Neurología
⭳ Abrir artículo (PDF)528.7 KBEste artículo fue revisado respecto a la Edición 3/2000. Ver esa versión →
Contenido del artículo
XVII ENFERMEDADES DEL SISTEMA NERVIOSO CENTRAL POR VIRUS LENTOS
- Demencia por virus de inmunodeficiencia humana
- ASPECTOS CLINICOS DE LA DVIH
- CARACTERISTICAS PATOLOGICAS DE LA DVIH
- PATOGENIA DE LA ENFERMEDAD DEL SNC INDUCIDA POR VIH
- TRATAMIENTO
- Mielopatía por HTLV-I
- Encefalopatías espongiformes trasmisibles
- Leucoencefalopatía multifocal progresiva
- Pancencefalitis esclerosante subaguda
XVII ENFERMEDADES DEL SISTEMA NERVIOSO CENTRAL POR VIRUS
LENTOS
DR. FRANCISCO GONZALEZ-SCARANO
En el sistema nervioso central pueden desarrollarse diversos padecimientos crónicos y progresivos como resultado de la infección por virus de varias familias, así como por agentes que aún no están clasificados del todo. El término infección por virus lentos se ha usado para describir estas condiciones, que tienen periodos de incubación largos y curso clínico progresivo y que causan disfunción neurológica severa o muerte. En este capítulo se revisan las más comunes, que son la demencia causada por virus de inmunodeficiencia humana (DVIH), la mielopatía asociada con virus linfotrópico de células T humanas tipo 1 (HTLV-I, por sus siglas en inglés, n. del t.) la enfermedad de Creutzfeld-Jakob (ECJ), la leucoencefalopatía multifocal progresiva (LMP) y la encefalitis asociada con virus del sarampión.
Demencia por virus de inmunodeficiencia humana
Los síndromes neurológicos primarios asociados con infección por VIH constituyen la complicación más compleja y enigmática de esta enfermedad.1,2 El VIH puede afectar el sistema nervioso central (SNC) y el periférico, pero la afección al SNC se ha estudiado en forma más extensa porque es progresiva y causa una demencia incapacitante. Además de la demencia por VIH (también conocida como complejo de demencia del síndrome de inmunodeficiencia adquirida), el VIH se ha asociado con degeneración de la médula espinal (mielopatía vacuolar) que ocasiona sintomatología motora y sensorial severa característica de la afección de la médula espinal. La DVIH es más común que la mielopatía vacuolar y se diagnostica clínicamente en hasta el 30 porciento de los pacientes infectados por VIH. Los estudios de autopsia han detectado cambios neuropatológicos en un gran porcentaje de pacientes, pero es frecuente la discordancia entre el síndrome clínico y los datos patológicos (ver adelante).
ASPECTOS CLINICOS DE LA DVIH
Se desarrolla una enfermedad febril con linfadenopatía y erupción en alrededor del 60 porciento de las personas infectadas por VIH. Durante este periodo de gran viremia algunos pacientes pueden tener problemas neurológicos, incluyendo neuropatías craneales, meningoencefalitis y, en ocasiones, coma. Aunque el síndrome primario por VIH es autolimitado, la diseminación al SNC puede causar meningitis mononuclear asintomática crónica en algunos pacientes.
La DVIH suele ocurrir en las etapas tardías de la infección por VIH, cuando los niveles bajos de células T CD4+ y un sistema inmunológico comprometido han causado infecciones oportunistas que afectan otros órganos y sistemas. Al inicio la DVIH puede presentarse como un deterioro cognoscitivo leve que es detectable solo por la administración de una batería de pruebas neuropsicológicas por psicólogos que tienen experiencia con esta condición en particular.3 Este deterioro cognoscitivo leve puede no interferir con el desempeño laboral o las actividades de la vida diaria. De hecho, se ha investigado en muchos estudios si es posible detectar afección subclínica del SNC en un número significativo de pacientes infectados por VIH que han sido objeto de muchos estudios. Aunque este aspecto puede resurgir de tiempo en tiempo, el consenso de los estudios bien controlados más extensos es que el deterioro cognitivo no es una característica del periodo relativamente asintomático entre la infección primaria y la disminución en la cuenta de células T CD4+,3 a pesar de que exista replicación viral en el tejido periférico.4,5
La DVIH es una demencia subcortical y el enlentecimiento psicomotor, la apatía y síntomas motores como la ataxia y la parálisis pueden preceder a la pérdida de memoria y al deterioro de la función del lenguaje. En sus fases finales [ver tabla 1], las personas con DVIH pueden estar en estado casi vegetativo.
El diagnóstico de DVIH depende de la exclusión de otras causas de demencia en un paciente infectado por VIH. Las alteraciones del líquido cefalorraquídeo (i.e., proteínas elevadas, disminución en la cuenta celular y síntesis intratecal de un anticuerpo a VIH) son características de la infección por VIH pero no son específicas de la DVIH, ni lo es la presencia de VIH en el LCR, ya que pueden cultivarse virus libres y asociados a células en el LCR de personas que no manifiestan síntomas del SNC. Aunque los estudios de resonancia magnética han indicado que la DVIH se asocia con atrofia global del cerebro, existe una gran cantidad de atrofia en muchas personas con síndrome de inmunodeficiencia adquirida (SIDA), por lo que este dato es más útil para la investigación que para el diagnóstico.
CARACTERISTICAS PATOLOGICAS DE LA DVIH
Los principales datos neuropatológicos asociados con la DVIH son (1) encefalitis por células gigantes multinucleadas (ECGM), (2) leucoencefalopatía (palidez de la mielina y otros cambios de la sustancia blanca), y (3) astrocitosis y quizá pérdida neuronal. La ECGM, que es el dato neuropatológico más específico en la DVIH, existe en alrededor del 25 porciento de las personas que se han diagnosticado clínicamente y se caracteriza por sincicios de microglia que pueden contener ácidos nucleicos específicos de VIH y antígenos. La ECGM es característicamente más prominente en los ganglio basales y otras regiones subcorticales. Existe palidez de la mielina, que como el nombre lo implica se define por menor captación de histoquímicos por la sustancia blanca del SNC, en el 33 porciento de los pacientes con DVIH, pero este es un dato menos específico que la ECGM y puede ser causada por alteraciones de la barrera hematoencefálica. En forma alternativa, la palidez de la sustancia blanca puede representar una infección de bajo nivel de los oligodendro-citos por VIH o quizá otro patógeno como el herpes virus humano tipo 6 (HVH-6).
La característica más importante de la neuropatología de la DVIH es la discordancia entre los datos histológicos y la enfermedad clínica. Por ejemplo, en un estudio no existió ECGM o palidez difusa en el 50 porciento de los pacientes, e incluso en casos con datos morfológicos, las alteraciones fueron inespecíficas en muchos pacientes.6 Esto implica que los síntomas asociados con la DVIH, aunque claramente asociada con infección viral de la microglia y la formación del sincicio de microglia, suele ser al final el resultado de un proceso en el que una infección leve de un tipo celular se amplifica por mecanismos desconocidos.
