Contenido del artículo
XII HEMOSTASIA Y SU REGULACION
- Formación del tapón plaquetario
- Cascada de la coagulación
- Mecanismos de control
- SISTEMA ANTITROMBINA III - HEPARAN SULFATO
- PROTEINA C Y PROTEINA S - SISTEMA DE TROMBOMODULINA
- INHIBIDOR DE LA VIA DEL FACTOR TISULAR
- PROSTACICLINA
- OXIDO NITRICO
- FIBRINOLISIS
- Generalidades sobre la coagulación
- Heterogeneidad de las células endoteliales y del lecho vascular - Hemostasia específica
- Producción de plaquetas y trombopoyetina
- Pruebas de coagulación y su uso
DR. LAWRENCE K. LEUNG
La hemostasia, que es el proceso de formación del coágulo sanguíneo, consiste en una serie de respuestas coordinadas ante la lesión vascular. Requiere de la actividad conjunta de las plaquetas, la cascada de la coagulación, el flujo sanguíneo y las fuerzas de cizalla, las células endoteliales y la fibrinolisis.
Formación del tapón plaquetario
Las plaquetas se activan en el sitio de lesión vascular para formar un tapón y detener la hemorragia. Entre los estímulos fisiológicos para las plaquetas se incluyen el difosfato de adenosina (ADP), la epinefrina, la trombina y la colágena. El ADP y la epinefrina son estímulos plaquetarios relativamente débiles, la trombina y la colágena son agonistas potentes. La activación de la trombina está mediada por los receptores activados de proteína G acoplada a proteasa (PAR, por sus siglas en inglés, n. el t.),1 específicamente los PAR-1 y PAR-4. La trombina separa el dominio externo del PAR para iniciar la señal transmembrana [ver figura 1].2 Existen también receptores específicos para ADP, epinefrina y colágena.
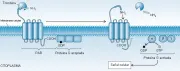
|
| Figura 1 |
| Activación de la trombina |
La activación plaquetaria consiste en cuatro procesos distintos: adhesión (depósito de plaquetas en la matriz subendotelial), agregación (cohesión de plaquetas), secreción (liberación de las proteínas de los gránulos ) y actividad procoagulante (aumento de la generación de trombina) [ver figura 2].
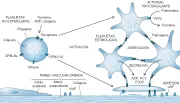
|
| Figura 2 |
| Activación plaquetaria |
ADHESION
La adhesión plaquetaria está mediada principalmente por la unión del complejo receptor de superficie plaquetaria de la glucoproteína (GP) Ib-IX-V a la proteína de adhesión factor de von Willebrand (vWF) en la matriz subendotelial.3 La deficiencia del complejo GPIb-IX-V o del vWF origina dos trastornos hemorragíparos congénitos respectivamente, la enfermedad de Bernard Soulier y la enfermedad de von Willebrand. Otras interacciones de adhesión (v.gr., la unión del receptor plaquetario de colágena GPIa-IIa a las fibrillas de colágena en la matriz) también contribuyen a la adhesión plaquetaria.4
La agregación plaquetaria ocurre por la unión del fibrinógeno al receptor de fibrinógeno plaquetario (i.e., el complejo GPIIb-IIIa). El GPIIb-IIIa (también denominado aIIbb3) es un miembro de la superfamilia de los receptores de proteínas de adhesión, denominadas integrinas y que se encuentran en muchos tipos diferentes de células. Es el receptor más abundante en la superficie de la plaqueta. El GPIIb-IIIa no fija fibrinógeno en las plaquetas no estimuladas. Después de la estimulación plaquetaria el GPIIb-IIIa sufre un cambio conformacional y se convierte de un receptor para fibrinógeno de baja afinidad en un receptor de alta afinidad en un proceso denominado señal de dentro hacia fuera. El fibrinógeno, una molécula divalente, sirve de puente a las plaquetas activadas [ver figura 3]. La porción citosólica del complejo GPIIb-IIIa activado se une al citoesqueleto plaquetario y puede mediar la diseminación plaquetaria y la retracción del coágulo (en un proceso denominado señal de fuera hacia adentro).5 La deficiencia congénita de GPIIb-IIIa o fibrinógeno causa la trombastenia de Glanzmann y la afibrinogenemia. La vía GPIIb-IIIa-fibrinógeno es la vía común final para la agregación plaquetaria. El bloqueo de esta vía es el mecanismo de acción de una clase importante de medicamentos antiplaquetarios.
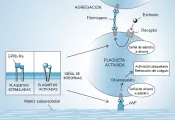
|
| Figura 3 |
| Mecanismo de agregación plaquetaria |
SECRECION PROTEICA
Después de la estimulación los gránulos plaquetarios liberan ADP y serotonina, que estimulan y reclutan más plaquetas, proteínas de adhesión como la fibronectina y la trombospondina, que reforzan y estabilizan los agregados plaquetarios, factor V, un componente de la cascada de la coagulación, tromboxano, que estimula la vasoconstricción, y factores de crecimiento como el factor de crecimiento derivado de plaquetas (FCDP), que estimula la proliferación de las células del músculo liso y media la reparación tisular. El FCDP puede también contribuir al desarrollo de ateroesclerosis y reoclusión después de la angioplastía coronaria.
