Hematología
⭳ Abrir artículo (PDF)1012.1 KBEste artículo fue revisado respecto a la Edición 3/2000. Ver esa versión →
Contenido del artículo
VII TRASTORNOS NO MALIGNOS DE LOS LEUCOSITOS
- Indicaciones de la presencia de un trastorno de una célula fagocítica
- Neutrófilos
- PRODUCCION DE NEUTROFILOS
- ESTRUCTURA DEL NEUTROFILO
- FUNCION DE LOS NEUTROFILOS
- Adherencia
- Quimiotaxis
- Reconocimiento e ingestión
- Degranulación
- Metabolismo oxidativo
- Destrucción bacteriana
- Respuestas a y producción de citocinas
- TRASTORNOS EN EL NUMERO DE NEUTROFILOS
- Neutropenia asociada con linfocitosis T-g
- Monocitos y macrófagos
- DESARROLLO DE MONOCITOS-MACROFAGOS
- FUNCION DE MONOCITOS Y MACROFAGOS
- SINDROMES HISTIOCITICOS
- Síndromes de histiocitosis de células de Langerhans
- Histiocitosis sinusal con linfadenopatía masiva
- Linfohistiocitosis hemofagocítica familiar
- Síndrome hemofagocítico asociado a infección
- ENFERMEDADES POR ALMACENAMIENTO LISOSOMAL
- Eosinófilos
- Basófilos
- Linfocitos
VII TRASTORNOS NO MALIGNOS DE LOS LEUCOCITOS
DR. HARVEY J. COHEN, PH.D.
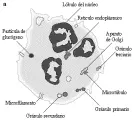
Los leucocitos, o glóbulos blancos, son las células que protegen al organismo contra la invasión por microrganismos. Existen dos tipos de leucocitos: los linfocitos, que son responsables de la producción de anticuerpos, la inmunidad mediada por células y la producción de moléculas secretadas que alteran la reactividad inmunológica, y los fagocitos, que son responsables de la ingestión de microrganismos y otros antígenos particulados. Los neutrófilos, los monocitos, los macrófagos y los eosinófilos y basófilos son todos fagocitos [ver figura 1]. Aunque suele referirse a los neutrófilos como granulocitos, todas las células fagocíticas contienen gránulos citoplásmicos característicos.
Pueden ocurrir trastornos no malignos tanto en el número como
función de los leucocitos. Los padecimientos del número y
función de las células fagocíticas y del número de
los linfocitos se analizan en esta sección, los trastornos de la
función linfocitaria se analizan en otro capítulo.
Indicaciones de la presencia de un trastorno de una célula fagocítica
Debido a que los fagocitos, especialmente los neutrófilos, representan la línea de defensa contra los microrganismos invasores, los trastornos en el número o función de estas células suelen causar una mayor susceptibilidad a la infección. Debe sospecharse un trastorno cuantitativo o cualitativo de las células fagocíticas siempre que un paciente tenga infecciones frecuentes, más severas o causadas por organismos no habituales, como Serratia marcescens, Pseudomonas cepacia y Aspergillus. Estas infecciones con frecuencia afectan la piel, los pulmones, el hígado y las mucosas. Las infecciones que requieren tratamiento antibiótico prolongado o que no parecen responder a los antibióticos adecuados sugieren también la existencia de un defecto en los fagocitos.
DETERMINACION DE LA CUENTA DE LEUCOCITOS
La cuenta de leucocitos es uno de los estudios más simples e importantes para evaluar al paciente con sospecha o propensión a una infección. Aunque este estudio es fácil de realizar en forma manual, la mayoría de los laboratorios emplean ya técnicas automatizadas de cuenta celular. Debe recordarse que aunque se usen técnicas manuales o automatizadas, se cuentan todas las células nucleadas, incluyendo a los eritrocitos nucleados. Por lo tanto, la cuenta verdadera de leucocitos (que casi siempre se proporciona con número de células en miles por milímetro cúbico de sangre) puede determinarse solo si primero se determina el porcentaje de eritrocitos nucleados y se corrige la cuenta con la siguiente fórmula:
La cuenta normal de leucocitos varía de 4,300 a 10,000/mm3, con un promedio de 7,000/mm3 [ver tabla 1].
DETERMINACION DE LA CUENTA ABSOLUTA DE NEUTROFILOS
La cuenta absoluta de neutrófilos se determina multiplicando el total de leucocitos por el porcentaje de neutrófilos maduros y formas en banda. Debido a que cada tipo de leucocito está regulado en forma independiente y tiene una función diferente en el organismo, es más adecuado usar la cuenta absoluta de neutrófilos que el porcentaje de neutrófilos para establecer las alteraciones.
Neutrófilos
Los neutrófilos son los fagocitos más abundantes en la circulación, pero realizan sus funciones más importantes en los tejidos, en donde actúan como primera línea de defensa contra las infecciones por bacterias y hongos. También causan el daño tisular que se observa en la infección y en procesos inflamatorios asociados con enfermedades como artritis o trastornos intestinales.
PRODUCCION DE NEUTROFILOS
Los neutrófilos y las otras células fagocíticas derivan de un progenitor común que también da origen a los eritrocitos, plaquetas y otros leucocitos.1 Esta célula progenitora está presente en la médula ósea y en concentraciones menores en sangre periférica. La proliferación y diferenciación de los neutrófilos es estimulada por dos citocinas relativamente específicas: el factor estimulador de colonias de granulocitos y macrófagos (FEC-GM) y el factor estimulador de colonias de granulocitos (FEC-G). El FEC-GM estimula una célula progenitora para diferenciarse en granulocitos (neutrófilos y eosinófilos) y monocitos-macrófagos.2 El FEC-G estimula a las células progenitoras para diferenciarse en neutrófilos3 [ver figura 2]. En la actualidad las técnicas recombinantes hacen posible administrar estos factores de crecimiento para estimular el crecimiento de las células fagocíticas en varios estados patológicos.
El ciclo de vida del neutrófilo incluye la fase en la médula ósea, en sangre y en los tejidos. La producción de neutrófilos en la médula ósea tarda alrededor de 10 a 14 días, y la médula ósea produce alrededor de 60 a 400 x 107 neutrófilos/kg/día. La mayoría del reservorio corporal de neutrófilos se encuentra en la médula ósea. El compartimiento mitótico, que contiene alrededor del 20 porciento del reservorio total, consiste en mieloblastos (los primeros precursores de neutrófilos que son reconocibles desde el punto de vista morfológico), los promielocitos y los mielocitos.5 El reservorio posmitótico contiene alrededor de 70 porciento de los neutrófilos del organismo y consiste en metamielocitos, bandas y neutrófilos maduros. En ocasiones se denomina a los neutrófilos y bandas de la médula ósea el compartimiento de almacén o la reserva medular.
La sangre periférica contiene menos de 10 porciento de los neutrófilos del organismo, que se dividen en proporción semejante entre el componente circulante y el de marginación. Al madurar, los neutrófilos pueden migrar de la médula ósea a la sangre en parte por la mayor capacidad de deformación de su membrana de superficie. La migración es resultado de interacciones complejas entre las células maduras y las células endoteliales que cubren la membrana basal de los sinusoides de la médula ósea. Estas interacciones están influidas por citocinas específicas y componentes del complemento.6,7 La liberación de neutrófilos de la médula ósea puede causar la duplicación o triplicación de la cuenta de neutrófilos tres a cinco horas después de que se presenta el estímulo apropiado.7 Las células del componente de marginación pueden entrar a la circulación en forma casi inmediata ante el efecto de epinefrina endógena o exógena, duplicando con rapidez la cuenta de neutrófilos.8 La demarginación es reversible, por lo general 30 a 60 minutos después de que se retira el estímulo. La vida media de los neutrófilos en sangre periférica es de alrededor de seis a 10 horas.4,5
La migración de los neutrófilos hacia los tejidos ocurre por diapedesis entre las células del endotelio capilar y por penetracion de la membrana basal como resultado de la estimulación por citocinas y otros factores (ver adelante). Por lo general se considera que los neutrófilos mueren por apoptosis, o muerte celular programada.8 Ciertas citocinas, como el FEC-G, el FEC-GM y el interferón, pueden retrasar la apoptosis y aumentar la supervivencia de los neutrófilos en los sitios de inflamación o infección.9 Existe cierta evidencia de que la lactoferrina producida por los gránulos específicos de los neutrófilos pueden causar inhibición de la producción de neutrófilos.
ESTRUCTURA DEL NEUTROFILO
Al madurar los precursores neutrófilos, ocurre condensación de la cromatina nuclear y los nucleolos se vuelven indistinguibles. Ocurre un proceso de segmentación nuclear, que causa la formación de un promedio de tres lóbulos nucleares por neutrófilo [ver figura 1]. El citoplasma del neutrófilo maduro contiene muy poco retículo endoplásmico, pero los neutrófilos son aún capaces de realizar cierta síntesis proteica (ver adelante). Las mitocondrias son escasas y el glucógeno es abundante. Estas observaciones se relacionan con el hecho de que el metabolismo energético del neutrófilo es principalmente glucolítico y anaeróbico, causando producción de lactato.
Los gránulos primarios, que contienen enzimas lisosomales y mieloperoxidasa (MPO)10 se producen en las fases de mieloblasto y promielocito del ciclo vital del neutrófilo, y se hacen aparentes al madurar las células. Los gránulos secundarios, que se producen sobre todo durante la fase de mielocito, son menos densos que los primarios y contienen lactoferrina, proteína fijadora de vitamina B12 y citocromo b558.11,12 Estos gránulos no se tiñen con Wright-Giemsa, un dato que permite distinguirlos de los eosinófilos y basófilos en el microscopio de luz. También se encuentran gránulos terciaros en los neutrófilos maduros, son más pequeños que los primarios y secundarios y contienen fosfatasas ácidas, arilsulfatasas y glucosaminoglucanos sulfatados, pero no MPO. Otras estructuras presentes en los neutrófilos son vesículas que pueden contener hidrolasas, como lactasas, fosfatasas alcalinas y componentes de la oxidasa de NADPH.13
El citoesqueleto del neutrófilo está compuesto de microtúbulos y microfilamentos que son indispensables para su función fagocítica. Los microtúbulos se encuentran cerca del aparato de Golgi y pueden ser factores importantes en el transporte de vesículas. Los microfilamentos, que consisten principalmente en polímeros de actina, se dispersan por todo el citoplasma. Los neutrófilos circulantes son esféricos y tienen algunas proyecciones citoplásmicas. Sin embargo, cuando se activan los neutrófilos los microfilamentos se mueven hacia la periferia y su distribución se vuelve asimétrica, causando una célula con aspecto polar.14
FUNCION DE LOS NEUTROFILOS
La principal función de los neutrófilos es responder con rapidez a la invasión del organismo por microbios al moverse hacia su localización, reconocerlos, engullirlos y finalmente destruirlos. Este proceso crítico de defensa del huésped, denominado fagocitosis, puede subdividirse en forma artificial en los siguientes componentes: adherencia, migración (o quimiotaxis), reconocimiento, ingestión, degranulación, metabolismo oxidativo y destrucción bacteriana [ver figura 3]. Además, en la actualidad se considera que la producción y respuestas de las citocinas son parte del proceso. Aunque estos componentes de la fagocitosis se analizarán por separado, muchos tienen características comunes, y los eventos no siempre ocurren en la secuencia descrita.
Adherencia
Los neutrófilos no contienen cilios, por lo que no tienen motilidad innata. Para que los neutrófilos se muevan hacia un sitio inflamatorio deben estar en contacto con la pared de un vaso. Alrededor del 50 porciento de los neutrófilos en la sangre se encuentran en el reservorio de marginación, fijos de modo laxo a las paredes capilares. El contacto entre los neutrófilos y las paredes de los vasos sanguíneos ocurre en forma aleatoria, pero puede aumentar si disminuye el flujo de los capilares. Este contacto permite la unión de baja afinidad de una glucoproteína de superficie del leucocito, como sialil-Lewisx (sLex), a una proteína de adherencia, como la P-selectina, en la superficie de las células endoteliales capilares15 [ver figura 4]. Las fuerzas de cizalla que actúan sobre el neutrófilo causan la unión y liberación alterna del complejo sLex-P-selectina, que a su vez produce que el neutrófilo ruede. La invasión bacteriana o la producción y liberación secundaria de citocinas causa mayor expresión en la P-selectina sobre la superficie de las células endoteliales, lo que permite que más neutrófilos se unan a las celulas endoteliales y se muevan de esta manera.16
Una familia de proteínas de superficie del neutrófilo llamada integrinas b2 facilitan la adhesión. Las tres proteínas de esta familia están formadas por una subunidad b idéntica y diferentes subunidades alfa (alfa1, alfa2 y alfa3). Cuando los neutrófilos son activados por quimioatrayentes, ocurre un cambio en el número17 y estructura18 de las integrinas, causando aumento en la afinidad de estas proteínas por las moléculas de adhesión intercelular (ICAM) sobre la superficie de las células endoteliales. Además, las citocinas estimulan con frecuencia la expresión de ICAM sobre la superficie de las células endoteliales.19 Estos procesos causan la unión de alta afinidad de neutrófilos activados a células endoteliales y la adhesion de las dos células.
Quimiotaxis
La quimiotaxis (movimiento dirigido), primera característica que permitió distinguir a los fagocitos de otras células, fue observada por Metchnikoff, y participa en la migración de los fagocitos desde el torrente sanguíneo hacia los sitios extravasculares de inflamación. Ocurre cuando los neutrófilos detectan ciertos ligandos, denominados quimiotácticos, en concentraciones muy bajas (v.gr., 10-10 a 10-9 mol/L) y siguen los gradientes de concentración hacia los sitios de generación o expresión de estas sustancias. Cuando estan fijos a las paredes capilares por integrinas b2 e ICAM endoteliales, los neutrófilos reptan a lo largo de las paredes y sufren diapedesis entre las células endoteliales. La diapedesis puede ocurrir porque estas uniones entre las células endoteliales son más laxas, quizá por la sustitución de ligandos de células endoteliales (v.gr., molécula de adhesión plaqueta-célula endotelial - 1[PECAM-1]) por ligandos neutrófilos análogos,20 y por la adhesión posterior de integrinas a las ICAM endoteliales. Los neutrófilos penetran en la membrana basal subendotelial para alcanzar el organismo invasor o el estímulo inflamatorio. Para permitir que los neutrófilos pasen a través de la membrana basal y se muevan hacia el tejido, enzimas proteolíticas e hidrolíticas que fueron liberadas de los gránulos de los neutrófilos en respuesta a los quimiotácticos digieren parte de la membrana basal.
Las interacciones reversibles de las actinas entre sí y con otras proteínas estructurales son responsables del movimiento de los neutrófilos.21 La actina constituye alrededor del 10 porciento del contenido proteico de los neutrófilos. Dentro de las células, los cambios en la configuración de la actina, la formación de redes ramificadas como resultado de la interacción de la actina con proteínas fijadoras y la conección de los polímeros de actina a la membrana celular provocan cambios característicos en la forma del neutrófilo que se asocian con activación y movimiento del mismo. Los filamentos de actina son polares, con un extremo principal que tiene gran afinidad por monómeros. El aumento en el calcio y el diacilglicerol intracelulares inhiben el ensamble y promueven el desensamble de las moléculas. Se ha sugerido que el movimiento de la miosina-1 a lo largo de los filamentos de actina empuja a la membrana celular hacia adelante.22
Reconocimiento e ingestión
En el sitio de infección o inflamación, los neutrófilos responden al estímulo a través de sus receptores opsonina de superficie, esto es, los receptores Fc y C3b. Los receptores Fc tienen una gran afinidad por la IgG en complejos con antígenos o en su forma polimerizada. Los receptores C3b reconocen moléculas de C3b que se han unido a partículas como resultado de la activación del complemento. Se desarrolla una vesícula fagocítica alrededor de las partículas opsonizadas por medio de seudópodos que las rodean y finalmente se fusionan. Este movimiento es análogo a la quimiotaxis. La interacción de las opsoninas con sus receptores respectivos causa también cambios en la forma de la célula, inicio de la degranulación y activación del estallido respiratorio asociado con la fagocitosis.
Degranulación
Los neutrófilos contienen muchos gránulos de diversos tipos [ver antes, Estructura de los neutrófilos]. Estos gránulos contienen tanto proteínas hidrolíticas (como proteasas, glucosidasas y fosfatasas) como proteínas bactericidas (como proteínas catiónicas, defensinas, lisozima, MPO y lactoferrina). Muchas de las proteínas pueden encontrarse en más de un tipo de gránulo.
Cuando un neutrófilo está en estado de reposo, los gránulos se encuentran en movimiento continuo y son separados de la membrana plasmática por la estructura submembrana de actina polimerizada. Cuando el neutrófilo se activa por interacción con quimiotácticos o partículas opsonizantes, la estructura submembrana cambia, permitiendo que los gránulos se pongan en contacto con la membrana plasmática. Ocurre fusión de las membranas de los gránulos con la membrana plasmática, liberando proteínas granulares. Las proteínas de la membrana de los gránulos se vuelven parte de la membrana plasmática y son componente importante de las integrinas (ver antes) y del sistema NADPH oxidasa (ver adelante). La activación de los receptores de opsoninas causa liberación de los gránulos principales, mientras que la activación por quimiotácticos produce principalmente liberación de proteasas e hidrolasas de los gránulos.
El proceso de degranulación es estimulado por el aumento en el calcio intracelular que ocurre con la activación del neutrófilo. Los eventos moleculares que causan fusión de la membrana de los gránulos probablemente incluyen a las proteínas citosólicas denominadas anexinas, que se fijan a fosfolípidos y promueven la fusión en respuesta a una mayor concentración de calcio y diacilglicerol.23
Metabolismo oxidativo
Los granulocitos en reposo son principalmente células aneróbicas que contienen pocas mitocondrias y que dependen de la glucólisis anaeróbica para la producción de trifosfato de adenosina (ATP).24 Aunque la quimiotaxis, la ingestión y la degranulación requieren energía, también se realizan en un medio anaeróbico. Sin embargo, la mayoría de los procesos de destrucción bacteriana requieren de consumo de oxígeno por el neutrófilo. De acuerdo con esto, la activación de la membrana del neutrófilo por partículas opsonizantes, y en menor grado por quimiotácticos, se asocia con un cambio rápido (en 15 segundos a un minuto) en el metabolismo de los neutrófilos, para consumir oxígeno. Dependiendo del estímulo, este proceso, denominado estallido respiratorio u oxidativo, dura 15 a 30 minutos.25
El estallido respiratorio ocurre como resultado de la activación de la oxidasa de NADPH en la membrana plasmática.26 Cuando el neutrófilo está en estadod e reposo, los componentes de esta oxidasa se localizan en la membrana plasmática, las membranas de los gránulos y el citosol. Las membranas contienen dos proteínas, gp91phox y p22phox,27,28 que forman un hueco hem; también contienen otras dos proteínas.29,30 El citosol contiene una proteína p47,31 y una p67,32 y proteínas que no se han caracterizado por completo aún. Cuando el neutrófilo se activa, las diversas proteínas del citosol se asocian, la proteína p47 sufre fosforilación y las proteínas se traslocan a la membrana plasmática.33,34 También las membranas de los gránulos se movilizan hacia la membrana plasmática. Estas traslocaciones producen un complejo que puede oxidar NADPH o NADP intracelular y reducir oxígeno en un electrón a superóxido en la superficie externa de la membrana plasmática. El NADPH se regenera a través de la vía de hexosa monofosfato, también conocida como la vía de la pentosa fosfato. La activación de la vía de hexosa monofosfato causa también la formación de bióxido de carbono.
En forma espontánea o en presencia de dismutasa de superóxido ocurre dismutación del superóxido. El peróxido de hidrógeno es metabolizado a través del ciclo del glutatión, requiriendo glutatión reducido, glutatión reductasa, glutatión peroxidasa y NADPH adicional. El peróxido de hidrógeno puede interactuar con superóxido en presencia de hierro para producir radicales hidróxilo y oxígeno simple, que son muy tóxicos. Además, el peróxido de hidrógeno y el cloruro se combinan en presencia de la MPO liberada en las vesículas fagocíticas para producir hipoclorito, que también es bactericida. Por último, en las vesículas fagocíticas se forman otras especies reactivas, como las cloraminas. Además de tener actividad bactericida, estos productos del estallido respiratorio pueden causar daño tisular cuando son liberados de los neutrófilos, y también contribuyen a la terminación del estallido respiratorio al inactivar a la NADPH oxidasa.
Destrucción bacteriana
Los productos del estallido respiratorio destruyen la integridad de la membrana de la bacteria, destruyendo así al organismo. Aunque la liberación de las proteínas bactericidas de los gránulos de los neutrófilos también es responsable de la muerte de algunas especies de bacterias, la mayoría de la destrucción parece ocurrir por actividad de la oxidasa de NADPH. El proceso de destrucción bacteriana por los neutrófilos es un evento intracelular, in vitro tarda 30 a 90 minutos.
