Reumatología
⭳ Abrir artículo (PDF)363.0 KBEste artículo fue revisado respecto a la Edición 3/2000. Ver esa versión →
Contenido del artículo
V ESCLERODERMIA Y PADECIMIENTOS RELACIONADOS
V ESCLERODERMIA Y PADECIMIENTOS RELACIONADOS
DR. DWIGHT R. ROBINSON
Esclerodermia
La esclerodermia, o esclerosis sistémica, se caracteriza por el depósito de tejido conjuntivo fibroso en la piel y, a menudo, en muchos otros sistemas orgánicos. Se puede acompañar de lesiones vasculares, en especial en la piel, pulmones y riñones. La evolución es variable, aunque por lo general progresa lentamente; no se conoce ningún tratamiento que altere el curso de la enfermedad. La esclerodermia se puede presentar de forma localizada y ser en apariencia compatible con una vida normal; sin embargo, la afección generalizada puede ser mortal.
PATOGENIA
La etiología y los mecanismos responsables de la esclerodermia se desconocen. Al parecer existe proliferación anormal del tejido conjuntivo fibroso, incluyendo de las fibras de colágena, en los tejidos afectados.1 Se observa una cantidad anormal de colágena, pero que parece ser cualitativamente normal, y no existe evidencia de trastornos en otras proteínas o polisacáridos del tejido conjuntivo. Los fibroblastos obtenidos de la piel afectada y que proliferan en cultivos celulares bajo ciertas condiciones experimentales tienden a producir más colágena que los fibroblastos de piel normal. Estas observaciones sugieren que la regulación de la síntesis de colágena por los fibroblastos está alterada en la esclerodermia. Aún más, los fibroblastos de la dermis con esclerodermia producen más cadenas a de colágena tipo V (A y B) que los fibroblastos de dermis normal.
Otros estudios experimentales han llevado a la conclusión de que pueden existir trastornos de la inmunidad celular en la esclerodermia. Las células T de pacientes con esta enfermedad muestran cierta hipersensibilidad a la colágena y a los extractos de piel in vitro. Sin embargo, esta hipersensibilidad podría ser secundaria, más que un fenómeno primario.
La importancia del fenómeno vascular en la esclerodermia (fenómeno de Raynaud, hipertensión pulmonar, afección esclerodermatosa del riñón y alteraciones capilares en la dermis) muestran la importancia de los cambios patológicos vasculares en esta enfermedad. La producción excesiva de cadenas a de colágena tipo V (A y B) sugiere un defecto en las células de origen vascular, apoyando el concepto de que los trastornos vasculares pueden ser de gran importancia en la esclerodermia.
MANIFESTACIONES CLINICAS
La esclerodermia se clasifica como difusa o limitada con base en su extensión cutánea y afección a órganos internos. La forma difusa se caracteriza por engrosamiento y fibrosis de la piel en la porción proximal de las extremidades y el tronco; es frecuente que se afecten el corazón, los pulmones, los riñones y el tubo digestivo por debajo del esófago. La esclerodermia limitada se manifiesta por afección cutánea de cara y manos, y la lesión visceral es mucho menos frecuente. La forma limitada tiene mejor pronóstico que la difusa, con excepción de los casos en que existe hipertensión pulmonar [ver tabla 1].
La esclerodermia ocurre también en una forma localizada en la que la enfermedad se limita a la piel, el tejido subcutáneo y el músculo, no se asocia con fenómeno de Raynaud, ni con acroesclerosis o afección visceral. Existen dos formas localizadas de esclerodermia: la morfea, que se manifiesta por placas de induración cutánea de tamaño variable, y la esclerodermia lineal, que se caracteriza por bandas de induración en la piel sobre la cara o una extremidad aislada. La esclerodermia lineal puede asociarse con atrofia muscular y afección del hueso subyacente. Suele afectar niños o adultos jóvenes y causar deterioro importante del crecimiento en la parte afectada. En la morfea las lesiones pueden persistir durante meses o años, seguido de mejoría o resolución [ver tabla 1].2-4
Piel
En las fases tempranas de la enfermedad la piel se engrosa y manifiesta un edema difuso de grado leve, sin fóvea. Posteriormente puede atrofiarse y al mismo tiempo perder sus anexos (i.e., pelo, glándulas cebáceas y glándulas sudoríparas). La piel también pierde sus pliegues y aparece acartonada, estirada y unida a las estructuras subyacentes. A menudo la afección cutánea es más prominente en las manos y en los dedos de las manos (esclerodactilia); con frecuencia también se afecta la cara. El estiramiento de la piel facial produce una disminución de las líneas cutáneas, así como disminución del tamaño de la boca, que adquiere un aspecto protrusivo. La induración de la piel llega a limitar la movilidad, en especial en los dedos de las manos, lo cual también puede ocasionar el desarrollo de contracturas en flexión [ver figura 1].3, 4 Con frecuencia la afección es más difusa, con induración de la piel de todo el organismo.
Otras alteraciones cutáneas pueden acompañar estos cambios. Las telangiectasias son frecuentes y llegan a ser muy numerosas; se caracterizan por áreas rojas, aplanadas, de uno a varios milímetros de diámetro, que están bien delimitadas de la piel adyacente [ver figura 2]. A menudo son más prominentes en la cara, manos y mucosa oral. La calcinosis, el depósito de cristales de hidroxiapatita en tejidos subcutáneos, puede estar limitada o diseminada ampliamente y, por lo general, se localiza alrededor de las cápsulas articulares. La ulceración cutánea sobre los depósitos de calcio produce la salida de un material blanquecino con una consistencia parecida a la pasta de dientes. Puede observarse hiperpigmentación melánica difusa sobre toda la superficie de la piel; a menudo, también se observan áreas de vitiligo.
Desde el punto de vista patológico la afección cutánea se caracteriza por aumento de los haces de colágena en la dermis y tejidos subcutáneos, causando pérdida de las estructuras accesorias y calcificación en algunos sitios [ver figura 3]. El examen del lecho vascular puede revelar cambios en las paredes de las arterias y las arteriolas, incluyendo fibrosis de la íntima y la media y estenosis de la luz de los vasos. Aunque ocurre obstrucción de las arterias de gran calibre, ésta es mucho menos frecuente que la de las arterias pequeñas.5 Estas lesiones vasculares parecen corresponder a las áreas de isquemia en la punta de los dedos, aunque también se llegan a desarrollar cambios semejantes en los vasos de otros órganos.
Los cambios patológicos en el sistema vascular, en especial en las extremidades, son frecuentes y por lo general se manifiestan en forma de fenómeno de Raynaud. Esta entidad consiste en episodios intermitentes de vasoespasmo de las pequeñas arterias, casi siempre en los dedos de las manos. El fenómeno se caracteriza por un cambio en tres fases de la coloración de los dedos de las manos, comenzando típicamente con palidez, seguido de cianosis y, al final, de coloración rojiza causada por hiperemia reactiva [ver figura 4]. Por lo general los episodios son desencadenados o exacerbados por el frío y mitigados al calentar la parte afectada; sin embargo, en casos severos puede resultar menos aparente la relación con la temperatura ambiente. La isquemia prolongada puede producir dolor intenso en las puntas de los dedos, ulceración e incluso gangrena.3, 4
Sistema musculoesquelético
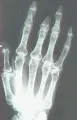
En la esclerodermia puede ocurrir una artritis inflamatoria leve que en general tiene una distribución simétrica y semeja a una forma leve de artritis reumatoide.3, 4 Las erosiones óseas yuxtarticulares son frecuentes, en especial sobre las articulaciones interfalángicas distales, pero el grado de destrucción casi siempre es menor al observado en la artritis reumatoide.6 A menudo se desarrollan contracturas en flexión de los dedos de las manos, que se relacionan más con fibrosis de los tendones y cápsulas articulares. Un hallazgo característico es la presencia de frotes de fricción y crepitación, apreciados mediante palpación o auscultación sobre los tendones y bursas y que se relacionan con los cambios fibróticos de los tejidos subyacentes. La fibrosis de éstas y otras estructuras del tejido conjuntivo puede contribuir a las contracturas fibrosas, sobre todo en las manos.