La procoagulación plaquetaria consiste en el ensamble de los complejos enzimáticos de la cascada de la coagulación en la superficie de la plaqueta. Es un ejemplo importante de la interrelación estrecha que existe entre la activación plaquetaria y la activación de la cascada de la coagulación.
La característica central de la cascada de la coagulación es la activación secuencial de una serie de proenzimas (cimógenos) a enzimas, generando al final fibrina y reforzando el trombo plaquetario. Otra característica, la amplificación, asegura una respuesta rápida para una hemostasia eficaz, pero demanda una regulación estrecha para evitar la trombosis exagerada.
La cascada de la coagulación suele dividirse en una vía intrínseca y una extrínseca [ver figura 4]. La vía intrínseca se inicia por la exposición de sangre a una superficie con carga negativa (v.gr., vidrio), mientras que la vía extrínseca se activa por el factor tisular o la tromboplastina. Ambas vías convergen en la activación del factor X, que después activa a la protrombina (factor II) a trombina, la enzima final de la cascada de la coagulación.
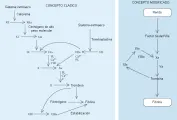
|
| Figura 4 |
| Cascada de la coagulación |
Aunque este punto de vista clásico de la cascada de la coagulación ha sido útil para interpretar los tiempos de coagulación, no es del todo exacto. Los pacientes que tienen deficiencia severa en el factor XII (así como muchos pacientes con deficiencia del factor XI) no tienen sangrado clínico, lo que indica que la parte inicial de la vía intrínseca (la fase de contacto) no es importante in vivo. Actualmente se ha establecido que la generación o exposición de factor tisular en el sitio de la herida es el principal evento fisiológico que inicia la coagulación [ver figura 4]. 6 El factor tisular funciona como un cofactor que se requiere en forma absoluta por el factor VII/factor VIIa para iniciar la coagulación. El factor VIIa activa el factor X en forma directa e indirecta por activación del factor IX. Esta vía dual de activación del factor X parece necesaria por la cantidad limitada de factor tisular generada in vivo y por la presencia de la vía inhibitoria del factor tisular (ver adelante) que, cuando forma un complejo con el factor Xa, inhibe al complejo factor tisular/factor VIIa.
Todos los procoagulantes se sintetizan en el hígado excepto el vWF, que se sintetiza en los megacariocitos y en las células endoteliales. Los procoagulantes dependientes de vitamina K son la protrombina, el factor VII, el factor IX y el factor X, y los anticoagulantes dependientes de vitamina K son la proteína C y la proteína S. Para cada uno de estos factores, la producción de residuos g-carboxiglutámicos por la carboxilación de residuos de ácido glutámico dependiente de la vitamina K actúa como una señal de reconocimiento que guía la modificación postraducción de la proteína requerida para su actividad biológica.7
INTERACCION ENTRE LAS PLAQUETAS ACTIVADAS Y LA CASCADA DE LA COAGULACION
Existe una estrecha interacción entre la cascada de la coagulación y la superficie plaquetaria activada in vivo. Cuando las plaquetas se activan se exponen lípidos aniónicos en su superficie y se libera factor V (almacenado en los gránulos plaquetarios) que se une a los lípidos aniónicos. El factor V en la superficie plaquetaria se activa a factor Va y actúa como un sitio de ensamble para la unión del factor Xa (enzima) y protrombina (sustrato) a los que se conoce como complejo protrombinasa. En el sitio de ensamble la generación de trombina por el complejo protrombinasa es alrededor de 300,000 veces más eficaz que la generación de trombina por el factor Xa en fase soluble y la protrombina sola, y el trombo plaquetario mantiene a la trombina relativamente fija. El factor Xa fijo al factor Va también está relativamente protegido de la inhibición por inhibidores circulantes como la antitrombina III (ver adelante). Un ensamble similar del complejo enzimático se aplica a la activación del factor X por el factor VIIIa (cofactor) y el factor IXa (la tenaza intrínseca). El resultado de estos procesos es una amplificación eficaz y fijación del coágulo.
La coagulación está controlada por diversos mecanismos: dilución de procoagulantes en la sangre circulante, remoción de factores activados a través del sistema retículoendotelial, en especial en el hígado y control por vías antitrombóticas naturales. Por lo menos seis sistemas de control diferentes modulan cada fase de la hemostasia y protegen contra la trombosis, la inflamación vascular y el daño tisular [ver tabla 1]. La antitrombina III, la proteína C, la proteína S y el inhibidor de la vía del factor tisular (IVFT) regulan en conjunto la cascada de la coagulación, la prostaciclina y el óxido nítrico modulan la reactividad vascular y plaquetaria, y la fibrinolisis elimina el coágulo de fibrina.
