendo el desarrollo de la tecnología de ADN recombinante en los años 70, ha hecho posible identificar los genes responsables de muchos padecimientos y comprender la patogenia de la enfermedad en términos celulares y moleculares precisos. Estos avances han permitido también el desarrollo de herramientas muy poderosas de diagnóstico y nuevos métodos de tratamiento. Además, en la actualidad es posible explorar la contribución genética a trastornos comunes como la enfermedad cardiovascular, la diabetes y el cáncer.
El médico requiere conocer cada vez más los patrones de trasmisión familiar de la enfermedad e identificar a los familiares con riesgo. La capacidad para interpretar los datos de las pruebas genéticas es una destreza cada vez más necesaria. Ningún área de la medicina está aislada de la influencia de los factores genéticos, y ningún médico puede no estar al tanto de la contribución de la genética a la salud y a la enfermedad.
Esta subsección se enfoca a la estructura y función del genoma humano y a la fisiopatología de los trastornos genéticos. Aunque el ámbito de la genética pertenece a la biología y el de las enfermedades genéticas a la medicina, existen temas comunes respecto a los mecanismos de la enfermedad que incluyen principalmente la expresión genética y su disrupción por mutaciones.
Estructura y función del genoma humano
El paradigma básico de la genética molecular, referido con frecuencia como el dogma central, es que la secuencia de aminoácidos de cada proteína está codificada por la secuencia de bases de su gen correspondiente.
ESTRUCTURA DEL ADN
El descubrimiento de la estructura del ADN por Watson y Crick en 1953 inició la era moderna de la biología molecular. La molécula de ADN consiste en un par de tiras o cadenas de azúcar-fosfato con una base (adenina A, guanina G, citosina C o timidina T) unida a cada azúcar desoxirribosa [ver figuras 1 y 2]. Las dos cadenas están unidas entre sí por puentes de hidrógeno entre las bases, formando una doble hélice. La adenina y la guanina son purinas heterocíclicas, la citosina y la timina son pirimidinas más pequeñas, de un solo anillo. La adenina siempre hace par con la timina y la citosina con la guanina, de modo que el ancho de la doble hélice es constante, 2 nm. Existe un fosfato terminal fijo en la posición 5' del azúcar en un extremo de cada cadena y un grupo hidroxilo libre en la posición 3' del azúcar en el extremo opuesto. Esta polaridad 5' a 3' es opuesta en las dos cadenas, de modo que las cadenas que componen la doble hélice tienen orientación antiparalela.
|
|

|
| Figura 3 |
| Proceso de replicación del ADN |
CODIGO Y EXPRESION GENETICA
En la década previa al descubrimiento de la estructura de doble hélice del ADN, Beadle y Tatum realizaron una serie de experimentos clásicos con el hongo Neurospora. Usando una cepa de Neurospora incapaz de sintetizar enzimas específicas por mutaciones genéticas, los investigadores demostraron que existe una corresponencia uno a uno entre el gen y la proteína específica. Esto originó la hipótesis denominada un gen-una enzima, y la noción de que la función de los genes consiste en codificar la estructura de las proteínas. Cuando se descubrió la estructura del ADN, fue claro que este código genético reside en la secuencia de bases del ADN.
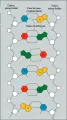
El mayor conocimiento del código genético y el mecanismo por el que su información es transferida del ADN a la proteína fue la siguiente tarea importante en la biología molecular, y la mayoría de las investigaciones realizadas en la década siguiente a la publicación del trabajo de Watson y Crick estaban dedicadas a ello. El proceso básico consiste en que la secuencia de bases de un gen es copiada a una molécula de ARN conocida como ARN mensajero (ARNm) [ver figura 4]. El ARNm es exportado del núcleo al citoplasma, en donde se une a los ribosomas. Aquí se ensambla la proteína por la formación sucesiva de uniones peptídicas entre aminoácidos que son colocados en posición por moléculas de ARN de transferencia (ARNt) que reconocen la secuencia de bases del ARNm. En efecto, las moléculas de ARNt leen el código.
|
| Figura 4 |
| Síntesis de la beta-globina |
La transcripción se refiere a la copia de la secuencia de bases de un gen al ARN. El ARN es copiado de una de las dos cadenas de ADN, llamada la cadena de codificación. la base uracilo sustituye a la timina en el ARN, o se inserta adenina en el ARN opuesto a timina en el ADN y las guaninas y citosinas se insertan opuestas entre sí. La enzima ARN polimerasa II cataliza la transcripción de los genes que codifican proteínas, a diferencia de los genes que codifican las moléculas de ARN que funcionan sin traducirse a proteína (i.e., ARNt y ARN ribosomal [ARNr], un constituyente de los ribosomas). la transcripción se inicia al unirse la polimerasa a una secuencia de bases dentro de una región del gen denominada promotora. La regulación de la expresión de los genes está mediada por proteínas denominadas factores de transcripción, que se unen a secuencias específicas en la región promotora. La transcripción es estimulada por otras secuencias, denominadas facilitadoras, que pueden localizarse hacia arriba o abajo del promotor. Una secuencia específica de bases en el extremo del gen marca el sitio donde termina la transcripción.
El ARNm se sintetiza en dirección 5' a 3'. Pronto después del inicio de la transcripción, la primera base del ARNm se une en forma covalente al residuo 7-metil-guanosina. La función de esta porción no se comprende del todo, pero parece ser importante para mantener la estabilidad del ARNm, sacarlo del núcleo e iniciar la traducción. El ARNm de longitud completa consiste en una secuencia no codificadora (conocida como la región 5' no traducida), la secuencia codificadora y una región 3' no traducida. Después de la transcripción la mayoría de los ARNm se modifican por la adición de 100 a 200 residuos de adenina, la cola poli (A). Una enzima rompe el ARNm en una secuencia AAUAAA en la región no traducida 3' y se agregan bases adenina sucesivas alrededor de 20 pares de bases hacia abajo de la señal AAUAAA. La cola sirve para mantener la estabilidad del ARNm y facilitar su salida del núcleo, y también parece participar en el inicio de la traducción.
Antes de salir, debe ocurrir un paso importante en el procesamiento del ARNm, la separación de los intrones. A diferencia de los genes procariocíticos, en los que existe una correspondencia uno a uno entre la secuencia de bases y la secuencia de aminoácidos de una proteína, la secuencia de codificación de los genes eucariotes, denominados exones, está interrumpida por secuencias no codificadoras denominadas intrones. Al inicio se transcribe toda la longitud del gen, incluyendo los intrones y los exones. Después se extraen los intrones y los exones se unen entre sí por medio de un complejo proceso mediado por enzimas que tiene lugar en el núcleo. Por último, el ARNm maduro es estraído al citoplasma para su traducción a proteína. La función de los intrones se desconoce, aunque se especula que pueden ser remanentes de partes arrastradas del genoma durante la evolución. La secuencia de bases de los exones tiende a estar muy conservada, pero las secuencias de base de los intrones no están sujetas a presión de selección, excepto las secuencias de bases al final de los intrones y en los sitios internos requeridos para su separación normal. Estos sitios son vulnerables a sufrir mutaciones que pueden alterar el proceso de separación (ver adelante).
El número de intrones en los genes varía mucho. Por ejemplo, el gen de la beta-globina incluye dos intrones y tres exones. El gen de la distrofina, una proteína del citoesqueleto muscular que se afecta en la distrofia muscular de Duchenne contiene 79 exones.1 Por lo general los intrones son mucho más largos que los exones, lo que indica que una porción significativa del ADN en el genoma no codifica información genética. Los tamaños de los genes varían con el número de exones e intrones desde algunos miles a varios cientos de miles de pares de bases. El gen más grande conocido es el de la distrofina, que incluye más de 2.5 millones de pares de bases.
El proceso de separación no produce un ARNm idéntico para cada gen en todos los tipos de células. Un transcrito puede dividirse en forma diferente en células diferentes y en momentos diferentes del desarrollo. Por lo tanto, en algunas células un exón específico es separado del ARNm junto con los intrones, en otras este exón es incluído en el ARNm. Además, pueden existir secuencias promotoras localizadas dentro de los genes que son reconocidas como sitios de inicio de la transcripción en ciertos tipos celulares, pero no en otros. Estos dos fenómenos permiten que una sola unidad de transcripción codifique diferentes formas de la misma proteína, aumentando en forma sustancial la diversidad de proteínas codificadas en el genoma. Este es el motivo por el que alrededor de 80,000 genes codifican para un número mucho mayor de proteínas.
