Gastroenterología
⭳ Abrir artículo (PDF)580.2 KBEste artículo es idéntico en la Edición 3/2000.
Contenido del artículo
XI ENFERMEDADES QUE PRODUCEN MALABSORCION Y MALADIGESTION
- Definición
- Etiología
- Manifestaciones clínicas
- Pruebas para investigar malabsorción
- MEDICION DE LA GRASA FECAL
- PRUEBA DE ABSORCION DE XILOSA
- ESTUDIOS DE IMAGEN
- BIOPSIA DEL INTESTINO DELGADO
- EVALUACION DE LA FUNCION EXOCRINA DEL PANCREAS
- PRUEBAS DE ABSORCION DE ACIDOS BILIARES
- Enfermedades del intestino delgado que producen malabsorción
- ENTEROPATIA SENSIBLE AL GLUTEN
- OTROS TRASTORNOS SEMEJANTES AL ESPRUE
- MALABSORCION SECUNDARIA A RESECCION MASIVA DEL INTESTINO DELGADO
- ENTERITIS POR RADIACION
- ENFERMEDAD DE WHIPPLE
- AFECCION INMUNOPROLIFERATIVA DEL INTESTINO DELGADO
- LINFANGIECTASIA INTESTINAL
- ABETALIPOPROTEINEMIA
- GASTROENTERITIS EOSINOFILICA
- ENFERMEDAD DE CROHN
- SINDROME DE ESTASIS (PROLIFERACION BACTERIANA)
- AMILOIDOSIS
- MASTOCITOSIS SISTEMICA
- Infestaciones parasitarias
- Pancreatitis crónica con insuficiencia exócrina
- Defectos combinados o múltiples en la digestión y la absorción
XI ENFERMEDADES QUE PRODUCEN MALABSORCION Y MALADIGESTION
DR. CHARLES M. MANSBACH II.
Definición
Típicamente malabsorción significa alteración en la absorción de grasa (esteatorrea), porque la medición de la absorción de grasa ha sido y aún es el mejor indicador de la normalidad del proceso global de absorción de nutrientes. Sin embargo, en ciertas condiciones la absorción de grasa puede ser normal y se absorben mal otras sustancias específicas, como hierro, calcio, sales biliares o, en algunos padecimientos hereditarios, aminoácidos específicos, disacáridos o monosacáridos.
Etiología
Al considerar las causas de malabsorción de grasas existen tres posibilidades generales: enfermedad del intestino delgado, afección hepática o de las vías biliares, e insuficiencia pancreática exócrina [ver tabla 1].
La afección del intestino delgado puede causar la presencia de cantidades moderadas de grasa en las heces (7 a 30 g/día con una dieta de 100 g de grasa). Los pacientes con enfermedad intestinal pueden perder proteínas (enteropatía perdedora de proteínas) a través de la mucosa intestinal, lo que causa una menor concentración de albúmina en suero. También pueden existir deficiencias de vitaminas liposolubles (i.e., vitaminas A, D, E y K). Los pacientes pueden no absorber bien la vitamina B12 por afección grave del íleon terminal o por resección del mismo (por lo general más de 60 cm). También puede absorberse mal el ácido fólico y puede ocurrir hipocalcemia e hipomagnesemia.
Los pacientes con enfermedad hepática o de las vías biliares suelen tener solo pequeños incrementos de grasa en las heces (7 a 15 g/día) y malabsorción de vitaminas liposolubles.
Los pacientes con insuficiencia exócrina del páncreas pueden tener hasta 80 g de grasa/día en las heces. La grasa que pueden absorber es resultado de la acción de las lipasas gástricas.
Manifestaciones clínicas
Los síntomas de la malabsorción son diversos. En el caso más obvio el paciente refiere pérdida de peso a pesar de un buen apetito. En estos casos existe un cambio claro en la calidad de las heces y aumento en el número de evacuaciones. La consistencia del excremento disminuye y, cuando existe exceso de grasa, las heces son fétidas y son difíciles de eliminar en el inodoro. Pueden aparecer gotas de aceite o una capa lípida en el agua. El exceso de gas en las heces causa que estas floten.1
Dependiendo de los constituyentes de la dieta que no se absorban, los pacientes pueden sufrir distensión abdominal, borgborismos, cólicos abdominales (intolerancia a la lactosa), equímosis fáciles (deficiencia de vitamina K), osteopenia o tetania (defciencia de vitamina D y malabsorción de calcio), deficiencia de hierro o ceguera nocturna (deficiencia de vitamina A). Los casos más complejos son aquellos en los que no se piensa en malabsorción por falta de cambio en la calidad de las heces.
La diarrea de la malabsorción se clasifica como osmótica y suele desaparecer durante el ayuno. En la malabsorción de grasas la diarrea es causada no solo por las partículas osmóticamente activas excesivas, sino también por los ácidos grasos, que estimulan la secreción de Cl- dependiente de monofosfato cíclico de adenosina (AMPc).
Varios datos físicos específicos de algunas enfermedades pueden acompañar al estado de malabsorción y ayudar en el diagnóstico. Por ejemplo, pueden existir cambios cutáneos de esclerodermia o dermatitis herpetiforme, así como signos de neuropatía diabética. Aunque la tirotoxicosis puede asociarse con exceso de grasa en el excremento, los pacientes con tirotoxicosis suelen comer en forma exagerada pero absorben un porcentaje normal de la grasa de la dieta (95 porciento) por lo que en realidad no sufren malabsorción.
Pruebas para investigar malabsorción
Las pruebas para investigar malabsorción incluyen la determinación de si existe un exceso de grasa en las heces [ver tabla 2]. La proteína es producida en grandes cantidades por el tubo digestivo, en especial el páncreas, lo que hace que la creatorrea sea difícil de interpretar. Los carbohidratos mal absorbidos que llegan al colon pueden ser metabolizados por las bacterias colónicas a ácidos grasos de cadena corta, que en parte se absorben en el colon. Esto hace que la medición cuantitativa de la absorción de carbohidratos sea poco exacta, aunque ocurre reducción en el pH de las heces, lo que indica la excreción de una cantidad excesiva de ácidos grasos de cadena corta.
MEDICION DE LA GRASA FECAL
La grasa fecal puede medirse en forma cualitativa y cuantitativa. La medición cualitativa de la grasa fecal usando la tinción Sudán II ha demostrado ser muy exacta,2 en especial si se excretan cantidades significativas de grasa. Como con muchas pruebas cualitativas, la destreza del observador es importante para el éxito de la prueba.
La medición cuantitativa de la grasa fecal es el parámetro con el que se comparan todas las otras pruebas. Es importante recordar que la prueba no puede realizarse a menos que el paciente sea capaz de ingerir por lo menos 80 g, y de preferencia 100 g, de grasa por día.
PRUEBA DE ABSORCION DE XILOSA
La capacidad del paciente para absorber el azúcar xilosa evalúa el área de absorción del intestino. A diferencia de la glucosa, la xilosa no se absorbe en forma activa por el intestino, sino por un proceso lento de difusión pasiva. Se administran 25 g por vía oral de xilosa y se recolecta la orina durante 5 horas. La excreción normal en orina es mayor de 4 g de xilosa en 5 horas. Para asegurar un flujo urinario adecuado el paciente debe beber 500 ml de agua después de la xilosa. Esta ingesta debe producir un volumen urinario de por lo menos 300 ml durante el periodo de recolección. Deben excretarse más de 4 g de xilosa. La excreción de xilosa puede ser baja en forma falsa en pacientes con menor función renal o con ascitis, ya que la xilosa se diluye en el líquido de ascitis. Para evitar este error se aconseja medir la concentración de xilosa en sangre. La xilosa no absorbida alcanza el colon y puede ser metabolizada por las bacterias locales a hidrógeno. El hidrógeno puede cuantificarse en la respiración, y se ha reportado que esta prueba es tan exacta como la medición de xilosa en el suero o la orina.
ESTUDIOS DE IMAGEN
La placa simple o el ultrasonido abdominal no suelen ser útiles en la mayoría de los casos de malabsorción. Sin embargo, el 30 porciento de los casos de pancreatitis crónica tienen calcificaciones visibles en la placa simple de abdomen. La detección de calcificaciones puede aumentar si se usa tomografía computada o ultrasonido. La TC o el ultrasonido pueden identificar conductos pancreáticos dilatados, otro signo característico de la pancreatitis crónica. La pancreatografía endoscópica retrógrada (PER) puede también ser útil cuando se observan cambios en los conductos indicativos de pancreatitis crónica.
La placa simple del intestino delgado puede ayudar en el diagnóstico de varias alteraciones. La presencia de divertículos del intestino delgado, de alteración en la peristalsis, como se observa en la esclerodermia, o de dismotilidad intestinal idiopática, puede apoyar la sospecha de proliferación bacteriana. El examen cuidadoso del íleon terminal puede identificar enfermedad de Crohn. Pueden identificarse estenosis en algunos pacientes con lesión por radiación o lesión causada por antinflamatorios no esteroides. La hipoalbuminemia que afecta al intestino delgado puede originar el llamado signo de pila de monedas.
BIOPSIA DEL INTESTINO DELGADO
Un patólogo experto puede ayudar en el diagnóstico de la enteropatía sensible al gluten (con o sin dermatitis herpetiforme), esprue hipogamaglobulinémico, esprue tropical, enfermedad de Whipple, enfermedad por complejo Mycobacterium avium, síndrome de estasis, amiloidosis y linfangiectasia intestinal.
EVALUACION DE LA FUNCION EXOCRINA DEL PANCREAS
Es necesario que se destruya más del 90 porciento de la función exócrina del páncreas antes de que ocurra malabsorción.3 La prueba más sensible requiere el paso de una sonda de doble luz.4 Se administra secretina o colecistoquinina (CKK) por vía intravenosa y se recolectan en forma separada las secreciones gástrica y duodenal. Cuando se administra la secretina se miden el volumen del líquido duodenal y la concentración de bicarbonato. Si se administra CKK se determina la actividad de la lipasa o la tripsina usando sustratos apropiados.
La prueba no invasiva de bentiromida se basa en la acción de la tripsina sobre la bentiromida para originar ácido aminobenzoico (PABA) y benzoil-tirosina [ver figura 1]. El PABA se absorbe con facilidad por el intestino y se excreta en la orina. En las personas sanas, cuando se ingieren 500 mg de bentiromida, el 57 porciento o más del PABA aparece en la orina en 6 horas. En los pacientes con pancreatitis crónica la cantidad de PABA excretado es significativamente menor, en promedio de 42 porciento. Usando la excreción de 57 porciento como límite, la sensibilidad es del 67 al 80 porciento y la especificidad es del 95%.5 El PABA puede también medirse en el plasma 120 minutos después de la ingestión de bentiromida, lo que aumenta la sensibilidad de la prueba.6 Las mediciones en plasma son útiles en los casos de menor excreción renal, que pueden ocurrir en ancianos. El PABA se identifica por colorimetría y, por lo tanto, otras arilaminas pueden interferir con su determinación (v.gr., acetaminofén, lidocaína, procainamida, sulfonamidas y diuréticos tiacídicos).7 En los casos en los que está alterada la absorción intestinal, como en el esprue, la absorción de PABA liberado puede disminuir, causando una recuperación en orina falsamente baja. Por desgracia, la prueba de bentiromida se vuelve positiva solo cuando la glándula está destruida en más del 90 porciento. De cualquier modo, al considerar el estudio de un paciente con esteatorrea la prueba de bentiromida puede ser útil porque se requiere una destrucción semejante de la glándula para provocar esteatorrea.
Aunque la gran mayoría de las proteasas y lipasas pancreáticas se almacenan en gránulos de cimógeno y son liberadas de la porción apical de la célula pancreática exócrina hacia el conducto pancreático, un pequeño porcentaje se fuga al intersticio de la glándula, es transportado en la circulación y puede ser medido (i.e., por un ensayo de tripsinógeno en suero). Debido a que el péptido activador de tripsina aún no es liberado y porque cualquier cantidad de tripsina activa se une con rapidez a la alfa1-antitripsina, la forma circulante libre de la tripsina es el tripsinógeno. En los pacientes con pancreatitis crónica e insuficiencia pancreática la concentración sérica de tripsinógeno es menor que en personas sanas (2 a 18 ng/ml, comparado con 29 a 79 ng/ml en gente sana).8 El nivel bajo de tripsinógeno en suero parece tener un mayor grado de especificidad para pancreattis crónica, aunque muy modesta sensibilidad.
PRUEBAS DE ABSORCION DE ACIDOS BILIARES
Los ácidos biliares se sintetizan a partir del colesterol en el hígado y requieren conjugarse con glicina o taurina antes de ser excretados hacia el intestino a través del colédoco. Los ácidos biliares conjugados solubilizan los productos de la hidrólisis de triacilgliceroles en micelas complejas, lo que facilita la absorción rápida de los lípidos de la dieta. Los ácidos biliares no se absorben en el intestino proximal con los lípidos de la dieta sino en el íleon distal. El reservorio de ácidos biliares recircula seis veces al día. Alrededor del 95 porciento de los ácidos biliares se reabsorben y recirculan en la circulación enterohepática cada día, y aparecen en las heces aproximadamente 0.5 g de ácidos biliares, lo que equivale a la tasa de síntesis hepática en condiciones estables. Si los ácidos biliares no se absorben en forma adecuada ocurre diarrea (enteropatía colerética). En ausencia completa de sales biliares los ácidos grasos se absorben en forma menos eficaz y aparecen hasta el 25 a 50 porciento de los lípidos ingeridos en el excremento. En los pacientes con diarrea idiopática o con diarrea después de resección ileal (> 30 cm), la malabsorción de sales biliares es una posibilidad etiológica. También los niños que tienen diarrea inexplicable pueden tener un defecto congénito del trasporte de ácidos biliares dependiente de sodio en el íleon terminal.9
Para probar la presencia de malabsorción de sales biliares existen dos métodos, no disponibles en todos los sitios. El primero es la prueba respiratoria con ácido 14C-glicocólico y la segunda es la pureba de absorción de ácido homocólico-taurina marcado con selenio 75 (75SeHCAT). En la primera [ver figura 2], se administra por vía oral una cantidad determinada de ácido 14C-glicocólico. Muchas bacterias son capaces de hidrolizar la unión amida y liberar la 14C-glicina; ésta es absorbida y se produce 14CO2 en el hígado o es metabolizada en la luz intestinal a 14CO2. En cualquier caso, aparece 14CO2 en cantidades mesurables en el aliento. El porcentaje de la dosis ingerida que se excreta en la respiración aumenta si la luz intestinal contiene más bacterias de lo normal o si llega un exceso de sales biliares al colon. Incluso un medicamento antisecretor gástrico puede aumentar la población de bacterias en el intestino a un nivel que cause una prueba del aliento anormal,10 como en el caso de la estasis intestinal. La utilidad de esta prueba es como indicador de malabsorción de ácidos biliares, por lo que es limitada. La prueba 75SeHCAT tiene mayor utilidad clínica por su estrecha correlación con la excreción de colato y la facilidad de medición de la retención de 75Se por la cámara gama de cuerpo total. Las personas normales retienen más del 19 porciento de una dosis administrada por vía oral de 75Se después de 7 días, mientras que los pacientes con disfunción significativa o resección de íleon retienen menos del 12 porciento.11
Enfermedades del intestino delgado que producen malabsorción
ENTEROPATIA SENSIBLE AL GLUTEN
La enteropatía sensible al gluten (ESG) se denominó alguna vez enfermedad celiaca en niños y esprue idiopático o no tropical en adultos. En 1960 se reconoció que estas enfermedades eran la misma, estaban causadas por la principal proteína del trigo, el gluten, y más específicamente, por su componente soluble en alcohol, la gliadina.12
Factores genéticos y etiológicos
La ESG es menos común en los Estados Unidos que en Europa occidental. Se asocia con los haplotipos DqA1*0501, DqB1*0201, HLA-B8 y DRw3. En grupos de gemelos monocigotos, la mayoría de los gemelos opuestos al caso índice no tienen ESG. Esto ha apoyado que existe otro factor desconocido (no genético) que es importante como causa de la enfermedad.