Otra complicación ósea es la osteólisis acra, que consiste en la resorción de las falanges terminales y tejidos blandos adyacentes con el consiguiente acortamiento de las puntas de los dedos [ver figura 5]. El mecanismo de este proceso, que puede ocurrir sin ulceración, es poco claro.
En el músculo esquelético puede desarrollarse una miopatía
parecida a la polimiositis, que se manifiesta sobre todo por debilidad y
atrofia de los grupos musculares proximales, en especial en la cintura
pélvica y escapular. La lesión histológica de fondo, las
elevaciones de las enzimas musculares séricas y los cambios
electromiográficos son parecidos a los encontrados en la polimiositis,
con frecuencia se observa fibrosis intersticial. La miopatía puede
mejorar con esteroides. Se considera que los pacientes que tienen signos de
esclerodermia, polimiositis y lupus eritematoso generalizado (LEG) tienen
enfermedad mixta del tejido conjuntivo.
Tubo digestivo
Muchas zonas del tubo digestivo pueden afectarse en la esclerodermia.3,4 El síndrome de Sjögren, que causa xeroftalmia y xerostomía, se presenta en una tercera parte de los pacientes con esclerodermia y suele asociarse con anticuerpos séricos contra los antígenos SS-A y SS-B (también denominados Ro y La, respectivamente).
Pueden ocurrir hipomotilidad y dilatación del esófago, del intestino delgado y del colon cuando el tejido conjuntivo fibrótico remplaza las capas de músculo liso. En estos casos no existe relación con disfunción de la inervación autonómica. Se presenta hipomotilidad del esófago, demostrable por cinefluoroscopía y estudios manométricos, en más del 90 porciento de los casos, muchos de ellos asintomáticos [ver figura 6]. La ausencia de ondas peristálticas normales en los dos tercios inferiores del esófago puede causar disfagia, la insuficiencia del esfínter gastroesofágico provoca esofagitis por reflujo y, en ocasiones, estenosis.
La función gástrica por lo general está conservada, pero la hipomotilidad del intestino delgado y grueso produce retardo del tránsito intestinal, dilatación, constipación o incluso obstrucción con grandes asas de intestino dilatadas. La presencia de malabsorción de grasas, que produce diarrea acuosa, se ha atribuido a proliferación en el intestino. El gas disecante dentro de la pared intestinal (neumomatosis intestinal) puede filtrarse hacia la cavidad peritoneal, simulando una perforación de víscera hueca. Se pueden desarrollar divertículos en colon, de boca amplia, característicos y patognomónicos de esclerodermia.
Pulmones
La esclerodermia se relaciona con una elevada incidencia de hipertensión pulmonar, aún en sujetos sin otros síntomas cardiopulmonares.3, 4 En estos individuos ocurre fibrosis de la íntima en las paredes de las arterias pulmonares. También se puede detectar una fibrosis pulmonar intersticial difusa, relacionada con estertores inspiratorios en las bases pulmonares, con síntomas o sin síntomas respiratorios. Sin embargo, la hipertensión pulmonar y la fibrosis intersticial difusa pueden ocurrir de modo independiente. Estas alteraciones pueden progresar a la insuficiencia pulmonar y al cor pulmonale. A pesar de que las manifestaciones pulmonares suelen ser crónicas, algunos pacientes desarrollan hipertensión pulmonar aguda e insuficiencia respiratoria rápidamente progresiva. En ocasiones, los enfermos con fibrosis pulmonar debida a esclerodermia tienen carcinoma de células alveolares, el cual también se relaciona con otras enfermedades que producen fibrosis pulmonar.
Corazón
La lesión cardiaca típica de la esclerodermia se caracteriza por remplazo extenso del músculo cardiaco con tejido fibroso, provocando arritmias, trastornos de la conducción e insuficiencia cardiaca congestiva.3,4 Ocurre disfunción miocárdica menos extensa en la mayoría de los pacientes con esclerodermia difusa. La fibrosis miocárdica se asocia con necrosis y contractura en banda, que es posible que sean causadas por lesión por reperfusión. También se ha sugerido que se presenta un trastorno vasoespástico reversible de las arterias coronarias de pequeño calibre, al que se ha denominado fenómeno de Raynaud miocárdico. En la mayoría de los pacientes con esclerodermia ocurren defectos reversibles inducidos por el ejercicio o defectos fijos que pueden observarse con el gamagrama con talio. Debido a que los angiogramas son normales, estos trastornos indican enfermedad de vasos intramiocárdicos de muy pequeño calibre.
Otras causas de insuficiencia cardiaca incluyen cor pulmonale e hipertensión secundaria a las manifestaciones renales. La pericarditis no es rara y se puede acompañar de dolor, derrame pericárdico y taponamiento cardiaco. Los pequeños derrames pericárdicos detectados mediante ecocardiografía tienen poca importancia clínica, pero los derrames grandes pueden indicar afección seria en otros órganos y tener un pronóstico desfavorable. La ecocardiografía puede proporcionar evidencias de hipertensión pulmonar, aunque esta alteración puede aparecer cuando los criterios ecocardiográficos están ausentes. Rara vez la esclerodermia afecta los grandes vasos, causando enfermedad oclusiva e insuficiencia arterial periférica.
Riñón
La proteinuria leve crónica y la hipertensión leve son efectos comunes de la esclerodermia. La complicación renal más importante es la hipertensión maligna, la cual produce insuficiencia renal oligúrica de rápida evolución, que a menudo es mortal. Desde el punto de vista patológico, se caracteriza por nefroesclerosis maligna, incluyendo necrosis de las paredes arteriales y áreas de infarto renal.3, 4
Otros órganos y sistemas
No es frecuente la afección de otros órganos en la esclerodermia difusa; sin embargo, existen varios informes de cirrosis biliar, en especial asociada con la variante CREST (calcinosis, fenómeno de Raynaud, trastornos de la motilidad esofágica, esclerodactilia y telangiectasias) de la esclerodermia. Algunos pacientes pueden no tener una o más de las manifestaciones características del síndrome.3,4
Aunque el sistema nervioso central no se afecta, puede ocurrir neuropatía periférica. La forma más común es la neuropatía de una o más de las tres ramas del trigémino, unilateral o bilateral, manifestada por dolor e hipoestesias progresivas. La neuralgia del trigémino a menudo se relaciona con miositis, títulos altos de anticuerpos a la ribonucleoproteína y otras características de la enfermedad mixta del tejido conjuntivo. Se han postulado como causas, lesiones vasculares y atrapamiento del nervio por tejido conjuntivo, pero el mecanismo exacto sigue siendo desconocido.