PATOGENIA DE LA ENFERMEDAD DEL SNC INDUCIDA POR VIH
La patogenia enigmática de la DVIH [ver figura 1] puede representar un nuevo paradigma para el desarrollo de problemas neurológicos por una infección viral. Primero, existe discordancia entre los datos clínicos extensos y el sustrato neuropatológico. Segundo, las células infectadas en forma más intensa son las de la microglia (los macrófagos residentes del SNC) más que las neuronas o neuroglia. Tercero, existe poca evidencia morfológica de alteraciones en las células efectoras del sistema nervioso (i.e., las neuronas).
La mayoría de los investigadores consideran que las alteraciones asociadas con el VIHD son causadas por la secreción de neurotoxinas por microglia infectada en forma crónica.7 Entre las posibles toxinas se encuentran proteínas virales como la gp120 (la glucoproteína de la cubierta de fijación del virus) y la tat (la transactivadora de la transcripción viral) y citocinas proinflamatorias como el factor de necrosis tumoral-alfa (FNT-alfa) y el factor activador de plaquetas.8 Todas estas sustancias han demostrado mediar efectos dañinos en cultivos de neuronas de varias especies de mamíferos, pero no existe prueba concluyente de que ninguno de ellos esté presente en el SNC infectado por VIH en niveles suficientemente altos como para causar esta magnitud de daño.
En forma alternativa, la infección de la nuroglia y neuronas de bajo nivel puede mediar la DVIH. Sin embargo, este tipo de infección tendría que ser muy limitado porque las técnicas convencionales (por ejemplo inmunohistoquímica e hibridización in situ) han dado principalmente resultados negativos respecto a la infección de astrocitos, oligodendrocitos y neuronas.9 Estudios recientes, que han usado la amplificación en cadena de polimerasa para ácidos nucleicos en cultivos de tejido, han sugerido que células diferentes a las de microglia pueden portar el genoma proviral del VIH.10,11 A pesar de ello, hasta que se confirmen estos datos por muchos investigadores, será prudente tomar una actitud expectante respecto al papel de otras células a demás de las de microglia en la medicación de la DVIH.
TRATAMIENTO
Varios reportes anecdóticos y piloto indican que la DVIH puede responder a tratamiento antiretroviral, pero los estudios bien controlados y extensos aún están en proceso. En vista de las cargas virales presentes en el cerebro,12 será crucial desarrollar estrategias para eliminar el VIH de las células de microglia. De otra manera, la afección del SNC puede frustrar los éxitos recientes para disminuir la carga viral en el plasma y el tejido linfoide.
Mielopatía por HTLV-I
La entidad conocida como mielopatía asociada a HTLV-I (MAH), que fue descrita en un principio en pacientes japoneses, es la misma enfermedad que la paraparesia espástica tropical (PET), observada en poblaciones del Caribe. Los estudios epidemiológicos han establecido con firmeza la asociación entre la infección por HTLV-I y el desarrollo de este síndrome neurológico lentamente progresivo. Sin embargo, la MAH es una complicación relativamente rara del HTLV-I, con una incidencia comparable a la de la leucemia de células T del adulto, otra condición rara asociada con HTLV-I.
La MAH se caracteriza por el desarrollo de signos de disfunción de la médula espinal, incluyendo paraparesia e incontinencia urinaria. Los cambios sensoriales son menos comunes. En personas seropositivas para el HTLV-I, el diagnóstico se basa en la exclusión de lesiones ocupativas por medio de una IRM de la médula espinal, que puede observarse atrófica. Además, las imágenes en T2 (T2 se refiere al tiempo de relajación giro-giro o transversa) pueden demostrar áreas de mayor intensidad de señal dentro de la médula espinal y, en ocasiones, por arriba del foramen magno. Estos datos recuerdan a los hallazgos radiológicos asociados con la esclerosis múltiple, aunque estas entidades son claramente distintas. El LCR de los pacientes con HAM puede demostrar aumento en las proteínas y bandas oligoclonales, indicativas de síntesis intratecal de anticuerpos. Pueden existir también en el LCR células que recuerdan a las de la leucemia de células T del adulto. Los pacientes con MAH suelen tener un curso progresivo, que cause parálisis de los miembros inferiores y, posiblemente, afección en miembros superiores. Sin embargo, la MAH rara vez es fatal.
Los estudios de autopsia han revelado niveles bajos de HTLV-I dentro de la médula espinal de los pacientes con MAH, indicando que la citólisis directa por la infección viral puede no ser la causa de la mielopatía.13 En general se considera que la patogenia se asocia con infiltración de la médula espinal por células T CD8+.14,15 Los pacientes infectados por HTLV-I en los que se desarrolla esta complicación tienen una respuesta citolítica de células T intensa a la proteína viral tax.16 Estas células T citolíticas pueden mediar la disfunción neurológica fijando células de neuroglia mínimamente infectadas con HTLV-I. En forma alternativa, un antígeno neural que sea parecido a la proteína viral tax podría desencadenar un ataque autoinmune.
Debido a la falta de evidencia de que la infección viral directa media la MAH, la mayoría de los estudios terapéuticos se han dirigido hacia la reducción del proceso autoinmune en el SNC. Hasta la fecha no existe un tratamiento establecido para la MAH, pero algunos pacientes mostraron mejoría de la función neurológica con dosis altas de esteroides intravenosos.
Encefalopatías espongiformes trasmisibles
Las encefalopatías espongiformes, un grupo de trastornos neurológicos de humanos y otros mamíferos, se caracterizan por (1) deterioro subagudo y progresivo de la función neurológica, afectando varias regiones del neuroeje (la disfunción cognoscitiva y motora son las más comunes, aunque pueden participar otros sistemas), (2) cambios neuropatológicos espongiformes en las áreas afectadas del SNC, (3) trasmisión experimental de la misma especie u otra relacionada, con periodos de incubación largos, típicamente de muchos meses o años, y (4) no evidencia de agentes trasmisibles convencionales como los virus.
En los humanos las encefalopatías espongiformes incluyen tanto las formas esporádica como genética de la ECJ, el síndrome de Gerstmann-Sträusler (SGS), la enfermedad de kuru de la tribu Fore de Nueva Guinea y el insomnio familiar fatal (IFF). Son enfermedades paralelas en otras especies el escrapie de las ovejas y la encefalopatía trasmisible de las chinchillas. Las enfermedades espongiformes se han trasmitido en forma experimental a ratones y cobayos de tejidos ovinos, a monos y chimpancés de tejidos humanos y en forma accidental a bovinos (encefalopatía espongiforme bovina [EEB] o enfermedad de las vacas locas) por el suplemento dietético con órganos de ovejas infectadas. Aunque existen barreras entre las especies, las enfermedades espongiformes suelen considerarse como causadas por procesos semejantes.
La ECJ y el escrapie han sido descritas desde hace muchos años cuando la trasmisión de la forma humana de la enfermedad se realizó por primera vez por inoculación intracerebral de primates con tejido de SNC de pacientes fallecidos por kuru, una encefalopatía espongiforme asociada con el endocanibalismo ritual en una tribu residente en las regiones montañosas remotas de Nueva Guinea.17 El kuru desapareció al cesar las prácticas de canibalismo, y la trasmisión de humano a humano de la ECJ y de otras encefalopatías espongiformes se limita en la actualidad a los raros casos de trasplantes accidentales de un órgano de una persona con la enfermedad o de exposición parenteral a los tejidos de ECJ por instrumentos contaminados.18
El diagnóstico de ECJ se basa en los datos clínicos. La característica principal es la demencia progresiva (que ocurre en semanas a meses, más que años, la que es más típica de la demencia de la enfermedad de Alzheimer). Este rápido deterioro cognitivo debe alertar al clínico ante el posible diagnóstico de ECJ. Además, los pacientes pueden referir insomnio, fatiga y problemas con la visión. En algunos casos las personas con ECJ pueden tener ceguera cortical. El examen neurológico muestra evidencia de afección de la vía piramidal y atrofia muscular ocasional. El mioclonus (una reacción brusca especialmente marcada después de un sobresalto) es un dato físico importante, pero su ausencia no descarta ECJ. Otros datos incluyen ataxia cerebelosa y crisis convulsivas.