Patogenia
Las causas de esteatorrea en la ESG son muchas. Las células que producen CCK están disminuidas en número o son tan defectuosas que la cantidad de CCK presente en la mucosa duodenal está muy reducida.12 Esto causa una menor cantidad de lipasa pancreática y ácidos biliares que llegan a la luz intestinal en respuesta a los lípidos de la dieta. Las células de las criptas intestinales son las principales células secretoras de fluído del intestino, por secreción de Cl- dependiente de AMPc con abundante agua asociada. En la ESG la porción de la cripta del complejo velloso está muy expandido, causando mayor secreción de agua. Debido a que las células de la punta de la vellosidad, que normalmente absorben el agua, están dañadas y disminuidas en número, la absorción de agua y electrolitos no es tan eficaz como la normal y el intestino se vuelve secretor.12 Esto reduce aún más la concentración de ácidos biliares en la luz intestinal, más de lo esperado solo por la menor liberación de CCK. La capacidad de los ácidos biliares para solubilizar los productos de la lipólisis depende de la presencia de sales biliares a una concentracón mayor de la concentración micelar crítica (CMC) de 1.4 mM.13 En condiciones normales, el intestino tiene una concentración posprandial de sales biliares de 10 mM.14 Los bordes en cepillo en la superficie de los enterocitos maduros se afectan mucho en la ESG. Además, las estructuras vellosas se aplanan. Estas dos condiciones causan reducción importante de la superficie en la que se absorben los lípidos. La severidad de la reducción del área de absorción puede calcularse por la prueba de absorción de D-xilosa. Los enterocitos en la superficie del intestino no son tan maduros como los enterocitos normales porque su frecuencia de recambio está muy aumentada. Es probable que esto reduzca la capacidad para procesar los lípidos en forma normal.
Diagnóstico
Manifestaciones clínicas Aunque la ESG puede iniciar en la infancia y responder a la supresión de gluten, los niños con la enfermedad sufren remisión en la adolescencia, incluso si ingieren una dieta que contenga gluten. Como adultos, estos pacientes, 25 o más de los cuales tuvieron síntomas en la infancia, pueden presentar molestias diversas, por lo común pérdida de peso, fatiga, cólicos abdominales, distensión, meteorismo y diarrea (esteatorrea), aunque puede no haber pérdida del apetito. En algunos la enfermedad tiene inicio insidioso y los síntomas son leves. Es solo después de que son tratados que estos pacientes se dan cuenta, en retrospectiva, lo enfermos que estaban. En estudios de población en los cuales la presencia de la enfermedad se estableció por biopsia intestinal, las personas con trastornos que justificaron la biopsia con frecuencia estaban asintomáticos, pero tenían menor estatura que los hermanos no afectados. Debido a que no existe ningún dato específico para el diagnóstico, en especial si no existe esteatorrea clínicamente evidente, el diagnóstico de la ESG puede retrasarse. Esto es especialmente cierto en pacientes que no tienen esteatorrea pero sí osteoporosis, equímosis fáciles como resultado de la deficiencia de vitamina K o anemia por deficiencia de hierro inexplicable.
Pruebas de laboratorio El diagnóstico depende de los datos en la biopsia del intestino delgado [ver figura 3]. Los datos clásicos incluyen atrofia vellosa parcial o completa, enterocitos de apariencia anormal en las puntas de las vellosidades, aumento de los linfocitos intraepiteliales, un infiltrado en la lámina propia que consiste principalmente de linfocitos y macrófagos, aumento en el tamaño de las criptas tanto vertical como horizontal, y aumento en el número de figuras mitóticas.12 Estas características, aunque típicas, no son patognomónicas. Para que el diagnóstico sea definitivo el paciente debe responder al tratamiento dietético. Puede esperarse mejoría sintomática en el 80 porciento de los pacientes en 1 mes, pero la mejoría histológica es mucho más lenta. Un 10 porciento de los pacientes no responde sino hasta los 2 meses, y el resto puede tardar hasta 2 años. Incluso bajo control dietético estricto, la biopsia puede no normalizarse. Lo más frecuente es que el paciente permanezca bien controlado mientras reciba la dieta, pero muchos pacientes rompen la dieta por situaciones diversas o creyendo que están curados. Esto causa en forma inevitable recurrencia de los síntomas, lo que confirma el diagnóstico.
Además de la biopsia intestinal, la presencia de anticuerpos contra
endomisio es la única otra prueba útil. Este anticuerpo
está presente en hasta el 95 porciento de los casos y rara vez en
sujetos controles.15 Otras pruebas de malabsorción, como la
prueba de absorción de D-xilosa o de grasas en las heces pueden ser
anormales. También puede existir reducción en los factores de la
coagulación, anemia causada por deficiencia de folato o hierro u
osteoporosis. Sin embargo, ninguno de estos datos es específico de
ESG.
Tratamiento
El tratamiento de la ESG consiste en una dieta estricta de eliminación de gluten, no trigo, centeno o cebada. La avena parece ser segura, pero suele evitarse durante la fase temprana, cuando se está evaluando la respuesta a la dieta. En ocasiones es difícil seguir la dieta porque muchos alimentos tienen un contenido oculto de gluten. Es importante la dieta con eliminación con gluten porque los pacientes que no reciben tratamiento tienen mayor posibilidad de desarrollar linfoma intestinal.16 Pueden ser útiles al respecto los grupos de apoyo, como el organizado por la Sociedad Celiaca en los Estados Unidos, en especial si la enfermedad es de diagnóstico reciente. La información sobre qué buscar en las etiquetas y recetas interesantes puede ser muy útil para mantener al paciente en el regimen dietético. Durante el periodo de prueba, incluso la cerveza, la ale y el whisky pueden contener suficiente gluten para sensibilizar al paciente, por lo que no deben consumirse. Después de evaluar la respuesta a la dieta pueden probarse estas bebidas, si se desea, para determinar si el paciente es sensible. Otro productos que no suele pensarse que contienen gluten pero así es incluyen los helados, las hostias de comulgar e incluso algunos medicamentos (como excipiente). A pesar de las restricciones, existen muchas opciones dietéticas para el paciente, incluyendo ciertos cereales, leche, queso, huevos, carne, pollo, pescado, chocolate y productos hechos de maíz, arroz o harina de papa.
Si el paciente no responde a la dieta, la causa más probable es que la dieta no ha sido suficientemente estricta. En este caso es útil que un dietista revise la historia dietética del paciente.12 Con menos frecuencia el paciente tendrá un síndrome de estasis o insuficiencia pancreática. Cuando se diagnostican estos problemas y se tratan con éxito, el paciente presenta buena respuesta a la dieta.
OTROS TRASTORNOS SEMEJANTES AL ESPRUE
Dermatitis herpetiforme y ESG
Muchos pacientes con dermatitis herpetiforme tienen ESG.17 En las rodillas, codos, hombros y glúteos aparecen lesiones ampollosas muy pruriginosas. La biopsia de piel en la dermatitis herpetiforme característicamente muestra depósitos de inmunoglobulina A (IgA). Al establecer la dieta sin gluten mejoran las lesiones tanto dermatológicas como intestinales, lo que indica una relación entre ambas. Sin embargo, las lesiones de la piel responden al tratamiento con dapsona y las intestinales no, lo que sugiere que también existen diferencias entre las dos.
Esprue tropical
El esprue tropical es una enfermedad de malabsorción que aparece en ciertas áreas del mundo, en especial los trópicos, entre la pbolación local y los turistas. Al considerar esta enfermedad, debe notarse que las biopsias intestinales obtenidas de residentes normales de estas regiones o de turistas que viven fuera de los grandes hoteles se clasificarían como anormales si se obtuvieran de personas residentes en los Estados Unidos o Europa. En estas pbolaciones la arquitectura vellosa puede ser más irregular, en la que varias vellosidades se fusionan en forma horizontal, mientras que otras parecen hojas o se aplanan y su lámina propia está infiltrada por células inflamatorias [ver figura 4], si se compara con las delgadas estructuras vellosas que se observan en los norteamericanos [ver figura 3b]. En dos poblaciones estudiadas con gran cuidado, el 5 a 13 porciento de los norteamericanos que vivieron en Puerto Rico durante 6 meses o más desarrollaron síntomas de esprue tropical. Los expatriados que han regresado a los Estados Unidos en forma permanente pueden desarrollar síntomas de esprue tropical incluso 10 años después de vivir ya en ese país.18 Los voluntarios de los Cuerpos de Paz enviados a Pakistan tienen lesiones demostrables en el intestino delgado y alteraciones funcionales que se normalizaron varios meses después de su retorno a los Estados Unidos.19 Los hindúes y pakistaníes que viven en los Estados Unidos pueden tardar más tiempo (hasta 4 años) en excretar cantidades normales de xilosa.20 No se conoce la causa exacta de estos cambios en el intestino delgado, pero se piensa que el síndrome de esprue tropical es ocasionado por una o más especies de bacterias coliformes, como especies de Klebsiella,21 que colonizan el tracto intestinal superior.
Los síntomas del esprue tropical difieren de los de la ESG. La pérdida de peso causada principalmente por anorexia es notable, lo mismo que la diarrea. La lengua escaldada (70 porciento), el edema en pies (25 porciento) y la deficiencia de folato y vitamina B12 (75 a 100 porciento) son mucho más comunes en el esprue tropical que en el no tropical.21 Los síntomas pueden ser muy severos, causando en ocasiones la muerte en áreas endémicas. Esto fue especialmente cierto en prisioneros de guerra alemanes e italianos apresados en la India durante la Segunda Guerra Mundial. Sin embargo, en la actualidad el pronóstico suele ser excelente para los pacientes que permanecen en los trópicos o regresan a los Estados Unidos. El tratamiento se inicia con ácido fólico (5 mg/día).21 Esto se asocia con rápida mejoría en el apetito y elimina la mayoría de los síntomas clínicos. En los pacientes con duración breve de los síntomas es suficiente con administrar el folato durante 6 meses a 1 año. Para los que tienen síntomas de mayor duración (más de 4 meses), deben agregarse antibióticos, como tetraciclinas (2 g/día por 1 año). La mayoría de los pacientes que regresan a los Estados Unidos recuperan peso con rapidez, incluso si los resultados de las pruebas de absorción o la biopsia intestinal no se normalizan.
Esprue colagenoso
El esprue colagenoso es una enfermedad rara y devastadora en la que existe una capa de colágena por debajo de los enterocitos del intestino delgado. Su relación con la colitis colagenosa no es clara, pero la característica histológica básica de depósito subepitelial de colágena es la misma. En el colon se deposita colágena tipo 6, pero el tipo de colágena depositada en el intestino delgado se desconoce. En el colon los síntomas suelen ser leves, pero el el intestino delgado son más severos. Esto puede ser porque la colágena constituye una barrera a la difusión, lo que impide que los nutrientes pasen hacia los capilares porta o los linfáticos. El diagnóstico se realiza por el cuadro histológico clásico de atrofia vellosa y depósito subepitelial de colágena. Sin embargo, si el diagnóstico pasa desapercibido y se piensa que el paciente tiene ESG por el aplanamiento de las estructuras vellosas, el paciente suele no responder a la dieta con eliminación de gluten. En estos casos el diagnóstico real del paciente se aclarará si la biopsia se envía a un patólogo especializado en enfermedades gastrointestinales. No existe un tratamiento específico para este padecimiento. El problema más común es la diarrea osmótica causada por el estado de malabsorción inducido por la enfermedad. En estos casos el paciente debe ser tratado como si tuviera síndrome del intestino corto. Algunos pacientes responden al tratamiento esteroideo. Pocos pacientes responden a esteroides y una dieta con eliminación de gluten, y cuando el paciente mejora suele poderse disminuir la dosis de esteroide en forma gradual.22
Esprue hipogamaglobulinémico
El tracto digestivo es el órgano linfoide más grande del organismo. El ambiente al que este sistema inmune es expuesto está lleno de antígenos extraños que deben ser enfrentados, identificados y, en caso necesario, atacados. Por lo tanto, no es sorprendente que pueda desarrollarse disfunción intestinal en los pacientes con inmunodeficiencia. Esto es especialmente cierto en el caso de la deficiencia de IgA porque esta es la inmunoglobulina más importante en el intestino. Algunos pacientes que tienen uno de los síndromes hipogamaglobulinémicos pueden tener malabsorción.23 Las biopsias de intestino no muestran células plasmáticas y son fácilmente distinguibles de las de los pacientes con enteropatía sensible al gluten, en las que estos tipos celulares son abundantes. Pueden existir también organismos Giardia lamblia. Los pacientes con deficiencia de IgA también suelen tener historia de infecciones respiratorias recurrentes22 que les distinguen de los pacientes con enteropatía sensible al gluten. La causa más común de malabsorción en este padecimiento es la giardiasis. Con frecuencia los síntomas intestinales mejoran si se administra metronidazol en dosis de 750 mg/día durante 10 días.
MALABSORCION SECUNDARIA A RESECCION MASIVA DEL INTESTINO DELGADO
La resección masiva del intestino delgado se usa para tratar varias enfermedades, incluyendo isquemia mesentérica, vólvulos y enfermedad de Crohn. Dependiendo de la cantidad de intestino resecado, los resultados pueden variar desde catastróficos a leves. La retención de la válvula ileocecal disminuye los síntomas. El íleon responde a la resección yeyunal con hiperplasia en forma mucho más eficaz de lo que el yeyuno responde a una resección ileal. Existen también funciones especializadas presentes en el íleon que no realiza el yeyuno, como el transporte de sales biliares y vitamina B12. El mantenimiento de un reservorio adecuado de ácidos biliares es importante para la absorción de grasa porque la menor superficie de absorción en estos pacientes hace necesario que la absorción de grasas sea lo más eficaz posible. En forma alternativa, el íleon puede realizar la mayoría de las funciones del yeyuno excepto por la absorción de ácido fólico, Ca2+ y Fe2+. Sin embargo, estos elementos pueden sustituirse con medicación adecuada.
Debido a que el intestino requiere de cierta área de superficie para que ocurra una absorción adecuada, la reducción del área a menos de un valor crítico suele causar malabsorción. Las manifestaciones clínicas del síndrome de intestino corto pueden incluir diarrea, esteatorrea, pérdida de peso, deficiencias de oligoelementos, hiponatremia e hipocalemia. Las proteínas requieren la mayor área de absorción.24 Por lo tanto, el tratamiento en estos pacientes depende de qué partes y cuánto del intestino se haya resecado. También se requieren vitaminas y minerales en el esquema terapéutico, dependiendo de la parte de intestino que falte. El tratamiento puede incluir varios alimentos de escasa cantidad al día, ingerir alimentos de absorción rápida, como los suplementos calóricos enlatados, picar o moler los alimentos, e ingerir triglicéridos de cadena media, que pueden absorberse en ausencia de sales biliares.24 Los alimentos ricos en ácidos grasos polinsaturados, como los aceites vegetales, se absorben con más facilidad que las carnes, que tienen grasas más saturadas. Por último, los suplementos dietéticos, como el Vivonex, son productos totalmente hidrolizados y se absorben con rapidez. Para disminuir el tránsito intestinal puede emplearse difenoxilato con atropina, loperamida o tintura de opio deodorizada. Un método alternativo consiste en hacer que el paciente beba una pequeña cantidad de aceite de girasol antes del alimento. El lípido pasa con rapidez al íleon (cuando está presente), el colon o ambos25 y elabora un péptido YY (PYY),26 que es el posible freno ileal, enlenteciendo el vaciamiento gástrico. Al probar diferentes dietas en los pacientes se podrá establecer la mejor dieta oral para ellos, intentando evitar que requieran de nutrición parenteral total (NPT), que es la alternativa menos deseable.