Respuestas a y producción de citocinas
Los factores de crecimiento que afectan la producción de neutrófilos, como el FEC-GM y el FEC-G, también afectan su función. Estas citocinas aumentan la supervivencia y aumentan la actividad de la oxidasa de NADPH dependiente de estímulo.35 También pueden afectar la quimiotaxis.36 El interferón alfa, el factor de necrosis tumoral-alfa (FNT-alfa), la interleucina-8 (IL-8) y otras citocinas que modifican la función de los neutrófilos en diversas maneras pueden tener importancia fisiológica.37 Aunque los neutrófilos contienen muy pocos ribosomas, pueden responder a los estímulos bacterianos sintetizando y secretando IL-1 y FNT-alfa.38 Además, pueden producir IL-6 y FEC-G en respuesta al FEC-GM.39 La IL-8, que es un quimiotáctico de neutrófilos, también aumenta en respuesta a estímulos inflamatorios, favoreciendo la quimiotaxis.40
TRASTORNOS EN EL NUMERO DE NEUTROFILOS
Neutrofilia
La neutrofilia, o granulocitosis, se define como una cuenta de neutrófilos mayor de dos desviaciones estándar del promedio. Para los adultos, la cuenta promedio de neutrófilos es de 4,400/mm3, y se define neutrofilia como una cuenta mayor de 7,700/mm3. En la práctica, ocurre neutrofilia clínicamente significativa (i.e., una respuesta que sugiera la presencia de inflamación, infección o un trastorno hematológico primario) cuando la cuenta de neutrófilos es mayor de 10,000/mm3. La neutrofilia se considera primaria cuando no es causada por ninguna otra enfermedad. Sin embargo, la mayoría de los casos de neutrofilia son secundarios, ocurriendo en respuesta a otros procesos patológicos. Una cuenta de leucocitos mayor de 30,000 a 50,000/mm3 con predominio de neutrófilos indica la presencia de una reacción leucemoide.
Neutrofilia primaria La neutrofilia primaria puede ocurrir en pacientes con padecimientos mieloproliferativos, como la leucemia mieloide crónica (LMC). La LMC se asocia con la presencia de cromosoma Philadelphia (Ph1) en las células de la médula ósea y baja actividad de la fosfatasa alcalina de los neutrófilos . Se han descrito neutrofilias hereditarias e idiopáticas, que son benignas y bastante raras. Además, se ha asociado neutrofila con algunas alteraciones congénitas, por ejemplo, los niños con síndrome de Down pueden tener una reacción leucemoide temporal41,42 que debe distinguirse de la leucemia congénita. La neutrofilia también se ha asociado con urticaria familiar por frío, incluye infiltración neutrofílica en la erupción y por lo general se presenta 10 horas después de la exposición al ambiente frío.43 Por último, se observa neutrofilia en pacientes con deficiencia de adhesión leucocitaria (DAL).44 Este trastorno se asocia con alteraciones en las integrinas de los neutrófilos o del antígeno sLex.
Neutrofilia secundaria La neutrofilia secundaria se observa con frecuencia en pacientes con infecciones bacterianas. Ocurre neutrofilia y aumento en la cuenta de bandas cuando se producen y liberan neutrófilos de la médula ósea en respuesta al factor estimulador de colonias generado por la endotoxina u otros productos bacterianos. La presencia de granulaciones tóxicas y de cuerpos de Döhle en los neutrófilos sugiere, pero no demuestra, la presencia de infección. La inflamación crónica, como la que ocurre con el tabaquismo o la necrosis tisular, puede producir una neutrofilia moderada que puede ser indistinguible de la asociada con infección.
La neutrofilia secundaria puede ser aguda o crónica. Los eventos estresantes agudos pueden causar neutrofilia45 cuando se liberan neutrófilos del compartimiento de marginación por la epinefrina endógena.46 También se ha asociado neutrofilia aguda con estados postictales y ejercicio.47 Ciertos medicamentos, como los esteroides y los agonistas beta-adrenérgicos pueden producir neutrofilia aguda al inducir la liberación de neutrófilos de la médula ósea48 y del compartimiento de marginación, respectivamente.
El litio produce neutrofilia crónica al estimular la producción de factores estimuladores de colonias. También puede ocurrir neutrofilia crónica en pacientes con neoplasias no hematológicas, como el cáncer de pulmón de células grandes,49 y en los que el cáncer ha metastatizado a la médula ósea.50 Las personas asplénicas y las que tienen bazos no funcionales (v.gr., pacientes con anemia de células falciformes) suelen tener neutrofilia crónica moderada.51 Además, las personas que tienen estimulación de la médula ósea secundaria a anemia hemolítica o trombocitopenia suelen tener neutrofilia crónica asociada.
Evaluación de la neutrofilia La neutrofilia se asocia con más frecuencia con inflamación crónica e infecciones bacterianas o de otro tipo. El tabaquismo es una de las causas más frecuentes de neutrofilia. Si no parece existir infección o si la neutrofilia tiene más de varias semanas, el clínico debe considerar la posibilidad de una enfermedad mieloproliferativa. La presencia de metamielocitos y mielocitos en sangre, esplenomegalia y baja actividad de fosfatasa alcalina en los neutrófilos sugieren LMC. Una actividad de fosfatasa alcalina en neutrófilos alta y los cuerpos de Döhle sugieren un proceso infeccioso.
Si la neutrofilia del paciente no puede atribuirse a infección, inflamación o medicamentos, está justificado realizar aspiración y biopsia de médula ósea, junto con análisis cromosómico de la médula ósea y cultivo en busca de agentes infecciosos. Los resultados de estos procedimientos permitirán al clínico hacer el diagnóstico de LMC u otra enfermedad mieloproliferativa, infección granulomatosa, trastorno inflamatorio o cáncer metastásico. Si no se determina la causa en una persona por lo demás sana, debe pensarse en el diagnóstico de neutrofilia idiopática o familiar. El clínico debe tener en cuenta que el 2.5 porciento de las personas normales tiene una cuenta de neutrófilos que está más allá del 95 porciento de intervalo de confianza para el valor normal.
Neutropenia
La neutropenia es de mayor preocupación que la neutrofilia, y quizá es un problema más serio porque los neutrófios son la primera línea de defensa contra los microrganismos invasores. La neutropenia se define como una cuenta de neutrófilos menor de dos desviaciones estándar por debajo del promedio normal, que equivale a menos de 1,800/mm3, aunque este valor debe modificarse a alrededor de 1,000/mm3 para los afroamericanos y los judíos Yemenitas.52,53 Aunque estos parámetros definen a la neutropenia clínicamente importante, no ocurren infecciones significativas a menos que la cuenta de neutrófilos sea menor de 500/mm3 en la mayoría de las personas o hasta menor de 200/mm3 en otros. En pacientes que tienen neutropenia causada por aumento en la destrucción de los neutrófilos, el grado de neutropenia no necesariamente correlaciona con un mayor riesgo de infección.
En pacientes con neutropenia aislada, las infecciones son poco frecuentes y afectan principalmente a la piel y los tejidos como la mucosa oral, pulmones y senos paranasales. En pacientes con neutropenia asociada con monocitopenia (v.gr., en pacientes con anemia aplástica, leucemia o mielodisplasia, y en los que acaban de recibir quimioterapia), las infecciones son muy frecuentes e incluyen sepsis y otras infecciones sistémicas. Además, las infecciones de la piel cuando existe neutropenia aislada se deben principalmente a estafilococos, mientras que cuando existen citopenias más severas y combinadas (v.gr., como después de recibir quimioterapia) participan más los organismos gram negativos. El tipo de infección que ocurre en un estado neutropénico también se relaciona con defectos concomitantes en las barreras físicas (v.gr., infecciones por Staphylococcus epidermidis en la piel y por Escherichia coli en la mucosa gastrointestinal) y está muy influenciada por el uso de antibióticos.
Fisiopatológicamente, la neutropenia puede ser causada por producción disminuida o ineficaz de neutrófilos en la médula ósea, secuestro de los mismos a lo largo de las paredes vasculares o mayor destrucción periférica. Sin embargo, con frecuencia coexiste más de un mecanismo. Al igual que en el caso de la neutrofilia, la neutropenia suele clasificarse como intrínseca (primaria) o adquirida (secundaria), dependiendo de si es causada por defectos conocidos o supuestos en los neutrófilos [ver tabla 2] o asociada con otras alteraciones. Las neutropenias adquiridas incluyen neutropenia asociada a infecciones, neutropenia inducida por medicamentos, neutropenias autoinmunes, isoinmunes y aloinmunes, neutropenia idiopática crónica y neutropenias asociadas con linfocitosis T-g, aplasia pura de leucocitos, síndrome de inmunodeficiencia adquirida (SIDA) y esplenomegalia. Debido a que las neutropenias adquiridas son mucho más comunes que las intrínsecas, se analizan con mayor detalle.
Neutropenia asociada a infección La neutropenia asociada a infcciones es la forma más común de neutropenia en los niños y con frecuencia se relaciona con infección viral. Comienza días después de inicio de la infección y, aunque suele resolverse en algunas semanas, dura hasta dos meses. La causa de esta forma de neutropenia incluye tanto menor producción como mayor destrucción de neutrófilos. El número de monocitos suele ser normal y, excepto en casos severos, la neutropenia no se asocia con una mayor susceptibilidad a infección bacteriana. Otras formas de neutropenia asociada a infecciones incluyen la neutropenia que se observa en pacientes con SIDA54 (ver adelante) y la asociada con infección grave por ciertas bacterias, como Salmonella y otros bacilos gram negativos y S. aureus. Además, en lactantes con sepsis puede desarrollarse neutropenia muy grave que puede requerir transfusiones de granulocitos.55
Neutropenia inducida por medicamentos La neutropenia inducida por medicamentos puede ser la forma más común de neutropenia en adultos.56 Se han implicado muchos fármacos [ver tabla 3]. Los mecanismos fisiopatológicos pueden incluir menor producción de neutrófilos,57 que se ejemplifica por el efecto dependiente de la dosis que se observa con las fenotiacinas, o reacción destructiva mediada por anticuerpos,58 como ocurre en el caso del propiltiouracilo. La recuperación suele ocurrir varios días después de suspendido el medicamento, en los casos de neutropenia causada por menor producción de neutrófilos, la recuperación casi siempre va precedida por monocitosis.59
Neutropenia autoinmune La neutropenia autoinmune ocurre como un fenómeno aislado o secundario a otros trastornos autoinmunes.60 También puede ser causada por infección61 o tratamiento con ciertos medicamentos.62 En pacientes con síndrome de Evans, se asocia con trombocitopenia inmune y en ocasiones con anemia hemolítica. Los pacientes con neutropenia autoinmune suelen tener neutropenia moderada a severa acompañada de monocitosis. La celularidad de la médula ósea está aumentada, con una disminución relativa en el número de precursores tardíos de neutrófilos. Se observa esplenomegalia en alrededor del 50 porciento de los pacientes. Se han detectado anticuerpos antineutrófilo específicos, por lo general de tipo IgG o IgM, en muchos pacientes con esta enfermedad, y en algunos pacientes se han demostrado complejos inmunes circulantes. También se han detectado anticuerpos antineutrófilo en pacientes con lupus eritematoso generalizado y síndrome de Felty.63 En estos últimos se han implicado a las células T supresoras en la mediación de la neutropenia.
Neutropenia isoinmune y aloinmune En neonatos puede ocurrir neutropenia moderada a severa secundario a la trasmisión de anticuerpos IgG de la madre al infante. La incidencia de neutropenia isoinmune se ha calculado en dos por 1,000 nacimintos.64 El niño puede sufrir sepsis o estar asintomático. Puede ser difícil distinguir la neutropenia isoinmue de la asociada con sepsis en el recién nacido. Se detectan anticuerpos antineutrófilo en el suero del niño y de la madre, pero con frecuencia muestran específicidad para los neutrófilos del padre y no de la madre.65 La neutropenia suele resolverse en 12 a 15 semanas. Puede también ocurrir neutropenia aloinmune en el recién nacido como resultado de la transferencia pasiva de anticuerpos de una madre con neutropenia autoinmune. La evolución clínica de la enfermedad es semejante a la de la neutropenia isoinmune.
Neutropenia asociada con linfocitosis T-g
Los pacientes con linfocitosis T-g (linfocitosis que incluye linfocitos granulares grandes) suelen tener neutropenia.66 Estos pacientes tienen 50 a 60 años de edad, padecen infecciones recurrentes y pueden tener historia de artritis reumatoide y esplenomegalia. La cuenta de linfocitos es alta, pero rara vez es mayor de 20,000/mm3. El examen de la médula ósea muestra una médula normocelular con mayor número de linfocitos y detención de la línea mieloide en la fase de mielocito. La evolución de la enfermedad suele ser tórpida, y los pacientes mueren por un trastorno linfoproliferativo o sepsis.67 La linfocitosis es de origen clonal y puede ser maligna. La infusión intravenosa de g-globulina disminuye la cuenta de linfocitos y aumenta el número de neutrófilos.68 El síndrome de Felty suele representar una variante de este padecimiento.
Neutropenia asociada con aplasia pura de leucocitos La aplasia pura de leucocitos es un padecimiento poco frecuente caracterizado por infecciones severas y neutropenia. En muchos casos se asocia con timoma. El examen de la médula ósea muestra ausencia casi total de precursores mieloides y un número normal de precursores eritroides y megacariocitos. La eliminación del timoma puede no ser suficiente para resolver la neutropenia. También puede observarse aplasia pura de leucocitos en pacientes que reciben tratamiento con ibuprofén. La ciclofosfamida, los esteroides, la ciclosporina y la g-globulina intravenosa han sido todos eficaces para este trastorno.
Neutropenia asociada con síndrome de inmunodeficiencia adquirida Se desarrolla neutropenia en la mayoría de los pacientes con SIDA, y alrededor del ocho porciento de las personas con prueba positiva para el virus de inmunodeficiencia humana (VIH) que no tienen SIDA presentan también cifras bajas de neutrófilos.54 El número de neutrófilos suele disminuir en forma gradual, siguiendo a la disminución en la cuenta de linfocitos T CD4+ por meses o años. La neutropenia suele ser leve o moderada (i.e., los pacientes tienen de 500 a 1,500 neutrófilos/mm3). La neutropenia severa en pacientes con infección por VIH suele ser causada por los tratamientos farmacológicos u otros factores de complicación. La médula ósea suele ser hipercelular y contiene un mayor número de linfocitos. Se observan cambios displásicos en los granulocitos. A pesar de la hipercelularidad de la médula ósea, se cree que la hematopoyesis ineficaz es la causa de la neutropenia.
Neutropenia idiopática crónica La neutropenia idiopática crónica suele asociarse con una cuenta de neutrófilos de 200 a 500 /mm3, monocitosis asociada y una médula ósea que muestra actividad mieloide normal o aumentada, pero escasas células mieloides maduras.69 La mayoría de los casos son adquiridos, pero algunos tienen patrón de herencia autosómico dominante. Los individuos con este trastorno suelen ser evaluados por vez primera como un hallazgo en una citología hemática de rutina al evaluar una infección. La evolución clínica del padecimiento es relativamente banigna. Los pacientes con neutropenia idiopática crónica suelen tener cuentas normales de neutrófilos durante las infecciones bacterianas, y pueden movilizar neutrófilos en respuesta al estímulo con prdnisona.69 La neutropenia idiopática crónica puede ser ocasionada por diversos factores y por lo general no requiere tratamiento porque las infecciones son raras y bien toleradas.
Neutropenia asociada con esplenomegalia La neutropenia puede asociarse con hiperesplenismo. En estos casos, casi nunca es tan severa como para causar infecciones. Se relaciona con marginación reversible de los neutrófilos en el bazo, por lo que no produce depleción corporal total de los mismos.
Evaluación y tratamiento de la neutropenia Cuando se encuentra que un paciente tiene neutropenia, debe realizarse una historia clínica (incluyendo antecedentes familiares) y examen fisico para determinar si la neutropenia se ha asociado con mayor incidencia o severidad de la infección. Además, se obtendrá una citología hemática completa para establecer si la neutropenia se asocia con alguna otra alteración hematológica y si los neutrófilos tienen una estructura normal. La mayoría de las neutropenias aisladas son ocasionadas por medicamentos o virus. Por lo tanto, si el paciente está asintomático, deben suspenderse los medicamentos y se vigilará al paciente hasta por dos meses. Si la neutropenia no se resuelve o si el paciente tiene síntomas e historia de infecciones importantes, debe realizarse un estudio de médula ósea que incluya examen morfológico y análisis cromosómico. Cuando esté justificado, puede ser útil investigar anticuerpos antineutrófilo, pruebas para determinar otros tipos de disfunción inmunológica (como cuentas de células T y subgrupos), y medición de las concentraciones de inmunoglobulinas.
Debe instruirse a los pacientes con neutropenia leve crónica y a los que tienen neutropenia severa a realizar buen cuidado de la piel y cavidad oral, administrando antibióticos adecuados cuando ocurran infecciones. Para los pacientes con neutropenia severa debe considerarse el tratamiento profiláctico con antibióticos, como trimetoprim-sulfametoxazol y el uso de FEC o FEC-GM. Los pacientes con neutropenia y monocitopenia tienen riesgo muy alto de infección, en especial de sepsis, y deben ser vigilados en forma estrecha. Se ha realizado trasplante de médula ósea en pacientes con neutropenia severa, pero el advenimiento de la terapia con citocinas ha disminuido la frecuencia con la que se realiza este procedimiento.
TRASTORNOS DE LA FUNCION DE LOS NEUTROFILOS
En pacientes que tienen infecciones recurrentes, severas o poco frecuentes asociadas a número normal de neutrófilos, debe considerarse la presencia de un trastorno funcional. Estos trastornos son causados por defectos en la adherencia, quimioteraxis, degranulación o metabolismo oxidativo de los neutrófilos [ver tabla 4].
El enfoque lógico para la evaluación de los pacientes con infecciones recurrentes, severas o poco frecuentes consiste en determinar primero si el paciente ha tenido realmente estas infeciones, si existe historia familiar de infección y en realizar un examen físico completo que incluya la búsqueda de evidencias de que existe una respuesta adecuada a la infección [ver figura 5]. La biometría hemática completa y el estudio de los granulocitos en un frotis puede mostrar neutropenia, deficiencia de gránulos específicos o síndrome de Chédiak-Higashi, o sugerir una alteración esplénica. Además, si existe anemia normocítica debe sospecharse deficiencia de deshidrogenasa de glucosa-6-fosfato (G6P) o de glutatión sintetasa. Es adecuado evaluar las inmunoglobulinas (IgG, IgM, IgA e IgE) y los componentes del complemento (C3 y CH50) para detectar deficiencia total o parcial aumento de IgE o consmo del complemento. Si todas estas mediciones son normales, se evaluará la función de los neutrófilos con nitroazul de tetrazolio (NBT), ensayos de producción de superóxido y de quimiotaxis. La prueba de NBT y el ensayo de superóxido pueden determinar si un paciente tiene enfermedad granulomatosa crónica (EGC), deficiencia severa de G6PD o un trastorno de la vía del glutatión. Los estudios quimotácticos pueden usarse para confirmar el síndrome de Chédiak-Higashi y defectos adquiridos. La deficiencia de adhesión leucocitaria I (DAL I) y II (DAL II) se diagnostica por citometría de flujo. Si los resultados de todas estas pruebas son normales, puede ser útil realizar estudios de ingestión con el suero y células del paciente y por medio de tinción de MPO. De esta manera pueden diagnosticarse todas las alteraciones funcionales de los neutrófilos conocidas, con frecuencia con ayuda de interconsultas especializadas y pruebas de laboratorio de experimentación.
Monocitos y macrófagos
Los monocitos y macrófagos tienen un papel muy importante en la homeostasis y el complemento, y aumentan la función de los neutrófilos en los mecanismos de defensa del huésped. Los monocitos y los macrófagos realizan funciones de mantenimiento en los tejidos, como remodelación ósea por osteoclastos después de una lesión70 y fagocitosis esplénica de los eritrocitos viejos,71 y participan en la regulación inmunológica, interactuando con células T y B durante la presentación de los antígenos.72 Además, los monocitos y los macrófagos ayudan a controlar las infecciones por micobacterias, parásitos, hongos y virus.73-76
DESARROLLO DE MONOCITOS-MACROFAGOS
Los monocitos se desarrollan a partir de células progenitoras en la médula ósea. La citocina responsable de dirigir estas células hacia el linaje monocito parece ser el factor estimulador de colonias de macrófagos (FEC-M), aunque también son importantes la IL-3 y el FEC-GM.77 Una vez que la célula progenitora se diferencía hacia el linaje monocítico, se desarrollan los monoblastos y los promonocitos.
El monocito maduro [ver figura 1] es liberado casi de inmadiato hacia la circulación. Como en el caso de los neutrófilos, los monocitos sanguíneos pueden encontrarse en reservorios circulantes y de marginación. Sin embargo, el número de monocitos en el reservorio de marginación es alrededor de 3.5 veces más grande que el circulante.78 El monocito permanece en la circulación ocho a 72 horas antes de migrar hacia los tejidos. Después de la migración la célula se vuelve más grande y se convierte en un macrófago tisular al adquirir ribosomas, lisosomas, mitocondrias, retículo endoplásmico e inclusiones electrodensas.