Las consecuencias hematológicas de la esclerodermia son raras. La anemia microangiopática es poco frecuente y no se ha dilucidado el mecanismo de producción.3, 4
ALTERACIONES DE LABORATORIO
En cerca del 95 porciento de los enfermos con esclerodermia se encuentran resultados positivos para anticuerpos antinucleares [ver tabla 1].7 Este alto grado de sensibilidad se basa en el uso de líneas celulares de carcinoma epitelial humano (HEp-2) en lugar de los sustratos convencionales como los cortes de riñón de ratón.
Algunos anticuerpos dirigidos contra ciertos antígenos son específicos de la esclerodermia, y cada tipo de anticuerpo se asocia con un patrón clínico particular de la enfermdad. Los anticuerpos específicos están dirigidos contra la topoisomerasa I, proteínas del centrómero, polimerasas ARN o componentes nucleares [ver tabla 2]. Los anticuerpos contra topoisomerasa I (Scl-70) y contra polimerasas ARN (por lo general polimerasa III) se observan en pacientes con esclerodermia difusa.7,8 Los anticuerpos anticentrómero se asocian con esclerodermia limitada o síndrome de CREST y los anticuerpos contra ribonucleoproteína nuclear (RNPn) se asocian con esclerodermia difusa y limitada. Los anticuerpos contra PM-1 y Ku son poco frecuentes. La presencia de estos anticuerpos suele asociarse con manifestaciones clínicas mixtas de polimiositis y esclerodermia. Alrededor de la mitad de los pacientes con esclerodermia localizada tienen anticuerpos contra histonas [ver tabla 2].9
Pueden aparecer diversas alteraciones parecidas a las encontradas en otras enfermedades del tejido conjuntivo. Puede ocurrir hiperglobulinemia leve, que casi siempre representa un aumento difuso en la IgG.3 En cerca de una cuarta parte de los casos se encuentran títulos bajos de actividad del factor reumatoide. Se han descrito alteraciones capilares en la dermis de los pliegues del lecho ungueal utilizando microscopía in vivo. Estos cambios son comunes en la esclerodermia y en la enfermedad mixta del tejido conjuntivo, pero rara vez se encuentran en pacientes con LEG, enfermedad de Raynaud o en individuos normales.
EVOLUCION CLINICA
Los pacientes con esclerodermia localizada tienen un buen pronóstico para la vida, debido a que las formas localizadas rara vez se relacionan con afección visceral.3, 4 El síndrome de CREST provoca afección limitada de la piel, con frecuencia sólo de las manos y la cara; el pronóstico para los pacientes con este síndrome en general es favorable, a menos que exista afección visceral. No obstante, incluso la variante limitada del síndrome de CREST tiende a tener un curso sin remisiones y lentamente progresivo.
Las manifestaciones y evolución de la esclerodermia difusa son muy variables, por lo que su velocidad de progresión es difícil de predecir. Con excepción de algunos de los cambios esclerodermatosos en la enfermedad mixta del tejido conjuntivo, el padecimiento rara vez remite completamente. Muchos enfermos siguen un curso indolente, con pocos cambios durante varios años; sin embargo, la progresión puede ser más rápida. La afección de vísceras como el corazón, el pulmón o en particular el riñón, indican un mal pronóstico. Dos características de la esclerodermia, la inflamación activa (manifestada por incremento de la velocidad de sedimentación globular) y la evidencia de enfermedad cardiopulmonar, renal o ambas, al año del diagnóstico, indican mal pronóstico.10 La supervivencia a cinco años para los pacientes con esclerodermia difusa es de alrededor del 50 porciento.
TRATAMIENTO
Por desgracia, ningún tratamiento para la esclerodermia ha demostrado ser eficaz.3, 4 La evaluación de los agentes terapéuticos propuestos se basa en los cambios en la textura o rigidez de la piel, pero estos indicadores son muy difíciles de cuantificar. Las determinaciones más objetivas, como las pruebas de función respiratoria en los pacientes con afección pulmonar, por lo general no son modificadas por ningún tratamiento farmacológico.
En general se acepta que los esteroides no tienen lugar en el tratamiento de la esclerodermia, ni los inmunosupresores o los medicamenos citotóxicos son eficaces.
Se ha reportado que la penicilamina es eficaz en el tratamiento del engrosamiento cutáneo y la fibrosis pulmonar, pero no ha sido estudiada en trabajos bien controlados. Los estudios controlados sobre el uso de colchicina no han confirmado los resultados alentadores de los primeros estudios no controlados. Los estudios no controlados realizados con inmunosupresores sugieren cierto beneficio de la azatioprina y el fluorouracilo (5-FU), pero un estudio controlado con clorambucil no demostró éxito. La ciclosporina causa citotoxicidad renal. Los resultados de los estudios preliminares con interferón gama y plasmaféresis no han sido alentadores. El tratamiento antiplaquetario con dipiridamol y aspirina no fue eficaz en un estudio controlado.
Aunque no se ha demostrado que ningún tratamiento modifique la evolución de la esclerodermia, el tratamiento de sostén parece ser útil para algunos aspectos del padecimiento. La evolución de la enfermedad puede alterarse en el caso de la crisis renal, que era antes la principal causa de muerte en pacientes con esclerodermia. El tratamiento de esta complicación grave con inhibidores de la enzima convertidora de angiotensina (ECA) y otros antihipertensivos potentes parece controlar la hipertensión y detener el deterioro de la función renal. Sin embargo, este tratamiento es más útil si se inicia antes de que los niveles de creatinina sérica excedan 3 mg/dl. En algunos pacientes continúa la progresión a insuficiencia renal a pesar del buen control de la presión arterial, requiriéndose diálisis y quizá trasplante renal.11
La fibrosis pulmonar es común en la esclerodermia y en la actualidad es la primera causa de muerte. Los estudios preliminares han demostrado que en pacientes con fibrosis pulmonar y alveolitis incipientes, la progresión puede detenerse por el tratamiento a largo plazo a base de ciclofosfamida por vía oral (1 a 2 mg/kg/día) y dosis bajas de prednisona (< 10 mg/día).12 La hipertensión pulmonar es una manifestación seria de esclerodermia y por lo general es difícil controlarla con vasodilatadores u otros agentes.
El fenómeno de Raynaud debe manejarse evitando la exposición al frío, y esto se aplica no solo a las extremidades sino a todo el organismo. Debe suspenderse el tabaquismo. El tratamiento con bloqueadores de los canales del calcio de acción prolongada o de antagonistas adrenérgicos alfa1, como el prazosín, puede ser benéfico. Las ulceraciones isquémicas en los dedos pueden ser difíciles de tratar y en ocasiones progresan a gangrena. Se ha reportado que los vasodilatadores experimentales, incluyendo la ketanserina, antagonista serotoninérgico, y el iloprost, análogo de prostaciclina, pueden ser útiles.13,14 La simpatectomía suele tener poco efecto sobre el fenómeno de Raynaud crónico y estable, y no se ha demostrado que influya en la evolución de la esclerodermia. Por lo tanto, el procedimiento debe reservarse para las crisis isquémicas.
La esofagitis, el reflujo esofágico y la estenosis se tratan por medio de elevación de la cabecera de la cama del paciente y administrando antiácidos, antagonistas de los receptores H2 u omeprazol. La motilidad esofágica puede aumentarse con el uso de metoclopramida. Cuando existe estenosis esofágica pueden requerirse dilataciones. Los estudios con seguimiento a corto plazo han demostrado que el análogo de somatostatina ocreótido y el agente procinético cisaprida, mejoran la motilidad intestinal y reducen la proliferación bacteriana en los pacientes con esclerodermia, pero no se conoce la eficacia a largo plazo de estos agentes.2,3,15,16 En ocasiones el síndrome de malabsorción puede responder a antibióticos de amplio espectro.