ENTERITIS POR RADIACION
La lesión del intestino es una secuela muy común de la administración de radioterapia como tratamiento oncológico. El sitio de lesión más común es el recto por estar fijo en su sitio y porque los tumores cervicouterinos y prostáticos son comunes y con frecuencia se tratan con radioterapia locorregional. La radiación del abdomen por otros tumores puede causar lesión del intestino delgado. La afección del intestino delgado es más común si el paciente tuvo una cirugía abdominal previa, que puede restringir el movimiento del intestino. El íleon terminal puede afectarse durante la irradiación pélvica. La lesión del intestino delgado puede causar malabsorción con esteatorrea.
ENFERMEDAD DE WHIPPLE
La enfermedad de Whipple es una enfermedad devastadora y rara causada por la bacteria Tropheryma whippelii.27 Es indispensable el diagnóstico exacto porque la mortalidad llega a ser del 100 porciento sin tratamiento.
Clásicamente, la enfermedad ocurre en un varón de edad media con artritis no deformante que suele iniciar años antes del inicio de los síntomas intestinales. Otras molestias incluyen fiebre, distensión abdominal, diarrea, pérdida de peso, linfadenopatía, hiperpigmentación de la piel y esteatorrea.28 Muchos pacientes expresan el isotipo HLA-B27. En ocasiones pueden no existir síntomas intestinales, incluso en algunos pacientes con afección del SNC.29 En un caso bien demostrado pero no habitual, no se identificó afección intestinal, incluso después de biopsias costosas en dos laboratorios, a pesar del hecho de que el paciente tenía por lo demás síntomas típicos de la enfermedad.30
El diagnóstico se basa en la identificación de macrófagos con tinción positiva ácido-Schiff (PAS), que contienen partículas falciformes.28 Con mucho, el sitio más común de biopsia positiva es el intestino. La lesión histológica muestra distensión de las vellosidades (en palillo de tambor) con macrófagos PAS positivos espumosos y dilatación linfática. En casos extremos puede observarse una superficie vellosa plana. Estos datos deben distinguirse en el contexto clínico adecuado de los de la enfermedad por complejo M. avium, en la que también se encuentran macrófagos PAS positivos. La tinción para bacilos ácido-alcohol resistentes debe distinguir entre ambos padecimientos. Puede existir afección del SNC en ausencia de síntomas neurológicos, en ocasiones asociada con macrófagos típicos y apoyada por la técnica más sensible de reacción en cadena de la polimerasa.31 En ocasiones se requiere una biopsia cerebral, que puede ser guiada por resonancia magnética. También puede ser útil la afección cardiaca y pulmonar.32
Debido a que la enfermedad es tan poco común, es difícil establecer un plan de tratamiento bien definido. El tratamiento originalmente propuesto era de penicilina y estreptomicina durante 2 semanas, seguido de tetraciclinas por 1 año. En la actualidad se administra trimetoprim-sulfametoxazol durante 1 año. Aunque los síntomas intestinales y sistémicos responden a cualquier tratamiento, el problema lo constituyen las manifestaciones del SNC. Por lo genral, en los pacientes que no tienen afección nerviosa al inicio, los síntomas a este sistema aparecen un año o más después del tratamiento de las molestias sistémicas e intestinales. Puede observarse demencia progresiva, pero los signos patognomónicos de la enfermedad neurológica, cuando existe, son miorritmia oculomasticatoria y miorritmia oculofacial-esquelética.33 Por lo tanto, se requieren antibióticos que crucen la barrera hemato-encefálica. Es interesante que la administración por un periodo corto de penicilina-estreptomicina es suficiente para bloquear los síntomas del SNC, mientras que incluso la administración prolongada de trimetoprim-sulfametoxazol puede causar en ocasiones manifestaciones neurológicas de la enfermedad de Whipple.34 La tetraciclina sola no erradica la afección neurológica y no debe administrarse, a pesar de que es eficaz para tratar los síntomas intestinales y sistémicos. Una característica importante que debe tenerse en cuenta es que en el 50 porciento de los pacientes el LCR puede contener macrófagos típicos o material RCP-positivo en ausencia de cualquier síntoma del SNC.31 Una vez que ocurre afección del SNC el tratamiento no suele ser útil, aunque puede presentarse cierta mejoría y detenerse la progresión de la enfermedad.
AFECCION INMUNOPROLIFERATIVA DEL INTESTINO DELGADO
La afección inmunoproliferativa del intestino delgado (AIPID), conocida antes como linfoma intestinal primario, es una condición rara en el mundo desarrollado en la que la lámina propia del intestino delgado se infiltra en forma intensa con linfocitos y los enterocitos suprayacentes son morfológicamente normales [ver figura 5]. En series de pacientes chinos, seis de 45 pacientes con linfoma intestinal tenían este padecimiento.35 Los pacientes presentaban malabsorción severa. Entre los pacientes sin AIPID, el 65 porciento tenía síntomas de dolor abdominal, pérdida de peso, una masa abdominal, obstrucción y perforación. La AIPID se asoció con enfermedad de cadenas pesadas alfa (de IgA), con paraproteínas en el suero, orina o líquido yeyunal. Afecta poblaciones de escasos recursos, principalmente en la segunda o tercera décadas de la vida, con predominio por el sexo masculino. Es un trastorno de las células B que afecta al tejido linfoide asociado a la mucosa (MALT). La duodenografía muestra pliegues engrosados y muchas elevaciones nodulares sin ulceración. El diagnóstico puede hacerse en la biopsia del intestino delgado en el 85 porciento de los casos.36 En las etapas tempranas de la enfermedad el proceso parece susceptible de tratamiento con antibióticos. Sin embargo, si se permite que progrese pueden desarrollarse formas más agresivas de linfoma.37
LINFANGIECTASIA INTESTINAL
La linfangiectasia intestinal suele ser un trastorno congénito en el que los linfáticos deformados impiden el transporte de quilomicrones de los enterocitos a los conductos linfáticos mesentéricos. Se adquiere un cuadro fisiopatológico semejante en ciertos casos de linfomas intestinales, enteritis granulomatosa, enteritis tuberculosa o enfermedad de Whipple, en la que se bloquea el drenaje linfático normal. Esto puede causar ascitis quilosa, quiluria o quilometrorrea en las personas afectadas.38 La enteropatía perdedora de proteínas y la linfopenia son características importantes. También existe esteatorrea moderada, y la excreción de grasa puede ser de 20 g/día. En la forma congénita de la enfermedd se observa linfedema de las piernas o de una pierna y un brazo. En la endoscopía pueden observarse vellosidades blancas, nódulos blancos y elevaciones submucosas.39 La aparicencia blanca de la mucosa es causada sin duda por el quilomicrón triacilglicerol retenido. El examen de rayos X con bario de doble contraste muestra protrusiones nodulares suaves y pliegues gruesos de la mucosa sin ulceración.40 Al examen histológico se observan linfáticos dilatados con vellosidades en forma de palillos de tambor, en ocasiones en pacientes asintomáticos, en quienes la evolución es benigna. El tratamiento se dirige a identificar algún proceso causal. En los pacientes con el trastorno congénito, en quienes no se espera mejoría de los linfáticos, suele ser útil administrar una dieta baja en grasas suplementada con triglicéridos de cadena media. Puede usarse la cirugía para eliminar áreas aisladas de disfunción linfática cuando pueden identificarse dichas áreas, o anastomosar un conducto linfático al sistema venoso. En ocasiones es útil la derivación de LeVeen.
ABETALIPOPROTEINEMIA
En la rara condición congénita de la abetalipoproteinemia, los pacientes no tienen quilomicronemia posprandial porque son incapaces de acoplar en forma adecuada la apolipoproteína B al quilomicrón en formación. A diferencia de las primeras teorías sobre la etiología de esta enfermedad, estos pacientes tienen un ARNm de apolipoproteína B que se transcribe en forma normal, a partir del cual la proteína se traduce de modo adecuado. Sin embargo, la apolipoproteína B no se secreta de la célula intestinal. El defecto en este padecimiento consiste en diversas mutaciones en el gen que codifica la proteína transportadora de triglicéridos microsomales.41 Esta proteína parecida a un chaperón, trasloca la apolipoproteína a través de la membrana del retículo endoplásmico.42 Sin este paso la apolipoproteína es degradada por las peptidasas citosólicas y microsomales. El resultado de este defecto es que tanto el intestino como el hígado son incapaces de producir y secretar sus lipoproteínas ricas en triacilglicerol, quilomicronaes y lipoproteínas de muy baja densidad. Esto causa un plasma que es pobre en triacilglicerol y colesterol. Al examen histológico los enterocitos se observan llenos de grasa. A pesar de este fenotipo, la cantidad de esteatorrea es de solo 20 g/día. Debido a que los quilomicrones no pueden transportar la grasa fuera del enterocito, se supone, aunque no se ha comprobado, que el 80 porciento de los lípidos que se absorben lo hacen por la vena porta.43
Además de los síntomas intestinales, estos pacientes tienen problemas neurológicos severos, que pueden ser causados en parte por deficiencia de ácidos grasos esenciales y en parte por la llegada de lípidos a los nervios o por interferencia con la síntesis local de lípidos. El resultado es una condición desmielinizante que causa ataxia sensorial por pérdida de sensaciones de posición y de vibración. Los síntomas son semejantes pero menos severos que los de la ataxia de Friedreich.44 Los pacientes pueden tener debilidad muscular y movimientos atetoides. También desarrollan retinitis pigmentosa, por lo general con pérdida leve de la agudeza visual pero conservación de la visión central. Además de las alteraciones neurológicas, los pacientes tienen acantocitos en la sangre. Los acantocitos son eritrocitos espiculados cuyo promedio de vida es cercano al normal pero que se ha demostrado tienen mayor propensión a traumatismos mecánicos en las pruebas in vitro.
La abetalipoproteinemia suele descubrirse en la infancia porque los pacientes no aumentan de peso y tienen esteatorrea. En los adultos la enfermedad se detecta por la combinación de datos neurológicos y oftalmológicos, la morfología de los eritrocitos, los niveles muy bajos de lípidos en plasma y la esteatorrea moderada. En la biopsia de intestino delgado se observan enterocitos llenos de lípidos incluso después de una noche de ayuno, lo que indica que el lípido absorbido no puede salir del enterocito.45 El tratamiento debe incluir vitamina E, así como otras vitaminas liposolubles y triglicéridos de cadena media para disminuir la esteatorrea.
GASTROENTERITIS EOSINOFILICA
La gastroenteritis eosinofílica es una enfermedad rara que se caracteriza por la presencia de infiltración eosinofílica de una o más porciones del tubo digestivo, desde el esófago al colon, junto con síntomas gastrointestinales. No existe una causa identificable del infiltrado eosinofílico, como una infestación parasitaria. Aunque los eosinófilos son constituyentes normales del tubo digestivo, en estos casos son mucho más numerosos de lo normal y más invasivos. Por ejemplo, la invasión eosinofílica de las criptas en el intestino delgado es característica de esta condición. Con frecuencia se observa eosinofilia periférica, pero esto no ocurre siempre.
La gastroenteritis eosinofílica puede dividirse en dos formas básicas: una masa tumoral de eosinófilos que producen una lesión de tipo granulomatoso y una forma infiltrativa más difusa. En el primer caso las lesiones suelen observarse en la porción distal del estómago, donde producen síntomas obstructivos, o en la porción proximal del estómago, el intestino delgado o el colon. Cuando las lesiones se encuentran en el intestino delgado o el colon el padecimiento debe diferenciarse de un linfoma o una enfermedad de Crohn.46 En el caso de enfermedad difusa que afecte el intestino delgado, la infiltración puede ser mucosa, con síntomas de enteropatía perdedora de proteínas o malabsorción. Si la infiltración se localiza principalmente en las capas musculares del intestino son comunes los síntomas obstructivos. Por último, la enfermedad puede encontrarse en el área subserosa del intestino, con ascitis eosinofílica resultante.47
No se sabe el motivo por el que los eosinófilos se congregan en el tubo digestivo en este padecimiento, pero evidencias recientes sugieren que, una vez activados, pueden producir citocinas que perpetúan el acúmulo de más eosinófilos. Estas citocinas son la interleucina-3 (IL-3), IL-5 y factor estimulador de colonias de granulocitos y macrófagos (FEC-GM), que se han identificado en los eosinófilos de pacientes pero no en los sujetos controles con colon irritable. Se ha sugerido la producción local de estas citocinas por el hallazgo de que las concentraciones séricas de IL-5 son normales en los pacientes con gastroenteritis eosinofílica, a diferencia de los pacientes con el síndrome hipereosinofílico, que tienen niveles aumentados de IL-5 en sangre.48
ENFERMEDAD DE CROHN
La enfermedad de Crohn, una enfermedad estenosante y fistulosa del intestino, puede alterar la absorción intestinal por dos mecanismos, disfunción ileal y síndrome de estasis [ver adelante, Síndrome de estasis (proliferación bacteriana)]. En el caso de la resección ileal o de afección ileal severa en la enfermedad de Crohn, el íleo no puede absorber sales biliares en forma normal. En este caso ocurre deficiencia posprandial de sales biliares en la porción superior del intestino, que se vuelve más severa mientras más tarde en el día se ingiera el alimento.49 Ocurre deficiencia posprandial de sales biliares a pesar de la respuesta del hígado de incrementar la síntesis de ácidos biliares ante la pérdida de ácidos biliares por la circulación enterohepática. El aumento en la síntesis de sales biliares no es adecuado porque cada vez que la vesícula se contrae en respuesta a un alimento, se pierde mayor parte del reservorio de sales biliares hacia el colon50 si se ha resecado una cantidad importante de íleon. Por lo tanto, el hígado no tiene tiempo para generar sales biliares suficientes para la absorción completa del alimento recién ingerido o el siguiente. A esto se ha denominado enteropatía colerética y puede ocurrir cuando se han resecado más de 30 cm de íleon terminal. El exceso de líquido en el colon es causado por secreción de Cl- dependiente de AMPc, específicamente por los ácidos biliares dihidroxilados quenodesoxicolato y desoxicolato, y no el ácido cólico trihidrolado.51 La pérdida de ácidos biliares hacia el colon y de la circulación enterohepática puede no asociarse con esteatorrea o esta ser mínima.52
Sin embargo, con la resección ileal más extensa, quizá de 100 cm o más, la diarrea no es causada por los ácidos biliares, sino por los ácidos grasos no absorbidos (esteatorrea).53 Por lo tanto, la diarrea asociada a la enfermedad de Crohn puede ser causada no solo por la enfermedad activa, sino también por la resección ileal. Esto es apoyado por el hecho de que ocurre diarrea cuando el paciente come después de la cirugía, un periodo en que la actividad de la enfermedad puede ser baja por la resección de la enfermedad activa, o por el hecho de que el paciente que no tenía diarrea antes de la cirugía o esta era mínima, puede tener diarrea más importante después.
Cuando la diarrea es causada por pérdida de ácidos biliares el tratamiento consiste en colestiramina.53 Esta resina se une en forma preferente a los ácidos biliares dihidroxilados, disminuyendo su concentración acuosa y su proporción en el reservorio total de ácidos biliares. Ambos efectos son benéficos. En el caso de resecciones ileales más extensas en que la esteatorrea es más importante, la colestiramina puede incluso provocar más diarrea porque reduce la concentración de ácidos biliares en el intestino delgado cuando se toman antes de los alimentos. En este caso se usan ácidos grasos de cadena media para sustituir a los ácidos grasos de cadena larga. Los resultados de esta estrategia no siempre son tan buenos como se esperaría. Debe también evaluarse la absorción de vitamina B12 en todos los pacientes con resección ileal y si se encuentra que la absorción es anormal debe administrarse esta vitamina por vía parenteral.
Algunos pacientes con enfermedad de Crohn sufren resección intestinal extensa y, por tanto, un síndrome de intestino corto. En forma semejante, los pacientes con numerosas fístulas enteroentéricas tienen síntomas de síndrome de intestino corto porque las fístulas causan que el quimo se brinque trayectos largos del intestino delgado. Ambos grupos de pacientes deben tratarse como si tuvieran síndrome de intestino corto.
Ahora que se ha clonado el transportador de ácidos biliares dependiente de sodio en los humanos, se están descubriendo defectos congénitos que causan malabsorción de ácidos biliares y diarrea.9 Esta puede ser la causa de la malabsorción primaria de ácidos biliares.