No pueden obtenerse datos diagnósticos específicos de la IRM o del estudio del LCR, pero el electroencefalograma puede demostrar formas trifásicas características. La ECJ siempre es fatal, generalmente en un periodo de dos años después del inicio.
HIPOTESIS DE LOS PRIONES
Estudios extensos han descartado la presencia de virus convencionales en pacientes con ECJ, lo que ha originado la hipótesis de que el padecimiento es ocasionado por un agente no convencional. La hipótesis de los priones, iniciada por Prusiner y colaboradores y ahora aceptada mundialmente (aunque aún no en forma universal), se basa en la presencia indudable de una forma patogénica resistente a proteasas de una proteína endógena (proteína prión o PrP) en el cerebro de todas las especies afectadas.19 La forma natural de la proteína, PrPc es una proteína glucosilada de función desconocida que contiene resortes largos (alfa-hélices). En los cerebros de los mamíferos afectados la PrPc parece convertirse en una estructura con una gran concentración de hojas con plegamiento ß, una conformación proteica que promueve la formación de cristales [ver figura 2]. La proteína con alteración en la conformación (PrPsc o PrP escrapie) se deposita en fibrillas de amiloide o placas en el cerebro enfermo [ver figura 3]. Estos depósitos proteináceos son característicos de la neuropatología de todas las encefalopatías espongiformes, aunque la magnitud del depósito varía entre los diversos padecimientos.
Las evidencias que apoyan la participación de la PrP en las enfermedades
espongiformes incluyen experimentos en los que ratones eliminaron el gen PrP
por ingeniería genética. Estos ratones se volvieron resistentes a
la infección cerebral por escrapie y no desarrollaron ningún
cambio espongiforme, incluso cuando se inyectaron dentro del cerebro grandes
concentraciones de tejido infectado por escrapie. Por el contrario, ratones
transgénicos modificados para expresar el gen PrP de otras especies,
incluyendo humanos, se volvieron especialmente susceptiles al tejido cerebral
enfermo obtenido de especies que aportaron el transgén.20 En
vista de que existen barreras entre especies para la trasmisión del
cambio espongiforme, estos últimos experimentos sugieren que el
transgén es responsable del proceso patológico.
¿Cómo es que una proteína sin función de ácido nucleico actúa como un agente infeccioso y codifica su propia replicación en el cerebro enfermo? Un esquema propone que una PrPsc con estructura alterada sirve como una semilla, que se combina con la PrPc del tejido normal [ver figura 2]. Esta interacción induce a la PrPc a cambiar por lo menos una porción de su estructura alfa-helicoidal en una estructura compuesta principalmente de hojas con plegamiento ß. Debido a que la PrPsc rica en plegamiento ß es menos soluble, la reacción se realiza en una sola dirección. El agregado inicial se expande por medio del reclutamiento de moléculas PrPc adicionales, produciendo un depósito insolubre que puede ser dañino para las neuronas y la neuroglia.
En este mecanismo se desconoce el origen de la PrPc que actúa como semilla. En una condición como la EEB, en la que la enfermedad parece trasmitirse por la alimentación de las vacas con tejidos infectados de ovejas, la proteína que actúa como semilla podría ser ingerida y trasmitirse de algún modo al cerebro desde el tejido periférico. Se sabe que el bazo y otros órganos linfoides pueden trasmitir el escrapie, por lo que las células linfoides circulantes en el SNC son los primeros candidatos (aunque esto no se ha demostrado) para esta neuroinvasión.
ENFERMEDADES GENETICAS ASOCIADAS CON MUTACIONES PrP
En los humanos las enfermedades espongiformes incluyen, además de la ECJ espontánea o trasmitida en forma accidental, formas genéticas de ECJ e incluso condiciones más raras como el SGS y el IFF.21,22 En el SGS, que se trasmite como un rasgo autosómico dominante, las personas afectadas desarrollan ataxia, seguida de demencia, los pacientes con IFF son incapaces de dormir. Todas estas condiciones pueden trasmitirse por incoulación de tejidos enfermos a primates no humanos susceptibles. Es notable que todas estas condiciones se han asociado con mutaciones en el gen PrP y en una serie de experimentos, los ratones transfectados con la mutación del SGS desarrollaron cambios espongiformes.21 Se considera que las mutaciones en el gen PrP promueven la conversión a un estado con estructura alterada, insoluble.
La existencia de enfermedades que simultáneamente son genéticas e infecciosas y la asociación de mutaciones en el gen PrP con estas enfermedades, proporcionan intensa evidencia para apoyar la hipótesis de los priones, aunque al inicio existió gran escepticismo al respecto porque viola el dogma central de la biología molecular.23 Un experimento que no se ha realizado aún es la síntesis in vitro de una proteína pura capaz de servir como semilla para el desarrollo de enfermedad espongiforme; muchos grupos participan en esta tarea.
Leucoencefalopatía multifocal progresiva
La LMP es una enfermedad de la sustancia blanca cerebral que se caracteriza por deterioro motor progresivo, pérdida de la visión, incontinencia y demencia eventual, causada por un virus ADn relacionado a los papovavirus conocido como JC. La LMP se describió por primera vez en pacientes con inmunodeficiencia causada por neoplasias hematológicas o por tratamiento para la prevención del rechazo de trasplantes. Sin embargo, fue un padecimiento poco frecuente hasta la aparición del SIDA. Estudios epidemiológicos indican que entre uno y cinco porciento de las personas infectadas por VIH desarrollarán eventualmente LMP. En muchos de estos casos el desarrollo de LMP marca el inicio de la inmunodeficiencia y es la enfermedad que define el SIDA. En otros casos constituye una complicación asociada con otras infecciones oportunistas.
El diagnóstico de LMP debe considerarse en cualquier persona con enfermedad progresiva subaguda que incluya alteraciones motoras y cognitivas y que tenga una inmunodeficiencia comprobada o probable (v.gr., neoplasia). La mayoría de los casos tienen datos de afección de la sustancia blanca, incluyendo ataxia, defectos visuales y debilidad. En casos raros ocurre LMP en personas por lo demás sanas. Debe sospecharse fuertemente LMP si la IRM muestra áreas confluentes de señales de mayor intensidad en las imágenes T2 [ver figura 4]. Estas áreas pueden ser hipointensas en las imágenes en T1 con respecto a los límites de sustancia gris-blanca. (T1 se refiere a tiempo de relajación giro-reja, o longitudinal). El LCR suele ser normal en la LMP, y cuando existe pleocitosis en el LCR, ésta es leve.