Fascitis eosinofílica
La fascitis eosinofílica3, 4 puede semejar en un principio a la esclerodermia. Se caracteriza por la presencia de dolor, tumefacción y sensibilidad en las extremidades, seguidos por induración de la piel y tejidos subcutáneos. También puede haber cierta limitación de la motilidad articular, pero no se observan el fenómeno de Raynaud ni otras manifestaciones de la esclerodermia.
Las alteraciones en las pruebas de laboratorio incluyen eosinofilia, que puede llegar a ser considerable, elevación de la velocidad de sedimentación e hiperglobulinemia. Los anticuerpos antinucleares y los resultados del factor reumatoide son negativos.
Las biopsias de las áreas afectadas muestran inflamación y engrosamiento desde la aponeurosis profunda a los tejidos subcutáneos. La piel tiene un aspecto normal, pero la aponeurosis profunda subyacente está infiltrada por linfocitos, células plasmáticas, histiocitos y, algunas veces, eosinofílicos.
La fascitis eosinofílica parece ser autolimitada o responde satisfactoriamente al tratamiento con dosis bajas de corticosteroides. La etiología sigue siendo desconocida, pero se han publicado varios casos posteriores a ejercicios muscular intenso.
Síndrome de mialgias y eosinofilia
En 1989 se describió e identificó un nuevo síndrome que consiste en eosinofilia y mialgias relacionadas con la ingestión de L-triptáfano.3, 4, 17-21 Este síndrome se caracteriza por la presencia de mialgias severas e incapacitantes y fatiga durante varias semanas. También pueden presentarse disnea y tos. La afección cutánea consiste en eritema variables y edema, así como cambios a la esclerodermia, por lo general sin las manifestaciones viscerales de la misma. Se pueden presentar infiltrados pulmonares intersticiales, hipoxia, hipertensión pulmonar y neumonitis por hipersensibilidad. La polineuropatía descrita toma la forma de una mononeuritis múltiple.
No existen pruebas de laboratorio diagnósticas. Los Centros para el Control y Prevención de las Enfermedades en los EUA mencionan tres criterios diagnósticos en su definición: una cuenta de eosinófilos mayor de 1,000 células/mm3, mialgia incapacitante o severa y la exclusión de otras causas posibles de los dos primeros criterios. La cuenta de eosinófilos puede llegar a 30,000/mm3. Los niveles de aminotransferasas y aldolasa pueden estar aumentados, reflejando daño hepático, y en ocasiones aumenta también la creantina cinasa, lo que indica afección muscular. Las biopsias revelan infiltrados inflamatorios que consisten en linfocitos, células plasmáticas, macrófagos y eosinófilos, acompañados de fibrosis de la dermis, tejido subcutáneo, fascia y músculo. También pueden observarse infiltrados inflamatorios del hígado.
Se han descrito alteraciones en el metabolismo del triptofano. El síndrome de mialgias y eosinofilia puede ser el resultado de un trastorno del metabolismo del L-triptosfano, que origina cantidades excesivas de este metabolito en personas susceptibles. También es posible que un contaminante tóxico añadido a las preparaciones de L-triptofano contribuya a la manifestación de la enfermedad.
Los resultados de un estudio epidemiológico indican que la epidemia fue causada por un contaminante o por alteración en un subgrupo del producto de un fabricante en Japón. La Food and Drug Administration de los EUA ha prohibido la distribución y venta de suplementos dietéticos a base de L-triptofano.
Enfermedades del tejido conjuntivo asociadas con cirugía estética
Dos procedimientos quirúrgicos estéticos se han asociado con el desarrollo subsecuente de enfermedades reumáticas: los implantes de mama de silicón y los implantes dérmicos de colágena.22 Algunas mujeres que fueron sometidas a mamoplastía de aumento con implantes de silicón han desarrollado manifestaciones de una enfermedad del tejido conjuntivo, sobre todo esclerodermia y menos, lupus eritematoso generalizado o artritis reumatoide. El hallazgo reumático más frecuente es la sinovitis leve y asimétrica. Se han detectado anticuerpos contra componentes mucleares por medio de técnicas de Western blot con más frecuencia en las pacientes sometidas a mamoplastía de aumento que en las poblaciones control.23,24
Se ha reportado polimiositis o dermatomiositis en varios pacientes que han recibido implantes de colágena bovina. En un estudio, la mayoría de los pacientes con miositis sufrieron reacciones de hipersensibilidad tardía a la colágena bovina y aumento en las concentraciones de anticuerpos contra colágena. Se ha encontrado que la incidencia de miositis es más alta en pacientes tratadas con colágena que en la población control.25
Estos reportes sugieren, aunque no demuestran, que existe una relación causal entre los implantes de silicón o colágena y las enfermedades del tejido conjuntivo. Se requieren de estudios epidemiológicos más detallados para determinar si existe esta relación causal.22
Bibliografía
DR. DWIGHT R. ROBINSON
Esclerodermia
La esclerodermia, o esclerosis sistémica, se caracteriza por el depósito de tejido conjuntivo fibroso en la piel y, a menudo, en muchos otros sistemas orgánicos. Se puede acompañar de lesiones vasculares, en especial en la piel, pulmones y riñones. La evolución es variable, aunque por lo general progresa lentamente; no se conoce ningún tratamiento que altere el curso de la enfermedad. La esclerodermia se puede presentar de forma localizada y ser en apariencia compatible con una vida normal; sin embargo, la afección generalizada puede ser mortal.
PATOGENIA
La etiología y los mecanismos responsables de la esclerodermia se desconocen. Al parecer existe proliferación anormal del tejido conjuntivo fibroso, incluyendo de las fibras de colágena, en los tejidos afectados.1 Se observa una cantidad anormal de colágena, pero que parece ser cualitativamente normal, y no existe evidencia de trastornos en otras proteínas o polisacáridos del tejido conjuntivo. Los fibroblastos obtenidos de la piel afectada y que proliferan en cultivos celulares bajo ciertas condiciones experimentales tienden a producir más colágena que los fibroblastos de piel normal. Estas observaciones sugieren que la regulación de la síntesis de colágena por los fibroblastos está alterada en la esclerodermia. Aún más, los fibroblastos de la dermis con esclerodermia producen más cadenas a de colágena tipo V (A y B) que los fibroblastos de dermis normal.
Otros estudios experimentales han llevado a la conclusión de que pueden existir trastornos de la inmunidad celular en la esclerodermia. Las células T de pacientes con esta enfermedad muestran cierta hipersensibilidad a la colágena y a los extractos de piel in vitro. Sin embargo, esta hipersensibilidad podría ser secundaria, más que un fenómeno primario.
La importancia del fenómeno vascular en la esclerodermia (fenómeno de Raynaud, hipertensión pulmonar, afección esclerodermatosa del riñón y alteraciones capilares en la dermis) muestran la importancia de los cambios patológicos vasculares en esta enfermedad. La producción excesiva de cadenas a de colágena tipo V (A y B) sugiere un defecto en las células de origen vascular, apoyando el concepto de que los trastornos vasculares pueden ser de gran importancia en la esclerodermia.