Debido a las estenosis presentes en algunos pacientes con enfermedad de Crohn, puede desarrollarse síndrome de estasis intestinal.
SINDROME DE ESTASIS (PROLIFERACION BACTERIANA)
El síndrome de estasis (o síndrome de proliferación bacteriana) ocurre cuando existe estasis intestinal que permite que las bacterias proliferen localmente. Muchos padecimientos pueden originar esta situación, destacando la diabetes, la esclerodermia, la diverticulosis intestinal, el asa aferente de la gastroyeyunostomía y la obstrucción intestinal causada por estenosis, adherencias o cáncer. Estas enfermedades pueden existir por años antes del desarrollo de los síntomas. Los síntomas pueden aparecer en un paciente por lo demás estable debido a la administración de un inhibidor de la bomba de protones o un opiáceo que reduce la motilidad intestinal aún más. Los síntomas son semejantes a los de otros estados de malabsorción e incluyen esteatorrea y anemia, El paciente puede tener deficiencia de vitamina B12 por varias causas, incluyendo unión de la vitamina a las bacterias54 y metabolismo bacteriano de la vitamina a metabolitos ineficaces. Los niveles de ácido fólico suelen ser altos debido a la producción bacteriana de folato.54 Los niveles de albúmina pueden ser bajos por la enteropatía perdedora de proteínas y siguen bajos a pesar de un tratamiento adecuado. El diagnóstico suele hacerse en un paciente con malabsorción y las características clínicas sugestivas. La diverticulosis intestinal (casi siempre yeyunal) puede no sospecharse hasta que se realizan radiografías del intestino delgado.
La disfunción intestinal en el síndrome de estasis parece causada por las glucosidasas bacterianas, que hidrolizan las moléculas de carbohidrato que forman la glucosilación extensa de las proteínas del borde en cepillo apical.56 Aunque en estas condiciones se observa deconjugación de los ácidos biliares, que en teoría puede causar menor solubilización de los productos de la hidrólisis de triglicéridos, los estudios han demostrado que, en realidad, la concentración de ácidos grasos en la fase acuosa del contenido intestinal posprandial es normal.57 Sin embargo, las micrografías electrónicas muestran que existe daño a los enterocitos debido a que los lípidos absorbidos y colectados en el retículo endoplásmico no progresan en forma normal hacia el aparato de Golgi.57
No es fácil establecer el diagnóstico de síndrome de estasis intestinal. La forma más exacta de hacerlo es pasar una sonda hacia el intestino y realizar cultivos cuantitativos del líquido en medio aeróbico y anaeróbico. En la mayoría de los casos se encontrarán más de 105 anaerobios. En forma alternativa, puede usarse la prueba no invasiva de hidrógeno en el aliento. Puede usarse como dato positivo el nivel de hidrógeno en reposo alto o el aumento rápido en el mismo en respuesta a un sustrato fermentable, como glucosa o lactulosa. Otra prueba en el aliento es la prueba de administración de 1 g de (14C)-D-xilosa, en la que se mide el 14CO2 del aliento.
El tratamiento se dirige a la corrección quirúrgica de los defectos, como un asa aferente que acumule bacterias o una fístula yeyunocólica. Si no existe esta opción, pueden emplearse dosis recurrentes de antibióticos. La tetraciclina da buenos resltados, 1 a 2 g/día durante 7 a 10 días u otro antibiótico que sea activo contra bacterias anaeróbicas. El paciente puede requerir varios tratamientos si los síntomas clínicos recurren.
AMILOIDOSIS
Con frecuencia se afecta el intestino en los pacientes con amiloidosis sistémica, en especial si tienen polineuropatía. En los pacientes mayores de 85 años el 36 porciento tiene afección intestinal con amiloide,58 aunque la mayoría son asintomáticos. Desde el punto de vista endoscópico puede observarse erosión de la mucosa, friabilidad o protrusiones polipoides.59 El diagnóstico se hace por biopsia intestinal de espesor completo o perioral. Si se realiza una biopsia perioral, ésta debe ser suficientemente profunda para que existan arterias visibles, de modo que pueda demostrarse el amiloide. Las arteriolas teñidas con rojo Congo que se vuelve verde manzana bajo luz polarizada confirman el diagnóstico. Las radiografías del intestino delgado pueden demostrar plicas intestinales edematosas, quizá con asas separadas de intestino. Si existe esteatorrea, ésta puede ser resultado de proliferación bacteriana causada por dismotilidad intestinal o menor absorción de ácidos biliares.60 No existe un tratamiento específico eficaz. Cuando existe proliferación bacteriana deben administrarse antibióticos apropiados.
MASTOCITOSIS SISTEMICA
En esta rara condición la piel (99 porciento de los casos), los huesos (9 porciento), el hígado (12 porciento), el bazo (11 porciento), los ganglios linfáticos y el tracto digestivo se afectan por la proliferación de células cebadas. Pueden observarse diarrea, dolor abdominal o ambos (23 porciento de los casos), úlcera péptica (4 porciento), prurito y rubor (36 porciento). En el 12 porciento de los pacientes existe cefalea, fatiga y malestar. Puede haber también disfunción cognitiva y se observa eosinofilia en el 12 a 50 porciento de los casos.61 Muchas de estas manifestaciones de la enfermedad son secundarias a histamina, que es liberada de las células cebadas. Esta liberación puede ser precipitada por alcohol, aspirina, narcóticos y antinflamatorios no esteroides, causando alteraciones episódicas de rubor, diarrea, dolor abdominal e hipotensión que puede progresar al síncope.61
El exceso de histamina (H2) se excreta en la orina en alrededor del 75 porciento de los pacientes, lo que le hace útil como prueba diagnóstica.61 La excreción urinaria de un metabolito de la prostaglandina D2 puede ser una prueba incluso mejor.62 Los estudios de radiografías del intestino delgado pueden mostrar pliegues engrosados o nodulaciones. Estos datos no son diagnósticos, pero indican un intestino delgado afectado.
La sobreproducción de ácido gástrico mediada por histamina puede causar úlcera péptica. En este caso, los bloqueadores H2 y los inhibidores de la bomba de protones son eficaces para controlar lo síntomas. En la piel la urticaria pigmentosa puede tratarse en forma eficaz con antagonistas de los receptores. Si persiste la diarrea puede administrarse cromolin sódico en dosis de 100 mg por vía oral cuatro veces por día.
Infestaciones parasitarias
Las infecciones por uncinarias y G. lamblia pueden causar malabsorción leve que rara vez tiene importancia clínica. La erradicación de estos parásitos cura el defecto de absorción. Estos padecimientos se analizan mejor en otros capítulos de la obra.
Pancreatitis crónica con insuficiencia exócrina
La malabsorción causada por pancreatitis se analiza en otro capítulo.
Defectos combinados o múltiples en la digestión y la absorción
ESTEATORREA POSGASTRECTOMIA
Una de las consecuencias de la cirugía gástrica es la esteatorrea. Esto es especialmente cierto en los pacientes con resección gástrica de tipo Billroth II con una gastroyeyunostomía. En esta operación el antro y una porción variable del cuerpo del estómago son resecados, el estómago se cierra y se crea una gastroyeyunostomía. Por lo tanto, el alimento brinca el duodeno y la porción más proximal del yeyuno, el sitio de máxima concentración de colecistoquinina y secretina, conjugación de folato y sitios activos de absorción de calcio y hierro. Alrededor de la mitad de esta población tiene esteatorrea, con 10 a 15 g de grasa/día. Esta parece deberse a que el alimento entra al yeyuno sin que los sitios sensibles a hormonas del duodeno reciban las señales apropiadas para su liberación. Por lo tanto, no ocurre la mezcla óptima del quimo con las enzimas pancreáticas y los ácidos biliares. El asa aferente, que drena el duodeno y el yeyuno proximal puede bloquearse y acumular bacterias como consecuencia, lo que provoca un síndrome de estasis. Debido a lo pequeño del estómago, estos pacientes no pueden tomar una comida tan grande como antes. Asociado a la esteatorrea, muchos pacientes se estabilizan a un menor peso del que tenían antes de la cirugía. También pueden encontrarse osteopenia y anemia por deficiencia de hierro. Las pérdidas constantes de pequeñas cantidades de sangre por la ostomía gástrica contribuyen al estado de deficiencia de hierro, que es la forma más común de anemia. También se encuentra deficiencia de folato secundaria a la incapacidad para generar una forma absorbible de folato (monoglutamil folato) a partir del folato heptaglutamil no absorbible (la forma más común en la dieta).63 Con menos frecuencia se observa deficiencia de vitamina B12 causada por hipoclorhidria y resección de las células parietales gástricas secretoras de factor intrínseco. No suele requerirse tratamiento para la esteatorrea porque esta no es clínicamente significativa. El hierro, el calcio o la vitamina B12 y el ácido fólico se sustituyen según esté indicado. Si el paciente tiene saciedad temprana pueden ser adecuado administrar varias comidas pequeñas al día.
Después de la cirugía pueden desarrollarse en algunos pacientes síntomas de ESG.64 Es posible que estos pacientes tengan ESG clínicamente silenciosa antes de la cirugía. La cirugía en sí causa esteatorrea moderada (10 a 15 g de grasa/día) en el 50 porciento de los casos, incluso en pacientes con intestino por lo demás normal. Sin embargo, en el paciente con ESG compensada, la cirugía es suficiente para causar sintomatología clínica. Por lo tanto, si los pacientes posgastrectomía muestran esteatorrea excesiva, se justifica una evaluación para determinar si existe ESG. La enfermedad inflamatoria del intestino que se desarrolla en los pacientes después de la gastrectomía puede ser un indicador de la presencia de ESG previamente silente.65
DIABETES MELLITUS Y MALABSORCION
La diarrea, una compliación común de la diabetes, tiene múltiples causas66 y puede provocar malabsorción . Las causas más comunes son proliferación bacteriana causada por la disfunción autonómica presente en esta condición con estasis intestinal y la enteropatía sensible al gluten. Con el uso de anticuerpos antiendomisio como escrutinio, se encontró enteropatía sensible al gluten en tres de 47 pacientes diabéticos (6 porciento), una incidencia mucho mayor de la esperada en la población general.67
Reconocimientos
Figura 1 Janet Betries.
Figura 2 Dana Burns-Pizer.
Bibliografía
DR. CHARLES M. MANSBACH II.
Definición
Típicamente malabsorción significa alteración en la absorción de grasa (esteatorrea), porque la medición de la absorción de grasa ha sido y aún es el mejor indicador de la normalidad del proceso global de absorción de nutrientes. Sin embargo, en ciertas condiciones la absorción de grasa puede ser normal y se absorben mal otras sustancias específicas, como hierro, calcio, sales biliares o, en algunos padecimientos hereditarios, aminoácidos específicos, disacáridos o monosacáridos.
Etiología
Al considerar las causas de malabsorción de grasas existen tres posibilidades generales: enfermedad del intestino delgado, afección hepática o de las vías biliares, e insuficiencia pancreática exócrina [ver tabla 1].
|
|||
|
La afección del intestino delgado puede causar la presencia de cantidades moderadas de grasa en las heces (7 a 30 g/día con una dieta de 100 g de grasa). Los pacientes con enfermedad intestinal pueden perder proteínas (enteropatía perdedora de proteínas) a través de la mucosa intestinal, lo que causa una menor concentración de albúmina en suero. También pueden existir deficiencias de vitaminas liposolubles (i.e., vitaminas A, D, E y K). Los pacientes pueden no absorber bien la vitamina B12 por afección grave del íleon terminal o por resección del mismo (por lo general más de 60 cm). También puede absorberse mal el ácido fólico y puede ocurrir hipocalcemia e hipomagnesemia.
Los pacientes con enfermedad hepática o de las vías biliares suelen tener solo pequeños incrementos de grasa en las heces (7 a 15 g/día) y malabsorción de vitaminas liposolubles.
Los pacientes con insuficiencia exócrina del páncreas pueden tener hasta 80 g de grasa/día en las heces. La grasa que pueden absorber es resultado de la acción de las lipasas gástricas.
Manifestaciones clínicas
Los síntomas de la malabsorción son diversos. En el caso más obvio el paciente refiere pérdida de peso a pesar de un buen apetito. En estos casos existe un cambio claro en la calidad de las heces y aumento en el número de evacuaciones. La consistencia del excremento disminuye y, cuando existe exceso de grasa, las heces son fétidas y son difíciles de eliminar en el inodoro. Pueden aparecer gotas de aceite o una capa lípida en el agua. El exceso de gas en las heces causa que estas floten.1
Dependiendo de los constituyentes de la dieta que no se absorban, los pacientes pueden sufrir distensión abdominal, borgborismos, cólicos abdominales (intolerancia a la lactosa), equímosis fáciles (deficiencia de vitamina K), osteopenia o tetania (defciencia de vitamina D y malabsorción de calcio), deficiencia de hierro o ceguera nocturna (deficiencia de vitamina A). Los casos más complejos son aquellos en los que no se piensa en malabsorción por falta de cambio en la calidad de las heces.
La diarrea de la malabsorción se clasifica como osmótica y suele desaparecer durante el ayuno. En la malabsorción de grasas la diarrea es causada no solo por las partículas osmóticamente activas excesivas, sino también por los ácidos grasos, que estimulan la secreción de Cl- dependiente de monofosfato cíclico de adenosina (AMPc).
Varios datos físicos específicos de algunas enfermedades pueden acompañar al estado de malabsorción y ayudar en el diagnóstico. Por ejemplo, pueden existir cambios cutáneos de esclerodermia o dermatitis herpetiforme, así como signos de neuropatía diabética. Aunque la tirotoxicosis puede asociarse con exceso de grasa en el excremento, los pacientes con tirotoxicosis suelen comer en forma exagerada pero absorben un porcentaje normal de la grasa de la dieta (95 porciento) por lo que en realidad no sufren malabsorción.
Pruebas para investigar malabsorción
Las pruebas para investigar malabsorción incluyen la determinación de si existe un exceso de grasa en las heces [ver tabla 2]. La proteína es producida en grandes cantidades por el tubo digestivo, en especial el páncreas, lo que hace que la creatorrea sea difícil de interpretar. Los carbohidratos mal absorbidos que llegan al colon pueden ser metabolizados por las bacterias colónicas a ácidos grasos de cadena corta, que en parte se absorben en el colon. Esto hace que la medición cuantitativa de la absorción de carbohidratos sea poco exacta, aunque ocurre reducción en el pH de las heces, lo que indica la excreción de una cantidad excesiva de ácidos grasos de cadena corta.
|
|||||||||||||||||||||
|
MEDICION DE LA GRASA FECAL
La grasa fecal puede medirse en forma cualitativa y cuantitativa. La medición cualitativa de la grasa fecal usando la tinción Sudán II ha demostrado ser muy exacta,2 en especial si se excretan cantidades significativas de grasa. Como con muchas pruebas cualitativas, la destreza del observador es importante para el éxito de la prueba.
La medición cuantitativa de la grasa fecal es el parámetro con el que se comparan todas las otras pruebas. Es importante recordar que la prueba no puede realizarse a menos que el paciente sea capaz de ingerir por lo menos 80 g, y de preferencia 100 g, de grasa por día.
PRUEBA DE ABSORCION DE XILOSA
La capacidad del paciente para absorber el azúcar xilosa evalúa el área de absorción del intestino. A diferencia de la glucosa, la xilosa no se absorbe en forma activa por el intestino, sino por un proceso lento de difusión pasiva. Se administran 25 g por vía oral de xilosa y se recolecta la orina durante 5 horas. La excreción normal en orina es mayor de 4 g de xilosa en 5 horas. Para asegurar un flujo urinario adecuado el paciente debe beber 500 ml de agua después de la xilosa. Esta ingesta debe producir un volumen urinario de por lo menos 300 ml durante el periodo de recolección. Deben excretarse más de 4 g de xilosa. La excreción de xilosa puede ser baja en forma falsa en pacientes con menor función renal o con ascitis, ya que la xilosa se diluye en el líquido de ascitis. Para evitar este error se aconseja medir la concentración de xilosa en sangre. La xilosa no absorbida alcanza el colon y puede ser metabolizada por las bacterias locales a hidrógeno. El hidrógeno puede cuantificarse en la respiración, y se ha reportado que esta prueba es tan exacta como la medición de xilosa en el suero o la orina.