|
||||||
|
SISTEMA ANTITROMBINA III - HEPARAN SULFATO
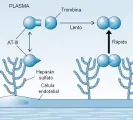
La antitrombina III (AT-III) es un inhibidor de las proteasas plasmáticas circulantes. Inhibe a la trombina y al factor Xa, las dos enzimas clave en la cascada de la coagulación. La AT-III también inhibe al factor XII activado y al factor XI. En ausencia del glucosaminoglucano heparina, la AT-III inhibe a la trombina y al factor Xa en forma relativamente lenta (la inhibición completa requiere algunos minutos). Cuando está presente, la heparina se une a un sitio específico en la AT-III, lo que causa un cambio conformacional en la molécula, que a su vez inhibe a la trombina en forma instantánea e irreversible. Este aumento en la inhibición de la trombina y del factor Xa es la base para el uso terapéutico de la heparina como anticoagulante. Los proteoglucanos heparán sulfato sobre la superficie luminal de las células endoteliales parecen activar a la AT-III en una forma semejante a la de la heparina [ver figura 5].8
|
| Figura 5 |
| Acción de la antitrombina III |
Por lo tanto, la superficie endotelial en condiciones normales está cubierta con una capa de AT-III que ya en sí está activada por el heparán sulfato endógeno. Debido a que 1 ml de sangre puede quedar expuesto hasta a 5,000 cm2 de superficie endotelial, el sistema AT-III-heparán sulfato tiene por objeto inactivar a cualquier trombina en la circulación general.
PROTEINA C Y PROTEINAS - SISTEMA DE TROMBOMODULINA
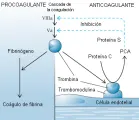
La trombomodulina es una proteína integral de la membrana que se encuentra en la superficie luminal del endotelio vascular en la microcirculación. La trombina sufre un cambio conformacional cuando se une a la trombomodulina, causando una modificación importante en la especificidad de sustrato: ya no precipita fibrinógeno ni activa plaquetas [ver figura 6]. Por otro lado, adquiere la capacidad para activar a la proteína C en el plasma.9 Se ha encontrado un receptor endotelial diferente para la proteína C que aumenta la activación de la misma por el complejo trombina-trombomodulina.10 La proteína C activada degrada al factor Va y al factor VIIIa, los dos cofactores responsables del ensamble de la protrombinasa y del complejo intrínseco tenaza en la cascada de la coagulación. La proteína S sirve como un cofactor para la proteína C activada. Las deficiencias de AT-III, proteína C y proteína S son causas importantes de un estado hipercoagulable.
|
| Figura 6 |
| Vía proteína C/proteína S |
La proteína C y la proteína S muestran ambos ciertas similitudes estructurales a los factores de coagulación dependientes de vitamina K (protrombina, factor VII, factor IX y factor X). La proteína S circula en dos formas: una forma libre, en la que es activa como anticoagulante, y una forma inactiva, fija, en la que constituye un complejo con la proteína de unión C4b del sistema del complemento. La proteína de unión C4b actúa como un reactante de fase aguda. El incremento resultante en los estados inflamatorios disminuye la actividad de la proteína S libre, aumentando la posibilidad de trombosis.
INHIBIDOR DE LA VIA DEL FACTOR TISULAR
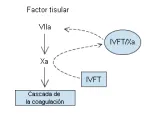
El inhibidor de la vía del factor tisular (IVFT) es un inhibidor tisular de las proteasas del plasma que se sintetiza en el endotelio microvascular. A diferencia de la AT-III, el IVFT tiene una concentración en plasma muy baja. El IVFT inhibe al factor Xa. El complejo IVFT/factor Xa se vuelve un inhibidor eficaz del factor tisular/factor VIIa, mediando así la retroalimentación de la inhibición del factor tisular/factor VIIa [ver figura 7]. Los estudios en animales han demostrado que la depleción del IVFT endógeno sensibilizará a los animales al desarrollo de coagulación intravascular diseminada inducida por el factor tisular o la endotoxina.11
|
| Figura 7 |
| Inhibidor de la vía del factor tisular |
El IVFT se sintetiza principalmente por el endotelio microvascular. Alrededor del 20 por ciento del IVFT circula en el plasma asociado con lipoproteínas, la mayoría de las cuales permanecen asociadas con la superficie endotelial, aparentemente unidas a los glucosaminoglucanos de la superficie celular. El nivel en plasma de IVFT aumenta en mucho después de la administración intravenosa de heparina. Esta liberación de IVFT endotelial puede contribuir a la eficacia antitrombótica de la heparina y de la heparina de bajo peso molecular. En la actualidad se realizan estudios clínicos con IVFT recombinante.12
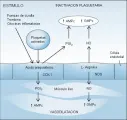
Ante una lesión celular, la fosfolipasa A2 libera de los fosfolípidos de la membrana ácido araquidónico, un ácido graso. La enzima prostaglandina endoperóxido H sintetasa-1 (PGHS-1) convierte el ácido araquidónico en endoperóxidos prostaglandinas y finalmente en tromboxano A2 (TXA2) en las plaquetas y prostaciclina (PGI2) en las células endoteliales. El TXA2 y la PGI2 tienen funciones opuestas. El TXA2 es un estimulador potente de la agregación plaquetaria y causa vasoconstricción, mientras que la PGI2 inhibe la agregación plaquetaria e induce vasodilatación. La PGI2 funciona al activar a la adenilato ciclasa, lo que causa un aumento en el monofosfato cíclico de adenosina (AMPc) intracelular [ver figura 8].