MANIFESTACIONES CLINICAS
La esclerodermia se clasifica como difusa o limitada con base en su extensión cutánea y afección a órganos internos. La forma difusa se caracteriza por engrosamiento y fibrosis de la piel en la porción proximal de las extremidades y el tronco; es frecuente que se afecten el corazón, los pulmones, los riñones y el tubo digestivo por debajo del esófago. La esclerodermia limitada se manifiesta por afección cutánea de cara y manos, y la lesión visceral es mucho menos frecuente. La forma limitada tiene mejor pronóstico que la difusa, con excepción de los casos en que existe hipertensión pulmonar [ver tabla 1].
|
||||||
|
La esclerodermia ocurre también en una forma localizada en la que la enfermedad se limita a la piel, el tejido subcutáneo y el músculo, no se asocia con fenómeno de Raynaud, ni con acroesclerosis o afección visceral. Existen dos formas localizadas de esclerodermia: la morfea, que se manifiesta por placas de induración cutánea de tamaño variable, y la esclerodermia lineal, que se caracteriza por bandas de induración en la piel sobre la cara o una extremidad aislada. La esclerodermia lineal puede asociarse con atrofia muscular y afección del hueso subyacente. Suele afectar niños o adultos jóvenes y causar deterioro importante del crecimiento en la parte afectada. En la morfea las lesiones pueden persistir durante meses o años, seguido de mejoría o resolución [ver tabla 1].2-4
Piel
En las fases tempranas de la enfermedad la piel se engrosa y manifiesta un edema difuso de grado leve, sin fóvea. Posteriormente puede atrofiarse y al mismo tiempo perder sus anexos (i.e., pelo, glándulas cebáceas y glándulas sudoríparas). La piel también pierde sus pliegues y aparece acartonada, estirada y unida a las estructuras subyacentes. A menudo la afección cutánea es más prominente en las manos y en los dedos de las manos (esclerodactilia); con frecuencia también se afecta la cara. El estiramiento de la piel facial produce una disminución de las líneas cutáneas, así como disminución del tamaño de la boca, que adquiere un aspecto protrusivo. La induración de la piel llega a limitar la movilidad, en especial en los dedos de las manos, lo cual también puede ocasionar el desarrollo de contracturas en flexión [ver figura 1].3, 4 Con frecuencia la afección es más difusa, con induración de la piel de todo el organismo.
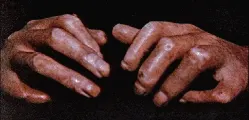
|
| Figura 1 |
| Efectos de la esclerodermia prolongada en manos |
Otras alteraciones cutáneas pueden acompañar estos cambios. Las telangiectasias son frecuentes y llegan a ser muy numerosas; se caracterizan por áreas rojas, aplanadas, de uno a varios milímetros de diámetro, que están bien delimitadas de la piel adyacente [ver figura 2]. A menudo son más prominentes en la cara, manos y mucosa oral. La calcinosis, el depósito de cristales de hidroxiapatita en tejidos subcutáneos, puede estar limitada o diseminada ampliamente y, por lo general, se localiza alrededor de las cápsulas articulares. La ulceración cutánea sobre los depósitos de calcio produce la salida de un material blanquecino con una consistencia parecida a la pasta de dientes. Puede observarse hiperpigmentación melánica difusa sobre toda la superficie de la piel; a menudo, también se observan áreas de vitiligo.

|
| Figura 2 |
| Telangiectasias: variante CREST |
Desde el punto de vista patológico la afección cutánea se caracteriza por aumento de los haces de colágena en la dermis y tejidos subcutáneos, causando pérdida de las estructuras accesorias y calcificación en algunos sitios [ver figura 3]. El examen del lecho vascular puede revelar cambios en las paredes de las arterias y las arteriolas, incluyendo fibrosis de la íntima y la media y estenosis de la luz de los vasos. Aunque ocurre obstrucción de las arterias de gran calibre, ésta es mucho menos frecuente que la de las arterias pequeñas.5 Estas lesiones vasculares parecen corresponder a las áreas de isquemia en la punta de los dedos, aunque también se llegan a desarrollar cambios semejantes en los vasos de otros órganos.
|
| Figura 3 |
| Calcinosis en articulaciones |
Los cambios patológicos en el sistema vascular, en especial en las extremidades, son frecuentes y por lo general se manifiestan en forma de fenómeno de Raynaud. Esta entidad consiste en episodios intermitentes de vasoespasmo de las pequeñas arterias, casi siempre en los dedos de las manos. El fenómeno se caracteriza por un cambio en tres fases de la coloración de los dedos de las manos, comenzando típicamente con palidez, seguido de cianosis y, al final, de coloración rojiza causada por hiperemia reactiva [ver figura 4]. Por lo general los episodios son desencadenados o exacerbados por el frío y mitigados al calentar la parte afectada; sin embargo, en casos severos puede resultar menos aparente la relación con la temperatura ambiente. La isquemia prolongada puede producir dolor intenso en las puntas de los dedos, ulceración e incluso gangrena.3, 4

|
| Figura 4 |
| Fenómeno de Raynaud: fase cianótica |
Sistema musculoesquelético
En la esclerodermia puede ocurrir una artritis inflamatoria leve que en general tiene una distribución simétrica y semeja a una forma leve de artritis reumatoide.3, 4 Las erosiones óseas yuxtarticulares son frecuentes, en especial sobre las articulaciones interfalángicas distales, pero el grado de destrucción casi siempre es menor al observado en la artritis reumatoide.6 A menudo se desarrollan contracturas en flexión de los dedos de las manos, que se relacionan más con fibrosis de los tendones y cápsulas articulares. Un hallazgo característico es la presencia de frotes de fricción y crepitación, apreciados mediante palpación o auscultación sobre los tendones y bursas y que se relacionan con los cambios fibróticos de los tejidos subyacentes. La fibrosis de éstas y otras estructuras del tejido conjuntivo puede contribuir a las contracturas fibrosas, sobre todo en las manos.
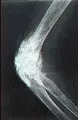
Otra complicación ósea es la osteólisis acra, que consiste en la resorción de las falanges terminales y tejidos blandos adyacentes con el consiguiente acortamiento de las puntas de los dedos [ver figura 5]. El mecanismo de este proceso, que puede ocurrir sin ulceración, es poco claro.
|
|
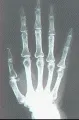
Tubo digestivo
Muchas zonas del tubo digestivo pueden afectarse en la esclerodermia.3,4 El síndrome de Sjögren, que causa xeroftalmia y xerostomía, se presenta en una tercera parte de los pacientes con esclerodermia y suele asociarse con anticuerpos séricos contra los antígenos SS-A y SS-B (también denominados Ro y La, respectivamente).
Pueden ocurrir hipomotilidad y dilatación del esófago, del intestino delgado y del colon cuando el tejido conjuntivo fibrótico remplaza las capas de músculo liso. En estos casos no existe relación con disfunción de la inervación autonómica. Se presenta hipomotilidad del esófago, demostrable por cinefluoroscopía y estudios manométricos, en más del 90 porciento de los casos, muchos de ellos asintomáticos [ver figura 6]. La ausencia de ondas peristálticas normales en los dos tercios inferiores del esófago puede causar disfagia, la insuficiencia del esfínter gastroesofágico provoca esofagitis por reflujo y, en ocasiones, estenosis.