ESTUDIOS DE IMAGEN
La placa simple o el ultrasonido abdominal no suelen ser útiles en la mayoría de los casos de malabsorción. Sin embargo, el 30 porciento de los casos de pancreatitis crónica tienen calcificaciones visibles en la placa simple de abdomen. La detección de calcificaciones puede aumentar si se usa tomografía computada o ultrasonido. La TC o el ultrasonido pueden identificar conductos pancreáticos dilatados, otro signo característico de la pancreatitis crónica. La pancreatografía endoscópica retrógrada (PER) puede también ser útil cuando se observan cambios en los conductos indicativos de pancreatitis crónica.
La placa simple del intestino delgado puede ayudar en el diagnóstico de varias alteraciones. La presencia de divertículos del intestino delgado, de alteración en la peristalsis, como se observa en la esclerodermia, o de dismotilidad intestinal idiopática, puede apoyar la sospecha de proliferación bacteriana. El examen cuidadoso del íleon terminal puede identificar enfermedad de Crohn. Pueden identificarse estenosis en algunos pacientes con lesión por radiación o lesión causada por antinflamatorios no esteroides. La hipoalbuminemia que afecta al intestino delgado puede originar el llamado signo de pila de monedas.
BIOPSIA DEL INTESTINO DELGADO
Un patólogo experto puede ayudar en el diagnóstico de la enteropatía sensible al gluten (con o sin dermatitis herpetiforme), esprue hipogamaglobulinémico, esprue tropical, enfermedad de Whipple, enfermedad por complejo Mycobacterium avium, síndrome de estasis, amiloidosis y linfangiectasia intestinal.
EVALUACION DE LA FUNCION EXOCRINA DEL PANCREAS
Es necesario que se destruya más del 90 porciento de la función exócrina del páncreas antes de que ocurra malabsorción.3 La prueba más sensible requiere el paso de una sonda de doble luz.4 Se administra secretina o colecistoquinina (CKK) por vía intravenosa y se recolectan en forma separada las secreciones gástrica y duodenal. Cuando se administra la secretina se miden el volumen del líquido duodenal y la concentración de bicarbonato. Si se administra CKK se determina la actividad de la lipasa o la tripsina usando sustratos apropiados.
La prueba no invasiva de bentiromida se basa en la acción de la tripsina sobre la bentiromida para originar ácido aminobenzoico (PABA) y benzoil-tirosina [ver figura 1]. El PABA se absorbe con facilidad por el intestino y se excreta en la orina. En las personas sanas, cuando se ingieren 500 mg de bentiromida, el 57 porciento o más del PABA aparece en la orina en 6 horas. En los pacientes con pancreatitis crónica la cantidad de PABA excretado es significativamente menor, en promedio de 42 porciento. Usando la excreción de 57 porciento como límite, la sensibilidad es del 67 al 80 porciento y la especificidad es del 95%.5 El PABA puede también medirse en el plasma 120 minutos después de la ingestión de bentiromida, lo que aumenta la sensibilidad de la prueba.6 Las mediciones en plasma son útiles en los casos de menor excreción renal, que pueden ocurrir en ancianos. El PABA se identifica por colorimetría y, por lo tanto, otras arilaminas pueden interferir con su determinación (v.gr., acetaminofén, lidocaína, procainamida, sulfonamidas y diuréticos tiacídicos).7 En los casos en los que está alterada la absorción intestinal, como en el esprue, la absorción de PABA liberado puede disminuir, causando una recuperación en orina falsamente baja. Por desgracia, la prueba de bentiromida se vuelve positiva solo cuando la glándula está destruida en más del 90 porciento. De cualquier modo, al considerar el estudio de un paciente con esteatorrea la prueba de bentiromida puede ser útil porque se requiere una destrucción semejante de la glándula para provocar esteatorrea.
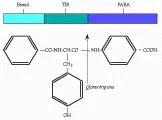
|
| Figura 1 |
| Bioquímica de la quimotripsina |
Aunque la gran mayoría de las proteasas y lipasas pancreáticas se almacenan en gránulos de cimógeno y son liberadas de la porción apical de la célula pancreática exócrina hacia el conducto pancreático, un pequeño porcentaje se fuga al intersticio de la glándula, es transportado en la circulación y puede ser medido (i.e., por un ensayo de tripsinógeno en suero). Debido a que el péptido activador de tripsina aún no es liberado y porque cualquier cantidad de tripsina activa se une con rapidez a la alfa1-antitripsina, la forma circulante libre de la tripsina es el tripsinógeno. En los pacientes con pancreatitis crónica e insuficiencia pancreática la concentración sérica de tripsinógeno es menor que en personas sanas (2 a 18 ng/ml, comparado con 29 a 79 ng/ml en gente sana).8 El nivel bajo de tripsinógeno en suero parece tener un mayor grado de especificidad para pancreattis crónica, aunque muy modesta sensibilidad.
PRUEBAS DE ABSORCION DE ACIDOS BILIARES
Los ácidos biliares se sintetizan a partir del colesterol en el hígado y requieren conjugarse con glicina o taurina antes de ser excretados hacia el intestino a través del colédoco. Los ácidos biliares conjugados solubilizan los productos de la hidrólisis de triacilgliceroles en micelas complejas, lo que facilita la absorción rápida de los lípidos de la dieta. Los ácidos biliares no se absorben en el intestino proximal con los lípidos de la dieta sino en el íleon distal. El reservorio de ácidos biliares recircula seis veces al día. Alrededor del 95 porciento de los ácidos biliares se reabsorben y recirculan en la circulación enterohepática cada día, y aparecen en las heces aproximadamente 0.5 g de ácidos biliares, lo que equivale a la tasa de síntesis hepática en condiciones estables. Si los ácidos biliares no se absorben en forma adecuada ocurre diarrea (enteropatía colerética). En ausencia completa de sales biliares los ácidos grasos se absorben en forma menos eficaz y aparecen hasta el 25 a 50 porciento de los lípidos ingeridos en el excremento. En los pacientes con diarrea idiopática o con diarrea después de resección ileal (> 30 cm), la malabsorción de sales biliares es una posibilidad etiológica. También los niños que tienen diarrea inexplicable pueden tener un defecto congénito del trasporte de ácidos biliares dependiente de sodio en el íleon terminal.9
Para probar la presencia de malabsorción de sales biliares existen dos métodos, no disponibles en todos los sitios. El primero es la prueba respiratoria con ácido 14C-glicocólico y la segunda es la pureba de absorción de ácido homocólico-taurina marcado con selenio 75 (75SeHCAT). En la primera [ver figura 2], se administra por vía oral una cantidad determinada de ácido 14C-glicocólico. Muchas bacterias son capaces de hidrolizar la unión amida y liberar la 14C-glicina; ésta es absorbida y se produce 14CO2 en el hígado o es metabolizada en la luz intestinal a 14CO2. En cualquier caso, aparece 14CO2 en cantidades mesurables en el aliento. El porcentaje de la dosis ingerida que se excreta en la respiración aumenta si la luz intestinal contiene más bacterias de lo normal o si llega un exceso de sales biliares al colon. Incluso un medicamento antisecretor gástrico puede aumentar la población de bacterias en el intestino a un nivel que cause una prueba del aliento anormal,10 como en el caso de la estasis intestinal. La utilidad de esta prueba es como indicador de malabsorción de ácidos biliares, por lo que es limitada. La prueba 75SeHCAT tiene mayor utilidad clínica por su estrecha correlación con la excreción de colato y la facilidad de medición de la retención de 75Se por la cámara gama de cuerpo total. Las personas normales retienen más del 19 porciento de una dosis administrada por vía oral de 75Se después de 7 días, mientras que los pacientes con disfunción significativa o resección de íleon retienen menos del 12 porciento.11
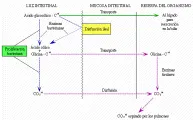
|
| Figura 2 |
| Prueba del aliento para ácidos biliares |
Enfermedades del intestino delgado que producen malabsorción
ENTEROPATIA SENSIBLE AL GLUTEN
La enteropatía sensible al gluten (ESG) se denominó alguna vez enfermedad celiaca en niños y esprue idiopático o no tropical en adultos. En 1960 se reconoció que estas enfermedades eran la misma, estaban causadas por la principal proteína del trigo, el gluten, y más específicamente, por su componente soluble en alcohol, la gliadina.12
Factores genéticos y etiológicos
La ESG es menos común en los Estados Unidos que en Europa occidental. Se asocia con los haplotipos DqA1*0501, DqB1*0201, HLA-B8 y DRw3. En grupos de gemelos monocigotos, la mayoría de los gemelos opuestos al caso índice no tienen ESG. Esto ha apoyado que existe otro factor desconocido (no genético) que es importante como causa de la enfermedad.
Patogenia
Las causas de esteatorrea en la ESG son muchas. Las células que producen CCK están disminuidas en número o son tan defectuosas que la cantidad de CCK presente en la mucosa duodenal está muy reducida.12 Esto causa una menor cantidad de lipasa pancreática y ácidos biliares que llegan a la luz intestinal en respuesta a los lípidos de la dieta. Las células de las criptas intestinales son las principales células secretoras de fluído del intestino, por secreción de Cl- dependiente de AMPc con abundante agua asociada. En la ESG la porción de la cripta del complejo velloso está muy expandido, causando mayor secreción de agua. Debido a que las células de la punta de la vellosidad, que normalmente absorben el agua, están dañadas y disminuidas en número, la absorción de agua y electrolitos no es tan eficaz como la normal y el intestino se vuelve secretor.12 Esto reduce aún más la concentración de ácidos biliares en la luz intestinal, más de lo esperado solo por la menor liberación de CCK. La capacidad de los ácidos biliares para solubilizar los productos de la lipólisis depende de la presencia de sales biliares a una concentracón mayor de la concentración micelar crítica (CMC) de 1.4 mM.13 En condiciones normales, el intestino tiene una concentración posprandial de sales biliares de 10 mM.14 Los bordes en cepillo en la superficie de los enterocitos maduros se afectan mucho en la ESG. Además, las estructuras vellosas se aplanan. Estas dos condiciones causan reducción importante de la superficie en la que se absorben los lípidos. La severidad de la reducción del área de absorción puede calcularse por la prueba de absorción de D-xilosa. Los enterocitos en la superficie del intestino no son tan maduros como los enterocitos normales porque su frecuencia de recambio está muy aumentada. Es probable que esto reduzca la capacidad para procesar los lípidos en forma normal.
Diagnóstico
Manifestaciones clínicas Aunque la ESG puede iniciar en la infancia y responder a la supresión de gluten, los niños con la enfermedad sufren remisión en la adolescencia, incluso si ingieren una dieta que contenga gluten. Como adultos, estos pacientes, 25 o más de los cuales tuvieron síntomas en la infancia, pueden presentar molestias diversas, por lo común pérdida de peso, fatiga, cólicos abdominales, distensión, meteorismo y diarrea (esteatorrea), aunque puede no haber pérdida del apetito. En algunos la enfermedad tiene inicio insidioso y los síntomas son leves. Es solo después de que son tratados que estos pacientes se dan cuenta, en retrospectiva, lo enfermos que estaban. En estudios de población en los cuales la presencia de la enfermedad se estableció por biopsia intestinal, las personas con trastornos que justificaron la biopsia con frecuencia estaban asintomáticos, pero tenían menor estatura que los hermanos no afectados. Debido a que no existe ningún dato específico para el diagnóstico, en especial si no existe esteatorrea clínicamente evidente, el diagnóstico de la ESG puede retrasarse. Esto es especialmente cierto en pacientes que no tienen esteatorrea pero sí osteoporosis, equímosis fáciles como resultado de la deficiencia de vitamina K o anemia por deficiencia de hierro inexplicable.
Pruebas de laboratorio El diagnóstico depende de los datos en la biopsia del intestino delgado [ver figura 3]. Los datos clásicos incluyen atrofia vellosa parcial o completa, enterocitos de apariencia anormal en las puntas de las vellosidades, aumento de los linfocitos intraepiteliales, un infiltrado en la lámina propia que consiste principalmente de linfocitos y macrófagos, aumento en el tamaño de las criptas tanto vertical como horizontal, y aumento en el número de figuras mitóticas.12 Estas características, aunque típicas, no son patognomónicas. Para que el diagnóstico sea definitivo el paciente debe responder al tratamiento dietético. Puede esperarse mejoría sintomática en el 80 porciento de los pacientes en 1 mes, pero la mejoría histológica es mucho más lenta. Un 10 porciento de los pacientes no responde sino hasta los 2 meses, y el resto puede tardar hasta 2 años. Incluso bajo control dietético estricto, la biopsia puede no normalizarse. Lo más frecuente es que el paciente permanezca bien controlado mientras reciba la dieta, pero muchos pacientes rompen la dieta por situaciones diversas o creyendo que están curados. Esto causa en forma inevitable recurrencia de los síntomas, lo que confirma el diagnóstico.
|
|
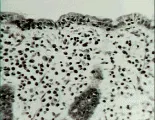
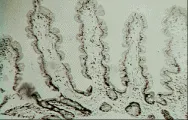
Tratamiento
El tratamiento de la ESG consiste en una dieta estricta de eliminación de gluten, no trigo, centeno o cebada. La avena parece ser segura, pero suele evitarse durante la fase temprana, cuando se está evaluando la respuesta a la dieta. En ocasiones es difícil seguir la dieta porque muchos alimentos tienen un contenido oculto de gluten. Es importante la dieta con eliminación con gluten porque los pacientes que no reciben tratamiento tienen mayor posibilidad de desarrollar linfoma intestinal.16 Pueden ser útiles al respecto los grupos de apoyo, como el organizado por la Sociedad Celiaca en los Estados Unidos, en especial si la enfermedad es de diagnóstico reciente. La información sobre qué buscar en las etiquetas y recetas interesantes puede ser muy útil para mantener al paciente en el regimen dietético. Durante el periodo de prueba, incluso la cerveza, la ale y el whisky pueden contener suficiente gluten para sensibilizar al paciente, por lo que no deben consumirse. Después de evaluar la respuesta a la dieta pueden probarse estas bebidas, si se desea, para determinar si el paciente es sensible. Otro productos que no suele pensarse que contienen gluten pero así es incluyen los helados, las hostias de comulgar e incluso algunos medicamentos (como excipiente). A pesar de las restricciones, existen muchas opciones dietéticas para el paciente, incluyendo ciertos cereales, leche, queso, huevos, carne, pollo, pescado, chocolate y productos hechos de maíz, arroz o harina de papa.
Si el paciente no responde a la dieta, la causa más probable es que la dieta no ha sido suficientemente estricta. En este caso es útil que un dietista revise la historia dietética del paciente.12 Con menos frecuencia el paciente tendrá un síndrome de estasis o insuficiencia pancreática. Cuando se diagnostican estos problemas y se tratan con éxito, el paciente presenta buena respuesta a la dieta.
OTROS TRASTORNOS SEMEJANTES AL ESPRUE
Dermatitis herpetiforme y ESG
Muchos pacientes con dermatitis herpetiforme tienen ESG.17 En las rodillas, codos, hombros y glúteos aparecen lesiones ampollosas muy pruriginosas. La biopsia de piel en la dermatitis herpetiforme característicamente muestra depósitos de inmunoglobulina A (IgA). Al establecer la dieta sin gluten mejoran las lesiones tanto dermatológicas como intestinales, lo que indica una relación entre ambas. Sin embargo, las lesiones de la piel responden al tratamiento con dapsona y las intestinales no, lo que sugiere que también existen diferencias entre las dos.