|
| Figura 8 |
| Función del óxido nítrico y la prostaciclina |
Cicloxigenasa- 1 y cicloxigenasa-2
La cicloxigenasa-1 (COX-1) es la isoforma constitutiva de la PGHS. Recientemente se ha descubierto una isoforma inducible, la cicloxigenasa-2 (COX-2). En condiciones normales no se detecta COX-2 en la mayoría de los tejidos; sin embargo puede ser inducida con rapidez en respuesta a factores de crecimiento, endotoxinas y citocinas en las células endoteliales y monocitos (aunque no en las plaquetas).13 La aspirina acetila e inhibe en forma irreversible tanto la COX-1 como la COX-2. Otros fármacos antinflamatorios no esteroideos (AINE) inhiben también ambas enzimas, aunque no en forma permanente. En la actualidad se dispone de inhibidores selectivos de COX-2 como una nueva generación de AINE.14
Debido a que la aspirina inhibe en forma irreversible la COX-1 y ya que las plaquetas no pueden producir nueva enzima, la exposición breve a la aspirina inhibirá en forma permanente la producción de TXA2 durante la vida de las plaquetas afectadas.
El óxido nítrico (NO) está formado por L-arginina en las células endoteliales. El NO estimula la guanilato ciclasa, causando aumento en el monofosfato cíclico de guanosina (GMPc) en las células blanco, provoca vasodilatación e inhibe la adhesión y agregación plaquetaria [ver figura 8]. 15 El NO es rápidamente destruido por la hemoglobina y, por tanto, funciona como una hormona local (i.e., parácrina). La infusión intravenosa de un análogo de arginina que bloquea la producción de NO causa un incremento inmediato y sustancial en la presión arterial. Este fenómeno sugiere que el NO se libera en forma continua y basal para regular el tono vascular (a diferencia de la producción de PGI2, cuya liberación es más de estímulo-respuesta). Existe un sinergismo significativo entre el NO y la PGI2. La formación de NO está catalizada por sintetasas de NO, que existen en diferentes isoformas en varios tejidos. Además de regular los eventos vasculares, el NO tiene un amplio rango de efectos biológicos (v.gr., función de neurotransmisión en el sistema nervioso central).
El activador del plasminógeno tisular (t-PA) se libera de las células endoteliales lesionadas cerca del sitio de la lesión vascular. El t-PA convierte el plasminógeno en plasmina. En forma análoga a la interacción de la AT-III con la trombina, que se acelera en presencia del heparán sulfato de la superficie de la célula endotelial, la generación de plasmina tiene lugar en condiciones óptimas en una superficie (en este caso el coágulo de fibrina). Tanto el t-PA como el plasminógeno se unen a la fibrina (a través del reconocimiento de residuos de lisina), lo que facilita la generación de plasmina y la fibrinolisis localizada [ver figura 9].
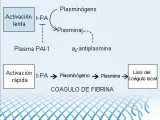
|
| Figura 9 |
| Vía plasminógeno/plasmina |
La plasmina escinde la cadena de fibrina polimerizada en múltiples sitios y libera productos de degradación de la fibrina. Uno de los principales productos es el dímero D, que consiste en dos dominios D de monómeros adyacentes de fibrina que han sufrido enlaces cruzados por el factor XIII activado [ver figura 10]. La plasmina tiene una especificidad de sustrato muy amplia y, además de la fibrina, rompe el fibrinógeno y diversas proteínas plasmáticas y factores de la coagulación. La plasmina unida al coágulo de fibrina está protegida de su inactivación, mientras que la plasmina liberada a la ciruclación es inactivada con rapidez por la a2-antiplasmina plasmática. Por lo tanto, se logra una fibrinolisis localizada, pero se evita la degradación inespecífica por la plasmina de las proteínas del plasma. En casos raros los pacientes tienen problemas hemorragíparos debido a una deficiencia congénita de la a2-antiplasmina.
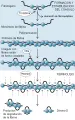
|
| Figura 10 |
| Transformación de fibrinógeno a fibrina |
La urocinasa es el segundo activador fisiológico del plasminógeno. Está presente en alta concentración en la orina. Aunque el t-PA es en gran parte responsable de iniciar la fibrinolisis intravascular, la urocinasa es el principal activador de la fibrinolisis en el compartimiento extravascular. La urocinasa es secretada por muchos tipos de células en la forma de prourocinasa, también denominada activador del plasminógeno tipo urocinasa de una sola cadena (scu-PA). La prourocinasa se convierte a urocinasa por acción de la plasmina. La urocinasa no tiene especificidad de la fibrina para convertir el plasminógeno en plasmina, mientras que la prourocinasa sí.
El principal inhibidor fisiológico del t-PA y el activador urocinasa plasminógeno (u-PA) es el inhibidor del activador del plasminógeno-1 (PAI-1, por sus siglas en inglés n. del t.).16 En las plaquetas se encuentran cantidades importantes de PAI-1. El PAI-1 también es liberado de las células endoteliales, y su deficiencia se asocia con diátesis hemorrágica, generalmente relacionada con traumatismos o cirugía.17 Un segundo inhibidor, el PAI-2, normalmente es secretado por los monocitos y durante el embarazo sus niveles aumentan mucho por su síntesis en la placenta. Aún no se ha establecido la importancia biológica del PAI-2.