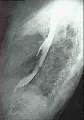
|
| Figura 6 |
| Cambios esofágicos en esclerodermia |
La función gástrica por lo general está conservada, pero la hipomotilidad del intestino delgado y grueso produce retardo del tránsito intestinal, dilatación, constipación o incluso obstrucción con grandes asas de intestino dilatadas. La presencia de malabsorción de grasas, que produce diarrea acuosa, se ha atribuido a proliferación en el intestino. El gas disecante dentro de la pared intestinal (neumomatosis intestinal) puede filtrarse hacia la cavidad peritoneal, simulando una perforación de víscera hueca. Se pueden desarrollar divertículos en colon, de boca amplia, característicos y patognomónicos de esclerodermia.
Pulmones
La esclerodermia se relaciona con una elevada incidencia de hipertensión pulmonar, aún en sujetos sin otros síntomas cardiopulmonares.3, 4 En estos individuos ocurre fibrosis de la íntima en las paredes de las arterias pulmonares. También se puede detectar una fibrosis pulmonar intersticial difusa, relacionada con estertores inspiratorios en las bases pulmonares, con síntomas o sin síntomas respiratorios. Sin embargo, la hipertensión pulmonar y la fibrosis intersticial difusa pueden ocurrir de modo independiente. Estas alteraciones pueden progresar a la insuficiencia pulmonar y al cor pulmonale. A pesar de que las manifestaciones pulmonares suelen ser crónicas, algunos pacientes desarrollan hipertensión pulmonar aguda e insuficiencia respiratoria rápidamente progresiva. En ocasiones, los enfermos con fibrosis pulmonar debida a esclerodermia tienen carcinoma de células alveolares, el cual también se relaciona con otras enfermedades que producen fibrosis pulmonar.
Corazón
La lesión cardiaca típica de la esclerodermia se caracteriza por remplazo extenso del músculo cardiaco con tejido fibroso, provocando arritmias, trastornos de la conducción e insuficiencia cardiaca congestiva.3,4 Ocurre disfunción miocárdica menos extensa en la mayoría de los pacientes con esclerodermia difusa. La fibrosis miocárdica se asocia con necrosis y contractura en banda, que es posible que sean causadas por lesión por reperfusión. También se ha sugerido que se presenta un trastorno vasoespástico reversible de las arterias coronarias de pequeño calibre, al que se ha denominado fenómeno de Raynaud miocárdico. En la mayoría de los pacientes con esclerodermia ocurren defectos reversibles inducidos por el ejercicio o defectos fijos que pueden observarse con el gamagrama con talio. Debido a que los angiogramas son normales, estos trastornos indican enfermedad de vasos intramiocárdicos de muy pequeño calibre.
Otras causas de insuficiencia cardiaca incluyen cor pulmonale e hipertensión secundaria a las manifestaciones renales. La pericarditis no es rara y se puede acompañar de dolor, derrame pericárdico y taponamiento cardiaco. Los pequeños derrames pericárdicos detectados mediante ecocardiografía tienen poca importancia clínica, pero los derrames grandes pueden indicar afección seria en otros órganos y tener un pronóstico desfavorable. La ecocardiografía puede proporcionar evidencias de hipertensión pulmonar, aunque esta alteración puede aparecer cuando los criterios ecocardiográficos están ausentes. Rara vez la esclerodermia afecta los grandes vasos, causando enfermedad oclusiva e insuficiencia arterial periférica.
Riñón
La proteinuria leve crónica y la hipertensión leve son efectos comunes de la esclerodermia. La complicación renal más importante es la hipertensión maligna, la cual produce insuficiencia renal oligúrica de rápida evolución, que a menudo es mortal. Desde el punto de vista patológico, se caracteriza por nefroesclerosis maligna, incluyendo necrosis de las paredes arteriales y áreas de infarto renal.3, 4
Otros órganos y sistemas
No es frecuente la afección de otros órganos en la esclerodermia difusa; sin embargo, existen varios informes de cirrosis biliar, en especial asociada con la variante CREST (calcinosis, fenómeno de Raynaud, trastornos de la motilidad esofágica, esclerodactilia y telangiectasias) de la esclerodermia. Algunos pacientes pueden no tener una o más de las manifestaciones características del síndrome.3,4
Aunque el sistema nervioso central no se afecta, puede ocurrir neuropatía periférica. La forma más común es la neuropatía de una o más de las tres ramas del trigémino, unilateral o bilateral, manifestada por dolor e hipoestesias progresivas. La neuralgia del trigémino a menudo se relaciona con miositis, títulos altos de anticuerpos a la ribonucleoproteína y otras características de la enfermedad mixta del tejido conjuntivo. Se han postulado como causas, lesiones vasculares y atrapamiento del nervio por tejido conjuntivo, pero el mecanismo exacto sigue siendo desconocido.
Las consecuencias hematológicas de la esclerodermia son raras. La anemia microangiopática es poco frecuente y no se ha dilucidado el mecanismo de producción.3, 4
ALTERACIONES DE LABORATORIO
En cerca del 95 porciento de los enfermos con esclerodermia se encuentran resultados positivos para anticuerpos antinucleares [ver tabla 1].7 Este alto grado de sensibilidad se basa en el uso de líneas celulares de carcinoma epitelial humano (HEp-2) en lugar de los sustratos convencionales como los cortes de riñón de ratón.
Algunos anticuerpos dirigidos contra ciertos antígenos son específicos de la esclerodermia, y cada tipo de anticuerpo se asocia con un patrón clínico particular de la enfermdad. Los anticuerpos específicos están dirigidos contra la topoisomerasa I, proteínas del centrómero, polimerasas ARN o componentes nucleares [ver tabla 2]. Los anticuerpos contra topoisomerasa I (Scl-70) y contra polimerasas ARN (por lo general polimerasa III) se observan en pacientes con esclerodermia difusa.7,8 Los anticuerpos anticentrómero se asocian con esclerodermia limitada o síndrome de CREST y los anticuerpos contra ribonucleoproteína nuclear (RNPn) se asocian con esclerodermia difusa y limitada. Los anticuerpos contra PM-1 y Ku son poco frecuentes. La presencia de estos anticuerpos suele asociarse con manifestaciones clínicas mixtas de polimiositis y esclerodermia. Alrededor de la mitad de los pacientes con esclerodermia localizada tienen anticuerpos contra histonas [ver tabla 2].9
|
|
|||||||||||||||||||||||||||||||||
* Requiere de líneas celulares de carcinoma epitelial humano (Hep-2). |
Pueden aparecer diversas alteraciones parecidas a las encontradas en otras enfermedades del tejido conjuntivo. Puede ocurrir hiperglobulinemia leve, que casi siempre representa un aumento difuso en la IgG.3 En cerca de una cuarta parte de los casos se encuentran títulos bajos de actividad del factor reumatoide. Se han descrito alteraciones capilares en la dermis de los pliegues del lecho ungueal utilizando microscopía in vivo. Estos cambios son comunes en la esclerodermia y en la enfermedad mixta del tejido conjuntivo, pero rara vez se encuentran en pacientes con LEG, enfermedad de Raynaud o en individuos normales.
EVOLUCION CLINICA
Los pacientes con esclerodermia localizada tienen un buen pronóstico para la vida, debido a que las formas localizadas rara vez se relacionan con afección visceral.3, 4 El síndrome de CREST provoca afección limitada de la piel, con frecuencia sólo de las manos y la cara; el pronóstico para los pacientes con este síndrome en general es favorable, a menos que exista afección visceral. No obstante, incluso la variante limitada del síndrome de CREST tiende a tener un curso sin remisiones y lentamente progresivo.