El diagnóstico definitivo de la LMP ha requerido de biopsia cerebral, pero estudios recientes han demostrado que puede detectarse el ADN del virus JC por amplificación con la reacción en cadena de la polimerasa (RCP) en el LCR de pacientes con LMP.24,25 La frecuencia de reacciones falsas positivas es baja, y se ha calculado que la RCP en LCR tiene una sensibilidad y especificidad mayor del 90 porciento.26 Se han atribuido casos ocasionales de mejoría espontánea de la LMP a tratamientos experimentales. Sin embargo, no se sabe si la reversión parcial de la inmunodeficiencia ha sido la responsable de la mejoría.
PATOLOGIA Y PATOGENIA
El examen patológico del cerebro con LMP muestra cambios dramáticos en la sustancia blanca subcortical. Los oligodendrocitos, que son las células responsables de la manufactura y mantenimiento de la mielina, son el principal blanco del virus JC. Por lo tanto, la citólisis oligodendrogial es el principal mecanismo patogénico en la LMP y causa desmielinización severa característica. El evento inicial en el desarrollo de la LMP parece ser la aparición de un foco microscópico de infección oligodendrogial, a partir del cual el virus se disemina en forma centrífuga para afectar grandes segmentos de la sustancia blanca. Los astrocitos, lo mismo que algunos oligodendrocitos, demuestran cambios morfológicos bizarros semejantes a los asociados con una neoplasia del SNC. Existe poca afección de la sustancia gris o las neuronas en la LMP, y la que ocurre puede reflejar citólisis de los oligodendrocitos adyacentes, quizá por el reclutamiento de microglia y macrófagos.27
VIRUS JC
La etiología viral de la LMP se sospechó por primera vez cuando Zu Rhein y colaboradores describieron micrografías electrónicas con partículas parecidas a poliomavirus en el núcleo de oligodendrocitos.28 El aislamiento viral se logró solo usando cultivos de cerebro fetal en experimentos de cocultivos, que demostraron que el virus JC está relacionado, aunque es claramente distinto, de otros poliomavirus. Un gran porcentaje de población adulta en los Estados Unidos (70 a 90 porciento) tiene anticuerpos contra virus JC, lo que indica que esta es una infección común, aunque no se han atribuido enfermedades a la infección primaria.
La evidencia de infección diseminada y el aislamiento del virus JC de la orina de algunos pacientes inmunosuprimidos son compatibles con la hipótesis de que la LMP representa la reactivación de un virus ADN latente. Sin embargo, no se ha identificado el órgano reservorio. La ausencia de ADN de virus JC en los cerebros de personas normales sugiere que el virus JC permanece latente en otros tejidos. Los linfocitos, los órganos linfoides y los riñones se consideran los candidatos más factibles porque el virus se ha detectado en estos tejidos por medio de RCP o se ha aislado en los mismos.29,30 Sin embargo, el virus JC aislado del tejido periférico de las personas no inmunosuprimidas (el virus arquetipo) difiere del aislado de pacientes con LMP en las regiones responsables de la regulación de la expresión viral.31 Una hipótesis intrigante, que puede explicar estos datos, es que el virus arquetipo es modificado por deleción o duplicación y así es convertido en una forma neurotrópica que es trasnportado hacia el cerebro en las células B infectadas.
No existe un tratamiento bien establecido para la LMP, pero enfoques experimentales han incluido análogos de nucleósidos, interferones, e inhibidores de la topoisomerasa. Algunos de estos se exploran en la actualidad en pacientes con SIDA.
Pancencefalitis esclerosante subaguda
La panencefalitis esclerosante subaguda (PEES) es una secuela rara de la infección por sarampión (uno de cada 100,000 casos). La panencefalitis progresiva por rubeola es una enfermedad semejante a la PEES causada por este virus.
Los pacientes con PEES han sufrido una infección no complicada por sarampión, con frecuencia a menor edad que el promedio, y desarrollan encefalitis progresiva varios años después. La PEES parece ser causada por una mutación no común en la proteína matriz (M) del virus de sarampión nativo. Esta mutación puede afectar el empaquetamiento del virus, lo que puede explicar las dificultades que tienen los investigadores para aislar el virus libre. Se ha supuesto que el virus que causa la PEES puede diseminarse solo por fusión entre una célula infectada y células adyacentes no infectadas. Esto puede explicar la persistencia viral en presencia de una respuesta humoral intensa.
La PEES puede también ser el resultado de la vacunación con una cepa viva y atenuada de sarampión. Sin embargo, la tasa de desarrollo es por lo menos 10 veces menor que la asociada con la infección por el virus nativo. En los Estados Unidos, en donde la prevalencia de infecciones por sarampión es menor de 30,000, los casos de PEES son raros, incluso en años de epidemia.
No existe un tratamiento establecido para la PEES y su evolución casi siempre es fatal.
DR. FRANCISCO GONZALEZ-SCARANO
En el sistema nervioso central pueden desarrollarse diversos padecimientos crónicos y progresivos como resultado de la infección por virus de varias familias, así como por agentes que aún no están clasificados del todo. El término infección por virus lentos se ha usado para describir estas condiciones, que tienen periodos de incubación largos y curso clínico progresivo y que causan disfunción neurológica severa o muerte. En este capítulo se revisan las más comunes, que son la demencia causada por virus de inmunodeficiencia humana (DVIH), la mielopatía asociada con virus linfotrópico de células T humanas tipo 1 (HTLV-I, por sus siglas en inglés, n. del t.) la enfermedad de Creutzfeld-Jakob (ECJ), la leucoencefalopatía multifocal progresiva (LMP) y la encefalitis asociada con virus del sarampión.
Demencia por virus de inmunodeficiencia humana
Los síndromes neurológicos primarios asociados con infección por VIH constituyen la complicación más compleja y enigmática de esta enfermedad.1,2 El VIH puede afectar el sistema nervioso central (SNC) y el periférico, pero la afección al SNC se ha estudiado en forma más extensa porque es progresiva y causa una demencia incapacitante. Además de la demencia por VIH (también conocida como complejo de demencia del síndrome de inmunodeficiencia adquirida), el VIH se ha asociado con degeneración de la médula espinal (mielopatía vacuolar) que ocasiona sintomatología motora y sensorial severa característica de la afección de la médula espinal. La DVIH es más común que la mielopatía vacuolar y se diagnostica clínicamente en hasta el 30 porciento de los pacientes infectados por VIH. Los estudios de autopsia han detectado cambios neuropatológicos en un gran porcentaje de pacientes, pero es frecuente la discordancia entre el síndrome clínico y los datos patológicos (ver adelante).
ASPECTOS CLINICOS DE LA DVIH
Se desarrolla una enfermedad febril con linfadenopatía y erupción en alrededor del 60 porciento de las personas infectadas por VIH. Durante este periodo de gran viremia algunos pacientes pueden tener problemas neurológicos, incluyendo neuropatías craneales, meningoencefalitis y, en ocasiones, coma. Aunque el síndrome primario por VIH es autolimitado, la diseminación al SNC puede causar meningitis mononuclear asintomática crónica en algunos pacientes.