FUNCION DE MONOCITOS Y MACROFAGOS
El sistema fagocito munonuclear incluye a los monoblastos, promonocitos, monocitos y macrófagos tisulares. Estos últimos son una población heterogénea de células con diferentes características morfológicas y funcionales dependiendo del tejido en el que resida. Los macrófagos alveolares, las células de Kuppfer, los osteoclastos, los macrófagos peritoneales y esplénicos y las células de Langerhans son todos macrófagos tisulares. Además de tener capacidad fagocítica, los monocitos y macrófagos tienen una función en otros aspectos de la respuesta inmunológica y en la regulación de la homeostasis.
Con excepción de los macrófagos alveolares, que dependen por completo del metabolismo aeróbico para la producción de energía, los monocitos y macrófagos son anaerobios facultativos.79 Al igual que la fagocitosis en los neutrófilos, la fagocitosis en los monocitos y macrófagos se asocia con un estallido oxidativo y estimulación de la vía hexosa monofosfato. La adhesión, quimiotaxis y activación de la fagocitosis son semejantes en ambos tipos de células, aunque los macrófagos son mejores que los neutrófilos para la fagocitosis, y realizan quimiotaxis con menos rapidez y eficacia. Los macrófagos son capaces de secretar más de 100 moléculas bien definidas, incluyendo citocinas, factores de crecimiento y ciertos reactantes de fase aguda.80 El macrófago también es capaz de tener actividad bactericida independiente de oxígeno que puede depender de la actividad lítica.81 Además, los macrófagos estimulados son capaces de producir óxico nítrico.82
Los monocitos y los macrófagos pueden presentar antígenos a las células T en asociación con las moléculas de clase II del complejo principal de histocompatibilidad (CPH). Esta asociación parece ocurrir dentro de los lisosomas de una célula mononuclear antes de que las moléculas de clase II se expresen en la superficie celular,83 parece incluir contacto intercelular directo y puede aumentarse por la liberación de citocinas como la IL-1. Los macrófagos pueden presentar el antígeno procesado a las células B para despertar una respuesta humoral.84 Los monocitos y los macrófagos participan en la citotoxicidad mediada por células dependiente e independiente de anticuerpos. La citotoxicidad incluye al metabolismo oxidativo, la producción de óxido nítrico y citocinas y la secreción de mediadores citotóxicos.
Los macrófagos tienen una función clave para metabolizar las proteínas de alto peso molecular, las glucoproteínas y otros materiales que participan en forma íntima en la destrucción de las células ancianas y muertas. También se requieren para la angiogénesis y la cicatrización de heridas, y son capaces de inducir neovascularización y proliferación de células endoteliales. Por último, los monocitos y macrófagos son capaces de secretar diversos factores de crecimiento hematopoyético, incluyendo FEC-GM, FEC-G, IL-1 y FEC-M.85
SINDROMES HISTIOCITICOS
Los síndromes histiocíticos son un grupo de trastornos malignos y no malignos en los que el macrófago es la célula principalmente alterada. Entre los trastornos malignos se incluyen la leucemia monocítica aguda, la histiocitosis maligna, también llamda reticulosis medular histiocítica y el linfoma histiocítico verdadero. Los trastornos no malignos, que se analizan en este capítulo, incluyen los síndromes de histiocitosis de células de Langerhans (HCL) y los síndromes hemofagocíticos, como la histiocitosis sinusal con linfadenopatía masiva, la linfohistiocitosis hemofagocítica familiar (LHF) y el síndrome hemofagocítico asociado a infección (SHAI).
Síndromes de histiocitosis de células de Langerhans
Clásicamente, los síndromes de HCL incluyen al granuloma eosinofílico solitario, al granuloma eosinofílico multifocal, a la enfermedad de Hand-Schüller-Christian y a la enfermedad de Letterer-Siwe. Estos padecimientos afectan sobre todo lactantes y niños pequeños, aunque se han descrito también en adultos jóvenes.86 Debido a que los síndromes de HCL representan una enfermedad continua que con frecuencia no puede designarse de este modo rígido y arbitrario, se han desarrollado otras clasificaciones que se relacionan con la edad en el momento del diagnóstico, la extensión de la enfermedad y la presencia de disfunción orgánica.87 Los signos y síntomas de los síndromes de HCL dependen de los órganos afectados. Pueden afectarse los huesos, la piel, los dientes, el tejido gingival, los oídos, los órganos endócrinos, los pulmones, el hígado, el bazo, los ganglios linfáticos y la médula ósea, y volverse disfuncionales por infiltración celular.
Los granulomas eosinofílicos tanto solitarios como multifocales se encuentran principalmente en niños mayores y adultos jóvenes. Al momento de la presentación estos pacientes suelen tener problema para aumentar de peso o tienen zonas de inflamación dolorosa por la infiltración de los tejidos sobre lesiones óseas marginales.88 La enfermedad de Hand-Schüller-Christian ocurre en niños pequeños (i.e., dos a cinco años de edad) y se manifiesta como defectos óseos con exoftalmos causados por una masa tumoral en la cavidad orbitaria, ausencia de dientes por la infiltración de la encía y afección de los huesos planos del cráneo, costillas, pelvis y escápula. También puede existir diabetes insípida asociada, que afecta hasta al 50 porciento de los pacientes con síndromes de HCL.89 Es común la otitis media crónica, causada por afección del hueso temporal. La forma más rara y severa de HCL es la enfemedad de Letterer-Siwe, que ocurre en forma típica en niños de menos de dos años. Los síntomas consisten en erupciones seborreicas, eccematosas, descamativas y pruriginosas que afectan el cuero cabelludo, los canales auditivos, el abdomen y el área intertriginosa del cuello y cara. Además existe secreción ótica, linfadenopatía, hepatoesplenomegalia y, en casos severos, disfunción hepática con hipoalbuminemia y menor síntesis de los factores de la coagulación. Puede ocurrir afección del sistema hematopoyético, incluyendo la anemia y la trombocitopenia, y daño grave al pulmón con hipoxia.
El pronóstico para el paciente con síndrome de HCL depende de tres factores: edad de inicio, número de órganos afectados y la presencia de disfunción de los órganos específicos.87 Un adolescente con un granuloma eosinofílico solitario en hueso tiene el mejor pronóstico, mientras que un niño con muchos órganos afectados el peor. El diagnóstico se realiza por estudio patológico de una lesión por medio de microscopía de luz y demostración de gránulos electrodensos en las células de Langerhans (gránulos de Birbeck) al microscopio electrónico o antígeno CD1 con la inmunotinción.
El tratamiento de la HCL en ocasiones no es necesario, cuando sí, la cirugía o la radioterapia local pueden ser curativas.90 Además, aunque los síndromes de HCL no son malignos, responden a agentes quimioterápicos.91 Debido a que el objetivo del tratamiento en los síndromes de HCL es administrar la mínima cantidad necesaria del tratamiento menos tóxico para controlar la enfermedad, el principio para el uso de los agentes quimioterápicos es comenzar con el agente más benigno y después pasar a agentes más tóxicos, tratando de nunca administrar un tratamiento peor que la enfermedad. Al inicio debe administrarse vinblastina intravenosa en dosis de 0.15mg/kg/semana, y la dosis debe aumentarse a 0.025 y 0.05 mg/kg/semana si no ocurre mejoría después de dos semanas. Si aún no existe mejoría, se agregará al esquema prednisona, 2 mg/kg/día, disminuyendo ambos medicamentos lo más rápido posible. Si este régimen fracasa, debe administrarse metotrexate por vía intramuscular, intravenosa u oral, en dosis de 10 mg/m2/semana y 6-mercaptopurina, 20 mg/m2/día, durante 14 a 21 días, y los medicamentos se disminuirán cuando el paciente mejore. Si estos esquemas no controlan por completo la enfermedad o si los efectos adversos asociados con su uso justifican su suspensión, se ha reportado que el etopósido puede ser benéfico.92
Histiocitosis sinusal con linfadenopatía masiva
La histiocitosis sinusal con linfadenopatía masiva se caracteriza clínicamente por linfadenopatía masiva benigna, con frecuencia crónica e indolora, que suele afectar los ganglios cervicales y con menos frecuencia los axilares, hiliares, peritraqueales o inguinales.93 En el 30 porciento de los pacientes existe enfermedad extranodal en las vías respiratorias, huesos, órbitas, piel, hígado y riñones.94 La enfermedad suele ser benigna, pero puede presentarse morbilidad importante e incluso la muerte si ocurre invasión tisular masiva del hígado, riñones, pulmones y otras estructuras críticas.95 Aunque el 80 porciento de los pacientes se diagnostican en la primera o segunda década de la vida, este padecimiento puede afectar también a los ancianos. Los pacientes suelen ser descendientes de africanos y la incidencia de esta enfermedad es más alta en Africa e India Occidental.
En pacientes con histiocitosis sinusal con linfadenopatía masiva, los ganglios linfáticos afectados suelen tener dilatación sinusoidal marcada e hiperplasia folicular con proliferación de histiocitos espumosos y células gigantes multinucleadas en los senos. La etiología de este padecimiento se desconoce y puede relacionarse con una inmunorregulación anormal.96 El tratamiento suele ser tanto innecesario como ineficaz. Al resolverse la enfermedd, la afección extranodal desaparece antes de la nodal. Los intentos de tratamiento deben reservarse para circunstancias especiales graves. Se han administrado radiación, esteroides, vinblastina y ciclofosfamida con grados variables de éxito.97
Linfohistiocitosis hemofagocítica familiar
La LHF es un padecimiento hereditario rápidamente fatal que se caracteriza por pancitopenia periférica con hiperplasia de la médula ósea, linfadenopatía sistémica, fiebre, disfunción hepática severa y hepatoesplenomegalia. Los pacientes tienen concentraciones bajas de fibrinógeno sin evidencia de coagulación intravascular diseminada. Los niños y los lactantes son los más afectados, y dos tercios de los pacientes tienen menos de tres meses de edad.
Los síntomas de presentación de la LHF incluyen palidez, irritabilidad, anorexia, diarrea, no incremento ponderal y una erupción maculopapular.98 La evolución clínica suele ser progresiva y dura un promedio de solo seis semanas. La mayoría de los pacientes mueren por infección, hemorragia o crisis convulsivas. Alrededor de la tercera parte de los pacientes tiene afección al sistena nervioso central. El estudio patológico del hígado muestra cambios grasos focales, necrosis e infiltración de linfocitos e histiocitos. La eritrofagocitosis y la hemofagocitosis son datos esenciales en el diagnóstico. Aunque la etiología de la LHF se desconoce, la inmunodeficiencia es un dato constante.
Por lo general los esteroides y los agentes citotóxicos son ineficaces en el tratamiento de la LHF. Se ha demostrado que el etopósido induce remisión clínica, pero el tratamiento definitivo requiere del uso de trasplante de médula ósea.99
Síndrome hemofagocítico asociado a infección
El SHAI se observa con frecuencia en personas inmunosuprimidas como consecuencia de una infección subyacente viral100 o bacteriana,101 o de tratamiento inmunosupresor.102 Aunque la evolución clínica del SHAI varía, se ha reportado una mortalidad del 40 porciento.100 En ausencia de una inmunodeficiencia subyacente, de historia familiar de SHAI o de consanguinidad, puede ser difícil distinguir entre el SHAI o la LHF. Desde el punto de vista patológico las enfermedades son muy semejantes. El SHAI, al igual que la LHF, puede responder a etopósido o puede resolverse en forma espontánea.
ENFERMEDADES POR ALMACENAMIENTO LISOSOMAL
Los monocitos y los macrófagos tienen una función importante en la remodelación tisular y la eliminación de detritos celulares viejos, y los lisosomas son los organelos que realizan estas funciones. Por lo tanto, las alteraciones enzimáticas que incluyen a los constituyentes lisosomales causan trastornos de almacenamiento relacionados con la función de los macrófagos. Estos padecimientos incluyen a las mucopolisacaridosis, las glucoproteinosis, las esfingolipidosis y las enfemedades por almacenamiento de lípidos neutros [ver tabla 5]. Se han descrito defectos enzimáticos para la mayoría de estos padecimientos, y el diagnóstico depende de demostrar la alteración enzimática en los macrófagos o histiocitos. La mayoría de estos defectos son causados por mutaciones puntiformes o rearreglos genéticos en un solo locus del gen que codifica para una hidrolasa lisosomal.103
Los dos tipos de tratamiento para las enfermedades por almacenamiento lisosomal disponibles en la actualidad son el trasplante celular y el tratamiento enzimático. Debido a que las células tronco de la médula ósea son pluripotenciales, estas células son las que se emplean en la mayoría de los trasplantes.104 Sin embargo, al parecer las enfermedades por almacenamiento lisosomal afectan otras células además de las derivadas dela médula ósea, por lo que pueden ser necesarios otros tipos de trasplante.105 En el pasado la enfermedad de Gaucher se trataba con trasplante de médula ósea, pero en la actualidad se maneja con alglucerasa, una glucocerebrosidasa con terminación a-manosil.106 Aunque no se ha establecido el esquema óptimo, se ha demostrado que la dosis inicial de 30 a 60 UI/kg es eficaz. Esta dosis puede repetirse cada dos semanas o puede calcularse la dosis mensual y administrarse infusiones más frecuentes. Alrededor del 10 a 15 porciento de los pacientes desarrollan anticuerpos contra la enzima. De estos pacientes, el 50 porciento sufre prurito y urticaria. Debido a que la mayoría de los pacientes que desarrolla anticuerpos lo hace en los primeros seis a nueve meses de tratamiento, se recomienda administrar la enzima bajo vigilancia médica durante alrededor de nueve meses antes de iniciar la atención domiciliaria. El uso de trasplante de médula ósea para otros trastornos de almacenamiento de lisosomas se considera aún experimental y ha dado resultados muy variables.107
Eosinófilos
Los eosinófilos pueden aumentar o suprimir las reacciones inflamatorias agudas y las respuestas mediadas por infecciones por helmintos, alergia y ciertos tumores.108 Al igual que los neutrófilos, los eosinófilos son capaces de fagocitar, pero principalmente son células secretoras. La mayoría de la funciones que realizan requieren de la lbieración del contenido de sus gránulos o de radicales de oxígeno generados por la membrana celular cuando ésta se activa. Los eosinófilos responden a agentes quimiotácticos específicos y a factores de crecimiento que permiten su acúmulo en sitios de inflamación.
ESTRUCTURA DE LOS EOSINOFILOS
Los gránulos de los eosinófilos contienen proteínas muy básicas que se tiñen en forma intensa con colorantes ácidos. Poseen una apariencia característica bajo el microscopio electrónico [ver figura 1]. Los gránulos consisten en un centro electrodenso rodeado de una matriz relativamente radiolúcida, y existe actividad de peroxidasa de eosinófilos (POE) en la matriz.109 El centro denso tiene una estrucutra cristaloide y contiene proteínas catiónicas eosinófilas (PCE), proteínas básicas principales (PBP) y neurotoxinas derivadas de eosinófilos.110 Las PBP son proteínas relativamente pequeñas capaces de causar daño importante a los esquistosómulas por unión y ruptura de sus membranas celulares. Además, aumentan la adherencia de los eosinófilos y los neutrófilos a los esquistosómulas.111 Las PCE también se unen a los esquistosómulas y son tóxicas para ellos.
FUNCION DE LOS EOSINOFILOS
Los eosinófilos responden a diversos factores quimiotácticos que les permiten penetrar en los tejidos y realizar sus funciones. Algunos de estos factores quimiotácticos (como C5a, péptidos que contienen N-formilmetionil y leucotrieno B4) estimulan tanto eosinófilos como neutrófilos. Sin embargo, varios estímulos quimiotácticos son muy específicos para los eosinófilos. Entre éstos se incluyen al factor activador de plaquetas (FAP), al factor quimiotáctico eosinófilo de anafilaxia y a varios factores derivados de parásitos. Las respuestas al FAP, uno de los activadores más potentes de los eosinófilos normales, incluyen quimiotaxis, adherencia, mayor unión de IgE, producción de superóxido, liberación de proteínas de los gránulos y síntesis de prostenoides.
Tanto la producción como la activación de los eosinófilos se afectan por el FEC-GM, la IL-5 y la IL-3.112 La exposición a dosis bajas de IL-5 activa en forma específica a los eosinófilos para responder a otros estimulantes.113 De hecho, aunque la IL-5 tiene un efecto específico y potente sobre el crecimiento de eosinófilos en cultivo, prácticamente no tiene efecto estimulador en el crecimiento de otras colonias mieloides. Además, los ratones transgénicos que expresan IL-5 muestran eosinofilia prolongada,114 y la administración de anticuerpos anti-IL-5 a ratones evita por completo que se desarrolle eosinofilia en respuesta a parásitos.115 Una vez activados, los eosinófilos presentes en ciertas condiciones patológicas asociadas con eosinofilia tienen mayor generación de especies reactivas del oxígeno, mayor utilización y transporte de glucosa, mayor consumo de oxígeno, menor carga en la superficie celular y activación de las fosfatasas ácidas en gránulos específicos.116 Estos cambios se acompañan de una reducción en la densidad celular.
Los eosinófilos aumentan la respuesta inmunológica a helmintos. Realizan esta función al unirse a la superficie de formas tanto larvarias como adultas, al dañar a las células blanco por mecanismos dependientes de oxígeno que son semejantes a los de los neutrófilos y al dañar superficies celulares al liberar proteínas de gránulos como PBP y PCE.117 Aunque la liberación de estas proteínas daña en forma semejante a las células tumorales, la interacción entre eosinófilos y las células tumorales se comprende menos bien. La presencia de eosinofilia en pacientes con enfermedad de Hodgkin parece depender de la producción de IL-5 por las células de Reed-Sternberg.118 Los eosinófilos contribuyen a la fibrosis de la enfermedad de Hodgkin tipo esclerosis nodular al producir factor de transformación de crecimiento-b1 (FTC-b1).
Los eosinófilos realizan una función inmunosupresora en las reacciones de hipersensibilidad inmediata. Las prostaglandinas E1 y E2 liberadas de las membranas de los eosinófilos activados son capaces de suprimir la degranulación de los basófilos.119 Otras sustancias de los gránulos, como la histaminasa y la fosfolipasa B, inactivan la histamina y el FAP de las células cebadas, respectivamente. Los eosinófilos median también el daño tisular a través de los efectos de las PBP, las PCE y el superóxido y los otros radicales reactivos del oxígeno aumentados por el sistema de peroxidasa de eosinófilos.120 El balance entre la estimulación eosinofílica y la inhibición de la inflamación es delicada y se rompe con facilidad.
TRASTORNOS EN EL NUMERO DE EOSINOFILOS
Eosinofilia
La evaluación del paciente con eosinofilia (una cuenta de eosinófilos > 700/mm3) es difícil porque la causa de este trastorno es múltiple y diversa. Las causas comunes de la eosinofilia secundaria incluyen trastornos alérgicos, infecciones causadas por parásitos u otros organismos, enfermedades dermatológicas, enfermedades pulmonares, enfermedades del tejido conectivo, neoplasias e inmunodeficiencias. También existen múltiples causas menos comunes, como gastroenteritis eosinofílica, enfermedad inflamatoria intestinal, hepatitis activa crónica, pancreatitis e hipopituitarismo. En ocasiones no se encuentra la causa de la eosinofilia incluso después de la evaluación exhaustiva del paciente.
Síndrome hipereosinofílico
El término síndrome hipereosinofílico (SHE) suele usarse para caraterizar la enfermedad para la que no se identifica la causa de la eosinofilia. Los criterios empleados para el SHE consisten en una cuenta de eosinófilos mayor de 1,500/mm3 durante más de seis meses, la ausencia de otro diagnóstico que explique la eosinofilia y signos o síntomas de infiltración de eosinófilos en los tejidos.121 El SHE suele distinguirse de trastornos malignos asociados con eosinofilia, como la leucemia aguda. También deben excluirse reacciones alérgicas, la exclusión de estas reacciones suele basarse en la historia, examen físico y revisión de los medicamentos actuales. Debido a que muchos medicamentos pueden generar una reacción alérgica asociada con eosinofilia, deben suspenderse todos los medicamentos que no sean indispensables antes de evaluar al paciente. Algunas enfermedades respiratorias alérgicas se asocian con eosinofilia, al parecer por liberación de PCE.122 Los pacientes con estas enfermedades pueden tener niveles elevados de IgE. Sin embargo, debe tenerse en cuenta que algunos pacientes con SHE pueden tener también elevación de esta inmunoglobulina.