La traducción, el proceso de ensamble de la proteína, tiene lugar en los ribosomas del citoplasma. La proteína es ensamblada como una cadena de aminoácidos, que se agregan uno por uno. Cada aminoácido es especificado por un triplete de bases en el ARNm (un codón) que corresponde a un triplete en el gen. El primer aminoácido siempre es metionina, codificado por el codón AUG (en algunas proteínas esta metionina es eliminada después). Existen 61 posibles codones que especifican 20 aminoácidos, de modo que muchos aminoácidos son determinados por más de un codón, y se dice que el código genético es degenerado [ver tabla 1]. La información del gen es traducido cuando cada codón en el ARNm correspondiente se une a la moléducla de ARNt que contiene una tripleta complementaria de bases (un anticodón). Cada ARNt está unido en forma covalente a un aminoácido específico. La maquinaria enzimática del ribosoma crea una unión peptídica entre los aminoácidos adyacentes atraídos hacia la cercanía del ribosoma al unirse los ARNt respectivos al ARNm. Los dobleces y modificación (v.gr., glucosilación) de la proteína comienzan incluso antes de que se termine la traducción. El codón final en una secuencia codificadora es un codón de final, sea UGA, UAA o UAG, que indica el final de la traducción y permite que la proteína sea liberada del ribosoma.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Es importante reconocer que una parte sustancial de los tres billones de pares de bases que constituyen el genoma humano no codifican. Además de los intrones, existen otros segmentos importantes del genoma que no codifican proteínas o ARN. Muchos de estos segmentos incluyen secuencias que se repiten muchas veces, y algunas tienen un papel estructural en la organización del genoma en sí. Las repeticiones de secuencias simples pueden contener miles de copias y tienden a acumularse en regiones cercanas a los centrómeros del cromosoma (ver adelante). Otras secuencias repetidas están dispersas en el genoma, y su función se desconoce.
GENOMA MITOCONDRIAL
Además de los genes en el núcleo celular, existe ADN fuera del núcleo, en la mitocondria. Cada mitocondria contiene múltiples copias de una molécula circular de ADN de doble cadena formada por 16,569 pares de bases.2 Aunque la mayoría de las proteínas en las mitocondrias son codificadas por el genoma nuclear, el genoma mitocondrial incluye las secuencias de codificación para 13 subunidades de proteínas que participan en la fosforilación oxidativa, un grupo completo de ARNt mitocondriales y el ARNr mitocondrial. El ADN mitocondrial se transcribe dentro de la mitocondria (usando una ARN polimerasa codificada por los genes nucleares) y es traducida en la mitocondria (usando ARNt y ARNr mitocondrial pero proteínas ribosomales codificadas por genes nucleares). Los genes mitocondriales, como los genes procarióticos de los que derivan, no tienen intrones. Las mutaciones en el ADN mitocondrial pueden alterar la fosforilación oxidativa, causando trastornos del metabolismo energético.
Todo el complemento del ADN mitocondrial en el huevo fertilizado es contribución del oocito, las mitocondrias de los espermatozoides se pierden en el momento de la fertilización. Los rasgos genéticos codificados en el ADN mitocondrial son, por lo tanto, trasmitidos de la madre a todos sus hijos (herencia materna). Este patrón de trasmisión se observa en trastornos energéticos que son resultado de mutaciones en el ADN mitocondrial.
Otra propiedad de la herencia del ADN mitocondrial importante en relación con la patogenia de los padecimientos mitocondriales consiste en la segregación de las mitocondrias en el momento de la mitosis.3 Cada mitocondria contiene múltiples copias de ADN y existen muchas mitocondrias en cada célula. Las mitocondrias se reparten al azar en las dos células hijas durante la mitosis, en contraste con la segregación ordenada de los genes del genoma nuclear. Cuando surge una mutación del ADN mitocondrial, suele ocurrir en una célula que contiene una mezcla de ADN mutante y nativo. La segregación pasiva de las mitocondrias durante la mitosis causa una distribución aleatoria de las moléculas de ADN mutante y nativo en las células hijas. Por azar algunas células pueden recibir un mayor número de mitocondrias con la mutación, mientras que otras pueden recibir muy pocas. Esto produce heteroplasmía, esto es, que la proporción de ADN mitocondrial mutado y nativo en los diversos tejidos es diferente, lo que puede causar diversos efectos clínicos en los miembros de la misma familia, a pesar de que compartan una mutación mitocondrial.
Estructura y tinción de los cromosomas
Aunque los genes están colocados en un orden lineal a lo largo de la cadena de ADN, el genoma humano se divide en 23 pares de moléculas de ADN denominadas cromosomas. Cada cromosoma consiste en una sola cadena continua de ADN que incluye varios miles de genes. En el núcleo el ADN existe como una estructura muy organizada acoplada con múltiples proteínas. La unidad básica de organización es el nucleosoma. El nucleosoma consiste en un centro de ocho proteínas histonas (dos copias cada una de H2A, H2B, H3 y H4) alrededor de las cuales se envuelve el ADN (con una longitud de alrededor de146 pares de bases). Los nucleosomas adyacentes están conectados como cuentas en un cordel por un ADN de unión de hasta 80 pares de bases de longitud. La fibra de cromatina que se forma tiene alrededor de 11 nm de grosor. Esta se dobla a su vez en una estructura semejante a un solenoide unida con histona H1 y otras proteínas no histonas. Esta estructura se desdobla durante la transcripción. Durante la división celular la cromatina se vuelve muy compacta y enrollada, condensándose para formar la estructura conocida como cromosoma mitótico.
La célula nucleada somática humana normal contiene 46 cromosomas, 23 heredados de cada padre. Existen 22 pares de cromosomas no sexuales, conocidos como autosomas, y dos cromosomas sexuales, un cromosoma X y uno Y en los varones y dos cromosomas X en las mujeres. La célula germinal contiene un grupo haploide de autosomas y uno de los cromosomas sexuales (un X en los oocitos y un X o un Y en los espermatozoides) Se usa una nomenclatura estándar para describir los cromosomas y las alteraciones cromosómicas.4 El cariotipo masculino normal se describe como 46,XY y el femenino normal como 46,XX.
Los cromosomas se observan con más facilidad en el estado muy compacto que ocurre durante la división celular. Las dos cadenas replicadas de ADN aparecen como dos cromátides unidas junto al centrómero. El centrómero divide al cromosoma en un brazo corto (p, por petit) y uno largo (q). La región centromérica contiene un bloque de material que se tiñe en forma muy intensa conocida como heterocromatina centromérica. Esta área consiste de muchas secuencias repetidas que no son genéticamente activas. Los extremos de los cromosomas se conocen como telómeros. Los telómeros incluyen secuencias especializadas que se requieren para la replicación del cromosoma.
La identificación de los cromosomas se facilita por patrones de bandeo específicos que se descubren por medio de protocolos especiales de tinción. Estos incluyen pretratamientos, como digestión con proteasa, antes de la tinción con Giemsa o colorante fluorescente. Cada par homólogo de cromosomas tiene un tamaño y forma característicos que se aprecia como una estructura fina reproducible y que se tiñe [ver figura 6]. Recientemente, la técnica de hibridización fluorescente in situ (FISH, por sus siglas en inglés, n. del t.) ha hecho posible teñir secuencias específicas del ADN en un cromosoma (ver adelante).

|
| Figura 6 |
| Cariotipo humano |
El análisis clínico del número y estructura de los cromosomas suele realizarse en las células en división. Estas células son principalmente linfocitos de sangre periférica que se estimulan para dividirse en cultivo por medio de lectina fitohemaglutinina. Los fibroblastos de piel, las células de líquido amniótico, las células de las vellosidades coriónicas o las células tumorales con división espontánea pueden también usarse para el análisis cromosómico.
La división de las células somáticas ocurre por el proceso de mitosis [ver figura 7]. La replicación del ADN ocurre durante la fase S (de síntesis) del ciclo celular, varias horas antes de que se inicie la división celular. La mitosis comienza con la disolución del núcleo y el comienzo de la condensación de los cromosomas (profase). Los cromosomas se alinean entre los dos polos del huso mitótico durante la metafase. Las cromátides se separan durante la anafase y se forma un núcleo en cada célula hija durante la telofase. La división del citoplasma por medio del proceso de citocinesis completa la mitosis.