La DVIH suele ocurrir en las etapas tardías de la infección por VIH, cuando los niveles bajos de células T CD4+ y un sistema inmunológico comprometido han causado infecciones oportunistas que afectan otros órganos y sistemas. Al inicio la DVIH puede presentarse como un deterioro cognoscitivo leve que es detectable solo por la administración de una batería de pruebas neuropsicológicas por psicólogos que tienen experiencia con esta condición en particular.3 Este deterioro cognoscitivo leve puede no interferir con el desempeño laboral o las actividades de la vida diaria. De hecho, se ha investigado en muchos estudios si es posible detectar afección subclínica del SNC en un número significativo de pacientes infectados por VIH que han sido objeto de muchos estudios. Aunque este aspecto puede resurgir de tiempo en tiempo, el consenso de los estudios bien controlados más extensos es que el deterioro cognitivo no es una característica del periodo relativamente asintomático entre la infección primaria y la disminución en la cuenta de células T CD4+,3 a pesar de que exista replicación viral en el tejido periférico.4,5
La DVIH es una demencia subcortical y el enlentecimiento psicomotor, la apatía y síntomas motores como la ataxia y la parálisis pueden preceder a la pérdida de memoria y al deterioro de la función del lenguaje. En sus fases finales [ver tabla 1], las personas con DVIH pueden estar en estado casi vegetativo.
|
||||||||||||||
|
El diagnóstico de DVIH depende de la exclusión de otras causas de demencia en un paciente infectado por VIH. Las alteraciones del líquido cefalorraquídeo (i.e., proteínas elevadas, disminución en la cuenta celular y síntesis intratecal de un anticuerpo a VIH) son características de la infección por VIH pero no son específicas de la DVIH, ni lo es la presencia de VIH en el LCR, ya que pueden cultivarse virus libres y asociados a células en el LCR de personas que no manifiestan síntomas del SNC. Aunque los estudios de resonancia magnética han indicado que la DVIH se asocia con atrofia global del cerebro, existe una gran cantidad de atrofia en muchas personas con síndrome de inmunodeficiencia adquirida (SIDA), por lo que este dato es más útil para la investigación que para el diagnóstico.
CARACTERISTICAS PATOLOGICAS DE LA DVIH
Los principales datos neuropatológicos asociados con la DVIH son (1) encefalitis por células gigantes multinucleadas (ECGM), (2) leucoencefalopatía (palidez de la mielina y otros cambios de la sustancia blanca), y (3) astrocitosis y quizá pérdida neuronal. La ECGM, que es el dato neuropatológico más específico en la DVIH, existe en alrededor del 25 porciento de las personas que se han diagnosticado clínicamente y se caracteriza por sincicios de microglia que pueden contener ácidos nucleicos específicos de VIH y antígenos. La ECGM es característicamente más prominente en los ganglio basales y otras regiones subcorticales. Existe palidez de la mielina, que como el nombre lo implica se define por menor captación de histoquímicos por la sustancia blanca del SNC, en el 33 porciento de los pacientes con DVIH, pero este es un dato menos específico que la ECGM y puede ser causada por alteraciones de la barrera hematoencefálica. En forma alternativa, la palidez de la sustancia blanca puede representar una infección de bajo nivel de los oligodendro-citos por VIH o quizá otro patógeno como el herpes virus humano tipo 6 (HVH-6).
La característica más importante de la neuropatología de la DVIH es la discordancia entre los datos histológicos y la enfermedad clínica. Por ejemplo, en un estudio no existió ECGM o palidez difusa en el 50 porciento de los pacientes, e incluso en casos con datos morfológicos, las alteraciones fueron inespecíficas en muchos pacientes.6 Esto implica que los síntomas asociados con la DVIH, aunque claramente asociada con infección viral de la microglia y la formación del sincicio de microglia, suele ser al final el resultado de un proceso en el que una infección leve de un tipo celular se amplifica por mecanismos desconocidos.
PATOGENIA DE LA ENFERMEDAD DEL SNC INDUCIDA POR VIH
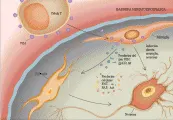
La patogenia enigmática de la DVIH [ver figura 1] puede representar un nuevo paradigma para el desarrollo de problemas neurológicos por una infección viral. Primero, existe discordancia entre los datos clínicos extensos y el sustrato neuropatológico. Segundo, las células infectadas en forma más intensa son las de la microglia (los macrófagos residentes del SNC) más que las neuronas o neuroglia. Tercero, existe poca evidencia morfológica de alteraciones en las células efectoras del sistema nervioso (i.e., las neuronas).
|
| Figura 1 |
| Patogenia de la demencia por VIH |
La mayoría de los investigadores consideran que las alteraciones asociadas con el VIHD son causadas por la secreción de neurotoxinas por microglia infectada en forma crónica.7 Entre las posibles toxinas se encuentran proteínas virales como la gp120 (la glucoproteína de la cubierta de fijación del virus) y la tat (la transactivadora de la transcripción viral) y citocinas proinflamatorias como el factor de necrosis tumoral-alfa (FNT-alfa) y el factor activador de plaquetas.8 Todas estas sustancias han demostrado mediar efectos dañinos en cultivos de neuronas de varias especies de mamíferos, pero no existe prueba concluyente de que ninguno de ellos esté presente en el SNC infectado por VIH en niveles suficientemente altos como para causar esta magnitud de daño.
En forma alternativa, la infección de la nuroglia y neuronas de bajo nivel puede mediar la DVIH. Sin embargo, este tipo de infección tendría que ser muy limitado porque las técnicas convencionales (por ejemplo inmunohistoquímica e hibridización in situ) han dado principalmente resultados negativos respecto a la infección de astrocitos, oligodendrocitos y neuronas.9 Estudios recientes, que han usado la amplificación en cadena de polimerasa para ácidos nucleicos en cultivos de tejido, han sugerido que células diferentes a las de microglia pueden portar el genoma proviral del VIH.10,11 A pesar de ello, hasta que se confirmen estos datos por muchos investigadores, será prudente tomar una actitud expectante respecto al papel de otras células a demás de las de microglia en la medicación de la DVIH.
TRATAMIENTO
Varios reportes anecdóticos y piloto indican que la DVIH puede responder a tratamiento antiretroviral, pero los estudios bien controlados y extensos aún están en proceso. En vista de las cargas virales presentes en el cerebro,12 será crucial desarrollar estrategias para eliminar el VIH de las células de microglia. De otra manera, la afección del SNC puede frustrar los éxitos recientes para disminuir la carga viral en el plasma y el tejido linfoide.
Mielopatía por HTLV-I
La entidad conocida como mielopatía asociada a HTLV-I (MAH), que fue descrita en un principio en pacientes japoneses, es la misma enfermedad que la paraparesia espástica tropical (PET), observada en poblaciones del Caribe. Los estudios epidemiológicos han establecido con firmeza la asociación entre la infección por HTLV-I y el desarrollo de este síndrome neurológico lentamente progresivo. Sin embargo, la MAH es una complicación relativamente rara del HTLV-I, con una incidencia comparable a la de la leucemia de células T del adulto, otra condición rara asociada con HTLV-I.