Las infecciones parasitarias, sobre todo las causadas por helmintos que invaden los tejidos como las filarias y Strongyloides, Trichinella, Schistosoma y Toxocara, con frecuencia presentan eosinofilia.123 Se recomienda estudiar múltiples muestras de excremento y aspirados de contenido del intestino delgado para descartar estos padecimientos, en especial en pacientes con diarrea o riesgo especial de infección (v.gr., los que viajan con frecuencia, están expuestos a animales y tienen inmunodeficiencias). Además, deben realizarse estudios serológicos, pruebas radiológicas y frotis de sangre periférica y médula ósea para excluir la presencia de enfermedades del tejido conectivo, síndromes linfoproliferativos ocultos y tumores sólidos, y neoplasias hematológicas, respectivamente.
El frotis sanguíneo de un paciente con SHE suele demostrar eosinófilos maduros normales de morfología típica. Sin embargo, se ha reportado la presencia de hipogranulación y vacuolas citoplásmicas.124 La cuenta total de leucocitos suele ser de 10,000 a 30,000/mm3, el 30 a 70 porciento de los cuales son eosinófilos. La leucocitosis progresiva con eosinofilia debe originar la posibilidad de que un paciente tenga SHE. La médula ósea suele ser hipercelular, y los eosinófilos que constituyen el 25 a 75 porciento de los elementos de la médula.124
La fibrosis en el SHE es rara, los cambios cardiacos son los que predominan en el espectro de las alteraciones clínicas asociadas con la enfermedad, y son la principal causa de morbimortalidad. Sin embargo, estos cambios cardiacos no son distinguibles de los encontrados en pacientes con eosinofilia secundaria. Los pacientes pueden tener insuficiencia cardiaca congestiva crónica, alteraciones valvulares y engrosamiento endocárdico biventricular característico y fibroso con trombos murales. La mayoría de los pacientes tienen engrosamiento de la pared ventricular izquierda. El ecocardiograma es el método más sensible para detectar estas alteraciones. Puede ocurrir también afección pulmonar, que causa fiebre, tos, pérdida de peso y sibilancias.
El enfoque diagnóstico de una persona con sospecha de SHE debe incluir una historia y examen físico completos, citología hemática y cuenta total de eosinófilos. Deberá estudiarse la posibilidad de eosinofilia secundaria por medio de cuantificación de IgE, muestras en heces para huevos y parásitos, aspirado duodenal, aspiración y biopsia de médula ósea, radiografía de tórax y ecocardiografía. Si el paciente tiene diagnóstico de SHE, la decisión de administrar tratamiento debe basarse en la presencia o ausencia de síntomas. Si no existen síntomas, no se instituirá tratamiento y el paciente se reevaluará en forma periódica. Si existen síntomas que afecten los pulmones o el corazón, debe administrarse prednisona en dosis de 1 mg/kg/día durante dos semanas, seguido de 1 mg/kg cada tercer día por tres meses. Si ocurre mejoría deberá suspenderse el tratamiento. Sin embargo, si el tratamiento no mejora los síntomas o la cuenta de eosinófilos, deberá administrarse hidroxiurea en dosis de 0.5 a 1.5 g/día para disminuir la cuenta de leucocitos a 10,000/mm3. Si este agente no produce respuesta clínica, puede administrarse otro como ciclofosfamida, interferón alfa, ciclosporina o etopósido.125-127 Si la enfermedad no responde a este tipo de tratamiento y el paciente tiene menos de 50 años, deberá considerarse realizar trasplante de médula ósea.
Basófilos
Los basófilos son mediadores clave de las reacciones de hipersensibilidad inmediata, asma, urticaria, rinitis alérgica y anafilaxia. Los basófilos son estimulados por los mediadores solubles, principalmente IgE, para liberar el contenido de los gránulos y metabolitos del ácido araquidónico de sus membranas plasmáticas. Se sabe menos sobre los basófilos que sobre el resto de los tipos de leucocitos por el menor número de estas células en el organismo. Las células cebadas están relacionadas, aunque son diferentes, a los basófilos.128 Ambos tipos de células derivan de una célula progenitora hematopoyética común en la médula ósea. Sin embargo, los basófilos expresan receptores para IL-2, IL-3 y moléculas de integrina, ninguno de los cuales se expresan en las células cebadas, mientras que la molécula de adhesión intercelular-1 (ICAM-1) y el producto c-kit sí se expresan en las células cebadas y no en los basófilos. No se ha observado hasta la fecha transformación de un tipo celular a otro.
ESTRUCTURA Y FUNCION DE LOS BASOFILOS
Los gránulos citoplásmicos de basófilos y células cebadas contienen glucosaminoglucanos sulfatados. En los basófilos normales, los glucosaminoglucanos sulfatados son principalmente heparina. Los glucosaminoglucanos sulfatados en los gránulos causan la intensa tinción de los basófilos.
La mayoría, si no es que toda, la histamina circulante en el organismo se sintetiza en el basófilo y se almacena dentro de los gránulos. La degranulación causa la liberación de histamina, que media muchos efectos de la hipersensibilidad inmediata y que, por su acción quimiotáctica potente de eosinófilos, lleva eosinófilos al sitio de degranulación. Otras sustancias que son liberadas al degranularse los basófilos incluyen factores quimiotácticos adicionales y varios metabolitos del ácido araquidónico, de los que el más importante en el leucotrieno C4.
Además, las membranas celulares de los basófilos contienen receptores de IgE de alta afinidad, y su número aumenta en personas alérgicas. La interacción entre los receptores de IgE y la inmunoglobulina causa degranulación anafiláctica. Cuando los receptores de IgE se reúnen después de interactuar con un estímulo apropiado, las membranas que limitan los gránulos citoplásmicos se fusionan con la membrana plasmática externa de la célula, causando liberación inmediata del contenido de los gránulos.129 La liberación de histamina causa síntomas clínicamente significativos; sin embargo, los esteroides pueden inhibir la liberación de histamina y de leucotrieno C4. Por ello, los basófilos regulan las reacciones alérgicas agudas a través de sus actividades secretoras de membrana.130
BASOFILIA
La basofilia (cuenta de basófilos > 150/mm3) se observa en trastornos mieloproliferativos como LMC, policitemia vera y metaplasia mieloide, después de esplenectomía, en algunas anemias hemolíticas y en la enfermedad de Hodgkin. La cuenta de basófilos también puede aumentar en pacientes con colitis ulcerativa y varicela. Aunque los basófilos y las células cebadas participan en las reacciones de hipersensibilidad inmediata y se observan basófilos en las áreas de dermatitis por contacto, no se ha observado basofilia en pacientes con estos trastornos.
Linfocitos
LINFOCITOSIS
La linfocitosis en adultos se define como una cuenta absoluta de linfocitos de más de 4,000/mm3.131 Sin embargo, debe tenerse en cuenta que las cuentas de linfocitos en los niños son mayores que en los adultos, y que pueden ser hasta de 20,000/mm3 en el primer año de vida. El frotis de cualquier paciente con linfocitosis debe examinarse con cuidado para determinar la morfología y diversidad de los linfocitos (v.gr., linfocitos reactivos, linfocitos granulares grandes, blastos o células manchadas).
La linfocitosis puede ser primaria o secundaria. La primaria, con frecuencia denominada enfermedad linfoproliferativa, es causada por alteración en la regulación de la producción de los linfocitos. Las linfocitosis primarias incluyen las leucemias (como la leucemia linfocítica crónica, la leucemia linfocítica aguda, la leucemia de células peludas y la leucemia de linfocitos granulares grandes), los linfomas y la linfocitosis monoclonal de células B [ver tabla 6].
Las linfocitosis secundarias, o reactivas, son condiciones que incluyen incrementos absolutos en los linfocitos causados por respuestas fisiológicas o fisiopatológicas a infección, inflamación, toxinas, citocinas o agentes desconocidos. La causa más frecuente de linfocitosis reactiva es la infección viral, y con frecuencia son responsables el virus Epstein-Barr, el citomegalovirus, el virus herpes simple, el virus varicela-zoster, el de rubeóla, el VIH, el adenovirus o alguno de los virus de la hepatitis. Otros patógenos que producen linfocitosis son Toxoplasma gondii y, en niños, Bordetella pertussis (que causa aumento en la cuenta de linfocitos hasta 70,000/mm3).132 La linfocitosis se asocia también con estrés y liberación subsecuente de epinefrina, como la observada en pacientes con colapso cardiovascular, choque séptico, crisis falciforme , estado epiléptico o traumatismos,133 y con cirugía mayor, reacciones medicamentosas e hipersensibilidad. Puede existir linfocitosis persistente en pacientes con enfermedades autoinmunes, sarcoidosis, hiperesplenismo y cáncer, así como en los fumadores crónicos.
LINFOCITOPENIA
La linfocitopenia se define como una cuenta total de linfocitos menor de 1,000/mm3.134 Debido a que el 80 porciento de los linfocitos adultos son células T, la mayoría de las causas de linfocitopenia son ocasionadas por reducción en la cuenta de linfocitos T. Con frecuencia el mecanismo de la linfocitopenia es desconocido, y la causa puede ser hereditaria o adquirida.
Las linfocitopenias hereditarias suelen ser ocasionadas por inmunodeficiencias congénitas. Estas enfermedades incluyen inmunodeficiencia combinada severa (v.gr., deficiencia de deaminasa de adenosina, deficiencia de fosforilasa de nucleótidos de purina y disgenesia reticular), ataxia telangiectasia, síndrome de Wiskott-Aldrich e hipoplasia del cartílago-cabello [ver tabla 7]. Además, algunas personas tienen linfocitopenia T CD4+ idiopática.135
Puede observarse linfocitopenia adquirida en pacientes con infecciones virales (como infección por VIH, hepatitis, influenza e infección por virus sincicial respiratorio), en los que tienen infecciones bacterianas (como fiebre tifoidea, neumonía, sepsis y tuberculosos) y en los que tienen anemia aplástica, enfermedades autoinmunes, enfermedad de Hodgkin, sarcoidosis, insuficiencia renal, enteropatías perdedoras de proteínas y ascitis quilosa. También la deficiencia de zinc y la ingesta crónica de alcohol se asocian con linfocitopenia. Por último, los agentes inmunosupresores como la globulina antitimocito, los esteroides, los agentes quimioterápicos y la radiación, también producen linfocitopenia.
Bibliografía
DR. HARVEY J. COHEN, PH.D.
Los leucocitos, o glóbulos blancos, son las células que protegen al organismo contra la invasión por microrganismos. Existen dos tipos de leucocitos: los linfocitos, que son responsables de la producción de anticuerpos, la inmunidad mediada por células y la producción de moléculas secretadas que alteran la reactividad inmunológica, y los fagocitos, que son responsables de la ingestión de microrganismos y otros antígenos particulados. Los neutrófilos, los monocitos, los macrófagos y los eosinófilos y basófilos son todos fagocitos [ver figura 1]. Aunque suele referirse a los neutrófilos como granulocitos, todas las células fagocíticas contienen gránulos citoplásmicos característicos.
|
|
|
|
Indicaciones de la presencia de un trastorno de una célula fagocítica
Debido a que los fagocitos, especialmente los neutrófilos, representan la línea de defensa contra los microrganismos invasores, los trastornos en el número o función de estas células suelen causar una mayor susceptibilidad a la infección. Debe sospecharse un trastorno cuantitativo o cualitativo de las células fagocíticas siempre que un paciente tenga infecciones frecuentes, más severas o causadas por organismos no habituales, como Serratia marcescens, Pseudomonas cepacia y Aspergillus. Estas infecciones con frecuencia afectan la piel, los pulmones, el hígado y las mucosas. Las infecciones que requieren tratamiento antibiótico prolongado o que no parecen responder a los antibióticos adecuados sugieren también la existencia de un defecto en los fagocitos.
DETERMINACION DE LA CUENTA DE LEUCOCITOS
La cuenta de leucocitos es uno de los estudios más simples e importantes para evaluar al paciente con sospecha o propensión a una infección. Aunque este estudio es fácil de realizar en forma manual, la mayoría de los laboratorios emplean ya técnicas automatizadas de cuenta celular. Debe recordarse que aunque se usen técnicas manuales o automatizadas, se cuentan todas las células nucleadas, incluyendo a los eritrocitos nucleados. Por lo tanto, la cuenta verdadera de leucocitos (que casi siempre se proporciona con número de células en miles por milímetro cúbico de sangre) puede determinarse solo si primero se determina el porcentaje de eritrocitos nucleados y se corrige la cuenta con la siguiente fórmula:
| Cuenta de leucocitos | = | No. de células nucleadas en sangre | x |
(100 -% de eritrocitos nucleados) _______________________________ |
| 100 |
La cuenta normal de leucocitos varía de 4,300 a 10,000/mm3, con un promedio de 7,000/mm3 [ver tabla 1].
|
||||||||||
* Para calcular el número de células /L, multiplique por 106. |
||||||||||
DETERMINACION DE LA CUENTA ABSOLUTA DE NEUTROFILOS
La cuenta absoluta de neutrófilos se determina multiplicando el total de leucocitos por el porcentaje de neutrófilos maduros y formas en banda. Debido a que cada tipo de leucocito está regulado en forma independiente y tiene una función diferente en el organismo, es más adecuado usar la cuenta absoluta de neutrófilos que el porcentaje de neutrófilos para establecer las alteraciones.
Neutrófilos
Los neutrófilos son los fagocitos más abundantes en la circulación, pero realizan sus funciones más importantes en los tejidos, en donde actúan como primera línea de defensa contra las infecciones por bacterias y hongos. También causan el daño tisular que se observa en la infección y en procesos inflamatorios asociados con enfermedades como artritis o trastornos intestinales.
PRODUCCION DE NEUTROFILOS
Los neutrófilos y las otras células fagocíticas derivan de un progenitor común que también da origen a los eritrocitos, plaquetas y otros leucocitos.1 Esta célula progenitora está presente en la médula ósea y en concentraciones menores en sangre periférica. La proliferación y diferenciación de los neutrófilos es estimulada por dos citocinas relativamente específicas: el factor estimulador de colonias de granulocitos y macrófagos (FEC-GM) y el factor estimulador de colonias de granulocitos (FEC-G). El FEC-GM estimula una célula progenitora para diferenciarse en granulocitos (neutrófilos y eosinófilos) y monocitos-macrófagos.2 El FEC-G estimula a las células progenitoras para diferenciarse en neutrófilos3 [ver figura 2]. En la actualidad las técnicas recombinantes hacen posible administrar estos factores de crecimiento para estimular el crecimiento de las células fagocíticas en varios estados patológicos.
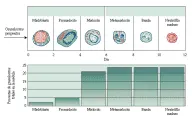
|
| Figura 2 |
| Proceso de maduración de los neutrófilos |
El ciclo de vida del neutrófilo incluye la fase en la médula ósea, en sangre y en los tejidos. La producción de neutrófilos en la médula ósea tarda alrededor de 10 a 14 días, y la médula ósea produce alrededor de 60 a 400 x 107 neutrófilos/kg/día. La mayoría del reservorio corporal de neutrófilos se encuentra en la médula ósea. El compartimiento mitótico, que contiene alrededor del 20 porciento del reservorio total, consiste en mieloblastos (los primeros precursores de neutrófilos que son reconocibles desde el punto de vista morfológico), los promielocitos y los mielocitos.5 El reservorio posmitótico contiene alrededor de 70 porciento de los neutrófilos del organismo y consiste en metamielocitos, bandas y neutrófilos maduros. En ocasiones se denomina a los neutrófilos y bandas de la médula ósea el compartimiento de almacén o la reserva medular.
La sangre periférica contiene menos de 10 porciento de los neutrófilos del organismo, que se dividen en proporción semejante entre el componente circulante y el de marginación. Al madurar, los neutrófilos pueden migrar de la médula ósea a la sangre en parte por la mayor capacidad de deformación de su membrana de superficie. La migración es resultado de interacciones complejas entre las células maduras y las células endoteliales que cubren la membrana basal de los sinusoides de la médula ósea. Estas interacciones están influidas por citocinas específicas y componentes del complemento.6,7 La liberación de neutrófilos de la médula ósea puede causar la duplicación o triplicación de la cuenta de neutrófilos tres a cinco horas después de que se presenta el estímulo apropiado.7 Las células del componente de marginación pueden entrar a la circulación en forma casi inmediata ante el efecto de epinefrina endógena o exógena, duplicando con rapidez la cuenta de neutrófilos.8 La demarginación es reversible, por lo general 30 a 60 minutos después de que se retira el estímulo. La vida media de los neutrófilos en sangre periférica es de alrededor de seis a 10 horas.4,5
La migración de los neutrófilos hacia los tejidos ocurre por diapedesis entre las células del endotelio capilar y por penetracion de la membrana basal como resultado de la estimulación por citocinas y otros factores (ver adelante). Por lo general se considera que los neutrófilos mueren por apoptosis, o muerte celular programada.8 Ciertas citocinas, como el FEC-G, el FEC-GM y el interferón, pueden retrasar la apoptosis y aumentar la supervivencia de los neutrófilos en los sitios de inflamación o infección.9 Existe cierta evidencia de que la lactoferrina producida por los gránulos específicos de los neutrófilos pueden causar inhibición de la producción de neutrófilos.
ESTRUCTURA DEL NEUTROFILO
Al madurar los precursores neutrófilos, ocurre condensación de la cromatina nuclear y los nucleolos se vuelven indistinguibles. Ocurre un proceso de segmentación nuclear, que causa la formación de un promedio de tres lóbulos nucleares por neutrófilo [ver figura 1]. El citoplasma del neutrófilo maduro contiene muy poco retículo endoplásmico, pero los neutrófilos son aún capaces de realizar cierta síntesis proteica (ver adelante). Las mitocondrias son escasas y el glucógeno es abundante. Estas observaciones se relacionan con el hecho de que el metabolismo energético del neutrófilo es principalmente glucolítico y anaeróbico, causando producción de lactato.
Los gránulos primarios, que contienen enzimas lisosomales y mieloperoxidasa (MPO)10 se producen en las fases de mieloblasto y promielocito del ciclo vital del neutrófilo, y se hacen aparentes al madurar las células. Los gránulos secundarios, que se producen sobre todo durante la fase de mielocito, son menos densos que los primarios y contienen lactoferrina, proteína fijadora de vitamina B12 y citocromo b558.11,12 Estos gránulos no se tiñen con Wright-Giemsa, un dato que permite distinguirlos de los eosinófilos y basófilos en el microscopio de luz. También se encuentran gránulos terciaros en los neutrófilos maduros, son más pequeños que los primarios y secundarios y contienen fosfatasas ácidas, arilsulfatasas y glucosaminoglucanos sulfatados, pero no MPO. Otras estructuras presentes en los neutrófilos son vesículas que pueden contener hidrolasas, como lactasas, fosfatasas alcalinas y componentes de la oxidasa de NADPH.13
El citoesqueleto del neutrófilo está compuesto de microtúbulos y microfilamentos que son indispensables para su función fagocítica. Los microtúbulos se encuentran cerca del aparato de Golgi y pueden ser factores importantes en el transporte de vesículas. Los microfilamentos, que consisten principalmente en polímeros de actina, se dispersan por todo el citoplasma. Los neutrófilos circulantes son esféricos y tienen algunas proyecciones citoplásmicas. Sin embargo, cuando se activan los neutrófilos los microfilamentos se mueven hacia la periferia y su distribución se vuelve asimétrica, causando una célula con aspecto polar.14
FUNCION DE LOS NEUTROFILOS
La principal función de los neutrófilos es responder con rapidez a la invasión del organismo por microbios al moverse hacia su localización, reconocerlos, engullirlos y finalmente destruirlos. Este proceso crítico de defensa del huésped, denominado fagocitosis, puede subdividirse en forma artificial en los siguientes componentes: adherencia, migración (o quimiotaxis), reconocimiento, ingestión, degranulación, metabolismo oxidativo y destrucción bacteriana [ver figura 3]. Además, en la actualidad se considera que la producción y respuestas de las citocinas son parte del proceso. Aunque estos componentes de la fagocitosis se analizarán por separado, muchos tienen características comunes, y los eventos no siempre ocurren en la secuencia descrita.
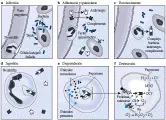
|
| Figura 3 |
| Respuesta del neutrófilo a la invasión bacteriana |
Adherencia
Los neutrófilos no contienen cilios, por lo que no tienen motilidad innata. Para que los neutrófilos se muevan hacia un sitio inflamatorio deben estar en contacto con la pared de un vaso. Alrededor del 50 porciento de los neutrófilos en la sangre se encuentran en el reservorio de marginación, fijos de modo laxo a las paredes capilares. El contacto entre los neutrófilos y las paredes de los vasos sanguíneos ocurre en forma aleatoria, pero puede aumentar si disminuye el flujo de los capilares. Este contacto permite la unión de baja afinidad de una glucoproteína de superficie del leucocito, como sialil-Lewisx (sLex), a una proteína de adherencia, como la P-selectina, en la superficie de las células endoteliales capilares15 [ver figura 4]. Las fuerzas de cizalla que actúan sobre el neutrófilo causan la unión y liberación alterna del complejo sLex-P-selectina, que a su vez produce que el neutrófilo ruede. La invasión bacteriana o la producción y liberación secundaria de citocinas causa mayor expresión en la P-selectina sobre la superficie de las células endoteliales, lo que permite que más neutrófilos se unan a las celulas endoteliales y se muevan de esta manera.16
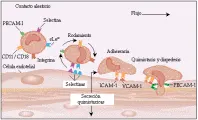
|
| Figura 4 |
| Salida de los neutrófilos de la sangre periférica |
Una familia de proteínas de superficie del neutrófilo llamada integrinas b2 facilitan la adhesión. Las tres proteínas de esta familia están formadas por una subunidad b idéntica y diferentes subunidades alfa (alfa1, alfa2 y alfa3). Cuando los neutrófilos son activados por quimioatrayentes, ocurre un cambio en el número17 y estructura18 de las integrinas, causando aumento en la afinidad de estas proteínas por las moléculas de adhesión intercelular (ICAM) sobre la superficie de las células endoteliales. Además, las citocinas estimulan con frecuencia la expresión de ICAM sobre la superficie de las células endoteliales.19 Estos procesos causan la unión de alta afinidad de neutrófilos activados a células endoteliales y la adhesion de las dos células.