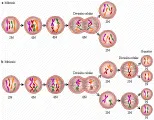
|
| Figura 7 |
| Mitosis y meiosis celular |
Las células germinales sufren un proceso de división especial denominado meiosis, en la que las células germinales primordiales diploides son reducidas a un estado haploide [ver figura 7]. La replicación cromosómica ocurre antes de la meiosis. Durante la profase de la primera división meiótica los cromosomas homólogos se aparean (sinapsis) y pueden intercambiar segmentos a través del proceso de recombinación (entrecruzamiento). Las copias mezcladas de genes específicos sin cambiar el orden genético global del cromosoma producen un incremento sustancial en la diversidad genética. La meiosis ocurre en un periodo de varios días en los espermatocitos. Sin embargo, los oocitos comienzan la meiosis durante la vida embrionaria y permanecen en la primera división meiótica de la profase hasta la ovulación (i.e., durante muchos años).
La primera división meiótica provoca la separación de los cromosomas homólogos en dos células hijas. Estas células sufren otra división completa, sin síntesis de ADN, para producir cuatro células germinales haploides. En el varón, estas cuatros células son los espermatozoides. En la mujer, la primera división meiótica produce un oocito primario y un primer cuerpo polar. La segunda división meiótica del oocito se completa solo después de la fertilización y produce un segundo cuerpo polar además del pronúcleo del oocito.
Se conoce como no disyunción al hecho de que las dos cromátides no pasen en forma independiente a las células hijas durante la mitosis o la meiosis. Esto provoca la producción de una célula con una copia extra de un cromosoma y una sin la copia. La fertilización de una célula germinal con un cromosoma extra da lugar a una trisomía, mientras que la fertilización de una célula germinal con un cromosoma ausente origina una monosomía. Las células monosómicas para algún autosoma generalmente no son viables, pero sí lo son las trisomías de los cromosomas 13, 18 y 21. Si ocurre no disyunción durante la mitosis durante el desarrollo embrionario, existirá una mezcla de células normales y trisómicas en el embrión (suponiendo que las células monosómicas no son viables). A esto se conoce como mosaicismo. Los efectos clínicos del mosaicismo suelen ser menos severos que los de las trisomías presentes en todas las células. Para algunas trisomías autosómicas, como la trisomía 8, el embrión es viable solo si la trisomía se presenta en forma de mosaico.
La causa de la nodisyunción se desconoce. Ejemplos poco frecuentes de asociación familiar de personas con trisomía sugieren la existencia de factores genéticos predisponentes.5 El único riesgo bien documentado para la nodisyunción es la edad materna. La frecuencia de no disyunción claramente aumenta en función de la edad materna, con un incremento constante a partir de la mitad de los 30 años.6 Los estudios sobre trisomía 21 indican que la nodisyunción ocurre con más frecuencia en la primera división meiótica materna.7 Esto puede ser una consecuencia del periodo tan prolongado de la meiosis I en la mujer. Si la sinapsis de un par de cromosomas homólogos se rompe durante este periodo, los dos cromosomas pueden separarse en forma aleatoria hacia las células hijas, causando no disyunción. Estudios sobre los efectos de la edad paterna no han revelado mas que, si acaso, una correlación muy débil con la no disyunción.8 Debido a la relación entre la edad materna y la frecuencia de trisomía, es práctica común ofrecer diagnóstico prenatal para detectar trisomías fetales a las mujeres que tendrán 35 o más años de edad en el momento del parto.
Proyecto genoma humano
Durante la mayor parte de este siglo, los genes se conocieron principalmente porque causaban fenotipos anormales. Con excepción de los genes de las moléculas de globina y algunas enzimas, las secuencias de los genes humanos y las proteínas que codifican seguían siendo un misterio. Además, el catálogo de rasgos genéticos humanos contenía solo algunos pocos miles de los alrededor de 80,000 genes que constituyen el genoma. El Proyecto genoma humano ha sido posible gracias al desarrollo de tecnologías que permiten aislar segmentos individuales del genoma y estudiarlos con detalle. En esta sección se revisan en forma breve estos adelantos técnicos dentro del contexto de su aplicación al estudio del genoma humano y la información que se está acumulado sobre la huella digital genética para el organismo humano.
ADELANTOS TECNOLOGICOS
La base para el estudio de los genes humanos a nivel molecular es la tecnología de ADN recombinante. Esta se usa para aislar fragmentos de ADN, cuyo tamaño varía de algunos cientos a más de un millón de pares de bases. Los fragmentos se recombinan con vectores de ADN que son capaces de tener replicación autónoma dentro de las células procarióticas. Esto permite obtener fragmentos en grandes cantidades para su análisis, incluyendo la determinación de secuencias nucléotidas.
La creación de moléculas recombinantes depende especialmente de un grupo de endonucleasas de restricción, que son enzimas que reconocen y rompen el ADN en sitios específicos (i.e., definidos por una secuencia especial de bases). Los sitios de reconocimiento con secuencia específica suelen consistir en cuatro a ocho nucleótidos y suelen manifestar simetría palindrómica [ver figura 8]. Incluso un solo cambio de base en el sitio de reconocimiento puede impedir la ruptura; por lo tanto, una endonucleasa de restricción específica cortará un segmento determinado de ADN en forma muy reproducible. En muchos casos las endonucleasas de restricción dejan fragmentos libres sin sus correspondientes pares de bases en los extremos de la molécula de ADN. Por lo tanto, dos fragmentos cortados con la misma enzima pueden formar moléculas híbridas que pueden unirse en forma covalente con una ADN ligasa. Las endonucleasas de restricción se aislan de las células bacterianas, en donde su función consiste en restringir el crecimiento viral al romper el ADN viral cuando éste entra en la célula. Estas enzimas reciben nombres cortos que corresponden a las bacterias de las que derivan (v.gr., EcoRI, que deriva de Escherichia coli).
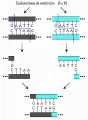
|
| Figura 8 |
| Endonucleasa de restricción EcoRI |
Un experimento básico de clonación del ADN consiste en generar fragmentos de ADN para ser clonados y ligar estos fragmentos a un vector de ADN [ver figura 9]. Se usan diferentes vectores, dependiendo del tamaño del ADN que va a clonarse. Los plásmidos bacterianos acomodan pequeños fragmentos de ADN, hasta 10,000 pares de bases (10kb). Los vectores bacteriófagos pueden clonar fragmentos tan largos como de 23 kb. Los cósmidos, que no se encuentran en la naturaleza, son vectores construidos que pueden comportarse como híbridos de los plásmidos y bacteriófagos y que pueden acomodar fragmentos de alrededor de 50 kb. Los cromosomas artificales de levaduras tienen la mayor capacidad de clonación, varios cientos de miles a un millón de pares de bases.
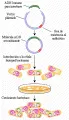
|
| Figura 9 |
| Clonación del ADN |
Pueden usarse varias fuentes de ADN para obtener el material para la clonación génica. El ADN genómico total puede romperse en pequeños fragmentos en forma mecánica o por medio de endonucleasas de restricción. Los fragmentos se insertan en forma aleatoria en un vector de clonación. Entonces se genera una librería genómica, en la que cada colonia bacteriana que resulta incluye un fragmento separado de ADN genómico. Otra fuente común de material para clonación es el llamado ADN complementario (ADNc). El ADNc se produce usando la enzima transcriptasa inversa para copiar el ARN a ADN. La transcriptasa inversa se aisla de virus tumorales ARN, en donde copia el genoma viral ARN en una doble cadena de ADN. Las secuencias del ADNc también pueden clonarse. Sin embargo, la biblioteca del ADNc no representa el ADN genómico total, sino solo los fragmentos que se expresan como ARN en un tipo celular determinado. Si el ADNc se forma a partir de un ARNm totalmente procesado, la secuencia clonada incluirá solo la secuencia codificadora y no los intrones. Las bibliotecas de ADNc son útiles para determinar la secuencia de codificación de un gen e identificar los transcritos que se elaboraron en un tipo celular específico.
El ADN clonado puede analizarse de diferentes maneras. Se han desarrollado técnicas químicas para la determinación rápida de la secuencia de bases. El enfoque más empleado, desarrollado por Sanger, Nicken y Coulson,9 se practica en la actualidad con un aparato automático capaz de secuenciar miles de bases por día.