Inhibidor activable de la fibrinolisis relacionado a trombina
La carboxipeptidasa plasmática es un inhibidor activable de la fibrinolisis relacionado a trombina (IAFT) recién descubierto [ver figura 11]. 18,19 Es el segundo sustrato fisiológico conocido para el complejo trombina-trombomodulina. Debe considerarse que después de que se forma el coágulo inicial de fibrina por la trombina en el sitio de la lesión vascular, la trombina se une a la trombomodulina en la superficie endotelial intacta cercana. La trombina unida a la trombomodulina causa la generación de proteína C activada, que bloquea la cascada de la coagulación y evita la generación excesiva de trombina. Al mismo tiempo, la trombina unida a la trombomodulina activa al IAFT, disminuyendo así la lisis del coágulo existente. Aún no se ha demostrado si la deficiencia de IAFT causa hemorragia y si el exceso en su actividad produce trombosis.
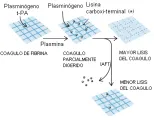
|
| Figura 11 |
| Inhibición de la fibrinolisis |
Generalidades sobre la coagulación
La cascada de la coagulación se inicia por exposición del factor tisular en una lesión vascular, lo que ocasiona la generación de trombina y el depósito de un coágulo de fibrina [ver figura 12]. En forma simultánea, el endotelio dañado libera t-PA, que convierte el plasminógeno en plasmina, que después lisa el coágulo. Ambas vías están controladas: el FT/factor VIIa está regulado por el complejo IVFT/factor Xa, y la trombina está regulada por las proteínas C y S. En forma semejante, la actividad del t-PA está controlada por el PAI-1. La trombina y la plasmina están bajo en control de sus inhibidores respectivos, la AT-III y la a2-antiplsmina. Cuando estas dos vías actúan en simetría coordinada, se produce un coágulo para detener el sangrado, con lisis posterior del mismo y remodelación tisular. La menor generación de trombina (como en la deficiencia de factor VIII) o la mayor producción de plasmina (como en la deficiencia de a2-antiplasmina) causan hemorragia. Por el contrario, la producción excesiva de trombina (como en la deficiencia de AT-III o proteína C) provoca trombosis.
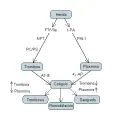
|
| Figura 12 |
| Remodelación del coágulo |
Heterogeneidad de las células endoteliales y del lecho vascular - Hemostasia específica
Aunque por lo general el endotelio se considera como un sistema característico y homogéneo, existen diferencias significativas entre las células endoteliales de arterias, venas y capilares en términos de morfología y susceptibilidad a enfermedades. Estudios recientes han demostrado grupos distintos de proteínas que caracterizan a las células endoteliales arteriales y venosas desde las primeras fases de la angiogénesis. Ephrin-B2, un ligando transmembrana de la familia Eph, existe en las células endoteliales arteriales, pero no en las venosas. Por el contrario, Eph-B4, un receptor tirosina cinasa para ephrin-B2, caracteriza a las venas, pero no a las arterias.20
También parece que el endotelio de diferentes lechos vasculares no es idéntico.21 Por ejemplo, el endotelio alto en las vénulas poscapilares de los ganglios linfáticos y las placas de Peyer regula la circulación de linfocitos de la sangre a los linfáticos y tejidos periféricos. En este endotelio venular alto se expresan en gran cantidad receptores específicos de proteínas de adhesión y proteínas de la matriz. El endotelio especializado que constituye en la barrera hematoencefálica es otro ejemplo.
Estas diferencias entre las células endoteliales arteriales y venosas y el endotelio específico del lecho vascular pueden explicar en parte las diferentes susceptibilidades para la trombosis. Por ejemplo, mientras que las deficiencias de AT-III y proteína C suelen asociarse con trombosis venosa profunda de las extremidades inferiores, la trombosis de las venas porta y hepática con frecuencia se asocia con enfermedades mieloproliferativas.22 En ambas condiciones el defecto subyacente es un estado hipercoagulable sistémico y existe una clara predisposición para que ocurran trombosis en lechos vasculares específicos. Por lo tanto, la trombosis clínica se atribuye a un desequilibrio entre el estímulo protrombótico sistémico y los mecanismos antitrombóticos locales.
Producción de plaquetas y trombopoyetina
Las plaquetas derivan de los megacariocitos, que surgen de células tronco mieloides pluripotenciales. La producción de plaquetas está controlada por una trombopoyetina que participa en la maduración final del megacariocito. La trombopoyetina, también conocida como ligando de c-Mpl, tiene múltiples acciones en el desarrollo del megacariocito.23 Comparte cierta homología estructural con la eritropoyetina y es producida principalmente por el hígado. Aumenta el tamaño y número de los megacariocitos, estimula la expresión de marcadores específicos de plaquetas y es un potente factor estimulador de colonias de megacariocitos. Aunque es claro que la trombopoyetina es un factor clave, el factor de células tronco (también denominado ligando kit), la interleucina-3 (IL-3), la IL-6 y la IL-11 participan también en el control de la megacariopoyesis.