Quimiotaxis
La quimiotaxis (movimiento dirigido), primera característica que permitió distinguir a los fagocitos de otras células, fue observada por Metchnikoff, y participa en la migración de los fagocitos desde el torrente sanguíneo hacia los sitios extravasculares de inflamación. Ocurre cuando los neutrófilos detectan ciertos ligandos, denominados quimiotácticos, en concentraciones muy bajas (v.gr., 10-10 a 10-9 mol/L) y siguen los gradientes de concentración hacia los sitios de generación o expresión de estas sustancias. Cuando estan fijos a las paredes capilares por integrinas b2 e ICAM endoteliales, los neutrófilos reptan a lo largo de las paredes y sufren diapedesis entre las células endoteliales. La diapedesis puede ocurrir porque estas uniones entre las células endoteliales son más laxas, quizá por la sustitución de ligandos de células endoteliales (v.gr., molécula de adhesión plaqueta-célula endotelial - 1[PECAM-1]) por ligandos neutrófilos análogos,20 y por la adhesión posterior de integrinas a las ICAM endoteliales. Los neutrófilos penetran en la membrana basal subendotelial para alcanzar el organismo invasor o el estímulo inflamatorio. Para permitir que los neutrófilos pasen a través de la membrana basal y se muevan hacia el tejido, enzimas proteolíticas e hidrolíticas que fueron liberadas de los gránulos de los neutrófilos en respuesta a los quimiotácticos digieren parte de la membrana basal.
Las interacciones reversibles de las actinas entre sí y con otras proteínas estructurales son responsables del movimiento de los neutrófilos.21 La actina constituye alrededor del 10 porciento del contenido proteico de los neutrófilos. Dentro de las células, los cambios en la configuración de la actina, la formación de redes ramificadas como resultado de la interacción de la actina con proteínas fijadoras y la conección de los polímeros de actina a la membrana celular provocan cambios característicos en la forma del neutrófilo que se asocian con activación y movimiento del mismo. Los filamentos de actina son polares, con un extremo principal que tiene gran afinidad por monómeros. El aumento en el calcio y el diacilglicerol intracelulares inhiben el ensamble y promueven el desensamble de las moléculas. Se ha sugerido que el movimiento de la miosina-1 a lo largo de los filamentos de actina empuja a la membrana celular hacia adelante.22
Reconocimiento e ingestión
En el sitio de infección o inflamación, los neutrófilos responden al estímulo a través de sus receptores opsonina de superficie, esto es, los receptores Fc y C3b. Los receptores Fc tienen una gran afinidad por la IgG en complejos con antígenos o en su forma polimerizada. Los receptores C3b reconocen moléculas de C3b que se han unido a partículas como resultado de la activación del complemento. Se desarrolla una vesícula fagocítica alrededor de las partículas opsonizadas por medio de seudópodos que las rodean y finalmente se fusionan. Este movimiento es análogo a la quimiotaxis. La interacción de las opsoninas con sus receptores respectivos causa también cambios en la forma de la célula, inicio de la degranulación y activación del estallido respiratorio asociado con la fagocitosis.
Degranulación
Los neutrófilos contienen muchos gránulos de diversos tipos [ver antes, Estructura de los neutrófilos]. Estos gránulos contienen tanto proteínas hidrolíticas (como proteasas, glucosidasas y fosfatasas) como proteínas bactericidas (como proteínas catiónicas, defensinas, lisozima, MPO y lactoferrina). Muchas de las proteínas pueden encontrarse en más de un tipo de gránulo.
Cuando un neutrófilo está en estado de reposo, los gránulos se encuentran en movimiento continuo y son separados de la membrana plasmática por la estructura submembrana de actina polimerizada. Cuando el neutrófilo se activa por interacción con quimiotácticos o partículas opsonizantes, la estructura submembrana cambia, permitiendo que los gránulos se pongan en contacto con la membrana plasmática. Ocurre fusión de las membranas de los gránulos con la membrana plasmática, liberando proteínas granulares. Las proteínas de la membrana de los gránulos se vuelven parte de la membrana plasmática y son componente importante de las integrinas (ver antes) y del sistema NADPH oxidasa (ver adelante). La activación de los receptores de opsoninas causa liberación de los gránulos principales, mientras que la activación por quimiotácticos produce principalmente liberación de proteasas e hidrolasas de los gránulos.
El proceso de degranulación es estimulado por el aumento en el calcio intracelular que ocurre con la activación del neutrófilo. Los eventos moleculares que causan fusión de la membrana de los gránulos probablemente incluyen a las proteínas citosólicas denominadas anexinas, que se fijan a fosfolípidos y promueven la fusión en respuesta a una mayor concentración de calcio y diacilglicerol.23
Metabolismo oxidativo
Los granulocitos en reposo son principalmente células aneróbicas que contienen pocas mitocondrias y que dependen de la glucólisis anaeróbica para la producción de trifosfato de adenosina (ATP).24 Aunque la quimiotaxis, la ingestión y la degranulación requieren energía, también se realizan en un medio anaeróbico. Sin embargo, la mayoría de los procesos de destrucción bacteriana requieren de consumo de oxígeno por el neutrófilo. De acuerdo con esto, la activación de la membrana del neutrófilo por partículas opsonizantes, y en menor grado por quimiotácticos, se asocia con un cambio rápido (en 15 segundos a un minuto) en el metabolismo de los neutrófilos, para consumir oxígeno. Dependiendo del estímulo, este proceso, denominado estallido respiratorio u oxidativo, dura 15 a 30 minutos.25
El estallido respiratorio ocurre como resultado de la activación de la oxidasa de NADPH en la membrana plasmática.26 Cuando el neutrófilo está en estadod e reposo, los componentes de esta oxidasa se localizan en la membrana plasmática, las membranas de los gránulos y el citosol. Las membranas contienen dos proteínas, gp91phox y p22phox,27,28 que forman un hueco hem; también contienen otras dos proteínas.29,30 El citosol contiene una proteína p47,31 y una p67,32 y proteínas que no se han caracterizado por completo aún. Cuando el neutrófilo se activa, las diversas proteínas del citosol se asocian, la proteína p47 sufre fosforilación y las proteínas se traslocan a la membrana plasmática.33,34 También las membranas de los gránulos se movilizan hacia la membrana plasmática. Estas traslocaciones producen un complejo que puede oxidar NADPH o NADP intracelular y reducir oxígeno en un electrón a superóxido en la superficie externa de la membrana plasmática. El NADPH se regenera a través de la vía de hexosa monofosfato, también conocida como la vía de la pentosa fosfato. La activación de la vía de hexosa monofosfato causa también la formación de bióxido de carbono.
En forma espontánea o en presencia de dismutasa de superóxido ocurre dismutación del superóxido. El peróxido de hidrógeno es metabolizado a través del ciclo del glutatión, requiriendo glutatión reducido, glutatión reductasa, glutatión peroxidasa y NADPH adicional. El peróxido de hidrógeno puede interactuar con superóxido en presencia de hierro para producir radicales hidróxilo y oxígeno simple, que son muy tóxicos. Además, el peróxido de hidrógeno y el cloruro se combinan en presencia de la MPO liberada en las vesículas fagocíticas para producir hipoclorito, que también es bactericida. Por último, en las vesículas fagocíticas se forman otras especies reactivas, como las cloraminas. Además de tener actividad bactericida, estos productos del estallido respiratorio pueden causar daño tisular cuando son liberados de los neutrófilos, y también contribuyen a la terminación del estallido respiratorio al inactivar a la NADPH oxidasa.
Destrucción bacteriana
Los productos del estallido respiratorio destruyen la integridad de la membrana de la bacteria, destruyendo así al organismo. Aunque la liberación de las proteínas bactericidas de los gránulos de los neutrófilos también es responsable de la muerte de algunas especies de bacterias, la mayoría de la destrucción parece ocurrir por actividad de la oxidasa de NADPH. El proceso de destrucción bacteriana por los neutrófilos es un evento intracelular, in vitro tarda 30 a 90 minutos.
Respuestas a y producción de citocinas
Los factores de crecimiento que afectan la producción de neutrófilos, como el FEC-GM y el FEC-G, también afectan su función. Estas citocinas aumentan la supervivencia y aumentan la actividad de la oxidasa de NADPH dependiente de estímulo.35 También pueden afectar la quimiotaxis.36 El interferón alfa, el factor de necrosis tumoral-alfa (FNT-alfa), la interleucina-8 (IL-8) y otras citocinas que modifican la función de los neutrófilos en diversas maneras pueden tener importancia fisiológica.37 Aunque los neutrófilos contienen muy pocos ribosomas, pueden responder a los estímulos bacterianos sintetizando y secretando IL-1 y FNT-alfa.38 Además, pueden producir IL-6 y FEC-G en respuesta al FEC-GM.39 La IL-8, que es un quimiotáctico de neutrófilos, también aumenta en respuesta a estímulos inflamatorios, favoreciendo la quimiotaxis.40
TRASTORNOS EN EL NUMERO DE NEUTROFILOS
Neutrofilia
La neutrofilia, o granulocitosis, se define como una cuenta de neutrófilos mayor de dos desviaciones estándar del promedio. Para los adultos, la cuenta promedio de neutrófilos es de 4,400/mm3, y se define neutrofilia como una cuenta mayor de 7,700/mm3. En la práctica, ocurre neutrofilia clínicamente significativa (i.e., una respuesta que sugiera la presencia de inflamación, infección o un trastorno hematológico primario) cuando la cuenta de neutrófilos es mayor de 10,000/mm3. La neutrofilia se considera primaria cuando no es causada por ninguna otra enfermedad. Sin embargo, la mayoría de los casos de neutrofilia son secundarios, ocurriendo en respuesta a otros procesos patológicos. Una cuenta de leucocitos mayor de 30,000 a 50,000/mm3 con predominio de neutrófilos indica la presencia de una reacción leucemoide.
Neutrofilia primaria La neutrofilia primaria puede ocurrir en pacientes con padecimientos mieloproliferativos, como la leucemia mieloide crónica (LMC). La LMC se asocia con la presencia de cromosoma Philadelphia (Ph1) en las células de la médula ósea y baja actividad de la fosfatasa alcalina de los neutrófilos . Se han descrito neutrofilias hereditarias e idiopáticas, que son benignas y bastante raras. Además, se ha asociado neutrofila con algunas alteraciones congénitas, por ejemplo, los niños con síndrome de Down pueden tener una reacción leucemoide temporal41,42 que debe distinguirse de la leucemia congénita. La neutrofilia también se ha asociado con urticaria familiar por frío, incluye infiltración neutrofílica en la erupción y por lo general se presenta 10 horas después de la exposición al ambiente frío.43 Por último, se observa neutrofilia en pacientes con deficiencia de adhesión leucocitaria (DAL).44 Este trastorno se asocia con alteraciones en las integrinas de los neutrófilos o del antígeno sLex.
Neutrofilia secundaria La neutrofilia secundaria se observa con frecuencia en pacientes con infecciones bacterianas. Ocurre neutrofilia y aumento en la cuenta de bandas cuando se producen y liberan neutrófilos de la médula ósea en respuesta al factor estimulador de colonias generado por la endotoxina u otros productos bacterianos. La presencia de granulaciones tóxicas y de cuerpos de Döhle en los neutrófilos sugiere, pero no demuestra, la presencia de infección. La inflamación crónica, como la que ocurre con el tabaquismo o la necrosis tisular, puede producir una neutrofilia moderada que puede ser indistinguible de la asociada con infección.
La neutrofilia secundaria puede ser aguda o crónica. Los eventos estresantes agudos pueden causar neutrofilia45 cuando se liberan neutrófilos del compartimiento de marginación por la epinefrina endógena.46 También se ha asociado neutrofilia aguda con estados postictales y ejercicio.47 Ciertos medicamentos, como los esteroides y los agonistas beta-adrenérgicos pueden producir neutrofilia aguda al inducir la liberación de neutrófilos de la médula ósea48 y del compartimiento de marginación, respectivamente.
El litio produce neutrofilia crónica al estimular la producción de factores estimuladores de colonias. También puede ocurrir neutrofilia crónica en pacientes con neoplasias no hematológicas, como el cáncer de pulmón de células grandes,49 y en los que el cáncer ha metastatizado a la médula ósea.50 Las personas asplénicas y las que tienen bazos no funcionales (v.gr., pacientes con anemia de células falciformes) suelen tener neutrofilia crónica moderada.51 Además, las personas que tienen estimulación de la médula ósea secundaria a anemia hemolítica o trombocitopenia suelen tener neutrofilia crónica asociada.
Evaluación de la neutrofilia La neutrofilia se asocia con más frecuencia con inflamación crónica e infecciones bacterianas o de otro tipo. El tabaquismo es una de las causas más frecuentes de neutrofilia. Si no parece existir infección o si la neutrofilia tiene más de varias semanas, el clínico debe considerar la posibilidad de una enfermedad mieloproliferativa. La presencia de metamielocitos y mielocitos en sangre, esplenomegalia y baja actividad de fosfatasa alcalina en los neutrófilos sugieren LMC. Una actividad de fosfatasa alcalina en neutrófilos alta y los cuerpos de Döhle sugieren un proceso infeccioso.
Si la neutrofilia del paciente no puede atribuirse a infección, inflamación o medicamentos, está justificado realizar aspiración y biopsia de médula ósea, junto con análisis cromosómico de la médula ósea y cultivo en busca de agentes infecciosos. Los resultados de estos procedimientos permitirán al clínico hacer el diagnóstico de LMC u otra enfermedad mieloproliferativa, infección granulomatosa, trastorno inflamatorio o cáncer metastásico. Si no se determina la causa en una persona por lo demás sana, debe pensarse en el diagnóstico de neutrofilia idiopática o familiar. El clínico debe tener en cuenta que el 2.5 porciento de las personas normales tiene una cuenta de neutrófilos que está más allá del 95 porciento de intervalo de confianza para el valor normal.
Neutropenia
La neutropenia es de mayor preocupación que la neutrofilia, y quizá es un problema más serio porque los neutrófios son la primera línea de defensa contra los microrganismos invasores. La neutropenia se define como una cuenta de neutrófilos menor de dos desviaciones estándar por debajo del promedio normal, que equivale a menos de 1,800/mm3, aunque este valor debe modificarse a alrededor de 1,000/mm3 para los afroamericanos y los judíos Yemenitas.52,53 Aunque estos parámetros definen a la neutropenia clínicamente importante, no ocurren infecciones significativas a menos que la cuenta de neutrófilos sea menor de 500/mm3 en la mayoría de las personas o hasta menor de 200/mm3 en otros. En pacientes que tienen neutropenia causada por aumento en la destrucción de los neutrófilos, el grado de neutropenia no necesariamente correlaciona con un mayor riesgo de infección.
En pacientes con neutropenia aislada, las infecciones son poco frecuentes y afectan principalmente a la piel y los tejidos como la mucosa oral, pulmones y senos paranasales. En pacientes con neutropenia asociada con monocitopenia (v.gr., en pacientes con anemia aplástica, leucemia o mielodisplasia, y en los que acaban de recibir quimioterapia), las infecciones son muy frecuentes e incluyen sepsis y otras infecciones sistémicas. Además, las infecciones de la piel cuando existe neutropenia aislada se deben principalmente a estafilococos, mientras que cuando existen citopenias más severas y combinadas (v.gr., como después de recibir quimioterapia) participan más los organismos gram negativos. El tipo de infección que ocurre en un estado neutropénico también se relaciona con defectos concomitantes en las barreras físicas (v.gr., infecciones por Staphylococcus epidermidis en la piel y por Escherichia coli en la mucosa gastrointestinal) y está muy influenciada por el uso de antibióticos.
Fisiopatológicamente, la neutropenia puede ser causada por producción disminuida o ineficaz de neutrófilos en la médula ósea, secuestro de los mismos a lo largo de las paredes vasculares o mayor destrucción periférica. Sin embargo, con frecuencia coexiste más de un mecanismo. Al igual que en el caso de la neutrofilia, la neutropenia suele clasificarse como intrínseca (primaria) o adquirida (secundaria), dependiendo de si es causada por defectos conocidos o supuestos en los neutrófilos [ver tabla 2] o asociada con otras alteraciones. Las neutropenias adquiridas incluyen neutropenia asociada a infecciones, neutropenia inducida por medicamentos, neutropenias autoinmunes, isoinmunes y aloinmunes, neutropenia idiopática crónica y neutropenias asociadas con linfocitosis T-g, aplasia pura de leucocitos, síndrome de inmunodeficiencia adquirida (SIDA) y esplenomegalia. Debido a que las neutropenias adquiridas son mucho más comunes que las intrínsecas, se analizan con mayor detalle.
|
|||||||||||||||||||||||||||||||||||||||||||||
AR- autosómico recesiva, AD- autosómico dominante, E- casos esporádicos, FEC-G- factor estimulador de colonias de granulocitos, BH- biometría hemática |
Neutropenia asociada a infección La neutropenia asociada a infcciones es la forma más común de neutropenia en los niños y con frecuencia se relaciona con infección viral. Comienza días después de inicio de la infección y, aunque suele resolverse en algunas semanas, dura hasta dos meses. La causa de esta forma de neutropenia incluye tanto menor producción como mayor destrucción de neutrófilos. El número de monocitos suele ser normal y, excepto en casos severos, la neutropenia no se asocia con una mayor susceptibilidad a infección bacteriana. Otras formas de neutropenia asociada a infecciones incluyen la neutropenia que se observa en pacientes con SIDA54 (ver adelante) y la asociada con infección grave por ciertas bacterias, como Salmonella y otros bacilos gram negativos y S. aureus. Además, en lactantes con sepsis puede desarrollarse neutropenia muy grave que puede requerir transfusiones de granulocitos.55
Neutropenia inducida por medicamentos La neutropenia inducida por medicamentos puede ser la forma más común de neutropenia en adultos.56 Se han implicado muchos fármacos [ver tabla 3]. Los mecanismos fisiopatológicos pueden incluir menor producción de neutrófilos,57 que se ejemplifica por el efecto dependiente de la dosis que se observa con las fenotiacinas, o reacción destructiva mediada por anticuerpos,58 como ocurre en el caso del propiltiouracilo. La recuperación suele ocurrir varios días después de suspendido el medicamento, en los casos de neutropenia causada por menor producción de neutrófilos, la recuperación casi siempre va precedida por monocitosis.59
|
||||
|
Neutropenia autoinmune La neutropenia autoinmune ocurre como un fenómeno aislado o secundario a otros trastornos autoinmunes.60 También puede ser causada por infección61 o tratamiento con ciertos medicamentos.62 En pacientes con síndrome de Evans, se asocia con trombocitopenia inmune y en ocasiones con anemia hemolítica. Los pacientes con neutropenia autoinmune suelen tener neutropenia moderada a severa acompañada de monocitosis. La celularidad de la médula ósea está aumentada, con una disminución relativa en el número de precursores tardíos de neutrófilos. Se observa esplenomegalia en alrededor del 50 porciento de los pacientes. Se han detectado anticuerpos antineutrófilo específicos, por lo general de tipo IgG o IgM, en muchos pacientes con esta enfermedad, y en algunos pacientes se han demostrado complejos inmunes circulantes. También se han detectado anticuerpos antineutrófilo en pacientes con lupus eritematoso generalizado y síndrome de Felty.63 En estos últimos se han implicado a las células T supresoras en la mediación de la neutropenia.
Neutropenia isoinmune y aloinmune En neonatos puede ocurrir neutropenia moderada a severa secundario a la trasmisión de anticuerpos IgG de la madre al infante. La incidencia de neutropenia isoinmune se ha calculado en dos por 1,000 nacimintos.64 El niño puede sufrir sepsis o estar asintomático. Puede ser difícil distinguir la neutropenia isoinmue de la asociada con sepsis en el recién nacido. Se detectan anticuerpos antineutrófilo en el suero del niño y de la madre, pero con frecuencia muestran específicidad para los neutrófilos del padre y no de la madre.65 La neutropenia suele resolverse en 12 a 15 semanas. Puede también ocurrir neutropenia aloinmune en el recién nacido como resultado de la transferencia pasiva de anticuerpos de una madre con neutropenia autoinmune. La evolución clínica de la enfermedad es semejante a la de la neutropenia isoinmune.