El ADN clonado, en una sola cadena, se unirá a una molécula de ADN o ARN que incluya una secuencia de bases complementaria. A esta unión se le conoce como hibridización. Por lo tanto, un fragmento clonado puede ser marcado con radioactividad o fluorescencia y usarse como una sonda para buscar secuencias homólogas en una muestra de ARN o de ADN de cualquier origen. El análisis de Southern blot es una técnica que consiste en separar los fragmentos de ADN de acuerdo por su tamaño por electroforesis, inmovilizando los diferentes segmentos en una membrana e hibridizándolos con un fragmento clonado y marcado.10 El análisis de Northern blot usa el mismo principio pero con ARN unidos a la membrana en lugar de ADN. La FISH usa el mismo principio, pero el ADN dentro de una célula o en un cromosoma es tratado sobre un portaobjetos de vidrio.
Otra técnica importante es la reacción en cadena de la polimerasa, que se usa para amplificar la cantidad de un fragmento de ADN por medio de ciclos repetidos de síntesis enzimática de ADN [ver figura 10]. Un par de pequeños oligonucleótidos sintéticos (20 a 30 bases) (denominados cebadores) se unen a las cadenas opuestas del fragmento de ADN blanco. Estos cebadores sirven como puntos de inicio para la replicación del ADN. El proceso incluye 20 a 30 ciclos de unión sucesiva de cebadores, su replicación y separación de los productos en cadenas simples. En cada ciclo los productos del ciclo previo sirven como un templete, causando un incremento exponencial en la cantidad de ADN formado por los cebadores. Esto permite la amplificación a gran escala del segmento de ADN, que entonces puede ser estudiado.

|
| Figura 10 |
| Reacción en cadena de la polimerasa |
MAPEO GENETICO
El esfuerzo para localizar cada uno de los genes humanos ha requerido la construcción de un mapa de alta resolución del genoma [ver figura 11]. Para este propósito se han generado fragmentos clonados del ADN para usarse como marcadores genéticos. Estos fragmentos incluyen segmentos polimórficos, secuencias de bases que varían de persona a persona. Estas secuencias se detectaron primero como fragmentos polimórficos de longitud restringida (FPLR),11 porque los cambios en la secuencia de bases alteran la capacidad de la endonucleasa de restricción para cortarlos en un sitio determinado. Los FPLR fueron analizados cortando el ADN por medio de una endonucleasa de restricción apropiada y realizando un estudio de Southern blot con una sonda para la región del polimorfismo. Se identificaron alelos como bandas de diferentes tamaños en el ensayo. Otro tipo de polimorfismo que se usa con más frecuencia en la actualidad en los estudios de mapeo genético es la repetición simple de la secuencia polimórfica. Es frecuente encontrar fragmentos de repeticiones simples, como el dinucleótido CACACA...., dentro y alrededor de los genes, y el número exacto de repeticiones suele ser polimórfica. Los alelos pueden detectarse usando RCP para amplificar un segmento de la región que contiene la repetición y después midiendo el tamaño de los productos por electroforesis.

|
| Figura 11 |
| Creación de un mapa genético |
Estas variantes en las secuencias de bases pueden servir como simples rasgos mendelianos y permitir que los alelos sean rastreados dentro de una familia para determinar si los alelos se segregan junto con un rasgo o trastorno genético. Si un marcador en particular se localiza cerca del rasgo que será mapeado, se segregará unto con el rasgo en una familia, y se dirá que el rasgo y el marcador están ligados entre sí. El entrecruzamiento entre el marcador y el locus del rasgo altera esta segregación. Mientras más cerca entre sí se encuentren en marcador y el rasgo, menor será la probabilidad de que se separen durante la recombinación genética. La distancia genética entre el rasgo y el marcador se mide como una función de la frecuencia de recombinación. La herramienta estadística empleada con más frecuencia es la calificación de lod (lod es el acrónimo para logaritmo de momios). Los datos provenientes de una familia se usan para calcular los momios relativos de que el marcador y el rasgo que se están mapeando estén ligados entre sí o no. La relación de momios se convierte a un logaritmo para facilitar el acúmulo de datos de múltiples familias al sumar los logaritmos en lugar de multiplicar la relación de momios. Por conveniencia, el marcador y el rasgo están ligados entre sí si la calificación lod es mayor de 3 (i.e., 1,000:1 momios favoreciendo la unión contra la no unión). Se excluye la unión si la calificación de lod es menor de -2 (i.e., 100:1 momios favoreciendo la no unión sobre la unión). En la actualidad el mapeo genético incluye marcadores polimórficos espaciados alrededor de cada 1 cM (centimorgan, que corresponde a la longitud del ADN entre marcadores dentro de los que existe un porciento de probabilidad de entrecruzamiento).12 Esta densidad es suficiente para asegurar que virtualmente cualquier rasgo genético puede ser mapeado si se cuenta con un ADN del pedigree de tamaño adecuado.
La identificación de los genes individuales requiere la correlación del mapa genético con un mapa físico del ADN, que es un mapa de las localizaciones de los marcadores identificables (v.gr., sitios de corte por las endonucleasas de restricción o marcadores de repetición CA).13 El mapa físico consiste en segmentos muy grandes del genoma clonados en vectores como los cromosomas artificiales de levaduras. El mapa físico se alínea con el mapa genético localizando los marcadores polimórficos a lo largo del ADN clonado. El mapa físico permite al investigador relacionar los segmentos clonados del ADN con un locus de interés que se ha colocado en el mapa genético. Para encontrar el gen en sí se requiere investigar las secuencias expresadas a lo largo del ADN clonado en la región y después examinar las secuencas buscando las propiedades esperadas en el gen blanco.14 En los casos en que se encuentran los genes responsables de la enfermedad, el proceso incluye buscar mutaciones en los genes de las personas afectados.
Durante la década pasada se han identificado varios genes con base en su localización dentro del genoma,15 lo que se conoce como genética reversa o, en forma más apropiada, clonación posicional. Esta técnica ha hecho posible clonar genes de trastornos cuya base bioquímica se desconoce. La identificación de los genes de padecimientos como la distrofia muscular de Duchenne,16 la fibrosis quística,17 y la neurofibromatosis tipo 118,19 por este método, ha facilitado el mayor conocimiento de su patogenia y, en consecuencia, de las observaciones clínicas. Antes de su descubrimiento por clonación posicional se ignoraba la existencia de las proteínas que participan en estos trastornos.
EL PROYECTO DEL GENOMA HUMANO Y LA BIOLOGIA HUMANA
El Proyecto del genoma humano va a la vanguardia de una revolución en el conocimiento de la biología humana que no dejará de abarcar a ningún área de la medicina. La identificación de todos los genes humanos permitirá estudiar en detalle la secuencia de los eventos responsables del desarrollo normal y revelará los mecanismos que ocasionan alteraciones en el desarrollo. También aclarará la patogenia de los trastornos genéticos. En la actualidad ya se están desarrollando nuevos métodos de diagnóstico basados en la detección de mutaciones en el ADN. El conocimiento de los mecanismos celulares de la enfermedad impulsará nuevos métodos de tratamiento que pueden incluir manipulación directa de genes, como la reparación de los genes defectuosos.
Fisiopatología genética
La variación en la secuencia de bases entre una persona y otra es la base de la diversidad genética en la población. La composición genética de un locus genético en particular se conoce como genotipo, y las manifestaciones físicas que resultan de la expresión de un gen en particular constituyen el fenotipo. Algunos cambios de bases no afectan la secuencia codificadora de las proteínas, o tienen un impacto mínimo en la estructura y función proteica. Otros cambios de bases pueden abolir por completo la síntesis de una proteína o alterar en forma sustancial sus propiedades químicas. Los efectos fenotípicos son también variables, siendo desde no detectables hasta manifestaciones de un trastorno genético severo o incluso mortal. La mayoría del conocimiento sobre la patología genética deriva del estudio de cambios en genes individuales que causan fenotipos obvios y severos o de cambios que pueden alterar grandes segmentos cromosómicos. Aunque clínicamente importantes, los trastornos estrictamente genéticos son poco frecuentes. Los factores genéticos contribuyen a la etiología de padecimientos mucho más comunes, pero estas enfermedades suelen ser resultado de la acción conjunta entre múltiples genes y factores ambientales. En esta sección se revisarán los mecanismos por los que los cambios genéticos influyen en la expresión genética y tienen efectos patológicos.