La MAH se caracteriza por el desarrollo de signos de disfunción de la médula espinal, incluyendo paraparesia e incontinencia urinaria. Los cambios sensoriales son menos comunes. En personas seropositivas para el HTLV-I, el diagnóstico se basa en la exclusión de lesiones ocupativas por medio de una IRM de la médula espinal, que puede observarse atrófica. Además, las imágenes en T2 (T2 se refiere al tiempo de relajación giro-giro o transversa) pueden demostrar áreas de mayor intensidad de señal dentro de la médula espinal y, en ocasiones, por arriba del foramen magno. Estos datos recuerdan a los hallazgos radiológicos asociados con la esclerosis múltiple, aunque estas entidades son claramente distintas. El LCR de los pacientes con HAM puede demostrar aumento en las proteínas y bandas oligoclonales, indicativas de síntesis intratecal de anticuerpos. Pueden existir también en el LCR células que recuerdan a las de la leucemia de células T del adulto. Los pacientes con MAH suelen tener un curso progresivo, que cause parálisis de los miembros inferiores y, posiblemente, afección en miembros superiores. Sin embargo, la MAH rara vez es fatal.
Los estudios de autopsia han revelado niveles bajos de HTLV-I dentro de la médula espinal de los pacientes con MAH, indicando que la citólisis directa por la infección viral puede no ser la causa de la mielopatía.13 En general se considera que la patogenia se asocia con infiltración de la médula espinal por células T CD8+.14,15 Los pacientes infectados por HTLV-I en los que se desarrolla esta complicación tienen una respuesta citolítica de células T intensa a la proteína viral tax.16 Estas células T citolíticas pueden mediar la disfunción neurológica fijando células de neuroglia mínimamente infectadas con HTLV-I. En forma alternativa, un antígeno neural que sea parecido a la proteína viral tax podría desencadenar un ataque autoinmune.
Debido a la falta de evidencia de que la infección viral directa media la MAH, la mayoría de los estudios terapéuticos se han dirigido hacia la reducción del proceso autoinmune en el SNC. Hasta la fecha no existe un tratamiento establecido para la MAH, pero algunos pacientes mostraron mejoría de la función neurológica con dosis altas de esteroides intravenosos.
Encefalopatías espongiformes trasmisibles
Las encefalopatías espongiformes, un grupo de trastornos neurológicos de humanos y otros mamíferos, se caracterizan por (1) deterioro subagudo y progresivo de la función neurológica, afectando varias regiones del neuroeje (la disfunción cognoscitiva y motora son las más comunes, aunque pueden participar otros sistemas), (2) cambios neuropatológicos espongiformes en las áreas afectadas del SNC, (3) trasmisión experimental de la misma especie u otra relacionada, con periodos de incubación largos, típicamente de muchos meses o años, y (4) no evidencia de agentes trasmisibles convencionales como los virus.
En los humanos las encefalopatías espongiformes incluyen tanto las formas esporádica como genética de la ECJ, el síndrome de Gerstmann-Sträusler (SGS), la enfermedad de kuru de la tribu Fore de Nueva Guinea y el insomnio familiar fatal (IFF). Son enfermedades paralelas en otras especies el escrapie de las ovejas y la encefalopatía trasmisible de las chinchillas. Las enfermedades espongiformes se han trasmitido en forma experimental a ratones y cobayos de tejidos ovinos, a monos y chimpancés de tejidos humanos y en forma accidental a bovinos (encefalopatía espongiforme bovina [EEB] o enfermedad de las vacas locas) por el suplemento dietético con órganos de ovejas infectadas. Aunque existen barreras entre las especies, las enfermedades espongiformes suelen considerarse como causadas por procesos semejantes.
La ECJ y el escrapie han sido descritas desde hace muchos años cuando la trasmisión de la forma humana de la enfermedad se realizó por primera vez por inoculación intracerebral de primates con tejido de SNC de pacientes fallecidos por kuru, una encefalopatía espongiforme asociada con el endocanibalismo ritual en una tribu residente en las regiones montañosas remotas de Nueva Guinea.17 El kuru desapareció al cesar las prácticas de canibalismo, y la trasmisión de humano a humano de la ECJ y de otras encefalopatías espongiformes se limita en la actualidad a los raros casos de trasplantes accidentales de un órgano de una persona con la enfermedad o de exposición parenteral a los tejidos de ECJ por instrumentos contaminados.18
El diagnóstico de ECJ se basa en los datos clínicos. La característica principal es la demencia progresiva (que ocurre en semanas a meses, más que años, la que es más típica de la demencia de la enfermedad de Alzheimer). Este rápido deterioro cognitivo debe alertar al clínico ante el posible diagnóstico de ECJ. Además, los pacientes pueden referir insomnio, fatiga y problemas con la visión. En algunos casos las personas con ECJ pueden tener ceguera cortical. El examen neurológico muestra evidencia de afección de la vía piramidal y atrofia muscular ocasional. El mioclonus (una reacción brusca especialmente marcada después de un sobresalto) es un dato físico importante, pero su ausencia no descarta ECJ. Otros datos incluyen ataxia cerebelosa y crisis convulsivas.
No pueden obtenerse datos diagnósticos específicos de la IRM o del estudio del LCR, pero el electroencefalograma puede demostrar formas trifásicas características. La ECJ siempre es fatal, generalmente en un periodo de dos años después del inicio.
HIPOTESIS DE LOS PRIONES
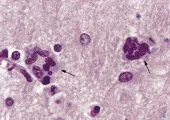
Estudios extensos han descartado la presencia de virus convencionales en pacientes con ECJ, lo que ha originado la hipótesis de que el padecimiento es ocasionado por un agente no convencional. La hipótesis de los priones, iniciada por Prusiner y colaboradores y ahora aceptada mundialmente (aunque aún no en forma universal), se basa en la presencia indudable de una forma patogénica resistente a proteasas de una proteína endógena (proteína prión o PrP) en el cerebro de todas las especies afectadas.19 La forma natural de la proteína, PrPc es una proteína glucosilada de función desconocida que contiene resortes largos (alfa-hélices). En los cerebros de los mamíferos afectados la PrPc parece convertirse en una estructura con una gran concentración de hojas con plegamiento ß, una conformación proteica que promueve la formación de cristales [ver figura 2]. La proteína con alteración en la conformación (PrPsc o PrP escrapie) se deposita en fibrillas de amiloide o placas en el cerebro enfermo [ver figura 3]. Estos depósitos proteináceos son característicos de la neuropatología de todas las encefalopatías espongiformes, aunque la magnitud del depósito varía entre los diversos padecimientos.
|
|
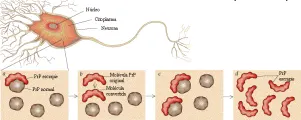
¿Cómo es que una proteína sin función de ácido nucleico actúa como un agente infeccioso y codifica su propia replicación en el cerebro enfermo? Un esquema propone que una PrPsc con estructura alterada sirve como una semilla, que se combina con la PrPc del tejido normal [ver figura 2]. Esta interacción induce a la PrPc a cambiar por lo menos una porción de su estructura alfa-helicoidal en una estructura compuesta principalmente de hojas con plegamiento ß. Debido a que la PrPsc rica en plegamiento ß es menos soluble, la reacción se realiza en una sola dirección. El agregado inicial se expande por medio del reclutamiento de moléculas PrPc adicionales, produciendo un depósito insolubre que puede ser dañino para las neuronas y la neuroglia.