Las manifestaciones y evolución de la esclerodermia difusa son muy variables, por lo que su velocidad de progresión es difícil de predecir. Con excepción de algunos de los cambios esclerodermatosos en la enfermedad mixta del tejido conjuntivo, el padecimiento rara vez remite completamente. Muchos enfermos siguen un curso indolente, con pocos cambios durante varios años; sin embargo, la progresión puede ser más rápida. La afección de vísceras como el corazón, el pulmón o en particular el riñón, indican un mal pronóstico. Dos características de la esclerodermia, la inflamación activa (manifestada por incremento de la velocidad de sedimentación globular) y la evidencia de enfermedad cardiopulmonar, renal o ambas, al año del diagnóstico, indican mal pronóstico.10 La supervivencia a cinco años para los pacientes con esclerodermia difusa es de alrededor del 50 porciento.
TRATAMIENTO
Por desgracia, ningún tratamiento para la esclerodermia ha demostrado ser eficaz.3, 4 La evaluación de los agentes terapéuticos propuestos se basa en los cambios en la textura o rigidez de la piel, pero estos indicadores son muy difíciles de cuantificar. Las determinaciones más objetivas, como las pruebas de función respiratoria en los pacientes con afección pulmonar, por lo general no son modificadas por ningún tratamiento farmacológico.
En general se acepta que los esteroides no tienen lugar en el tratamiento de la esclerodermia, ni los inmunosupresores o los medicamenos citotóxicos son eficaces.
Se ha reportado que la penicilamina es eficaz en el tratamiento del engrosamiento cutáneo y la fibrosis pulmonar, pero no ha sido estudiada en trabajos bien controlados. Los estudios controlados sobre el uso de colchicina no han confirmado los resultados alentadores de los primeros estudios no controlados. Los estudios no controlados realizados con inmunosupresores sugieren cierto beneficio de la azatioprina y el fluorouracilo (5-FU), pero un estudio controlado con clorambucil no demostró éxito. La ciclosporina causa citotoxicidad renal. Los resultados de los estudios preliminares con interferón gama y plasmaféresis no han sido alentadores. El tratamiento antiplaquetario con dipiridamol y aspirina no fue eficaz en un estudio controlado.
Aunque no se ha demostrado que ningún tratamiento modifique la evolución de la esclerodermia, el tratamiento de sostén parece ser útil para algunos aspectos del padecimiento. La evolución de la enfermedad puede alterarse en el caso de la crisis renal, que era antes la principal causa de muerte en pacientes con esclerodermia. El tratamiento de esta complicación grave con inhibidores de la enzima convertidora de angiotensina (ECA) y otros antihipertensivos potentes parece controlar la hipertensión y detener el deterioro de la función renal. Sin embargo, este tratamiento es más útil si se inicia antes de que los niveles de creatinina sérica excedan 3 mg/dl. En algunos pacientes continúa la progresión a insuficiencia renal a pesar del buen control de la presión arterial, requiriéndose diálisis y quizá trasplante renal.11
La fibrosis pulmonar es común en la esclerodermia y en la actualidad es la primera causa de muerte. Los estudios preliminares han demostrado que en pacientes con fibrosis pulmonar y alveolitis incipientes, la progresión puede detenerse por el tratamiento a largo plazo a base de ciclofosfamida por vía oral (1 a 2 mg/kg/día) y dosis bajas de prednisona (< 10 mg/día).12 La hipertensión pulmonar es una manifestación seria de esclerodermia y por lo general es difícil controlarla con vasodilatadores u otros agentes.
El fenómeno de Raynaud debe manejarse evitando la exposición al frío, y esto se aplica no solo a las extremidades sino a todo el organismo. Debe suspenderse el tabaquismo. El tratamiento con bloqueadores de los canales del calcio de acción prolongada o de antagonistas adrenérgicos alfa1, como el prazosín, puede ser benéfico. Las ulceraciones isquémicas en los dedos pueden ser difíciles de tratar y en ocasiones progresan a gangrena. Se ha reportado que los vasodilatadores experimentales, incluyendo la ketanserina, antagonista serotoninérgico, y el iloprost, análogo de prostaciclina, pueden ser útiles.13,14 La simpatectomía suele tener poco efecto sobre el fenómeno de Raynaud crónico y estable, y no se ha demostrado que influya en la evolución de la esclerodermia. Por lo tanto, el procedimiento debe reservarse para las crisis isquémicas.
La esofagitis, el reflujo esofágico y la estenosis se tratan por medio de elevación de la cabecera de la cama del paciente y administrando antiácidos, antagonistas de los receptores H2 u omeprazol. La motilidad esofágica puede aumentarse con el uso de metoclopramida. Cuando existe estenosis esofágica pueden requerirse dilataciones. Los estudios con seguimiento a corto plazo han demostrado que el análogo de somatostatina ocreótido y el agente procinético cisaprida, mejoran la motilidad intestinal y reducen la proliferación bacteriana en los pacientes con esclerodermia, pero no se conoce la eficacia a largo plazo de estos agentes.2,3,15,16 En ocasiones el síndrome de malabsorción puede responder a antibióticos de amplio espectro.
Fascitis eosinofílica
La fascitis eosinofílica3, 4 puede semejar en un principio a la esclerodermia. Se caracteriza por la presencia de dolor, tumefacción y sensibilidad en las extremidades, seguidos por induración de la piel y tejidos subcutáneos. También puede haber cierta limitación de la motilidad articular, pero no se observan el fenómeno de Raynaud ni otras manifestaciones de la esclerodermia.
Las alteraciones en las pruebas de laboratorio incluyen eosinofilia, que puede llegar a ser considerable, elevación de la velocidad de sedimentación e hiperglobulinemia. Los anticuerpos antinucleares y los resultados del factor reumatoide son negativos.
Las biopsias de las áreas afectadas muestran inflamación y engrosamiento desde la aponeurosis profunda a los tejidos subcutáneos. La piel tiene un aspecto normal, pero la aponeurosis profunda subyacente está infiltrada por linfocitos, células plasmáticas, histiocitos y, algunas veces, eosinofílicos.
La fascitis eosinofílica parece ser autolimitada o responde satisfactoriamente al tratamiento con dosis bajas de corticosteroides. La etiología sigue siendo desconocida, pero se han publicado varios casos posteriores a ejercicios muscular intenso.
Síndrome de mialgias y eosinofilia
En 1989 se describió e identificó un nuevo síndrome que consiste en eosinofilia y mialgias relacionadas con la ingestión de L-triptáfano.3, 4, 17-21 Este síndrome se caracteriza por la presencia de mialgias severas e incapacitantes y fatiga durante varias semanas. También pueden presentarse disnea y tos. La afección cutánea consiste en eritema variables y edema, así como cambios a la esclerodermia, por lo general sin las manifestaciones viscerales de la misma. Se pueden presentar infiltrados pulmonares intersticiales, hipoxia, hipertensión pulmonar y neumonitis por hipersensibilidad. La polineuropatía descrita toma la forma de una mononeuritis múltiple.
No existen pruebas de laboratorio diagnósticas. Los Centros para el Control y Prevención de las Enfermedades en los EUA mencionan tres criterios diagnósticos en su definición: una cuenta de eosinófilos mayor de 1,000 células/mm3, mialgia incapacitante o severa y la exclusión de otras causas posibles de los dos primeros criterios. La cuenta de eosinófilos puede llegar a 30,000/mm3. Los niveles de aminotransferasas y aldolasa pueden estar aumentados, reflejando daño hepático, y en ocasiones aumenta también la creantina cinasa, lo que indica afección muscular. Las biopsias revelan infiltrados inflamatorios que consisten en linfocitos, células plasmáticas, macrófagos y eosinófilos, acompañados de fibrosis de la dermis, tejido subcutáneo, fascia y músculo. También pueden observarse infiltrados inflamatorios del hígado.