MUTACION
Un cambio en la estructura de un gen o un grupo de genes se conoce como mutación. El más sutil de estos cambios en el ADN es la alteración de una sola base, que sin embargo puede tener importantes efectos sobre la expresión de la proteína correspondiente. El extremo opuesto incluye cambios que afectan a grandes bloques de genes o cromosomas enteros.
Los cambios cromosómicos incluyen poliploidía (uno o más juegos extra completos de cromosomas), aneuploidía (ganancia o pérdida de uno o más cromosomas individuales), traslocaciones (intercambio de segmentos entre dos o más cromosomas) y deleción, duplicación, inserción o inversión dentro de un solo cromosoma. Algunos cambios cromosómicos alteran la estructura del cromosoma pero no causan ganancia o pérdida del material genético. Pueden no tener efectos fenotípicos y existen como variantes en la población general. En casos raros el sitio de ruptura de un cromosoma para un rearreglo balanceado puede interrumpir un gen, causando el fenotipo de un trastorno de un solo gen. Por ejemplo, algunas personas con distrofia muscular de Duchenne ligada al X tienen rearreglos en el cromosoma X que alteran el gen de la distrofina.20
En la mayor parte de los casos, las ganancias o pérdidas de grandes bloques de material cromosómico son letales y producen un aborto. Solo una minoría de los infantes llega a término, aunque con alteraciones importantes del desarrollo tanto físico como neurológico. Las alteraciones cromosómicoas ejercen sus efectos fenotípicos al modificar el balance en el nivel de expresión genética. Al tener tres copias de un gen existen niveles más altos de transcripción, lo que, por lo menos en algunos genes, altera la función. Se han hecho intentos por mapear los efectos fenotípicos asociados con ganancia o pérdida de regiones cromosómicas específicas. Incluso los fenotipos complejos como el del síndrome de Down, que se debe a la trisomía 21, se han analizado de esta forma.21 Al completar el mapa genético de cada cromosoma, se identificarán los genes asociados con los fenotipos particulares y podrán estudiarse los mecanismos por los que los genes causan los problemas del desarrollo.
Las deleciones cromosómicas que son visibles al microscopio incluyen más de un millón de pares de base, lo que equivale a docenas de genes. Con FISH pueden detectarse deleciones sutiles que no se observan con microscopía de luz.22 Estas deleciones pueden producir fenotipos característicos conocidos como síndromes de microdeleción. Muchos de estos síndromes se atribuyen a los efectos acumulativos de deleción de los genes contiguos. Por ejemplo, en el síndrome WAGR (por tumor de Wilms, aniridia, alteraciones genitourinarias o gonadoblastoma y retraso mental), cada componente del síndrome es resultado de la deleción de un gen en particular.23 Pueden ocurrir diversas manifestaciones del síndrome dependiendo de la magnitud de una deleción en particular. Otros síndromes puede ser también resultado de la deleción de un solo gen. Por ejemplo, en algunos casos el fenotipo complejo del síndrome de Alagille (asociado con atresia biliar intrahepática, estenosis pulmonar y anomalías faciales) es causado por deleción parcial del cromosoma 20, y en otros casos la enfermedad ocurre por mutaciones dentro de un solo gen en esta región cromosómica.24-26
El estudio de las deleciones cromosómicas ha revelado un mecanismo importante de la regulación genética, conocido como impresión genómica, en el que existe expresión desequilibrada de la copia materna y paterna de un gen [ver figura 12].27 Solo un subgrupo de genes sufren impresión genómica. En éstos solo se expresa la copia materna o paterna, y la huella o impresión puede cambiar en cada generación. La deleción de un gen de este tipo causará diversos efectos fenotípicos, dependiendo cual copia se pierda. La pérdida de la secuencia expresada puede tener efectos fenotípicos importantes, mientras que la deleción de la secuencia no expresada puede no tener consecuencias hasta que el gen pasa a la siguiente generación, cuando quizá deberá expresarse.
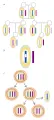
|
| Figura 12 |
| Loci impresos |
El síndrome de Prader-Willi y el síndrome de Angelman son dos trastornos causados por cambios en la expresión de genes impresos.28 Ambos síndromes causan deterioro del desarrollo, pero por lo demás son muy diferentes. Los signos y síntomas del síndrome de Prader-Willi incluyen una apariencia facial característica y un trastorno de la alimentación que causa obesidad. En el síndrome de Angelman existen características dismórficas, movimientos mal coordinados y crisis convulsivas. Ambos síndromes se asocian con deleciones en la región proximal del brazo largo del cromosoma 15; la deleción de las secuencias paternas causa el síndrome de Prader-Willi, mientras que la deleción de las secuencias maternas produce el síndrome de Angelman.29 Un solo gen es responsable de muchos de los efectos del síndrome de Angelman,30,31 pero el gen o genes responsables del síndrome de Prader-Willi no ha sido identificado aún.
Algunos casos de síndrome de Prader-Willi o de Angelman no son causados por deleción sino por un fenómeno conocido como disomía unipaterna. En estos casos las dos copias del cromosoma 15 se heredan del mismo padre. Debido a que existen genes impresos tanto paternos como maternos en el cromosoma 15, las dos copias del cromosoma 15 que derivan del mismo padre provocan la falta de expresión de algunos genes impresos y, en consecuencia, el síndrome de Prader-Willi o de Angelman. Ocurre disomía unipaterna cuando un cromosoma trisómico sufre un evento secundario de no disyunción, restableciendo la disomía para el cromosoma pero produciendo una disomía unipaterna si las dos copias derivan del mismo padre. No todos los cromosomas tienen genes impresos, pero los que los tienen originan alteraciones fenotípicas cuando ocurre disomía unipaterna.32
Las mutaciones dentro de genes individuales se analizan mejor desde el punto de vista del impacto sobre la función del gen [ver figura 13]. La mutación genética puede causar reducción o ausencia de la transcripción, producción de un producto truncado, duplicación o deleción de aminoácidos en la proteína o sustitución de un aminoácido por otro dentro de la proteína. Las mutaciones pueden ocurrir dentro de los exones o los intrones. Las mutaciones en los intrones causan efectos fenotípicos al alterar la separación del ARN.
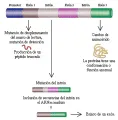
|
| Figura 13 |
| Efectos de las mutaciones en la expresión del gen |
Las mutaciones en la región promotora causan niveles aberrantes de transcripción genética. Es posible que el promotor se pierda o que los cambios de una sola base inhiban la unión de la polimerasa de ARN o de las moléculas reguladoras. Las mutaciones en el promotor son responsables de algunos casos de talasemia beta: los cambios en la secuencia ocasionan una transcripción deficiente del gen de beta-globina.33 Las mutaciones del promotor pueden también causar expresión constitutiva de un gen. Estas mutaciones se han demotrado en procariocitos. La activación de un gen por alteración en la secuencia promotora es un mecanismo importante de algunos cambios genéticos somáticos que originan neoplasias (ver adelante).
Las mutaciones que crean nuevos codones de terminación o alteran el marco de lectura de la transcripción provocan la producción de un producto truncado. Los nuevos codones de terminación pueden originarse por el cambio de una sola base en una tripleta. Por ejemplo, la tripleta CGA codifica al aminoácido arginina. El cambio de C a T produce un codón de terminación TGA. Este es un tipo especialmente frecuente de mutación. Las bases de citosina seguidas por guanina (dinucleótidos CpG) suelen estar metiladas , y la deaminación de la citosina metilada origina timina.
El desplazamiento del marco de lectura se produce por la inserción de material en el exón o deleción de material de un exón en un múltiplo diferente a tres bases. La alteración del marco de lectura produce una secuencia sin sentido que inevitablemente genera un codón de terminación. La inserción, deleción o duplicación de una o dos base produce desplazamiento del marco de lectura. Esto puede ocurrir también cuando todos los exones o grupos de exones sufren deleción o se duplican. Los exones no necesariamente terminan en la tercera posición de un codón, pero el marco de lectura se conserva mientras el siguiente exón continúe en la posición en que se quedó el exón previo.34 Cuando un exón o grupo de ellos sufre deleción, los dos exones que limitan la porción perdida serán yuxtapuestos en el ARNm. Si el marco de lectura se conserva, la proteína será más corta, pero por lo demás se traducirá en su totalidad. Si el marco de lectura no se conserva, se producirá una proteína truncada. Ocurre un proceso semejante si un exón o grupo de exones se duplica, la yuxtaposición de los exones en el ARNm puede o no conservar el mismo marco de lectura. Las mutaciones de los intrones pueden causar que se brinquen exones (ver adelante) y provocar desplazamiento del marco de lectura por el mismo mecanismo.