Neutropenia asociada con linfocitosis T-g
Los pacientes con linfocitosis T-g (linfocitosis que incluye linfocitos granulares grandes) suelen tener neutropenia.66 Estos pacientes tienen 50 a 60 años de edad, padecen infecciones recurrentes y pueden tener historia de artritis reumatoide y esplenomegalia. La cuenta de linfocitos es alta, pero rara vez es mayor de 20,000/mm3. El examen de la médula ósea muestra una médula normocelular con mayor número de linfocitos y detención de la línea mieloide en la fase de mielocito. La evolución de la enfermedad suele ser tórpida, y los pacientes mueren por un trastorno linfoproliferativo o sepsis.67 La linfocitosis es de origen clonal y puede ser maligna. La infusión intravenosa de g-globulina disminuye la cuenta de linfocitos y aumenta el número de neutrófilos.68 El síndrome de Felty suele representar una variante de este padecimiento.
Neutropenia asociada con aplasia pura de leucocitos La aplasia pura de leucocitos es un padecimiento poco frecuente caracterizado por infecciones severas y neutropenia. En muchos casos se asocia con timoma. El examen de la médula ósea muestra ausencia casi total de precursores mieloides y un número normal de precursores eritroides y megacariocitos. La eliminación del timoma puede no ser suficiente para resolver la neutropenia. También puede observarse aplasia pura de leucocitos en pacientes que reciben tratamiento con ibuprofén. La ciclofosfamida, los esteroides, la ciclosporina y la g-globulina intravenosa han sido todos eficaces para este trastorno.
Neutropenia asociada con síndrome de inmunodeficiencia adquirida Se desarrolla neutropenia en la mayoría de los pacientes con SIDA, y alrededor del ocho porciento de las personas con prueba positiva para el virus de inmunodeficiencia humana (VIH) que no tienen SIDA presentan también cifras bajas de neutrófilos.54 El número de neutrófilos suele disminuir en forma gradual, siguiendo a la disminución en la cuenta de linfocitos T CD4+ por meses o años. La neutropenia suele ser leve o moderada (i.e., los pacientes tienen de 500 a 1,500 neutrófilos/mm3). La neutropenia severa en pacientes con infección por VIH suele ser causada por los tratamientos farmacológicos u otros factores de complicación. La médula ósea suele ser hipercelular y contiene un mayor número de linfocitos. Se observan cambios displásicos en los granulocitos. A pesar de la hipercelularidad de la médula ósea, se cree que la hematopoyesis ineficaz es la causa de la neutropenia.
Neutropenia idiopática crónica La neutropenia idiopática crónica suele asociarse con una cuenta de neutrófilos de 200 a 500 /mm3, monocitosis asociada y una médula ósea que muestra actividad mieloide normal o aumentada, pero escasas células mieloides maduras.69 La mayoría de los casos son adquiridos, pero algunos tienen patrón de herencia autosómico dominante. Los individuos con este trastorno suelen ser evaluados por vez primera como un hallazgo en una citología hemática de rutina al evaluar una infección. La evolución clínica del padecimiento es relativamente banigna. Los pacientes con neutropenia idiopática crónica suelen tener cuentas normales de neutrófilos durante las infecciones bacterianas, y pueden movilizar neutrófilos en respuesta al estímulo con prdnisona.69 La neutropenia idiopática crónica puede ser ocasionada por diversos factores y por lo general no requiere tratamiento porque las infecciones son raras y bien toleradas.
Neutropenia asociada con esplenomegalia La neutropenia puede asociarse con hiperesplenismo. En estos casos, casi nunca es tan severa como para causar infecciones. Se relaciona con marginación reversible de los neutrófilos en el bazo, por lo que no produce depleción corporal total de los mismos.
Evaluación y tratamiento de la neutropenia Cuando se encuentra que un paciente tiene neutropenia, debe realizarse una historia clínica (incluyendo antecedentes familiares) y examen fisico para determinar si la neutropenia se ha asociado con mayor incidencia o severidad de la infección. Además, se obtendrá una citología hemática completa para establecer si la neutropenia se asocia con alguna otra alteración hematológica y si los neutrófilos tienen una estructura normal. La mayoría de las neutropenias aisladas son ocasionadas por medicamentos o virus. Por lo tanto, si el paciente está asintomático, deben suspenderse los medicamentos y se vigilará al paciente hasta por dos meses. Si la neutropenia no se resuelve o si el paciente tiene síntomas e historia de infecciones importantes, debe realizarse un estudio de médula ósea que incluya examen morfológico y análisis cromosómico. Cuando esté justificado, puede ser útil investigar anticuerpos antineutrófilo, pruebas para determinar otros tipos de disfunción inmunológica (como cuentas de células T y subgrupos), y medición de las concentraciones de inmunoglobulinas.
Debe instruirse a los pacientes con neutropenia leve crónica y a los que tienen neutropenia severa a realizar buen cuidado de la piel y cavidad oral, administrando antibióticos adecuados cuando ocurran infecciones. Para los pacientes con neutropenia severa debe considerarse el tratamiento profiláctico con antibióticos, como trimetoprim-sulfametoxazol y el uso de FEC o FEC-GM. Los pacientes con neutropenia y monocitopenia tienen riesgo muy alto de infección, en especial de sepsis, y deben ser vigilados en forma estrecha. Se ha realizado trasplante de médula ósea en pacientes con neutropenia severa, pero el advenimiento de la terapia con citocinas ha disminuido la frecuencia con la que se realiza este procedimiento.
TRASTORNOS DE LA FUNCION DE LOS NEUTROFILOS
En pacientes que tienen infecciones recurrentes, severas o poco frecuentes asociadas a número normal de neutrófilos, debe considerarse la presencia de un trastorno funcional. Estos trastornos son causados por defectos en la adherencia, quimioteraxis, degranulación o metabolismo oxidativo de los neutrófilos [ver tabla 4].
|
|||||||||||||||||||||||||||||||||||||||||||||||||
AR-autosómico recesiva, E-casos esporádicos, sLex-sialil-Lewisx, NBT- nitroazul de tetrazolio |
El enfoque lógico para la evaluación de los pacientes con infecciones recurrentes, severas o poco frecuentes consiste en determinar primero si el paciente ha tenido realmente estas infeciones, si existe historia familiar de infección y en realizar un examen físico completo que incluya la búsqueda de evidencias de que existe una respuesta adecuada a la infección [ver figura 5]. La biometría hemática completa y el estudio de los granulocitos en un frotis puede mostrar neutropenia, deficiencia de gránulos específicos o síndrome de Chédiak-Higashi, o sugerir una alteración esplénica. Además, si existe anemia normocítica debe sospecharse deficiencia de deshidrogenasa de glucosa-6-fosfato (G6P) o de glutatión sintetasa. Es adecuado evaluar las inmunoglobulinas (IgG, IgM, IgA e IgE) y los componentes del complemento (C3 y CH50) para detectar deficiencia total o parcial aumento de IgE o consmo del complemento. Si todas estas mediciones son normales, se evaluará la función de los neutrófilos con nitroazul de tetrazolio (NBT), ensayos de producción de superóxido y de quimiotaxis. La prueba de NBT y el ensayo de superóxido pueden determinar si un paciente tiene enfermedad granulomatosa crónica (EGC), deficiencia severa de G6PD o un trastorno de la vía del glutatión. Los estudios quimotácticos pueden usarse para confirmar el síndrome de Chédiak-Higashi y defectos adquiridos. La deficiencia de adhesión leucocitaria I (DAL I) y II (DAL II) se diagnostica por citometría de flujo. Si los resultados de todas estas pruebas son normales, puede ser útil realizar estudios de ingestión con el suero y células del paciente y por medio de tinción de MPO. De esta manera pueden diagnosticarse todas las alteraciones funcionales de los neutrófilos conocidas, con frecuencia con ayuda de interconsultas especializadas y pruebas de laboratorio de experimentación.

|
| Figura 5 |
| Evaluación de la infección recurrente |
Monocitos y macrófagos
Los monocitos y macrófagos tienen un papel muy importante en la homeostasis y el complemento, y aumentan la función de los neutrófilos en los mecanismos de defensa del huésped. Los monocitos y los macrófagos realizan funciones de mantenimiento en los tejidos, como remodelación ósea por osteoclastos después de una lesión70 y fagocitosis esplénica de los eritrocitos viejos,71 y participan en la regulación inmunológica, interactuando con células T y B durante la presentación de los antígenos.72 Además, los monocitos y los macrófagos ayudan a controlar las infecciones por micobacterias, parásitos, hongos y virus.73-76
DESARROLLO DE MONOCITOS-MACROFAGOS
Los monocitos se desarrollan a partir de células progenitoras en la médula ósea. La citocina responsable de dirigir estas células hacia el linaje monocito parece ser el factor estimulador de colonias de macrófagos (FEC-M), aunque también son importantes la IL-3 y el FEC-GM.77 Una vez que la célula progenitora se diferencía hacia el linaje monocítico, se desarrollan los monoblastos y los promonocitos.
El monocito maduro [ver figura 1] es liberado casi de inmadiato hacia la circulación. Como en el caso de los neutrófilos, los monocitos sanguíneos pueden encontrarse en reservorios circulantes y de marginación. Sin embargo, el número de monocitos en el reservorio de marginación es alrededor de 3.5 veces más grande que el circulante.78 El monocito permanece en la circulación ocho a 72 horas antes de migrar hacia los tejidos. Después de la migración la célula se vuelve más grande y se convierte en un macrófago tisular al adquirir ribosomas, lisosomas, mitocondrias, retículo endoplásmico e inclusiones electrodensas.
FUNCION DE MONOCITOS Y MACROFAGOS
El sistema fagocito munonuclear incluye a los monoblastos, promonocitos, monocitos y macrófagos tisulares. Estos últimos son una población heterogénea de células con diferentes características morfológicas y funcionales dependiendo del tejido en el que resida. Los macrófagos alveolares, las células de Kuppfer, los osteoclastos, los macrófagos peritoneales y esplénicos y las células de Langerhans son todos macrófagos tisulares. Además de tener capacidad fagocítica, los monocitos y macrófagos tienen una función en otros aspectos de la respuesta inmunológica y en la regulación de la homeostasis.
Con excepción de los macrófagos alveolares, que dependen por completo del metabolismo aeróbico para la producción de energía, los monocitos y macrófagos son anaerobios facultativos.79 Al igual que la fagocitosis en los neutrófilos, la fagocitosis en los monocitos y macrófagos se asocia con un estallido oxidativo y estimulación de la vía hexosa monofosfato. La adhesión, quimiotaxis y activación de la fagocitosis son semejantes en ambos tipos de células, aunque los macrófagos son mejores que los neutrófilos para la fagocitosis, y realizan quimiotaxis con menos rapidez y eficacia. Los macrófagos son capaces de secretar más de 100 moléculas bien definidas, incluyendo citocinas, factores de crecimiento y ciertos reactantes de fase aguda.80 El macrófago también es capaz de tener actividad bactericida independiente de oxígeno que puede depender de la actividad lítica.81 Además, los macrófagos estimulados son capaces de producir óxico nítrico.82
Los monocitos y los macrófagos pueden presentar antígenos a las células T en asociación con las moléculas de clase II del complejo principal de histocompatibilidad (CPH). Esta asociación parece ocurrir dentro de los lisosomas de una célula mononuclear antes de que las moléculas de clase II se expresen en la superficie celular,83 parece incluir contacto intercelular directo y puede aumentarse por la liberación de citocinas como la IL-1. Los macrófagos pueden presentar el antígeno procesado a las células B para despertar una respuesta humoral.84 Los monocitos y los macrófagos participan en la citotoxicidad mediada por células dependiente e independiente de anticuerpos. La citotoxicidad incluye al metabolismo oxidativo, la producción de óxido nítrico y citocinas y la secreción de mediadores citotóxicos.
Los macrófagos tienen una función clave para metabolizar las proteínas de alto peso molecular, las glucoproteínas y otros materiales que participan en forma íntima en la destrucción de las células ancianas y muertas. También se requieren para la angiogénesis y la cicatrización de heridas, y son capaces de inducir neovascularización y proliferación de células endoteliales. Por último, los monocitos y macrófagos son capaces de secretar diversos factores de crecimiento hematopoyético, incluyendo FEC-GM, FEC-G, IL-1 y FEC-M.85
SINDROMES HISTIOCITICOS
Los síndromes histiocíticos son un grupo de trastornos malignos y no malignos en los que el macrófago es la célula principalmente alterada. Entre los trastornos malignos se incluyen la leucemia monocítica aguda, la histiocitosis maligna, también llamda reticulosis medular histiocítica y el linfoma histiocítico verdadero. Los trastornos no malignos, que se analizan en este capítulo, incluyen los síndromes de histiocitosis de células de Langerhans (HCL) y los síndromes hemofagocíticos, como la histiocitosis sinusal con linfadenopatía masiva, la linfohistiocitosis hemofagocítica familiar (LHF) y el síndrome hemofagocítico asociado a infección (SHAI).
Síndromes de histiocitosis de células de Langerhans
Clásicamente, los síndromes de HCL incluyen al granuloma eosinofílico solitario, al granuloma eosinofílico multifocal, a la enfermedad de Hand-Schüller-Christian y a la enfermedad de Letterer-Siwe. Estos padecimientos afectan sobre todo lactantes y niños pequeños, aunque se han descrito también en adultos jóvenes.86 Debido a que los síndromes de HCL representan una enfermedad continua que con frecuencia no puede designarse de este modo rígido y arbitrario, se han desarrollado otras clasificaciones que se relacionan con la edad en el momento del diagnóstico, la extensión de la enfermedad y la presencia de disfunción orgánica.87 Los signos y síntomas de los síndromes de HCL dependen de los órganos afectados. Pueden afectarse los huesos, la piel, los dientes, el tejido gingival, los oídos, los órganos endócrinos, los pulmones, el hígado, el bazo, los ganglios linfáticos y la médula ósea, y volverse disfuncionales por infiltración celular.
Los granulomas eosinofílicos tanto solitarios como multifocales se encuentran principalmente en niños mayores y adultos jóvenes. Al momento de la presentación estos pacientes suelen tener problema para aumentar de peso o tienen zonas de inflamación dolorosa por la infiltración de los tejidos sobre lesiones óseas marginales.88 La enfermedad de Hand-Schüller-Christian ocurre en niños pequeños (i.e., dos a cinco años de edad) y se manifiesta como defectos óseos con exoftalmos causados por una masa tumoral en la cavidad orbitaria, ausencia de dientes por la infiltración de la encía y afección de los huesos planos del cráneo, costillas, pelvis y escápula. También puede existir diabetes insípida asociada, que afecta hasta al 50 porciento de los pacientes con síndromes de HCL.89 Es común la otitis media crónica, causada por afección del hueso temporal. La forma más rara y severa de HCL es la enfemedad de Letterer-Siwe, que ocurre en forma típica en niños de menos de dos años. Los síntomas consisten en erupciones seborreicas, eccematosas, descamativas y pruriginosas que afectan el cuero cabelludo, los canales auditivos, el abdomen y el área intertriginosa del cuello y cara. Además existe secreción ótica, linfadenopatía, hepatoesplenomegalia y, en casos severos, disfunción hepática con hipoalbuminemia y menor síntesis de los factores de la coagulación. Puede ocurrir afección del sistema hematopoyético, incluyendo la anemia y la trombocitopenia, y daño grave al pulmón con hipoxia.
El pronóstico para el paciente con síndrome de HCL depende de tres factores: edad de inicio, número de órganos afectados y la presencia de disfunción de los órganos específicos.87 Un adolescente con un granuloma eosinofílico solitario en hueso tiene el mejor pronóstico, mientras que un niño con muchos órganos afectados el peor. El diagnóstico se realiza por estudio patológico de una lesión por medio de microscopía de luz y demostración de gránulos electrodensos en las células de Langerhans (gránulos de Birbeck) al microscopio electrónico o antígeno CD1 con la inmunotinción.
El tratamiento de la HCL en ocasiones no es necesario, cuando sí, la cirugía o la radioterapia local pueden ser curativas.90 Además, aunque los síndromes de HCL no son malignos, responden a agentes quimioterápicos.91 Debido a que el objetivo del tratamiento en los síndromes de HCL es administrar la mínima cantidad necesaria del tratamiento menos tóxico para controlar la enfermedad, el principio para el uso de los agentes quimioterápicos es comenzar con el agente más benigno y después pasar a agentes más tóxicos, tratando de nunca administrar un tratamiento peor que la enfermedad. Al inicio debe administrarse vinblastina intravenosa en dosis de 0.15mg/kg/semana, y la dosis debe aumentarse a 0.025 y 0.05 mg/kg/semana si no ocurre mejoría después de dos semanas. Si aún no existe mejoría, se agregará al esquema prednisona, 2 mg/kg/día, disminuyendo ambos medicamentos lo más rápido posible. Si este régimen fracasa, debe administrarse metotrexate por vía intramuscular, intravenosa u oral, en dosis de 10 mg/m2/semana y 6-mercaptopurina, 20 mg/m2/día, durante 14 a 21 días, y los medicamentos se disminuirán cuando el paciente mejore. Si estos esquemas no controlan por completo la enfermedad o si los efectos adversos asociados con su uso justifican su suspensión, se ha reportado que el etopósido puede ser benéfico.92
Histiocitosis sinusal con linfadenopatía masiva
La histiocitosis sinusal con linfadenopatía masiva se caracteriza clínicamente por linfadenopatía masiva benigna, con frecuencia crónica e indolora, que suele afectar los ganglios cervicales y con menos frecuencia los axilares, hiliares, peritraqueales o inguinales.93 En el 30 porciento de los pacientes existe enfermedad extranodal en las vías respiratorias, huesos, órbitas, piel, hígado y riñones.94 La enfermedad suele ser benigna, pero puede presentarse morbilidad importante e incluso la muerte si ocurre invasión tisular masiva del hígado, riñones, pulmones y otras estructuras críticas.95 Aunque el 80 porciento de los pacientes se diagnostican en la primera o segunda década de la vida, este padecimiento puede afectar también a los ancianos. Los pacientes suelen ser descendientes de africanos y la incidencia de esta enfermedad es más alta en Africa e India Occidental.
En pacientes con histiocitosis sinusal con linfadenopatía masiva, los ganglios linfáticos afectados suelen tener dilatación sinusoidal marcada e hiperplasia folicular con proliferación de histiocitos espumosos y células gigantes multinucleadas en los senos. La etiología de este padecimiento se desconoce y puede relacionarse con una inmunorregulación anormal.96 El tratamiento suele ser tanto innecesario como ineficaz. Al resolverse la enfermedd, la afección extranodal desaparece antes de la nodal. Los intentos de tratamiento deben reservarse para circunstancias especiales graves. Se han administrado radiación, esteroides, vinblastina y ciclofosfamida con grados variables de éxito.97
Linfohistiocitosis hemofagocítica familiar
La LHF es un padecimiento hereditario rápidamente fatal que se caracteriza por pancitopenia periférica con hiperplasia de la médula ósea, linfadenopatía sistémica, fiebre, disfunción hepática severa y hepatoesplenomegalia. Los pacientes tienen concentraciones bajas de fibrinógeno sin evidencia de coagulación intravascular diseminada. Los niños y los lactantes son los más afectados, y dos tercios de los pacientes tienen menos de tres meses de edad.
Los síntomas de presentación de la LHF incluyen palidez, irritabilidad, anorexia, diarrea, no incremento ponderal y una erupción maculopapular.98 La evolución clínica suele ser progresiva y dura un promedio de solo seis semanas. La mayoría de los pacientes mueren por infección, hemorragia o crisis convulsivas. Alrededor de la tercera parte de los pacientes tiene afección al sistena nervioso central. El estudio patológico del hígado muestra cambios grasos focales, necrosis e infiltración de linfocitos e histiocitos. La eritrofagocitosis y la hemofagocitosis son datos esenciales en el diagnóstico. Aunque la etiología de la LHF se desconoce, la inmunodeficiencia es un dato constante.
Por lo general los esteroides y los agentes citotóxicos son ineficaces en el tratamiento de la LHF. Se ha demostrado que el etopósido induce remisión clínica, pero el tratamiento definitivo requiere del uso de trasplante de médula ósea.99
Síndrome hemofagocítico asociado a infección
El SHAI se observa con frecuencia en personas inmunosuprimidas como consecuencia de una infección subyacente viral100 o bacteriana,101 o de tratamiento inmunosupresor.102 Aunque la evolución clínica del SHAI varía, se ha reportado una mortalidad del 40 porciento.100 En ausencia de una inmunodeficiencia subyacente, de historia familiar de SHAI o de consanguinidad, puede ser difícil distinguir entre el SHAI o la LHF. Desde el punto de vista patológico las enfermedades son muy semejantes. El SHAI, al igual que la LHF, puede responder a etopósido o puede resolverse en forma espontánea.