En este mecanismo se desconoce el origen de la PrPc que actúa como semilla. En una condición como la EEB, en la que la enfermedad parece trasmitirse por la alimentación de las vacas con tejidos infectados de ovejas, la proteína que actúa como semilla podría ser ingerida y trasmitirse de algún modo al cerebro desde el tejido periférico. Se sabe que el bazo y otros órganos linfoides pueden trasmitir el escrapie, por lo que las células linfoides circulantes en el SNC son los primeros candidatos (aunque esto no se ha demostrado) para esta neuroinvasión.
ENFERMEDADES GENETICAS ASOCIADAS CON MUTACIONES PrP
En los humanos las enfermedades espongiformes incluyen, además de la ECJ espontánea o trasmitida en forma accidental, formas genéticas de ECJ e incluso condiciones más raras como el SGS y el IFF.21,22 En el SGS, que se trasmite como un rasgo autosómico dominante, las personas afectadas desarrollan ataxia, seguida de demencia, los pacientes con IFF son incapaces de dormir. Todas estas condiciones pueden trasmitirse por incoulación de tejidos enfermos a primates no humanos susceptibles. Es notable que todas estas condiciones se han asociado con mutaciones en el gen PrP y en una serie de experimentos, los ratones transfectados con la mutación del SGS desarrollaron cambios espongiformes.21 Se considera que las mutaciones en el gen PrP promueven la conversión a un estado con estructura alterada, insoluble.
La existencia de enfermedades que simultáneamente son genéticas e infecciosas y la asociación de mutaciones en el gen PrP con estas enfermedades, proporcionan intensa evidencia para apoyar la hipótesis de los priones, aunque al inicio existió gran escepticismo al respecto porque viola el dogma central de la biología molecular.23 Un experimento que no se ha realizado aún es la síntesis in vitro de una proteína pura capaz de servir como semilla para el desarrollo de enfermedad espongiforme; muchos grupos participan en esta tarea.
Leucoencefalopatía multifocal progresiva
La LMP es una enfermedad de la sustancia blanca cerebral que se caracteriza por deterioro motor progresivo, pérdida de la visión, incontinencia y demencia eventual, causada por un virus ADn relacionado a los papovavirus conocido como JC. La LMP se describió por primera vez en pacientes con inmunodeficiencia causada por neoplasias hematológicas o por tratamiento para la prevención del rechazo de trasplantes. Sin embargo, fue un padecimiento poco frecuente hasta la aparición del SIDA. Estudios epidemiológicos indican que entre uno y cinco porciento de las personas infectadas por VIH desarrollarán eventualmente LMP. En muchos de estos casos el desarrollo de LMP marca el inicio de la inmunodeficiencia y es la enfermedad que define el SIDA. En otros casos constituye una complicación asociada con otras infecciones oportunistas.
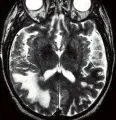
El diagnóstico de LMP debe considerarse en cualquier persona con enfermedad progresiva subaguda que incluya alteraciones motoras y cognitivas y que tenga una inmunodeficiencia comprobada o probable (v.gr., neoplasia). La mayoría de los casos tienen datos de afección de la sustancia blanca, incluyendo ataxia, defectos visuales y debilidad. En casos raros ocurre LMP en personas por lo demás sanas. Debe sospecharse fuertemente LMP si la IRM muestra áreas confluentes de señales de mayor intensidad en las imágenes T2 [ver figura 4]. Estas áreas pueden ser hipointensas en las imágenes en T1 con respecto a los límites de sustancia gris-blanca. (T1 se refiere a tiempo de relajación giro-reja, o longitudinal). El LCR suele ser normal en la LMP, y cuando existe pleocitosis en el LCR, ésta es leve.
|
| Figura 4 |
| Patología de la demencia por VIH |
El diagnóstico definitivo de la LMP ha requerido de biopsia cerebral, pero estudios recientes han demostrado que puede detectarse el ADN del virus JC por amplificación con la reacción en cadena de la polimerasa (RCP) en el LCR de pacientes con LMP.24,25 La frecuencia de reacciones falsas positivas es baja, y se ha calculado que la RCP en LCR tiene una sensibilidad y especificidad mayor del 90 porciento.26 Se han atribuido casos ocasionales de mejoría espontánea de la LMP a tratamientos experimentales. Sin embargo, no se sabe si la reversión parcial de la inmunodeficiencia ha sido la responsable de la mejoría.
PATOLOGIA Y PATOGENIA
El examen patológico del cerebro con LMP muestra cambios dramáticos en la sustancia blanca subcortical. Los oligodendrocitos, que son las células responsables de la manufactura y mantenimiento de la mielina, son el principal blanco del virus JC. Por lo tanto, la citólisis oligodendrogial es el principal mecanismo patogénico en la LMP y causa desmielinización severa característica. El evento inicial en el desarrollo de la LMP parece ser la aparición de un foco microscópico de infección oligodendrogial, a partir del cual el virus se disemina en forma centrífuga para afectar grandes segmentos de la sustancia blanca. Los astrocitos, lo mismo que algunos oligodendrocitos, demuestran cambios morfológicos bizarros semejantes a los asociados con una neoplasia del SNC. Existe poca afección de la sustancia gris o las neuronas en la LMP, y la que ocurre puede reflejar citólisis de los oligodendrocitos adyacentes, quizá por el reclutamiento de microglia y macrófagos.27
VIRUS JC
La etiología viral de la LMP se sospechó por primera vez cuando Zu Rhein y colaboradores describieron micrografías electrónicas con partículas parecidas a poliomavirus en el núcleo de oligodendrocitos.28 El aislamiento viral se logró solo usando cultivos de cerebro fetal en experimentos de cocultivos, que demostraron que el virus JC está relacionado, aunque es claramente distinto, de otros poliomavirus. Un gran porcentaje de población adulta en los Estados Unidos (70 a 90 porciento) tiene anticuerpos contra virus JC, lo que indica que esta es una infección común, aunque no se han atribuido enfermedades a la infección primaria.
La evidencia de infección diseminada y el aislamiento del virus JC de la orina de algunos pacientes inmunosuprimidos son compatibles con la hipótesis de que la LMP representa la reactivación de un virus ADN latente. Sin embargo, no se ha identificado el órgano reservorio. La ausencia de ADN de virus JC en los cerebros de personas normales sugiere que el virus JC permanece latente en otros tejidos. Los linfocitos, los órganos linfoides y los riñones se consideran los candidatos más factibles porque el virus se ha detectado en estos tejidos por medio de RCP o se ha aislado en los mismos.29,30 Sin embargo, el virus JC aislado del tejido periférico de las personas no inmunosuprimidas (el virus arquetipo) difiere del aislado de pacientes con LMP en las regiones responsables de la regulación de la expresión viral.31 Una hipótesis intrigante, que puede explicar estos datos, es que el virus arquetipo es modificado por deleción o duplicación y así es convertido en una forma neurotrópica que es trasnportado hacia el cerebro en las células B infectadas.
No existe un tratamiento bien establecido para la LMP, pero enfoques experimentales han incluido análogos de nucleósidos, interferones, e inhibidores de la topoisomerasa. Algunos de estos se exploran en la actualidad en pacientes con SIDA.
Pancencefalitis esclerosante subaguda
La panencefalitis esclerosante subaguda (PEES) es una secuela rara de la infección por sarampión (uno de cada 100,000 casos). La panencefalitis progresiva por rubeola es una enfermedad semejante a la PEES causada por este virus.