Esprue tropical
El esprue tropical es una enfermedad de malabsorción que aparece en ciertas áreas del mundo, en especial los trópicos, entre la pbolación local y los turistas. Al considerar esta enfermedad, debe notarse que las biopsias intestinales obtenidas de residentes normales de estas regiones o de turistas que viven fuera de los grandes hoteles se clasificarían como anormales si se obtuvieran de personas residentes en los Estados Unidos o Europa. En estas pbolaciones la arquitectura vellosa puede ser más irregular, en la que varias vellosidades se fusionan en forma horizontal, mientras que otras parecen hojas o se aplanan y su lámina propia está infiltrada por células inflamatorias [ver figura 4], si se compara con las delgadas estructuras vellosas que se observan en los norteamericanos [ver figura 3b]. En dos poblaciones estudiadas con gran cuidado, el 5 a 13 porciento de los norteamericanos que vivieron en Puerto Rico durante 6 meses o más desarrollaron síntomas de esprue tropical. Los expatriados que han regresado a los Estados Unidos en forma permanente pueden desarrollar síntomas de esprue tropical incluso 10 años después de vivir ya en ese país.18 Los voluntarios de los Cuerpos de Paz enviados a Pakistan tienen lesiones demostrables en el intestino delgado y alteraciones funcionales que se normalizaron varios meses después de su retorno a los Estados Unidos.19 Los hindúes y pakistaníes que viven en los Estados Unidos pueden tardar más tiempo (hasta 4 años) en excretar cantidades normales de xilosa.20 No se conoce la causa exacta de estos cambios en el intestino delgado, pero se piensa que el síndrome de esprue tropical es ocasionado por una o más especies de bacterias coliformes, como especies de Klebsiella,21 que colonizan el tracto intestinal superior.
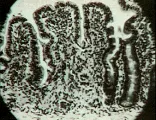
|
| Figura 4 |
| Esprue tropical: biopsia |
Los síntomas del esprue tropical difieren de los de la ESG. La pérdida de peso causada principalmente por anorexia es notable, lo mismo que la diarrea. La lengua escaldada (70 porciento), el edema en pies (25 porciento) y la deficiencia de folato y vitamina B12 (75 a 100 porciento) son mucho más comunes en el esprue tropical que en el no tropical.21 Los síntomas pueden ser muy severos, causando en ocasiones la muerte en áreas endémicas. Esto fue especialmente cierto en prisioneros de guerra alemanes e italianos apresados en la India durante la Segunda Guerra Mundial. Sin embargo, en la actualidad el pronóstico suele ser excelente para los pacientes que permanecen en los trópicos o regresan a los Estados Unidos. El tratamiento se inicia con ácido fólico (5 mg/día).21 Esto se asocia con rápida mejoría en el apetito y elimina la mayoría de los síntomas clínicos. En los pacientes con duración breve de los síntomas es suficiente con administrar el folato durante 6 meses a 1 año. Para los que tienen síntomas de mayor duración (más de 4 meses), deben agregarse antibióticos, como tetraciclinas (2 g/día por 1 año). La mayoría de los pacientes que regresan a los Estados Unidos recuperan peso con rapidez, incluso si los resultados de las pruebas de absorción o la biopsia intestinal no se normalizan.
Esprue colagenoso
El esprue colagenoso es una enfermedad rara y devastadora en la que existe una capa de colágena por debajo de los enterocitos del intestino delgado. Su relación con la colitis colagenosa no es clara, pero la característica histológica básica de depósito subepitelial de colágena es la misma. En el colon se deposita colágena tipo 6, pero el tipo de colágena depositada en el intestino delgado se desconoce. En el colon los síntomas suelen ser leves, pero el el intestino delgado son más severos. Esto puede ser porque la colágena constituye una barrera a la difusión, lo que impide que los nutrientes pasen hacia los capilares porta o los linfáticos. El diagnóstico se realiza por el cuadro histológico clásico de atrofia vellosa y depósito subepitelial de colágena. Sin embargo, si el diagnóstico pasa desapercibido y se piensa que el paciente tiene ESG por el aplanamiento de las estructuras vellosas, el paciente suele no responder a la dieta con eliminación de gluten. En estos casos el diagnóstico real del paciente se aclarará si la biopsia se envía a un patólogo especializado en enfermedades gastrointestinales. No existe un tratamiento específico para este padecimiento. El problema más común es la diarrea osmótica causada por el estado de malabsorción inducido por la enfermedad. En estos casos el paciente debe ser tratado como si tuviera síndrome del intestino corto. Algunos pacientes responden al tratamiento esteroideo. Pocos pacientes responden a esteroides y una dieta con eliminación de gluten, y cuando el paciente mejora suele poderse disminuir la dosis de esteroide en forma gradual.22
Esprue hipogamaglobulinémico
El tracto digestivo es el órgano linfoide más grande del organismo. El ambiente al que este sistema inmune es expuesto está lleno de antígenos extraños que deben ser enfrentados, identificados y, en caso necesario, atacados. Por lo tanto, no es sorprendente que pueda desarrollarse disfunción intestinal en los pacientes con inmunodeficiencia. Esto es especialmente cierto en el caso de la deficiencia de IgA porque esta es la inmunoglobulina más importante en el intestino. Algunos pacientes que tienen uno de los síndromes hipogamaglobulinémicos pueden tener malabsorción.23 Las biopsias de intestino no muestran células plasmáticas y son fácilmente distinguibles de las de los pacientes con enteropatía sensible al gluten, en las que estos tipos celulares son abundantes. Pueden existir también organismos Giardia lamblia. Los pacientes con deficiencia de IgA también suelen tener historia de infecciones respiratorias recurrentes22 que les distinguen de los pacientes con enteropatía sensible al gluten. La causa más común de malabsorción en este padecimiento es la giardiasis. Con frecuencia los síntomas intestinales mejoran si se administra metronidazol en dosis de 750 mg/día durante 10 días.
MALABSORCION SECUNDARIA A RESECCION MASIVA DEL INTESTINO DELGADO
La resección masiva del intestino delgado se usa para tratar varias enfermedades, incluyendo isquemia mesentérica, vólvulos y enfermedad de Crohn. Dependiendo de la cantidad de intestino resecado, los resultados pueden variar desde catastróficos a leves. La retención de la válvula ileocecal disminuye los síntomas. El íleon responde a la resección yeyunal con hiperplasia en forma mucho más eficaz de lo que el yeyuno responde a una resección ileal. Existen también funciones especializadas presentes en el íleon que no realiza el yeyuno, como el transporte de sales biliares y vitamina B12. El mantenimiento de un reservorio adecuado de ácidos biliares es importante para la absorción de grasa porque la menor superficie de absorción en estos pacientes hace necesario que la absorción de grasas sea lo más eficaz posible. En forma alternativa, el íleon puede realizar la mayoría de las funciones del yeyuno excepto por la absorción de ácido fólico, Ca2+ y Fe2+. Sin embargo, estos elementos pueden sustituirse con medicación adecuada.
Debido a que el intestino requiere de cierta área de superficie para que ocurra una absorción adecuada, la reducción del área a menos de un valor crítico suele causar malabsorción. Las manifestaciones clínicas del síndrome de intestino corto pueden incluir diarrea, esteatorrea, pérdida de peso, deficiencias de oligoelementos, hiponatremia e hipocalemia. Las proteínas requieren la mayor área de absorción.24 Por lo tanto, el tratamiento en estos pacientes depende de qué partes y cuánto del intestino se haya resecado. También se requieren vitaminas y minerales en el esquema terapéutico, dependiendo de la parte de intestino que falte. El tratamiento puede incluir varios alimentos de escasa cantidad al día, ingerir alimentos de absorción rápida, como los suplementos calóricos enlatados, picar o moler los alimentos, e ingerir triglicéridos de cadena media, que pueden absorberse en ausencia de sales biliares.24 Los alimentos ricos en ácidos grasos polinsaturados, como los aceites vegetales, se absorben con más facilidad que las carnes, que tienen grasas más saturadas. Por último, los suplementos dietéticos, como el Vivonex, son productos totalmente hidrolizados y se absorben con rapidez. Para disminuir el tránsito intestinal puede emplearse difenoxilato con atropina, loperamida o tintura de opio deodorizada. Un método alternativo consiste en hacer que el paciente beba una pequeña cantidad de aceite de girasol antes del alimento. El lípido pasa con rapidez al íleon (cuando está presente), el colon o ambos25 y elabora un péptido YY (PYY),26 que es el posible freno ileal, enlenteciendo el vaciamiento gástrico. Al probar diferentes dietas en los pacientes se podrá establecer la mejor dieta oral para ellos, intentando evitar que requieran de nutrición parenteral total (NPT), que es la alternativa menos deseable.
ENTERITIS POR RADIACION
La lesión del intestino es una secuela muy común de la administración de radioterapia como tratamiento oncológico. El sitio de lesión más común es el recto por estar fijo en su sitio y porque los tumores cervicouterinos y prostáticos son comunes y con frecuencia se tratan con radioterapia locorregional. La radiación del abdomen por otros tumores puede causar lesión del intestino delgado. La afección del intestino delgado es más común si el paciente tuvo una cirugía abdominal previa, que puede restringir el movimiento del intestino. El íleon terminal puede afectarse durante la irradiación pélvica. La lesión del intestino delgado puede causar malabsorción con esteatorrea.
ENFERMEDAD DE WHIPPLE
La enfermedad de Whipple es una enfermedad devastadora y rara causada por la bacteria Tropheryma whippelii.27 Es indispensable el diagnóstico exacto porque la mortalidad llega a ser del 100 porciento sin tratamiento.
Clásicamente, la enfermedad ocurre en un varón de edad media con artritis no deformante que suele iniciar años antes del inicio de los síntomas intestinales. Otras molestias incluyen fiebre, distensión abdominal, diarrea, pérdida de peso, linfadenopatía, hiperpigmentación de la piel y esteatorrea.28 Muchos pacientes expresan el isotipo HLA-B27. En ocasiones pueden no existir síntomas intestinales, incluso en algunos pacientes con afección del SNC.29 En un caso bien demostrado pero no habitual, no se identificó afección intestinal, incluso después de biopsias costosas en dos laboratorios, a pesar del hecho de que el paciente tenía por lo demás síntomas típicos de la enfermedad.30
El diagnóstico se basa en la identificación de macrófagos con tinción positiva ácido-Schiff (PAS), que contienen partículas falciformes.28 Con mucho, el sitio más común de biopsia positiva es el intestino. La lesión histológica muestra distensión de las vellosidades (en palillo de tambor) con macrófagos PAS positivos espumosos y dilatación linfática. En casos extremos puede observarse una superficie vellosa plana. Estos datos deben distinguirse en el contexto clínico adecuado de los de la enfermedad por complejo M. avium, en la que también se encuentran macrófagos PAS positivos. La tinción para bacilos ácido-alcohol resistentes debe distinguir entre ambos padecimientos. Puede existir afección del SNC en ausencia de síntomas neurológicos, en ocasiones asociada con macrófagos típicos y apoyada por la técnica más sensible de reacción en cadena de la polimerasa.31 En ocasiones se requiere una biopsia cerebral, que puede ser guiada por resonancia magnética. También puede ser útil la afección cardiaca y pulmonar.32
Debido a que la enfermedad es tan poco común, es difícil establecer un plan de tratamiento bien definido. El tratamiento originalmente propuesto era de penicilina y estreptomicina durante 2 semanas, seguido de tetraciclinas por 1 año. En la actualidad se administra trimetoprim-sulfametoxazol durante 1 año. Aunque los síntomas intestinales y sistémicos responden a cualquier tratamiento, el problema lo constituyen las manifestaciones del SNC. Por lo genral, en los pacientes que no tienen afección nerviosa al inicio, los síntomas a este sistema aparecen un año o más después del tratamiento de las molestias sistémicas e intestinales. Puede observarse demencia progresiva, pero los signos patognomónicos de la enfermedad neurológica, cuando existe, son miorritmia oculomasticatoria y miorritmia oculofacial-esquelética.33 Por lo tanto, se requieren antibióticos que crucen la barrera hemato-encefálica. Es interesante que la administración por un periodo corto de penicilina-estreptomicina es suficiente para bloquear los síntomas del SNC, mientras que incluso la administración prolongada de trimetoprim-sulfametoxazol puede causar en ocasiones manifestaciones neurológicas de la enfermedad de Whipple.34 La tetraciclina sola no erradica la afección neurológica y no debe administrarse, a pesar de que es eficaz para tratar los síntomas intestinales y sistémicos. Una característica importante que debe tenerse en cuenta es que en el 50 porciento de los pacientes el LCR puede contener macrófagos típicos o material RCP-positivo en ausencia de cualquier síntoma del SNC.31 Una vez que ocurre afección del SNC el tratamiento no suele ser útil, aunque puede presentarse cierta mejoría y detenerse la progresión de la enfermedad.
AFECCION INMUNOPROLIFERATIVA DEL INTESTINO DELGADO
La afección inmunoproliferativa del intestino delgado (AIPID), conocida antes como linfoma intestinal primario, es una condición rara en el mundo desarrollado en la que la lámina propia del intestino delgado se infiltra en forma intensa con linfocitos y los enterocitos suprayacentes son morfológicamente normales [ver figura 5]. En series de pacientes chinos, seis de 45 pacientes con linfoma intestinal tenían este padecimiento.35 Los pacientes presentaban malabsorción severa. Entre los pacientes sin AIPID, el 65 porciento tenía síntomas de dolor abdominal, pérdida de peso, una masa abdominal, obstrucción y perforación. La AIPID se asoció con enfermedad de cadenas pesadas alfa (de IgA), con paraproteínas en el suero, orina o líquido yeyunal. Afecta poblaciones de escasos recursos, principalmente en la segunda o tercera décadas de la vida, con predominio por el sexo masculino. Es un trastorno de las células B que afecta al tejido linfoide asociado a la mucosa (MALT). La duodenografía muestra pliegues engrosados y muchas elevaciones nodulares sin ulceración. El diagnóstico puede hacerse en la biopsia del intestino delgado en el 85 porciento de los casos.36 En las etapas tempranas de la enfermedad el proceso parece susceptible de tratamiento con antibióticos. Sin embargo, si se permite que progrese pueden desarrollarse formas más agresivas de linfoma.37
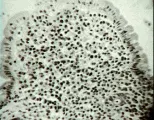
|
| Figura 5 |
| Linfoma intestinal primario: biopsia |
LINFANGIECTASIA INTESTINAL
La linfangiectasia intestinal suele ser un trastorno congénito en el que los linfáticos deformados impiden el transporte de quilomicrones de los enterocitos a los conductos linfáticos mesentéricos. Se adquiere un cuadro fisiopatológico semejante en ciertos casos de linfomas intestinales, enteritis granulomatosa, enteritis tuberculosa o enfermedad de Whipple, en la que se bloquea el drenaje linfático normal. Esto puede causar ascitis quilosa, quiluria o quilometrorrea en las personas afectadas.38 La enteropatía perdedora de proteínas y la linfopenia son características importantes. También existe esteatorrea moderada, y la excreción de grasa puede ser de 20 g/día. En la forma congénita de la enfermedd se observa linfedema de las piernas o de una pierna y un brazo. En la endoscopía pueden observarse vellosidades blancas, nódulos blancos y elevaciones submucosas.39 La aparicencia blanca de la mucosa es causada sin duda por el quilomicrón triacilglicerol retenido. El examen de rayos X con bario de doble contraste muestra protrusiones nodulares suaves y pliegues gruesos de la mucosa sin ulceración.40 Al examen histológico se observan linfáticos dilatados con vellosidades en forma de palillos de tambor, en ocasiones en pacientes asintomáticos, en quienes la evolución es benigna. El tratamiento se dirige a identificar algún proceso causal. En los pacientes con el trastorno congénito, en quienes no se espera mejoría de los linfáticos, suele ser útil administrar una dieta baja en grasas suplementada con triglicéridos de cadena media. Puede usarse la cirugía para eliminar áreas aisladas de disfunción linfática cuando pueden identificarse dichas áreas, o anastomosar un conducto linfático al sistema venoso. En ocasiones es útil la derivación de LeVeen.