ENFERMEDADES POR ALMACENAMIENTO LISOSOMAL
Los monocitos y los macrófagos tienen una función importante en la remodelación tisular y la eliminación de detritos celulares viejos, y los lisosomas son los organelos que realizan estas funciones. Por lo tanto, las alteraciones enzimáticas que incluyen a los constituyentes lisosomales causan trastornos de almacenamiento relacionados con la función de los macrófagos. Estos padecimientos incluyen a las mucopolisacaridosis, las glucoproteinosis, las esfingolipidosis y las enfemedades por almacenamiento de lípidos neutros [ver tabla 5]. Se han descrito defectos enzimáticos para la mayoría de estos padecimientos, y el diagnóstico depende de demostrar la alteración enzimática en los macrófagos o histiocitos. La mayoría de estos defectos son causados por mutaciones puntiformes o rearreglos genéticos en un solo locus del gen que codifica para una hidrolasa lisosomal.103
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Los dos tipos de tratamiento para las enfermedades por almacenamiento lisosomal disponibles en la actualidad son el trasplante celular y el tratamiento enzimático. Debido a que las células tronco de la médula ósea son pluripotenciales, estas células son las que se emplean en la mayoría de los trasplantes.104 Sin embargo, al parecer las enfermedades por almacenamiento lisosomal afectan otras células además de las derivadas dela médula ósea, por lo que pueden ser necesarios otros tipos de trasplante.105 En el pasado la enfermedad de Gaucher se trataba con trasplante de médula ósea, pero en la actualidad se maneja con alglucerasa, una glucocerebrosidasa con terminación a-manosil.106 Aunque no se ha establecido el esquema óptimo, se ha demostrado que la dosis inicial de 30 a 60 UI/kg es eficaz. Esta dosis puede repetirse cada dos semanas o puede calcularse la dosis mensual y administrarse infusiones más frecuentes. Alrededor del 10 a 15 porciento de los pacientes desarrollan anticuerpos contra la enzima. De estos pacientes, el 50 porciento sufre prurito y urticaria. Debido a que la mayoría de los pacientes que desarrolla anticuerpos lo hace en los primeros seis a nueve meses de tratamiento, se recomienda administrar la enzima bajo vigilancia médica durante alrededor de nueve meses antes de iniciar la atención domiciliaria. El uso de trasplante de médula ósea para otros trastornos de almacenamiento de lisosomas se considera aún experimental y ha dado resultados muy variables.107
Eosinófilos
Los eosinófilos pueden aumentar o suprimir las reacciones inflamatorias agudas y las respuestas mediadas por infecciones por helmintos, alergia y ciertos tumores.108 Al igual que los neutrófilos, los eosinófilos son capaces de fagocitar, pero principalmente son células secretoras. La mayoría de la funciones que realizan requieren de la lbieración del contenido de sus gránulos o de radicales de oxígeno generados por la membrana celular cuando ésta se activa. Los eosinófilos responden a agentes quimiotácticos específicos y a factores de crecimiento que permiten su acúmulo en sitios de inflamación.
ESTRUCTURA DE LOS EOSINOFILOS
Los gránulos de los eosinófilos contienen proteínas muy básicas que se tiñen en forma intensa con colorantes ácidos. Poseen una apariencia característica bajo el microscopio electrónico [ver figura 1]. Los gránulos consisten en un centro electrodenso rodeado de una matriz relativamente radiolúcida, y existe actividad de peroxidasa de eosinófilos (POE) en la matriz.109 El centro denso tiene una estrucutra cristaloide y contiene proteínas catiónicas eosinófilas (PCE), proteínas básicas principales (PBP) y neurotoxinas derivadas de eosinófilos.110 Las PBP son proteínas relativamente pequeñas capaces de causar daño importante a los esquistosómulas por unión y ruptura de sus membranas celulares. Además, aumentan la adherencia de los eosinófilos y los neutrófilos a los esquistosómulas.111 Las PCE también se unen a los esquistosómulas y son tóxicas para ellos.
FUNCION DE LOS EOSINOFILOS
Los eosinófilos responden a diversos factores quimiotácticos que les permiten penetrar en los tejidos y realizar sus funciones. Algunos de estos factores quimiotácticos (como C5a, péptidos que contienen N-formilmetionil y leucotrieno B4) estimulan tanto eosinófilos como neutrófilos. Sin embargo, varios estímulos quimiotácticos son muy específicos para los eosinófilos. Entre éstos se incluyen al factor activador de plaquetas (FAP), al factor quimiotáctico eosinófilo de anafilaxia y a varios factores derivados de parásitos. Las respuestas al FAP, uno de los activadores más potentes de los eosinófilos normales, incluyen quimiotaxis, adherencia, mayor unión de IgE, producción de superóxido, liberación de proteínas de los gránulos y síntesis de prostenoides.
Tanto la producción como la activación de los eosinófilos se afectan por el FEC-GM, la IL-5 y la IL-3.112 La exposición a dosis bajas de IL-5 activa en forma específica a los eosinófilos para responder a otros estimulantes.113 De hecho, aunque la IL-5 tiene un efecto específico y potente sobre el crecimiento de eosinófilos en cultivo, prácticamente no tiene efecto estimulador en el crecimiento de otras colonias mieloides. Además, los ratones transgénicos que expresan IL-5 muestran eosinofilia prolongada,114 y la administración de anticuerpos anti-IL-5 a ratones evita por completo que se desarrolle eosinofilia en respuesta a parásitos.115 Una vez activados, los eosinófilos presentes en ciertas condiciones patológicas asociadas con eosinofilia tienen mayor generación de especies reactivas del oxígeno, mayor utilización y transporte de glucosa, mayor consumo de oxígeno, menor carga en la superficie celular y activación de las fosfatasas ácidas en gránulos específicos.116 Estos cambios se acompañan de una reducción en la densidad celular.
Los eosinófilos aumentan la respuesta inmunológica a helmintos. Realizan esta función al unirse a la superficie de formas tanto larvarias como adultas, al dañar a las células blanco por mecanismos dependientes de oxígeno que son semejantes a los de los neutrófilos y al dañar superficies celulares al liberar proteínas de gránulos como PBP y PCE.117 Aunque la liberación de estas proteínas daña en forma semejante a las células tumorales, la interacción entre eosinófilos y las células tumorales se comprende menos bien. La presencia de eosinofilia en pacientes con enfermedad de Hodgkin parece depender de la producción de IL-5 por las células de Reed-Sternberg.118 Los eosinófilos contribuyen a la fibrosis de la enfermedad de Hodgkin tipo esclerosis nodular al producir factor de transformación de crecimiento-b1 (FTC-b1).
Los eosinófilos realizan una función inmunosupresora en las reacciones de hipersensibilidad inmediata. Las prostaglandinas E1 y E2 liberadas de las membranas de los eosinófilos activados son capaces de suprimir la degranulación de los basófilos.119 Otras sustancias de los gránulos, como la histaminasa y la fosfolipasa B, inactivan la histamina y el FAP de las células cebadas, respectivamente. Los eosinófilos median también el daño tisular a través de los efectos de las PBP, las PCE y el superóxido y los otros radicales reactivos del oxígeno aumentados por el sistema de peroxidasa de eosinófilos.120 El balance entre la estimulación eosinofílica y la inhibición de la inflamación es delicada y se rompe con facilidad.
TRASTORNOS EN EL NUMERO DE EOSINOFILOS
Eosinofilia
La evaluación del paciente con eosinofilia (una cuenta de eosinófilos > 700/mm3) es difícil porque la causa de este trastorno es múltiple y diversa. Las causas comunes de la eosinofilia secundaria incluyen trastornos alérgicos, infecciones causadas por parásitos u otros organismos, enfermedades dermatológicas, enfermedades pulmonares, enfermedades del tejido conectivo, neoplasias e inmunodeficiencias. También existen múltiples causas menos comunes, como gastroenteritis eosinofílica, enfermedad inflamatoria intestinal, hepatitis activa crónica, pancreatitis e hipopituitarismo. En ocasiones no se encuentra la causa de la eosinofilia incluso después de la evaluación exhaustiva del paciente.
Síndrome hipereosinofílico
El término síndrome hipereosinofílico (SHE) suele usarse para caraterizar la enfermedad para la que no se identifica la causa de la eosinofilia. Los criterios empleados para el SHE consisten en una cuenta de eosinófilos mayor de 1,500/mm3 durante más de seis meses, la ausencia de otro diagnóstico que explique la eosinofilia y signos o síntomas de infiltración de eosinófilos en los tejidos.121 El SHE suele distinguirse de trastornos malignos asociados con eosinofilia, como la leucemia aguda. También deben excluirse reacciones alérgicas, la exclusión de estas reacciones suele basarse en la historia, examen físico y revisión de los medicamentos actuales. Debido a que muchos medicamentos pueden generar una reacción alérgica asociada con eosinofilia, deben suspenderse todos los medicamentos que no sean indispensables antes de evaluar al paciente. Algunas enfermedades respiratorias alérgicas se asocian con eosinofilia, al parecer por liberación de PCE.122 Los pacientes con estas enfermedades pueden tener niveles elevados de IgE. Sin embargo, debe tenerse en cuenta que algunos pacientes con SHE pueden tener también elevación de esta inmunoglobulina.
Las infecciones parasitarias, sobre todo las causadas por helmintos que invaden los tejidos como las filarias y Strongyloides, Trichinella, Schistosoma y Toxocara, con frecuencia presentan eosinofilia.123 Se recomienda estudiar múltiples muestras de excremento y aspirados de contenido del intestino delgado para descartar estos padecimientos, en especial en pacientes con diarrea o riesgo especial de infección (v.gr., los que viajan con frecuencia, están expuestos a animales y tienen inmunodeficiencias). Además, deben realizarse estudios serológicos, pruebas radiológicas y frotis de sangre periférica y médula ósea para excluir la presencia de enfermedades del tejido conectivo, síndromes linfoproliferativos ocultos y tumores sólidos, y neoplasias hematológicas, respectivamente.
El frotis sanguíneo de un paciente con SHE suele demostrar eosinófilos maduros normales de morfología típica. Sin embargo, se ha reportado la presencia de hipogranulación y vacuolas citoplásmicas.124 La cuenta total de leucocitos suele ser de 10,000 a 30,000/mm3, el 30 a 70 porciento de los cuales son eosinófilos. La leucocitosis progresiva con eosinofilia debe originar la posibilidad de que un paciente tenga SHE. La médula ósea suele ser hipercelular, y los eosinófilos que constituyen el 25 a 75 porciento de los elementos de la médula.124
La fibrosis en el SHE es rara, los cambios cardiacos son los que predominan en el espectro de las alteraciones clínicas asociadas con la enfermedad, y son la principal causa de morbimortalidad. Sin embargo, estos cambios cardiacos no son distinguibles de los encontrados en pacientes con eosinofilia secundaria. Los pacientes pueden tener insuficiencia cardiaca congestiva crónica, alteraciones valvulares y engrosamiento endocárdico biventricular característico y fibroso con trombos murales. La mayoría de los pacientes tienen engrosamiento de la pared ventricular izquierda. El ecocardiograma es el método más sensible para detectar estas alteraciones. Puede ocurrir también afección pulmonar, que causa fiebre, tos, pérdida de peso y sibilancias.
El enfoque diagnóstico de una persona con sospecha de SHE debe incluir una historia y examen físico completos, citología hemática y cuenta total de eosinófilos. Deberá estudiarse la posibilidad de eosinofilia secundaria por medio de cuantificación de IgE, muestras en heces para huevos y parásitos, aspirado duodenal, aspiración y biopsia de médula ósea, radiografía de tórax y ecocardiografía. Si el paciente tiene diagnóstico de SHE, la decisión de administrar tratamiento debe basarse en la presencia o ausencia de síntomas. Si no existen síntomas, no se instituirá tratamiento y el paciente se reevaluará en forma periódica. Si existen síntomas que afecten los pulmones o el corazón, debe administrarse prednisona en dosis de 1 mg/kg/día durante dos semanas, seguido de 1 mg/kg cada tercer día por tres meses. Si ocurre mejoría deberá suspenderse el tratamiento. Sin embargo, si el tratamiento no mejora los síntomas o la cuenta de eosinófilos, deberá administrarse hidroxiurea en dosis de 0.5 a 1.5 g/día para disminuir la cuenta de leucocitos a 10,000/mm3. Si este agente no produce respuesta clínica, puede administrarse otro como ciclofosfamida, interferón alfa, ciclosporina o etopósido.125-127 Si la enfermedad no responde a este tipo de tratamiento y el paciente tiene menos de 50 años, deberá considerarse realizar trasplante de médula ósea.
Basófilos
Los basófilos son mediadores clave de las reacciones de hipersensibilidad inmediata, asma, urticaria, rinitis alérgica y anafilaxia. Los basófilos son estimulados por los mediadores solubles, principalmente IgE, para liberar el contenido de los gránulos y metabolitos del ácido araquidónico de sus membranas plasmáticas. Se sabe menos sobre los basófilos que sobre el resto de los tipos de leucocitos por el menor número de estas células en el organismo. Las células cebadas están relacionadas, aunque son diferentes, a los basófilos.128 Ambos tipos de células derivan de una célula progenitora hematopoyética común en la médula ósea. Sin embargo, los basófilos expresan receptores para IL-2, IL-3 y moléculas de integrina, ninguno de los cuales se expresan en las células cebadas, mientras que la molécula de adhesión intercelular-1 (ICAM-1) y el producto c-kit sí se expresan en las células cebadas y no en los basófilos. No se ha observado hasta la fecha transformación de un tipo celular a otro.
ESTRUCTURA Y FUNCION DE LOS BASOFILOS
Los gránulos citoplásmicos de basófilos y células cebadas contienen glucosaminoglucanos sulfatados. En los basófilos normales, los glucosaminoglucanos sulfatados son principalmente heparina. Los glucosaminoglucanos sulfatados en los gránulos causan la intensa tinción de los basófilos.
La mayoría, si no es que toda, la histamina circulante en el organismo se sintetiza en el basófilo y se almacena dentro de los gránulos. La degranulación causa la liberación de histamina, que media muchos efectos de la hipersensibilidad inmediata y que, por su acción quimiotáctica potente de eosinófilos, lleva eosinófilos al sitio de degranulación. Otras sustancias que son liberadas al degranularse los basófilos incluyen factores quimiotácticos adicionales y varios metabolitos del ácido araquidónico, de los que el más importante en el leucotrieno C4.
Además, las membranas celulares de los basófilos contienen receptores de IgE de alta afinidad, y su número aumenta en personas alérgicas. La interacción entre los receptores de IgE y la inmunoglobulina causa degranulación anafiláctica. Cuando los receptores de IgE se reúnen después de interactuar con un estímulo apropiado, las membranas que limitan los gránulos citoplásmicos se fusionan con la membrana plasmática externa de la célula, causando liberación inmediata del contenido de los gránulos.129 La liberación de histamina causa síntomas clínicamente significativos; sin embargo, los esteroides pueden inhibir la liberación de histamina y de leucotrieno C4. Por ello, los basófilos regulan las reacciones alérgicas agudas a través de sus actividades secretoras de membrana.130
BASOFILIA
La basofilia (cuenta de basófilos > 150/mm3) se observa en trastornos mieloproliferativos como LMC, policitemia vera y metaplasia mieloide, después de esplenectomía, en algunas anemias hemolíticas y en la enfermedad de Hodgkin. La cuenta de basófilos también puede aumentar en pacientes con colitis ulcerativa y varicela. Aunque los basófilos y las células cebadas participan en las reacciones de hipersensibilidad inmediata y se observan basófilos en las áreas de dermatitis por contacto, no se ha observado basofilia en pacientes con estos trastornos.
Linfocitos
LINFOCITOSIS
La linfocitosis en adultos se define como una cuenta absoluta de linfocitos de más de 4,000/mm3.131 Sin embargo, debe tenerse en cuenta que las cuentas de linfocitos en los niños son mayores que en los adultos, y que pueden ser hasta de 20,000/mm3 en el primer año de vida. El frotis de cualquier paciente con linfocitosis debe examinarse con cuidado para determinar la morfología y diversidad de los linfocitos (v.gr., linfocitos reactivos, linfocitos granulares grandes, blastos o células manchadas).
La linfocitosis puede ser primaria o secundaria. La primaria, con frecuencia denominada enfermedad linfoproliferativa, es causada por alteración en la regulación de la producción de los linfocitos. Las linfocitosis primarias incluyen las leucemias (como la leucemia linfocítica crónica, la leucemia linfocítica aguda, la leucemia de células peludas y la leucemia de linfocitos granulares grandes), los linfomas y la linfocitosis monoclonal de células B [ver tabla 6].
|
|
VEB-virus Epstein Barr, CMV-citomegalovirus, VIH-virus de inmunodeficiencia humana, VHS-virus del herpes simple, VVZ-virus varicela zoster |
Las linfocitosis secundarias, o reactivas, son condiciones que incluyen incrementos absolutos en los linfocitos causados por respuestas fisiológicas o fisiopatológicas a infección, inflamación, toxinas, citocinas o agentes desconocidos. La causa más frecuente de linfocitosis reactiva es la infección viral, y con frecuencia son responsables el virus Epstein-Barr, el citomegalovirus, el virus herpes simple, el virus varicela-zoster, el de rubeóla, el VIH, el adenovirus o alguno de los virus de la hepatitis. Otros patógenos que producen linfocitosis son Toxoplasma gondii y, en niños, Bordetella pertussis (que causa aumento en la cuenta de linfocitos hasta 70,000/mm3).132 La linfocitosis se asocia también con estrés y liberación subsecuente de epinefrina, como la observada en pacientes con colapso cardiovascular, choque séptico, crisis falciforme , estado epiléptico o traumatismos,133 y con cirugía mayor, reacciones medicamentosas e hipersensibilidad. Puede existir linfocitosis persistente en pacientes con enfermedades autoinmunes, sarcoidosis, hiperesplenismo y cáncer, así como en los fumadores crónicos.
LINFOCITOPENIA
La linfocitopenia se define como una cuenta total de linfocitos menor de 1,000/mm3.134 Debido a que el 80 porciento de los linfocitos adultos son células T, la mayoría de las causas de linfocitopenia son ocasionadas por reducción en la cuenta de linfocitos T. Con frecuencia el mecanismo de la linfocitopenia es desconocido, y la causa puede ser hereditaria o adquirida.
Las linfocitopenias hereditarias suelen ser ocasionadas por inmunodeficiencias congénitas. Estas enfermedades incluyen inmunodeficiencia combinada severa (v.gr., deficiencia de deaminasa de adenosina, deficiencia de fosforilasa de nucleótidos de purina y disgenesia reticular), ataxia telangiectasia, síndrome de Wiskott-Aldrich e hipoplasia del cartílago-cabello [ver tabla 7]. Además, algunas personas tienen linfocitopenia T CD4+ idiopática.135
|
|
|
Puede observarse linfocitopenia adquirida en pacientes con infecciones virales (como infección por VIH, hepatitis, influenza e infección por virus sincicial respiratorio), en los que tienen infecciones bacterianas (como fiebre tifoidea, neumonía, sepsis y tuberculosos) y en los que tienen anemia aplástica, enfermedades autoinmunes, enfermedad de Hodgkin, sarcoidosis, insuficiencia renal, enteropatías perdedoras de proteínas y ascitis quilosa. También la deficiencia de zinc y la ingesta crónica de alcohol se asocian con linfocitopenia. Por último, los agentes inmunosupresores como la globulina antitimocito, los esteroides, los agentes quimioterápicos y la radiación, también producen linfocitopenia.