Los pacientes con PEES han sufrido una infección no complicada por sarampión, con frecuencia a menor edad que el promedio, y desarrollan encefalitis progresiva varios años después. La PEES parece ser causada por una mutación no común en la proteína matriz (M) del virus de sarampión nativo. Esta mutación puede afectar el empaquetamiento del virus, lo que puede explicar las dificultades que tienen los investigadores para aislar el virus libre. Se ha supuesto que el virus que causa la PEES puede diseminarse solo por fusión entre una célula infectada y células adyacentes no infectadas. Esto puede explicar la persistencia viral en presencia de una respuesta humoral intensa.
La PEES puede también ser el resultado de la vacunación con una cepa viva y atenuada de sarampión. Sin embargo, la tasa de desarrollo es por lo menos 10 veces menor que la asociada con la infección por el virus nativo. En los Estados Unidos, en donde la prevalencia de infecciones por sarampión es menor de 30,000, los casos de PEES son raros, incluso en años de epidemia.
No existe un tratamiento establecido para la PEES y su evolución casi siempre es fatal.
Bibliografía
- Price RW, BJ: The AIDS dementia complex. J Infect Dis 158:1079, 1988 [PMID 3053922]
- McArthur JC, Selnes OA, Glass JD, et al: HIV dementia, incidence, and risk factors. HIV, AIDS and the Brain. Price RW, Perry SW, Eds. Raven Press, New York, 1994, p 251
- McArthur JC, Cohen BA, Selnes OA, et al: Low prevalence of neurological and neuropsychological abnormalities in otherwise healthy HIV-1 infected persons: results from the multicenter AIDS cohort study. Ann Neurol 26:601, 1989 [PMID 2817836]
- Embretson J, Zupancic M, Ribas JL, et al: Massive infection of helper lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature 362:359, 1993 [PMID 8096068]
- Mellors JW, Rinaldo CR, Gupta P, et al: Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science 272:1167, 1996 [PMID 8638160]
- Glass JD, Fedor H, Wesselingh SL, et al: Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol 38:755, 1995 [PMID 7486867]
- Achim CL, Wang R, Miners DK, et al: Brain viral burden in HIV infection. J Neuropathol Exp Neurol 53:284, 1994
- Epstein LG, Gendelman HE: Human immunodeficiency virus type 1 infection of the nervous system: pathogenetic mechanisms. Ann Neurol 33:429, 1993 [PMID 8498818]
- Wiley CA, Schrier RD, Nelson JA, et al: Cellular localization of human immuno deficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc Natl Acad Sci USA 83:7089, 1986 [PMID 3018755]
- Bagasra O, Lavi E, Bobroski L, et al: Cellular reservoirs of HIV-1 in the central nervous system of infected persons: identification by the combination of in situ polymerase chain reaction and immunohistochemistry. AIDS 10:573, 1996 [PMID 8780811]
- Takahashi K, Wesselingh SL, Griffin DE, et al: Localization of HIV-1 in human brain using polymerase chain reaction/in situ hybridization and immunohistochemistry. Ann Neurol 39:705, 1996 [PMID 8651642]
-
Johnson RT, Glass JD, McArthur JC, et al: Quantitation
of human immunodeficiency virus in brains of demented and nondemented patients
with AIDS. Ann Neurol 39:392, 1996
-
Kuroda Y, Matsui M, Kikuchi M, et al: In situ demonstration
of the HTLV-I genome in the spinal cord of a patient with HTLV-I myelopathy.
Neurology 44: 2295, 1994 [PMID
7991115]
-
Jacobson S: Human T-lymphotropic virus, type-I myelopathy:
an immunopathologically mediated chronic progressive disease of the central
nervous system. Curr Opin Neurol 8:179, 1995
-
Hollsberg P, Hafler DA: What is the pathogenesis of
human T-cell lymphotro pic virus type I-associated myelopathy/tropical
spastic paraparesis (editorial)? Ann Neurol 37:143, 1995
-
Elovaara I, Koenig S, Brewah AY, et al: High HTLV-I
specific precursor cytotoxic T cell (CTL) responses to tax in HTLV-I infected
patients with neurological disease. J Immunol 156:3874, 1993
-
Brown P, Gibbs CJ, Rodgers-Johnson P, et al: Human
spongiform encephalopathy: the NIH series of 300 cases of experimentally
transmitted disease. Ann Neurol 35:513, 1994 [PMID
8179297]
-
Brown P, Cervenakova L, Goldfarb LG, et al: Iatrogenic
Creutzfeldt-Jakob disease: an example of the interplay between ancient
genes and modern medicine. Neurology 44:291, 1994 [PMID
8309577]
-
Prusiner SB: Molecular biology of prion disease. Science
252:1515, 1991
-
Telling GC, Scott M, Hsiao KK, et al: Transmission
of Creutzfeldt-Jakob disease from humans to transgenic mice expressing
chimeric human-mouse prion protein. Proc Natl Acad Sci USA 91:9936, 1994
-
Hsiao K, Baker HF, Crow TJ, et al: Linkage of a prion
protein missense variant to Gerstmann-Straussler syndrome. Nature 338:342,
1989 [PMID
2564168]
-
Chapman J, Arlazoroff A, Goldfarb LG, et al: Fatal
insomnia in a case of familial Creutzfeldt-Jakob disease with the codon
200Lys mutation. Neurology 46:758, 1996
-
Parchi P, Castellani R, Capellari S, et al: Molecular
basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease.
Ann Neurol 39:767, 1996 [PMID
8651649]
-
Weber T, Turner RW, Frye S, et al: Progressive multifocal
leukoencephalopathy diagnosed by amplification of JC virus-specific DNA
from cerebrospinal fluid. AIDS 8:49, 1994 [PMID
8011236]
-
Gibson PE, Knowles WA, Hand JF, et al: Detection of
JC virus DNA in the cerebrospinal fluid of patients with progressive multifocal
leukoencephalopathy. J Med Virol 39:278, 1993
-
McGuire D, Barhite S, Hollander H, et al: Virus DNA
in cerebrospinal fluid of human immunodeficiency virus-infected patients:
predictive value for progressive multifocal leukoencephalopathy. Ann Neurol
37:395, 1995 [PMID
7695239]
-
Major EO, Amemiya K, Tornatore CS, et al: Pathogenesis
and molecular biology of progressive multifocal leukoencephalopathy, the
JC virus-induced demyelinating disease of the human brain. Clin Microbiol
Rev 5:49, 1992 [PMID
1310438]
-
ZuRhein GM, Chou S-M: Particles resembling papova viruses
in human cerebral demyelination disease. Science 148:1477, 1965
-
Tornatore C, Berger JR, Houff SA, et al: Detection
of JC virus DNA in peripheral lymphocytes from patients with and without
progressive multifocal leukoencephalopathy. Ann Neurol 31:454, 1992 [PMID
1316734]
-
Dubois V, Lafon M-E, Ragnaud J-M, et al: Detection
of JC virus DNA in the peripheral blood leukocytes of HIV-infected patients.
AIDS 10:353, 1996 [PMID
8728037]
- Daniel AM, Swenson JJ, Mayreddy RPR, et al: Sequences within the early and late promoters of archetype JC virus restrict viral DNA replication and infectivity. Virology 216:90, 1996