ABETALIPOPROTEINEMIA
En la rara condición congénita de la abetalipoproteinemia, los pacientes no tienen quilomicronemia posprandial porque son incapaces de acoplar en forma adecuada la apolipoproteína B al quilomicrón en formación. A diferencia de las primeras teorías sobre la etiología de esta enfermedad, estos pacientes tienen un ARNm de apolipoproteína B que se transcribe en forma normal, a partir del cual la proteína se traduce de modo adecuado. Sin embargo, la apolipoproteína B no se secreta de la célula intestinal. El defecto en este padecimiento consiste en diversas mutaciones en el gen que codifica la proteína transportadora de triglicéridos microsomales.41 Esta proteína parecida a un chaperón, trasloca la apolipoproteína a través de la membrana del retículo endoplásmico.42 Sin este paso la apolipoproteína es degradada por las peptidasas citosólicas y microsomales. El resultado de este defecto es que tanto el intestino como el hígado son incapaces de producir y secretar sus lipoproteínas ricas en triacilglicerol, quilomicronaes y lipoproteínas de muy baja densidad. Esto causa un plasma que es pobre en triacilglicerol y colesterol. Al examen histológico los enterocitos se observan llenos de grasa. A pesar de este fenotipo, la cantidad de esteatorrea es de solo 20 g/día. Debido a que los quilomicrones no pueden transportar la grasa fuera del enterocito, se supone, aunque no se ha comprobado, que el 80 porciento de los lípidos que se absorben lo hacen por la vena porta.43
Además de los síntomas intestinales, estos pacientes tienen problemas neurológicos severos, que pueden ser causados en parte por deficiencia de ácidos grasos esenciales y en parte por la llegada de lípidos a los nervios o por interferencia con la síntesis local de lípidos. El resultado es una condición desmielinizante que causa ataxia sensorial por pérdida de sensaciones de posición y de vibración. Los síntomas son semejantes pero menos severos que los de la ataxia de Friedreich.44 Los pacientes pueden tener debilidad muscular y movimientos atetoides. También desarrollan retinitis pigmentosa, por lo general con pérdida leve de la agudeza visual pero conservación de la visión central. Además de las alteraciones neurológicas, los pacientes tienen acantocitos en la sangre. Los acantocitos son eritrocitos espiculados cuyo promedio de vida es cercano al normal pero que se ha demostrado tienen mayor propensión a traumatismos mecánicos en las pruebas in vitro.
La abetalipoproteinemia suele descubrirse en la infancia porque los pacientes no aumentan de peso y tienen esteatorrea. En los adultos la enfermedad se detecta por la combinación de datos neurológicos y oftalmológicos, la morfología de los eritrocitos, los niveles muy bajos de lípidos en plasma y la esteatorrea moderada. En la biopsia de intestino delgado se observan enterocitos llenos de lípidos incluso después de una noche de ayuno, lo que indica que el lípido absorbido no puede salir del enterocito.45 El tratamiento debe incluir vitamina E, así como otras vitaminas liposolubles y triglicéridos de cadena media para disminuir la esteatorrea.
GASTROENTERITIS EOSINOFILICA
La gastroenteritis eosinofílica es una enfermedad rara que se caracteriza por la presencia de infiltración eosinofílica de una o más porciones del tubo digestivo, desde el esófago al colon, junto con síntomas gastrointestinales. No existe una causa identificable del infiltrado eosinofílico, como una infestación parasitaria. Aunque los eosinófilos son constituyentes normales del tubo digestivo, en estos casos son mucho más numerosos de lo normal y más invasivos. Por ejemplo, la invasión eosinofílica de las criptas en el intestino delgado es característica de esta condición. Con frecuencia se observa eosinofilia periférica, pero esto no ocurre siempre.
La gastroenteritis eosinofílica puede dividirse en dos formas básicas: una masa tumoral de eosinófilos que producen una lesión de tipo granulomatoso y una forma infiltrativa más difusa. En el primer caso las lesiones suelen observarse en la porción distal del estómago, donde producen síntomas obstructivos, o en la porción proximal del estómago, el intestino delgado o el colon. Cuando las lesiones se encuentran en el intestino delgado o el colon el padecimiento debe diferenciarse de un linfoma o una enfermedad de Crohn.46 En el caso de enfermedad difusa que afecte el intestino delgado, la infiltración puede ser mucosa, con síntomas de enteropatía perdedora de proteínas o malabsorción. Si la infiltración se localiza principalmente en las capas musculares del intestino son comunes los síntomas obstructivos. Por último, la enfermedad puede encontrarse en el área subserosa del intestino, con ascitis eosinofílica resultante.47
No se sabe el motivo por el que los eosinófilos se congregan en el tubo digestivo en este padecimiento, pero evidencias recientes sugieren que, una vez activados, pueden producir citocinas que perpetúan el acúmulo de más eosinófilos. Estas citocinas son la interleucina-3 (IL-3), IL-5 y factor estimulador de colonias de granulocitos y macrófagos (FEC-GM), que se han identificado en los eosinófilos de pacientes pero no en los sujetos controles con colon irritable. Se ha sugerido la producción local de estas citocinas por el hallazgo de que las concentraciones séricas de IL-5 son normales en los pacientes con gastroenteritis eosinofílica, a diferencia de los pacientes con el síndrome hipereosinofílico, que tienen niveles aumentados de IL-5 en sangre.48
ENFERMEDAD DE CROHN
La enfermedad de Crohn, una enfermedad estenosante y fistulosa del intestino, puede alterar la absorción intestinal por dos mecanismos, disfunción ileal y síndrome de estasis [ver adelante, Síndrome de estasis (proliferación bacteriana)]. En el caso de la resección ileal o de afección ileal severa en la enfermedad de Crohn, el íleo no puede absorber sales biliares en forma normal. En este caso ocurre deficiencia posprandial de sales biliares en la porción superior del intestino, que se vuelve más severa mientras más tarde en el día se ingiera el alimento.49 Ocurre deficiencia posprandial de sales biliares a pesar de la respuesta del hígado de incrementar la síntesis de ácidos biliares ante la pérdida de ácidos biliares por la circulación enterohepática. El aumento en la síntesis de sales biliares no es adecuado porque cada vez que la vesícula se contrae en respuesta a un alimento, se pierde mayor parte del reservorio de sales biliares hacia el colon50 si se ha resecado una cantidad importante de íleon. Por lo tanto, el hígado no tiene tiempo para generar sales biliares suficientes para la absorción completa del alimento recién ingerido o el siguiente. A esto se ha denominado enteropatía colerética y puede ocurrir cuando se han resecado más de 30 cm de íleon terminal. El exceso de líquido en el colon es causado por secreción de Cl- dependiente de AMPc, específicamente por los ácidos biliares dihidroxilados quenodesoxicolato y desoxicolato, y no el ácido cólico trihidrolado.51 La pérdida de ácidos biliares hacia el colon y de la circulación enterohepática puede no asociarse con esteatorrea o esta ser mínima.52
Sin embargo, con la resección ileal más extensa, quizá de 100 cm o más, la diarrea no es causada por los ácidos biliares, sino por los ácidos grasos no absorbidos (esteatorrea).53 Por lo tanto, la diarrea asociada a la enfermedad de Crohn puede ser causada no solo por la enfermedad activa, sino también por la resección ileal. Esto es apoyado por el hecho de que ocurre diarrea cuando el paciente come después de la cirugía, un periodo en que la actividad de la enfermedad puede ser baja por la resección de la enfermedad activa, o por el hecho de que el paciente que no tenía diarrea antes de la cirugía o esta era mínima, puede tener diarrea más importante después.
Cuando la diarrea es causada por pérdida de ácidos biliares el tratamiento consiste en colestiramina.53 Esta resina se une en forma preferente a los ácidos biliares dihidroxilados, disminuyendo su concentración acuosa y su proporción en el reservorio total de ácidos biliares. Ambos efectos son benéficos. En el caso de resecciones ileales más extensas en que la esteatorrea es más importante, la colestiramina puede incluso provocar más diarrea porque reduce la concentración de ácidos biliares en el intestino delgado cuando se toman antes de los alimentos. En este caso se usan ácidos grasos de cadena media para sustituir a los ácidos grasos de cadena larga. Los resultados de esta estrategia no siempre son tan buenos como se esperaría. Debe también evaluarse la absorción de vitamina B12 en todos los pacientes con resección ileal y si se encuentra que la absorción es anormal debe administrarse esta vitamina por vía parenteral.
Algunos pacientes con enfermedad de Crohn sufren resección intestinal extensa y, por tanto, un síndrome de intestino corto. En forma semejante, los pacientes con numerosas fístulas enteroentéricas tienen síntomas de síndrome de intestino corto porque las fístulas causan que el quimo se brinque trayectos largos del intestino delgado. Ambos grupos de pacientes deben tratarse como si tuvieran síndrome de intestino corto.
Ahora que se ha clonado el transportador de ácidos biliares dependiente de sodio en los humanos, se están descubriendo defectos congénitos que causan malabsorción de ácidos biliares y diarrea.9 Esta puede ser la causa de la malabsorción primaria de ácidos biliares.
Debido a las estenosis presentes en algunos pacientes con enfermedad de Crohn, puede desarrollarse síndrome de estasis intestinal.
SINDROME DE ESTASIS (PROLIFERACION BACTERIANA)
El síndrome de estasis (o síndrome de proliferación bacteriana) ocurre cuando existe estasis intestinal que permite que las bacterias proliferen localmente. Muchos padecimientos pueden originar esta situación, destacando la diabetes, la esclerodermia, la diverticulosis intestinal, el asa aferente de la gastroyeyunostomía y la obstrucción intestinal causada por estenosis, adherencias o cáncer. Estas enfermedades pueden existir por años antes del desarrollo de los síntomas. Los síntomas pueden aparecer en un paciente por lo demás estable debido a la administración de un inhibidor de la bomba de protones o un opiáceo que reduce la motilidad intestinal aún más. Los síntomas son semejantes a los de otros estados de malabsorción e incluyen esteatorrea y anemia, El paciente puede tener deficiencia de vitamina B12 por varias causas, incluyendo unión de la vitamina a las bacterias54 y metabolismo bacteriano de la vitamina a metabolitos ineficaces. Los niveles de ácido fólico suelen ser altos debido a la producción bacteriana de folato.54 Los niveles de albúmina pueden ser bajos por la enteropatía perdedora de proteínas y siguen bajos a pesar de un tratamiento adecuado. El diagnóstico suele hacerse en un paciente con malabsorción y las características clínicas sugestivas. La diverticulosis intestinal (casi siempre yeyunal) puede no sospecharse hasta que se realizan radiografías del intestino delgado.
La disfunción intestinal en el síndrome de estasis parece causada por las glucosidasas bacterianas, que hidrolizan las moléculas de carbohidrato que forman la glucosilación extensa de las proteínas del borde en cepillo apical.56 Aunque en estas condiciones se observa deconjugación de los ácidos biliares, que en teoría puede causar menor solubilización de los productos de la hidrólisis de triglicéridos, los estudios han demostrado que, en realidad, la concentración de ácidos grasos en la fase acuosa del contenido intestinal posprandial es normal.57 Sin embargo, las micrografías electrónicas muestran que existe daño a los enterocitos debido a que los lípidos absorbidos y colectados en el retículo endoplásmico no progresan en forma normal hacia el aparato de Golgi.57
No es fácil establecer el diagnóstico de síndrome de estasis intestinal. La forma más exacta de hacerlo es pasar una sonda hacia el intestino y realizar cultivos cuantitativos del líquido en medio aeróbico y anaeróbico. En la mayoría de los casos se encontrarán más de 105 anaerobios. En forma alternativa, puede usarse la prueba no invasiva de hidrógeno en el aliento. Puede usarse como dato positivo el nivel de hidrógeno en reposo alto o el aumento rápido en el mismo en respuesta a un sustrato fermentable, como glucosa o lactulosa. Otra prueba en el aliento es la prueba de administración de 1 g de (14C)-D-xilosa, en la que se mide el 14CO2 del aliento.
El tratamiento se dirige a la corrección quirúrgica de los defectos, como un asa aferente que acumule bacterias o una fístula yeyunocólica. Si no existe esta opción, pueden emplearse dosis recurrentes de antibióticos. La tetraciclina da buenos resltados, 1 a 2 g/día durante 7 a 10 días u otro antibiótico que sea activo contra bacterias anaeróbicas. El paciente puede requerir varios tratamientos si los síntomas clínicos recurren.
AMILOIDOSIS
Con frecuencia se afecta el intestino en los pacientes con amiloidosis sistémica, en especial si tienen polineuropatía. En los pacientes mayores de 85 años el 36 porciento tiene afección intestinal con amiloide,58 aunque la mayoría son asintomáticos. Desde el punto de vista endoscópico puede observarse erosión de la mucosa, friabilidad o protrusiones polipoides.59 El diagnóstico se hace por biopsia intestinal de espesor completo o perioral. Si se realiza una biopsia perioral, ésta debe ser suficientemente profunda para que existan arterias visibles, de modo que pueda demostrarse el amiloide. Las arteriolas teñidas con rojo Congo que se vuelve verde manzana bajo luz polarizada confirman el diagnóstico. Las radiografías del intestino delgado pueden demostrar plicas intestinales edematosas, quizá con asas separadas de intestino. Si existe esteatorrea, ésta puede ser resultado de proliferación bacteriana causada por dismotilidad intestinal o menor absorción de ácidos biliares.60 No existe un tratamiento específico eficaz. Cuando existe proliferación bacteriana deben administrarse antibióticos apropiados.
MASTOCITOSIS SISTEMICA
En esta rara condición la piel (99 porciento de los casos), los huesos (9 porciento), el hígado (12 porciento), el bazo (11 porciento), los ganglios linfáticos y el tracto digestivo se afectan por la proliferación de células cebadas. Pueden observarse diarrea, dolor abdominal o ambos (23 porciento de los casos), úlcera péptica (4 porciento), prurito y rubor (36 porciento). En el 12 porciento de los pacientes existe cefalea, fatiga y malestar. Puede haber también disfunción cognitiva y se observa eosinofilia en el 12 a 50 porciento de los casos.61 Muchas de estas manifestaciones de la enfermedad son secundarias a histamina, que es liberada de las células cebadas. Esta liberación puede ser precipitada por alcohol, aspirina, narcóticos y antinflamatorios no esteroides, causando alteraciones episódicas de rubor, diarrea, dolor abdominal e hipotensión que puede progresar al síncope.61
El exceso de histamina (H2) se excreta en la orina en alrededor del 75 porciento de los pacientes, lo que le hace útil como prueba diagnóstica.61 La excreción urinaria de un metabolito de la prostaglandina D2 puede ser una prueba incluso mejor.62 Los estudios de radiografías del intestino delgado pueden mostrar pliegues engrosados o nodulaciones. Estos datos no son diagnósticos, pero indican un intestino delgado afectado.
La sobreproducción de ácido gástrico mediada por histamina puede causar úlcera péptica. En este caso, los bloqueadores H2 y los inhibidores de la bomba de protones son eficaces para controlar lo síntomas. En la piel la urticaria pigmentosa puede tratarse en forma eficaz con antagonistas de los receptores. Si persiste la diarrea puede administrarse cromolin sódico en dosis de 100 mg por vía oral cuatro veces por día.
Infestaciones parasitarias
Las infecciones por uncinarias y G. lamblia pueden causar malabsorción leve que rara vez tiene importancia clínica. La erradicación de estos parásitos cura el defecto de absorción. Estos padecimientos se analizan mejor en otros capítulos de la obra.
Pancreatitis crónica con insuficiencia exócrina
La malabsorción causada por pancreatitis se analiza en otro capítulo.