Se han descrito alteraciones en el metabolismo del triptofano. El síndrome de mialgias y eosinofilia puede ser el resultado de un trastorno del metabolismo del L-triptosfano, que origina cantidades excesivas de este metabolito en personas susceptibles. También es posible que un contaminante tóxico añadido a las preparaciones de L-triptofano contribuya a la manifestación de la enfermedad.
Los resultados de un estudio epidemiológico indican que la epidemia fue causada por un contaminante o por alteración en un subgrupo del producto de un fabricante en Japón. La Food and Drug Administration de los EUA ha prohibido la distribución y venta de suplementos dietéticos a base de L-triptofano.
Enfermedades del tejido conjuntivo asociadas con cirugía estética
Dos procedimientos quirúrgicos estéticos se han asociado con el desarrollo subsecuente de enfermedades reumáticas: los implantes de mama de silicón y los implantes dérmicos de colágena.22 Algunas mujeres que fueron sometidas a mamoplastía de aumento con implantes de silicón han desarrollado manifestaciones de una enfermedad del tejido conjuntivo, sobre todo esclerodermia y menos, lupus eritematoso generalizado o artritis reumatoide. El hallazgo reumático más frecuente es la sinovitis leve y asimétrica. Se han detectado anticuerpos contra componentes mucleares por medio de técnicas de Western blot con más frecuencia en las pacientes sometidas a mamoplastía de aumento que en las poblaciones control.23,24
Se ha reportado polimiositis o dermatomiositis en varios pacientes que han recibido implantes de colágena bovina. En un estudio, la mayoría de los pacientes con miositis sufrieron reacciones de hipersensibilidad tardía a la colágena bovina y aumento en las concentraciones de anticuerpos contra colágena. Se ha encontrado que la incidencia de miositis es más alta en pacientes tratadas con colágena que en la población control.25
Estos reportes sugieren, aunque no demuestran, que existe una relación causal entre los implantes de silicón o colágena y las enfermedades del tejido conjuntivo. Se requieren de estudios epidemiológicos más detallados para determinar si existe esta relación causal.22
- LeRoy EC: Pathogenesis of systemic sclerosis. Arthritis and Allied Conditions. A Textbook of Rheumatology, 12th ed., Vol 2. McCarty DJ, Koopman WJ, Eds. Lea & Febiger, Philadelphia, 1993, p 1293
- Falanga V, Medsger TA Jr, Reichlin M, et al: Linear scleroderma: clinical spectrum, prognosis, and laboratory abnormalities. Ann Intern Med 104:849, 1986
- Medsger TA Jr: Systemic sclerosis (scleroderma), localized forms of scleroderma, and calcinosis. Arthritis and Allied Conditions. A Textbook of Rheumatology, 12th ed., Vol 2. McCarty DJ, Koopman WJ, Eds. Lea & Febiger, Philadelphia, 1993, p 1253
- Seibold JR: Scleroderma. Textbook of Rheumatology, 4th ed., Vol 2. Kelley WN, Harris ED Jr, Ruddy S, et al, Eds. WB Saunders Co, Philadelphia, 1993, p 113
- Youssef P, Englert H, Bertouch J: Large vessel occlusive disease associated with CREST syndrome and scleroderma. Ann Rheum Dis 52:464, 1993
- Baron M, Lee P, Keystone EC: The articular manifestations of progressive systemic sclerosis (scleroderma). Ann Rheum Dis 41:147, 1982
- Craft J, Hardin JA: Antinuclear antibodies. Textbook of Rheumatology, 4th ed., Vol 2. Kelley WN, Harris ED Jr, Ruddy S, et al, Eds. WB saunders Co, Philadelphia, 1993, p 164
- Okano Y, Steen VD, Medsger TA Jr: Autoantibody reactive with RNA polymerase III in systemic sclerosis. Ann Intern Med 119:1005, 1993
- Sato S, Ihn H, Soma Y, et al: Antihistone antibodies in patients with localized scleroderma. Arthritis Rheum 36:1137, 1993
- Bulpitt KJ, Clements PJ, Lachenbruch PA, et al: Early undifferentiated connective tissue disease: III. Outcome and prognostic indicators in early scleroderma (systemic sclerosis). Ann Intern Med 118:602, 1993
- Steen VD, Costantino JP, Shapiro AP, et al: Outcome of renal crisis in systemic sclerosis: relation to availability of angiotensin converting enzyme (ACE) inhibitors. Ann Intern Med 113:352, 1990
- Silver RM, Warrick JH, Kinsella MB, et al: Cyclophosphamide and low-dose prednisone therapy in patients with systemic sclerosis (scleroderma) with interstitial lung disease. J Rheumatol 20:838, 1993
- Seibold JR, Jageneau AHM: Treatment of Raynaud's phenomenon with ketanserin, a selective antagonist of the serotonin2 (5-HT2) receptor. Arthritis Rheum 27:139, 1984
- Wigley FM, Seibold JR, Wise RA, et al: Intravenous iloprost treatment of Raynaud's phenomenon and ischemic ulcers secondary to systemic sclerosis. J RheumatoI 19:1407, 1992
- Soudah HC, Hasler WL, Owyang C: Effect of octreotide on intestinal motility and bacterial overgrowth in scleroderma. N Engl J Med 325:1461, 1991
- Toskes PP: Hope for the treatment of intestinal scleroderma. N Engl J Med 325:1508, 1991
- Kilbourne EM, Swygert LA, Philen RM, et al: Interim guidance of the eosinophilia-myalgia syndrome (editorial). Ann Intern Med 112:85, 1990
- Hertzman PA, Blevins WL, Mayer J, et al: Association of the eosinophilia-myalgia syndrome with the ingestion of tryptophan. N Engl J Med 322:869, 1990
- Silver RM, Heyes MP, Maize JC, et al: Scleroderma, fasciitis, and eosinophilia associated with the ingestion of tryptophan. N Engl J Med 322:874, 1990
- Slutsker L, Hoesly FC, Miller L, et al: Eosinophilia-myalgia syndrome associated with exposure to tryptophan from a single manufacturer. JAMA 264:213, 1990
- Freundlich B: Eosinophilia-myalgia syndrome. Textbook of Rheumatology, 4th ed., Vol 2. Kelley WN, Harris ED Jr, Ruddy S, et al, Eds. WB Saunders Co, Philadelphia, 1993, p 1150
- Hochberg MC: Cosmetic surgical procedures and connective tissue disease: the Cleopatra syndrome revisited. Ann Intern Med 118:981, 1993
- Bridges AJ, Conley C, Wang G, et al: A clinical and immunologic evaluation of women with silicone breast implants and symptoms of rheumatic disease. Ann Intern Med 118:929, 1993
- Spiera H, Kerr LD: Scleroderma following silicone implantation: acumulative experience of 11 cases. J RheumatoI 20:958, 1993
- Cukier J, Beauchamp RA, Spindler JS, et al: Association between bovine collagen dermal implants and a dermatomyositis or a polymyositis-Iike syndrome. Ann Intern Med 118:920, 1993