Los megacariocitos sufren endomitosis, en la que la división nuclear ocurre sin división celular y es seguida de fusión nuclear, para originar una célula con un contenido cromosómico 8n, 16n o 32n. El citoplasma del megacariocito cambia para formar una serie de cintas delgadas y cilíndricas que eventualmente se fragmentan en pequeñas piezas de megacariocitos, llamadas proplaquetas, que se liberan hacia la circulación. El volumen del megacariocito correlaciona con la ploidía y madurez citoplásmica, los megacariocitos más grandes producen el mayor número de plaquetas. Las plaquetas grandes se denominan megatrombocitos y se observan en la sangre periférica en los estados trombocitopénicos, en especial en la púrpura trombocitopénica idiopática. Estos megatrombocitos probablemente son proplaquetas jóvenes y son responsables del incremento en el volumen plaquetario promedio que ocurre durante la respuesta a o la recuperación de la trombocitopenia aguda.
Las plaquetas que entran a la circulación sobreviven alrededor de 8.5 a 10 días y tienen una vida media de alrededor de 4 días. Alrededor del 30 a 40 por ciento de las plaquetas están presentes en un lecho esplénico que se intercambia en forma libre con la circulación. Cuando surge la necesidad de plaquetas la producción puede aumentar siete a ocho veces. Debido a que no existe un reservorio medular de plaquetas esperando ser liberadas, los mayores requerimientos de plaquetas pueden tardar algunos días en ser satisfechos. Las plauqetas tienen receptores para la trombopoyetina y la remueven del plasma, la masa plaquetaria funciona como un regulador importante de trombopoyetina.24 En casos de hipoplasia de megacariocitos y trombocitopenia se metaboliza poca trombocpoyetina y el nivel plasmático de la misma aumenta, causando mayor producción de megacariocitos y plaquetas. En el caso de trombocitosis ocurre aumento en el metabolismo de la trombopoyetina, lo que disminuye el nivel de trombopoyetina en el plasma y, por tanto, la producción de plaquetas.
Pruebas de coagulación y su uso
PRUEBAS PARA LA CASCADA DE COAGULACION
La mayoría de las pruebas de coagulación miden el tiempo requerido para que la fibrina del plasma forme bandas, que pueden detectarse por medios ópticos o eléctricos. Su prolongación puede representar reducción en la concentración de un factor, factores inactivos o la presencia de inhibidores.
Tiempo parcial de tromboplastina (TPT) El TPT, en ocasiones denominado TPT activado (TPTa), mide el sistema de coagulación intrínseco. Se agrega una superficie con carga negativa (v.gr., caolín y sílice), seguido de cefalina, al plasma total para activar a los factores XII y XI. El TPT es más sensible a alteraciones y deficiencias en la secuencia de la cascada de la coagulación antes de la activación del factor X. El TPT también es muy sensible a la acción de la heparina. Se usa para vigilar y ajustar el tratamiento anticoagulante con heparina regular, pero no con heparinas de bajo peso molecular.
Tiempo de protrombina (TP) El tiempo de protrombina es una prueba para el sistema extrínseco. Detecta deficiencias en el fibrinógeno, factor II (protrombina), factor V, factor VII y factor X. Se agrega factor tisular al plasma total, causando formación de fibrina, normalmente en 9 a 12 segundos. Los resultados suelen reportarse usando la relación normalizada internacional (INR, por sus siglas en inglés, n. del t.). El INR se calcula por medio de la siguiente ecuación:
INR = (Log TP del paciente / Log TP control)C
en donde C representa el índice de sensibilidad internacional (ISI). De esta manera la tromboplastina usada en un laboratorio individual, con su ISI específico, se calibra contra una tromboplastina estándar de referencia y el TP se reporta como un INR.25 La presencia del anticoagulante lúpico puede también interferir con el TP.26
Tiempo de dilución con veneno de víbora de Russell (TDVVR) El veneno de víbora de Russell contiene una enzima que activa el factor X; por lo tanto, el TDVVR mide la vía común de la cascada de la coagulación. Es sensible a la presencia de anticoagulante semejante a lupus, que inhibe al complejo de fosfolípidos dependientes de protrombinasa.
Tiempo de trombina (TT) El TT se usa para investigar alteraciones en la conversión de fibrinógeno a fibrina. Puede estar prolongado por hipofibrinogenemia, fibrinógeno anormal (disfibrinógeno) o la presencia de inhibidores (v.gr., productos de degradación de la fibrina) que interfieren con la polimerización de la fibrina. Los factores clínicos asociados con frecuencia con el TT prolongado son la hepatopatía severa, la coagulación intravascular diseminada y el tratamiento con heparina.
Tiempo de reptilasa (TR) La reptilasa en una enzima semejante a la trombina que convierte el fibrinógeno en fibrina. El TR se prolonga en condiciones semejantes a las del TT prolongado, con una diferencia significativa: la reptilasa no es inhibida por el complejo AT-III-heparina. Por lo tanto, el TR no se prolonga con la heparina. Un tiempo prolongado de trombina y un TR normal sugieren efecto de heparina.
Fibrinopéptido A (FPA) La trombina activa al fibrinógeno al separar dos moléculas de los fibrinopéptidos A y B, dejando una molécula del monómero de fibrina. La medición del FPA en la sangre puede usarse como un índice de la actividad de la trombina in vivo. Sin embargo, debido a que la cascada de la coagulación puede activarse durante la toma de muestra de sangre, deben tomarse precauciones en la medición e interpretación de los niveles de FPA.