- Ogawa M: Differentiation and proliferation of hematopoietic stem cells, Blood 81:2844,1993
- Kaushansky K, O'Hara PJ, Berliner K, et al: Genomic cloning characterization and multilineage growth-promoting activity of human granulocyte-macrophage colony-stimulating factor, Proc Natl Acad Sci USA 83:3101,1986
- O'Hara A, Suca T, Saito M, et al: Effect of recombinant human granulocyte colony stimulating factor on hemopoietic cells in serum-free culture. Exp HematoI 15:695, 1987
- Warner HR, Athens JW: An analysis of granulocyte kinetics in blood and bone marrow. Ann NY Acad Sci 113:523,1964
- Boggs DR : The kinetics of neutrophil leukocytes in health and disease. semin HematoI 3:359, 1967
- Harlan JM: Leukocyte-endothelial interactions. Blood 65:513,1985
- Boxer L, Allen J, Baehner R: Diminished polymorphonuclear leukocyte adherence? function dependent upon release of cyclic AMP by endothelial cells after stimulation by epinephrine, J Clin lnvest 66.268, 1980
- Colotta R, Re F, Polentarutti N, et al: Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 80:2012,1992
- Broxmeyer HE, Gentile P, Cooper S, et al : Functional activities of acidic isoferritins and lactoferrin in vitro and in vivo. Blood Cells 10:397,1984
- Bainton DF, Ullyot JL, Farquhar MG: The development of neutrophilic polymorphonuclear leukocytes in human bone marrow: origin and content of azurophil and specific granules. J Exp Med 134:907,1971
- Pryzwansky KB, Rausch PG, spitznagel JK, et al: Immunocytochemical distinction between primary and secondary granule formation in developing human neutrophils: correlations with Romanowsky stains. Blood 53:179,1979
- Jesaitis AJ, Beuscher ES, Harrison D, et al: Ultrastructural localization of cytochrome b in the membranes of resting and phagocytosing human granulocytes.J Clin Invest 85:821,1990
- Borregaard N, Miller LJ, Springer TA: Chemoattractant-regulated mobilization of a novel intracellular compartment Ín human neutrophils. Science 237:1204,1987
- Coates TD, Watts RG, Hartman R, et al: Relationship of F-actin distribution to development of polar shape in human polymorphonuclear neutrophils. J Cell BioI 117:765, 1992
- Lorant DE, Topham MK, Whatley RE, et al: Inflammatory roles of P-selectin. J CIin Invest 92:559,1993
- Springer TA, Lasky LA: Cell adhesion: sticky sugars for selectins. Nature 349:196,1991
- Carlos TM, Harlan JM: Membrane proteins involved in phagocyte adherence to endothelium. Immunol Rev 114:5,1990
- Lo SK, Lee S, Ramos RA, et al: Endothelial-Ieukocyte adhesion molecule 1 stimulates the adhesive activity of leukocyte integrin CR3 (CD11b/CD18, Mac-l,[alpha]m [beta]2) on human neutrophils. J Exp Med 173:1493,1991
- Osborn L: Leukocyte adhesion to endothelium duringin inflammation. Cell 62:3, 1990
- MulIer WA, Weigl SA, Deng X, et al: PECAM-1 is required from transendothelial migration of leukocytes. J Exp Med 178:449,1993
- Stossel TP, Chaponnier C, Ezzell RM, et al: Nonmuscle actinbinding proteins. Annu Rev Cell Biol 1:353, 1985
- Lehrer RI, Ganz T, Selsted ME: Oxygen-independent bactericidal systems: mechanisms and disorders. Hematol Oncol Clin North Am 2:159,1988
- Francis JW, Balazovich KJ, Smolen JE, et al: Human neutrophil annexin I promotes granule aggregation and modulates Ca2+- dependent membrane infusion. J CIin Invest 90:537, 1992
- Sbarra AJ, Kamovsky ML: The biochemical basis of phagocytosis: 1. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem 234:1355,1959
- Whitin JC, Cohen HJ: Disorders of respiratory burst termination. Hematol Oncol Clin North Am 2:289,1988
- Babior BM: The respiratory burst oxidase. Adv Enzymol Relat Areas MoI BioI 65:49, 1992
- Parkos CA, Allen RA, Cochrane CG, et al: Purified cytochrome b from human granulocyte plasma embrane is cornposed of two polypeptides with relative molecular weights of 91,000 and 22,000. J Clin Invest 80:732,1987
- Parkos CA, Dinnauer MC, Walker LE, et al: Primary structure and unique expression of the 22-kilodalton light chain of human neutrophil cytochrome b. Proc Natl Acad Sci USA 85:3319,1988
- Quinn MT, MulIen ML, Jesaitis AJ, et al: subcellular distribution of the Rap 1 A protein in human neutrophils: colocalization and cotranslocation with cytochrome b559. Blood 79:1563,1992
- Segal AW, West I, Wientjes F, et al: Cytochrome b-245 is a flavocytOChrome containing FAD and the NADPH-bÍndÍng site of the microbicidal oxidase of phagocytes (pt 3). Biochem J 284:781, 1992
- Volpp BD, Nauseef WM, Donelson JE, et al: Cloning of the cDNA and functional expression of the 47-kilodalton cytosolic cornponent of the hurnan neutrophil respiratory burst oxidase. Proc Natl Acad Sci USA 86:7195,1989
- Leto TL, Lomax KJ, Volpp BD, et al: Cloning of a 67-kD neutrophil oxidase factor with similarity to a noncatalytic region of p60c-src Science 248:727, 1990
- Tyagi SR, Neckelrnann N, Uhlinger DJ, et al: Cell-free translocation of recombinant p47-phox, a component of the neutrophil NADPH oxidase: effects .of guanosine 5'-0-(3-thiotriphosphate), diacylglycerol, and an anionic amphiphile. Biochernistry 31:2765, 1992
- Rotrosen D, Leto TL: Phosphorylation of neutrophil 47-kDa cytosolic oxidase factor: translocation to membrane is associated with distinct phosphorylation events. J Biol Chem 265:19910,1990
- Roilides E, Walsh TJ, Pizzo PA, et al: Granulocyte colonystimulating factor enhances the phagocytic and bactericidal activity of normal and defective human neutrophils. J Infect Dis 163:579, 1991
- Yong K, Addison IE, Johnson B, et al: Role of leucocyte integrins in phagocytic responses to ranulocyte-macrophage colony- stimulating factor (GM-CsF): in vitro and in vivo studies on leucocyte adhesion deficient neutrophils. Br J Haematol 77:150, 1991
- Ferrante A: Activation of neutrophiIs by interleukins-1 and -2 and tumornecrosis factors. Immunol Ser 57:417,1992
- Dubravec DB, Spriggs DR, Mannick JA, et al: Circulating human peripheral blood granulocytes synthesize and secrete tumor necrosis factor alpha. Proc Natl Acad Sci USA 87:6758, 1990
- Lindemann A, Riedel D, Oster W, et al: Granulocyte-macrophage colony-stimulating factor induces cytokine secretion by human polymorphonuclear leukocytes. J Clin Invest 83:1308,1989
- Bazzoni F, Cassatella MA, Rossi F, et al: Phagocytosing neutrophiIs produce and release high amounts of the neutrophil-activating peptide l/interleukin 8. J Exp Med 173:771,1991
- Emery J, Gordon R, Rendle-short J, et al: Congenital amegakaryocytic thrombocytopenia with congenital deformities and a leukemoid blood picture in the newborn. Blood 12:567,1957
- Dignan P, Mauer A, Frantz C: Phocomelia with congenital hypoplastic thrombocytopenia and myeloid leukemoid reactions. J Pediatr 70:561,1967
- Doeglas HM, Bleumink E: FamiIial cold urticaria: clinical findings. Arch DermatoI110:382, 1974
- Fischer A, Lisowska-Grospierre B, Anderson D, et al: Leukocyte adhesion deficiency: molecular basis and functional consequences. Immunodefic Rev 1:39,1988
- Christensen RD, Rothstein G: Pitfalls in the interpretation of leukocyte counts of newbom infants. Am J Clin Pathol 72:608, 1979
- Mulroy J, Mickell J, Tong T, et al: Postictal pulmonary edema in chiIdren. Neurology 35:403,1985
- Altman A, Palmer C, Baehner R: Juvenile "chronic granulocytic" leukemia: a panmyelopathy with prominent monocytic and circulating monocyte colony forrning cells. Blood 43:341,1974
- Dale D, Fauci A, Guerry D, et al: Comparison of agents producing a neutrophilic leukocytosis in man. J Clin Invest 56:808, 1975
- Ascensao J, Oken M, Ewing S, et al: Leukocytosis and large cell lung cancer: a frequent association. Cancer 60:903,1987
- Eichenhorn M, Van-slyck E: Marked mature neutrophilic leukocytosis: a leukemoid variant associated with malignancy. Am J Med Sci 284:32,1982
- McBride J, Dacie J, shapley R: The effect of splenectomy on the leukocyte count. Br J HaematoI 14:225, 1968
- Caramihai E, Karayalcin G, Aballi A, et al: Leukocyte count differences in healthy white and black chiIdren 1 to 5 years of age. J Pediatr 86:252, 1975
- Shoenfeld Y, Weinberger A, Avishar R, et al: Familial leukopenia among Yemenite Jews. Isr J Med Sci 14:1271,1978
- Fronteira M, Myers A: Peripheral blood and bone marrow abnormalities in the acquired immunodeficiency syndrome. West J Med 147:157,1987
- Christensen RD, Rothstein G: Exhaustion of mature marrow neutrophils in neonates with sepsis. J Pediatr 96:316,1980
- Vincent PC: Drug-induced aplastic anemia and agranulocytosis. Drugs 31:52,1986
- Levitt L: Pure white-cell aplasia: antibody-mediated autoimmune inhibitionof granulopoiesis. N Engl J Med 308:1141,1983
- Boxer L: Immune neutropenias: clinical and biological irnplications. Am J Pediatr Hematol OncoI 3:89, 1981
- Heimpel H: Drug-induced agranulocytosis. Medical Toxicology and Adverse Drug Experience 3:449, 1988
- Cines D, Passero F, Guerry D, et al: Granulocyte.associated IgG in neutropenic disorders. Blood 59:124, 1982
- Stevens D, Everett E, Boxer L, et al: Infectious mononucleosis with severe neutropenia and opsonic antineutrophil activity. South Med J 72:519,197
- Logue G, Shimm D: Autoimmune granulocytopenia. Annu Rev Med 31:191,1980
- Spivak JL: Felty's syndrome: an analytical review. Johns Hopkins Medical JournaI 141:156, 1977
- Hanada T, Shin R, Husio M, et al : Intravenous gammaglobulin in the treatment of isoimmune neonatal neutropenia. Eur I Pediatr 148:218, 1988
- Levine D, Madyastha P: lsoimmune neonatal neutropenia. Am J PerinatoI3:231,1986
- Berliner N: T gamma lymphocytosis and T cell chronic leukemias. Hematol Oncol Clin North Am 4:473,1990
- Loughran TP Jr, Kadin M, starkebaum G, et al: Leukemia of large granular lymphocytes: association with clonal chromosomal abnormalities and autoimmune neutropenia, thrombocytopenia, and hemolytic anemia. Ann Intern Med 102:169,1985
- Ikeda H, Ozawa T, Takahashi H, et al. himmunological and hematological changes during high.dose immunoglobulin therapy in an infant with autoimmune neutropenia. Eur I Pediatr 146:412,1987
- Dale D, Guerry D, Wewerka I, et al: Chronic neutropenia. Medicine (Baltimore) 58:128,1979
- Loutit JF, Nisbet NW: The origin of osteoclasts. Immunobiology 161:193,1982
- Simon GT, Burke Js: Electron microscopy of the spleen: III. Erythroleukophagocytosis. Am J PathoI58:451, 1970
- Unanue ER, Al!en PM: The basis for the immunoregulatory role of macrophages and other accessory cel!s. Science 36:551, 1987
- Collins FM. Cellular mechanisms of anti.mycobacterial immunity. Adv Exp Med BioI162:157, 1983
- Trischmann TM: Natural and acquired resistance to Trypanosoma cruzi. Adv Exp Med BioI 162:365, 1983
- Domer JE, Carrow EW: Immunity to fungal infections. Adv Exp Med BioI 162:383, 1983
- Rager-Zisman B, Brosnan CF, Bloom BR: Macrophage oxidative metabolism: a defense mechanism against virus infection? Adv Exp Med BioI 162:489, 1983
- Ralph P, Warren MK, Ladner MB, et al: Molecular and biological properties of human macrophage growth factor, CSF-1 (pt 1). Cold Spring Harb Symp Quant BioI 51:679, 1986
- Meuret G, Hoffman G: Moriocyte kinetic studies in normal and disease states. Br J HaematoI 24:275, 1973
- Simon LM, Robin ED, Phillips JR, et al: Enzymatic basis for bioenergetic differences of alveolar versus peritoneal macrophages and enzyme regulation by molecular O2.J Clin Invest 59:443,1977
- Nathan CF: Secretory products of macrophages. J Clin Invest 79:319,1987
- Young LH, Liu CC, Joag S, et al: How lymphocytes kill. Annu Rev Med 41:45,1990
- Stuehr DJ, Nathan CF: Nitric oxide, a macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J Exp Med 169:1543,1989
- Peters PJ, Neefjes JJ, Oorschot V, et al: Segregation of MHC class II molecules from MHC class I molecules in the Golgi complex for tran5port to lysosomal compartments. Nature 349:669,1991
- Kosco MH: Antigen presentation to B cells. Curr Opin Immunol 3:336,1991
- Rappolee DA, Werb Z: Macrophage.derived growth factors. Macrophages and Macrophage Activation. Russell sW, Gordon S, Eds. springer-Verlag. Berlin, 1992, p 87
- Cheyne C: Histiocytosis X. J Bone Joint Surg Br 53:366,1971
- Komp DM, Herson J, starling KA, et al: A staging system for histiocytosls X: a Southwest Oncology Group study. Cancer 47:798,1981
- Bunch WG: Orthopedic and rehabllitatlon aspects of eosinophillc granuloma. Am J Pediatr Hematol OncoI 3:151, 1981
- Dunger DB, Broadben V, Yeoman E, et al: The frequency and natural h¡story of diabetes Insipidus in children with Langerhanscell histiocytosis. N Engl J Med 321:1157,1989
- Greenberger Js, Cassady JR, Jaffe N, et al: Radiation therapy in patients with hlstiocytosis: management of diabetes insipidus and bone ]esions. Int J Radiat Oncol Biol Phys 5:1749, 1979
- Starlil'g KA. Chemotherapy of histiocytosis. Am J Pediatr Hematol Oncol 3:157, 1981J
- Ceci A, DeTerlizzi M, Colella R, el al: Etoposide in recurrent childlhood Langerhans ceII histiocylosis: an Italian cooperative study. Cancer 62:2528,1988
- Rosai J, Dorfman RF: sinus histiocytosis with massive lymphadenopathy: a pseudolymphomatous benign disorder: ana]ysis of 34 cases. Cancer 30:1174,1972
- Foucar E, Rosai J, Dorfman RF: Sinus histiocytosls wlth massive Iymphadenopathy. Arch OtolaryngoI 104:687, 1978
- Foucar E, Rosai J, Dorfman R: Sinus hlstiocytosis wlth masslve lymphadenopathy (Rosal-Dorfman disease): review of the entity. Semln Diagn PathoI 7:19, 1990
- Olsen EA, Crawford JR, Vollmer RT: Sinus histiocytosls with masslve Iymphadenopathy: case report and revlew of a multisystemic disease with cutaneous infiltrates. J Am Acad DermatoI 18:1322,1988
- Suarez CR, Zeller WP, Silberman S, et al: slnus histiocytosis with massive Iymphadenopathy: remission with chemotherapy. Am J Pediatr Hematol OncoI 3:235, 1983
- Perry MC, Harrison EG, Burger EO, et al: Familial erythrophagocytic lymphohist¡ocytosis: report of two cases and cllnicopathologic review. Cancer 38:209,1976
- Ware R, Friedman Hs, Klnney TR: Familial erythrophagocytic Iymphohistiocytosls: late relapse despite continuous high-dose VP 16 chemotherapy. Med Pediatr Oncol 18:27,1990
- Risdall RJ, McKenn RW, Nesbit ME, et al: Virus-associated hemophagocytic syndrome: a benign hlstiocytic proliferation distinct from malignant histiocytosis. Cancer 44:993,1979
- Arya S, Hong R, Gilbert EF: Reactive hemophagocytic syndrome. Pediatr PatlhoI 3:129, 1985
- Suster S, Hilsenbeck S, Rywlin AM: Reactive histiocytic hyperplasia wilh hemophagocytosis in hemopoietic organs. Hum PalhoI 19:705, 1988
- Beutler E, Grabowski GA: Glucosylceramide lipidoses: Gaucher disease. The Metabolic and Molecular Bases of Inherited Disease, 7th ed. Scriver CR, Beaudet AL, sly Ws, et al, Eds. McGraw-Hlll, New York, 1994, p 2641
- Tsal P, Lipton JM, Sahdev I, et al: Allogenic bone marrow transplantation in severe Gaucher disease. Pediatr Res 31:503,1992
- Snyder EY, Deitcher DL, Walsh C, et al: Multipotent neural cell Iines can engraft and participate in development of mouse cerebellum. Cell68:33, 1992
- Pastores G, sibille A, Grabowskí GA: Enzyme therapy In Gaucher disease type 1: dosage efficacy and adverse effects in thirty-three patients treated for six to twenty.four months. Blood 82:408,1993
- Krivit W, shapiro EG: Bone marrow transplantation for storage diseases. Treatment of Genetic Diseases. Desnlck RJ, Ed. Churchill Livingstone, New York,1991, p 203
- Gleich GJ, Loegering DA: Immunobiology of eosinophils, Annu Rev ImmunoI 2:429, 1984
- Ackerman sJ, Loegerlng DA, Venge P, et al: Distinctive cationlc proteins of the human eosinophil granule: major basic protein, eosinophil cationic protein and eosinophil.derived neurotoxin, J ImmunoI 131:2977,1983
- Lewis DM, Lewls JC, Loegering DA, et al: Localization of the guinea pig eosinophil major basic proteln to the core of the granule. J Cell Biol 77:702, 1978
- Butterworth AE, Vadas MA, Wassom DL, et al: Interactions between human eosinophils and Schlstosoma mansoni II. The mechanlsm of irreversible eosinophll adherence. J Exp Med 150:14S6, 1979
- Clutterbuck EJ, Hirst EM, Sanderson CJ: Human interleukln.5 (lL. 5) reguIates the production of eosinophils in human bone marrow cultures: comparison and interaction with IL-1, IL-3, IL-6 and GMCSF, Blood 73:1504,1989
- Carlson M, Peterson C, Venge P: The influence of IL-3, IL-5, and GM-CSF on normal human eosinophil and neutrophil C3b-mduced degranulation, Allergy 48:437,1993
- Dent LA, Strath M, Mellor AL, et al: Eosinophilia in transgenic mice expressing interleukin 5, J Exp Med 172:1425,1990
- Coffman RL, Seymour BW, Hudak S, et al: Antibody to interleukin-5 inhibits helminth-induced eosinophilia in mice, Science 245'308,1989
- Vadas M, David JR, Butterworth A, et al: A new method for the purification of human eosinophils and neutrophils, and a comparison of the ability of these cells to damage schistosomula of Schistosoma mansoni, J ImmunoI122:1228, 1979
- Caulfield JP, Korman G, Butterworth AE, et al: The adherence of human neutrophils and eosinophils to schistosomula evidence for membrane fusion between cells and parasites, J Cell Biol 86:46, 1980
- Samoszuk M, Nansen L: Detection of interleukin-5 messenger RNA in Reed-Sternberg cells of Hodgkin's disease with eosinophilia, Blood 75:13,1990
- Hubscher T: Role of the eosinophil in the allergic reactions: II, Release of prostaglandins from human eosinophilic leukocytes, J ImmunoI 114:1389,1975
- Dri P, Cramer R, Spessotto P, et al: Eosinophil activation on biologic surfaces: production of O2- in response to physiologic soluble stimuli is differentially modulated by extracellular matrix components and endothelial cells, J ImmunoI 147:613, 1991
- Chusid M, Dale D, West B, et al : The hypereosinophilic syndrome, Medicine (Baltimore) 54:1,1975
- Sur S, Adolphson C, Gleich G: Eosinophils, Allergy: Principies and Practice, Middleton E, Reed CE, Ellis EF, et al, Eds, CV Mosby Co, St, Louis, 1993, p 169
- Warren KS: Diseases due to helminths: introduction, Principies and Practice of Infectious Diseases, 3rd ed, Mandell GL, Douglas RG Jr, Bennett JE, Eds, Churchill Livingstone, New York, 1990, p 2134
- Fauci A, Harley J, Roberts W, et al: The idiopathic hypereosinophilic syndrome, Ann Intern Med 7:78,1982
- Zielinski RM, Lawrence WD' Interferon-alpha for the hypereosinophilic syndrome, Ann Intern Med 113:716,1990
- Zabel P, Schlaak M: Cyclosporin for hypereosinophilic syndrome, Ann HematoI 62:230, 1991
- Smit AJ, van Essen LH, de Vries EGE: Successfullong-term control of idiopathic hypereosinophilic syndrome with etoposide, Cancer 67:2826, 1991
- Denburg JA: Basophil and mast cell lineage in vitro and in vivo. Blood 79:846,1992
- Marone G: Control mechanisms of mediator release in human basophils and mast cells.lmmunol lnvest 17:707,1988
- Stain C, Stockinger H, Scharf M, et al: Human blood basophils display a unique phenolype including activation-linked membrane structures. Blood 70:1872,1987
- Macintyre EA, Lynch DC: Lymphocytosis: is it leukemia and when to treat. Postgrad Med J 64:42,1988
- Kubic VL, Kubic fYr , Brunning RD: The morphologic and immunophenotypic assessment of the lymphocytosis accompanying Bordetella pertussis infection. Am J Clin PathoI95:809, 1991
- Teggatz JR, Parkin J, Peterson L: Transient alypical lymphocytosis in patients with emergency medical conditions. Arch Pathol Lab Med 111:712,1987
- Miale JB: Laboratory Medicine-Hematology, 6th ed. CV Mosby Co, St. Louis, 1982
- Smith DK, Neal JJ, Holmberg SD: Unexplained opportunistic infections and CD4+ T -lymphocytopenia without HIV infection. N Engl J Med 328:373, 1993