Las inserciones o deleciones de múltiples aminoácidos o las sustituciones de los mismos pueden causar una proteína estructuralmente aberrante. Sin embargo, debido a la degeneración del códico genético, algunas sustituciones de base no cambian del todo la secuencia de aminoácidos; a éstas se les denomina mutaciones silenciosas. Otras sustituciones de base causan la incorporación de aminoácidos con propiedades químicas semejantes a los nativos, por lo que no existe impacto, o este es mínimo, sobre la estructura o función de la proteína; éstas se denominan mutaciones conservadoras. Ni las mutaciones silenciosas ni las conservadoras están sujetas a presión selectiva y pueden existir como variantes de secuencias en la población general (polimorfismos). Los cambios de base que causan sustitución de aminoácidos con diferentes propiedades químicas pueden alterar en forma importante las propiedades de una proteína (v.gr., un aminoácido con carga, como la arginina, es sustituido por uno no polar, como la leucina). Con algunas sustituciones de aminoácidos la proteína se vuelve inestable y es degradada en la célula. Con otras se produce una proteína intacta pero sin actividad enzimática o sin que pueda realizar un papel estructural normal.
Las mutaciones dentro de los intrones pueden tener consecuencias al interferir con la formación del ARN. Aunque la secuencia de bases exacta de un intrón no está muy conservada, si se conservan un grupo de bases en los extremos 5' y 3' (donador y aceptor del empalme, respectivamente), ya que esta es la región dentro del intrón necesaria para el empalme normal. La región donadora siempre comienza con la secuencia GT, seguida de un grupo más variable pero generalmente conservado de bases (una secuencia de consenso). La región aceptora termina con AG, precedida de una secuencia de consenso. Las mutaciones en la región donadora o aceptora pueden causar que todo un exón sea excluido en la transcripción al ARNm (exón brincado) o pueden ocasionar la inserción de cierto material del intrón en la molécula madura de ARNm. Esto último ocurre si se presenta un donador o aceptor denominado críptico dentro del intrón. Esta es una secuencia que puede servir como donadora o aceptora si la secuencia real se altera. El brinco de un exón tiene el mismo impacto que su deleción. La inserción de un intrón suele causar desplazamiento del marco de lectura o producir un codón de terminación que provoca el fin prematuro de la traducción.
En los años recientes se ha descubierto un mecanismo adicional de mutación genética. Este es la expansión de repeticiones de tripletas, que es responsable de varios trastornos neurológicos.35 Esta mutación ocurre en genes que incluyen segmentos de una tripleta de bases que se repite múltiples veces. La expansión de la repetición puede ocurrir en la región 5' no traducida, como en el gen FNR1, que participa en el síndrome del X frágil (retraso mental ligado al X), en la región 3' no traducida, como en el gen de la distrofia miotónica, en la secuencia codificada, como en el gen de la enfermedad de Huntington, o en un intrón, como en el gen de la ataxia de Friederich. El número exacto de copias de la tripleta puede diferir de persona a persona en la población, pero suele ser estable de generación en generación. La expansión del número de repeticiones por arriba de un umbral causa la expresión genética anormal. Los trastornos de repetición de tripletas sufren un fenómeno conocido como anticipación, en el que el padecimiento se vuelve más severo o tiene un inicio más temprano de una generación a la siguiente.36 La tripleta expandida es inestable y, por lo tanto, puede expanderse de generación en generación. La severidad clínica del padecimiento tiende a relacionarse con el tamaño de la región expandida. Hasta el momento todos los padecimientos identificados que resultan de la expansión repetida de tripletas afectan principalmente el sistema nervioso. Sin embargo, aún debe establecerse el rango completo de efectos fenotípicos de estos trastornos.
FISIOPATOLOGIA DE LA ENFERMEDAD GENETICA
Las consecuencias fisiológicas de alterar la cantidad o calidad de una proteína específica dependen de la función normal de la misma. Debido a que se han identificado más de decenas de miles de proteínas diferentes, cada vez se aclara más el repertorio completo de la función proteica.
En vista de que los humanos son organismos diploides, cada individuo tiene dos copias de cada gen (excepto para los genes de los cromosomas X y Y en los varones). Las dos copias de un gen en particular se denominan alelos. Con respecto a un cambio particular de secuencia de bases los dos alelos pueden ser idénticos, caso en el que el individuo se denomina homocigoto, o pueden diferir, caso en el que se le considera heterocigoto. En algunos casos ambos alelos tienen mutaciones, pero estas son diferentes. A esto se le conoce como herocigocidad compuesta.
Los primeros trastornos genéticos humanos fueron descrito por Archibald Garrod en la primera década del siglo XX. Los errores congénitos del metabolismo (término acuñado por Garrod) son causados por deficiencias enzimáticas que resultan de la mutación de los genes que codifican las enzimas o los cofactores que catalizan las reacciones químicas. Los efectos fenotípicos son consecuencia de la deficiencia de la producción de la reacción, el acúmulo de sustratos o ambos. La mayoría de los errores congénitos del metabolismo se heredan en forma recesiva, esto es, ambos alelos deben ser mutados para producir el efecto fenotípico. Esto se debe a la naturaleza catalítica de las enzimas. Debido a que solo pequeñas cantidades de proteína activa son suficientes para producir una reacción adecuada, los portadores heterocigotos de una mutación enzimática suelen ser fenotípicamente normales. Virtualmente cualquier tipo de mutación, sea que produzca deficiencia del producto o altere la secuencia de aminoácidos de una enzima, puede causar un error congénito del metabolismo.
Las mutaciones de los genes que codifican las proteínas estructurales pueden causar efectos fenotípicos en un estado heterocigoto y se dice que se heredan en forma dominante. Las proteínas estructurales son las que se secretan hacia la matriz extracelular, como la colágena, la elastina y la fibrilina, así como las que tienen un papel estructural dentro de la célula, como la globina. La mayoría de las proteínas estructurales participan en interacciones complejas proteína-proteína. Con frecuencia ocurren dos tipos de mutaciones en los padecimientos asociados con las proteínas estructurales. El primer tipo consiste en mutaciones que causan deficiencia del producto, como deleciones, mutaciones en el promotor y mutaciones que causan un péptido truncado no funcional. Por ejemplo, las deleciones provocan algunos tipos de talasemia, en especial las talasemias-alfa. Las mutaciones con deficiencia del producto suelen ocasionar un fenotipo leve en los heterocigotos. La deficiencia de una proteína estructural puede debilitar un tejido o disminuir el estado funcional del sistema, pero si cierta proteína normal se produce a partir del otro alelo el fenotipo será relativamente leve.
Por el contrario, las mutaciones que alteran la secuencia de aminoácidos de una proteína estructural suelen tener un efecto más severo, incluso en el estado heterocigoto, porque pueden alterar las interacciones proteína-proteína. Puede producirse una proteína estructural mutante en cantidades normales y afectar la integridad tisular. Estas mutaciones se conocen como dominantes negativas. Incluyen, por ejemplo, las mutaciones de la colágena que causan la osteogénesis imperfecta (huesos brillantes con fracturas múltiples). La colágena tipo I es una triple hélice con dos cadenas alfa (I)1 y una alfa (I)2. La mutación de un alelo para una cadena alfa (I)1 causa que tres cuartas partes de las moléculas de colágena tipo I sean anormales, lo que ocasiona una forma severa de osteogénesis imperfecta.37
Una tercera clase de trastornos es causada por mutación de genes que codifican proteínas que participan en las señales intra o extracelulares. Estas incluyen hormonas peptídicas, receptores de membrana, canales iónicos, proteínas del citoesqueleto, la cascada de proteínas que envía las señales recibidas en la membrana celular y factores de transcripción. Esta clase heterogénea de proteínas está sujeta a una amplia variedad de mutaciones. La alteración en la secuencia de aminoácidos puede ocasionar reducción en la función o activación constitutiva. Un ejemplo interesante es el gen ret, que codifica una tirosina cinasa de membrana.38 Algunas mutaciones del ret ocasionan activación constitutiva del receptor, lo que origina división celular aberrante en ausencia del ligando. Este tipo de mutación causa que la proteína ret funcione como un oncogén (un gen cuya actividad contribuye a las propiedades neoplásicas de una célula canderosa) y causa el trastorno de herencia dominante conocido como neoplasia endócrina múltiple tipo II. Por el contrario, un grupo diferente de mutaciones del ret causa inactivación del receptor. Cuando ambos alelos han mutado ocurre diferenciación aberrante de las células de la cresta neural en el intestino y se presenta la enfermedad de Hirschprung.