Defectos combinados o múltiples en la digestión y la absorción
ESTEATORREA POSGASTRECTOMIA
Una de las consecuencias de la cirugía gástrica es la esteatorrea. Esto es especialmente cierto en los pacientes con resección gástrica de tipo Billroth II con una gastroyeyunostomía. En esta operación el antro y una porción variable del cuerpo del estómago son resecados, el estómago se cierra y se crea una gastroyeyunostomía. Por lo tanto, el alimento brinca el duodeno y la porción más proximal del yeyuno, el sitio de máxima concentración de colecistoquinina y secretina, conjugación de folato y sitios activos de absorción de calcio y hierro. Alrededor de la mitad de esta población tiene esteatorrea, con 10 a 15 g de grasa/día. Esta parece deberse a que el alimento entra al yeyuno sin que los sitios sensibles a hormonas del duodeno reciban las señales apropiadas para su liberación. Por lo tanto, no ocurre la mezcla óptima del quimo con las enzimas pancreáticas y los ácidos biliares. El asa aferente, que drena el duodeno y el yeyuno proximal puede bloquearse y acumular bacterias como consecuencia, lo que provoca un síndrome de estasis. Debido a lo pequeño del estómago, estos pacientes no pueden tomar una comida tan grande como antes. Asociado a la esteatorrea, muchos pacientes se estabilizan a un menor peso del que tenían antes de la cirugía. También pueden encontrarse osteopenia y anemia por deficiencia de hierro. Las pérdidas constantes de pequeñas cantidades de sangre por la ostomía gástrica contribuyen al estado de deficiencia de hierro, que es la forma más común de anemia. También se encuentra deficiencia de folato secundaria a la incapacidad para generar una forma absorbible de folato (monoglutamil folato) a partir del folato heptaglutamil no absorbible (la forma más común en la dieta).63 Con menos frecuencia se observa deficiencia de vitamina B12 causada por hipoclorhidria y resección de las células parietales gástricas secretoras de factor intrínseco. No suele requerirse tratamiento para la esteatorrea porque esta no es clínicamente significativa. El hierro, el calcio o la vitamina B12 y el ácido fólico se sustituyen según esté indicado. Si el paciente tiene saciedad temprana pueden ser adecuado administrar varias comidas pequeñas al día.
Después de la cirugía pueden desarrollarse en algunos pacientes síntomas de ESG.64 Es posible que estos pacientes tengan ESG clínicamente silenciosa antes de la cirugía. La cirugía en sí causa esteatorrea moderada (10 a 15 g de grasa/día) en el 50 porciento de los casos, incluso en pacientes con intestino por lo demás normal. Sin embargo, en el paciente con ESG compensada, la cirugía es suficiente para causar sintomatología clínica. Por lo tanto, si los pacientes posgastrectomía muestran esteatorrea excesiva, se justifica una evaluación para determinar si existe ESG. La enfermedad inflamatoria del intestino que se desarrolla en los pacientes después de la gastrectomía puede ser un indicador de la presencia de ESG previamente silente.65
DIABETES MELLITUS Y MALABSORCION
La diarrea, una compliación común de la diabetes, tiene múltiples causas66 y puede provocar malabsorción . Las causas más comunes son proliferación bacteriana causada por la disfunción autonómica presente en esta condición con estasis intestinal y la enteropatía sensible al gluten. Con el uso de anticuerpos antiendomisio como escrutinio, se encontró enteropatía sensible al gluten en tres de 47 pacientes diabéticos (6 porciento), una incidencia mucho mayor de la esperada en la población general.67
Figura 1 Janet Betries.
Figura 2 Dana Burns-Pizer.
Bibliografía
-
Levitt MD, Duane WC: Floating stools: fatus versus
fat. N Engl J Med 286:973, 1972 [PMID
5015442]
-
Drummy GD, Benson JA Jr, Jones CM: Microscopical examination
of the stool for steatorrhea. N Engl J Med 264:85, 1961
-
DiMagno EP, Go VLW, Summerskill WHJ: Relation between
pancreatic enzyme outputs and malabsorption in severe pancreatic insufficiency.
N Engl J Med 288:813, 1973 [PMID
4693931]
-
Dreiling DA, Janowitz HD: The measurement of pancreatic
secretory function. The Exocrine Pancreas. De Reuck AVS, Cameron MP, Eds.
Ciba Foundation Symposium. Little, Brown & Co, New York, 1961, p 225
-
Kato H, Nakao A, Kishimoto W, et al: 13C-labeled
trioctanoin breath test for exocrine pancreatic function test in patients
after pancreatoduodenectomy. Am J Gastroenterol 88:64, 1993
-
Lang C: Value of serum PABA as a pancreatic function test.
Gut 25:508, 1984
-
Bando N, Ogawa T, Tsuji H: Enzymatic method for selective
determination of 4 aminobenzoic acid in urine. Clin Chem 36:1937, 1990
[PMID
2242573]
-
Jacobson DG, Curlington C, Connery K, et al: Trypsin-like
immunoreactivity as a test for pancreatic insufficiency. N Engl J Med 310:1307,
1984 [PMID
6717495]
-
Oelkers P, Kirby LC, Heubi JE, et al: Primary bile
acid malabsorption caused by mutations in the ileal sodium-dependent bile
acid transporter gene. J Clin Invest 99: 1880, 1997 [PMID
9109432]
-
Shindo K, Yamazaki R, Koide K, et al: Alteration of
bile acid metabolism by cimetidine in healthy humans. J Investig Med 44:462,
1996 [PMID
8952227]
-
Nyhlin H, Merrick MV, Eastwood MA, et al: Evaluation
of ileal function using 23-selena-25-homotaurocholate, a g-labeled
conjugated bile acid. Gastroenterology 84:63, 1983 [PMID
6847855]
-
Make M, Collin P: Coeliac disease. Lancet 349:1755,
1997
-
Hofmann AF: The function of bile salts in fat absorption.
Biochem J 89:57, 1963
-
Mansbach CM II, Cohen RS, Leff PB: Isolation and properties
of the mixed micelles present in intestinal content during fat digestion
in man. J Clin Invest 56:781, 1975
-
Volta V, Molinaro N, deFranceschi L, et al: IgA antiendomysial
antibodies on human umbilical cord tissue for celiac disease screening.
Dig Dis Sci 40:1902, 1995
-
Holmes GKT, Prior P, Lane MR, et al: Malignancy in
coeliac disease: effect of a gluten-free diet. Gut 30:333, 1989
-
Gawkrodger DJ, Vestey JP, O'Mahouny S: Dermatitis herpetiformis
and established coeliac disease. Br J Dermatol 129:694, 1993 [PMID
8286252]
-
Klipstein FA, Falaiye JM: Tropical sprue in expatriates
from the tropics living in the continental United States. Medicine (Baltimore)
48:475, 1969 [PMID
4951235]
-
Lindenbaum J, Gerson CD, Kent TH: Recovery of small
intestinal structure and function after residence in the tropics. Ann Intern
Med 74:218, 1971 [PMID
5545229]
-
Gerson CD, Kent TH, Saha JR, et al: Recovery of small
intestinal structure and function after residence in the tropics. Ann Intern
Med 75:41, 1971 [PMID
5091566]
-
Haghighi P, Wolf PL: Tropical sprue and subclinical
enteropathy: a vision for the nineties. Crit Rev Clin Lab Sci 34:313, 1997
[PMID
9288443]
-
McCashland TM, Donovan JP, Strobach RS, et al: Collagenous
enterocolitis: a manifestation of gluten-sensitive enteropathy. J Clin
Gastroenterol 15:45, 1992 [PMID
1500661]
-
Hermaszewski RA, Webster AD: Primary hypogammaglobulinaemia:
a survey of clinical manifestations and complications. Q J Med 86:31, 1993
[PMID
8438047]
-
Ladefoged K, Hessov I, Jarnum S: Nutrition in short-bowel
syndrome. Scand J Gastroenterol Suppl 216:122, 1996
-
Lin HC, Zhao X-T, Wang L: Fat absorption is not complete
by midgut but is dependent on load of fat. Am J Physiol 271(1 Pt 1):G62,
1996
-
Lin HC, Zhao X-T, Wong H: Fat-induced ileal brake in
the dog depends on peptide YY. Gastroenterology 110:1491, 1996 [PMID
8613054]
-
Redman DA, Schmidt TM, McDermott RP, et al: Identification
of the uncultured bacillus of Whipple's disease. N Engl J Med 327:393,
1992
-
Durand DV, Lecomte C, Cathebras P, et al: Whipple disease:
clinical review of 52 cases. The SNFMI Research Group on Whipple Disease.
Medicine (Baltimore) 76:170, 1997
-
Dobbins WO III: HLA antigens in Whipple's disease.
Arthritis Rheum 30:102, 1987
-
Mansbach CM II, Shelburne J, Stevens RD, et al: Lymph
node bacilliform bodies morphologically resembling those of Whipple's disease
in a patient without intestinal involvement. Ann Intern Med 89:64, 1978
-
Von Herbay A, Ditton H-J, Schumacher F, et al: Whipple's
disease: staging and monitoring by cytology and polymerase chain reaction
analysis of cerebrospinal fluid. Gastroenterology 113:434, 1997 [PMID
9247461]
-
Kelly CA, Egan M, Rawlinson J: Whipple's disease presenting
with lung involvement. Thorax 51:343, 1996 [PMID
8779149]
-
Louis ED, Lynch T, Kaufmann P, et al: Diagnostic guidelines
in central nervous system Whipple's disease. J Ann Neurol 40:561, 1996
-
Feurle GE, Marth T: An evaluation of antimicrobial
treatment for Whipple's disease: tetracycline versus trimethoprim-sulfamethoxazole.
Dig Dis Sci 39:1642, 1994 [PMID
7519538]
-
Shih LY, Liaw SJ, Dunn P, et al: Primary small-intestinal
lymphomas in Taiwan: immunoproliferative small-intestinal disease and nonimmunoproliferative
small-intestinal disease. J Clin Oncol 12:1375, 1994 [PMID
8021727]
-
Halphen M, Najjar T, Jaafoura H, et al: Diagnostic
value of upper intestinal fiber endoscopy in primary small intestinal lymphoma:
a prospective study by the Tunisian-French Intestinal Lymphoma Group. Cancer
58:2140, 1986 [PMID
3756829]
-
Khojasteh A, Haghighi P: Immunoproliferative small
intestinal disease: portrait of a potentially preventable cancer from the
Third World. Am J Med 89:483, 1990 [PMID
2145762]
-
Fox C, Lucani G: Disorders of the intestinal mesenteric
lymphatic system. Lymphology 26:61, 1993
-
Aoyagi K, Iida M, Yao T, et al: Characteristic endoscopic
features of intestinal lymphangiectasia: correlation with histological
findings. Hepatogastroenterology 44:133, 1997 [PMID
9058131]
-
Aoyagi K, Iida M, Yao T, et al: Intestinal lymphangiectasia:
value of double-contrast radiographic study. Clin Radiol 49:814, 1994 [PMID
7955851]
-
Sharp D, Blinderman L, Combs KA, et al: Cloning and
gene defects in microsomal triglyceride transfer protein associated with
abetalipoproteinaemia. Nature 365:65, 1993 [PMID
8361539]
-
Gordon DA, Jamil H, Gregg RE, et al: Inhibition of
the microsomal triglyceride transfer protein blocks the step of apolipoprotein
B lipoprotein assembly but not the addition of bulk core lipids in the
second step. J Biol Chem 271:33047, 1996 [PMID
8955151]
-
Mansbach CM II, Dowell RF, Pritchett D: Portal transport
of absorbed lipids in the rat. Am J Physiol 261:G530, 1991 [PMID
1887899]
-
Isselbacher KJ, Scheig R, Plotkin GR, et al: Congenital
b-lipoprotein deficiency: an hereditary disorder
involving a defect in the absorption and transport of lipids. Medicine
(Baltimore) 43:347, 1964
-
Ways PO, Parmentier CM, Kayden HJ, et al: Studies on
the absorptive defect for triglyceride in abetalipoproteinemia. J Clin
Invest 46:35, 1967 [PMID
6018748]
-
Salmon PR, Paulley JW: Eosinophilic granuloma of the
gastrointestinal tract. Gut 8:8, 1967 [PMID
4289737]
-
Klein NC, Hargrove RL, Sleisenger MH, et al: Eosinophilic
gastroenteritis. Medicine (Baltimore) 49:299, 1970 [PMID
5426746]
-
Desreumaux P, Blogot F, Seguy D, et al: Interleukin
3, granulocyte-macrophage colony-stimulating factor, and interleukin 5
in eosinophilic gastroenteritis. Gastroenterology 110:768, 1996 [PMID
8608886]
-
Van Deest BW, Fordtran JS, Morawski SG, et al: Bile
salt and micellar fat concentration in proximal small bowel contents of
ileectomy patients. J Clin Invest 47:1314, 1968 [PMID
5653211]
-
Low-Beer TS, Wilkins RM, Lack L, et al: Effect of one
meal on enterohepatic circulation of bile salts. Gastroenterology 67:490,
1974 [PMID
4851672]
-
Merhjian HS, Phillips SF, Hofmann AF: Colonic secretion
of water and electrolytes induced by bile acids: perfusion studies in man.
J Clin Invest 50:1569, 1971
-
Mansbach CM II, Newton DF, Stevens RD: Fat digestion
in patients with bile acid malabsorption but minimal steatorrhea. Dig Dis
Sci 25:353, 1980
-
Hofmann AF, Poley JR: Role of bile acid malabsorption
in pathogenesis of diarrhea and steatorrhea in patients with ileal resection:
I. Response to cholestyramine or replacement of dietary long chain triglyceride
by medium chain triglyceride. Gastroenterology 62:918, 1972
-
Gianella RA, Broitman SA, Zamcheck N: Vitamin B12
uptake by intestinal microorganisms: mechanisms and relevance to syndromes
of bacterial overgrowth. J Clin Invest 50:1100, 1971
-
Hoffbrand AV, Tabaqchali S, Booth CC, et al: Small
intestinal bacterial flora and folate status in gastrointestinal disease.
Gut 12:27, 1971 [PMID
4993410]
-
Riepe S, Goldstein J, Alpers DH: Effect of secreted
Bacteroides proteases on human intestinal brush border hydrolases.
J Clin Invest 66:314, 1980 [PMID
6995483]
-
Ament ME, Shimoda SS, Saunders DR, et al: Pathogenesis
of steatorrhea in three cases of small intestinal stasis syndrome. Gastroenterology
63:728, 1972 [PMID
4627991]
-
Rocken C, Saeger W, Linke RP: Gastrointestinal amyloid
deposits in old age: report of 110 consecutive autopsical patients and
98 retrospective bioptic specimens. Pathol Res Pract 190:641, 1994 [PMID
7808962]
-
Tada S, Iida M, Yao KK, et al: Endoscopic features
in amyloidosis of the small intestine: clinical and morphologic differences
between chemical types of amyloid protein. Gastrointest Endosc 40:45, 1994
[PMID
8163134]
-
Suhr O, Danielsson A, Steen L: Bile acid malabsorption
caused by gastrointestinal motility dysfunction? An investigation of gastrointestinal
disturbances in familial amyloidosis with polyneuropathy. Scand J Gastroenterol
27:201, 1992 [PMID
1502482]
-
Golkar L, Bernhard JD: Mastocytosis. Lancet 349:1379,
1997 [PMID
9149712]
-
Morrow JD, Guzzo C, Lazarus G, et al: Improved diagnosis
of mastocytosis by measurements of the major urinary metabolite of prostaglandin
D2. J Invest Dermatol 104:937, 1995
-
Rosenberg IH: Folate absorption and malabsorption.
N Engl J Med 293:1303, 1975
-
Bai J, Moran C, Martinez C: Celiac sprue after surgery
of the upper gastrointestinal tract: report of 10 patients with special
attention to diagnosis, clinical behavior, and follow-up. J Clin Gastroenterol
13:521, 1991 [PMID
1744387]
-
Kitis G, Holmes GTK, Cooper BT: Association of coeliac
disease and inflammatory bowel disease. Gut 21:636, 1980 [PMID
7429328]
-
Valdovinos MA, Camilleri M, Zimmerman BR: Chronic diarrhea
in diabetes mellitus: mechanisms and an approach to diagnosis and treatment.
Mayo Clin Proc 68:691, 1993 [PMID
8350642]
-
Rensch MJ, Merenich JA, Lieberman M, et al: Gluten-sensitive
enteropathy in patients with insulin-dependent diabetes mellitus. Ann Intern
Med 124:564, 1996 [PMID
8597319]