Fibrinógeno El nivel de fibrinógeno en plasma puede medirse por métodos antigénicos o, con más frecuencia, por pruebas de coagulación. Los resultados se reportan en mg/dl.
Dímero-D y productos de degradación de fibrina-fibrinógeno (PDF) Los PDF se producen por la degradación por la plasmina del fibrinógeno y del coágulo de fibrina [ver figura 9]. El dímero-D es liberado por la degradación mediada por plasmina de la fibrina polimerizada. La ruptura del fibrinógeno o del monómero de fibrina por la plasmina no produce dímero-D. Por lo tanto, el aumento del dímero-D es una medida específica del depósito de fibrina intravascular y de la degradación de la plasmina característicos de la coagulación intravascular diseminada. La prueba de dímero-D ha sustituido con mucho a la prueba de PDF.
Factor XIII El factor XIII es el único factor de la coagulación cuya actividad no se evalúa en el TP o TPT por que el punto final de ambas pruebas es la formación de polímeros de fibrina, independientemente de si estos polímeros tienen enlaces cruzados covalentes causados por el factor XIII activado. Debe sospecharse deficiencia de factor XIII en cualquier niño que tenga hemorragia significativa después de la circuncisión o, más raramente, en un paciente adulto con hemorragia inexplicable.
Plasminógeno y a2-antiplasmina La activación del sistema plasminógeno-plasmina puede inferirse por los datos de un TT prolongado, un nivel bajo de fibrinógeno en plasma y un nivel alto de dímero-D. Otra prueba burda usada para medir la activación de plasminógeno-plasmina es el tiempo de lisis de euglobina. Sin embargo, la sensibilidad y especificidad de esta prueba no está bien definida. Durante la trombosis y fibrinolisis extensa se consumen tanto el plasminógeno como la a2-antiplasmina (el inhibidor fisiológico de la plasmina). La medición directa de los niveles en plasma de plasminógeno y a2-antiplasmina en ocasiones es útil para evaluar la extensión de la fibrinolisis y la necesidad de sustituir estas proteínas plasmáticas usando plasma fresco congelado.
PRUEBAS DE PLAQUETAS Y DE FUNCION PLAQUETARIA
Evaluación del frotis de sangre periférica Este examen proporciona información rápida y definitiva para confirmar o investigar la cuenta de plaquetas. En condiciones normales existen 8 a 12 plaquetas por campo de alto poder (magnificación 1,000x), lo que corresponde a una cuenta de plaquetas de 150,000 a 300,000/µl. El frotis muestra también la granularidad de las plaquetas y si existen megatrombocitos.
Tiempo de sangrado Esta prueba mide principalmente la función plaquetaria. Se usa una lanceta estándar para realizar una incisión en el antebrazo. Normalmente el tiempo de sangrado es normal con cuentas de plaquetas mayores de 100,000/µl y se prolonga si el número es menor. Un tiempo de sangrado prolongado con plaquetas de más de 100,000/µl implica disfunción. El tiempo de sangrado es difícil de estandarizar y un tiempo normal no predice la seguridad de un procedimiento quirúrgico o el riesgo de hemorragia.27 No debe usarse como prueba general de escrutinio en el preoperatorio. Puede ser útil para investigar enfermedad de von Willebrand o ciertos trastornos de la función plaquetaria.
Agregometría plaquetaria Los agregómetros de plaquetas son equipos fotométricos para registrar la trasmisión de luz a través de una suspensión de plaquetas. Cuando las plaquetas se agregan la luz pasa a través de la suspensión con más facilidad. Para probar la agregación se añaden concentraciones diluidas de agonistas de plaquetas (v.gr., ADP, epinefrina, colágena y ristocetina) al plasma citrado rico en plaquetas. Con los agonistas débiles, como el ADP y la epinefrina, la onda inicial de agregación va seguida de una onda secundaria. La onda secundaria refleja la inducción de la reacción de liberación de las plaquetas, en la que el contenido de los gránulos se libera para aumentar la agregación plaquetaria. Se observa una onda secundaria subóptima cuando existen defectos de almacenamiento de plaquetas, en los que el contenido de los gránulos está disminuido o la actividad liberadora está alterada. Este último trastorno se asocia con ingesta de aspirina o trombocitopatía relacionada a uremia. Los pacientes con enfermedad de von Willebrand tendrán una respuesta de agregación plaquetaria subóptima a la ristocetina, pero una respuesta normal a otros agonistas.
PRUEBAS DE INHIBIDORES DE LA HEMOSTASIA
Estudios de mezcla El tiempo de coagulación (v.gr., TPT de 60 segundos [normal de 28 a 30 segundos]) puede prolongarse por una deficiencia de un factor de la coagulación o por un inhibidor. Generalmente un inhibidor es un anticuerpo dirigido contra un factor de la coagulación específico o contra un complejo fosfolípido-proteína, el llamado anticoagulante lúpico. En los estudios de mezcla, se coloca un volumen de plasma del paciente con un volumen igual de plasma normal. La mezcla resultante proporcionará por lo menos el 50 por ciento de un factor deficiente y corregirá la alteración. Si el problema es causado por un inhibidor, la mezcla resultante presentará aún un tiempo de coagulación prolongado. Siempre debe hacerse un estudio de mezcla cuando existe un tiempo de coagulación prolongado.