Los diferentes efectos producidos por las mutaciones en el gen ret ilustran el principio de la heterogeneidad genética. La heterogeneidad en el locus génico se refiere a mutaciones de diferentes genes que originan un fenotipo similar, con frecuencia indistinguible. El trastorno esclerosis tuberosa (asociado con displasia cortical, tumores renales, máculas hipopigmentadas y rabdomiomas cardiacos) puede ser causado por mutaciones en alguno de dos loci (TSC1 o TSC2).39 La heterogeneidad del locus génico puede reflejar el hecho de que múltiples proteínas participan en vías comunes del metabolismo celular. La otra forma de heterogeneidad genética es la heterogeneidad alélica. Aquí diferentes tipos de mutaciones del mismo gen causan uno o más fenotipos diferentes. La fibrosis quística, asociada con secreciones espesas, enfermedad pulmonar crónica y malabsorción) es causada por mutación en el gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR, por sus siglas en inglés, n. del t.). Una deleción de tres pares de bases denominada ?F508 es responsable de alrededor del 70 porciento de las mutaciones CFTR en la población blanca, y más de 600 mutaciones diferentes causan el 30 porciento restante de los casos.40 El caso del gen ret muestra que trastornos completamente diferentes pueden ser causados por mutaciones diferentes del mismo gen. Los mismo es verdad para las mutaciones de los genes del receptor de factor de crecimiento de fibroblastos.41 Diferentes mutaciones del gen que codifica al receptor 2 del factor de crecimiento de fibroblastos (FGFR2, por sus siglas en inglés, n. del t.) pueden originar el síndrome de Apert (asociado con craniosinostosis y sindactilia), el síndrome de Crouzon (asociado con craniosinostosis y órbitas poco profundas) o el síndrome de Pfeiffer (asociado con craniosinostosis, pulgares anchos y primeros ortejos grandes). El síndrome de Pfeiffer puede ser causado también por mutaciones en el FGFR1. El descubrimiento de las bases genéticas para los trastornos clínicos con frecuencia pone en duda los esquemas antiguos de clasificación clínica y demuestra que fenotipos que se superponen pueden ser causados por mutaciones de genes que codifican proteínas con funciones que también se superponen.
La importancia médica de la mutación genética se aplica no solo a los trastornos hereditarios, sino también a mutaciones somáticas que se adquieren después de la concepción. Es probable que se desconozca el rango completo de los efectos fenotípicos causados por las mutaciones somáticas, pero mucho se ha aprendido sobre mutaciones que originan las propiedades fenotípicas de las células neoplásicas. Los genes cuyos productos contribuyen a las neoplasias se conocen como oncogenes.42 Los oncogenes dominantes son activados por mutaciones que afectan su intensidad de transcripción, estabilidad proteica o actividad biológica. Algunos mecanismos de la activación de los oncogenes no ocurren en los genes relacionados con las enfermedades hereditarias. Estos mecanismos incluyen traslocaciones que yuxtaponen oncogenes con promotores activos y amplificación del gen, en la que un gen o grupo de genes se replica decenas a cientos de veces en una célula.
Los oncogenes recesivos también se conocen como genes supresores de tumores. Estos genes ejercen su efecto solo cuando han mutado ambas copias, causando ausencia del producto. Las mutaciones de los genes supresores de tumores suelen ser deleciones o mutaciones truncadas que eliminan la actividad de la proteína. La pérdida de la actividad de un gen supresor de tumores puede influir en la etiología de un cáncer. Las personas que nacen con una mutación heterocigota tienen un mayor riesgo de cáncer porque el paso que limita hacia la aparición de la neoplasia será la pérdida de la copia restante del gen. En algunos casos las mutaciones heterocigotas pueden asociarse con efectos fenotípicos, principalmente alteraciones congénitas.
Trastornos genéticos en la población
Todos los humanos comparten la carga de la enfermedad genética, pero el espectro de padecimientos no es el mismo en todas las poblaciones. El estudio de las influencias sobre la frecuencia de la enfermedad genética en las diferentes poblaciones es el tema de la genética de población. Estas influencias incluyen mutación, selección, migración y el fenómeno conocido como el efecto de fundador (ver adelante).
Algunos genes están sometidos a una tasa relativamente alta de mutaciones nuevas. Por ejemplo, la prevalencia de neurofibromatosis tipo 1 es de alrededor de uno por 3,500 personas y el 50 porciento de los casos se debe a una mutación nueva.43 Los trastornos que se originan de una mutación nueva se distribuyen en todo el mundo, sin predilección racial o étnica. Aunque la frecuencia elevada de mutaciones debe originar un aumento gradual en la frecuencia del padecimiento, se llega a alcanzar un equilibrio entre los nuevos casos que se originan por mutación y la pérdida de alelos mutantes de la población como resultado de la selección (v.gr., muertes tempranas, infertilidad o menor capacidad para reproducirse).
Si la tasa de mutación es baja, el trastorno genético será poco frecuente, pero las fuerzas de selección o el efecto de fundador pueden causar una frecuencia aberrantemente alta en poblaciones específicas. En algunas circunstancias la selección puede favorecer a un alelo mutante sobre el alelo nativo (normal). El mejor ejemplo lo constituyen las mutaciones de la globina responsables de la anemia de células falciformes o de la talasemia. La heterocigosidad para las variantes de la globina tienden a conferir una resistencia relativa al paludismo.33 Esto preserva las mutaciones en estado heterocigoto en ciertas partes del mundo en donde el paludismo es prevalente. La ventaja de los heterocigotos también se ha propuesto como un mecanismo para mantener las mutaciones de la fibrosis quística, en este caso para mantener una resistencia relativa a la pérdida de líquido causada por la toxina del cólera.44
El efecto de fundador es otra manera por la que mutaciones específicas son confinadas a poblaciones específicas. Este ocurre cuando un pequeño número de individuos fundan una población nueva que permanece relativamente aislada. Los alelos recesivos que son raros en la población de origen pueden volverse más prevalentes en los fundadores de la nueva población. Se piensa que este es el mecanismo responsable de la prevalencia de alelos para enfermedad de Tay-Sachs, de Canavan y de Gaucher en los judíos Ashkenazi. El efecto de fundador puede explicar también porqué mutaciones de globina específicas (v.gr., celulas falciformes en Africa y beta-talasemia en la región del Mediterráneo) predominan en diferentes regiones del llamado cinturón del paludismo.
El reconocimiento de la predilección étnica para la enfermedad genética es importante en la práctica clínica. Las frecuencias de portadores para las enfermedades recesivas pueden variar mucho en las diferentes poblaciones. Las personas cuyos antecedentes étnicos les confieren un mayor riesgo de ser portadores de un trastorno específico deben someterse a escrutinio para determinar el riesgo de tener un hijo afectado. En muchos de estos padecimientos es posible realizar diagnóstico prenatal.
Conclusión
El siglo XX será recordado en la historia de la medicina como la era en la que se comprendieron las bases moleculares para el desarrollo normal y la función celular. Los cambios en la integridad del programa genético, sean heredados o adquiridos somáticamente, son la raíz o contribuyen en virtualmente todos los aspectos de la patología humana. Al aclararse la estructura y función del genoma humano, podrá contarse con las herramientas para definir la patogenia molecular de esencialmente todos los aspectos de la enfermedad. Será tarea de las siguientes generaciones de científicos médicos el integrar esta información en un cuadro de función normal y anormal, y es seguro suponer que la práctica médica se modificará en mucho al surgir nuevos conocimientos.
Figuras 1 a 3 George Kelvin.
Figuras 4,5,7,9,11,12 y 13 Dimitry Schidlovsky.
Figura 6 Micrografía cortesía del Dr. Jorge J. Yunis, Department of Laboratory Medicine and Pathology, University of Minnesota Medical School.
Figura 8 Talar Agasyan.
Figura 10 Tom Moore.
Bibliografía
-
Koenig M, Monaco AP, Kunkel LM: The complete sequence of
dystrophin predicts a rod-shaped cytoskeletal protein. Cell 53:219, 1988
[PMID
3282674]
-
Johns DR: Seminars in medicine of the Beth Israel Hospital,
Boston: mitochondrial DNA and disease. N Engl J Med 333:638, 1995
-
Shoffner JM, Lott MT, Lezza AMS, et al: Myoclonic epilepsy
and ragged-red fiber disease (MERRF) is associated with a mitochondrial
DNA tRNA(Lys) mutation. Cell 61: 931, 1990 [PMID
2112427]
-
ISCN 1995: An International System for Cytogenetic Nomenclature.
Mitelman F, Ed. S. Karger, Basel, 1995
-
Krishna Murthy DS, Farag TI: Recurrent regular trisomy-21
in two Bedouin families: parental mosaicism versus genetic predisposition.
Ann Genet 38:217, 1995 [PMID
8629809]
-
Hook EB, Cross PK, Schreinemachers MS: Chromosomal abnormality
rates at amniocentesis and in live-born infants. JAMA 249:2034, 1983 [PMID
6220164]
-
Antonarakis SE: Parental origin of the extra chromosome in
trisomy 21 as indicated by analysis of DNA polymorphisms. Down Syndrome
Collaborative Group. N Engl J Med 324:872, 1991
-
Hook E: Issues in analysis of data on paternal age and 47,+21:
implications for genetic counseling for Down syndrome. Hum Genet 77:303,
1987
-
Sanger F, Nicklen S, Coulson AR: DNA sequencing with chain-termination
inhibitors. Proc Natl Acad Sci USA 74:5463, 1977 [PMID
271968]
-
Southern EM: Detection of specific sequences among DNA fragments
separated by gel electrophoresis. J Mol Biol 98:503,1975
-
Botstein D, White RL, Skolnick M, et al: Construction of
a genetic linkage map in man using restriction fragment length polymorphisms.
Am J Hum Genet 32:314, 1980 [PMID
6247908]
-
Murray JC, Buetow KH, Weber JL, et al: A comprehensive human
linkage map with centimorgan density. Cooperative Human Linkage Center
(CHLC). Science 265:2049, 1994
-
Hudson TJ, Stein LD, Gerety SS, et al: An STS-based map of
the human genome. Science 270:1945, 1995 [PMID
8533086]
-
Schuler GD, Boguski MS, Stewart EA, et al: A gene map of
the human genome. Science 274:540, 1996
-
Orkin SH: Reverse genetics and human disease. Cell 47:845,
1986
-
Monaco AP, Neve RL, Colletti-Feener C, et al: Isolation of
candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature
323:646, 1986 [PMID
3773991]
-
Riordan JR, Rommens JM, Kerem B, et al: Identification of
the cystic fibrosis gene: cloning and characterization of complementary
DNA. Science 245:1066, 1989 [PMID
2475911]
-
Cawthon RM, Weiss R, Xu G, et al: A major segment of the
neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point
mutations. Cell 62:193, 1990 [PMID
2114220]
-
Wallace MR, Marchuk DA, Andersen LB, et al: Type 1 neurofibromatosis
gene: identification of a large transcript disrupted in three NF1 patients.
Science 249:181, 1990 [PMID
2134734]
-
Cockburn DJ, Munro EA, Craig IW, et al: Mapping of X chromosome
translocation breakpoints in females with Duchenne muscular dystrophy with
respect to exons of the dystrophin gene. Hum Genet 90:407, 1992 [PMID
1483697]
-
Korenberg JR, Chen X-N, Schipper R, et al: Down syndrome
phenotypes: the consequences of chromosomal imbalance. Proc Natl Acad Sci
USA 91:4997, 1994 [PMID
8197171]
-
Dewald G, Stallard R, Bader PI, et al: Toward quality assurance
for metaphase FISH: a multi-center experience. Am J Med Genet 64:539, 1996
[PMID
8870919]
-
Rose EA, Glaser T, Jones C, et al: Complete physical map
of the WAGR region of 11p13 localizes a candidate Wilms' tumor gene. Cell
60:405, 1990
-
Li LH, Krantz ID, Deng Y, et al: Alagille syndrome is caused
by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet
16:243, 1997
-
Oda T, Elkahloun AG, Pike BL, et al: Mutations in the human
Jagged1 gene are responsible for Alagille syndrome. Nat Genet 16:235, 1997
[PMID
9207787]
-
Rand EB, Spinner NB, Piccoli DA, et al: Molecular analysis
of 24 Alagille syndrome families identifies a single submicroscopic deletion
and further localizes the Alagille region within 20p12. Am J Hum Genet
57:1068, 1995 [PMID
7485156]
-
Hall JG: Genomic imprinting: review and relevance to human
disease. Am J Hum Genet 46:857, 1990
-
Cassidy SB: Conference report: first international scientific
workshop on Prader-Willi syndrome and other chromosome 15q deletion disorders.
Am J Med Genet 42:220, 1992
-
Knoll JHM, Lalande M: Cytogenetic and molecular studies in
the Prader-Willi and Angelman syndromes: an overview. Am J Med Genet 46:2,
1993
-
Kishino T, Lalande M, Wagstaff J: UBE3A/E6-AP mutations cause
Angelman syndrome. Nat Genet 15:70, 1997 [PMID
8988171]
-
Matsuura T, Sutcliffe JS, Fang P, et al: De novo truncating
mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome.
Nat Genet 15:74, 1997 [PMID
8988172]
-
Ledbetter DH, Engel E: Uniparental disomy in humans: development
of an imprinting map and its implications for prenatal diagnosis. Hum Mol
Genet 4:1757, 1995 [PMID
8541876]
-
Orkin SH: Disorders of hemoglobin synthesis: the thalassemias.
The Molecular Basis of Blood Diseases. Stamatoyannopoulos G, Nienhuis AW,
Leder P, et al, Eds. WB Saunders Co, Philadelphia, 1987, p 106
-
Monaco AP, Bertelson CJ, Liechti-Gallati S, et al: An explanation
for the phenotypic differences between patients bearing partial deletions
of the DMD locus. Genomics 2:90, 1989
-
Ashley CT Jr, Warren ST: Trinucleotide repeat expansion and
human disease. Ann Rev Genet 29:703, 1995
-
McInnis MG: Anticipation: an old idea in new genes. Am J
Hum Genet 59:973, 1996
-
Prockop DJ, Baldwin CT, Constantinou CD: Mutations in type
I procollagen genes that cause osteogenesis imperfecta. Adv Hum Genet 19:105,
1990 [PMID
2193488]
-
Eng C: Seminars in medicine of the Beth Israel Hospital,
Boston: the RET proto-oncogene in multiple endocrine neoplasia type 2 and
Hirschsprung's disease. N Engl J Med 335:943, 1996
-
Povey S, Burley MW, Attwood J, et al: Two loci for tuberous
sclerosis: one on 9q34 and one on 16p13. Ann Hum Genet 58:107, 1994 [PMID
7979156]
-
Kerem B, Rommens JM, Buchanan JA, et al: Identification of
the cystic fibrosis gene: genetic analysis. Science 245:1073, 1989 [PMID
2570460]
-
Cohen MM Jr: Craniosynostoses: phenotypic/molecular correlations.
Am J Med Genet 56:334, 1995
-
Weinberg RA: Oncogenes, antioncogenes, and the molecular
bases of multistep carcinogenesis. Cancer Res 49:3713, 1989
-
Carey JC, Baty BJ, Johnson JP, et al: The genetic aspects
of neurofibromatosis. Ann NY Acad Sci 486:45, 1986 [PMID
3105404]
-
Gabriel SE, Brigman KN, Koller BH, et al: Cystic fibrosis
heterozygote resistance to cholera toxin in the cystic fibrosis mouse model.
Science 266:107, 1994 [PMID
7524148]
-
Yunis JJ: Chromosomes and cancer: new nomenclature and future
directions. Hum Pathol 12:500, 1981
-
Recombinant DNA, 2nd ed. Watson JD, Gilman M, Witkowski J,
et al. Scientific American Books, New York, 1992
-
Mullis KB: The unusual origin of the polymerase chain reaction.
Sci Am 262(4): 56, 1990