Antitrombina-III Existen bioensayos e inmunoensayos disponibles para evaluar la actividad de la AT-III. Los dos tipos de estudios son necesarios porque los pacientes pueden tener niveles normales de AT-III con las pruebas inmunológicas, pero anormales con las pruebas funcionales.
Proteína C y proteína S Estas proteínas se miden con métodos funcionales e inmunológicos. Debido a que las proteínas C y S son dependientes de vitamina K, su medición puede complicarse en pacientes que toman warfarina. Es mejor medir la proteína C o S cuando el paciente ha estado sin warfarina durante 3 a 4 semanas.
Figuras 1, 2, 3, 5, 6 y 8 a 11 Seward Hung.
Figuras 4, 7 y 12 Marcia Kammerer.
Bibliografía
- Coughlin SR: How thrombin 'talks' to cells: molecular mechanisms and roles in vivo. Arterioscler Thromb Vasc Biol 18:514, 1998
- Kahn ML, Zheng YW, Huang W, et al: A dual thrombin receptor system for platelet activation. Nature 394:690, 1998 [PMID 9716134 ]
- Clemetson KJ: Platelet GPIb-V-IX complex. Thromb Haemost 78:266, 1997
- Sixma JJ, Zanten HV, Huizinga EG, et al: Platelet adhesion to collagen: an update. Thromb Haemost 78:434, 1997 [PMID 9198192 ]
- Shattil SS, Kashiwagi H, Pampori N: Integrin signaling: the platelet paradigm. Blood 91:2645, 1998
- Rapaport SI, Rao LVM: The tissue factor pathway: how it has become a "prima ballerina". Thromb Haemost 74:7, 1995 [PMID 8578528 ]
- Furie B, Bouchard BA, Furie BC: Vitamin K-dependent biosynthesis of g-carboxyglutamic acid. Blood 93:1798, 1999 [PMID 10068650 ]
- Marcum JA, McKenney JB, Rosenberg RD: Acceleration of thrombin-antithrombin complex formation in rat hindquarters via heparinlike molecules bound to the endothelium. J Clin Invest 74:341, 1986
- Esmon CT: The roles of protein C and thrombomodulin in the regulation of blood coagulation. J Biol Chem 264:4743, 1989
- Esmon CT, Ding W, Yasuhiro K, et al: The protein C pathway: new insights. Thromb Haemost 78:70, 1997 [PMID 9198130 ]
- Morten S, Bendz B: Tissue factor pathway inhibitor: clinical deficiency states. Thromb Haemost 78:467, 1997
- Bajaj MS, Bajaj SP: Tissue factor pathway inhibitor: potential therapeutic applications. Thromb Haemost 78:471, 1997 [PMID 9198199 ]
- Smith WL, Garavito RM, DeWitt DL: Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem 271:33157, 1996 [PMID 8969167 ]
- Hawkey CJ: COX-2 inhibitors. Lancet 353:307, 1999
- Moncada S, Higgs A: The L-arginine-nitric oxide pathway. N Engl J Med 329:2002, 1993 [PMID 7504210 ]
- Van Meijer M, Pannekoek H: Structure of plasminogen activator inhibitor 1 (PAI-1) and its function in fibrinolysis: an update. Fibrinolysis 9:263, 1995
- Fay WP, Parker AC, Condrey LR, et al: Human plasminogen activator inhibitor-1 (PAI-1) deficiency: characterization of a large kindred with a null mutation in the PAI-1 gene. Blood 90:204, 1997 [PMID 9207454 ]
- Bajzar L, Morser J, Nesheim M: TAFI, or plasma procarboxypeptidase B, couples coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem 271:16603, 1996 [PMID 8663147 ]
- Broze GJ, Higuchi DA: Coagulation-dependent inhibition of fibrinolysis: role of carboxypeptidase-U and the premature lysis of clots from hemophilic plasma. Blood 88:3815, 1996 [PMID 8916945 ]
- Wang HU, Chen ZF, Anderson DJ: Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93:741, 1998 [PMID 9630219 ]
- Garlanda C, Dejana E: Heterogeneity of endothelial cells: specific markers. Arterioscler Thromb Vasc Biol 17:1193, 1997 [PMID 9261246 ]
- Dilawari JB, Bambery P, Chawla Y, et al: Hepatic outflow obstruction (Budd-Chiari syndrome): experience with 177 patients and a review of the literature. Medicine (Baltimore) 73:21, 1994 [PMID 8309360 ]
- Kaushansky K: Thrombopoietin. N Engl J Med 339:746, 1998
- Kuter DJ, Rosenberg RD: The reciprocal relationship of thrombopoietin (c-Mpl ligand) to changes in the platelet mass during busulfan-induced thrombocytopenia in the rabbit. Blood 85:2720, 1995 [PMID 7742532 ]
- Hirsh J, Dalen JE, Anderson DR, et al: Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 114(suppl):445S, 1998 [PMID 9822057 ]
- Moll S, Ortel TL: Monitoring warfarin therapy in patients with lupus anticoagulants. Ann Intern Med 127:177, 1997 [PMID 9245222 ]
- The bleeding time (editorial). Lancet 337:1447, 1991